
Abstract
Increases in the activated state of microglia, the main neuroimmune cells, are widely reported in the brains of patients with neurological and psychiatric disorders. Microglia transform from the resting to the activated state by sensing their environment, aided by a variety of ion channels. To examine the effect of ion channels on microglial phenotypes, we conducted a systematic review of immunohistochemical analyses of these neuroimmune cells in animal models following administration of ion channel antagonists, compared to control conditions. A systematic search of the PubMed and Web of Science electronic databases using the PRISMA and WHO methodologies for systematic reviews yielded 15 original peer-reviewed studies. The majority (13 out of 15) of these studies reported a decrease in microglial activated state after ion signaling pharmacological blockade. The studies provide evidence that acute administration of ion channel antagonists leads to a reduction in microglial activation in rodent brains in the models for epilepsy, Parkinson’s disease, inflammation, pain, ischemia, and brain and spinal cord injury. Future research should explore microglial-specific druggable targets for neurological and psychiatric disorders. The investigation of acute and chronic administration of ion channel antagonists in microglial phenotypes in primates and the development of microglia-like cells derived from human stem cells could be valuable sources in this direction.
Similar content being viewed by others

Introduction
Microglia, the resident immune cells of the central nervous system (CNS), can differentiate into distinct phenotypes, including resting and activated cells1. In vivo resting microglial cells exhibit a ramified morphology, characterized by several branched processes arising from an elongated and flattened cell body. Microglial cells undergo shape changes following activation, i.e., cells transform from ramified to ameboid morphology in response to neuronal injury and during inflammation or infection1. Activated microglia are capable of proliferation, migration, and antigen presentation and release a variety of substances that can be either neuroprotective or neurotoxic1.
Evidence indicates that changes in microglial morphology depend on the cells sensing the environment by repeatedly extending and retracting their processes, but the factors regulating microglial surveillance are unknown2. Microglia interact with their environment with the aid of a complicated ensemble of transporters and ion channels3. The latter include purinergic metabotropic P2Y receptors and ionotropic P2X receptors, the transient receptor potential (TRP) channels such as TRPC6, and the K+ channels kir 2.1, KV 1.3 and KCa 3.11,4. The variety of channels expressed by microglia shows complex spatiotemporal patterns according to changes in the immune cells’ microenvironment, which may contribute to the different phenotypes expressed by microglia5. Furthermore, these various molecules play different roles in microglial function. For instance, K+ channels such as kir, KV, and KCa act by regulating microglial membrane potential6. TRP channels are associated with microglial activation1,7, and the purinergic receptors P2X and P2Y are related to microglial surveillance and phagocytic activity8. Additionally, all the channels may maintain the resting potential of microglia and thus contribute to microglial ramification and continuous surveillance of the brain via process motility1.
In recent decades, an increasingly compelling body of evidence has emerged linking microglial activation to neurological and psychiatric disorders. Broadly, this evidence stems from the observations that microglial surveillance plays an important role in monitoring synaptic function and determining brain connectivity9,10. During postnatal development, synapses that are to be pruned become tagged with complement molecules and are thus removed by microglia11. Disruption of this system may lead to altered CNS connectivity, generating excess excitatory synapses that may be involved in the pathogenesis of various disorders, such as epilepsy12 and autism13. Furthermore, microglial cells release different molecules that are potentially implicated in an excitatory-inhibitory imbalance, which may also contribute to the pathogenesis of psychiatric and neurological disorders14. Based on the above, we review the evidence of current preclinical literature on ion signaling in microglial phenotypes, providing evidence for the role of ion channels in microglial state and identifying gaps in the literature to inform future research.
Our primary outcome
-
Does ion channel pharmacological blockade modify microglial phenotype?
Our secondary outcomes
-
How does interaction between microglial ion channels occur?
-
Can ion signaling contribute to the development of neurological and psychiatric disorders?
Materials and methods
The systematic search was conducted in PubMed and Web of Science, covering articles published up to 31 December 2017. The search protocol was developed based on Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) and World Health Organization (WHO) Review Protocol Template Guidelines where applicable for this systematic review, as provided in Supplementary Materials Section 1. We also manually checked references cited in the systematically searched articles. To avoid publication bias, non-english language studies and gray literature (for example, conference abstracts) were included. A broad but highly structured search strategy was used, based on the PICOS framework. The study population was microglia, the intervention/exposure was ionic channel antagonism, comparison was with absence of ion channel blockade, outcome was resting versus activated microglia, and study design included any type of design. Keywords for the search included various combinations of terms for microglia and the nervous system, including both historical and contemporary ion channel names. A full list of terms used for the search strategy can be found in Supplementary Materials Section 2.
Study selection
Studies were selected for data extraction and analysis based on the following inclusion criteria: (1) ion channel antagonists administered in vivo and (2) studies evaluating microglial activated or resting state. Exclusion criteria were (1) studies that lacked a baseline condition or control group, (2) studies that did not report original data, (3) studies without immunohistochemical analysis of microglia, (4) studies evaluating genetic ionic channel deficiencies, (5) studies that investigated only in vitro ion channel blockade. Due to the highly reactive nature of microglia, which substantially alters their morphology and functional properties when exposed to culture conditions15, in vitro studies should be interpreted with caution.
Data extraction
A standard data extraction template adapted from the Cochrane checklist of items (Supplementary Materials Section 3) was used. As the type of outcome reporting was extremely heterogeneous, results were reported as higher, lower, or unchanged for ion channel antagonism relative to control conditions as identified. Meta-analyses and other summary statistics were not used because of the wide variation between studies in assessment techniques and brain regions examined.
Quality assessment
This systematic review was performed according to preferred reporting items for systematic reviews and meta-analyses (PRISMA) guidelines16 (Supplementary Materials Section 4). Quality assessment used SYRCLES Bias Tool17 (Supplementary Materials Section 5).
Results
The literature search identified 6362 potentially relevant articles for initial screening. Duplicates (N = 1475) were identified using a function in Endnote and confirmed by manual screening of the titles. A total of 4605 studies were excluded from initial assessment of titles and abstracts. In all, 282 abstracts were classified for possible inclusion, and their full texts were obtained. In all, 267 papers were excluded from further analysis. In all, 282 full texts were reviewed, of which 15 met the inclusion criteria for our systematic review (see Fig. 1 for a PRISMA diagram of the literature search). Table 1 summarizes the included studies. Additional data regarding the included studies can be found in Supplementary Materials Section 6. Reasons for the exclusion of studies can be found in Supplementary Material Section 7.

PRISMA Diagram
Microglial ion channel antagonists in epilepsy animal models
Three studies assessed the effect of ion signaling blockade on microglial phenotypes in animal models for epilepsy. Two studies evaluated P2X718,19, while only one investigated TRPC620. These studies were performed on the hippocampus of corialactone and pilocarpine seizure models and showed an increase in microglial channels after induction of status epilepticus. Furthermore, all of the studies demonstrated that P2X7 and TRPC6 antagonism were related to a decrease in microglial activated state.
Microglial ion channel antagonists in ischemic animal models
The majority of studies (4 out of 6) on ischemic models associated a decrease in microglial activation with purinergic channel blockade. Of these, three evaluated P2X7 channels21,22,23, and two of the three demonstrated a reduction in microglial activation21,22. Although the investigation of reactive blue 2 (RB2) in P2X7 blockade showed an increase in reactive microglia, a protective effect against the severity of post ischemic neurological impairment was seen in RB2-treated rats23. Studies evaluating P2X4 and P2Y12 antagonists also demonstrated a reduction in the activated state of CNS-resident immune cells24,25.
Regarding K+ channels in ischemic models, it was shown that K+ channel blockade by glibenclamide did not modify microglial state26. However, glibenclamide decreased neurological deficits and reduced neuronal death in preclinical studies26.
Microglial ion channel antagonists and inflammation in animal models
One study assessed ion channel blockade and microglial cell function in an inflammatory model27 and demonstrated a time-dependent increase in P2X7, to a maximum level at 12 h, after injection of lipopolysaccharide (LPS). Furthermore, P2X7 antagonism with oxATP pointed to a decrease in microglial activated state27.
Animal models for microglial ion channel blockade in traumatic brain injury and spinal cord injury
There was only one study evaluating P2X7 blockade and microglial activation in traumatic brain injury (TBI)28. According to the study, purinergic antagonism attenuated microglial activated state and improved neurobehavioral outcomes after TBI. Moreover, P2X7 blockade decreased IL-1β expression and p38 phosphorylation, increasing the survival of neurons in the injured cerebral cortex28.
In a study on P2X4 antagonism in microglia after spinal cord injury (SCI), Zhou et al. found that dexmedetomidine, a highly selective α2 adrenergic agonist with sedative properties, decreased microglial activation and reversed mechanical hyperalgesia29. In addition, spared nerve injury rats presented high levels of p38 and brain-derived neurotropic factor (BDNF) expression in the dorsal horn compared to controls, downregulated by dexmedetomidine treatment29.
Microglial ion channel antagonists in pain models
One study assessed P2X7 blockade in microglial phenotype in preclinical pain models30. This channel expression was increased in the ipsilateral spinal cord after nerve injury. Temporal evolution in P2X7 levels in the dorsal horn of the spinal cord and the difference in P2X7 levels matched the emergence of mechanical allodynia and thermal hypersensitivity. The study showed that nerve injury-induced mechanical allodynia and thermal hypersensitivity was reversed by intrathecal administration of Brilliant Blue G (BBG)30.
Microglial ion channel antagonists in animal models for Parkinson’s disease
In animal models for Parkinson’s disease, P2X7 and kir 2.1 blockade decreased microglial activation31,32. In rats treated with BBG, a P2X7 antagonist, p38-MAPK activation was reversed, microglial activation was attenuated, and a reduction in the loss-of-dopaminergic neurons was observed in the substantia nigra31. Increases in neuron survival was also demonstrated by estrogenic blockade of kir 2.1. Estrogen incremented anti-apoptotic genes and pro-survival PI3K-Akt signaling32.
Discussion
Our review shows that pharmacological blockade of ionic signaling in microglia in preclinical models for epilepsy, ischemia, Parkinson, pain, TBI, SCI, and inflammation modify microglial phenotypes when compared to controls, thereby decreasing microglial activation. There is evidence of reduced microglial activation following blockade of purinergic channels, such as P2X7, P2X4, and P2Y1218,19,24,25,27,28,29,30,31. Specifically, the majority (8 out of 9) of studies investigating P2X7 channels in preclinical models for epilepsy, ischemia, TBI, SCI, Parkinson, pain, and inflammation demonstrated a decrease in microglial activated state18,19,21,22,27,28,30,31. However, there was an increase in microglial activation in an ischemic animal model following P2X7 blockade23. Variations between studies in the range of doses of ion channel antagonists and the time elapsed from the last drug treatment to microglial evaluation may explain the differences between studies. All the P2X4 and P2Y12 studies showed a decline in immune cell activation after blockade of these channels (3 out of 3 studies)24,25,29.
Studies with animal models for epilepsy and Parkinson’s disease showed a reduction in microglial activated state after TRPC6 and kir 2.1 blockade20,32. However, in an ischemic animal model, K+-channel antagonism with glibenclamide showed no differences in microglial state26. This variety in the results may occur since glibenclamide does not reach significant concentrations in the brain unless the plasma concentration is extremely high33. The drug binds to plasma proteins, which impairs its entry into the brain, as plasma proteins are unable to cross the blood–brain barrier33. Additionally, glibenclamide appears to be removed from the brain rapidly by a highly efficient efflux system when injected directly into the CNS33.
Taken together, these findings indicate that ion channel blockade elicits a significant decrease in microglial activated state in preclinical models for epilepsy, ischemia, TBI, SCI, inflammation, Parkinson, and pain. Nevertheless, further in vivo studies are needed to determine the effects of ion channel antagonists on microglial function (see Box 1 for suggested future directions).
General methodological considerations
Importantly, despite substantial evidence that microglial dysfunction is part of the core pathology of many neurological and psychiatric disorders, evidence remains limited for a direct causal link between a specific and druggable target in microglial cells and a specific disease. Under physiological conditions, microglia reside in the brain parenchyma, so a drug targeting microglia needs to be able to enter the CNS1,34. In recent years, various ion channel antagonists have been developed as potential drugs. However, most of these compounds were not designed to enter the CNS and would therefore lack the properties to be effective in CNS disorders mediated by microglial channels34.
Additionally, microglial cells are highly plastic. Therefore, it is not always clear whether the molecule targeted by a drug is indeed expressed by the microglia34. For example, various P2 receptors have been suggested as drug targets in microglia. However, the expression pattern of P2 receptors strongly depends on the cell phenotype35. For example, microglia may lose P2Y12 receptors in the disease state, whereas P2X4 and P2X7 expression is often increased in microglia in disease35,36,37. Thus, the spatial-temporal effect of microglial channel antagonists still needs to be tested directly (Box 1).
General limitations of the selected studies were that the strain- and sex-specific effects of acute and repeated ion channel blockade on microglial function have still not been tested directly (Box 1). There is evidence for sexual dimorphism in microglial function and a sex bias in CNS disorders with microglial pathology37.
In an attempt to produce more homogeneous outcomes and better understand how pharmacological blockade of microglial ion channels alters microglial state, our search strategy only included studies that evaluated microglia in the activated state (ameboid morphology) or resting state (ramified morphology) as evidenced by microglial immunohistochemical analysis. Moreover, studies that evaluated only in vitro ion channel blockade were also excluded. This is justified due to the highly reactive nature of microglia, which substantially alters their morphology and functional properties when exposed to culture conditions15. The two restrictions significantly reduced the number of studies included in this review.
Our review also included different types of microglial ion channels. Although we found that ion channel blockade was associated with a decrease in microglial activated state in 13 of 15 studies, caution is necessary when interpreting this result. Importantly, microglia have different types of ion channels that play a variety of roles1. It is thus plausible that some ion channel antagonists decrease microglial function while others increase it. Our search criteria yielded studies showing that P2X7, P2X4, P2Y12, kir 2.1, and TRPC6 blockade were related to a decrease in microglial activated state. However, future studies should be performed on different types of ion channels and their association with microglial phenotypes. Furthermore, although our review demonstrated that ionic signaling blockade decreases microglial activation, the majority of studies were conducted in rodent models. Thus, extrapolation of microglial ion channel modulation to primates should be treated with caution, and further studies are needed (Box 1).
Hypothesis on the effects of ion channel antagonists on microglial function
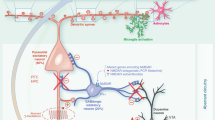
The underlying mechanism of ion channel action in microglia remains to be fully established. However, several lines of evidence indicate that the mechanism involves Ca+2 signaling38,39,40. Microglial activation occurs via a cascade of extracellular and intracellular signaling events beginning with cell surface receptor and ligand-gated ion channel activation followed by short-term and long-term changes in intracellular Ca+240. These Ca+2 signals can cause release of other factors in autocrine-paracrine feedback loops and are necessary for various subsequent cellular responses in microglia39,40 (Fig. 2).

a Extrusion of Ca+2 from the cytosol is accomplished by Ca+2 uptake into the ER via SERCA and Ca+2 accumulation in mitochondria. A significant Ca+2 influx and downstream activation of Ca+2-dependent intracellular pathways, among which the phosphorylation of p38 MAP kinase or the activation of the PI3K/Akt pathway, are directly involved in microglial functions such as synthesis and release of BDNF and chemotactic responses. b Activation of PI3K and PLC accompanied by a rise in intracellular Ca+2, subsequently triggers phosphorylation and hence activation of Akt. This results in the enhanced expression and activation of integrins, promoting adhesion of microglial processes to the extracellular matrix, causing reorganization of the actin cytoskeleton and leading to chemotaxis. c BDNF rapidly downregulates KCC2 through TrkB, causing a deficiency in synaptic inhibition
Microglia contain at least two types of intracellular Ca+2 stores: the mitochondria and the endoplasmic reticulum (ER)1. The main pathway for generation of intracellular Ca+2 signaling is associated with inositol 1,4,5-trisphosphate (InsP3) receptors on the ER membrane1,40. Stimulation of G protein-coupled metabotropic receptors results in the activation of phospholipase C (PLC), production of two secondary messengers, including diacylglycerol and InsP3, and Ca+2 release from the ER1. Importantly, ER depletion activates store-operated Ca+2 entry (SOCE), known as a capacitive Ca+2 influx, mediated by plasmalemmal channels such as calcium release-activated Ca+2 (CRAC) channels and/or transient receptor potential (TRP) channels1. In addition, STIM1, one of the ER membrane proteins, senses the filling state of ER Ca+2 and delivers the ER to the plasma membrane, where it directly activates Orai1/CRAC channels, thereby facilitating the reuptake of Ca+2 to ER through the endoplasmic reticulum Ca+2-ATPases (SERCA). Ca+2 concentration in the ER is precisely controlled by SERCA1. The influx of Ca+2 through ion channels plays an important role in many inflammatory processes, including microglial activation40.
In response to an injury, various extracellular signals may activate microglia and upregulate ion channels. For instance, proinflammatory cytokines upregulate kir, TRPC6, and KCa expression, which may cause hyperpolarization of microglial cells41. Additionally, not only cytokines but also ATP can cause an increase in purinergic receptors, which are highly Ca+2 permeable1. A significant Ca+2 influx and the downstream activation of Ca+2-dependent intracellular pathways, among which the phosphorylation of p38 MAP kinase or the activation of the PI3K/Akt pathway, are directly involved in microglial functions such as synthesis and release of BDNF and chemotactic responses1.
Under pathological conditions, one of the first activities at the injury site is ATP release, activating microglia40. ATP binds to P2X7, a “sensor of danger”, driving resting microglial cells into the activated form, forming large pores and allowing Ca+2 entry40. Ca+2 entry is also increased by the inward rectifier K currents (ikir), an early marker for activated microglia4. Furthermore, extracellular ATP induces membrane ruffling and microglial chemotaxis mediated by the Gi/o protein-coupled P2Y12 receptor1. P2Y12 receptor-mediated activation of the PI3K pathway and increased Akt phosphorylation are required for microglial chemotaxis in response to ATP1.
Microglia exert phagocytic activity at the site of injury. Microglial phagocytosis can initiate respiratory burst, a key function of activated microglia that produces toxic reactive oxygen species42. The K+-channels are involved in controlling microglial respiratory burst42, whereas P2X7 channels are associated with NADPH-oxidase and consequently with oxidative stress1. Phagocytic activity of microglial cells is felt to be involved in synapse removal during development and potentially in pruning synapses in the postnatal brain, which is associated with different psychiatric and neurological disorders9,11. Microglial phagocytosis is intimately associated with cytokine modulation and may contribute to synaptic plasticity43.
Microglial action on synaptic plasticity
Microglia may regulate synaptic plasticity by modulating Cl− gradient in neurons through microglial BDNF release and purinergic signaling44. The purinergic receptors P2X7, P2Y12, and P2X4 are related to p38 MAP kinase, which is directly involved in BDNF synthesis and release45. BDNF release occurs through Ca+2-regulated exocytosis, and regulation of BDNF synthesis involves the p38-MAPK signaling pathway46. BDNF rapidly downregulates K+- Cl− cotransporter 2 (KCC2) through receptor tyrosine kinase B (TrkB)46. KCC2 activity maintains a low intracellular Cl− concentration, a prerequisite for effective GABA/Gly-mediated inhibition in the nervous system47. Thus, since microglial BDNF release decreases KCC2 activity, there is a deficiency in the inhibition of synapses, increasing neuronal hyperexcitability. However, microglial channels could also be linked to neuronal hyperexcitability in a different way: BDNF secreted following stimulation of microglial purinergic receptors may induce phosphorylation of NR1 subunit of NMDA neuronal receptors. This may contribute to neuronal hyperexcitability48.
Implications for neurological and psychiatric disorders
Microglial activation is related to a variety of neurological and psychiatric disorders49. This activated state is related to an excitatory-inhibitory imbalance, generating oxidative stress50, modifying BDNF release51, and changing molecular signaling52,53. Additionally, in the brain, Cl− homeostasis and the reduction of KCC2 activity has been associated with several neurological and psychiatric disorders, such as epilepsy, neuropathic pain, autism spectrum disorders, affective disorders, and schizophrenia54,55,56,57. Several lines of evidence accumulated during the last decade have indeed demonstrated that the increase in excitability in these pathological conditions can be largely explained by a loss of inhibition, and KCC2 has been recognized as an important molecular target underlying this loss55,56.
It is thus plausible that Ca+2 entry through microglial ion channels may activate microglia and influence MAPK and AKT pathways. This could alter microglial phagocytosis functions, modify BDNF release, and increase neuronal hyperexcitability through KCC2 inhibition, contributing to the pathogenesis of mental disorders. Furthermore, first-line treatments for neurological and psychiatric disorders have potential inhibitory effects on microglia. To name a few, antipsychotics such as haloperidol, risperidone, olanzapine, clozapine, and chlorpromazine inhibit microglial proton currents, suppressing reactive oxygen production, acting as anti-inflammatory modulators58. Proton currents are also inhibited by the SSRIs paroxetine and sertraline, for example, attenuating the mobilization of intracellular Ca+2 59. Aripiprazole also downregulates microglial Ca+2 signaling59, while carbamazepine suppresses microglial activation via p38-MAPK. Additionally, these drugs interfere with neurotransmitters, such as dopamine, serotonin, acetylcholine, and norepinephrine. This could contribute to further alterations in microglial phenotypes. Norepinephrine inhibits microglial inflammatory reactions through activation of cyclic AMP and suppression of downstream MAPK60. Dopamine and serotonin could interfere with microglial chemotaxis, and acetylcholine may alter Ca+2 signaling60. These effects could additionally contribute to a decrease in microglial activated state in neurological and psychiatric disorders.
Calcium-channel blockers might also contribute to a decrease in microglial activated state61,62, providing potential utility in the treatment of neurological and psychiatric disorders63,64,65. In fact, calcium-channel blockers have been used successfully to treat absence seizures63, and are emerging as potential therapeutic targets for pathologies, such as Parkinson disease, pain, mood disorders, and anxiety63. For instance, centrally acting calcium-channel blockers targeting Ca2+ channels of dopaminergic neurons might decrease risk of Parkinson disease66, the calcium-channel blocker isradipine could be efficacious for bipolar disorder65, and the drug nifedipine might have an antiepileptic effect67. Nevertheless, clinical trials with calcium-channel blockers in neurological and psychiatric disorders present inconsistent results64. One possible reason for this is the pharmacokinetic variability among different calcium-channel blockers68. Dihydripyridines, such as isradipine, have more favorable brain–blood barrier penetration and binding to calcium-channel compared to other calcium-channel blockers, such as diltiazem and verapamil68. Therefore, although there is evidence supporting the utility of calcium channel blockers in a variety of psychiatric and neurological disorders, what remains to be understood is how to develop new types of calcium channel inhibitors that specifically target calcium channels that are involved in pathophysiological processes, while sparing those that contribute to normal physiological function63. This requires an extensively understanding of how calcium channels participate in the function of specific brain circuits that are implicated in pathophysiology and how these channels may be dysregulated in pathological states63. This represents an enormous challenge in finding compounds that effectively cross the blood–brain barrier, have high affinity and target selectivity63.
Clinical trials with drugs targeting-specific ion channels are currently underway. Some P2X7 receptor antagonists have been developed as potential drugs. For example, AZD905669 and CE-224,53570 have been tested in clinical trials in rheumatoid arthritis, while GSK148216071 was developed to target peripheral pain. These compounds were not designed to enter the CNS and would therefore most likely lack the properties to be effective in nervous system disorders that could be associated with microglial channels. Nevertheless, as our understanding of the role of ion channels on microglia in human diseases grows, the development of therapeutics targeting-specific microglial molecules will probably occur. However, some challenges remain for the development of microglial channel antagonists for use in humans, such as target specificity, CNS penetrance, and differences between animal and human microglia. New research strategies such as the development of microglia-like cells derived from human stem cells might help overcome the current limitations (Box 1).
Conclusion
The administration of ion channel antagonists leads to a decrease in microglial activation in different preclinical models. These findings suggest that ion channels have convergent pathways that potentially contribute to differentiation of microglial phenotypes and functions, which could be altered in a variety of neurological and psychiatric disorders. Further studies are required to explore whether the same antagonists’ effects are seen in primate brain microglia and to investigate-specific druggable targets in microglial cells.
References
Kettenmann, H., Hanisch, U. K., Noda, M. & Verkhratsky, A. Physiology of microglia. Physiol. Rev. 91, 461–553 (2011).
Nimmerjahn, A., Kirchhoff, F. & Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 (2005).
Schilling, T. & Eder, C. Ion channel expression in resting and activated microglia of hippocampal slices from juvenile mice. Brain Res. 1186, 21–28 (2007).
Nguyen, H. M. et al. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 65, 106–121 (2017).
O’Hare Doig, R. L. et al. Specific ion channels contribute to key elements of pathology during secondary degeneration following neurotrauma. BMC Neurosci. 18, 62 (2017).
Gueguinou, M. et al. KCa and Ca(2+) channels: the complex thought. Biochim. Biophys. Acta 1843, 2322–2333 (2014).
Liu, N. et al. NF-kappaB dependent up-regulation of TRPC6 by Abeta in BV-2 microglia cells increases COX-2 expression and contributes to hippocampus neuron damage. Neurosci. Lett. 651, 1–8 (2017).
Burnstock, G. An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology 104, 4–17 (2016).
Schafer, D. P. et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012).
Tremblay, M. E., Lowery, R. L. & Majewska, A. K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8, e1000527 (2010).
Stevens, B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007).
Zhang, B., Zou, J., Han, L., Rensing, N. & Wong, M. Microglial activation during epileptogenesis in a mouse model of tuberous sclerosis complex. Epilepsia 57, 1317–1325 (2016).
Zhan, Y. et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 17, 400–406 (2014).
Villegas-Llerena, C., Phillips, A., Garcia-Reitboeck, P., Hardy, J. & Pocock, J. M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 36, 74–81 (2016).
Hellwig, S., Heinrich, A. & Biber, K. The brain’s best friend: microglial neurotoxicity revisited. Front. Cell Neurosci. 7, 71 (2013).
Moher, D., Liberati, A., Tetzlaff, J. & Altman, D. G. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 6, e1000097 (2009).
Hooijmans, C. R. et al. SYRCLE’s risk of bias tool for animal studies. BMC Med. Res. Methodol. 14, 43 (2014).
Choi, H. K. et al. The roles of P2X7 receptor in regional-specific microglial responses in the rat brain following status epilepticus. Neurol. Sci. 33, 515–525 (2012).
Huang, C. et al. Inhibition of P2X7 receptor ameliorates nuclear factor-Kappa B mediated neuroinflammation induced by status epilepticus in rat hippocampus. J. Mol. Neurosci. 63, 173–184 (2017).
Lee, S. K., Kim, J. E., Kim, Y. J., Kim, M. J. & Kang, T. C. Hyperforin attenuates microglia activation and inhibits p65-Ser276 NFkappaB phosphorylation in the rat piriform cortex following status epilepticus. Neurosci. Res. 85, 39–50 (2014).
Yu, Q. et al. Block of P2X7 receptors could partly reverse the delayed neuronal death in area CA1 of the hippocampus after transient global cerebral ischemia. Purinergic. Signal. 9, 663–675 (2013).
Chu, K. et al. Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J. Neuroinflamm. 9, 69 (2012).
Melani, A. et al. P2X7 receptor modulation on microglial cells and reduction of brain infarct caused by middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab. 26, 974–982 (2006).
Wixey, J. A., Reinebrant, H. E., Carty, M. L. & Buller, K. M. Delayed P2X4R expression after hypoxia-ischemia is associated with microglia in the immature rat brain. J. Neuroimmunol. 212, 35–43 (2009).
Gelosa, P. et al. Microglia is a key player in the reduction of stroke damage promoted by the new antithrombotic agent ticagrelor. J. Cereb. Blood Flow Metab. 34, 979–988 (2014).
Ortega, F. J. et al. ATP-dependent potassium channel blockade strengthens microglial neuroprotection after hypoxia-ischemia in rats. Exp. Neurol. 235, 282–296 (2012).
Choi, H. B., Ryu, J. K., Kim, S. U. & McLarnon, J. G. Modulation of the purinergic P2X7 receptor attenuates lipopolysaccharide-mediated microglial activation and neuronal damage in inflamed brain. J. Neurosci. 27, 4957–4968 (2007).
Liu, X. et al. Inhibition of P2X7 receptors improves outcomes after traumatic brain injury in rats. Purinergic Signal. 13, 529–544 (2017).
Zhou, T. T., Wu, J. R., Chen, Z. Y., Liu, Z. X. & Miao, B. Effects of dexmedetomidine on P2X4Rs, p38-MAPK and BDNF in spinal microglia in rats with spared nerve injury. Brain Res. 1568, 21–30 (2014).
He, W. J. et al. Spinal P2X(7) receptor mediates microglia activation-induced neuropathic pain in the sciatic nerve injury rat model. Behav. Brain Res. 226, 163–170 (2012).
Wang, X. H., Xie, X., Luo, X. G., Shang, H. & He, Z. Y. Inhibiting purinergic P2X7 receptors with the antagonist brilliant blue G is neuroprotective in an intranigral lipopolysaccharide animal model of Parkinson’s disease. Mol. Med. Rep. 15, 768–776 (2017).
Wu, S. Y. et al. Estrogen ameliorates microglial activation by inhibiting the Kir2.1 inward-rectifier K(+) channel. Sci. Rep. 6, 22864 (2016).
Lahmann, C., Kramer, H. B. & Ashcroft, F. M. Systemic administration of glibenclamide fails to achieve therapeutic levels in the brain and cerebrospinal fluid of rodents. PLoS ONE 10, e0134476 (2015).
Biber, K., Moller, T., Boddeke, E. & Prinz, M. Central nervous system myeloid cells as drug targets: current status and translational challenges. Nat. Rev. Drug Discov. 15, 110–124 (2016).
Koizumi, S., Ohsawa, K., Inoue, K. & Kohsaka, S. Purinergic receptors in microglia: functional modal shifts of microglia mediated by P2 and P1 receptors. Glia 61, 47–54 (2013).
Inoue, K. Purinergic systems in microglia. CMLS 65, 3074–3080 (2008).
Crain, J. M., Nikodemova, M. & Watters, J. J. Expression of P2 nucleotide receptors varies with age and sex in murine brain microglia. J. Neuroinflamm. 6, 24 (2009).
Ikeda, M. et al. Ca(2+) spiking activity caused by the activation of store-operated Ca(2+) channels mediates TNF-alpha release from microglial cells under chronic purinergic stimulation. Biochim. Biophys. Acta 1833, 2573–2585 (2013).
Korvers, L. et al. Spontaneous Ca(2+) transients in mouse microglia. Cell Calcium 60, 396–406 (2016).
Farber, K. & Kettenmann, H. Functional role of calcium signals for microglial function. Glia 54, 656–665 (2006).
Deftu, A. F., Ristoiu, V. & Suter, M. R. Intrathecal administration of CXCL1 enhances potassium currents in microglial cells. Pharmacology 101, 262–268 (2018).
Khanna, R., Roy, L., Zhu, X. & Schlichter, L. C. K+channels and the microglial respiratory burst. Am. J. Physiol. Cell Physiol. 280, C796–C806 (2001).
Riazi, K. et al. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc. Natl Acad. Sci. USA 105, 17151–17156 (2008).
Coull, J. A. et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021 (2005).
Trang, T., Beggs, S., Wan, X. & Salter, M. W. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J. Neurosci. 29, 3518–3528 (2009).
Rivera, C. et al. BDNF-induced TrkB activation down-regulates the K(+)–Cl(−) cotransporter KCC2 and impairs neuronal Cl(−) extrusion. J. Cell Biol. 159, 747–752 (2002).
Rivera, C. et al. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255 (1999).
Caldeira, M. V. et al. BDNF regulates the expression and traffic of NMDA receptors in cultured hippocampal neurons. Mol. Cell Neurosci. 35, 208–219 (2007).
Salter, M. W. & Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027 (2017).
Hassan, W. et al. Association of oxidative stress with psychiatric disorders. Curr. Pharm. Des. 22, 2960–2974 (2016).
Autry, A. E. & Monteggia, L. M. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 64, 238–258 (2012).
Naziroglu, M. & Demirdas, A. Psychiatric disorders and TRP channels: focus on psychotropic drugs. Curr. Neuropharmacol. 13, 248–257 (2015).
Chen, H. M. et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Transl. Psychiatry 4, e375 (2014).
Tao, R. et al. Transcript-specific associations of SLC12A5 (KCC2) in human prefrontal cortex with development, schizophrenia, and affective disorders. J. Neurosci. 32, 5216–5222 (2012).
Di Cristo, G., Awad, P. N., Hamidi, S. & Avoli, M. KCC2, epileptiform synchronization, and epileptic disorders. Prog. Neurobiol. 162, 1–16 (2018).
Ford, A. et al. Engagement of the GABA to KCC2 signaling pathway contributes to the analgesic effects of A3AR agonists in neuropathic pain. J. Neurosci. 35, 6057–6067 (2015).
Corradini, I. et al. Maternal immune activation delays excitatory-to-inhibitory gamma-aminobutyric acid switch in offspring. Biol. Psychiatry 83, 680–691 (2018).
Shin, H., Kim, J. & Song, J.-H. Clozapine and olanzapine inhibit proton currents in BV2 microglial cells. Eur. J. Pharmacol. 755, 74–79 (2015).
Horikawa, H. et al. Inhibitory effects of SSRIs on IFN-γ induced microglial activation through the regulation of intracellular calcium. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 1306–1316 (2010).
Kato, T. A. et al. Neurotransmitters, psychotropic drugs and microglia: clinical implications for psychiatry. Curr. Med. Chem. 20, 331–344 (2013).
Huang, B. R. et al. Anti-neuroinflammatory effects of the calcium channel blocker nicardipine on microglial cells: implications for neuroprotection. PLoS ONE 9, e91167 (2014).
Hashioka, S., Klegeris, A. & McGeer, P. L. Inhibition of human astrocyte and microglia neurotoxicity by calcium channel blockers. Neuropharmacology 63, 685–691 (2012).
Zamponi, G. W. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug Discov. 15, 19–34 (2016).
Cipriani, A. et al. A systematic review of calcium channel antagonists in bipolar disorder and some considerations for their future development. Mol. Psychiatry 21, 1324–1332 (2016).
Ostacher, M. J. et al. Pilot investigation of isradipine in the treatment of bipolar depression motivated by genome-wide association. Bipolar Disord. 16, 199–203 (2014).
Ritz, B. et al. L-type calcium channel blockers and Parkinson’s disease in Denmark. Ann. Neurol. 67, 600–606 (2010).
Damasceno, D., Ferreira, A., Doretto, M. & Almeida, A. Anticonvulsant and antiarrhythmic effects of nifedipine in rats prone to audiogenic seizures. Braz. J. Med. Biol. Res. 45, 1060–1065 (2012).
Casamassima, F. et al. L-type calcium channels and psychiatric disorders: a brief review. AJMG 153b, 1373–1390 (2010).
Keystone, E. C., Wang, M. M., Layton, M., Hollis, S. & McInnes, I. B. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Rheum. Dis. 71, 1630–1635 (2012).
Stock, T. C. et al. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J. Rheumatol. 39, 720–727 (2012).
Ali, Z. et al. Pharmacokinetic and pharmacodynamic profiling of a P2X7 receptor allosteric modulator GSK1482160 in healthy human subjects. Br. J. Clin. Pharmacol. 75, 197–207 (2013).
Acknowledgements
The authors wish to thank Fatima Azevedo for her assistance in the elaboration of Fig. 2.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Quagliato, L.A., Nardi, A.E. The role of convergent ion channel pathways in microglial phenotypes: a systematic review of the implications for neurological and psychiatric disorders. Transl Psychiatry 8, 259 (2018). https://doi.org/10.1038/s41398-018-0318-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-018-0318-0
This article is cited by
-
Spinal Microglia and Astrocytes: Two Key Players in Chronic Visceral Pain Pathogenesis
Neurochemical Research (2022)
-
Maternal immune activation generates anxiety in offspring: A translational meta-analysis
Translational Psychiatry (2021)
