
Abstract
The environmental sources of microbial aerosols and processes by which they are emitted into the atmosphere are not well characterized. In this study we analyzed microbial cells and biological ice nucleating particles (INPs) in smoke emitted from eight prescribed wildland fires in North Florida. When compared to air sampled prior to ignition, samples of the air–smoke mixtures contained fivefold higher concentrations of microbial cells (6.7 ± 1.3 × 104 cells m−3) and biological INPs (2.4 ± 0.91 × 103 INPs m−3 active at temperatures ≥ −15 °C), and these data significantly positively correlated with PM10. Various bacteria could be cultured from the smoke samples, and the nearest neighbors of many of the isolates are plant epi- and endophytes, suggesting vegetation was a source. Controlled laboratory combustion experiments indicated that smoke emitted from dead vegetation contained significantly higher numbers of cells, INPs, and culturable bacteria relative to the green shrubs tested. Microbial viability of smoke aerosols based on formazan production and epifluorescent microscopy revealed no significant difference in the viable fraction (~80%) when compared to samples of ambient air. From these data, we estimate each fire aerosolized an average of 7 ± 4 × 109 cells and 2 ± 1 × 108 biological INPs per m2 burned and conclude that emissions from wildland fire are sources of viable microbial aerosols to the atmosphere.
Similar content being viewed by others


Introduction
Biological particles that are aerosolized from surface ecosystems include pollen, fungi, protozoa, algae, bacteria, archaea, and viruses as well as biological detritus [1]. Numerous studies have examined biological dissemination in the atmosphere [2,3,4,5] and demonstrated the importance of bioaerosol transport to human, animal, and plant health (e.g. [6,7,8]). The properties of certain bioaerosols may allow them to affect meteorological processes by acting as cloud condensation or ice nuclei, thereby influencing cloud cover, precipitation formation, and the Earth’s energy budget (e.g. [9,10,11,12]). Despite the varied roles of bioaerosols in environmental health, biological dispersion, and the land–atmosphere system, their ecological sources and emission mechanisms remain poorly understood [13]. The aerial habitats of plants (i.e., the phyllosphere) harbor large quantities of microorganisms (106–107 cm−2 [14]), and globally, represents a major atmospheric source of bioaerosols [2, 15]. Although plant-associated microbes are known to be aerosolized via rain splash [16] and wind [17,18,19,20], little is known of other emission mechanisms from vegetation and data to constrain atmospheric fluxes of bioaerosols are limited [13].
Wildland fires are globally important aerosol sources, emit an average of 82.4 Tg of particulate matter (PM2.5) per year [21], and could serve as significant atmospheric sources of bioaerosols [22,23,24,25,26,27]. Although natural and prescribed fires in the USA have combusted 41.4 Tg yr−1 of biomass fuels over the last decade [28], few data have been available to assess if smoke aerosols transport viable microbes [23,24,25, 29]. Wildland fire emissions are also known to be sources of ice nucleating particles (INPs) [30], which have implications to meteorological processes during their transport through mixed-phase clouds. INPs documented in prescribed fire smoke from Georgia, USA [31] initiated freezing at warm temperatures (−5 °C) typically attributed to biogenic particles or certain bacteria [32, 33]. Considering that INPs affect the production of precipitation (enhancing or suppressing rainfall), cloud albedo, and cloud lifetime [33], advancing understanding of the mechanisms that influence INP abundance in the atmosphere is highly relevant to deciphering their contributions to cloud microphysical processes.
The objectives of this study were to determine if wildland fire smoke is an atmospheric source of viable microbes and biological INPs and estimate the scale of their emissions. To accomplish this, we sampled air at sites in North Florida prior to and during prescribed wildland fires and performed laboratory experiments to test three hypotheses. Hypothesis 1 is that total and viable microbial aerosols are at increased abundances in air sampled during fires when compared to that prior to burning. If true, Hypothesis 2 predicts that a portion of the smoke aerosols emitted during burning are warm temperature INPs (i.e., active at temperatures > −15 °C) and biological in origin. Since fuel type and active growth or decomposition stage affects smoke aerosol production [34] and microbial biomass [35,36,37,38], Hypothesis 3 tested that the combustion of dead fuels emits more viable microbes as compared to live fuel types. The data collected from the eight prescribed fires examined in this study indicate they were atmospheric sources of viable microbial cells and biological INPs. We connect discussion of our results to emerging perspectives on the global scale of microbial emissions to and transport in the atmosphere, with specific emphasis on the ecological, epidemiological, and meteorological significance of wildland fire as a microbial aerosol source.
Methods
Site description
The Ordway-Swisher Biological Station (OSBS) is located in Melrose, Florida and consists of ~38 km2 of natural and altered landcovers (Fig. S1). Natural fire regimes are maintained in the sandhill communities at OSBS by prescribed burns of individual units every 2–3 years. Consumption of understory vegetation such as leaf litter, downed woody debris, bunch grasses (e.g., Aristida spp., Sporobolus junceus, Sorghastrum secundum, Andropogon spp., and Schizachyrium scoparium [39]), and runner oak (Quercus pumila) are viewed as outcomes that meet the ecological objectives of a prescribed burn at OSBS. Meteorological data for OSBS was obtained from the Melrose Bay Station located in Putnam County, Florida. Environmental data (e.g., fuel moisture at time of ignition) were obtained from the OSBS real-time meteorological observation network station 102 (ordway-swisher.ufl.edu/Met.aspx), which is located within 5 km of the study sites (Fig. S1).
Aerosol measurements and collection
During each fire, aerosol data and samples were collected 2 m above ground level at three downwind locations in close proximity to each fire (i.e., within ~40 m of the flaming front). Two sampler types were used to provide replicate measurements. An ARA N-FRM volumetric sampler equipped with a Real-Time Particulate Profiler (ARA Instruments) was used to measure the mass concentration of particulate matter ≤10 µm in aerodynamic diameter (PM10; µg m−3) every 5 min. Whatman® Nuclepore™ track-etched polycarbonate filters (1 µm pore, Cat. No. 111110) that were housed within an inline filter holder were used to capture particles at a flow rate of 16.5 L min−1. In addition, two continuous volumetric samplers with wind orientation (miniature Cyclone, Burkard Manufacturing Co. Ltd) were used to sample aerosols during the prescribed burns (flow rate of 16.5 L min−1). Autoclaved Fisherbrand® 1.5 mL microcentrifuge tubes (Cat. No. 05-408-129) were used to collect the aerosols obtained by the miniature Cyclone samplers. Capture efficiency approached 100% for particles >1 µm for all sampling devices.
To characterize the aerosol properties (i.e., mass concentration, microbial cell and INP number concentration, and microbial viability) associated with wildland fire, air samples were collected in 30-min intervals immediately prior to and during the burning of each unit at OSBS. Sample collection initiated 30 min prior to ignition and continued during each fire’s ~2 h duration. The samples obtained in the field were kept chilled (at ~4 °C) after collection and during the <1 h transport to a laboratory at the University of Florida.
Laboratory combustion experiments
Prior to ignition of unit I-3a (Fig. S1) on 25 June 2018, aseptic sampling of the major surface and ground fuel sources included the following: live specimens of (1) Q. pumila (runner oak) and (2) Serenoa repens (saw palmetto), and (3) the organic soil horizon (Oi, Oe, Oa) that contained senesced and partially decomposed bunch grasses. The fuel samples were frozen, shipped overnight to the University of Idaho, and stored at −80 °C until they were processed.
Combustion experiments were performed in the Idaho Fire Initiative for Research and Education (IFIRE) laboratory at the University of Idaho (Moscow, Idaho). Replicate samples of each fuel were combusted on a custom burn table that consisted of a sterilized aluminum pan to hold the fuels, which rests on kiln bricks atop a 1.5 × 2.1 m metal table. A 50 cm diameter ignition ring made of perforated copper tubing plumbed with propane gas was used for ignition; it surrounded the fuels but was not in contact with them. Additional detail on the IFIRE laboratory is provided in the Supplemental methods.
Aerosols were collected using the ARA N-FRM, positioned 1.5 m above the fuel bed, and sampling was conducted during the duration of each burn (average sampling time of 3 min per sample, see Supplemental methods). A Biostage 200 impactor (SKC, Inc.) was used to directly collect microbial aerosols for culturing by sampling for 3 min at a flow rate of 14.1 L min−1 onto 90 cm petri-dishes containing trypticase soy (TSA) or potato dextrose (PDA) agar. Following incubation of the inoculated media at 25 °C for 6 days, the number of colony-forming units (CFUs) was counted.
Enumeration of microbial cells and INPs
To conduct multiple assays on each air sample, each filter was placed in a sterile 50 mL conical tube (Corning, Cat. No. 14-959-49A) and particles were suspended by shaking for 15 min in 30 mL of 1× phosphate buffered saline (PBS) that had previously been clarified by passing through a 0.2 µm syringe filter (Thermo Fisher Scientific, Cat. No. 42225-PV). The suspended material was combined with an additional 30 mL of PBS to a final volume of 60 mL. Aerosols collected into the microcentrifuge tubes were suspended by vortexing in 1 mL of PBS for 20 s and then mixed with 59 mL of PBS.
To enumerate DNA-containing cells, three technical replicates of 10 mL from each suspended sample were fixed with sodium-buffered formalin to a final concentration of 4% (v/v) upon return to the laboratory and the samples were stored at 4 °C [40]. The preserved samples were subsequently filtered onto black 0.22 µm polycarbonate Isopore™ filters (Millipore, Cat. No. GTTP04700). The cells collected on the filters were stained by adding 1.5 mL of 25× SYBR™ Gold (Life Technologies Corp., Cat. No. S-11494) that was diluted in Tris/borate/EDTA buffer, the samples were incubated in the dark for 15 min, and the staining solution was removed from the filter by applying vacuum. The filters were then rinsed with 10 mL of Tris/Borate/EDTA buffer, removed from the filter holder, and mounted to a glass microscope slide. A 4 µL drop of antifade solution (1:1 of PBS and glycerol with 0.1 g of phenylenediamine) was added to the filter surface before applying the coverslip. For each filter, the cells in 60 random fields of view (area of 1.8 × 104 µm2 each) were visualized and enumerated using a Nikon ECLIPSE Ni epifluorescence microscope. The calculated number of cells per filter, aerosol sampling time, and flow rate were used to estimate the cell concentration per cubic meter of air.
Immersion freezing assays were conducted to determine the concentration of INPs > 1 µm that catalyze freezing at temperatures between −4 and −15 °C. Fifty microliter aliquots of the PBS-suspended particulates were transferred into each well of a 96-well plate. Measurements for each aerosol sample are based on three technical replicates. The plates were sealed with adhesive film and placed at −4 °C within a refrigerated ethylene glycol bath. The number of wells that froze as the temperature decreased in half degree intervals was recorded to a temperature of −15 °C. The concentration of total INPs active at each temperature was calculated based on the proportion of unfrozen wells and the sample volume, according to Vali [41]. To estimate the concentration of INPs that were biological in origin, the heat lability of the INPs was determined. Since mild heat treatments do not alter the ice nucleating activities of mineral INPs, heat-labile INPs are commonly used to infer INPs that are proteinaceous or other heat sensitive biomolecules [42, 43]. These measurements were done by conducting an immersion freezing assay on identical samples that were incubated at 100 °C for 15 min. Blanks consisting of PBS were prepared and analyzed in parallel with the samples to serve as procedural controls.
Bacterial isolation and identification
To culture microorganisms from aerosols sampled prior to and during the burning of unit I-3a (Fig. S1) on 1 March 2019, material collected into microcentrifuge tubes by the miniature Cyclone samplers was suspended by vortexing in 1 mL of PBS for 20 s and combined with 5 mL of PBS. One hundred microliter of this suspension was spread plated in triplicate onto Reasoner’s 2A (R2A), TSA, and PDA. Procedural blanks prepared in the field were used to inoculate the same media, and the controls were incubated in the same manner as the samples.
After incubation at 25 °C for 3 days, the number of CFUs in the samples and controls were enumerated. Bacterial colonies that formed on the R2A spread plates were differentiated based on size, pigmentation, and morphology; select colonies were picked and isolated using a standard three-phase streak plating technique. Genomic DNA from pure cultures of each isolate was extracted using the PureLink™ Microbiome DNA Purification Kit (Thermo Fisher Scientific, Cat. No. A2970). A portion of the 16S rRNA gene (27F and 1492R primers [44, 45]) from each isolate was amplified, sequenced, and analyzed (see Supplementary methods).
Estimates of microbial viability using CTC
5-Cyano-2,3-ditolyl tetrazolium chloride (CTC; Biotium, Cat. No. 10063) was used to detect cells with functional electron transport chains in the samples. Thirty-minute intervals of aerosol sampling (495 L of air, Burkard sampler) before and during the burning of two units (H-8a and H-8b) were conducted to collect material for the viability measurements. Procedural blanks were prepared in the field, stored with the samples, and processed in parallel. After return to the laboratory, the samples and procedural blanks were suspended by vortexing in 1 mL of PBS for 20 s. Each sample was divided into three technical replicates (0.33 mL each), amended with 1.6 mL of R2A broth containing 5 mM CTC, and incubated aerobically without shaking at 30 °C. After incubation for 24 h, each sample was filtered onto black 0.22 µm polycarbonate Isopore™ filters (Millipore, Cat. No. GTTP04700), counterstained with 10 µg mL−1 4′,6-diamidino-2-phenylindole (DAPI, Biotium Cat. No. 40043) and affixed to glass microscope slides (see Section “Enumeration of microbial cells and INPs”). Cells that reduced CTC and accumulated a red fluorescent formazan product were inferred to be viable. The CTC- and DAPI-stained cells were enumerated as described in Section “Enumeration of microbial cells and INPs”.
During the 24 h incubation period, growth was assumed to be exponential. The number of generations was calculated based on the equation for exponential growth: N = N0 × 2n, where N is the concentration of cells after incubation (DAPI-stained count at time = 24 h), N0 is the initial total cell concentration (DAPI count at time = 0 h), and n is the number of generations. Based on the calculated number of generations and the CTC count at time = 24 h, the growth equation was subsequently used to estimate the initial concentration of viable cells at time = 0 h.
Modeling smoke production and bioaerosol emissions
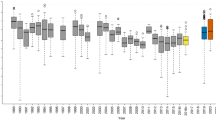
The First Order Fire Effects Model (FOFEM version 6.5 [46]) was used to estimate total smoke production (in µg m−2) for each unit area burned at the OSBS. Model inputs were parameterized for each unit based on burn history and frequency following Kreye et al. [47] and are detailed in Supplemental Table 1. Duff moisture content was categorized as dry or moderate based on days without rain and rainfall amounts following Kreye et al. [48]. To calculate the total number of cells and biological INPs emitted per m2 for each prescribed burn, FOFEM estimates of particulate matter ≤ 10 µm in diameter (g m−2) were multiplied by the number of bioaerosols emitted per gram of particulate matter (Fig. 1b).

Total DNA-containing cells (a) and cell concentration per mass of PM10 (b) are shown for samples collected prior to ignition (i.e., ambient) and during the prescribed burns (n = 8). The bottom panels contain the concentrations of cells (c) and CFUs (d) per mass of PM10 in smoke from the laboratory burns of litter and duff (Grass/Soil O horizon), runner oak, or saw palmetto. Boxes represent the interquartile range, the middle horizontal line is the median, and the whiskers represent minimum and maximum values. Outliers are 1.5 interquartile ranges below the first quartile and above the third quartile. Asterisks indicate level of significance as follows: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; not significant (ns) p > 0.05.
Statistical analysis
Two-tailed independent Student’s t test and analysis of variance (ANOVA) were used to compare concentration means for PM10, cells, CFUs, and INPs. All statistical analyses were performed in RStudio with R version 3.6.0 (R Core Team [49]). Plots were designed in R using ggplot2 [50] and ggtree [51] packages.
Results
Environmental conditions and air quality
The OSBS units burned during this study were distributed throughout the reserve (Fig. S1) and the average area burned per unit was 0.29 ± 0.07 km2 (±standard error of the mean, n = 8; Table 1). To allow moisture from morning condensation on the fuel to dry, each burn was ignited at ~10:00 local time. Relative humidity and temperature at OSBS were similar among burns at 63 ± 3.7% and 23 ± 1.9 °C, respectively (Table 1). Wind speed was relatively uniform between observations (average of 3.6 ± 0.6 m s−1) and background values were not significantly different from measurements during the prescribed burns (p > 0.05), suggesting that large indirect effects associated with wind speed could be excluded. On average, PM10 was 1.1 ± 0.47 × 103 µg m−3 and the concentration was not significantly different among the burns (ANOVA, p > 0.05; n = 8), but was significantly higher (t-test, p < 0.05; Fig. S2) than air sampled prior to ignition (1.0 ± 0.067 × 102 µg m−3).
The average air temperature during combustion experiments in the IFIRE laboratory was 10.5 ± 0.3 °C and relative humidity was 62.2 ± 0.9%. Fuel beds were loosely packed and influenced by ambient cooling air, with maximum temperatures across burns measured at the fuel bed, 30, 100, and 150 cm heights of 572, 106, 62, and 49 °C, respectively. Average temperatures at the fuel bed and 30 cm during combustion were not significantly different among fuel types (ANOVA, p > 0.05). The live fuels (runner oak and saw palmetto) produced higher concentrations of PM10 as compared to the dead fuels (Soil O horizon and senesced grasses) and were several orders of magnitude higher than PM10 values in ambient air (2.3 µg m−3 of PM10). An average of 4.0 × 103, 1.0 × 104, and 3.5 × 104 µg m−3 of PM10 was measured during the burning of the soil O horizon and senescent grasses, runner oak, and saw palmetto, respectively.
Measurement of microbial aerosols
The concentrations of DNA-containing aerosols collected on the 1 µm pore size filters during burns (6.7 ± 1.3 × 104 cells m−3) were fivefold higher than those in air sampled immediately prior to ignition (1.3 ± 0.13 × 104 cells m−3; Fig. 1a). Procedural controls did not contain any DNA-containing cells. Cell concentrations observed during each of the burns were not statistically different (ANOVA, p > 0.05; n = 8). In samples of smoke–air mixtures, cell concentration positively correlated with average PM10 concentration (Pearson’s r = 0.55, p < 0.01), which was consistent with microscopic observations that showed many cells in the samples were attached to large particles (4 to ~70 µm diameter; Fig. S3). Due to variations in wind direction and fire intensity among individual burns, cell abundance was divided by the PM10 mass concentration (Fig. S2) to facilitate comparison of proportions among the samples. This indicates that the smoke-derived aerosols contained threefold higher cell concentrations per µg of PM10 than those in ambient air (Fig. 1b). This agreed well with results from the laboratory experiments, which also demonstrated that four to fivefold higher cell concentrations per mass of particulate matter were emitted from the burning of the soil O horizon and senescent grasses when compared to live specimens of saw palmetto and runner oak (Fig. 1c).
INP aerosols
Similar to the microbial cell data, the cumulative concentration of total and biological INPs > 1 µm and active at −15 °C in the sampled aerosols were fivefold higher than air sampled before each burn (Fig. 2a). Procedural controls did not contain any INPs active at temperatures ≥ −15 °C. The fraction of INPs active at −15 °C in the smoke that were inferred to be biological (80%) was nearly twice that observed in samples of ambient air (43%). Based on total cell concentration and assuming all INPs were DNA-containing cells, we estimate that ~4% of microbial aerosols were INPs at temperatures ≥ −15 °C.

a Cumulative concentrations of total and biological INPs sampled in air prior to and during each prescribed burn (n = 6). b Cumulative concentrations of total and biological INPs per mass of PM10 sampled in smoke from laboratory experiments that burned senesced grass and soil O horizon (GS), runner oak (RO), and saw palmetto (SP); n = 3.
The results of the laboratory burns were comparable to the field data and significantly higher concentrations of total INPs active at −15 °C were observed when compared to ambient air (t-test, p < 0.05; Fig. S4A). Biological INP concentrations during burning were also higher; however, this was only significant for the senescent grass/soil O horizon fuel (t-test, p < 0.05, Fig. S4B). When the proportion of INPs per mass of PM10 are examined, the data follow trends similar to cell concentration in terms of fuel type (Fig. 2b), with 8- to 15-fold more total and biological INPs emitted per microgram of particulate matter during the burning of the grass/soil O horizon as compared to live fuels (Fig. 2b). Of the live fuels, burned runner oak emitted three- and sevenfold more total and biological INPs per microgram of particulate matter as compared to saw palmetto.
It is important to note that a potential caveat of our experimental design is that aqueous suspensions of the collected particulates were created to apportion samples and conduct the various analyses. Since the possibility cannot be excluded that aqueous suspension promoted particle disaggregation, our INP concentration data (per cubic meter of air) may be an overestimation relative to INP aerosols at the emission source.
Viability of the microbial aerosols
Aerosol particles >1 µm collected at OSBS burn unit H-8a/b (Fig. S1) were suspended and incubated in R2A with CTC to assess the fraction of viable cells in the air before and during a prescribed burn. Preliminary experiments were conducted that incubated samples for 30 min, 1 h, and 3 h, which resulted in CTC-stained cells that were too few to count. Therefore, the incubations were extended to 24 h. During the incubation, the concentration of cells in the smoke and ambient samples increased by ~50-fold. Procedural controls did not contain any CTC-stained cells. The populations in the smoke samples had an average doubling time of 4.5 ± 0.51 h, whereas those from ambient air doubled every 3.8 ± 0.26 h, but these differences were not statistically different (t-test, p > 0.05). Based on the number of cells stained with CTC after 24 h of incubation and inferred doubling times, the initial concentration of viable cells in the samples was estimated to be 1.3 ± 0.41 × 104 and 2.6 ± 0.50 × 105 viable cells m−3 for ambient air and smoke–air mixtures, respectively. The initial proportions of cells with functional electron transport chains when compared to the total (DAPI count at t = 0 h), was not significantly different between smoke and ambient samples and indicated that ~80% of the cells were viable (Fig. 3). Since the incubation media and organic electron donors used in this experiment (R2A) may not provide suitable substrates for all microorganisms in the samples analyzed, our results should be viewed as conservative estimates of the viable microbial aerosol concentration.

The CTC data represent inferred initial concentrations of viable cells based on the number of formazan-accumulating cells after for 24 h and the assumption of exponential growth, as described in the text. DAPI (n = 3, burn units H-8a/b) and SYBR Gold (n = 3, burn units H-8a/b) data represent total cell concentrations. Stars depict significance level between corresponding measurements.
Smoke aerosol samples from burn units H-8a and H-8b that were spread plated onto R2A and incubated formed 27 ± 3 colonies per plate, which corresponds to air concentrations of 3.2 ± 0.4 × 103 CFU m−3. Smoke aerosol samples plated onto TSA and PDA had an average of 19 ± 1 and 24 ± 1 colonies per plate and 2.3 ± 0.1 × 103 and 2.9 ± 0.1 × 103 CFU m−3, respectively. In contrast, the number of colonies per R2A, TSA, and PDA plate for ambient aerosol samples (12 ± 2) was below the standard level of quantification for dilution plating but represented 1.4 ± 0.2 × 103 CFU m−3. Media inoculated with samples from the procedural blanks did not result in any colony formation after incubation for 72 h at 25 °C. Based on the total number of microbial cells measured in these samples (5.1 ± 1.4 × 104 cells m−3), a maximum of ~6% were culturable using standard media and techniques.
In the laboratory experiments, the average concentration of CFUs m−3 in the smoke–air mixtures (mean of 1.1 ± 0.36 × 103 CFUs m−3) was significantly higher (t-test, p < 0.01) than that of ambient air (7.7 ± 2.4 × 101 CFUs m−3) when burning dead fuels, but not for the green saw palmetto and runner oak shrubs (t-test, p > 0.05). CFU and cell concentrations that were divided by PM10 values followed a similar pattern regarding fuel type, with significantly more cells (Fig. 1c) and CFUs (Fig. 1d) per microgram of particulate matter in smoke emitted from burned dead fuels as compared to the live fuels.
Identification of bacteria cultured from the aerosol samples
Actinobacteria were cultured from samples collected during two fires (ten isolates) and in air collected immediately prior to ignition (five isolates), whereas the five proteobacterial isolates recovered were only observed in smoke samples (Fig. 4). Many of the bacteria isolated were closely related (>97% 16S rRNA gene identity) to taxa common in soil (e.g., Nocardioides cavernae) and inhabiting plant surfaces (e.g., Microbacterium phyllosphaerae and Microbacterium foliorum). Three of the isolates had 94–96% (C_07A, C_06A, C_30S) similarity to their nearest neighbors and may represent new actinobacterial species. Based on the environmental source of their nearest neighbors, 68% (15/22) of the isolates from smoke samples were closely related (>98% 16S rRNA gene identity) to plant-associated bacteria, with a quarter of these being most closely related to taxa isolated from root nodules (e.g., Bosea caraganae MH633716) (Fig. 4).

Triangle and square symbols indicate type strains that were originally isolated from phyllosphere, plant tissue, or root nodule sources, Bootstrap values are shown as a percentage of 1000 replicates. The phylogenetic tree is based on maximum likelihood and the scale bar represents 0.03 substitutions per site. GenBank accession numbers are shown in parentheses.
Estimation of bioaerosol emissions
According to outputs from the FOFEM model, the fires examined during this study emitted 16 ± 2.1 g of PM10 per m2 (Table 2). The FOFEM PM10 emission estimates (i.e., g PM10 m−2) were multiplied by the cell and biological INP concentration per g of PM10 to calculate emissions per area burned. Based on data from the eight prescribed burns sampled at OSBS, we estimate that 7.3 ± 3.9 × 109 cells and 1.6 ± 0.94 × 108 biological INPs (cumulative concentration at −15 °C) were aerosolized per m2 (Table 2). When the total area of the units are considered (2.6 ± 0.85 × 105 m2, Table 1), we infer that each fire aerosolized a total of 1.6 ± 0.84 × 1015 cells and 5.0 ± 3.4 × 1013 INPs.
Discussion
The results of this study are in accord with previous investigations that have concluded biomass burning is a microbial aerosol emission source [23,24,25]. Although survival during the aerosolization processes associated with high temperature combustion is not axiomatic, there are several possible explanations for the enrichment and viability of bioaerosols observed in wildland fire smoke. First, wildfires generate convective air currents that are expected to enhance the aerosolization of microbes from proximal plants and soils [24]. Since convection creates atmospheric pressure gradients, equilibration with local regions of higher pressure advects aerosols toward the fire, entraining them into the vertical column of rising air. Hence, elevated concentrations of microbes associated with the smoke may not necessarily be sourced from the immediate region of combustion. However, it is also possible that microbes are directly aerosolized in association with incompletely combusted biomass, as suggested by Rajput et al. [23]. This possibility is supported by numerous observations demonstrating that wildfires emit high concentrations of organic particulate matter aerosols [52, 53], partially combusted foliage [54], and unburned plant material [55]. Convective air currents that promote advection of microbes from sources outside the combustion zone were largely controlled for in the laboratory experiments performed during this study. This together with microscopic observations (Fig. S3) suggests that the elevated concentrations of microbes documented in the smoke samples were directly emitted in association with incompletely combusted plant and soil material.
Particulate matter is a commonly used tracer of smoke aerosols [31] and its concentration provides indications of the amount and type of fuel burned [34, 56]. Previous studies have reported positive correlations between PM10 and cell concentration [33, 57] similar to those we have documented for smoke aerosols (Pearson’s r = 0.65, p < 0.01). Concentrations of airborne cells (i.e., those retained on 1 µm-pore size filters) prior to each fire averaged 1.3 × 0.13 ± 104 cells m−3 and were similar to those reported in wetlands of the Southeastern USA (~2 × 104 cells m−3 [58]). Smoke–air mixtures contained fivefold higher cell concentrations (6.7 ± 1.3 × 104 cells m−3; Fig. 1a) than ambient air and were one to two orders of magnitude higher than those documented in proximity to other important point sources such as sewage plants [59]. When cell abundance is adjusted to the PM10 data (Fig. 1b), there were statistically larger (t-test, p < 0.05) numbers of cells (>1 µm) per microgram of PM10 in smoke–air mixtures (340 ± 108 cells µg−1) as compared to ambient air (127 ± 8 cells µg−1), indicating the contribution of a point source. INP concentrations in the smoke also positively correlated with PM10 (Pearson’s r = 0.42, p = 0.05), whereas those sampled in ambient air did not (Pearson’s r = −0.16, p > 0.05). Laboratory experiments showed that smoke from the combustion of dead fuels contained 2- to 15-fold more cells, CFUs, and biological INPs compared to live fuel types (Figs. 1c, d; 2b). This is likely due to differences in abundance at the source as decomposing plant material typically harbors orders of magnitude more INPs (105–109 at −15 °C) and microbial cells (108–109) per gram than living leaves (~104 INPs g−1; 104–106 cells g−1) [35,36,37,38].
Despite the fact that heat exposure might be expected to affect the viability of microbial aerosols in wildfire smoke, ~80% of DNA-containing cells in the smoke samples demonstrated active respiratory activity when provided with metabolic substrates (Fig. 3). This proportion is comparable to viability estimates of cloud water (72–95%) [60] and is in the high range of observations for the near-surface atmosphere (19–83%) [61]. We also observed that a relatively high fraction of total cells in the smoke samples were culturable (~6%) and this proportion did not differ from values in air sampled prior to ignition. The nearest phylogenetic neighbors to the majority of bacteria isolated inhabit phyllosphere or soil ecosystems, with 23% (5/22) of the actinobacterial and proteobacterial smoke isolates being most closely related to endophytic species that were isolated from within plant tissues (Fig. 4). It has been suggested [24] that microbial species capable of surviving aerosolization from wildland fires might require adaptations that increase their tolerance to environmental stress (e.g., formation of spores or cysts [62,63,64]). However, none of the bacteria isolated (Fig. 4) belong to taxonomic groups known to possess such survival strategies. This coupled with detection of heat-labile INPs (i.e., biological INPs; Fig. 2) in the smoke samples implies that the processes aerosolizing microbes during combustion may not be as biologically damaging as presumed. Possibly, microbes that exist in biofilms on leaves or colonize internal plant tissues may be afforded a degree of protection from heating and desiccation (e.g. [65]) associated with combustion and aerosolization processes.
We measured cumulative concentrations of total and biological INPs active at −15 °C in smoke samples that were three- and fivefold higher per m3, respectively, than in ambient air (Fig. 2a). Despite observing higher values in smoke, the INP concentrations we report may overestimate actual abundances in air since disaggregation of particulates in aqueous solution for the immersion freezing test cannot be excluded. Nevertheless, the INP concentration values observed are comparative to those reported by McCluskey et al. [31] (~1 × 103 INP m−3 active at −15 °C) for smoke from a prescribed burn in Georgia (USA). The fraction of microbial cells inferred to be INPs at −15 °C (4%) is higher than similar estimates of ~1% for air in a North American forest ecosystem [9] and precipitation from the Southeastern USA [66]. Together, these findings imply that wildland fires may contribute to increasing atmospheric concentrations of relatively rare cloud-active aerosols that affect the formation and number of ice crystals in mixed-phase clouds, precipitation formation, cloud lifetime, and radiative forcing [67,68,69]. This contention is supported by polarization lidar analysis of smoke plumes from boreal fires that observed particles inducing atmospheric ice nucleation at −15 °C [70], which is a freezing temperature that cannot be readily explained without invoking biological INPs [42, 43]. Furthermore, nearly twice the fraction of INPs in the smoke were inferred to be biological when compared to ambient conditions (80% and 43%, respectively). These results collectively provide strong support for smoke aerosols serving as direct sources of biological INPs to the atmosphere.
Based on modeled simulations of smoke production, the estimated magnitude of bioaerosol emissions from the prescribed fires examined averaged 7.2 ± 3.9 × 109 cells and 1.6 ± 0.95 × 108 biological INPs per m2. For comparison, the value for cell emission is 360- to 3600-fold higher than concentrations used to model microbial transport over North America (i.e., 2–20 × 106 m−2 [2]). Convective transport of smoke and water vapor into the atmosphere can promote cloud development and influence the microphysical processes that form ice in clouds (e.g. [71]). If intense wildland fires transport large quantities of biological INPs to cloud altitudes, these bioaerosols may contribute to suppressing local and regional precipitation by freezing cloud droplets and eliminating the vapor source for ice crystal growth (e.g. [71,72,73]); however, these bioaerosols could also exert a positive influence that may in some way counteract the action of other smoke-derived aerosols. Consequently, numerical modeling efforts that simulate dispersion transport, cloud microphysical processes, and mesoscale atmospheric phenomena are required to assess the meteorological effects of individual fires and their aerosols (e.g., bioaerosols versus other fire-produced aerosols).
Our results demonstrate wildland fire emissions are sources of viable microbes and highly efficient biological INPs to the atmosphere. The high-frequency of burning at our study site results in relatively low-intensity fires, and future investigations should examine biomass burns of larger intensity and differing fuel composition to discern how these variables affect the magnitude of bioaerosol emission and scale of transport. Since high intensity fires can inject aerosols into the stratosphere [74, 75, 76], transport within smoke plumes may provide a mechanism to disperse the entrained bioaerosols globally. Given that ~4% of land surfaces are burned each year (1.8 × 108 m−2 in the USA, 10-year average, National Interagency Fire Center) and that wildfire occurrence is expected to increase in the future [77], the meteorological and environmental impacts of fire-emitted bioaerosols warrant more detailed investigation. Various studies have shown that phytopathogens are transported large distances in the atmosphere by wind [78, 79], and there is also evidence for their transport with smoke [25, 26]. Our results imply that wildland fire smoke is a point source for the aerial dissemination of plant-associated bacteria (Fig. 4), and potentially, pathogenic species as well. An improved understanding of the ecological sources, characteristics, and meteorological effects of microbes transported in smoke is particularly relevant to communities in proximity to frequent wildland fires and where fire incidence is predicted to increase in the future (e.g., Western US forests, Southern and Southeastern Australia, Amazon forests, and Indonesia [80,81,82,83]).
References
Amato P, Brisebois E, Draghi M, Duchane C, Fröhloch-Nowoisky J, Huffman JA, et al. Main biological aerosols, specificities, abundance, and diversity. In: Delort A-M, Amato P, editors. Microbiology of aerosols. 1st ed. John Wiley & Sons, Inc.; Hoboken, New Jersey, USA. 2018. p. 3–11.
Burrows SM, Butler T, Jöckel P, Tost H, Kerkweg A, Pöschl U, et al. Bacteria in the global atmosphere—part 2: modeling of emissions and transport between different ecosystems. Atmos Chem Phys. 2009;9:9281–97.
Wilkinson DM, Koumoutsaris S, Mitchell EAD, Bey I. Modelling the effect of size on the aerial dispersal of microorganisms. J Biogeogr. 2012;39:89–97.
Bryan NC, Christner BC, Guzik TG, Granger DJ, Stewart MF. Abundance and survival of microbial aerosols in the troposphere and stratosphere. ISME J. 2019;13:2789–99.
Amato P, Joly M, Schaupp C, Attard E, Möhler O, Morris CE, et al. Survival and ice nucleation activity of bacteria as aerosols in a cloud simulation chamber. Atmos Chem Phys. 2015;15:6455–65.
Brown JKM, Hovmøller MS. Aerial dispersal of pathogens on the global and continental scales and its impact on plant disease. Science. 2002;297:537–41.
Douwes J, Thorne P, Pearce N, Heederik D. Bioaerosol health effects and exposure assessment: progress and prospects. Ann Occup Hyg. 2003;47:187–200.
Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, et al. Emerging fungal threats to animal, plant and ecosystem health. Nature. 2012;484:186–94.
Huffman JA, Prenni AJ, DeMott PJ, Pöhlker C, Mason RH, Robinson NH, et al. High concentrations of biological aerosol particles and ice nuclei during and after rain. Atmos Chem Phys. 2013;13:6151–64.
Möhler O, Demott PJ, Vali G, Levin Z. Microbiology and atmospheric processes: the role of biological particles in cloud physics. Biogeosciences. 2007;1059–71.
Morris CE, Conen F, Alex Huffman J, Phillips V, Pöschl U, Sands DC. Bioprecipitation: a feedback cycle linking Earth history, ecosystem dynamics and land use through biological ice nucleators in the atmosphere. Glob Change Biol. 2014;20:341–51.
Sands DC, Langhans VE, Scharen AL, de Smet G. The association between bacteria and rain and possible resultant meteorological implications. J Hung Meteorol Ser. 1982;86:148–52.
Burrows SM, Elbert W, Lawrence MG, Pöschl U. Bacteria in the global atmosphere—part 1: review and synthesis of literature data for different ecosystems. Atmos Chem Phys. 2009;9:9263–80.
Lindow SE, Brandl MT. Microbiology of the phyllosphere. Appl Environ Microbiol. 2003;69:1875–83.
Lindemann J, Constantinidou HA, Barchet WR, Upper CD. Plants as sources of airborne bacteria, including ice nucleation-active bacteria. Appl Environ Microbiol. 1982;44:1059–63.
Butterworth J, McCartney HA. The dispersal of bacteria from leaf surfaces by water splash. J Appl Bacteriol. 1991;71:484–96.
Wickman HH. Deposition, adhesion, and release of bioaerosols. Atmos Microb Aerosols. 1994;5:99–165.
Aylor DE, Parlange JY. Ventilation required to entrain small particles from leaves. Plant Physiol. 1975;56:97–9.
Lighthart B, Shaffer BT, Marthi B, Ganio LM. Artificial wind-gust liberation of microbial bioaerosols previously deposited on plants. Aerobiologia. 1993;9:189–96.
Jones AM, Harrison RM. The effects of meteorological factors on atmospheric bioaerosol concentrations—a review. Sci Total Environ. 2004;326:151–80.
Andreae MO, Merlet P. Emission of trace gases and aerosols from biomass burning. Glob Biogeochem Cycles. 2001;15:955–66.
Yang Y, Chan C, Tao J, Lin M, Engling G, Zhang Z, et al. Observation of elevated fungal tracers due to biomass burning in the Sichuan Basin at Chengdu City, China. Sci Total Environ. 2012;431:68–77.
Rajput P, Anjum MH, Gupta T. One year record of bioaerosols and particles concentration in Indo-Gangetic Plain: implications of biomass burning emissions to high-level of endotoxin exposure. Environ Pollut. 2017;224:98–106.
Kobziar LN, Pingree MRA, Larson H, Dreaden TJ, Green S, Smith JA. Pyroaerobiology: the aerosolization and transport of viable microbial life by wildland fire. Ecosphere. 2018;9:e02507.
Mims SA, Mims IIIFM. Fungal spores are transported long distances in smoke from biomass fires. Atmos Environ. 2004;38:651–5.
Bonde MR, Prescott JM, Matsumoto TT, Peterson GL. Possible dissemination of teliospores of Tilletia-indica by the practice of burning wheat stubble. Am Phytopathol Soc. 1987;77:639.
Hu W, Wang Z, Huang S, Ren L, Yue S, Li P, et al. Biological aerosol particles in polluted regions. Curr Pollut Rep. 2020;6:65–89.
Urbanski SP, Reeves MC, Corley RE, Silverstein RP, Hao WMM. Contiguous United States wildland fire emission estimates during 2003-2015 | Rocky Mountain Research Station. Earth Syst Sci Data. 2018;10:2241–74.
Kobziar LN, Pingree MRA, Watts AC, Nelson KN, Dreaden TJ, Ridout M. Accessing the Life in Smoke: A New Application of Unmanned Aircraft Systems (UAS) to Sample Wildland Fire Bioaerosol Emissions and Their Environment. 2019;15.
Petters MD, Parsons MT, Prenni AJ, DeMott PJ, Kreidenweis SM, Carrico CM, et al. Ice nuclei emissions from biomass burning. J Geophys Res. 2009;114:D07209.
McCluskey CS, DeMott PJ, Prenni AJ, Levin EJT, McMeeking GR, Sullivan AP, et al. Characteristics of atmospheric ice nucleating particles associated with biomass burning in the US: prescribed burns and wildfires. J Geophys Res Atmos. 2014;119:10458–70.
Maki LR, Galyan EL, Chang-Chien M-M, Caldwell DR. Ice nucleation induced by Pseudomonas syringae. Appl Environ Microbiol. 1974;28:456–59.
DeMott PJ, Prenni AJ, Liu X, Kreidenweis SM, Petters MD, Twohy CH, et al. Predicting global atmospheric ice nuclei distributions and their impacts on climate. Proc Natl Acad Sci USA. 2010;107:11217–22.
Urbanski S. Wildland fire emissions, carbon, and climate: emission factors. For Ecol Manag. 2013;317:51–60.
Schnell RC, Vali G. Atmospheric ice nuclei from decomposing vegetation. Nature. 1972;236:163–5.
Beattie GA, Lindow SE. The secret life of foliar bacterial pathogens on leaves. Ann Rev Phytopathol. 1995;33:145–72.
Baldrian P. Forest microbiome: diversity, complexity and dynamics. FEMS Microbiol Rev. 2016;41:fuw040.
Vali G, Christensen M, Fresh RW, Galyan EL, Maki LR, Schnell RC. Biogenic ice nuclei—2. Bacterial Sources. J Atmos Sci. 1976;33:1565–70.
FNAI. Guide to the natural communities of Florida: 2010 edition. Tallahassee; Florida Natural Areas Inventory. 2010.
Moore RA, Hanlon R, Powers C, Schmale DG, Christner BC. Scavenging of sub-micron to micron-sized microbial aerosols during simulated rainfall. Atmosphere. 2020;11:80.
Vali G. Quantitative evaluation of experimental results an the heterogeneous freezing nucleation of supercooled liquids. J Atmos Sci. 1971;28:402–9.
Murray BJ, O’Sullivan D, Atkinson JD, Webb ME. Ice nucleation by particles immersed in supercooled cloud droplets. Chem Soc Rev. 2012;41:6519.
DeMott PJ, Prenni AJ. New directions: need for defining the numbers and sources of biological aerosols acting as ice nuclei. Atmos Environ. 2010;44:1944–5.
Turner S, Pryer KM, Miao VPW, Palmer JD. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J. Eukaryot. Microbiol. 1999;46:327–338.
Lane DJ. 16S/23S rRNA sequencing. In: Nucleic acid techniques in bacterial systematics. (Eds Stackebrandt E and Goodfellow M) John Wiley and Sons: New York, NY; 1991.
Reinhardt ED, Keane RE, Brown JK. First order fire effects model: FOFEM 4.0, user’s guide. Missoula; U.S. Department of Agriculture, Forest Service, Intermountain Research Station. 1997.
Kreye JK, Varner JM, Kobziar LN. Long-duration soil heating resulting from forest floor duff smoldering in longleaf pine ecosystems. For Sci. 2020;66:1–13.
Kreye JK, Brewer NW, Morgan P, Varner JM, Smith AMS, Hoffman CM, et al. Fire behavior in masticated fuels: a review. Ecol Manag. 2014;314:193–207.
R Core Team. A language and environment for statistical computing. R Core Team; Vienna, Austria. 2018.
Wickham H. ggplot2: elegant graphics for data analysis. Springer-Verlag: New York; 2016.
Yu G, Smith DK, Zhu H, Guan Y, Lam TT-Y. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8:28–36.
Radzi bin Abas M, Oros DR, Simoneit BRT. Biomass burning as the main source of organic aerosol particulate matter in Malaysia during haze episodes. Chemosphere. 2004;55:1089–95.
Rajput P, Sarin M, Sharma D, Singh D. Characteristics and emission budget of carbonaceous species from post-harvest agricultural-waste burning in source region of the Indo-Gangetic Plain. Tellus B Chem Phys Meteorol. 2014;66:21026.
Reid JS, Koppmann R, Eck TF, Eleuterio DP. A review of biomass burning emissions part II: intensive physical properties of biomass burning particles. Atmos Chem Phys. 2005;5:799–825.
Hungershoefer K, Zeromskiene K, Iinuma Y, Helas G, Trentmann J, Trautmann T, et al. Modelling the optical properties of fresh biomass burning aerosol produced in a smoke chamber: results from the EFEU campaign. Atmos Chem Phys. 2008;8:3427–39.
Robertson KM, Hsieh YP, Bugna GC. Fire environment effects on particulate matter emission factors in Southeastern US pine-grasslands. Atmos Environ. 2014;99:104–11.
Santos-Burgoa C, Rosas I, Yela A. Occurrence of airborne enteric bacteria in Mexico city. Aerobiologia. 1994;10:39–45.
Ziemba LD, Beyersdorf AJ, Chen G, Corr CA, Crumeyrolle SN, Diskin G, et al. Airborne observations of bioaerosol over the Southeast United States using a Wideband Integrated Bioaerosol Sensor. J Geophys Res Atmos. 2016;121:8506–24.
Yousefi V, Rama DBK. Monitoring of air for microbial and metal contamination at selected sites in the vicinity of Johannesburg, South Africa. Sci Total Environ. 1992;116:159–67.
Bauer H, Kasper-Giebl A, Löflund M, Giebl H, Hitzenberger R, Zibuschka F, et al. The contribution of bacteria and fungal spores to the organic carbon content of cloud water, precipitation and aerosols. Atmos Res. 2002;64:109–19.
Hara K, Zhang D. Bacterial abundance and viability in long-range transported dust. Atmos Environ. 2012;47:20–5.
Vela GR. Survival of Azotobacter in dry soil. Appl Microbiol. 1974;28:77–9.
Henriques AO, Moran CP. Structure and assembly of the bacterial endospore coat. Methods. 2000;20:95–110.
Julien B, Kaiser AD, Garza A. Spatial control of cell differentiation in Myxococcus xanthus. Proc Natl Acad Sci USA. 2000;97:9098–103.
Almatroudi A, Tahir S, Hu H, Chowdhury D, Gosbell IB, Jensen SO, et al. Staphylococcus aureus dry-surface biofilms are more resistant to heat treatment than traditional hydrated biofilms. J Hosp Infect. 2018;98:161–7.
Joyce R, Lavender H, Farrar J, Werth JT, Weber CF, D’Andrilli J, et al. Characterization and source identification of biological ice nucleating particles deposited year-round in subtropical precipitation. Appl Environ Microbiol. 2019;85:1–21.
Rosenfeld D, Yu X, Liu G, Xu X, Zhu Y, Yue Z, et al. Glaciation temperatures of convective clouds ingesting desert dust, air pollution and smoke from forest fires. Geophys Res Lett. 2011;38:1–5.
Koren I, Kaufman YJ, Remer LA, Martins JV. Measurement of the effect of Amazon Smoke on inhibition of cloud formation. Science (80-). 2004;303:1342–5.
Lohmann U. A glaciation indirect aerosol effect caused by soot aerosols. Geophys Res Lett. 2002;29:1052.
Sassen K, Khvorostyanov VI. Cloud effects from boreal forest fire smoke: evidence for ice nucleation from polarization lidar data and cloud model simulations. Environ Res Lett. 2008;3:025006.
Rosenfeld D, Rudich Y, Lahav R. Desert dust suppressing precipitation: a possible desertification feedback loop. Proc Natl Acad Sci USA. 2001;98:5975–80.
Andreae MO, Rosenfeld D, Artaxo P, Costa AA, Frank GP, Longo KM, et al. Smoking rain clouds over the Amazon. Science (80-). 2004;303:1337–42.
Rosenfeld D. TRMM observed first direct evidence of smoke from forest fires inhibiting rainfall. Geophys Res Lett. 1999;26:3105–8.
Peterson DA, Campbell JR, Hyer EJ, Fromm MD, Kablick GP, Cossuth JH, et al. Wildfire-driven thunderstorms cause a volcano-like stratospheric injection of smoke. npj Clim Atmos Sci. 2018;1:1–8.
Yu P, Toon OB, Bardeen CG, Zhu Y, Rosenlof KH, Portmann RW, et al. Black carbon lofts wildfire smoke high into the stratosphere to form a persistent plume. Science (80-). 2019;365:587–90.
Val Martin M, Kahn R, Tosca M. A Global analysis of wildfire smoke injection heights derived from space-based multi-angle imaging. Remote Sens. 2018;10:1609.
Shukla PR, Skea J, Buendia EC, Masson-Delmotte V, Pörtner H-O, Roberts DC, et al. Climate change and land: an IPCC special report on climate change, desertification, land degradation, sustainable land management, food security, and greenhouse gas fluxes in terrestrial ecosystems. The Intergovernmental Panel on Climate Change. 2019.
Aylor DE. Spread of plant disease on a continental scale: role of aerial dispersal of pathogens. Ecology. 2003;84:1989–97.
Nagarajan S, Singh DV. Long-distance dispersion of rust pathogens. Annu Rev Phytopathol. 1990;28:139–53.
Abatzoglou JT, Williams AP. Impact of anthropogenic climate change on wildfire across western US forests. Proc Natl Acad Sci USA. 2016;113:11770–5.
Sharples JJ, Cary GJ, Fox-Hughes P, Mooney S, Evans JP, Fletcher MS, et al. Natural hazards in Australia: extreme bushfire. Clim Change. 2016;139:85–99.
Fernandes K, Verchot L, Baethgen W, Gutierrez-Velez V, Pinedo-Vasquez M, Martius C. Heightened fire probability in Indonesia in non-drought conditions: the effect of increasing temperatures. Environ Res Lett. 2017;12:054002.
Le Page Y, Morton D, Hartin C, Bond-Lamberty B, Pereira JMC, Hurtt G, et al. Synergy between land use and climate change increases future fire risk in Amazon forests. Earth Syst Dyn. 2017;8:1237–46.
Acknowledgements
We thank J. Davis for introducing us to research activities at OSBS and are indebted to the OSBS burn crew, which included A. Rappe, N. Burmester, L. Huey, J. Perry, and many others. We also thank A. Smith for use of the Idaho Fire Initiative for Research and Education laboratory and C. Trucco for burn assistance. This work was supported by awards (to BCC) from the Dean of Research in the Institute of Food and Agricultural Sciences (as part of the University of Florida’s OSBS Jumpstart Award Program) and from USDA/NIFA (McIntire-Stennis Project FLA-MCS-005671/project accession no. 005671). Partial support was also provided by NSF award 1241161/1643288 (to BCC), and grant no. IDAZ-MS-0115/project accession no. 1009933 from the USDA/NIFA (to LNK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Moore, R.A., Bomar, C., Kobziar, L.N. et al. Wildland fire as an atmospheric source of viable microbial aerosols and biological ice nucleating particles. ISME J 15, 461–472 (2021). https://doi.org/10.1038/s41396-020-00788-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-00788-8
This article is cited by
-
Survival of Icelandic airborne microbes towards simulated atmospheric stress factors
Extremophiles (2023)
-
Soot Aerosols from Wheat Stubble Burning Lead to Ice Nucleation and Heavy Rainfall Over Arid Rajasthan, India
Water, Air, & Soil Pollution (2023)
-
Wildland fire smoke alters the composition, diversity, and potential atmospheric function of microbial life in the aerobiome
ISME Communications (2022)
