
Abstract
Schizophrenia has been associated with a range of genetic and environmental risk factors. Here we explored a link between two risk factors that converge on a shared neurobiological pathway. Recent genome-wide association studies (GWAS) have identified risk variants in genes that code for L-type voltage-gated calcium channels (L-VGCCs), while epidemiological studies have found an increased risk of schizophrenia in those with neonatal vitamin D deficiency. The active form of vitamin D (1,25(OH)2D) is a secosteroid that rapidly modulates L-VGCCs via non-genomic mechanisms in a range of peripheral tissues, though its non-genomic effects within the brain remain largely unexplored. Here we used calcium imaging, electrophysiology and molecular biology to determine whether 1,25(OH)2D non-genomically modulated L-VGCCs in the developing prefrontal cortex, a region widely implicated in schizophrenia pathophysiology. Wide-field Ca2+ imaging revealed that physiological concentrations of 1,25(OH)2D rapidly enhanced activity-dependent somatic Ca2+ levels in a small subset of neurons in the developing PFC, termed vitamin D-responsive neurons (VDRNs). Somatic nucleated patch recordings revealed a rapid, 1,25(OH)2D-evoked increase in high-voltage-activated (HVA) Ca2+ currents. Enhanced activity-dependent Ca2+ levels were mediated by L-VGCC but not associated with any changes to Cacna1c (L-VGCC pore-forming subunit) mRNA expression. Since L-VGCC activity is critical to healthy neurodevelopment, these data suggest that suboptimal concentrations of 1,25(OH)2D could alter brain maturation through modulation of L-VGCC signalling and as such may provide a parsimonious link between epidemiologic and genetic risk factors for schizophrenia.
Similar content being viewed by others


Introduction
Schizophrenia is a poorly understood group of mental disorders with a lifetime prevalence of 0.7%1. The disorder is associated with both common and rare genetic variants, as well as a range of environmental exposures. Recent genome-wide association studies have identified genetic variants of L-type voltage-gated calcium channel (L-VGCC) subunits that are associated with an increased risk for schizophrenia (CACNA1C, CACNB2, CACNA2D1)2. Evidence from whole-exome sequencing3 and copy number variant studies4 have also implicated L-VGCC-related genes as risk factors for schizophrenia. While these risk variants do not result in gross dysfunction in L-VGCCs, it is thought that they may contribute to subtle changes in the developmental expression or activity of L-VGCC subunits5, potentially leading to neurodevelopmental abnormalities. Indeed, modulation of L-VGCC activity during brain development has been shown to alter the extent and complexity of dendritic arborisation, as well as the developmental maturation and migration of neurons in both the hippocampus6,7 and cortex8. Moreover, deletion of Cacna1c during embryonic development, but not in adulthood, resulted in altered adult behavioural phenotypes relevant to schizophrenia9. Taken together, these findings support the hypothesis that factors that alter L-VGCC activity in the developing brain may contribute to risk of schizophrenia.
There is also a growing body of epidemiological evidence implicating developmental vitamin D deficiency as a risk factor for schizophrenia. Epidemiological studies have demonstrated that winter/spring season of birth is associated with increased risk for schizophrenia—vitamin D deficiency is most prevalent during these seasons10. There is also an increased risk of schizophrenia in dark-skinned migrants to Nordic and northern European countries, groups known to be at increased risk of vitamin D deficiency11,12. Neonatal vitamin D status has also been directly linked to an increased risk for schizophrenia. A case–control study (n = 848) found that neonates with vitamin D deficiency had a twofold increased risk of being diagnosed with schizophrenia in later life (Incidence Rate Ratio = 2.1; 95% confidence intervals (CIs) 1.3–3.5)13. This finding was recently replicated using a larger sample (n = 2602; Incidence Rate Ratio = 1.44; 95% CIs 1.12–1.85)14. The biological plausibility of vitamin D deficiency as a risk factor for schizophrenia is supported by the expression of the vitamin D receptor (VDR) and 25-hydroxyvitamin D-1α-hydroxylase (an enzyme required for 1,25-dihydroxyvitamin D (1,25(OH)2D) production) in the human brain15. Further, rodent experiments have demonstrated that transient developmental vitamin D deficiency is associated with persistent neurochemical and behavioural changes that involve biological pathways of interest to schizophrenia, including the dopamine system16,17.
To date, this research has been based on the assumption that the biological consequences of developmental vitamin D deficiency operated via the classical genomic pathways. For example, vitamin D deficiency impacts on neuronal proliferation and differentiation through transcriptional activity, which is mediated by VDR forming a heterodimer with the retinoic acid X receptor and then binding to response elements in the genome18,19,20,21,22. Long-term 1,25(OH)2D treatment has also been shown to confer neuroprotective effects following insult, which correlated with the downregulation of both L-VGCC mRNA expression and L-VGCC surface expression23,24. However, despite the documented non-genomic effects of 1,25(OH)2D on L-VGCC-dependent calcium influx in several tissues including bone25, muscle26 and pancreas27,28, its non-genomic effects within the brain remain largely unexplored. To our knowledge, only a single study has implicated 1,25(OH)2D with rapid changes to Ca2+ signalling in the brain, using liquid scintillation spectrometry to reveal 1,25(OH)2D-induced slice uptake of 45Ca2+ that was dependent on L-type, but not N-type, VGCC activity29. Based on the convergence of these genetic and environmental risk factors for schizophrenia upon L-VGCC-related mechanisms, we investigated the non-genomic effects of 1,25(OH)2D on neuronal L-VGCC activity in the developing brain using both functional and molecular approaches.
Material and methods
All experimental and animal care procedures were in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and approved by the University of Queensland Animal Ethics Committee.
Slice preparation
Balb/c mice (P8–P12) of both sexes were deeply anaesthetised using isoflurane anaesthesia, decapitated, and the brain rapidly removed and submerged in ice-cold oxygenated artificial cerebrospinal fluid (ACSF) containing (in mM): 87 NaCl, 2.5 KCl, 25 NaHCO3, 25 glucose, 50 sucrose, 4 MgCl2, 0.5 CaCl2, and 1.2 NaH2PO4, pH 7.4 (95% O2 and 5% CO2). Coronal slices (300 μm) containing prefrontal cortex (PFC) were prepared for both nucleated patch recordings and Ca2+ imaging protocols using a Vibratome (VT1000S, Leica) and incubated at 34 °C for 30 min in ACSF, containing (in mM): 118 NaCl, 2.5 KCl, 25 NaHCO3, 10 glucose, 1.3 MgCl2, 2.5 CaCl2, and 1.2 NaH2PO4, pH 7.4 (95% O2 and 5% CO2). PFC neurons were visualised using an upright microscope (Olympus BX50WI, Japan) and infrared differential interference contrast optics at high magnification.
Calcium imaging
Following 25-min recovery at 34 °C, slices were loaded with Texas Red Hydrazide (TxRed; 1.8 μM; ThermoFisher Scientific; Ex/Em 582/602) for 5 min, before being transferred into standard ACSF and equilibrated to room temperature for at least 30 min. Slices were then transferred to a small volume (3 mL) incubation chamber made in-house for Cal-520 AM loading (AAT Bioquest; Ex/Em 492/514) at either room temperature or 34 °C (PhysioSuite, Kent Scientific) for 2–3 h, containing (in mM): 118 NaCl, 2.5 KCl, 25 NaHCO3, 10 glucose, 1.3 MgCl2, 2.5 CaCl2, and 1.2 NaH2PO4, 0.01 Cal-520, 0.03% pluronic F-127, pH 7.4 (95% O2 and 5% CO2). Slices were incubated in L-VGCC blocker nifedipine (nif; 10 μM) for >30 min prior to imaging where indicated and were imaged in alternation with bath application of 1,25(OH)2D alone. Following loading, slices were transferred to a submerged imaging chamber perfused with oxygenated ACSF maintained at 25 ± 1 °C (TC-324B, Warner) and secured with a platinum harp strung with parallel nylon threads. Cell depolarisation was evoked using electric field stimulation (efs) generated by two parallel platinum electrodes, separated by a distance of 5 mm, with an isolated stimulator (DS2A, Digitimer Ltd; square pulse, 1 ms, 0.05 Hz, 20–40 V). Both the imaging and stimulation systems were controlled by Metafluor (version 7.10, Molecular Devices) through a 74HC08 AND gate box (Scitech). Fluorescence imaging of the developing PFC was performed using a Polychrome V monochromator (TILL Photonics) combined with an sCMOS camera (pco.edge 5.5, PCO) and ×40 water-immersion objective, which captured a visual field of approximately 411 μm × 338 μm. Wide-field Cal-520 (50 ms) and TxRed (30 ms) fluorescent images were acquired at 0.1 Hz (2 × 2 binning), with every second of Cal-520 exposure time-locked to efs (0.05 Hz). Cal-520 and TxRed fluorescent images were collected using a multi-band dichroic and filter set (FITC/TxRed-A, Semrock; Supplementary Fig. 1a–c). TxRed images were collected to monitor for changes in z-depth via in-plane astrocytic processes (<3 μm), and manual depth corrections were made as required. Imaging sessions were excluded from the data set when changes in z-depth were observed (movement of the imaged z-plane towards or away from the objective lens). Instantaneous ΔF/F (0.05 Hz) was calculated for individual somatic regions of interest (ROIs) as fluorescence during field stimulation (F2) minus the immediately preceding fluorescence at rest (unstimulated, F1), divided by the fluorescence at rest (F1, Fig. 1a). Single-cell ROI fluorescence levels were extracted offline from these acquired temporal z-stacks using Fiji. Background subtraction (rolling ball, radius 50 pixels) and XY image stabilisation were applied using built-in plugins. Instantaneous ΔF/F analysis of wide-field images was automated using custom MATLAB scripts and converted to txt files that were baselined (flat or extrapolated sloping function) and quantified using the Axograph software (Axograph X, version 1.7.2). ROI data met signal-to-noise inclusion criteria if bath application of 1,25(OH)2D induced a percentage change in the instantaneous ΔF/F that exceeded three standard deviations of >5 min pre-1,25(OH)2D baseline ΔF/F amplitude. 1,25(OH)2D was bath applied at the physiological concentration of 0.1 nM30 from a 1-mM stock solution (dimethyl sulfoxide (DMSO)). Where specified, imaging sessions were conducted in the presence of synaptic blockers (synblock), which included 6-cyano-7-nitroquinoxaline-2-3-dione (10 μM; Tocris), D-2-amino-5-phosphonopentanoic acid (50 μM; Tocris) and picrotoxin (100 μM; Sigma-Aldrich); the L-VGCC agonist Bay K8644 (BAYK; 2.5 μM; Sigma-Aldrich); or the L-VGCC antagonist nif (10 μM; Sigma-Aldrich). The duration of an imaging session baseline was dependent on the applied pharmacology: ACSF 10–20 min, synblock 20 min, nif 20 min31,32 (Supplementary Fig. 1d). Neurons that ceased to respond to electrical stimulation following bath application of synblock were excluded from the total cell count imaged per slice. Only electrically responsive, TxRed-negative cells were included in vitamin D-responsive neuron (VDRN) subsets. VDRN slice Responding % was calculated from the total number of Cal-520-positive cells in focus per imaged area (TxRed-positive and TxRed-negative) to avoid errors stemming from inaccurate TxRed staining. As such, VDRN Proportion Responsive (%) statistics may underestimate the percentage of imaged neurons that were VDRNs.

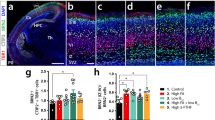
a Schematic of field stimulation configuration and analysis. b Representative PFC region bulk-loaded with Cal-520, zoomed in (white box) to show difference between unstimulated (c) and stimulated (d) fluorescence intensity in electrically responsive cells. e Bath application of L-VGCC agonist Bay K8644 evoked increased Ca2+ ΔF/F in single neurons within 3 min. f Bay K8644 evoked an increase in Ca2+ ΔF/F in almost half the imaged neurons (average ΔF/F trace, n = 38), while application of equimolar DMSO (107-fold dilution, equivalent to dilution of 1,25(OH)2D stock) produced a negligible effect on Ca2+ ΔF/F (1/144 cells, 0.7%). g The majority of neurons showed no change in Ca2+ ΔF/F following bath application of 1,25(OH)2D (average ΔF/F trace, n = 427). h Raw (left) and analysed (right) Ca2+ imaging data from a single representative VDRN (directly responsive). i Average ΔF/F trace of all detected VDRN (n = 53). j 1,25(OH)2D induced a smaller change in the mean single cell Ca2+ ΔF/F compared to Bay K8644 (p < 0.0001). k Range of response times to 1,25(OH)2D was significantly larger compared to Bay K8644 (p < 0.0001). l Representative Cal-520 bulk-loaded slice (imaged in the presence of synaptic blockers) labelling detected VDRN with orange arrow heads. m Neither the percentage of VDRNs detected per imaged slice (p = 0.6) nor from the total pool of imaged neurons (n) was significantly altered by the presence (orange) or absence (blue) of synaptic blockers. Data = Mean ± SEM (unpaired two-tailed t test with Welch’s correction, ****p < 0.0001)
Nucleated patch recordings
High-voltage-activated (HVA) Ca2+ channel currents were recorded in nucleated patch configuration33 using barium as the charge carrier. Nucleated patches were extracted from the soma of visually identified L2/3 PFC neurons (series resistance <15 MΩ). Briefly, negative pressure (10–15 kPa; Series 475 MARK III Digital Manometer, Dwyer Instruments, IN, USA)34 was applied to the patch pipette in whole-cell configuration, drawing the nucleus towards the pipette tip as it was slowly withdrawn from the soma, to produce an outside-out patch of somatic membrane internally scaffolded by the nucleus. Following isolation of the nucleated patch, negative pipette pressure was reduced to 3–5 kPa for the remainder of the experiment, and extracellular Ca2+ was exchanged for Ba2+ (5–10 mM), containing (in mM): 115.5/110.5 NaCl, 2.5 KCl, 25 NaHCO3, 10 glucose, 1.3 MgCl2, 5/10 BaCl2, and 1.2 NaH2PO4, pH 7.4 (95% O2 and 5% CO2). VGCC currents were isolated pharmacologically by bath application of tetradotoxin (1 μM) and 4-aminopyridine (5 mM) to block voltage-gated sodium and voltage-gated potassium channels, respectively, which were only applied once the nucleated patch was isolated from the remnant (anucleated) neuron and situated in the external ACSF (input resistance range typically 1–5 GΩ). Recording pipettes were fabricated from borosilicate glass that was pulled and fire-polished to a tip resistance between 3 and 6 MΩ (GC150F, 1.5 mm, Harvard Apparatus, UK) when filled with caesium-based internal solution, containing (in mM): 120 CsMeSO4, 10 TEACl, 10 HEPES, 4 Mg2ATP, 0.3 Na3GTP, 20 phosphocreatine, and 0.3 EGTA (pH 7.3 with CsOH; osmolarity ~295–300 mOsm/kg). VGCC currents were evoked with a 50-ms depolarising pulse from −80 to 0 mV every 20 s (0.05 Hz). Current–voltage (I–V) relationships were investigated by 50-ms step depolarisations from −90 to +40 mV in 10-mV increments from a holding potential of −80 mV. Linear leak and capacitive currents were subtracted online by a P/N protocol (15 repetitions of voltage step −80 to −100 mV). 1,25(OH)2D was applied at the physiological concentration of 0.1 nM30 from a 1-mM stock solution (DMSO). Recordings were made using a MultiClamp 700B amplifier (Molecular Devices) with Sutter manipulators (MP-285). Recordings were filtered at 6 kHz and digitised at 10–50 kHz using an ITC-18 board (InstruTech, Port Washington, NY), attached to an iMac. Recordings were acquired and analysed offline using Axograph software (Axograph X, version 1.4.4).
Statistics
Calcium imaging statistics were evaluated using unpaired two-tailed t test with Welch’s correction (unequal variances t test). All data were analysed using Prism 7 (GraphPad Software) and are reported as mean ± SEM. Additional details and experimental design are included in Supplementary Methods.
Results
Simultaneous measurement of single-cell VGCC activity from a population of neurons
Conventional methods for the functional investigation of VGCC activity have low spatial resolution, in that they involve single-cell recording configurations in order to provide sufficient voltage control. To locate and quantify the effects of 1,25(OH)2D on VGCCs in the brain, we developed a high-throughput imaging method that allowed the simultaneous measure of single-cell activity-dependent cytosolic Ca2+ from a population of neurons (Fig. 1a–d). This method combines wide-field Ca2+ fluorescence imaging with field stimulation in brain slices, thereby retaining cortical layer organisation (Fig. 1a). Cytosolic Ca2+ levels were visualised using the fluorescence indicator Cal-520 (50-ms exposure; 0.1 Hz; Fig. 1b), with every second exposure time-locked to field stimulation (F2; 0.05 Hz; Fig. 1c, d). This approach evoked stable activity-dependent increases in cytosolic Ca2+ in postnatal PFC neurons (P8–P12) throughout the imaged area, for extended imaging durations (>1 h). Bath application of dihydropyridine (DHP) L-VGCC agonist BAYK (2.5 μM) confirmed sufficient sensitivity to detect changes in L-VGCC activity (Fig. 1e), resolving changes to ΔF/F as low as 7% (59 ± 6%, n = 38; Fig. 1f). BAYK increased Ca2+ ΔF/F within 3 min of application (2.3 ± 0.05 min), which was consistent with the effect of BAYK on single-channel L-VGCC recordings35. Further, BAYK evoked an increased Ca2+ ΔF/F in 47.5% of the imaged neurons (Fig. 1f), consistent with L-VGCC protein expression in the PFC9. Application of equimolar DMSO (107-fold dilution, equivalent to dilution of 1,25(OH)2D stock) evoked negligible effect on Ca2+ ΔF/F measured from imaged neurons (1/144 DMSO-responsive neuron, 0.7%; Fig. 1f).
1,25(OH)2D enhanced activity-dependent cytosolic Ca2+ levels in a subset of neurons
Using the approach described above, 1,25(OH)2D (0.1 nM) was bath applied to brain slices containing PFC. In order to identify neurons directly responsive to 1,25(OH)2D, calcium imaging was conducted in either the presence (colour-coded orange) or absence (colour-coded blue) of synblock (Supplementary Fig. 1d). While the majority of electrically responsive neurons were non-responsive to 1,25(OH)2D (Non-VDRN, Fig. 1g), a subset of PFC neurons rapidly responded to 1,25(OH)2D application with significantly enhanced activity-dependent Ca2+ levels (Fig. 1h; Supplementary Fig. 2), termed VDRNs. In the presence of synblock (orange), cytosolic Ca2+ ΔF/F levels increased by as much as 66.5% (24 ± 2%, n = 53; Fig. 1i, j) as early as 2 min following bath application, with the majority of VDRNs responding within 10 min (6.7 ± 0.5 min, n = 53; Fig. 1k). Notably, both the change in Ca2+ ΔF/F levels (p < 0.0001; Fig. 1j) and the range of response times (p < 0.0001; Fig. 1k) were significantly different compared to neurons responding to the direct-binding L-VGCC agonist BAYK. Application of 1,25(OH)2D in the absence of synblock did not change the 1,25(OH)2D-induced increase in ΔF/F (29 ± 4%, n = 26; p = 0.17) nor the onset of response in detected VDRNs (7.6 ± 1 min, n = 26; p = 0.43), suggesting 1,25(OH)2D did not enhance Ca2+ ΔF/F levels through calcium-permeable AMPA or N-methyl-d-aspartate receptors. Mapping directly responsive VDRNs onto their respective Ca2+-imaged areas revealed the scattered nature of VDRN soma localisation throughout the cortical layers of the PFC (Fig. 1l, orange arrows). There was also no significant difference (p = 0.6) between the proportion of cells that responded with increased Ca2+ ΔF/F levels in either the presence (7.3 ± 2%, n = 7 slices, Fig. 1m; 53/730 cells, 7.3%, Fig. 1n) or absence (slice mean = 5.7 ± 2%, n = 5 slices, Fig. 1m; 26/428 cells, 6.1%, Fig. 1n) of synblock, suggesting that VDRNs did not drive enhanced Ca2+ ΔF/F levels in local postsynaptic non-VDRN. In summary, wide-field calcium imaging revealed that 1,25(OH)2D directly enhanced activity-dependent Ca2+ levels in a subset of neurons scattered throughout the PFC, within minutes of application.
1,25(OH)2D enhanced HVA Ca2+ channel currents in a subset of PFC neurons
Fluorescent Ca2+ indicators cannot differentiate Ca2+ released intracellularly from Ca2+ that enters a cell through channels in the plasma membrane. In order to test whether 1,25(OH)2D evoked changes in voltage-gated Ca2+ influx, we recorded HVA Ca2+ currents in nucleated patch configuration from layer 2/3 PFC neurons. Consistent with the above Ca2+ imaging findings, a subset of recorded neurons showed rapidly (3 ± 1 min onset, n = 5; Fig. 2a) increased voltage-gated Ca2+ currents (33 ± 5%, n = 5; Fig. 2a–d) in response to 1,25(OH)2D (0.1 nM; peak response by 8 ± 2 min, n = 5). Following 1,25(OH)2D application, currents were entirely blocked by the broad-spectrum Ca2+ channel blocker cadmium (1 μM; Fig. 2c) to confirm that only HVA Ca2+ currents were evoked. Typical of L-VGCC currents, and despite the inclusion of high phosphocreatine in the recording pipette, the majority of nucleated patch recordings showed rundown in HVA Ca2+ current amplitudes36,37 (Fig. 2a, d, white circles).

a Baseline VGCC current amplitudes recorded in nucleated patch configuration (averaged in b, black trace) rapidly increased (averaged in b, blue trace) during bath application of 0.1 nM 1,25(OH)2D in a subset of PFC neurons (above), while the majority of recorded neurons were insensitive (below). Inset, representative and schematic examples of nucleated patch configuration following extraction from whole-cell configuration. c VGCC I–V recorded pre- (black) and post-1,25(OH)2D application (blue), abolished by cadmium (1 μM, grey). d 1,25(OH)2D evoked a mean increase in the recorded HVA Ca2+ current amplitudes of 33 ± 5% (blue circles, n = 5), while the majority of cells (open circles) demonstrated peak current amplitude rundown typical of L-VGCC channels (−29 ± 5%; n = 16). Data = Mean ± SEM
Enhanced activity-dependent Ca2+ levels mediated by L-type VGCCs
Since 1,25(OH)2D enhanced HVA Ca2+ influx in a subset of PFC neurons (Fig. 2), and 1,25(OH)2D is known to modulate L-type VGCC activity in peripheral tissues, we considered whether 1,25(OH)2D enhanced activity-dependent Ca2+ levels by rapidly modulating L-VGCC activity. We first assessed whether VDRN expressed functional L-VGCCs by bath applying L-VGCC-specific agonist BAYK (2.5 μM38) following 1,25(OH)2D application. While BAYK evoked a rapid Ca2+ ΔF/F increase in approximately half the total imaged neurons (Fig. 1h), the vast majority of VDRNs (48/53, 91%; Fig. 3a) showed a significant increase in Ca2+ ΔF/F following BAYK application. Conversely, approximately 12% of BAYK-responsive neurons were VDRNs (53/438 neurons; 12.7 ± 3%, n = 7 slices). In the presence of 1,25(OH)2D, BAYK evoked a similar increase in Ca2+ ΔF/F (72 ± 6%, n = 48, p = 0.15; Fig. 3b) compared to application of BAYK alone (59 ± 6%, n = 38; Fig. 1f), suggesting no facilitative or depressive interaction between the two molecules.

a The majority of VDRNs showed an increased Ca2+ ΔF/F in response to L-VGCC agonist Bay K8644, suggesting functional L-VGCC expression. b In the presence of 1,25(OH)2D, Bay K8644 increased VDRN Ca2+ ΔF/F in a manner comparable to Bay K8644 alone (Fig. 1i) and was attenuated by preincubation in L-VGCC antagonist nifedipine (p < 0.0001). Pre-incubation of imaged slices in nifedipine significantly reduced both the proportion of detected VDRN (c p = 0.01) and magnitude of detected Ca2+ ΔF/F changes in response to 1,25(OH)2D (d p < 0.0001). Data = Mean ± SEM (Unpaired two-tailed t test with Welch’s correction, *p < 0.05, ****p < 0.0001)
We next tested whether 1,25(OH)2D enhanced activity-dependent Ca2+ levels in VDRN through modulation of L-VGCC activity. Slices were preincubated with L-VGCC-specific blocker nif (10 μM; >30 min), and baseline calcium imaging was conducted in the presence of nif for a further 20 min (electrically stimulated 60 times at 0.05 Hz; Supplementary Fig. 1d) to maximise the state and depolarisation dependency of DHP antagonism efficacy31,32. Significantly, preincubation in nif almost entirely abolished 1,25(OH)2D-induced changes to Ca2+ ΔF/F, attenuating both the proportion of 1,25(OH)2D-responsive neurons (8/675, 1.2%, n = 9 slices; p = 0.01; Fig. 3c) and the magnitude of detected Ca2+ ΔF/F changes (10.7 ± 1%, n = 8 cells, n = 9 slices; p < 0.0001; Fig. 3d). Interestingly, preincubation in nif attenuated BAYK-induced increases in VDRN Ca2+ ΔF/F to a similar extent (14 ± 3%, n = 4 cells, n = 9 slices; p < 0.0001; Fig. 3b), suggesting that residual 1,25(OH)2D-induced changes to Ca2+ ΔF/F (Fig. 3d) resulted from the incomplete antagonism of nif32,38,39. These findings suggest that 1,25(OH)2D enhanced activity-dependent Ca2+ levels in a distinct subset of developing PFC neurons through rapid modulation of L-VGCC activity. Further, this effect was not associated with changes to Cacna1c mRNA levels (Supplementary Fig. 3), suggesting a non-genomic mechanism.
Discussion
Vitamin D deficiency, a prevalent exposure in many parts of the world40, reduces the expected (basal) concentration of 1,25(OH)2D41. While developmental vitamin D deficiency is associated with a range of altered brain outcomes19, the assumption to date has been that these properties were mediated via classical genomic pathways (i.e., involving the nuclear receptor VDR). Here we provide evidence that 1,25(OH)2D also affects brain function via rapid, non-genomic mechanisms. Physiological levels of 1,25(OH)2D rapidly enhanced Ca2+ influx through L-VGCCs in a small subset of neurons in the PFC, leading to elevated intracellular calcium levels during neural activity (Fig. 4). We propose that suboptimal concentrations of this secosteroid during critical periods of brain development may result in altered L-VGCC function in VDRNs. Since L-VGCC activity is critical to healthy neurodevelopment7, vitamin D deficiency may disrupt the excitability and maturation of VDRNs, with possible consequences for local circuit integration, function, and information processing. This non-genomic mechanism may represent a link between two independent genetic and epidemiologic risk factors for schizophrenia.

VDRNs responded rapidly to 1,25(OH)2D treatment with increased Ca2+ influx mediated by L-VGCCs. Through their modulation of L-VGCC, optimal levels of 1,25(OH)2D may contribute to the healthy development of VDRNs within the maturing cortical circuit, such as through neurite extension, neuronal excitability, and gene expression
1,25(OH)2D non-genomically modulated L-VGCC activity in a subset of PFC neurons
We show with single-cell resolution that 1,25(OH)2D rapidly enhanced activity-dependent Ca2+ ΔF/F through L-VGCCs in a subset of developing PFC neurons. Changes to L-VGCC-mediated Ca2+ influx can be evoked through (1) modulation of single-channel conductance or gating, such as through DHP agonists and antagonists38,42 and/or (2) regulation of the number of channels present in the plasma membrane via protein trafficking. Interestingly, the kinetics of this effect were different to those evoked by L-VGCC agonist BAYK, where 1,25(OH)2D induced a smaller mean change to Ca2+ ΔF/F, with a slower mean response time, and was not observed in all neurons that expressed L-VGCCs. This difference likely reflects an alternative mechanism of action compared to BAYK, which binds directly to the pore-forming α1 subunit of L-VGCCs to enhance channel gating. Further, this finding indicates that only a subset of L-VGCC-expressing neurons contained the machinery required for initiation of 1,25(OH)2D-evoked effects, which may suggest the involvement of specific auxiliary L-VGCC subunits43,44 or second messenger signalling pathways. Indeed, a previous study showed that non-genomic effects of 1,25(OH)2D in neurons are dependent on Ca2+/calmodulin-dependent protein kinase II (CaMKII) and phosphoinositide 3-kinase (PI3K)29, where both CaMKII and P13K have previously been shown to promote trafficking of L-VGCC neurons through the β2 subunit45,46. The propensity of these second messengers to facilitate L-VGCC activity through the β2 subunit is noteworthy here, since variants of the β2 gene (CACNB2) have also been associated with an increased risk for schizophrenia2,3. In order to determine whether 1,25(OH)2D enhances L-type VGCC Ca2+ influx in VDRNs by increasing CaV1.2 surface expression levels, future studies will require live imaging of CaV1.2 channel trafficking with single-cell resolution. Future studies should also determine: (1) the identity and role of VDRNs in the PFC, (2) whether altered L-VGCC activity during developmental vitamin D deficiency affects VDRN properties and circuit function, and (3) the distribution of VDRNs outside the PFC and at other stages of development.
L-VGCCs, brain development and schizophrenia
L-VGCCs are highly expressed in the developing brain and are widely known to play a critical role in neuronal development7, neuronal excitability, synaptic plasticity, homoeostatic plasticity and the transduction of neuronal activity into gene expression47. L-VGCC activity plays a critical role in mediating cortical neurite extension and radial migration8, and both ionic and conformational signalling in L-VGCC are required to drive neuronal gene expression48. Indeed, disruption to L-VGCC function in hippocampal parvalbumin interneurons during development significantly reduced cell number and dendritic arbor complexity7. L-VGCCs are also critical in the regulation of basal and burst firing activity in dopaminergic neurons of the ventral tegmental area49, which are considered responsible for the control of basal dopamine levels in downstream areas50, and are also the target of significant PFC projections51. Notably, both parvalbumin interneurons and the dopamine system are implicated in current hypotheses in schizophrenia research52. Critically, disruption to L-VGCC activity has also been implicated in behavioural phenotypes of interest to psychiatry. A recent transgenic animal study suggested that deletion of Cacna1c in forebrain glutamatergic neurons during embryonic development, but not adulthood, was associated with hyperactivity, impaired hippocampal synaptic plasticity, cognitive impairment and reduced sociability9. These findings support the hypothesis that factors influencing L-VGCC activity in the developing brain may contribute to risk of schizophrenia.
In addition to schizophrenia3, variants in L-VGCC-related genes such as CACNA1C and CACNB2 have also been linked to a range of other mental disorders53. In particular, variants in L-VGCC-related genes have been linked to the risk of autism-related phenotypes54. Recent studies based on the Generation R birth cohort have found that gestational vitamin D deficiency is also associated with an increased risk of both autism-related traits and autism-spectrum disorder55,56. These findings are in keeping with the expectation that genetic and environmental risk factors are often shared across a range of psychiatric phenotypes.
Genetic and epidemiological studies have provided important insights into the aetiology of schizophrenia; however, it is rare that discoveries from these fields converge on a shared neurobiological pathway. Indeed, studies that can combine genetics, epidemiology and basic neuroscience are considered better able to triangulate on causal mechanisms57. This approach led us to investigate the links between two established risk factors for schizophrenia: risk variants in L-VGCC genes and developmental vitamin D deficiency. Using functional approaches, we have demonstrated that vitamin D rapidly enhanced L-VGCC activity in a subset of PFC neurons during brain development. We propose that optimal concentrations of 1,25(OH)2D contribute to the normal (expected) development and maturation of VDRNs through the non-genomic modulation of L-VGCC activity. Conversely, we propose that suboptimal concentrations of 1,25(OH)2D may disrupt developmentally critical L-VGCC-dependent processes within the immature cortical circuit. In other words, vitamin D deficiency may produce a transient channelopathy-like state, in that the activity of L-VGCCs is altered during critical periods of neurodevelopment. However, unlike genetic diseases that affect L-VGCC activity, vitamin D deficiency is preventable with supplementation. Unravelling the underlying mechanism, as well as the identity and role these neurons play within the circuit, may provide further insight into the aetiology and pathophysiology of schizophrenia.
References
McGrath, J., Saha, S., Chant, D. & Welham, J. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol. Rev. 30, 67–76 (2008).
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Purcell, S. M. et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190 (2014).
Glessner, J. T. et al. Strong synaptic transmission impact by copy number variations in schizophrenia. Proc. Natl Acad. Sci. USA 107, 10584–10589 (2010).
Yoshimizu, T. et al. Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Mol. Psychiatry 20, 162–169 (2015).
Redmond, L., Kashani, A. H. & Ghosh, A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34, 999–1010 (2002).
Jiang, M. & Swann, J. W. A role for L-type calcium channels in the maturation of parvalbumin-containing hippocampal interneurons. Neuroscience 135, 839–850 (2005).
Kamijo, S. et al. A critical neurodevelopmental role for L-type voltage-gated calcium channels in neurite extension and radial migration. J. Neurosci. 38, 5551–5566 (2018).
Dedic, N. et al. Cross-disorder risk gene CACNA1C differentially modulates susceptibility to psychiatric disorders during development and adulthood. Mol. Psychiatry 23, 533–543 (2018).
Holick, M. F. Vitamin D deficiency. N. Engl. J. Med. 357, 266–281 (2007).
Cantor-Graae, E. & Selten, J. P. Schizophrenia and migration: a meta-analysis and review. Am. J. Psychiatry 162, 12–24 (2005).
Davies, G., Welham, J., Chant, D., Torrey, E. F. & McGrath, J. A systematic review and meta-analysis of Northern Hemisphere season of birth studies in schizophrenia. Schizophr. Bull. 29, 587–593 (2003).
McGrath, J. J. et al. Neonatal vitamin D status and risk of schizophrenia: a population-based case-control study. Arch. Gen. Psychiatry 67, 889–894 (2010).
Eyles, D. W. et al. The association between neonatal vitamin D status and risk of schizophrenia. Sci. Rep. 8, 17692 (2018).
Eyles, W., Smith, S., Kinobe, R., Hewison, M. & McGrath, J. The receptor for vitamin D and the enzyme responsible for its synthesis are widely distributed in human brain. Int. J. Neuropsychopharmacol. 7, S264–S264 (2004).
Cui, X., Groves, N. J., Burne, T. H. J., Eyles, D. W. & McGrath, J. J. Low vitamin D concentration exacerbates adult brain dysfunction. Am. J. Clin. Nutr. 97, 907–908 (2013).
Kesby, J. P., Burne, T. H., McGrath, J. J. & Eyles, D. W. Developmental vitamin D deficiency alters MK 801-induced hyperlocomotion in the adult rat: An animal model of schizophrenia. Biol. Psychiatry 60, 591–596 (2006).
Luan, W. et al. Developmental vitamin D (DVD) deficiency reduces Nurr1 and TH expression in post-mitotic dopamine neurons in rat mesencephalon. Mol. Neurobiol. 55, 2443–2453 (2017).
Cui, X., Pelekanos, M., Burne, T. H., McGrath, J. J. & Eyles, D. W. Maternal vitamin D deficiency alters the expression of genes involved in dopamine specification in the developing rat mesencephalon. Neurosci. Lett. 486, 220–223 (2010).
Kesby, J. P. et al. Developmental vitamin D deficiency alters multiple neurotransmitter systems in the neonatal rat brain. Int. J. Dev. Neurosci. 62, 1–7 (2017).
Eyles, D. W. et al. Developmental vitamin D deficiency causes abnormal brain development. Psychoneuroendocrinology 34(Suppl 1), S247–S257 (2009).
Eyles, D., Brown, J., Mackay-Sim, A., McGrath, J. & Feron, F. Vitamin D3 and brain development. Neuroscience 118, 641–653 (2003).
Brewer, L. D., Porter, N. M., Kerr, D. S., Landfield, P. W. & Thibault, O. Chronic 1alpha,25-(OH)2 vitamin D3 treatment reduces Ca2+ -mediated hippocampal biomarkers of aging. Cell Calcium 40, 277–286 (2006).
Brewer, L. D. et al. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J. Neurosci. 21, 98–108 (2001).
Li, B., Chik, C. L., Taniguchi, N., Ho, A. K. & Karpinski, E. 24,25(OH)2 vitamin D3 modulates the L-type Ca2+ channel current in UMR 106 cells: involvement of protein kinase A and protein kinase C. Cell calcium 19, 193–200 (1996).
Morelli, S., de Boland, A. R. & Boland, R. L. Generation of inositol phosphates, diacylglycerol and calcium fluxes in myoblasts treated with 1,25-dihydroxyvitamin D3. Biochem. J. 289(Pt 3), 675–679 (1993).
Kajikawa, M. et al. An insulinotropic effect of vitamin D analog with increasing intracellular Ca2+ concentration in pancreatic beta-cells through nongenomic signal transduction. Endocrinology 140, 4706–4712 (1999).
Hii, C. S. & Ferrante, A. The non-genomic actions of vitamin D. Nutrients 8, 135 (2016).
Zanatta, L. et al. 1alpha,25-dihydroxyvitamin D(3) mechanism of action: modulation of L-type calcium channels leading to calcium uptake and intermediate filament phosphorylation in cerebral cortex of young rats. Biochim. Biophys. Acta 1823, 1708–1719 (2012).
Zanatta, L. et al. 1alpha,25-Dihydroxyvitamin D(3) signaling pathways on calcium uptake in 30-day-old rat Sertoli cells. Biochemistry 50, 10284–10292 (2011).
Bean, B. P. Nitrendipine block of cardiac calcium channels: high-affinity binding to the inactivated state. Proc. Natl Acad. Sci. USA 81, 6388–6392 (1984).
Helton, T. D., Xu, W. & Lipscombe, D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J. Neurosci. 25, 10247–10251 (2005).
Sather, W., Dieudonne, S., MacDonald, J. F. & Ascher, P. Activation and desensitization of N-methyl-D-aspartate receptors in nucleated outside-out patches from mouse neurones. J. Physiol. 450, 643–672 (1992).
Martina, M. & Jonas, P. Functional differences in Na+ channel gating between fast-spiking interneurones and principal neurones of rat hippocampus. J. Physiol. 505(Pt 3), 593–603 (1997).
Nowycky, M. C., Fox, A. P. & Tsien, R. W. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 316, 440–443 (1985).
Forscher, P. & Oxford, G. S. Modulation of calcium channels by norepinephrine in internally dialyzed avian sensory neurons. J. Gen. Physiol. 85, 743–763 (1985).
Almog, M. & Korngreen, A. Characterization of voltage-gated Ca(2+) conductances in layer 5 neocortical pyramidal neurons from rats. PLoS ONE 4, e4841 (2009).
Xu, W. & Lipscombe, D. Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 21, 5944–5951 (2001).
Fox, A. P., Nowycky, M. C. & Tsien, R. W. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J. Physiol. 394, 149–172 (1987).
Palacios, C. & Gonzalez, L. Is vitamin D deficiency a major global public health problem? J. Steroid Biochem. Mol. Biol. 144 (Pt A), 138–145 (2014).
Zittermann, A., Ernst, J. B., Birschmann, I. & Dittrich, M. Effect of vitamin D or activated vitamin D on circulating 1,25-dihydroxyvitamin D concentrations: a systematic review and metaanalysis of randomized controlled trials. Clin. Chem. 61, 1484–1494 (2015).
Grabner, M., Wang, Z., Hering, S., Striessnig, J. & Glossmann, H. Transfer of 1,4-dihydropyridine sensitivity from L-type to class A (BI) calcium channels. Neuron 16, 207–218 (1996).
Pichler, M. et al. Beta subunit heterogeneity in neuronal L-type Ca2+ channels. J. Biol. Chem. 272, 13877–13882 (1997).
McEnery, M. W. et al. Differential expression and association of calcium channel subunits in development and disease. J. Bioenerg. Biomembr. 30, 409–418 (1998).
Viard, P. et al. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat. Neurosci. 7, 939–946 (2004).
Grueter, C. E. et al. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol. Cell 23, 641–650 (2006).
Nanou, E. & Catterall, W. A. Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron 98, 466–481 (2018).
Li, B., Tadross, M. R. & Tsien, R. W. Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 351, 863–867 (2016).
Liu, Y. et al. Cav1.2 and Cav1.3 L-type calcium channels regulate dopaminergic firing activity in the mouse ventral tegmental area. J. Neurophysiol. 112, 1119–1130 (2014).
Philippart, F. et al. Differential somatic Ca2+ channel profile in midbrain dopaminergic neurons. J. Neurosci. 36, 7234–7245 (2016).
Murugan, M. et al. Combined social and spatial coding in a descending projection from the prefrontal cortex. Cell 171, 1663.e16–1677.e16 (2017).
Chung, D. W., Fish, K. N. & Lewis, D. A. Pathological basis for deficient excitatory drive to cortical parvalbumin interneurons in schizophrenia. Am. J. Psychiatry 173, 1131–1139 (2016).
Cross-disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013).
Li, J. et al. Schizophrenia related variants in CACNA1C also confer risk of autism. PLoS ONE 10, e0133247 (2015).
Vinkhuyzen, A. A. E. et al. Gestational vitamin D deficiency and autism-related traits: the Generation R Study. Mol. Psychiatry 23, 240–246 (2018).
Vinkhuyzen, A. A. E. et al. Gestational vitamin D deficiency and autism spectrum disorder. Br. J. Psychiatry Open 3, 85–90 (2017).
Munafo, M. R. & Davey Smith, G. Robust research needs many lines of evidence. Nature 553, 399–401 (2018).
Acknowledgements
This project was supported by NHMRC Project grants (APP 1007677, APP 1099709), a John Cade Fellowship (APP1056929) and a Rebecca L Cooper Medical Research Foundation grant (APP 10843). J.J.M. was supported by a Niels Bohr Professorship from the Danish National Research Foundation. V.A. was supported by grants from the Australian Research Council (DP170102402), John T. Reid Charitable Trusts and the Clem Jones Centre for Ageing Dementia Research. S.E.J. is a University of Queensland International Scholar. These funding agencies were not involved in any aspect of the study. We thank Gavin Bird and Karin Bird for general support related to this field of research, Luke Hammond and Christian Loebbe for assistance with microscopy and Ca2+ imaging, Suzy Alexander and staff of the QBI Animal House for assistance with the animal studies and Jack Dou and Hilary Yong for assistance with neuronal culture studies.
Author information
Authors and Affiliations
Contributions
All authors designed research; H.G., V.A., X.C., M.C.T. and S.E.J. performed research; H.G., V.A. and X.C. analysed data; H.G., X.C., V.A. and J.J.M. wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gooch, H., Cui, X., Anggono, V. et al. 1,25-Dihydroxyvitamin D modulates L-type voltage-gated calcium channels in a subset of neurons in the developing mouse prefrontal cortex. Transl Psychiatry 9, 281 (2019). https://doi.org/10.1038/s41398-019-0626-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-019-0626-z
This article is cited by
-
Tremendous Fidelity of Vitamin D3 in Age-related Neurological Disorders
Molecular Neurobiology (2024)
-
Vitamin D and schizophrenia: 20 years on
Molecular Psychiatry (2021)
