
Abstract
Plant microbiomes are shaped by forces working at different spatial scales. Environmental factors determine a pool of potential symbionts while host physiochemical factors influence how those microbes associate with distinct plant tissues. These scales are seldom considered simultaneously, despite their potential to interact. Here, we analyze epiphytic microbes from nine Hibiscus tiliaceus trees across a steep, but short, environmental gradient within a single Hawaiian watershed. At each location, we sampled eight microhabitats: leaves, petioles, axils, stems, roots, and litter from the plant, as well as surrounding air and soil. The composition of bacterial communities is better explained by microhabitat, while location better predicted compositional variance for fungi. Fungal community compositional dissimilarity increased more rapidly along the gradient than did bacterial composition. Additionally, the rates of fungal community compositional dissimilarity along the gradient differed among plant parts, and these differences influenced the distribution patterns and range size of individual taxa. Within plants, microbes were compositionally nested such that aboveground communities contained a subset of the diversity found belowground. Our findings indicate that both environmental context and microhabitat contribute to microbial compositional variance in our study, but that these contributions are influenced by the domain of microbe and the specific microhabitat in question, suggesting a complicated and potentially interacting dynamic.
Similar content being viewed by others
Introduction
Plants harbor communities of microorganisms that influence their biology, including phenology [1], water conductance [2], niche occupancy and range expansion [3,4,5], and competitive ability [6]. Nearly all plant traits are likely affected by microbial partners in some way. Our understanding of symbiotic microbial functions, however, has outpaced our understanding of how plants and their microbes form relationships that persist in nature. This disjuncture stems from the sheer complexity of microbial communities, compounded by assembly patterns governed by interacting processes at multiple ecological scales, from the landscape level to variation among plant tissues within an individual plant [7, 8].
At large scales, abiotic environmental factors can influence plant microbiome composition [9]. Plant-associated microbial communities can change across elevation gradients [10,11,12,13,14] or in response to soil properties (e.g., organic carbon, soil pH, nitrogen content [15], and land-use history [16]). Even in the absence of obvious environmental clines, geographic distance coupled with presumed dispersal limitation can alter microbiome composition [17,18,19]. Host identity [20] and genotype [8, 21, 22] can also covary with environmental factors, which can influence microbial composition as well [23].
Within plants, distinct microbial communities associate with different plant tissues [24,25,26], and this factor can be more predictive of plant microbial composition than location, even over broad geographic areas. For instance, a root microbiome can be more similar to another root microbiome several hundred kilometers away than to a leaf microbiome on the same individual plant [23]. The assembly processes governing within-plant microbial assembly is not merely a recapitulation of dispersal- or environmentally mediated dissimilarity at a smaller scale. While large environmental clines can result in compositional turnover (i.e., replacement of species between communities) [27], microbiomes within individual plants tend to be compositionally nested such that apical parts (e.g., leaves, flowers) house a subset of the microbial species associated with subterranean plant parts, which themselves are most species-rich [19]. This nestedness pattern indicates that, although compositionally distinct, microbiomes of different plant tissues within a plant are neither entirely independent of each other nor of the larger environmental species pool. Therefore, the microbial composition of a given plant part may be partially attributed to the local environment [28], as well as to strong selective forces from the host plant itself [29].
Despite a large and growing body of literature examining plant microbial assembly at a landscape or within-plant scales [23, 28, 30,31,32], few studies have considered how environmental context at the landscape-scale relates to within-plant microhabitat composition, or how residency within plant microhabitats relates to microbial distributions at landscape scales. A more complete understanding of plant microbiome assembly might simultaneously consider whether environmental factors circumscribe a local pool of microbial symbionts [33,34,35] and the extent to which that local pool is secondarily partitioned within plants [24,25,26, 29, 30]. A true interaction between scales might work in the opposite direction as well because factors at small spatial scales can influence distribution at larger scales. For example, a microbe’s specificity for different plant tissues might affect the availability and density of habitat and therefore a microbe’s prevalence over space or along an environmental gradient.
Our field site in Hawai’i is uniquely provisioned to address questions about plant microbial assembly processes at multiple scales. In fewer than 6 km, the main rivers of Waimea Valley plunge from a mid-elevation rainforest, over cascading waterfalls, into a protected estuary and finally the ocean. This short distance spans steep gradients in elevation and precipitation, with an annual rainfall differential of nearly one meter from our highest to lowest sites. Along this gradient exists a diversity of connected terrestrial habitats located in close proximity, where an entire watershed is contained within a small, relatively isolated, area. Although plant communities change rapidly along this gradient, Hibiscus tiliaceus trees are found throughout, enabling us to focus on a single plant species and thus disentangle host identity from large variations in abiotic conditions within a distance plausibly connected by microbial dispersal.
In this study, we asked how plant microbial compositional variance is partitioned between factors at large and small scales. We expected a strong environmental gradient to correlate with microbial community differentiation across sampling locations, especially among those communities not directly associated with plant tissues. Among plant-associated communities, we expected that strong tissue-level association would reduce community turnover along the abiotic gradient. We hypothesized that microbes occupying a greater number of habitats would occupy greater spans of the gradient due to greater environmental tolerance and higher density of available habitat. Finally, we assessed the nature of the linkages among plant parts by investigating patterns of nestedness between belowground communities and apical plant parts, and how these linkages differ among environmental contexts. This system serves as a unique example of how biotic and abiotic determinants can overlap to contribute to variation in microbial community composition within a watershed system encompassing a steep natural gradient.
Materials and methods
Site description and sampling
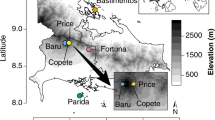
Within 5 h on the morning of January 31, 2019, we collected microbial samples from, and adjacent to, nine mature healthy Hibiscus tiliaceus L. (Malvaceae; Hawaiian: hau) individuals within Waimea Valley on the north shore of the island of Oʻahu, Hawai’i (Fig. 1). This broad-leafed medium-sized tree is indigenous to tropical Indo-Pacific, but has attained a pantropical distribution via human transport [36] and was possibly introduced to the Hawaiian Islands by Polynesian settlers [37]. Sample trees were located along a transect that paralleled a perennial stream. Locations of H. tiliaceus ranged from 0 to 700 m elevation, spanning 1174–2026 mm annual rainfall over a distance of ~5.5 km (Fig. 1). Rainfall, elevation, and distance from the shore are highly collinear, so we refer to this continuous variable cline as the “environmental gradient”. For each plant, we sampled epiphytic microbes from standardized surface areas (200 cm2) of leaves, petioles, axils, stems, roots and adhering soil, and litter with a sterile cotton swab (one per microhabitat). We sampled surface microbes because (1) doing so let us standardize sampling area across these microhabitat types; (2) it mitigates known issues of co-amplification of chloroplasts (16S) and plant host (ITS) DNA when microbial:plant biomass ratios are low; and (3) plant surfaces represent important interfaces between plants and their environments where gas, water, and nutrient exchange occurs. To additionally sample the soil microbiome, we dug a 5-cm hole with a sterilized corer ~1 m from the canopy edge to avoid sampling the rhizosphere, and swabbed the sides and bottom of the hole. For 2 weeks prior to collecting the above samples, we also deployed an air sampler modified from Quesada et al. [38] at each of our H. tiliaceus sites, which collected aerial microbes on rotating sterilized glass slides lined with microtiter plate sealing film (Thermo Fisher Scientific, Waltham, MA). After collection, we immediately transferred all swabs and film to a microcentrifuge tube with 1 mL lysis buffer and garnet homogenization beads (Qiagen NV, Venlo, Netherlands; see Supplementary Methods for details). In total, we collected 72 biological samples (9 sites by 8 microhabitats). For negative controls, we exposed sterile swabs dipped in lysis buffer to the air for ~20 s. and processed them using the same protocols as the biological samples. A single air sampling film that we did not expose to the environment served as an air sampler negative control. Hereafter, we refer to the five plant tissue types (leaves, petioles, axils, stems, and roots) as well as the air, litter, and soil collectively as “microhabitats”, which we treat as a categorical variable.

Circle radius indicates the amount of annual precipitation, with shaded gradient indicating elevation.
DNA extraction and library preparation
We extracted and purified DNA using the KingFisher™ Flex™ System (Thermo Fisher Scientific, Waltham, MA, USA) with a MagAttract PowerSoil KF Kit (Qiagen NV, Venlo, Netherlands) following the manufacturer’s protocol, but increased the volume of elution buffer to 125 µL. To survey bacteria, we amplified the V4 region of the 16S ribosomal RNA gene with the primers 515F and 806R, and for fungi, we amplified the ribosomal internal transcribed spacer 1 (ITS1) using the forward ITS1F and reverse ITS2 primer schemes [39, 40]. This primer pair has the advantage of high fungal fidelity, which is useful when assessing host-associated organisms, although this comes at the cost of known mismatches in the priming region of some fungal lineages [41]. Both library preparations involved an 8-base-pair index, a gene-specific primer, and an adaptor construct concatenated on the same oligomer. PCR protocols entailed an initial phase of 95° C for 3 min using Hot Start Taq polymerase (New England BioLabs Inc., Ipswich, MA, USA), followed by 35 cycles at 50 °C and 52 °C annealing temperatures for 16S and ITS amplicons, respectively. We then cleaned and normalized PCR products using the Just-a-Plate™ 96 PCR Purification and Normalization Kit (Charm Biotech, San Diego, CA, USA). We separately pooled bacteria and fungi amplicons and checked the quality of libraries with a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). The Advanced Studies in Genomics, Proteomics, and Bioinformatics Laboratory at the University of Hawai’i–Mānoa sequenced the libraries in two runs using separate 300-bp paired-end Illumina MiSeq v3 reagent kits (Illumina Inc., San Diego, CA, USA). Sequence data are available at the Sequence Read Archive as PRJNA543421.
Data processing and analyses
Because of fundamental differences between ITS and 16S loci, we used different processing pipelines. We processed fungal FASTQ files using ITSxpress [42] to extract ITS1 regions of variable lengths from adjacent conserved ribosomal subunit genes. We used the FASTX-Toolkit [43] to filter sequences by quality scores, and to discard reads that met at least one of the following conditions: (1) 10% or more of their bases contained a q-score lower than 25; (2) they contained an “N” nucleotide; or (3) their length was less than 20 bp. We used VSEARCH [44] to identify and remove chimeras. The DADA2 package [45] corrected sequencing errors and binned amplicon sequence variants (ASVs; see Callahan et al. [46] for discussion on ASVs). We used CONSTAX [47] with the UNITE 8.0 database to assign fungal taxonomy via consensus of three separate classification algorithms. Finally, we used the LULU algorithm [48] to collapse putative within-genome ribotype variants into a single ASV.
We used DADA2 to process 16S reads. First, we truncated reads at position 210 (190 for the reverse read) and discarded these if they contained at least one base below quality 2 or a number of expected errors above 3. We used DADA2’s default parameters to denoise the data, and merged reads if they overlapped by at least 20 bases, allowing for one mismatch at most. We used MOTHUR [49] along with the Silva database v132 to filter and annotate sequences. We removed potential chimeras with VSEARCH as implemented in MOTHUR, and assigned bacteria taxonomy via the MOTHUR functions classify.seqs() and classify.otus(). As with the ITS pipeline, we corrected bacteria ASVs with LULU. Finally, we used the R package decontam [50] to identify and remove putative contaminants (33 bacteria and 4 fungi) based on their prevalence in the extraction and PCR negative controls.
The processed dataset contained 15,649 bacterial and 12,558 fungal ASVs, belonging to 96 and 35 classes, respectively. We amalgamated ASV read abundance data, taxonomic assignments, and sample metadata using the R package phyloseq [51], and appended tree location climate information from the University of Hawai’i–Mānoa’s Climate of Hawai’i data portal [52] using the R package raster [53].
To test the extent to which either location along the gradient or microhabitat explained the composition of bacteria or fungi, we used permutational multivariate analysis of variance (PERMANOVA) [54] on a Bray–Curtis dissimilarity matrix of Hellinger transformed data with 10,000 permutations in the R package vegan [55]. We performed an additional PERMANOVA to test the influence of ground type (i.e., above- or belowground) habitats on bacteria and fungi community composition. We used the same distance matrices in a series of Mantel tests examining the correlation between community dissimilarity and geographic distance (i.e., the pairwise distance between tree locations) for each microhabitat separately and combined, using Benjamin–Hochberg corrected P values to account for Type 1 errors due to multiple hypothesis testing [56]. Because geographic distance is inextricably correlated with gradient variables (such as elevation, precipitation, and solar irradiance), this measure is a proxy for the combined effects of dispersal limitation and environmental dissimilarity. To examine the extent to which deviations among compositions of communities of bacteria and fungi were coordinated, we tested the correlation between Bray–Curtis distance matrices for fungi and bacteria ASVs using Mantel.
For each site and all sites combined, we used the R packages vegan and bipartite [57, 58] to evaluate nestedness in two ways: (1) a temperature statistic (T) in which 0° is perfectly nested and 100° is perfectly random based on pairwise compositional differences [59]; and (2) the Nestedness metric based on Overlap and Decreasing Fill (NODF) [60]. Significance values were calculated by comparing empirical nestedness temperature against the distribution of 1000 randomized null communities, in which marginal values were maintained. Furthermore, we assessed relationships between nestedness metrics and gradient location (distance from shore) using Spearman correlations. To account for differences in sampling depth, these analyses used sample counts randomly down-sampled (rarefied) to the same depth per sample (bacteria: 26,406; fungi: 20,000). To assess the extent to which within-sample microbial richness was captured given our sequencing depth, we calculated rarefaction curves on each sample (Supplementary Fig. S1).
To examine the relationships between local ASV abundances, geographic ranges, and number of occupied microhabitats, we used regression analyses. We calculated local abundance as the mean sequence abundance per sample of an ASV was present, omitting nulls. The maximum distance between locations where we detected an ASV along the transect gave us its range; ASVs detected at a single location had a range of zero. We determined an ASV’s habitat occupancy as the sum of microhabitats where we detected an ASV. Although these calculations may include dead cells and thus overestimate empirical ranges or habitat breadths of viable cells, we believe these errors will be propagated randomly throughout the dataset and are nevertheless preferable to arbitrary sequence abundance cutoffs. A two-tailed t test assessed the difference of mean range sizes between distributions of fungi and bacteria.
To provide an assessment of bacterial and fungal taxa circumscribed at more comparable taxonomic levels, we repeated the range distribution, PERMANOVA and Mantel analysis using a dataset, in which ASVs were binned by genus (presented in the Supplementary Information). Although this provides additional data to evaluate differences and similarities between these domains, we caution that intrinsic differences between reproductive strategies and taxonomic conventions (among other issues) make direct comparisons between fungi and bacteria problematic. Scripts, data, and markdown documents necessary to reproduce all analyses are accessible on GitHub (github.com/cbwall/Waimea-plant-microbiomes).
Results
PERMANOVA tests indicated that communities were significantly differentiated by microhabitat type and that this factor best predicted bacterial community variance (Table 1, R2 = 0.336, P < 0.001; Supplementary Table S1). In contrast, site location was the most predictive factor in the analysis of fungal composition (R2 = 0.255, P < 0.001). At the class level, communities of bacteria from above- and belowground samples were compositionally distinct (Supplementary Fig. S2), whereas fungal classes were uniformly distributed among microhabitats (Supplementary Fig. S3). At the ASV scale, however, compositional differences were more readily apparent among bacteria samples from different microhabitats and between fungal samples from below- and aboveground (Fig. 2 and Supplementary Table S2).

a Bacterial communities cluster by above- and belowground habitats. b Fungal communities also cluster somewhat by aboveground and belowground parts, albeit with higher dispersion in the belowground communities and more overlap with the aboveground parts compared to communities of bacteria.
There was no evidence for a distance decay relationship (i.e., compositional dissimilarity over distance) among bacteria communities, except for those associated with roots or when the entire microbiome of the site was considered together (Table 2). Conversely, communities of fungi, when aggregated by either site or microhabitat, demonstrated significant distance decay patterns over the environmental gradient. Fungal communities from different microhabitats showed dissimilar levels of variation across the environmental gradient. This suggests that some plant parts host a more conserved microbial community than others. Mantel correlations of subterranean fungal samples (roots and soil) were highest, while there were no significant distance decay patterns among fungal communities on axils and leaves (Table 2). Fungal communities that were not directly associated with a plant (soil and air) showed strong distance decay of similarity patterns. Patterns were similar when we examined taxa at the genus-scale, although correlation values were generally lower than those measured at the scale of ASVs (Supplementary Table S3).
Although fungal and bacterial compositional dissimilarities were best explained by different factors, their between-sample dissimilarities were correlated. In other words, pairwise fungal and bacterial community composition of the same biological samples, as assessed by Bray–Curtis dissimilarity, was significantly more correlated than would be expected by chance (Mantel r = 0.417, P < 0.001; Supplementary Fig. S4).
Communities of both bacteria and fungi were significantly nested by microhabitat (Fig. 3), with belowground samples containing much of the species diversity found within the other microhabitat communities. Each had a moderate nestedness temperature (bacteria: T = 38.8°, P = 0.001, NODF = 39.04%; fungi: T = 43.1°, P = 0.001, NODF = 34.76%). We detected no nestedness with regard to location along the gradient (e.g., microbial communities in drier locations were not subsets of those in wetter locations).

Each vertical line represents an ASV’s presence among microhabitats. Both (a) bacterial and (b) fungal display moderate nestedness with most of the microbial diversity contained in the subterranean microhabitats. In a perfectly nested model (T = 0°) ASVs would be constrained to the left of the black line.
The range size of bacterial and fungal ASVs differed significantly (t = 32.062, df = 13235.00, P < 0.001), with the mean range of bacteria (x̄ = 2078.0 m, SD = 2044.9 m) being nearly twice the range of fungi (x̄ = 1170.2 m, SD = 1845.1 m; Fig. 4). As expected, the range sizes of microbial genera were larger than those of ASVs, but bacteria (x̄ = 3757.5 m, SD = 2096.5 m) remained significantly more widespread than fungi (x̄ = 3046.9 m, SD = 2270.3 m; t = 5.729, df = 869.79, P < 0.001). For both bacteria and fungi, an ASV’s range size correlated positively with the number of microhabitats in which an ASV occurred, although this relationship was stronger for fungi (R2 = 0.451, P < 0.001) than bacteria (R2 = 0.262, P < 0.001). Fungal ASV local abundance scarcely correlated with range size (R2 = 0.012, P < 0.001), and not at all for bacteria (P = 0.554).

Boxplots show range spans of (a) bacterial and (b) fungal genera and ASVs, depicting the interquartile range, including median, and whiskers showing 95th percentiles. Scatterplots show the relationship between each (c) bacterial and (d) fungal ASV’s range and the number of microhabitat types in which it occurs, with lines depicting the least-squares fit of the data. The relationship between range span and log-transformed ASV abundance for (e) bacteria and (f) fungi per occupied microhabitat, revealing no significance for bacteria but a weak trend for fungi.
Discussion
Here, we demonstrate that environmental context and microhabitat shape the plant microbiome in domain-specific and potentially synergistic ways. In our study, fungal communities are determined more by location along the environmental gradient than by microhabitat type. Nonetheless, fungal communities had a high variation of distance decay patterns among microhabitat types, suggesting links between a fungal community’s distribution at large and small scales. This correlation is mirrored in the distribution of individual microbes: microhabitat breadth was a significant predictor of gradient range sizes of microbes. Bacteria community compositional variance, in contrast, was better explained by microhabitat than by location, and there was little indication that communities associated with different microhabitats demonstrated different rates of compositional dissimilarity along the gradient. The pairwise dissimilarity between communities of fungi and communities of bacteria was significantly correlated among biological samples, and both were compositionally nested within plants, suggesting that at least some compositional patterns apply universally to members of both domains.
Effect of microhabitat on microbial distribution
Similar to previous studies [19, 28, 61], we show that microhabitat type significantly predicts microbial community composition. This was the most discriminant factor for bacteria communities (Table 1). At the ASV level, both bacterial and fungal communities are distinguishable primarily by whether they are located above- or belowground (Fig. 2 and Supplementary Table S2). This distinction is influenced by the taxonomic scale considered. While this division between above and belowground composition was apparent at the class level for bacteria (Supplementary Fig. S2), nearly all fungal classes were uniformly distributed across the eight microhabitats Supplementary (Supplementary Fig. S3).
Surface microbes may have patterns of microhabitat niche breadth that are distinct from those residing within plants. Previous studies showed that different variables drive epiphytic versus endophytic microbes [23]. Because surface communities contain a portion of microbes only transiently associated with hosts, more persistent endophytic microbes might demonstrate higher fidelity than would epiphytes. In a survey of endophytic fungi associated with grassland plants, Wearn et al. [29] found most taxa were associated with single plant tissue, and that those associated with more than one were rare, implying a link between specialization and endophytic habitat [30]. Nevertheless, we found that microhabitat predicted a significant and canonically large proportion of compositional variance (bacteria R2 = 0.336; fungi R2 = 0.192), suggesting that specialization is not just restricted to endophytes.
The wide niche breadth of some microbes is reflected in patterns of compositional nesting, with aboveground communities comprising a subset of the ASVs present belowground (Fig. 3). This pattern is replicated at all locations throughout our environmental gradient and is consistent with previous studies on other plant species [19, 28, 61]. Although the causes of these vertically stratified patterns are undetermined, soils may inoculate plants with generalist microbes during germination, followed by additional non-random factors (e.g., desiccation, dispersal limitation) that prune the original microbial community over time and distance from the ground [19, 61]. An alternative explanation might be simply that rainfall and gravity disperse microbes downwards where they persist in high proportions. Intriguingly, rhizosphere communities, not soil, are basal in the nestedness hierarchy, congruent with previous studies [19, 27]. This might reflect this habitat’s unique mix of diverse soil- and plant-associated microbial communities in a resource-rich and heterogeneous environment.
Links between microhabitat and environmental gradient
In contrast to communities of bacteria, fungi demonstrated strong differences among communities assorted with microhabitat type with regards to compositional dissimilarity along the environmental gradient (Table 2 and Supplementary Table S3). Our results show that subterranean fungal communities demonstrated the steepest dissimilarity slopes, which could result from greater heterogeneity among subsurface microhabitats [61,62,63] or more limited dispersal among soil-bound microbes [64] as compared to aboveground counterparts. The absence of distance decay patterns among foliar microbiomes is not surprising given the high compositional variance of these consortia compared with those on other plant surfaces [19, 65] even within plant individuals [26]. We originally suspected that communities on plant tissues might demonstrate less compositional turnover than free-living samples [27], and that leaf litter communities might be intermediate, and our data partially supported this hypothesis. Leaf and axil-associated fungal communities were not correlated with the gradient, whereas litter fungal communities were. However, petiole and stem tissue fungal communities were also correlated, indicating that merely being associated with a plant does not, in and of itself, buffer a microbial community from additional environmental determinants. However, both free-living fungal communities were strongly influenced by the gradient. Fungal communities in air, for example, which should be the least constrained by dispersal limitation but, maximally influenced by the abiotic environment, showed a strong distance decay pattern (Table 2 and Supplementary Table S3).
Furthermore, the correlation between an ASV’s range size and the number of microhabitats it occupies (Fig. 4) implies a relationship between habitat specialization and environmental (in)tolerance, such that niche breadth within a plant is predictive of ecological breadth across the entire gradient. Some environmental gradients across Waimea Valley might be replicated at a small scale within a plant (moisture being the most obvious), so this relationship might reflect environmental filtering for certain traits (e.g., desiccation tolerance). Phylogenetics and physiology can dictate microbial biogeography on a global scale [66]. Alternatively, taxa with broad niches have a higher probability of establishing after dispersal as they are more likely to find a suitable microhabitat. In our study, the most widespread bacterium (an ASV identified as a member of the Rokubacteria) and the most widespread fungus (an ASV identified as a member of the Geminibasidiomycetes) are known as habitat generalists, with the latter being both heat and desiccation-tolerant (Supplementary Figs. S5 and S6 [67, 68]).
The weak (fungi) or non-significant (bacteria) relationship between ASV abundance and microhabitat occupancy suggests that the correlation between niche breadth at small and large scales are not solely attributable to numerically dominant taxa. Despite having wider ranges, generalists are not, on average, more abundant than less generalist taxa, suggesting that neither generalists nor more specialized taxa dominate the communities in our study system (Fig. 4).
Differences and similarities between bacteria and fungi
Several factors might explain why the environmental gradient was the best determinant of fungal communities, but secondary among bacteria. Dispersal limitation [9, 69], environmental dissimilarity [70], host genetic clines [8, 21] or some combination of these or other factors may affect bacteria and fungi differently. Of the relatively few studies that concurrently examine bacteria and fungi, fungal communities tend to be more influenced by geographic distance. Coleman-Derr et al. [23] found that agave hosts in southern California and Mexico shared a smaller percentage of fungi than bacteria (18.2% and 72.2%, respectively). A study on Populus deltoides roots in Tennessee and North Carolina revealed that geography explained more variance for epiphytic fungal communities than for bacterial ones, although this trend was not the same for endophytes [71].
Notably, differing rates of evolution between marker genes might partially explain the contrasting patterns we found between fungi and bacteria. Because the ITS gene is less phylogenetically conserved than the adjacent small subunit rRNA gene, the ITS circumscribes taxa at a finer scale than does 16S. ASVs derived from 16S and ITS sequences, therefore, may not be equivalent in terms of evolutionary divergence. We partially account for these differences by corroborating our results with taxa circumscribed at the genus level, as opposed to sequence similarity. However, taxonomic concepts are not directly comparable between domains with different reproductive strategies, so attributing the different patterns we detected to biology versus taxonomic scale is challenging.
Notwithstanding differences in spatial patterns and plant part specialization, however, the community compositional turnover of fungi was correlated with that of bacteria (Supplementary Fig. S2). Regardless of whether this relationship reflects interdomain microbial biotic interactions or merely coordinated response to environmental cues, we find it remarkable that despite billions of years of evolutionary divergence, organisms’ distributions are similarly constrained by being small and plant-associated.
Conclusion
Our results contribute important baseline observational data within the constrained natural setting of Waimea Valley to determine which factors and scales predict the distribution of tree-associated bacteria and fungi along a steep, but short, environmental gradient. We show that microhabitat type (i.e., soil, litter, plant parts, air) was better at describing bacterial communities, whereas fungal community variance was more correlated with location along the environmental gradient, which differed based on microhabitat considered. A microbe’s habitat breadth within sites generally correlated with range breadth among sites along the gradient. Communities from both microbial domains were strongly nested such that aboveground microbes represented a subset of subterranean ones. While we are limited in our ability to apply causal interpretations to these patterns, given that our data were observational, our results are consistent with interacting assembly processes working at the landscape and within-plant scales. With these data in hand, we can devise manipulative experimental systems to directly model and parameterize hierarchical models of plant symbiont assembly processes, and address their underlying mechanisms such as dispersal abilities, and environmental or microhabitat specificity.
References
Zahn G, Amend AS. Foliar fungi alter reproductive timing and allocation in Arabidopsis under normal and water-stressed conditions. Fungal Ecol. 2019;41:101–6.
Arnold AE, Engelbrecht BMJ. Fungal endophytes nearly double minimum leaf conductance in seedlings of a neotropical tree species. J Trop Ecol. 2007;23:369–72.
Taghavi S, Garafola C, Monchy S, Newman L, Hoffman A, Weyens N, et al. Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl Environ Microbiol. 2009;75:748–57.
Berg G, Köberl M, Rybakova D, Müller H, Grosch R, Smalla K. Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiol Ecol. 2017;93:fix050. https://doi.org/10.1093/femsec/fix050.
Choudoir MJ, Barberan A, Menninger HL, Dunn RR, Fierer N. Variation in range size and dispersal capabilities of microbial taxa. Ecology. 2017;99:322–34.
Klironomos JN. Feedback with soil biota contributes to plant rarity and invasiveness in communities. Nature. 2002;417:67–70.
Dini-Andreote F, Raaijmakers JM. Embracing community ecology in plant microbiome research. Trends Plant Sci. 2018;23:467–9.
Beilsmith K, Thoen MPM, Brachi B, Gloss AD, Khan MH, Bergelson J. Genome-wide association studies on the phyllosphere microbiome: embracing complexity in host-microbe interactions. Plant J Cell Mol Biol. 2019;97:164–81.
Peay KG, Kennedy PG, Talbot JM. Dimensions of biodiversity in the Earth mycobiome. Nat Rev Microbiol. 2016;14:434–47.
Wang J, Soininen J, He J, Shen J. Phylogenetic clustering increases with elevation for microbes. Environ Microbiol Rep. 2012;4:217–26.
Zimmerman NB, Vitousek PM. Fungal endophyte communities reflect environmental structuring across a Hawaiian landscape. Proc Natl Acad Sci USA. 2012;109:13022–7.
Yang Y, Gao Y, Wang S, Xu D, Yu H, Wu L, et al. The microbial gene diversity along an elevation gradient of the Tibetan grassland. ISME J. 2014;8:430–40.
Shen C, Ni Y, Liang W, Wang J, Chu H. Distinct soil bacterial communities along a small-scale elevational gradient in alpine tundra. Front Microbiol. 2015;6:582.
Yao F, Yang S, Wang Z, Wang X, Ye J, Wang X, et al. Microbial taxa distribution is associated with ecological trophic cascades along an elevation gradient. Front Microbiol. 2017;8:2071.
Na X, Xu TT, Li M, Ma F, Kardol P. Bacterial diversity in the rhizosphere of two phylogenetically closely related plant species across environmental gradients. J Soils Sediment. 2017;17:122–32.
Jangid K, Williams MA, Franzluebbers AJ, Schmidt TM, Coleman DC, Whitman WB. Land-use history has a stronger impact on soil microbial community composition than aboveground vegetation and soil properties. Soil Biol Biochem. 2011;43:2184–93.
Talbot JM, Bruns TD, Taylor JW, Smith DP, Branco S, Glassman SI, et al. Endemism and functional convergence across the North American soil mycobiome. Proc Natl Acad Sci USA. 2014;111:6341–6.
Oono R, Rasmussen A, Lefèvre E. Distance decay relationships in foliar fungal endophytes are driven by rare taxa: distance decay in fungal endophytes. Environ Microbiol. 2017;19:2794–805.
Amend AS, Cobian GM, Laruson AJ, Remple K, Tucker SJ, Poff KE, et al. Phytobiomes are compositionally nested from the ground up. PeerJ. 2019;7:e6609.
Fitzpatrick CR, Copeland J, Wang PW, Guttman DS, Kotanen PM, Johnson MTJ. Assembly and ecological function of the root microbiome across angiosperm plant species. Proc Natl Acad Sci USA. 2018;115:E1157–65.
Bálint M, Tiffin P, Hallström B, O’Hara RB, Olson MS, Fankhauser JD, et al. Host genotype shapes the foliar fungal microbiome of balsam poplar (Populus balsamifera). PLoS ONE. 2013;8:e53987.
Bálint M, Bartha L, O’Hara RB, Olson MS, Otte J, Pfenninger M, et al. Relocation, high-latitude warming and host genetic identity shape the foliar fungal microbiome of poplars. Mol Ecol. 2015;24:235–48.
Coleman‐Derr D, Desgarennes D, Fonseca‐Garcia C, Gross S, Clingenpeel S, Woyke T, et al. Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. N. Phytol. 2016;209:798–811.
Lindow SE, Brandl MT. Microbiology of the phyllosphere. Appl Environ Microbiol. 2003;69:1875–83.
Redford AJ, Bowers RM, Knight R, Linhart Y, Fierer N. The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ Microbiol. 2010;12:2885–93.
Leff JW, Del Tredici P, Friedman WE, Fierer N. Spatial structuring of bacterial communities within individual Ginkgo biloba trees. Environ Microbiol. 2015;17:2352–61.
Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551:457–63.
de Souza RSC, Okura VK, Armanhi JSL, Jorrín B, Lozano N, da Silva MJ, et al. Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Sci Rep. 2016;6:28774.
Wearn JA, Sutton BC, Morley NJ, Gange AC. Species and organ specificity of fungal endophytes in herbaceous grassland plants. J Ecol. 2012;100:1085–92.
Beckers B, Op De Beeck M, Weyens N, Boerjan W, Vangronsveld J. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome. 2017;5:25.
Desgarennes D, Garrido E, Torres-Gomez MJ, Peña-Cabriales JJ, Partida-Martinez LP. Diazotrophic potential among bacterial communities associated with wild and cultivated Agave species. FEMS Microbiol Ecol. 2014;90:844–57.
Fonseca-García C, Coleman-Derr D, Garrido E, Visel A, Tringe SG, Partida-Martínez LP. The cacti microbiome: interplay between habitat-filtering and host-specificity. Front Microbiol. 2016;7:150.
Lindström ES, Langenheder S. Local and regional factors influencing bacterial community assembly. Environ Microbiol Rep. 2012;2012:1–9.
Kembel SW, O’Connor TK, Arnold HK, Hubbell SP, Wright SJ, Green JL. Relationships between phyllosphere bacterial communities and plant functional traits in a neotropical forest. Proc Natl Acad Sci USA. 2014;111:13715–20.
Frank AC, Saldierna Guzmán JP, Shay JE. Transmission of bacterial endophytes. Microorganisms. 2017;5:70.
Wu Z, Raven PH, Hong D. Hibiscus tiliaceus. eFloras. St. Louis, MO: Missouri Botanical Garden & Cambridge, MA: Harvard University Herbaria; 2019. p. 287–8.
Motooka P, Castro L, Nelson D, Nagai G, Ching L. Weeds of Hawaiʻi’s pastures and natural areas: an identification and management guide. Honolulu: University of Hawaiʻi Press; 2014.
Quesada T, Hughes J, Smith K, Shin K, James P, Smith J. A low-cost spore trap allows collection and real-time PCR quantification of airborne Fusarium circinatum spores. Forests. 2018;9:586.
Smith DP, Peay KG. Sequence depth, not PCR replication, improves ecological inference from next generation DNA sequencing. PLoS ONE. 2014;9:e90234.
Walters W, Hyde ER, Berg-Lyons D, Ackermann G, Humphrey G, Parada A, et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems. 2016;1:e00009–15.
Bellemain E, Carlsen T, Brochmann C, Coissac E, Taberlet P, Kauserud H. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol. 2010;10:189.
Rivers AR, Weber KC, Gardner TG, Liu S, Armstrong SD. ITSxpress: software to rapidly trim internally transcribed spacer sequences with quality scores for marker gene analysis. F1000Research. 2018;7:1418.
Hannon GJ. FASTX-Toolkit: FASTQ/A short-reads pre-processing tools. 2010. http://hannonlab.cshl.edu/fastx_toolkit/.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11:2639–43.
Gdanetz K, Benucci GMN, Vande Pol N, Bonito G. CONSTAX: a tool for improved taxonomic resolution of environmental fungal ITS sequences. BMC Bioinforma. 2017;18:538.
Frøslev TG, Kjøller R, Bruun HH, Ejrnæs R, Brunbjerg AK, Pietroni C, et al. Algorithm for post-clustering curation of DNA amplicon data yields reliable biodiversity estimates. Nat Commun. 2017;8:1188.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41.
Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217.
Giambelluca TW, Shuai X, Barnes ML, Alliss RJ, Longman RJ, Miura T, et al. Evapotranspiration of Hawai’i. 2014. Final report submitted to the U.S. Army Corps of Engineers—Honolulu District, and the Commission on Water Resource Management, State of Hawai’i.
Hijmans RJ. raster: geographic data analysis and modeling. R package v 2.9-5. 2019. https://CRAN.R-project.org/package=raster.
Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46.
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. vegan: community ecology package. R package v 2.5-5. 2019. https://CRAN.R-project.org/package=vegan.
Guillot G, Rousset F. Dismantling the Mantel tests. Methods Ecol Evol. 2013;4:336–44.
Dormann CF, Gruber B, Fruend J. Introducing the bipartite package: analysing ecological networks. R N. 2008;8:8–11.
Dormann CF, Fründ J, Blüthgen N, Gruber B. Indices, graphs and null models: analyzing bipartite ecological networks. Open Ecol J. 2009;2:7–24.
Atmar W, Patterson BD. The measure of order and disorder in the distribution of species in fragmented habitat. Oecologia. 1993;96:373–82.
Almeida‐Neto M, Guimarães P, Guimarães PR, Loyola RD, Ulrich W. A consistent metric for nestedness analysis in ecological systems: reconciling concept and measurement. Oikos. 2008;117:1227–39.
Zarraonaindia I, Owens SM, Weisenhorn P, West K, Hampton-Marcell J, Lax S, et al. The soil microbiome influences grapevine-associated microbiota. mBio. 2015;6:e02527–14. https://mbio.asm.org/content/6/2/e02527-14.
Zinger L, Amaral-Zettler LA, Fuhrman JA, Horner-Devine MC, Huse SM, Welch DBM, et al. Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS ONE. 2011;6:e24570.
Matthew W, Fraser DBG, Pauline F, Grierson BL, Kendrick GaryA. Metagenomic evidence of microbial community responsiveness to phosphorus and salinity gradients in seagrass sediments. Front Microbiol. 2018;9:1703.
Peay KG, Schubert MG, Nguyen NH, Bruns TD. Measuring ectomycorrhizal fungal dispersal: macroecological patterns driven by microscopic propagules. Mol Ecol. 2012;21:4122–36.
Massoni J, Bortfeld-Miller M, Jardillier L, Salazar G, Sunagawa S, Vorholt JA. Consistent host and organ occupancy of phyllosphere bacteria in a community of wild herbaceous plant species. ISME J. 2020;14:245–58.
Treseder KK, Maltz MR, Hawkins BA, Fierer N, Stajich JE, McGuire KL. Evolutionary histories of soil fungi are reflected in their large-scale biogeography. Ecol Lett. 2014;17:1086–93.
Nguyen HDT, Chabot D, Hirooka Y, Roberson RW, Seifert KA. Basidioascus undulatus: genome, origins, and sexuality. IMA Fungus. 2015;6:215–31.
Becraft ED, Woyke T, Jarett J, Ivanova N, Godoy-Vitorino F, Poulton N, et al. Rokubacteria: genomic giants among the uncultured bacterial phyla. Front Microbiol. 2017;8:2264.
Meiser A, Bálint M, Schmitt I. Meta-analysis of deep-sequenced fungal communities indicates limited taxon sharing between studies and the presence of biogeographic patterns. N Phytol. 2014;201:623–35.
Crowther TW, Maynard DS, Crowther TR, Peccia J, Smith JR, Bradford MA. Untangling the fungal niche: the trait-based approach. Front Microbiol. 2014;5:579.
Shakya M, Gottel N, Castro H, Yang ZK, Gunter L, Labbé J, et al. A multifactor analysis of fungal and bacterial community structure in the root microbiome of mature Populus deltoides trees. PLoS ONE. 2013;8:e76382.
Acknowledgements
We greatly appreciated the help of Josie Hoh and Chad Durkin at Hiʻipaka LLC. Kirsten Cannoles and Sean Swift for their assistance in the laboratory, Niranjan Gunasekara for his help setting up air samplers and during fieldwork, and Cédric Arisdakessian and Mahdi Belcaid for developing the bioinformatic pipeline used. We also appreciate the efforts of three anonymous reviewers whose insights and suggestions greatly improved this manuscript. We also acknowledge the generous support of Illumina Corporation, Michele Langner, and the ASGPB for sequencing.
Funding
This work was supported by a grant from the W.M. Keck Foundation to ASA.
Author information
Authors and Affiliations
Contributions
ASA planned and designed research; all authors conducted fieldwork, analyzed data, and contributed to the paper content; VNSS prepared next-generation sequencing libraries and constructed air samplers; CBW and JB curated codes; ASA, JB, and CBW interpreted the results; and JB, CBW, MSC, RLR, and ASA wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Bernard, J., Wall, C.B., Costantini, M.S. et al. Plant part and a steep environmental gradient predict plant microbial composition in a tropical watershed. ISME J 15, 999–1009 (2021). https://doi.org/10.1038/s41396-020-00826-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-00826-5
This article is cited by
-
Plant sex affects plant-microbiome assemblies of dioecious Populus cathayana trees under different soil nitrogen conditions
Microbiome (2022)
-
Grass species identity shapes communities of root and leaf fungi more than elevation
ISME Communications (2022)
-
Plant hosts may influence arbuscular mycorrhizal fungal community composition in mangrove estuaries
Mycorrhiza (2021)
