
Similar content being viewed by others
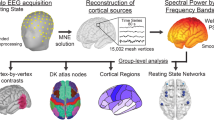
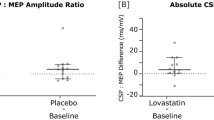
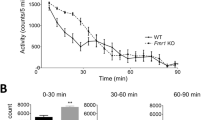
This article is cited by
-
Neurophysiological assessment of cortical activity in DEPDC5- and NPRL3-related epileptic mTORopathies
Orphanet Journal of Rare Diseases (2023)
-
Association of lipid rafts cholesterol with clinical profile in fragile X syndrome
Scientific Reports (2022)
-
Assessment of cortical inhibition depends on inter individual differences in the excitatory neural populations activated by transcranial magnetic stimulation
Scientific Reports (2022)
-
Channelopathies in fragile X syndrome
Nature Reviews Neuroscience (2021)
-
Novel fragile X syndrome 2D and 3D brain models based on human isogenic FMRP-KO iPSCs
Cell Death & Disease (2021)
