
Abstract
Larger volumes of sea ice have been thawing in the Central Arctic Ocean (CAO) during the last decades than during the past 800,000 years. Brackish brine (fed by meltwater inside the ice) is an expanding sympagic habitat in summer all over the CAO. We report for the first time the structure of bacterial communities in this brine. They are composed of psychrophilic extremophiles, many of them related to phylotypes known from Arctic and Antarctic regions. Community structure displayed strong habitat segregation between brackish ice brine (IB; salinity 2.4–9.6) and immediate sub-ice seawater (SW; salinity 33.3–34.9), expressed at all taxonomic levels (class to genus), by dominant phylotypes as well as by the rare biosphere, and with specialists dominating IB and generalists SW. The dominant phylotypes in IB were related to Candidatus Aquiluna and Flavobacterium, those in SW to Balneatrix and ZD0405, and those shared between the habitats to Halomonas, Polaribacter and Shewanella. A meta-analysis for the oligotrophic CAO showed a pattern with Flavobacteriia dominating in melt ponds, Flavobacteriia and Gammaproteobacteria in solid ice cores, Flavobacteriia, Gamma- and Betaproteobacteria, and Actinobacteria in brine, and Alphaproteobacteria in SW. Based on our results, we expect that the roles of Actinobacteria and Betaproteobacteria in the CAO will increase with global warming owing to the increased production of meltwater in summer. IB contained three times more phylotypes than SW and may act as an insurance reservoir for bacterial diversity that can act as a recruitment base when environmental conditions change.
Similar content being viewed by others


Introduction
A recent survey of the global ocean microbiome excluded only one major oceanic region owing to absence of data—the Arctic Ocean [1]. This illustrates that the Arctic Ocean is heavily under-sampled, in particular, the permanently ice-covered Central Arctic Ocean (CAO), which comprises the ~ 3.3 million km2 large marine ecosystem (LME) around the geographic North Pole (Fig. 1). The reason for the data absence for the CAO is obviously the difficulty of accessing this remote cold area for on-site research, which requires an icebreaker.

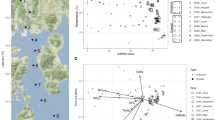
Map showing the geographical positions of the 12 sampling stations in the Central Arctic Ocean (CAO). White circles indicate the eight stations where ice brine (IB) samples were collected and green circles indicate the four stations where immediate sub-ice seawater (SW) samples were collected. Station 12 is located at the geographic North Pole. The background map was extracted from the International Bathymetric Chart of the Arctic Ocean (IBCAO) Version 3.0 [120]. The borders of the CAO large marine ecosystem (white line) were defined by the PAME Working Group of the Arctic Council [121]
Although the CAO is less productive than other oligotrophic oceanic regions not covered by ice, it is not a biological desert [2, 3]. Heterotrophic bacteria and the rest of the microbial loop are active components of the biological communities in the CAO and bacterial production is high relative to primary production [4, 5]. Detailed studies of bacterial community structure in the sympagic and pelagic systems of the CAO, targeting the 16S rRNA gene or the metagenome, are only a handful: three studies include samples from the water column [6,7,8] and two studies include samples from melt ponds, melted ice cores as well as surface sea water (SW) [9, 10]. These five previous studies together include only six samples from the sympagic habitat and 27 samples from the pelagic habitat in the 3.3 million km2 large CAO. Similar ice and SW studies carried out in the more nutrient-rich Arctic shelf LMEs, mainly from coastal sites in the Pacific Arctic region and around Svalbard, are about six times as many [11, 12].
The Arctic region is warming faster than the rest of the globe [13] and both the summer and winter sea ice extents have constantly been breaking low records during the last decades [14,15,16]. The summer ice has decreased at an estimated rate of ~1 million km2 (~13.2%) per decade over the period 1979 to 2017 (www.nasa.gov; accessed 18 January 2018). Simultaneously, the sea ice has become thinner [17,18,19], at an estimated loss rate of ~3100 km3 (~13.5%) per decade over the period 1979 to 2017 (psc.apl.uw.edu; accessed 18 January 2018). Climate models for the Arctic region predict a further decline of the summer sea-ice cover, with estimations down to < 1 million km2 within the coming 30 years depending on which political decisions are made [15, 20, 21].
Today, most of the CAO is still ice-covered in summer but the entire area is subject to increased melting, and the melt season is being prolonged at a rate of 5 days per decade since 1979 [22]. Melt ponds on the ice and open-water areas between ice sheets increase in abundance and size [23], and thick perennial ice is replaced by annual ice [24]. Seasonally, the sea ice shrinks from mid-March to mid-September, and seasonal differences in the habitat of sympagic microbes in the CAO become more pronounced because of the stronger ice-melt in summer with global warming [25]. Different microhabitats are formed during the lifetime of sea ice [11, 26], but the main habitat for psychrophilic microbes are the brine channels that run like blood vessels within the solid ice matrix. When sea ice forms, salts are concentrated in a liquid fraction in the brine channels, together with trapped gases, organic matter and microbes, and brine salinity can be up to approximately six times that of SW [27, 28]. When sea ice melts, the brine volume increases dramatically [29], open pathways for brine drainage and exchange of matter and gases between air, ice cover and SW emerge, and brine salinity drops from hypersaline to low-salinity brackish.
Larger volumes of sea ice have been thawing in summer during the last decades than during the past 800,000 years [30]. Brackish brine (fed by meltwater inside the ice) is an expanding sympagic (ice-associated) habitat for microbes in summer all over the CAO and the flux of sympagic bacteria and their metabolic products to the pelagic zone is enhanced as well. The brine is brackish through mixing of meltwater with saline winter brine and/or diffusion of salts from the underlying SW through increased porosity (permeability) of the solid ice [28]. These processes are expected to culminate if the North Pole area would become ice-free in summer as predicted in climate scenarios [20]. In this paper, we report for the first time the structure of the bacterial communities in brackish summer brine in the CAO and follow the flux of brine bacteria to the sub-ice SW for all taxonomic levels (class to genus), and for dominant phylotypes as well as for the rare biosphere. In a meta-analysis relating our new 16S rRNA data to the five previous studies carried out in the CAO [6,7,8,9,10], we detected patterns in the bacterial community structure in the sympagic and pelagic habitats of the oligotrophic CAO.
Materials and methods
Sample collection
Ice brine (IB) and immediate sub-ice SW samples were collected from eight and four stations, respectively, between 7 August and 3 September 2012 during the Lomrog III expedition with the Swedish icebreaker RV Oden (Fig. 1, Table S1). The 12 stations were located at 85–90 °N and covered an area of ca. 250,000 km2 of the CAO and about half of the Amundsen Basin. To avoid microbial contamination from the icebreaker the sampling stations were reached by helicopter. Shortly after sampling the record low Arctic sea-ice minimum ever, 3.4 million km2, was reached (www.nasa.gov; accessed 18 January 2017). Therefore, the sea ice was only 1.3–1.7 m thick at our stations and in such a melting stage that basically all the brine water was immediately drained from the cores when lifting them up from the ice.
Ice and snow depth were measured after a first hole was made through the sea ice with a Kovacs ice-core drill of 9 cm in diameter. Water temperature and salinity were measured directly in the field with an YSI Pro30™ handheld conductivity meter. IB samples were taken from a second hole drilled until half a metre above the ice SW interface and at least 10 m away from the first hole, i.e., far enough away to ensure that the brine was not disturbed by drainage from the first hole. The second hole immediately filled up with brine water and 20 L were pumped up from the bottom of the hole in < 5 min with a hand-operated membrane pump connected to a 3-m long tube of 25 mm in diameter with a 200 µm net at the end to exclude larger organisms. During pumping it was continuously checked that the brine salinity remained stable, i.e., that it was not influenced by freshwater from melt ponds from above or by SW from below. SW samples were pumped up from the first hole ~30 cm below the ice-water interface in the same way as the IB samples. The ice cores were used by another research group studying sea-ice light attenuation and coloured dissolved organic matter absorption [31].
For DNA analyses 5–11 L of water were pre-filtered through 8 µm Millipore® polycarbonate membrane filters and collected on 0.22 µm Millipore® Sterivex™ filters using a Cole Palmer System™ peristaltic pump Model No. 7553-70. All samples were filtered and stored at −80 °C within 3 hours after field sampling. The rest of each 20-L water sample was used for analyses of basic sample characteristics, such as bacterial cell density, nutrients and Chla (chlorophyll-a) (Table S1), according to the methods described in the Supplementary Information.
DNA and phylogenetic analyses
DNA was extracted from the 12 samples using a modification of a phenol:chloroform:IAA protocol and the 16S rRNA gene was amplified and sequenced on a MiSeq® Illumina sequencer as described in the Supplementary Information. Two IB samples, IB12 at the North Pole and IB23 at the marginal ice zone, were sequenced deeper than the other 10 samples. All sequences from this study are available in the NCBI SRA (Sequence Read Archive) under accession number PRJNA326760.
The obtained 16S rRNA gene sequences were de-multiplexed by index to separate them by sample. A quality report was made for each sample and then the sequences were filtered using ‘Prinseq’ (http://prinseq.sourceforge.net) applying a cutoff of minimal Phred Quality Score of 30. For taxonomic identification, operational taxonomic units (OTUs) were created using a workflow for de novo OTU picking in QIIME (v.1.9.1) [32], producing an OTU mapping file, by clustering sequences based on a similarity threshold of 97%, against the GreenGenes 16S rRNA gene database [33], after which a representative set of sequences was picked in order to keep one sequence per OTU. Sampling efficiency was evaluated by individual-based rarefaction curves [34].
The representative sequences corresponding to each OTU were aligned using PyNAST [35] and taxonomic assignment was made with the UCLUST algorithm [36]. OTU sequences clustered with one or two reads, i.e., singletons and doubletons, respectively, were removed from the analysis. As cyanobacterial and chloroplast sequences are highly similar, all OTUs identified as Cyanobacteria were aligned with16S rRNA gene sequences of Cyanobacteria obtained from SILVA ribosomal RNA database project (https://www.arb-silva.de), and chloroplast sequences that were initially classified as Cyanobacteria were discarded [37].
Phylogenetic trees were constructed using the ‘common’ OTUs in the data set, defined as those OTUs with relative abundance (RA) ≥ 0.01% across all samples. Community analyses were made for different taxonomic levels (bacterial classes, families and OTUs) and separately for the ‘common’ OTUs and the ‘rare biosphere’ OTUs, the latter defined as OTUs with RA < 0.01% across all samples [38, 39]. Further details of DNA extraction, 16S rRNA amplification, high-throughput sequencing protocol, data capture and phylogenetic analysis are described in the Supplementary Information.
Bacterial community analyses
To characterise the IB and SW habitats, a Kruskal–Wallis test was performed to test for differences in 12 abiotic and biotic variables between the two habitats. For evaluating community structure at a higher taxonomic level, the OTUs were assigned to bacterial class and the percentage OTUs within each class was calculated for each sample. A separate analysis was made for the ‘rare biosphere’ OTUs. To put our results into context with bacterial communities in other habitats of the CAO ecosystem we performed a meta-analysis by including the available published data on the structure of sympagic and pelagic bacterial communities in the CAO [6,7,8,9,10].
Community analyses were carried out using the ‘vegan’ package [40] in the software environment ‘R’ [41]. To prevent bias owing to sampling depth in OTU diversity, the samples were rarefied to 7000 sequences prior to all community analyses. Hierarchical cluster analysis based on Bray–Curtis dissimilarity was performed for the 26 most abundant OTUs (defined as RA ≥ 1.0% across all samples). The influence of environmental factors in shaping bacterial community structure was evaluated with redundancy analysis, using a Monte Carlo permutation test based upon 999 permutations. A Mantel test [42], based on 999 permutations was used to assess the effect of geographic distance among sampling stations on community structure. To explore the structure of the sympagic bacterial metacommunity by network analysis, probabilistic graphical models were constructed as described in the Supplementary Information.
Results
Abiotic and biotic variables
The IB and SW habitats were closely connected in space, but salinity, water temperature, and dissolved inorganic phosphorus (DIP), dissolved inorganic silicon (DSi), particulate organic carbon (POC) and Chla concentrations were significantly different (Tables 1 and S1). IB salinity varied between 2.4 and 9.6, whereas SW salinity was that of SW with weak dilution from the brine at some stations (33.3–34.9). Average water temperature was higher in IB (− 0.53 °C) than in SW (− 1.45 °C), which reflects the difference in the freezing point of water at the respective salinities. DIP and DSi concentrations were lower in IB than in SW but not in dissolved inorganic nitrogen (DIN). NH4+ concentrations were very low (~ 0.1 µM) in all samples. Lower Chla concentration in IB compared with SW indicates lower abundance of primary producers in the brine and higher POC concentrations in IB compared with SW indicates higher abundance of organic matter in the brine. Bacterial cell density (generally lower in IB) showed no significant differences between IB and SW owing to large variability between sampling stations, especially for IB.
16S iTag sequencing data
Altogether, sequencing of the 12 samples yielded 839,405 reads and 7150 OTUs (Table S2), of which 2715 were unique OTUs; 180 (6.6%) of the unique OTUs were ‘common’ (RA ≥ 0.01%) and 2535 (93.4%) belonged to the ‘rare biosphere’ (RA < 0.01%). Rarefaction curves (Figure S1) show that the sequencing effort did not capture the complete diversity in any of the samples, not even in the samples IB12 and IB23 that were sequenced with a higher coverage. However, three out of the four SW curves flattened out earlier than the IB curves.
OTU diversity
Of the 2715 unique OTUs recovered in this study, 1912 (70.4%) were only found in IB, 280 (10.3%) were only found in SW, whereas 523 (19.3%) occurred in both habitats. Within-class diversity differed between the habitats: OTU richness was higher in IB for Betaproteobacteria (IB 36%, SW 4%), Flavobacteriia (IB 16%, SW 11%) and Actinobacteria (IB 11%, SW 2%), and it was higher in SW for Gammaproteobacteria (SW 56%, IB 29%), Alphaproteobacteria (SW 15%, IB 5%) and Deltaproteobacteria (SW 2%, IB 0.2%).
It may be suspected that the three times higher OTU richness in the IB is biased by the two deep-sequenced IB samples from the North Pole (IB12) and the marginal ice zone (IB23) and by the fact that twice as many IB samples as SW samples were analysed (Table S2). To evaluate such a possible bias, we tested how OTU richness differed between IB and SW in relation to the number of reads (Figure S2) and found that: (1) Comparing four IB samples with different numbers of sequences showed that the higher the number of sequences yielded, the more OTUs were recovered. (2) Comparing the four SW samples showed the same but the proportion of OTUs in the deepest sequenced sample was lower. (3) When increasing the number of reads in IB samples by 25% the number of OTUs increased by 33% but when increasing the number of reads in SW samples by 100% the number of OTUs remained ~ 320. (4) IB and SW samples with a similar number of reads again showed that IB was inhabited by more OTUs than SW relative to the number of reads.
Phylogeny
Phylogenetic diversity was higher in IB (14.05) than in SW (10.63). The mean pairwise distance was lower in IB (2.9) than in SW (4.9), indicating that the IB communities consisted of more closely related taxa than the SW communities. The positive values of the mean pairwise distance in both habitats denote over-dispersion (large phylogenetic variability) in general.
Of the 180 ‘common’ OTUs, 161 belonged to three dominant phyla: 56 clades of Proteobacteria (109 OTUs, RA = 65%), 20 clades of Bacteroidetes (44 OTUs, RA = 21%), and 6 clades of Actinobacteria (8 OTUs, RA = 13%), and these were used for constructing phylogenetic trees (Fig. 2 and S3). The remaining 19 OTUs (RA together 0.7%) belonged to nine other phyla and they were not used in the analyses. When including reference sequences from other studies, we found that many of our OTUs best matched sequences from both the Arctic and Antarctic polar regions (e.g., Colwellia, Flavobacterium, Glaciecola, Halomonas, Polaribacter, Polaromonas, Pseudoalteromonas, Psychromonas). The source habitat of the reference sequences—sea ice or SW—normally agreed with the OTUs from our habitats. The recorded marine sequences, e.g., SAR11, showed closest relationship with Arctic SW references rather than with SAR11 references from tropical and subtropical seas.

Phylogenetic tree based on 161 OTUs with relative abundance ≥ 0.01% across all samples belonging to the phyla Proteobacteria, Bacteroidetes and Actinobacteria. The outer coloured ring denotes taxonomy (classes). The relative abundances of the OTUs per sampling station are indicated in shades of black for ice brine (IB) and in shades of red for the immediate sub-ice seawater (SW) with the sampling station order (from inside to outside the circle): SW4, SW6, SW11, SW20, IB7, IB12, IB14, IB15, IB16, IB18, IB21, IB23. Bootstrap values (ranging from 0.8 to 1.0) are indicated by dot size. A more detailed phylogenetic tree, including OTU names and numbers, is provided in Figure S3
Halomonas and Shewanella were the two genera of the Gammaproteobacteria with the highest representation among the 161 OTUs: 7 and 6, respectively (Fig. 2 and S3a). Other dominant Gammaproteobacteria were AO-5 (Alteromonas), AO-10331 (Glaciecola), AO-6488 (Psychrobacter), AO-3408 and AO-18446 (both Balneatrix), AO-36354 (ZD0405) and AO-37058 (SAR92 clade). We also detected an unknown cluster of five gammaproteobacterial OTUs (AO-10798, AO-22585, AO-25005, AO-29964, AO-31465). Major groups within the Alphaproteobacteria, were related to the Roseobacter RCA cluster (including nine OTUs of the genera Loktanella, Octadecabacter, Planktomarina and Sulfitobacter) and the SAR11 clade (four OTUs). Whereas the SAR11 clade was almost exclusively restricted to SW, the RCA cluster predominated in IB, except for AO-19648 (Planktomarina) that was more abundant in SW samples. Among the Betaproteobacteria, Polaromonas (three OTUs), BAL58 (two OTUs), OM43 clade (three OTUs) and AO-8052 (with unknown affiliation) were the most abundant clades.
Major clades within the class Flavobacteriia (Fig. 2 and S3b) were Flavobacterium, Polaribacter and the NS5 marine group (with four, four and five OTUs, respectively). We also detected an unknown cluster consisting of five OTUs in the class Chitinophaghia (AO-4553, AO-5084, AO-8387, AO-13329, AO-21833), which—based on their phylogenetic affiliation—might belong to the genus Lewinella.
Despite that the Actinobacteria were represented by several clades, only one OTU (AO-30973) best matching with Candidatus Aquiluna was highly abundant (Fig. 2 and S3c).
Community composition
A major difference between IB and SW was that Actinobacteria and Betaproteobacteria were abundant in IB but rare in SW (Fig. 3a). Whereas Gammaproteobacteria dominated in SW (79% of the total reads), the classes Gammaproteobacteria, Flavobacteriia, Actinobacteria, and Betaproteobacteria were more evenly distributed in IB (34, 25, 19 and 14%, respectively). Alphaproteobacteria were almost equally represented in IB and SW with lower abundances (6 and 8%, respectively).

Community composition in ice brine and immediate sub-ice seawater in the Central Arctic Ocean (CAO). The eight ice brine samples are indicated with prefix IB followed by sampling station number and the four immediate sub-ice seawater samples are indicated with prefix SW followed by sampling station number. a Relative OTU abundance (% of the total reads) of bacterial classes. The category 0.01–0.50% includes 154 OTUs of the less-common classes Deltaproteobacteria, Bacteroidetes (Saprospiria, Sphingobacteriia, Cytophagia), Verrucomicrobia (Opitutae, Verrucomicrobiae), Marinimicrobia (AB16, also known as SAR406), Acidobacteria (Acidimicrobiia), Firmicutes (Bacilli) and Cyanobacteria (Nostocaceae). b Hierarchical cluster dendrogram for the 26 most abundant OTUs (with relative abundance ≥ 1.0% across all samples) based on Bray–Curtis dissimilarity. The colours in the heat map illustrate the relative abundance of each OTU per sampling station. The sampling stations in a are arranged according to the clustering results in b
A cluster analysis based on the 26 most abundant OTUs (RA ≥ 1.0%) showed that community composition was habitat-dependent: all eight IB samples fell into one cluster and all four SW samples into another (Fig. 3b). There was no pattern with salinity within the IB habitat, whereas the two samples with the lowest salinity in the SW habitat (most influenced by brine water) clustered together. Only three of these 26 OTUs were abundant in both habitats (RA ≥ 1.0% within a habitat): two Gammaproteobacteria (AO-22161 Halomonas and AO-11040 Shewanella) and one Flavobacteriia (AO-29203 Polaribacter) (Table S3). The most abundant OTUs typical of IB were AO-30973 (Candidatus Aquiluna, Actinobacteria) and AO-4702 (Flavobacterium, Flavobacteriia) with 19 and 14% of all OTUs in IB, respectively. The most abundant OTUs typical of SW were the Gammaproteobacteria AO-3408 (Balneatrix) and AO-36354 (ZD0405) with 22 and 9% of all OTUs in SW, respectively.
Redundancy analysis (Figure S4) showed a significant association between bacterial community composition and the 10 abiotic and biotic variables tested in combination (salinity, temperature, DIN, DIP, DSi, POC, PON (particulate organic nitrogen), POP (particulate organic phosphorus), bacterial density and Chla concentration; Table S1). However, salinity explained most of the variance in community composition and was the only variable with a significant effect on community composition by its own. A Mantel test [42] showed that there was no significant influence of the geographical distances among sampling stations on bacterial community composition (R = −0.02; p = 0.56).
The rare biosphere
We found a clear segregation between the IB and SW habitats not only for the 180 ‘common’ OTUs, but also for the 2535 ‘rare biosphere’ OTUs (Fig. 4). Most of them (1888) were detected only in IB, 66% of which belonged to the four families Comamonadaceae and Oxalobacteriaceae (both Betaproteobacteria), Flavobacteraceae (Flavobacteriia), and Microbacteriaceae (Actinobacteria). Of the 267 ‘rare biosphere’ OTUs that were detected only in SW, 46% belonged to five other families: Halomonadaceae, Oceanospirillaceae and Alteromonadaceae (all three Gammaproteobacteria), and Rhodospirillaceae and Pelagibacteraceae (both Alphaproteobacteria). The 380 ‘rare biosphere’ OTUs that overlapped between IB and SW had affiliations more similar to those detected only in SW than to those detected only in IB. The same calculations but based on the total number of sequences of ‘rare biosphere’ OTUs (abundance) instead of the number of unique rare biosphere OTUs (richness) showed very similar patterns for nearly all the families.

Richness and abundance of the rare biosphere. The 2535 rare biosphere OTUs (relative abundance < 0.01%) were subdivided into those that occurred either in ice brine (IB) or in the immediate sub-ice seawater (SW) or in both habitats (IB+SW). a Venn diagram showing OTU richness. b The % richness (% of the total number of unique rare biosphere OTUs) and % abundance (% of the total number of sequences of rare biosphere OTUs) at the family level
Network analysis
The first network analysis, based on all 2715 OTUs, yielded high modularity (Fig. 5a, Table S4). Most of the modules contained OTUs representing ‘habitat specialists’ (found in one of the habitats, IB or SW), whereas the remaining modules contained ‘habitat generalists’ (found in both habitats). The network presented a higher number of IB modules compared with SW modules. A striking feature of this network is that most modules have high taxonomic assortativity (i.e., neighbours are closely related taxa). The biotic and abiotic variables incorporated in the network reconstruction depicted that these variables, with exception of DIN, PON and POP (only one or no connections), were associated with generalists (Fig. 5a). Salinity, temperature and DIP were the factors with most connections, depicting these variables as those with the highest influence in differentiating the communities between the two habitats in the analysis.

Network analysis of the sympagic metacommunity in the Central Arctic Ocean (CAO). a Network reconstructed by using all 2715 OTUs. b Network reconstructed by using only the 180 OTUs with relative abundance ≥ 0.01% across all samples. The red rectangle in a marks a module that mainly contains taxa that occurred in both ice brine (IB) and in the immediate sub-ice seawater (SW), most of which correspond to the abundant and widely distributed taxa in b. Vertices (circles) represent OTUs and edges (lines) represent non-random significant associations among OTUs. The colour of the circles denotes taxonomy and the circles around the circles denote habitat. White squares represent biotic and biotic variables; the numbers in brackets after the variables present the number of connections of the respective variable for networks a and b, respectively. DIN, PON and POP yielded no or only one connection and are not shown in the networks
The second network analysis, based on the 180 ‘common’ OTUs (Fig. 5b, Table S4), was characterised by lower modularity and higher connectivity than the first network, and was composed mainly of habitat generalists. One pronounced habitat generalist module in Fig. 5a nearly corresponded to the whole network in Fig. 5b, whereas the habitat specialist modules in Fig. 5a corresponded mainly to the low-abundant taxa that were absent from the second analysis. However, the patterns of connectance for biotic and abiotic variables did not differ between the two networks. For the second network we explored the number of edges for each node (OTU) and identified 28 ‘hubs’, i.e., nodes with a number of links that greatly exceeds the average (here defined as the OTUs accounting for > 60% of all edges in the analysis). All these hubs were most abundant in SW and 19 of them belonged to the orders Rickettsiales, Methylophilales, Flavobacteriales and Oceanospirillales (Table S5).
Meta-analysis of bacterial communities in the CAO
The meta-analysis for the oligotrophic CAO showed that there were large differences in bacterial community composition between the different sympagic habitats (melt ponds, IB, melted ice cores, immediate sub-ice SW) as well as between sympagic and pelagic habitats (Fig. 6a). Gammaproteobacteria and Bacteroidetes (mainly Flavobacteriia) dominated in the four sympagic habitats while Alphaproteobacteria, and to a lesser extent Gammaproteobacteria, dominated in SW. The sympagic habitats showed an increase in Gammaproteobacteria, and a decrease in Bacteroidetes, in a sequence from melt ponds, via IB and ice cores to the immediate sub-ice SW. Striking were the high abundances of Betaproteobacteria and Actinobacteria in the IB as these two groups were nearly absent from the other sympagic habitats as well as from the water column. Deltaproteobacteria were basically restricted to the water column with highest abundance in the sub-surface SW (depth > 30 m), whereas Bacteroidetes were more abundant in surface SW (depth 1–30 m).

Meta-analysis of bacterial community structure in sympagic and pelagic habitats in the Central Arctic Ocean (CAO). a Relative abundances of the dominant bacterial classes. The Bacteroidetes consisted mainly of Flavobacteriia. When Archaea were included in the published data they were excluded and the relative abundances were adapted to include only Bacteria. Cyanobacteria were excluded from Bowman et al. [9] with reference to Bowman [58]. b Overlap of the dominant bacterial genera/clades. The OTUs that were identified to the genus level and with relative abundance ≥ 1.0% were selected, normalised to 100% for each habitat and plotted together. Data from ice brine and immediate sub-ice seawater: present study, melt ponds: Rapp [10], melted ice cores: Bowman et al. [9], Rapp [10]. Surface seawater (depth ~ 1–30 m): Bowman et al. [9], Rapp [10], Li et al. [8]
Most abundant genera were abundant in only one or two habitats in the CAO (Fig. 6b). Polaribacter (Flavobacteriia) was the only genus that was abundant in all four sympagic habitats as well as in surface SW. Another genus of the Flavobacteriia, Flavobacterium, was abundant in melt ponds, IB and melted ice cores but not in the immediate sub-ice SW or in surface SW. Typical for the brackish IB were Candidatus Aquiluna (Actinobacteria), Loktanella (Alphaproteobacteria), BAL58 and Polaromonas (Betaproteobacteria), and Halomonas, Alteromonas and Shewanella (Gammaproteobacteria). Of these, Halomonas and Shewanella were able to at least temporarily survive the high SW salinity as they remained abundant in the immediate sub-ice SW (but not in the whole surface SW layer). Another gammaproteobacterium, Psychrobacter, occurred in the brine but was much more abundant in ice cores. The SW influence in the immediate sub-ice SW was characterised by the Gammaproteobacteria Balneatrix, ZD405 and SAR92, the alphaproteobacterium SAR11, and the NS5 marine group of the Flavobacteriia. Obligate marine taxa (not abundant in the immediate sub-ice SW) were Roseobacter, Cytophaga and the SAR116 clade.
Discussion
Sampling microbial communities from melting sea ice
There is a substantial sampling enigma for sympagic microbial communities because (1) part of the ‘pelagic’ fraction of the community (in the liquid brine phase) is lost during sampling and slice-cutting ice cores—especially in summer when the ice is in a melting phase, and (2) the rest of the community, dominated by benthic microbes (associated with ice surfaces), is changed by osmotic shocks and other processes, whereas melting the slices of solid ice in the laboratory [43, 44]. Sampling only the brine water—as we did—has the advantage that the organisms remain relatively undisturbed because they can be filtered/preserved/analysed immediately but has the drawback that the attached component of the community—especially diatoms—is left out [29, 45]. As our aim was to study the melting effect, sampling brine was more appropriate for our study than sampling cores.
The fact that part of the sympagic microbial community is lost during ice-core sampling has always been a problem, especially in the marginal ice zone in summer, but it is a growing issue as sea-ice melting is accelerating everywhere in the CAO through global warming [18, 23, 46]. We recommend that, if the aim is to assess the complete sympagic microbial community, both ice-core and brine samples should be taken if an ice core visibly loses part of its brine. Brine can be taken from a hole drilled until half a metre above the ice SW interface. In summer, such a hole will immediately fill up with brine water and 20 L can be pumped up in < 5 min with a simple hand pump. Salinity should be monitored, whereas pumping as it must remain stable, i.e., not become influenced by freshwater from melt ponds from above or SW from below. From the length and diameter of the ice core, its total volume can be calculated, related to the volume of water of the melted ice core, and the community data (biomass, species composition, salinity, etc.) can be compensated for brine loss. This is not ideal, e.g., the lower 0.5 m IB is not included and brine channels can be partly air-filled. However, sampling both cores and brine will provide a more complete record of the true microbial community living in the sea ice.
Community structure—sampling area and salinity
Sampling area and environmental heterogeneity underlie differences in bacterial community structure [47, 48]. As our sampling stations covered a large area (ca. 250,000 km2) it might be expected that community composition would vary between the stations. However, differences in community composition within the IB and the SW samples were small, whereas they were large between IB and SW samples. This is a strong indication that our samples are representative for their respective habitats with different salinities (IB and SW) and that the sympagic habitat of the ultra-oligotrophic Amundsen Basin is a highly uniform environment. Many other studies have, similar to our results, identified salinity as the principal environmental driver for microbial community structure in aquatic environments [49,50,51,52], but the spatial salinity gradient is never so short and steep as at the ice SW interface of a deep ocean basin. In our study the meltwater from the ice cover only had a marginal effect on the salinity of the immediate sub-ice SW, but in shallower coastal waters of the Arctic shelf LMEs salinities down to 25 have been measured at 7 m depth during sea-ice melt, which affected pelagic bacterial community structure [53].
Community structure—bacterial classes
Known global patterns of pelagic bacterial distributions at the class level are valid also for the CAO as shown by the meta-analysis based on our new data and those of Bano & Hollibaugh [7], Galand et al. [6], Bowman et al. [9], Rapp [10] and Li et al. [8]. First, Alphaproteobacteria and Gammaproteobacteria are the two dominant bacterial classes in the pelagic zone [1, 54,55,56]. Second, other abundant classes are Flavobacteriia (phylum Bacteroidetes) in the photic zone and Deltaproteobacteria in deeper water [1, 54, 57]. Third, sea ice habitats are typically dominated by Gammaproteobacteria and Flavobacteriia [12, 58, 59].
A new observation is that the brackish brine in the oligotrophic CAO contains high diversity (OTU richness) as well as high abundances of Actinobacteria (19%) and Betaproteobacteria (15%). These two bacterial classes were very rare or absent from melt ponds, ice cores and SW in our meta-analysis. Neither have they been reported as abundant in the more nutrient-rich Arctic shelf LMEs [11, 60]. We propose that Actinobacteria and Betaproteobacteria are indicators of melting sea ice as they are generally more abundant in fresh than in marine waters [50, 61], and they have been reported in a few studies from the Arctic marginal ice zone in melt ponds, the upper ice layer and surface SW mixed with meltwater [62,63,64,65].
We expect that the pattern we found in the meta-analysis for the CAO, following the flux of meltwater through the ice down to the immediate sub-ice SW—with Flavobacteriia dominating in melt ponds, Flavobacteriia and Gammaproteobacteria in ice cores, Flavobacteriia, Gamma- and Betaproteobacteria and Actinobacteria in brackish IB, and Alphaproteobacteria in SW—is typical for the oligotrophic CAO given the large geographical area we covered. In contrast, in the Arctic shelf LMEs the dominant classes in sea ice habitats are highly variable. This variability is probably related to different nutritional status of the parent water, which determines the bacterial community composition in the ice cover when the ice forms [66]. For example, the Chukchi Sea is one of the most productive seas on Earth [67]. Some ice-core studies in the Arctic shelf LMEs report dominance of Gammaproteobacteria [68, 69], whereas others report dominance of Alphaproteobacteria [53, 70] or more or less equal abundances of Gammaproteobacteria, Alphaproteobacteria and Flavobacteriia [64, 70]. This variability seems to be independent of the age of the ice (perennial or annual) and more related to the proximity of land, which strongly influences trophic state.
Similarly, bacterial community composition in the SW of the Arctic shelf LMEs is quite variable at the class level [12, 71]. The differences may partly be attributed to environmental variability on temporal (seasonal) or spatial (coastal vs. open ocean) scales, or if the sea was ice-covered or not, but often there does not seem to be any clear pattern. In some surveys of Arctic shelf LMEs it was found that, contrary to temperate regions, Gammaproteobacteria and Bacteroidetes were more often detected in Arctic SW than Alphaproteobacteria [60, 72, 73]. Other studies report that Alphaproteobacteria dominated in winter, both in ice cores and surface SW, and that this was caused by high abundances of SAR11 clade phylotypes [74, 75]. Members of the SAR11 clade belong to the most abundant and ubiquitous SW microbes in the world ocean [76] and are adapted to a wide range of salinities [77, 78]. Winter communities have not yet been studied in the CAO, but SAR11 phylotypes were rare in our summer IB samples (0.04%) but abundant in the immediate sub-ice SW (3%). In agreement with our results, SAR11 phylotypes were reported to be low-abundant in summer ice cores (0.2%; [9]) and abundant in surface SW (10–32%; [9, 10]). As no winter data exist for the CAO, future studies will reveal if SAR11 phylotypes are dominant in the winter sympagic communities of the CAO as has been reported for the Canadian Arctic coast [74]. It is possible that SAR11 phylotypes get entrapped in the brine channels in autumn when the ice is formed, persist there through winter, are released to the sea during spring brine drainage, and then replaced by typical summer ice communities dominated by Gammaproteobacteria and Flavobacteriia.
Are Cyanobacteria absent?
According to most previous studies carried out in the Arctic Ocean [12, 71, 79], as well as in Antarctic seas [80], Cyanobacteria seem to be practically absent from sea ice and surface SW. Initially we detected various sequences that were classified as Cyanobacteria, but these were later identified as chloroplasts. Confusion between cyanobacteria and phototrophic eukaryotic chloroplast sequences are a common feature in 16S rRNA gene analysis [9, 10, 58, 64, 68, 81]. After correcting our data for eukaryotic chloroplasts, only two cyanobacterial OTUs were left and they belonged to the subclasses Nostocophycideae (0.09%, one OTU, 709 sequences) and Synechococcophycideae (0.0003%, one OTU, four sequences). This would corroborate the hypothesis that cyanobacteria are rare in the polar seas.
However, low temperature should not be a restriction for the occurrence of Cyanobacteria [82], e.g., coccoid picocyanobacteria have been reported in several studies from Arctic coastal regions [71, 83,84,85,86]. It has been argued that the Cyanobacteria reported from the Arctic Ocean represent allochthonous influences from land [87]. However, previously we have recorded high cyanobacterial nifH gene diversity (orders Nostocales, Oscillatoriales, Chroococcales) in Arctic IB and SW at 82 °N in the northern Fram Strait at a station located centrally between Greenland and Svalbard [88], i.e., not close to land. Furthermore, a cyanobacterial nifH phylotype related to Nodularia sp. has been reported from sea ice in the CAO [2]. Thus, it cannot be excluded that autochthonous Cyanobacteria occur in the CAO and that they can be detected by metagenomics but not by all primers used in 16S rRNA gene analysis.
Community structure—dominant phylotypes
Many psychrophilic bacteria occurring in the Arctic Ocean are known to be > 97% identical to Antarctic ones, thus displaying bipolar distributions [57, 63, 89]. In agreement with this we found in our phylogenetic analysis both Arctic and Antarctic phylotypes, mainly among the brackish brine bacteria (e.g., Colwellia, Flavobacterium, Glaciecola, Polaribacter, Polaromonas, Pseudoalteromonas and Psychromonas), but also for some SW bacteria (e.g., Sphingomonas and Sulfitobacter).
Relatively few abundant phylotypes were responsible for the patterns we observed at the class level. Three phylotypes (classified as Halomonas, Shewanella and Polaribacter) were abundant in both the brine and in the immediate sub-ice SW, which implies that they at least initially survived the salinity shock from brackish to marine in the continuous brine flow from the ice to the SW during summer ice-melt. Of these three genera only Polaribacter has been reported as abundant in the surface SW of the CAO [8,9,10]. In ice cores from the CAO, Halomonas and Shewanella were extremely rare, with one read each of 12,352 reads [9]. The genus Halomonas consists of salt-tolerant bacteria [90] and has rarely been reported from the Arctic region [69], whereas more records exist from Antarctica [11, 91]. Shewanella has been found in the Arctic region more often, mainly in sediments [92], but also in snow in the CAO [81].
Particularly Flavobacterium, Polaribacter and Nonlabens seem to be typical of melt ponds [10] and may partly penetrate into the brine channels from there. However, Flavobacterium and Nonlabens, as well as the gammaproteobacterium Psychrobacter, were abundant in both brine (this study) as well as in ice cores [9, 10], which suggests that they may partly be associated with the solid ice surface, especially Psychrobacter, which seems to be more abundant in ice cores than in brine.
The genomic information of Candidatus Aquiluna, dominant in our IB samples, suggests that this actinorhodopsin-carrying photoheterotroph [93], could be of relevance in the CAO. Candidatus Aquiluna was initially characterised as a freshwater bacterium by Hahn [94] but was later reported from many different environments [93]. In the Arctic region it has been found in surface ice and SW near Svalbard [69, 93] and the Chukchi Sea [62], but not in the CAO and also not in a study targeting phototrophic bacteria in the Beaufort Sea [95].
BAL58 and Polaromonas, the dominant Betaproteobacteria in our brine samples, are known from different habitats elsewhere. BAL58 was isolated from the brackish Baltic Sea [96], and Polaromonas, a globally widely distributed psychrophilic genus commonly found in air and snow, has been reported from melt ponds on pack ice near Svalbard [63]. In the immediate sub-ice SW, the brine bacteria were mixed with marine bacteria from the SW below, especially the Gammaproteobacteria Balneatrix and ZD0405, which both were found to be abundant in the CAO surface SW in August–September 2012 by Rapp [10]. Phylotypes of Balneatrix have also been reported as dominants in SW from both the Arctic Chukchi Sea [62] and Antarctica [97].
Candidatus Aquiluna, BAL58, and Polaromonas formed, together with some other psychrophilic phylotypes, classified as Glaciecola, Alteromonas and Loktanella, the core bacteria in the brine that were not abundant in the immediate sub-ice SW. This suggests that the phylotypes we found in the brackish brine are unable to survive the salinity shock when they meet the SW while they otherwise are cold-adapted [98, 99] and perhaps adapted to low-nutrient concentrations such as BAL58 [96] or able to degrade polysaccharides as some members of Glaciecola [100]. Furthermore, sea ice brine channels normally contain higher DOM concentrations than SW [101], which selects for bacteria that can exploit these conditions; e.g., Polaromonas strains were shown to have an affinity for simple organic acids that can result from photodegradation of humic and fulvic substances [102]. Nutrient starvation, or other stress conditions that may emerge in the enclosed brine environment, may also be met by survival strategies such as the use of light for producing energy through the light harvesting pigment, actinorhodopsin in Candidatus Aquiluna [103, 104] or by forming dormant cells as found in Polaromonas [105]. However, in our study we did not explore clade-specific activity and we cannot conclude which specific strategies are coupled to the phylotypes uncovered in our samples.
Community structure—the rare biosphere
Our finding that the habitat segregation between IB and SW was expressed at all taxonomic levels (class to genus) and by dominant phylotypes as well as by the rare biosphere confirms the hypothesis that the rare biosphere is not randomly distributed but reflects the same environmental selection as the dominant phylotypes [38]. For example, the families Flavobacteraceae and Microbacteriaceae were abundant among the rare phylotypes in the brine, as well as their dominant relatives Flavobacterium and Candidatus Aquiluna in the whole community. Similarly, the families Halomonadaceae and Oceanospirillaceae were abundant among the rare phylotypes in the immediate sub-ice SW, as well as their dominant relatives Halomonas and Balneatrix in the whole community. It has been suggested that the similarity between the phylogeny of rare and abundant phylotypes signifies that most rare phylotypes (99% of which always are rare) are adapted to and active in their environment in a way similar to that of the abundant phylotypes [6]. On the other side, there is also the possibility that a fraction of the rare biosphere detected in high-throughput sequencing studies of environmental samples is composed of dead microbes [106]. The latter might occur in ice habitats where microbes can get trapped in the ice, although turn-over of organic material is probably fast with the high bacterial densities occurring here. Another aspect is that there is a risk to overestimate the diversity of the rare biosphere when sequencing the 16S rRNA [107]. However, the latter cannot explain the higher diversity (OTU richness) we discovered in our IB samples because the SW samples, with lower diversity, were treated simultaneously and in the same way.
The high diversity among the rare biosphere brine bacteria may be regarded as an ‘insurance potential’ [108], a reservoir of phylotypes that maintains psychrophilic bacterial biodiversity, both for sympagic and pelagic phylotypes. The metabolic versatility of bacteria may enable some of them to respond promptly to fine-scale environmental changes and suddenly form blooms when conditions become favourable [109]. This ‘seedbank’ theory, including dormancy [110], seems to fit with the sea ice habitat of the CAO. Habitat selection occurred at the ice SW interface for families that are common in SW such as Halomonadaceae, Shewanellaceae, Flavobacteriaceae, Oceanospirillaceae and Rhodospirillaceae (with many phylotypes occurring both in IB and SW) and against families that are typical of the sea-ice habitat such as Comamonadaceae, Oxalobacteraceae and Microbacteriaceae (many phylotypes only in IB).
Specialist bacteria populate the brine
On the one hand, our observation that the IB bacteria were more closely related to each other than the SW bacteria were to each other may be attributed to extreme (psychrophilic) adaptation inside the ice. On the other hand, the high richness of bacterial phylotypes in the brine compared with SW could be explained by the high environmental heterogeneity of the sea-ice habitat with many different microhabitats, from brine channels populated by free-living microbes to different forms of solid ice for attachment [28]. In addition, within the ice there is large variability in environmental conditions in space and time. This includes physico-chemical factors such as salinity, nutrients, and irradiation, and interactions with other microbes such as archaea, protists, and viruses [11, 26]. Environmental heterogeneity interacts with species niche breadth to influence species co-occurrence patterns [111], and a metacommunity affected by high environmental heterogeneity results in a network with high modularity where modules constitute groups of species with similar niche requirements [112]. This can explain why our network analysis without the rare biosphere phylotypes had lower modularity compared with the network including all 2715 phylotypes, indicating that the rare phylotypes had narrower niches and consequently only co-occurred with a restricted number of other phylotypes. Co-occurrence networks of specialists are typically separate from those of generalists, i.e., specialists tend to co-occur with other specialists, but not with generalists, and vice versa [111]. This suggests that the brine communities consisted more of specialists, psychrophilic extremophiles also adapted to brackish and other specific environmental conditions, whereas the immediate sub-ice SW contained more generalists.
The bacterial hubs (OTUs with many connections to other OTUs) in our networks were mainly represented by phylotypes with low abundance in the IB and higher abundance in the immediate sub-ice SW. These are taxa with a wide range of environmental tolerances and consequently a wide distribution or with many metabolic dependencies [112, 113]. Although most of these bacterial hubs were not highly abundant in our samples, they may have a strong impact in shaping communities and the functioning of the ecosystem [114, 115]. The hubs in our analysis belonged to metabolically important marine orders such as the Rickettsiales, Oceanospirillales, Flavobacteriales, and Methylophilales, that despite low abundances may have key roles in organising the communities both in and under the sea ice. For example, Methylophilales, known to use not only reduced carbon compounds such as methane as a source for their growth, but also multi-carbon compounds that do not contain carbon bonds, such as dimethyl ether and dimethylamine, are having a relevant role in cold Arctic waters [116, 117].
Conclusions
In the changing Arctic region the perennial sea ice cover is shrinking, whereas melting habitats, such as melt ponds and brackish IB, are expanding in space and time. This creates fundamental habitat shifts for microbial communities in summer. Based on our results, we expect that the roles of freshwater and brackish psychrophilic Actinobacteria, Betaproteobacteria and Flavobacteriia in the upper zone of the CAO ecosystem will increase in concert with global warming. This may have unexpected consequences that range from altered metabolic functions, such as increased photoheterotrophy by Candidatus Aquiluna and its relatives [93], increased use of DOM from brine drainage as a substrate by Polaromonas and its relatives [102] and emerging infections by Flavobacterium and Polaribacter and their relatives of ice-associated fish [118], i.e., the polar cod Boreogadus saida in the CAO [119]. To achieve a better understanding of the consequences of the fast ecosystem changes in the CAO, future studies should concentrate on identifying the many still-unknown metabolic functions of the psychrophilic microbial communities, how they are related to biogeochemical cycles and how they are affected by the changing environment.
References
Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, et al. Structure and function of the global ocean microbiome. Science. 2015;348:1261359.
Fernández-Méndez M, Turk-Kubo KA, Buttigieg PL, Rapp JZ, Krumpen T, Zehr JP, Boetius A. Diazotroph diversity in the sea ice, melt ponds, and surface waters of the Eurasian Basin of the Central Arctic Ocean. Front Microbiol. 2016;7:1884.
Gosselin M, Levasseur M, Wheeler PA, Horner RA, Booth BC. New measurements of phytoplankton and ice algal production in the Arctic Ocean. Deep Sea Res II. 1997;44:1623–44.
Rich J, Gosselin M, Sherr E, Sherr B, Kirchman DL. High bacterial production, uptake and concentration of dissolved organic matter in the Central Arctic Ocean. Deep Sea Res II. 1997;44:1645–63.
Wheeler PA, Gosselin M, Sherr E, Thibault D, Kirchman DL, Benner R, Whitledge TE. Active cycling of organic carbon in the Central Arctic Ocean. Nature. 1996;380:697–9.
Galand PE, Potvin M, Casamayor EO, Lovejoy C. Hydrography shapes bacterial biogeography of the deep Arctic Ocean. ISME J. 2010;4:564–76.
Bano N, Hollibaugh JT. Phylogenetic composition of bacterioplankton assemblages from the Arctic Ocean. Appl Environ Microbiol. 2002;68:505–18.
Li Y, Wang Z, Lin X. Microbial community structure of Arctic seawater as revealed by pyrosequencing. Acta Oceanol Sin. 2016;35:78–84.
Bowman JS, Rasmussen S, Blom N, Deming JW, Rysgaard S, Sicheritz-Ponten T. Microbial community structure of Arctic multiyear sea ice and surface seawater by 454 sequencing of the 16S RNA gene. ISME J. 2012;6:11–20.
Rapp JZ. Bacterial diversity in sea ice, melt ponds, water column, ice algal aggregates and deep-sea sediments of the Central Arctic Ocean. Bremen, Germany: AWI; 2014. p. 99.
Deming JW, Collins RE. Sea ice as a habitat for bacteria, archaea and viruses. In:Thomas DN, editor. Sea Ice. 3rd ed. Oxford: John Wiley & Sons; 2017. p. 326–51.
Pedrós-Alió C, Potvin M, Lovejoy C. Diversity of planktonic microorganisms in the Arctic Ocean. Progr Oceanogr. 2015;139:233–43.
Overland J, Walsh J, Kattsov V. Trends and feedbacks. In: Snow, water, ice and permafrost in the Arctic (SWIPA). Arctic Monitoring and Assessment Programme (AMAP): Oslo, Norway; 2017. p. 9–23.
Ding Q, Schweiger A, L’Heureux M, Battisti DS, Po-Chedley S, Johnsen NA, et al. Influence of high-latitude atmospheric circulation changes on summertime Arctic sea ice. Nat Clim Change. 2017;7:289–95.
IPCC. Climate Change 2014 – Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change [Pachauri RK, Meyer LA, editors]. Geneva, Switzerland: IPCC, 2015. p.151.
Barnhart KR, Miller CR, Overeem I, Kay JE. Mapping the future expansion of Arcticopen water. Nat Clim Change. 2015;6:280–5.
Schweiger A, Lindsay R, Zhang J, Steele M, Stern H, Kwok R. Uncertainty in modeled Arctic sea ice volume. J Geophys Res. 2011;116:C00D06.
Laxon SW, Giles KA, Ridout AL, Wingham DJ, Willatt R, Cullen R, et al. CryoSat-2 estimates of Arctic sea ice thickness and volume. Geophys Res Lett. 2013;40:732–7.
Perovich DK, Richter-Menge JA. Regional variability in sea ice melt in a changing Arctic. Philos Trans R Soc A. 2015;373:20140165.
Screen JA, Williamson D. Ice-free Arctic at 1.5 °C? Nat Clim Change. 2017;7:230–1.
Duarte CM, Lenton TM, Wadhams P, Wassmann P. Abrupt climate change in the Arctic. Nat Clim Change. 2012;2:60–2.
Stroeve JC, Markus T, Boisvert L, Miller J, Barrett A. Changes in Arctic melt season and implications for sea ice loss. Geophys Res Lett. 2014;41:1216–25.
Wang C, Granskog MA, Hudson SR, Gerland S, Pavlov AK, Perovich DK, et al. Atmospheric conditions in the Central Arctic Ocean through the melt seasons of 2012 and 2013: impact on surface conditions and solar energy deposition into the ice-ocean system. J Geophys Res. 2016;121:1043–58.
Maslanik J, Stroeve J, Fowler C, Emery W. Distribution and trends in Arctic sea ice age through spring 2011. Geophys Res Lett. 2011;38:L13502.
Haine TWN, Martin T. The Arctic-Subarctic sea ice system is entering a seasonal regime: implications for future Arctic amplification. Sci Rep. 2017;7:4618.
Arrigo KR. Sea ice as a habitat for primary producers. In:Thomas DN, editor. Sea Ice. 3rd ed. Oxford: John Wiley & Sons; 2017. p. 352–69.
Eicken H, Bock C, Wittig R, Miller H, Poertner HO. Magnetic resonance imaging of sea-ice pore fluids: methods and thermal evolution of pore microstructure. Cold Reg Sci Technol. 2000;31:207–25.
Petrich C, Eicken H. Overview of sea ice growth and properties. In:Thomas DN, editor. Sea Ice. 3rd ed. Oxford: John Wiley & Sons; 2017. p. 1–41.
Stoecker DK, Gustafson DE, Baier CT, Black MMD. Primary production in the upper sea ice. Aquat Microb Ecol. 2000;21:275–87.
Reid PC, Johns DG, Edwards M, Starr M, Poulin M, Snoeijs P. A biological consequence of reducing Arctic ice cover: arrival of the Pacific diatom Neodenticula seminae in the North Atlantic for the first time in 800 000 years. Glob Change Biol. 2007;13:1910–21.
Lund-Hansen LC, Markager S, Hancke K, Stratman T, Rysgaard S, Ramløv H, et al. Effects of sea-ice light attenuation and CDOM absorption in the water below the Eurasian sector of Central Arctic Ocean (>88°N). Polar Res. 2015;34:23978.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010a;7:335–6.
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–8.
Gotelli NJ, Entsminger GL. Swap and fill algorithms in null model analysis: rethinking the knight’s tour. Oecologia. 2001;129:281–91.
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010b;26:266–7.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nuclei Acids Res. 2013;41:D590–D596.
Lynch MDJ, Neufeld JD. Ecology and exploration of the rare biosphere. Nat Rev Microbiol. 2015;13:217–29.
Pedrós-Alió C. Dipping into the rare biosphere. Science. 2007;315:192–3.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, et al. (2013). Vegan: community Ecology Package. R package version 2.0-7. (http://cran.r-project.org/package=vegan).
R Development Core Team. R: A Language and Environment for Statistical Computing Vienna. Austria: the R Foundation for Statistical Computing; 2014. ISBN: 3-900051-07-0, Available online at http://www.R-project.org/#.
Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27:209–20.
Rintala JM, Piiparinen J, Blomster J, Majaneva M, Müller S, Uusikivi J, Autio R. Fast direct melting of brackish sea-ice samples results in biologically more accurate results than slow buffered melting. Polar Biol. 2014;37:1811–22.
Garrison R, Buck KR. Organism losses during ice melting: a serious bias in sea-ice community studies. Polar Biol. 1986;6:237–9.
Stoecker DK, Gustafson DE, Black MMD, Baier CT. Population dynamics of microalgae in the upper land-fast sea ice at a snow-free location. J Phycol. 1998;34:60–69.
Harada N. Review: potential catastrophic reduction of sea ice in the western Arctic Ocean: its impact on biogeochemical cycles and marine ecosystems. Glob Planet Change. 2016;136:1–17.
Fuhrman JA, Cram JA, Needham DM. Marine microbial community dynamics and their ecological interpretation. Nat Rev Microbiol. 2015;13:133–46.
Horner-Devine MC, Bohannan BJM. Phylogenetic clustering and overdispersion in bacterial communities. Ecology. 2006;87:S100–S108.
Dupont CL, Larsson J, Yooseph S, Ininbergs K, Goll J, Asplund-Samuelsson J, et al. Functional tradeoffs underpin salinity-driven divergence in microbial community composition. PLoS ONE. 2014;9:e89549.
Herlemann DPR, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ, Andersson AF. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011;5:1571–9.
Herlemann DPR, Lundin D, Andersson AF, Labrenz M, Jürgens K. Phylogenetic signals of salinity and season in bacterial community composition across the salinity gradient of the Baltic Sea. Front Microbiol. 2016;7:1883.
Lozupone CA, Knight R. Global patterns in bacterial diversity. PNAS. 2007;104:11436–40.
Han D, Kang I, Ha HK, Kim HC, Kim OS, Lee BY, et al. Bacterial communities of surface mixed layer in the Pacific sector of the western Arctic Ocean during sea-ice melting. PLoS ONE. 2014;9:e86887.
Zinger L, Amaral-Zettler LA, Fuhrman JA, Horner-Devine MC, Huse SM, Mark Welch DB. Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS ONE. 2011;6:e24570.
Ladau J, Sharpton TJ, Finucane MM, Jospin G, Kembel SW, O’Dwyer J, et al. Global marine bacterial diversity peaks at high latitudes in winter. ISME J. 2013;7:1669–77.
Marteinsson VB, Groben R, Reynisson E, Vannier P. Biogeography of marine microorganisms. In:Stal LJ, Cretoiu MS, editor. The Marine Microbiome. Switzerland: Springer International Publishing; 2016. p. 187–207.
Ghiglione JF, Galand PE, Pommier T, Pedrós-Alió C, Maas EW, Bakker K, et al. Pole-to-pole biogeography of surface and deep marine bacterial communities. PNAS. 2012;109:17633–8.
Bowman JS. The relationship between sea ice bacterial community structure and biogeochemistry: a synthesis of current knowledge and known unknowns. Elem Sci Anthr. 2015;3:000072.
Boetius A, Anesio AM, Deming JW, Mikucki JA, Rapp JZ. Microbial ecology of the cryosphere: sea ice and glacial habitats. Nat Rev Microbiol. 2015;13:677–90.
Malmström RR, Straza TRA, Cottrell MT, Kirchman DL. Diversity, abundance, and biomass production of bacterial groups in the western Arctic Ocean. Aquat Microb Ecol. 2007;47:45–55.
Kirchman DL, Dittel AI, Malmstrom RR, Cottrell MT. Biogeography of major bacterial groups in the Delaware estuary. Limnol Oceanogr. 2005;50:1697–706.
Zeng YX, Zhang F, He JF, Lee SH, Qiao ZY, Yu Y, et al. Bacterioplankton community structure in the Arctic waters as revealed by pyrosequencing of 16S rRNA genes. Antonie Van Leeuwenhoek. 2013;103:1309–19.
Brinkmeyer R, Knittel K, Jurgens J, Weyland H, Amann R, Helmke E. Diversity and structure of bacterial communities in Arctic versus Antarctic pack ice. Appl Environ Microbiol. 2003;69:6610–9.
Hatam I, Lange B, Beckers J, Haas C, Lanoil B. Bacterial communities from Arctic seasonal sea ice are more compositionally variable than those from multi-year sea ice. ISME J. 2016;10:2543–52.
Larose C, Berger S, Ferrari C, Navarro E, Dommergue A, Schneider D, Vogel TM. Microbial sequences retrieved from environmental samples from seasonal Arctic snow and meltwater from Svalbard, Norway. Extremophiles. 2010;14:205–12.
Eronen-Rasimus E, Kaartokallio H, Lyra C, Autio R, Kuosa H, Dieckmann GS, et al. Bacterial community dynamics and activity in relation to dissolved organic matter availability during sea-ice formation in a mesocosm experiment. Microbiol Open. 2014;3:139–56.
Sambrotto RN, Goering JJ, McRoy CP. Large yearly production of phytoplankton in the western Bering Strait. Science. 1984;225:1147–50.
Yergeau E, Michel C, Tremblay J, Niemi A, King TL, Wyglinski J, et al. Metagenomic survey of the taxonomic and functional microbial communities of seawater and sea ice from the Canadian Arctic. Sci Rep. 2017;7:42242.
Eronen-Rasimus E, Piiparinen J, Karkman A, Lyra C, Gerland S, Kaartokallio H. Bacterial communities in Arctic first-year drift ice during the winter/spring transition. Environ Microbiol Rep. 2016;8:527–35.
Hatam I, Charchuk R, Lange B, Beckers J, Haas C, Lanoil B. Distinct bacterial assemblages reside at different depths in Arctic multiyear sea ice. FEMS Microbiol Ecol. 2014;90:115–25.
Boeuf D, Humily F, Jeanthon C. Diversity of Arctic pelagic Bacteria with an emphasis on photoheterotrophs: a review. Biogeosciences. 2014;11:3309–22.
Kelllogg CTE, Deming JW. Comparison of free-living, suspended particle, and aggregate-associated bacterial and archaeal communities in the Laptev Sea. Aquat Microb Ecol. 2009;57:1–18.
Monier A, Findlay HS, Charvet S, Lovejoy C. Late winter under ice pelagic microbial communities in the high Arctic Ocean and the impact of short-term exposure to elevated CO2 levels. Front Microbiol. 2014;5:490.
Collins RE, Rocap G, Deming JW. Persistence of bacterial and archaeal communities in sea ice through an Arctic winter. Environ Microbiol. 2010;12:1828–41.
Kirchman DL, Cottrell MT, Lovejoy C. The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ Microbiol. 2010;12:1132–43.
Giovannoni SJ. SAR11 bacteria: the most abundant plankton in the oceans. Annu Rev Mar Sc. 2017;9:231–55.
Herlemann DPR, Woelk J, Labrenz M, Jürgens K. Diversity and abundance of “Pelagibacterales” (SAR11) in the Baltic Sea salinity gradient. Syst Appl Microbiol. 2014;37:601–4.
Logares R, Bråte J, Heinrich F, Shalchian-Tabrizi K, Bertilsson S. Infrequent transitions between saline and fresh waters in one of the most abundant microbial lineages (SAR11). Mole Biol Evol. 2010;27:347–57.
Lovejoy C, Galand PE, Kirchman DL. Picoplankton diversity in the Arctic Ocean and surrounding seas. Mar Biodiv. 2011;41:5–12.
Koh EY, Cowie ROM, Simpson AM, O’Toole R, Ryan KG. The origin of cyanobacteria in Antarctic sea ice: marine or freshwater? Environ Microbiol Rep. 2012;4:479–83.
Hauptmann AL, Stibal M, Bœlum J, Sicheritz-Pontén T, Brunak S, Bowman JS, et al. Bacterial diversity in snow on North Pole ice floes. Extremophiles. 2014;18:945–51.
Jungblut AD, Lovejoy C, Vincent WF. Global distribution of cyanobacterial ecotypes in the cold biosphere. ISME J. 2010;4:191–202.
Cottrell MT, Kirchman DL. Photoheterotrophic microbes in the Arctic Ocean in summer and winter. Appl Environ Microbiol. 2009;75:4958–66.
Gradinger R, Ikävalko J. Organism incorporation into newly forming Arctic sea ice in the Greenland Sea. J Plankton Res. 1998;20:871–86.
Huang S, Wilhelm SW, Harvey HR, Taylor K, Jiao N, Chen F. Novel lineages of Prochlorococcus and Synechococcus in the global oceans. ISME J. 2012;6:285–97.
Nelson RJ, Ashjian CJ, Bluhm BA, Conlan KE, Gradinger RR, Grebmeier JM, et al. Biodiversity and biogeography of the lower trophic taxa of the Pacific Arctic region: sensitivities to climate change. In: Grebmeier JM, Maslowski W, editors. The Pacific Arctic Region: Ecosystem Status and Trends in a Rapidly Changing Environment. Dordrecht: Springer Science+Business Media; 2014. p. 269–336.
Waleron M, Waleron K, Vincent WF, Wilmotte A. Allochthonous inputs of riverine picocyanobacteria to coastal waters in the Arctic Ocean. FEMS Microbiol Ecol. 2007;59:356–65.
Díez B, Bergman B, Pedrós-Alió C, Antó M, Snoeijs P. High cyanobacterial nifH gene diversity in Arctic seawater and sea ice brine. Environ Microbiol Rep. 2012;4:360–6.
Sul WJ, Oliver TA, Ducklow HW, Amaral-Zettler LA, Sogin ML. Marine bacteria exhibit a bipolar distribution. PNAS. 2013;110:2342–7.
Vreeland RH, Litchfield CD, Martin EL, Elliot E. Halomonas elongata, a new genus and species of extremely salt-tolerant bacteria. Int J Syst Bacteriol. 1980;30:485–95.
Celussi M, Balestra C, Fabbro C, Crevatin E, Cataletto B, Fonda Umani S, et al. Organic-matter degradative potential of Halomonas glaciei isolated from frazil ice in the Ross Sea (Antarctica). FEMS Microbiol Ecol. 2008;65:504–12.
Li WKW. From cytometry to macroecology: a quarter century quest in microbial oceanography. Aquat Microb Ecol. 2009;57:239–51.
Kang I, Lee K, Yang SJ, Choi A, Kang D, Lee YK, et al. Genome sequence of “Candidatus Aquiluna” sp. Strain IMCC13023, a marine member of the Actinobacteria isolated from an Arctic fjord. J Bacteriol. 2012;194:3550–1.
Hahn MW. Description of seven candidate species affiliated with the phylum Actinobacteria, representing planktonic freshwater bacteria. Int J Syst Evol Microbiol. 2009;59:112–7.
Boeuf D, Cottrell MT, Kirchman DL, Lebaron P, Jeanthon C. Summer community structure of aerobic anoxygenic phototrophic bacteria in the western Arctic Ocean. FEMS Microbiol Ecol. 2013;85:417–32.
Simu K, Hagström Å. Oligotrophic bacterioplankton with a novel single-cell life strategy. Appl Environ Microbiol. 2004;70:2445–51.
Moreno-Pino M, De la Iglesia R, Valdivia N, Henríquez-Castilo C, Galán A, Díez B, Trefault N. Variation in coastal Antarctic microbial community composition at sub-mesoscale: spatial distance or environmental filtering? FEMS Microbiol Ecol. 2016;92:fiw088.
Bosi E, Fondi M, Orlandini V, Perrin E, Maida I, de Pascale D, et al. The pangenome of (Antarctic) Pseudoalteromonas bacteria: evolutionary and functional insights. BMC Genomics. 2017;18:93.
Qin Q, Xie B, Yu Y, Shu Y, Rong J, Zhang Y, et al. Comparative genomics of the marine bacterial genus Glaciecola reveals the high degree of genomic diversity and genomic characteristic for cold adaptation. Environ Microbiol. 2014;16:1642–53.
Klippel B, Lochner A, Bruce DC, Davenport KW, Detter C, Goodwin LA, et al. Complete genome sequence of the marine cellulose- and xylan-degrading bacterium Glaciecola sp. strain 4H-3–7+YE-5. J Bacteriol. 2011;193:4547–8.
Norman L, Thomas DN, Stedmon CA, Granskog MA, Papadimitriou S, Krapp RH, et al. The characteristics of dissolved organic matter (DOM) and chromophoric dissolved organic matter (CDOM) in Antarctic sea ice. Deep Sea Res II. 2011;58:1075–91.
Gawor J, Grzesiak J, Sasin-Kurowska J, Borsuk P, Gromadka R, Górniak D, et al. Evidence of adaptation, niche separation and microevolution within the genus Polaromonas on Arctic and Antarctic glacial surfaces. Extremophiles. 2016;20:403–13.
Sharma AK, Zhaxybayeva O, Papke RT, Doolittle WF. Actinorhodopsins: proteorhodopsin-like gene sequences found predominantly in non-marine environments. Environ Microbiol. 2008;10:1039–56.
Pinhassi J, DeLong EF, Béjà O, González JM, Pedrós-Alió C. Marine bacterial and archaeal ion-pumping rhodopsins: genetic diversity, physiology, and ecology. Microbiol Mol Biol Rev. 2016;80:929–54.
Darcy JL, Lynch RC, King AJ, Robeson MS, Schmidt SK. Global distribution of Polaromonas phylotypes - evidence for a highly successful dispersal capacity. PLoS ONE. 2011;6:e23742.
Cangelosi GA, Meschke JS. Dead or alive: molecular assessment of microbial viability. Appl Environ Microbiol. 2014;80:5884–91.
Kunin V, Engelbrektson A, Ochman H, Hugenjoltz P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol. 2010;12:118–23.
Matias MG, Combe M, Barbera C, Mouquet N. Ecological strategies shape the insurance potential of biodiversity. Front Microbiol. 2013;3:431.
Alonso-Sáez L, Sánchez O, Gasol JM, Balagué V, Pedrós-Alió C. Winter-to-summer changes in the composition and single-cell activity of near-surface Arctic prokaryotes. Environ Microbiol. 2008;10:2444–54.
Lennon JT, Jones SE. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev. 2011;9:119–30.
Barberán A, Bates ST, Casamayor EO, Fierer N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012;6:343–51.
Bar-Massada A. Complex relationships between species niches and environmental heterogeneity affect species co-occurrence patterns in modelled and real communities. Proc R Soc B. 2015;282:20150927.
Zelezniak A, Andrejev S, Ponomarova O, Mende DR, Bork P, Patil KR. Metabolic dependencies drive species co-occurrence in diverse microbial communities. PNAS. 2015;112:6449–54.
Agler MT, Ruhe J, Kroll S, Morhenn C, Kim ST, Weigel D, et al. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016;14:e1002352.
Comte J, Lovejoy C, Crevecoeur S, Vincent WF. Co-occurrence patterns in aquatic bacterial communities across changing permafrost landscapes. Biogeoscienes. 2016;13:175–90.
Verdugo J, Damm E, Snoeijs P, Díez B, Farías L. Climate relevant trace gases (N2O and CH4) in the Eurasian Basin (Arctic Ocean). Deep Sea Res I. 2016;117:84–94.
Eyice Ö, Namura M, Chen Y, Mead A, Samavedam S, Schäfer H. SIP metagenomics identifies uncultivated Methylophilaceae as dimethylsulphide degrading bacteria in soil and lake sediment. ISME J. 2015;9:2336–48.
Loch TP, Faisal M. Emerging flavobacterial infections in fish: a review. J Adv Res. 2015;6:283–300.
David C, Lange B, Krumpen T, Schaafsma F, van Franeker JA, Flores H. Under-ice distribution of polar cod Boreogadus saida in the Central Arctic Ocean and their association with sea-ice habitat properties. Polar Biol. 2016;39:981–94.
Jakobsson, M., Mayer, L., Coakley B, Dowdeswell JA, Forbes S, et al. (2012). The International Bathymetric Chart of the Arctic Ocean (IBCAO) Version 3.0. Geophysical Research Letters 39:L12609.
PAME. Large Marine Ecosystems (LMEs) of theArctic area: Revision of the Arctic LME map 15th of May 2013. 2nd edn. Akureyri, Iceland: PAME International Secretariat; 2013. p. 19.
Acknowledgements
We warmly thank the Swedish Polar Research Secretariat (www.polar.se) and the crew of RV Oden for their assistance with sampling and further help with practical matters before and during the LOMROG III cruise. We thank Lars Cresten Lund-Hansen and Brian K. Sorrell for drilling holes in the ice and Brenda Riquelme for laboratory assistance. This work was supported by grants from the Swedish research foundations VR (SWEDARCTIC 2011-2015), FORMAS (2012-1459) and the Carl Trygger Foundation for Scientific Research to PSL. Analyses and travels of BD and LF were supported by grants from the Chilean research foundations CONICYT (FONDAP 15110009 and DPI20140044 to BD, Postdoctorado 2014 N° 3140422 to BFG) and INACH (15-10 to BD, FP_03-13 to BFG, MT_01-12 to CS), CONICYT (Master Program) to CS, FONDECYT (1120719) to LF, and PFB-023 and ICM P05-002 to PM. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Fernández-Gómez, B., Díez, B., Polz, M.F. et al. Bacterial community structure in a sympagic habitat expanding with global warming: brackish ice brine at 85–90 °N. ISME J 13, 316–333 (2019). https://doi.org/10.1038/s41396-018-0268-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-018-0268-9
This article is cited by
-
Stochastic Processes Dominate in the Water Mass–Based Segregation of Diazotrophs in a High Arctic Fjord (Svalbard)
Microbial Ecology (2023)
-
Comparison of Deep-Sea Picoeukaryotic Composition Estimated from the V4 and V9 Regions of 18S rRNA Gene with a Focus on the Hadal Zone of the Mariana Trench
Microbial Ecology (2022)
-
Exploration of the Types of Rarity in the Arctic Ocean from the Perspective of Multiple Methodologies
Microbial Ecology (2022)
-
Impact of a bacterial consortium on the soil bacterial community structure and maize (Zea mays L.) cultivation
Scientific Reports (2021)
-
MinION sequencing from sea ice cryoconites leads to de novo genome reconstruction from metagenomes
Scientific Reports (2021)
