
Abstract
Acidobacteriota are widespread and often abundant in marine sediments, yet their metabolic and ecological properties are poorly understood. Here, we examined metabolisms and distributions of Acidobacteriota in marine sediments of Svalbard by functional predictions from metagenome-assembled genomes (MAGs), amplicon sequencing of 16S rRNA and dissimilatory sulfite reductase (dsrB) genes and transcripts, and gene expression analyses of tetrathionate-amended microcosms. Acidobacteriota were the second most abundant dsrB-harboring (averaging 13%) phylum after Desulfobacterota in Svalbard sediments, and represented 4% of dsrB transcripts on average. Meta-analysis of dsrAB datasets also showed Acidobacteriota dsrAB sequences are prominent in marine sediments worldwide, averaging 15% of all sequences analysed, and represent most of the previously unclassified dsrAB in marine sediments. We propose two new Acidobacteriota genera, Candidatus Sulfomarinibacter (class Thermoanaerobaculia, “subdivision 23”) and Ca. Polarisedimenticola (“subdivision 22”), with distinct genetic properties that may explain their distributions in biogeochemically distinct sediments. Ca. Sulfomarinibacter encode flexible respiratory routes, with potential for oxygen, nitrous oxide, metal-oxide, tetrathionate, sulfur and sulfite/sulfate respiration, and possibly sulfur disproportionation. Potential nutrients and energy include cellulose, proteins, cyanophycin, hydrogen, and acetate. A Ca. Polarisedimenticola MAG encodes various enzymes to degrade proteins, and to reduce oxygen, nitrate, sulfur/polysulfide and metal-oxides. 16S rRNA gene and transcript profiling of Svalbard sediments showed Ca. Sulfomarinibacter members were relatively abundant and transcriptionally active in sulfidic fjord sediments, while Ca. Polarisedimenticola members were more relatively abundant in metal-rich fjord sediments. Overall, we reveal various physiological features of uncultured marine Acidobacteriota that indicate fundamental roles in seafloor biogeochemical cycling.
Similar content being viewed by others


Introduction
Bacteria of the phylum Acidobacteriota (also known as “Acidobacteria”) are highly diverse and inhabit a vast array of environments on Earth, yet the properties of various Acidobacteriota lineages remain poorly understood [1,2,3,4,5,6]. Knowledge regarding the functions and ecology of Acidobacteriota is biased to isolates and genomes obtained from soils, where they are especially prevalent and often dominate microbial communities [3, 4]. Soil-derived Acidobacteriota are generally known as aerobic heterotrophs that utilize various carbohydrates including polysaccharides like chitin or cellulose [3, 7, 8]. Some Acidobacteriota known from other environments have unique physiological properties, such as the ability to reduce iron [9], perform phototrophy [9, 10], or exhibit thermophilic lifestyles [11]. Members of Acidobacteriota subdivisions 1 and 3 from peatland and permafrost soils have the potential to dissimilate inorganic and/or organic sulfur compounds [2, 12]. In comparison to terrestrial Acidobacteriota, very little is known about Acidobacteriota in marine systems.
Acidobacteriota 16S rRNA genes or genomes are frequently detected in marine environments including ocean waters, marine sponges, hydrothermal vents, or sediments [13,14,15,16,17]. Studies of 16S rRNA genes in marine sediments showed that Acidobacteriota are widespread and reach relative abundances in amplicon libraries of up to 23% [18,19,20,21,22,23]. This suggests they play important roles in microbial community functioning and biogeochemical processes, although our knowledge regarding their specific roles in sediments remains limited. A recent stable isotope probing study showed some Acidobacteriota in deep-sea sediments are capable of fixing nitrogen [24]. Acidobacteriota were also shown to be active under sulfidic conditions in 18O–H2O incubations with estuarine sediment, by stable isotope-labeling of their 16S rRNA and dissimilatory sulfite reductase (dsrB) genes [6]. One novel Acidobacteriota metagenome-assembled genome (MAG) (Ca. Guanabacteria) encoded genes for the CO dehydrogenase/CO-methylating acetyl-CoA synthase complex and heterodisulfide reductases, indicating a possible anaerobic lifestyle [25].
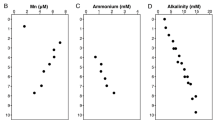
Marine sediments are a massive global habitat for microorganisms [26], with average cell densities of microorganisms up to 109 cells per cm3 in surface sediments of organic-rich sediments [27]. Substantial amounts of organic matter are processed in marine sediments, which makes them a critical component of marine and global biogeochemical cycles [28]. Marine sediments are often stratified with respect to redox states, whereby oxygen is typically depleted within millimeters to centimetres below the surface at sites where organic inputs are relatively high [29]. Vast expanses of sediments are therefore anoxic, and many microorganisms survive via anaerobic lifestyles, such as fermentation, or respiration of nitrate, metals, sulfate or CO2. Sulfate is abundant in sediments and is used by sulfite/sulfate-reducing microorganisms (SRMs) as an electron acceptor for anaerobic respiration. Sulfate reduction is estimated to facilitate ~29% of organic matter degradation in marine sediments globally [26, 28]. The sulfur-cycle is therefore a major driver of microbial life and biogeochemical cycling in the seafloor, and therefore understanding the microorganisms that catalyze sulfur cycling is of great importance.
Because sulfate reduction is a major process in marine sediments, the activities, distributions, and diversity of SRMs have been relatively well studied [28, 30, 31]. Members of the Desulfobacterota (formerly “Deltaproteobacteria”) are known as abundant SRMs in marine sediments, playing key roles in anaerobic food webs by utilizing fermentation products released by primary degraders of organic matter [32,33,34]. They are also represented by various isolates, and many have been subject to genomic and physiological studies [35]. Surveys of functional marker genes for sulfite/sulfate reducers in marine sediments, i.e., of dsrAB, have repeatedly shown that dsrAB from the phylum Desulfobacterota are typically the dominant dsrAB-harboring group in marine sediments, but importantly, that several other lineages of uncultivated dsrAB-harboring organisms are also abundant and prevalent [36]. Recently, some dsrAB sequences in marine sediments have been inferred to belong to Acidobacteriota [6, 37], although nothing is known about the metabolic properties, genomes, or the sulfur-dissimilating pathways of the organisms that harbor these genes. Identifying and understanding these undescribed dsrAB-harboring microorganisms is therefore critical for understanding the microbial groups that drive sulfur cycling in marine sediments.
In this study, we aimed to gain insights into the metabolic potential of uncultured Acidobacteriota lineages in marine sediments, as well as their diversity and distributions. We therefore recovered metagenome-assembled genomes (MAGs) from abundant Acidobacteriota populations present in marine fjord sediments of Svalbard, and predicted their metabolic features. Focus was placed on MAGs from the class Thermoanaerobaculia of the Acidobacteriota, which represent a newly described lineage of dsrAB-harboring organisms that may be important sulfur cycling bacteria in marine sediments. These analyses were complemented with comparative genomics, incubation experiments, transcript analyses, and analyses of Acidobacteriota distributions in Svalbard sediments and publicly available datasets, together revealing they may play various roles in sedimentary biogeochemical cycles, and that they are a prominent group of sulfur-dissimilating organisms.
Materials and methods
Sample collection
Marine sediments were collected from Smeerenburgfjorden, Kongsfjorden and Van Keulenfjorden, of Svalbard, Norway, in July 2016 and/or June 2017 with the vessel “MS Farm”. Extensive biogeochemical data for these sites is available from previous studies [38,39,40,41,42]. Maps of sample locations are presented in Michaud et al. [41]. From Smeerenburgfjorden, individual core samples were taken from three stations: station GK (79°38.49N, 11°20.96E), station J (79°42.83N, 11°05.10E), and station GN (79°45.01N, 11°05.99E), with the station J cores being taken in both 2016 and 2017. Duplicate core samples from Van Keulenfjorden were taken from sites AC (77°32.260′N, 15°39.434′E) and AB (77°35.249′N, 15°05.121′E). A sample was also taken from Kongsfjorden station F (78°55.075′N, 12°15.929′E) [43]. For molecular biological analyses, samples were taken with HAPS [44] or Rumohr corers [45]. Details of core subsampling procedures are provided in the Supplementary Information.
Microcosm incubations with tetrathionate additions
To examine tetrathionate reductase gene expression, a sediment incubation experiment was performed. A sediment slurry was prepared inside an anoxic glove box (nitrogen atmosphere containing 2% hydrogen and 10% CO2), from samples collected in 2017 from 5 to 10 cmbsf at Station J, Smeerenburgfjorden. Sediments had been stored at 4 °C for 6 months prior to the experiment. Anoxic artificial seawater [46] containing 28 mM sulfate was well-mixed with a 2:1 ratio with sediment. Autoclaved serum bottles (250 ml) that were left in the anoxic glove box overnight prior to the experiment to remove traces of oxygen, were filled with 30 ml of sediment slurry. All microcosms received a small amount of organic material to boost heterotrophic activity, i.e., yeast extract (0.22 mg ml−1) (Oxoid). All experiments were set up in triplicates. The experimental treatments included additions of: (i) tetrathionate (500 µM final), or (ii) “no substrate” controls. All microcosms were sealed with autoclaved butyl rubber stoppers. The experiment was incubated at 4 °C for 8 days, and samples were taken at the start of the experiments, day 1 and day 8. Additional tetrathionate (to make 500 µM additional) was spiked into the microcosms on day 5. Subsampling was done inside the anoxic glove box with microcosms placed on ice-pads to reduce warming of the samples. Samples (250 µl) were taken for DNA/RNA-based analyses, kept on the ice-pads in the anoxic glove box and transferred immediately to dry-ice outside the glove box, and stored at −80 °C.
Nucleic acid extractions and reverse transcription
For amplicon-based analyses, DNA and RNA was extracted from the sediment core samples (~500 µl) and microcosm samples (~250 µl) using the RNeasy PowerSoil Total RNA Kit (Qiagen) according to the manufacturer’s instructions. Additionally, a phenol/chloroform based extraction method [47], was used to extract total nucleic acids from sediment samples from station J sampled in July 2016 (Supplementary Information). Eluted nucleic acids were stored in molecular biology grade water at −80 °C. Aliquots for DNA-based analyses were used as eluted, while aliquots for RNA-based analyses were DNase-treated using the TURBO DNA-free kit (Thermo Fisher), followed by reverse transcription of the RNA to cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher) according to the manufacturer’s instructions. To test if any DNA remained in the RNA samples after the DNase digestion step, control samples were processed as above except the RevertAid M-MuLV Reverse Transcriptase was excluded. These controls were checked for DNA by PCR using 16S rRNA gene targeting primers (described below).
Sediment samples from 2016 were used for metagenome sequencing. DNA was extracted by the Vienna group from 3 to 5 mL of sediment from varying depths or microcosms derived from station J, Smeerenburgfjorden, and 18 centimeters below seafloor (cmbsf) from station AC of Van Keulenfjorden (Supplementary Table 1) using the DNeasy PowerSoil Kit (Qiagen) according to the manufacturer’s protocol. DNA was also extracted by the Knoxville group from 2 g of sample spanning 0–5 cmbsf from site AB of Van Keulenfjorden and site F of Kongsfjorden (Supplementary Table 1), using the RNeasy PowerSoil Kit (Qiagen) with DNA elution following the manufacturer’s protocol.
Metagenome sequencing and genome binning
DNA libraries were prepared from individual samples (detailed in Supplementary Information) and sequenced using 2 × 150 bp paired-end mode with a HiSeq 3000 (Illumina) instrument at the Biomedical Sequencing Facility (BSF), Vienna. Metagenomic libraries were generated from the combined extracts from the first 5 cm (spanning 0–5 cm downcore) in sites AB and F in the Center for Environmental Biotechnology, Knoxville, using HiSeq (Illumina), 2 × 250 bp in paired-end mode [43]. Sequencing output summaries are provided in Supplementary Table 1.
Sequence reads were quality filtered, trimmed, and normalized as described in the Supplementary Information. Processed reads from each sample were assembled separately using IDBA-UD (version 1.1.1) [48] with default settings and the following options: --min_contig 500 --pre_correction. Reads from site F (Kongsfjorden) were assembled via metaSPAdes (version 3.11) [49] with kmer sizes set to 21, 33, 55, 77, 99, and 127 to find the best assembly. All other samples were assembled using metaSPAdes on the KBase server [50] with the default parameters and following options: minimum contig length of 1000 bp, and kmer sizes of 21, 33, and 55. All samples were also assembled using Megahit [51] on the KBase server using default parameters. KBase Narratives are available/searchable from user “jbuongio”.
Coverage profiles of assembled unbinned contigs were acquired by mapping trimmed reads (not normalized) to assemblies using BWA [52] and SAMtools [53]. Contigs from each assembly were then binned into MAGs using MetaBat2 (using each binning strategy) (version 2.12.1) [54], CONCOCT (version 0.4.1) [55], and MaxBin2 (version 2.2.4) [56]. MAG collections derived from each binning strategy, from all respective assemblies, were then aggregated using DasTool (version 1.1.0) (Supplementary Fig. 1A) [57]. Finally, all MAGs were dereplicated using dRep (version 1.4.3) [58], with the options: an average nucleotide identity (ANI) of 98% was used as cutoff to dereplicate MAGs from the secondary ANI comparison [59], and MAGs >50% complete and <10% contamination were retained. Estimations of completeness and degree of contamination of MAGs were obtained by CheckM (version 1.0.7) [60]. Read mapping to compare relative abundances of read recruitment to MAGs was performed using BBMap [61], with the default settings and “minid” of 0.99 for the minimum identity threshold. Taxonomic affiliations of MAGs were determined with GTDB-Tk [62]. ANI comparisons of MAGs were obtained using JSpeciesWS server based on BLASTN (“ANIb”) [63] and ANIcalculator [64].
Gene annotations and in silico analyses of inferred proteins
Calling of genes and automatic annotations were obtained using the RAST server with default settings and the “classic” annotation pipeline [65]. Functions of predicted proteins of interest were manually checked after searches with BLASTP [66] against the NCBI-nr and SWISS-PROT databases [67] (>25% identity), and the Conserved Domain Database (CDD) [68] (default expect value of 0.01). Functions were together manually assessed by considering various features including: (i) proteins had >25% amino acid identity to biochemically characterized proteins present in SWISS-PROT databases, (ii) considering if functional domains were detected by CDD, (iii) if genes for proteins being examined were present in gene clusters with functionally related proteins (e.g., of same enzymatic complex and/or biochemical pathways [69, 70]), (iv) after being evaluated using literature searches and the MetaCyc database [71], and (v) in certain cases, phylogenetic comparisons between related protein sequences (described below). Methods for further annotations and protein sequence analyses and for gene content comparisons among MAGs are described in the Supplementary Information.
MiSeq amplicon sequencing and sequence analyses
For amplification of bacterial and archaeal 16S rRNA genes or transcripts (cDNA) from Smeerenburgfjorden sediments, the primers 515F (5′-GTGYCAGCMGCCGCGGTAA-3′) [72] and 806R (5′-GGACTACNVGGGTWTCTAAT-3′) [73] including a 5′-head sequence for 2-step PCR barcoding [74], were used (further details in Supplementary Information). Triplicate PCRs were performed in the first-step PCR, and then pooled before the second barcoding PCR. Slight variants of these PCR primers 515F and 806R [75] for 16S rRNA genes were used in amplicon sequencing profiling of sediments from Van Keulenfjorden in a previous study, although a standard “one-step PCR” approach was used [40]. Amplicon pools were extracted from the raw sequencing data using the FASTQ workflow in BaseSpace (Illumina) with default parameters. Demultiplexing was performed with the python package demultiplex (Laros JFJ, github.com/jfjlaros/demultiplex) allowing one mismatch for barcodes and two mismatches for linkers and primers. DADA2 [76] was used for demultiplexing amplicon sequencing variants using a previously described standard protocol [77]. FASTQ reads 1 and 2 were trimmed at 220 nt and 150 nt, respectively, with allowed expected errors of 2. Taxonomy was assigned to 16S rRNA gene/transcript sequences based on SILVA taxonomy (release 138) using the naïve Bayesian classification method as implemented in mothur [78]. Amplicon sequence datasets were analysed with the Rhea pipeline [79] implemented in R (https://www.r-project.org/).
Primers DSR-1762Fmix and DSR-2107Rmix, including a 5′-head sequence for barcoding, were used for amplification of dsrB-genes or -transcripts (cDNA) [80] (further details in Supplementary Information). Triplicate PCRs were performed in the first-step PCR, and then pooled before the second barcoding PCR. Raw reads were then processed as previously described [74, 80], into dsrB operational taxonomic units (OTUs) with >99% identity. Classification of amplicon-derived DsrB sequences was performed using a combined phylogenetic and naïve Bayesian classification approach as previously described [80].
Methods for analyses of publicly available 16S rRNA gene and dsrAB sequence datasets are described in the Supplementary Information.
Quantitative reverse-transcription PCR
RT-qPCR assays targeting the octaheme cytochrome tetrathionate reductase (otr) and dsrB genes of MAG AM3-C were performed using the newly-designed primers TetraC-C-F (5′-CACCACGACCTGTCTCGG-3′) and TetraC-C-R (5′-CCCCCTGGAGTTCTTGGT-3′), and Acido-dsrB-F (5′-GGAGAACTATGGGAAGTGGG-3′) and Acido-dsrB-R (5′-GTTGAGGCAGCACGCGTA-3′). Primers 1329-B-F (5′-AACCTTTGGGCGATTTCTCG-3′) and 1329-B-R (5′-GAGAGAGTGGCAACGTGAAC-3′) targeting the DNA-directed RNA polymerase alpha subunit gene of MAG AM3-C were used to examine expression of a housekeeping gene. Details of RT-qPCR assay conditions are presented in the Supplementary Information. Relative-fold changes of target gene transcripts were calculated relative to the housekeeping gene of the alpha subunit of DNA-directed RNA polymerase by the 2–ΔCt calculation as described by Schmittgen and Livak [81].
Phylogenetic analyses
A phylogenomic maximum-likelihood tree was created using the IQ-TREE web-server with automatic substitution model selection and ultrafast bootstrapping (1000×) [82] using an alignment of concatenated protein sequences derived from single copy marker genes retrieved from CheckM [60]. The tree was visualized with iTol [83]. Phylogenetic analysis of 16S rRNA was performed in ARB [84] using the SILVA database release 138 [85], and dsrAB sequences were also analysed using ARB using previously described database [36, 80] (Supplementary Information). Phylogenetic analyses of all other protein sequences were performed using the IQ-TREE web-server with automatic substitution model selection and ultrafast bootstrapping (1000×) [82]. For the Complex-Iron-Sulfur-Molybdoenzyme (CISM) tree, query protein sequences were added to a previous alignment of CISM protein sequences [86], using MAFFT using the “add full length sequences” option (--add) [87]. All other protein sequence alignments were made de novo with MUSCLE [88] within Mega6 [89].
Sequence and MAG accessions
Metagenomic sequence reads from Van Keulenfjorden and Kongsfjorden samples are available under NCBI-Genbank Bioproject PRJNA493859. Metagenomic sequence reads, and 16S rRNA gene and dsrB sequence reads from Smeerenburgfjorden samples are available under NCBI-Genbank Bioproject PRJNA623111. MAGs are available under NCBI-Genbank Bioproject PRJNA623111, with Biosample accessions SAMN15691661-SAMN15691666.
Results
Recovery of novel Acidobacteriota genomes from marine sediments
Metagenomic sequencing and genome binning was performed from DNA extracted and sequenced from sediments originating from three fjords from Svalbard, Norway (Supplementary Table 1). Our genome binning strategy based on multiple assemblies and multiple binning algorithms recovered more MAGs with higher completeness, as compared to applying multiple binning approaches based on single assembly approaches (Supplementary Fig. 1A, B). From the dereplicated MAGs (n = 97), four represented populations of the phylum Acidobacteriota and were chosen for in-depth analyses.
Phylogenomic analyses showed three MAGs (AM1, AM2, and AM3-A) affiliated with GTDB family “FEB-10” of the class Thermoanaerobaculia (“subdivision 23”) (Fig. 1). We included two additional MAGs in our analyses, i.e., AM3-B and AM3-C, that were highly similar to the AM3-A MAG (>98% ANI), but were classified as redundant during MAG dereplication. We studied them in further detail because: (i) the AM3 MAGs represent the most abundant Thermoanaerobaculia species based on sequence coverage (Supplementary Table 2), (ii) they encoded enzymes of interest not present in MAG AM3-A (Fig. 2A), and (iii) were more complete than MAG AM3-A (Table 1). Comparisons of ANI values suggested the Thermoanaerobaculia MAGs represent three distinct species (<95% ANI) (Supplementary Table 3) [90], all from a novel genus for which we propose the name Candidatus Sulfomarinibacter (Supplementary Table 4). The MAG AM3-C represents the type species Ca. Sulfomarinibacter kjeldsenii (Supplementary Table 4). MAG AM4 represents the type species of another novel genus affiliated with the GTDB class “Mor1” (“subdivision 22”) (Fig. 1), and for which we propose the name Ca. Polarisedimenticola svalbardensis (Table 1 and Supplementary Table 4).

Maximum-likelihood tree of concatenated protein sequences from MAGs and genomes. Single marker genes were retrieved with CheckM. Highlighted in blue are MAGs obtained in this study. Highlighted in purple are dsrAB-containing MAGs obtained from the NCBI database from the class Thermoanaerobaculia. The genus Ca. Acidiflorens is represented by the most complete MAG (GCA_003166525.1) from the corresponding study [12]. Our phylogenomic analysis showed that one MAG that was previously assigned to Ca. Aminicenantes (GCA_004524955.1), recovered from the Bothnian Sea [146], is affiliated with the newly proposed family Ca. Sulfomarinibacteraceae. Black dots indicate dsrAB-containing genomes/MAGs. Bootstrap values with >90% are indicated with filled black circles on nodes. Nitrospina gracilis 3/211 (GCA 000341545.2) was used as an outgroup. The scale bar represents 10% sequence divergence.

A Presence–absence of genes for sulfur-dissimilating enzymes, and metabolic models of (B) Ca. Sulfomarinibacter kjeldsenii MAG AM3-C, and (C) Ca. Polarisedimenticola svalbardensis MAG AM4 suggest different fundamental niches of the two species in marine sediments. GH glycoside hydrolase, RDH reductive dehalogenase homologous enzyme, Ack acetate kinase, Pta phosphotransacetylase, PFL pyrivate-fomate lyase, FDH formate dehydrogenase, Hdr heterodisulfide reductase, NUO NADH dehydrogenase, Otr Tetrathionate reductase, NosZ nitrous oxide reductase, Sat sulfate adenylyltransferase, Apr adenylylsulfate reductase, Qmo quinone-interacting membrane oxidoreductase complex, Dsr dissimilatory sulfate reductase, Nap Periplasmic nitrate reductase, Psr poylsulfide reductase, Sdh Sulfhydrogenase complex, and TusA sulfur-carrier protein.
Marine Acidobacteriota encode the full dissimilatory sulfate reduction pathway
Together, the gene content of the Ca. Sulfomarinibacter suggests they encode a complete canonical dissimilatory sulfate reduction pathway (Fig. 2A, B and Supplementary Table 5). This includes enzymes required for sulfate activation to APS (Sat) and reduction of APS to sulfite (AprAB, QmoABC), and further reduction of sulfite to sulfide (DsrAB, DsrC, DsrMKJOP, and DsrN) (Fig. 2A, B and Supplementary Table 5). Acidobacteriota dsr were also found on scaffolds (up to 20 kb) that were not binned into MAGs, yet had highly similar genes and therefore derive from closely related populations, e.g., >99% dsrB nucleotide identity (Fig. 3 and Supplementary Table 5). The unbinned acidobacteriotal contig “ThM_scaffold_807” harbored all dsr on one contig (Fig. 3). The predicted DsrC had two conserved cysteine residues critical for respiratory functioning (Supplementary Fig. 2) [91]. Similar to Acidobacteriota MAGs from peatlands and permafrost [2, 12], the marine Acidobacteriota encoded both DsrL and DsrD proteins. DsrL acts as a NAD(P)H:acceptor oxidoreductase for DsrAB [92], while the function of DsrD has not been proven, it is possibly a transcriptional regulator [93]. The DsrL sequences were phylogenetically related to group “DsrL-2” from Desulfurella amilsiiI (38% amino acid identity), peatland Acidobacteriota and other subsurface bacteria, and were phylogenetically distinct from group “DsrL-1” of sulfur-oxidizing aerobes (Supplementary Fig. 3A) [94]. The DsrL had conserved YRR-motifs in the NAD(P)H substrate-binding domains that are present in the DsrL-2 group, and absent in DsrL-1 of sulfur-oxidizing aerobes (Supplementary Fig. 3B) [94].

Scaffold names in blue were retrieved from this study. Scaffold names in purple were derived from best BLASTP hits to sequences from this study. Ca. Sulfotelmatomonas gaucii SbA5 was retrieved from Hausmann et al. [2]. Green: dsr, dark red: other genes, and orange: hypothetical genes. Shaded blue lines indicate degree of sequence similarity as determined by tBLASTx within EasyFig [148], which depicts sequence identity similarities among protein sequences.
The DsrAB sequences from the novel Acidobacteriota MAGs and unbinned metagenomic contigs are phylogenetically affiliated with the “Uncultured family-level lineage 9” within the “Environmental supercluster 1”, which is part of the “reductive, bacterial-type DsrAB branch” in the DsrAB tree [36] (Fig. 4). Sequences of “lineage 9” are primarily derived from marine sediments [36]. This lineage is closely related to the “Uncultured family-level lineage 8” that harbors DsrAB sequences from peatland- and permafrost-derived Acidobacteriota of subdivisions 1 and 3 [2, 12]. We also identified several “lineage 9” dsrA and/or dsrB sequences in Acidobacteriota MAGs from public databases that derived from marine or groundwater environments (Fig. 4). Herein, we refer to this clade as the “Thermoanaerobaculia Dsr lineage”.

Blue leaves in the DsrAB tree represent MAGs or contigs identified in this study. Red leaves represent the most abundant acidobacteriotal amplicon-derived DsrB sequences identified in this study. Purple leaves represent sequences from MAGs retrieved from public databases. The DsrAB sequences were added to the consensus tree from Müller et al. [36] in ARB. SD, pertaining to “subdivisions” of Acidobacteriota. Numbers in parentheses are Genbank accessions. The scale bar represents 10% sequence divergence.
Marine Acidobacteriota use tetrathionate and potentially also other sulfur-cycle intermediates
Several Ca. Sulfomarinibacter MAGs encoded c-type cytochromes annotated as octaheme tetrathionate reductases (Otr), which was supported by phylogenetic analysis (Supplementary Fig. 4) [95]. The Otr were predicted to be periplasmic and may enable respiration with tetrathionate, a sulfur compound of intermediate oxidation state (“sulfur-cycle intermediate” (SCI)) [31] (Fig. 2B). To provide support that the Otr catalyses tetrathionate reduction and that this occurs within in situ-like conditions, we conducted anoxic microcosm experiments with Svalbard sediments with additions of tetrathionate (500 µM) versus controls without any additions (all contained 28 mM sulfate). We hypothesized that additions of tetrathionate, which has a relatively high redox potential (+198 ± 4 mV versus standard hydrogen electrode (SHE)) [96], would trigger increased expression of otr because it is a favorable electron acceptor over sulfate. We thus examined expression of otr and dsrB of Ca. Sulfomarinibacter MAG AM3-C by RT-qPCR analysis of reverse-transcribed mRNA, from multiple times points after tetrathionate additions. Relative expression levels of these transcripts were determined in comparison to the housekeeping gene for DNA-directed RNA polymerase alpha subunit, in order to account for potential changes in cell numbers over time. This showed that transcription of otr was upregulated (1.8-fold) in response to tetrathionate at day 1 versus controls (although not significantly), and was significantly upregulated (p < 0.0488) 36-fold at day 8. Additionally, the transcription of dsrB appeared lower at both days in tetrathionate-amended microcosms versus controls (0.48–0.63-fold), although not significantly (Fig. 5).

Relative expression was determined by rt-qPCR. Expression of otr was significantly higher at day 8 (p = 0.0488) as determined using a two-tailed T-test, and is indicated by an asterisk. Center lines indicate medians; box limits indicate 25th and 75th percentiles as determined by R software; and whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles.
“YTD gene clusters” encoding sulfur-trafficking rhodonase-like proteins [97] were identified among Ca. Sulfomarinibacter MAGs. Genes for YedE-related permease-like proteins, a DsrE2-like protein, a rhodonase-domain containing sulfur-carrier TusA, and two conserved hypothetical proteins, were present in the YTD gene clusters (Supplementary Table 5). The TusA sulfurtransferase had conserved Cys‐Pro‐X‐Pro sulfane sulfur‐binding domains (Supplementary Fig. 5A). The TusA were phylogenetically most closely related to various TusA from anaerobic Desulfobacterota that are capable of reducing and/or disproportionating inorganic sulfur compounds such as elemental sulfur, sulfite, and/or thiosulfate (Supplementary Fig. 5B). Together, this suggested Ca. Sulfomarinibacter are capable of internal trafficking of sulfur, and may use it to reduce and/or disproportionate inorganic sulfur compounds of intermediate redox states.
The marine Acidobacteriota MAGs encoded several Complex-Iron-Sulfur-Molybdoenzyme (CISM) enzymes that may catalyse redox reactions of sulfur compounds. The Ca. Sulfomarinibacter MAG AM3-A encoded a putative tetrathionate reductase (TtrA) (Supplementary Fig. 6), and also had an adjacent TtrB (FeS protein) encoded. A ttrC encoding a membrane anchor was missing, but the ttrAB were situated on the end of the contig and therefore ttrC may have been present in DNA that either was not sequenced or on a contig that was not binned. The Ttr complex may provide an additional means to reduce tetrathionate.
Ca. P. svalbardensis MAG AM4 had genes for a CISM subunit A enzyme that phylogenetically affiliated with the polysulfide/thiosulfate reductase clade (“Psr”) (Supplementary Fig. 6). Subunits for PsrABC were encoded in a gene cluster, where the terminal reductase PsrA had a TAT-leader peptide for export from the cytoplasm, PsrB had FeS domains for electron transfer between PsrA and PsrC, and the PsrC subunit was predicted to be membrane-bound. This suggested a periplasm location and that the complex may play a role in respiration of sulfur/polysulfide or thiosulfate. Selenite reductases (SrrA) also phylogenetically affiliate with the polysulfide/thiosulfate reductase clade, but conserved rhodonase-like proteins encoded in the gene neighborhood of SrrA are thought to be indicative of selenite-reducing organisms [98], but were absent near psrABC in Ca. P. svalbardensis MAG AM4.
Ca. P. svalbardensis MAG AM4 also harbored a gene cluster encoding four subunits of a sulfhydrogenase complex (Fig. 2A, B and Supplementary Table 5). Similar to the characterized sulfhydrogenase from Pyrococcus furiosus, this included two NiFe hydrogenase subunits, as well as two subunits of anaerobic sulfite reductases [99,100,101]. These complexes can use elemental sulfur or polysulfides as electron sinks when available [101], or act in reverse as hydrogen-evolving hydrogenases during fermentative growth [102].
Marine Acidobacteriota may respire additional electron acceptors including metals
All MAGs had gene clusters encoding multi-heme c-type cytochromes with predicted periplasmic or extracellular locations, as well as associated predicted β-barrel proteins (Fig. 2A, B, and Supplementary Table 5). In known metal-reducing and/or -oxidizing bacteria, extracellular, and periplasmic cytochromes can insert into outer-membrane traversing β-barrel proteins, and transfer electrons through the complexes to/from metals [103, 104]. These gene clusters were syntenous among the MAGs and Thermoanaerobaculum aquaticum (Supplementary Fig. 7A), a related hot spring-derived isolate that can anaerobically reduce iron- and manganese-oxides [11]. We therefore propose these cytochromes are likely candidates for facilitating the reduction of metal-oxides by Thermoanaerobaculum aquaticum, because no other predicted extracellular cytochromes are encoded by its genome. It is therefore probable that the similar cytochromes encoded by our marine MAGs may also perform this function.
The Ca. P. svalbardensis MAG AM4 encoded two additional cytochrome-c proteins with similarity to metal-reducing outer-membrane cytochromes (OmcS) from known metal-reducing bacteria, i.e., various Desulfuromonadia (formerly Desulfuromonadales) such as Geobacter and Geopsychrobacter spp. (Supplementary Table 6) [105, 106]. These cytochromes had six heme-binding sites like characterized OmcS, and were also clustered among genes for predicted periplasmic cytochromes and β-barrel proteins (Supplementary Fig. 7B). They could therefore also potentially exchange electrons with metal-oxides (or other insoluble substrates such as humic-like substances, or other cells) (Fig. 2B, C).
The marine Acidobacteriota MAGs also encoded the potential to reduce various other electron acceptors. Both groups have potential for oxygen reduction through cytochromes (Fig. 2B, C), although we speculate Ca. Sulfomarinibacter may reduce oxygen as a defense mechanism, because they otherwise encode various features of strict anaerobes (Supplementary Information). Ca. Sulfomarinibacter also encode type-II nitrous oxide reductases (Fig. 2B and Supplementary Fig. 8) and reductive dehalogenases (Fig. 2B and Supplementary Fig. 9), suggesting nitrous oxide and organohalides could be reduced by these enzymes, respectively. Ca. P. svalbardensis encodes periplasmic nitrate reductases, although no other genes for enzymes for other steps of denitirification (Fig. 2C and Supplementary Table 5). A putative arsenate reductase is also encoded by Ca. P. svalbardensis (Supplementary Fig. 6 and Supplementary Table 4).
Additional energy conserving mechanisms among marine Acidobacteriota
Electron-bifurcating heterodisulfide reductase complexes were only encoded in Ca. Sulfomarinibacter MAGs (Fig. 2B and Supplementary Table 5). These complexes enable flavin-based redox balancing and formation of low-potential electron carriers (i.e., ferredoxin and/or flavodoxin), and are common among strict anaerobes [107, 108]. A high-molecular-weight cytochrome c3-type protein with a predicted periplasmic location was encoded in Ca. Sulfomarinibacter MAG AM1 (Supplementary Table 5). These typically act as periplasmic redox hubs to link electron flows between the periplasm and cytoplasm in SRM [109]. All Acidobacteriota MAGs recovered in this study encoded NADH-ubiquinone oxidoreductase (Nuo) complexes required for energy conservation via respiration (Fig. 2B, C and Supplementary Table 5). Genes for additional sodium-dependent Nuo complexes were also present (Fig. 2B and Supplementary Table 5). Apart from the potential for respiration, some Acidobacteriota MAGs from both Ca. Sulfomarinibacter and Ca. P. svalbardensis MAG AM4 encoded acetate kinase and phosphate acetyltransferase for fermentation via acetogenesis, or which may act in reverse to facilitate acetate consumption (Fig. 2B, C and Supplementary Table 5).
Marine Acidobacteriota use diverse nutrient and electron sources
The Ca. Sulfomarinibacter AM3 MAGs encoded predicted cellulase A enzymes with signal peptides for export from the cytoplasm (Fig. 2B). They were phylogenetically affiliated with cellulase A from various anaerobic degraders of cellulose and/or plant-derived polysaccharides (Supplementary Fig. 10). A cellobiose phosphorylase was encoded in Ca. S. kjeldsenii MAG AM3-C, and had relatively high amino acid identity (63%) to a characterized cellobiose phosphorylase from Thermotoga neapolitana [110]. These enzymes catalyse phosphorolysis of cellobiose to ɑ-D-glucose 1-phosphate (G1P) and D-glucose, thereby saving an ATP before entering glycolysis, and are typically used by anaerobic cellulose-degraders [111]. This suggests these organisms have the capacity to anaerobically degrade cellulose, a derivative of cellulose, or a structurally similar compound. Overall, the marine Ca. Sulfomarinibacter MAGs encoded few genes for glycoside hydrolases or other carbohydrate active enzymes, i.e., 0.47–0.75% of protein encoding genes encoded glycoside hydrolases (further detailed in Supplementary Information) (Supplementary Table 7). The Ca. P. svalbardensis MAG AM4 also encoded few glycoside hydrolases (0.54% of protein encoding genes), with none predicted to be exported to the extracellular environment, and a single endo-1,4-beta-xylanase predicted to be periplasmic (Supplementary Table 5).
Genes for cyanophycinases among Ca. Sulfomarinibacter MAGs indicated they may utilize the storage compound cyanophycin as a nutrient (Fig. 2B and Supplementary Table 5). The cyanophycinases had Secretion-signal peptides (Sec-) for export from the cytoplasm, indicating they act on an external substrate and not an internally stored compound. Accordingly, no genes for cyanophycin synthetases were found. An isoaspartyl dipeptidase was encoded in Ca. S. kjeldsenii MAG AM3-C, which may enable utilization of the products released by the cyanophycinase, i.e., a dipeptide of aspartate and arginine (Supplementary Table 5). The capacity to catabolically degrade aspartate and arginine was also encoded (Supplementary Table 5).
The Ca. Sulfomarinibacter MAG AM3-C may degrade extracellular proteins using two predicted secreted proteases, as well as adjacently encoded peptidases predicted to be membrane-bound (Fig. 2B and Supplementary Table 5). The Ca. P. svalbardensis MAG AM4 harbored numerous genes for proteases/peptidases (n = 7) that were predicted to be secreted, strongly indicating these bacteria use proteins as nutrients (Fig. 2C and Supplementary Table 5).
Membrane-bound NiFe uptake-hydrogenases were encoded by both Ca. Sulfomarinibacter and Ca. P. svalbardensis MAGs (Fig. 2B, C and Supplementary Table 5). These may be important for oxidizing environmental hydrogen. The Ca. Sulfomarinibacter MAGs encoded “type-1c” NiFe hydrogenases typically found in obligate anaerobes and that are thought to be oxygen sensitive (Supplementary Fig. 11) [112]. The Ca. P. svalbardensis MAG AM4 encoded a “type-1d” NiFe hydrogenase, which are typically found in aerobes and facultative anaerobes (Supplementary Fig. 11) [112]. Inspection of best BLASTP hits from the NCBI-nr database to the Ca. Sulfomarinibacter NiFe hydrogenase sequences identified various sequences previously shown to be expressed in tidal flat sediments [113]. Formate dehydrogenases encoded among MAGs of both Ca. Sulfomarinibacter and Ca. P. svalbardensis also suggested formate may be used as an electron donor (Fig. 2B, C and Supplementary Table 5).
Genome comparisons reveal distinct properties between novel genera and adaptations to marine environments
To compare unique proteins between Ca. Sulfomarinibacter and Ca. P. svalbardensis, and therefore potential functional differences, we performed reciprocal BLASTP analyses of protein sequence complements from each genus (Supplementary Table 8, and further detailed in the Supplementary Information). In this section, we describe unique proteins encoded in multiple copies (>2 SD above the mean, excluding hypotheticals/unknowns) among each set of unique protein sequences. We hypothesized these might be of special importance for the organisms if they are present in multiple copies (Supplementary Table 8). The whole list of unique proteins are provided in Supplementary Table 8. Among proteins unique to Ca. Sulfomarinibacter were: (i) various mobile element proteins (transposases), (ii) rubrerythrins that are known to play roles in detoxification pathways for reactive oxygen species (e.g., H2O2) in anaerobes [114], (iii) heterodisulfide reductase subunits (described above), (iv) sulfatases that may play a role in removing sulfate groups from organics [115], (v) hydrolases (beta-lactamase super-family) that may play various roles, including hydrolyzing antibiotic compounds. Among those unique to Ca. P. svalbardensis were various: (i) peptidases/proteases, (ii) toxin-related proteins, (iii) c-type cytochromes, and (iv) tetratricopeptide-repeat-containing proteins that often function in protein–protein interactions [116]. Overall, these comparisons highlight many differences in protein content among the two genera, likely reflecting distinct physiologies and the large phylogenetic distances between the two genera.
Comparisons of encoded protein content of the marine Acidobacteriota MAGs with seven dsr-harboring Acidobacteriota MAGs from peatland soil [2] suggested the marine Acidobacteriota encoded unique adaptations to marine settings (Supplementary Information) (Supplementary Fig. 12). These included various predicted transporters/symporters and pumps for ions (e.g., sodium and potassium) and metals/metalloids (e.g., zinc and arsenic) that were unique to the marine MAGs. Genes for a sodium-translocating NADH-quinone oxidoreductase complex, which are used by various marine microorganisms to support respiration and cellular homeostasis [117], were only present in marine MAGs. Symporters for the osmolytes proline, glutamate and glycine, were also only present in marine MAGs.
Acidobacteriota are prevalent, active, and diverse in marine sediments
Amplicon sequencing of 16S rRNA genes and transcripts was performed from cores of Smeerenburgfjorden sediments (Fig. 6A–D and Supplementary Fig. 13). General features of these sediments include: (i) high sulfate reduction rates that peak near 100 nmol SO4–2 cm−3 d−1 around 5–6 cmbsf and slowly decline with depth, (ii) iron reduction in the top 5 cm of sediments of station J and GN, (iii) higher amounts and deeper accumulation of dissolved iron (>250 µM) from dissimilatory iron reduction in sediments of station GK, and (iv) free sulfide accumulates below 10 cm in station J and GN sediments [38, 41, 113]. Overall, the sequencing results revealed Acidobacteriota had an average relative abundance of 4.5 ± 2.2% (Fig. 6A and Supplementary Fig. 13). Thermoanaerobaculia-affiliated sequences were the most dominant of any Acidobacteriota (Fig. 6B), and reached the most abundant (11%) genus-level clade of Bacteria at 31 cmbsf in Station J (sampled in 2016). The same clade was on average the fourth most abundant genus-level clade in the same core (averaged 4.5 ± 2.8%). 16S rRNA transcripts of Thermoanaerobaculia-affiliated sequences were below 0.5% relative abundances in the surface sediments (0–1 cmbsf) of Smeerenburgfjorden cores (Fig. 6B). At station GK, Acidobacteriota 16S rRNA transcripts reached 6% relative abundance at 15 cmbsf (Fig. 6B).

A Relative abundances of phylum Acidobacteriota 16S rRNA genes or transcripts. B Relative abundances of class Thermoanaerobaculia 16S rRNA genes or transcripts. C Relative abundances of class Ca. Polarisedimenticolia 16S rRNA genes or transcripts. D Relative abundances of Ca. Sulfomarinibacter ASV_2257 16S rRNA genes or transcripts. E Relative abundances of Thermoanaerobaculia dsrB genes or transcripts. Relative abundances for genes or transcripts are depicted in red or blue, respectively. Smeerenburgfjorden stations GK, J, and GN were sampled in June 2017, and J16 was sampled in July 2016. Replicate cores from Van Keulenfjorden stations AB and AC are derived from Buongiorno et al. [40].
We also examined Acidobacteriota 16S rRNA genes from metal-rich Van Keulenfjorden sediment cores (Fig. 6A–D), from a previously published study [40]. Sediments from these cores are characterized by very high concentrations of dissolved iron (up to >500 µM) and manganese (>100 µM) that persist over tens of centimeters of depth, while exhibiting low sulfate reduction rates (<12 nmol SO4–2 cm−3 d−1) [38, 41]. The sequence analyses showed Ca. Polarisedimenticolia related sequences were the most prominent Acidobacteriota, reaching 1.5%, and averaging 1.1 ± 0.21% of communities in four cores (Fig. 6C). Members of the Thermoanaerobaculia were in much lower abundances (0.3 ± 0.2% average overall), although they reached 1.1% in deeper sections of core AB (Fig. 6B).
Mapping of metagenomic reads to the Acidobacteriota MAGs supported the general distribution trends from 16S rRNA amplicon analyses, i.e., that Thermoanaerobaculia were abundant in Smeerenburgfjorden sediments and Ca. Polarisedimenticolia were more abundant in Van Keulenfjorden sediments (Supplementary Table 2, and further detailed in Supplementary Information).
Phylogenetic analysis of 16S rRNA genes from Smeerenburgfjorden sediments revealed diverse Acidobacteriota sequences (Supplementary Fig. 14). Examination of the taxonomy of Acidobacteriota 16S rRNA sequences derived from marine sediments in the SILVA database also revealed that Thermoanaerobaculia (subdivision 23) and Ca. Polarisedimenticolia (subdivision 22) are among the most prominent Acidobacteriota lineages in marine sediments (Supplementary Fig. 15, and further detailed in Supplementary Information).
Analyses of 16S rRNA gene sequences among publically available datasets derived from diverse locations further showed members of the Ca. Sulfomarinibacter and Ca. Polarisedimenticolia are prevalent in marine sediments worldwide (Fig. 7A, B). Among the 40 16S rRNA gene study datasets where either Ca. Sulfomarinibacter and/or Ca. Polarisedimenticolia were identified, they averaged 1.6% and 1.2% of sequences, respectively. Notably, Ca. Sulfomarinibacter sequences were absent from sediments of abyssal and hadal trenches [114,115,116], and from sediments underlying mid-oceanic sites/oceanic gyres [117, 118], i.e., sediments with low organic inputs and where oxygen typically penetrates centimeters to tens-of-centimeters into sediments [119, 120]. Only Ca. Polarisedimenticolia sequences were identified at several of these deep water sites (Fig. 7B). Analyses of sequence data from surface sediments (0–2 cmbsf) of the North Sea that exhibit different permeability properties [121], revealed Ca. Sulfomarinibacter sequences were in very low relative abundances in the most permeable sites (<0.06%), yet reached a maximum of 0.7% in the impermeable sediments (Fig. 7D). In contrast, Ca. Polarisedimenticolia reached up to 2% relative abundances in the permeable sediments (Fig. 7D). These two groups therefore appear to have contrasting tolerances for oxygen.

Global maps depicting presence and relative abundances of: A Ca. Sulfomarinibacter 16S rRNA genes; B Ca. Polarisedimenticola 16S rRNA genes; C Thermoanaerobaculia dsrA/B genes. D Relative abundances of Ca. Sulfomarinibacter and Ca. Polarisedimenticola 16S rRNA genes in sediments from the North Sea that exhibit a permeability gradient (Probandt et al. [136]), and that were reanalysed in this study. Relative abundances presented from clone library-based studies were calculated from all sequences belonging to each study. Relative abundances presented from amplicon sequencing-based studies (Short Read Archives) were derived from the highest relative abundance found in any individual sample per study. As exception was that an average relative abundance of Thermoanaerobaculia dsrB was calculated from all dsrB sequences from the Northwestern Gulf of Mexico study (23 surface sediment samples) [147]. List of studies/datasets used are listed https://doi.org/10.6084/m9.figshare.14369657.v1.
Acidobacteriota actively express dsrB and are prominent dsrAB-harboring bacteria in marine sediments
Sequencing of dsrB genes and transcripts from Smeerenburgfjorden sediments revealed that Acidobacteriota dsrB averaged 13 ± 6.6% of all dsrB (DNA-derived) sequences, and 4 ± 2% of dsrB-transcripts (cDNA-derived) (Fig. 6E and Supplementary Fig. 16). Acidobacteriota dsrB sequences were the second most abundant group after Desulfobacterota dsrB, which dominated the sediments and averaged 75 ± 6% in relative abundance (Supplementary Fig. 16). Acidobacteriota dsrB reached a maximum of 19% at station GK and 31% at station J. The most abundant Acidobacteriota dsrB-OTU-17 was 100% identical (over 321 nucleotides) to dsrB from Ca. Sulfomarinibacter AM3-B MAG (Fig. 4). Amplicon-derived DsrB sequences that affiliated with the DsrB from marine Acidobacteriota MAGs were phylogenetically diverse and spread through-out the “Thermoanaerobaculia Dsr clade” (Fig. 4).
Analyses of publically available dsrAB sequence datasets (n = 24) revealed sequences of the Thermoanaerobaculia dsr-lineage were widespread (Fig. 7C) and averaged 15% of all dsrAB sequences analysed (n = 14,077 classified sequences). They reached a maximum of 54.3% in hydrothermally influenced sediments of the Guaymas Basin [118]. From all datasets analysed, sequences from the Thermoanaerobaculia dsr-lineage were the second most abundant dsrAB amplified from marine sediments after sequences of the Desulfobacterota.
Description of novel Acidobacteriota Candidatus taxa
Based on their unique phylogeny, predicted metabolic properties, CARD-FISH visualized cells of Thermoanaerobaculia (thin rods present in three different sites, see Supplementary Fig. 17) and relatively complete MAGs, we propose the following new Candidatus taxa of Acidobacteriota (Supplementary Table 4):
class Thermoanaerobaculia (subdivision 23)
order Thermoanaerobaculales
fam. nov. Ca Sulfomarinibacteraceae (GTDB family FEB-10)
gen. nov. Ca. Sulfomarinibacter
sp. nov. Ca. Sulfomarinibacter kjeldsenii sp. nov. MAG AM3-C
Ca. Sulfomarinibacter sp. MAG AM1
Ca. Sulfomarinibacter sp. MAG AM2
class nov. Ca. Polarisedimenticolia (GTDB class Mor1, subdivision 22)
ord nov. Ca. Polarisedimenticolales (GTDB order Mor1)
fam. nov. Ca. Polarisedimenticolaceae (GTDB family Mor1)
gen. nov. Ca. Polarisedimenticola
sp. nov. Ca. Polarisedimenticola svalbardensis MAG AM4.
Discussion
This study provides the first insights into the genomes and metabolic potential of abundant Thermoanaerobaculia from marine sediments, and new insights into the metabolisms of Ca. Polarisedimenticolia (Acidobacteriota subdivision 22 or GTDB class Mor1). Most notably, we revealed that MAGs from both of the major lineages of Acidobacteriota from marine sediments have capabilities to dissimilate various inorganic sulfur compounds.
Genes for the full dissimilatory sulfate reduction pathway provided the first direct link between genomes of marine sediment Acidobacteriota and DsrAB sequences of the previously undescribed “Uncultured family-level lineage 9” clade (here named “Thermoanaerobaculia Dsr lineage”). In addition to being abundant and actively transcribed in Svalbard sediments as shown here, our analysis of dsrAB sequences from various sediment sites around the world further revealed Acidobacteriota are the next most abundant dsr-harboring lineage outside of Desulfobacterota in marine sediments in general. Together, this indicates Acidobacteriota are a widespread and prominent group of inorganic sulfur-dissimilating microorganisms in marine sediments, and therefore likely make significant contributions to the sulfur-cycle in global marine sediments.
While enzymes of the dissimilatory sulfate reduction pathway are widely used for anaerobic reduction of sulfite/sulfate [35], some organisms can use them in reverse for the oxidation of reduced sulfur compounds [122], or for disproportionation of sulfur compounds [123, 124]. Because no enzymes are currently known that distinguish these different metabolisms, discerning sulfur metabolisms based on genomic data requires careful interpretation [123, 124]. For instance, the Ca. Sulfomarinibacter MAGs encode DsrL, which was previously thought to be exclusively found in sulfur-oxidizing bacteria [92]. However, recent work showed DsrL can function in a reductive manner in biochemical assays [92, 94], and was highly expressed during reductive sulfur- and thiosulfate-respiration by Desulfurella amilsii [92, 125]. The DsrL of Ca. Sulfomarinibacter contained putative NADP(H)-binding domain structures that may enable coupling of NADPH as electron donor to sulfite reduction [94], as well as phylogenetic relatedness with DsrL of Desulfurella amilsii. Together, this indicates the DsrL of Ca. Sulfomarinibacter has potential to facilitate a reductive pathway.
The Ca. Sulfomarinibacter MAGs encoded rhodonase-like TusA and DsrE2, which act as sulfur-trafficking proteins in reverse-Dsr harboring sulfur-oxidizing bacteria, i.e., they help deliver sulfur to DsrABC for oxidation [126]. Interestingly, the “YTD gene clusters” that encode these enzymes are also common in genomes of anaerobic elemental sulfur-reducing and/or -disproportionating bacteria that have Dsr, and are suggested to be genetic indicators for disproportionation potential among these anaerobes [97]. The TusA proteins from Ca. Sulfomarinibacter were most closely related to TusA from various anaerobic sulfur-reducing and -disproportionating Desulfobacterota (Supplementary Fig. 5A). This suggested Ca. Sulfomarinibacter could reduce and/or disproportionate elemental sulfur, or possibly other sulfur compounds that can be trafficked by TusA, like thiosulfate [127]. Indeed, the ability to disproportionate sulfur compounds is common among sulfate-reducing Desulfobacterota [128]. Elemental sulfur is often the most abundant sulfur-cycle intermediate (SCI) in marine sediments [129], and was measured in sediments from Smeerenburgfjorden up to 0.15 wt% of total sulfur [39]. Overall, the gene content of Ca. Sulfomarinibacter MAGs indicated flexible dissimilatory sulfur metabolisms that may be dictated by and/or switch under different biogeochemical and redox conditions.
Results indicated Ca. Sulfomarinibacter likely use the dissimilatory sulfate reduction pathway in a reductive direction in most depths of the sediments studied. Firstly, Acidobacteriota were relatively abundant and expressed dsrB in deeper (>15–75 cmbsf), strictly anoxic sediment layers of Smeerenburgfjorden. These sediments depths lack electron acceptors that could sustain these abundant populations growing via biological oxidation of sulfides, i.e., oxygen, nitrate or oxidized metals [31, 130]. In Station J sediments, oxygen and nitrate are depleted within millimeters-to-centimetres of the surface [131, 132], and sulfide oxidation facilitated by Fe(III) is negligible [41]. An alternative possibility is that cryptic biogeochemical cycling could sustain sulfide oxidation, i.e., fast consumption and production of low concentrations of sulfides and oxidants [133]. Nevertheless, it remains unproven whether biological sulfide oxidation occurs in deep sediments that lack measurable concentrations of required oxidants [28]. On the other hand, the relative abundances of Acidobacteriota peaked in subsurface zones around 5 cmbsf in Station J sediments, where sulfate reduction rates also peak [41, 134]. In another study, Acidobacteriota 16S rRNA gene relative abundances were also highly correlated with sulfate reduction rates in sediments from Greenland [37]. These associations therefore point toward an active role in the reduction and/or disproportionation of sulfur compounds of various oxidation states by Ca. Sulfomarinibacter in marine sediments.
Our results also suggested marine Acidobacteriota have potential to reduce various inorganic sulfur compounds independent of the Dsr pathway. Our tetrathionate-amended microcosm experiment suggested Ca. Sulfomarinibacter use tetrathionate as an electron acceptor via cytochromes because genes for otr increased expression after tetrathionate was added to microcosms (Fig. 5). Although some expression of otr was determined in the control at day 1, possibly as a result of expression in the starting sediments, expression of otr was nearly undetectable in controls at day 8, but remained high in tetrathionate-amended microcosms. Overall, these results support the roles of these enzymes in tetrathionate reduction under in situ-like conditions. This is noteworthy because these enzymes were only previously shown to perform this function during biochemical assays [135], i.e., their utilization under in situ-like conditions was unknown. The ability to utilize SCI, e.g., tetrathionate or elemental sulfur/polysulfides/thiosulfate, could be important in sediment zones where SCI might be generated from sulfides reacting with available oxidants [41].
The Ca. Sulfomarinibacter MAGs indicated they could respire oxygen using terminal cbb3- or aa3-type cytochromes, although we speculate these may instead be used for defense against oxygen. This is because they encoded many characteristics of obligate anaerobes, and they appear to prefer subsurface and impermeable sediments where oxygen is absent or scarce (Supplementary Discussion). We also hypothesize that the different redox metabolisms of Ca. Sulfomarinibacter and Ca. Polarisedimenticola may explain their different abundances among Svalbard fjord sediments with different biogeochemical properties (Supplementary Discussion). That is, the Ca. Sulfomarinibacter may be adapted to low redox environments, and are thus more abundant in the reduced (visibly black), sulfidic subsurface sediments of Smeerenburgfjorden. In comparison, the Ca. P. svalbardensis MAG had additional genes to utilize high-potential electron acceptors such as oxygen, nitrate and oxidized metals, and these organisms may thus be better adapted to the more high redox, metal-rich sediments of Van Keulenfjorden (visibly reddish-orange). This was also supported by our reanalyses of 16S rRNA genes from a sediment permeability gradient in the North Sea [136], which showed Ca. Sulfomarinibacter prefer impermeable sediments where oxygen penetration is limited, while the Ca. Polarisedimenticola thrive in permeable sediments where oxygen is available.
If members of the Ca. Sulfomarinibacter are indeed SRM, a question arises regarding how they co-exist with dominant sulfate-reducing Desulfobacterota populations, as both can apparently use hydrogen, acetate or formate as substrates. However, we identified genes for use of several organic substrates that may enable Ca. Sulfomarinibacter to occupy a distinct nutrient niche. Complex carbohydrates such as cellulose (or structurally similar compounds) could be used. Carbohydrates are not used by most known isolated Desulfobacterota SRM [35]. Plant-derived molecules could stem from terrestrial run-off, which is a major source of organic carbon to arctic sediments [137, 138] and to coastal marine systems in general [139]. Additionally, various marine algae are known to produce cellulose [140]. The predicted ability to utilize cyanophycin could also facilitate a unique nutrient niche. Cyanophycin is a multi-L-arginyl-poly-L-aspartic acid, commonly produced by cyanobacteria as a storage compound [141, 142]. Indeed, few organisms are known to use cyanophycin anaerobically [143], and no anaerobes are known from marine sediments.
Ca. Polarisedimenticola svalbardensis appeared to have a high propensity for the degradation of proteins, which was indicated by a suite of predicted secreted peptidases. A related Mor1 Acidobacteriota genome (GCA_001664505.1) (Fig. 1) was recovered as a bacterial co-inhabitant of a cyanobacterial enrichment culture from seawater, suggesting it used organic material/necromass from the primary-producing cyanobacterium [144]. Ca. Polarisedimenticola may therefore contribute to protein degradation in marine sediments, where proteinaceous organics comprise a large proportion (~10%) of available organic matter [145].
In summary, the genome-encoded dissimilatory sulfur metabolisms, the high abundances and activity of Ca. Sulfomarinibacter in the sulfidic zones of Svalbard sediments, as well as prominence in global marine sediments, suggests these novel Acidobacteriota of the class Thermoanaerobaculia (subdivision 23) are important players in the biogeochemical sulfur cycles of marine sediments. Our data also indicated that Ca. Sulfomarinibacter thrive largely via anaerobic metabolisms with the capability to use various other electron acceptors with different redox potentials, including biogeochemically relevant metal-oxides. Additionally, we show that Ca. Polarisedimenticola svalbardensis, a member of a different class of Acidobacteriota (subdivision 22), has the genetic potential for protein degradation and for metabolisms driven by high redox potential electron acceptors such as oxygen, nitrate and metal-oxides.
References
Kuske CR, Ticknor LO, Miller ME, Dunbar JM, Davis JA, Barns SM, et al. Comparison of soil bacterial communities in rhizospheres of three plant species and the interspaces in an arid grassland. Appl Environ Microbiol. 2002;68:1854–63.
Hausmann B, Pelikan C, Herbold CW, Köstlbacher S, Albertsen M, Eichorst SA, et al. Peatland Acidobacteria with a dissimilatory sulfur metabolism. ISME J. 2018;12:1729–42.
Kielak AM, Barreto CC, Kowalchuk GA, van Veen JA, Kuramae EE. The ecology of acidobacteria: moving beyond genes and genomes. Front Microbiol. 2016;7:744.
Eichorst SA, Trojan D, Roux S, Herbold C, Rattei T, Woebken D. Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ Microbiol. 2018;20:1041–63.
LaPara TM, Nakatsu CH, Pantea L, Alleman JE. Phylogenetic analysis of bacterial communities in mesophilic and thermophilic bioreactors treating pharmaceutical wastewater. Appl Environ Microbiol. 2000;66:3951–9.
Coskun ÖK, Özen V, Wankel SD, Orsi WD. Quantifying population-specific growth in benthic bacterial communities under low oxygen using H218O. ISME J. 2019;13:1546–59.
Ward NL, Challacombe JF, Janssen PH, Henrissat B, Coutinho PM, Wu M, et al. Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl Environ Microbiol. 2009;75:2046–56.
Challacombe JF, Eichorst SA, Hauser L, Land M, Xie G, Kuske CR. Biological consequences of ancient gene acquisition and duplication in the large genome of Candidatus Solibacter usitatus Ellin6076. PLoS ONE. 2011;6:e24882.
Coates JD, Ellis DJ, Gaw CV, Lovley DR. Geothrix fermentans gen. nov., sp. nov., a novel Fe(III)-reducing bacterium from a hydrocarbon-contaminated aquifer. Int J Syst Evol Microbiol. 1999;49:1615–22.
Garcia Costas AM, Liu Z, Tomsho LP, Schuster SC, Ward DM, Bryant DA. Complete genome of Candidatus Chloracidobacterium thermophilum, a chlorophyll-based photoheterotroph belonging to the phylum Acidobacteria. Environ Microbiol. 2012;14:177–90.
Losey NA, Stevenson BS, Busse H-J, Sinninghe Damsté JS, Rijpstra WIC, Rudd S, et al. Thermoanaerobaculum aquaticum gen. nov., sp. nov., the first cultivated member of Acidobacteria subdivision 23, isolated from a hot spring. Int J Syst Evol Microbiol. 2013;63:4149–57.
Woodcroft BJ, Singleton CM, Boyd JA, Evans PN, Emerson JB, Zayed AAF, et al. Genome-centric view of carbon processing in thawing permafrost. Nature. 2018;560:49–54.
Fukunaga Y, Kurahashi M, Yanagi K, Yokota A, Harayama S. Acanthopleuribacter pedis gen. nov., sp. nov., a marine bacterium isolated from a chiton, and description of Acanthopleuribacteraceae fam. nov., Acanthopleuribacterales ord. nov., Holophagaceae fam. nov., Holophagales ord. nov. and Holophagae classis nov. in the phylum ‘Acidobacteria’. Int J Syst Evol. 2008;58:2597–601.
Yilmaz P, Yarza P, Rapp JZ, Glöckner FO. Expanding the world of marine bacterial and archaeal clades. Front Microbiol. 2015;6:1524.
Quaiser A, Zivanovic Y, Moreira D, López-García P. Comparative metagenomics of bathypelagic plankton and bottom sediment from the Sea of Marmara. ISME J. 2011;5:285–304.
O’Connor-Sánchez A, Rivera-Domínguez AJ, Santos-Briones C, de L, López-Aguiar LK, Peña-Ramírez YJ, et al. Acidobacteria appear to dominate the microbiome of two sympatric Caribbean Sponges and one Zoanthid. Biol Res. 2014;47:67.
Zhou Z, Liu Y, Xu W, Pan J, Luo Z-H, Li M. Genome- and community-level interaction insights into carbon utilization and element cycling functions of hydrothermarchaeota in hydrothermal sediment. mSystems. 2020;5:e00795–19.
Polymenakou PN, Lampadariou N, Mandalakis M, Tselepides A. Phylogenetic diversity of sediment bacteria from the southern Cretan margin, Eastern Mediterranean Sea. Syst Appl Microbiol. 2009;32:17–26.
Kielak AM, van Veen JA, Kowalchuk GA. Comparative analysis of acidobacterial genomic fragments from terrestrial and aquatic metagenomic libraries, with emphasis on acidobacteria subdivision 6. Appl Environ Microbiol. 2010;76:6769–77.
Orcutt BN, Sylvan JB, Knab NJ, Edwards KJ. Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol Mol Biol Rev. 2011;75:361–422.
Wang Y, Sheng H-F, He Y, Wu J-Y, Jiang Y-X, Tam NF-Y, et al. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl Environ Microbiol. 2012;78:8264–71.
Choi H, Koh H-W, Kim H, Chae J-C, Park S-J. Microbial community composition in the marine sediments of Jeju Island: next-generation sequencing surveys. J Microbiol Biotechnol. 2016;26:883–90.
Conte A, Papale M, Amalfitano S, Mikkonen A, Rizzo C, De Domenico E, et al. Bacterial community structure along the subtidal sandy sediment belt of a high Arctic fjord (Kongsfjorden, Svalbard Islands). Sci Total Environ. 2018;619–620:203–11.
Kapili BJ, Barnett SE, Buckley DH, Dekas AE. Evidence for phylogenetically and catabolically diverse active diazotrophs in deep-sea sediment. ISME J. 2020;14:971–83.
Tschoeke DA, Coutinho FH, Leomil L, Cavalcanti G, Silva BS, Garcia GD, et al. New bacterial and archaeal lineages discovered in organic rich sediments of a large tropical Bay. Mar Genom. 2020;54:100789.
Bowles MW, Mogollón JM, Kasten S, Zabel M, Hinrichs K-U. Global rates of marine sulfate reduction and implications for sub-sea-floor metabolic activities. Science. 2014;344:889–91.
Parkes RJ, John Parkes R, Cragg BA, Wellsbury P. Recent studies on bacterial populations and processes in subseafloor sediments: a review. Hydrogeol J. 2000;8:11–28.
Jørgensen BB, Findlay AJ, Pellerin A. The biogeochemical sulfur cycle of marine sediments. Front Microbiol. 2019;10:849.
Revsbech NP, Barker Jorgensen B, Blackburn TH. Oxygen in the Sea Bottom Measured with a microelectrode. Science. 1980;207:1355.
Anantharaman K, Hausmann B, Jungbluth SP, Kantor RS, Lavy A, Warren LA, et al. Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 2018;12:1715–28.
Wasmund K, Mußmann M, Loy A. The life sulfuric: microbial ecology of sulfur cycling in marine sediments. Environ Microbiol Rep. 2017;9:323–44.
Jørgensen BB. Mineralization of organic matter in the sea bed—the role of sulphate reduction. Nature. 1982;296:643–5.
Müller AL, Pelikan C, de Rezende JR, Wasmund K, Putz M, Glombitza C, et al. Bacterial interactions during sequential degradation of cyanobacterial necromass in a sulfidic arctic marine sediment. Environ Microbiol. 2018;20:2927–40.
Finke N, Vandieken V, Jørgensen BB. Acetate, lactate, propionate, and isobutyrate as electron donors for iron and sulfate reduction in Arctic marine sediments, Svalbard. FEMS Microbiol Ecol. 2007;59:10–22.
Rabus R, Venceslau SS, Wöhlbrand L, Voordouw G, Wall JD, Pereira IAC. A post-genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. Adv Microb Physiol. 2015;66:55–321.
Müller AL, Kjeldsen KU, Rattei T, Pester M, Loy A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 2015;9:1152–65.
Pelikan C, Jaussi M, Wasmund K, Seidenkrantz M-S, Pearce C, Kuzyk ZZA, et al. Glacial runoff promotes deep burial of sulfur cycling-associated microorganisms in marine sediments. Front Microbiol. 2019;10:2558.
Wehrmann LM, Formolo MJ, Owens JD, Raiswell R, Ferdelman TG, Riedinger N, et al. Iron and manganese speciation and cycling in glacially influenced high-latitude fjord sediments (West Spitsbergen, Svalbard): evidence for a benthic recycling-transport mechanism. Geochim Cosmochim Acta. 2014;141:628–55.
Wehrmann LM, Riedinger N, Brunner B, Kamyshny A, Hubert CRJ, Herbert LC, et al. Iron-controlled oxidative sulfur cycling recorded in the distribution and isotopic composition of sulfur species in glacially influenced fjord sediments of west Svalbard. Chem Geol. 2017;466:678–95.
Buongiorno J, Herbert LC, Wehrmann LM, Michaud AB, Laufer K, Røy H, et al. Complex microbial communities drive iron and sulfur cycling in arctic fjord sediments. Appl Environ Microbiol. 2019;85:e00949–19.
Michaud AB, Laufer K, Findlay A, Pellerin A, Antler G, Turchyn AV, et al. Glacial influence on the iron and sulfur cycles in Arctic fjord sediments (Svalbard). Geochim Cosmochim Acta. 2020;280:423–40.
Jørgensen BB, Laufer K, Michaud AB, Wehrmann LM. Biogeochemistry and microbiology of high Arctic marine sediment ecosystems-—case study of Svalbard fjords. Limnol Oceanogr. 2020;30:85.
Buongiorno J, Sipes K, Wasmund K, Loy A, Lloyd KG. Woeseiales transcriptional response to shallow burial in Arctic fjord surface sediment. PLOS ONE. 2020;15:e0234839.
Kanneworff E, Nicolaisen W. The ‘Haps’ a frame-supported bottom corer. Ophelia. 1972;10:119–28.
Meischner D, Rumohr J. A light-weight, high-momentum gravity corer for subaqueous sediments. Senckenberg Marit. 1974;6:105–17.
Graue J, Engelen B, Cypionka H. Degradation of cyanobacterial biomass in anoxic tidal-flat sediments:a microcosm study of metabolic processes and community changes. ISME J. 2012;6:660–9.
Angel R, Claus P, Conrad R. Methanogenic archaea are globally ubiquitous in aerated soils and become active under wet anoxic conditions. ISME J. 2012;6:847–62.
Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–8.
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–34.
Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, et al. KBase: the United States department of energy systems biology knowledgebase. Nat Biotechnol. 2018;36:566–9.
Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359
Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, et al. Binning metagenomic contigs by coverage and composition. Nat Methods. 2014;11:1144–6.
Wu Y-W, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32:605–7.
Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol. 2018;3:836–43.
Olm MR, Brown CT, Brooks B, Banfield JF. dRep:a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11:2864–8.
Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 2016;7:13219.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Bushnell B, Rood J, Singer E. BBMerge-accurate paired shotgun read merging via overlap. PLOS ONE. 2017;12:e0185056.
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36:1925–7.
Richter M, Rosselló-Móra R, Oliver Glöckner F, Peplies J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016;32:929–31.
Varghese NJ, Mukherjee S, Ivanova N, Konstantinidis KT, Mavrommatis K, Kyrpides NC, et al. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 2015;43:6761–71.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genom. 2008;9:75.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Boeckmann B, Bairoch A, Apweiler R, Blatter M-C, Estreicher A, Gasteiger E, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–70.
Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017;45:D200–3.
Ogata H, Fujibuchi W, Goto S, Kanehisa M. A heuristic graph comparison algorithm and its application to detect functionally related enzyme clusters. Nucleic Acids Res. 2000;28:4021–8.
Tamames J, Casari G, Ouzounis C, Valencia A. Conserved clusters of functionally related genes in two bacterial genomes. J Mol Evol. 1997;44:66–73.
Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014;42:D459–71.
Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol. 2016;18:1403–14.
Apprill A, McNally S, Parsons R, Weber L. Minor revision to V4 region SSU rRNA 806 R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat Microb Ecol. 2015;75:129–37.
Herbold CW, Pelikan C, Kuzyk O, Hausmann B, Angel R, Berry D, et al. A flexible and economical barcoding approach for highly multiplexed amplicon sequencing of diverse target genes. Front Microbiol. 2015;6:731.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16 S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 2011;108:4516–22.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Callahan BJ, Sankaran K, Fukuyama JA, McMurdie PJ, Holmes SP. Bioconductor Workflow for Microbiome Data Analysis: from raw reads to community analyses. F1000 Res. 2016;5:1492.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41.
Lagkouvardos I, Fischer S, Kumar N, Clavel T. Rhea: a transparent and modular R pipeline for microbial profiling based on 16S rRNA gene amplicons. PeerJ. 2017;5:e2836.
Pelikan C, Herbold CW, Hausmann B, Müller AL, Pester M, Loy A. Diversity analysis of sulfite- and sulfate-reducing microorganisms by multiplex dsrA and dsrB amplicon sequencing using new primers and mock community-optimized bioinformatics. Environ Microbiol. 2016;18:2994–3009.
Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8.
Trifinopoulos J, Nguyen L-T, von Haeseler A, Minh BQ. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016;44:W232–5.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, et al. ARB: a Software environment for sequence data. Nucleic Acids Res. 2004;32:1363–71.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6.
Duval S, Ducluzeau A-L, Nitschke W, Schoepp-Cothenet B. Enzyme phylogenies as markers for the oxidation state of the environment:the case of respiratory arsenate reductase and related enzymes. BMC Evol Biol. 2008;8:206.
Katoh K, Misawa K, Kuma K-I, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66.
Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004;5:113.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Barco RA, Garrity GM, Scott JJ, Amend JP, Nealson KH, Emerson D. A genus definition for bacteria and archaea based on a standard genome relatedness index. Mbio. 2020;11:e02475–19.
Santos AA, Venceslau SS, Grein F, Leavitt WD, Dahl C, Johnston DT, et al. A protein trisulfide couples dissimilatory sulfate reduction to energy conservation. Science. 2015;350:1541–5.
Löffler M, Feldhues J, Venceslau SS, Kammler L, Grein F, Pereira IAC, et al. DsrL mediates electron transfer between NADH and rDsrAB in Allochromatium vinosum. Environ Microbiol. 2019;22:783–95.
Mizuno N, Voordouw G, Miki K, Sarai A, Higuchi Y. Crystal structure of dissimilatory sulfite reductase D (DsrD) protein–possible interaction with B- and Z-DNA by its Winged-Helix Motif. Structure. 2003;11:1133–40.
Löffler M, Wallerang KB, Venceslau SS, Pereira IAC, Dahl C. The iron-sulfur flavoprotein DsrL as NAD(P)H: acceptor oxidoreductase in oxidative and reductive dissimilatory sulfur metabolism. Front Microbiol. 2020;11:2560.
Mowat CG, Rothery E, Miles CS, McIver L, Doherty MK, Drewette K, et al. Octaheme tetrathionate reductase is a respiratory enzyme with novel heme ligation. Nat Struct Mol Biol. 2004;11:1023–4.
Kurth JM, Dahl C, Butt JN. Catalytic protein film electrochemistry provides a direct measure of the tetrathionate/thiosulfate reduction potential. J Am Chem Soc. 2015;137:13232–5.
Umezawa K, Kojima H, Kato Y, Fukui M. Disproportionation of inorganic sulfur compounds by a novel autotrophic bacterium belonging to Nitrospirota. Syst Appl Microbiol. 2020;43:126110.
Wells M, McGarry J, Gaye MM, Basu P, Oremland RS, Stolz JF. Respiratory Selenite Reductase from Bacillus selenitireducens Strain MLS10. J Bacteriol. 2019;201:e00614–8.
Ma K, Schicho RN, Kelly RM, Adams MW. Hydrogenase of the hyperthermophile Pyrococcus furiosus is an elemental sulfur reductase or sulfhydrogenase: evidence for a sulfur-reducing hydrogenase ancestor. Proc Natl Acad Sci USA. 1993;90:5341–4.
Bryant FO, Adams MW. Characterization of hydrogenase from the hyperthermophilic archaebacterium, Pyrococcus furiosus. J Biol Chem. 1989;264:5070–9.
Ma K, Weiss R, Adams MW. Characterization of hydrogenase II from the hyperthermophilic archaeon Pyrococcus furiosus and assessment of its role in sulfur reduction. J Bacteriol. 2000;182:1864–71.
van Haaster DJ, Silva PJ, Hagedoorn P-L, Jongejan JA, Hagen WR. Reinvestigation of the steady-state kinetics and physiological function of the soluble NiFe-hydrogenase I of Pyrococcus furiosus. J Bacteriol. 2008;190:1584–7.
Edwards MJ, White GF, Lockwood CW, Lawes MC, Martel A, Harris G, et al. Structural modeling of an outer membrane electron conduit from a metal-reducing bacterium suggests electron transfer via periplasmic redox partners. J Biol Chem. 2018;293:8103–12.
Edwards MJ, White GF, Butt JN, Richardson DJ, Clarke TA. The crystal structure of a biological insulated transmembrane molecular wire. Cell. 2020;181:665–73.
Mehta T, Coppi MV, Childers SE, Lovley DR. Outer membrane c-type cytochromes required for Fe(III) and Mn(IV) oxide reduction in Geobacter sulfurreducens. Appl Environ Microbiol. 2005;71:8634–41.
Tang H-Y, Holmes DE, Ueki T, Palacios PA, Lovley DR. Iron corrosion via direct metal-microbe electron transfer. Mbio. 2019;10:10:e00303–19.
Yan Z, Wang M, Ferry JG. A ferredoxin- and F420H2-dependent, electron-bifurcating, heterodisulfide reductase with homologs in the domains bacteria and archaea. Mbio. 2017;8:e02285–16.
Buckel W, Thauer RK. Flavin-based electron bifurcation, a new mechanism of biological energy coupling. Chem Rev. 2018;118:3862–86.
Pereira IAC, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS. A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front Microbiol. 2011;2:69.
Yernool DA, McCarthy JK, Eveleigh DE, Bok JD. Cloning and characterization of the glucooligosaccharide catabolic pathway beta-glucan glucohydrolase and cellobiose phosphorylase in the marine hyperthermophile Thermotoga neapolitana. J Bacteriol. 2000;182:5172–9.
Kim SK, Himmel ME, Bomble YJ. Expression of a cellobiose phosphorylase from Thermotoga maritima in Caldicellulosiruptor bescii improves the phosphorolytic pathway and results in a dramatic Increase in cellulolytic activity. Appl Environ Microbiol. 2018;84:e02348–17.
Søndergaard D, Pedersen CNS, Greening C. HydDB: a web tool for hydrogenase classification and analysis. Sci Rep. 2016;6:34212.
Dyksma S, Pjevac P, Ovanesov K, Mussmann M. Evidence for H2 consumption by uncultured Desulfobacterales in coastal sediments. Environ Microbiol. 2018;20:450–61.
Sztukowska M, Bugno M, Potempa J, Travis J, Kurtz DM Jr. Role of rubrerythrin in the oxidative stress response of Porphyromonas gingivalis. Mol Microbiol. 2002;44:479–88.
Kertesz MA. Riding the sulfur cycle-metabolism of sulfonates and sulfate esters in Gram-negative bacteria. FEMS Microbiol Rev. 2000;24:135–75.
Cerveny L, Straskova A, Dankova V, Hartlova A, Ceckova M, Staud F, et al. Tetratricopeptide repeat motifs in the world of bacterial pathogens: role in virulence mechanisms. Infect Immun. 2013;81:629–35.
Verkhovsky MI, Bogachev AV. Sodium-translocating NADH:quinone oxidoreductase as a redox-driven ion pump. Biochim Biophys Acta. 2010;1797:738–46.
Dhillon A, Teske A, Dillon J, Stahl DA, Sogin ML. Molecular characterization of sulfate-reducing bacteria in the Guaymas Basin. Appl Environ Microbiol. 2003;69:2765–72.
Jochum LM, Chen X, Lever MA, Loy A, Jørgensen BB, Schramm A, et al. Depth distribution and assembly of sulfate-reducing microbial communities in marine sediments of Aarhus Bay. Appl Environ Microbiol. 2017;83:e01547–17.
Petro C, Zäncker B, Starnawski P, Jochum LM, Ferdelman TG, Jørgensen BB, et al. Marine deep biosphere microbial communities assemble in near-surface sediments in Aarhus Bay. Front Microbiol. 2019;10:758.
Marshall IPG, Ren G, Jaussi M, Lomstein BA, Jørgensen BB, Røy H, et al. Environmental filtering determines family-level structure of sulfate-reducing microbial communities in subsurface marine sediments. ISME J. 2019;13:1920–32.
Loy A, Duller S, Baranyi C, Mussmann M, Ott J, Sharon I, et al. Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur-oxidizing prokaryotes. Environ Microbiol. 2009;11:289–99.
Thorup C, Schramm A, Findlay AJ, Finster KW, Schreiber L. Disguised as a sulfate reducer: growth of the deltaproteobacterium desulfurivibrio alkaliphilus by sulfide oxidation with nitrate. mBio. 2017;8:e00671–17.
Kjeldsen KU, Schreiber L, Thorup CA, Boesen T, Bjerg JT, Yang T, et al. On the evolution and physiology of cable bacteria. Proc Natl Acad Sci USA. 2019;116:19116–25.
Florentino AP, Pereira IAC, Boeren S, van den Born M, Stams AJM, Sánchez-Andrea I. Insight into the sulfur metabolism of Desulfurella amilsii by differential proteomics. Environ Microbiol. 2019;21:209–25.
Dahl C. Cytoplasmic sulfur trafficking in sulfur-oxidizing prokaryotes. IUBMB Life. 2015;67:268–74.
Liu L-J, Stockdreher Y, Koch T, Sun S-T, Fan Z, Josten M, et al. Thiosulfate transfer mediated by DsrE/TusA homologs from acidothermophilic sulfur-oxidizing archaeon Metallosphaera cuprina. J Biol Chem. 2014;289:26949–59.
Slobodkin AI, Slobodkina GB. Diversity of sulfur-disproportionating microorganisms. Microbiology. 2019;88:509–22.
Zopfi J, Ferdelman TG, Fossing H. Distribution and fate of sulfur intermediates-sulfite, tetrathionate, thiosulfate, and elemental sulfur-in marine sediments. In: Amend JP, Edwards KJ, Lyons TW, editors. Sulfur biogeochemistry: past and present. 379. Colorado, USA: Geological Society of America Special Paper; 2004. p. 97–116.
Henkel JV, Dellwig O, Pollehne F, Herlemann DPR, Leipe T, Schulz-Vogt HN. A bacterial isolate from the Black Sea oxidizes sulfide with manganese(IV) oxide. Proc Natl Acad Sci USA. 2019;116:12153–5.
Jørgensen BB, Dunker R, Grünke S, Røy H. Filamentous sulfur bacteria, Beggiatoa spp., in arctic marine sediments (Svalbard, 79°N). FEMS Microbiol Ecol. 2010;73:500–13.
Canion A, Overholt WA, Kostka JE, Huettel M, Lavik G, Kuypers MMM. Temperature response of denitrification and anaerobic ammonium oxidation rates and microbial community structure in Arctic fjord sediments. Environ Microbiol. 2014;16:3331–44.
Kappler A, Bryce C. Cryptic biogeochemical cycles: unravelling hidden redox reactions. Environ Microbiol. 2017;19:842–6.
Vandieken V, Finke N, Jørgensen BB. Pathways of carbon oxidation in an Arctic fjord sediment (Svalbard) and isolation of psychrophilic and psychrotolerant Fe(III)-reducing bacteria. Mar Ecol Prog Ser. 2006;322:29–41.
Buckley A, MacGregor B, Teske A. Identification, expression and activity of candidate nitrite reductases from orange Beggiatoaceae, Guaymas Basin. Front Microbiol. 2019;10:644.
Probandt D, Knittel K, Tegetmeyer HE, Ahmerkamp S, Holtappels M, Amann R. Permeability shapes bacterial communities in sublittoral surface sediments. Environ Microbiol. 2017;19:1584–99.
Benner R, Benitez-Nelson B, Kaiser K, Amon RMW. Export of young terrigenous dissolved organic carbon from rivers to the Arctic Ocean. Geophys Res Lett. 2004;31:L05305.
Opsahl S, Benner R, Amon RMW. Major flux of terrigenous dissolved organic matter through the Arctic Ocean. Limnol Oceanogr. 1999;44:2017–23.
Burdige DJ. Burial of terrestrial organic matter in marine sediments: a re-assessment. Global Biogeochem Cycles. 2005;19:GB4011.1–7.
Lakshmi DS, Trivedi N, Reddy CRK. Synthesis and characterization of seaweed cellulose derived carboxymethyl cellulose. Carbohydr Polym. 2017;157:1604–10.
Simon RD. Cyanophycin Granules from the Blue-Green Alga Anabaena cylindrica: a reserve material consisting of copolymers of aspartic acid and arginine. Proc Natl Acad Sci USA. 1971;68:265–7.
Simon RD, Weathers P. Determination of the structure of the novel polypeptide containing aspartic acid and arginine which is found in Cyanobacteria. Biochim Biophys Acta. 1976;420:165–76.
Obst M, Krug A, Luftmann H, Steinbüchel A. Degradation of cyanophycin by Sedimentibacter hongkongensis strain KI and Citrobacter amalonaticus strain G isolated from an anaerobic bacterial consortium. Appl Environ Microbiol. 2005;71:3642–52.
Cummings SL, Barbé D, Leao TF, Korobeynikov A, Engene N, Glukhov E, et al. A novel uncultured heterotrophic bacterial associate of the cyanobacterium Moorea producens JHB. BMC Microbiol. 2016;16:198.
Burdige DJ. Preservation of organic matter in marine sediments:controls, mechanisms, and an imbalance in sediment organic carbon budgets? Chem Rev. 2007;107:467–85.
Rasigraf O, Helmond NAGM, Frank J, Lenstra WK, Egger M, Slomp CP, et al. Microbial community composition and functional potential in Bothnian Sea sediments is linked to Fe and S dynamics and the quality of organic matter. Limnol Oceanogr. 2020;65:e00169.
Sánchez-Soto MF, Cerqueda-García D, Alcántara-Hernández RJ, Falcón LI, Pech D, Árcega-Cabrera F, et al. Assessing the diversity of benthic sulfate-reducing microorganisms in Northwestern Gulf of Mexico by illumina sequencing of dsrB gene. Microb Ecol. 2020;81:908–21.
Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27:1009–10.
Acknowledgements
This research was supported by the Austrian Science Fund (FWF grants P29426 to KW and P25111-B22 to AL) and the MetaBac Research Platform of the University of Vienna. We thank Captain Stig Henningsen of MS Farm during Svalbard expeditions. We particularly thank Bo Barker Jørgensen, Alexander Michaud, and Susann Henkel for organizing the 2016 and 2017 Svalbard expeditions, and all members of Svalbard expeditions for help with sample collection, especially Claus Pelikan. We thank the Biomedical Sequencing Facility (BSF) Vienna for sequencing of metagenome samples, the Joint Microbiome Facility (JMF) of the Medical University of Vienna and the University of Vienna, and Microsynth for sequencing amplicons. We specifically thank Jasmin Schwarz, Gudrun Kohl, and Petra Pjevac from the JMF for assisting with amplicon sequencing. We thank Marc Mussman and Stefan Dyksma for providing the hydrogenase database. We are grateful to Bernhard Schink for help with Latin naming of taxa.
Author information
Authors and Affiliations
Contributions
KW and AL conceived the study. KW, JB, KGL, and AL collected samples. MF and KW performed microcosm experiments. MF performed DNA/RNA extractions and PCRs for amplicon sequencing. KW, MF, and JB performed bioinformatic analyses of metagenomic and amplicon data, with support from CH, BH, and TR. MF and BH analysed amplicon sequencing data. MF designed and performed RT-qPCR experiments. KW and MF interpreted genomic data. MF and KW performed CARD-FISH. MF, KW, and AL wrote the paper, with contributions from all authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Flieder, M., Buongiorno, J., Herbold, C.W. et al. Novel taxa of Acidobacteriota implicated in seafloor sulfur cycling. ISME J 15, 3159–3180 (2021). https://doi.org/10.1038/s41396-021-00992-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-021-00992-0
This article is cited by
-
Oxygen respiration and polysaccharide degradation by a sulfate-reducing acidobacterium
Nature Communications (2023)
-
Chemolithoautotroph distributions across the subsurface of a convergent margin
The ISME Journal (2023)
-
Replicated life-history patterns and subsurface origins of the bacterial sister phyla Nitrospirota and Nitrospinota
The ISME Journal (2023)
-
Genome-resolved metagenomics of Venice Lagoon surface sediment bacteria reveals high biosynthetic potential and metabolic plasticity as successful strategies in an impacted environment
Marine Life Science & Technology (2023)
-
Polychaete Bioturbation Alters the Taxonomic Structure, Co-occurrence Network, and Functional Groups of Bacterial Communities in the Intertidal Flat
Microbial Ecology (2023)
