
Abstract
The interaction between airway microbiome and host in chronic obstructive pulmonary disease (COPD) is poorly understood. Here we used a multi-omic meta-analysis approach to characterize the functional signature of airway microbiome in COPD. We retrieved all public COPD sputum microbiome datasets, totaling 1640 samples from 16S rRNA gene datasets and 26 samples from metagenomic datasets from across the world. We identified microbial taxonomic shifts using random effect meta-analysis and established a global classifier for COPD using 12 microbial genera. We inferred the metabolic potentials for the airway microbiome, established their molecular links to host targets, and explored their effects in a separate meta-analysis on 1340 public human airway transcriptome samples for COPD. 29.6% of differentially expressed human pathways were predicted to be targeted by microbiome metabolism. For inferred metabolite–host interactions, the flux of disease-modifying metabolites as predicted from host transcriptome was generally concordant with their predicted metabolic turnover in microbiome, suggesting a synergistic response between microbiome and host in COPD. The meta-analysis results were further validated by a pilot multi-omic study on 18 COPD patients and 10 controls, in which airway metagenome, metabolome, and host transcriptome were simultaneously characterized. 69.9% of the proposed “microbiome-metabolite–host” interaction links were validated in the independent multi-omic data. Butyrate, homocysteine, and palmitate were the microbial metabolites showing strongest interactions with COPD-associated host genes. Our meta-analysis uncovered functional properties of airway microbiome that interacted with COPD host gene signatures, and demonstrated the possibility of leveraging public multi-omic data to interrogate disease biology.
Similar content being viewed by others


Introduction
Chronic obstructive pulmonary disease (COPD) is one of the most prevalent respiratory diseases and is characterized by impaired lung function as a consequence of airway inflammation, small airway obliteration, and alveolar destruction [1, 2]. The composition of airway microbiome in COPD has been well characterized. Increasing evidence suggests that airway microbial communities differ between health and COPD [3, 4], shift during episodes of disease exacerbations [5,6,7] and associate with airway inflammation [6]. It is thought that airway microbiome dysbiosis contributes to airway inflammation, immune dysregulation, and susceptibility to infection [8, 9].
Despite advances, our understanding of the precise role of airway microbiome in COPD pathogenesis remains limited. Several factors contribute to this knowledge gap. First, a clear understanding of COPD-associated changes in the airway microbiome is lacking, partly due to the incongruence in the findings between previous airway microbiome studies [10]. Second, the functional capacity of the airway microbiome remains unknown. This is of importance, as the microbiome interacts with host through metabolites that serve as ligands for host receptors. To date, most studies have characterized taxonomic composition of the airway microbiome through 16S rRNA gene-based amplicon sequencing, yet few studies have used metagenomic [11] or meta-transcriptomic [12] sequencing to explore the functional properties of airway microbiome in COPD. While recent studies have highlighted a link between individual microbial metabolites and inflammation [13, 14], the broader community-level landscape on how airway microbiome generates metabolites that modulate host immunity in COPD remains unexplored.
The ever-increasing availability of public microbiome and host multi-omic datasets presents an opportunity to synthesize existing knowledge to uncover microbiome–host interactions. Meta-analysis has demonstrated its power to identify reproducible disease-associated microbiome signatures at an unprecedented scale [15,16,17]. Here we applied a large-scale, multi-omic meta-analysis approach to characterize the functional signature of airway microbiome in COPD. We retrieved all publicly available COPD airway microbiome datasets, including 16S rRNA gene (n = 1640) and metagenomic (n = 26) data and analyzed them using a standardized pipeline. We identified disease-associated microbiome shifts across datasets using statistical meta-analysis to enhance statistical power against study-specific biases. We inferred a microbiome-metabolite catalog and its molecular links to host targets. Host–microbiome interactions were then validated in a separate meta-analysis on all public COPD airway host transcriptomic datasets, to identify interactions that are associated with disease signatures. Finally, the proposed host–microbiome interaction links were validated in an independent multi-omic characterization of COPD patients and healthy controls. We demonstrated the value of the meta-analysis approach in understanding host–microbiome interactions and interrogating disease biology.
Material and methods
Microbiome datasets collection and processing
Detailed information on dataset collection, processing, and meta-analysis are provided in the supplementary document. All public COPD lung microbiome datasets were retrieved from National Center for Biotechnology Information (NCBI) by literature search in PubMed and dataset search in SRA using the term “COPD microbiome”. Literature citations were filtered based on criteria detailed in the supplementary document. The datasets covered multiple lower airway sites (sputum, bronchoalveolar lavage (BAL), bronchial brushing). Only datasets on sputum samples from stable COPD patients and/or controls were included. Both 16S rRNA gene and metagenomic datasets were included and analyzed separately. The raw sequencing data were downloaded from SRA or using links in the publications.
All 16S rRNA gene datasets were processed using a standardized pipeline in QIIME 2.0 [18]. For each dataset, demultiplexed sequencing reads were denoised to generate amplicon sequence variants (ASVs) using DADA2 algorithm [19]. Additional parameters were used to denoise 454 data, according to DADA2 protocol. A custom Naive Bayes classifier was trained on the Greengenes 13_8 99% operational taxonomic units (OTUs) to assign taxonomy for each dataset. PICRUSt2 analysis with ‘stratified’ mode was performed using ASVs as input [20]. For metagenomic data, raw sequencing reads were quality filtered using cutadapt v1.18 [21] and human reads were filtered by aligning reads to human genome GRCh37/hg19 using Bowtie2 [22]. The remaining reads were subject to microbial taxon and gene identifications using MetaPhlAn2 [23] and HUMAnN2 [24].
For 16S rRNA gene datasets that involved controls, differentially abundant taxa and inferred genes were identified for each dataset using a generalized linear model [25]. Demographic factors such as age, gender and smoking history were included as covariates in the model, whenever possible. The summary statistics (fold-change and P value) of each taxa in each dataset were retrieved for a random effect statistical meta-analysis using the MetaDE package in R [26]. We chose to use the combined effect size method for meta-analysis, which generates more conservative and biologically consistent results than the p value combination method [27, 28]. As an alternative, we also conducted batch-effect adjustment for the 16S rRNA gene case-control datasets using percentile normalization in Gibbons et al. [29], and combined the batch-adjusted datasets for statistical analysis.
For random forest analysis, microbiome relative abundances were arcsine-square root-transformed and scaled to unit variance. Random forest analysis was performed for the significant genera in the meta-analysis using Weka v3.8.3 [30]. Cross-validations were performed within dataset using sevenfold cross-validation, and between datasets using the leave-one-study-out strategy. Datasets were further split at the country-level, to perform leave-one-country-out cross-validation across five countries.
Metabolic inference of COPD airway microbiome
Genes with congruent direction of changes in COPD versus controls in PICRUSt2 analysis and in metagenomic dataset were retained for metabolic inference. For each gene, we retrieved its associated enzymatic reaction from the MetaCyc [31] database using enzyme Commission (EC) number as query. Metabolic substrates and products were derived from the enzymatic reactions based on the reaction equation and its reversibility. Compounds without PubChem or ChEBI IDs and without known structures were excluded from further analysis.
The metabolite–host interaction was obtained from STITCH v5.0 [32], a database for interactions between chemical and proteins that integrates various resources including PubChem, ChEMBL, and Reactome. Interactions with confidence score >0.9 and with known functional effects (activation or inhibition) were retained. Tissue specificity of host genes was examined in the GTEx portal [33]. Pathway enrichment analysis was performed using MetaBase R v6.30.68780 (Clarivate Analytics, FDR P < 0.01, enriched genes ≥ 10).
The relative turnover of metabolites in COPD versus controls was calculated using the predicted relative metabolomic turnover (PRMT) method as described previously [34, 35]. Briefly, a stoichiometric matrix describing the quantitative relationship between microbial genes and metabolites was constructed based on metabolic reactions in the MetaCyc database [31]. The resulting matrix composing of m metabolites and n bacterial genes was then used for multiplication with a vector containing log2 fold-change of the n bacterial genes in COPD versus controls in the meta-analysis, to generate a vector of PRMT scores for m metabolites. It is important to note that the PRMT scores do not predict the net abundance of a metabolite in a dataset per se but only its relative turnover between different groups (here COPD versus controls).
To identify significant taxonomic contributors for the inferred metabolites, we performed a leave-one-genus-out (LOGO) analysis. Briefly, the PRMT score of each metabolite was recalculated when each genus was excluded one at a time from the microbiome data. The relative contribution of each genus to the metabolite was calculated as the deviation of the PRMT score when removing that genus, normalized by the standard deviation of PRMTs calculated when removing all genera one at a time (similar as a z-score index). A taxa was considered as significant contributor to a metabolite if the absolute z-score was greater than 2.0 (equivalent to P < 0.05).
Host transcriptome datasets collection and processing
The public COPD airway host transcriptomic datasets were retrieved from NCBI Gene Expression Omnibus database and were filtered based on the criteria as detailed in the supplement. Raw microarray data were processed using a standardized pipeline to generate gene-set-level expression matrix data [36]. For RNA-Seq data, the quality-filtered sequencing reads were mapped to the human reference genome GRCh37/hg19 using STAR [37]. The gene count and FPKM matrices were generated using Subread [38]. We used the log2-transformed FPKM data to ensure consistency on downstream analytical methods with microarray datasets. Inter-study batch-effect adjustment was performed using ComBat prior to meta-analysis according to previous studies [28, 39]. Differentially expressed genes (DEGs) between COPD and controls were identified using limma package in R [40]. The random effect meta-analysis was performed using combined effect size method using MetaDE in R [26].
A metabolite can have disease-promoting or disease-ameliorating effect through influencing the expression of its targets in disease. For instance, a disease-promoting metabolite could achieve its role through collectively activating genes that were upregulated in disease (i.e., pro-inflammatory genes), and/or inhibiting genes downregulated in disease (i.e., anti-inflammatory genes). Accordingly, the disease-modifying effect of an inferred metabolite on host transcriptome can be assessed using the average fold-change of all its host targets in the transcriptome meta-analysis, adjusted by activating or inhibitory effects of the metabolite to the targets, using the equation:
where EF is the predicted disease-promoting effect (or disease-alleviating effect if it is negative) of a metabolite on host transcriptome, ES is the combined effect size of each of its host targets in the meta-analysis, and m and n are the number of genes activated or inhibited by the metabolite, respectively.
Multi-omic analysis on independent COPD cohort
Induced sputum samples were collected from 18 stable COPD patients and 10 healthy controls in the First Affiliated Hospital of Guangzhou Medical University. The study was approved by the ethics committee of the First Affiliated Hospital of Guangzhou Medical University (No. 2017–22). Patients with antibiotic usage within 4 weeks were excluded. Bacterial genomic DNA was extracted from selected sputum plugs using Qiagen DNA Mini kit (along with negative controls) and subject to metagenomic sequencing using Illumina NovaSeq. The remaining sputum was subject to sputum cell and supernatant isolation according to previous protocol [41]. RNA was extracted from sputum cell using Qiagen RNease Mini kit for RNA-Seq using Illumina NovaSeq. Two-hundred microliters of sputum supernatant was subject to untargeted metabolomic characterization using Xevo G2-XS QTOF (Waters, UK).
The raw sequencing reads for metagenome and host transcriptome were processing using the same analytical pipeline in the meta-analysis. For metabolome, ion features (monoisotopic m/z, charge, retention time, peak size) were extracted using Progenesis QI v2.2. Low quality ions or ions with relative standard deviation greater than 30% were filtered from downstream analysis [42]. Identification of metabolites was performed by searching HMDB [43], METLIN [44], and KEGG databases [45]. Metabolomic data analysis was performed using MetaboAnalystR [46]. The microbial contributors to metabolome were analyzed using the paired metagenome and metabolome data using MIMOSA2 [47].
Results
Taxonomic and functional profiles of COPD airway microbiome
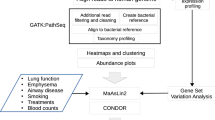
The overall strategy of the multi-omic meta-analysis is shown in Fig. 1. To begin, we searched PubMed using the term “COPD microbiome”, which returned 231 publications (accessed January 2020, Table S1). We restricted the sample type to sputum which included the largest number of studies and samples. This resulted in 11 16S rRNA gene and one metagenomic datasets. We obtained two additional 16S rRNA gene and one metagenomic datasets from the Sequence Read Archive (SRA) database. Although unpublished, the two 16S rRNA gene datasets showed comparable quality control statistics with published datasets (Table S2), indicating their reasonable data quality to be included for downstream analyses. Altogether, the search yielded a total of 13 16S rRNA gene and two metagenomic datasets, comprising of 1517 COPD and 123 control samples from 16S rRNA gene datasets and 16 COPD and 10 control samples from metagenomic datasets, together from 10 countries across Europe, Asia, and America (Fig. 1a and Table 1).

a Geographical distribution of collection sites for 1666 airway microbiome samples (1640 samples from 16S rRNA gene datasets and 26 samples from metagenomic datasets) and 1340 host transcriptome samples included in the meta-analysis. b Flowchart of the integrative meta-analysis for microbiome and host transcriptome datasets, as well as the independent multi-omic cohort validation. Each data analysis step is shown in the gray box, with the analysis method and software described within. In the steps of dataset collection and processing, n represents the number of studies/datasets involved in each step. The output of key steps is shown in the pink box.
We profiled microbial composition for the 16S rRNA gene datasets using DADA2 algorithm [19]. ASVs were generated for each dataset separately. Notably, these datasets were confounded with different sample origins, hypervariable regions and sequencing platforms. To assess data heterogeneity, we first performed 99% close-reference clustering for ASVs for each dataset so they can be compared with each other. Principal coordinate analysis on all samples based on 99% OTUs showed a clear separation of samples by study (PERMANOVA R2 = 0.45, P < 0.001, Fig. S1), indicating study-specific batch effects. Among all confounding factors, hypervariable region showed the strongest association with variation of microbial composition (Canonical correspondence analysis (CCA), F = 5.629, P = 0.0035, Fig. S2), followed by country (F = 3.243, P = 0.019) and sequencing platform (F = 2.781, P = 0.026). To alleviate impact of these batch effects, previous studies chose to perform microbiome meta-analysis at the genus-level, which sacrificed the sensitivity in detecting finer-level variation in exchange for less data heterogeneity [15, 48]. A total of 490 genera were identified across all samples, of which 304 genera were shared by at least two datasets and were retained for further analysis. 26 genera had an average relative abundance greater than 0.001 across all samples (Fig. 2a). Among the 26 genera, Streptococcus, Haemophilus, Veillonella, Prevotella, Neisseria, Rothia, Capnocytophaga, Actinomyces, Leptotrichia and Fusobacterium also had average relative abundance >0.001 across all 13 16S rRNA gene datasets. Nineteen genera had an average relative abundance >0.001 across 12 out of 13 16S rRNA gene datasets (except for Moraxella, Lactobacillus, Campylobacter, Oribacterium, Treponema, Stenotrophomonas and Ralstonia). Streptococcus, Veillonella, Rothia, Prevotella, Haemophilus, and Actinomyces were core genera present in at least 90% of all samples. Examining distribution of each genus in each dataset revealed a clustering of datasets largely by hypervariable regions (Fig. S3), indicating different regions of 16S rRNA gene surveyed may contribute to the divergence of taxa observed between studies. The cross-dataset heterogeneity was alleviated when analyzed at the genus-level, as indicated both in PERMANOVA and CCA results (Figs. S1, 2). Hypervariable region remained of borderline significance in the genus-level CCA (F = 2.318, P = 0.047).

a Heatmap showing COPD microbiome composition at the genus-level for all 16S rRNA gene datasets. The 26 genera with average relative abundance greater than 0.001 across all samples were shown. The number of COPD samples were indicated in the parenthesis besides each dataset identifier. b The relative abundances in COPD versus controls for the 12 genera significant in the meta-analysis, along with their log2 fold-changes in the two case-control 16S rRNA gene datasets, combined effect sizes, and the log2 fold-changes in the case-control metagenomic dataset. The 12 genera showed congruent direction of changes in COPD versus controls among all case-control studies. c Receiver operative characteristic (ROC) curves for the random forest models in classifying COPD and controls using within-dataset and between-dataset cross-validations. The area under curve (AUC) for each prediction was shown in the lower right corner of the plot. d The positive correlation between the fold-changes of 1124 genes in COPD versus controls in the two 16S rRNA gene datasets as inferred by PICRUSt2. The subset of 759 genes that also had congruent direction of changes in COPD versus controls in the metagenomic dataset were highlighted in dark red.
The vast majority of samples (1640 out of 1666 samples, 98.4%) belonged to the 16S rRNA gene datasets. For 16S rRNA gene datasets, we performed functional inference using PICRUSt2. This resulted in 2341 EC gene families (referred as PICRUSt2-inferred genes). For the 26 samples from the metagenomic datasets, we performed functional profiling using HUMAnN2 [24]. A leaner catalog of 1236 gene families were obtained, of which 1124 genes overlapped with the PICRUSt2-inferred genes (Table S3). The 1124 genes were involved in a diverse range of functional processes and covered 289 of 305 pathways predicted by PICRUSt2 (Fig. S4), indicating its functional diversity. Of the 1124 genes, 299 were core genes present in 90% of COPD metagenomic samples (Table S3). The relative abundances of genes in the metagenomic datasets were correlated with their corresponding abundances inferred from 16S rRNA gene datasets, irrespective of them being core or accessory genes (Spearman’s R ≥ 0.60, Fig. S5), indicating congruent functional profiling between the two different types of data.
Meta-analysis on the airway microbiome in COPD versus controls
For the two 16S rRNA gene datasets that included controls (SRP066375 and SRP136124, Table 1), we performed a statistical meta-analysis to identify bacterial taxa and genes consistently altered in COPD versus controls across datasets. The two datasets had the same sequencing platform and hypervariable region, further eliminating confounding effects. We identified significant microbial taxa in COPD versus controls within each dataset, adjusting demographic factors such as country, age, gender, and smoking history as covariates whenever available. We then pooled evidence of differential abundance across datasets by combining effect size using random effect statistical meta-analysis. Instead of pooling raw data directly, such meta-analysis technique by pooling summary statistics from each dataset is more robust to the between-study data heterogeneity [49]. A total of 12 genus-level taxa were significant in the meta-analysis (false discovery rate (FDR)-adjusted P < 0.05, Table S4). Haemophilus, Streptococcus, Moraxella, and Lactobacillus were enriched in COPD, while Selenomonas, Leptotrichia, Dialister, Porphyromonas, Peptostreptococcus, Campylobacter, Catonella, and Prevotella were depleted (Fig. 2b). These taxa were consistent in direction of changes across the two 16S rRNA gene datasets, as well as in the metagenome dataset (Fig. 2b). As an alternative, we also performed batch-effect correction for the two datasets using the percentile normalization method in Gibbons et al. [29] and combined the batch-corrected data for multivariate analysis. Nineteen genera were significant using the combined dataset (FDR P < 0.05, Table S4), including 11 of the 12 genera identified by random effect meta-analysis (except for Moraxella which had an FDR P value of 0.08). However, the precise combined fold-change of genera can no longer be assessed here due to information loss in percentile conversion.
The 12 genera noted above had an area under curve (AUC) of 0.773 and 0.828 in classifying COPD from controls for each dataset alone using random forest with within-dataset cross-validation (Fig. 2c). The AUCs were 0.804 and 0.729 when applying between-dataset cross-validation in which classifier was trained on one dataset and validated on the other (Fig. 2c). This indicated the generalizability of these markers when extrapolated across datasets. The two datasets included COPD patients and controls from five different countries. Applying a leave-one-country-out cross-validation yielded an average AUC of 0.723 (range: 0.648–0.804, Fig. S6).
We next sought to identify functional genes altered in COPD versus controls. We restricted our analysis to the 1124 gene families present both in PICRUSt2 inference and metagenomic datasets. We first performed a multivariate analysis to identify differentially abundant genes in COPD within each case-control study, adjusting for demographic covariates. The 1124 genes showed an overall correlation in their PICRUSt2-inferred fold-changes in COPD versus controls between the two 16S rRNA gene datasets, with 891 genes having the same direction between datasets (Fig. 2d and Table S5). Of the 891 genes, 759 genes further showed the same direction of fold-changes in the case-control metagenomic dataset (Fig. 2d and Table S5). These 759 genes with congruent alternations across all 16S rRNA gene and metagenomic case-control studies were retained, and their combined effect sizes in the random effect meta-analysis of all three datasets (two 16S rRNA gene and one metagenome) were used as proxy for their associations with COPD. Of the 759 genes, 17 genes in xenobiotic, carbohydrate, peptidoglycan, and amino acid metabolism were significantly enriched in COPD versus controls in the meta-analysis (Fig. S7, Table S5, FDR P < 0.05), whereas 9 genes in biosynthesis of secondary metabolites, lipid, and fatty acid metabolism were significantly depleted. Haemophilus, Streptococcus, and Moraxella were top contributors to the 17 enriched genes, consistent with their increased abundances in COPD. On the other hand, commensal genera such as Veillonella and Prevotella that were decreased in COPD, were top contributors to the 9 depleted genes (Fig. S7).
Metabolic potential of the COPD airway microbiome
To infer the metabolic potential of the COPD airway microbiome, we mapped the 759 microbial genes onto the MetaCyc database. This step yielded a total of 575 possible metabolites with known structures as products from enzymatic reactions. Surveying all literatures resulted in two COPD airway metabolomic studies with 294 metabolites reported in their supplementary data [14, 50]. Two-hundred eleven out of 575 inferred metabolites (36.7%) were observed in the list of 294 metabolites. Three hundred forty-six metabolite–target interactions with known functional effects were further obtained between 67 metabolites and 244 human host genes in the STITCH database, using a confidence score of 0.9 (“highest confidence” group, Fig. 3a and Table S6). Forty-six of these 67 metabolites (68.7%) were observed in the public metabolomic data (Table S6).

a Sankey diagram delineating all interaction links between microbial genera, metabolic reactions, inferred metabolites, and host targets. Metabolic reactions were grouped by the MetaCyc pathway categories. Metabolites were grouped by their classes in PubChem. Host genes were grouped by the enriched pathways and only genes in the top pathways (FDR P < 1e−8) are shown. Pathways were highlighted in red and in asterisks if they overlapped with the 54 pathways significantly enriched for differentially expressed genes in the host transcriptome meta-analysis. b The inferred metabolites ranked by their PRMT scores. The 20 metabolites with at least 5 interactions with host targets were shown for display purpose. The metabolite was highlighted in asterisks if it was present in the public COPD airway metabolomic data. c The biosynthesis pathways for palmitate and nitric oxide. Each metabolite was colored by its PRMT score and each corresponding gene was colored by its most predominant microbial contributor. d Heatmap showing significant microbial contributors to the 20 metabolites in (b) in the leave-one-genus-out (LOGO) analysis (absolute z-score > 2.0, P < 0.05). The z-score of each species to each metabolite was indicated in the heatmap. A positive z-score means that the biosynthesis or degradation of the metabolite by the taxa contributes to its relative enrichment in COPD, whereas a negative z-score means the biosynthesis or degradation of the metabolite by the taxa contributes to its depletion in COPD. The microbial genera were colored by their corresponding phyla. The PRMT score was shown for each metabolite beside the heatmap.
We adapted the PRMT method by Larsen et al. [34] to estimate the relative turnover of metabolites in COPD versus controls, based on the combined effect sizes of their associated genes in the microbiome meta-analysis. PRMT method utilizes a stoichiometric matrix describing the quantitative relationships between microbial genes and metabolites, to provide an estimate on the impact of the metagenome on the accumulation or depletion of metabolites. For the 67 metabolites with host interactors, palmitate and N-acetyl-d-mannosamine had the highest PRMT scores thus were inferred to be most COPD-enriched, while d-aspartate had the lowest PRMT score (Table S6 and Fig. 3b). Extension to the entire pathways further revealed consistent alternations of metabolites across some pathways. For instance, six of eight metabolites in palmitate pathway were inferred to be elevated in COPD (Fig. 3c). All metabolites in nitric oxide pathway except for nitrate were predicted to be COPD-depleted (Fig. 3c). Calculating the PRMT scores based on fold-changes in the metagenomic data alone indicated highly consistent results with those based on the combined effect sizes in the meta-analysis of both 16S rRNA gene and metagenome data (Fig. S8).
We identified key microbial drivers to metabolites through a LOGO analysis. The relative contribution of each genus to each metabolite was estimated by assessing the deviation of PRMT scores that resulted from removing it from the analysis (as a z-score index). A total of 25 genera were considered as significant contributors to the 67 metabolites (absolute z-score>2.0, P < 0.05, Fig. S9), among which Streptococcus, Haemophilus, and Veillonella contributed to most number of metabolites (Fig. 3d). For palmitate that was predicted to be COPD-enriched, Rothia contributed most to its enrichment (z = 7.52). On the other hand, Prevotella contributed most to the depletion of d-aspartate in COPD (z = −4.50).
The 244 host targets of microbial metabolites exhibited a high degree of lung tissue specificity. Among all human tissues, lung had the highest average expression levels for the 244 genes (GTEx, Fig. S10). Functional enrichment of the 244 genes revealed 75 significant MetaBase pathways (FDR P < 0.01, enriched genes>10, Table S7). Many top pathways were related to inflammatory signaling such as IL-8, CCL2, TNF-alpha and RAGE signaling in respiratory diseases such as asthma and COPD (Fig. 3a). This suggests that the identified host–microbiome interactions could have a plausible role in COPD pathogenesis.
Assessing metabolic effects in human transcriptome meta-analysis
To assess whether the 244 host targets are implicated in COPD host response, we performed a separate meta-analysis on all public host airway transcriptome datasets in COPD. In total, we included 19 host airway transcriptome datasets with samples from 745 stable COPD patients and 595 controls across multiple anatomical sites (lung, bronchial brushings, alveolar macrophage, small or large airways, sputum and BAL) (Tables 1 and S8). To assess anatomical-site heterogeneity, we conducted a meta-analysis on datasets from each site separately. Study-specific batch effect was adjusted using Combat prior to random effect meta-analysis (Fig. S11). An averaged 73.2% of DEGs from each site were also present in at least one other sites (Fig. S12a). At the pathway level, 63 MetaBase pathways were shared by at least four of five sites, many of which were related to immune processes (Fig. S12b and Table S9). These results suggest that despite site heterogeneity, there are common immune-related host transcriptome signatures that can be potentially revealed by a combined random effect meta-analysis.
To quantify potential impacts of microbial metabolites on host gene expression, we calculated the average fold-change of their host targets in the combined transcriptomic meta-analysis, adjusted by the activating or inhibitory effects of the metabolites. We found that for most metabolites, their disease-promoting or alleviating effects on host transcriptome were concordant with their predicted relative turnover in COPD airway microbiome (Spearman’s R = 0.459, Fig. 4a). For example, palmitate was inferred to be elevated with PRMT score of 4.34 in the COPD microbiome. Mirroring this observation, eight of ten genes activated by palmitate were significantly upregulated in COPD host transcriptome, collectively with a disease-modifying score of 2.89. In contrast, d-aspartate was predicted to be decreased in COPD versus controls (PRMT = −4.31). In concordance, it showed a potential disease-ameliorating effect by activating two genes (GRIN2A and GRIA1) significantly downregulated in COPD (disease-modifying score = −3.89). Our results suggest a synergistic response between microbiome and host in COPD.

a Scatterplot showing the predicted disease-modifying effects of metabolites on host transcriptome (x-axis) and their PRMT scores in COPD airway microbiome (y-axis). The direction of disease-modifying effects were concordant with that of the PRMT scores for 44 out of 58 metabolites (Spearman’s R = 0.507, P < 1e−4). Metabolites were colored in red if they had concordant disease-promoting effects, in blue if they had concordant disease-ameliorating effects, and in gray if they had discordant effects. The size of the circles in the plot are proportional to the number of predicted host interactors for the metabolites. b Heatmap for the 41 meta-DEGs that were linked to at least one microbial metabolites. Only metabolites linked to at least two host genes were shown for display purpose. Each link between a metabolite and a gene indicate their interactions colored by activation or inhibition as obtained from the STITCH database. Metabolites were colored similarly as in (a). The number of samples were indicated in the parenthesis besides each sample type.
Meta-analysis on the combined datasets identified 474 and 490 host genes significantly up- or downregulated in COPD versus controls (referred as meta-DEGs, log2 fold-change>1, FDR P < 0.05, Fig. S12c, Table S10). The meta-DEGs were enriched for 54 host pathways (FDR P < 0.01). Of them, 16 pathways (29.6%) overlapped with pathways for the 244 gene set, indicating these pathways could be modulated by microbiome metabolites (Table S10). Fifty meta-DEGs overlapped with the 244 gene set and were linked to 37 metabolites, which presumably contained disease-specific metabolite–host interaction links (Fig. 4b, Table 2). A total of 19 genera were significant microbial contributors to these 37 metabolites (absolute z-score>2.0), which together constituted a total of 296 “microbiome-metabolite–host” interaction links to be further investigated (Table S11).
Validation of host-microbiome interaction in independent multi-omic cohort
To validate the “microbiome-metabolite–host” interaction links inferred from the meta-analysis, we conducted a pilot multi-omic study on 18 stable COPD patients and 10 healthy controls. Sputum metagenome, metabolome and host transcriptome were simultaneously characterized, resulting in a profile of 218 genera, 1511 microbial EC gene families, 2021 metabolites and 19,142 host genes (Fig. 5a). Of them, 9 genera, 61 microbial genes, 33 metabolites, and 469 host genes were significantly altered in COPD versus controls (FDR P < 0.1, Table S12). For the 12 signature genera in the meta-analysis, they all showed consistent direction of changes in COPD versus controls in our cohort, and Streptococcus and Prevotella were statistically significant (FDR P < 0.1). The 1511 microbial genes included 1074 of the 1124 genes in the meta-analysis, indicating high specificity of this gene catalog. The 469 host DEGs were enriched for 60 pathways (FDR P < 0.01), of which 238 genes (50.7%) and 32 pathways (53.3%) overlapped with the 964 meta-DEGs, suggesting overall congruent transcriptomic signatures. Among all 575 inferred metabolites in the meta-analysis, 320 (55.7%) were detected in the metabolome. The detection rate varied across categories of metabolites, with the highest rates for vitamins and lowest for inorganic compounds (Fig. 5b). For the 37 metabolites targeting meta-DEGs, 34 (91.9%) were present in the metabolome (Table S13). Twenty-six of them further exhibited the same direction of changes in COPD versus controls with their PRMT scores, indicating accuracy of PRMT scoring in inferring trends of metabolic turnover (Fig. 5c, Spearman’s R = 0.581, Table S13). Among them, xylulose-5-P, d-mannopyranose, palmitate, and L-ornithine were significant (FDR P < 0.1).

a A schematic illustration for the sample processing steps to simultaneously obtain metagenome, metabolome and host transcriptome from sputum samples. b The proportion of the 575 inferred metabolites detected in the actual metabolome in each category. The number of inferred metabolites in each category were indicated in the parenthesis. c Scatterplot showing the overall concordance between the PRMT scores of the 31 metabolites and their log2 fold-changes in COPD versus controls in the metabolome. The sizes of the circles in the plot are proportional to the number of predicted host interactors for the metabolites. Metabolites were colored similarly in Fig. 4. Significant metabolites in COPD versus controls (FDR P < 0.1) were highlighted in asterisks. d The 91 “microbiome-metabolite–host” interaction links that were validated in the COPD multi-omic cohort and involved at least one metabolite or host targets significant in COPD versus controls (FDR P < 0.1). The microbial genera were colored by their corresponding phyla. The metabolites and host targets were colored by their direction of changes in COPD versus controls with the strength of the color representing their absolute fold-change. The links between metabolites to host targets were colored by the activation or inhibition effects and the strength of the links are proportional to the absolute Spearman correlation coefficient between the paired metabolome and host transcriptome. The strength of the links between microbial taxa and metabolites are proportional to the contribution scores of the genus to the metabolite between the paired metagenome and metabolome in MIMOSA2 analysis. The significant metabolites and host targets were highlighted in asterisks (FDR P < 0.1). e The scatterplot for the top three metabolite–host target correlations in (d). Samples were colored by COPD or controls. The distribution of metabolites and host targets in COPD and controls were shown in the boxplots. The significant genera, metabolites and host targets were highlighted in asterisks (FDR P < 0.1). For significant genus-level contributors to the metabolites, their contributions to variance in MIMOSA2 analysis were shown on the right side.
We investigated the 296 inferred “microbiome-metabolite–host” links from meta-analysis in our multi-omic data. We excluded links involving two gaseous metabolites, nitric oxide and nitrous oxide, which were impossible to be observed in our samples. This resulted in 272 links. For the links between metabolites and host targets, we examined their correlation patterns in paired metabolome and host transcriptome profiles. In total, 78.1% (57 out of 73) of the metabolite-target pairs showed correlation patterns (positive or negative) in the multi-omic data in agreement with the predicted mode of action from meta-analysis (activation or inhibition, Table S14). To validate microbial contributors for metabolites, we performed a MIMOSA2 analysis for paired metagenome and metabolome data. MIMOSA2 utilized the PRMT metrics to estimate the metabolic potential scores from metagenome and correlate with the actual abundance of metabolites in the metabolome [47]. It then decomposed the model fit into contribution from each microbial taxa, similar as our LOGO analysis. For 85.2% (104 out of 122) of the microbiome-metabolite pairs, the microbial genera were also among the contributors to the variation of the metabolites in the MIMOSA2 analysis, supporting their metabolic associations.
Collectively, 190 of the 272 (69.9%) proposed “microbiome-metabolite–host” links in the meta-analysis were validated in our pilot multi-omic data, involving 16 microbial genera, 27 metabolites, and 37 host genes. Among them, 91 interaction links further involved metabolites or host genes significantly altered in COPD versus controls in the multi-omic data (Fig. 5d, Tables 3, S14, FDR P < 0.1), suggesting these interactions were likely disease-specific. The strongest associations were butyrate-SOD2, followed by homocysteine-MMP9 and palmitate-ACSL1 (Fig. 5e). Fusobacterium, Streptococcus, and Rothia were top microbial contributors to these metabolites respectively (Fig. 5e).
Discussion
Here we present a large-scale, integrative meta-analysis on public multi-omic datasets to characterize the functional signatures of airway microbiome in COPD. The statistical meta-analysis enabled us to identify microbial taxa that had subtle but consistent changes in COPD versus controls that may not be significant in analysis of each individual study alone [29]. The reproducibility of the 12 genera in segregating COPD patients versus controls across-countries implicates the possibility of a global microbiome classifier for COPD diagnostics. The vast majority of COPD airway microbiome datasets in the public databases are 16S rRNA gene-based, from which microbial gene contents were inferred using PICRUSt analysis. PICRUSt analysis is capable of inferring core microbial functions in a community but may not be well resolved for accessory genes resulting from genomic variations within genus or species, and thus tends to overestimate the gene pool. To mitigate this risk, we generated COPD airway microbial gene catalog by overlapping genes predicted by PICRUSt2 with those present in metagenomic datasets. These genes covered a diverse range of microbial pathways, and largely overlapped with the metagenomic genes in our pilot cohort, adding credibility for their presence in human airways. It is further reassuring that for the majority of PICRUSt2-inferred genes consistently up- or downregulated in COPD in 16S rRNA gene datasets, they also had the same direction of changes in the metagenomic data. These microbial genes with congruent disease associations were retained for metabolic inferences.
The multi-omic meta-analysis revealed a synergistic response between microbiome and host in COPD. This was demonstrated by an impressive proportion of overlap between microbiome and host signatures at the pathway level, as well as a concordance between the disease-modifying effects of metabolites and their predicted turnover in the microbiome. About 70% of the proposed host–microbiome interactions were further validated in the independent multi-omic cohort. Based on these findings, possible mechanistic routes of host–microbiome interactions that were co-perturbed in disease may be uncovered. In COPD, members of Proteobacteria, Actinobacteria, and Firmicutes are main contributors to the biosynthesis of palmitate, homocysteine, and urate that are postulated to have disease-promoting effects. Palmitate is a known pro-inflammatory agent associated with enhanced inflammation [51] and oxidative stress [52] by activating inflammation-related enzymes such as ACSL1 [53], and was reported to be increased in COPD airways in previous studies [13, 50] as well as in our cohort. Homocysteine, reported as elevated in COPD and related to its severity [54], may achieve its role through activating pro-inflammatory agent MMP9 via ERK1/2 pathway [55]. Urate could also have pro-inflammatory effects via activation of CCL2 and NLRP3 inflammasome [56]. On the other hand, metabolites such as butyrate, glutathione, aspartate, and glutamate, which tended to be enriched in Firmicutes and Bacteroidetes, could have disease-ameliorating effects via protecting SOD2-mediated oxidative stress [57], blunting CXCL1-induced neutrophilic recruitment [58], controlling NLRP3 inflammasome-mediated IL-1β signaling [59], and suppressing TLR4 and NOD signaling pathways [60, 61].
The main strength of this study is the development of a comprehensive statistical framework to identify host–microbiome interaction links leveraging the power of public omic datasets through meta-analysis, and to validate these links in the independent multi-omic data. We demonstrated that there were common microbiome and host signatures that can be revealed from public datasets when analyzed properly, and those signatures that showed coherent associations with disease across omics may contain biologically meaningful links that can provide insights into potential mechanisms of host–microbiome interaction. This is helpful for generating testable hypotheses in particular in areas such as COPD airway microbiome where paired multi-omics data remained scarce. More importantly, the meta-analysis framework is set to provide a quantitative context for host–microbiome interaction to facilitate biological interpretation and prioritization for the associations observed in the multi-omic data. This analysis is the first to integrate disease-centric public omics data at the host-microbiome interface and highlights the importance of making raw data and associated patient metadata available to enable more comprehensive meta-analysis.
It is important to recognize that the sequential inference from microbial taxa, genes, metabolites to host targets, as employed in our meta-analysis, came with inherent uncertainties. For example, the microbial taxa to genes inference can have uncertainties originated from a series of steps including 16S rRNA gene sequence placement, phylogenetic hidden state prediction, and gene content prediction, due to sequencing errors, incomplete nature of reference database, insufficient resolution of 16S rRNA gene sequence in species or strain-level discrimination and gene content divergence at these finer taxonomic levels. Assessing uncertainties in microbial gene-metabolite inference would then require an in-depth understanding of the expression of microbial genes in the ecosystems and their regulatory mechanisms (i.e. with meta-transcriptome data), the protein biosynthesis pattern (i.e. with meta-proteome data), and the enzyme activities in catalyzing processes of metabolite production. In light of these uncertainties, the multi-omic inference using meta-analysis can only be regarded as a hypothesis-generating tool that is subject to extensive validation using independent omics data before consideration for experimental exploitation. We note that 91.9% of metabolites inferred to interact with COPD-associated host genes were identified in our metabolomic data, possibly reflecting their central roles in host-microbiome interactions and disease pathophysiology. This suggests that utilizing host gene signature may increase the confidence of microbiome metabolic prediction.
There are some limitations to our study. First, we integrated microbiome and host transcriptome datasets across studies, to enhance statistical power and identify disease-associated signatures robust to inter-study variation. It is important to note that, while we tried to account for data heterogeneity, the microbiome data are not directly comparable across studies due to confounded effects of experimental procedures, amplification regions and sequencing platforms. As there is currently no optimal way to correct for inter-study batch effects for microbiome data [29, 62], we chose to limit our microbiome analysis to the genus-level according to previous studies of the same kind [15, 48], which effectively alleviated certain heterogeneity. In the statistical meta-analysis, instead of directly pooling raw datasets, we pooled summary statistics from each individual dataset into a random effect model, which is a more statistically conservative approach but effective in mitigating data heterogeneity [49]. Second, despite efforts to integrate multi-omic data in the public domain, the current study may still be under-powered, in particular due to the lack of COPD airway metagenomic and metabolomic datasets to generate highly robust disease signature. Therefore we considered the results of this study encouraging but preliminary in fully capturing the diversity and functionality of the airway microbiome especially given the heterogeneity nature of COPD. Third, in our analysis, only two 16S rRNA gene and one metagenomic studies with publicly available data had a case-control design, which somewhat limited the power of meta-analysis. The field of COPD airway microbiome has shifted from cross-sectional case-control profiling to longitudinal follow-up of microbiome during disease progression, to gain insights into microbiome’s temporal variability and possible cause–effect relationship with disease [63]. We argue that additional large-scale, case-control studies on COPD airway microbiome, preferably with a multi-omic focus, are still needed to boost power for a robust identification of disease-associated microbiome and host signatures. Last, our analysis was limited to the bacterial microbiome, yet mycobiome and virome are key members of airway microbial community whose roles are only beginning to be elucidated.
In summary, our multi-omic meta-analysis identified functional signatures of COPD airway microbiome and uncovered novel microbiome-metabolite–immunity interactions that could be implicated in COPD pathogenesis. Our analysis demonstrates the possibility of leveraging integrated meta-analysis on public multi-omic datasets to interrogate disease biology. Results in this study may provide hypotheses for future mechanistic studies toward understanding airway host–microbiome interactions in COPD.
Data availability
The processed public 16S rRNA gene, metagenomics, and host transcriptome data are available on Figshare (https://doi.org/10.6084/m9.figshare.12199436). The raw multi-omic data for the pilot cohort has been deposited in the Chinese National Gene Bank Nucleotide Sequence Archive (CNSA) under accession code CNP0000837.
Code availability
The key computer codes are on GitHub (https://github.com/wangzlab/COPD_metaanalysis/) or in the supplementary document.
References
Lopez AD, Shibuya K, Rao C, Mathers CD, Hansell AL, Held LS, et al. Chronic obstructive pulmonary disease: current burden and future projections. Eur Respir J. 2006;27:397–412.
Taraseviciene-Stewart L, Douglas IS, Nana-Sinkam PS, Lee JD, Tuder RM, Nicolls MR, et al. Is alveolar destruction and emphysema in chronic obstructive pulmonary disease an immune disease? Proc Am Thorac Soc. 2006;3:687–90.
Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS ONE. 2012;7:e47305.
Einarsson GG, Comer DM, McIlreavey L, Parkhill J, Ennis M, Tunney MM, et al. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax. 2016;71:795–803.
Huang YJ, Sethi S, Murphy T, Nariya S, Boushey HA, Lynch SV. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52:2813–23.
Wang Z, Bafadhel M, Haldar K, Spivak A, Mayhew D, Miller BE, et al. Lung microbiome dynamics in COPD exacerbations. Eur Respir J. 2016;47:1082–92.
Wang Z, Singh R, Miller BE, Tal-Singer R, Van Horn S, Tomsho L, et al. Sputum microbiome temporal variability and dysbiosis in chronic obstructive pulmonary disease exacerbations: an analysis of the COPDMAP study. Thorax. 2018;73:331–8.
Budden KF, Shukla SD, Rehman SF, Bowerman KL, Keely S, Hugenholtz P, et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir Med. 2019;7:907–20.
Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet. 2014;384:691–702.
Cui L, Morris A, Huang L, Beck JM, Twigg HL 3rd, von Mutius E, et al. The microbiome and the lung. Ann Am Thorac Soc. 2014;11:S227–232.
Castaner O, Goday A, Park YM, Lee SH, Magkos F, Shiow STE, et al. The gut microbiome profile in obesity: a systematic review. Int J Endocrinol. 2018;2018:4095789.
Ren L, Zhang R, Rao J, Xiao Y, Zhang Z, Yang B, et al. Transcriptionally active lung microbiome and its association with bacterial biomass and host inflammatory status. mSystems. 2018;3:e00199–18. https://doi.org/10.1128/mSystems.00199-18.
Segal LN, Clemente JC, Tsay JC, Koralov SB, Keller BC, Wu BG, et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol. 2016;1:16031.
Segal LN, Clemente JC, Wu BG, Wikoff WR, Gao Z, Li Y, et al. Randomised, double-blind, placebo-controlled trial with azithromycin selects for anti-inflammatory microbial metabolites in the emphysematous lung. Thorax. 2017;72:13–22.
Duvallet C, Gibbons SM, Gurry T, Irizarry RA, Alm EJ. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun. 2017;8:1784.
Wirbel J, Pyl PT, Kartal E, Zych K, Kashani A, Milanese A, et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat Med. 2019;25:679–89.
Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat Med. 2019;25:667–78.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al-Ghalith GA, et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr. 2018;6:e27295v2.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–21.
Marcel M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12:902–3.
Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol. 2012;8:e1002358.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Wang X, Kang DD, Shen K, Song C, Lu S, Chang LC, et al. An R package suite for microarray meta-analysis in quality control, differentially expressed gene analysis and pathway enrichment detection. Bioinformatics. 2012;28:2534–6.
Marot G, Foulley JL, Mayer CD, Jaffrezic F. Moderated effect size and P-value combinations for microarray meta-analyses. Bioinformatics. 2009;25:2692–9.
Zhou G, Stevenson MM, Geary TG, Xia J. Comprehensive transcriptome meta-analysis to characterize host immune responses in helminth infections. PLoS Negl Trop Dis. 2016;10:e0004624.
Gibbons SM, Duvallet C, Alm EJ. Correcting for batch effects in case-control microbiome studies. PLoS Comput Biol. 2018;14:e1006102.
Frank E, Hall M, Witten I, The WEKA Workbench. Online appendix for “Data mining: practical machine learning tools and techniques”. 4th ed. San Francisco, CA: Morgan Kaufmann; 2016.
Caspi R, Foerster H, Fulcher CA, Hopkinson R, Ingraham J, Kaipa P, et al. MetaCyc: a multiorganism database of metabolic pathways and enzymes. Nucleic Acids Res. 2006;34:D511–516.
Szklarczyk D, Santos A, von Mering C, Jensen LJ, Bork P, Kuhn M. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016;44:D380–384.
Consortium GT. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–5.
Larsen PE, Collart FR, Field D, Meyer F, Keegan KP, Henry CS, et al. Predicted Relative Metabolomic Turnover (PRMT): determining metabolic turnover from a coastal marine metagenomic dataset. Micro Inf Exp. 2011;1:4.
Noecker C, Eng A, Srinivasan S, Theriot CM, Young VB, Jansson JK, et al. Metabolic model-based integration of microbiome taxonomic and metabolomic profiles elucidates mechanistic links between ecological and metabolic variation. mSystems. 2016;1:e00013–15. https://doi.org/10.1128/mSystems.00013-15.
Wang Z, Arat S, Magid-Slav M, Brown JR. Meta-analysis of human gene expression in response to Mycobacterium tuberculosis infection reveals potential therapeutic targets. BMC Syst Biol. 2018;12:3.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Liao Y, Smyth GK, Shi W. The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Bafadhel M, McCormick M, Saha S, McKenna S, Shelley M, Hargadon B, et al. Profiling of sputum inflammatory mediators in asthma and chronic obstructive pulmonary disease. Respiration. 2012;83:36–44.
Dunn WB, Broadhurst D, Begley P, Zelena E, Francis-McIntyre S, Anderson N, et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc. 2011;6:1060–83.
Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vazquez-Fresno R, et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018;46:D608–D617.
Guijas C, Montenegro-Burke JR, Domingo-Almenara X, Palermo A, Warth B, Hermann G, et al. METLIN: a technology platform for identifying knowns and unknowns. Anal Chem. 2018;90:3156–64.
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34.
Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, et al. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018;46:W486–94.
Noecker C, Chiu HC, McNally CP, Borenstein E. Defining and evaluating microbial contributions to metabolite variation in microbiome-metabolome association studies. mSystems. 2019;4:e00579–19. https://doi.org/10.1128/mSystems.00579-19.
Sze MA, Schloss PD. Looking for a signal in the noise: revisiting obesity and the microbiome. mBio. 2016;7:e01018–16. https://doi.org/10.1128/mBio.01018-16.
Ramasamy A, Mondry A, Holmes CC, Altman DG. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 2008;5:e184.
Michaeloudes C, Kuo CH, Haji G, Finch DK, Halayko AJ, Kirkham P, et al. Metabolic re-patterning in COPD airway smooth muscle cells. Eur Respir J. 2017;50:1700202. https://doi.org/10.1183/13993003.00202-2017.
Zhou BR, Zhang JA, Zhang Q, Permatasari F, Xu Y, Wu D, et al. Palmitic acid induces production of proinflammatory cytokines interleukin-6, interleukin-1beta, and tumor necrosis factor-alpha via a NF-kappaB-dependent mechanism in HaCaT keratinocytes. Mediators Inflamm. 2013;2013:530429.
Sato Y, Fujimoto S, Mukai E, Sato H, Tahara Y, Ogura K, et al. Palmitate induces reactive oxygen species production and beta-cell dysfunction by activating nicotinamide adenine dinucleotide phosphate oxidase through Src signaling. J Diabetes Investig. 2014;5:19–26.
Kanter JE, Kramer F, Barnhart S, Averill MM, Vivekanandan-Giri A, Vickery T, et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc Natl Acad Sci USA. 2012;109:E715–724.
Seemungal TA, Lun JC, Davis G, Neblett C, Chinyepi N, Dookhan C, et al. Plasma homocysteine is elevated in COPD patients and is related to COPD severity. Int J Chron Obstruct Pulmon Dis. 2007;2:313–21.
Moshal KS, Sen U, Tyagi N, Henderson B, Steed M, Ovechkin AV, et al. Regulation of homocysteine-induced MMP-9 by ERK1/2 pathway. Am J Physiol Cell Physiol. 2006;290:C883–891.
Crane JK, Mongiardo KM. Pro-inflammatory effects of uric acid in the gastrointestinal tract. Immunol Investig. 2014;43:255–66.
Biljak VR, Rumora L, Cepelak I, Pancirov D, Popovic-Grle S, Soric J, et al. Glutathione cycle in stable chronic obstructive pulmonary disease. Cell Biochem Funct. 2010;28:448–53.
Trompette A, Gollwitzer ES, Pattaroni C, Lopez-Mejia IC, Riva E, Pernot J, et al. Dietary fiber confers protection against flu by shaping Ly6c(-) patrolling monocyte hematopoiesis and CD8(+) T cell metabolism. Immunity. 2018;48:992–1005.e1008.
Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013;23:201–12.
Wang H, Liu Y, Shi H, Wang X, Zhu H, Pi D, et al. Aspartate attenuates intestinal injury and inhibits TLR4 and NODs/NF-kappaB and p38 signaling in weaned pigs after LPS challenge. Eur J Nutr. 2017;56:1433–43.
Qin Q, Xu X, Wang X, Wu H, Zhu H, Hou Y, et al. Glutamate alleviates intestinal injury, maintains mTOR and suppresses TLR4 and NOD signaling pathways in weanling pigs challenged with lipopolysaccharide. Sci Rep. 2018;8:15124.
Wang Y, LeCao KA. Managing batch effects in microbiome data. Brief Bioinform. 2019. https://doi.org/10.1093/bib/bbz105.
Ditz B, Christenson S, Rossen J, Brightling C, Kerstjens HAM, van den Berge M, et al. Sputum microbiome profiling in COPD: beyond singular pathogen detection. Thorax. 2020;75:338–44.
Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFC1310600) funded to RC, and the National Natural Science Foundation of China (31970112) and the Science and Technology Foundation of Guangdong Province (2019A1515011395) funded to ZW. Funders had no role in study design, collection, analysis and interpretation of data, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
ZW conceived and designed the study. ZW, ZY and HL acquired data, developed data analysis workflows and interpreted the data. YY, ZL, FW collected clinical samples. BEM, RTS, MRS, CEB provided clinical insights to data interpretation. XY and JL assisted in the statistical analysis. ZW drafted the article. BEM, RTS, MRS, HZ, CEB, JRB, MW, WS, and RC provided critical revisions to the article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
BEM, RTS and JRB were employees and shareholders in GlaxoSmithKline PLC at the time of this study. Other authors have no conflict of interest to declare.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, Z., Yang, Y., Yan, Z. et al. Multi-omic meta-analysis identifies functional signatures of airway microbiome in chronic obstructive pulmonary disease. ISME J 14, 2748–2765 (2020). https://doi.org/10.1038/s41396-020-0727-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0727-y
This article is cited by
-
Bacterial interactome disturbance in chronic obstructive pulmonary disease clinical stability and exacerbations
Respiratory Research (2024)
-
Lung microbiome: new insights into the pathogenesis of respiratory diseases
Signal Transduction and Targeted Therapy (2024)
-
Butyrate inhibits iILC2-mediated lung inflammation via lung-gut axis in chronic obstructive pulmonary disease (COPD)
BMC Pulmonary Medicine (2023)
-
The airway microbiome mediates the interaction between environmental exposure and respiratory health in humans
Nature Medicine (2023)
-
Lung microbiome and cytokine profiles in different disease states of COPD: a cohort study
Scientific Reports (2023)
