
Abstract
Lakes play a pivotal role in ecological and biogeochemical processes and have been described as “sentinels” of environmental change. Assessing “lake health” across large geographic scales is critical to predict the stability of their ecosystem services and their vulnerability to anthropogenic disturbances. The LakePulse research network is tasked with the assessment of lake health across gradients of land use on a continental scale. Bacterial communities are an integral and rapidly responding component of lake ecosystems, yet large-scale responses to anthropogenic activity remain elusive. Here, we assess the ecological impact of land use on bacterial communities from over 200 lakes covering more than 660,000 km2 across Eastern Canada. In addition to community variation between ecozones, land use across Eastern Canada also appeared to alter diversity, community composition, and network structure. Specifically, increasing anthropogenic impact within the watershed lowered diversity. Likewise, community composition was significantly correlated with agriculture and urban development within a watershed. Interaction networks showed decreasing complexity and fewer keystone taxa in impacted lakes. Moreover, we identified potential indicator taxa of high or low lake water quality. Together, these findings point to detectable bacterial community changes of largely unknown consequences induced by human activity within lake watersheds.
Similar content being viewed by others

Introduction
Lake ecosystems have garnered a large amount of attention from researchers and policy makers in recent years as “sentinels” of climate change and human impacts [1] owing to their central importance in biogeochemical cycles [2]. This role arises from their pivotal position within watersheds from which they receive nutrients and environmental contaminants. Consequently, many anthropogenic activities in the watershed threaten the ecosystem and freshwater services provided by lakes. For example, intense agricultural activity, and specifically the use of fertilizers within watersheds, has been connected to lake eutrophication [3, 4], in conjunction with the bloom of certain species of cyanobacteria [5], decreases in oxygen conditions [6], and increased methane emissions [7], while watershed urban development has been connected to overall water degradation caused by road salt runoff [8, 9] and phosphorus export [10]. However, many important human impacts with significant consequences for ecosystem function are likely undetected and therefore cannot be addressed by policy or monitoring programs.
Bacterial communities mediate essential biogeochemical processes within lakes. Recent advances in microbial molecular ecology have demonstrated high taxonomical and functional diversity of lake bacteria and links between shifts in community composition and activity and ecosystem-level responses [11, 12]. Given the high bacterial diversity and ability to respond rapidly to changing environments, these communities may be powerful indicators of environmental stressors (e.g., excess nutrient loading, acidification, salinification, metal contamination). However, little is known about the variability of freshwater bacterial communities across large spatial gradients of land use (but see [13]).
Canada is responsible for the stewardship of more than 1,000,000 lakes, which make up 20% of the world’s freshwater stocks and provide the main source of drinking water for many major Canadian cities [14, 15]. While there is no one standard definition of lake “health” [16], we have previously described it as the departure of the ecological state of a lake from the pristine state; the further removed from the pristine state the less healthy a lake is [15]. While large-scale assessments of lake health have been conducted elsewhere [17, 18], no such survey exists for Canadian freshwaters. In this context, the LakePulse network has embarked upon a continental-scale assessment of lake health across Canada in relation to anthropogenic activity as determined from satellite imaging and lake environmental variables collected in situ. In this study, we connect the bacterial community structure to watershed agriculture, forestry, pasture, and urban development in more than 200 lakes covering over 660,000 km2 in Eastern Canada (Fig. 1a).

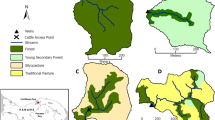
a Map of Eastern Canada showing 210 sampled lakes colored by their human impact index (HII) and underlying regional land use. b–e Detailed maps for the four ecozones studied here. Lakes are numbered by increasing HII values within each ecozone. Stacked bar charts surrounding each ecozone map show the relative abundance of phyla for each lake (sorted by HII value). The five most common phyla across all lakes are colored according to the legend, all other phyla are shown in dark gray.
Lake bacterial communities are shaped by a multitude of current and past selective pressures as well as processes of drift, and the lakes sampled in the context of this study are located in vastly different ecozones with diverse geographies and geological histories [19]. Moreover, land use is not independent of geography (e.g., agriculture is more intense in certain regions of Eastern Canada). Bearing this in mind, we designed the study in order to sample lakes with varying degrees of human impact in their watershed to investigate potential human-mediated community changes beyond the scope of spatial variation due to geography and geology [15].
We aim to untangle the underlying proximate environmental variables that cause the community shifts observed, as well as describe how anthropogenic impacts change bacterial interaction networks. Lastly, we use the scope of our dataset to investigate the relative strengths of different processes of community assembly such as drift and selection. We found that bacterial community diversity, richness, and composition correlated with watershed anthropogenic activity, specifically agriculture and urban development, which themselves were related to variation in lake salinity. Moreover, highly impacted watersheds harbored more fragmented bacterial communities with fewer keystone taxa, which could affect ecosystem function and resilience.
Methods
Lake selection
Lakes were selected across Eastern Canada as described in detail in [15]. Briefly, lakes were picked in a random sampling design with ecozone, lake size and human impact index (HII) as stratifying factors. Canadian ecozones (represented by AH: Atlantic Highlands, AM: Atlantic Maritime, BS: Boreal Shield, and MP: Mixedwood Plains, all in in south-eastern Canada) represent regions with distinct geological, climatic and ecological features [19]. For each lake within 1 km of a road the watershed was delineated as described in [15] and briefly in the Supplementary methods section. Independence of the watersheds of all lakes was confirmed. Within the watershed, each pixel was assigned a human impact value between 0 and 1 depending on their category (urban development/road: 1; mines/oil: 1, agriculture: 1, pasture: 0.5, forestry (recent clear-cuts [20]): 0.5, natural landscapes: 0) and the average HII for the lake calculated across the watershed. Across the ecozones of Eastern Canada, the mines/oil category was found to be negligible in magnitude (only five lakes total with >1% mining within their watershed and only one lake with >5% mining) and was thus not explicitly considered in the analyses of specific land use classes herein. Overall, nearly 80,000 watersheds were delineated, and lakes were chosen for sampling such that they represented different ecozones, sizes, and HIIs [15].
Surface water sampling
The surface water of 220 lakes (Fig. 1a) was sampled during the time of maximum summer lake stratification between July and September 2017 at the deepest point of each lake (as measured on site using a sonar). The depth of the euphotic zone was determined as twice the Secchi disk depth and an integrated epilimnion sampler (detail in the Supplementary methods section) was used to sample to that depth, or to a depth of 2 m, whichever was shallower. Surface water samples mostly comprised the lake epilimnion (change of temperature along the depth of the sampling tube <1 °C), except in ~2% of cases in which we also sampled the upper edge of the metalimnion in stratified or partially stratified lakes (Supplementary Table 1). Water samples were immediately transferred and stored in the cold and dark until subsequent processing with a maximum delay of 3 h. Water samples were filtered on site through a Durapore 0.22 µm membrane filter (Sigma-Aldrich, St. Louis, USA) using a Gast Pressure pump (Fisher Scientific, Quebec, Canada) and the filters immediately frozen at −80 °C until analysis. We aimed to filter 500 ml of water for each sample but stopped filtration at a lower volume if the filter clogged. If the filter showed no color at 500 ml, we filtered larger volumes until a coloration of the filter was observed. For each lake we used the same water sample to determine dissolved organic and inorganic carbon (DOC and DIC (mg/l), Supplementary material), total nitrogen (TN (mg/l) [21]), concentrations of the ions magnesium, potassium, sodium [22], chloride, and sulfate [23] (all in mg/l). The average concentration of dissolved oxygen (DO, mg/ml) over the same sampling depth was measured by averaging the values from a multiparameter probe profile (RBR Maestro3 profile, RBR Ltd., Ottawa, Canada; with a fast oxygen probe Rinko III, JFE Advantech Co., Nishinomiya, Japan) over that depth.
DNA extraction and sequencing
DNA was extracted from filters with PowerWater kits (Mobio Technologies Inc., Vancouver, Canada) and the V4 region of the 16S rRNA gene PCR-amplified using the standard primers U515_F and E786_R before sequencing on an Illumina MiSeq machine. DNA extraction and sequencing succeeded for 212 of the 220 samples. Details of PCR and sequencing reactions can be found in the Supplementary material. Sequences are deposited under accession number PRJEB38100 in the European Nucleotide Archive (www.ebi.ac.uk).
Sequence data processing
Read files were demultiplexed using idemp [24] and Illumina adapters removed with cutadapt [25]. Reads were processed using the DADA2 package in R [26]: Reads were trimmed, merged, chimeras removed, and taxonomy of amplicon sequence variants (ASVs) assigned to the genus level using the freshwater-specific Fresh Train taxonomic database [27] or, if no classification with the freshwater-specific database was possible, the SILVA database (version v128align) [28] to obtain an ASV table. Further analysis of the ASV table was done using the phyloseq package in R [29]. ASVs from chloroplasts were removed before rarifying the data to a common read depth of 15,000 reads after removing two lakes with <15,000 reads. A phylogeny of the remaining 11,510 ASVs was created using FastTree [30]. Bacterial diversity (Shannon–Weaver and Inverse Simpson indices) or richness (Chao1 index) of each lake was calculated as implemented within the phyloseq package.
Processing of environmental and spatial data
To describe the complex dataset in a concise way and include both spatial and environmental effects we used a dimension reduction approach that included both spatial eigenvectors and environmental variables. To describe spatial effects, we calculated Moran’s eigenvector maps (MEMs) based on distances between lakes (in km) (adespatial R package; [31]). MEMs allow the modeling of complex geographic patterns of higher order (e.g., non-linear) than simple distance–decay relationships. For example, MEMs can model “plateaus” where lakes are quite similar even though they are far apart in space (as could occur within a homogenous ecozone for example), as well as describe spatial decay over varying scales. We visually inspected the MEM eigenvalue distribution and chose the first six MEMs to describe spatial structure within our system as they encompass most of the spatial variation. Maps of the first six MEMs are shown in Supplementary Fig. 1. We extracted the six MEMs’ coordinates and combined them with data on the lake area, lake depth, DOC, DIC, TN, DO, magnesium, potassium, sodium, chloride, and sulfate. Lakes with missing data for any of these variables were removed, leaving 167 lakes.
To reduce dimensionality and ensure independence between environmental and spatial variables a PCA (after scaling of the environmental variables) was performed for the remaining lakes (rda function, vegan R package [32]). The first seven principal component axes (PCs) were chosen for further analysis as they represented more variation than the mean. Factor loadings for these PCs are shown in Supplementary Table 2. We considered factor loadings ≥|0.43| to be significant [33]. Ecozone, HII, and size class membership, as well as environmental data for each lake can be found in Supplementary Table 1.
Linear modeling of diversity and community composition analyses
We first tested the overall impact of geographic variation (in the form of either ecozone or longitude and latitude) on bacterial diversity and richness using the anova and lm functions as implemented in R [34]. To examine whether there was an impact of human alteration of watersheds on bacterial diversity and richness, we modeled the impact of the HII variable in conjunction with ecozones, as well as percent land use types (see Supplementary Table 1) in the watershed on different bacterial diversity and richness measures using linear models, generalized least squares models (GLS models; nlme R package [35]) and linear mixed models (LMM; lme4 package [36]). LMM additionally contained ecozone as a random effect, but in most cases either fit worse than the corresponding GLS model or failed to converge.
To further explore the connection between HII, environmental and spatial factors, and bacterial diversity and richness, we performed structural equation modeling (SEM) (lavaan R package [37]). We considered the impact of HII onto the first seven environmental and spatial PCs, as well as the impact of the PCs and HII directly onto ranked diversity. In addition, we modeled the impact of land use classes on the environmental PCs directly (Supplementary material). Models were only considered to fit better than the null model if the chi-square p value was >0.05 and the comparative fit index >0.95.
We investigated geographic variation in beta diversity using PERMANOVA tests (adonis function, vegan R package [32]) with either ecozone or longitude and latitude as explanatory variables. To investigate the impact of the environmental and spatial factors (coded as PCs) on community composition, we utilized a PERMANOVA test [32]. In addition, we performed forward selection distance-based redundancy analysis (db-RDA) to determine how MEMs and watershed land use impact community composition (capscale function; vegan R package [32]). In db-RDA, taxa are first transformed into synthetic, uncorrelated variables and then explanatory variables are added one after another to the model to assess their ability to explain variation within the response matrix. In addition, we performed a corresponding NMDS analysis (function metaMDS [32]) and performed a post hoc fitting of land use and spatial variables.
Networks of lake bacterial communities
To examine whether human alteration within watersheds qualitatively influences the structure of bacterial interactions, we performed network analysis. Lakes were assigned to a low (HII 0–0.1, 91 lakes), medium (HII > 0.1 and ≤0.5, 96 lakes), or highly impacted (HII > 0.5, 23 lakes) class. Within each network dataset, we excluded ASVs that were not present in at least 10% of the samples, resulting in 826 ASVs in low-HII lakes, 775 ASVs in medium-HII, and 764 ASVs in high-HII lakes. We followed a combinatorial approach to determine significant connections between ASVs using both the maximum information criterion (MIC) [38] and sparse inverse covariance estimation (spiec.easi) [39] as detailed in the Supplementary material. To determine significant co-occurrence patterns using MIC, we bootstrapped matrices 999 times and recalculated MIC values. We obtained quasi p values by counting the number of permutated MIC values larger than the observed “real” MIC value and dividing this number by 999. Quasi p values were corrected for multiple testing using the false discovery rate (fdr) method as implemented in R and only edges with a corrected p value below 0.05 retained.
We determined order influence within high, moderate, and low-impact networks by dividing the total number of edges of each order by the number of nodes belonging to the order (excluding orders only represented by a single ASV or absent in any of the networks) [40]. Moreover, we dropped all ASVs that could not be identified to the order level.
Indicator species of lake health
High or low-HII environments may be characterized by the presence of specific taxa which can thus serve as indicators of environmental quality. We investigated potential indicator species of high and low-impact lakes based on abundance changes of taxa in pairs of lakes that were geographically similar but varied with respect to their HII. The log-ratio of ASV abundance change between paired lake communities was compared to control lake pairs (which were pairs of geographically similar lakes that were either both low or both highly impacted). To control for geographic variation as much as possible, all lakes compared were chosen from within the same ecozone. In addition, we screened all ASVs for rank changes of at least 33% between paired impacted and pristine lakes as described in detail in the Supplementary material.
Community assembly processes of lake bacterial communities
We defined generalist and specialist taxa within our dataset as follows: ASVs present in 158 lakes or more (at least 75% of the dataset) were considered generalists, while ASVs present in ten lakes or less (5%) and with a total relative abundance higher than 2% across all lakes were considered specialists [41]. We investigated distance–decay curves between community dissimilarity (determined as the Bray–Curtis dissimilarity as calculated using the vegdist function in vegan [32], and the distance between the lakes (in km)) using Mantel tests with 9999 permutations (p values corrected using the “holm” method as implemented in R [34]).
We followed the null model method as proposed by Stegen et al. [42] to determine quantitative estimates for the strength of selection, dispersal limitation, and ecological drift in the lake communities on a subsetted dataset in which rare taxa (abundance <500 across all lakes) were removed (679 ASVs remaining) as detailed in the Supplementary methods.
Results
Bacterial diversity patterns across geography and land use gradients
An overview of the relative abundances of the five most common phyla across all lakes sorted by increasing HII values, as well as the spatial location of lakes are shown in Fig. 1. We first investigated the impact of geographic variation on bacterial diversity and richness. Geographic variation coded as longitude and latitude did not significantly impact Shannon–Weaver diversity or the inverted Simpson index (all p > 0.05, slopes and effect sizes in Supplementary Table 3), but did impact Chao1 richness (latitude: slope: 19.702, p = 0.0047; longitude: slope: −3.459, p = 0.0399, R2-estimate 0.612, Cohan’s f = 0.0652). Correspondingly, ecozone did not significantly impact Shannon–Weaver or inverted Simpson measures, though ecozone was nearly significant in the case of the inverted Simpson index (Supplementary Fig. 2A, B, Shannon–Weaver index: p = 0.23, R2-estimate 0.0205, Cohan’s f = 0.021; inverted Simpson index: p = 0.0525, R2-estimate 0.0366, Cohan’s f = 0.038). In contrast, ecozone significantly impacted Chao1 richness measures (p = 0.0009, R2-estimate 0.0766, Cohan’s f = 0.093, Supplementary Fig. 2C).
Next, we investigated the relationship between the HII, ecozone variation, and bacterial diversity and richness (all model details in Supplementary Table 3) of lake communities. Overall, we detected a significantly negative impact of HII, but not ecozone on Shannon–Weaver diversity (p values: 0.035, 0.226, R2-estimate 0.0416, Cohan’s f = 0.0434). In contrast, neither HII nor ecozone were significant for the inverted Simpson index (though ecozone was marginal: p values: 0.292, 0.0524, R2-estimate 0.0419, Cohan’s f = 0.0437), and only ecozone impacted the Chao1 index significantly (p values: 0.414, 0.0009, R2-estimate 0.0796, Cohan’s f = 0.0865; Supplementary Table 3).
Land use is not evenly distributed across ecozones (Fig. 1) and we therefore investigated whether differences in land use could be correlated with the patterns observed. Modeling Shannon–Weaver diversity as a function of different land use classes, we found the percentage of forestry (defined as forest cuts within the last 6 years) within a watershed to have a significantly positive impact (slope: 1.26, p = 0.0066), while the percentage of urban development had a nearly significant negative impact (slope: −0.67, p = 0.0565, total model: R2-estimate 0.137, Cohan’s f = 0.158). We obtained similar results using the other diversity or richness indices (Supplementary Table 3).
To further investigate the relationship between HII and diversity and to try to untangle geographic from anthropogenic effects, we performed SEM to investigate the relationship between HII and the first seven PCs (including the spatial MEMs as well as environmental variables) and in turn the impact of the PCs onto ranked Shannon–Weaver diversity (Fig. 2) and other diversity or richness measures (Supplementary Table 4). The SEM (χ2 = 5.684, DF = 21, p = 1.00, CFI = 1.00) showed a significant positive impact of PC2 (p < 0.001) and PC6 (p = 0.006) on Shannon–Weaver diversity. PC2 is negatively loaded with lake depth, while PC6 carries the spatial eigenvector MEM2. PC6 was in turn significantly positively impacted by the HII variable (p = 0.031). In addition, we detected a nearly significant negative impact of PC1 (p = 0.056) on diversity. While PC1 did not carry significant loadings, it is strongly negatively loaded with the ions Mg+2, K+, Ca+2, and Cl−. PC1 was strongly positively impacted by the HII variable (p < 0.001). Lastly, the HII variable also directly impacted ranked diversity (p = 0.008). All parameter estimates, exact p values, and effect sizes of this and other models can be found in Supplementary Table 4.

All paths were tested, but only those showing significant (p < 0.05, solid lines) or nearly significant (p < 0.1, dashed lines) correlations are shown. Positive interactions and loadings are shown in black, negative interactions in gray. Entries into PC boxes indicate significant factor loadings, except for PC1, where no environmental factor had a significant loading. Positive factor loadings are shown in black, negative ones in grey. Instead we report the highest loading non-significant factors for this PC. Numbers associated with arrows show the correlations between each explanatory and response variable.
Thus, HII was associated with increased lake ion concentrations, which in turn led to a decrease in bacterial diversity (Fig. 2). Moreover, diversity was impacted or correlated with lake depth. The link between HII, PC6 (loaded with a spatial eigenvector), and diversity may indicate correlation with an unmeasured environmental variable. Lastly, HII also negatively impacted diversity independent of the environmental PCs considered here, suggesting unknown environmental or spatial variables.
To understand the cause of the impact of the HII variable, we examined the underlying land use classes and investigated their relationship with ranked diversity and the seven environmental PCs in an additional SEM (Supplementary methods, Supplementary Fig. 3). We found PC1 to be significantly positively associated with agriculture, urban development, and pasture within the watershed. Similar results were obtained when using the ranked Inverse Simpson index or Chao1 richness as dependent variables in SEMs (Supplementary Table 5).
Community composition across geography and land use gradients
We investigated the impact of geographic variation on community structure using PERMANOVA tests. Both longitude and latitude significantly impacted community composition but explained only a small amount of the variation observed (latitude: p = 0.001, R2 = 0.02; longitude: p = 0.001, R2 = 0.04). Similarly, a model with ecozone as an explanatory variable significantly correlated with community composition, but only explained ~8.5% of the variation observed (p = 0.001, R2 = 0.085).
To decompose geographical from anthropogenic effects, we conducted PERMANOVA with the first seven environmental and spatial PCs as explanatory variables. All PCs except PC7 were found to significantly impact community composition (all p < 0.05). PC1, carrying ion concentration loadings, showed the highest R2 value in the model (0.062), but all factors included in the model only explained a relatively small amount of the total variation (~18%, Cohan’s f = 0.217).
We utilized db-RDA to select watershed-scale variables impacting community composition. A full model containing the spatial eigenvectors, lake morphometric parameters (lake depth and area), and land use data was found to be significant (p = 0.001), so we proceeded with forward selection. Overall, a model containing the land use classes: natural landscapes, forestry, agriculture, pasture, and urban development in the watershed, as well as the first four MEMs, and lake depth was selected as the best model (Fig. 3). However, the model only explained ~15% of the variation observed (Cohan’s f = 0.175). An NMDS analysis showed qualitatively similar results (Supplementary Fig. 4).

Arrows show the significant environmental variables after forward selection. Point shapes indicate the ecozone of origin for each lake and coloring along a gradient from light blue to dark red indicates increasing human impact index values for each lake.
Land use impact on community interactions
To determine how anthropogenic impact may alter the structure of bacterial interactions, we constructed networks of high, moderate, and low-HII lake communities. These co-occurrence networks describe the tendency of specific taxa to either co-occur or exclude each other in lakes of a specific HI class compared to randomized interactions. Low-HII lake communities showed significant co-occurrence patterns for 179 nodes (taxa) with a total of 193 edges (co-occurrence connections). Moderate impact communities had significant co-occurrence networks for 145 nodes and 163 edges, while high impact communities had 220 nodes and 174 edges. Networks were more fragmented under high human impact (low: 24 components, moderate: 13 components, high: 59 components) and the clustering coefficient as well as the centralization value decreased with increasing HII (Fig. 4). Both low and moderate impact lake communities were characterized by ASVs having similar average numbers of neighbors (low: 2.16, medium: 2.25) and keystone taxa (with over five connections each: low: 13, moderate: 11), whereas ASVs in highly impacted lake communities had on average fewer neighbors (1.58). Lastly, highly impacted lakes only had two keystone taxa (taxa with >5 edges, Fig. 4). Keystone species mostly belonged to common, cosmopolitan freshwater taxa and comprised nine ASVs belonging to the order Acidimicrobiales, six ASVs belonging to the Burkholderiales and five ASVs belonging to the Frankiales (Supplementary Table 6).

Nodes are colored by their degree (from yellow representing only a single connection over green to dark blue indicating a large number of connections). Smaller visualizations under the main network show network fragments in the dataset.
We determined whether specific bacterial orders varied in their influence in high, moderate, or low-HII lakes by dividing their number of total edges by the number of nodes of a given order. We identified 17 orders, which were represented in all three sets of networks (Supplementary Table 7). Four of these orders changed consistently between low, moderate, and high lake networks. Burkholderiales ASVs, Frankiales ASVs, and Rhizobiales ASVs had decreasing influence from low to moderate to high impacted networks, while verrucomicrobial OPB35 ASVs’ influence increased. Furthermore, Acidimicrobiales and Planctomycetales ASVs were of relatively low influence in high impact networks, but of higher influence in moderate and low impacted networks.
We investigated potential indicator species, i.e., species that can be used as ecological indicators for specific environmental conditions due to their niche preferences, for high or low-impact lakes using log-ratio and rank abundance changes of ASVs. Overall, no strong indicator patterns emerged, demonstrating the high variability of freshwater communities under both pristine and impacted conditions. Log-ratio changes in treatment (pristine–impacted), but not control (pristine–pristine or impacted–impacted), lake pairs were detected for six ASVs in AH, two in AM, nine in BS, and ten in MP. In total, 26 ASVs showed significant log-ratio changes, but these ASVs rarely overlapped between ecozones. Taking into account ASV taxonomy, we found ASVs belonging to the Burkholderiales betI-A clade and of the cyanobacterial Synechococcales to be associated with low-impact lakes across three and two ecozones, respectively (Supplementary Table 8).
Ninety-five ASVs were found to show significant rank abundance changes between high and low-impact lakes, but not between control lake pairs (17 ASVs in AH, 19 in AM, 46 in BS, 25 in MP, Supplementary Table 9). Specific taxa were associated with changing lake conditions across ecozones. For example, the Burkholderiales betI-A clade (represented by six ASVs) was associated with low-impact lakes in all four ecozones, except for one ASV, which was a high impact indicator in AH.
Evolutionary and ecological processes shaping lake bacterial communities
We did not detect any low-occupancy (present in <5% of lakes), high-abundance (total abundance across all lakes more than 2%) specialists within our dataset. In contrast, we identified 25 generalists, which were present in 75% or more of lakes. The most common generalist lineage was the actinobacterial acI lineage (9 ASVs), followed by the betaproteobacterial betI lineage (5 ASVs). Other lineages represented included actinobacterial acIV (2 ASVs), betaproteobacterial betIV (2 ASVs), and Bacteroides bacI (2 ASVs) (Supplementary Table 10).
Overall, we found bacterial communities to be less similar the further apart in space they were sampled (Mantel test, p < 0.001, Supplementary Fig. 5A). Specifically, across all communities investigated, lakes sampled up to ~350 km apart were significantly more similar than expected by chance (Mantel correlogram analysis of spatial correlation values as a function of distance, p = 0.004, Supplementary Fig. 5B). To determine the causes for this structure, we performed a quantitative analysis of the community assembly processes shaping lake communities to estimate the relative strength of selection and drift processes in our dataset. Following [42], we utilized two measures to quantify the strength of selection and drift within our system. β-mean-nearest taxon distance (βMNTD) quantifies the phylogenetic distance between each ASV in a community and its closest neighbor in a different community. High values of βMNTD may indicate heterogeneous (or diversifying) selection while low values may indicate homogeneous (or purifying) selection. βMNTD is expressed as the deviation from a null model (based on randomly shuffling species identities and abundances) as the β-nearest taxon index (βNTI). βNTI values lower than −2 indicate lower than expected phylogenetic turnover (e.g., due to homogeneous selection), while βNTI values above two indicate higher than expected phylogenetic turnover (e.g., heterogeneous selection) [42]. Secondly, we assessed the magnitude of drift in our system using an extended Raup–Crick (RC) index, which takes into account relative abundances of ASVs. A null model of RC values was constructed by calculating the Bray–Curtis dissimilarity of 999 probabilistically assembled communities based on ASV prevalence and relative abundance. The deviation between the empirical and permutated RC values was standardized to vary between −1 and 1 and is referred to as RCBray. RCBray between −0.95 and 0.95 indicate that the observed turnover is consistent with the action of drift, while values higher than 0.95 or lower than −0.95 indicate dispersal limitation, or, respectively, homogenizing dispersal [42].
Firstly, we determined whether we could detect a phylogenetic signal within our data (i.e., whether closely related species had similar niches) by testing for a correlation of phylogenetic distance, based on the cophenetic distance from a phylogenetic tree of the 16S rRNA amplicons, with ecological distance (differences in abundance patterns expressed as Bray–Curtis dissimilarity). Overall, we found a significant decay of phylogenetic distance with ecological distance (Mantel test: p < 0.05, Supplementary Fig. 5C). Specifically, and as expected, we detected a phylogenetic signal over relatively short phylogenetic distances (cophenetic distance > 0.365), indicating that the βNTI, which indicates the impact of selection on community composition, is an appropriate metric in our dataset to measure phylogenetic turnover [42].
Within the 21,945 community comparisons possible with our data, we found selection to be responsible for 12.3% of the observed community turnover (βNTI > |2|). Most selection increased community similarity (homogeneous selection βNTI < −2: 9.6%), while only 2.7% of community turnover was consistent with heterogeneous selection (βNTI > 2). Of the remaining 87.7% variation, 16% was governed by dispersal limitation (βNTI <| 2| and RCBray > 0.95), whereas 39.1% was attributable to homogenizing dispersal (βNTI < |2| and RCBray < 0.95). This leaves 32.6% of community turnover attributable to ecological drift (e.g., stochastic processes) alone (Supplementary Fig. 5D). Environmental PCs associated with either selection or drift processes are described in the Supplementary results section.
Discussion
In our dataset, encompassing over 200 lakes located across Eastern Canada, we sampled across large spatial scales and lakes are thus characterized by diverging geological histories and large-scale geographic variation, which are roughly represented by the ecozone concept [15]. This large-scale spatial variation had a relatively small, but significant impact on lake bacterial communities both with respect to alpha diversity and richness measures, as well as community composition. The present study was designed in order to determine the impact of land use (and specifically land use associated with human activities within the watershed) on lake bacterial communities across Eastern Canada. However, land use is not independent of geography, requiring us to separate, as much as possible, general geographic effects from those that may be caused by anthropogenic impact. Overall, we were able to detect correlations between watershed anthropogenic activity and surface water bacterial community composition. Despite a large fraction of unexplained variation in the dataset, we found that models containing land use explained more variation than those only containing factors encoding geographic variation. Thus, human-mediated land use appeared to be related to lake bacterial communities above and beyond the effects of geographic variation alone, but the large amount of unexplained variation in our dataset warrants further investigation.
Human impact on bacterial diversity and community composition
The HII variable was found to be associated with surface water communities with significantly lower diversity. Specifically, high-HII values were associated with high chloride, calcium, magnesium, and potassium values, which in turn were associated with reduced diversity. Salinity has been shown to be a major factor structuring bacterial diversity in a range of environments [43,44,45], but the effect of salinity on diversity in lake systems is less clear [46, 47]. Importantly, these findings point to a significant impact of salt contamination on the bacterial community even at relatively low levels of salinity (and well below the chronic pollution thresholds for chloride ions at 230 mg/l) and thus suggests that bacterial ecosystem services in lakes may be vulnerable to salt contamination.
Even though our sampling design does not allow us to separate natural variation in salinity from salt contamination, PC1 was found to be impacted by agriculture, pasture, and urban development within the watershed, indicating road salt (including rock salt (NaCl) and other de-icers such as potassium chloride, calcium chloride, and magnesium chloride) as one likely source for the environmental variables loaded onto the PC. Chloride concentrations in streams and lakes have been previously shown to be driven by the application of road salt [48, 49] and even relatively low road cover within a watershed (~1%) has been linked to increased chloride concentrations within lakes [9].
Another possible source for some of the ions loading onto PC1 are fertilizers, including potassium from potash and calcium as part of phosphate and nitrate-based fertilizers. Most studies focus on the impact of nitrogen and phosphate in fertilizers on bacterial blooms and phytoplankton communities [50,51,52], but the impact of fertilizer cations is less clear. However, calcium has been shown to influence the bioavailability of phosphorous in lakes [53].
Community composition was also structured by landscape-scale variables. Lake communities were impacted by nearly every factor explored here, ranging from lake morphometry to most land use classes and spatial components. However, although most factors that we included in our models were significant, our models were only able to explain a small proportion of lake community composition overall (~9–18%). Thus, despite the inclusion of over 30 partially nested variables in the models, we acknowledge that we still fall short of describing the majority of the prokaryotic community diversity observed in this system.
Human impact on bacterial interaction networks
Overall, anthropogenic activity is associated with shifts in bacterial community composition. To relate these shifts to community stability and functionality, we investigated how communities in lakes with high, moderate, or low human impacted watersheds were altered in their interactions and co-occurrences. As we were interested in overall community structure rather than specific group interactions (e.g., [54]), we treated all interactions (positive and negative) as undirected, to facilitate analysis. As all lake communities belonging to a specific impact class were used to calculated occurrence patterns within the low, moderate, or high HII network, we are not able to replicate the networks themselves within each broad impact category. Thus, all comparisons between networks are qualitative, while co-occurrence links within networks are those deemed significant across all lakes within the category. Significant co-occurrence within a network may indicate that taxa are directly interacting but may also show taxa as co-occurring due to overall niche similarity (or competitive exclusion in the case of negative co-occurrence). While lake networks constructed from communities from low and moderately impacted watersheds were similar, we detected altered structure and topology in networks from highly impacted lakes. Even though communities from highly impacted lakes had the highest number of taxa involved in significant co-occurrences (220 taxa), the number of significant connections did not scale up proportionally, resulting in, on average, less neighbors per node in these communities than in moderate or low impacted communities. In turn, this resulted in less highly connected keystone taxa and thus a more fragmented network. This higher number of fragments correlates with a lower clustering coefficient (which indicates the average number of actual three-node connections that each node has compared to the possible number of three-node connections). Likewise, highly impacted networks showed lower network centralization. Higher levels of centralization (i.e., average number of nodes connected to each node or how “star-shaped” a network structure is) have been linked to increased system stability in root microbiomes [40] and lake systems [55].
Interestingly, moderate HII communities seemed to be only marginally impacted with respect to their co-occurrence network in comparison with low-impact communities. This finding may indicate a buffering effect in which microbiomes are able to maintain most ecosystem functions despite shifts in the underlying taxa performing them, for example via functional redundancy. The altered pattern in high impacted community networks may indicate that once a threshold is reached, the system’s buffering capacities are exhausted, and that specifically the loss of highly connected and influential keystone taxa may lead to cascading effects of community fragmentation [55]. Alternatively, the patterns could indicate that while low and moderately impacted lakes are quite similar to each other, highly impacted lakes may be overall more variable or that, depending on the exact nature of the anthropogenic impact, multiple high impact communities may exist.
Ecological and evolutionary processes structuring lake communities
In addition to investigating the effect of watershed anthropogenic activity, the large spatial scale of the study allowed us to assess the ecological and evolutionary processes structuring lake bacterial communities in Eastern Canada in general, as has been previously done in a range of bacterial communities including aquifers [42] and the ocean [56]. Firstly, we investigated whether we could identify generalist and specialist taxa within our dataset [41]. We were able to identify 25 generalist taxa with an occupancy of over 75% in our dataset, including well-known cosmopolitan freshwater lineages such as the actinobacterial acI clade ASVs, betaproteobacterial betI ASVs, and an alphaproteobacterial LD12 ASV. In contrast, we were unable to identify any specialist (low occupancy but relatively high abundance) ASVs, indicating that taxa have either low abundance or wider distributions.
We also utilized the dataset to investigate patterns in community assembly. In accordance with the absence of specialists, we found heterogenizing selection to be only responsible for a very small amount of community variation. Rather, lake ecosystems seemed to be extensively connected by homogenizing dispersal and successful lineages were thus able to establish themselves in other communities (homogenizing selection). Consequently, dispersal limitation was not found to be an important factor structuring lake bacterial communities and instead, ecological drift processes were prevalent. We are not aware of any comparable study of community assembly in lakes, but it was noticeable that, in our system, selection was a much less powerful force shaping communities than in subsurface or marine ecosystems [42, 56].
Caveats and conclusion
Data obtained within the first sampling season of the LakePulse project allows an unprecedented insight into how bacterial communities vary across landscapes in relation to geography and anthropogenic impact. However, the large-scale nature of the project and the sampling effort involved also lead to several caveats. Due to technical difficulties we were unable to measure pH and total phosphorus directly for a large quantity of the lakes and were thus not able to include these variables in our models. pH has been previously shown to be a key driver of bacterial community structure in a range of environments [57,58,59]. pH is a complex variable and correlated to some degree with the concentrations of ions such as chloride and ammonium within the water, lake geography and morphometry, phytoplankton biomass [60, 61], and time of day [62]. Likewise, phosphate concentrations have been shown to impact lake bacterial community compositions [63, 64] and sediments [65] but were not taken into account in this model. As phosphate, in combination with nitrate and ammonia, is often used in agricultural settings, we expect some positive correlation between phosphate and nitrogen-species but are unable to directly investigate its role in bacterial community diversity. Nutrient availability is strongly linked with chlorophyll-a concentrations, which have been used to predict lake nutrient status and link eutrophication to land use [66,67,68]. We suspect that at least some of the unexplained variation in our system is connected to these missing environmental variables. Likewise, we limited our analysis to only include abiotic factors as explanatory variables of bacterial community structure. However, biotic interactions are not only likely to be altered by environmental factors, as shown here via network analysis, but can also exert strong biological control mechanisms, for example via anticompetitor compounds [69], predator-prey, or mutualistic relationships across trophic levels. Future work within the project is aimed at incorporating bacterial diversity into a food web framework utilizing phytoplankton and zooplankton data also collected from the lakes, as well as deepening our functional understanding of lake communities using metagenomics. Moreover, current sampling efforts are underway that will allow us to include more lakes to extend our frameworks to all of Canada, including regions of extremely high human impact and associated environmental decay due to agriculture and mining.
In conclusion, we used an extensive dataset of lake bacterial communities to model the impact of anthropogenic activity across vast areas of Eastern Canada. Human impact, and specifically variables related to urban and agricultural development within watersheds, appeared to have an effect on lake communities and showed that when high-intensity human activities alter more than about 50% of a watershed, fragmentation of bacterial communities may be observed which may ultimately be tied to the decline of the ecosystem services provided by them.
References
Adrian R, O’Reilly CM, Zagarese H, Baines SB, Hessen DO, Keller W, et al. Lakes as sentinels of climate change. Limnol Oceanogr. 2009;54:2283–97.
Tranvik LJ, Downing JA, Cotner JB, Loiselle SA, Striegl RG, Ballatore TJ, et al. Lakes and reservoirs as regulators of carbon cycling and climate. Limnol Oceanogr. 2009;54:2298–314.
Arbuckle KE, Downing JA. The influence of watershed land use on lake N: P in a predominantly agricultural landscape. Limnol Oceanogr. 2001;46:970–5.
Taranu ZE, Gregory-Eaves I. Quantifying relationships among phosphorus, agriculture, and lake depth at an inter-regional scale. Ecosystems. 2008;11:715–25.
Heisler J, Glibert PM, Burkholder JM, Anderson DM, Cochlan W, Dennison WC, et al. Eutrophication and harmful algal blooms: a scientific consensus. Harmful Algae. 2008;8:3–13.
Scavia D, David Allan J, Arend KK, Bartell S, Beletsky D, Bosch NS, et al. Assessing and addressing the re-eutrophication of Lake Erie: Central basin hypoxia. J Gt Lakes Res. 2014;40:226–46.
Bastviken D, Cole J, Pace M, Tranvik L. Methane emissions from lakes: dependence of lake characteristics, two regional assessments, and a global estimate. Glob Biogeochem Cycles. 2004;18:1–12.
Novotny EV, Murphy D, Stefan HG. Increase of urban lake salinity by road deicing salt. Sci Total Environ. 2008;406:131–44.
Dugan HA, Bartlett SL, Burke SM, Doubek JP, Krivak-Tetley FE, Skaff NK, et al. Salting our freshwater lakes. Proc Natl Acad Sci USA. 2017;114:4453–8.
Hobbie SE, Finlay JC, Janke BD, Nidzgorski DA, Millet DB, Baker LA. Contrasting nitrogen and phosphorus budgets in urban watersheds and implications for managing urban water pollution. Proc Natl Acad Sci. 2017;114:4177–82.
Shade A, Kent AD, Jones SE, Newton RJ, Triplett EW, McMahon KD. Interannual dynamics and phenology of bacterial communities in a eutrophic lake. Limnol Oceanogr. 2007;52:487–94.
Kara EL, Hanson PC, Hu YH, Winslow L, McMahon KD. A decade of seasonal dynamics and co-occurrences within freshwater bacterioplankton communities from eutrophic Lake Mendota, WI, USA. ISME J. 2013;7:680–4.
Marmen S, Blank L, Al-Ashhab A, Malik A, Ganzert L, Lalzar M, et al. The role of land use types and water chemical properties in structuring the microbiome of a connected lake system. Front Microbiol. 2020;11:1–16.
Environment Canada Whole organism responses and intersex severity in rainbow darter (Etheostoma caeruleum) following exposures to municipal wastewater in the Grand River basin, ON, Canada. Part A, Municipal Water Use Rep. 2011;159:2011–301.
Huot Y, Brown CA, Potvin G, Antoniades D, Baulch HM, Beisner BE, et al. The NSERC Canadian Lake Pulse Network: a national assessment of lake health providing science for water management in a changing climate. Sci Total Environ. 2019;695:133668.
Lu Y, Wang R, Zhang Y, Su H, Wang P, Jenkins A, et al. Ecosystem health towards sustainability. Ecosyst Heal Sustain. 2015;1:1–15.
Hering D, Borja A, Carvalho L, Feld CK. Assessment and recovery of European water bodies: Key messages from the WISER project. Hydrobiologia 2013;704:1–9.
U.S. Environmental Protection Agency. National Lake Assessment: a collaborative survey of the Nation’s Lakes. Washington, DC: EPA 841-R-09-001; 2009.
Ecological Stratification Working Group. A national ecological framework for Canada. Urbana-Champaign, Illinois: Ecological Stratification Working Group; 1996.
Glaz P, Gagné JP, Archambault P, Sirois P, Nozais C. Impact of forest harvesting on water quality and fluorescence characteristics of dissolved organic matter in eastern Canadian Boreal Shield lakes in summer. Biogeosciences. 2015;12:6999–7011.
Patton C, Kryskalla J. Methods of analysis by the U.S. Geological Survey National Water Quality Laboratory—evaluation of alakline digestion as an alternative to kjedahl digestion for determination of total and dissolved nitrogen and phosphorous. Denver, Colorado: Water-Resources Investigations Report 03; 2003.
U.S. Environmental Protection Agency. Method 200.7: determination of metals and trace elements in water and wastes by inductively coupled plasma-atomic emission spectrometry. Cincinatti, Ohio: U.S. Environmental Protection Agency; 1994.
U.S. Environmental Protection Agency. Method 300.1: determination of inorganic anions in drinking water by ion chromatography. Cincinatti, Ohio; 1997.
Wu Y. Barcode Demultiplex for Illumina I1, R1, R2 fastq.gz files. 2014.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Rohwer RR, Hamilton JJ, Newton RJ, McMahon KD. TaxAss: leveraging a custom freshwater database achieves fine-scale taxonomic resolution. mSphere. 2018;3:1–14.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013;41:590–6.
McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217.
Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490.
Dray S, Dufour A-B. The ade4 Package: implementing the duality diagram for ecologists. J Stat Softw. 2007;22:1–20.
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, Mcglinn D, et al. Vegan: community ecology package. 2016. https://cran.r-project.org; https://github.com/vegandevs/vegan.
Hair J, Tatham R, Anderson R, Black W. Multivariate data analysis. 5th ed. London: Prentice-Hall; 1998.
R Development Core Team T. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2009.
Pinheiro J, Bates D, DebRoy S, Sarkar D, R Development Core Team T. nlme: linear and nonlinear mixed effect models. R package version. 3.1-141; 2019.
Bates D, Mächler M, Bolker BM, Walker SC. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:1–51.
Rosseel Y. Lavaan: an R package for structural equation modeling. J Stat Softw. 2012;48:1–37.
Albanese D, Filosi M, Visintainer R, Riccadonna S, Jurman G, Furlanello C. Minerva and minepy: a C engine for the MINE suite and its R, Python and MATLAB wrappers. Bioinformatics. 2013;29:407–8.
Kurtz ZD, Müller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput Biol. 2015;11:e1004226.
Banerjee S, Walder F, Büchi L, Meyer M, Held AY, Gattinger A, et al. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 2019;13:1722–36.
Barberán A, Bates ST, Casamayor EO, Fierer N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012;6:343–51.
Stegen JC, Lin X, Fredrickson JK, Chen X, Kennedy DW, Murray CJ, et al. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013;7:2069–79.
Benlloch S, López-López A, Casamayor EO, Øvreås L, Goddard V, Daae FL, et al. Prokaryotic genetic diversity throughout the salinity gradient of a coastal solar saltern. Environ Microbiol. 2002;4:349–60.
Abed RMM, Kohls K, De Beer D. Effect of salinity changes on the bacterial diversity, photosynthesis and oxygen consumption of cyanobacterial mats from an intertidal flat of the Arabian Gulf. Environ Microbiol. 2007;9:1384–92.
Lozupone CA, Knight R. Global patterns in bacterial diversity. Proc Natl Acad Sci USA. 2007;104:11436–40.
Wu QL, Zwart G, Schauer M, Kamst-Van Agterveld MP, Hahn MW. Bacterioplankton community composition along a salinity gradient of sixteen high-mountain lakes located on the Tibetan Plateau, China. Appl Environ Microbiol. 2006;72:5478–85.
Wang J, Yang D, Zhang Y, Shen J, van der Gast C, Hahn MW, et al. Do patterns of bacterial diversity along salinity gradients differ from those observed for macroorganisms? PLoS ONE. 2011;6:e27597.
Kelly VR, Lovett GM, Weathers KC, Findlay SEG, Strayer DL, Burns DJ, et al. Long-term sodium chloride retention in a rural watershed: legacy effects of road salt on streamwater concentration. Environ Sci Technol. 2008;42:410–5.
Corsi SR, Graczyk DJ, Geis SW, Booth NL, Richards KD. A fresh look at road salt: aquatic toxicity and water-quality impacts on local, regional, and national scales. Environ Sci Technol. 2010;44:7376–82.
Levine SN, Schindler DW. Influence of nitrogen to phosphorus supply ratios and physicochemical conditions on cyanobacteria and phytoplankton species composition in the Experimental Lakes Area, Canada. Can J Fish Aquat Sci. 1999;56:451–66.
Stockner JG, Shortreed KS. Response of Anabaena and Synechococcus to manipulation of nitrogen: phosphorus ratios in a lake fertilization experiment. Limnol Oceanogr. 1988;33:1348–61.
Thad Scott J, McCarthys MJ. Nitrogen fixation may not balance the nitrogen pool in lakes over timescales relevant to eutrophication management. Limnol Oceanogr. 2010;55:1265–70.
Håkanson L, Blenckner T, Bryhn AC, Hellström SS. The influence of calcium on the chlorophyll-phosphorus relationship and lake Secchi depths. Hydrobiologia. 2005;537:111–23.
Eiler A, Heinrich F, Bertilsson S. Coherent dynamics and association networks among lake bacterioplankton taxa. ISME J. 2012;6:330–42.
Peura S, Bertilsson S, Jones RI, Eiler A. Resistant microbial cooccurrence patterns inferred by network topology. Appl Environ Microbiol. 2015;81:2090–7.
Logares R, Tesson SVM, Canbäck B, Pontarp M, Hedlund K, Rengefors K. Contrasting prevalence of selection and drift in the community structuring of bacteria and microbial eukaryotes. Environ Microbiol. 2018;20:2231–40.
Lindström ES, Kamst-Van Agterveld MP, Zwart G. Distribution of typical freshwater bacterial groups is associated with pH, temperature, and lake water retention time. Appl Environ Microbiol. 2005;71:8201–6.
Lauber CL, Hamady M, Knight R, Fierer N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol. 2009;75:5111–20.
Xiong J, Liu Y, Lin X, Zhang H, Zeng J, Hou J, et al. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ Microbiol. 2012;14:2457–66.
Findlay DL, Kasian SEM. Phytoplankton community responses to acidification of lake 223, experimental lakes area, northwestern Ontario. Water Air Soil Pollut. 1986;30:719–26.
Findlay DL, Kasian SEM. The effect of incremental pH recovery on the Lake 223 phytoplankton community. Can J Fish Aquat Sci. 1996;53:856–64.
Maberly SC. Diel, episodic and seasonal changes in pH and concentrations of inorganic carbon in a productive lake. Freshw Biol. 2008;35:579–98.
Tong Y, Lin G, Ke X, Liu F, Zhu G, Gao G, et al. Comparison of microbial community between two shallow freshwater lakes in middle Yangtze basin, East China. Chemosphere. 2005;60:85–92.
Romina Schiaffino M, Unrein F, Gasol JM, Massana R, Balagué V, Izaguirre I. Bacterial community structure in a latitudinal gradient of lakes: the roles of spatial versus environmental factors. Freshw Biol. 2011;56:1973–91.
Zeng J, Yang L, Li J, Liang Y, Xiao L, Jiang L, et al. Vertical distribution of bacterial community structure in the sediments of two eutrophic lakes revealed by denaturing gradient gel electrophoresis (DGGE) and multivariate analysis techniques. World J Microbiol Biotechnol. 2009;25:225–33.
Canfield DE, Bachmann RW. Prediction of total phosphorus concentrations, chlorophyll a, and Secchi depths in natural and artificial lakes. Can J Fish Aquat Sci. 1981;38:414–23.
Meeuwig JJ, Peters RH. Circumventing phosphorus in lake management: a comparison of chlorophyll a predictions from land-use and phosphorus-loading models. Can J Fish Aquat Sci. 1996;53:1795–806.
Yang L, Lei K, Meng W, Fu G, Yan W. Temporal and spatial changes in nutrients and chlorophyll-α in a shallow lake, Lake Chaohu, China: an 11-year investigation. J Environ Sci (China). 2013;25:1117–23.
Kraemer SA, Soucy JPR, Kassen R. Antagonistic interactions of soil pseudomonads are structured in time. FEMS Microbiol Ecol. 2017;93:1–9.
Acknowledgements
This work was supported by the genome Quebec and Genome Canada-funded ATRAPP Project (Algal blooms, Treatment, Risk Assessment, Prediction and Prevention) (awarded to BJS), by the NSERC Canadian LakePulse network (Strategic Partnership network NETG 479720-15), by NSERC Discovery Grant #6693-2016 and by the NSERC Canadian Research Chair #230456 (DW) and FQRNT and NSERC/CREATE-GRIL fellowships (NBDC). We thank the coordinators and field team members of the LakePulse 2017 sampling campaign for their efforts. We also would like to thank members of the network, and specifically B. Beisner and V. Fugere, for helpful discussions during the manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kraemer, S.A., Barbosa da Costa, N., Shapiro, B.J. et al. A large-scale assessment of lakes reveals a pervasive signal of land use on bacterial communities. ISME J 14, 3011–3023 (2020). https://doi.org/10.1038/s41396-020-0733-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0733-0
This article is cited by
-
A genome catalogue of lake bacterial diversity and its drivers at continental scale
Nature Microbiology (2023)
-
Shotgun metagenomes from productive lakes in an urban region of Sweden
Scientific Data (2023)
-
Phylogenetic diversity and functional potential of the microbial communities along the Bay of Bengal coast
Scientific Reports (2023)
-
Plastic pollution fosters more microbial growth in lakes than natural organic matter
Nature Communications (2022)
