
Abstract
Under the same selection pressures, two genetically divergent populations may evolve in parallel toward the same adaptive solutions. Here, we hypothesized that magnetotaxis (i.e., magnetically guided chemotaxis) represents a key adaptation to micro-oxic habitats in aquatic sediments and that its parallel evolution homogenized the phenotypes of two evolutionary divergent clusters of freshwater spirilla. All magnetotactic bacteria affiliated to the Magnetospirillum genus (Alphaproteobacteria class) biomineralize the same magnetic particle chains and share highly similar physiological and ultrastructural features. We looked for the processes that could have contributed at shaping such an evolutionary pattern by reconciling species and gene trees using newly sequenced genomes of Magnetospirillum related bacteria. We showed that repeated horizontal gene transfers and homologous recombination of entire operons contributed to the parallel evolution of magnetotaxis. We propose that such processes could represent a more parsimonious and rapid solution for adaptation compared with independent and repeated de novo mutations, especially in the case of traits as complex as magnetotaxis involving tens of interacting proteins. Besides strengthening the idea about the importance of such a function in micro-oxic habitats, these results reinforce previous observations in experimental evolution suggesting that gene flow could alleviate clonal interference and speed up adaptation under some circumstances.
Similar content being viewed by others


Introduction
Magnetotaxis couples the biomineralization of ferrimagnetic nanoparticles in organelles called magnetosomes, to a complex system of aero-chemotaxis for guiding the locomotion of magnetotactic bacteria (MTB) parallel to the Earth’s magnetic field lines [1]. This function is particularly well described in a group of freshwater MTB of the Alphaproteobacteria class represented by the Magnetospirillum genus (MTBMag). Magnetotaxis is assumed to facilitate vertical navigation toward their optimal niches located at the oxic-anoxic transition zone [2]. Our knowledge of the genetic basis, ecology and biophysical processes involved in magnetite magnetosome biogenesis was built mainly from the study of two model strains, Magnetospirillum gryphiswaldense strain MSR-1 [3] and Magnetospirillum magneticum strain AMB-1 [4,5,6]. Since their first observation, numerous other spirilla were isolated and affiliated to the same group based on phenotypic, physiological and morphological features, among which the ability to form magnetosomes [7,8,9,10,11]. As for many prokaryotes, the increasing biodiversity assessment and the development of molecular typing revealed the polyphyletism of magnetotaxis in freshwater Rhodospirillaceae (Alphaproteobacteria) [8, 12]. Members of two genera with different lifestyles: Phaeospirillum and Dechlorospirillum are actually more closely related to some Magnetospirillum lineages than what some Magnetospirillum lineages are to each other [7, 8, 12, 13]. The genus Phaeospirillum contains spiral-shaped, phototrophic, purple nonsulphur bacterial species [14], while Dechlorospirillum, now affiliated to the Magnetospirillum genus based on phylogenetic analyses, is represented by non-MTB only [15, 16].
The polyphyletism of magnetotaxis and the lack of a clear correlation between genetic clusters and ecology created some confusion in the classification of these organisms that remains partially resolved [17]. Today, we are still unable to state whether or not magnetotactic Magnetospirillum form their own genera [8, 18] because their specific ecological boundaries have not been identified yet and they do not form a monophyletic group [8, 12]. Current data support the existence of two genetically distinct groups represented by strains MSR-1 and AMB-1, respectively [8]. All magnetotactic strains affiliated to the Magnetospirillum genus fix carbon dioxide through the Calvin-Benson-Bassham cycle with the presence of a form II ribulose-1,5-bisphosphate carboxylase/oxygenase gene in their genome [8, 19], and all seem to fix atmospheric nitrogen [20, 21]. Other shared features of described magnetotactic Magnetospirillum species relate to biomineralization. They form a single chain of biomineralized cuboctahedral crystals of magnetite, have a bipolarly flagellated helical shape and a respiratory form of metabolism that uses organic acids as a source of carbon and electrons [3]. Moreover, the magnetite synthesis occurs only at very low levels of oxygen or under anaerobic conditions when nitrate is the alternative terminal electron acceptor to oxygen [22,23,24,25]. Their metabolic specificities are less obvious to pinpoint, maybe because some traits are variable within species or because they were insufficiently characterized. It seems that their requirement for energy source and carbon source may vary according to the strain. For example, while most species are facultative anaerobes that utilize nitrate as an alternative terminal electron acceptor to oxygen, M. magnetotacticum is an obligate microaerophile that requires oxygen even when growing with nitrate [22] and M. gryphiswaldense can grow autotrophically using reduced sulfur compounds as electron donor [26].
This high degree of phenotypic homogeneity of magnetic spirilla suggests an adaptation to similar environments that exert important constraints on their metabolism and magnetotaxis. From an evolutionary perspective, the common genetic determinism to all MTB indicates a single emergence of magnetotaxis in prokaryotes. Magnetosome biogenesis is encoded by unique genes (referred as the mam and/or mms genes) that are, for most of them, clustered into operons within a specific genomic region [27,28,29]. Compiling studies show both evidence of vertical inheritance of these genes [12, 30, 31] and multiple acquisitions by horizontal gene transfer (HGT) in some lineages in MTB of the Alphaproteobacteria class [32,33,34] with duplication events [34, 35]. These observations raise questions on the evolutionary mechanisms shaping the adaptation to this ecological niche and the phenotypic maintenance at such level of phylogenetic distance between two evolutionarily distinct lineages.
Selection contributes to divergent evolution and different ecological speciation in the presence of reproductive isolation [36]. However, similar environments in different locations may promote parallel or convergent evolution of traits and maintain a high degree of phenotypic similarity in independent and divergent populations [37]. Tracking the genetic parallelism involved in parallel ecological speciation may help to better understand the mechanisms of adaptive evolution. Because similar niches may exert the same degree of evolutionary constraint on traits, the same de novo beneficial mutations are likely to be repeatedly selected and fixed in populations [37,38,39]. In microorganisms, other evolutionary forces than mutation, like homologous recombination or orthologous replacement mediated by HGT generate genetic variation [40,41,42]. In prokaryotes, the contribution of these mechanisms in parallel evolution specifically has been less documented [43], although HGT and homologous recombination could promote the repeated exchange of beneficial alleles in genetically distant organisms, purge the genic variability to maintain phenotypic and ecological cohesiveness in an ecotype [44]. In laboratory-controlled experiments, recombination has been shown to alleviate clonal interference and accelerate adaptation of Escherichia coli populations under some circumstances [45]. In natura, two divergent populations inhabiting the same habitat are more likely to exhibit weaker barrier to gene flow than do different ecotypes, regardless the genetic distance [42, 46]. Our understanding of these processes leading to ecological differentiation in bacteria suffers from the lack of niches and habitats studied. Indeed, our vision has been built mainly from studies on pathogens [44]. But a broader investigation of environmental niches and ecotypes may help to map bacterial diversity onto ecology involving other traits than those related to cell surface, DNA binding and pathogenicity-related functions [47].
Here, we investigated the evolutionary mechanisms involved in the parallel evolution of magnetotaxis in two distinct groups of magnetotactic Magnetospirillum species. We particularly tested the hypothesis that recombination sensus lato could be involved in the maintenance of highly homogenous phenotypes in two divergent lineages keeping them adapted to very similar niches. We studied the biology and genome of newly isolated freshwater magnetotactic and nonmagnetotactic Magnetospirillum related species. By comparing gene contents and inferring phylogenies based on whole genomes and magnetosomes associated genes, we showed that ancestors of these groups exchanged their magnetosome gene clusters (MGCs) several times over the course of magnetotactic Magnetospirillum diversification, which might have been at the origin of the maintenance of a high degree of global genomic conservation and phenotypic similarity. Our results showed that repeated gene exchanges of entire operons involved in sensing and motility may contribute to the parallel evolution of species facing similar environmental constraints, which likely accelerate their adaptation to microoxic conditions.
Materials and methods
Genomes collection and growth of bacteria
Draft and closed genomes of all Rhodospirillaceae available in October 2017 were downloaded from the public repository database at NCBI and uploaded into the MicroScope platform for their automatic and manual annotation [48]. Sequencing and assembling of Magnetospirillum magneticum AMB-1 [49], M. gryphiswaldense MSR-1 [50], M. magnetotacticum MS-1 [51], M. caucaseum SO-1 [52], M. marisnigri SP-1, M. aberrantis SpK [53], M. bellicus VDY [54], Magnetospirillum sp. XM-1 [55], Magnetospirillum sp. ME-1 [56], P. fulvum MGU-K5 [57], and P. molischianum DSM120 [58] were described previously. Draft genome assemblies of nonmagnetotactic Magnetospirillum strains CP2, CP3, VDY, and WD, and those of Phaeospirillum strains DSM 114, DSM 115, DSM 117, and DSM 13234 were provided by the Joint Genome Institute (www.jgi.doe.gov) and described in supplementary Table S1. This list was completed with five genomes sequenced in this study. Genomes of magnetotactic strains LM-1, SS-4, UT-4, LM-5, and CP-1 were sequenced and assembled following the exact procedure described in [34] based on genomic DNA obtained from axenic cultures. Their sampling sites, the isolation procedure and culture medium were described in [8], with the exception of strain CP-1 reported here for the first time. Strain CP-1 was isolated using a similar procedure as previously described [8], from the freshwater river Couze Pavin, Auvergne, France (45°30′35.0″N, 2°54′12.6″E). Chemolithoautotrophic growth experiments were carried out on strains AMB-1, SS-4, CP-1, MSR-1, UT-4, LM-1, and LM-5 using a similar semisolid growth medium as previously described [8] but removing sodium acetate and sodium sulfide and adding after autoclave 3 ml of 40% sodium thiosulfate (Na2S2O3 · 5H2O). Thiosulfate was deemed positive for the support of growth if three conditions were met: (i) a band of cells formed in the tube, (ii) the band after growth was significantly thicker than the control containing no thiosulfate and (iii) cells continued to grow in the same medium in three successive transfers. The draft genome sequences partially annotated of LM-1, SS-4, UT-4, LM-5, and CP-1 were submitted to the European Nucleotide Archive and carry the accession numbers PRJEB35448, PRJEB35447, PRJEB35446, PRJEB35445, and PRJEB35444, respectively.
Light and electron microscopy
Motility and magnetotactic behavior of the different Magnetospirillum strains were analysed and recorded under the light microscope Leica LMD6000 equipped with the camera Leica DMC 4500. Transmission electron microscopes (TEM) was used to analyse the ultrastructure of Magnetospirillum strains directly deposited onto TEM copper grids coated with a carbon film. Magnetosomes shape and organization were visualized with a Tecnai G2 BioTWIN (FEI Company, Eindhoven, Netherlands) equipped with a CCD camera (Megaview III, Olympus Soft imaging Solutions GmbH, Münster, Germany) with an accelerating voltage of 100 kV.
Comparative genomics
Evolutionary relationships between Magnetospirillum-related species and their closest members of the Rhodospirillaceae family were investigated using whole genome sequences. Clusters of orthologous were defined using the OrthoMCL clustering algorithm implemented in GET_HOMOLOGUES open-source software [59]. We considered any cluster of orthologous proteins for which pairwise BLASTP alignments had an expectation value E < 10−5 and a minimal protein coverage of 50%.
Genomes and encoded proteomes of Magnetospirillum species and their closest relatives were compared with several complementary algorithms to define sub-groups/species based on their nucleotidic/amino acid composition: (i) Average nucleotide identity (ANI) and average amino acid identity (AAI) values were calculated using the ANI/AAI-Matrix online service (http://enve-omics.ce.gatech.edu/g-matrix/) [60] using reciprocal best hits (http://enve-omics.ce.gatech.edu/), (ii) digital DNA-DNA hybridization (dDDH) values were determined using the Genome-to-Genome Distance Calculator (2.1; https://ggdc-test.dsmz.de/ggdc.php) using the recommended BLAST + alignment and formula 2 (identities/HSP length) [61] and (iii), percentages of conserved proteins (POCP) [62] were calculated using the script runPOCP.sh [63].
Metabolic pathways were predicted with the microscope platform [64] using the PathoLogic algorithm [65], which computes an initial set of pathways by comparing genome annotations to a collection of microbial Pathway/Genome Databases MicroCyc derived from the metabolic reference database MetaCyc [66]. For each MicroCyc pathway, the tool gives the completion value referring to the number of reactions in a given strain/total number of reactions in the same pathway defined in the MetaCyc database.
The presence of conjugative system was checked in all genomes using MacSyFinder software and the TXSScan module [67, 68], which uses HMM profiles for efficient genomic detection of bacterial secretion systems encountered in diderm-LPS bacteria, and their discrimination from homologous machineries.
Statistical analyses
Statistical analyses were performed in R version 3.5.1 [69]. Averages of AAI and POCP values estimated from the pairwise comparison of genomes sets were compared either by the Student’s t test or by the nonparametric Mann–Whitney U test (if the underlying assumptions of the first test were not satisfied). Differences were considered significant when P values were below 0.05.
A principal component analysis (PCA) was performed using the ade4 package [70] to compare metabolic pathways of the 23 Magnetospirillum relatives using a reduced matrix of MicroCyc pathway completion values generated with the MicroScope platform. The reduced matrix consisted of the 99 pathways whose completion was variable between groups among the 410 nonredundant pathways detected in at least one genome.
Phylogenetic analyses
For phylogenetic analyses, only clusters with single-copy sequences were taken into consideration and in-paralogs were excluded. All complete gene outliers were removed with Phylo-MCOA software [71]. For the species tree,7 amino acids (AA) sequences of 839 orthologous proteins were aligned independently using MAFFT [72] and alignments were trimmed using BMGE [73] setting block length to 3, and concatenated into a final alignment of 269,968 AA positions among which 151,377 were polymorphic. The maximum likelihood tree was built from the concatenated sequences with IQ-TREE [74] using a partition model; the substitution model for each protein was selected by ModelFinder [75] with the Bayesian Information Criterion. The statistical support of the branches was estimated by the ultrafast bootstrapping approach implemented in IQ-TREE applying 1000 replicates. A previous study suggesting that strain LM-1 was a new freshwater MTBMag genus [8], we included this strain in the external group for phylogenetics and comparative genomics.
Evolution of genes encoding for proteins involved in magnetosome biogenesis was studied by reconciling binary gene trees built from gene-by-gene alignments of mam genes or their concatenation with the nonbinary species tree under the duplication-transfer-loss (DTL) parsimony algorithm implemented in Notung 2.9 software [76]. The algorithm captures gene duplication (D), transfer (T) and loss (L) driving tree incongruence and infers all optimal solutions to finally report the complete and temporally feasible event histories giving the data.
Results
Species of magnetotactic Magnetospirillum do not form a monophyletic group at the whole genome level
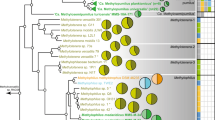
We built a phylogenetic framework to study the evolution of magnetotaxis in MTBMag at the whole genome scale using draft genomes of freshwater magnetotactic and non-MTB close to Magnetospirillum gryphiswaldense MSR-1 or M. magneticum AMB-1 (Table S1). A first phylogenetic tree based on all proteins shared by strains representing the Rhodospirillaceae diversity and related other Alphaproteobacteria confirmed that all 23 Magnetospirillum relatives in this study cluster together at the genome level and positioned strain LM-1 as the closest freshwater strain of this group (Fig. S1). Based on this phylogeny and what we currently know about the biology of strain LM-1 [8], this strain represents a new genus. Whereas Magnetospirillum species exhibit a spirillum shape with flagella at both poles and synthesize cuboctahedral crystals, strain LM-1 displays a vibrio shape with a single polar flagellum and synthesizes elongated prismatic crystals resembling those of Magnetovibrio blakemorei strain MV-1 [8]. Despite these morphological similarities between MV-1 and LM-1, the proposed LM-1 genus is much closer to freshwater Rhodospirillaceae MTB than to marine Rhodospirillaceae MTB. Thus, we propose the name Candidatus Magneticavibrio boulderlitore (the magnetic vibrio isolated from the shore of boulder beach in Lake Mead) [8]. We further used strain LM-1 as a control and external outgroup to Magnetospirillum related taxa in this study.
The phylogeny of Magnetospirillum related species only was then reconstructed based on 839 orthologous proteins selected after all complete gene outliers were removed [71]. The tree topology was partially congruent with those of the trees built from 16S rRNA sequences of some strains in previous studies [8, 17]. For example here, the strain UT-4 clusters with the MSR-1 clade (Fig. 1a), while it was supposed to be ancestral to all MTBMag. The strain CP-1, isolated here from the Couze Pavin River, France, clusters with the ancestor of all AMB-1 related strains. Together, all Magnetospirillum related species form four monophyletic groups (namely SpK, MSR-1, MGU-K5, and AMB-1 clades), among which two: MSR-1 and AMB-1, are composed of MTB only. The nonmagnetotactic MGU-K5 clade (Phaeospirillum species) shares a more recent ancestor with the AMB-1 clade composed of strains ME-1, XM-1, SO-1, AMB-1, SS-4, SP-1, and CP-1. The SpK clade (formerly Dechlorospirillum including M. aberrantis) is composed of non-MTB only (CP2, CP3, VDY, WD, and SpK) and shares a more recent ancestor with the MSR-1 clade composed of magnetotactic strains LM-5, BB-1, MSR-1, and UT-4. These two polyphyletic groups regarding magnetotaxis relate to each other with a direct common ancestor.

a Phylogeny of Magnetospirillum species rooted with strains representing other Rhodospirillaceae species (gray). The Maximum-likelihood tree was drawn to scale and branches length represents the number of base substitutions per site. Black circles represent nodes supported by 100% of the replicates. Names in bold represent MTB strains. Strains related by branches with the same color represented different clades distinguished here, and could be considered as different genera according to the average amino-acid identity (AAI) (Fig. S2) and their lifestyle. b Histograms drawn to scale showing the number of orthologous proteins specific to a group of bacteria that are shared by all members of this group (black bars represent 100 proteins). The first, second and third histograms give the number of proteins specific to (1) each clade, (2) the MSR-1/SpK group vs. AMB-1/Phaeospirillum group, and (3) MTB groups vs. non-MTB, respectively. c Pairwise comparison of percentages of conserved proteins (POCP) (Fig. S3) and average amino-acid identities (AAI) between the strains of the four groups ordered according to the phylogenetic tree presented in panel a. Circles and colors are proportional to the values that represent averages and standard errors calculated from the pairwise comparisons within each group. All the values are significantly different from each other (Student t test, n = 23, P < 0.05). d Principal component analysis of the metabolic networks of strains predicted with the MicroCyc resource implemented in MicroScope with the projection of ellipses and gravity centers of classes representing the phylogenetic groups.
Lifestyle and environment maintain genetic relatedness and phenotypic homogeneity in two divergent magnetotactic lineages
The intriguing polyphyletism of magnetotaxis in MTBMag and the high degree of phenotypic divergence of species of the MGU-K5 and SpK clades raised questions about the definition of Magnetospirillum as a genus. By calculating several indexes that are generally used to refine taxonomic boundaries for prokaryotes like the AAI, ANI, dDDH or POCP, we investigated the support of their classification at the genome level (Fig. S2–S5). The comparison of genomes pairwise within and between monophyletic groups confirmed the existence of four clades for which AAI values ranged from 64 to 70% (Fig. 1c). These values justify a new genus description for each group according to previous large metadata analyses assuming a significant ecological differentiation [60]. Here, it was interesting to observe higher POCP values between all magnetotactic species than between these species and their genetically closer nonmagnetotactic species, which showed that MTB lifestyle fostered the maintenance of similar genome contents (Fig. 1c). On the basis of their phenotype, different magnetotactic species studied here and previously were difficult to distinguish using light and electron microscopy approaches [3, 4, 7,8,9,10,11, 26, 77]; they share all very similar magnetotactic behaviors, motilities, cell ultrastructures and magnetosome morphology/organization (Fig. S6). Experimental attempts to find a different physiology specific to one of the two groups failed too. According to the literature, chemolithoautophy with thiosulfate as electron donor was potentially the only difference between AMB-1 and MSR-1 [26].
We tested this hypothesis in silico by comparing whole genomes and noticed that some genes involved in the thiosulfate-oxidizing Sox enzyme system were specific to the MSR-1 clade (Table S2). Testing chemolithoautotrophy with thiosulfate as electron donor for strains of both clades, we showed that not all members of the MSR-1 group, such as strain LM-5, were able to use thiosulfate as energy source, while none of the cultured species tested of the AMB-1 group grow in such conditions. No other clear interpretation could be made about the biological differences between magnetotactic Magnetospirillum lineages. We thus looked for other genomic evidences of clear biological boundaries by reconstructing the metabolic pathways. PCA built from the matrix of pathway complementation (Fig. 1d, Table S3), confirmed that Phaeospirillum species do not cluster with the other lineages and that AMB-1 clade is metabolically closer to that of MSR-1, than MGU-K5 and SpK clades. Searching pathways that participate the most to the variability for both PCA axis, we confirmed that MGU-K5 relatives use light-harvesting complexes with the impossibility to denitrify, degrade alanine or produce vitamin B12. Differences between the three other groups were much less significant and were associated, for example, to creatinine degradation, cyclopropane fatty acids, spermidine and preQ0 metabolite biosynthesis. In the absence of significant ecological differentiation for the nonphotosynthetic clades, there is no support for the creation of two genera for AMB-1 and MSR-1 clades.
Conservation of mam genes composition and synteny supports magnetotaxis was functionally constrained by similar selective pressures
Draft versions of the MGCs were reconstructed for the new genomes, and despite the incompleteness of some operons, several observations were validated. First, the feoABm, mms6, mamGFDC, mamAB, and mamXY gene clusters encoding proteins involved in magnetosome biogenesis were found in all genomes with a very high degree of synteny conservation within operons (Fig. 2). Most of the strains carry an additional mamJOE-like gene cluster downstream mamV and mamW. If present, this cluster of paralogs was followed by several orthologous proteins localized upstream mamXY operon, among which some are specific to MTBMag. For instance, a homolog of the ferric uptake regulation protein Fur is often present in the magnetosomes gene cluster and in M. gryphiswaldense MSR-1, a homolog that was previously shown to affect magnetite biomineralization, is present outside the MGC [78]. With the exception of LM-5, whose MGC organization is almost identical to that of the AMB-1 cluster, strains of the MSR-1 group harbor a slightly different structure. The apparent global conservation of the MGC in Magnetospirillum spp. could support their orthology in freshwater MTBMag. However, we found several genomic features, typical of mobile regions and genomic islands that could be responsible for genomic instability, recombination and HGT. Transposases, integrases and recombinases (e.g., homologs to y4qJ, members of the IS110/IS3/IS407 family) were found in the vicinity or within the MGCs of several strains (e.g., AMB-1, ME-1, UT-4, MSR-1), some of which are phage-related or even involved in a functional conjugation system (e.g., virB6, virB8, virB9 of Type 4 Secretion System) [79, 80]. It should be noted the presence of two copies of mamJ in the mamAB operon of strain LM-1. Although the percentage of similarity of its encoded protein is low compared with MamJ copies of members of the Magnetospirillum genus, it shows that this gene is not restricted to the magnetotactic spirilla from freshwater. In strains MSR-1 and AMB-1, MamJ was shown to interact with MamK filament and thus involved in magnetosome chain formation [81, 82].

Sequences are organized according to species phylogeny. Homologous genes are symbolized by the same letters and their relative 5′-3′ orientation compared with each other’s is given by the bottom line color (red or black). A vertical blue dotted line between two genes symbolizes a truncation (contig edge) while a grey slash denotes that two genes are related through a continuous genomic region of one or more genes. Genes are next to each other if they are not separated by a slash. The additional paralogous mam genes (the mamJOE-like operon or a duplicated genes set of mms6, mmsF, mag1, and mag3 for example) are associated with a quotation mark. Paralogous mam genes were found on very short contigs outside the MGC in CP-1 and were not shown on this figure (mamOBQ and feoABm for example). Genes h1 to h4, m1 to m3, and furm represent unknown homologous genes conserved in many MGCs, the mag genes previously described [34] and a homolog of the ferric uptake regulator fur specific to the MGC, respectively. The accession number of these genes in the reference genome AMB-1 are BAE49759, BAE49764, BAE49812, BAE49814, BAE49823, BAE49824, BAE49825, and BAE4981, respectively.
Operons involved in magnetotaxis were acquired and lost repeatedly in the course of the Magnetospirillum evolution
The evolutionary history of magnetotaxis was investigated by reconstructing phylogenies based on known magnetosome-associated proteins described in MSR-1 and AMB-1 genomes that are shared by all strains used in this study. We checked the automatic annotation manually gene by gene for rare ambiguous cases. Gene orthology was then tested by comparing gene and species trees topologies. Trees reconstructed from each of the 31 mam genes/proteins conserved in all Magnetospirillum draft and closed genomes were consistent with each other within putative operons. However, some nodes were not always well statistically supported because some AA and DNA sequences were almost identical between genomes. Indeed, for numerous magnetosome-associated proteins, AA sequences identity percentage was up to 25% higher than the average AAI value. For example, MamH sequences were 100% identical between AMB-1, ME-1, and SP-1 while the AAI values of their genomes compared pairwise were between 75 and 87%. Unless a strong negative selection pressure purified polymorphism over Magnetospirillum speciation history, such result was congruent with allelic exchange. Trees topologies built from the concatenation of Mam proteins and genes were incongruent with that of the species evolutionary history (Fig. 3). They showed magnetotaxis has a common origin, but does not share the same ancestry in Magnetospirillum and has been regularly transferred between ancestors by homologous recombination and/or HGT events. The mms6/mamGFDC and mamAB operons had the same history (Fig. S7) so data were combined to build one tree (Fig. 3b). This history was different for the mamXY operon (including here the mag genes) (Fig. 3c). The feoAB evolution could not be totally resolved because the polymorphism was too low and the degree of site saturation too high (Fig. 3d). From these trees, the most obvious incongruences between the species tree and magnetotaxis trees were for the relationships between strains LM-5 and CP-1 with the rest of the lineages, even if conflicting signals were globally observed for all strains. Indeed, we confirmed at the whole genome level that although strain LM-5 belongs to the MSR-1 group, its magnetosome genes were most closely related to species of the AMB-1 group [8]. For strain CP-1, isolated in this study, it is the opposite; CP-1 belongs to the AMB-1 group but its MGC shared a more recent ancestry with bacteria of the MSR-1 group.

a Species tree from Fig. 1a. Gene trees built from the concatenation of Mam proteins present in b mms6, mamGFDC and mamAB operons, c the mamXY operon (including here the mag1, 2, and 3 genes) and d the feoABm operon. Strains names and external branches are colored according to the subgroups of the first panel to facilitate the visualization of relationships in gene trees. Gene trees were rooted with the other magnetotactic Alphaproteobacteria of the Fig. 1a including strain LM-1.
To go further, we inferred the most parsimonious scenarios explaining such evolutionary histories of the magnetosome genes by reconciling these gene trees and the species tree under the DTL model implemented in Notung 2.9 software [83] (Fig. 4). Under such a model, the ancestor of the four monophyletic groups of magnetotactic and nonmagnetotactic species had a magnetotactic ancestor. Magnetotaxis was then lost in the SpK and MGU-K5 groups (Figs. 4 and S8). The MGCs of the UT-4 and CP-1 strains represent the most ancestral forms of the MGC in the MSR-1 group and AMB-1 group, respectively. However, an ancient HGT event replaced the whole MGC of the most recent MSR-1 group species by an ancestral form that has emerged anciently first in the AMB-1 clade. This event was followed by other similar events of orthologs replacement during the Magnetospirillum diversification within and between groups. Importantly, several paralogs of the mamQ, mamR, and mamB genes seem to have been horizontally transferred together with the mamAB operon during the AMB-1 clade diversification. These genes are identical or nearly identical in the closest lineages of the AMB-1 clade and have likely emerged recently. Further analyses using a larger dataset will be needed to test the evolutionary relationships of these paralogs with the mamJOE-like gene cluster. This later likely emerged from duplication of mamJOE in the ancestor of all Magnetospirillum spp. of AMB-1 group (Fig. 4). The mamQRB paralogs present in AMB-1 closest relatives seem to have emerged from a different and more recent duplication event than that linking the mamMOBQ paralogs in CP-1, SP-1, and SS-4.

Events of gene duplication (D), loss (L) and transfer (T) were inferred using the duplication-transfer-loss (DTL) model of Notung 2.9 software [76] by reconciling the tree based on the concatenation of shared mam AA sequences (thin cladogram, Fig. 3) with the species tree (thick grey cladogram, Fig. 1). Colors symbolize divergent clades or lineages of the species tree. The color of the internal branches shows the ancestry of the genes encoding for magnetosome-associated proteins. For horizontal gene transfer events, the name of the donor lineage associated is given. The black solid lines represent the vertical inheritance of the genes while the black dotted lines represent their loss. D* denotes a putative duplication event that could have occurred in the ancestor of AMB-1 clade and led to the emergence of both ancestors of the mamJOE-like cluster.
Discussion
Determining the evolutionary forces shaping genomic polymorphism allows to identify key features involved in microbial adaptation and to resolve their evolutionary histories. In prokaryotes, genetic parallelism was identified by the detection of repeated emergences of the same de novo beneficial mutations [37,38,39]. Here, we showed that multiple allelic exchanges of whole operons mediated the parallel evolution of magnetotaxis in two evolutionarily distinct groups of bacteria. We thus provide evidence that another force than mutation can participate in the maintenance of highly similar phenotypes within a group of phylogenetically distant prokaryotes sharing the same ecological niche. Parallel evolution of distantly related populations is favored when they experience similar selection pressures [37, 43]. Freshwater magnetotactic species cluster into two genomic genera for which it was difficult, even impossible, to identify biological and ecological boundaries. Although, these two species clades represented by AMB-1 and MSR-1 strains, could represent two different genera and carry on different names, we thus believe that the absence of these boundaries impedes the amending of the Magnetospirillum genus name of either the AMB-1 or MSR-1 groups.
Here, these results raise several questions about the role of magnetotaxis with microbial adaptation to habitats at the oxic-anoxic interfaces of freshwater sediments. A part of MTBMag similarity could not be associated to phylogenetic inertia solely, but more likely to adaptation of similar environmental constraints [84]. Did magnetotaxis trigger the ecological cohesiveness of these divergent lineages? Indeed, magnetotaxis is known to optimize bacterial motility along with redox and chemical gradients [2, 85]. Repeated acquisition of beneficial alleles could thus speed up microbial adaptation to microoxic settings by optimizing the sensing or facilitating magnetosome formation in specific conditions. Instead, it is also possible that these narrow and unstable niches selected few specialized catabolic reactions on which magnetosome biogenesis depends on [86,87,88,89]. In that case, magnetotaxis selection would arise secondarily from divergent populations already ecologically cohesive that exploit the same redox reactions and carbon sources. According to the theory, this cohesiveness arises when few adaptive solutions exist to face the environment, which in turn fosters the genetic parallelism or even convergence [37]. Gene flow and allelic exchange are known to allow the recipient bacteria with pre-existing adaptations to invade a new niche. However, in this specific situation, it can also improve its performance in its current niche [90]. Given our data, this second scenario is the most parsimonious explanation, but in any case, they both support that magnetotaxis is an important adaptation to oxic-anoxic interfaces.
For years, studies compiled evidences of the MGC transferability between MTB and non-MTB. Adaptive transfer of genes is limited to those that can be transferred as a functional unit, provide a niche-transcending adaptation, and are compatible with the architecture and physiology of other organisms [42]. To make the exchange possible, genes must fit on a chromosomal segment short enough to be transferred or they must fit on a mobile extrachromosomal element [41]. The MGC totally fits with all these requirements. First, the heterologous expression of mam operons in foreign species with compatible physiology led to the formation of magnetosomes [91]. Then, these genes are surrounded by mobile elements from type IV secretion systems involved in conjugation, transposases with a high degree of conservation and phage elements [29, 92]. Interestingly, numerous similar mobile elements are surrounding the magnetosome islet, observed so far in the genome of M. Magneticum AMB-1 only [93, 94]. This genomic region harbors few mam genes homologs, whose acquisition has been related to an HGT event from the Ca. Etaproteobacteria strain MC-1. However, this scenario is unlikely because of their homology with Magnetospirillum mam genes. Data rather support that the AMB-1 islet is the remnant of one or several independent ancient events (including duplication and/or HGT) that occurred in the early time of Magnetospirillum groups emergence. Currently, the lack of sequence conservation and the dramatic evolutionary acceleration of Mam homologs in the islet could result from a neofunctionalization and prevent to fully resolve their history in the light of the current data.
So far, the contribution of gene flow and allelic exchange to magnetotaxis evolution was formally evidenced for ancestors of closely related bacterial genera only [34]. The current evolutionary scenario proposes that these forces did not contribute to magnetotaxis evolution at larger taxonomic scales [30, 31]. However, this unexpected scenario relies on big assumptions, which led to other authors to question it and to state that HGT contributed in the early steps of phyla emergences [33]. Only new genomes of deep branching lineages and proper analyses will resolve MTB evolution that very likely involve mutation, recombination and duplication events. The parallel evolution of magnetotaxis observed in the Magnetospirillum clades may have occurred at larger taxonomic scales between MTB donors and nonmagnetotactic recipient lineages with compatible physiologies and sharing the same ecological niches. The successful transformation of Rhodospirillum rubrum [91] with the MSR-1 MGC showed that such event is possible. In most aquatic habitats, oxic-anoxic interface niche represents a tiny layer of ∼100 µm in thickness where microaerophilic microorganisms thrive due to the overlap of reduced and oxidized chemical species. This layer is a very coveted zone and thus a hot spot for microorganismal adaptation where adaptive solutions may be reduced [95].
Multiple genome sequencing projects revealed years after years the multi-faceted evolution of magnetotaxis with evidences of both vertical and horizontal inheritance [17, 31,32,33,34]. Despite the polyphyletism of magnetotaxis at the phylum level, some authors argued for a vertical inheritance of the MGC followed by multiple and independent losses in non-MTB clades. Here, we showed further evidence that genetic exchanges may explain this polyphyletism at the genus level. Because our sample was limited to few strains collected worldwide over a large time scale, we undoubtedly underestimated the number of events, including potential duplications [35]. Repeated de novo mutation might have also contributed in some extent to the parallel evolution of the function, but our approach was not suitable to identify such events. In the case of magnetotaxis, recombination between distantly related bacteria could represent a more parsimonious and rapid solution for adaptation compared with independent and repeated de novo mutations. The reason comes from the MGCs architecture: magnetosome biogenesis relies on a set of operons involving tens of interacting proteins that certainly coevolved to optimize the function [5]. Thus, any mutation optimizing the function of a single gene would not necessarily optimize the function itself and could even decrease it if other beneficial mutations do not occur at the same time. Selection acting at the operon level rather than on genes individually, homologous recombination of entire operons could rapidly purge the diversity and spread the beneficial mutations in the population. It is important to further test these hypotheses using MTBMag and experimental evolution approaches. Such study would definitively quantify the relative role of gene flow compared with mutation in speeding up adaptation when this later involves large operons of interacting genes and functions as complex as biomineralization.
References
Bazylinski DA, Frankel RB. Magnetosome formation in prokaryotes. Nat Rev Microbiol. 2004;2:217–30.
Klumpp S, Lefèvre CT, Bennet M, Faivre D. Swimming with magnets: from biological organisms to synthetic devices. Phys Rep. 2019;789:1–54.
Schleifer KH, Schüler D, Spring S, Weizenegger M, Amann R, Ludwig W, et al. The genus Magnetospirillum gen. nov. description of Magnetospirillum gryphiswaldense sp. nov. and transfer of Aquaspirillum magnetotacticum to Magnetospirillum magnetotacticum comb. nov. Syst Appl Microbiol. 1991;14:379–85.
Matsunaga T, Sakaguchi T, Tadakoro F. Magnetite formation by a magnetic bacterium capable of growing aerobically. Appl Microbiol Biotechnol. 1991;35:651–5.
Uebe R, Schüler D. Magnetosome biogenesis in magnetotactic bacteria. Nat Rev Microbiol. 2016;14:621–37.
Grant CR, Wan J, Komeili A. Organelle formation in bacteria and archaea. Annu Rev Cell Dev Biol. 2018;34:217–38.
Schüler D, Spring S, Bazylinski DA. Improved technique for the isolation of magnetotactic spirilla from a freshwater sediment and their phylogenetic characterization. Syst Appl Microbiol. 1999;22:466–71.
Lefèvre CT, Schmidt ML, Viloria N, Trubitsyn D, Schüler D, Bazylinski DA. Insight into the evolution of magnetotaxis in magnetospirillum spp., based on mam gene phylogeny. Appl Environ Microbiol. 2012;78:7238–48.
Wang Y, Lin W, Li J, Zhang T, Li Y, Tian J, et al. Characterizing and optimizing magnetosome production of Magnetospirillum sp. XM-1 isolated from Xi’an City Moat, China. FEMS Microbiol Lett. 2015;362:fnv167.
Dziuba M, Koziaeva V, Grouzdev D, Burganskaya E, Baslerov R, Kolganova T, et al. Magnetospirillum caucaseum sp. nov., Magnetospirillum marisnigri sp. nov. and Magnetospirillum moscoviense sp. nov., freshwater magnetotactic bacteria isolated from three distinct geographical locations in European Russia. Int J Syst Evol Microbiol. 2016;66:2069–77.
Ke L, Chen Y, Liu P, Liu S, Wu D, Yuan Y, et al. Characteristics and optimised fermentation of a novel magnetotactic bacterium, Magnetospirillum sp. ME-1. FEMS Microbiol Lett. 2018;365:14.
Wang Y, Casaburi G, Lin W, Li Y, Wang F, Pan Y. Genomic evidence of the illumination response mechanism and evolutionary history of magnetotactic bacteria within the Rhodospirillaceae family. BMC Genomics. 2019;20:407.
Lefèvre CT, Bazylinski DA. Ecology, diversity, and evolution of magnetotactic bacteria. Microbiol Mol Biol Rev. 2013;77:497–526.
Gonçalves RP, Bernadac A, Sturgis JN, Scheuring S. Architecture of the native photosynthetic apparatus of Phaeospirillum molischianum. J Struct Biol. 2005;152:221–8.
Thrash JC, Ahmadi S, Torok T, Coates JD. Magnetospirillum bellicus sp. nov., a novel dissimilatory perchlorate-reducing alphaproteobacterium isolated from a bioelectrical reactor. Appl Environ Microbiol. 2010;76:4730–7.
Gorlenko VM, Dziuba MV, Maleeva AN, Panteleeva AN, Kolganova TV, Kuznetsov BB. Magnetospirillum aberrantis sp. nov., a new freshwater bacterium with magnetic inclusions. Mikrobiologiia. 2011;80:679–90.
Lin W, Zhang W, Zhao X, Roberts AP, Paterson GA, Bazylinski DA, et al. Genomic expansion of magnetotactic bacteria reveals an early common origin of magnetotaxis with lineage-specific evolution. ISME J. 2018;1:1508–19.
Burgess JG, Kawaguchi R, Sakaguchi T, Thornhill RH, Matsunaga T. Evolutionary relationships among Magnetospirillum strains inferred from phylogenetic analysis of 16S rDNA sequences. J Bacteriol. 1993;175:6689–94.
Bazylinski DA, Dean AJ, Williams TJ, Long LK, Middleton SL, Dubbels BL. Chemolithoautotrophy in the marine, magnetotactic bacterial strains MV-1 and MV-2. Arch Microbiol. 2004;182:373–87.
Bazylinski DA, Blakemore RP. Nitrogen fixation (acetylene-reduction) in Aquaspirillum-magnetotacticum. Curr Microbiol. 1983;9:305–8.
Bazylinski DA, Dean AJ, Schüler D, Phillips EJ, Lovley DR. N2-dependent growth and nitrogenase activity in the metal-metabolizing bacteria, Geobacter and Magnetospirillum species. Environ Microbiol. 2000;2:266–73.
Bazylinski DA, Blakemore RP. Denitrification and assimilatory nitrate reduction in Aquaspirillum magnetotacticum. Appl Environ Microbiol. 1983;46:1118–24.
Blakemore RP, Short KA, Bazylinski DA, Rosenblatt C, Frankel RB. Microaerobic conditions are required for magnetite formation within Aquaspirillum magnetotacticum. Geomicrobiol J. 1985;4:53–71.
Schüler D, Baeuerlein E. Dynamics of iron uptake and Fe3O4 biomineralization during aerobic and microaerobic growth of Magnetospirillum gryphiswaldense. J Bacteriol. 1998;180:159–62.
Heyen U, Schüler D. Growth and magnetosome formation by microaerophilic Magnetospirillum strains in an oxygen-controlled fermentor. Appl Microbiol Biotechnol. 2003;61:536–44.
Geelhoed JS, Kleerebezem R, Sorokin DY, Stams AJM, van Loosdrecht MCM. Reduced inorganic sulfur oxidation supports autotrophic and mixotrophic growth of Magnetospirillum strain J10 and Magnetospirillum gryphiswaldense. Environ Microbiol. 2010;12:1031–40.
Grünberg K, Wawer C, Tebo BM, Schüler D. A large gene cluster encoding several magnetosome proteins is conserved in different species of magnetotactic bacteria. Appl Environ Microbiol. 2001;67:4573–82.
Grünberg K, Müller E-C, Otto A, Reszka R, Linder D, Kube M, et al. Biochemical and proteomic analysis of the magnetosome membrane in Magnetospirillum gryphiswaldense. Appl Environ Microbiol. 2004;70:1040–50.
Ullrich S, Kube M, Schübbe S, Reinhardt R, Schüler D. A hypervariable 130-kilobase genomic region of Magnetospirillum gryphiswaldense comprises a magnetosome island which undergoes frequent rearrangements during stationary growth. J Bacteriol. 2005;187:7176–84.
Lefèvre CT, Trubitsyn D, Abreu F, Kolinko S, Almeida LGP, Vasconcelos ATR, et al. Monophyletic origin of magnetotaxis and the first magnetosomes. Environ Microbiol. 2013;15:2267–74.
Lin W, Paterson GA, Zhu Q, Wang Y, Kopylova E, Li Y, et al. Origin of microbial biomineralization and magnetotaxis during the Archean. Proc Natl Acad Sci USA. 2017;114:2171–6.
Jogler C, Kube M, Schübbe S, Ullrich S, Teeling H, Bazylinski DA, et al. Comparative analysis of magnetosome gene clusters in magnetotactic bacteria provides further evidence for horizontal gene transfer. Environ Microbiol. 2009;11:1267–77.
Wang S, Chen Y. Origin of magnetotaxis: vertical inheritance or horizontal transfer? Proc Natl Acad Sci. 2017;114:E5016–8.
Monteil CL, Perrière G, Menguy N, Ginet N, Alonso B, Waisbord N, et al. Genomic study of a novel magnetotactic Alphaproteobacteria uncovers the multiple ancestry of magnetotaxis. Environ Microbiol. 2018;20:4415–30.
Du H, Zhang W, Zhang W, Zhang W, Pan H, Pan Y, et al. Magnetosome Gene Duplication as an Important Driver in the Evolution of Magnetotaxis in the Alphaproteobacteria. mSystems. 2019;4:e00315–9.
Rundle HD, Nosil P. Ecological speciation. Ecol Lett. 2005;8:336–52.
Conte GL, Arnegard ME, Peichel CL, Schluter D. The probability of genetic parallelism and convergence in natural populations. Proc R Soc B Biol Sci. 2012;279:5039–47.
Orr HA. The probability of parallel evolution. Evolution. 2005;59:216–20.
Gompel N, Prud’homme B. The causes of repeated genetic evolution. Dev Biol. 2009;332:36–47.
Maynard Smith J, Smith NH, O’Rourke M, Spratt BG. How clonal are bacteria? Proc Natl Acad Sci USA. 1993;90:4384–8.
Lawrence JG, Retchless AC. The interplay of homologous recombination and horizontal gene transfer in bacterial speciation. In: Gogarten MB, Gogarten JP, Olendzenski LC (eds.). Horizontal gene transfer: Genomes in Flux. 2009. Humana Press, Totowa, NJ, pp 29–53.
Wiedenbeck J, Cohan FM. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol Rev. 2011;35:957–76.
Bailey SF, Blanquart F, Bataillon T, Kassen R. What drives parallel evolution?: How population size and mutational variation contribute to repeated evolution. BioEssays N. Rev Mol Cell Dev Biol. 2017;39:1–9.
Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP. The bacterial species challenge: making sense of genetic and ecological diversity. Science. 2009;323:741–6.
Barrick JE, Lenski RE. Genome dynamics during experimental evolution. Nat Rev Genet. 2013;14:827–39.
Cohan FM. Bacterial species and speciation. Syst Biol. 2001;50:513–24.
Nakamura Y, Itoh T, Matsuda H, Gojobori T. Biased biological functions of horizontally transferred genes in prokaryotic genomes. Nat Genet. 2004;36:760–6.
Vallenet D, Calteau A, Cruveiller S, Gachet M, Lajus A, Josso A, et al. MicroScope in 2017: an expanding and evolving integrated resource for community expertise of microbial genomes. Nucleic Acids Res. 2017;45:D517–28.
Matsunaga T, Okamura Y, Fukuda Y, Wahyudi AT, Murase Y, Takeyama H. Complete genome sequence of the facultative anaerobic magnetotactic bacterium Magnetospirillum sp. strain AMB-1. DNA Res. 2005;12:157–66.
Wang X, Wang Q, Zhang W, Wang Y, Li L, Wen T, et al. Complete genome sequence of Magnetospirillum gryphiswaldense MSR-1. Genome Announc. 2014;2:e00171–14.
Smalley MD, Marinov GK, Bertani LE, DeSalvo G. Genome sequence of Magnetospirillum magnetotacticum Strain MS-1. Genome Announc. 2015;3:e00233–15.
Grouzdev DS, Dziuba MV, Sukhacheva MS, Mardanov AV, Beletskiy AV, Kuznetsov BB, et al. Draft genome sequence of Magnetospirillum sp. Strain SO-1, a freshwater magnetotactic bacterium isolated from the Ol’khovka River, Russia. Genome Announc. 2014;2:e00235–14.
Dzyuba MV, Mardanov AV, Beletskii AV, Kolganova TV, Sukhacheva MV, Shelenkov AA, et al. Reconstruction of iron metabolism pathways of bacteria Magnetospirillum aberrantis SpK spp. based on sequenced genome analysis. Dokl Biol Sci. 2012;444:202–5.
Melnyk RA, Engelbrektson A, Clark IC, Carlson HK, Byrne-Bailey K, Coates JD. Identification of a perchlorate reduction genomic island with novel regulatory and metabolic genes. Appl Environ Microbiol. 2011;77:7401–4.
Wang Y, Zhang T, Lin W, Zhang B, Cai Y, Yang C, et al. Complete genome sequence of Magnetospirillum sp. Strain XM-1, isolated from the Xi’an City Moat, China. Genome Announc. 2016;4:e01171–16.
Ke L, Liu P, Liu S, Gao M. Complete genome sequence of Magnetospirillum sp. ME-1, a novel magnetotactic bacterium isolated from East Lake, Wuhan, China. Genome Announc 2017;5:e00485–17.
Ivanovsky RN, Keppen OI, Lebedeva NN, Beletsky AV, Mardanov AV, Grouzdev DS. Draft genome sequence of the anoxygenic phototrophic bacterium Phaeospirillum fulvum MGU-K5. Genome Announc. 2017;5:e00895–17.
Duquesne K, Prima V, Ji B, Rouy Z, Médigue C, Talla E, et al. Draft genome sequence of the purple photosynthetic bacterium Phaeospirillum molischianum DSM120, a particularly versatile bacterium. J Bacteriol. 2012;194:3559–60.
Contreras-Moreira B, Vinuesa P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol. 2013;79:7696–701.
Rodriguez-R LM, Konstantinidis KT. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Inc. 2016;4:e1900v1.
Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinforma. 2013;14:60.
Qin Q-L, Xie B-B, Zhang X-Y, Chen X-L, Zhou B-C, Zhou J, et al. A proposed genus boundary for the Prokaryotes based on genomic insights. J Bacteriol. 2014;196:2210–5.
Pantiukh K, Grouzdev D. POCP-matrix calculation for a numberof genomes. 2017; https://doi.org/10.6084/m9.figshare.4577953.v1.
Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, et al. MicroScope-an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 2013;41:D636–47.
Karp PD, Latendresse M, Caspi R. The pathway tools pathway prediction algorithm. Stand Genom Sci. 2011;5:424–9.
Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012;40:D742–53.
Abby SS, Néron B, Ménager H, Touchon M, Rocha EPC. MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PloS One. 2014;9:e110726.
Abby SS, Rocha EPC. Identification of protein secretion systems in bacterial genomes using MacSyFinder. Methods Mol Biol Clifton NJ. 2017;1615:1–21.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/.
Dray S, Dufour A-B. The ade4 package: implementing the duality diagram for ecologists. J Stat Softw. 2007;22:1–20.
de Vienne DM, Ollier S, Aguileta G. Phylo-MCOA: a fast and efficient method to detect outlier genes and species in phylogenomics using multiple co-inertia analysis. Mol Biol Evol. 2012;29:1587–98.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Criscuolo A, Gribaldo S. BMGE (block mapping and gathering with entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10:210.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9.
Stolzer M, Lai H, Xu M, Sathaye D, Vernot B, Durand D. Inferring duplications, losses, transfers and incomplete lineage sorting with nonbinary species trees. Bioinformatics. 2012;28:i409–15.
Koziaeva VV, Dziuba MV, Ivanov TM, Kuznetsov BB, Skryabin KG, Grouzdev DS. Draft Genome Sequences of Two Magnetotactic Bacteria, Magnetospirillum moscoviense BB-1 and Magnetospirillum marisnigri SP-1. Genome Announc. 2016;4:e00814–16.
Uebe R, Voigt B, Schweder T, Albrecht D, Katzmann E, Lang C, et al. Deletion of a fur-like gene affects iron homeostasis and magnetosome formation in Magnetospirillum gryphiswaldense. J Bacteriol. 2010;192:4192–204.
Guglielmini J, Néron B, Abby SS, Garcillán-Barcia MP, de la Cruz F, Rocha EPC. Key components of the eight classes of type IV secretion systems involved in bacterial conjugation or protein secretion. Nucleic Acids Res. 2014;42:5715–27.
Cury J, Touchon M, Rocha EPC. Integrative and conjugative elements and their hosts: composition, distribution and organization. Nucleic Acids Res. 2017;45:8943–56.
Scheffel A, Gruska M, Faivre D, Linaroudis A, Plitzko JM, Schüler D. An acidic protein aligns magnetosomes along a filamentous structure in magnetotactic bacteria. Nature. 2006;440:110–4.
Murat D, Quinlan A, Vali H, Komeili A. Comprehensive genetic dissection of the magnetosome gene island reveals the step-wise assembly of a prokaryotic organelle. Proc Natl Acad Sci USA. 2010;107:5593–8.
Chen K, Durand D, Farach-Colton M. NOTUNG: a program for dating gene duplications and optimizing gene family trees. J Comput Biol J Comput Mol Cell Biol. 2000;7:429–47.
Boratyński Z, Brito JC, Campos JC, Cunha JL, Granjon L, Mappes T, et al. Repeated evolution of camouflage in speciose desert rodents. Sci Rep. 2017;7:3522.
Bennet M, McCarthy A, Fix D, Edwards MR, Repp F, Vach P, et al. Influence of magnetic fields on magneto-aerotaxis. PLoS ONE. 2014;9:e101150.
Wang K, Ge X, Bo T, Chen Q, Chen G, Liu W. Interruption of the denitrification pathway influences cell growth and magnetosome formation in Magnetospirillum magneticum AMB-1. Lett Appl Microbiol. 2011;53:55–62.
Li Y, Katzmann E, Borg S, Schüler D. The periplasmic nitrate reductase Nap is required for anaerobic growth and involved in redox control of magnetite biomineralization in Magnetospirillum gryphiswaldense. J Bacteriol. 2012;194:4847–56.
Li Y, Raschdorf O, Silva KT, Schüler D. The terminal oxidase cbb3 functions in redox control of magnetite biomineralization in Magnetospirillum gryphiswaldense. J Bacteriol. 2014;196:2552–62.
Li Y, Sabaty M, Borg S, Silva KT, Pignol D, Schüler D. The oxygen sensor MgFnr controls magnetite biomineralization by regulation of denitrification in Magnetospirillum gryphiswaldense. BMC Microbiol. 2014;14:153.
Cohan FM, Koeppel AF. The origins of ecological diversity in prokaryotes. Curr Biol. 2008;18:R1024–34.
Kolinko I, Lohße A, Borg S, Raschdorf O, Jogler C, Tu Q, et al. Biosynthesis of magnetic nanostructures in a foreign organism by transfer of bacterial magnetosome gene clusters. Nat Nanotechnol. 2014;9:193–7.
Schübbe S, Kube M, Scheffel A, Wawer C, Heyen U, Meyerdierks A, et al. Characterization of a spontaneous nonmagnetic mutant of Magnetospirillum gryphiswaldense reveals a large deletion comprising a putative magnetosome island. J Bacteriol. 2003;185:5779–90.
Rioux J-B, Philippe N, Pereira S, Pignol D, Wu L-F, Ginet N. A second actin-like MamK protein in Magnetospirillum magneticum AMB-1 encoded outside the genomic magnetosome island. PloS One. 2010;5:e9151.
Abreu N, Mannoubi S, Ozyamak E, Pignol D, Ginet N, Komeili A. Interplay between two bacterial actin homologs, MamK and MamK-Like, is required for the alignment of magnetosome organelles in Magnetospirillum magneticum AMB-1. J Bacteriol. 2014;196:3111–21.
Brune A, Frenzel P, Cypionka H. Life at the oxic-anoxic interface: microbial activities and adaptations. FEMS Microbiol Rev. 2000;24:691–710.
Acknowledgements
This work was supported by projects from the French National Research Agency (ANR Tremplin-ERC: ANR-16-TERC-0025-01 and ANR PRC: ANR-18-CE31-0003). Genome sequencing was funded thanks to the Appel à Projets en Génomique Environnementale—CNRS (DJ/ST/IP/2013/D-112). We are grateful to the INRA MIGALE bioinformatics platform (http://migale.jouy.inra.fr) for providing computational resources. We acknowledge the Institut de Radioprotection et de Sûreté Nucléaire (IRSN) at CEA Cadarache for the access of the transmission electron microscope Tecnai G2 BioTWIN.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Monteil, C.L., Grouzdev, D.S., Perrière, G. et al. Repeated horizontal gene transfers triggered parallel evolution of magnetotaxis in two evolutionary divergent lineages of magnetotactic bacteria. ISME J 14, 1783–1794 (2020). https://doi.org/10.1038/s41396-020-0647-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0647-x
This article is cited by
-
Silent gene clusters encode magnetic organelle biosynthesis in a non-magnetotactic phototrophic bacterium
The ISME Journal (2023)
-
McaA and McaB control the dynamic positioning of a bacterial magnetic organelle
Nature Communications (2022)
-
Magnetotactic bacteria and magnetofossils: ecology, evolution and environmental implications
npj Biofilms and Microbiomes (2022)
