
Abstract
The phyla Nitrospirota and Nitrospinota have received significant research attention due to their unique nitrogen metabolisms important to biogeochemical and industrial processes. These phyla are common inhabitants of marine and terrestrial subsurface environments and contain members capable of diverse physiologies in addition to nitrite oxidation and complete ammonia oxidation. Here, we use phylogenomics and gene-based analysis with ancestral state reconstruction and gene-tree–species-tree reconciliation methods to investigate the life histories of these two phyla. We find that basal clades of both phyla primarily inhabit marine and terrestrial subsurface environments. The genomes of basal clades in both phyla appear smaller and more densely coded than the later-branching clades. The extant basal clades of both phyla share many traits inferred to be present in their respective common ancestors, including hydrogen, one-carbon, and sulfur-based metabolisms. Later-branching groups, namely the more frequently studied classes Nitrospiria and Nitrospinia, are both characterized by genome expansions driven by either de novo origination or laterally transferred genes that encode functions expanding their metabolic repertoire. These expansions include gene clusters that perform the unique nitrogen metabolisms that both phyla are most well known for. Our analyses support replicated evolutionary histories of these two bacterial phyla, with modern subsurface environments representing a genomic repository for the coding potential of ancestral metabolic traits.
Similar content being viewed by others


Introduction
Approximately 13% of Earth’s biomass—and 80% of all bacterial and archaeal biomass—is estimated to be located within the subsurface [1, 2]. Recent advances in field technologies have allowed for expansive sampling of this biomass in the terrestrial and marine subsurface [3]. Many of these subsurface environments have been suggested as possible locations for the origins of life or to retain signatures of early evolutionary history [4,5,6,7,8,9]. Analysis of nucleic acids from subsurface biomass has allowed for a broader understanding of the characteristics of microorganisms that inhabit these habitats [10,11,12,13,14]. Several groups of primarily subsurface-inhabiting Bacteria and Archaea are presumed to have retained ancient traits due to the environments being analogous to early-Earth, in some cases isolated from the surface world on geologic timescales [15, 16].
The phyla Nitrospirota and Nitrospinota both share nitrite-oxidizing metabolisms and have long been considered to be sister phyla [17,18,19]. Confirming this evolutionary relationship, recent systematic reconstructions of the tree of life have placed these two phyla as direct relatives [20,21,22,23]. Both phyla have cosmopolitan distributions and are present in a large variety of environments, including deep terrestrial and marine subsurface environments. For example, the more well-studied groups of the Nitrospirota (class Nitrospiria) and Nitrospinota (class Nitrospinia) are commonly detected in marine environments, activated sludge, soil, drinking-water, and waste-water treatment plants [24,25,26,27,28,29,30,31,32,33]. These taxa are known for their nitrite oxidization and complete ammonia oxidation “comammox” metabolisms [24,25,26,27,28,29,30,31,32,33]. By contrast, the Thermodesulfovibrionia class of Nitrospirota is not common in surface environments but have frequently been sampled from in marine and terrestrial subsurface aquifers [11, 12, 34,35,36,37,38]. Members of the Thermodesulfovibrionia class have different physiologies than the Nitrospiria class, including hydrogen oxidation, sulfate reduction, nitrate reduction and sulfur disproportionation [36,37,38,39,40].
Though the shared trait of nitrite oxidation has long been known, a broader comparison of these sister phyla has not yet been performed. Here we explore and compare the functional characteristics of these phyla along their evolutionary histories in order to fill that knowledge-gap. We use phylogenomic, functional, and gene-tree-based methods to establish the connection of basal clades subsurface environments and reveal patterns of metabolic expansion driven by a combination of vertical evolution and horizontal gene transfer. These analyses document a partially replicated evolutionary history of these sister phyla which demonstrates how multiple modes of evolution can shape closely related phyla that occupy similar niches.
Materials and methods
Genomic dataset collection, curation, and quality control
This study used publicly available genome assemblies as well as newly generated datasets (Supplemental Methods). Existing publicly available genome assemblies were downloaded from the National Center for Biotechnology Investigation (NCBI) and the Integrated Microbial Genomes (IMG) database of the U.S. Department of Energy’s Joint Genome Institute in June 2021. The Genome Taxonomy Database (GTDB) website (release 202) [41] was used to access lists of NCBI assembly accession numbers for the following GTDB-assigned phyla: Nitrospinota, Nitrospinota_A (now called Tectomicrobia), Nitrospinota_B, Nitrospirota, Nitrospirota_A (Leptospirilla). The IMG assemblies were found using the same GTDB taxonomy classifier using the search function on the IMG website. IMG metagenome assemblies that were designated as “public” and “published” were also downloaded for these phyla. Duplicate entries between IMG and NCBI were manually removed.
New single-cell amplified genomes (SAGs) from several field sites that were recently made public were used. These include subsurface hydrothermal fluids in September 2018 from the marine serpentinizing Lost City hydrothermal vent field (NCBI BioProject PRJNA779602, Supplemental Table 1), collected in April 2021 from a continental fracture fluids of the Death Valley Regional Flow System (Amaragosa Valley, USA), via the Inyo-BLM1 well (NCBI BioProject PRJNA853307, Supplemental Table 2), and deeper sequenced SAGs collected in 2015 from the of the Atlantis Massif that hosts Lost City, originally described in reference [13] (NCBI BioProject PRJNA825747, Supplemental Table 3). Detailed information on the generation of these new SAGs is available in the Supplemental Methods file.
Quality control of the assemblies was performed using the CheckM qa workflow (v 1.07) to remove genomes with <50% genome completion and >10% sequence contamination, leaving genomes that fall within the MIMAG categories “medium” (>50% completion, <10% contamination) and “high” (>90% completion, <5% contamination) [42, 43]. These resulting genomes were dereplicated with dRep, using default parameters, to remove nearly-identical assemblies [44]. All genomes were then classified using the GTDB-tk classifier tool (v1.5.0, r202) [41] (Supplemental File 1). In the methodology described below, polyphyletic groups that were once considered a part of Nitrospirota and Nitrospinota (i.e., Nitrospirota_A (Leptospirilla), Nitrospinota_A (Tectomicrobia), and Nitrospinota_B) were included only in the phylogenomic trees. These groups were not included in the gene cluster based functional analyses. All code to recreate these processes are available at https://github.com/ts-dangelo/bioinformatic_scripts_python and outlined in Supplemental Fig. 1.
Phylogenomic analysis
Phylogenies of the individual phyla were constructed using the PhyloPhlan pipeline with the Bac120 conserved marker-protein database R202) [41, 45]. The PFAM and TIGRFAM protein files for the Bac120 database contain 218–248 k sequences per file and are too large for the memory requirements of Diamond [46]. Therefore, each protein family in the Bac120 database was randomly subset to 1000 sequences per family. For comparison, the default PhyloPhlan marker protein database contains 337–1344 sequences per protein family. The subsampled version of the Bac120 database was used to create a custom PhyloPhlan database using the command phylophlan_setup_database. The default PhyloPhlan pipeline was run with the -min_num_markers flag set to 12, using the default parameters for the --diversity high and –accurate settings (additional details in Supplemental Info) [45]. The alignments of individual marker genes were concatenated into one file and used as input for IQ-TREE (v2.0.3) using the parameters -m TEST -bb 1000, where ModelFinder was used to choose the most appropriate model by the Bayesian Information Criteria (BIC) [47, 48]. Desulfobacterota_D (Dadabacteria) and class Thermodesulfobacteria (Phylum Desulfobacterota) were used as outgroups. Polyphyletic phyla (Nitrospirota_A; Leptospirilla, Nitrospinota_A (Tectomicrobia), and Nitrospinota_B) were included in their respective phylogenies. Relative Evolutionary Divergence (RED) scaling was used to display appearance of certain metabolic traits in relative time along the phylogeny of the investigated phyla [41].
Gene clustering, gene tree production, and gene reconciliation
Open reading frames were identified in genome assemblies by Prodigal (v2.6.3), using the default parameters of anvi-gen-contigs-database in the Anvi’o analysis pipeline (v7) [49, 50]. The amino acid sequences for all assemblies were clustered into gene clusters using Diamond and the Markov Cluster Algorithm (MCL) with an inflation parameter of 1.2, after blast-hits were filtered using the MINBIT parameter of 0.5 [46, 51, 52]. The resulting gene clusters were exported from Anvi’o and assembly × gene cluster count matrices for each phylum were created from the data using a custom Python script. Matrices were pruned to only contain gene clusters present in at least four genomes and then converted to presence/absence. These matrices were used to hierarchically cluster genomes by gene content using Ward’s linkage method. Gene clusters were annotated with the eggNOG database (version 5.0) using the eggNOG emapper (version 2) using default parameters [53]. In addition, KOFAMSCAN was used to annotate gene clusters. The default thresholds of the “exec_annotation -f mapper” command were used [54]. Consensus annotation for each gene cluster was created by tallying the annotations assigned by eggNOG and KOFAMSCAN for each sequence in a given gene cluster and choosing the most frequent annotation as the consensus annotation, respectively (Supplemental Files 2–7).
Gene trees were constructed for each gene cluster by aligning the gene cluster amino-acid file with MAFFT (v7.490, options –retree 2), trimming the alignments with TRIMAL (v1.2, -automated1 -resoverlap 0.55 -seqoverlap 0.6) and constructing trees with IQ-TREE (v2.0.3, using ModelFinder to identify the most appropriate model via BIC and 1000 UltraFast non-parametric bootstraps (UFboot)) [47, 48, 55, 56], similar to other recent analyses [57, 58]. To calculate the location of the gene originations of enriched gene clusters (described below), gene trees were reconciled against the phylogenomic tree (species tree) using the standard workflow of GeneRax [59]. Gene trees were constructed per phylum, as described above, and were reconciled to the phylogenomic trees of the individual phylum (the phylogenetic relationships of gene clusters of particular interest were investigated in detail, described below). The phylogenomic trees used for reconciliation methods were rooted using the Minimum Ancestor Deviation method (MAD) [60]. This was done to circumvent dataset size and complications of including an outgroup for this data analysis. Testing showed that trees rooted with MAD have nearly identical topologies as outgroup rooted trees, with minor differences only occurring at nodes that were not bootstrap supported (UFboot >95%, Supplemental Figs. 2, 3).
Broader relationships of gene cluster of interest (nxrA, dsrA) were investigated. All GenBank amino acid sequences annotating to nxrA nitrite oxidoreductase were downloaded and the gene cluster sequences were aligned with the GenBank sequences using MAFFT (--auto) and the alignments were trimmed using trimAL (-automated1) [55, 56]. A phylogeny for dsrA was made using the RefSeq-quality Bacterial sequences in TIGRFAM02064. Phylogenies were constructed using IQ-TREE as described above. The nxrA tree was rooted with tetrathionate reductase (ttrA) from Desulfobacterota and the dsrA was rooted on the Firmicutes clade, as done elsewhere [61].
Statistical analyses
Gene clusters that were differentially distributed in the major delineations identified by hierarchical clustering (mainly corresponding to the taxonomic class) were identified using the proportional generalized linear model incorporated into the Anvi’o package [50]. Enriched gene clusters were filtered by the heuristics of: (1) being present in a given clade more than expected by chance, (2) being above a significant q value threshold, and (3) further filtered to the gene clusters occurring in less than 10% of the genomes of the other clade besides the focal clade. These heuristics were used to focus on genes that are highly prevalent in a given clade. To determine statistical differences between genome properties (estimated length and coding density) analysis of variance (ANOVA) was performed using the f_oneway command in the SciPy Python library [62].
Ancestral state reconstruction (ASR) facilitated by MrBayes (v3.2.7a) was used to reconstruct the approximate traits of the respective ancestors of the phyla Nitrospirota and Nitrospinota. The gene clusters in the given phyla were prevalence filtered to only include clusters present in >10% of the genomes. The presence or absence of a gene cluster was treated as a binary state variable and the MAD-rooted phylogeny of the phyla was used to estimate the probability of the state of a given gene cluster at the internal nodes of the tree using the MrBayes Markov Chain Monte Carlo (MCMC) sampler with 500,000 MCMC samples [63, 64]. Gene clusters having a greater than 0.5 posterior probability (pp) at the root node were interpreted as potentially present in the ancestral relative of the given phyla. These ancestral state reconstruction results were compared to the gene-tree species-tree reconciliation results, with the assumption that gene clusters likely present at the root of the phylogenomic tree would have their origination event deep within the tree, at or close to the root node. The habitat-type where genomes were sampled were coded as variables to infer the probability that the ancestral node of the given phyla occupied a particular environment type using MrBayes (v3.2.7a). The NCBI BioSamples that produced the given assemblies were aggregated in sample-type groups using the “isolation source” metadata on the NCBI website. The classifications are outlined in columns “P” and “Q” of Supplemental Data 1.csv.
Results
Phylogenetic and gene content clustering patterns
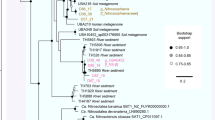
The quality-controlled public data and newly released SAG data contained 367 and 57 assemblies belonging to the phyla Nitrospirota and Nitrospinota, respectively (Supplemental Fig. 4). Phylogenomic analysis of Nitrospirota shows that Thermodesulfovibrio, RBG-16-64-22, and UBA9217 are earlier branching, basal classes while Nitrospiria is a later-branching class within the Nitrospirota phylogeny (Fig. 1, Supplemental Figs. 2, 3). Likewise, phylogenomic analysis of Nitrospinota shows classes UBA7883 and UBA9942 as earlier branching basal groups, with Nitrospinia as a later branching class. The earlier branching classes in both of these phyla tend to have smaller genomes with higher coding density (% of total base pairs contained within open reading frames) (ANOVA P < 0.05 except for Nitrospinota, where early-branching genome lengths had a smaller mean, but a non-significant p value; Fig. 2).

The trees are oriented so the middle of the image contains the outgroups for both trees. Phylogenetic trees were produced used the Bac120 marker set (min 12 genes) using the PhyloPhlan pipeline to create the alignments and IQ-TREE using the LG + F + G4 model chosen by ModelFinder using the Bayesian Information Criterion (BIC) and 1000 ultra-fast non-parametric bootstraps. Heatmaps of gene cluster absence (empty) or present in at least 10% of the genomes in the given phyla (filled) are displayed below the trees. Rows of gene clusters are ordered by hierarchical clustering using wards linkage. Blocks of genes enriched in the Nitrospinia or Nitrospiria are highlighted with the green boxes, and purple used to highlight the enriched gene clusters in the basal classes of the given phyla. Hierarchical clustering of genomes solely by gene content produced similar groupings as the phylogenies (Supplementary Figure 5).

Estimated genome length y-axis scale is 106 base pairs, and coding density y-axis is as percent. Red asterics denote results that show significant differences between metrics in the clades by a ANOVA (p value < 0.05). For Nitrospirota the p values for genome length and coding density are 3.63e−20 and 3.25e−20, respectively. For Nitrospinota the p values for genome length and coding denisty are 0.13 and 0.0011, respectively.

The size of the circles represents the percentage of the enriched gene clusters subset (basal enriched or later-branching enriched) that had its initial origination event at the given node. Gene clusters enriched in basal clades of both phyla show overlap with gene clusters of the Last Common Ancestor (LCA) identified by Ancestral State Reconstruction and have their originations at the root or in basal nodes close to the root (purple, both sides). Gene clusters enriched in later-branching clades do not overlap with LCA gene clusters and have most of their originations at the base of classes Nitrospiria and Nitrospinia, respectively (green, both sides).
Hierarchical clustering of these phyla by gene content also shows distinct separation of the class Nitrospiria from the classes Thermodesulfovibrio, RBG-16-64-22, and UBA9217 of the Nitrospirota (Fig. 1, Supplemental Fig. 5). Similar gene content clustering is apparent with class Nitrospinia distinct from basal classes UBA7883/UBA9942 in Nitrospinota. The later-branching classes of both phyla contain more enriched gene clusters than the basal classes. In the Nitrospirota there are 1127 gene clusters enriched in the Nitrospiria and 486 gene clusters enriched in Thermodesulfovibrio/UBA9217/RBG-16-64-22. In Nitrospinota there are 871 gene clusters enriched in the Nitrospinia and 412 gene clusters enriched in classes UBA7883 and UBA9942 (Fig. 1, Supplemental Fig. 5).
Classification of the sampling sites that produced these assemblies using NCBI metadata shows that most members of the earlier branching classes in both Nitrospirota and Nitrospinota (i.e., Thermodesulfovibrio, RBG-16-64-22, UBA9217, UBA7883, UBA9942) were sampled from marine or terrestrial subsurface aquifers. The later-branching classes of Nitrospirota contain a mixture of subsurface and surface inhabitants while later-branching Nitrospinota (Nitrospinia) are comprised mainly of assemblies sampled from marine environments, including deep water layers (Supplemental File 1, Supplemental Figs. 6, 7, 8). Ancestral state reconstruction (ASR) using broad environmental categorization suggests that the ancestral nodes of both Nitrospirota and Nitrospinota have high posterior probability of resembling assemblies sampled from terrestrial subsurface aquifers (99% pp, 87% pp, respectively; Supplementary File 1, Supplemental Figs. 6, 7, 8).
Patterns of enriched gene clusters
In the Nitrospirota phylum, of the 4598 gene clusters present in >10% of the assemblies, there were 823 gene clusters identified by ancestral state reconstruction (ASR) to have >0.5 pp at the root node. Gene-tree reconciliation methods show that the majority of these gene clusters (84%) had their originations between the root and the three deepest nodes of the tree (Fig. 3), indicating good agreement between methods. Of these gene clusters identified by ASR, 187 also belonged to the 485 gene clusters identified as enriched in the early branching basal classes Thermodesulfovibrio/RBG-16-64-22/UBA9217 by the proportional GLM test (38%). These gene clusters have their origination events at basal nodes in the phylogeny (Fig. 3). None of the 1127 gene clusters enriched in the later branching Nitrospiria class were identified by ASR to have >0.5 pp at the root node. Gene-tree species-tree reconciliation shows that the gene clusters enriched in Nitrospiria do not originate until early nodes in the class Nitrospiria and order Nitrospirales (Fig. 3).
In the Nitrospinota, of the 3901 gene clusters present in >10% of the assemblies, there were 761 gene clusters with >0.5 pp at the root node identified by ASR, and 88% of these gene clusters had their origination event at the root or between the root and the three deepest nodes of the tree (Fig. 3). Of these gene clusters, 212 also belonged to the 412 gene clusters enriched in the basal Nitrospinota classes (51.4%). Reconciliation methods show these gene clusters originated near the base of the phylogeny (Fig. 3). In contrast, none of the ancestral gene clusters overlapped with significantly enriched gene clusters in the class Nitrospinia, the same pattern seen in Nitrospirota. Reconciliation methods indicate that the majority of these gene clusters have their original speciation event at one of the four deepest nodes in the class Nitrospinia (Fig. 3).
Energy production and conversion traits inferred from LCA gene clusters
The putative traits of the last common ancestor (LCA) of the Nitrospirota depict an organism that uses one-carbon compounds as electron sources (Fig. 4). Genes for formate oxidation (fdhA) and carbon monoxide oxidation (cooS/acsA) are present as the only genes indicative of an electron source in the dataset. The carbon monoxide dehydrogenase complex cooS/acsA exists as part of the Carbonyl branch of the Wood-Ljungdahl Pathway (WLP) with other components of the methyl-transferase module of the WLP (acsCD) [65]. The rnf complex is present, which allows for the production of NADH and a proton gradient from reduced ferredoxin produced from CO oxidation [65, 66]. The electron bifurcating complex etfAB is also present, which participates in FAD and ferredoxin recycling [67, 68]. All components of the NADH dehydrogenase nuo operon and one subunit of the cytochrome bc1 complex are present for proton-motive force production. The majority of the dissimilatory sulfate reduction pathway is observed (qmoAB/AprAB/dsrABMKOP). Additional sources of proton-motive force may be produced by the pyrophosphate-hydrolysis powered proton pump hppA. Energy conservation is performed via an F-type ATPase. Gene clusters annotating as the methyl branch of the WLP that contains the enzymes to integrate Acetyl-CoA metabolism into Glycine/Serine biosynthesis are present in the LCA dataset [69]. The oxygen detoxification genes super-oxide dismutase sodA and rubredoxin are present in the LCA gene clusters (Fig. 4).

Gene symbols in red are present, gene symbols in gray are genes that are a part of the given pathway depicted, but are not present or are not above the 0.5 > pp threshold for inclusion in the LCA gene cluster set.
The traits of the LCA of Nitrospinota depict an ancestor that uses sulfur compounds and hydrogen as electron sources (Fig. 4). The genes for subunits of subgroup 1b NiFe hydrogenase are present (hyaABCD). The same gene clusters for dsrAB present in the LCA of Nitrospirota are present in the LCA of Nitrospinota. A phylogenetic analysis of this dsrA gene cluster with RefSeq representatives of TIGRFAM02064 (dsrA) shows Nitrospinota and Nitrospirota RBG-16-64-22 dsrA sequences forming a clade close to sequences from sulfide oxidizing Alphaproteobacteria [61, 70] (Supplementary Figure 9). The sulfide oxidation via reverse dissimilatory sulfate reduction (rDSR) accessory protein dsrL is present in the LCA gene clusters [71]. In addition, present are sulfur-metabolism gene clusters annotated as the membrane-bound sulfite dehydrogenase (soeA/dmsA) responsible for sulfite oxidation to sulfate with oxygen [72, 73]. A proton-motive force is generated by NADH dehydrogenase and an electron transport chain (Fig. 4). Oxygen is used as a terminal electron acceptor via cytochrome c oxidase (ctaCDE). Similar to the Nitrospirota LCA dataset, the same F-type ATPase is present along with hppA. Core carbon anabolism/catabolism is performed by the TCA cycle, which has been described in representatives of this phylum [17].
Common and contrasting metabolic properties of early and late branching clades
In both phyla, early branching clades have metabolic capabilities that are absent in later branching clades (Figs. 3, 5). Gene clusters with annotations as 2-oxoacid oxidoreductases (korABC/oorABC), involved in the rTCA cycle and also playing roles in ferredoxin cycling, one and two-carbon compound metabolisms, and low-potential electron transfers are only present in the basal clades of Nitrospirota and Nitrospinota (Fig. 5) [74]. Gene clusters involved in the WLP are present in the class Thermodesulfovibrio and absent in class Nitrospiria of Nitrospirota and completely absent in Nitrospinota (Fig. 5). Citrate-synthase (CS/gltA) and the other components of the TCA/rTCA cycle are present in the class Nitrospiria of the Nitrospirota and present throughout the Nitrospinota (Fig. 5) [75, 76].

The rows are ordered by hierarchical clustering of presence/absence patterns in Nitrospirota using Ward’s Linkage. Functional differences and similarities between the two phyla can be noted, namely the use of WLP in early-branching Nitrospirota and the shared use of the rTCA cycle in Nitrospirales and Nitrospinota. The purpled shaded boxes denote gene clusters with annotations involving C1 metabolisms, the red shaded boxes denote kor/oor annotated gene clusters likely involved in low redox-potential ferredoxin cycling present in basal groups of both phyla.
In both phyla, gene clusters involved in nitrogen fixing nif operon are enriched in the early-branching classes, but absent in the later branching classes (Fig. 6). Although they did not pass the thresholds used for inclusion into the putative LCA gene cluster dataset, many gene clusters involved in dissimilatory nitrate reduction processes are present in Thermodesulfovibrio genomes (Fig. 6) [39, 40]. Terminal oxidases are also sporadically present in Thermodesulfovibrio (Supplementary Figure 10). Unique cytochromes and other genes known to be involved in manganese oxidation are present solely in order SBBL01 (also referred Ca. Manganitrophaceae), which have been analyzed in detail recently (Fig. 8, Supplementary Figure 10) [77, 78].

The rows are ordered by hierarchical clustering of presence/absence patterns in Nitrospirota using Ward’s Linkage. Functional differences and similarities between the two phyla can be noted. The shaded boxes denote functions of interest that are discussed in the text.
Gene clusters involved in nitrite-based metabolisms and comammox metabolisms are absent in basal lineages of both phyla but are present in both later branching Nitrospiria and Nitrospinia classes (Fig. 6). Both phyla have the same enriched gene clusters for nitrite oxidoreductase (nxrAB) (Fig. 7). The phylogeny of the nxrA gene cluster is largely monophyletic (Fig. 7A). Additional phylogenetic analysis of this gene cluster with nxrA sequences from GenBank suggest that these sequences from both phyla have transfer histories with the Planctomycetota (Fig. 7B) [19]. Gene-tree reconciliation methods indicate acquisition of these genes by the phyla early within the later branching Nitrospiria/Nitrospinia classes (Fig. 8). Gene clusters with annotations ammonia monooxygenase and hydroxylamine dehydrogenase (pmo-amoABC, hoa), involved in comammox metabolism, originate at the base of the Nitrospiraceae family (Figs. 6, 8).

A Displays the phylogeny of the gene cluster rooted at the branch separating the majority of the Nitrospirota sequences from the Nitrospinota sequences. The LG + G4 model was used, as chosen by the B.I.C by ModelFinder. A highly divergent sequence from GCA_016212295 (Nitrospinota) was removed from the left panel for ease of visualization. This sequence can be seen as a diverged relative of Nitrotoga nxrA sequences on the right panel. B Displays gene cluster GC_00000823 sequences with all nxrA from GenBank. Respiratory narG from Desulfobacterota was included as well as tetrathionate reductase A (ttrA) from Desulfobacterota, that was used as an outgroup. The LG + I + G4 model was used, as chosen by the B.I.C by ModelFinder. The sequence from Nitrospirota assembly GCA_003354025, which branches closer to the Nitrospinota in the left panel is within the Planctomycetota on the right panel.

A The phylogeny of Nitrospirota scaled with Relative Evolutionary Divergence (RED) (x axis), as used to denote particular taxonomic ranks by GTDB. The branches are colored in the same fashion as Fig. 1. The taxonomic groups are colored by boxes denoting the main metabolisms of those groups as defined in panel (C). B RED scaled phylogeny of Nitrospinota. Gene cluster originations, as determined by gene reconciliation with Generax, of gene clusters of interest are denoted by colored asterisks and arrows. C Schematic interpretation of the data in panels (A) and (B). Sulfate respiring Nitrospirota originate first. The second-branching groups of Nitrospirota perform reverse dissimilatory sulfate reduction (rDSR), while Nitrospinota using this metabolism likely arises around the same time. After rDSR in Nitrospirota, groups performing manganese oxidation form. The last acquired metabolisms in both phyla are the nitrogen-based metabolisms involving nitrite oxidation genes and comammox genes.
Discussion
Shared evolutionary traits of Nitrospirota and Nitrospinota
Recent systematic reconstructions of Bacterial phylogeny and evolution place the Nitrospirota and Nitrospinota as direct relatives [20,21,22,23]. This relationship has long been considered due to their shared nitrite-oxidizing metabolisms and the observation that orthologous proteins from Nitrospina gracilis of the Nitrospinota have Nitrospira of the Nitrospirota as a most frequent neighbor [17]. Our analysis demonstrates that these two sister phyla share several traits throughout their histories besides nitrite oxidation. These include the primarily subsurface-inhabitation of basal clades that use sulfur-based metabolisms, followed by expanded metabolic capabilities in later branching clades. These changes appear driven by genome expansion and a combination of gene gain and loss. The phylum Nitrospirota contains a more diverse set of metabolic capabilities than Nitrospinota and the metabolic capabilities of the LCA gene clusters of the two phyla suggest that Nitrospirota is older (i.e., Formate, CO oxidization, WLP) [7, 8].
Gene clusters identified as enriched in the basal clades overlap with gene clusters likely to be present in the LCA (Figs. 3, 4). These results suggest that the extant subsurface-inhabiting members share many traits with the ancestors of these phyla. In contrast to the basal clades, gene clusters identified as enriched in the later-branching clades show no overlap with the LCA gene clusters. Gene-tree reconciliation techniques show that these gene clusters originated at the base of the later-branching classes (Fig. 3). The genomes of these later branching classes are larger (Fig. 2), suggesting these phyla have undergone genome expansion. This is exemplified by the acquisition of notable nitrogen-cycling genes from other phyla (Figs. 7, 8). It is interesting to note that Dadabacteria (a relative to Nitrospirota and Nitrospinota [20, 21]) presents an opposing pattern, where early branching clades contain many genomes from subsurface organisms that have larger genomes than later-branching marine clades [79]. This suggests that genome expansion and streamlining patterns are influenced by the particular metabolisms and niche occupation of a given group of organisms. This has been noted in Thaumarchaeota and Cyanobacteria, where lateral gene transfer and duplication are associated with occupation of terrestrial niches while gene loss is more prevalent in clades that live in marine environments [57, 80, 81].
Ancestral metabolisms inferred from LCA gene clusters
Ancestral state reconstruction of the traits with a >0.5 pp at the root of Nitrospirota depict a physiology very different from the more well-studied order Nitrospirales [18, 19, 26, 28,29,30, 32, 33]. The gene cluster annotations suggest a C1-compound-based metabolism that utilizes formate and CO and reduces sulfate as an electron acceptor (Fig. 4). Formate and CO are used as electron sources for deeply-branching bacteria and archaea [15, 16] and they can be produced abiotically in the hydrothermal environments were many of these genomes were sampled from [4, 5]. A representative of the class Thermodesulfovibrio which was isolated from the terrestrial subsurface can perform sulfate reduction with hydrogen [35]. Although hydrogen oxidation genes were not present in the LCA dataset based above the >0.5 pp threshold used, genes with these annotations are present in the subsurface-enriched subset of gene clusters (Supplemental Data File 2). The same is true of several gene clusters involved with dissimilatory nitrate reduction, which has been documented in members of the Thermodesulfovibrio (Fig. 6) [39, 40].
A phylogeny of the gene cluster that annotates as dsrA with sequences from TIGR02064 has a similar topology to a study concluding that some Nitrospirota and Nitrospinota use dsr genes in reverse to oxidize hydrogen sulfide [61]. The presumed oxidative dsrA sequences from Nitrospirota and Nitrospinota form a clade, which contains sequences from Ca. Magnetaquicoccus inordinatus, that is basal to most sulfide oxidizing Alphaproteobacteria [70]. This topology suggests Nitrospirota/Nitrospinota oxidative dsrA sequences share a history with sequences belonging to a sulfide-oxidizing Proteobacterial ancestor (Supplementary Figure 9). A gene cluster annotating as dsrL, which acts as an accessory protein involved in rDSR in Allochomatium vinosu is present in the Nitrospinota LCA and but only 7/367 (0.27%) of Nitrospirota genomes [71]. Four of these Nitrospirota dsrL sequences belong to genomes in the clade containing class RBG-16-64-22 and one from an assembly from the early-branching Nitrospiria order HDB-SIOI813. The assemblies in order HDB-SIOI813 contain incomplete dsr operons and other sulfur-metabolism-related genes such as aprAB and sat (Fig. 6). These gene content patterns suggest these groups perform rDSR or metabolize other sulfur-cycle intermediates, which has been observed in other species with these genes, such as Desulfurivibrio alkaliphilus and some Acidobacteria [82,83,84] (Figs. 6, 8).
Time-calibrated phylogenies of Bacterial evolution suggest the basal node of the close relatives of Nitrospirota—Acidobacteria and Desulfobacterota—originated just prior to, or around the time of the great oxidation event (GOE) [22, 23, 85,86,87]. The time of origin of these phyla closely coincides with a proliferation of oxygen-utilizing enzymes on the Bacterial tree [87]. An analysis of the Cyanobacteria, using Bayesian molecular clocks, calibrated with microfossils, suggest that sodA did not appear in this phylum until after the GOE [88]. Thus, the existence of sodA in the Nitrospirota LCA gene cluster set suggests this phylum originated after there was appreciable oxygen on Earth [87]. Gene clusters annotated as terminal oxidases are sporadically present in the Thermodesulfovibrio (Supplementary Figure 10). None of these gene clusters passed the thresholds for the enriched or LCA datasets used in this analysis, but gene reconciliation methods place the origination of one of these gene clusters at the root of the Nitrospirota tree (GC_00005908 – cbb-3 type cytochrome oxidase) (Supplementary Figure 10). Recently, the co-occurrence of the WLP and facultative aerobic respiration in Thermodesulfovibrio assemblies sampled from the terrestrial subsurface has been reported [38]. It appears the co-occurrence of these metabolic strategies in this phylum could be a widespread trait inherited from an ancestor which evolved during the period of oxygen accumulation on Earth [87].
Metabolic expansion and progression
These analyses demonstrate that there is a metabolic progression throughout the history of Nitrospirota that is partially replicated in Nitrospinota (Fig. 6). The Thermodesulfovibrio and UBA9217 are primarily sulfate reducers that utilize C1 compounds and hydrogen as electron sources, although other metabolisms such as sulfur disproportionation, sulfur oxidation, and nitrate reduction are documented (Figs. 4–6) [35, 37,38,39,40]. After these groups is the class RBG 16-64-22 and Nitrospiria order HDB-SIOI813, which contain the dsr genes for rDSR and other sulfur-intermediate metabolisms [61, 71] (Fig. 6). Next is the Nitrospirota order SBBL01, containing recently described manganese-oxidizers [77, 78]. Genomes in this clade contain unique cytochromes involved in manganese oxidation that have been discussed in detail (Fig. 8, Supplementary Figure 10) [78].
The nxr genes responsible for nitrite-based metabolisms, originated early in the order Nitrospirales and were likely transferred from the Planctomycetota (Figs. 3, 6–8) [19]. The genes responsible for comammox (pmo-amoABC K10944-46, hoa K10535) originated at the base of the family Nitrospiraceae and are most closely related to order Nitrosomonadales (Nitrosomonadacea in GTDB) according to the taxonomy of the best eggNOG seed ortholog, which has been previously reported [26, 30]. These observations suggest Nitrospirota gained its comammox abilities by multiple gene acquisitions from different bacterial phyla. These patterns show Nitrospirota progressing from sulfate reduction to other sulfur-compound metabolisms, and then manganese oxidation, nitrite oxidation, and comammox (Figs. 6, 8).
The Nitrospinota show a partial replication of the metabolic progression of the Nitrospirota (Fig. 8C). The basal Nitrospinota classes UBA7883 and UBA9442 likely use rDSR and sulfur intermediates as an electron source, the same as Nitrospirota orders RBG-16-64-22 and HDB-SIOI813 (Figs. 4, 8) [61]. The phylogeny of the dsrA gene cluster shows that Nitrospinota dsrA sequences (and Nitrospirota class RBG-16-64-22) share a common evolutionary origin that is different to the reductive dsrA sequences in Thermodesulfovibrio (Supplementary Figure 10). In addition, a gene cluster involved in sulfite oxidation (soeA) is present in the LCA of Nitrospinota, and the soeB subunit is enriched in the basal classes, indicating the metabolisms of sulfur-cycle intermediates by basal Nitrospinota (Figs. 6, 8 Supplemental Data Files 6, 8) [71,72,73]. The basal classes of Nitrospinota and intermediate branching groups of Nitrospirota encode genes for the use of oxygen as a terminal electron acceptor (Fig. 4, Supplementary Figure 10). This is parsimonious with the interpretation that Nitrospinota originated in a more oxygenated environment than the basal groups of Nitrospirota. These shared sulfur oxidation metabolisms among basal Nitrospinota groups and intermediate-branching Nitrospirota groups might suggest a similar time of origin. This is supported by the dsrA phylogeny, which shows oxidative Nitrospirota and Nitrospinota dsrA sequences originating from the same common ancestor (Supplementary Figure 10) [61].
Following these sulfur-cycling metabolisms, both phyla acquire the nxrAB genes needed for nitrite oxidation (Figs. 6–8). The phylogenetic analysis of the nxrA gene cluster GC_00000823 with GenBank nxrA demonstrates that Nitrospinota nxrA sequences are also closely related to nxrA sequence in Planctomycetota (Fig. 7) [19]. Interestingly, the Nitrospirota sequences forms a bootstrap-supported monophyletic clade branching next to the Planctomycetota, while the Nitrospinota sequences form a second bootstrap supported monophyletic clade branching after the initial split between Nitrospirota/Planctomycetota (Fig. 7). This suggests that the nxr genes in Nitrospirota and Nitrospinota were transferred from Planctomycetota at two different times. These independent acquisitions of nxrA suggests the order Nitrospirales existed prior to the transfer of nxrA to order Nitrospinales. This scenario is similar to multiple independent gene acquisitions that define the lineage specific metabolisms of Thermoplasmatota order Lutacidiplasmatales [89]. The vertical relationship of oxidative dsrA sequences, the independent horizontal acquisitions of nxrA and similar gene loss/gain patterns depict an entangled and partially replicated evolution in the staggered history of these sister phyla.
Conclusions
Here we demonstrate that the ancestral metabolisms of early branching clades for the sister phyla Nitrospirota and Nitrospinota are markedly different than the later branching groups that have received much attention due to their ecological prominence, especially in the marine environment, and unique nitrogen-based metabolisms. Despite some differences in particular metabolic functions, the similar evolutionary histories of Nitrospirota and Nitrospinota demonstrate how multiple modes of evolution can shape closely related phyla that occupy similar ecological niches. These data demonstrate that gene loss, de novo origination, or lateral acquisition of new genes is a replicated pattern in later-branching clades of phyla whose extant subsurface-inhabiting members resemble ancestral lineages that initially evolved in a primordial habitat.
Data availability
Supplemental Data file 1 and Supplemental Data file 2 contain the NCBI and IMG Assembly IDs and NCBI BioSample accessions for all assemblies used in this study. Newly generated SAG data used in this study are available under NCBI BioProject IDs PRJNA825747 (Atlantis Massif), PRJNA779602 (Lost City Hydrothermal Field), PRJNA853307 (BLM1 Inyo-1), and PRJNA842252 (Juan De Fuca). All data processing scripts used to perform this analysis are available here: https://github.com/ts-dangelo/bioinformatic_scripts_python.
References
Bar-On YM, Phillips R, Milo R. The biomass distribution on Earth. Proc Natl Acad Sci USA. 2018;115:6506–11. https://doi.org/10.1073/pnas.1711842115.
Magnabosco C, Lin LH, Dong H, Bomberg M, Ghiorse W, Stan-Lotter H, et al. The biomass and biodiversity of the continental subsurface. Nat Geosci. 2018;11:707–17. https://doi.org/10.1038/s41561-018-0221-6.
Magnabosco C, Biddle JF, Cockell CS, Jungbluth SP, Twing KI. Biogeography, ecology, and evolution of deep life. Deep Carbon. 2019:524–55. https://doi.org/10.1017/9781108677950.017.
Martin W, Baross J, Kelley D, Russell MJ. Hydrothermal vents and the origin of life. Nat Rev Microbiol. 2008;6:805–14. https://doi.org/10.1038/nrmicro1991.
Schrenk MO, Brazelton WJ, Lang SQ. Serpentinization, carbon, and deep life. Rev Mineral Geochem. 2013;75:575–606. https://doi.org/10.2138/rmg.2013.75.18.
Li L, Wing BA, Bui TH, McDermott JM, Slater GF, Wei S, et al. Sulfur mass-independent fractionation in subsurface fracture waters indicates a long-standing sulfur cycle in Precambrian rocks. Nat Commun. 2016;7:13252. https://doi.org/10.1038/ncomms13252.
Weiss MC, Sousa FL, Mrnjavac N, Neukirchen S, Roettger M, Nelson-Sathi S, et al. The physiology and habitat of the last universal common ancestor. Nat Microbiol. 2016;1:1–8. https://doi.org/10.1038/nmicrobiol.2016.116.
Weiss MC, Neukirchen S, Roettger M, Mrnjavac N, Nelson-Sathi S, Martin WF, et al. Reply to ‘Is LUCA a thermophilic progenote?’. Nat Microbiol. 2016;1:1–2. https://doi.org/10.1038/nmicrobiol.2016.230.
Telling J, Voglesonger K, Sutcliffe CN, Lacrampe-Couloume G, Edwards E, Sherwood Lollar B. Bioenergetic constraints on microbial hydrogen utilization in Precambrian deep crustal fracture fluids. Geomicrobiol J. 2018;35:108–19. https://doi.org/10.1080/01490451.2017.1333176.
Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 2016;7:13219. https://doi.org/10.1038/ncomms13219.
Jungbluth SP, Amend JP, Rappé MS. Metagenome sequencing and 98 microbial genomes from Juan de Fuca Ridge flank subsurface fluids. Sci Data. 2017;4:1–11. https://doi.org/10.1038/sdata.2017.37.
Probst AJ, Castelle CJ, Singh A, Brown CT, Anantharaman K, Sharon I, et al. Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO2 concentrations. Environ Microbiol. 2017;19:459–74. https://doi.org/10.1111/1462-2920.13362.
Goordial J, D’angelo T, Labonté JM, Poulton NJ, Brown JM, Stepanauskas R, et al. Microbial diversity and function in shallow subsurface sediment and oceanic lithosphere of the Atlantis Massif. mBio. 2021;12:e00490–21. https://doi.org/10.1128/mbio.00490-21.
Chivian D, Brodie EL, Alm EJ, Culley DE, Dehal PS, DeSantis TZ, et al. Environmental genomics reveals a single-species ecosystem deep within Earth. Science. 2008;322:275–8. https://doi.org/10.1126/science.1155495.
Carr SA, Jungbluth SP, Eloe-Fadrosh EA, Stepanauskas R, Woyke T, Rappé MS, et al. Carboxydotrophy potential of uncultivated Hydrothermarchaeota from the subseafloor crustal biosphere. ISME J. 2019;13:1457–68. https://doi.org/10.1038/s41396-019-0352-9.
Becraft ED, Lau Vetter MC, Bezuidt OK, Brown JM, Labonté JM, Kauneckaite-Griguole K, et al. Evolutionary stasis of a deep subsurface microbial lineage. ISME J. 2021;15:2830–42. https://doi.org/10.1038/s41396-021-00965-3.
Lücker S, Nowka B, Rattei T, Spieck E, Daims H. The genome of Nitrospina gracilis illuminates the metabolism and evolution of the major marine nitrite oxidizer. Front Microbiol. 2013;4:27. https://doi.org/10.3389/fmicb.2013.00027.
Koch H, Lücker S, Albertsen M, Kitzinger K, Herbold C, Spieck E, et al. Expanded metabolic versatility of ubiquitous nitrite-oxidizing bacteria from the genus Nitrospira. Proc Natl Acad Sci USA. 2015;112:11371–6. https://doi.org/10.1073/pnas.1506533112.
Lücker S, Wagner M, Maixner F, Pelletier E, Koch H, Vacherie B, et al. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc Natl Acad Sci USA. 2010;107:13479–84. https://doi.org/10.1073/pnas.1003860107.
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, et al. A new view of the tree of life. Nat Microbiol. 2016;1:1–6. https://doi.org/10.1038/nmicrobiol.2016.48.
Castelle CJ, Banfield JF. Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell. 2018;172:1181–97. https://doi.org/10.1016/j.cell.2018.02.016.
Martinez-Gutierrez CA, Aylward FO. Phylogenetic signal, congruence, and uncertainty across bacteria and archaea. Mol Biol Evol. 2021;38:5514–27. https://doi.org/10.1093/molbev/msab254.
Moody ER, Mahendrarajah TA, Dombrowski N, Clark JW, Petitjean C, Offre P, et al. An estimate of the deepest branches of the tree of life from ancient vertically evolving genes. Elife. 2022;11:e66695. https://doi.org/10.7554/eLife.66695.
Van Kessel MA, Speth DR, Albertsen M, Nielsen PH, Op den Camp HJ, Kartal B, et al. Complete nitrification by a single microorganism. Nature. 2015;528:555–9. https://doi.org/10.1038/nature16459.
Pachiadaki MG, Sintes E, Bergauer K, Brown JM, Record NR, Swan BK, et al. Major role of nitrite-oxidizing bacteria in dark ocean carbon fixation. Science 2017;358:1046–51. https://doi.org/10.1126/science.aan8260.
Palomo A, Pedersen AG, Fowler SJ, Dechesne A, Sicheritz-Pontén T, Smets BF. Comparative genomics sheds light on niche differentiation and the evolutionary history of comammox Nitrospira. ISME J. 2018;12:1779–93. https://doi.org/10.1038/s41396-018-0083-3.
Sun X, Kop LF, Lau MC, Frank J, Jayakumar A, Lücker S, et al. Uncultured Nitrospina-like species are major nitrite oxidizing bacteria in oxygen minimum zones. ISME J. 2019;13:2391–402. https://doi.org/10.1038/s41396-019-0443-7.
Sakoula D, Koch H, Frank J, Jetten MS, van Kessel MA, Lücker S. Enrichment and physiological characterization of a novel comammox Nitrospira indicates ammonium inhibition of complete nitrification. ISME J. 2021;15:1010–24. https://doi.org/10.1038/s41396-020-00827-4.
Daims H, Lebedeva EV, Pjevac P, Han P, Herbold C, Albertsen M, et al. Complete nitrification by Nitrospira bacteria. Nature. 2015;528:504–9. https://doi.org/10.1038/nature16461.
Pinto AJ, Marcus DN, Ijaz UZ, Bautista-de Lose Santos QM, Dick GJ, Raskin L. Metagenomic evidence for the presence of comammox Nitrospira-like bacteria in a drinking water system. mSphere. 2016;1:e00054–15. https://doi.org/10.1128/mSphere.00054-15.
Mueller AJ, Jung MY, Strachan CR, Herbold CW, Kirkegaard RH, Wagner M, et al. Genomic and kinetic analysis of novel Nitrospinae enriched by cell sorting. ISME J. 2021;15:732–45. https://doi.org/10.1038/s41396-020-00809-6.
Poghosyan L, Koch H, Lavy A, Frank J, van Kessel MA, Jetten MS, et al. Metagenomic recovery of two distinct comammox Nitrospira from the terrestrial subsurface. Environ Microbiol. 2019;21:3627–37. https://doi.org/10.1111/1462-2920.14691.
Palomo A, Dechesne A, Pedersen AG, Smets BF. Genomic profiling of Nitrospira species reveals ecological success of comammox Nitrospira. Microbiome. 2022;10:204. https://doi.org/10.1186/s40168-022-01411-y.
Burstein D, Sun CL, Brown CT, Sharon I, Anantharaman K, Probst AJ, et al. Major bacterial lineages are essentially devoid of CRISPR-Cas viral defence systems. Nat Commun. 2016;7:1–8. https://doi.org/10.1038/ncomms10613.
Frank YA, Kadnikov VV, Lukina AP, Banks D, Beletsky AV, Mardanov AV, et al. Characterization and genome analysis of the first facultatively alkaliphilic Thermodesulfovibrio isolated from the deep terrestrial subsurface. Front Microbiol. 2016;7:2000. https://doi.org/10.3389/fmicb.2016.02000.
Mullin SW, Wanger G, Kruger BR, Sackett JD, Hamilton-Brehm SD, Bhartia R, et al. Patterns of in situ mineral colonization by microorganisms in a ~60 C deep continental subsurface aquifer. Front Microbiol. 2020;11:536535. https://doi.org/10.3389/fmicb.2020.536535.
Umezawa K, Kojima H, Kato Y, Fukui M. Corrigendum to “Dissulfurispira thermophila gen. nov., sp. nov., a thermophilic chemolithoautotroph growing by sulfur disproportionation, and proposal of novel taxa in the phylum Nitrospirota to reclassify the genus Thermodesulfovibrio” [Syst. Appl. Microbiol. 44 (2021) 126184]. Syst Appl Microbiol. 2022;45:126323. https://doi.org/10.1016/j.syapm.2022.126323.
Rogers TJ, Buongiorno J, Jessen GL, Schrenk MO, Fordyce JA, de Moor JM, et al. Chemolithoautotroph distributions across the subsurface of a convergent margin. ISME J. 2023;17:140–50. https://doi.org/10.1038/s41396-022-01331-7.
Zhang W, Wang Y, Liu L, Pan Y, Lin W. Identification and genomic characterization of two previously unknown magnetotactic Nitrospirae. Front Microbiol. 2021;12:690052. https://doi.org/10.3389/fmicb.2021.690052.
Arshad A, Dalcin Martins P, Frank J, Jetten MS, Op den Camp HJ, et al. Mimicking microbial interactions under nitrate‐reducing conditions in an anoxic bioreactor: enrichment of novel Nitrospirae bacteria distantly related to Thermodesulfovibrio. Environ Microbiol. 2017;19:4965–77. https://doi.org/10.1111/1462-2920.13977.
Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018;36:996–1004. https://doi.org/10.1038/nbt.4229.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. https://doi.org/10.1101/gr.186072.114.
Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TB, et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35:725–31. https://doi.org/10.1038/nbt.3893.
Olm MR, Brown CT, Brooks B, Banfield JF. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11:2864–8. https://doi.org/10.1038/ismej.2017.126.
Asnicar F, Thomas AM, Beghini F, Mengoni C, Manara S, Manghi P, et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun. 2020;11:2500. https://doi.org/10.1038/s41467-020-16366-7.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60. https://doi.org/10.1038/nmeth.3176.
Kalyaanamoorthy S, Minh BQ, Wong TK, Von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9. https://doi.org/10.1038/nmeth.4285.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–4. https://doi.org/10.1093/molbev/msaa015.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Integrated nr database in protein annotation system and its localization. Nat Commun. 2010;6:1–8. https://doi.org/10.3389/fgene.2015.00348.
Eren AM, Kiefl E, Shaiber A, Veseli I, Miller SE, Schechter MS, et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat Microbiol. 2021;6:3–6. https://doi.org/10.1038/s41564-020-00834-3.
Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–84. https://doi.org/10.1093/nar/30.7.1575.
Benedict MN, Henriksen JR, Metcalf WW, Whitaker RJ, Price ND. ITEP: an integrated toolkit for exploration of microbial pan-genomes. BMC Genomics. 2014;15:1–1. https://doi.org/10.1186/1471-2164-15-8.
Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, Von Mering C, et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol. 2017;34:2115–22. https://doi.org/10.1093/molbev/msx148.
Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020;36:2251–2. https://doi.org/10.1093/bioinformatics/btz859.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. https://doi.org/10.1093/molbev/mst010.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3. https://doi.org/10.1093/bioinformatics/btp348.
Sheridan PO, Raguideau S, Quince C, Holden J, Zhang L. Thames Consortium, et al. Gene duplication drives genome expansion in a major lineage of Thaumarchaeota. Nat Commun. 2020;11:5494. https://doi.org/10.1038/s41467-020-19132-x.
Jaffe AL, Thomas AD, He C, Keren R, Valentin-Alvarado LE, Munk P, et al. Patterns of gene content and co-occurrence constrain the evolutionary path toward animal association in Candidate Phyla Radiation Bacteria. mBio. 2021;12:e00521–21. https://doi.org/10.1128/mBio.00521-21.
Morel B, Kozlov AM, Stamatakis A, Szöllősi GJ. GeneRax: a tool for species-tree-aware maximum likelihood-based gene family tree inference under gene duplication, transfer, and loss. Mol Biol Evol. 2020;37:2763–74. https://doi.org/10.1093/molbev/msaa141.
Tria FD, Landan G, Dagan T. Phylogenetic rooting using minimal ancestor deviation. Nat Ecol Evol. 2017;1:0193. https://doi.org/10.1038/s41559-017-0193.
Anantharaman K, Hausmann B, Jungbluth SP, Kantor RS, Lavy A, Warren LA, et al. Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 2018;12:1715–28. https://doi.org/10.1038/s41396-018-0078-0.
Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020;17:261–72. https://doi.org/10.1038/s41592-019-0686-2.
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42. https://doi.org/10.1093/sysbio/sys029.
Heritage S MBASR: Workflow-simplified ancestral state reconstruction of discrete traits with MrBayes in the R environment. BioRxiv. 2021. https://doi.org/10.1101/2021.01.10.426107.
Adam PS, Borrel G, Gribaldo S. Evolutionary history of carbon monoxide dehydrogenase/acetyl-CoA synthase, one of the oldest enzymatic complexes. Proc Natl Acad Sci USA. 2018;115:E1166–73. https://doi.org/10.1073/pnas.1807540115.
Westphal L, Wiechmann A, Baker J, Minton NP, Müller V. The Rnf complex is an energy-coupled transhydrogenase essential to reversibly link cellular NADH and ferredoxin pools in the acetogen Acetobacterium woodii. J Bacteriol. 2018;200:e00357–18. https://doi.org/10.1128/JB.00357-18.
Demmer JK, Pal Chowdhury N, Selmer T, Ermler U, Buckel W. The semiquinone swing in the bifurcating electron transferring flavoprotein/butyryl-CoA dehydrogenase complex from Clostridium difficile. Nat Commun. 2017;8:1577. https://doi.org/10.1038/s41467-017-01746-3.
Poudel S, Dunham EC, Lindsay MR, Amenabar MJ, Fones EM, Colman DR, et al. Origin and evolution of flavin-based electron bifurcating enzymes. Front Microbiol. 2018;9:1762. https://doi.org/10.3389/fmicb.2018.01762.
Zhuang WQ, Yi S, Bill M, Brisson VL, Feng X, Men Y, et al. Incomplete Wood–Ljungdahl pathway facilitates one-carbon metabolism in organohalide-respiring Dehalococcoides mccartyi. Proc Natl Acad Sci USA. 2014;111:6419–24. https://doi.org/10.1073/pnas.1321542111.
Koziaeva V, Dziuba M, Leão P, Uzun M, Krutkina M, Grouzdev D. Genome-based metabolic reconstruction of a novel uncultivated freshwater magnetotactic coccus “Ca. Magnetaquicoccus inordinatus” UR-1, and proposal of a candidate family “Ca. Magnetaquicoccaceae”. Front Microbiol. 2019;10:2290. https://doi.org/10.3389/fmicb.2019.02290.
Löffler M, Feldhues J, Venceslau SS, Kammler L, Grein F, Pereira IA, et al. DsrL mediates electron transfer between NADH and rDsrAB in Allochromatium vinosum. Environ Microbiol. 2020;22:783–95. https://doi.org/10.1111/1462-2920.14899.
Dahl C, Franz B, Hensen D, Kesselheim A, Zigann R. Sulfite oxidation in the purple sulfur bacterium Allochromatium vinosum: identification of soeABC as a major player and relevance of soxYZ in the process. Microbiology 2013;159:2626–38. https://doi.org/10.1099/mic.0.071019-0.
Boughanemi S, Infossi P, Giudici-Orticoni MT, Schoepp-Cothenet B, Guiral M. Sulfite oxidation by the quinone-reducing molybdenum sulfite dehydrogenase soeABC from the bacterium Aquifex aeolicus. Biochim Biophys Acta Bioenerg 2020;1861:148279. https://doi.org/10.1016/j.bbabio.2020.148279.
Gibson MI, Chen PY, Drennan CL. A structural phylogeny for understanding 2-oxoacid oxidoreductase function. Curr Opin Struct Biol. 2016;41:54–61. https://doi.org/10.1016/j.sbi.2016.05.011.
Nunoura T, Chikaraishi Y, Izaki R, Suwa T, Sato T, Harada T, et al. A primordial and reversible TCA cycle in a facultatively chemolithoautotrophic thermophile. Science. 2018;359:559–63. https://doi.org/10.1126/science.aao3407.
Mall A, Sobotta J, Huber C, Tschirner C, Kowarschik S, Bačnik K, et al. Reversibility of citrate synthase allows autotrophic growth of a thermophilic bacterium. Science. 2018;359:563–7. https://doi.org/10.1126/science.aao2410.
Yu H, Leadbetter JR. Bacterial chemolithoautotrophy via manganese oxidation. Nature. 2020;583:453–8. https://doi.org/10.1038/s41586-020-2468-5.
Yu H, Chadwick GL, Lingappa UF, Leadbetter JR. Comparative genomics on cultivated and uncultivated freshwater and marine “Candidatus Manganitrophaceae” species implies their worldwide reach in manganese chemolithoautotrophy. mBio. 2022;13:e03421–21. https://doi.org/10.1128/mbio.03421-21.
Graham ED, Tully BJ. Marine Dadabacteria exhibit genome streamlining and phototrophy-driven niche partitioning. ISME J. 2021;15:1248–56. https://doi.org/10.1038/s41396-020-00834-5.
Braakman R, Follows MJ, Chisholm SW. Metabolic evolution and the self-organization of ecosystems. Proc Natl Acad Sci USA 2017;114:E3091–100. https://doi.org/10.1073/pnas.1619573114.
Chen MY, Teng WK, Zhao L, Hu CX, Zhou YK, Han BP, et al. Comparative genomics reveals insights into cyanobacterial evolution and habitat adaptation. ISME J. 2021;15:211–27. https://doi.org/10.1038/s41396-020-00775-z.
Flieder M, Buongiorno J, Herbold CW, Hausmann B, Rattei T, Lloyd KG, et al. Novel taxa of Acidobacteriota implicated in seafloor sulfur cycling. ISME J. 2021;15:3159–80. https://doi.org/10.1038/s41396-021-00992-0.
Hausmann B, Pelikan C, Herbold CW, Köstlbacher S, Albertsen M, Eichorst SA, et al. Peatland Acidobacteria with a dissimilatory sulfur metabolism. ISME J. 2018;12:1729–42. https://doi.org/10.1038/s41396-018-0077-1.
Thorup C, Schramm A, Findlay AJ, Finster KW, Schreiber L. Disguised as a sulfate reducer: growth of the deltaproteobacterium Desulfurivibrio alkaliphilus by sulfide oxidation with nitrate. mBio. 2017;8:e00671–17. https://doi.org/10.1128/mBio.00671-17.
Battistuzzi FU, Feijao A, Hedges SB. A genomic timescale of prokaryote evolution: insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evolut Biol. 2004;4:1–4. https://doi.org/10.1186/1471-2148-4-44.
Marin J, Battistuzzi FU, Brown AC, Hedges SB. The timetree of prokaryotes: new insights into their evolution and speciation. Mol Biol Evol. 2016;34:437–46. https://doi.org/10.1093/molbev/msw245.
Jabłońska J, Tawfik DS. The evolution of oxygen-utilizing enzymes suggests early biosphere oxygenation. Nat Ecol Evol. 2021;5:442–8. https://doi.org/10.1038/s41559-020-01386-9.
Boden JS, Konhauser KO, Robbins LJ, Sánchez-Baracaldo P. Timing the evolution of antioxidant enzymes in Cyanobacteria. Nat Commun. 2021;12:4742. https://doi.org/10.1038/s41467-021-24396-y.
Sheridan PO, Meng Y, Williams TA, Gubry-Rangin C. Recovery of Lutacidiplasmatales archaeal order genomes suggests convergent evolution in Thermoplasmatota. Nat Commun. 2022;13:4110. https://doi.org/10.1038/s41467-022-31847-7.
Acknowledgements
The authors thank the science parties and crews of the following oceanographic expeditions for their help to collect the samples used for new single-cell genome data used in this study: expeditions AT18-07 (Juan de Fuca 2011), particularly chief scientists C. Geoff Wheat, Andrew Fisher, and Mike Rappe), AT42-01 (Lost City 2018, particularly co-chief scientists Susan Lang and William Brazelton), and IODP Expedition 357 in 2015 to Atlantis Massif (particularly co-chief scientist Gretchen Früh-Green). Samples from the Juan de Fuca Ridge flank observatories were collected with the consent of the Government of Canada, as reviewed by Global Affairs Canada; other marine subsurface samples originated from international waters and did not require permission for collection. Inyo-BLM 1 was sampled under scientific research permit DEVA-2013-SCI-0069 from the U.S. National Park Service (NPS). We thank Richard Friese, Josh Hoines, and Kevin Wilson of the NPS along with Alisa Lembke and the Inyo County Planning Commission (CA, USA) for site access and Great Basin Drilling (Greg Daun, president) and Jamieson Walker of the Nuclear Waste Repository Program Office for design and deployment of the pumping system at Inyo-BLM 1. We also thank Ali Saidi-Mehrabad, Molly Devlin, etc. for their pivotal support in sample collection from the Inyo-BLM 1 well in 2021. We thank the staff of the Single Cell Genomics Center at the Bigelow Laboratory for Ocean Science for SAG data generation. Funding for field work for new sample collection was provided by the U.S. National Science Foundation (award OCE-1536702 to Susan Lang for Lost City 2018, award OCE-1031808 to Andrew Fisher for AT18-07, and award OIA-1826734 to RS, DM, and BNO for Inyo-BLM1 sampling). Funding for genomic analyses was provided in part from the NSF (award OCE-173017 to BNO and award award OIA-1826734 to RS and BNO), from the NASA Exobiology program (80NSSC19K0466 to BNO), and from the Center for Dark Energy Biosphere Investigation (C-DEBI; subaward to BNO from OCE-0939654).
Author information
Authors and Affiliations
Contributions
TD conceived of the analysis with input from JG and BNO. TD performed all bioinformatic and statistical analysis. TD, JG, ML, JM, DM, and BO participated in field work and sample collection. RS oversaw genome sequencing. TD wrote the paper with input from all authors. BNO, RS, and DM secured funding for the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
D’Angelo, T., Goordial, J., Lindsay, M.R. et al. Replicated life-history patterns and subsurface origins of the bacterial sister phyla Nitrospirota and Nitrospinota. ISME J 17, 891–902 (2023). https://doi.org/10.1038/s41396-023-01397-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-023-01397-x
