
Abstract
Microorganisms play a key role in arsenic (As) biogeochemistry, transforming As species between inorganic and organic forms and different oxidation states. Microbial As methylation is enhanced in anoxic paddy soil, producing primarily dimethylarsenic (DMAs), which can cause rice straighthead disease and large yield losses. DMAs can also be demethylated in paddy soil, but the microorganisms driving this process remain unclear. In this study, we showed that the enrichment culture of methylotrophic methanogens from paddy soil demethylated pentavalent DMAs(V) efficiently. DMAs(V) was reduced to DMAs(III) before demethylation. 16S rRNA gene diversity and metagenomic analysis showed that Methanomassiliicoccus dominated in the enrichment culture, with Methanosarcina and Methanoculleus also being present. We isolated Methanomassiliicoccus luminyensis CZDD1 and Methanosarcina mazei CZ1 from the enrichment culture; the former could partially demethylate trivalent DMAs(III) but not DMAs(V) and the latter could demethylate neither. Addition of strain CZDD1 to the enrichment culture greatly accelerated DMAs(V) demethylation. Demethylation of DMAs(V) in the enrichment culture was suppressed by ampicillin, suggesting the involvement of bacteria. We isolated three anaerobic bacterial strains including Clostridium from the enrichment culture, which could produce hydrogen and reduce DMAs(V) to DMAs(III). Furthermore, augmentation of the Methanomassiliicoccus-Clostridium coculture to a paddy soil decreased DMAs accumulation by rice and alleviated straighthead disease. The results reveal a synergistic relationship whereby anaerobic bacteria reduce DMAs(V) to DMAs(III) for demethylation by Methanomassiliicoccus and also produce hydrogen to promote the growth of Methanomassiliicoccus; enhancing their populations in paddy soil can help alleviate rice straighthead disease.
Similar content being viewed by others


Introduction
Arsenic (As) is a ubiquitous contaminant in the environment. Arsenic exists in the environment in various chemical species differing in the toxicity to organisms [1]. Microorganisms play a key role in the transformation of As species, such as reduction and oxidation between the pentavalent [As(V)] and trivalent [As(III)] oxidation states, and methylation and demethylation of As compounds [1]. Arsenic methylation is catalyzed by As(III) S-adenosylmethionine methyltransferases (ArsMs) in microorganisms and AS3MT in higher eukaryotes [1,2,3,4], where arsenite is methylated stepwise to produce mono-, di- and tri-methylated As compounds. Arsenic methylation is enhanced under anoxic conditions, such as submerged paddy soils, likely because the substrate arsenite becomes more available and the ability to methylate arsenite is more widespread in anaerobes, such as sulfate-reducing bacteria and fermentative bacteria [5,6,7,8]. In paddy soils and rice grain, dimethylarsenic (DMAs) is the most prevalent methylated As species [5, 9,10,11]. Arsenic in DMAs and MMAs can be present in either the pentavalent [DMAs(V) and MMAs(V)] or trivalent [DMAs(III) and MMAs(III)] oxidation state, with the trivalent form being much more toxic than the pentavalent form [12, 13]. Both DMAs(III) and MMAs(III) have been found in anoxic paddy soil and rice plants [14,15,16], although both, especially the former, are unstable when exposed to air [17]. Excessive accumulation of DMAs in rice plants can cause a physiological disorder called straighthead disease, resulting in spikelet sterility and large yield losses [18,19,20,21]. The toxicity may be attributed to DMAs(III) rather than DMAs(V) [14]. The straighthead disease is prevalent in many rice growing regions, especially after upland soil is converted to paddy and in the upland–paddy rotation system [22, 23]; the reasons for this prevalence are unknown, but differences in microbial community are likely involved.
Opposite to the process of As methylation, methylated As compounds can be demethylated by some microorganisms. It has been shown that MMAs(III) is demethylated by the dioxygenase C−As lyase ArsI in some bacteria [24, 25]. DMAs can also be demethylated, although the mechanism remains unclear. Typically, DMAs accumulates in paddy soil initially after flooding of the soil, followed by gradual disappearance due to demethylation [5, 6, 26]. Thus, the concentration of DMAs in paddy soil and the level of DMAs accumulated in rice plants depend on not only As methylation but also demethylation of DMAs. Based on the experiments of inhibition of methanogenesis and 13C-labeled DMAs, we have previously shown that some methanogens likely mediate demethylation of DMAs in anoxic paddy soil [6]. However, the methanogens that can demethylate DMAs have not been identified. This knowledge could be highly useful for developing strategies to prevent or alleviate rice straighthead disease.
The objective of the present study was to identify methanogens that can demethylate DMAs in paddy soil. We showed that the H2-dependent methylotrophic methanogen Methanomassiliicoccus can demethylate DMAs(III) but not DMAs(V). We also found that anaerobic bacteria reduce DMAs(V) to DMAs(III) for the demethylation by Methanomassiliicoccus and also provide H2 for the growth of Methanomassiliicoccus. We further showed that augmentation of the Methanomassiliicoccus-Clostridium coculture decreased DMAs accumulation in rice plants and alleviated straighthead disease.
Materials and methods
Enrichment cultures of methanogens
Methanogenic enrichment cultures were constructed by inoculating 0.5 g of an arsenic contaminated (total As 86 mg kg−1) paddy soil collected from Chenzhou (CZ), Hunan province in China into 5 ml pre-reduced medium (Table S1) and incubated for 30 days at 30 °C. Three types of methanogenic substrates were used, including H2/CO2 (80%/20%, 0.1 MPa), 20 mM acetate and 20 mM methanol. A negative control without methanogenic substrates was included. DMAs(V) (80 μM) was added to all enrichment cultures. The methanol enrichment culture was transferred for 14 generations using the previous generation as inoculant (10% V:V). Methanol (20 mM) and DMAs(V) (20 μM) were added to each generation. In some experiments, DMAs(III) (20 μM) was used instead of DMAs(V). DMAs(III) was synthesized from DMAs(V) according to the procedure described elsewhere [27]. In one experiment, 2-bromoethanesulfonic acid (BES, 10 mM) and ampicillin (100 mg l−1) were added separately to the methanol enrichment culture to inhibit methanogens and bacteria, respectively. At different time points, enrichment cultures were sampled for determination of As species, the compositions of bacteria and archaea, and the mtaB gene abundance. Methane and hydrogen (H2) in the headspace were quantified using gas chromatograph (Agilent 8860 GC system, USA). The mass balance of As species was determined in the first-generation methanol enrichment culture at the end of the experiment by measuring the As species in the solution and sorbed by the soil residues, as well as volatile As in the headspace. Volatile As was trapped using chemotraps with silica gels impregnated with 10% (w/v) AgNO3 and quantified as described previously [7]. Arsenic species in the soil residues were extracted with 50 mM NH4H2PO4 and determined as described below.
Design of specific primers for mtaB gene of Methanomassiliicoccus
MtaB (methanol: 5-hydroxy-benzimidazolyl-cobamide methyltransferase) could be used for phylogenetic analysis, because the MtaB sequences of Methanomassiliicoccus are not intermixed with members of Methanosarcinales and form robustly supported monophyletic groups [28]. Sixteen mtaB gene sequences of Methanomassiliicoccus and three of Methanosarcina were downloaded from GeneBank in NCBI and from assembled contigs of metagenomic sequencing of enrichment cultures in the present study. Multiple sequence alignments of all sequences were performed using the CLUSTALW program. Sequences of mtaB were conserved among the Methanomassiliicoccus group, but distinct from mtaB in Methanosarcina (Fig. S1A). Therefore, we designed a pair of primers (P3/P4) specifically targeting the Methanomassiliicoccus mtaB genes (Table S2). The specificity of the primers was verified using the genomic DNA of Methanomassiliicoccus luminyensis CZDD1, Methanomassiliicoccus luminyensis B10 and Methanosarcina mazei CZ1 as templates. Using the P3/P4 primers, mtaB in Methanomassiliicoccus luminyensis CZDD1 and B10 was successfully amplified, but not from Methanosarcina mazei CZ1 (Fig. S1B). A single PCR product as indicated by agarose gel electrophoresis was amplified from both paddy soil and enrichment culture (Fig. S1C). The primer specificity was further verified on a clone library of mtaB gene amplified from paddy soil. Among the 70 clones sequenced, 94% were identified by BLAST on the NCBI (https://blast.ncbi.nlm.nih.gov/) to be Methanomassiliicoccus and the remainder was unclassified (Fig. S1D).
DNA extraction and quantitative real-time PCR
Total DNA in different generations of enrichment cultures were extracted using a Power Soil DNA isolation Kit (QIAGEN, Germany). DNA concentration was determined by using a NanoDrop 2000C spectrophotometer (Thermo Scientific, Wilmington, USA). The absolute abundances of mtaB genes were quantified using the primers P3/P4 (Table S2) and conducted with a real-time PCR detection system (Bio-Rad CFX96, USA). Bacterial 16S rRNA gene abundance was quantified using the primers P5/P6 (Table S2).
High-throughput sequencing and metagenomic assembly
Bacterial and archaeal community structures in enrichment cultures were analyzed by using high-throughput sequence based on bacterial and archaeal 16S rRNA genes. The V4–V5 regions of bacterial and archaeal 16S rRNA were amplified using the primers P5/P6 and P7/P8, respectively (Table S2). The amplicons were purified, quantified and sequenced on a Mieseq platform (Illumina; PE250 mode) (Shanghai Biozeron Biotech. Co., LTD). Raw sequences were quality filtered and assembled. The sequences were processed using Quantitative Insights Into Microbial Ecology (QIIME, V1.9.0 http://qiime.org/scripts/assign_taxonomy.html). Effective sequences were grouped into operational taxonomic units (OTUs) based on similarity score of 97% using UPARSE (version 7.1 http://drive5.com/uparse/). Representative sequences were selected and the taxonomic classification of representative sequences for each OTU were identified by uclust algorithm (http://www.drive5.com/usearch/manual/uclust_algo.html) against SILVA(SSU138.1) 16S rRNA database based on the confidence threshold of 80% [29]. Total genomic DNA in sixth generation of enrichment cultures was extracted and used for metagenomic analysis. The quality of DNA was determined using Qubit (Thermo Fisher Scientific, Waltham, MA) and Nanodrop (Thermo Fisher Scientific, Waltham, MA). DNA libraries were constructed and sequenced on the Illumina Novaseq 6000 sequencer using the pair-end technology (PE 150). The raw sequencing reads of three enriched samples were processed with MetaWRAP v1.2.2, and the integrated pipeline for metagenomic data analysis. Read_qc module in meta-WRAP was used to filter raw paired-end reads. Clean reads were assembled individually using MEGAHIT v1.1.3 with default parameters, and short contigs (<1000 bp) were removed. The initial metagenome-assembled genomes (MAGs) were recovered according to the Multiple contig binning methods [CONCOCT v1.0.0, MetaBAT2 v2.12.1 [30] and MaxBin2 v2.2.6 [31]] applied in binning module of MetaWRAP. Final MAGs were obtained after three replicate MAG sets were merged and refined using the bin_refinement module in metaWRAP. Taxonomy prediction of the selected MAGs was classified using GTDB-Tk v1.0.2 [32] (classify_wf workflow, default parameter). Functional annotation was predicted using Prokka v1.13 [33] and KofamKOALA [34].
Isolation and identification of methylotrophic methanogens and bacteria
Single strains of methanogens and bacteria were isolated by using the Hungate rolling tube technique [35]. Briefly, the methanol enrichment culture (the 6th generation) was diluted in a 10-fold series from 10−1 to 10−7. Each 500 μl dilution was injected into anaerobic tubes containing 5 ml of agar medium and incubated for 30 days, with or without H2 supplementation. Methanol (20 mM), H2 (±0.1 MPa), and ampicillin (100 mg l−1) were added into the tubes. After 30 days incubation, methanogenic colonies were picked into liquid medium in a glovebox filled with 100% N2. For isolation of bacteria, 20 mM methanol and 10 mM BES were applied. After culture for 2 weeks, bacterial colonies were picked into fresh liquid medium and purified to obtain pure cultures. The methanogenic and bacterial isolates were identified based on the similarity of 16S rRNA gene sequences, which were amplified using the primers P9/P10 and P11/P12, respectively (Table S2). Seventeen 16S rRNA sequences of methylotrophic methanogens from NCBI database closely related to the isolates obtained in the present study were selected as reference sequences and used for constructing the polygenic tree using neighbor-joining algorithm with software MEGA 11.0. The tree topology was estimated by bootstraps based on 1000 replications. Twenty reference sequences of bacteria closely related to the bacterial isolates obtained in the present study were downloaded, and a phylogenetic tree of bacterial isolates was also established as above.
Demethylation of DMAs by the monocultures of Methanomassiliicoccus luminyensis CZDD1 and Methanosarcina mazei CZ1 or the coculture of Methanomassiliicoccus-Clostridium
Strains Methanomassiliicoccus luminyensis CZDD1 and Methanosarcina mazei CZ1 were isolated in the present study. Strain CZDD1 was cultured in a medium with exogenously added H2 (0.1 MPa), 20 mM methanol, ampicillin (100 mg l−1), DMAs(V) (5 μM) or DMAs(III) (0.5, 1 or 5 μM). Strain CZ1 was cultured in a medium containing 20 mM methanol with or without DMAs(V) (5 μM). In a separate experiment, the two strains were cultured to the mid-log phase and inoculated into the methanol enrichment culture (1st generation) with the addition of 20 μM DMAs(V). A control without inoculation was included. Each experiment was replicated three times. After culture for 36 days at 30 °C, CH4 in the headspace and As species in the culture medium were determinated.
Three anaerobic bacteria, Clostridium malenominatum CZB5, Tissierella carlieri CZB10 and Clostridium subterminale CZB11, were isolated from the methanol enrichment culture. These strains were cultured in a medium with 20 μM DMAs(V) (Table S1) at 30 °C. Syntrophic cocultures between methanogenic strain Methanomassiliicoccus luminyensis CZDD1 and bacterial strain Clostridium malenominatum CZB5, Tissierella carlieri CZB10 and Clostridium subterminale CZB11 were constructed. The bacterial strains were precultured to the mid-log phase. Methanogenic and bacterial strains were mixed in equal volume in a medium with 20 mM methanol, DMAs(V) (20 μM) or DMAs(III) (1 μM) and incubated at 30 °C. Methane and H2 in the headspace, and As species in the culture medium were determined. In addition, 13C-labeled DMAs(V) (5 μM) was synthesized according to Chen et al. [6]. 13C-DMAs(V) (5 µM) was added to the Methanomassiliicoccus-Clostridium coculture. 13C/12C isotope ratios of methane in the headspace were determined using GC - IRMS (Thermo Fisher Scientific, Germany).
Soil incubation
A paddy soil was collected from Tancheng (TC), Shangdong province, China, where straighthead disease was observed in rice crops [19]. Twenty g soil (<2 mm) were placed into 100 ml serum bottles, to which 40 ml deionized water was added. The bottles were flushed with N2 for 30 min, sealed using rubber stoppers, secured with aluminum crimp caps and incubated at 30 °C in the darkness inside a glovebox (100% N2). Soil was incubated for 4 days when DMAs concentration in the soil solution reached a peak. Subsequently, 5 ml inoculants of Methanomassiliicoccus luminyensis CZDD1, Clostridium malenominatum CZB5, or the Methanomassiliicoccus-Clostridium coculture were added to the soil. A control without inoculant was included. Each treatment was replicated in three bottles. Inoculants were precultured to the mid-log phase before being introduced to the soil. Soil solution was collected at different time points during 15 days of incubation for the determination of As species [25].
Pot experiment with rice
TC soil weighing 1.8 kg was placed into a 2 l plastic cup. Compound fertilizers (0.18 g, N: P2O5: K2O = 15: 15: 15) were mixed into the soil. The soil was flooded with a 3–5 cm layer of standing water above the soil surface for 1 week. One rice seedling (cultivar Jingliangyouhuazhan) was transplanted into each pot. Three treatments were established including control, the addition of Clostridium malenominatum CZB5 or the Methanomassiliicoccus-Clostridium coculture (150 ml kg−1 soil). Each treatment was replicated in three pots. The inoculants were injected into the soil at about 5 cm below the soil surface at the late tilling stage of rice growth. Rice was grown in a greenhouse with 16 h light (30 °C)/8 h dark (22 °C) photoperiod. After inoculation for 1, 7 and 30 days, soil was collected and total DNA was extracted. The abundances of mtaB and methyl-coenzyme reductase M coding gene (mcrA) were quantified. At rice maturity, the symptoms of straighthead disease and seed setting rate of rice were recorded. Arsenic species in the husks were extracted using 1% HNO3 and determined by using HPLC-ICP-MS [19].
Demethylation of DMAs in different paddy soils
Six paddy soils and six upland soils (Table S3) were incubated under flooded conditions. Twenty grams of each soil were placed into a 100 ml serum bottle, to which 40 ml ultra-pure water was added. The bottles was purged with N2 for 30 min, sealed using rubber stoppers, secured with aluminum crimp caps and preincubated at 25 °C in the darkness. Each soil was replicated in 3 bottles. After incubation for 14 days, 40 μmol kg−1 DMAs(V) was added into each bottle inside an anaerobic glovebox. All soils were incubated for another 14 days. Before and after incubation, As species in solution and soil solid phase were determined. Arsenic species in the solid phase were extracted by using 50 mM NH4H2PO4 (M:V = 1:10) for 12 h and centrifuged [36]. Supernatant was collected and used for As species analysis. After incubation, a portion of soil was collected for DNA extraction. The absolute abundance of mcrA for total methanogens, mtaB specific for Methanomassiliicoccus were quantified by Real-Time PCR.
Analysis of As species
Two methods were used for As species analysis. To test DMAs demethylation by enrichment cultures and isolates, As species in the culture medium were oxidized to their corresponding pentavalent forms using H2O2 (1 ml culture with 200 μl 30% H2O2), which stabilized the As species during sample preparation and quantification. Arsenic species were determined by using high performance liquid chromatography–inductively coupled plasma mass spectrometry (HPLC-ICP-MS, NexION 300X, PerkinElmer, USA), using an anion exchange column (Hamilton PRP 100) and a phosphate/nitrate mobile phase (8.5 mM NH4H2PO4 and 8.5 mM NH4NO3, pH 6.0) [19]. Arsenic species in rice husks from the pot experiment were extracted using 1% HNO3 and determined by HPLC-ICP-MS [19]. To determine DMAs(III) and MMAs(III), which are unstable in oxic conditions, As species in the culture medium were preserved with 8 mM diethyldithiocarbamic acid diethylammonium (DDDC) and quantified by HPLC-ICP-MS using a C18 column (Atlantis DC18, Waters) and a mobile phase containing 5 mM tetrabutylammonium hydroxide (TBAH), 3 mM malonic acid and 5% methanol [14, 37].
Results
Demethylation of DMAs(V) by enrichment cultures of methylotrophic methanogens
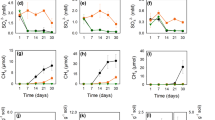
To investigate which methanogenic pathway (hydrogenotrophic, acetoclastic and methylotrophic) is responsible for the demethylation of DMAs(V), we determined the demethylation activities of the methanogenic enrichment cultures from CZ paddy soil amended with either H2/CO2, acetate or methanol. During 30 days of culture, the enrichment culture amended with methanol produced the highest methane yield, followed by acetate and H2/CO2 (Fig. 1A). The control also produced some CH4, likely because the first generation of enrichment culture contained some soil residues that could be utilized by methanogens. DMAs(V) (80 μM) added to the methanol enrichment culture was completely demethylated (Fig. 1B). In contrast, the H2/CO2 enrichment culture did not enhance DMAs(V) demethylation compared with the control; both groups demethylated 28.5–35.2% of the added DMAs(V) after 30 days. In the acetate enrichment culture, 48.3% of the DMAs(V) added was demethylated. Concurrent with DMAs(V) demethylation was the production of MMAs(V); the amount of MMAs(V) in the solution accounted for 13.8–40.5% of the DMAs(V) demethylated (Fig. 1C and Table S4). The concentration of inorganic As (iAs), the final product of demethylation, also increased in the methanol enrichment culture (Fig. S2). Some of the iAs and MMAs(V) could be sorbed by the soil residue present in the enrichment cultures, which contained 0.5 g soil in 5 ml culture solution. We determined the mass balance in the methanol enrichment culture at the end of the experiment by measuring As species in the solution and the soil residues, as well as volatile As in the headspace (Table S4). Volatile As, MMAs(V) and iAs in both the solution and the solid phases accounted for 0.04%, 62% and 37%, respectively, of the DMAs(V) added initially, indicating a near complete recovery. These results suggest that the methylotrophic methanogens facilitate DMAs(V) demethylation, whereas the hydrogenotrophic methanogenic pathway does not contribute to DMAs(V) demethylation.

A Production of methane; B demethylation of DMAs(V); C production of MMAs of different methanogen enrichment cultures. D HPLC-ICP-MS chromatograms showing demethylation of DMAs(V) and production of MMAs(V) by different generations of methanol enrichment culture, after oxidation with H2O2. E The composition of methanogens in the 3rd, 5th and 6th generations of methanol enrichment cultures. Data in (A–C) are means ± SD (n = 3). Data in (E) are means of three replicates (n = 3).
The methanol enrichment culture was successively subcultured for 14 generations. Each generation of the subculture retained the ability to produce methane (data not shown) and to demethylate DMAs(V) (Fig. 1D). Sequencing of archaeal 16S rRNA in the 3rd, 5th and 6th generations of subculture showed that two genera of methylotrophic methanogens, Methanomassiliicoccus and Methanosarcina, were enriched, with Methanomassiliicoccus being consistently the most abundant (20–72% of total archaea) in each generation of the enrichment and Methanosarcina exhibiting varied abundance (Fig. 1E). The hydrogenotrophic methanogen Methanoculleus was also present in the enrichment (7–33%).
Isolation of methylotrophic methanogens
We performed metagenomic analysis of the 6th generation of the methanol enrichment culture and obtained 17 methanogenic MAGs, among which 8 belonged to Methanomassiliicoccales with 66.8–100% genome completeness and <2.4% contamination (Fig. S3). The other nine MAGs were clustered to hydrogenotrophic Methanomicrobiales, with >95.9% genome completeness and 0.21–4.76% contamination.
We isolated two methane-producing pure strains from the 6th generation of the methanol enrichment culture. Based on 16S rRNA gene homolog (Fig. 2A), strain CZ1 was identified as Methanosarcina mazei at 99.4% similarity with that of Methanosarcina mazei FA9604c, and strain CZDD1 as Methanomassiliicoccus luminyensis having 99.08% similarity with that of Methanomassiliicoccus luminyensis B10 isolated from human feces. We grew Methanosarcina mazei CZ1 in methanol and Methanomassiliicoccus luminyensis CZDD1 in methanol supplemented with H2 as the latter is a H2-dependent methylotrophic methanogen [38]. Methanosarcina mazei CZ1 produced more CH4 than Methanomassiliicoccus luminyensis CZDD1 (Fig. 2B).

A Phylogenetic relationship of Methanomassiliicoccus luminyensis CZDD1 and Methanosarcina mazei CZ1 with other known methylotrophic methanogens. B Methane production by strains Methanomassiliicoccus luminyensis CZDD1 and Methanosarcina mazei CZ1 cultured with 20 mM methanol and 20 μM DMAs(V), supplemented with H2 for strain CZDD1. Data in (B) are means ± SD (n = 3).
Methanomassiliicoccus luminyensis CZDD1 accelerated DMAs(V) demethylation in the methanol enrichment culture
We tested whether Methanosarcina mazei CZ1 or Methanomassiliicoccus luminyensis CZDD1 could demethylate DMAs(V) in pure culture with methanol (20 mM) and DMAs(V) (20 µM) as the substrates. H2 (0.1 MPa) was also provided for CZDD1. Over a period of 30 days, while methane was produced by both strains, no DMAs(V) was demethylated by either strain (Fig. S4).
The lack of DMAs(V) demethylation by the pure culture of Methanosarcina mazei CZ1 or Methanomassiliicoccus luminyensis CZDD1 could be because demethylation requires synergistic actions from other microorganisms. To test this hypothesis, we added pregrown culture of either strains to the 1st generation of the methanol enrichment culture. The addition of Methanosarcina mazei CZ1 greatly increased methane production compared with the control, whereas the addition of Methanomassiliicoccus luminyensis CZDD1 only marginally enhanced methane production (Fig. 3A). The addition of Methanomassiliicoccus luminyensis CZDD1 accelerated the demethylation of DMAs(V) and the production of MMAs(V) markedly compared with the control, whereas the addition of Methanosarcina mazei CZ1 suppressed DMAs(V) demethylation (Fig. 3B, C). Using the information of the genome sequence of Methanomassiliicoccus luminyensis CZDD1 and fifteen mtaB sequences of other strains and contigs belonging to Methanomassiliicoccus, we designed a specific primer pair targeting the mtaB gene for quantification (see Materials and Methods). The addition of Methanomassiliicoccus luminyensis CZDD1 increased the mtaB copy number by 5.7–32.2 fold compared with the control (Fig. 3D). In contrast, the addition of Methanosarcina mazei CZ1 decreased the mtaB copy number by 30.4–68.7% (p < 0.05). Analysis of archaeal 16S rRNA showed that the addition of Methanomassiliicoccus luminyensis CZDD1 significantly increased its relative abundance by 95%, but decreased the abundance of Methanosarcina and Methanoculleus (Fig. S5). These results suggest that Methanomassiliicoccus luminyensis CZDD1, but not Methanosarcina mazei CZ1, accelerated DMAs(V) demethylation. In fact, the addition of Methanosarcina mazei CZ1 appeared to suppress the growth of Methanomassiliicoccus luminyensis CZDD1, resulting in a suppression of DMAs(V) demethylation.

The effects of addition of Methanomassiliicoccus luminyensis CZDD1 or Methanosarcina mazei CZ1 to methanol enrichment culture on (A) methane production, (B) demethylation of DMAs(V), (C) production of MMAs(V), and (D) the absolute abundance of Methanomassiliicoccus specific mtaB. Data are means ± SD (n = 3). Different letters in (D) indicates significant difference at p < 0.05 between treatments (Tukey’s test).
Trivalent DMAs(III) is the substrate for demethylation
In the experiments described above, DMAs(V) was added to the enrichment cultures or pure cultures of methanogens. It is possible that DMAs(V) needs to be reduced to DMAs(III) before demethylation, as has been shown for bacterial demethylation of MMAs(V) [39]. We have recently shown that DMAs(III) is present in anoxic paddy soils and some anaerobes are able to reduce DMAs(V) to DMAs(III) [14]. To test whether DMAs(V) is reduced to DMAs(III) prior to demethylation, we determined As species transformation on day 0, 14 and 30 after the addition of DMAs(V) to the 14th generation of the methanol enrichment culture, using a protocol that preserves and quantify DMAs(III) [14, 37]. After 14 days, DMAs(V) was partially reduced to DMAs(III) and a trace amount of MMAs(III) was produced (Fig. 4A). After 30 days, both DMAs(V) and DMAs(III) had disappeared and MMAs(III) was produced quantitatively, suggesting that reduction of DMAs(V) to DMAs(III) precedes demethylation. In a further experiment, we found that DMAs(III) was demethylated much faster than DMAs(V) by the 6th generation of the methanol enrichment culture; by day 21, 92% of the added DMAs(III) was demethylated, compared with 31% of the added DMAs(V) (Fig. 4B, C). These results suggest that reduction of DMAs(V) to DMAs(III) may be the rate-limiting step for its demethylation.

A HPLC-ICP-MS chromatograms showing As species in the methanol enrichment culture with 20 μM DMAs(V) during incubation. Trivalent As species were preserved with DDDC and separated by a C18 column before ICP-MS determination. B Demethylation rate of DMAs(V) and DMAs(III) by the methanol enrichment culture. C MMAs production in the methanol enrichment culture amended with DMAs(V) or DMAs(III). D Production of MMAs in the monoculture of Methanomassiliicoccus luminyensis CZDD1 supplemented with 0.5, 1 or 5 μM DMAs(III). Data in (B–D) are means ± SD (n = 3).
We next tested whether pure cultures of Methanomassiliicoccus luminyensis CZDD1 and Methanosarcina mazei CZ1 could demethylate DMAs(III). After 30 days of culture with the addition of 0.5–5 μM DMAs(III), Methanomassiliicoccus luminyensis CZDD1 demethylated 1–7% of the added DMAs(III) to MMAs (Fig. 4D), indicating a weak ability to demethylate DMAs(III). In contrast, Methanosarcina mazei CZ1 could not demethylate DMAs(III) (data not shown).
The presence of bacteria enhances demethylation of DMAs(V) by methanogens
Because Methanomassiliicoccus strains are obligate H2-dependent methylotrophic methanogens [38] and H2 is produced by some anaerobic bacteria [40], we examined whether bacteria play a role in DMAs(V) demethylation. As expected, the addition of BES, a specific inhibitor for methanogenic methyl-coenzyme M reductase, almost completely inhibited demethylation of DMAs(V) in the methanol enrichment culture (Fig. 5A). The addition of ampicillin, an antibiotic inhibiting most bacteria, produced a strong inhibiting effect on core genera of bacteria in the enrichment culture, including Clostridium sensu stricto 16, Hydrogenispora, Clostridium sensu stricto 13, and Alkalibaculum (Fig. S6). Ampicillin also inhibited DMAs(V) demethylation in the enrichment culture substantially (Fig. 5A). The addition of ampicillin decreased the concentration of H2 produced in the headspace of the enrichment culture by 93.7% (Fig. 5B). Sequencing of 16S rRNA in the enrichment culture showed that the addition of ampicillin decreased the abundance of Methanomassiliicoccus but increased the abundance of Methanosarcina (Fig. 5C). The abundance of hydrogenotrophic Methanoculleus was also decreased slightly by ampicillin, although its abundance was very low due to the use of methanol as the substrate to enrich methylotrophic methanogens. Quantification of the mtaB gene specific to Methanomassiliicoccus further confirmed the suppressive effect of ampicillin (Fig. 5D). The effect of ampicillin on the abundance of Methanomassiliicoccus in the enrichment culture was likely to be indirect, because it did not affect methane production by Methanomassiliicoccus luminyensis CZDD1 in pure culture during a time course of 48 days (Fig. S7).

A Effect of BES (5 mM) or ampicillin (Amp, 100 mg l−1) on DMAs(V) demethylation by the methanol enrichment culture. Effect of Amp addition on (B) H2 production, (C) the composition of methanogens, (D) the absolute abundance of Methanomassiliicoccus specific mtaB, and (E) reduction of DMAs(V) to DMAs(III). The methanol enrichment cultures were amended with 20 μM DMAs(V). Data in (B) and (D) are means ± SD (n = 3). Arsenic species in (A) were oxidized by H2O2 to their corresponding pentavalent forms and separated via an anion-exchange column. Arsenic species in (D) were preserved with DDDC and separated via a C18 column. * in (B) and (D) indicates significant difference at p < 0.05 between the control and +Amp treatments (Tukey’s test).
In addition to producing H2 for methanogens, bacteria could also contribute to the reduction of DMAs(V) to DMAs(III), thus enhancing DMAs(V) demethylation. Indeed, the addition of ampicillin decreased the reduction of DMAs(V) to DMAs(III) in the methanol enrichment culture after incubation for 14 days (Fig. 5E). These results support the notion that the presence of some bacteria enhances DMAs(V) demethylation driven by some methylotrophic methanogens, such as those in the Methanomassiliicoccus genus, likely through enhancing their growth and the reduction of DMAs(V) to DMAs(III).
Isolation of bacteria capable of H2 production and DMAs(V) reduction
To further examine the role of bacteria in DMAs(V) demethylation, we isolated three bacteria, Clostridium malenominatum CZB5, Clostridium subterminale CZB11, and Tissierella carlieri CZB10, from the methanol enrichment culture (Fig. S8). All three bacterial strains produced H2 (Fig. S9). Among the 145 bacterial MAGs constructed from metagenomic analysis of the methanol enrichment culture, 72 MAGs harbored Fe-Fe or Ni-Fe hydrogenase coding genes likely involved in H2 production (Fig. S3). All three bacterial strains, especially Clostridium malenominatum CZB5 and Tissierella carlieri CZB10, were able to reduce DMAs(V) to DMAs(III) (Fig. S10). For comparison, the ability to reduce DMAs(V) was relatively weak for Methanosarcina mazei CZ1 and negligible for Methanomassiliicoccus luminyensis CZDD1 (Fig. S10). Furthermore, all three bacterial strains could not demethylate DMAs(V) or DMAs(III) (data not shown).
Synergistic effect between Methanomassiliicoccus luminyensis CZDD1 and Clostridium malenominatum CZB5 on DMAs(V) demethylation
We tested whether coculture of Methanomassiliicoccus luminyensis CZDD1 with Clostridium malenominatum CZB5 could enable the former to demethylate DMAs(V). In the absence of exogenous H2, the coculture produced methane, reaching a peak on day 14 (Fig. S11A), suggesting that Methanomassiliicoccus luminyensis CZDD1 could utilize H2 produced by Clostridium malenominatum CZB5. Compared with the monoculture of Methanomassiliicoccus luminyensis CZDD1 supplemented with H2, Methanomassiliicoccus luminyensis CZDD1 grew much faster (measured by methane production) when cocultured with Clostridium malenominatum CZB5 (Fig. S11B). In contrast to monoculture of Methanomassiliicoccus luminyensis CZDD1, which did not demethylate DMAs(V), the coculture produced 0.08 μM MMAs from DMAs(V) demethylation after 30-day incubation (Fig. 6A). To confirm that the methyl group from DMAs(V) was converted to CH4, 13C-DMAs(V) was supplemented to the coculture. Compared with the control of adding non-label 12C-DMAs(V), methane produced by the coculture supplemented with13C-DMAs(V) was significantly enriched with 13C (Fig. 6B), indicating that 13C-DMAs(V) was demethylated to produce 13CH4.

A Production of MMAs in Methanomassiliicoccus-Clostridium coculture amended with 5 μM DMAs(V). B δ13C in CH4 produced by Methanomassiliicoccus-Clostridium coculture amended with 5 μM 12C-DMAs(V) or 13C-DMAs(V). Data are means ± SD (n = 3).
Augmentation of Methanomassiliicoccus luminyensis CZDD1 accelerates DMAs demethylation in paddy soil, decreases DMAs accumulation in rice and the incidence of straighthead disease
We tested whether the addition of Methanomassiliicoccus luminyensis CZDD1, Clostridium malenominatum CZB5, or the coculture of the two strains could accelerate the demethylation of DMAs produced by soil. TC paddy soil with a high As methylation potential [41] was preincubated under anoxic conditions for 4 days to allow the production of DMAs, to which inoculants of CZDD1, CZB5 or the coculture of the two strains were added. In the control, DMAs in the soil solution disappeared gradually during the following 15 days (Fig. S12). Augmentation of Methanomassiliicoccus luminyensis CZDD1 accelerated the disappearance of DMAs with concurrent increases in the concentrations of MMAs and iAs, whereas augmentation of Clostridium malenominatum CZB5 had no such effect. Augmentation of the coculture of Methanomassiliicoccus-Clostridium accelerated the disappearance of DMAs slightly faster than that of the Methanomassiliicoccus luminyensis CZDD1 monoculture (Fig. S12).
We grew rice in a pot experiment using TC paddy soil. At the late tillering stage, 150 ml of the Clostridium malenominatum CZB5 monoculture or the Methanomassiliicoccus-Clostridium coculture were added to the soil, with addition of uninoculated medium as the control. At the rice maturity stage, 73.5% of the rice grains from the control group showed distorted husks, a symptom characteristic of the straighthead disease. The augmentation of the Methanomassiliicoccus-Clostridium coculture decreased the percentage of rice grains with distorted husks to 24.3% (Fig. 7A). Meanwhile, the addition of coculture increased the seed setting rate from 10.3 to 52.7% (Fig. 7B) and decreased the concentration of DMAs in rice husks by 78.2% (Fig. 7C). In contrast, the addition of the Clostridium malenominatum CZB5 monoculture had no significant effect. Quantification of mtaB in the soil collected between 1–30 days after inoculation showed 1.9–3.1 fold higher gene copies in the treatment augmented with the Methanomassiliicoccus-Clostridium coculture than the control or the treatment with the Clostridium malenominatum CZB5 monoculture (Fig. 7D). These results indicate that augmentation of the Methanomassiliicoccus-Clostridium coculture decreased DMAs accumulation by rice plants and alleviated the straighthead disease, and the effect can be attributed to Methanomassiliicoccus.

The effects of coculture and monoculture of Clostridium malenominatum CZB5 addition on (A) the percentage of rice grain with straighthead symptoms, (B) seed setting rate, (C) DMAs concentration in rice husks, and (D) the absolute abundance of Methanomassiliicoccus specific mtaB. Data are means ± SD (n = 3). Different letters indicate significant difference at p < 0.05 between treatments (Tukey’s test).
Variation in the DMAs(V) demethylation activity among paddy and upland soils
Long-term cultivation of paddy rice is likely to increase the population of methanogens in the soil [42], possibly leading to higher activity of DMAs(V) demethylation. We selected six paddy soils and six upland soils in an incubation experiment to measure demethylation of exogenous DMAs(V). After flooded incubation for 14 days, the exogenously added DMAs(V) (0.6 μmol kg−1 soil) was nearly completely demethylated in all six paddy soils, whereas substantial proportions (40–100%) of the added DMAs(V) was not demethylated in the six upland soils (Fig. 8). The abundances of both mcrA and mtaB genes were higher in the paddy soils than in the upland soil, with 3.4–2414.4- and 2.4–3247.8-fold differences between the two soil groups, respectively (Fig. 8). There were significant correlations between the percentage of DMAs(V) demethylation and the copy number of mcrA or mtaB when the data from all soils were combined, but there were no significant correlations in the separate groups of paddy or upland soils (Fig. S13).

Differences between paddy and upland soils in the demethylation rate of DMAs(V), the absolute abundance of methanogen mcrA gene and Methanomassiliicoccus specific mtaB gene. Data are means ± SD (n = 3). Soil information is given in Table S3.
Discussion
We have previously shown that methanogens are likely involved in the demethylation of DMAs in anoxic paddy soils, based on the evidence from experiments using methanogenesis inhibitor and 13C-labeled DMAs [6]. However, the identity of methanogens responsible for DMAs demethylation was unknown. Consistent with our previous study [6], we found that BES completely inhibited DMAs demethylation. We further show that methylotrophic, rather than hydrogenotrophic and aceticlastic, methanogens are likely responsible for DMAs demethylation (Fig. 1). This is not surprising as methylotrophic methanogens can transfer methyl group from methylated compounds to cognate corrinoid protein and to coenzyme M, forming methyl-CoM that is subsequently reduced to methane [43, 44]. Other methylated compounds, such as methanol, methylated amines, methylated sulfide, dimethylselenide and methoxylated aromatic compounds, can also serve as substrates for methylotrophic methanogens [43, 45, 46].
Methanomassiliicoccus and Methanosarcina were the dominant genera (Fig. 1) in the methanol enrichment culture. Eight MAGs of Methanomassiliicoccus were also assembled from metagenomic analysis of the methanol enrichment culture. From the enrichment culture, we isolated two methylotrophic strains, Methanomassiliicoccus luminyensis CZDD1 and Methanosarcina mazei CZ1. Methanomassiliicoccus luminyensis CZDD1 is the first methanogen isolate in the order of Methanomassiliicoccales from paddy soils. Previously, Methanomassiliicoccus luminyensis B10 was isolated from human feces [38]. Strains of Methanosarcina mazei have been isolated from paddy soils [47] and wetland before [48]. Both Methanomassiliicoccus luminyensis CZDD1 and Methanosarcina mazei CZ1 are able to produce methane from methanol, although H2 is also required as the electron doner for the former as it lacks the Wood–Ljungdahl pathway [49, 50]. Methanomassiliicoccus luminyensis is known to be a H2-dependent methylotrophic methanogen, which uses H2 to reduce the methyl group to produce methane [50, 51]. We found that the pure culture of neither Methanomassiliicoccus luminyensis CZDD1 nor Methanosarcina mazei CZ1 could utilize DMAs(V), but the former could demethylate DMAs(III) to some extent (Fig. 6). When added to the methanol enrichment culture, Methanomassiliicoccus luminyensis CZDD1 greatly enhanced DMAs(V) demethylation, whereas Methanosarcina mazei CZ1 suppressed DMAs(V) demethylation likely by competing with the former (Fig. 3). Our study also establishes DMAs(III), not DMAs(V), as the substrate for demethylation (Fig. 4). This finding is similar to MMAs(III), rather than MMAs(V), being the substrate for bacterial demethylation [25, 39]. These findings suggest that the bond between the methyl group and trivalent As(III) is weaker and more prone to demethylation than that with pentavalent As(V). Although Methanomassiliicoccus luminyensis CZDD1 could not reduce DMAs(V), it could demethylate DMAs(III) that is reduced from DMAs(V) by other microorganisms. The ability to reduce DMAs(V) to DMAs(III) appears to be common among anaerobic bacteria [14]. Indeed, three anaerobic bacteria isolated from the methanol enrichment culture could reduce DMAs(V) to DMAs(III) (Fig. S10), and the coculture of Methanomassiliicoccus luminyensis CZDD1 with Clostridium malenominatum CZB5 enables the demethylation of DMAs(V) by the former. Moreover, the addition of ampicillin to the methanol enrichment culture suppressed bacteria-mediated reduction of DMAs(V), resulting in an inhibition of DMAs(V) demethylation driven by methylotrophic methanogens (Fig. 5). Compared with the enrichment culture, demethylation of DMAs(III) by Methanomassiliicoccus luminyensis CZDD1 or of DMAs(V) by the coculture of Methanomassiliicoccus luminyensis CZDD1 and Clostridium malenominatum CZB5 was relatively slow, suggesting the presence of other methanogens with a greater demethylation ability in the enrichment culture. Another possible explanation is that the concentration of methanol in the enrichment culture medium was three orders of magnitude higher than that of DMAs, which could competitively inhibit the uptake and utilization of DMAs by Methanomassiliicoccus luminyensis CZDD1. Competitive inhibition was observed between dimethylsulfide and dimethylselenide for demethylation by methylotrophic methanogens [46].
Apart from reduction of DMAs(V), anaerobic bacteria could also produce hydrogen for the growth of some H2-dependent methanogens, such as Methanomassiliicoccus sp. Hydrogen is produced by the [FeFe] or [NiFe] hydrogenases in some anaerobic bacteria during carbohydrate fermentation [52, 53]. The interspecies transfer of H2 between bacteria and methanogens helps sustain the growth in syntrophic methanogenic communities [54,55,56]. In the present study, genes coding for [FeFe] or [NiFe] hydrogenases are widely present in the bacterial MAGs of the methanol enrichment culture (Fig. S3). Three bacterial isolates from the methanol enrichment culture are all capable of producing H2 and promoting methane production in a coculture with Methanomassiliicoccus luminyensis CZDD1 (Figs. S9 and S11). In addition, ampicillin suppressed the production of H2 in the methanol enrichment culture as well as the abundance of Methanomassiliicoccus sp. (Fig. 5). These results support a synergistical relationship between Methanomassiliicoccus sp. and some anaerobic bacteria, with bacteria providing H2 for growth of Methanomassiliicoccus sp. and reducing DMAs(V) prior to demethylation.
We further demonstrate that the addition of a coculture of Methanomassiliicoccus luminyensis CZDD1 and Clostridium malenominatum CZB5 to paddy soil decreased DMAs accumulation by rice plants and, as a result, alleviated straighthead disease (Fig. 7). This effect was produced by the methanogen as the addition of Clostridium malenominatum CZB5 alone had no such effect. These results suggest that DMAs demethylation in paddy soil can be enhanced by augmentation of Methanomassiliicoccus. In contrast, suppression of methanogenesis in paddy soil by BES was found to increase the severity of straighthead disease in rice [57].
In anoxic paddy soil, DMAs(V) can be demethylated completely to arsenite [6, 26]. In the present study, however, we found that Methanomassiliicoccus sp. only partially demethylates DMAs(III), producing MMAs(III) as the main product. Further demethylation of MMAs(III) requires other microorganisms. It has been shown that MMAs(III) can be further demethylated to arsenite by bacteria harboring the C−As lyase enzyme ArsI [25]. In anoxic paddy soil, some denitrifying bacteria possessing ArsI are able to demethylate MMAs(III) to arsenite [25]. Thus, complete demethylation of DMAs(V) in paddy soil requires concerted actions of multiple microorganisms, from bacteria mediating DMAs(V) reduction, methylotrophic methanogens such as Methanomassiliicoccus sp. demethylating DMAs(III), and bacteria possessing ArsI for further demethylation of MMAs(III). Demethylation of DMAs(III) by methylotrophic methanogens appears to be the rate-limiting step, because MMAs(III) does not persist in anoxic paddy soil [6, 25]. Compared with paddy soils, upland soils have a smaller population of methylotrophic methanogens and also demethylate DMAs(V) at a slower rate (Fig. 8). In fact, rice straighthead disease is often prevalent when upland soils are converted to paddy fields; the disease rarely occurs in old paddy soils [22, 23]. A possible explanation is that newly converted paddy soils have low populations of methylotrophic methanogens to demethylate DMAs.
In conclusion, we have identified Methanomassiliicoccus luminyensis CZDD1 from paddy soil as a methanogen capable of demethylating DMAs(III). Anaerobic bacteria also play a role by reducing DMAs(V) to DMAs(III) and generating H2 for the growth of H2-dependent methylotrophic methanogens. Augmentation of Methanomassiliicoccus–Clostridium coculture enhanced DMAs demethylation in paddy soil, which could provide an effective strategy for controlling rice straighthead disease caused by DMAs accumulation.
Data availability
The raw sequences of 16S rRNA have been deposited in the NCBI SRA database under the accession numbers PRJNA909841 for bacteria and PRJNA909967 and PRJNA1003412 for archaea in enrichment cultures. The MAG genomic sequences are deposited in the NCBI Genome database under the BioProject ID PRJNA1004092. The sequencing data of 16S rRNA of methanogenic isolates are under the NCBI GenBank accession OQ554539 and OQ566822, the accession number for bacterial isolates are OQ548091, OQ566864 and OQ560997.
References
Zhu YG, Yoshinaga M, Zhao FJ, Rosen BP. Earth abides arsenic biotransformations. Annu Rev Earth Planet Sci. 2014;42:443–67.
Qin J, Rosen BP, Zhang Y, Wang G, Franke S, Rensing C. Arsenic detoxification and evolution of trimethylarsine gas by a microbial arsenite S-adenosylmethionine methyltransferase. Proc Natl Acad Sci USA. 2006;103:2075–80.
Qin J, Lehr CR, Yuan C, Le XC, McDermott TR, Rosen BP. Biotransformation of arsenic by a Yellowstone thermoacidophilic eukaryotic alga. Proc Natl Acad Sci USA. 2009;106:5213–7.
Lin S, Shi Q, Nix FB, Styblo M, Beck MA, Herbin-Davis KM, et al. A novel S-adenosyl-L-methionine:arsenic(III) methyltransferase from rat liver cytosol. J Biol Chem. 2002;277:10795–803.
Zhao FJ, Harris E, Yan J, Ma J, Wu L, Liu W, et al. Arsenic methylation in soils and its relationship with microbial arsM abundance and diversity, and as speciation in rice. Environ Sci Technol. 2013;47:7147–54.
Chen C, Li L, Huang K, Zhang J, Xie WY, Lu Y, et al. Sulfate-reducing bacteria and methanogens are involved in arsenic methylation and demethylation in paddy soils. ISME J. 2019;13:2523–35.
Mestrot A, Uroic MK, Plantevin T, Feldmann J, Meharg AA. Quantitative and qualitative trapping of arsines deployed to assess loss of volatile arsenic from paddy soil. Environ Sci Technol. 2009;43:8270–75.
Viacava K, Qiao J, Janowczyk A, Poudel S, Jacquemin N, Meibom KL, et al. Meta-omics-aided isolation of an elusive anaerobic arsenic-methylating soil bacterium. ISME J. 2022;16:1740–49.
Zhu YG, Sun GX, Lei M, Teng M, Liu YX, Chen NC, et al. High percentage inorganic arsenic content of mining impacted and nonimpacted Chinese rice. Environ Sci Technol. 2008;42:5008–13.
Meharg AA, Williams PN, Adomako E, Lawgali YY, Deacon C, Villada A, et al. Geographical variation in total and inorganic arsenic content of polished (White) rice. Environ Sci Technol. 2009;43:1612–7.
Chen H, Tang Z, Wang P, Zhao FJ. Geographical variations of cadmium and arsenic concentrations and arsenic speciation in Chinese rice. Environ Pollut. 2018;238:482–90.
Moe B, Peng H, Lu X, Chen B, Chen LWL, Gabos S, et al. Comparative cytotoxicity of fourteen trivalent and pentavalent arsenic species determined using real-time cell sensing. J Environ Sci. 2016;49:113–24.
Styblo M, Del Razo LM, Vega L, Germolec DR, LeCluyse EL, Hamilton GA, et al. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch Toxicol. 2000;74:289–99.
Chen C, Yu Y, Wang YJ, Gao AX, Yang BY, Tang Z, et al. Reduction of dimethylarsenate to highly toxic dimethylarsenite in paddy soil and rice plants. Environ Sci Technol. 2023;57:822–30.
Li RY, Stroud JL, Ma JF, McGrath SP, Zhao FJ. Mitigation of arsenic accumulation in rice with water management and silicon fertilization. Environ Sci Technol. 2009;43:3778–83.
Chen J, Yoshinaga M, Rosen BP. The antibiotic action of methylarsenite is an emergent property of microbial communities. Mol Microbiol. 2019;111:487–94.
Gong Z, Lu X, Cullen WR, Chris Le X. Unstable trivalent arsenic metabolites, monomethylarsonous acid and dimethylarsinous acid. J Anal Spectrom. 2001;16:1409–13.
Tang Z, Wang Y, Gao AX, Ji YC, Yang BY, Wang P, et al. Dimethylarsinic acid is the causal agent inducing rice straighthead disease. J Exp Bot. 2020;71:5631–44.
Gao AX, Chen C, Zhang H, Yang B, Yu Y, Zhang W, et al. Multi-site field trials demonstrate the effectiveness of silicon fertilizer on suppressing dimethylarsenate accumulation and mitigating straighthead disease in rice. Environ Pollut. 2022;316:120515.
Zheng MZ, Li G, Sun GX, Shim H, Cai C. Differential toxicity and accumulation of inorganic and methylated arsenic in rice. Plant Soil. 2013;365:227–38.
Limmer MA, Wise P, Dykes GE, Seyfferth AL. Silicon decreases dimethylarsinic acid concentration in rice grain and mitigates straighthead disorder. Environ Sci Technol. 2018;52:4809–16.
Meharg AA, Zhao FJ. Arsenic & rice. Dordrecht: Springer; 2012.
Iwamoto R. Straighthead of rice plants effected by functional abnormality of thiol- compound metabolism. Memo Tokyo Univ Agric. 1969;13:62–80.
Yoshinaga M, Rosen BP. A C·As lyase for degradation of environmental organoarsenical herbicides and animal husbandry growth promoters. Proc Natl Acad Sci USA. 2014;111:7701–6.
Chen C, Shen Y, Li YH, Zhang WW, Zhao FJ. Demethylation of the antibiotic methylarsenite is coupled to denitrification in anoxic paddy soil. Environ Sci Technol. 2021;55:15484–94.
Jia Y, Huang H, Zhong M, Wang FH, Zhang LM, Zhu YG. Microbial arsenic methylation in soil and rice rhizosphere. Environ Sci Technol. 2013;47:3141–8.
Burrows GJ, Turner EE. A new type of compound containing arsenic. J Chem Soc Trans. 1920;117:1373–83.
Borrel G, O’Toole PW, Harris HM, Peyret P, Brugere JF, Gribaldo S. Phylogenomic data support a seventh order of methylotrophic methanogens and provide insights into the evolution of methanogenesis. Genome Biol Evol. 2013;5:1769–80.
Amato KR, Yeoman CJ, Kent A, Righini N, Carbonero F, Estrada A, et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013;7:1344–53.
Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165.
Wu YW, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32:605–7.
Parks DH, Chuvochina M, Chaumeil PA, Rinke C, Mussig AJ, Hugenholtz P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol. 2020;38:1079–86.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9.
Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020;36:2251–2.
Hungate RE. The roll-tube method for cultivation of strict anaerobes. In: Methods in Microbiology. Vol. 3B, ed. J.R. Norris & D. W. Ribbons, Academic Press, London, 1969. p. 117–32.
Wenzel WW, Kirchbaumer N, Prohaska T, Stingeder G, Adriano DC. Arsenic fractionation in soils using an improved sequential extraction procedure. Anal Chim Acta. 2001;436:309–23.
Wang Z, Zhou J, Lu X, Gong Z, Le XC. Arsenic speciation in urine from acute promyelocytic leukemia patients undergoing arsenic trioxide treatment. Chem Res Toxicol. 2004;17:95–103.
Dridi B, Fardeau ML, Ollivier B, Raoult D, Drancourt M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int J Syst Evol Microbiol. 2012;62:1902–07.
Yoshinaga M, Cai Y, Rosen BP. Demethylation of methylarsonic acid by a microbial community. Environ Microbiol. 2011;13:1205–15.
Thauer RK. Hydrogenases and the global H2 cycle. Eur J Inorg Chem. 2011;7:919–21.
Chen C, Yang BY, Shen Y, Dai J, Tang Z, Wang P, et al. Sulfate addition and rising temperature promote arsenic methylation and the formation of methylated thioarsenates in paddy soils. Soil Biol Biochem. 2021;154:108129.
Liu Y, Dong Y, Wang P, Hussain Q, Ge T, Wang J. Distribution of methane production and methanogenic archaeal community structure across soil particle size fractions along a rice chronosequence. J Soil Water Conserv. 2019;74:235–46.
Liu Y, Whitman WB. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci. 2008;1125:171–89.
Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008;6:579–91.
Mayumi D, Mochimaru H, Tamaki H, Yamamoto K, Yoshioka H, Suzuki Y, et al. Methane production from coal by a single methanogen. Science. 2016;354:222–5.
Oremland RS, Zehr JP. Formation of methane and carbon dioxides from dimethylselenide in anoxic sediments and by a metanogenic bactrium. Appl Environ Microbiol. 1986;52:1031–36.
Tonouchi A, Nishizaki Y, Tohyama H, Takeda K. Cloning of a gene encoding acetate kinase from Methanosarcina mazei 2-P isolated from a Japanese paddy field soil. Curr Microbiol. 2002;45:390–3.
Wang H, Byrne JM, Liu P, Liu J, Dong X, Lu Y. Redox cycling of Fe(II) and Fe(III) in magnetite accelerates aceticlastic methanogenesis by Methanosarcina mazei. Environ Microbiol Rep. 2020;12:97–109.
Borrel G, Parisot N, Harris HM, Peyretaillade E, Gaci N, Tottey W, et al. Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genomics. 2014;15:679–701.
Bueno de Mesquita CP, Wu DY, Tringe SG. Methyl-based methanogenesis: an ecological and genomic review. Microbiol Mol Biol Rev. 2023;87:e0002422.
Evans PN, Boyd JA, Leu AO, Woodcroft BJ, Parks DH, Hugenholtz P, et al. An evolving view of methane metabolism in the archaea. Nat Rev Microbiol. 2019;17:219–32.
Lubitz W, Ogata H, Rudiger O, Reijerse E. Hydrogenases. Chem Rev. 2014;114:4081–148.
Li QS, Wang R, Ma ZY, Zhang XM, Jiao JZ, Zhang ZG, et al. Dietary selection of metabolically distinct microorganisms drives hydrogen metabolism in ruminants. ISME J. 2022;16:2535–46.
Stams AJ, Plugge CM. Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat Rev Microbiol. 2009;7:568–77.
Boone DR, Johnson RL, Liu Y. Diffusion of the interspecies electron carriers H2 and formate in methanogenic ecosystems, and implications in the measurement of KM for H2 or formate uptake. Appl Environ Microbiol. 1989;55:1735–41.
Stams AJ, de Bok FA, Plugge CM, van Eekert MH, Dolfing J, Schraa G. Exocellular electron transfer in anaerobic microbial communities. Environ Microbiol. 2006;8:371–82.
Chen C, Yang BY, Gao AX, Li LY, Dong XZ, Zhao FJ. Suppression of methanogenesis in paddy soil increases dimethylarsenate accumulation and the incidence of straighthead disease in rice. Soil Biol Biochem. 2022;169:108689.
Acknowledgements
We thank Ms. Zhu Tang and Dr. Jun Luo for technical assistance in arsenic speciation analysis. This work was supported by National Natural Science Foundation of China (41930758, 92251302, 42007288, 32070061), and the Fundamental Research Funds for the Central Universities (XUEKEN2023042, XUEKEN2023038).
Author information
Authors and Affiliations
Contributions
CC, LL, XD and FJZ designed the study. CC and LL performed the experiment and analyzed data. YW contributed to the design of the research. CC, LL, XD and FJZ wrote the manuscript. All authors agreed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, C., Li, L., Wang, Y. et al. Methylotrophic methanogens and bacteria synergistically demethylate dimethylarsenate in paddy soil and alleviate rice straighthead disease. ISME J 17, 1851–1861 (2023). https://doi.org/10.1038/s41396-023-01498-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-023-01498-7
