
Abstract
The ability to measure microbial fitness directly in natural conditions and in interaction with other microbes is a challenge that needs to be overcome if we want to gain a better understanding of microbial fitness determinants in nature. Here we investigate the influence of the natural microbial community on the relative fitness of the North American populations SpB, SpC and SpC* of the wild yeast Saccharomyces paradoxus using DNA barcodes and a soil microcosm derived from soil associated with oak trees. We find that variation in fitness among these genetically distinct groups is influenced by the microbial community. Altering the microbial community load and diversity with an irradiation treatment significantly diminishes the magnitude of fitness differences among populations. Our findings suggest that microbial interactions could affect the evolution of yeast lineages in nature by modulating variation in fitness.
Similar content being viewed by others
Introduction
Microorganisms are powerful model systems to address evolutionary and ecological questions that relate to adaptation to biotic and abiotic conditions [1, 2]. Since most of the information on microbial adaptation comes from experimental evolution [3], little is known about adaptation and fitness variation of microbes in natural environments, especially for microbes that are not involved in host–parasite or host–pathogen interactions [4, 5]. Experiments with synthetic communities are beginning to emerge to study microbial interactions at the community scale [6,7,8]. However, these models typically include only one strain of each species or even genera. Thus, understanding how interactions with other microbes affect microbial species and population fitness in natural environments has yet to be addressed [9]. More specifically, it remains difficult to estimate the fitness of individual genotypes in the presence of other microbial species in the same experiment, and it is also challenging to identify which species are relevant when trying to approximate natural conditions.
To overcome these limitations, microcosms can be used as proxies for natural habitats to measure and compare the fitness of different genotypes in the laboratory. Microcosms mimic the composition of natural environments, but simplify their complexity while increasing control over the experiments, allowing us to better understand natural processes [10, 11]. To investigate microorganism adaptation and evolution in microcosms, several studies have used soil or leaf resuspended in water before sterilization [12,13,14]. However, such studies are limited to the ecological factors considered in the experiment that includes abiotic factors and occasionally biotic ones when competitors are added to the sterile substrate [13, 15]. Further research is needed to test whether and how abiotic and biotic factors affect microbial population fitness to better understand for instance if microbial populations are locally adapted in nature.
The wild yeast Saccharomyces paradoxus is one of the best-studied non-domesticated fungi and the closest known relative to Saccharomyces cerevisiae, the eukaryotic model in genetics and cell biology [16, 17]. Isolates of S. paradoxus are mostly found on the bark and exudates of deciduous trees and their surrounding soil [18, 19]. Recent studies have confirmed a closer association with oak than with other tree species, and showed that Saccharomyces yeasts are more abundant on oak leaf litter and soil than on bark [20, 21]. Recent technologies have been developed to study and follow S. paradoxus in its natural habitat. These methods exploit natural polymorphisms in local populations as trackable genetic markers [22, 23]. Studies using these tools concluded that low dispersal could promote local adaptation in the S. paradoxus environment, which could include adaptation to the local microbial communities. Although several studies have shown the capacity of Saccharomyces to grow on nutrient-poor and close-to-natural substrates using sterilized oak infusion [24] or maple sap [25], the extent of fitness variation in their soil habitat is still unknown, even less so the role of the other microbial species inhabiting these soils. In vitro studies have shown that yeasts undergo various interactions with other microbes, such as competition, predation and cooperation, on tree bark and in soil [24, 26]. For instance, a recent study by Kowallik et al. [24] examined the effect of other microbial species isolated from the bark of oak trees on the growth of S. paradoxus. Their results show that fungal and bacterial species present on the bark can positively and negatively interact with S. paradoxus, suggesting that microbial interactions could affect the performance of this budding yeast in nature.
Genome sequencing and large-scale surveys recently revealed the presence of distinct S. paradoxus lineages that have only partially overlapping geographic ranges within North America [18, 27, 28]. The SpB lineage has a southwestern distribution, whereas SpC has a northeastern distribution. The introgressed lineage SpC* has an overlapping distribution between those of its parental lineages, SpB and SpC [28]. Local temperature has been identified as the ecological factor that best predicts the geographical distribution of S. paradoxus [29]. This observation is supported by a correlation of growth rate in liquid cultures with temperatures that correspond to average temperature of the strain geographical origin, suggesting local adaptation [30]. Finally, Filteau et al. [25] also revealed a correlation between growth rate and geographical origin. This result suggests a variation in performance among lineages, for instance with SpB strains performing better than SpC strains on maple sap in laboratory conditions [25]. S. paradoxus’ spatial distribution in North America therefore offers the opportunity to investigate the relative importance of dispersal, drift and local adaptation in microbial evolution. Most importantly and still unexplored, the collections of natural isolates that are genetically trackable make it possible to explore the potential role of biotic interactions in this model species.
Here we investigate variation in fitness of the North American lineages of S. paradoxus in a natural substrate, taking into account biotic interactions with its complex microbiota. Our objective was to determine whether the microbiota influenced the relative fitness of lineages of S. paradoxus. As S. paradoxus has been shown to interact with its microbial community [24], some strains or lineages could be better adapted to interact with other microbes. A manipulation of the soil microbial community properties would thus be expected to impact the relative fitness of S. paradoxus lineages if these interact with microbes. To reach this objective, we performed a competition assay using 159 strains from lineages SpB, SpC and SpC* in a natural soil sampled in proximity of an oak tree. In a parallel assay, we manipulated the natural microbial community of the same soil with an irradiation treatment, altering the microbial load and diversity. Strain fitness was estimated using DNA barcodes that individually identify the strains and allow for their quantification through time by deep barcode sequencing [31]. Each barcoded strain was originally sampled at a precise location in North America and from a specific substrate. This information allowed us to estimate and compare the change in the frequency of the barcoded strains in experimental soils, and evaluate the influence of microbial community, geographical and substrate of origin on strain-relative fitness values.
Materials and methods
Soil sampling
Soil samples were collected around an oak tree (Quercus rubra) in Québec City (46.782454N, −71.267844W) on 27 August 2018, in an urban woodland (Supplementary Fig. S1). The samples were taken from 0.5 to 1 m from the trunk, from a lateral area of 50 cm and a depth of 10 cm. We used a sterile spatula to collect the soil and to transfer it into sterile 50-ml tubes (≈30 g of soil).
Three tubes were treated by gamma irradiation with a resultant dose of 16 kGy (GammaCell 220 Cobalt 60 Irradiation Unit, at Université Laval, Québec City, Canada), and three tubes were conserved at room temperature in parallel for the same period of 72 h [32]. The remaining tubes were conserved at −80 °C for further use. The post-radiation microbial biomass was analyzed by dilution plating and inspection of the plates for any microbial growth after 2 days of incubation at 25 °C. In all, 100-μl aliquots were spread in three replicates of yeast peptone dextrose (YPD) medium petri dishes (with 10 g/L of yeast extract, 20 g/L of tryptone and 20 g/L of glucose) and LB medium (with 5 g/L of yeast extract, 10 g/L of tryptone and 10 g/L of NaCl) that were incubated for 48 h (Supplementary Fig. S2). Irradiated and natural soils were then conserved at 4 °C until the start of the competition experiment.
Yeast pool preparation
Selected strains from the S. paradoxus barcoded collection were pooled as in Bleuven et al. [31] with the following modifications: the set of SpB-barcoded strains was first pooled together in an intermediate pool, and SpC and SpC* strains in two other intermediate pools. The three pools were combined into a single one to obtain a mix of the four lineages. The resulting pool was adjusted to a concentration equivalent to 50 optical density unit (OD595) by concentrating the cells and measuring OD using a plate reader (Infinite F200 Pro Tecan, Zürich, Switzerland). The culture media was removed after a quick centrifugation and replaced with sterile water. DNA from the initial pool was extracted following the protocol described in Bleuven et al. [31]. Strains used in this study are listed in Supplementary Table S1.
Competition experiment in natural and irradiated soils
For each treatment, natural and irradiated, six tubes containing 15 g of soil were prepared from the same batch of initial soil. Three of the six tubes were inoculated with 2 ml of the S. paradoxus pool solution at 10 OD595, while the remaining three tubes were inoculated with sterile water, to reach concentrations similar to what was used in Anderson et al. [23], but higher than naturally occurring in similar environment, as for example, oak leaf litter [20]. However, this concentration is required for such experiments to be performed, at least with the current tools available. Soil samples were incubated statically for 21 days at 25 °C in non-hermetically closed tubes. Each tube was weighed every 5 days; the reduction in weight due to evaporation over the preceding time was compensated by the addition of sterile water.
At day 0 after inoculation and day 21, colony-forming unit estimation was conducted on selective media to visually track the barcoded strains in inoculated soil samples and the absence of barcoded strains in the non-inoculated soils (see Supplementary Table S2). In a 15-ml plastic tube, about 0.25 g of soil was diluted in 6 ml of sterile water. After a quick vortex, 100 μl of dilution suspension was spread onto petri dishes with YPD medium (10 g/L of yeast extract, 20 g/L of tryptone and 20 g/L of glucose), YPD supplemented with 250 μg/mL of hygromycin B and 12.5 μg/mL of chloramphenicol (HPH + Chlo) and YPD supplemented with 100 μg/mL of nourseothricin and 12.5 μg/mL of chloramphenicol (NAT + Chlo). Petri dishes were incubated at 30 °C for 2 days (Fig. 2 and Supplementary Fig. S3).
The HO locus, at which the barcodes were inserted, was amplified by PCR on isolated barcoded yeast colonies (n = 47, for each experimental soil) as in Bleuven et al. [31] to verify the heterozygous genotype of strains compared with control diploid barcoded strains from the collection (Supplementary Fig. S4, oligonucleotides are listed in Supplementary Table S3). At day 21 of incubation, DNA was extracted in two replicates from the six tubes inoculated with S. paradoxus strains (natural and irradiated soil) using the DNeasy® PowerSoil® Kit (Qiagen, Hilden, Germany). DNA was extracted once for irradiated soil and natural non-inoculated soil, without barcoded strains. DNA samples were conserved at −20 °C.
Metabarcoding amplicon sequencing and analysis
We sequenced the bacterial 16S rDNA V3–V4 region and the fungal ITS2 region (identified as 16S and ITS henceforth) from the soil samples to determine the species composition of the microbial communities. We sequenced two DNA amplicons from extractions of the original soil conserved at −80 °C, two extractions of the inoculated and non-inoculated natural soils at day 5 and day 21. This sampling scheme allowed us to compare the microbial community before, during and at the end of the experiment, and examine the variation in the microbial community composition caused by the irradiation. Amplification of ITS and 16S regions, equimolar pooling and sequencing on the Illumina Miseq were performed at the Plateforme d’analyses génomiques (IBIS, Université Laval, Quebec City, Canada) as described in Supplementary Information. The oligonucleotide sequences used for amplification are listed in Supplementary Table S3.
The 16S and ITS amplicon sequencing was performed in two independent runs. Raw sequences were first demultiplexed by the IBIS platform following the Illumina default parameters, resulting in 659,768 and 637,864 sequences for fungi and bacteria, respectively. For the assignment of amplicons to the operational taxonomic unit (OTU), several steps were carried out based on the Dada2 package version 1.5.0 [33] in R version 3.1.2 as described in Supplementary Information. To compare samples relative abundance, a rarefaction was performed using the function rarefy_even_depth () in the package phyloseq with 20,000 reads per library for the bacterial 16S and 40,000 for the ITS2 region [34]. We calculated Shannon diversity indices to investigate whether samples differ according to treatment and sampling time using the vegan package [35]. Relative phylum and genus abundances were calculated based on the number of OTUs of the phylum and genus divided by total number of OTUs within sample (Supplementary Table S4).
Yeast barcode sequencing and analysis
Forward and reverse primers were used for multiplex sequencing with Ion Torrent technology using predefined indexes [36]. Each DNA extraction of natural and irradiated inoculated soil sample was amplified in two independent PCR amplifications with two distinct indexes as technical replicates. Each 50 μL of PCR reaction mix sample contained 1 ×PCR buffer, 0.2 mM of dNTPs, 1.5 mM of MgCl2, 0.03 U of DNA polymerase (Bioshop, Burlington, Canada), 6.25 ng of genomic DNA and 0.2 μM of each primer. PCR amplification was conducted in a MasterCycle ProS (Eppendorf, Hamburg, Germany) with the following conditions: 3 min at 94 °C, 10 cycles of 30 s at 94 °C, 30 s at 52 °C and 15 s at 72 °C, followed by 20 cycles of 30 s at 94 °C, 30 s at 75 °C and 15 s at 72 °C, and a final extension of 30 s at 72 °C. PCR products were pooled and purified using the KAPA Hyper Prep Kit from Illumina (KAPA Biosystems, Wilmington, USA), and were sequenced at the IBIS sequencing platform on an Ion Proton instrument (Thermo-Fisher Scientific, Waltham, USA) according to the manufacturer’s instructions.
Sequencing quality control was performed using FastQC [37], and Trimmomatic version 0.36 [38] was used to filter out reads that did not have the expected amplicon length. Barcode sequencing data were analyzed using a custom Python script (Jupyter Notebook available on Github: https://github.com/Landrylab/Bleuvenetal2019). This resulted in a total of ~39 million reads with a valid library and strain assignment across the entire dataset. Following validation of the barcodes [39], only strains barcoded with both HPH and NAT resistance cassettes, and that had more than 100 reads in the initial pool in each replicate, were considered for further analysis (see Supplementary Table S1 for strains considered).
Library preparation, sequencing and biological variability generate experimental errors. We filtered the outliers to limit the noise from the PCR indexing, using a filtering threshold: we calculated the differences between the indexing replicate of each barcode, and the barcodes with a difference larger or smaller than the mean ± 2.5 × sd were removed (Supplementary Fig. S5C).
We calculated the relative fitness of each barcoded strain of the pool, where each genotype occurred in two biological replicates with either a NAT or HPH resistance cassette. First, to obtain the relative frequency of each copy, we used the mean of the multiplexing replicates and then the mean of their extraction replicates (Fig. 1c). Second, relative fitness (ω) was calculated with the following equation: \(\omega = {\mathrm{log}}_{2}( {\frac{{P_{{\mathrm{final}}}}}{{P_{{\mathrm{initial}}}}}})\) where Pfinal is the frequency of the strain at the final time of the experiment (day 21) and Pinitial is the frequency of the strain in the initial pool. Frequencies were calculated as the number of barcode reads divided by the total number of reads in the library considered. This gives us an estimate of the relative (ranks) fitness of each individual strains (see Jupyter Notebook available on Github: https://github.com/Landrylab/Bleuvenetal2019 for simulations of fitness estimates).

a Geographic distribution of SpB (red), SpC (blue) and SpC* (purple) lineages in North America. The sampling site in Québec City is indicated by the black arrow and the white circle. b The soil sample was splitted in two treatments: incubated at room temperature for 72 h (natural soil) and treated with gamma irradiation for 72 h (irradiated soil). The initial pool, containing the barcoded SpB (n = 114), SpC (n = 30) and SpC* (n = 15) strains, was used to inoculate the experimental soils in three biological replicates. Three biological replicates of non-inoculated soils were also included as negative controls. DNA was extracted from the initial pool, and at day 21 for barcode quantification, while sequencing of ITS and 16S was performed on the original soil, at day 5 and 21. Experimental soils, inoculated and non-inoculated, were incubated at 25 °C for 21 days. c For each inoculated soil of both treatments, two DNA extractions and two indexing PCR were performed on each DNA sample at day 21. d To calculate the relative fitness of each NAT and HPH copies of the individual strain, the mean of the ratio of the frequencies of each indexing PCR replicate within each DNA extraction was calculated, and the overall mean for each biological replicate was calculated. Yellow circle: indexing PCR replicate 1, green circle: indexing PCR replicate 2, grey star: two DNA extraction replicates (a, b).
To estimate the relative fitness of each lineage, the relative fitness averaged of all strains belonging to each lineage was calculated. The normality criterion of the data was not met (Shapiro–Wilk test, p value < 0.01). The variance in relative fitness was non-homogeneous between the treatments for SpB, SpC and SpC* (Levine’s test, p value < 0.001). To test how the lineages and strains differ in relative fitness between the two treatments (irradiated and natural soil) and between substrate of origin (soil or non-soil), we used Kruskal–Wallis and Dunn post-hoc tests with correction for multiple testing (FDR method). We examined the interaction between lineage and treatment, and between each genotype within the lineages and treatment, using fitness rank-based ANOVA with the raov function from the Rfit package [40, 41]. Spearman’s correlation was used to analyze the effect of distance of the original sampling site from the study site on strains performance. All statistical analyses and data handling were performed using RStudio version 1.0.153 from R Core [42].
Results
To investigate the influence of the soil microbial community on S. paradoxus fitness, we measured the relative abundance of barcoded North American strains through time by competitive assay [31]. Two types of soil microcosms were produced from the soil collected at the base of an oak tree (Supplementary Fig. S1): from the sample, a fraction, labelled natural soil, was kept at room temperature for 72 h, and another fraction was treated with gamma irradiation for 72 h to reduce microbial viability (Fig. 1). No growth was detected on the petri dishes inoculated with the irradiated soil following irradiation (Supplementary Fig. S2), suggesting that the living biomass was significantly decreased. We also plated the soils inoculated with the S. paradoxus barcoded collection to assess the persistence of barcoded strains throughout the experiments. The number of inoculated barcoded strains appeared to remain similar in the experimental soil from the initial to the final time of the experiment (21 days). This result indicates that overall populations are maintained at a stable density throughout the experiments. Because the data may not fulfil the criteria for using standard analysis of variance, we used non-parametric analyses and considered fitness ranks within an experiment as measure of relative fitness for our analyses. Because fitness values are relative within a given treatment, the overall effect of the treatment on fitness cannot be assessed. The inoculated strains were maintained throughout the experiment in both diploid (27–56%) and sporulated forms (44–73%) (Fig. 2 and Supplementary Figs. S3–S4).

a Colonies of S. paradoxus strains isolated from experimental soils directly after inoculation and after 21 days of experiment were counted. Results were expressed as colony forming units (CFU) per gram of soil. For each soil, counts were measured on HPH + Chl and NAT + Chl medium to select for the barcoded strains and eliminate bacteria. A Kruskal–Wallis test was used to compare log10 CFU/g of soil between the days 0 and 21. P values are shown on the plot for each treatment. b Comparison of the percentage of heterozygous strains between the original strains from the collection and the strains from the experimental soils at day 21. Strains in the initial pools were all heterozygous for the barcodes (HO/Barcode) as expected, as shown by the control. At the end of the experiment, nearly half of the isolated strains (n = 47 for each condition) are homozygous (Barcode/Barcode), indicating that at least some inoculated strains went through sporulation during the course of the experiment.
Identification of the experimental soil microbial community
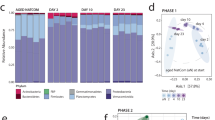
While plate counts of soils before the inoculation of the barcoded strains were negative for the irradiated soil (Supplementary Fig. S2), counts throughout the experiment showed that the irradiated soils were not entirely sterile (Supplementary Fig. S3). To determine the composition of the microbial community that could interact with the inoculated S. paradoxus strains and the potential change in community composition obtained by irradiation treatment, we sequenced the bacterial 16S and fungal ITS regions in the experimental soil of the two treatments at different time points (the original soil before treatment, at day 5 and at day 21). Each of the dominant phyla and genera identified is represented by more than one OTU (Fig. 3).

Relative abundance and richness proportion of fungi and bacteria phyla in the initial soil before treatment (day 0), and in natural (dark brown) and irradiated soils (light brown) at day 5 and 21. Numbers in parenthesis indicate the replicate considered when relevant. Diversity is quantified by the Shannon index with the blue bars.
The fungal community consisted of 670 OTUs when considering all samples combined, ranging from 176 to 227 OTUs per sample (Supplementary Table S4). The dominant phylum in richness was Ascomycota (mean richness proportion: 67 ± 13.8%) followed by Basidiomycota (25 ± 13.4%). The richness of Ascomycota was higher in natural soils (75 ± 5.32%) than in irradiated soils where Ascomycota (46 ± 1.04%) and Basidiomycota (45 ± 0.77%) were in similar proportions (Fig. 3). The relative abundance of the two phyla was similar in natural soils (47 ± 7.5% for Ascomycota and 50 ± 7.94% for Basidiomycota), but Basidiomycota had higher relative abundance in irradiated soils (62 ± 16.5%) than Ascomycota (24 ± 2.69%). The irradiated soils were dominated in richness and relative abundance by a basidiomycetous yeast of the genus Apiotrichum and a Mortierella fungus from the Mortierellomycota phylum. The natural soils were mostly dominated by Penicillium (Ascomycota) and Russula (Basidiomycota) (Fig. 3).
The bacterial community consisted of 1565 OTUs when considering all samples, ranging from 344 to 540 OTUs in each sample (Supplementary Table S4). Phylum-level analysis showed high relative abundance and richness of Actinobacteria, Proteobacteria and Acidobacteria (Fig. 3). The phyla Acidobacteria, Proteobacteria and Actinobacteria are known to be among the most dominant in forest soils [36]. Genus-level analysis showed that all the soils were largely dominated either by Acidothermus (Actinobacteria) or Ktedonobacter (Chloroflexi) (Fig. 3).
Diversities of the fungal and bacterial communities appeared stable throughout the experiment (Fig. 3). The bacterial community was more diverse, with a higher Shannon index in all soils (H’ = 5.11–5.73) compared with the fungal community (H’ = 3.97–4.44). In both groups, the irradiated soils presented lower Shannon index than natural soils after 5 and 21 days (Fig. 3). The range of Shannon index values and the difference between bacterial and fungal communities are consistent with several studies of forest soils [43, 44]. This suggests that the soil sampled for our study is representative of forest soils in terms of bacterial and fungal community composition and diversity. Furthermore, these analyses suggest that the total biomass was not only reduced by irradiation, but also modified in terms of taxonomic composition, at least for the dominant taxa.
Effect of the microbial community on relative fitness
Using barcode sequencing, the ratio of the barcode frequency at day 21 over the frequency in the initial pool was used as a proxy for the relative fitness of each strain within a given soil (Supplementary Material). The strains of the three main lineages showed significant differences in their relative fitness. In the natural soil, SpB displayed a higher fitness than both SpC and SpC* (Dunn test, p value ≤ 0.001) and SpC had higher fitness than SpC* (Dunn test, p value = 0.0044) (Fig. 4a). In the irradiated soil, the SpB and SpC relative fitness was not significantly different, while SpC* had a lower fitness than both SpB and SpC (Fig. 4b, Dunn test, p value ≤ 0.02). This observation was similar whether we considered the two biological replicates separately (NAT and HPH barcodes, Fig. 4), or using the average of the relative fitness of the two copies (Supplementary Fig. S6).

Each biological replicate (NAT and HPH barcodes) was considered separately. a Relative fitness of SpB, SpC and SpC* strains in the natural soil. SpB performs better than both SpC and SpC*, while SpC* has a lower fitness than both SpB and SpC (Kruskal–Wallis and Dunn post-hoc tests, p values on plot). b In the irradiated soil, SpB and SpC present no significant difference in relative fitness, while SpC* still performed more poorly (Kruskal–Wallis and Dunn post-hoc tests, p values shown on plot). c Average rank of the three lineages in natural and irradiated soils. While a statistically significant Lineage × Treatment interaction was detected (Supplementary Table S5, Section A), the rank of the lineages did not change across the environment. d Details of the individual strain ranks in natural and irradiated soils within each lineage. SpB is shown in red, SpC in blue and SpC* in purple. In each case, we detect a significant Genotype × Treatment interaction within lineage using ranked fitness values (p value < 0.05) (Supplementary Table S5, Section B).
When considering the copies separately, a significant Lineage × Treatment interaction was detected (rank-based ANOVA, p value < 0.002) caused by a change in magnitude of the difference among lineages but no change in rank (Fig. 4c, Supplementary Table S5). Then, we performed a more conservative analysis; we used the average of the relative fitness of the two biological replicates and ranked each strain within each treatment and replicate. The Lineage × Treatment interaction is marginally significant, but supports the analysis above (rank-based ANOVA [40], p value = 0.07, Supplementary Table S5). This shows that the relative fitness of the three lineages depends on the composition and biomass of the natural microbial community.
To examine whether the relative fitness of individual strains was also affected by the microbial community, we considered each individual genotype within lineage. Using the same conservative method, we ranked each strain within each treatment, replicate and lineage. Within each lineage, we tested if there was a significant Genotype × Treatment interaction using a rank-based ANOVA. We found a significant interaction in the three lineages (rank-based ANOVA, p value < 0.05, Supplementary Table S5, Fig. 4d). These results show that genetic variation within a lineage led to fitness variation in the soil, and that relative fitness of strains within lineage is significantly affected by the soil and community composition.
Geographical location and substrate of origin does not influence relative fitness
Each barcoded strain was sampled at a precise location in North America. This gives us an opportunity to explore the possibility of adaptation to specific conditions even if the experimental design does not allow to conclusively test for local adaptation. Under the local adaptation hypothesis, one would expect for instance a negative correlation between relative fitness and distance from the sampling site of the sites at which strains were isolated. However, we find that the contribution of the distance of the strain original location to the soil sampling site to fitness variation within each lineage in natural soil is not significant and positive when significant: relative fitness is not negatively correlated with the distance from the sampling site (SpB Spearman’s rho = 0.1, p value = 0.002; SpC Spearman’s rho = −0.05, p value = 0.45; SpC* Spearman’s rho = −0.03, p value = 0.72). In addition, we tested the pairwise differences in relative fitness between the strains from different locations in SpB and SpC. While no specific pattern emerged from SpB analysis, in SpC, among all comparisons, the strains from the closest locations (46.81388 N) to the soil sampled showed significant differences with several other locations in the natural soil (p value < 0.02, Dunn test, Supplementary Table S6).
We compared the performance of the barcoded collection in the natural and irradiated soils with previous experimental conditions that comprise variation in carbon and nitrogen sources, chemical compounds and incubation temperatures [31, 45] (Supplementary Fig. S7). The clustering indicates that the relative fitness of the strains in natural and irradiated soil conditions is more similar to each other than to the laboratory conditions previously tested. Most importantly, the analysis indicates that the relative fitness of the two SpC strains from the closest locations to the soil sampled is often higher than the rest of the SpC strains in the previous experimental conditions. This result suggests that the highest fitness value of these two strains in the soil microcosm does not reflect local adaptation but rather a general advantage (Supplementary Fig. S7).
Then, we compared the relative fitness of strains from soil and non-soil origin in natural soil and within each lineage. The substrate of origin presents no significant effect on the relative fitness of the strains in our experimental design (Kruskal–Wallis test, p value > 0.1 for SpB, SpC and SpC*), which does not support a model in which strains sampled from the soil would be better adapted to this substrate. The graphical representation of the S. paradoxus strains relative fitness in natural and irradiated soils associated with their location and substrate of origin highlights the absence of a clear effect of these factors (Supplementary Fig. S8).
Discussion
We measured the relative fitness of S. paradoxus strains from natural SpB, SpC and SpC* lineages in a microcosm representing a natural soil and a natural soil with an altered microbial community. We tested for the effect of an alteration of the microbial community on strain relative fitness. We found that the magnitude of fitness differences was attenuated in the soil with an altered microbial community.
One factor to consider in our experiments is the density of cells used for the assays. S. paradoxus is thought to occur at very low density in nature [20] and the cell density used in our experimental soils was much higher than observed in nature. Therefore, the relative fitness measured could also have been influenced by density- and frequency-dependent effects if strains interact with each other. However, density dependence and diversity dependent effects in S. paradoxus have not yet been documented. In addition, frequency-dependent effects and interactions among strains are difficult to track in experimental settings, such as the one used here, which is common to all studies comparing fitness of genotypes in a single pool [46,47,48]. Further technological developments may be needed to examine whether these types of effects are common and important.
Our observations show that strains could persist in different viable states, vegetative and sporulated. The large proportion of sporulated cells gives some indications about the fact that the soil is poor in nutrients or that specific molecules may trigger sporulation. Further investigations on the soil nutrient composition could contribute to better understand the dynamics of spore formation in S. paradoxus, and if there are differences among lineages. In addition, if mating occurred, it most likely occurred within asci [49], thereby preserving the association between strain genotypes and their barcodes. Tracking barcoded strains during our experiment may have provided information about the survival component of fitness rather than solely growth rate in the soil since cell density remained constant during the experiment. This difference in the estimated fitness of strains could limit the comparisons with other studies in which growth was used to infer fitness in continuously renewed liquid media [31], or by measuring the colony growth on solid media [45].
Differences in relative fitness between lineages and effect of the microbial community
The better performance of SpB in our microcosms is consistent with previous studies in most conditions from artificial media and maple sap [25, 45]. In addition, SpB and SpC were shown to perform differently on various nitrogen sources, for example, allantoin and allantoate [25]. These molecules are natural nitrogen sources found in plants exudate [50]. This difference in nitrogen metabolism could be linked to the difference in performance in the natural soil, where nitrogen oligotrophy is an important ecological adaptation in yeast [51]. Furthermore, the fact that SpB was shown to perform better at 25 °C, temperature at which our experiment was conducted, could also have influenced our results [28, 30]. Moreover, in nature, soils are subjected to temperature variation throughout the year, and testing several temperatures as well as fluctuations in further studies could help to understand the natural S. paradoxus population dynamics [52]. This may reflect another limitation in our study, that is, we used one soil sample taken at one time point, thus limiting the study of fluctuating selection in time and space [53].
In our study, gamma irradiation was preferred to the more commonly used autoclaving because prior research has shown that the former reduces the chemical and physical alteration of soil properties [32, 54, 55]. The drawback is that autoclaving better eliminates DNA molecules and microorganisms than gamma irradiation, which makes DNA amplification still possible even though no bacterial or fungal growth is detected [32]. However, microorganisms can persist in a viable but non-cultivable state, as the plate counts after several days seem to indicate, along with the changes in relative proportions of the most abundant genera over the experiment. Irradiated soils were therefore not sterile. Our choice was motivated by the idea of ensuring similar abiotic conditions in the different experimental soils. The dominant phyla in our soils are those frequently occurring in forest soil [43, 56] with Ascomycota and Basidiomycota that coexist in soil with a diversity of organisms, and are involved in many types of interactions [26].
The interactions of one microbe with other populations or species are predicted to have strong evolutionary consequences [9, 57, 58]. We find that the microbial community appears to influence the magnitude of fitness variation between lineages and also influence the fitness ranking of the strains within the three lineages. Irradiation of the original soil led to a change in the microbial community composition and diversity and biomass reduction. The fungal community was the most impacted with a change in dominant genus between natural and irradiated soils. Thus, potential biotic interactions were clearly modified, and the resulting community impacted the S. paradoxus strain fitness. The change in the community composition could also have impacted diversity-dependant effects in the S. paradoxus lineages. Species that could have positively affected SpB or negatively affected SpC and/or SpC* relative fitness in natural soil could have been reduced in irradiated soil. The effect of the variation in the microbial community across the treatments could be explained by direct or indirect interactions. For example, S. paradoxus strain fitness could be impacted by the effect of competition or differential access to public goods [59]. The degradation of organic compounds present in the soil by the microbial community could benefit S. paradoxus and facilitate the access to nutrients. In addition, diffusible and volatile compounds emission from bacteria and fungi are important actors of microbial interactions in soil [60, 61]. These molecules can serve as nutrient sources and mediate various interaction, such as inhibition as well as stimulation of growth [62]. Microbial diffusible compounds are produced by several bacterial and fungal genera found in our soil, such as Clostridium, Flavobacterium, Methylobacterium, Streptomyces, Penicillium and Trichoderma [60, 63]. The presence of such compounds could have impacted differentially the fitness of the SpC, SpC* and SpB strains. The SpB lineage could display an advantage in using soil nutrients, in coping with microbial interactions and thus, be more adapted to the natural soil environment. As a future perspective, analysis of soil compounds and the isolation of the dominant microbial species would help test the biotic effect on each lineage. One interesting finding related to the evolution and divergence of the SpC and SpB lineages is that SpB outperforms SpC in most conditions tested in the laboratory and here in the natural soil, making it difficult to understand why it does not outcompete SpC in its range. Our results show that these differences were reduced when grown in a natural soil with an altered microbial community. This suggests that interaction with other microbes may play a role in shaping the ecology of these three lineages.
Influence on relative fitness of geographical and substrate origin
Although our experimental design does not allow to test for local adaptation directly, it allowed to examine whether strains could be adapted in terms of geographical origin and in terms of substrate of origin. In the first case, one would expect that strains isolated closer to the soil sampling site would show higher fitness than other strains. In the second case, one would expect strains that were originally isolated from soil would outperform strains originally isolated from other substrates. First, we detected a higher relative fitness for the two SpC strains that were originally the closest to the sampling site compared with the others. Given that the only signal comes from two particular strains that also performed better in other conditions, these strains may have been found in the site nearby due to stochastic processes [64] rather than due to their fitness advantage. Second, we did not find evidence that strains could be adapted to their local substrates. Indeed, no significant differences were observed between strains isolated from the soil when grown in the soil and strains isolated from another substrate. This result is consistent with previous studies in which the adaptation of S. paradoxus to the substrate of origin was not detected by reciprocal transplant experiment [22], and with the difficulty to detect local adaptation in free-living microorganisms [14, 65, 66]. Our choice of experimental substrate was mainly driven by the higher abundance of S. paradoxus strains in oak-associated soil than in other substrates. The presence of yeast in soil could also be explained by dispersal due to insects or climatic conditions, and therefore masking the optimal substrate for wild yeasts. Additional work in the field may be needed to further examine these questions.
Data availability
Raw data of barcode sequencing and microbial diversity are available at Bioproject PRJNA529380 at http://www.ncbi.nlm.nih.gov/bioproject/.
References
Jessup CM, Kassen R, Forde SE, Kerr B, Buckling A, Rainey PB, et al. Big questions, small worlds: microbial model systems in ecology. Trends Ecol Evolution. 2004;19:189–97.
Rainey PB, Buckling A, Kassen R, Travisano M. The emergence and maintenance of diversity: insights from experimental bacterial populations. Trends Ecol Evolution. 2000;15:243–7.
Bleuven C, Landry CR. Molecular and cellular bases of adaptation to a changing environment in microorganisms. Proc Biol Sci. 2016;283:20161458.
Rúa MA, Lamit LJ, Gehring C, Antunes PM, Hoeksema JD, Zabinski C, et al. Accounting for local adaptation in ectomycorrhizas: a call to track geographical origin of plants, fungi, and soils in experiments. Mycorrhiza. 2018;28:187–95.
Vos M, Birkett PJ, Birch E, Griffiths RI, Buckling A. Local adaptation of bacteriophages to their bacterial hosts in soil. Science. 2009;325:833.
Gutiérrez N, Garrido D. Species deletions from microbiome consortia reveal key metabolic interactions between gut microbes. mSystems. 2019;4:e00185–19.
Venturelli OS, Carr AV, Fisher G, Hsu RH, Lau R, Bowen BP, et al. Deciphering microbial interactions in synthetic human gut microbiome communities. Mol Syst Biol. 2018;14:e8157.
Wolfe BE, Button JE, Santarelli M, Dutton RJ. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell. 2014;158:422–33.
Barraclough TG. How do species interactions affect evolutionary dynamics across whole communities? Annu Rev Ecol, Evolution, Syst. 2015;46:25–48.
Fraser LH, Keddy P. The role of experimental microcosms in ecological research. Trends Ecol Evolution. 1997;12:478–81.
Benton TG, Solan M, Travis JMJ, Sait SM. Microcosm experiments can inform global ecological problems. Trends Ecol Evolution. 2007;22:516–21.
Belotte D, Curien JB, Maclean RC, Bell G. An experimental test of local adaptation in soil bacteria. Evolution. 2003;57:27–36.
Lawrence D, Fiegna F, Behrends V, Bundy JG, Phillimore AB, Bell T, et al. Species interactions alter evolutionary responses to a novel environment. PLOS Biol. 2012;10:e1001330.
Kraemer SA, Kassen R. Patterns of local adaptation in space and time among soil bacteria. Am Naturalist. 2015;185:317–31.
Fiegna F, Moreno-Letelier A, Bell T, Barraclough TG. Evolution of species interactions determines microbial community productivity in new environments. ISME J. 2015;9:1235–45.
Replansky T, Koufopanou V, Greig D, Bell G. Saccharomyces sensu stricto as a model system for evolution and ecology. Trends Ecol Evolution. 2008;23:494–501.
Botstein D, Fink GR. Yeast: an experimental organism for 21st century biology. Genetics. 2011;189:695–704.
Charron G, Leducq J-B, Bertin C, Dubé AK, Landry CR. Exploring the northern limit of the distribution of Saccharomyces cerevisiae and Saccharomyces paradoxus in North America. FEMS Yeast Res. 2014;14:281–8.
Sampaio JP, Gonçalves P. Natural populations of Saccharomyces kudriavzevii in Portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl Environ Microbiol. 2008;74:2144–52.
Kowallik V, Greig D. A systematic forest survey showing an association of Saccharomyces paradoxus with oak leaf litter. Environ Microbiol Rep. 2016;8:833–41.
Alsammar HF, Naseeb S, Brancia LB, Gilman RT, Wang P, Delneri D. Targeted metagenomics approach to capture the biodiversity of Saccharomyces genus in wild environments. Environ Microbiol Rep. 2019;11:206–14.
Boynton PJ, Stelkens R, Kowallik V, Greig D. Measuring microbial fitness in a field reciprocal transplant experiment. Mol Ecol Resour. 2017;17:370–80.
Anderson JB, Kasimer D, Xia W, Schröder NCH, Cichowicz P, Lioniello S, et al. Persistence of resident and transplanted genotypes of the undomesticated yeast Saccharomyces paradoxus in forest soil. mSphere. 2018;3:e00211–8.
Kowallik V, Miller E, Greig D. The interaction of Saccharomyces paradoxus with its natural competitors on oak bark. Mol Ecol. 2015;24:1596–610.
Filteau M, Charron G, Landry CR. Identification of the fitness determinants of budding yeast on a natural substrate. ISME J. 2016;11:959–71.
Botha A. The importance and ecology of yeasts in soil. Soil Biol Biochem. 2011;43:1–8.
Kuehne HA, Murphy HA, Francis Chantal A, Sniegowski PD. Allopatric divergence, secondary contact, and genetic isolation in wild yeast populations. Curr Biol. 2007;17:407–11.
Leducq J-B, Nielly-Thibault L, Charron G, Eberlein C, Verta J-P, Samani P, et al. Speciation driven by hybridization and chromosomal plasticity in a wild yeast. Nat Microbiol. 2016;1:15003.
Robinson HA, Pinharanda A, Bensasson D. Summer temperature can predict the distribution of wild yeast populations. Ecol Evol. 2016;6:1236–50.
Leducq J-B, Charron G, Samani P, Dubé AK, Sylvester K, James B, et al. Local climatic adaptation in a widespread microorganism. Proc R Soc B. 2014;281:20132472.
Bleuven C, Dubé AK, Nguyen GQ, Gagnon-Arsenault I, Martin H, Landry CR. A collection of barcoded natural isolates of Saccharomyces paradoxus to study microbial evolutionary ecology. MicrobiologyOpen. 2019;8:e00773.
Berns AE, Philipp H, Narres HD, Burauel P, Vereecken H, Tappe W. Effect of gamma-sterilization and autoclaving on soil organic matter structure as studied by solid state NMR, UV and fluorescence spectroscopy. Eur J Soil Sci. 2008;59:540–50.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217.
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. Vegan: community ecology package. 2017; Rpackage version 2.4-5. https://CRAN.R-project.org/package=vegan.
Faircloth BC, Glenn TC. Not all sequence tags are created equal: designing and validating sequence identification tags robust to indels. PLoS ONE. 2012;7:e42543.
Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Smith AM, Heisler LE, Mellor J, Kaper F, Thompson MJ, Chee M, et al. Quantitative phenotyping via deep barcode sequencing. Genome Res. 2009;19:1836–42.
Kloke JD, Mckean JW. Rfit: rank-based estimation for linear models. R J. 2012;4:57–64.
Hettmansperger TP, McKean JW Robust. Nonparametric statistical methods. 2nd ed. Boca Raton, FL: CRC Press; 2010.
RC Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2017. https://www.R-project.org/.
Nacke H, Thürmer A, Wollherr A, Will C, Hodac L, Herold N, et al. Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and Grassland soils. PLoS ONE. 2011;6:e17000.
Urbanová M, Šnajdr J, Baldrian P. Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol Biochem. 2015;84:53–64.
Eberlein C, Hénault M, Fijarczyk A, Charron G, Bouvier M, Kohn LM, et al. Hybridization is a recurrent evolutionary stimulus in wild yeast speciation. Nat Commun. 2019;10:923.
Li Y, Venkataram S, Agarwala A, Dunn B, Petrov DA, Sherlock G, et al. Hidden complexity of yeast adaptation under simple evolutionary conditions. Curr Biol. 2018;28:515–25.e6.
Maclean CJ, Metzger BPH, Yang J-R, Ho W-C, Moyers B, Zhang J. Deciphering the genic basis of yeast fitness variation by simultaneous forward and reverse genetics. Mol Biol Evolution. 2017;34:2486–502.
Payen C, Sunshine AB, Ong GT, Pogachar JL, Zhao W, Dunham MJ. High-throughput identification of adaptive mutations in experimentally evolved yeast populations. PLOS Genet. 2016;12:e1006339.
Tsai IJ, Bensasson D, Burt A, Koufopanou V. Population genomics of the wild yeast Saccharomyces paradoxus: quantifying the life cycle. Proc Natl Acad Sci. 2008;105:4957–62.
Streeter JG. Allantoin and allantoic acid in tissues and stem exudate from field-grown soybean plants. Plant Physiol. 1979;63:478–80.
Yurkov A. Yeasts in forest soils. In: Buzzini P, Lachance M-A, Yurkov A, editors. Yeasts in natural ecosystems: diversity. Cham: Springer International Publishing; 2017. p. 87–116.
Castaño C, Lindahl BD, Alday JG, Hagenbo A, Martínez de Aragón J, Parladé J, et al. Soil microclimate changes affect soil fungal communities in a Mediterranean pine forest. New Phytol. 2018;220:1211–21.
Ketola T, Saarinen K. Experimental evolution in fluctuating environments: tolerance measurements at constant temperatures incorrectly predict the ability to tolerate fluctuating temperatures. J Evolut Biol. 2015;28:800–6.
Wigley K, Wakelin SA, Moot DJ, Hammond S, Ridgway HJ. Measurements of carbon utilization by single bacterial species in sterile soil: insights into Rhizobium spp. J Appl Microbiol. 2016;121:495–505.
McNamara NP, Black HIJ, Beresford NA, Parekh NR. Effects of acute gamma irradiation on chemical, physical and biological properties of soils. Appl Soil Ecol. 2003;24:117–32.
Kaiser K, Wemheuer B, Korolkow V, Wemheuer F, Nacke H, Schöning I, et al. Driving forces of soil bacterial community structure, diversity, and function in temperate grasslands and forests. Sci Rep. 2016;6:33696.
Meyer JR, Kassen R. The effects of competition and predation on diversification in a model adaptive radiation. Nature. 2007;446:432–5.
Pekkonen M, Ketola T, Laakso JT. Resource availability and competition shape the evolution of survival and growth ability in a bacterial community. PLoS ONE. 2013;8:e76471.
Richards TA, Talbot NJ. Horizontal gene transfer in osmotrophs: playing with public goods. Nat Rev Microbiol. 2013;11:720–7.
Effmert U, Kalderás J, Warnke R, Piechulla B. Volatile mediated interactions between bacteria and fungi in the soil. J Chem Ecol. 2012;38:665–703.
Schmidt R, Cordovez V, de Boer W, Raaijmakers J, Garbeva P. Volatile affairs in microbial interactions. ISME J. 2015;9:2329–35.
Mackie AE, Wheatley RE. Effects and incidence of volatile organic compound interactions between soil bacterial and fungal isolates. Soil Biol Biochem. 1999;31:375–85.
Schulz S, Dickschat JS. Bacterial volatiles: the smell of small organisms. Nat Prod Rep. 2007;24:814–42.
Dini-Andreote F, Stegen JC, van Elsas JD, Salles JF. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc Natl Acad Sci USA. 2015;112:E1326–32.
Kraemer SA, Boynton PJ. Evidence for microbial local adaptation in nature. Mol Ecol. 2017;26:1860–76.
Fox JW, Harder LD. Using a “time machine” to test for local adaptation of aquatic microbes to temporal and spatial environmental variation. Evolution. 2015;69:136–45.
Acknowledgements
We thank Alexandre K. Dubé, François Rouleau and Julien Ponchart for discussion, and Souhir Marsit and Anna Fijarczyk for comments on the paper. This project was supported by a NSERC Discovery grant to CRL. CRL holds the Canada Research Chair in Evolutionary Cell and Systems Biology.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bleuven, C., Nguyen, G.Q., Després, P.C. et al. Competition experiments in a soil microcosm reveal the impact of genetic and biotic factors on natural yeast populations. ISME J 14, 1410–1421 (2020). https://doi.org/10.1038/s41396-020-0612-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-0612-8
