
Abstract
Sulfoquinovose (SQ) is one of the most abundant organosulfur compounds in the biosphere, and its biosynthesis and degradation can represent an important contribution to the sulfur cycle. To data, in marine environments, the microorganisms capable of metabolising SQ have remained unidentified and the sources of SQ are still uncertain. Herein, the marine Roseobacter clade bacteria (RCB) Dinoroseobacter shibae DFL 12 and Roseobacter denitrificans OCh 114 were found to grow using SQ as the sole source of carbon and energy. In the presence of SQ, we identified a set of highly up-regulated proteins encoded by gene clusters in these two organisms, of which four homologues to proteins in the SQ monooxygenase pathway of Agrobacterium fabrum C58 may confer the ability to metabolise SQ to these marine bacteria. The sulfite released from SQ desulfonation by FMN-dependent SQ monooxygenase (SmoC) may provide bacteria with reduced sulfur for assimilation, while proteins associated with sulfite production via assimilatory sulfate reduction were significantly down-regulated. Such SQ catabolic genes are restricted to a limited number of phylogenetically diverse bacterial taxa with the predominate genera belonging to the Roseobacter clade (Roseobacteraceae). Moreover, transcript analysis of Tara Oceans project and coastal Bohai Sea samples provided additional evidence for SQ metabolism by RCB. SQ was found to be widely distributed in marine phytoplankton and cyanobacteria with variable intracellular concentrations ranging from micromolar to millimolar levels, and the amounts of SQ on particulate organic matter in field samples were, on average, lower than that of dimethylsulfoniopropionate (DMSP) by one order of magnitude. Together, the phototroph-derived SQ actively metabolised by RCB represents a previously unidentified link in the marine sulfur cycle.
Similar content being viewed by others


Introduction
SQ is a sulfonated hexose compound that is present in higher plants, algae and photosynthetic bacteria [1]. SQ can be produced via microbial acyl- and glucoside hydrolases acting on sulfoquinovosyl diacylglycerol (SQDG), a sulfolipid that can partially compensate for phosphatidylglycerol deficiency in photosynthetic membranes under phosphate-limiting conditions [2, 3]. Up to 10 billion tons annually, SQ is one of the most abundant organic sulfur compounds in the biosphere [4, 5], and represents a significant fraction of the sulfur cycle.
Within the natural organic sulfur cycle, microorganisms generally decompose sulfur-containing organic compounds in order to grow, yielding sulfur that is further incorporated into living organisms or released into the environment [6, 7]. The bacterial catabolism of SQ has attracted considerable attention because of its significance to the global sulfur flux and the regulation of SQ metabolism in the intestinal track [4, 8, 9]. The existence of microbial SQ degradation was first observed in soil [10]. In addition to the abundant SQ-utilizing bacterial strains from soil environments, cultured isolates have been recovered from plants, sewage sludge, as well as fresh- and hypersaline water, and even the human intestine [8, 9, 11,12,13,14]. In parallel to classic glycolysis, bacteria can convert SQ to the three-carbon organosulfonates 2,3-dihydroxypropane-1-sulfonate (DHPS) and sulfolactate via analogous reactions through three pathways, namely the sulfoglycolytic Entner–Doudoroff (sulfo-ED), the sulfoglycolytic Embden-Meyerhof-Parnas (sulfo-EMP) and its variant (sulfo-EMP2) pathways [8, 11, 14]. The sulfo-EMP pathway is commonly found in Gram-negative intestinal bacteria and the sulfo-EMP2 pathway extends the sulfo-EMP to Gram-positive bacteria [8, 15]. The sulfo-ED pathway was frequently found in freshwater and soil bacteria [11]. Sulfolactate and DHPS can also be produced in aerobic and anaerobic bacteria, respectively, via the transaldolase-dependent sulfoglycolysis (sulfo-transaldolase) pathway [12]. Anaerobic bacteria were also found to carry out transketolase-dependent sulfoglycolysis (sulfo-transketolase) pathway for fermenting SQ, producing isethionate [14]. The sulfonate sulfur derived from these pathways could be further converted into sulfite or sulfide by other bacteria to complete a fully degradative process for SQ [13, 16,17,18,19]. Recently, a novel pathway (sulfoquinovose monooxygenase pathway, sulfo-SMO) has been identified as a prevalent SQ metabolic pathway across rhizobial strains, especially in the genera Agrobacterium and Rhizobium [20]. The sulfo-SMO pathway represents the only known complete degradation pathway for SQ within a single bacterial species, where the desulfonation of SQ into sulfite by a monooxygenase occurs at the first step and the subsequent two reduction reactions result in the formation of glucose for bacterial growth [20].
Marine eukaryotic phytoplankton and prokaryotic cyanobacteria have the ability to synthesize SQDG and thus have been proposed as the sources of SQ in the ocean [21]; however, there is no evidence for the presence of free-form SQ in their metabolites. Moreover, the known SQ metabolic pathways have not yet been reported in phytoplankton and cyanobacteria [21]. Accordingly, marine microorganisms can be considered metabolic specialists, having evolved the capacity to catabolize SQ similar to microorganisms in other ecosystems, but to date, the identity of marine SQ-degrading microorganisms remain unclear and the likely relevance of SQ in ecosystem functioning poorly understood. This work identifies such a missing module of the marine organic sulfur cycle by revealing the SQ degradation pathway in marine bacteria as well as the microbial catabolic potential and natural occurrence of SQ in the ocean.
Materials and methods
Chemical synthesis of SQ and DHPS
SQ was synthesized chemically using a previously published protocol [22]. The structure of SQ was verified by 1H NMR (Fig. S1) and mass spectrometry (Fig. S2). DHPS was synthesized chemically and verified elsewhere [17].
SQ: 1H NMR (400 MHz, D2O): δ = 5.24 (d, J = 2.28 Hz, 1H),4.70 (d, J = 5.28 Hz, 1H), 4.25 (t, J = 6.36 Hz, 1H), 3.82 (t, J = 6.24 Hz, 1H), 3.75 (t, J = 6.32 Hz, 1H), 3.58 (dd, J = 2.44, 6.48 Hz, 1H), 3.52 (dd, J = 6.12,12.32), 3.43 (d, J = 9.84 Hz, 2H), 3.30 (dd, J = 5.96, 11.56 Hz, 3H), 3.10 (m, 2H) ppm.
Cultivation
The strains D. shibae DFL 12 (DSM 16493), R. denitrificans OCh 114 (DSM 7001) and R. pomeroyi DSS-3 (DSM 15171) were obtained from German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). D. shibae DFL 12 and R. denitrificans OCh 114 were grown in modified Silicibacter basal medium (pH 7.2) [23] supplemented with SQ (6 mM), glucose (6 mM) or SQ (3 mM) plus glucose (3 mM). R. pomeroyi DSS-3 was grown in modified Silicibacter basal medium (pH 7.2) [23] supplemented with SQ (6 mM), yeast (0.25 g L-1) or SQ (6 mM) plus yeast (0.25 g L-1). The cultures (100 mL) were incubated in acid-cleaned and pre-steriled flasks at 28 °C with a shake speed of 160 rpm. At each sampling timepoint, triplicate biological replicates and triplet blank control samples were taken. Samples were taken at intervals to monitor the growth with OD600 and the concentrations of SQ, sulfite and sulfate.
Thalassiosira pseudonana 220, Navicula pelliculosa 350, Phaeodactylum tricornutum 267, Emiliania huxleyi CS369, E. Huxleyi 2090, E. Huxleyi 1516, Isochrysis galbana 079, I. galbana 321, Phaeocystis globose 124, Amphidinium carterae Hulburt 279, Alexandrium tamarense 260, Chlorella sp. 309, Chlorella sp. 410, Prochlorococcus sp. 9301, Prochlorococcus sp. 9312, Synechorococcus sp. XM24, Synechorococcus sp. 1333, Synechorococcus sp. YX04-3 were obtained from Center for Collections of Marine Algae (Xiamen University, China). Triplicate axenic phytoplankton cultures were grown in acid-cleaned and sterilized flasks in an incubator. Diatoms, dinoflagellates and haptophytes were grown under the following conditions: light of 100 μE·m-2·s-1 and temperature of 20 °C. Diatoms were incubated in f/2 media supplemented with Na2SiO3. Dinoflagellates and haptophytes were incubated in f/2 media [24] without Na2SiO3. Prochlorococcus and Synechococcus were grown on Pro99 media [25] and SN media [26], respectively, with light of 50 μE·m-2·s-1 and temperature of 20 °C. Samples of phytoplankton cultures at plateau phase were collected to analyze SQ and DHPS concentrations. Cell abundance of diatoms, haptophytes, cholorophyta and Synechococcus were measured using an Accuri C6 flow cytometer (BD Biosciences, New Jersey, USA). Cell abundance of Prochlorococcus was measured using FACSAria flow cytometer (BD Biosciences). Cell counts for dinoflagellates were measured using microscopy. Cell volumes of diatoms, haptophytes, cholorophyta and dinoflagellates (Table S1) were based on cell dimensions directly observed under the microscope and calculated using a previously reported method [27]. Cell volumes of cyanobacteria were based on previously reported values of cell dimension and size [28, 29].
Study sites and sample collection
Samples were collected from five sites (A18: 38.42° N, 119.42° E; A31: 38.75° N, 120.42° E; A35: 39.42° N, 119.70° E; A42: 37.75° N, 119.40° N; A45: 38.00 ° N, 120.42° E) in the Bohai Sea, China, on August 18–30, 2018 (Fig. S3). Seawater samples were collected using 10-L Niskin bottles mounted on a rosette sampler (model 1018, General Oceanics, Florida, USA). Seawater samples were collected from three depths (surface, ~3 m below the surface; middle; and bottom, ~2 m above the bottom), with the depths dependent on the sampling sites. Seawater (0.7–3 L) was pre-filtered through a nylon screen (200 μm), then filtered via a polycarbonate filter (47 mm, 3 μm, Merck Millipore, New Jersey, USA) to collect eukaryotic phytoplankton cells. After collection, the filters were immediately frozen by liquid nitrogen, and stored at −80 °C until further extraction. Approximately 0.5 L of seawater was filtered through precombusted (480 °C for 5 h) GF/F glass fiber filter membrane (47 mm, 0.75 μm, Whatman, USA), and the filters were subsequently stored at −20 °C until the measurement of particular organic carbon (POC) with an Elemental Analyzer (Carlo-Erba, Milano, Italy) [30].
Samples were collected from the SS1 station (14.00 ° N, 116.00 ° E) in the South China Sea, on July 13–14, 2019. Seawater was sampled using 10 L Niskin bottles mounted on rosette sampler (SBE-911 plus, SEA-BIRD, WA, USA). Sampling depth was ~5 m below the surface. Seawater (200 L) was pre-filtered via a polycarbonate filter (144 mm, 3 μm, Merck Millipore), then concentrated to 750 mL using crossflow filters (Sartocon, Göttingen, Germany) equipped with a Sartocon Hydrosart Cassette (0.6 m2, 0.2 µm, Sartocon). Concentrated seawater (100 mL) was taken to filter through a 0.2 μm polycarbonate membrane (Merck Millipore). The filters were immediately frozen at −20 °C until further extraction of metabolites. The solution of 50% glutaraldehyde was added to the concentrated seawater (1:100 v/v) and then frozen at −20 °C until further analysis. The cell abundance of Prochlorococcus within seawater was determined by FACSAria flow cytometer (BD Biosciences) [31].
Seawater samples were collected from a harbor in the Xiamen coastal sea (24.53 ° N, 118.18 ° E) using an acid-cleaned plastic bucket. Sampling depth was ~0.5 m below the surface. Seawater (7 L) was filtered via polycarbonate membrane (144 mm, 3 μm, Merck Millipore) followed by a 0.2 μm polycarbonate membrane (Merck Millipore). The 0.2 μm filters were immediately frozen at −20 °C until further extraction of intercellular SQ for Synechococcus in field sample. The solution of 50% glutaraldehyde was added to the 3 μm-filtered seawater (1:100 v/v) and then frozen at −20 °C until further analysis. The cell abundance of Synechococcus within seawater was determined by FACSAria flow cytometer (BD Biosciences) [31]. The 0.2 μm-filtered seawater was used for the extraction of dissolved SQ.
Extraction and sample preparation of metabolites in culture samples
For determination of metabolites in the medium supernatant, 10 μL of supernatant, from 100 μL of medium that had been centrifuged at 12,000 rpm for 15 min, was added to 990 μL of an extraction solvent, composed of acetonitrile, methanol and water (2:2:1 v/v/v) and stored at −20 °C for 2 h. The sample was centrifuged at 12,000 rpm for 5 min at 4 °C, and 100 μL of that supernatant was transferred to 1 mL glass vial (CNW, Düsseldorf, Germany).
For determination of intracellular metabolites from phytoplankton and cyanobacteria, cultures were filtered thru a 0.2 μm polycarbonate membrane (Merck Millipore). The filters were cut into pieces with dissecting scissors rinsed with water in 10% formic acid. The filter pieces were transferred to bead beating tubes and 1 mL of cool extraction solvent and approximately 500 μL of silica beads were added. The samples were shaken on a FastPrep-24 Homogenizer (MP Biomedicals, CA, USA) for 40 s and then frozen for 30 min. This step was repeated for a total of two times. The samples were centrifuged at 12,000 rpm for 5 min at 4 °C to remove precipitates. The 100 μL of supernatant was transferred to a 1 mL glass vial.
Extraction and sample preparation of metabolites in field samples
The intracellular metabolites of phytoplankton cells were extracted by acetonitrile:methanol:water (2:2:1 v/v/v, −20 °C) solution. Filter samples were homogenized using the FastPrep-24 Homogenizer (MP Biomedicals), followed by incubation at −20 °C for 2 h. After centrifugation, the supernatant was filtered through a hydrophilic polyvinylidene fluoride membrane filter (13 mm, 0.2 μm, JINTENG, Tianjin, China), and then stored at −80 °C until UPLC/MS analysis.
For determination of dissolved SQ in field samples, a 90-mL seawater sample was filtered through a polycarbonate membrane (47 mm, 0.2 μm, Merck Millipore) and then freeze-dried and redissolved in 6 mL acetonitrile. The precipitate was discarded after centrifuging at 5000 g for 10 min. The supernatant was transferred to a 40 mL brown glass vial and dried under a gentle stream of nitrogen at room temperature and redissolved in 100 µL of a mixture of acetonitrile and water (9:1 v/v). The sample was stored at −80 °C until HPLC/MS analysis after centrifuging at 8000 g for 10 min.
HPLC/MS analysis
The concentration of dissolved SQ in sea water was quantified using a Q Exactive high performance liquid chromatography system (Thermo Fisher Scientific, Waltham, USA) fitted with a ZIC-HILIC column (3.5 μm, 200 Å, 150 × 2.1 mm; Merck), which was connected to an orbitrap mass spectrometer (Thermo Fisher Scientific). Electrospray ionization was performed in negative mode ionization with the following parameters: capillary temperature 320 °C, spray voltage 3500 V, sheath gas flow 40 arbitrary units and aux gas flow 10 arbitrary units. The HPLC conditions were as follows: the column was equilibrated with 90% solvent A (100% acetonitrile) and 10% solvent B (10 mM NH4Ac) for 10 min. The gradient was comprised of an increase from 10% to 35% solvent B over 25 min, then held at 35% for 10 min, 35% to 10% over 0.5 min, and finally held at 10% for 10 min. The flow rate was 0.25 mL/min and the injection volume was 2 µL. Data analyses were performed with the Xcalibur software version 2.0.3 (Thermo Fisher Scientific). Calibration curve for synthetic SQ as standard was used to determine SQ concentrations. The detection limit was 10 nM for SQ with a signal-to-noise ratio of three.
UPLC/MS analysis
The concentrations of SQ, DHPS, DMSP, sulfolactate and isethionate in culture and on particulate organic matter were quantified by ultra-high performance liquid chromatography-mass spectrometry analysis. Metabolite concentrations were generated using an Agilent 6460 triple quadrupole mass spectrometer (Agilent, CA, USA) coupled to an Agilent 1290 Infinity II series UHPLC System (Agilent), with an UHPLC BEH Amide column (1.7 μm, 2.1 mm × 100 mm, Waters, MA, USA). The determination parameters were set up as described in [17]. Agilent MassHunter Work Station Software B.08.00 (Agilent) was used for MRM data acquisition and processing. Calibration curves for individual analytes were generated to determine metabolite concentrations. The detection limits were 0.61 nM for SQ, 0.08 nM for DHPS, 0.02 nM for DMSP, 0.30 nM for sulfolactate and 0.30 nM for isethionate with signal-to-noise ratios of three. Taurine-d4 was used as isotopically labelled internal standard.
The quantification for sulfite
Sulfite of culture supernatant was quantified colorimetrically by the fuchsin sulfite assay [20, 32], which requires solution A (4.0 mg of basic fuchsin was dissolved in 8.25 mL of deionized water at 0 °C, prior to the addition of 1.25 mL of 98% H2SO4), solution B (0.32 mL of 36% formaldehyde added into 9.68 mL deionized water at 0 °C) and solution C (1 mL solution A diluted with 7 mL deionized water, prior to addition of 1 mL solution B). Sample (36 μL) was mixed with solution C (174 μL). The mixture was incubated for 10 min at room temperature and the absorbance at 570 nm was measured using a SynergyH1 Multi-Mode Reader (BioTek, Winooski, Vermont). Calibration curve for a Na2SO3 standard was used to determine sulfite concentrations. The detection limit was calculated to 1.4 μM for sulfite with the standard deviation method [33].
The quantification for sulfate
Sulfate of culture supernatant was quantified using the ICS 5000 ion chromatography system (Dionex, Thermo Fisher Scientific) with IonPac AS15 column (9 μm, 4 mm × 250 mm, Dionex). The mobile phase was deionized water. Suppressor hydroxide was 36 mM and the current was 90 mA. The flow rate was 1.0 mL/min and the injection volume was 20 µL. Calibration curve for a Na2SO4 standard was used to determine sulfate concentrations. The detection limit of sulfate was 7.0 μM with a signal-to-noise ratio of three.
Proteomic analysis
D. shibae DFL 12 was grown in modified Silicibacter basal medium with SQ (6 mM) as the sole carbon source. The control group medium was glucose (3 mM) or acetate (9 mM) as the sole carbon source. Cell pellets were harvested after centrifugation at 12,000 rpm for 15 min. Lysis buffer (8 M urea, 1% protease inhibitor, Cocktail) was used to suspend the cell pellets and the mixture was sonicated three times on ice to lyse cells. Cell debris were removed by centrifugation at 12,000 g at 4 °C for 10 min. Concentration of extracted protein was determined with BCA Protein Assay Kit (Bio-Rad, CA, USA). Trichloroacetic acid was slowly added to the protein solution at a final concentration of 20%. The sample solution was placed at 4 °C for 2 h after vortexing and then centrifuged at 4500 g for 5 min. The supernatant was discarded. The protein precipitates were washed three times with cold acetone. Final concentration of 200 mM triethylamine borane was added to the dry protein precipitates followed by sonication to resuspend the protein pellets. Trypsin (1/50 w/w) was added to the sample solution to digest the protein overnight. The digested protein solution was reduced with dithiothreitol, 5 mM final concentration, for 30 min at 56 °C followed by incubation with 11 mM iodoacetamide at room temperature, in the dark, for 15 min. Digested peptides were desalted using a C18 SPE column (7 mm, volume 3 mL, Sigma, Missouri, USA) and vacuum dried. Peptides were then dissolved in solvent A (0.1% formic acid, 2% acetonitrile). The gradient was comprised of an increase from 6% to 24% solvent B (0.1% formic acid in 100% acetonitrile) over 70 min, 24% B to 35% B over 14 min, then climbing to 80% B in 3 min, and held at 80% B for 3 min, at a constant flow rate of 0.45 μL/min. Peptides were then subjected to capillary ion source followed by mass spectrometry (MS/MS) on a tims TOF Pro (Bruker, Billerica, USA) coupled online to an UPLC system with the spray voltage of 1.75 kV and MS/MS survey scan of 100–1700 m/z. Data acquisition was in the PASEF mode. The resulting MS/MS data were searched using MaxQuant (v1.6.15.0) against the genome sequences of D. shibae DFL 12 downloaded from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/). The protease specificity was set to trypsin/P. Two miscleavage events were allowed. The first search and main search of MS tolerance were both set at 20 ppm. MS/MS tolerance was set at 20 ppm. Carbamidomethylation of cysteine was set as a fixed modification. Oxidation of methionine as well as acetylation of protein N-termini were set as variable modifications. A false discovery rate (FDR) of 1.0% was set for protein and peptide-spectrum matching identifications. Differential expression analysis was performed using LFQ intensities, which were transformed to Log2, and the value of Log2FC were used to generate heatmap using GraphPad Prism 8.0.2 (GraphPad Software, San Diego, USA). The same protocol was used for the proteomic analysis of R. denitrificans OCh 114.
Bioinformatic analysis
The sulfur metabolism-related protein sequences in D. shibae DFL 12 (FMN-dependent SQ monooxygenase, SmoC, Dshi_2757; DHPS dehydrogenase, HpsN, Dshi_2034; DMSP demethylase, DmdA, Dshi_2320; L-cysteate sulfolyase, CuyA, Dshi_1439; phosphoadenylylsulfate reductase, CysH, Dshi_1152; sulfite oxidase, SoeB, Dshi_1237; sulfolactate sulfolyase, SuyB, Dshi_2038; sulfoacetaldehyde acetyltransferase, Xsc, Dshi_2045), taurine dioxygenase in Escherichia coli K12 (TauD, Eco06DRAFT_02825) and SQ dehydrogenase in Pseudomonas putida SQ1 (SqED, PpSQ1_00090) were BLASTp against the marine prokaryote genome databases MAR [34] with an E-value of 1e − 5. The candidate amino acid sequences of these genes were then submitted to similarity searches against UniRef90 (E-value, 1e − 5) [35] and were manually examined in terms of annotations. MAFFT [36] was used to align retrieved sequences with the query sequences that possessed known three-dimensional structures (Protein Data Bank ID: SmoC, 7OH2; DmdA, 3TFH; CuyA, 4D8T; CysH, 2GOY; TauD, 6EDH) [20, 37,38,39,40]. Those sequences containing conserved amino acid residues within the catalytic domains of enzymes were retained (Fig. S4a–e). AlphaFold2 was used to predict the structures of proteins [41, 42]. The structure of the lowest-scoring sequence had highly similar folds to the given protein structure of query sequence with the template modeling score > 0.5 measured by US-align [43, 44] (Fig. S4). IQ-TREE [45] was used to determine phylogenetic relationships between homologous sequences using the maximum likelihood method and 1000 bootstraps (Fig. S4). Thus, the minimum bit score of sequences was selected as a cut-off value of an alignment against the Tara Oceans database [46] and Bohai Sea metatranscriptomes [30] (143 for SmoC, 110 for DmdA, 139 for CuyA, 103 for CysH, 104 for TauD, 167 for HpsN, 276 for SoeB, 147 for SuyB, 160 for Xsc, 51 for SqED). The abundance results represented the homologs fraction in the total gene set for each sample. The abundance of eukaryotes and cyanobacteria were obtained from Tara Oceans OTU 18 S V9 version 2 database [47]. The potential key proteins of other SQ degradation pathways (sulfo-ED, sulfo-EMP, sulfo-EMP2, sulfo-transaldolase and sulfo-transketolase) were BLASTp against the Ocean Microbiomics Database [48], which included the metagenomic data (Tara Oceans, Malaspina, Biogeotraces, HOT and BATS) [49,50,51] and publicly available genome data (GORG SAGs, the MAR databases and manually-curated MAGs) [34, 52, 53]. The genomes that contained homologs (sequence identities > 40%, simultaneously) of all key proteins for SQ degradation pathways were retained for further analysis. Protein structures were displayed using Pymol 2.4 (Schrodinger, NY, USA). Violin plot was generated by GraphPad Prism 9 (GraphPad Software). Correlation analysis and graph creation were performed by OriginPro (OriginLab, MA, USA). The geographical pattern of smoC in the global oceans was visualized with Tableau 2022 (Tableau Software, Seattle, USA). A phylogenetic tree for the distribution of candidate sulfo-SMO genes in publicly available marine bacterial genomes using the concatenated alignment of 39 single-copy orthologous genes shared by bacterial strains was constructed by the Maximum Likelihood method using MEGA 6.0 with LG + F model plus a gamma distribution with eight categories [54]. Bootstrap resampling was performed for 1000 replications. Phylogenetic trees were visualized with Evolview v3 [55].
Heterologous expression and purification of proteins
The genes smoABC of D. shibae DFL 12 were synthesized by Sangon Biotech (Sangon, Shanghai, China). The synthesized genes were reconstructed into vectors pColdII followed by transformation into Escherichia coli BL21 (DE3), which were grown in LB media (50 μg/mL ampicillin) with shaking at 200 rpm and 37 °C until OD600 reached 0.8. The culture was cooled to 16 °C and isopropyl β-D-thiogalactopyranoside was added to the culture with a final concentration of 500 μM. The culture was maintained at 200 rpm of shaking at 16 °C for 16 h. Cells were harvested by 12,000 g of centrifugation for 30 min at 4 °C, followed by resuspension with 10 mM Tris-HCl buffer (pH 8.0, 150 mM NaCl). The suspension was subjected to sonication to disrupt the cells. Cell debris was removed by 12,000 g of centrifugation for 20 min at 4 °C. The supernatant was filtered through a 0.45 μm membrane (Merck Millipore) and then loaded onto AKTA pure (Cytiva, MA, USA) couple with a HisTrap HP column (5 mL, Cytiva). The product fraction was eluted with imidazole-containing (50–500 mM) elution buffer (10 mM Tris-HCl, 150 mM NaCl, pH 8.0) at a flow rate of 1 mL/min. The collected product fraction was further subjected to a desalting column (5 mL, Cytiva) with elution buffer to remove imidazole.
Enzyme assay and product analysis
For SmoA, a 200 μL reaction mixture containing 10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM NADPH, 100 μM FMN and 57 μM SmoA was incubated at 30 °C for 20 min. The absorbance at 340 nm was monitored by SynergyH1 Multi-Mode Reader (BioTek, Winooski, Vermont) in real time. For verifying the enzymatic function of SmoB, a 200 μL reaction mixture containing 10 mM Tris-HCl pH 8.0, 150 mM NaCl, 3 mM NADPH, 20 μM FMN, 11 μM SmoA and 2 μM SmoC was incubated at 30 °C for 3 h. SmoC and SmoA in the reaction mixture were removed by an ultrafiltration tube (1.5 mL, 3000 Dalton, Millipore), followed by the additions of 2 μM SmoB and 0.5 mM NADPH and allowed to continue to incubate for 1 h at 30 °C. After the addition of 990 μL acetonitrile into 10 μL final reaction mixture, the sample was subjected to LC-MS. The LC-MS protocol has been described in the HPLC/MS analysis section. For SmoC, a 200 μL reaction mixture containing 10 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM NADPH, 20 μM FMN, 5 mM SQ, 11 μM SmoA and 10 μM SmoC was incubated at 30 °C for 1 h. The 50 μL reaction mixture was taken to detect for the generation of sulfite in the acidic fuchsin assay [56]. The reaction mixture was derivatized with 2, 4-dinitrophenylhydrazine (DNPH) before LC-MS analyses [19]. LC-MS analysis was performed using a UPLC BEH C18 column (1.7 μm, 2.1 mm × 50 mm, Waters, MA, USA) with the following protocol: The column was equilibrated with 95% solvent A (0.1% formic acid) and 5% solvent B (100% acetonitrile) for 10 min. The gradient was comprised of an increase from 5% to 50% solvent B over 40 min, then an increase to 95% solvent B for 5 min, held at 95% for 5 min, then decreased from 95% to 5% over 1 min, and finally held at 5% for 10 min. The flow rate was 0.2 mL/min and the injection volume was 2 µL. Data analyses were performed with the Xcalibur software version 2.0.3 (Thermo Fisher Scientific).
Results and Discussion
Bacterial culture work
A marine Roseobacter clade bacterium, Dinoroseobacter shibae DFL 12 [57] was found to have the ability to degrade SQ (Fig. 1). The potential SQ catabolic products DHPS, sulfolactate and isethionate were not detected in culture supernatant. Instead, SQ was utilized completely by D. shibae DFL 12 for growth with the concomitant release of sulfite and sulfate into the culture medium (Fig. 1). D. shibae DFL 12 exhibited a significantly lower growth rate with SQ as the sole organic substrate compared to that in glucose and acetate (p < 0.01, t-test) (Fig. S5), suggesting that SQ was a less-preferred substrate for this organism. The bacterium Ruegeria pomeroyi DSS-3 displayed a relatively slow-growth rate with either sulfonate DHPS or isethionate as the sole organic substrate as well, but growth was faster when more-labile substrates were co-present with the less-labile sulfonates [58]. Similarly, the maximum growth rate and biomass for D. shibae DFL 12 grown on SQ plus glucose were both significantly higher than when grown on SQ alone (p < 0.01, t-test) (Fig. 1C, E). Meanwhile, the maximum SQ consumption rates for D. shibae DFL 12 in SQ plus glucose medium was higher than that in SQ only medium (Fig. 1D); however, the specific SQ degradation rates showed no significant difference (Fig. 1F), suggesting that the enhancement of utilization of SQ in SQ plus glucose relative to only SQ was most likely due to the increased biomass with growth on glucose (Fig. 1A, B, F, and S4). The simultaneous availability of multiple organic substrates derived from algae enables heterotrophic microorganisms to grow relatively fast, which would presumably be the case within a phycosphere that harbors various labile resources as well as less-labile substrates, such as sulfonates [58].

D. shibae DFL 12 utilization of SQ (A) and SQ plus glucose (B) for growth. Optical density (green) of D. shibae DFL 12, concentration of SQ in medium (orange), change in concentration of sulfate in medium (purple) and change in concentration of sulfite in medium (black), with respect to time. Maximum growth rate (C) and maximum biomass (E) for D. shibae DFL 12 grown on SQ and SQ plus glucose. Maximum (D) and specific (relative to total biomass) (F) SQ consumption rates for D. shibae DFL 12 grown on SQ and SQ plus glucose. Error bars represent the standard deviation (n = 3). P values were calculated based on t-test to indicate the significance between different groups.
Comparative proteomic analysis and gene functional verification
Compared to cells grown on glucose and acetate, one cluster of significantly up-regulated proteins in D. shibae DFL 12 were clearly discerned during growth with SQ, (locus tag, Dshi_2752-2759; fold-change > 3, q-value < 0.0001, Multiple t-test) (Fig. 2A; Table S2). The gene cluster was comprised of seven consecutive genes that had previously been annotated in the Integrated Microbial Genomes database [59] as alkanesulfonate monooxygenase, a short-chain oxidoreductase, a flavin reductase, a transcriptional factor, and TRAP dicarboxylate transporter components (Fig. 3). Among which, four encoded proteins were homologous to SmoABCD as encoded by a gene cluster in the soil bacterium A. fabrum C58 for SQ catabolism (Fig. 3) [20]. Intriguingly, two encoded proteins (Dshi_2756 and 2757; up-regulated 40- to 56-fold) shared substantial amino acid sequence similarity to the well characterized FMN-dependent SQ monooxygenase (SmoC) and 6-oxo-glucose oxidoreductase (SmoB) (78% and 67%, respectively) that mediate the conversion of SQ to sulfite and glucose in the sulfo-SMO pathway [20]. Another Roseobacter clade bacterium R. denitrificans OCh 114 [60] possesses the same gene contents as D. shibae DFL 12 but arranged in the opposite order (Fig. 3), allowing the use of SQ as a sole organic substrate (Fig. S6). Comparative proteomics of R. denitrificans OCh 114 grown on SQ, acetate and glucose showed that the two encoded proteins (locus tag, RD1_2310-2311) homologous to SmoC and SmoB in A. fabrum C58 were significantly up-regulated (6- to 16-fold) (Figs. 3, 2B; Table. S2). The transcriptional regulator (SmoD, locus tag, RD1_2309) in R. denitrificans OCh 114 was only identified during growth on SQ compared to acetate (Fig. 2B; Table. S2). A clear distinction between the gene clusters of these two RCB and A. fabrum C58 was the presence of dctPMQ that encods TRAP transporters in D. shibae DFL 12 and R. denitrificans OCh 114, instead of smoEFGH that encods a putative ABC-type SQ transporter in A. fabrum C58 (Fig. 3). Proteins encoded by dctM (locus tag, Dshi_2753) and dctQ (locus tag, Dshi_2754) were only identified during D. shibae DFL 12 growth on SQ (Fig. 2A; Table S2), and DctP (encoded by RD1_2315, up-regulated 1.4- to 24-fold) and DctM (encoded by RD1_2314, not detected in acetate group) of R. denitrificans OCh 114 were significantly up-regulated in the SQ group compared to the glucose and acetate groups (Fig. 2B; Table S2). Moreover, the homologues of TRAP transporter genes dctPMQ also exist in other organisms that harbor predicted smoABCD gene cluster and are adjacent to these SQ catabolic genes (Fig. S7). This information suggests that dctPMQ are probably associated with SQ uptake in D. shibae DFL 12 and R. denitrificans OCh 114 and further characterization will be required. Thus, these two RCB contained the entire set of genes necessary to specify the catabolism of SQ. Together, the gene clusters Dish_2752-2758 in D. shibae DFL 12 and RD1_2309-2315 in R. denitrificans OCh 114 were likely important for the growth of these two RCB strains on SQ.

D. shibae DFL 12 (A) and R. denitrificans OCh 114 (B) grown on SQ vs glucose and SQ vs acetate. Since each treatment was performed in triplicate, results are shown as average values. Corrected P-values (q-values) were calculated based on Multiple t-test using FDR approach (5%). Significance: *q < 0.05; **q < 0.01.

Gene functions are represented by different colors.  smoA, gene encoding flavin reductase;
smoA, gene encoding flavin reductase;  smoB, gene encoding oxidoreductase;
smoB, gene encoding oxidoreductase;  smoC, gene encoding FMN-dependent SQ monooxygenase;
smoC, gene encoding FMN-dependent SQ monooxygenase;  smoD, gene encoding GntR-like transcriptional regulator;
smoD, gene encoding GntR-like transcriptional regulator;  smoEFGH, genes encoding ABC transporters for SQGro (glycer-1-yl α-sulfoquinovoside) uptake;
smoEFGH, genes encoding ABC transporters for SQGro (glycer-1-yl α-sulfoquinovoside) uptake;  dctPMQ, genes encoding predicted TRAP dicarboxylate transporters for SQ uptake. Homologous genes are connected by colored bars between the genomes. The percentage values on the genes dctP, dctM and dctQ indicate the amino acid similarities between D. shibae DFL 12 and R. denitrificans OCh 114. The percentage values on the genes smoA, smoB, smoC and smoD indicate the amino acid similarities to the A. fabrum C58 proteins.
dctPMQ, genes encoding predicted TRAP dicarboxylate transporters for SQ uptake. Homologous genes are connected by colored bars between the genomes. The percentage values on the genes dctP, dctM and dctQ indicate the amino acid similarities between D. shibae DFL 12 and R. denitrificans OCh 114. The percentage values on the genes smoA, smoB, smoC and smoD indicate the amino acid similarities to the A. fabrum C58 proteins.
To verify the catalytic capacity of the predicted SQ metabolic enzymes of D. shibae DFL 12, an in vitro enzymatic assay was performed. SQ was incubated with SmoC, SmoA, FMN, and NADPH. The release of sulfite was detected by Fuchsin assay (Fig. S8) [14]. DNPH derivatization of reaction mixture generated a mass peak with negative ion m/z 357.0688 on LC-MS, which was consistent with DNPH-6-dehydroglucose (Fig. S9) [14], demonstrating the formation of 6-oxo-glucose. Incubation of SmoB with SQ, SmoA, SmoC, FMN and NADPH led to the appearance of [M + Na]+ ion peak at m/z 203.0524, which was identical to the glucose standard (Fig. S10), proving the formation of glucose. A decrease of absorbance at 340 nm over time after incubation of SmoA with NADPH and FMN (Fig. S11) indicated that FMN was catalyzed by SmoA. These results demonstrate that the enzymes encoded by smoACB in D. shibae DFL 12 indeed convert SQ to glucose.
The other striking result was comparative proteomic profiling of D. shibae DFL 12 and R. denitrificans OCh 114, which both showed that growth with SQ strongly down regulated three key proteins of the bacterial sulfate assimilation pathway. Sulfate adenylyltransferases (Sat, encoded by Dshi_2656 in D. shibae DFL 12 and RD1_B0030 in R. denitrificans OCh 114), which transforms sulfate into an organic intermediate, were down-regulated by 3- and 5-fold in D. shibae DFL 12 (1- and 3-fold in R. denitrificans OCh 114) compared to growth in glucose and acetate, respectively (Table S2). The severely down-regulated phosphoadenylylsulfate reductase (CysH, encoded by Dshi_1152, not detected in SQ-grown D. shibae DFL 12; encoded by RD1_2968, down-regulated by 9- and 91-fold in SQ-grown R. denitrificans OCh 114) and sulfite reductase (CysI, encoded by Dshi_1153, down-regulated by 16- and 60-fold in D. shibae DFL 12; encoded by RD1_2966, down-regulated by 50- and 71-fold in R. denitrificans OCh 114) reduced the conversion of organic intermediate product into sulfide, which is then used for the synthesis of sulfur-containing amino acids, while the expression of the protein involved in last step of cysteine biosynthesis remained stable (Table S2). This regulation pattern was identical in the presence of the phytoplankton metabolite DHPS [17], which is indicative of the close interaction between sulfonate metabolism and sulfate assimilation. When bacteria utilize sulfonate-derived sulfite for sulfur incorporation, it may save energy, at the equivalent of 2-3 ATP per molecule, compared to sulfate assimilation, showing that organosulfonates serve as a more economical sulfur source than sulfate for bacteria. Accordingly, the sulfate assimilation pathway was down-regulated when bacteria were offered reduced sulfur compounds, similar to the manner of sulfur demand-driven regulation in plant sulfate assimilation [61]. In contrast, as the sulfite anion is a reactive and toxic compound that can cause damage to vital cell components, such as DNA and protein, its level must be carefully controlled [62]. Bacterial sulfite oxidases were up-regulated in the presence of both SQ (Fig. 2) and DHPS catabolism [17], as a means of generating energy via sulfite oxidation but also for detoxifying excess amounts of sulfite.
The distribution of sulfo-SMO pathway in RCB
In contrast to D. shibae DFL 12 and R. denitrificans OCh 114, R. pomeroyi DSS-3 [63] failed to utilize SQ as a sole carbon source (Fig. S12), and no such SQ catabolic genes were found within its genome. Besides D. shibae DFL 12 and R. denitrificans OCh 114, around 1/3 of currently sequenced marine RCB (recently reclassified as Roseobacteraceae of the Rhodobacterales) [64] genomes (75 of 248 genomes) contained at least three genes that encode SmoABC enzymes required for complete SQ degradation (Fig. 4). The genetic capability for SQ degradation was also present in a limited number of marine relatives of the alphaproteobacterial Rhodobacteraceae of the Rhodobacterales (11 species), Rhizobiales (10 species), the SAR116 clade (2 species) and three gammaproteobacterial species (Fig. 4).

The tree was based on the 39 single-copy orthologous genes shared by bacterial strains that possess predicted smoABC. The different colors represent different taxa.  , Roseobacteraceae;
, Roseobacteraceae;  , Rhodobacteraceae;
, Rhodobacteraceae;  , Rhizobiales. White dots indicate that the species have the genetic potential of metabolising not only SQ, but also harbor the candidate genes for complete DHPS degradation pathways.
, Rhizobiales. White dots indicate that the species have the genetic potential of metabolising not only SQ, but also harbor the candidate genes for complete DHPS degradation pathways.
Marine bacteria are able to catabolize the ubiquitous algal metabolites DMSP and DHPS via DMSP demethylation (dmdA-mediated) [65] and three DHPS dehydrogenation reactions (encoded by hpsN, hpsO and hpsP) [17], respectively. These pathways have been found among a wider range of bacterial taxa than SQ degradation, with genes encoding proteins experimentally verified in the Roseobacter clade [17] and the SAR11 clade [66, 67]. The majority of candidate SQ-degrading RCB (61 of 75 strains) had predicted hpsN genes. More than half of the candidate SQ-degrading RCB contained predicted dmdA (51 of 75 strains) and (54 of 75 strains) harbored at least one predicted sulfonate desulfonation pathway that involved the metabolism of sulfolactate, isethionate and cysteate (mediated by suyAB, xsc and cuyA, respectively) (Fig. 4; Table S3). The ability to catabolize multiple naturally occurring organosulfur species might provide an advantage for RCB by allowing them to obtain a steady source of reduced sulfur in various marine environments. However, a total of 11 candidate SQ degraders lacked the capabilities of DMSP demethylation, DHPS dehydrogenation and sulfonates (sulfolactate, isethionate and cysteate) desulfonation (Table S3), suggesting that SQ is a resource that cannot be ignored for members of the Roseobacter clade.
Metatranscriptomes
5We further investigated the expression patterns of smoC, which encodes an enzyme for SQ desulfonation, across the Tara Oceans stations and Bohai Sea. All 186 of the available metatranscriptomic samples from the Tara Oceans contained smoC transcripts, showing a widespread occurrence of gene expression (Table S4). Tara Oceans’ smoC comprised on average 0.001% of the total prokaryotic metatranscriptomic reads, with levels almost one order of magnitude lower than that of the dmdA gene (on average 0.023%) and hpsN gene (on average 0.013%) which encodes a key enzyme for DHPS catabolism (Table S4) [16]. We also found wide expression of smoC from costal samples in the Bohai Sea (Fig. 5; Table S6). Similar to Tara Oceans, smoC transcripts was on average 0.001% of the total prokaryotic transcripts in Bohai Sea, and the reads of hpsN and dmdA were 2- to 41-fold and 5- to 42-fold higher than that of smoC, respectively (Table S6). dmdA and hpsN are not only highly abundant organosulfur compound degrading genes in the marine environment, but are also widespread and expressed among diverse marine bacterial taxa including the Roseobacter clade, SAR11 and Gammaproteobacteria. Similarly, the abundance of smoC transcripts in each sample was also significantly lower than those of genes involved in the desulfonation of sulfoacetaldehyde (xsc) and sulfolactate (suyB) that are known to be vital components of a sulfonate-based network between phytoplankton and heterotrophic bacteria (p < 0.0001, one-way analysis of variance (ANOVA) (Fig. 5) [67]. Those genes were identified within members of the Alpha-, Gamma- and Deltaproteobacteria [7], representing a wider range of taxonomic origins than smoC. In most (144 of 186 samples) smoC-containing metatranscriptomic samples of the Tara Oceans project, the majority of smoC transcripts were from members of the Roseobacter clade (Table S4), and the majority of the total smoC transcripts in each sample from Bohai Sea belonged to taxonomic relatives of the Roseobacter, suggesting that RCB were the predominate microorganisms carrying and actively expressing smoC in the ocean. This taxonomically limited distribution pattern in marine SQ catabolism suggests that SQ might be used for targeted feeding of specific RCB and enhance associations between ubiquitous RCB and phytoplankton especially in the SQ-abundant phycosphere. The homologous genes for other SQ degradation pathways (sulfo-EMP, sulfo-EMP2, sulfo-ED, sulfo-transaldolase and sulfo-transketolase) were not currently found in marine cultivated and uncultivated bacterial genomes, except that the five candidate genes for the sulfo-ED pathway were simultaneously detected in few bacterial genomes from Tara Oceans (Table S7). The transcripts abundance of the predicted SqED gene were on average lower than that of smoC in Tara Oceans by almost one order of magnitude (Fig. 5; Table S4). These results indicate that the sulfo-SMO pathway and its primary carrier RCB currently appear to be the most prevalent known SQ metabolic pathway and SQ consumers, respectively, in the marine pelagic environment.

Black dots represent the data from Bohai Sea. Relative abundance of genes transcripts in each sample was estimated with RPKM (reads per kilobase per million mapped reads). smoC, FMN-dependent SQ monooxygenase; SqED, SQ dehydrogenase; hpsN, DHPS dehydrogenase; suyB, sulfolactate sulfolyase; cuyA, L-cysteate sulfolyase; xsc, sulfoacetaldehyde acetyltransferase; tauD, taurine dioxygenase; soeB, sulfite oxidase; dmdA, DMSP demethylase; cysH, phosphoadenylylsulfate reductase. Statistical differences in relative abundance by analysis of variance (ANOVA). Black lines connect genes that are not significantly different, otherwise, significantly different (p < 0.001).
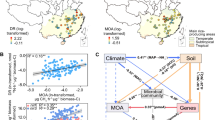
The highest levels of smoC abundance and expression were detected at the deep chlorophyll maximum layer of the Arctic Ocean (Table S4). Both dmdA and hpsN transcript abundances displayed significant positive correlation with chlorophyll a concentrations, as was smoC (Spearman’s r = 0.506, p < 0.001), while cysH showed a negative correlation (r = −0.438, p < 0.001) (Fig. 6), suggesting that marine phytoplankton provided exogenous reduced sulfur for bacterial assimilation. Furthermore, dmdA and hpsN transcript abundances were both positively correlated with diatoms, haptophytes and chlorophyta, while cysH displayed an inverse correlation (Fig. 6). Similarly, the expression level of smoC exhibited a strong positive correlation with diatoms (r = 0.582, p < 0.001) and chlorophyta (r = 0.595, p < 0.001) and a weak positive correlation with haptophytes (r = 0.149, p < 0.01) (Fig. 6). Diatoms are particularly prevalent at high latitudes and in upwelling zones [68], while haptophytes, such as the cosmopolitan Phaeocystis, are the main primary producers in polar marine environments [69]. Higher levels of smoC transcript abundance were observed in the polar waters of the Arctic and Southern Oceans, and the coastal waters of Chile-Peru Current compared to other Tara Oceans stations (Fig. 7; Table S4), indicating a clear regional pattern for SQ catabolism. Moreover, RCB from high latitudes (60–90 °N and 60–90 °S) contributed a higher proportion of the smoC gene transcripts (average 80% of smoC transcripts) than that from low latitudes (30 °S-30 °N) (average 60% of smoC transcripts) (Table S5), suggesting the relatively higher bacterial catabolism potential for SQ in the polar pelagic ocean.

smoC, FMN-dependent SQ monooxygenase; hpsN, DHPS dehydrogenase; suyB, sulfolactate sulfolyase; cuyA, L-cysteate sulfolyase; xsc, sulfoacetaldehyde acetyltransferase; tauD, taurine dioxygenase; soeB, sulfite oxidase; dmdA, DMSP demethylase; cysH, phosphoadenylylsulfate reductase. Color represents the Spearman’s r. Circle size represents statistical significance. Empty space indicates no significant correlation values (p ≥ 0.05).

The size of the cycle represents the relative abundance of smoC transcripts in each sample. Color bar represents the percent of smoC transcripts from RCB.
Cultured plankton and environmental metabolomics
The metabolite SQ was present at micromolar to millimolar cellular concentrations in all pure cultures of phytoplankton and cyanobacteria species investigated in this study, showing that SQ has both marine eukaryotic and bacterial origins (Table 1). The diatom Navicula pelliculosa was a high SQ producer with a median intracellular concentration in excess of 1 mM, which was comparable to the concentrations of the two cytosolic structural analogues sulfolactate and DHPS (0.9 ± 0.5 mM and 6.6 ± 1.3 mM, respectively). The other diatoms Thalassiosira pseudonana (44.5 ± 13.0 μM) and Phaeodactylum tricornutum (62.8 ± 19.5 μM) maintained low intracellular concentrations of SQ, which were around 153- and 128-fold lower than cytosolic DHPS concentrations, respectively. Sulfolactate also co-occurred with SQ and DHPS in the cytosol of T. pseudonana (113.9 ± 23.6 μM) and P. tricornutum (70.6 ± 15.3 μM) with SQ concentrations showing similar levels (Table 1). SQ production (97–527 μM) in the three haptophyte Emiliania huxleyi were lower 22- to 87-fold than cytosolic DHPS concentrations, which were more than the co-occurred isethionate concentrations by approximately one order of magnitude (Table 1). The sulfolactate cytosolic concentrations (4–15 μM) were similar to the levels of isethionate cytosolic concentrations (12–24 μM) in dinoflagellates (Table 1), which contained substantially lower micromolar concentrations of SQ than the reported hundreds of millimolar concentrations of the organosulfur compound DMSP in these organisms [70, 71]. Sulfolactate and isethionate were also detected in cholorophyta and cyanobacteria, all of which could produce SQ. In conjunction with the active sulfonate conversions in the surface ocean [67], SQ is likely an integral part of the sulfonate network in marine algae. Dissolved SQ was detected in the culture supernatants from all of these photosynthetic plankton with concentrations of up to 31.2 ± 10.9 μM in diatom culture supernatant and 80.2 ± 65.7 μM in Prochlorococcus culture supernatant. These findings suggested that phytoplankton and cyanobacteria mediated SQ production could be a common and important process in marine surface waters. Consistent with this hypothesis, SQ was detected at multiple coastal sites in the Bohai Sea. Particulate (size > 3 μm) SQ concentrations were up to 1.90 ± 0.76 μmol/g POC with an average of 0.88 ± 0.20 μmol/g POC in the water column (Fig. 8), which were ~5-fold lower than DHPS (8.43 ± 4.37 μmol/g POC) and ~10-fold lower than DMSP (10.46 ± 6.95 μmol/g POC) on particles that represent the major fraction of total DMSP in most sea waters [72]. Particulate sulfolactate (0.40 ± 0.36 μmol/g POC) and isethionate (0.48 ± 0.17 μmol/g POC) were both detected in these coastal sites at averages of 29.6% and 64.0% of the particulate SQ concentrations, respectively (Fig. 8). Dissolved SQ in natural seawater (0.2 ± 0.1 nM) was detected (Fig. S13), which was approximately 4-fold lower than the DHPS concentration in the dissolved organic matter pool (0.7 ± 0.5 nM) [16], suggesting that unbound SQ was released into seawater by photosynthetic organisms.

Sites A18, A31, A35, A42 and A45 were analyzed. The organosulfur concentrations were normalized by the concentrations of particulate organic carbon (POC). Error bars represent the standard deviation (n = 3). Asterisks indicate significant difference between SQ and different compounds (Two-way ANOVA; *p < 0.05; **p < 0.01; ***p < 0.001).
SQ concentrations were lower by one order of magnitude compared to DMSP in field samples (Fig. 8). Based on DMSP production that exceeds 103 Tg per year in the oceans, the annual particulate SQ production was estimated at teragram amounts. We generated a crude estimate of the annual SQ production in marine cyanobacteria at about 1.3 Tg, based on the total number of cyanobacterial cells (approximately Prochlorococcus 3 × 1027 cells and Synechococcus 7 × 1026 cells, respectively) [73], in conjunction with the SQ contents of single Prochlorococcus and Synechococcus cells in the marine environment, which were 1.7 ± 1.0 amol cell-1 and 0.5 ± 0.1 amol cell-1, respectively.
The amount of SQDG was higher than SQ by two orders of magnitude in T. pseudonana, making up ~9% of the cellular sulfur pool [67], suggesting that phytoplankton contained considerable amounts of SQ in a combined form. Normally, the SQDG degradation processes occurs in the gut and soil environments through glucoside hydrolase from bacteria, and also possibly from fungal, protist and animal origins [4]. Marine SQDG decomposition can occur as particles sink to deeper waters [74]; however, no defined enzyme has yet been linked to the conversion of SQDG to SQ in the ocean. Meanwhile, our results showed that marine algae and cyanobacteria could directly produce SQ and release it into the environment. RCB have no homologs to known SQDG degradation enzymes, but after release of photosynthetic organism-derived SQ into the seawater through viral lysis and active/passive exudation, it can be turned over by RCB through the monooxygenase pathway. We cannot exclude the possibility of the direct uptake and catabolism of SQDG in marine bacterial cells as bacteria can cleave complex carbohydrate compounds through the use of diverse glycosidases [75]. It has been shown that marine cyanobacteria and phytoplankton show a decline in phospholipids with a marked accumulation of SQDG in phosphorus-depleted environments [2, 3]. SQDG was often observed throughout the entire water column in the oligotrophic ocean ranging from 1-4% of organic carbon in sinking particles [74], and in some cases was comparable to the contribution of DMSP [72, 76, 77]. Given this high proportion of SQDG in marine phototrophic organisms and the role that they play in carbon and sulfur export, SQ transformation may be of global significance in the ocean.
Conclusion
Since the identification of the first SQ-degrader thirty years ago, it has been shown that SQ catabolism is widely present in various environments; however, little known is about this process in the ocean. Our study revealed that marine RCB, such as D. shibae DFL 12 and R. denitrificans OCh 114, not only used the recently identified monooxygenase pathway to catabolize SQ as carbon and energy source, but also the reduced sulfur for assimilation. This pathway was associated primarily with the members of the Roseobacter clade. The ubiquitous gene expression for SQ desulfonation throughout the global ocean indicates this molecule likely plays an important and yet overlooked role in the marine sulfur cycle. Furthermore, SQ was found to be widespread in marine phytoplankton and cyanobacteria. The prevalence of SQ in the marine environment and the ability of SQ metabolism in specialized species highlight the importance of RCB as major players in ocean biogeochemical organosulfur transformations.
Data availability
Proteomic data have been uploaded to the National Omics Data Encyclopedia (NODE, https://www.biosino.org/node/) database with the accession number OEP003145.
References
Snow AJD, Burchill L, Sharma M, Davies GJ, Williams SJ. Sulfoglycolysis: Catabolic pathways for metabolism of sulfoquinovose. Chem Soc Rev. 2021;50:13628–45.
Van Mooy BAS, Rocap G, Fredricks HF, Evans CT, Devol AH. Sulfolipids dramatically decrease phosphorus demand by picocyanobacteria in oligotrophic marine environments. Proc Natl Acad Sci USA 2006;103:8607–12.
Wu J, Sunda W, Boyle EA, Karl DM. Phosphate depletion in the western North Atlantic. Ocean Sci 2000;289:759–62.
Goddard-Borger ED, Williams SJ. Sulfoquinovose in the biosphere: occurrence, metabolism and functions. Biochem J. 2017;474:827–49.
Harwood JL, Nicholls RG. The plant sulpholipid- a major component of the sulphur cycle. Biochem Soc Trans. 1979;7:440–7.
Moran MA, Durham BP. Sulfur metabolites in the pelagic ocean. Nat Rev Microbiol. 2019;17:665–78.
Tang K. Chemical diversity and biochemical transformation of biogenic organic sulfur in the ocean. Front Mar Sci. 2020;7:68.
Denger K, Weiss M, Felux AK, Schneider A, Mayer C, Spiteller D, et al. Sulphoglycolysis in Escherichia coli K-12 closes a gap in the biogeochemical sulphur cycle. Nature 2014;507:114–7.
Hanson BT, Kits KD, Loffler J, Burrichter AG, Fiedler A, Denger K, et al. Sulfoquinovose is a select nutrient of prominent bacteria and a source of hydrogen sulfide in the human gut. ISME J. 2021;15:2779–91.
Strickland TC, Fitzgerald JW. Mineralization of sulfur in sulfoquinovose by forest soils. Soil Biol Biochem. 1983;15:347–9.
Felux AK, Spiteller D, Klebensberger J, Schleheck D. Entner-Doudoroff pathway for sulfoquinovose degradation in Pseudomonas putida SQ1. Proc Natl Acad Sci USA 2015;112:E4298–E305.
Frommeyer B, Fiedler AW, Oehler SR, Hanson BT, Loy A, Franchini P, et al. Environmental and intestinal phylum Firmicutes bacteria metabolize the plant sugar sulfoquinovose via a 6-deoxy-6-sulfofructose transaldolase pathway. Iscience. 2020;23:101510.
Roy AB, Hewlins MJE, Ellis AJ, Harwood JL, White GF. Glycolytic breakdown of sulfoquinovose in bacteria: A missing link in the sulfur cycle. Appl Environ Microbiol. 2003;69:6434–41.
Liu J, Wei Y, Ma K, An J, Liu X, Liu Y, et al. Mechanistically diverse pathways for sulfoquinovose degradation in bacteria. ACS Catal. 2021;11:14740–50.
Zhang S, Li Z, Yan Y, Zhang C, Li J, Zhao B. Bacillus urumqiensis sp. nov., a moderately haloalkaliphilic bacterium isolated from a salt lake. Int J Syst Evol Microbiol. 2016;66:2305–12.
Durham BP, Sharma S, Luo H, Smith CB, Amin SA, Bender SJ, et al. Cryptic carbon and sulfur cycling between surface ocean plankton. Proc Natl Acad Sci USA 2015;112:453–7.
Chen X, Liu L, Gao X, Dai X, Han Y, Chen Q, et al. Metabolism of chiral sulfonate compound 2,3-dihydroxypropane-1-sulfo-nate (DHPS) by Roseobacter bacteria in marine environment. Environ Int. 2021;157:106829.
Liu J, Wei Y, Lin L, Teng L, Yin J, Lu Q, et al. Two Radical-dependent mechanisms for anaerobic degradation of the globally abundant Organosulfur Compound Dihydroxypropanesulfonate. Proc Natl Acad Sci USA 2020;117:15599.
Xing M, Wei Y, Zhou Y, Zhang J, Lin L, Hu Y, et al. Radical-mediated C-S bond cleavage in C2 sulfonate degradation by anaerobic bacteria. Nat Commun. 2019;10:1609.
Sharma M, Lingford JP, Petricevic M, Snow AJD, Zhang Y, Jarva MA, et al. Oxidative desulfurization pathway for complete catabolism of sulfoquinovose by bacteria. Proc Natl Acad Sci USA 2022;119:e2116022119.
Scholz SS, Serif M, Schleheck D, Sayer MDJ, Cook AM, Kupper FC. Sulfoquinovose metabolism in marine algae. Bot Mar. 2021;64:301–12.
Abayakoon P, Epa R, Petricevic M, Bengt C, Mui JWY, van der Peet PL, et al. Comprehensive synthesis of substrates, intermediates, and products of the sulfoglycolytic Embden-Meyerhoff-Parnas pathway. J Org Chem. 2019;84:2901–10.
Denger K, Smits THM, Cook AM. L-Cysteate sulpho-lyase, a widespread pyridoxal 5 ‘-phosphate-coupled desulphonative enzyme purified from Silicibacter pomeroyi DSS-3. Biochem J. 2006;394:657–64.
Guillard RRL. Culture of Phytoplankton for Feeding Marine Invertebrates. Smith WL, Chanley MH, (eds): Springer US; 1975. Boston, MA. pp 29–60.
Moore LR, Coe A, Zinser ER, Saito MA, Sullivan MB, Lindell D, et al. Culturing the marine cyanobacterium Prochlorococcus. Limnol Oceanogr Methods. 2007;5:353–62.
Waterbury J, Watson S, Valois F, Franks D. Biological and ecological characterization of the marine unicellular cyanobacterium Synechococcus. Platt T, Li WKW, (eds). Department of Fisheries and Oceans, Ottawa 1986. pp 71–120.
Olenina I, Hajdu S, Edler L, Andersson A, Wasmund N, Busch S, et al. Biovolumes and size-classes of phytoplankton in the Baltic Sea. HELCOM Balt Sea Environ Proc. 2006;106:144.
Zheng Q, Wang Y, Lu J, Lin W, Chen F, Jiao N. Metagenomic and metaproteomic insights into photoautotrophic and heterotrophic interactions in a Synechococcus culture. mbio 2020;11:e03261–19.
Partensky F, Hess WR, Vaulot D. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol Mol Biol Rev. 1999;63:106–27.
Han Y, Zhang M, Chen X, Zhai W, Tan E, Tang K. Transcriptomic evidences for microbial carbon and nitrogen cycles in the deoxygenated seawaters of Bohai Sea. Environ Int. 2022;158:106889.
Li WKW. Primary production of prochlorophytes, cyanobacteria, and eukaryotic ultraphytoplankton - measurements from flow cytometric sorting. Limnol Oceanogr. 1994;39:169–75.
Denger K, Ruff A, Rein U, Cook AM. Sulphoacetaldehyde sulpho-lyase (EC 4.4.1.12) from Desulfonispora thiosulfatigenes: purification, properties and primary sequence. Biochem J. 2001;357:581–6.
Ismail R, Lee HY, Mahyudin NA, Abu, Bakar F. Linearity study on detection and quantification limits for the determination of avermectins using linear regression. J Food Drug Anal. 2014;22:407–12.
Klemetsen T, Raknes IA, Fu J, Agafonov A, Balasundaram SV, Tartari G, et al. The MAR databases: development and implementation of databases specific for marine metagenomics. Nucleic Acids Res. 2018;46:D692–D9.
Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007;23:1282–8.
Rozewicki J, Li S, Amada KM, Standley DM, Katoh K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019;47:W5–W10.
Schuller DJ, Reisch CR, Moran MA, Whitman WB, Lanzilotta WN. Structures of dimethylsulfoniopropionate-dependent demethylase from the marine organism Pelagabacter ubique. Protein Sci. 2012;21:289–98.
Bharath SR, Bisht S, Harijan RK, Savithri HS, Murthy MR. Structural and mutational studies on substrate specificity and catalysis of Salmonella typhimurium D-cysteine desulfhydrase. PLoS One. 2012;7:e36267.
Chartron J, Carroll KS, Shiau C, Gao H, Leary JA, Bertozzi CR, et al. Substrate Recognition, Protein Dynamics, and Iron-Sulfur Cluster in Pseudomonas aeruginosa Adenosine 5′-Phosphosulfate Reductase. J Mol Biol. 2006;364:152–69.
Davis KM, Altmyer M, Martinie RJ, Schaperdoth I, Krebs C, Bollinger JM Jr, et al. Structure of a Ferryl Mimic in the Archetypal Iron(II)- and 2-(Oxo)-glutarate-Dependent Dioxygenase, TauD. Biochemistry 2019;58:4218–23.
Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: Making protein folding accessible to all. Nat Methods. 2022;19:679–82.
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596:583–9.
Zhang C, Shine M, Pyle AM, Zhang Y. US-align: universal structure alignments of proteins, nucleic acids, and macromolecular complexes. Nat Methods. 2022;19:1109–15.
Xu J, Zhang Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 2010;26:889–95.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2014;32:268–74.
Villar E, Vannier T, Vernette C, Lescot M, Cuenca M, Alexandre A, et al. The Ocean Gene Atlas: exploring the biogeography of plankton genes online. Nucleic Acids Res. 2018;46:W289–W95.
Vernette C, Henry N, Lecubin J, de Vargas C, Hingamp P, Lescot M. The Ocean barcode atlas: A web service to explore the biodiversity and biogeography of marine organisms. Mol Ecol Resour. 2021;21:1347–58.
Paoli L, Ruscheweyh H-J, Forneris CC, Hubrich F, Kautsar S, Bhushan A, et al. Biosynthetic potential of the global ocean microbiome. Nature 2022;607:111–8.
Sunagawa S, Acinas SG, Bork P, Bowler C, Acinas SG, Babin M, et al. Tara Oceans: towards global ocean ecosystems biology. Nat Rev Microbiol. 2020;18:428–45.
Acinas SG, Sánchez P, Salazar G, Cornejo-Castillo FM, Sebastián M, Logares R, et al. Deep ocean metagenomes provide insight into the metabolic architecture of bathypelagic microbial communities. Commun Biol. 2021;4:604.
Biller SJ, Berube PM, Dooley K, Williams M, Satinsky BM, Hackl T, et al. Marine microbial metagenomes sampled across space and time. Sci Data. 2018;5:180176.
Pachiadaki MG, Brown JM, Brown J, Bezuidt O, Berube PM, Biller SJ, et al. Charting the Complexity of the Marine Microbiome through Single-Cell Genomics. Cell 2019;179:1623–35.
Delmont TO, Quince C, Shaiber A, Esen ÖC, Lee STM, Rappé MS, et al. Nitrogen-fixing populations of Planctomycetes and Proteobacteria are abundant in surface ocean metagenomes. Nat Microbiol. 2018;3:804–13.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Subramanian B, Gao S, Lercher MJ, Hu S, Chen W-H. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019;47:W270–W5.
Xing M, Wei Y, Zhou Y, Zhang J, Lin L, Hu Y, et al. Radical-mediated C-S bond cleavage in C2 sulfonate degradation by anaerobic bacteria. Nat Commun. 2019;10:1609.
Biebl H, Allgaier M, Tindall BJ, Koblizek M, Lunsdorf H, Pukall R, et al. Dinoroseobacter shibae gen. nov., sp nov., a new aerobic phototrophic bacterium isolated from dinoflagellates. Int J Syst Evol Microbiol. 2005;55:1089–96.
Fu H, Uchimiya M, Gore J, Moran MA. Ecological drivers of bacterial community assembly in synthetic phycospheres. Proc Natl Acad Sci USA 2020;117:3656–62.
Chen I-MA, Chu K, Palaniappan K, Ratner A, Huang J, Huntemann M, et al. The IMG/M data management and analysis system v.7: content updates and new features. Nucleic Acids Res. 2022. https://doi.org/10.1093/nar/gkac976.
Shiba T. Roseobacter litoralis gen. nov., sp. nov., and Roseobacter denitrificans sp. nov., aerobic pink-pigmented bacteria which contain bacteriochlorophyll a. Syst Appl Microbiol. 1991;14:140–5.
Kopriva S, Calderwood A, Weckopp SC, Koprivova A. Plant sulfur and big data. Plant Sci. 2015;241:1–10.
Simon J, Kroneck PMH. Microbial sulfite respiration. Adv Micro Physiol. 2013;62:45–117.
Gonzalez JM, Covert JS, Whitman WB, Henriksen JR, Mayer F, Scharf B, et al. Silicibacter pomeroyi sp nov and Roseovarius nubinhibens sp nov., dimethylsulfoniopropionate-demethylating bacteria from marine environments. Int J Syst Evol Microbiol. 2003;53:1261–9.
Liang KYH, Orata FD, Boucher YF, Case RJ. Roseobacters in a sea of poly- and paraphyly: whole genome-based taxonomy of the family Rhodobacteraceae and the proposal for the split of the “Roseobacter clade” into a novel family, Roseobacteraceae fam. nov. Front Microbiol. 2021;12:683109.
Howard EC, Sun S, Biers EJ, Moran MA. Abundant and diverse bacteria involved in DMSP degradation in marine surface waters. Environ Microbiol. 2008;10:2397–410.
Howard EC, Henriksen JR, Buchan A, Reisch CR, Buergmann H, Welsh R, et al. Bacterial taxa that limit sulfur flux from the ocean. Science. 2006;314:649–52.
Durham BP, Boysen AK, Carlson LT, Groussman RD, Heal KR, Cain KR, et al. Sulfonate-based networks between eukaryotic phytoplankton and heterotrophic bacteria in the surface ocean. Nat Microbiol. 2019;4:1706–15.
Smetacek V. Diatoms and the ocean carbon cycle. Protist 1999;150:25–32.
Stoecker DK, Lavrentyev PJ. Mixotrophic plankton in the polar seas: A pan-Arctic review. Front Mar Sci. 2018;5:292.
Turner SM, Malin G, Liss PS, Harbour DS, Holligan PM. The seasonal-variation of dimethyl sulfide and dimethylsulfoniopropionate concentrations in nearshore waters. Limnol Oceanogr. 1988;33:364–75.
Belviso S, Kim S-K, Rassoulzadegan F, Krajka B, Nguyen BC, Mihalopoulos N, et al. Production of dimethylsulfonium propionate (DMSP) and dimethylsulfide (DMS) by a microbial food web. Limnol Oceanogr. 1990;35:1810–21.
Simo R, Pedros-Alio C, Malin G, Grimalt JO. Biological turnover of DMS, DMSP and DMSO in contrasting open-sea waters. Mar Ecol Prog Ser. 2000;203:1–11.
Flombaum P, Gallegos JL, Gordillo RA, Rincon J, Zabala LL, Jiao N, et al. Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc Natl Acad Sci USA 2013;110:9824–9.
Gasparovic B, Penezic A, Frka S, Kazazic S, Lampitt RS, Holguin FO, et al. Particulate sulfur-containing lipids: Production and cycling from the epipelagic to the abyssopelagic zone. Deep Sea Res Part I Oceanogr Res Pap. 2018;134:12–22.
Zhan P, Tang K, Chen X, Yu L. Complete genome sequence of Maribacter sp T28, a polysaccharide-degrading marine flavobacteria. J Biotechnol. 2017;259:1–5.
Van Mooy BAS, Fredricks HF. Bacterial and eukaryotic intact polar lipids in the eastern subtropical South Pacific: Water-column distribution, planktonic sources, and fatty acid composition. Geochim Cosmochim Acta. 2010;74:6499–516.
Popendorf KJ, Tanaka T, Pujo-Pay M, Lagaria A, Courties C, Conan P, et al. Gradients in intact polar diacylglycerolipids across the Mediterranean Sea are related to phosphate availability. Biogeosciences 2011;8:3733–45.
Acknowledgements
We would like to thank Prof Spencer J. Williams (University of Melbourne, Australia) for providing the SQ standard. We also thank Prof Kunshan Gao (Xiamen University, China) and Center for Collections of Marine Algae (Xiamen University, China) for providing algae strains.
Funding
This study was supported by the National Natural Science Foundation of China project (92251306), the National Key Research and Development Program of China (2020YFA0608300), and the NSFC project (42276120, 42076160, 42188102).
Author information
Authors and Affiliations
Contributions
KT conceived the study, analyzed data, and wrote the manuscript. LL performed the experiments, analyzed data, wrote and edited the manuscript. XC sampled, performed the experiments, analyzed data, and wrote the manuscript. JY performed the experiments. XM performed the experiments. YuH sampled, and analyzed data. YajieH performed the experiments. All authors have approved the submitted final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, L., Chen, X., Ye, J. et al. Sulfoquinovose is a widespread organosulfur substrate for Roseobacter clade bacteria in the ocean. ISME J 17, 393–405 (2023). https://doi.org/10.1038/s41396-022-01353-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-022-01353-1
This article is cited by
-
A Broad-Spectrum α-Glucosidase of Glycoside Hydrolase Family 13 from Marinovum sp., a Member of the Roseobacter Clade
Applied Biochemistry and Biotechnology (2024)
