
Abstract
Propionate is a key intermediate in anaerobic digestion processes and often accumulates in association with perturbations, such as elevated levels of ammonia. Under such conditions, syntrophic ammonia-tolerant microorganisms play a key role in propionate degradation. Despite their importance, little is known about these syntrophic microorganisms and their cross-species interactions. Here, we present metagenomes and metatranscriptomic data for novel thermophilic and ammonia-tolerant syntrophic bacteria and the partner methanogens enriched in propionate-fed reactors. A metagenome for a novel bacterium for which we propose the provisional name ‘Candidatus Thermosyntrophopropionicum ammoniitolerans’ was recovered, together with mapping of its highly expressed methylmalonyl-CoA pathway for syntrophic propionate degradation. Acetate was degraded by a novel thermophilic syntrophic acetate-oxidising candidate bacterium. Electron removal associated with syntrophic propionate and acetate oxidation was mediated by the hydrogen/formate-utilising methanogens Methanoculleus sp. and Methanothermobacter sp., with the latter observed to be critical for efficient propionate degradation. Similar dependence on Methanothermobacter was not seen for acetate degradation. Expression-based analyses indicated use of both H2 and formate for electron transfer, including cross-species reciprocation with sulphuric compounds and microbial nanotube-mediated interspecies interactions. Batch cultivation demonstrated degradation rates of up to 0.16 g propionate L−1 day−1 at hydrogen partial pressure 4–30 Pa and available energy was around −20 mol−1 propionate. These observations outline the multiple syntrophic interactions required for propionate oxidation and represent a first step in increasing knowledge of acid accumulation in high-ammonia biogas production systems.

Similar content being viewed by others

Introduction
Through anaerobic digestion, waste is efficiently converted to a combustible gas, comprising methane (CH4) and carbon dioxide (CO2). This is one of the most sustainable options for renewable energy production when accounting for the additional benefits, such as abated greenhouse gas emissions, efficient waste management, recovery of nutrients and substitution of synthetic and mineral fertiliser when using the residue as bio-fertiliser [1]. The anaerobic degradation process involves a series of microbial degradation steps engaging different anaerobic microorganisms, often operating at near thermodynamic equilibrium [2, 3]. In the process, complex organic materials are initially hydrolysed to sugars, amino acids and long-chain fatty acids that are further fermented to volatile fatty acids (VFA), alcohols and smaller amounts of hydrogen (H2). In the following anaerobic oxidation step, VFA longer than acetate, such as propionate and butyrate, are degraded to acetate, H2 and CO2, which in a terminal step are converted to CH4 and CO2 by methanogenic archaea. Although the anaerobic digestion process is generally operated in continuous/semi-continuous mode, disturbances can occur during operation, triggered, for instance, by a change in feeding composition/rate, temperature fluctuations, trace element deficiency or ammonia toxicity [4]. A pervasive consequence, particularly with ammonia-induced perturbations if the anaerobic digester is fed protein-rich substrates, is accumulation of propionate and acetate, which exposes the process disturbance [5, 6]. At high ammonia levels, acetate accumulation is often caused by inactivation of acetate-cleaving methanogens, opening up an opportunity for development of an alternative acetate-degrading community involving a synergy between syntrophic acetate oxidisers (SAOB) and hydrogenotrophic methanogens (HM) [7]. The exact cause of propionate accumulation at high ammonia concentrations is poorly understood, but may relate to suppression of ammonia-sensitive propionate-degrading communities [6, 8, 9] and a temporal lapse in establishment of ammonia-tolerant propionate degraders [10].
Propionate is degraded through interactions between syntrophic propionate-oxidising bacteria (SPOB) and hydrogen- and/or formate-utilising HM. Acetate is subsequently degraded by aceticlastic methanogens in low ammonia conditions [11, 12] or by SAOB and hydrogen- and/or formate-utilising HM in high-ammonia conditions [13]. The SPOB characterised to date are from the families Peptococcaceae and Syntrophobacteraceae, which all degrade propionate using the methylmalonyl-CoA pathway or the dismutating pathway (only Smithella) [10]. SPOB candidates include members of the phylum Cloacimonadota found in both mesophilic and thermophilic biogas processes [14, 15], ‘Candidatus Propionivorax syntrophicum’ discovered in a mesophilic wastewater treatment plant [16] and the only known ammonia-tolerant SPOB, ‘Candidatus Syntrophopropionicum ammoniitolerans’ identified from a mesophilic biogas process [13]. The majority of these SPOB originate from biogas reactors, clearly demonstrating that syntrophic propionate oxidation (SPO) is a distinct feature of the anaerobic digestion process. However, although high-ammonia anaerobic digestion has been widely studied from a process perspective, there are indications that several acid-degrading microorganisms with key roles in that process have not been identified, isolated or characterised [10, 13]. Consequently, the reciprocal communal interactions within and between such communities, i.e. between SAOB, SPOB and HM, are currently underexplored. In particular, current understanding of SPO in thermophilic and high-ammonia biogas processes and the cross-species interactions enabling stepwise conversion of propionate to methane is limited.
In the present study, a thermophilic SPOB community in high-ammonia conditions was enriched and its molecular exchange and interaction network with SAOB and HM were analysed. The microbial communities were explored through both 16S rRNA gene amplicon sequencing and whole metagenome sequencing, and their physiological activity during propionate and acetate degradation was characterised using metatranscriptomics. Chemical monitoring of batch trials of propionate- and acetate-supplemented cultures was performed to study the effects of intermediate product formation and consumption on propionate- and acetate-degradation kinetics.
Materials and methods
Continuously fed reactor set-up and operation
Four identical laboratory-scale continuously stirred (80 rpm) tank reactors (Belach Bioteknik) with working volume 1.1 L were operated at 52 °C. Separate pairs of reactors were designated for propionate (RP1, RP2) and acetate (RA1, RA2) enrichment. The reactors were continuously fed with anoxic, sterile bicarbonate-buffered basal medium (BM) [17] supplemented with 8.9 g L−1 ammonium chloride (3 g L−1 NH4+-N) and 0.1 M (9.6 g L−1) sodium propionate (RP1, RP2) or 0.1 M (8.2 g L−1) sodium acetate (RA1, RA2). The initial pH of the BM was 7.3 and the reactors were inoculated with 1 L sterile BM and 0.1 L sludge from a thermophilic and high-ammonia large-scale biogas digester [18] (Table S1) under flushing with N2. The four reactors were fed with peristaltic pumps (Belach Bioteknik) at a dilution rate of 26 µL min−1, resulting in a hydraulic retention time of 28 days. Concentrations of short-chain VFA (acetate, propionate, butyrate, isobutyrate, valerate, isovalerate, capronate and isocapronate) were analysed using high performance liquid chromatography (HPLC), and methane content of the gas was determined by gas chromatography (GC) as described previously [19]. Volume of gas produced was measured continuously using µFlow (2 mL resolution, BioProcess Control). The VFA and gas production were monitored weekly for the first 320 days and thereafter occasionally to confirm the stable conditions till the end of the experiment at day 630.
Batch experiment set-up
To evaluate the kinetics of the cultures enriched in the different reactors, anaerobic batch assays were prepared by transferring 0.5 L of the enrichment culture from the continuously propionate- and acetate-fed reactors after 630 days of operation directly to sterile anaerobic serum bottles (1 L) under constant N2 flushing. After a few days of acclimatisation at 52 °C without stirring, sodium propionate or sodium acetate was added to the batch culture, with the intention to reach a final concentration of 30 mM (due to variation in VFA levels in the culture transferred from the reactors, the initial acid concentration range after addition in the batch assays was 21–37 mM propionate and 34–80 mM acetate). Duplicate batch assays were prepared for each set-up and reactor, giving a total of 12 batch assays (B01-B12). Incubation was at 52 °C (pH 8.5 ± 0.1) at a free ammonia level of 1.2 g NH3 L-1. Cultures without addition of propionate or acetate served as negative controls. Determination of gas composition (H2, CH4, CO2) and analyses of liquid samples (acetate, propionate) were conducted. Hydrogen partial pressure was measured as described elsewhere [20]. The concentrations of acetate, propionate and formate after cessation of methane formation (after 50–86 days) were determined by nuclear magnetic resonance spectroscopy as described in Supplementary note 1.
Thermodynamic calculations
For the thermodynamic calculations, the chemical reactions were evaluated using equations 1–3 (Supplementary note 2). Doubling time (td) was estimated from specific methane production rate (µCH4/P/A) as td,CH4/P/A = ln2/µCH4/P/A, where µCH4/P/A was calculated from the slope of logarithmic methane, propionate or acetate change during exponential acid degradation and methane production.
DNA extraction, 16S rRNA gene amplicon sequencing, qPCR and data analysis
Total DNA was extracted from samples taken from reactors and batch assay on 19 and 8–10 occasions, respectively. The preparation of samples for MiSeq paired-end (2 × 300 bp) (Illumina) 16S rRNA gene amplicon sequencing is described in Supplementary note 3. Illumina adapters and primer sequences were trimmed using BBMap (v38.61b) [21]. Generation of amplicon sequence variants (ASV), abundance table and taxonomic assignment of ASVs were performed using the package dada2 (v1.22.0) [22] in R (v4.1.3) [23]. Taxonomic classification of the ASVs was carried out using the GTDB taxonomic training dataset v202 formatted for DADA2 [24]. A phyloseq object was created using abundance and taxonomy tables and community structure was visualised using the package phyloseq (v1.38.0) [25] in RStudio (v2022.02.3 + 492) [26]. Quantitative PCR (qPCR) was performed to determine 16S rRNA gene copy number of methanogens in the orders Methanomicrobiales and Methanobacteriales, using primers and conditions as described previously [27, 28].
Metagenomic sequencing, assembly, binning and functional analysis
Whole metagenome sequencing on samples from the parallel propionate-fed digesters (RP1 and RP2) withdrawn at day 115 was performed using HiSeq (Illumina), with 2 × 150 bp pair-end reads (Eurofins, Germany). For long read sequencing, genomic high molecular weight DNA extraction, sample preparation and sequencing using a MinION device (Oxford Nanopore Technologies) were performed as described in Supplementary note 3. Hybrid genome assembly was performed using flye (v2.8) [29, 30], racon (v1.4.13) [31] and medaka (v1.0.3) [32]. Refinement of subsequent long read assembly was done with short reads using multiple polishing rounds with Pilon (v1.23) [33]. Metagenomic binning was done using metaWRAP pipeline [34] and quality of resulting bins was accessed using CheckM (v1.0.18) [35]. Taxonomic classification of metagenome assembled genomes (MAGs) was performed with GTDB-tk (v1) [36], using the GTDB database (R202) [37]. Functional annotation of MAGs was done using Bakta (v1.4.1) [38] for bacterial and Prokka (v1.14.6) [39] for archaea (see Supplementary note 4 for details).
Annotated MAGs were manually validated for the genes involved in the methylmalonyl-CoA pathway and hydrogenase gene sequences extracted from the MAGs were manually classified against hydrogenase database HydDB (accessed June 2022) [40]. Digital DNA-DNA hybridisation (dDDH) between retrieved MAGs and genomes of closest relatives were determined using the GGDC (v3.0) [41]. Whole-genome average nucleotide identity (ANI) and average amino acid identity (AAI) were calculated using Pyani (blast+ algorithm) (v0.2.10) [42] and CompareM [43], respectively. Visualisation of ANI and AAI results was performed in RStudio with the package ggplot2. Species trees for the methanogens and SPOBs were created by the STAG method implemented in Orthofinder (v2.5.4) [44, 45]. Visualisation and annotation of all phylogenetic trees was performed with package ggplot2 (v3.3.2) [46] and Figtree (v1.4.3) [47].
RNA extraction and analysis of transcriptomics data
For RNA extraction, 50 mL volume of microbial culture were withdrawn from the batch assays (propionate B01 and B03, acetate B09) during the exponential phase of acetate and propionate degradation. Each culture sample was anaerobically transferred to a Falcon tube and immediately centrifuged at 5000 g and 4 °C for 10 min. The cell pellet was dissolved in 1 mL Trizol and 0.2 mL chloroform, and total RNA was extracted using the Quick-RNA Fecal/Soil Microbe Microprep Kit with an additional DNase I depletion step [48]. A two-step ribosomal RNA depletion protocol was employed using pan-prokaryotes riboPOOL probes and Dynabeads (MyOne Streptavidin C1, Invitrogen #65001) according to the manufacturer’s protocol [49]. Ribosomal RNA-depleted RNA samples were used for paired-end (2 × 75 bp) MiSeq sequencing (Illumina, v3 chemistry) on the SNP&SEQ platform [50]. The raw RNA sequence data were processed with BBMap (v38.61b) for quality control and removal of ribosomal RNA reads. Quantification (transcripts per million, TPM) of the filtered reads was performed by mapping against the MAGs using Salmon (v1.6.0) [51]. Quantification results were used for differential gene expression and other analyses in Rstudio, with the packages DESeq2 (v1.37.0) [52], ggplot2 and pheatmap (v1.0.12) [53]. All values used and represented in heatmaps are based on TPM counts (Supplementary data).
Results and discussion
Reactor performance revealed temporal changes in propionate degradation rate
The four propionate- and acetate-fed reactors used in the study produced biogas with an average methane content of 62–70% (Table S2). The pH was 8.1–8.3, resulting in an ammonia-nitrogen level of 0.7–0.9 g NH3 L-1. This free ammonia level is well above the threshold at which many microorganisms are inhibited, frequently causing reductions in overall methane production and accumulation of VFAs even in ammonia-adapted biogas processes [54].
In agreement with previous findings for thermophilic acetate-fed reactors [19], acetate content remained stable at around 0.7–0.9 g L−1 in the acetate-fed reactors RA1 and RA2. However, VFA degradation in the propionate-fed reactors was less stable. Propionate fluctuations (0.8–3.9 g L−1) were especially pronounced in reactor RP2 during the latter stages of operation (days 200–320, Fig. S1). As a result of changes in propionate level, acetate level in RP2 fluctuated from below detection to 3.5 g L−1 throughout the operating period. Other VFAs analysed were not detected above the detection limit of 0.2 g L−1 in the reactors. Comparing thermophilic reactor performance with that in previous mesophilic enrichment study [13], revealed less efficient propionate removal in thermophilic than in the mesophilic propionate reactors (Table S4). One reason for this may be the somewhat higher pH and associated higher ammonia levels in the thermophilic than in the mesophilic reactors.
Batch assays and thermodynamics
For reasons unclear, it was not possible to initiate propionate-degrading activity in batch assays by preparing culture media and inoculating with 5–20% (v/v) of culture from the continuously fed reactors, a procedure routinely applied with success for mesophilic propionate-degrading cultures (unpublished data) originating from analogous mesophilic reactor experiment [13]. Propionate and acetate-degradation rates were therefore analysed by adding these VFAs to batches consisting of undiluted cultures from the continuously fed reactors. The degradation rates of propionate and acetate, H2 level and methane production rate were similar in the duplicate batches originating from the same reactor, but the degradation rate of propionate differed between the RP1 and RP2 communities (Fig. S2). The communities originating from RP1 (batches B01-B02) had the highest propionate-degradation rate and degraded the added propionate within 25 days. The rate of propionate degradation in the RP2 batches was considerably lower, and up to 100 days were required for complete propionate degradation. In batches B01-B02 propionate was consumed and methane formed according to the expected stoichiometry of 1.75 mol methane per mol propionate, whereas in batches B03-B04 the methane yield was slightly higher than expected (Table S3). Hydrogen levels were relatively similar in the propionate-fed batch cultures, foremost ranging between 3.5 and 12 Pa (Table S5), and were in line with levels previously reported for thermophilic acetate-oxidising cultures [55, 56], but somewhat higher than values reported for mesophilic propionate-oxidising communities [20, 57].
The underlying cause of the slower propionate degradation of the RP2 community compared with the RP1 community is unknown. However, the relatively similar acetate-degradation rates and H2 partial pressures in all communities (Table S5) indicate that the slower propionate degradation in RP2 was not because of low activity of the acetate-degrading community or slow removal of H2 in this reactor. Thus, the SPOB in RP2 were most likely inhibited by another factor. Furthermore, the relatively rapid propionate degradation in the RP1 batches (B01-B02) resulted in formation of 2–3 g acetate L−1 before the SAOB initiated acetate degradation (Fig. S2). The accumulated acetate and the calculated rate of propionate and acetate degradation in the present study also indicated that the SPOB community was able to degrade propionate at a rate that exceeded the acetate-degrading capacity of SAOB (Fig. S2, Table S5). These results support previous observation of a peak in acetate concentration due to rapid propionate degradation in thermophilic conditions [58]. Similar elevations in acetate levels following propionate degradation have been observed in high-ammonia mesophilic reactors after VFA pulsing and in dairy manure digesters [59, 60]. However, in several studies of mesophilic and low-ammonia processes with aceticlastic methanogens as the main acetate degrader, the acetate concentration has remained at low levels despite degradation of 1–4 g propionate L−1 [16, 61,62,63]. One reason for the disparity between high- and low-ammonia conditions is possibly that acetate above a certain concentration may be required before initiation of acetate degradation by SAOB. Alternatively, less efficient HM activity at higher ammonia levels [64] increases the H2 and/or formate levels, impeding the activity of SPOB and SAOB. This emphasises the need to support the activity of both syntrophic bacteria and HM in order to obtain a stable process [13, 57, 63, 65,66,67]. In the continuously fed reactors in the present study, the relatively constant flow of acetate formed from the propionate was manageable by the SAOB community, in maintaining acetate level at <2.5 g L−1 (Fig. S1). This is important, since acetate at >4.8 g L−1 (80 mM) has been shown to severely restrict propionate oxidation [68].
The ∆G values calculated from measured parameters from the batch experiment varied somewhat between the different species, but for the SPOB the ∆G value for conversion of propionate to acetate and H2 fluctuated around −20 mol−1 propionate. For acetate oxidation to CO2 and H2 by the SAOB, the ΔG value was −10 to −30 kJ mol−1 acetate. For HM, the ΔG value was similar to that of propionate in both series of batches, although during the later stages of propionate degradation of the batches with slow propionate degradation (B03-B04) the ΔG values for SPOB were more favourable than those for HM (Fig. S3A). In the acetate fed batches, ΔG was consistently more favourable for HM than for SAOB. This agrees well with results previously obtained for mesophilic SAOB cultures, where the HM also obtained more energy than the SAOB (−20 and −10 kJ mol−1, respectively) [20]. Due to the dual syntrophy in the propionate-fed systems, the outcome changed when energy distribution was evaluated per mole of propionate mineralised to CH4 (Table S6). HM then gained most of the energy, as 1.75 moles of methane were generated for each mole of propionate mineralised. The dual syntrophy underpinning propionate degradation in these systems makes H2 one of the central intermediates, as it can be produced by both SAOB and SPOB, and low H2 levels are beneficial for both. For acetate, the interdependency is more complicated, i.e. the SPOB benefit from low levels, while the SAOB benefit from high levels. In the batch experiment, the average hydrogen levels were slightly lower in the propionate-fed batches than in the acetate-fed batches, irrespective of the acetate level (Figs. S3B, C). To gain further thermodynamic insights into the microbe interplay in the dual syntrophy and to unravel why and when the hydrogen scavenger operates at lower hydrogen levels in syntrophy with SPOB than with SAOB, future studies should monitor growth of the syntrophs and the methanogen under a set of constant H2 levels.
Microbial community structure (16S rRNA gene) in enrichment reactors and batch assays
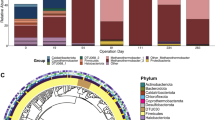
Microbial community structure based on 16S rRNA gene amplicon sequencing most strikingly revealed enrichment of the family Pelotomaculaceae only in the propionate-fed reactors (RP1, RP2; 2–48%) and not in the acetate-fed reactors (RA1, RA2) (Fig. 1). Pelotomaculaceae harbours many known [10] and proposed SPOB [13, 16]. Major families observed throughout the experimental period in both the propionate- and acetate-fed reactors were Acetomicrobiaceae (3–13%), Campylobacteraceae (20–50%), Ch115 (5–20%) and Thermacetogeniaceae (2–34%). (Fig. 1). These five families were equally dominant in all batch assays except for family Pelotomaculaceae, which was specifically higher in relative abundance in the assays inoculated from the propionate-fed continuous reactors (B01-B08, Fig. S4). The batch assays from the propionate-fed reactors prepared with acetate as growth substrate (B05-B08) showed declining relative abundance (<5%) of Pelotomaculaceae, whereas its relative abundance was higher (3–50%) in batches fed propionate (B01-B04, Fig. S4). The enrichment of Pelotomaculaceae in propionate-fed continuous reactors (RP1, RP2) and its consistent presence at high relative abundances in propionate-based batch assays (B01-B04) indicate that members of this family were involved in SPO under the high-ammonia thermophilic conditions in the present study. The 16S rRNA gene sequencing indicated presence of two methanogenic species belonging to the genera Methanoculleus and Methanothermobacter in both acetate- and propionate-fed communities (Fig. 1, S4). These methanogenic genera have previously been suggested to be partners of a thermophilic Pelotomaculum sp. growing in low-ammonia conditions [69]. The absence of aceticlastic methanogens in the propionate-degrading community in the present study demonstrates the importance of the SAOB for acetate removal in high-ammonia conditions. Further detailed information of the sequencing result is given in Supplementary note 5.

Bubble plot showing percentage relative abundance (>2%) of microbial communities at family level in the acetate-fed (RA1, RA2) and propionate-fed (RP1, RP2) enrichment reactors.
Metagenomic binning and metatranscriptomics-based functional analysis
Nine good-quality MAGs were obtained and based on taxonomic annotation and genomic content indicating putative involvement in syntrophic interactions, four of these MAGs were chosen for detailed analyses (Table S7). For instance, the MAGs affiliating to Campylobacteraceae and Ch115, highly abundant in the enrichment cultures, were shown to lack several of the crucial genes required for SAO/SPO-activity and were not included in further analyses. The four MAGs of interest for syntrophic acid degradation and their functional activities and their pathways are described in detail below.
Novel SPOB of family Pelotomaculaceae ‘Candidatus Thermosyntrophopropionicum ammoniitolerans’
A high-quality MAG (MAG4, Table S7) classified to the family Pelotomaculaceae [phylum Bacillota_B, class Desulfotomaculia, order Desulfotomaculales] was recovered in metagenomic sequencing of samples from the propionate-fed reactors. No MAG with similar classification was recovered from the acetate-fed reactors. In a phylogenetic assessment based on whole-genome sequencing, MAG4 clustered together with ‘Ca. Propionivorax syntrophicum’, which further sub-clustered under SPOBs in the family Pelotomaculaceae (Fig. S5). Moreover, in an assessment based on 16S rRNA gene retrieval from MAGs, MAG4 showed a relationship to Pelotomaculum spp. and ‘Ca. Syntrophopropionicum ammoniitolerans’ (Fig. S6).
Comparison of MAG4 with available genomes of Pelotomaculaceae spp. and other known or proposed SPOBs revealed similarities below recommended cut-offs for delineating a new species (i.e. 70% dDDH, 95% ANI, 60% coverage) [70, 71]. MAG4 had highest similarities with ‘Ca. Propionivorax syntrophicum’ (dDDH of 42%, ANI of 90% and AAI of 89%). However, the genome assembly of ‘Ca. Propionivorax syntrophicum’ is of low quality (74.7% completeness), lack the 16S rRNA gene sequence and is considerably smaller (2.0 Mbp) than MAG4 (3.2 Mbp), which obstruct a complete and accurate comparison. The taxonomic analysis against other SPOB revealed highest dDDH with Desulfofundulus thermobenzoicus (24%), AAI with P. thermopropionicum (71%) and ANI with Pelotomaculum schinkii and Pelotomaculum thermopropionicum (74%) (<45% coverage) (Fig. S7). These results indicate that this bacterium will form a novel genus when isolated and characterised and we propose the provisional name ‘Candidatus Thermosyntrophopropionicum ammoniitolerans’.
Methylmalonyl-CoA pathway
MAG4 harboured and expressed a complete set of genes required for propionate degradation through the methylmalonyl-CoA (MMC) pathway (Figs. 2, 3, S8), which strongly suggest that this bacterium can perform syntrophic propionate oxidation in high-ammonia, thermophilic biogas systems. MAG4 expressed CoA-transferase and carboxyltransferase. This indicates that, as in the thermophilic and mesophilic SPOB P. thermopropionicum and P. schinkii [72, 73], MAG4 coupled the two first endergonic steps, propionate activation (step P1 in Fig. 2) and propionyl-CoA carboxylation (P2), with the downstream and exergonic steps forming acetate (P11) and pyruvate (P9), respectively. Other genes encoding enzymes involved in the MMC pathway expressed by MAG4 include methylmalonyl-CoA epimerase, methylmalonyl-CoA mutase and succinate-CoA synthetase (P3-P5). To catalyse the energetically most unfavourable step in the MMC pathway, the oxidation of succinate to fumarate (P6), MAG4 expressed a gene encoding the membrane-bound succinate dehydrogenase/fumarate reductase complex, requiring reducing power via reverse electron transport. Genes encoding fumarate hydratase catalyses the conversion of fumarate to malate (P7) and malate dehydrogenase catalyses the conversion of malate into oxaloacetate (P8) were expressed. MAG4 also contained one Fe-S-containing hydrolyase which was annotated as fumarase/fumarate hydratase in P. thermopropionicum (BAF59538.1). The gene encoding pyruvate carboxylase in step P9 (Fig. 2), i.e. conversion of oxaloacetate to pyruvate, was not found in MAG4. However, the gene encoding methylmalonyl-CoA carboxyltransferase for step P2 (Figs. 2, 3) was expressed and this enzyme has been found to catalyse the conversion of oxaloacetate into pyruvate (KEGG reaction: R00930). For conversion of pyruvate to acetyl-CoA (P10), MAG4 encoded pyruvate:ferredoxin flavodoxin oxidoreductase.

Visualisation of the molecular exchange anchored interplay and metabolic pathways employed by the multiple syntrophic bacteria and their methanogenic partner during syntrophic propionate degradation under thermophilic and high-ammonia conditions. The figure highlighted the cooperation of syntrophic propionate oxidising bacteria (SPOB, MAG4) via acetate assimilation by syntrophic acetate oxidising bacteria (SAOB, MAG9). These SPOB and SAOB further obligately establish formate or hydrogen pivoted syntrophic network to circumvent the reducing potential which is used by hydrogenotrophic methanogen (MAG1) to reduce carbon dioxide and generate methane.

Metatranscriptomics expression profile of the methylmalonyl CoA (MMC) pathway of propionate metabolism (based on transcripts per million (TPM) counts) in propionate versus acetate batch assay (B01, B03 and B09) for the novel syntrophic propionate-oxidising bacteria (SPOB) `Candidatus Thermosyntrophopropionicum ammoniitolerans´ (MAG4). The numerical values with the enzyme name denote the step in the MMC pathway. The values on heatmap represented are the aggregated TPM counts of all copies and subunits for respective gene present and expressed in the metagenome assembled genome.
In MAG4, most of the genes coding for the MMC pathway enzymes were found to be clustered together (except the genes for steps P1 and P6). The gene for propionate-CoA transferase (PCT) was not found in MAG4 (and is also absent in P. thermopropionicum). Instead, MAG4 expressed genes for two other CoA-transferases (acyl/acetate transferase, glutaconate-CoA transferase) that are homologous to PCT and have also been suggested to activate propionate [72, 74, 75]. These CoA-transferases genes were clustered in an operon fashion and highly expressed compared with other flanking genes in MAG4. MAG4 also expressed MMC pathway-associated genes, viz. formate and sodium/solute (propionate) transporters, formate dehydrogenase, methylmalonyl-CoA decarboxylase etc. (Figs. 2, 3). A gene encoding an uncharacterised protein likely involved in propionate catabolism (32% similarity to an uncharacterised protein (BAF60599.1) found in the P. thermopropionicum genome (AP009389.1)) was expressed by MAG4 (Fig. 3). This gene showed 100% similarity and query coverage (Blastx) to MmgE/PrpD family protein (NLW37044.1) belonging to a Peptococcaceae bacterium MAG (JAAYEO000000000.1). The PrpD family is involved in propionate oxidation to pyruvate in E. coli [76], but this protein has still not been annotated or characterised in other anaerobic bacteria. The expression of this gene by MAG4 indicate that this protein might be involved in propionate degradation by an as yet unknown mechanism.
Hydrogen/formate production and energy conservation systems
In SPO, hydrogenases or formate dehydrogenases catalyse electron transfer from NADH or reduced ferredoxin (Fd, generated from substrate oxidation) to the final electron acceptors H+ and CO2 [12, 77]. MAG4 expressed genes for both hydrogenases and formate dehydrogenases (Fig. 3). More specifically, expression of genes for cytoplasmic [FeFe] electron-bifurcating- (HndAC) ([FeFe] group A3), membrane [Fe]-bound [NiFe] and iron-only hydrogenases ([FeFe] group A4) was revealed (Fig. S9). The latter has been shown to couple the endergonic formation of H2 from NADH to its exergonic formation from Fdred [78]. In MAG4, the gene encoding Hnd was found to be flanked by expressed genes for formate dehydrogenase (FDH) and a formate transporter (FdhC) (Fig. 3, S8). It has been speculated that two FDH are needed for syntrophic growth on propionate, one for fixing CO2 by the reductive Wood-Ljungdahl pathway (the membrane-bound FDH1) and one for removal of reducing equivalents as format (the cytoplasmic FDH2) [79, 80]. For P. thermopropionicum, four types of FDH have been found [73] and these can be differentially and independently up- or down-regulated [81]. MAG4 expressed four FDH types (Fig. 3, S8), indicating the possibility that in MAG4, FDH could be utilising reducing equivalents (together with electron transfer/bifurcating flavoproteins and electron transport Rnf complex) to form H2 [82]. Further, formate transporter (transmembrane FocA) could be assisting in mediation of H2/formate-dependent electron sharing or electron bifurcation [83] between MAG4 (as SPOB) (Fig. 3, S9) and the HM, as also reported previously [16].
Genome and transcriptomic analysis of candidate SAOB
For SAO, a distinct candidate (MAG9) was identified by genomic and transcriptomic analysis in both propionate- and acetate-degrading reactor communities. MAG9 was classified to genus DTU068 (95% dDDH similarity with place holder species sp001513545 in the phylum Firmicutes, class Moorellia, family Thermacetogeniaceae, Fig. S10). The taxonomic placement of MAG9 and the transcriptomic data in the propionate- and acetate-fed batch assays strongly indicate that this species represents a novel thermophilic SAOB, related to the mesophilic SAOB Syntrophaceticus schinkii (Figs. S11, S12). However, the genome sequence of MAG9 had high contamination (~15%) thus MAG quality was not sufficiently high for proposal of a provisional name for this species.
MAG9 harboured and expressed a complete set of genes for the Wood-Ljungdahl pathway (WLP) in both propionate and acetate cultures (Fig. 4). The genome revealed a cluster of several WLP genes (steps A3-A6, A12 and A9 in Fig. 2) but for acetate activation (A1-A2) the genes were located separately and the transcriptome data indicated that MAG9 activates acetate through the ATP-consuming acetate kinase. Acetate can potentially also be activated through an ATP-independent aldehyde ferredoxin oxidoreductase followed by oxidation of acetaldehyde to acetyl-CoA, as postulated to be used by Thermacetogenium phaeum to balance the overall ATP budget [84]. Even though MAG9 encoded aldehyde ferredoxin oxidoreductase, the low transcript level of the encoding gene compared with the gene for acetate kinase indicates that, as seen in S. schinkii [85] MAG9 consumes ATP in this first step and forms acetyl-CoA using phosphate acetyltransferase. For the carbonyl branch, CO-methylating acetyl-CoA synthetase and carbon monoxide dehydrogenase were expressed (A4, Figs. 2, 4), whereas expression of corrinoid methyltransferases (A3), methylene tetrahydrofolate (THF) reductase (A5), methylene THF dehydrogenase/cyclohydrolase (A6, A7), formyl THF synthetase (A8) and format dehydrogenase (A9) indicated their importance in operation of the methyl branch (Figs. 2, 4).

Metatranscriptomics expression profile of the Wood-Ljungdahl pathway and other genes of relevance for acetate metabolism (based on transcripts per million (TPM) counts) in propionate versus acetate batch assay (B01, B03 and B09) for the novel syntrophic acetate-oxidising bacteria (SAOB) MAG9 and for MAG5 belonging to the genus Acetomicrobium. The numerical values with the enzyme name denote the step in the pathway. The values on heatmap represented are the aggregated TPM counts of all copies and subunits for respective gene present and expressed in the metagenome assembled genome.
In the direction of acetate oxidation, the methylene THF reductase (A5) releases electrons at a redox potential too low to be used directly for NAD+ reduction [86]. For T. phaeum, this has been proposed to be solved by electron transfer to a methyl-viologen-reducing hydrogenase subunit D (MvrD), followed by heterodisulphide reductase (HdrABC) and further to a quinone, which in turn is re-oxidised by formate dehydrogenase [84]. The gene expression seen for MvhD, HdrABC and NAD-quinone oxidoreductase, the four-iron-four-sulphur (4Fe-4S) cluster and 4Fe-4S ferredoxin by MAG9 indicate that a similar path is followed by this ammonia-tolerant SAOB (Fig. S12). Furthermore, expression of hydrogenase Fe-S, which was found to be encoded next to the genes for step A6 (Figs. 2, 4), indicates importance of electron transport and proton translocation. However, since the pathway proposed for T. phaeum requires establishment of a proton gradient from ATP hydrolysis (reverse electron transport), MAG9 metabolism would not generate enough ATP to drive acetate activation. Thus, further research is needed to confirm the metabolic route used by this and other SAOB. Similarly to S. schinkii and T. phaeum, [84, 85] MAG9 expressed hydrogenase EchCE, formate dehydrogenase and Ni-Fe hydrogenases in both acetate- and propionate-fed batches. MAG9 also expressed a complete set of genes for NADH-quinone oxidoreductase (Fig. S12).
Additional active bacterium in the syntrophic consortia
A MAG (MAG5) belonging to the genus Acetomicrobium (76% [sourmash] and with 84% [dDDH] similarity to GCA_012518015.1) was present and showed activity in both acetate- and propionate-degrading cultures. This species expressed genes encoding the reductive glycine pathway (rGlyP), including the glycine cleavage system, the glycine reductase complex, pyruvate synthase and associated proteins (Fig. 4, S13). A detailed discussion regarding the role of MAG5 is given in Supplementary note 6, with the conclusion that considering the wide range of substrates used by members of Acetomicrobium [87,88,89] and that continuing cultivation of the syntrophic community in the present study demonstrated decreased abundance of Acetomicrobium on omitting yeast extract in the growth media (data not shown), MAG5 most likely fermented compounds included in the yeast extract or grew oxidatively using cysteine as electron acceptor.
Genome and transcriptomics analysis of methanogen
The 16S rRNA gene sequencing analysis of the reactor microbial communities demonstrated higher abundance of Methanothermobacter_NA than Methanoculleus spp. (Fig. S14). The qPCR analyses demonstrated that Methanomicrobiales sp. decreased from 107-8 to 105 gene copies L−1 over the course of operation of the continuously fed reactors. Species belonging to Methanobacteriales were present at relatively stable levels over time, varying between 105 and 107 gene copies L−1 in all reactors (Table S8). However, in the batch assays, 16S rRNA gene amplicon sequencing analysis showed that Methanothermobacter_NA and ‘Ca. Methanoculleus thermohydrogenotrophicum’ were often present in similar relative abundance in batches from RP1. In batches from RP2, ‘Ca. Methanoculleus thermohydrogenotrophicum’ was the only dominant species (Fig. S15). This is in agreement with the qPCR results demonstrating higher abundance of Methanobacteriales in batches with faster propionate degradation (B01-B02, 106 gene copies L-1) than in batches with slower propionate degradation (B03-B04, 104 gene copies L−1) (Table S8). Methanomicrobiales sp. were present at 106 gene copies L−1 in all batch assays. Taxonomic profiling using the custom krakan2 [90] database (Supplementary note 4) with the metagenomics data revealed that ‘Ca. Methanoculleus thermohydrogenotrophicum’ was the most dominant methanogen and that Methanothermobacter sp. was higher in relative abundance in reactor RP1 compared to RP2 (Fig. S16).
Metatranscriptomics results were in agreement with the 16S rRNA gene sequencing results from batch assays (Fig. S15), showing a high number of 16S rRNA transcripts mapped to Methanoculleus and Methanothermobacter, with a particularly high number of reads mapping to Methanothermobacter (default kraken2 database) (Fig. S17, Table S6) in the B01 having higher propionate degradation rate. Although Methanothermobacter was among the dominant methanogenic genera in the 16S rRNA gene sequencing, metagenomics and metatranscriptomics analyses (Figs. S14–S17), we were unfortunately not able to recover a MAG belonging to the genus Methanothermobacter, making it difficult to reveal the activity of this methanogen in the syntrophic cultures. However, a high-quality MAG (MAG1) was recovered and it showed relationship to ‘Ca. Methanoculleus thermohydrogenotrophicum’ (78% dDDH) (GCA_001512375.1) (Figs. S18, S19, Table S6). As reported for the ammonia-tolerant mesophilic Methanoculleus bourgensis [91], methanogenesis pathway genes were found to be clustered together in MAG1 (Fig. S20). Metatranscriptomic data revealed expression of genes by MAG1 coding the transferases, reductases and dehydrogenases needed for hydrogenotrophic metabolism, including methyl CoM-reductase and formylmethanofuran dehydrogenase (Fig. S20). Moreover, MAG1 expressed genes for the V-type ATP synthetase and alcohol dehydrogenases (Fig. S21). The latter has been found previously in Methanoculleus genomes, indicating a trait of using alcohol as electron donor [92]. As in other thermophilic Methanoculleus [69], the electron-bifurcating hydrogenases (coenzyme F420) (FrhABDG) and formate dehydrogenases (FdhABD) were expressed by MAG1 (Fig. S21). MAG1 also expressed genes for hyp type (HypABCDE), which encode proteins for expression and maturation of hydrogenases (Fig. S21). For the metabolic process and biosynthesis, MAG1 encoded and expressed acetate/acyl-CoA ligase (Fig. S21), which uses ATP for activation of acetate to acetyl-CoA. Metatranscriptomics quantification indicated almost identical expression pattern in MAG1 in both the propionate- and acetate-fed batch experiments. Overall, expression of the methanogenesis pathway and associated genes in MAG1 did not appear to give any specific differences in propionate versus acetate treatments that could reveal its partnership with SPOB or SAOB (Figs. S20, S21).
Expression of other genes potentially related to a syntrophic lifestyle
Low energy gain is a well-known challenge and bottleneck for the thermodynamically constrained syntrophic interactions in microbial communities. Hence, it is reasonable to believe that strategies to reduce energy investment in cell metabolism is important for the species involved. To shed light on how the thermophilic ammonia-tolerant syntrophic communities in this study acclimatised to energy scarcity, particular attention was paid to activities with potential to increase energy gain and facilitate interspecies interactions.
For energy production, the candidate syntrophs, MAG4 and MAG9, both expressed the F0F1-type ATP synthase complex (Figs. S9, S12), as reported for known SAOB (S. schinkii and T. phaeum) [84, 85] and SPOB members of Peptococcaceae and Syntrophobacteraceae [10]. Both MAG4 and MAG9 express ATP synthase subunit C to a higher extent than the other subunits. Subunit C has been shown to be crucial for ion translocation that leverages the proton/sodium motive force across the cell membrane and prevents ion leakage [93, 94]. It has also been reported that the number of protons translocated is proportional to the number of subunit C [95] and that regulation of ATP synthase operon is proportional to ATP generation [96,97,98]. This suggests that this type of proton translocation mechanism could also be involved in the bioenergetics of SAO and SPO communities and efficient energy conserving ATP synthesis near thermodynamic equilibrium [97, 98]. Moreover, it has been suggested for the SPOB P. thermopropionicum, P. schinkii and S. fumaroxidans that presence of a reverse electron transfer mechanism, menaquinone loop and higher number of expressed genes encoding hydrogenases and formate dehydrogenase (and associated higher enzymatic and cellular activity) could provide more metabolic agility and flexibility in the case of varying hydrogen or formate consumption by the syntrophic methanogenic partner [72, 98, 99]. In MAG4 and MAG9, these genes are encoded as either more than one copy in the genome, or were highly expressed (or both), which further indicates that the proteins encoded by these genes play a critical role in the complex syntrophic interactions among acetate-/propionate-degrading and methanogenic communities at the thermodynamic borderline (Figs. 3, 4, S9, S12, Table S6).
For initiation of syntrophic oxidation, the SPOB and SAOB need to transport the substrate across the cell membrane, which can be done actively using a transport system or through passive diffusion [10]. The candidate SPOB MAG4 expressed sodium/solute transporter and both MAG4 and the candidate SAOB MAG9 expressed MFS transporter proteins for putative propionate intake (Figs. S9, S12). MFS transporters are broad-spectrum transport systems involved in uniport, symport or antiport of various cellular metabolites, sugars and organic acids [100] and have been reported to play a role in tolerance to high levels of acetate and propionate, for example in Acetobacter spp., E. coli and P. putida [101, 102]. Moreover, a gene belonging to the oxalate/formate antiporter (OFA) family of MFS transporters was located in the operon together with CoA-transferases and showed higher expression by MAG4 in the culture with faster propionate oxidisation relative to the culture with slower propionate degradation (B01 vs. B03, Fig. S9). This suggests that MFS transporters maybe responsible for acetate/propionate or formate transport in MAG4.
The role of sulphur compounds in metabolic cooperation
The transcriptome data revealed activity related to sulphur metabolism by the candidate SPOB (Fig. S9), including Hdr and CoA-disulphide reductase (cdr). The Hdr gene complex has previously been found to assist in electron confurcation in S. fumaroxidans [99]. Moreover, MAG4 expressed genes for dissimilatory sulphite reductase (dsrC) and anaerobic sulphite reductase (asrAB), which are the key determinants for sulphur reduction-based energy conservation in sulphate-reducing bacteria [99, 103]. In MAG4, these sulphite reductase genes are encoded next to expressed putative NADH:ubiquinone oxidoreductase (nfC) and a mvhD, which are likely involved in electron transport phosphorylation or hydrogenase activities. Other important genes involved in dissimilatory sulphate reduction (adenylylsulphate reductase (aprAB), sulphate adenylyltransferase (sat), pyrophosphatase (ppaX), ABC-type sulphate transporter) are also encoded by MAG4, but were not highly expressed under the conditions investigated here. However, presence of all the genes required for sulphate reduction in MAG4 strongly indicates that this bacterium has the ability to respire sulphate if available. The ability of MAG4 to perform sulphate reduction and the exact mechanisms involved warrant further investigation, since this would improve understanding of another thermophilic SPOB, P. thermopropionicum. It is suggested to be a sulphate/thiosulphate/sulphite reducer [104], even though it has been described as unable to utilise sulphate due to absence of aprB and dsrAB genes [105, 106].
Similar sulphur metabolism potential as observed for MAG4 was observed for ‘Ca. Propionivorax syntrophicum’, possibly as a step in a series of reactions for sulphate-reducing metabolism, and a complex of hdr, rnfC and dsr, which could provide reduced ferredoxin for H2/formate production and also for low-energy metabolism [16, 99]. However, the cultivation medium in the present study contained no sulphate or sulphite, contradicting the suggestion that the enzymes are involved in metabolism of these sulphur compounds. Instead, the cultivation medium included Na2S, cysteine, yeast extract and sulphur-containing vitamins, i.e. biotin, thioctic acid and thiamine, which might give rise to hydrogen sulphide and other sulphur compounds [107, 108]. Many bacteria can also produce different di- and trisulphides [109, 110]. Furthermore, as discussed above, MAG5 was taxonomically related to species that can reduce cysteine to sulphide, indicating similar activity by MAG5 in the enrichment culture. MAG4 also expressed cysteine desulfurase (icsS), which is responsible for sulphur activation in the cysteine degradation pathway. However, it might also be involved in formation of amino acids, as observed for the SAOB Schnuerera ultunensis, in which this gene is associated with production of alanine and sulphane/persulphide sulphur intermediates from cysteine degradation [111,112,113]. Apart from cysteine serving as a reducing agent in the medium, it may also mediate the electron carrier in SPO and subsequent methanogenesis [114]. Protein trisulphides are of interest in the present context since they are involved in sulphur reduction machinery-based energy conservation. The DsrC associated protein trisulphide (metacycM:DsrC-trisulphides) can act as a key intermediate in the reversible redox reaction producing and consuming sulphite [103, 104]. Expression of asrAB and dsrC is a key determinant for sulphur reduction-based energy conservation in sulphate-reducing organisms [103]. Considering that several sulphate-reducing organisms have been found to establish syntrophic interactions in environments where sulphur is absent or limited [115], sulphate-reducing metabolic potential of MAG4 is further indicated. The genes for the sulphate-reducing pathway, together with different hydrogenases, could possibly also be involved in low-energy metabolism rather than sulphate reduction [99].
Mobility and other features with potential to facilitate interspecies cooperation
MAG4 and MAG9 contained six and 23 motility associated proteins, respectively (Figs. S22, S23). One of the pilus-associated proteins in MAG4 is PilT (type IV pili) (Figs. S22, S23), which has been found to be associated with twitching motility, cellular adhesion, pilus retraction and sequence-specific DNA uptake [116, 117]. Further, MAG4, MAG5 and MAG9 all expressed TIGR00282 family metallophosphoesterase proteins which were similar to YmdB, characterised for its role in nanotube and biofilm formation and intercellular molecular exchange in Bacillus subtilis [118,119,120,121]. Different types of flagellar and pilus-related proteins are known to play a role in initiating cellular contact, biofilm formation and establishing syntrophy. For instance, P. thermopropionicum FliD is used to establish contact with the partner methanogen M. thermautotrophicus and to synchronise their metabolism [122, 123]. Considering the absence of the FliD gene in MAG4, it is likely that if this candidate SPOB uses direct interspecies electron transfer (DIET), it is employing a mechanism somewhat different from those characterised previously. Further, MAG4 and MAG9 expressed genes for cysteine synthase/O-acetylserine sulfhydrolase (CysK) and stage 0 sporulation protein (Spo0A) (Fig. S23), which can be involved in biofilm formation, as suggested for Vibrio fischeri (CysK) [124] and B. subtilis and Clostridium difficile (Spo0A) [125, 126]. This perhaps explains the absence of motility-related (flagella) proteins and strongly suggests that MAG4 and MAG9 use pilus appendages for physically establishing deep physical contact with each other and the syntrophic methanogenic partner when present in close proximity (PilT-mediated) or for biofilm formation or nanotube communication and intercellular molecular exchange [118, 127, 128]. This feature would resemble that in P. thermopropionicum, which is characterised for biofilm and nanowire formation and interspecies electron and hydrogen sharing when growing in syntrophy with Methanothermobacter [123, 129, 130]. Cross-cellular communication, signalling and quorum sensing is another important concept intrinsic to syntrophic associations. Quorum sensing and signalling mechanism-related genes were expressed in all MAGs (Fig. S23). These genes, together with other associated genes, e.g. for motility, signalling, biofilm formation have been shown to be involved in DIET, intercellular metabolite exchange and communications [131]. The exact mechanism of the cooperation (e.g. establishment of nanotubes, use of flagellar or pilar assemblies) used by the syntrophic bacteria and the methanogens warrants further investigation.
Stress response
Expression of genes related to stress response, viz. chaperones (DnaJ, DnaK, ClpB, different chaperonins), heat shock protein (Hsp20) and hyperosmotic response (GrpE), are important for stress tolerance [132, 133], was seen for MAG1 MAG4 and MAG9 (Fig. S24). Ammonia tolerance and resistance is a physiological phenomenon rather than a genetic property. Several complex mechanisms, i.e. osmo-tolerance, ionic membrane transport, molecular chaperones. etc. impart physiological resistance to evade metabolic deterioration under ammonia stress [132,133,134,135]. These stress genes have been found to be upregulated under acetate/acetic acid stress in E. coli [101]. Expression of stress-related proteins in MAG1, MAG4 and MAG9 could be due to the thermophilic temperature and high ammonia concentration applied in this study (Fig. S24). The transcriptomic response of stress-related genes of SPOB has not been characterised, so the exact role of these stress-regulated genes in MAG4 requires further investigation. Several genes in the candidate SPOB (MAG4), SAOB (MAG9) and the HM (MAG1), as also discussed above, were found to be present and expressed in an operon-like fashion (e.g. MMC, WLP, HM pathway genes, CoA-transferases, hydrogenases/dehydrogenases). The clustering of genes in SPOB (Pelotomaculaceae family, reviewed elsewhere [10]) has been proposed to enable energetically advantageous coordinated expression of series of genes, since it requires less transcriptional machinery [73, 136]. Similar to SPOB, the present results also indicate that the coordinated expression of series of genes energetically beneficial to ammonia-tolerant SAOB and methanogens.
Conclusions
Use of a long-term enrichment approach to increase the abundance of an ammonia-tolerant syntrophic propionate-degrading community made it possible to identify key species and their metabolic activities, and to distinguish activities potentially related to the syntrophic lifestyle. Two novel ammonia-tolerant and thermophilic syntrophic species were identified, and we propose the name ‘Candidatus Thermosyntrophopropionicum ammoniitolerans’ for the SPOB. Batch cultivation, 16S rRNA gene analyses (sequencing and expression) and qPCR analysis indicated that Methanothermobacter could be crucial for syntrophic methanogenesis from propionate. Similar dependence for acetate degradation was not observed, indicating that the SAOB cooperated well with the Methanoculleus sp. also present in the syntrophic communities. Transcriptome data revealed activity related to sulphur metabolism, intercellular contact and molecular exchange by pili/flagellar appendages and nanotubes by the candidate SPOB, which can be crucial for efficient interdependent metabolism in a thermodynamically unfavourable environment. An additional bacterial species in the syntrophic community displayed activity for the reductive glycine pathway, but the wide substrate span of related bacterium and decreased abundance during cultivation without yeast extract suggest that this species is not directly involved in acid degradation. Thus, caution is needed when claiming that the reductive glycine pathway can be operated in the oxidative direction by SAOB to oxidise acetate. A deeper understanding of important syntrophic players and their mutualistic interactions under high-ammonia conditions is key for optimal design of anaerobic processes degrading protein-rich biomass. Future work should focus on identifying and characterising the functional interactions of ammonia-tolerant, thermophilic VFA-oxidising and methanogenic syntrophic communities as a model, which would be a milestone in metabolic modelling and systems biological approaches to anaerobic digester systems. With enhanced understanding of syntrophic synergy and coupled metabolic networks, industrial reactor operation can be steered to obtain higher efficiency and productivity of the methanogenic process.
Data availability
The data availability is described in Supplementary Note 7.
References
WBA. The contribution of anaerobic digestion and biogas towards achieving the UN Sustainable Development Goals. World Biogas Association. 2018. World Biogas Association (WBA), London; Available at: https://www.worldbiogasassociation.org/wp-content/uploads/2018/12/WBA_SDG_Biogas_Report.pdf.
De Vrieze J, Verstraete W. Perspectives for microbial community composition in anaerobic digestion: from abundance and activity to connectivity. Environ Microbiol. 2016;18:2797–809.
Şengör SS, Ginn TR, Brugato CJ, Gikas P. Anaerobic microbial growth near thermodynamic equilibrium as a function of ATP/ADP cycle: the effect of maintenance energy requirements. Biochem Eng J. 2013;81:65–72.
Westerholm M, Schnürer A. Microbial responses to different operating practices for biogas production systems. In: Banu R (ed). Anaerobic Digestion. IntechOpen. 2019. https://www.intechopen.com/chapters/65614.
Gallert C, Winter J. Propionic acid accumulation and degradation during restart of a full-scale anaerobic biowaste digester. Bioresour Technol. 2008;99:170–8.
Zhang C, Yuan Q, Lu Y. Inhibitory effects of ammonia on syntrophic propionate oxidation in anaerobic digester sludge. Water Res. 2018;146:275–87.
Westerholm M, Moestedt J, Schnürer A. Biogas production through syntrophic acetate oxidation and deliberate operating strategies for improved digester performance. Appl Energy. 2016;179:124–35.
Li Y, Zhang Y, Kong X, Li L, Yuan Z, Dong R, et al. Effects of ammonia on propionate degradation and microbial community in digesters using propionate as a sole carbon source. J Chem Technol Biotechnol. 2017;92:2538–45.
Wang H-Z, Yan Y-C, Gou M, Yi Y, Xia Z-Y, Nobu MK, et al. Response of propionate-degrading methanogenic microbial communities to inhibitory conditions. Appl Biochem Biotechnol. 2019;189:233–48.
Westerholm M, Calusinska M, Dolfing J. Syntrophic propionate-oxidizing bacteria in methanogenic systems. FEMS Microbiol Rev. 2022;46:fuab057.
Kutscha R, Pflügl S. Microbial upgrading of acetate into value-added products - examining microbial diversity, bioenergetic constraints and metabolic engineering approaches. Int J Mol Sci. 2020;21:8777.
Stams AJM, Plugge CM. Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat Rev Microbiol. 2009;7:568–77.
Singh A, Schnürer A, Westerholm M. Enrichment and description of novel bacteria performing syntrophic propionate oxidation at high ammonia level. Environ Microbiol. 2021;23:1620–37.
Pelletier E, Kreimeyer A, Bocs S, Rouy Z, Gyapay G, Chouari R, et al. “Candidatus Cloacamonas Acidaminovorans”: genome sequence reconstruction provides a first glimpse of a new bacterial division. J Bacteriol. 2008;190:2572–9.
Dyksma S, Gallert C. Candidatus Syntrophosphaera thermopropionivorans: a novel player in syntrophic propionate oxidation during anaerobic digestion. Environ Microbiol Rep. 2019;11:558–70.
Hao L, Michaelsen TY, Singleton CM, Dottorini G, Kirkegaard RH, Albertsen M, et al. Novel syntrophic bacteria in full-scale anaerobic digesters revealed by genome-centric metatranscriptomics. ISME J. 2020;14:906–18.
Westerholm M, Roos S, Schnürer A. Syntrophaceticus schinkii gen. nov., sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from a mesophilic anaerobic filter. FEMS Microbiol Lett. 2010;309:100–4.
Westerholm M, Liu T, Schnürer A. Comparative study of industrial-scale high-solid biogas production from food waste: process operation and microbiology. Bioresour Technol. 2020;304:122981.
Westerholm M, Müller B, Singh A, Karlsson Lindsjö O, Schnürer A. Detection of novel syntrophic acetate-oxidizing bacteria from biogas processes by continuous acetate enrichment approaches. Micro Biotechnol. 2018;11:680–93.
Westerholm M, Dolfing J, Schnürer A. Growth characteristics and thermodynamics of syntrophic acetate oxidizers. Environ Sci Technol. 2019;53:5512–20.
Bushnell B. BBMap: A Fast, Accurate, Splice-Aware Aligner. https://sourceforge.net/projects/bbmap/. 2014.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria; http://www.r-project.org. 2021.
Alishum A, Greenfield P, Seersholm F, Christophersen C. DADA2 formatted 16S rRNA gene sequences for both bacteria & archaea. Zenodo. https://doi.org/10.5281/zenodo.4735821. 2021.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217.
RStudio Team. RStudio: Integrated Development Environment for R. RStudio, PBC., http://www.rstudio.com. 2020.
Westerholm M, Dolfing J, Sherry A, Gray ND, Head IM, Schnürer A. Quantification of syntrophic acetate‐oxidizing microbial communities in biogas processes. Environ Microbiol Rep. 2011;3:500–5.
Yu Y, Lee C, Kim J, Hwang S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng. 2005;89:670–9.
Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37:540–6.
Lin Y, Yuan J, Kolmogorov M, Shen MW, Chaisson M, Pevzner PA. Assembly of long error-prone reads using de Bruijn graphs. Proc Natl Acad Sci USA. 2016;113:E8396–E8405.
Vaser R, Sović I, Nagarajan N, Šikić M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27:737–46.
Oxford Nanopore Technologies. Medaka: a tool to create consensus sequences and variant calls from nanopore sequencing data. https://github.com/nanoporetech/medaka/. 2020.
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963.
Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6:158.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36:1925–7.
Parks DH, Chuvochina M, Chaumeil P-A, Rinke C, Mussig AJ, Hugenholtz P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol. 2020;38:1079–86.
Schwengers O, Jelonek L, Dieckmann MA, Beyvers S, Blom J, Goesmann A. Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Micro Genom. 2021;7:000685.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9.
Søndergaard D, Pedersen CNS, Greening C. HydDB: a web tool for hydrogenase classification and analysis. Sci Rep. 2016;6:34212.
Meier-Kolthoff JP, Carbasse JS, Peinado-Olarte RL, Göker M. TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022;50:D801–7.
Pritchard L, Glover RH, Humphris S, Elphinstone JG, Toth IK. Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal Methods. 2016;8:12–24.
Parks D. CompareM: a software toolkit which supports performing large-scale comparative genomic analyses. https://github.com/dparks1134/CompareM. 2020.
Emms DM, Kelly S. STAG: species tree inference from all genes. bioRxiv - Prepr Serv Biol. 2018;1:1–29.
Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238.
Wickham H. ggplot2, 2nd ed. Use R!. Springer International Publishing, http://link.springer.com/10.1007/978-3-319-24277-4. 2016.
Rambaut A. FigTree: a graphical viewer of phylogenetic trees. 2009. Institute of Evolutionary Biology, Department of Molecular Evolution, Phylogenetics and Epidemiology, The university of Edinburgh; v1.4.4; Date released: 2018-11-26; http://tree.bio.ed.ac.uk/software/figtree/.
Zymo Research. Quick-RNATM Fecal/Soil Microbe Microprep. Zymo Research, Cat. no. R2040, v2.0.2. 2022;2:1–13. Available at: https://files.zymoresearch.com/protocols/_r2040_quick-rna_fecal-soil_microbe_microprep_kit.pdf.
SitoolsBiotech. Pan-Prokaryote riboPOOL. Cat. no. dp-K024-000003, v1-6. 2019;1:1–15: Available at: https://www.sitoolsbiotech.com/pdf/riboPOOL-Protocol_v1-6.pdf.
NGI. The SNP&SEQ Technology Platform. National Genomics Infrastructure, Uppsala University; https://snpseq.medsci.uu.se/. 2022.
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Kolde R. Pheatmap: pretty heatmaps. R Pacakage Version. 2012. Comprehensive R Archive Network (CRAN); https://cran.r-project.org/package=pheatmap.
Capson-Tojo G, Moscoviz R, Astals S, Robles Á, Steyer J-P. Unraveling the literature chaos around free ammonia inhibition in anaerobic digestion. Renew Sustain Energy Rev. 2020;117:109487.
Lee MJ, Zinder SH. Hydrogen partial pressures in a thermophilic acetate-oxidizing methanogenic coculture. Appl Environ Microbiol. 1988;54:1457–61.
Hattori S, Luo H, Shoun H, Kamagata Y. Involvement of formate as an interspecies electron carrier in a syntrophic acetate-oxidizing anaerobic microorganism in coculture with methanogens. J Biosci Bioeng. 2001;91:294–8.
Scholten JCM, Conrad R. Energetics of syntrophic propionate oxidation in defined batch and chemostat cocultures. Appl Environ Microbiol. 2000;66:2934–42.
Zhao J, Westerholm M, Qiao W, Yin D, Bi S, Jiang M, et al. Impact of temperature and substrate concentration on degradation rates of acetate, propionate and hydrogen and their links to microbial community structure. Bioresour Technol. 2018;256:44–52.
Bonk F, Popp D, Weinrich S, Sträuber H, Kleinsteuber S, Harms H, et al. Ammonia inhibition of anaerobic volatile fatty acid degrading microbial communities. Front Microbiol. 2018;9:02921.
Khafipour A, Jordaan EM, Flores-Orozco D, Khafipour E, Levin DB, Sparling R, et al. Response of microbial community to induced failure of anaerobic digesters through overloading with propionic acid followed by process recovery. Front Bioeng Biotechnol. 2020;8:604838.
Han Y, Green H, Tao W. Reversibility of propionic acid inhibition to anaerobic digestion: inhibition kinetics and microbial mechanism. Chemosphere. 2020;255:126840.
Li Y, Sun Y, Li L, Yuan Z. Acclimation of acid-tolerant methanogenic propionate-utilizing culture and microbial community dissecting. Bioresour Technol. 2018;250:117–23.
Lee C, Kim J, Shin SG, Hwang S. Monitoring bacterial and archaeal community shifts in a mesophilic anaerobic batch reactor treating a high-strength organic wastewater. FEMS Microbiol Ecol. 2008;65:544–54.
Wang H, Zhang Y, Angelidaki I. Ammonia inhibition on hydrogen enriched anaerobic digestion of manure under mesophilic and thermophilic conditions. Water Res. 2016;105:314–9.
Kim J, Jung H, Lee C. Shifts in bacterial and archaeal community structures during the batch biomethanation of Ulva biomass under mesophilic conditions. Bioresour Technol. 2014;169:502–9.
Kim M, Ahn Y-H, Speece R. Comparative process stability and efficiency of anaerobic digestion; mesophilic vs. thermophilic. Water Res. 2002;36:4369–85.
Gallert C, Bauer S, Winter J. Effect of ammonia on the anaerobic degradation of protein by a mesophilic and thermophilic biowaste population. Appl Microbiol Biotechnol. 1998;50:495–501.
Kaspar HF, Wuhrmann K. Product inhibition in sludge digestion. Micro Ecol. 1977;4:241–8.
Chen Y-T, Zeng Y, Wang H-Z, Zheng D, Kamagata Y, Narihiro T, et al. Different interspecies electron transfer patterns during mesophilic and thermophilic syntrophic propionate degradation in chemostats. Micro Ecol. 2020;80:120–32.
Varghese NJ, Mukherjee S, Ivanova N, Konstantinidis KT, Mavrommatis K, Kyrpides NC, et al. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 2015;43:6761–71.
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91.
Hidalgo‐Ahumada CAP, Nobu MK, Narihiro T, Tamaki H, Liu W, Kamagata Y, et al. Novel energy conservation strategies and behaviour of Pelotomaculum schinkii driving syntrophic propionate catabolism. Environ Microbiol. 2018;20:4503–11.
Kato S, Kosaka T, Watanabe K. Substrate-dependent transcriptomic shifts in Pelotomaculum thermopropionicum grown in syntrophic co-culture with Methanothermobacter thermautotrophicus. Micro Biotechnol. 2009;2:575–84.
Kosaka T, Uchiyama T, Ishii S, Enoki M, Imachi H, Kamagata Y, et al. Reconstruction and regulation of the central catabolic pathway in the thermophilic propionate-oxidizing syntroph Pelotomaculum thermopropionicum. J Bacteriol. 2006;188:202–10.
Lindenkamp N, Schürmann M, Steinbüchel A. A propionate CoA-transferase of Ralstonia eutropha H16 with broad substrate specificity catalyzing the CoA thioester formation of various carboxylic acids. Appl Microbiol Biotechnol. 2013;97:7699–709.
Brock M, Maerker C, Schütz A, Völker U, Buckel W. Oxidation of propionate to pyruvate in Escherichia coli. Eur J Biochem. 2002;269:6184–94.
Sieber JR, McInerney MJ, Gunsalus RP. Genomic insights into syntrophy: the paradigm for anaerobic metabolic cooperation. Annu Rev Microbiol. 2012;66:429–52.
Schut GJ, Adams MWW. The Iron-Hydrogenase of Thermotoga maritima Utilizes Ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J Bacteriol. 2009;191:4451–7.
de Bok FAM, Hagedoorn P-L, Silva PJ, Hagen WR, Schiltz E, Fritsche K, et al. Two W-containing formate dehydrogenases (CO2-reductases) involved in syntrophic propionate oxidation by Syntrophobacter fumaroxidans. Eur J Biochem. 2003;270:2476–85.
de Bok FAM, Roze EHA, Stams AJM. Hydrogenases and formate dehydrogenases of Syntrophobacter fumaroxidans. Antonie Van Leeuwenhoek. 2002;81:283–91.
Liu P, Lu Y. Concerted metabolic shifts give new insights into the syntrophic mechanism between propionate-fermenting Pelotomaculum thermopropionicum and hydrogenotrophic Methanocella conradii. Front Microbiol. 2018;9:01551.
Schuchmann K, Chowdhury NP, Müller V. Complex Multimeric [FeFe] Hydrogenases: biochemistry, physiology and new opportunities for the hydrogen economy. Front Microbiol. 2018;9:02911.
Wang S, Huang H, Kahnt J, Thauer RK. Clostridium acidurici electron-bifurcating formate dehydrogenase. Appl Environ Microbiol. 2013;79:6176–9.
Keller A, Schink B, Müller N. Alternative pathways of acetogenic ethanol and methanol degradation in the thermophilic anaerobe Thermacetogenium phaeum. Front Microbiol. 2019;10:1–15.
Manzoor S, Bongcam-Rudloff E, Schnürer A, Müller B. Genome-guided analysis and whole transcriptome profiling of the mesophilic syntrophic acetate oxidising bacterium Syntrophaceticus schinkii. PLoS One. 2016;11:e0166520.
Bertsch J, Öppinger C, Hess V, Langer JD, Müller V. Heterotrimeric NADH-oxidizing methylenetetrahydrofolate reductase from the acetogenic bacterium Acetobacterium woodii. J Bacteriol. 2015;197:1681–9.
Rees GN, Patel BKC, Grassia GS, Sheehy AJ. Anaerobaculum thermoterrenum gen. nov., sp. nov., a novel, thermophilic bacterium which ferments citrate. Int J Syst Bacteriol. 1997;47:150–4.
Hania WB, Bouanane-Darenfed A, Cayol J-L, Ollivier B, Fardeau ML. Reclassification of Anaerobaculum mobile, Anaerobaculum thermoterrenum, Anaerobaculum hydrogeniformans as Acetomicrobium mobile comb. nov., Acetomicrobium thermoterrenum comb. nov. and Acetomicrobium hydrogeniformans comb. nov., respectively, and emendation of the genus Acetomicrobium. Int J Syst Evol Microbiol. 2016;66:1506–9.
Menes RJ, Muxí L. Anaerobaculum mobile sp. nov., a novel anaerobic, moderately thermophilic, peptide-fermenting bacterium that uses crotonate as an electron acceptor, and emended description of the genus Anaerobaculum. Int J Syst Evol Microbiol. 2002;52:157–64.
Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257.
Manzoor S, Schnürer A, Bongcam-Rudloff E, Müller B. Complete genome sequence of Methanoculleus bourgensis strain MAB1, the syntrophic partner of mesophilic acetate-oxidising bacteria (SAOB). Stand Genom Sci. 2016;11:80.
Berk H, Thauer RK. Function of coenzyme F420-dependent NADP reductase in methanogenic archaea containing an NADP-dependent alcohol dehydrogenase. Arch Microbiol. 1997;168:396–402.
Stock D, Gibbons C, Arechaga I, Leslie AGW, Walker JE. The rotary mechanism of ATP synthase. Curr Opin Struct Biol. 2000;10:672–9.
Nakamoto RK, Baylis Scanlon JA, Al-Shawi MK. The rotary mechanism of the ATP synthase. Arch Biochem Biophys. 2008;476:43–50.
Nakanishi-Matsui M, Futai M. Stochastic rotational catalysis of proton pumping F-ATPase. Philos Trans R Soc B Biol Sci. 2008;363:2135–42.
Zhou J, He Q, Hemme CL, Mukhopadhyay A, Hillesland K, Zhou A, et al. How sulphate-reducing microorganisms cope with stress: lessons from systems biology. Nat Rev Microbiol. 2011;9:452–66.
Hoehler T. Apparent minimum free energy requirements for methanogenic Archaea and sulfate-reducing bacteria in an anoxic marine sediment. FEMS Microbiol Ecol. 2001;38:33–41.
Müller N, Worm P, Schink B, Stams AJM, Plugge CM. Syntrophic butyrate and propionate oxidation processes: from genomes to reaction mechanisms. Environ Microbiol Rep. 2010;2:489–99.
Sedano-Núñez VT, Boeren S, Stams AJM, Plugge CM. Comparative proteome analysis of propionate degradation by Syntrophobacter fumaroxidans in pure culture and in coculture with methanogens. Environ Microbiol. 2018;20:1842–56.
Saier MH. Molecular phylogeny as a basis for the classification of transport proteins from bacteria, Archaea and Eukarya. In: Poole RK (ed). Advances in Microbial Physiology. 1998. Academic Press, pp 81–136.
Trček J, Mira NP, Jarboe LR. Adaptation and tolerance of bacteria against acetic acid. Appl Microbiol Biotechnol. 2015;99:6215–29.
Ma C, Mu Q, Xue Y, Xue Y, Yu B, Ma Y. One major facilitator superfamily transporter is responsible for propionic acid tolerance in Pseudomonas putida KT2440. Micro Biotechnol. 2021;14:386–91.
Santos AA, Venceslau SS, Grein F, Leavitt WD, Dahl C, Johnston DT, et al. A protein trisulfide couples dissimilatory sulfate reduction to energy conservation. Science (80-). 2015;350:1541–5.
Venceslau SS, Stockdreher Y, Dahl C, Pereira IAC. The “bacterial heterodisulfide” DsrC is a key protein in dissimilatory sulfur metabolism. Biochim Biophys Acta - Bioenerg. 2014;1837:1148–64.
Visser M, Worm P, Muyzer G, Pereira IAC, Schaap PJ, Plugge CM, et al. Genome analysis of Desulfotomaculum kuznetsovii strain 17T reveals a physiological similarity with Pelotomaculum thermopropionicum strain SIT. Stand Genom Sci. 2013;8:69–87.
Imachi H. Pelotomaculum thermopropionicum gen. nov., sp. nov., an anaerobic, thermophilic, syntrophic propionate-oxidizing bacterium. Int J Syst Evol Microbiol. 2002;52:1729–35.
Ames JM. Volatile Sulfur Compounds in Yeast Extracts. Sulfur Compounds in Foods. 1994. American Chemical Society, pp 12–147.
Morfee TD, Liska BJ. Elemental sulfur: degradation product of thiamine in model systems. J Dairy Sci. 1972;55:123–5.
Seefeldt KE, Weimer BC. Diversity of sulfur compound production in lactic acid bacteria. J Dairy Sci. 2000;83:2740–6.
Schöller C, Molin S, Wilkins K. Volatile metabolites from some gram-negative bacteria. Chemosphere. 1997;35:1487–95.
Wei Y, Wu Y, Zhang L, Zhou Z, Zhou H, Yan X. Genome recovery and metatranscriptomic confirmation of functional acetate-oxidizing bacteria from enriched anaerobic biogas digesters. Environ Pollut. 2020;265:114843.
Schnürer A, Schink B, Svensson BH. Clostridium ultunense sp. nov., a mesophilic bacterium oxidizing acetate in syntrophic association with a hydrogenotrophic methanogenic bacterium. Int J Syst Bacteriol. 1996;46:1145–52.
Dos Santos PC. B. subtilis as a model for studying the assembly of Fe–S clusters in gram-positive bacteria. Methods Enzymol. 2017:185–212.
Zhuang L, Ma J, Tang J, Tang Z, Zhou S. Cysteine-accelerated methanogenic propionate degradation in paddy soil enrichment. Micro Ecol. 2017;73:916–24.
Martins M, Pereira IAC. Sulfate-reducing bacteria as new microorganisms for biological hydrogen production. Int J Hydrog Energy. 2013;38:12294–301.
Bertrand JJ, West JT, Engel JN. Genetic analysis of the regulation of type IV Pilus function by the Chp chemosensory system of Pseudomonas aeruginosa. J Bacteriol. 2010;192:994–1010.
Wolfgang M, Lauer P, Park H, Brossay L, Hébert J, Koomey M. PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Mol Microbiol. 1998;29:321–30.
Baidya AK, Bhattacharya S, Dubey GP, Mamou G, Ben-Yehuda S. Bacterial nanotubes: a conduit for intercellular molecular trade. Curr Opin Microbiol. 2018;42:1–6.
Dubey GP, Malli Mohan GB, Dubrovsky A, Amen T, Tsipshtein S, Rouvinski A, et al. Architecture and characteristics of bacterial nanotubes. Dev Cell. 2016;36:453–61.
Zhang J, Wang H, Xie T, Huang Q, Xiong X, Liu Q, et al. The YmdB protein regulates biofilm formation dependent on the repressor SinR in Bacillus cereus 0–9. World J Microbiol Biotechnol. 2020;36:165.
Diethmaier C, Newman JA, Kovacs AT, Kaever V, Herzberg C, Rodrigues C, et al. The YmdB phosphodiesterase is a global regulator of late adaptive responses in Bacillus subtilis. J Bacteriol. 2014;196:265–75.
Shimoyama T, Kato S, Ishii S, Watanabe K. Flagellum mediates symbiosis. Science (80-). 2009;323:1574.
Ishii S, Kosaka T, Hori K, Hotta Y, Watanabe K. Coaggregation facilitates interspecies hydrogen transfer between Pelotomaculum thermopropionicum and Methanothermobacter thermautotrophicus. Appl Environ Microbiol. 2005;71:7838–45.
Singh P, Brooks JF, Ray VA, Mandel MJ, Visick KL. CysK plays a role in biofilm formation and colonization by Vibrio fischeri. Appl Environ Microbiol. 2015;81:5223–34.
Dawson LF, Valiente E, Faulds-Pain A, Donahue EH, Wren BW. Characterisation of Clostridium difficile Biofilm Formation, a Role for Spo0A. PLoS One. 2012;7:e50527.
Hamon MA, Lazazzera BA. The sporulation transcription factor Spo0A is required for biofilm development in Bacillus subtilis. Mol Microbiol. 2002;42:1199–209.
Benomar S, Ranava D, Cárdenas ML, Trably E, Rafrafi Y, Ducret A, et al. Nutritional stress induces exchange of cell material and energetic coupling between bacterial species. Nat Commun. 2015;6:6283.
Pande S, Shitut S, Freund L, Westermann M, Bertels F, Colesie C, et al. Metabolic cross-feeding via intercellular nanotubes among bacteria. Nat Commun. 2015;6:6238.
Gorby YA, Yanina S, McLean JS, Rosso KM, Moyles D, Dohnalkova A, et al. Electrically conductive bacterial nanowires produced by Shewanella oneidensis strain MR-1 and other microorganisms. Proc Natl Acad Sci. 2006;103:11358–63.
Hayes CS, Low DA. Signals of growth regulation in bacteria. Curr Opin Microbiol. 2009;12:667–73.
Yin Q, Gu M, Hermanowicz SW, Hu H, Wu G. Potential interactions between syntrophic bacteria and methanogens via type IV pili and quorum-sensing systems. Environ Int. 2020;138:105650.
Hill C, Cotter PD, Sleator RD, Gahan CGM. Bacterial stress response in Listeria monocytogenes: jumping the hurdles imposed by minimal processing. Int Dairy J. 2002;12:273–83.
de Macario EC, Macario AJL. Molecular biology of stress genes in methanogens: Potential for bioreactor technology. Adv Biochem Engineering/biotechnol. 2003;81:95–150.
Sayavedra-Soto L, Ferrell R, Dobie M, Mellbye B, Chaplen F, Buchanan A, et al. Nitrobacter winogradskyitranscriptomic response to low and high ammonium concentrations. FEMS Microbiol Lett. 2015;362:1–7.
Csonka LN. Physiological and genetic responses of bacteria to osmotic stress. Microbiol Rev. 1989;53:121–47.
Kato S, Watanabe K. Ecological and evolutionary interactions in syntrophic methanogenic consortia. Microbes Environ. 2010;25:145–51.
Acknowledgements
We thank Simon Isaksson for assistance in operating the reactors and Professor (Emeritus) Aharon Oren for Latin assistance for the candidatus name.
Funding
Funding was provided by HR2020 ERC - Grant European Research Council (grant number 948138), the Swedish Research Council (Formas, grant number 2018-00928) and the Swedish University of Agricultural Sciences. Open access funding provided by Swedish University of Agricultural Sciences.
Author information
Authors and Affiliations
Contributions
MW conceived and designed the study and supervised the project; AbS, MW operated the reactors; AbS, AnS, MW conducted the batch experiments; MW conducted 16S rRNA gene, qPCR and metagenomic analyses; AbS conducted the metatranscriptomic analyses, analysed the 16S rRNA gene sequencing, metagenomic and metatranscriptomic data and generated the figures visualizing the results; JD conducted thermodynamic calculations; AbS, AnS, JD, MW interpreted the data; AbS, MW wrote the manuscript with input from all coauthors; MW, AnS contributed to funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, A., Schnürer, A., Dolfing, J. et al. Syntrophic entanglements for propionate and acetate oxidation under thermophilic and high-ammonia conditions. ISME J 17, 1966–1978 (2023). https://doi.org/10.1038/s41396-023-01504-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-023-01504-y
