
Abstract
Organohalide pollutants are prevalent in coastal regions due to extensive intervention by anthropogenic activities, threatening public health and ecosystems. Gradients in salinity are a natural feature of coasts, but their impacts on the environmental fate of organohalides and the underlying microbial communities remain poorly understood. Here we report the effects of salinity on microbial reductive dechlorination of tetrachloroethene (PCE) and polychlorinated biphenyls (PCBs) in consortia derived from distinct environments (freshwater and marine sediments). Marine-derived microcosms exhibited higher halotolerance during PCE and PCB dechlorination, and a halotolerant dechlorinating culture was enriched from these microcosms. The organohalide-respiring bacteria (OHRB) responsible for PCE and PCB dechlorination in marine microcosms shifted from Dehalococcoides to Dehalobium when salinity increased. Broadly, lower microbial diversity, simpler co-occurrence networks, and more deterministic microbial community assemblages were observed under higher salinity. Separately, we observed that inhibition of dechlorination by high salinity could be attributed to suppressed viability of Dehalococcoides rather than reduced provision of substrates by syntrophic microorganisms. Additionally, the high activity of PCE dechlorinating reductive dehalogenases (RDases) in in vitro tests under high salinity suggests that high salinity likely disrupted cellular components other than RDases in Dehalococcoides. Genomic analyses indicated that the capability of Dehalobium to perform dehalogenation under high salinity was likely owing to the presence of genes associated with halotolerance in its genomes. Collectively, these mechanistic and ecological insights contribute to understanding the fate and bioremediation of organohalide pollutants in environments with changing salinity.

Similar content being viewed by others


Introduction
A variety of halogenated organic chemicals (e.g., organochlorine pesticides and chlorinated solvents) are manufactured in massive amounts for agricultural and industrial applications [1, 2]. The extensive historical use and improper disposal of these compounds have led to their prevalent contamination on earth, which is likely to remain a long-term environmental issue for the foreseeable future [1, 3, 4]. Coastal regions which are home to nearly 40–50% of global human populations [5] are particularly vulnerable to severe contamination of coastal sediments, water, and biota by diverse anthropogenic organohalide pollutants [6, 7]. For example, polychlorinated biphenyls (PCBs) have been detected in the Venice Lagoon, which provides recreational and critical transportation services to residents [8,9,10]. This contamination poses ongoing risks to public health and ecosystems, such as the bioaccumulation of PCBs in humans via consumption of fish from contaminated marine environments [11]. Therefore, it is of vital importance to elucidate the fate and improve remediation of organohalide pollutants in coastal environments.
The marine environment is highly saline, with an average salinity of ~35 g/L in seawater, contrasting with the low salinity of freshwater and typical terrestrial soil and sediments (salinity <0.5 g/L) [12]. Salinity gradients form naturally along the transition from the land to the ocean due to freshwater runoff and seawater intrusion in coastal regions [12,13,14]. In the contexts of global sea level rise and climate change, it has been predicted that more groundwater and soil in coastal regions will encounter salinity alteration (i.e., elevating salinity in coastal freshwater systems and decreasing salinity in coastal marine systems) [13]. This salinity alteration will challenge the survival and metabolic activities of microorganisms in these systems [15, 16], for example, the inhibitory effects of elevating salinity on biological nitrogen cycling [17, 18], methanogenesis [19], and biotransformation of organic pollutants [20]. However, the responses of microbial reductive dehalogenation of organohalide pollutants performed by organohalide-respiring bacteria (OHRB) to changes of salinity remain largely unknown, with contradictory observations in limited studies [21,22,23]. For example, microbial reductive dechlorination activity of 1,2,3,4-tetrachlorodibenzo-p-dioxin in microcosms established with different estuarine sediments tended to decrease with increased in situ salinity in the sediments [21], whereas sediment microbiota from saline wetlands showed higher debromination activity of polybrominated diphenyl ethers than those from freshwater ponds in another study [22]. Separate studies also reported microbial reductive dechlorination of PCBs by sediment microbiota from the Venice Lagoon under high salinity (~30 g/L), but these reports did not analyze how PCB dechlorination activity changed with salinity [8,9,10, 24]. Therefore, more evidence is needed to clarify the impacts of salinity on microbial reductive dehalogenation. Additionally, mechanisms underlying the varying dehalogenation activity with salinity gradient remain to be elucidated. Furthermore, salinity has been reported as a critical environmental factor controlling microbial community structure and assembly in various ecosystems [14, 25,26,27], but its impacts on microbial ecology in dehalogenating cultures has not been investigated.
In this study, the responses of microbial reductive dechlorination of PCE and PCBs to different salinities (i.e., 2.5 − 50 g/L) were investigated in consortia derived from distinct environments (i.e., freshwater and marine sediments). The influence of salinity on succession, co-occurrence networks, and assembly of the dechlorinating microbial communities were elucidated by 16 S rRNA gene amplicon sequencing-based microbial community analyses. The microbial activities affected by high salinity in the dechlorinating consortia were revealed by multiple lines of evidence, including monitoring of dechlorination-supporting metabolic activities, growth quantification of functional populations, and in vivo and in vitro tests of microbial reductive dechlorination. Lastly, a halotolerant dechlorinating culture was obtained by consecutive subculturing of the marine microcosm via acclimation under high salinity, which was subjected to metagenomic analyses to elucidate the genetic mechanisms underlying halotolerance of dehalogenating microbial populations.
Materials and methods
Cultivation of microbial cultures
Dehalococcoides mccartyi strains MB and CG1 were cultivated in 160 mL serum bottles containing 100 mL anaerobic mineral salt medium spiked with PCE (final nominal concentration 0.5 mM), acetate (10 mM), and hydrogen (33.4 kPa) as previously described [28,29,30]. Sediment microcosms GY and F were derived from a non-saline freshwater sediment (0.2 g/L salinity) at an e-waste recycling site and a saline marine sediment (35 g/L salinity) in the East China Sea, respectively, which were cultivated in the same anaerobic mineral salt medium spiked with PCE (0.5 mM) and lactate (5 mM) [31, 32]. To investigate the influence of salinity on microbial reductive dehalogenation, the salinity of the anaerobic mineral salt medium was adjusted using NaCl (2.5, 5, 10, 15, 20, 25, 35, and 50 g/L). PCE (0.5 mM) or Aroclor1260 (80.65 µM) was separately spiked into different serum bottles as the model organohalide pollutants owing to their environmental prevalence and distinct structures [33, 34]. Subsequently, D. mccartyi strains (CG1 and MB) and microcosms (GY and F) were separately inoculated (5% v/v) into 160 mL serum bottles containing 100 mL of the same anaerobic mineral salt medium amended with different concentrations of NaCl.
To obtain halotolerant dechlorinating cultures, the marine microcosm F which showed PCE dechlorination activity under high salinity (i.e., 35 and 50 g/L) was consecutively sub-cultured under high salinity (i.e., 35 and 50 g/L). Autoclaved cultures spiked with PCE or Aroclor1260 were setup as abiotic control. All cultures were set up with triplicates and incubated at 30 ± 1 °C without shaking.
In vitro enzymatic assays of microbial reductive dechlorination
To investigate the effects of salinity on the metabolic activity of reductive dehalogenases (RDases), in vitro enzymatic activity assays were performed for D. mccartyi CG1 and the marine microcosm F owing to their better dechlorination performance. The in vitro assays were conducted as previously described [35]. Briefly, cells were harvested from 500 mL cultures of D. mccartyi CG1 and microcosm F grown on 0.5 mM PCE by centrifugation at 15 000 g and 4°C for 15 min. The collected cells were re-suspended in 10 mM tris-HCl solution and mechanically disrupted by sonication. The in vitro assays were performed in 20 mL serum bottles containing 4 mL tris-HCl (100 mM) buffered solution (pH=7.0) with titanium-citrate (20 mM) and methyl viologen (20 mM). Salinity in the in vitro assay solution was adjusted to 2.5 − 75 g/L using NaCl accordingly. PCE (0.5 mM) was spiked to in vitro assay bottles. Finally, 100 µL crude cell extracts were spiked to the serum bottles. Assay bottles were incubated at 30 ± 1 °C in the dark without shaking.
Chemical analyses
PCBs were extracted and measured on a gas chromatograph (GC; 6890 N; Agilent, Santa Clara, CA, USA) equipped with an electron capture detector and a DB-5 capillary column (30 m × 0.32 mm × 0.25 μm; J&W Scientific, Folsom, CA, USA) as previously described [36]. Chlorinated ethenes were measured by injecting 200 µL headspace gas to a GC (7890 A; Agilent) equipped with a flame ionization detector and a GS-GasPro column (30 m × 0.32 mm × 0.25 μm; J&W Scientific) [29]. Average chlorine per PCB (Cl/PCB) or chlorinated ethene (Cl/CE) were calculated and used to demonstrate dechlorination activity by including all dechlorination steps [37]. The time-course changes of Cl/PCB and Cl/CE were fitted to the first order equation to estimate the dechlorination rate constant k [38]. Hydrogen was detected with a GC (7890 A; Agilent) equipped with a thermal conductivity detector and a Varian PLT-5A column (30 m × 0.530 mm × 0.25 μm; Omega Scientific, Singapore). Short-chain volatile fatty acids including lactate and acetate were measured on a HPLC system (Agilent 1260) equipped with a UV detector (210 nm) and a Rezex ROA-Organic Acid H+ (8%) HPLC column (30 cm × 7.8 mm × 8 µm; Phenomenex, Singapore) [39].
Quantitative PCR and amplicon sequencing
For quantitative PCR and amplicon sequencing, cell pellets were harvested from 1 mL samples by centrifugation at 15 000 g and 4 °C for 15 min and stored at −80 °C until further processing. Genomic DNA extraction from the pure culture of Dehalococcoides and microcosms was performed using the DNeasy Blood & Tissue kit (QIAGEN, Hilden, Germany) and the DNeasy PowerSoil Pro Kit (QIAGEN), respectively, according to the manufacturer’s instructions. Quantitative PCR (qPCR) was conducted on an ABI 7500 Fast real-time PCR system with SYBR Green reporter (ABI, Foster City, USA) to quantify Dehalococcoides, Dehalobacter, Dehalogenimonas and Dehalobium using genus-specific primers targeting their 16 S rRNA genes (Table S1) [36, 40]. The standard curves were obtained by 10-fold serial dilution of plasmids containing target genes at concentrations ranging from 102 to 107 copies/µL. Quality of amplification was evaluated by melt-curves. Nuclease-free water was used as the negative control.
The universal primers U515F/U909R were used to amplify the V4-V5 regions of 16 S rRNA genes for amplicon sequencing [36]. The sequencing service on a Novaseq 6000 platform (Illumina, San Diego, CA, USA) was provided by Novogene AIT (Singapore) [35]. QIIME2 (v2021.4.0) was used to perform microbial community analysis [41]. Briefly, the demultiplexed sequencing data was quality filtered and denoised, followed by chimera removal and identification of amplicon sequence variants (ASVs) with DADA2 [42]. MAFFT and RAxML were used to align ASVs and construct the phylogeny, respectively [43, 44]. Taxonomic classification of ASVs was performed using the QIIME2 feature-classifier plugin trained against the SILVA 138 database [45, 46]. Microbial community diversity was analyzed using the q2-diversity plugin [42]. Ordination of distances between samples as determined by Bray-Curtis dissimilarity was done using principal coordinate analysis (PCoA) [47].
Network and assembly analyses of microbial community
Cooccurrence networks of microbial populations inferred from 16 S rRNA gene amplicon sequencing were analyzed for microcosms GY and F exposed to different salinities; genera with an average relative abundance of >0.01% in all samples were included. These genera accounted for 99.7 ± 0.3% of the entire microbial community in any sample. The samples were divided into four salinity groups based on the Venice classification system [48], namely: Group 1 (2.5 and 5 g/L salinity; n = 8), Group 2 (10 and 15 g/L; n = 8), Group 3 (20 and 25 g/L; n = 8), and Group 4 (35 and 50 g/L; n = 8). Individual networks were constructed for each group using the R packages VEGAN v2.5-7 [49], igraph v1.2.6 [50] and Hmisc v4.5-0 [51]. Correlations among different genera were evaluated using Spearman’s correlation coefficient with cutoff values of >|0.6| and p values < 0.01 [52]. To reduce false-positive correlations, p values were adjusted by the Benjamini–Hochberg method. Visualization of the constructed networks was performed with Gephi v0.9.2 [53].
Both stochastic and deterministic processes contribute to community assembly [54]. To infer the proportion of each process in governing community assembly, the normalized stochasticity ratio (NST) was estimated using the NST package as previously described [54]. Briefly, NST was calculated by normalizing the difference of the theoretical extreme values of selection stress under completely deterministic assembly (DSS) and the observed selection strength (SS) to the difference of the theoretical extreme values of SS under completely deterministic (DSS) and stochastic (TSS) assembly, namely \(NST = \frac{{DSS - SS}}{{DSS - TSS}}\) [54]. The Bray–Curtis metric was used for NST estimation and 1000 times randomization was run during null model (the PF null model) analyses. Because all microbial cultures were derived from the same parent microcosms, they were considered from the same regional species pool. That is, the regional species pool was taken to comprise all taxa detected in all samples and the abundance of each taxon was assigned by a random draw of individuals with probabilities proportional to the regional relative abundances of the taxa [54]. Additionally, 16 S rRNA genes have been reported to link microbes with their fitness under different salinities [14, 55, 56]. A cutoff value of 50% was set as the boundary value for NST. Stochastic processes are predicted to play more important roles in community assembly when NST is >50%, whereas deterministic processes overwhelm stochastic processes when NST is <50%.
Metagenome sequencing, binning, assembly, and annotation
Cell pellets were harvested (15000 × g, 15 min and 4 °C) from 100 mL enrichment cultures cultivated in medium with different salinities (5, 35 and 50 g/L NaCl) and amended with PCE (0.5 mM) and lactate (5 mM). A genomic DNA buffer kit with a 100-gauge Genomic-tip (QIAGEN) was used to extract genomic DNA according to the manufacturer’s instructions. Metagenomic sequencing was performed on a Novaseq 6000 platform (Illumina) in Novogene AIT (Singapore). Residual sequencing adapters, low quality, and short (<150 bp) reads were removed using BBDuk; cleaned reads were evaluated using FastQC and assembled using metaSPAdes v3.15.5 [57]. Assembled contigs were polished with Pilon v1.24 [58] and annotated with the DIAMOND search algorithm against the EggNOG v5.0 database [59] using emapper v2.1.5 [60]. Polished contigs were binned using CONCOCT v1.1.0 [61], metaBAT v2.15 [62], and maxBin2 v2.2.7 [63]. Bins identified by each binning algorithm were automatically compared and curated using DASTool v1.1.5 [64]. The relative abundance and quality of each bin were evaluated with CheckM 1.2.2 [65]. Bins were taxonomically characterized by BUSCO v5.4.4 [66] based on the presence of lineage-specific markers in the current BUSCO database (*_odb10); 16 S rRNA gene sequences were identified using RNAmmer v1.2 [67]. Presence and abundance of specific CDS in the assembled metagenome were determined using HT-seq v2.02 [68] and abundances of CDS were TPM-normalized to facilitate cross-sample comparison.
Results
Similar responses of microbial reductive dechlorination to elevating salinity in consortia derived from distinct environments
To determine the responses of microbial reductive dechlorination by non-saline terrestrial microbiota to elevating salinity, microcosm GY derived from a freshwater sediment in an e-waste recycling area was exposed to elevating salinity from 2.5 to 50 g/L, mimicking the salination phenomenon in coastal freshwater systems. Results showed that the rate of PCE dechlorination fitted best to the first-order rate equation. The dechlorination rate was relatively constant at salinity between 5 and 15 g/L, decreased sharply at 25 g/L, and was negligible at and beyond 35 g/L (Fig. 1a and S1). Additionally, the rate of dechlorination at moderate salinity (5–20 g/L) was higher than that at 2.5 g/L. In the active PCE-dechlorinating microcosm GY, PCE was sequentially dechlorinated to dichloroethenes (DCEs; Fig. S2). The ratio of trans-DCE to cis-DCE produced from PCE dechlorination in microcosm GY changed in the range of 0.46–0.75 under different salinities, with more trans-DCE being produced at low salinity (Fig. S3), suggesting the succession of different PCE-dechlorinating populations with increasing salinity. Similarly, the dechlorination rate constant of PCBs was relatively stable at salinity 5–15 g/L and dramatically decreased at 20 g/L, the value of which was negligible at salinity 35–50 g/L (Fig. 1c). The average chlorine per PCB (Cl/PCB) decreased from 6.44 ± 0.01 to 6.19 ± 0.04 and 6.36 ± 0.03 in microcosm GY exposed to a salinity of 2.5 g/L and 25 g/L, respectively, after 360 days incubation (Fig. S4), with majority of chlorines removed from the para- and meta-positions of PCBs (Fig. S5). Correspondingly, the PCB dechlorination rate constant decreased from 0.044 ± 0.002 year−1 to 0.009 ± 0.001 year−1 when salinity increased from 2.5 g/L to 25 g/L (Fig. 1c). These results showed that elevating salinity inhibited microbial reductive dechlorination in microcosms derived from non-saline terrestrial sediment in this study.

The dechlorination rate constants (k) of PCE and PCBs in (a, c) terrestrial-derived microcosms GY and (b, d) marine-derived microcosm F under different salinities. Error bars are the standard deviations of triplicates.
Because marine microorganisms may have naturally adapted to saline environments, we hypothesized that marine microbiota may catalyze PCE/PCB dechlorination at higher salinity than the non-saline terrestrial microbiota. To test this hypothesis, the marine microcosm F, capable of dechlorinating PCE and PCBs, was also exposed to different salinities. The marine microcosm F exhibited higher tolerance to high salinity, with PCE dechlorination proceeding even under the highest salinity tested in this study (50 g/L), which was not observed in microcosm GY (Fig. S6). However, PCE dechlorination rate constant decreased from 0.040 ± 0.005 d−1 to 0.006 ± 0.001 d−1 when salinity increased from 5 g/L to 25 g/L (Fig. 1b). The ratio of trans-DCE to cis-DCE in microcosm F also varied with salinity in the range of 0.40 − 1.70, with more trans-DCE produced under low salinity (<25 g/L; Fig. S3). Similarly, PCB dechlorination in microcosm F showed higher tolerance to high salinity. In contrast to the complete inhibition of PCB dechlorination in microcosm GY when salinity was >25 g/L, PCB dechlorination was still observed in microcosm F even under a high salinity of 35 g/L (Fig. S4). Different from the para- and meta-dechlorination of PCBs in microcosm GY, chlorines were preferentially removed from the meta- and ortho-positions of PCBs in microcosm F (Fig. S5), indicating distinct PCB-dechlorinating populations. Collectively, these results indicate that high salinity inhibited microbial reductive dechlorination in both the freshwater and marine sediment microbiota evaluated in this study. Yet, sustained PCE and PCB dechlorination by microbiota in microcosms derived from marine environments demonstrated higher halotolerance of marine-derived cultures relative to those derived from freshwater environments.
Salinity changes the succession, assembly, and co-occurrence networks of dechlorinating microbial communities
To gain more insight into the influence of changing salinity on microbial ecology in the PCE/PCB-dechlorinating cultures, 16 S rRNA gene amplicon sequencing-based analyses were performed. Strong negative correlations (r = −0.84, p < 0.001) between microbial diversity and salinity were observed in the freshwater microcosm GY, whereas there was no significant correlation between microbial diversity and salinity in the marine microcosm F (Fig. 2b and S7). PCoA showed that the dechlorinating microbial communities were separated by organohalide substrates, the origin of microbiota, and salinity in the dimensions of PC1, PC2, and PC3, explaining 29.3%, 18.5%, and 12.2% of the community variance, respectively (Fig. 2c and S8). However, it should be noted that the change of microbial community in microcosm GY in the dimension PC3 with salinity was more dramatic and significant than that in the microcosm F (Fig. S8c). These results indicate that salinity exhibits more profound influence on both the diversity and structure of microbial communities in the freshwater microcosm GY than the marine microcosm F. This is likely because a greater fraction of microbial populations in the marine microcosm which could naturally tolerate a wider range of salinity tend to be less affected by salinity. Microbial co-occurrence network analysis showed that the complexity of networks initially increased with elevating salinity, peaked in intermediate salinity (Group 2), but substantially decreased when salinity further increased (Group 3 and Group 4), indicating weakened microbial networks under high salinity (Fig. 3a, b). It is known that both deterministic (e.g., environmental filtering and biological interactions) and stochastic processes (e.g., random birth-death events, probabilistic dispersal, and ecological drift) could contribute to community assembly [54]. The low NST values (<30%) demonstrated that deterministic processes governed assembly of the dechlorinating microbial communities (Fig. 3c). Additionally, the NST values were lower in microbial communities under high salinity (i.e., Group 3 and 4) than that under comparatively lower salinity (i.e., Group 1 and 2), indicating that high salinity was an environmental filter driving community assembly to be more deterministic.

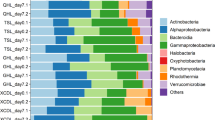
a Taxonomic compositions, (b) correlations between α-diversity and salinity (8 salinity concentrations and 2 samples at each salinity for each microcosm), (c) principal coordinates analysis of microbial communities in the soil microcosm GY and marine microcosm F under different salinities based on the Bray-Curtis dissimilarity metrics. The p values were obtained from ANOVA tests. The numbers besides the spheres represent the salinity in the microcosms (g/L).

a Co-occurrence networks (colored by modularity class) and (b) their topological properties of microbial communities in microcosms under different salinities. The number of connections (i.e., degree) between nodes are proportional to the size of each node. c The normalized stochasticity ratio (NST) of microbial communities under different salinities. Eight samples were included in each group during the network and community assembly analyses.
Detailed taxonomic analyses showed that Dehalococcoides and/or Dehalobium (informally named) were the dominant PCE/PCB dechlorinating populations, the relative abundance of which were at least two orders of magnitudes higher than that of other putative non-obligate OHRB (e.g., Geobacter, Sulfurospirillum, and Desulfitobacterium; Fig. 2a and Table S2). A shift of dominant obligate OHRB populations with elevating salinity was observed in microcosm F (Fig. 2a). The relative abundance of Dehalococcoides decreased with elevating salinity, whereas the relative abundance of Dehalobium increased from <0.3% to 6.2–13.6% when salinity increased from 2.5 g/L to 35–50 g/L in the active microcosms F, suggesting osmo-tolerance of Dehalobium. Besides OHRB, other microbial populations that may benefit or compete with PCE/PCB dechlorinating populations were also affected by salinity, the enrichment or extinction of which depended on organohalide substrate and source of microbiota. For example, Pelolinea, Cloacimonadales and Veillonellales-Selenomonadales were replaced by Desulfosarcinaceae when salinity was increased in microcosm F fed with PCBs; the relative abundance of both Mesotoga and Synergistaceae also increased as salinity increased. In microcosm F fed with PCE, the microbial community became dominated by Clostridium_sensu_stricto_7 at higher salinity, whereas the abundance of Lentimicrobium was markedly reduced.
High salinity directly suppresses growth of OHRB rather than nutrients providers
Dehalococcoides and/or Dehalobium have been identified as the dominant PCE/PCB dechlorinating bacteria in both microcosms, which exclusively use acetate and hydrogen as organic carbon and electron donor for dechlorination. Therefore, salinity may affect microbial reductive dechlorination by controlling the availability of acetate and hydrogen produced from acidogenesis and hydrogenesis or/and directly disturbing the viability of OHRB cells in microcosms supplied with lactate. To further elucidate the mechanisms underlying the decreased dechlorination performance with elevating salinity, volatile fatty acids and hydrogen were monitored in the PCE-dechlorinating microcosms GY and F. Lactate was rapidly fermented to acetate and hydrogen (Fig. S9). Acetate and hydrogen accumulated at higher concentrations in most PCE-dechlorinating microcosms GY and F under high salinity, suggesting that acidogenesis and hydrogenesis were not the limiting processes that hindered microbial reductive dechlorination under high salinity.
The cell abundance of obligate OHRB were quantified by qPCR to determine whether the growth of OHRB was directly inhibited under high salinity. Dehalococcoides was the dominant obligate OHRB in both microcosms when salinity was <35 g/L, but Dehalobium became the dominant one in active microcosm F under higher salinity (≥35 g/L; Table S3), in line with the 16 S rRNA gene amplicon sequencing results. Although the cell abundance of Dehalococcoides increased during PCE and PCB dechlorination in most active dechlorinating microcosms (Fig. 4), high salinity inhibited growth of Dehalococcoides, reflected by decreased maximum cell abundance and increased doubling time with elevating salinity (Table S4). For example, the doubling time of Dehalococcoides increased from 3.6 d and 9.5 d at 2.5 g/L salinity to 13.2 d and 30.7 d at 25 g/L salinity in microcosms GY dechlorinating PCE and PCBs, respectively. Nevertheless, the cell abundance of Dehalobium increased and outcompeted that of Dehalococcoides in microcosms F under the salinity of 35–50 g/L, indicating the predominant role of Dehalobium in dechlorinating PCE and PCBs under high salinity. These results showed that high salinity directly inhibited growth of OHRB, particularly Dehalococcoides, whereas syntrophic microorganisms sustained their metabolic activities to provide sufficient acetate and hydrogen for OHRB even under high salinity.

Changes in the cell abundance of Dehalococcoides and Dehalobium in microcosms (a, c) GY and (b, d) F during dechlorination of PCE and PCBs under different salinities. The values were the average of duplicates. Dehalobium was absent in microcosm GY. “×” indicates not detected.
Functionality of RDases in Dehalococcoides is not affected by high salinity
In vivo tests using D. mccartyi strains CG1 and MB were performed to further investigate the direct inhibitory effects of high salinity on Dehalococcoides for dechlorination in the defined pure cultures, which were provided with excessive amounts of acetate (carbon source) and hydrogen (electron donor) required by Dehalococcoides during microbial reductive dechlorination. Consistent with the observations in microcosms, high salinity (>10 g/L) inhibited PCE dechlorination in both pure cultures, e.g., increasing salinity from 2.5 to 20 g/L decreased the PCE dechlorination rate constant from 0.090 ± 0.003 d−1 and 0.054 ± 0.004 d−1 to 0.052 ± 0.002 d−1 and 0.019 ± 0.004 d−1 in D. mccartyi strains CG1 and MB, respectively (Fig. 5a, b, S10, and S11). PCE dechlorination was completely inhibited when salinity was >20 g/L although all essential substrates (e.g., acetate and hydrogen) were supplied at excessive amounts. Growth of strains CG1 and MB was also inhibited under high salinity (Fig. 5c, d), suggesting direct inhibition of high salinity on Dehalococcoides growth.

The dechlorination rate constant k and cell growth during PCE dechlorination in D. mccartyi strain (a, c) CG1 and (b, d) MB exposed to different salinities. The values were the average of duplicates. “×” indicates not detected.
RDases are the enzymes that directly cleave carbon-halogen bonds during microbial reductive dehalogenation. To clarify whether the inhibition of high salinity on Dehalococcoides was due to disruption of RDases or other cellular components, in vitro tests of PCE dechlorination were conducted under different salinities using crude cell extracts of PCE-grown cells. PCE dechlorination continued in the in vitro tests of strain CG1 and microcosm F under all the tested salinities (2.5–75 g/L; Fig. 6 and S12). The PCE dechlorination rate constant did not show obvious decrease even when salinity increased to 75 g/L in the in vitro tests (Fig. 6). These results indicate that RDases are intrinsically halotolerant, and high salinity most likely disrupted other unidentified cellular components in Dehalococcoides cells to inhibit dechlorination, which needs further investigation in future studies. The halotolerant property of RDases also provides an opportunity to acclimate dechlorinating microbiota to adapt to high salinity.

Dechlorination of PCE in the in vitro tests using crude cell extracts of (a) D. mccartyi CG1 and (b) microcosm F.
Acclimation of marine OHRB to high salinity and genomic insights
Owing to the higher halotolerance, marine microcosm F was continuously transferred under high salinity to investigate whether it could further adapt to high salinity with better dechlorination performance. PCE dechlorination rate increased by over 5 times in microcosm F under the salinity of 35 and 50 g/L after a single sub-culturing compared with the initial microcosm F before subculturing, reaching 0.021 ± 0.002 d−1 and 0.018 ± 0.001 d−1, respectively (Fig. 7a). High PCE dechlorination activity was maintained during the subsequent 3 rounds of sub-culturing under high salinity and was comparable with that under lower salinity of 5 g/L (i.e., 0.029 ± 0.002, 0.032 ± 0.006, and 0.027 ± 0.005 d−1 under the salinity of 5, 35 and 50 g/L, respectively; p > 0.05 based on Two-Sample t test). The ratio of trans-DCE to cis-DCE also stabilized during sub-culturing under different salinities, indicating stable compositions of PCE-dechlorinating populations in microcosms F under each salinity (Fig. 7b). Dehalococcoides and Dehalobium were retained as the dominant obligate OHRB under low (5 g/L) and high salinity (35 and 50 g/L), respectively, in the enrichment cultures after 4 times subculturing (Fig. 7c). For example, the cell abundance of Dehalobium was over 3 orders of magnitude higher than that of other OHRB in the enriched F culture under a salinity of 35 and 50 g/L. The rapid adaption of marine OHRB to high salinity implies that the marine environment could be an ideal niche to obtain halotolerant dehalogenating consortia.

The changes of (a) PCE dechlorination rate constant and (b) trans-DCE to cis-DCE ratio during continuous subculturing of microcosm F under different salinities. c Cell abundance of known obligate OHRB in the enrichment culture F after 4 times subculturing as quantified by qPCR. The values were the average of duplicates.
Metagenomic sequencing was performed for different sub-cultures to gain insights into the genetic mechanisms underlying increased halotolerance in the enrichments. Diverse genes involved in synthesis and transport of osmolytes (ectoine, glycine betaine, glucosylglycerol, proline and trehalose), sodium efflux and potassium uptake were detected (Fig. 8). A majority (83.0%) of these genes were detected at higher abundance in subcultures under high salinity (35 and 50 g/L) than that under low salinity (5 g/L; Fig. 8a). A more detailed analysis of metagenome bins representing Dehalococcoides and Dehalobium genomes was performed to identify potential genes that could contribute to the observed halotolerance in the latter. The metagenome of each high-salinity subculture contained one bin representing a draft genome of Dehalobium, whereas a Dehalococcoides genome was recovered from the subculture under low salinity. The Dehalobium genomes obtained from the different cultures shared high average nucleotide identity (ANI; 99.99%) and were of similar size (1.38−1.55 Mbp) and GC content (42.4–43.6%; Table S5). Each Dehalobium genome harbored 13 putative RDase genes, all of which are distinct from all other reported RDase genes (Table S6). Genes involved in proline synthesis (proA/B/C and ocd), glycine betaine/proline transport (proV/W/X), sodium efflux (nhaA) and potassium uptake (trkA/G/H) were also identified in the Dehalobium genomes (Fig. 8b), which may sustain the metabolic activity of Dehalobium under high salinity. These genes were detected in all Dehalobium genomes recovered from different subcultures that have been cultivated for different periods and under different salinities, suggesting that they were likely present in the genomes before enrichment and were not acquired (e.g., via horizontal gene transfer) during cultivation. Contrarily, one or more genes responsible for sodium efflux and proline synthesis and transport were absent in the Dehalococcoides genome, in line with the susceptibility of Dehalococcoides to high salinity. Besides halotolerance genes, metagenomic analyses showed that the genomes of diverse microorganisms contained genes responsible for fermentation of lactate to acetate and hydrogen (e.g., lactate dehydrogenase and acetate kinase genes) as well as cobalamin synthesis (e.g., glutamyl-tRNA reductase, uroporphyrin-III C-methyltransferase and porphobilinogen synthase genes; Table S7), which were absent in Dehalococcoides and Dehalobium genomes. This suggests potential microbial interactions and cross-feeding between OHRB and these syntrophic partners. Additionally, genomes of some putative fermenters (e.g., Clostridium and Desulfobacteraceae) harbored at least one set of genes associated with halotolerance, which was consistent with efficient fermentation of lactate to acetate and hydrogen under high salinity.

a The abundance of genes associated with halotolerance in different sub-cultures. b Presence and absence of genes associated with halotolerance in Dehalococcoides (Dhc) and Dehalobium (Dbium) genomes recovered from different subcultures. The full name of genes and their presence in each genome were provided in Table S7. SA5-T9, 9th subculturing under 5 g/L salinity; SA35-T4, 4th subculturing under 35 g/L salinity; SA35-T10, 10th subculturing under 35 g/L salinity; SA50-T4, 4th subculturing under 50 g/L salinity.
Discussion
OHRB-mediated bioremediation has been successfully applied to cleanup non-saline organohalide-contaminated sites in situ, where salinity is generally low (<0.5 g/L) [69,70,71]. Thus far, most bioremediation practices used commercial organohalide-dehalogenating consortia (e.g., KB-1 and SDC-9) containing Dehalococcoides derived from non-saline environments [69, 72]. To achieve effective bioremediation of organohalide-contaminated sites with salinity gradient such as coastal sediments, knowledge about the influence of salinity on microbial reductive dehalogenation is needed. Our findings showed that Dehalococcoides, whether derived from non-saline terrestrial or saline marine environments, are susceptible to high salinity; environmentally relevant high salinity (>20 g/L) severely inhibited PCE and PCB dechlorination. This result is consistent with inhibition of microbial reductive dechlorination of 1,2,3,4-tetrachlorodibenzo-p-dioxin by elevating salinity in estuarine sediments in a previous study [21]. This implies that bioremediation employing currently available commercial consortia, which have been demonstrated to be effective in remediating non-saline sites, may lead to uncertain performance at contaminated sites with high salinity. Nonetheless, this study showed that the marine consortia containing both Dehalococcoides and Dehalobium could better adapt to changing salinity, where Dehalococcoides and Dehalobium functioned to dechlorinate organohalides under low and high salinity, respectively. This was consistent with the reported capability of Dehalobium obtained from estuarine sediments to dechlorinate PCBs and 2,3,7,8-tetrachlorodibenzo-p-dioxin under high salinity [73, 74]. The succession of different OHRB populations with elevating salinity also suggests salinity as an important factor that can be manipulated to selectively enrich specific dechlorinating bacteria. It is notable that the marine consortia tended to remove a higher proportion of ortho-chlorines from PCBs when relative abundance of Dehalobium and salinity increased, indicating a potential role for Dehalobium in ortho-dechlorination of PCBs. From the marine consortia, a halotolerant and competent dechlorinating culture dominated by Dehalobium was obtained, suggesting that marine and other saline environments (e.g., saline wetlands and lakes) could serve as excellent niches to obtain halotolerant OHRB for bioremediation application in saline sites. However, isolation of the Dehalobium species from our marine consortia has not been achieved despite continuous efforts. This could be likely attributed to the inability of Dehalobium to grow axenically, and Dehalobium required Desulfovibrio in a co-culture for growth exclusively on organohalide respiration [73]. Nevertheless, we successfully constructed 3 high-quality draft genomes of Dehalobium via metagenomic sequencing, being the first draft genomes from members of this informally named genus. The obtained Dehalobium genomes harbored 13 putative RDase genes that showed low similarity (38.5−72.5%) to the currently known ones [75].
Aiming to elucidate the microbial activity affected by high salinity in the dechlorinating consortia, our comprehensive analyses showed that high salinity directly inhibited growth of OHRB, particularly Dehalococcoides, whereas the fermentation of lactate to acetate and hydrogen proceeded efficiently under high salinity. Additionally, it was revealed that RDases were intrinsically halotolerant, and high salinity likely disrupted other cellular components in Dehalococcoides. Contrarily, Dehalobium genomes were found to harbor genes for synthesis and transport of osmolytes, sodium efflux and potassium uptake, indicating that Dehalobium may survive under high salinity by employing both the “salt-in” strategy via accumulation of salts and the “compatible solute” strategy by synthesizing compatible organic compounds in cells [76]. However, our results cannot exclude other possible strategies for Dehalobium to survive under high salinity. For example, to maintain the functions of enzymes, the halotolerant OHRB may have also evolved to have a highly acidic proteome with a high excess of negatively charged amino acids, which may make the proteins more soluble and prevent protein aggregation under high salt conditions [77]. Future studies will further investigate the biomolecular mechanisms underlying halotolerance of Dehalobium via multi-omics approaches. Besides biomolecular evolution, physical shielding by large aggregates and biofilms may also help microorganisms survive under environmental stress (high salinity in this study) [78, 79]. In the environment, OHRB usually form a syntrophic relationship with other microorganisms (e.g., fermenters, methanogens, and sulfate-reducing bacteria) [80, 81]. The extensive microbial interactions frequently lead to the formation of large aggregate or thick biofilm [82, 83], which was also observed in our microcosms (Fig. S13). However, it remains to be unraveled how such aggregates and biofilms help OHRB survive under osmotic stress.
Understanding interactions and assembly in microbial communities as well as their relationship with environmental factors are fundamental goals in ecology [54, 84, 85]. Salinity has been reported as a critical environmental factor governing structure, function, and assembly of microbial communities in coastal wetlands, rivers, and salinized soil, where natural salinity gradient exists [14, 25, 26, 55]. But the influence of salinity on ecology in dechlorinating microbial community remains unknown. Our results showed that salinity exhibited profound impacts on the taxonomic compositions, diversity, assembly, and co-occurrence networks of dechlorinating microbial communities. Although the exact microbial populations responding to elevating salinity depended on the origins of microbiota and spiked organohalide substrates, the diversity and structure of microbial community showed similar responses to changing salinity in dehalogenating cultures fed with different substrates and derived from different environments. This suggests that the overall microbial community diversity and structure, rather than specific microbial populations, are more general and predictable responses to environmental factors (i.e., salinity in this study) [14]. However, the extent of responses of microbial diversity and structure to salinity may vary between microbiota from different environments. For example, microbial diversity in the microcosm derived from the non-saline terrestrial sediment decreased faster with elevating salinity than that derived from saline marine sediment, consistent with the observations in a prior study [25]. This implies that dechlorinating cultures originating from non- or low-saline habitats may encounter larger disturbance in microbial community when used for the bioremediation of saline sites. The initially stable microbial community linked with high dechlorination performance is more likely to be destroyed; this further highlights the necessity to develop halotolerant dechlorinating cultures for application in bioremediation of sites with salinity gradient. Accompanying with decreasing microbial diversity under elevating salinity, stochastic processes become less important in community assembly. By contrast, deterministic processes played increasingly overwhelming roles under high salinity [54]. This could be attributed to stronger environmental filtering (i.e., high osmotic stress). Collectively, our study provides new mechanistic and ecological insights into the impacts of salinity on microbial reductive dechlorination and the underlying microbial communities. This information, together with the obtained halotolerant dechlorinating consortia, contribute to bioremediation of organohalide-contaminated sites with salinity gradient.
Data availability
The 16 S rRNA gene amplicon sequencing and metagenomic sequencing data are archived at National Center for Biotechnology Information under accession number PRJNA836174.
References
Xu G, Zhao X, Zhao S, Chen C, Rogers MJ, Ramaswamy R, et al. Insights into the occurrence, fate, and impacts of halogenated flame retardants in municipal wastewater treatment plants. Environ Sci Technol. 2021;55:4205–26.
Jones KC. Persistent organic pollutants (POPs) and related chemicals in the global environment: some personal reflections. Environ Sci Technol. 2021;55:9400–12.
He H, Li Y, Shen R, Shim H, Zeng Y, Zhao S, et al. Environmental occurrence and remediation of emerging organohalides: A review. Environ Pollut. 2021;290:118060.
Zhang Y, Xi B, Tan W. Release, transformation, and risk factors of polybrominated diphenyl ethers from landfills to the surrounding environments: A review. Environ Int. 2021;157:106780.
Creel L. Ripple effects: population and coastal regions. DC: Population Reference Bureau Washington; 2003.
Fu J, Fu K, Chen Y, Li X, Ye T, Gao K, et al. Long-range transport, trophic transfer, and ecological risks of organophosphate esters in remote areas. Environ Sci Technol. 2021;55:10192–209.
Meng J, Hong S, Wang T, Li Q, Yoon SJ, Lu Y, et al. Traditional and new POPs in environments along the Bohai and Yellow Seas: an overview of China and South Korea. Chemosphere 2017;169:503–15.
Fava F, Zanaroli G, Young LY. Microbial reductive dechlorination of pre-existing PCBs and spiked 2,3,4,5,6-pentachlorobiphenyl in anaerobic slurries of a contaminated sediment of Venice Lagoon (Italy). FEMS Microbiol Ecol. 2003;44:309–18.
Zanaroli G, Perez-Jimenez JR, Young LY, Marchetti L, Fava F. Microbial reductive dechlorination of weathered and exogenous co-planar polychlorinated biphenyls (PCBs) in an anaerobic sediment of Venice Lagoon. Biodegradation. 2006;17:121–9.
Nuzzo A, Negroni A, Zanaroli G, Fava F. Identification of two organohalide-respiring Dehalococcoidia associated to different dechlorination activities in PCB-impacted marine sediments. Micro Cell Fact. 2017;16:127.
Domingo JL, Bocio A. Levels of PCDD/PCDFs and PCBs in edible marine species and human intake: a literature review. Environ Int. 2007;33:397–405.
Thorslund J, Bierkens MFP, Oude Essink GHP, Sutanudjaja EH, van Vliet MTH. Common irrigation drivers of freshwater salinisation in river basins worldwide. Nat Commun. 2021;12:4232.
Jasechko S, Perrone D, Seybold H, Fan Y, Kirchner JW. Groundwater level observations in 250,000 coastal US wells reveal scope of potential seawater intrusion. Nat Commun. 2020;11:3229.
Zhang G, Bai J, Tebbe CC, Zhao Q, Jia J, Wang W, et al. Salinity controls soil microbial community structure and function in coastal estuarine wetlands. Environ Microbiol. 2021;23:1020–37.
Marshall IPG, Karst SM, Nielsen PH, Jørgensen BB. Metagenomes from deep Baltic Sea sediments reveal how past and present environmental conditions determine microbial community composition. Mar Genomics. 2018;37:58–68.
Wood JM. Bacterial responses to osmotic challenges. J Gen Physiol. 2015;145:381–8.
Meng Y, Yin C, Zhou Z, Meng F. Increased salinity triggers significant changes in the functional proteins of ANAMMOX bacteria within a biofilm community. Chemosphere 2018;207:655–64.
Li W, Li H, Liu Y-D, Zheng P, Shapleigh JP. Salinity-aided selection of progressive onset denitrifiers as a means of providing nitrite for Anammox. Environ Sci Technol. 2018;52:10665–72.
De Vrieze J, Christiaens MER, Walraedt D, Devooght A, Ijaz UZ, Boon N. Microbial community redundancy in anaerobic digestion drives process recovery after salinity exposure. Water Res. 2017;111:109–17.
Alva VA, Peyton BM. Phenol and catechol biodegradation by the haloalkaliphile Halomonas campisalis: influence of pH and salinity. Environ Sci Technol. 2003;37:4397–402.
Dam HT, Häggblom MM. Impact of estuarine gradients on reductive dechlorination of 1,2,3,4-tetrachlorodibenzo-p-dioxin in river sediment enrichment cultures. Chemosphere 2017;168:1177–85.
Wang YF, Zhu HW, Wang Y, Zhang XL, Tam NFY. Diversity and dynamics of microbial community structure in different mangrove, marine and freshwater sediments during anaerobic debromination of PBDEs. Front Microbiol. 2018;9:952.
Fu QS, Barkovskii AL, Adriaens P. Microbial dechlorination of dioxins in estuarine enrichment cultures: effects of respiratory conditions and priming compound on community structure and dechlorination patterns. Mar Environ Res. 2005;59:177–95.
Zanaroli G, Balloi A, Negroni A, Daffonchio D, Young LY, Fava F. Characterization of the microbial community from the marine sediment of the Venice lagoon capable of reductive dechlorination of coplanar polychlorinated biphenyls (PCBs). J Hazard Mater. 2010;178:417–26.
Rath KM, Fierer N, Murphy DV, Rousk J. Linking bacterial community composition to soil salinity along environmental gradients. ISME J. 2019;13:836–46.
Rath KM, Maheshwari A, Rousk J. Linking microbial community structure to trait distributions and functions using salinity as an environmental filter. mBio. 2019;10:e01607–19.
Herlemann DP, Labrenz M, Jurgens K, Bertilsson S, Waniek JJ, Andersson AF. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011;5:1571–9.
Cheng D, He J. Isolation and characterization of Dehalococcoides sp. strain MB, which dechlorinates tetrachloroethene to trans-1,2-dichloroethene. Appl Environ Microbiol. 2009;75:5910–8.
Wang S, Chng KR, Wilm A, Zhao S, Yang KL, Nagarajan N, et al. Genomic characterization of three unique Dehalococcoides that respire on persistent polychlorinated biphenyls. Proc Natl Acad Sci USA. 2014;111:12103–8.
Xu G, Zhao S, Chen C, Zhao X, Ramaswamy R, He J. Dehalogenation of polybrominated diphenyl ethers and polychlorinated biphenyls catalyzed by a reductive dehalogenase in Dehalococcoides mccartyi strain MB. Environ Sci Technol. 2022;56:4039–49.
Xu G, He J. Resilience of organohalide-detoxifying microbial community to oxygen stress in sewage sludge. Water Res. 2022;224:119055.
Zhao S, Ding C, Xu G, Rogers MJ, Ramaswamy R, He J. Diversity of organohalide respiring bacteria and reductive dehalogenases that detoxify polybrominated diphenyl ethers in e-waste recycling sites. ISME J. 2022;16:2123–31.
Lu Q, Liang Y, Fang W, Guan KL, Huang C, Qi X, et al. Spatial distribution, bioconversion and ecological risk of PCBs and PBDEs in the surface sediment of contaminated urban rivers: a nationwide study in China. Environ Sci Technol. 2021;55:9579–90.
Schaefer CE, Lavorgna GM, Haluska AA, Annable MD. Long-term impacts on groundwater and reductive dechlorination following bioremediation in a highly characterized trichloroethene DNAPL source area. Ground Water Monit R. 2018;38:65–74.
Xu G, Lu Q, Yu L, Wang S. Tetrachloroethene primes reductive dechlorination of polychlorinated biphenyls in a river sediment microcosm. Water Res. 2019;152:87–95.
Xu G, Zhao X, Zhao S, He J. Acceleration of polychlorinated biphenyls remediation in soil via sewage sludge amendment. J Hazard Mater. 2021;420:126630.
Bedard DL, Van Dort H, Deweerd KA. Brominated biphenyls prime extensive microbial reductive dehalogenation of Aroclor 1260 in Housatonic river sediment. Appl Environ Microbiol. 1998;64:1786–95.
Lu Q, Zou X, Liu J, Liang Z, Shim H, Qiu R, et al. Inhibitory effects of metal ions on reductive dechlorination of polychlorinated biphenyls and perchloroethene in distinct organohalide-respiring bacteria. Environ Int. 2020;135:105373.
Ding C, Alvarez-Cohen L, He J. Growth of Dehalococcoides mccartyi species in an autotrophic consortium producing limited acetate. Biodegradation 2018;29:487–98.
Freeborn RA, West KA, Bhupathiraju VK, Chauhan S, Rahm BG, Richardson RE, et al. Phylogenetic analysis of TCE-dechlorinating consortia enriched on a variety of electron donors. Environ Sci Technol. 2005;39:8358–68.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6:90.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D6.
Bray JR, Curtis JT. An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr. 1957;27:325–49.
Alves AS, Adão H, Patrício J, Neto JM, Costa MJ, Marques JC. Spatial distribution of subtidal meiobenthos along estuarine gradients in two southern European estuaries (Portugal). J Mar Biol Assoc UK. 2009;89:1529–40.
Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14:927–30.
Csardi G, Nepusz T. The igraph software package for complex network research. Inter J Complex Syst. 2006;1695:1–9.
Harrell FE, Jr. Hmisc: Harrell miscellaneous. R package version 4.5-0 ed 2021.
Deng Y, Zhang P, Qin Y, Tu Q, Yang Y, He Z, et al. Network succession reveals the importance of competition in response to emulsified vegetable oil amendment for uranium bioremediation. Environ Microbiol. 2016;18:205–18.
Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media. Menlo Park, CA, USA: AAAI Press; 2009.
Ning D, Deng Y, Tiedje JM, Zhou J. A general framework for quantitatively assessing ecological stochasticity. Proc Natl Acad Sci USA. 2019;116:16892–8.
Zhang K, Shi Y, Cui X, Yue P, Li K, Liu X, et al. Salinity is a key determinant for soil microbial communities in a desert ecosystem. mSystems. 2019;4:e00225–18.
Campbell BJ, Kirchman DL. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J. 2013;7:210–20.
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–34.
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963.
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309–d14.
Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol. 2021;38:5825–9.
Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, et al. Binning metagenomic contigs by coverage and composition. Nat Methods. 2014;11:1144–6.
Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019;7:e7359.
Wu Y-W, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2015;32:605–7.
Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol. 2018;3:836–43.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 2021;38:4647–54.
Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–8.
Putri GH, Anders S, Pyl PT, Pimanda JE, Zanini F. Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics. 2022;38:2943–5.
Vainberg S, Condee CW, Steffan RJ. Large-scale production of bacterial consortia for remediation of chlorinated solvent-contaminated groundwater. J Ind Microbiol Biotechnol. 2009;36:1189–97.
Scheutz C, Durant ND, Dennis P, Hansen MH, Jørgensen T, Jakobsen R, et al. Concurrent ethene generation and growth of Dehalococcoides containing vinyl chloride reductive dehalogenase genes during an enhanced reductive dechlorination field demonstration. Environ Sci Technol. 2008;42:9302–9.
Xu G, Ng HL, Chen C, Zhao S, He J. Efficient and complete detoxification of polybrominated diphenyl ethers in sediments achieved by bioaugmentation with Dehalococcoides and microbial ecological insights. Environ Sci Technol. 2022;56:8008–19.
Duhamel M, Mo K, Edwards EA. Characterization of a highly enriched Dehalococcoides-containing culture that grows on vinyl chloride and trichloroethene. Appl Environ Microbiol. 2004;70:5538–45.
May HD, Miller GS, Kjellerup BV, Sowers KR. Dehalorespiration with polychlorinated biphenyls by an anaerobic ultramicrobacterium. Appl Environ Microbiol. 2008;74:2089–94.
Lee M, Liang G, Holland SI, O’Farrell C, Osborne K, Manefield MJ. Dehalobium species implicated in 2,3,7,8-tetrachlorodibenzo-p-dioxin dechlorination in the contaminated sediments of Sydney Harbour Estuary. Mar Pollut Bull. 2022;179:113690.
Molenda O, Puentes Jacome LA, Cao X, Nesbo CL, Tang S, Morson N, et al. Insights into origins and function of the unexplored majority of the reductive dehalogenase gene family as a result of genome assembly and ortholog group classification. Environ Sci Process Impacts. 2020;22:663–78.
Gunde-Cimerman N, Plemenitaš A, Oren A. Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol Rev. 2018;42:353–75.
Edbeib MF, Aksoy HM, Kaya Y, Wahab RA, Huyop F. Haloadaptation: insights from comparative modeling studies between halotolerant and non-halotolerant dehalogenases. J Biomol Struct Dyn. 2020;38:3452–61.
Yin W, Wang Y, Liu L, He J. Biofilms: the microbial “protective clothing” in extreme environments. Int J Mol Sci. 2019;20:3423.
Xu G, Zheng X, Lu Y, Liu G, Luo H, Li X, et al. Development of microbial community within the cathodic biofilm of single-chamber air-cathode microbial fuel cell. Sci Total Environ. 2019;665:641–8.
Yuan J, Li S, Cheng J, Guo C, Shen C, He J, et al. Potential role of methanogens in microbial reductive dechlorination of organic chlorinated pollutants in situ. Environ Sci Technol. 2021;55:5917–28.
Wang S, Chen C, Zhao S, He J. Microbial synergistic interactions for reductive dechlorination of polychlorinated biphenyls. Sci Total Environ. 2019;666:368–76.
Liang Y, Lu Q, Liang Z, Liu X, Fang W, Liang D, et al. Substrate-dependent competition and cooperation relationships between Geobacter and Dehalococcoides for their organohalide respiration. ISME Commun. 2021;1:23.
Kruse S, Turkowsky D, Birkigt J, Matturro B, Franke S, Jehmlich N, et al. Interspecies metabolite transfer and aggregate formation in a co-culture of Dehalococcoides and Sulfurospirillum dehalogenating tetrachloroethene to ethene. ISME J. 2021;15:1794–809.
Sutherland WJ, Freckleton RP, Godfray HCJ, Beissinger SR, Benton T, Cameron DD, et al. Identification of 100 fundamental ecological questions. J Ecol. 2013;101:58–67.
Deng Y, Jiang Y-H, Yang Y, He Z, Luo F, Zhou J. Molecular ecological network analyses. BMC Bioinforma. 2012;13:113.
Acknowledgements
This study was supported by the Ministry of Education, Singapore under Academic Research Fund Tier 2 under project No.: MOE-00003301 and Tier 1 under Project No.: R-302-000-239-114.
Author information
Authors and Affiliations
Contributions
GX and JH designed the study. GX and XZ performed the experiments. GX and MR analyzed the data. GX wrote the manuscript with the help of SZ. GX, SZ, MR and JH contributed to the revision and finalization of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xu, G., Zhao, X., Zhao, S. et al. Salinity determines performance, functional populations, and microbial ecology in consortia attenuating organohalide pollutants. ISME J 17, 660–670 (2023). https://doi.org/10.1038/s41396-023-01377-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-023-01377-1
