
Abstract
BET proteins, which influence gene expression and contribute to the development of cancer, are epigenetic interpreters. Thus, BET inhibitors represent a novel form of epigenetic anticancer treatment. Although preliminary clinical trials have shown the anticancer potential of BET inhibitors, it appears that these drugs have limited effectiveness when used alone. Therefore, given the limited monotherapeutic activity of BET inhibitors, their use in combination with other drugs warrants attention, including the meaningful variations in pharmacodynamic activity among chosen drug combinations. In this paper, we review the function of BET proteins, the preclinical justification for BET protein targeting in cancer, recent advances in small-molecule BET inhibitors, and preliminary clinical trial findings. We elucidate BET inhibitor resistance mechanisms, shed light on the associated adverse events, investigate the potential of combining these inhibitors with diverse therapeutic agents, present a comprehensive compilation of synergistic treatments involving BET inhibitors, and provide an outlook on their future prospects as potent antitumor agents. We conclude by suggesting that combining BET inhibitors with other anticancer drugs and innovative next-generation agents holds great potential for advancing the effective targeting of BET proteins as a promising anticancer strategy.
Similar content being viewed by others
Introduction
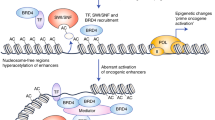
BET inhibitors are novel targeted medications in the research phase that regulate epigenetic modifications in the therapy of malignant tumors.1 Numerous investigations have demonstrated in the past few years that epigenetic modifications perform an essential function in tumor formation. Inhibitors targeting these epigenetic modification-related proteins can suppress overexpressed oncogenes, thus acting as potential antitumor agents. BET is a family of proteins with bromodomains (BRDs), which are distinguished by the presence of conserved BD1 and BD2 sequences at their N-terminals, as well as an extraterminal (ET) structure at the C-terminal (Fig. 1). They identify acetylated residues on histone H3 and H4 and have a stronger affinity when there are multiple acetylated fragments of 1–5 amino acids.2 BRD2, BRD3, BRD4, and BRDT are the four members of the BET proteins. Among them, the most powerful and well-researched BET protein is BRD4, known as the “reader” of lysine acetylation. It acts as a regulator of transcription factors that specifically bind to acetylated histone tails, recruit tumor-associated target genes, and act on the promoter and/or enhancer regions.3 Throughout the genome, the initiation of transcription for numerous genes is blocked by the interaction between ribonucleic acid polymerase II (RNA Pol II) and the site where transcription begins. RNA Pol II is released when BRD4 binds to an acetylated histone located at the site where transcription begins. The principle is that upon binding to active acetylated chromatin, it can replace the protein HEXIM1/7SK (small nuclear ribonucleoproteins), which inhibits its activity by binding with the positive transcriptional elongation factor (P-TEFb) through the BD2 sequence. As a result, RNA Pol II undergoes serine phosphorylation, which transforms it into an active, elongated state.4,5 A complex including BRD4 and various proteins acts as an intermediate and interacts with RNA Pol II, linking the enhancer to Pol II activation (Fig. 2a). Therefore, The BRD4 protein serves as a pivotal transcriptional regulator and is implicated in the control of gene expression for several super-enhancer-associated genes, such as the prominent oncogene c-MYC.6 This suggests that the manipulation of BET family proteins might have significant potential as a viable strategy for cancer treatment.

Structural and functional classification of human bromodomain proteins. The figure depicts the functional roles of the nine subclasses of human bromodomain proteins and provides a brief overview of the constituent proteins of each class and their structures. Modified from Zaware368

Basis and mechanism of BET inhibitor action. a The mechanism by which BRD4 promotes transcriptional elongation. BRD4 can regulate gene transcription in the nucleus through the recruitment of transcription factors. P-TEFb is a complex of cyclin T1 and CDK9 that binds to BRD4 and promotes serine phosphorylation of the C-terminal domain of RNA pol II, driving the transcription process. During gene transcription, BRD4 binds to P-TEFb, displacing the protein HEXIM1/7SK small nuclear ribonucleoprotein, which represses its activity, and converts it to an active elongated state. In the case of the transcription factor c-MYC, for example, BRD4 activates transcriptional initial response genes and is involved in cell cycle, cell proliferation, and apoptosis processes. b Schematic representation of the mechanism of action of BET inhibitors. The BET protein recognizes the acetylated lysine on BRD4 and acts as a backbone to recruit mediators, transcription factors, and P-TEFb to the promoter or enhancer, phosphorylating RNA Pol II at the target gene promoter or enhancer to drive the transcription process. BET inhibitors reduce BRD4 levels at the promoter and enhancer at a genome-wide level in tumor cells, and this inhibition is more pronounced at the super-enhancer compared with at the normal enhancer, targeting oncogenes more efficiently and specifically
In this paper, we review the biological properties and structural features associated with BET proteins, as well as the advancements achieved with the investigation of BET inhibitors. The initial topic of discussion pertains to the preclinical foundation for the therapeutic targeting of BET proteins in the context of cancer. Subsequently, a review is conducted on the many small-molecule inhibitors of BET proteins that have been investigated thus far. The review places particular emphasis on inhibitors that selectively target the BD1 and BD2 domains, as well as proteolysis targeting chimera (PROTAC) degraders. We review the early findings from clinical studies with BET inhibitors used alone as well as in tandem with various other medicines and discuss the potential for these drugs to serve as antineoplastics in the future. We also highlight the emerging issues of therapeutic resistance and adverse drug events and offer reasonable solutions to these problems. We offer an extensive review of studies involving this novel protein inhibitor in conjunction with a wide variety of anti-cancer therapies. This article underscores the potential of targeting BET proteins for addressing various diseases and enhancing human health.
Bromodomains: the epigenetic modification readers
Bromodomain development and discovery
Initially discovered in the Brahma gene of Drosophila melanogaster in 1992, bromodomains are 110-amino-acid protein modules and possess the ability to identify acetylated lysine residues present in histones and other proteins.3,7,8 Lysine acetylation (Kac) is a significant and prevalent post-transcriptional modification of proteins that plays a crucial role in the control of gene transcription, metabolic processes, and cell signaling.9,10,11,12 As scaffolding for the construction of macromolecules that modify the accessibility of chromatin for transcription factors and permit recruiting and the activation of RNA polymerases, BRDs influence transcriptional control by detecting acetylation marks on histones.2,13
Subsequent investigations have shown the significant involvement of BET proteins, particularly BRD4, in the pathogenesis and advancement of cancer. This discovery generated interest in utilizing BET proteins for cancer therapy.14,15,16,17,18,19 In a 2008 patent (PCT/JP2008/073864), BET inhibitors were initially described as agents that exhibit antitumor activity by diminishing the interaction between BD-containing proteins and acetylated histones. Two seminal articles revealed the cancer-fighting and inflammation-reducing effects of BET inhibitors in 2010.1,20 The subsequent identification of an increasing number of BET inhibitors, which showed substantial preclinical anticancer efficacy in an extensive number of solid and hematological tumors, led to the initiation of clinical trials for a variety of cancer therapies. The purpose of these studies was to evaluate the feasibility of combining BET inhibitors with other therapies in terms of effectiveness, security, and potential. They have also shown efficacy in treating other conditions, including inflammatory and cardiovascular diseases. Over the years, research on BET proteins, their biological functions, diseases, and targeted therapies has increased exponentially, providing a deeper understanding of their role and therapeutic potential. Ongoing research persists in investigating novel applications and combination therapies involving BET inhibitors, providing promising prospects for enhanced treatments in the future.
Structure of bromodomains
The human genome has a total of 61 BRDs, which are found throughout 46 distinct proteins.3 The proteins under consideration are characterized by their globular folding pattern, whereby they exhibit four α-helical tufts referred to as αZ, αA, αB, and αC.2 Two interhelical loops, ZA and BC, are present, which together provide a joining region for acetyl-lysine at one extremity of the helical bundle.2,21 While the ZA loop connects the B and Z helices, the BC loop binds A and C. A highly conserved asparagine residue in the BC loop creates a hydrogen bond that links the amide nitrogen and the acetyl-lysine carbonyl oxygen.2,22 The conserved tyrosine and asparagine residues are of paramount importance in the recognition of lysine acetylation. BRDs consist of a very conserved structural domain at the N-terminus accompanied by a divergent structural domain at the C-terminus, and this modular structure makes bromodomains the functional unit of protein interaction.23 Asparagine is present in 48 of the 61 BRDs in the human genome, whereas aspartic acid, tyrosine, and threonine are present in the other 13 BRDs. Those BRDs that include asparagine are considered “typical”, whereas the others are “atypical”.24,25
The BET family of proteins contains the BRD proteins BRD2, BRD3, BRD4, and BRDT, which form subfamily V of the BRD family (Fig. 1). BET differs from other BRDs by having two highly conserved tandem bromodomains (BD1 and BD2) at the N-terminal end and an extraterminal structural domain located at the C-terminal end. BRD4 and BRDT possess an extra C-terminal domain (CTD).20,26,27 The N-terminal structural domains, namely BD1 and BD2, serve as binding sites for acetylated lysine. The ET domain facilitates the recruitment of transcriptional cofactors and enhances the process of transcription. Additionally, the C-terminal domain plays a role in the recruitment of P-TEFb.2,28
Functions of bromodomains
Bromodomain proteins play crucial roles in several physiological processes and participate in interactions between proteins as well as between proteins and nucleic acids.2 Based on their cellular roles, BRDs may be categorized into nine primary groups (Fig. 1).3,29 These are, I. Histone lysine acetylation23,30,31,32; II. Histone lysine methylation23; III. Chromatin remodeling33,34,35; IV. Replication-coupled chromatin assembly36,37; V. Transcriptional activation29,38,39; VI. Protein sumoylation, ubiquitination, degradation21,40,41; VII. Promyelocytic leukemia (PML) nuclear bodies, transcriptional regulation39,42,43; VIII. Transcriptional repression, DNA repair44,45; IX. Cell shape control, insulin regulation.46 Among bromodomain proteins, the BET family has received the greatest attention and research, with BRD4 being the most characterized member.
BRD2
BRD2, formerly named RING3, is the earliest mammalian BET protein that has been functionally acknowledged.47 It operates as an atypical protein kinase, mostly localized inside the nucleus.48,49 Its primary role involves the activation of E2F1 and E2F2 proteins, hence facilitating the production of proteins essential for the G1/S phase transition.47,48 The protein in question has an affinity for the H3 and H4 domains of histones that have undergone acetylation. Its primary function involves the modulation of transcriptional activity via the recruitment of various transcription factors, co-activators, and repressors.50 During chromatin remodeling, BRD2 recruits many proteins, such as histone deacetylases.51,52 Research has shown that BRD2 is at its peak during the development and closure of the neural tube, and it is essential for both neurogenesis and embryogenesis.53,54 Researchers have noted in recent years that BRD2 is associated with improvements in insulin signaling as well as metabolic diseases.55
BRD3
The gene BRD3, alternatively called ORFX or FSHRG2, has the ability to interact with acetylated lysine residues found in the GATA1 transcription factor.56 This interaction plays a crucial role in regulating the expression of genes specific to the red lineage and megakaryocytes.56,57 It has been shown to be increased in activated lymphocytes, which points to the possibility of it playing a role in adaptive immunity.58 People who suffer with osteoarthritis and rheumatoid arthritis have also had this protein found in the synovial tissue, particularly in macrophages.59,60 This observation strongly indicates a potential significance of this molecule in the development of immune disorders. BRD3 contributes to the G1/S transition of dividing cells by controlling the transcription of the cell cycle protein D1.49
BRD4
BRD4, previously identified as MCAP, FSGRG4, or Hunk1, is extensively found in almost all types of tissues, mainly in the nucleus, and shares 80% of its amino acid sequence with BRD2.49,61,62 BRD4 is an important cell cycle regulatory protein that binds to chromosomes during mitosis and performs a crucial function in the regulation of the cell cycle, embryonic development, and maintaining genome stability.16,19,62,63,64 BRD4 plays a role in the activation of the P-TEFb complex by phosphorylating CDK9, which is the complex’s active motif. The phosphorylation of RNA Pol II is a consequence of this activation, thus regulating both transcription initiation and elongation.65,66,67 BRD4 may operate as a transcriptional repressor in addition to being an activator of transcription, according to recent research.66 The BRD4 protein has kinase activity, and its extraterminal domain has the capability to interact with SWI-SNF and CHD2 proteins, two proteins that are involved in ATP-dependent chromatin remodeling.28,68 The role of BRD4 in the appropriate activation of that nonhomologous end-joined pathway is of significant importance. Its influence on the DNA damage repair system relies on its capacity to establish a platform connecting components of histone modifications and DNA repair mechanisms.69,70 BRD4 significantly contributes to telomere maintenance through the recruitment of telomerase and telomerase-associated complexes to the telomeric ends.71,72 This recruitment facilitates the extension of telomeres and stabilizes them through the accumulation of acetylated chromatin at the telomeric termini.71,72
BRDT
BRDt is a bromodomain protein that exhibits specificity for the testis, being expressed only within this particular organ.73,74 Studies conducted on animals have demonstrated that the absence of the BRDt protein leads to infertility, low sperm count, and abnormalities in sperm morphology.75 Cell cycle protein A1 is a crucial regulating gene in the male germline that is required for sperm cells to enter the first meiotic division, and the expression of cell cycle protein A1 is initiated and regulated by BRDt.76,77
Mechanism and targets of action of BET inhibitors in tumors
BRD proteins exert a significant influence on regulating many critical oncogenes, including MYC, inside cancer cells. The specific targeting and modulation of key pathways in tumor progression, including the JAK/STAT and NF-κB signaling pathways, could be achieved by the inhibition of BRD proteins. BET inhibitors work by displacing BRD4 out of super-enhancer regions, which has a significant impact on vital pathways in cancerous cells. Super-enhancers, as a large set of enhancer clusters, can significantly regulate gene expression, occupying areas of up to 50 kb, far exceeding the coverage of several hundred bases of normal enhancers.78,79 In cancer, super-enhancers are mainly found in oncogenes and genes associated with tumor progression.78,79
Numerous genes’ expressions are regulated by BET inhibitors
Malignancies arise from genetic mutations and are associated with abnormal epigenetic regulation of chromatin. Histone modifications are important for the expression of genes. Abnormal histone acetylation can affect cell differentiation and apoptosis, which can result in the increase of transcription for pro-oncogenic growing genes. BRD4 plays a regulatory role in transcriptional elongation, mainly by binding to P-TEFb, which promotes RNA Pol II to become phosphorylated in order to aid in the transcriptional elongation of genes. The key regulatory mechanism of BRD4 is the promotion of c-MYC transcription.4,5,80,81 The analysis of genetic mutations in primary, metastatic, or recurrent ovarian cancers has shown that c-MYC gains are potential targets of BET inhibitors.82,83 Additionally, BRD4 plays a significant role in several cellular processes, such as cell differentiation, signal transmission, control of proto-oncogenes, modulation of the cellular cycle, and other pathophysiological processes through non-histone acetylation modifications. This suggests that the BD of BRD4 selectively inhibits the transcription of genes relevant to malignancy through binding acetylated histones as well as interacting with acetylated lysine residues from various proteins (Fig. 2b).84
Furthermore, BET inhibitors have other regulatory targets than c-MYC. Previous studies have reported numerous BET inhibitor-regulated genes in different cancers.81,85,86 BET inhibitors primarily function in hematologic malignancies by suppressing the transcriptional expression of BCL2 and c-MYC, two proto-oncogenes.4,80
BET inhibitors mainly regulate the signaling pathway known as NF-κB in non-small-cell lung cancer by targeting BRD4.87 By preventing BRD4 and the androgen receptor from directly interacting, BET inhibitors in prostate cancer slow the development of tumors and lower the expression of genes that are targets of androgen.88 Research on breast cancer indicates that BET inhibitors primarily prevent BRD4 from attaching to the BR-α promoter.89 In ovarian cancer studies, BET inhibitors have shown different mechanisms of action depending on the subtype of ovarian cancer and the genetic alterations present in the tumors. Utilizing cytology and ovarian cancer xenograft (PDX) models, Zhang et al. demonstrated that for sensitive ovarian cancer cells, BET inhibitors specifically suppressed FoxM1 and its subsequent targets by cell cycle block, while c-MYC expression was just temporarily downregulated and then returned to baseline levels. In patients with homologous recombination repair defects, Yang et al. observed that BET inhibitors directly act on BRCA1 and RAD51 to reduce the efficiency of homologous recombination, leading to reduced PARP-induced repair of DNA-damaged tumor cells, which results in apoptosis.90 In addition, studies have shown that BRD4 can interact with CtIP to reduce CtIP expression, and independent of BRCA1/2, RAS, or BRAF mutations, multiple tumor cell lines are more sensitive to PARP inhibitors.91 Moreover, Villar-Prados et al. demonstrated, through an advanced drug target screening platform (TPT) and an experimental approach, that NOTCH3 is significantly upregulated in ovarian carcinoma, which was shown to be closely linked to cell proliferation, chemoresistance, and apoptosis. BET inhibitors have the capability to selectively modulate the expression of NOTCH3 by targeting BRD4 and affect its downstream effector genes.92 Liu et al. showed that BET inhibitors can upregulate the pro-apoptotic factor BIM (BCL2L11) (of the BCL2 family), cause tumor cells to undergo apoptosis via the mitochondrial apoptotic pathway, and inhibit tumor cell invasion and migration through the STAT3 pathway by downregulating its phosphorylation. They also found that inhibition of STAT3 affects the immune microenvironment by increasing CD3+ and CD8+ lymphocytes and promoting tumor cell immunogenic death.93 Recent research indicates that tumor-associated macrophages are crucial to the development of ovarian cancer, with high expression of BRD4 upregulating CSF1 to promote macrophage proliferation and differentiation. BET inhibitors significantly inhibit CSF1 production by tumor cells, which could affect TAM-mediated cancer progression.94
JAK/STAT pathway targeted by BET inhibitors
JAK/STAT signaling is a crucial process in cell proliferation and differentiation, facilitated by extracellular cytokine-activated receptors. It is thought that disruption of this pathway plays a significant role in the development of malignant tumors.95 Through forming a heterodimer with the interleukin-7 receptor (IL7R), CRLF2 promotes proliferating and inhibits apoptosis in B-cell ALL cells through signaling through JAK2, JAK1, and STAT5.96 JQ1, a BET inhibitor, downregulates the transcription of IL7R, displaces BRD4 out of the IL7R promoter, and also suppresses the activation of JAK2 as well as STAT5.96 OTX015, a BET inhibitor, was found to negatively regulate intermediaries of the JAK/STAT signaling pathway in in vitro and in vivo experimental studies of mature B-cell lymphomas.97 Consequently, BET inhibitors present a promising therapeutic approach for disorders that are dependent on JAK/STAT signaling.
BET inhibitors target the NF-κB pathway
The dysregulation of NF-κB activity plays a significant role in the development and advancement of malignancy.98 The phosphorylation of the IκB kinase (IKK) complex constitutes a crucial step in the activation of the typical NF-κB pathway.99,100 Research into ABC DLBCL has shown that BET inhibitors reduce IKK activity; therefore, these inhibitors hinder the subsequent NF-κB-driven transcription processes and induce ABC DLBCL cell death.101 JQ1, a BET inhibitor, prevents BRD4 from binding to Ac-Lys310, hence suppressing NF-κB activation and NF-κB-related transcription.87,102 A study on melanoma found that BRD4 regulates SPP1 expression via NFKB2 and BET inhibitors stop the growth of carcinoma via the noncanonical NF-κB/SPP1 pathway.103 These results imply that BET inhibitors might be a useful therapeutic strategy for the management of malignancies caused by NF-κB.
Small-molecule BET protein inhibitors
BRD recruits several additional proteins to active genes that induce transcription by recognizing acetylated lysine residues in histones and transcribed proteins.23,104 The main way that BET proteins regulate transcription is by interacting with acetylated chromatin.105 Because of their significant significance in the pathophysiology of several diseases, BET proteins, a unique category of BD proteins, have the potential to be effective therapeutic targets. Numerous small-molecule compounds were formulated with the specific purpose of targeting BET proteins, presenting potential treatments for cancer, inflammation, cardiovascular diseases, and autoimmune disorders. These compounds hold promise for diverse cancer types, especially when employed alongside other small-molecule inhibitors and epigenetic modulators, yielding promising outcomes in certain cases. Nonetheless, the obstacles of pharmaceutical resistance and side effects persist, impeding the progress of BET inhibitors in clinical applications.106,107,108,109,110,111 The pursuit of enhancing medication effectiveness and minimizing drug-related adverse effects has led to the development of novel selective molecules. This section presents a detailed examination of several classes of BET inhibitors, elucidating their prospective applications and potential for novel drug development.
Non-selective BET inhibitors
A 2008 patent (PCT/JP2008/073864) first detailed the antitumor activity of BET inhibitors, which involves reducing the interaction of BRD proteins with acetylated histones. Two landmark investigations carried out in 2010 demonstrated that BET inhibitors have anticancer and anti-inflammatory properties.1,20 Subsequently, more new BET inhibitors have been discovered, exhibiting significant preclinical anticancer efficacy in many types of solid and hematological tumors.23,112,113
JQ1, regarded as a pan-BET inhibitor with similar inhibiting effects against BD1 as well as BD2, was among the earliest BET inhibitors to become available.1 Differential scanning fluorometry revealed that JQ1 had the highest binding affinity for BRD4 among 41 human BD-containing proteins.1 By interacting with BRD4’s acetylation lysine binding region and removing BRD4 from nuclear chromatin, it induces differentiation and suppresses tumor development in BRD4-dependent carcinomas.1 In animal models, JQ1 has also shown promising pharmacodynamic and antitumor.1,114 Considering that JQ-1 has a brief in vivo half-life and hence modest translational prospective,1 numerous BET inhibitors, including OTX015, i-BET762, and NHWD-870, were recently developed and clinically investigated to evaluate their safety and effectiveness among individuals with hematologic malignancies or solid tumors (Table 1).
OTX015 is a small-molecule inhibitor of BRD2, BRD3, and BRD4 with a structure comparable to that of JQ1, and an important advancement is its oral administration.97,115,116 In a 2016 clinical trial, the effects of OTX015 on AML and ALL were examined.117 Exposure to OTX015 caused a potent decrease in BRD2, BRD4, as well as c-MYC, along with an increase in HEXIM1 protein levels, whereas BRD3 expression remained unchanged.118 These changes suggest that OTX015 may lead to cell cycle arrest, apoptosis, and growth suppression.119 Additionally, research conducted in vitro has shown that OTX015 has promising synergistic effects with several anticancer drugs, particularly mTOR and BTK inhibitors.97
I-BET is a synthetic that “mimicks” acetylated histones to disrupt chromatin complexes that are essential to the transcription of important genes associated with inflammation in activated macrophages.20 Previous studies have demonstrated that it provides a safeguard against lipopolysaccharide-induced endotoxic shock and bacterial-induced sepsis.20 I-BET762 is an orally bioavailable BET inhibitor with pan-affinity for BET proteins, primarily targeting BRD2, BRD3, and BRD4.20,120 Studies have demonstrated that I-BET762 acts mainly through the downregulation of MYC and IRF4, as well as the upregulation of HEXIM1.121
NHWD-870, a recently developed BET inhibitor, demonstrates notable efficacy in suppressing the proliferation of multiple cancers by downregulating the expression of macrophage CSF1 in tumor cells. In comparison to three prominent BET inhibitors currently in clinical development, namely BMS-986158, OTX015, and GSK525762, NHWD-870 exhibits greater potency, as evidenced by a cytometric assay.94 Currently, NHWD-870 is undergoing Phase I clinical trials for the treatment of various kinds of cancer (CXHL200250). Through controlling SPINK6, NHWD-870 has been shown in recent studies to significantly diminish melanoma invasion and metastasis both in vivo and in vitro,122 and NHWD-870 in combination with cytarabine demonstrated synergistic effects against melanoma both in vivo and in vitro.123 NHWD-870 has also shown excellent therapeutic potential in the treatment of osteosarcoma.124 NHWD870 exhibited a favorable contraceptive effect in in vivo animal experiments, and upon discontinuation of the drug, fertility can be restored to its baseline. This suggests a potential future role for NHWD870 as a male contraceptive. Nonetheless, its safety and efficacy require further investigation in preclinical studies.125
Notably, the overall remission rate (complete/partial remission or stable disease status) for pan-BET inhibitors is less than 30% in most clinical trials based on current data.117,126,127,128 This low response rate might have something to do with the intermittent dosing schedule, whose purpose was originally designed to reduce targeted side effects such as thrombocytopenia.117,126,127 The primary challenge encountered in the development of pan-BET inhibitors is balancing the enhancement of drug efficacy with the reduction of side effects. Consequently, an important direction of BET inhibitor research is advancing the development of more potent or selective BET inhibitors.
Selective BD1 inhibitors
While the sequence similarity of BD1 and BD2 exceeds 75%, the similarity of their structural domains is merely 38%.129 The lack of specificity of pan-BET inhibitors towards BRD proteins and BD1 as well as BD2 has resulted in the occurrence of several undesirable effects. Consequently, enhancing selectivity by inhibiting either BD1 or BD2 exclusively may offer a potential solution to boost specificity, avoid adverse effects, and improve the clinical efficacy of BET inhibitors.23
MS436, the earliest identified small nanomolar BD1 inhibitor, has a selectivity that is ten times higher towards BRD4-BD1 compared to BRD4-BD2.130 The selectivity of MS436 towards BD1 is associated with the hydrogen bonding interactions between MS436 and certain amino acid residues, including Pro82, Gln85, Lys91, and Asn140.130 In mouse macrophages, MS436 effectively inhibited the transcriptional activity of BRD4 in the NF-κB-driven generation of the pro-inflammatory cytokines IL6 and nitric oxide.130 The subsequent inhibitor of this series, MS402, is nine-times more specific for BD1 than BD2.131 It effectively inhibits and mitigates colitis generated by T-cell metastasis in mice by impeding the excessive proliferation of Th17 cells.131
Olinone has a 100-fold higher BD1 selectivity than BD2, and Olinone promotes mouse oligodendrocyte progenitor differentiation, whereas JQ1 inhibits oligodendrocyte progenitor differentiation, suggesting that selective regulation of individual BDs enhances cellular regenerative activity in aging and neurodegeneration.132
GSK789, being a highly selective BET-BD1 inhibitor, exhibits selectivity for BD1 that is approximately 1000 times better than that of BD2. Comparable to the antiproliferative activities of pan-BET inhibitors, it maintains robust anti-inflammatory and immunomodulatory activity in vitro.133
Selective BD2 inhibitors
RVX-208 was the earliest known BD2-selective BET inhibitor to induce a more specific expression profile compared with the gene expression altered in response to JQ1.134 The limited range of genes impacted by RVX-208 diminishes its efficacy as an antitumor medication, but because of its anti-inflammatory effects, it is well tolerated as a treatment for cardiovascular disease and type 2 diabetes.135,136,137,138
ABBV-744 exhibits a remarkable degree of selectivity towards BD2 in BRD2, BRD3, and BRD4 and has an affinity for BD2 that is hundreds of times higher than that for BD1.139,140 In contrast to RVX-208, which has limited efficacy as an anticancer agent, treatment with ABBV-744 produces potent antiproliferative effects in AML and prostate cancer.139,140,141
SJ432 is a novel tetrahydroquinoline-like BD2 selective inhibitor that exhibits higher effectiveness than JQ1 in neuroblastoma models in vivo through sustained inhibition of expression of the driver oncoprotein c-MYC.142 This suggests that BD2-selective molecules may retain efficacy while mitigating several kinds of undesirable toxicity linked to excessive dosages of BET inhibitors, providing a strong rationale for the development of BD2-selective BET inhibitors in cancer therapy.
BY27 is a compound that selectively inhibits BD2 and demonstrates promising anticancer properties.143 It has been found to effectively inhibit cancer growth in mice while exhibiting lower toxicity compared to pan-BET inhibitors.143 The unique structural features of BY27 make it a promising candidate for the development and synthesizing of future BD2 selective inhibitors.143
However, there is ongoing debate surrounding the notion of specifically focusing on BD2 as a target for cancer therapy. Some researchers propose that selectively inhibiting BD1, while excluding BD2, is adequate to elicit the desired antitumor effects of pan-BET inhibitors.141 This stands in contrast to various BD2-selective BET inhibitors demonstrating potent anticancer activity, necessitating further investigation to reconcile these disparities. Nonetheless, it remains a fact that BD1 and BD2 may play different functions in the proliferation of diverse tissue types. Nevertheless, all the BD2-selective inhibitors have the ability to mitigate some of the undesired toxicity associated with pan-BET inhibitors.138,139,141,142,144 This suggests that the development of BET inhibitors that selectively target either the BD1 or BD2 domains might be a viable approach to improving their safety profile.
BET degraders: PROTACs
BET inhibitors prevent E3 ligase-mediated identification of the degradation region between BD1 and BD2, resulting in attenuated ubiquitination-dependent degradation of BET proteins, which causes frequent feedback upregulation of BET proteins themselves and leads to back-skipping of c-MYC expression.97,145 BET degraders offer a promising strategy for targeting BET proteins to overcome drug resistance by promoting BET protein breakdown to ensure a sustainable block of their function. The strategy is called PROTAC (Fig. 3).146,147 Target protein binder, E3 ligase recruitment element, and chemical linker make up the bifunctional small molecule known as PROTAC.148,149,150,151 The ligands situated on the two termini of PROTAC exhibit recognition capabilities towards the E3 ubiquitin ligase and the target protein, respectively, forming a ternary complex.152,153 The formation of the ternary complex facilitates the spatial closeness between the two proteins, leading to the polyubiquitination of the target protein, which subsequently undergoes destruction inside the cell via the action of the proteasome.152,153 Currently, cell inhibitor of apoptosis protein, cereblon ligand, murine double minute 2 ligand, and VHL ligand are the major E3 ubiquitin ligases used in PROTAC.154

Schematic diagram of proteolysis targeting chimera (PROTAC). The PROTAC molecule consists of a target protein binder, an E3 ligase recruitment element, and a chemical linker. The ligands at either end of PROTAC recognize E3 ubiquitin ligase, and the target protein forms a ternary complex (target protein/PROTAC/E3 ligase). E3 uses the E2 ubiquitin-binding enzyme to transfer ubiquitin to the surface of the target protein, following which the proteasome recognizes the polyubiquitination signal and degrades the target protein, while the PROTAC molecule breaks down and participates in the next degradation cycle
dBET1, the first BET degrader reported in 2015, was developed utilizing a thalidomide derivative and JQ1 as an E3 ligase handle, efficiently and selectively degraded multiple BET family members expressed in leukemic cells, and significantly reduced MYC levels.147 dBET1 significantly reduces BRD4 within an hour and completely degrades it in two, whereas JQ1 or thalidomide, as a single treatment, had no effect.147 As PROTACs completely deplete BRD4 protein levels and subsequent downregulation of MYC, they exhibit higher in vitro action than BET inhibitors.155,156,157 Although JQ1 is also used as a BD recruitment scaffold, the BET degrader MZ1 exhibits selectivity towards BRD4, suggesting that target selectivity might be improved by the introduction of the E3 ubiquitin ligase VHL.158 This is due to MZ1’s ternary crystal structure, which is linked to the VHL ligand via polyethylene glycol chains, allowing the development of particular interactions between molecules within the ternary complex, causing BRD4 to degrade selectively.159 MYC, CDK4, cyclin D1, and BTK are all significantly downregulated in a mantle cell lymphoma (MCL) cell line when exposed to BET degraders ARV-825 and ARV-771.155 ARV-771 has also shown antitumor efficacy in AML and CRPC models.160,161
Although PROTACs hold great promise for drug development, off-targeting, cell permeability, and stability are still significant challenges for clinical applications. One potential problem to overcome is the difficulty of synthesizing hybrid molecules, a process that involves optimizing the size and composition of linkers as well as E3 ligase ligands. Additionally, the complete loss of BET proteins may result in potential adverse effects due to their critical role in physiological states. Hence, once these challenges are overcome, PROTACs are expected to become an important antitumor treatment, ushering in a new era of biopharmaceutical innovation.
Clinical activity of BET inhibitors
A few single or combination BET inhibitors have been formulated for the therapeutic intervention of hematologic malignancies, NUT midline cancers, and various solid tumors (Tables 1 and 2). Most BET inhibitors are only in Phase I or II, with several having shown mixed outcomes in clinical trials. In this section, we examine published results from BET inhibitor clinical trials and delve into the mechanistic underpinnings of select combinations under investigation.
Clinical trial results of BET inhibitor monotherapy
In 2014, a Phase 1 dose-escalation trial for OTX015 among individuals with advanced acute leukemia was the first to report the clinical activity of a BET inhibitor.162 In 46 individuals with CRPC, NMC, and NSCLC, birabresib (OTX015) was the subject of a Phase Ib research to assess its safety, effectiveness, and pharmacokinetics (NCT02259114).163 Birabresib 80 mg one day, administered continuously, is the recommended Phase II dosage for individuals with solid tumors. In the future, research on birabresib should take into account intermittent dosage to potentially reduce the toxicity of long-term therapy since the medication has a favorable safety profile and dose-proportional exposure.
A recent Phase I/II study on molibresib monotherapy for relapsed or refractory hematologic malignancies (NCT01943851) involving 111 patients found that molibresib therapy was tolerated, although gastrointestinal and thrombocytopenic toxicities limited its use. The most frequently occurring side effects included diarrhea (50%), nausea (46%), and thrombocytopenia (40%). A total of 13% of the patients had an objective remission rate, with six of them experiencing full remission and seven experiencing partial remission.164
In 2019, individuals with relapsed/refractory solid tumors participated in a first-in-human trial involving mivebresib (ABBV-075).165 Mivebresib was administered to 14 individuals with solid tumors during dosage escalation, subsequent to an expanded cohort of 49 individuals with prostate cancer. The most frequent side effects linked to mivebresib that occurred as a result of therapy were dysgeusia (49%), thrombocytopenia (48%), fatigue (26%), and nausea (25%). Of the 61 patients evaluable for dose escalation, 26 (43%) had stable disease and 35 (57%) had progressive disease. The median progression-free survival was 1.8 months (95% CI: 1.8–1.9).
A Phase 1/2a open-label study (NCT02419417) evaluated the safety, tolerability, pharmacokinetics, and pharmacodynamics of BMS-986158.166 For the purpose of dose escalation, three BMS-986158 dosimeters were used, namely A, B, and C. Dosimeter A was administered for a duration of 5 days, followed by 2 days of rest, with a dosage range of 0.75–4.5 mg. Dosimeter B was administered for a duration of 14 days, followed by 7 days of rest, with a dosage range of 2.0–3.0 mg. Dosimeter C was administered for a duration of 7 days, followed by 14 days of rest, with a dosage range of 2.0–4.5 mg. A total of 83 patients were included in the study, and the most treatment-associated side effects observed were diarrhea (43%), followed by thrombocytopenia (39%), with lower rates of treatment-related adverse events in schedules A (72%) and C (72%) compared with schedule B (100%). Stable disease was seen in 12 individuals (26.1%) after treatment with schedule A, in 3 individuals (37.5%) following treatment with schedule B, and in 9 individuals (31.0%) following treatment with schedule C. The study demonstrated that Plan A dosing had acceptable tolerability, preliminary anticancer efficacy, and dose-proportional pharmacokinetics.
A first-in-human Phase I study of 64 patients with relapsed/refractory lymphoma reported on the safety, pharmacokinetics, pharmacodynamics, and preliminary clinical activity of CPI-0610 (NCT01949883).167 Pelabresib (CPI-0610) was found to be a selectivity and potency small-molecule inhibitor of BET proteins at a maximum tolerated dose (MTD) of 225 mg once daily for 14 days with a 7-day break, with a clear pharmacokinetic/pharmacodynamic relationship and a manageable clinical safety profile. Pelabresib was able to inhibit BET target genes in a dose-dependent manner, with inhibition of IL8 and CCR1 mRNA at doses above 120 and 170 mg, respectively.
A Phase 1 study was conducted in patients with NMC, other solid tumors, or DLBCL with MYC dysregulation using a 21-day or 14-day cycle of subcutaneous RO6870810 injections, respectively (NCT01987362).168 Fatigue (42%), decreased appetite (35%), and injection site erythema (35%) were the most common treatment-emergent adverse events. This study demonstrated the safety, favorable pharmacokinetics, evidence of target engagement, and preliminary single-agent activity of RO6870810.
The safety and efficacy of ZEN-3694 plus enzalutamide in metastatic CRPC (mCRPC) were evaluated in a Phase Ib/IIa study.169 Of the 75 patients enrolled, 30 (40.0%) were resistant to abiraterone, 34 (45.3%) to enzalutamide, and 11 (14.7%) to both. Fourteen patients (18.7%) experienced grade ≥3 toxic reactions, including three patients with thrombocytopenia (4%). The trial findings indicate that the combination of ZEN-3694 and enzalutamide exhibits satisfactory tolerability and promising effectiveness in individuals diagnosed with mCRPC who have developed resistance to inhibitors targeting androgen signaling.
Results from clinical trials of BET inhibitors in combination with other drugs
In relapsed/refractory AML patients, a Phase 1 research examined the security and effectiveness of the BET inhibitor mivebresib (ABBV-075) as monotherapy (MIV-mono) or in combination with venetoclax (MIV-Ven). Of the 44 treated patients, 19 received MIV-mono and 25 received MIV-Ven. The study reported that the most common treatment-emergent adverse events associated with mivebresib were dysesthesia (74%), decreased appetite (42%), and diarrhea (42%), in the MIV-mono group, and decreased appetite (44%), vomiting (44%), and nausea (40%), in the MIV-Ven group. In the MIV-mono group, one patient achieved complete remission with incomplete recovery of blood counts, and 15 patients experienced resistance to the drug. In the MIV-Ven group, two patients were in complete remission, two were in partial remission, two had no morphological leukemic status, one was aplastic, and twelve were drug-resistant. The research findings indicated that mivebresib exhibited favorable tolerability and demonstrated anti-leukemic efficacy, both when administered as a standalone treatment and when used in conjunction with venetoclax.127
Patients with JAKi-naïve myelofibrosis received pelabresib (CPI-0610) with ruxolitinib in a Phase 2 MANIFEST trial (NCT02158858).170,171 Eighty-four patients received ≥1 dose of pelabresib and ruxolitinib. The combination of pelabresib and ruxolitinib demonstrated improved splenic and symptomatic responses, as well as improved myelofibrosis outcomes, in both ruxolitinib-naïve and formerly treated individuals. Furthermore, this combination therapy exhibited a favorable safety profile. Currently, the Phase 3 MANIFEST-2 trial is underway to assess the efficacy of pelabresib in combination with ruxolitinib specifically in myelofibrosis individuals who have not responded to previous JAKi therapy (NCT04603495).
The results of a Phase 1b dose-escalation study of a novel subcutaneous BET inhibitor, RO6870810, in combination with the BCL2 inhibitors venetoclax and rituximab in relapsed/refractory DLBCL were recently reported (NCT03255096).172 Dose-limiting toxicities included grade 3 febrile neutropenia, grade 4 diarrhea, and hypomagnesemia with the RO6870810 + venetoclax combination, and grade 3 hyperbilirubinemia and grade 4 diarrhea with the addition of rituximab. The most common grade 3 and 4 side effects were neutropenia (28%), anemia (23%), and thrombocytopenia (23%).
Potential mechanisms for combination therapy in clinical trials
A potential mechanism for the combination of the BET inhibitor ABBV-075 and the BCL2 inhibitor venetoclax (NCT02391480) in patients with AML is that venetoclax monotherapy increases MCL1 protein levels, whereas combination therapy with ABBV-075 results in a reduction in both MCL1 and Bcl-xL levels and together induces tumor cell apoptosis.173,174
The mechanism underlying the synergistic effect of the BET inhibitor AZD5153 with the PARP inhibitor olaparib (NCT03205176) in the treatment of ovarian cancer is that AZD5153 enhances the sensitivity of cells to olaparib and destabilizes genetic material by downregulating PTEN expression, thereby reversing acquired resistance.175 Through PAX5 and B-cell antigen receptor mechanisms essential for ABC DLBCL, AZD5153 enhances the efficacy of the Bruton tyrosine kinase inhibitor acalabrutinib in ABC DLBCL cell lines (NCT03527147).176
The safety and effectiveness of the combination of ZEN003694 and abemaciclib were evaluated in a Phase I clinical study (NCT05372640), including patients with NUT cancer or other solid tumors. The potential mechanism by which this combination was found to be effective in estrogen receptor (ER)-positive breast cancer was that ZEN003694 treatment reversed the significant overexpression of CDK6 and CCND1 that leads to abemaciclib resistance.177
Adverse events associated with BET inhibitors
While exploring the clinical activity of BET inhibitors, the adverse events related to BET inhibitors require more attention. According to current studies, nausea/vomiting, thrombocytopenia, fatigue, diarrhea, dysgeusia, anemia, decreased appetite, and hyperglycemia are the most common adverse effects of BET inhibitors.117,120,126,163,165,178,179,180 Most of these adverse effects also occur with other anticancer drugs and are tolerated by patients; however, some can have serious consequences and must be treated with caution.
Thrombocytopenia is the most common and serious hematologic adverse effect of dose-limiting toxicity observed clinically with BET inhibitors,117,120,126,165,178 and in severe cases, may lead to coagulation failure or even uncontrollable bleeding. This side effect may be related to BET inhibitors interfering with the transcription factor GATA1,57,181,182,183 which is known to have an essential function in maintaining red blood cell function.184,185 In clinical trials, BET inhibitor-induced thrombocytopenia was found to be reversed by stopping the therapy for one week after two consecutive weeks of oral OTX015.117,126 While it is generally seen that platelet counts tend to recover within a week after discontinuing the medication, this intermittent dosing may reduce drug efficacy and contribute to acquired drug resistance. Another concern associated with increasing BET inhibitor doses to consistently achieve effective plasma drugs concentrations, there may be an increased risk of other dose-limiting toxicities. One approach worth exploring is that combining a BET inhibitor with an antiplatelet-destructive agent may ameliorate this adverse effect.
Studies have demonstrated that BET inhibitor therapy can induce the transactivation of viral genomes, such as the EB virus and Kaposi’s sarcoma-associated herpesvirus.186,187,188,189 Furthermore, there is a concern that latent human immunodeficiency virus (HIV) could potentially be reactivated as a result of such therapeutic intervention. HIV destroys the individual’s immune system via weakening CD4-positive helper T cells, leaving the host susceptible to pathogenic infections and malignancies.190 BRD4 can silence the HIV genome by competing with Tat, a P-TEFb-related viral transactivator, and by inducing CDK9 phosphorylation at threonine 29, which inactivates CDK9.65,191,192,193 BRD2 can silence the HIV genome by introducing the repressor complex E2F1/p50 heterodimer into the HIV promoter or by interacting with the architectural/insulator protein CCCTC-binding factor to form a transcriptional boundary.194,195 Treatment with BET inhibitors deregulates the HIV genome by targeting BRD4 and BRD2, leading to the transactivation of HIV. Therefore, it is important to monitor for potential reactivation of viral infections during BET inhibitor treatment.
For male patients using BET inhibitors, testicular atrophy is a concerning adverse effect. As BRDT performs a vital function in spermatogenesis and this role is mainly mediated by BD1, and BRDT and the BD1 structural domain of BRD4 share 81% homology, even modest dosages of pan-BET inhibitors cause testicular atrophy in male mice.75,196 This testicular toxicity induced by JQ1 was more pronounced in mature male mice compared to young males, suggesting a possible inhibition of spermatogenesis, an inhibition that appeared to be reversible without affecting hormone levels, a finding that suggests a potential application of BET inhibitors as male contraceptives.196 However, whether sperm inhibition by BET inhibitors can be sufficient for contraception, whether testicular function can be fully restored after discontinuation of the drug, and whether fetal malformations can be caused if defective or incompletely restored sperm complete the fertilization process and produce offspring after use of the drug are questions that need to be addressed in further research.
Data from in vivo studies suggest that BRD4 inhibition can also induce a variety of important, albeit reversible, phenotypes, including alopecia, skin hyperplasia, and small intestinal stem cell deficiency.197 Furthermore, BET inhibitors also have therapeutic effects on non-tumor cells, raising concerns about collateral damage to healthy tissues from their usage as anticancer drugs. BET inhibitors may affect cardiomyocytes’ BRD4-dependent transcriptional programs, which might help prevent heart failure brought on by hypertrophy of the cardiomyocytes; however, this also highlights the risk of potential adverse cardiac effects.198,199,200
Preclinical data suggest that synergistic therapy may reduce overlapping toxicity issues by enhancing cytotoxicity and enabling every drug to be administered with a lower dosage. As an example, treatment of lymphoma cells with a combination of the BET inhibitor RVX2135 and the histone deacetylase (HDAC) inhibitors vorinostat or panobinostat resulted in apoptosis at doses of each drug that induced only cell cycle arrest when used as monotherapy.201 The overall survival of mice that had a transplantation of 2749 lymphoma cells was shown to be significantly enhanced when treated with a combination of RVX2135 and vorinostat, demonstrating a significant synergistic effect.201 In addition, vorinostat is effective at lower doses, suggesting that toxicity from the combination may be reduced if the dose of individual drugs can be reduced without compromising the efficacy.201 In many cases, the therapeutic activity of BET inhibitors in conjunction with other pharmaceutical agents may be attained at non-cytotoxic dosages, suggesting that using lower doses than when these drugs are administered alone may avoid excessive toxicity in humans. These issues are explored in depth in the subsequent section. Notably, not all combination therapies achieve this considerable effect. For example, when combining BET inhibitors and mTOR inhibitors, thrombocytopenia is a common adverse effect of these two groups of medicines; therefore, the problem of superimposed side effects from both drugs must be considered when using them in combination.44
Resistance to BET inhibitors
Although BET inhibitors have demonstrated efficacy against various cancers, resistance to these inhibitors remains a significant issue in clinical treatment. Tumor or cancer cell insensitivity to BET inhibitors may be due to either primary (or inherent) or acquired resistance resulting from secondary adaptation to treatment. Therefore, exploring the mechanisms underlying resistance to BET inhibitors is essential for optimizing their clinical efficacy.
Several studies have investigated the mechanisms responsible for resistance to BET inhibitors in different cancers. For instance, a 2015 study on leukemia found that in human and mouse leukemia cells, enhanced Wnt/β-catenin signaling partly contributed to BET inhibitor resistance. However, negative regulation of this process led to a return of susceptibility to BET inhibition, as demonstrated both in vitro and in vivo.202 In the same year, Rathert et al. also demonstrated that Wnt signaling was a driver and potential biomarker of BET resistance in leukemia, and they suggested that rewiring of the transcriptional program is a key mechanism that promotes resistance to BET inhibitors.203 CRISPR-mediated loss-of-function screens have emerged as a robust methodology for the unbiased identification of potential candidates linked to various biological phenotypes.204,205,206 In a combinatorial CRISPR screen of KMT2A rearranged leukemia cell lines, deficiency of the gene encoding SPOP was found to cause significant BET inhibitor resistance, which was subsequently proven in a xenograft model.207 In a study on TNBC, resistance to BET inhibitors was closely associated with hyperphosphorylation of MED1 and BRD4, which was associated with the decreased activity of protein phosphatase 2A in resistant cells.208 BRD4 as well as LSD1/NuRD complexes co-localize at super-enhancers in TNBC, and these complexes limit the over-activation of gene clusters associated with drug-resistance function, including GNA13 and PDPK1.209 As a result of the BRD4/LSD1/NuRD complex’s decommissioning due to prolonged treatment or PELI1 instability against LSD1, resistance to JQ1 and other medicinal substances developed.209 This suggests that treating TNBC by jointly targeting BRD4 and PELI1 may address the drug-resistance problem to some extent. TAMs are often involved in mediating the resistance of cancerous cells to platinum-based chemotherapy, anti-VEGF/VEGFR treatment, and radiotherapy,210,211,212,213 and one study discovered that TNBC-stimulated TAMs activate NF-κB signaling by upregulating IKBKE expression to increase breast carcinoma cell resistance to BET inhibitors.214 BRD4 hyperphosphorylation, which is associated with CDK9 kinase activity, has also been found in NMC and other cancers. This indicates that BRD4 hyperphosphorylation may be another important mechanism involved in resistance to BET inhibitors.215
These findings establish a theoretical foundation for synergizing BET inhibitors with other pharmaceutical agents to enhance their clinical effectiveness. Further investigation is required to have a better understanding of the processes through which medication resistance arises and the steps that work to avoid it. These studies will help develop more effective methods for treating patients who have developed drug resistance and will improve the administration of these medications in combination treatment.
Combination treatment of BET inhibitors
Although resistance to BET inhibitors is a significant impediment when it comes to clinical use, preclinical studies have shown that resistance to a single-target drug can be overcome when used in combination with other drugs, providing a bright future for BET inhibitors. Based on preclinical data, combining a BET inhibitor with another drug can produce synergistic effects in a variety of ways. There are currently several clinical trials that are investigating the use of BET inhibitors in conjunction with other medications (Table 2). The concurrent administration of BET inhibitors and other pharmaceutical agents also contributes to the mitigation of potential drug toxicity, because of their higher efficacy at lower drug doses.
Combination with chemotherapy agents
Multiple studies have shown evidence of the synergistic effects of BET inhibitors in combination with cisplatin in ovarian malignancies as well as mouse xenograft models, including those that are resistant to platinum-based treatments and relapsing models.216,217,218,219 Aldehyde dehydrogenase (ALDH)-positive cells are recognized contributors to tumor progression and relapse following initial chemotherapy response.220 This may stem from the differential impact of BET inhibitors, which suppress ALDH activity and ALDH1A1 expression, in contrast to the typical effect of cisplatin.217 Furthermore, ovarian cancer cells have exhibited a synergistic response through ALDH1A1 downregulation when combined with platinum-based chemotherapeutic agents.221 In addition, it has been demonstrated that BET inhibitors downregulate the expression of Bcl-2 and Survivin, increasing the sensitivity of ovarian cancer cells to cisplatin.218,222 Furthermore, the predominant mechanism of action is thought to be DNA damage induction,223 and BET inhibitors can decrease the expression and functionality of BRCA1 and RAD51 via inhibiting BRD4 and impair the reporting activity of homologous recombination (HR), thus making HR-proficient ovarian tumor cells more sensitive to DNA damage.216,224 Taken together, these findings present a compelling justification for the administration of BET inhibitors when combined with cisplatin for the treatment of HR-proficient ovarian carcinoma.
TNBC is distinguished by the lack of clearly defined molecular targets, and its genomic heterogeneity poses limitations on the available treatment choices for chemotherapy. The use of platinum-based drugs is increasingly being recognized as a viable approach for the development of effective therapeutics targeting TNBC.225,226,227,228 Studies have shown that BET inhibitors can downregulate BRCA1 and RAD51 expression and impair homologous recombination-mediated DNA damage repair, thereby inducing the functional creation of a BRCAness phenotype in TNBC cells.224,229 Findings conducted in vitro have shown that TNBC cells with BRCAness are more sensitive to platinum salts. Clinical trials have also shown enhanced efficacy of carboplatin in TNBC populations with BRCA1/2 mutations.224,229,230 Furthermore, it has been shown that BET inhibitors have cytotoxic effects in combination with chemotherapy in in vitro tests on TNBC cells. JQ1 has been shown to have a synergistic interaction with mitotic drugs, such as docetaxel and vinorelbine, as well as with DNA-damaging agents like cisplatin and carboplatin.230
In a study on colorectal cancer, the concurrent administration of BET inhibitors together with 5-fluorouracil or oxaliplatin demonstrated a noteworthy enhancement in the effectiveness of treatment in colorectal cancer cell.231,232 Recently, the mechanisms underlying these findings have also been explored. Death receptor 5 (DR5) is an essential constituent of the extrinsic apoptotic pathway in colorectal cancer cells.233 Studies have shown that depletion of BRD4 and treatment with BET inhibitors significantly induce DR5 expression.232 The induction of DR5 is regulated by endoplasmic reticulum stress and CHOP, which is critical for the chemical sensitization and apoptotic effects of BET inhibitors.232 Additionally, increased DR5 induction is also responsible for enhancing the sensitivity of colorectal malignancies containing SPOP mutations to BET inhibition.232 The BRD4 E3 ubiquitin ligase subunit SPOP is important for regulating this process.234 Combining BET inhibitors with chemotherapy effectively inhibited cancer development in a DR5-dependent manner in a colorectal cancer xenograft model; moreover, enhanced DR5 induction and apoptosis could effectively suppress the growth of patient-derived xenograft tumors.232
In summary, the use of BET inhibitors in conjunction with chemotherapy is a highly effective approach to cancer treatment that can improve outcomes, minimize toxicity, and prevent drug resistance (Fig. 4a). However, it is important to note that this combo treatment may potentially raise the possibility of treatment-related adverse events. Therefore, the selection of the optimal combination therapy regimen requires a comprehensive evaluation of the patient’s individual characteristics, disease status, and tumor type.

BET inhibitors synergize with a variety of antitumor agents. a BET inhibitors in combination with chemotherapeutic agents re-sensitize chemotherapy-resistant tumor cells. The figure summarizes the combinations of BET inhibitors and chemotherapeutic agents that are currently used in multiple tumors. b BET inhibitors combined with PARP inhibitors re-sensitize tumor cells resistant to PARP inhibitors. A combination of two drugs currently applied to several tumors is presented in the figure. c Inhibition of BET proteins upregulates Rab8A expression, which upregulates PD-L1 expression at the cell membrane, thereby sensitizing PD-L1 blockade. d Combined inhibition of BET and BCL-2 re-sensitizes tumor cells to BCL-2 inhibitors. Multiple drug combinations demonstrated synergistic effects in a variety of tumors
Combination with PARP inhibitors
In preclinical animal models of ovarian carcinoma with HR proficiency, BET inhibitors have demonstrated an ability to heighten tumor sensitivity to PARP inhibitors.224 This outcome could potentially stem from BET inhibitors diminishing HR activity while intensifying the DNA damage response prompted by PARP inhibitors. BET inhibitors can impact the transcription of pivotal HR genes like RAD51 and BRCA1.224 Additional research has demonstrated that BET and PARP inhibitors exhibit synergistic activity against BRCA1/2 wild-type ovarian malignancies. This can be attributed to the downregulation of DNA topoisomerase II binding protein 1 (TOPBP1) and mitosis inhibitor protein kinase WEE1 by BET inhibitors.235 Prior to mitosis, at G2/M cell cycle checkpoints, WEE1 is essential in stopping DNA repair.236 Whereas TOPBP1 is crucial for DNA replication and damage signaling.237 Downregulation of TOPBP1 renders cells highly sensitive to PARP inhibitors.238,239 Hence, directing interventions toward WEE1 and TOPBP1 may make BRCA1/2 carcinoma cells sensitive to PARP inhibitors.
BET inhibitors enhance DNA damage induced by PARP inhibitors.224 Investigations conducted in a breast carcinoma model found that the combined use of JQ1 and olaparib markedly inhibited cancer development, which was related to the inhibition of BRCA1 and RAD51 expression by BET inhibitors.224,229
As part of a study to better understand their underlying mechanisms, researchers assessed the antiproliferative activity of the PARP inhibitor olaparib and the BET inhibitor JQ1 on PDAC in vitro and in vivo.240 The findings indicated that the combined application of JQ1 and olaparib exerted synergistic effects in vitro and were more effective in the independently derived PDX model in vivo than either drug alone.240 This could be attributed to JQ1’s ability to decrease the association between BRD4 and BRD2 with Ku80 and RAD51 promoter sites, and data from short hairpin RNA (shRNA) suggested that the expression of Ku80 and RAD51 in PDAC cell lines was dependent on BRD4 and BRD2.240 These findings offer a novel strategy of combining BET inhibitors and PARP inhibitors as a promising approach for treating PDAC and may lead to more strong countermeasures against this kind of cancer (Fig. 4b).
Furthermore, researchers evaluated the effects of combining BET inhibitors (JQ1 or I-BET762) with PARP inhibitors (olaparib or veliparib) on the cholangiocarcinoma (CCA) cell lines, as well as the effectiveness of JQ1 and olaparib in a CCA xenograft model.241 The results demonstrated that the efficacy of each combination therapy surpassed that of each medicine administered alone.241 Mechanistically, downregulation of the BET inhibitor molecular targets BRD2 or BRD4 using shRNA rendered CCA cells sensitive to BET inhibitors as a single drug and in combination with a PARP inhibitor.241 These results provide valuable information on the potential of combining BET inhibitors and PARP inhibitors as a novel approach to treating CCA. Moreover, a synergistic effect has been observed between BET inhibitors and PARP inhibitors.242
Although the combination of BET inhibitors and PARP inhibitors has shown promising prospects in antitumor therapy, there are also potential risks and limitations. For example, combined use may increase the toxicity of the treatment, which could lead to immune system suppression and liver damage. Therefore, while the combination of BET inhibitors and PARP inhibitors is a promising antitumor treatment approach, further research is needed to validate and refine its effectiveness.
Combination with immune checkpoint inhibitors
It has been identified that BET inhibitors are suppressors of CD274 (encoding PD-L1), acting at the transcriptional level via the inhibition of BRD4. As a result, there is a downregulation of PD-L1 expression on the surfaces of both tumor cells and immune cells.243,244,245 The combination of BET inhibitors and immune checkpoint inhibitors has demonstrated more effective synergistic effects than monotherapy, both in vivo and in vitro.244 Furthermore, evidence supporting the effectiveness of combination treatment was provided by the development of a mathematical model to simulate the combined action of BET and immune checkpoint inhibitors in breast cancer.246
BET inhibitors were shown to reduce BRD4 binding to the promoter of the encoded PD-L1 gene, hence downregulating PD-L1 expression levels in a mouse model of Eμ-myc transgenic lymphoma.244,247,248 This has led to the idea of combining BET inhibitors with immune checkpoint inhibitors, such as anti-PD-1 antibodies, to target the PD-1/PD-L1 axis. Combining these two types of inhibitors produced a synergistic response in mice harboring MYC-driven lymphomas, indicating an interaction between BET inhibitors and PD-1/PD-L1 immune checkpoints.244
Combining JQ1 with anti-PD-L1 antibodies to treat liver cancer in transgenic mice could strongly inhibit tumor progression.249 The effectiveness of this combination therapy is associated with BET inhibitor-induced upregulation of Rab8A expression, which can upregulate PD-L1 expression on the cell membrane and sensitize PD-L1 blockade (Fig. 4c).249 Overall, this combination therapy promotes the anticancer effect of anti-PD-L1 antibodies and enhances the liver’s anticancer immune response. In a mouse model of primary liver cancer bearing a heart xenograft, researchers explored a combination immunotherapy strategy employing JQ1 alongside the anti-PD-L1 antibody.250 The utilization of a combined immunotherapy strategy significantly inhibited the advancement of primary liver cancer without accelerating xenograft rejection.250 This presents an innovative and effective treatment strategy for solid organ transplant recipients.
Significant synergistic effects between BET inhibitors and PD-L1 inhibitors have been demonstrated in colorectal cancer,251 NSCLC,252 and prostate cancer.253 The combination of BET inhibitors and immune checkpoint inhibitors is a promising antitumor treatment approach, providing more treatment options for cancer patients. However, combination therapy also has potential risks and limitations. For example, it may increase the toxicity of treatment, leading to immunosuppression and liver damage. Further research is necessary to verify its safety and efficacy and to determine the optimal approach for combination therapy.
Combination with PI3K inhibitors
Research has shown that BET inhibitors have the ability to enhance the susceptibility of ovarian cancer cells to phosphoinositide 3-kinase (PI3K) inhibitors and inhibit the reactivation of the PI3K pathway subsequent to PI3K inhibitor therapy.254 Furthermore, it has been shown that BET inhibitors possess the ability to block receptor tyrosine kinases (RTKs) and the subsequent signaling pathways they activate. However, ovarian cancer cell lines can acquire resistance to BET inhibitors through adaptive reprogramming.84 The development of such resistance mechanisms primarily depends on the reactivation of the signaling pathway. Therefore, combination therapy involving BET inhibitors and kinase inhibitors may be a potential strategy for mitigating BET inhibitor resistance and improving the efficacy of BET inhibitor therapy. The synergistic impact of combining ponatinib with BET inhibitors has proven effective in ovarian cancer treatment. The heightened anticancer potency achieved through the combination of ponatinib and BRD4-targeted agents is related to the reduction of MYC expression inside cancer cells.255 One compelling study has shown that BET/PI3K combination therapy has synergistic effects in tumor organoid models derived from patients with ovarian clear cell carcinoma, offering a potential treatment option for this rare subtype of chemoresistant ovarian cancer.256 Nevertheless, further preclinical research is necessary to verify the effectiveness and security of this approach.
Research has indicated that persistent mTORC1 activation diminishes sensitivity to PI3K inhibitors, while inhibiting mTORC1 enhances PI3K inhibitor efficacy and prolongs resistance delays.257,258 Resistance to PI3K inhibitors frequently occurs via the mechanism of feedback activation of RTKs, such as the AKT and mTOR pathways (Fig. 5a).259,260,261,262,263 RTKs belonging to the epidermal growth factor receptor and insulin receptor families have been identified as direct targets of BRD4 in multiple cancer models.254 In these models, the inhibition of BET proteins can effectively regulate the PI3K signaling pathway in breast cancer cells.254 This regulation is achieved by preventing the recruitment of BRD4 to the regulatory regions of these receptors, thereby blocking the feedback activation of the RTKs.254 The combination inhibition of PI3K/BET in breast cancer cells demonstrated an effectively curbed reactivation of AKT and mTORC1, re-sensitizing the cells to PI3K inhibitors.254 This combination therapy’s effectiveness was also substantiated through an in vivo mice model of breast cancer.254 Researchers studying breast cancer cells discovered that combining therapy with JQ1 and the dual PI3K/mTOR inhibitor BEZ235 could prevent chromatin structure alterations, prevent malignancy cells from developing resistance, and cause xenograft regression in vivo as well as in vitro cell death.264 Significantly, this combination also exhibited remarkable synergy in hepatocellular carcinoma265 and colorectal cancer,266 showcasing the effectiveness of BET inhibitors in countering tumor resistance to dual PI3K/mTOR inhibition. This discovery offers an innovative and effective treatment strategy for cancer therapy. Notably, this combination treatment can also inhibit the development of drug-tolerant persisters, making therapy more durable and effective.264

BET inhibitors have synergistic antitumor effects with multiple signaling pathway inhibitors. a BET inhibitors block the feedback activation of RTKs, thereby re-sensitizing tumor cells to PI3K inhibitors. The figure shows PI3K inhibitors that have now been used in combination with BET inhibitors. b Combined inhibition of mTOR and BET cooperatively promotes tumor cell apoptosis, and synergistic effects of the two drugs have been found for a variety of drug combinations. c Mechanism of the synergistic effect of BET inhibitors with lapatinib and AKT inhibitors. In HER2 breast cancer cells, lapatinib inhibits kinases, leading to the activation of FOXO, which in turn translocates to the nucleus and recruits MLL2 and GCN5 to their target genes to add active histone markers, including H3K4m3 and histone acetylation. The modified histones can further recruit BRD4 protein to the target gene, thereby promoting Myc gene transcription to reduce the sensitivity of cancer cells to lapatinib.When cells are treated with AKT inhibitors, FOXO is dephosphorylated and translocates to the nucleus, while dephosphorylated SirT6 loses its ability to bind FOXO, resulting in increased FOXO acetylation. Acetylated FOXO interacts with BRD4 to form a transcriptional activation complex with P-TEFb and RNA polymerase II at the CDK6 gene promoter, activating CDK6 transcription and leading to resistance of breast cancer cells to AKT inhibitors. BET inhibitors target the BRD4 protein to affect Myc and CDK6 transcription, thereby restoring tumor cell sensitivity to lapatinib and AKT inhibitors
Activation of PI3K performs an essential function in the pathogenesis of lymphomas driven by the MYC oncogene, making it a prospective target for therapeutic intervention.267,268,269 Several studies have investigated the effectiveness of combined treatment, including BET and PI3K inhibitors, in various lymphomas, such as DLBCL97,101,270 Burkitt’s lymphoma (BL),270,271 MCL,272,273 marginal zone lymphoma,273 chronic lymphocytic leukemia (CLL),273,274 peripheral T-cell lymphoma,273 and mouse MYC-stimulated lymphoma cells.275 Combination therapies involve multiple mechanisms of action. First, BET inhibitors can improve the antiproliferative effect of PI3K inhibitors by increasing GSK3β S9 phosphorylation levels and β-catenin abundance through the downregulation of the E2 ubiquitin-conjugating enzymes UBE2C and UBE2T.270 Second, PI3K inhibitors upregulate various constituents within the B-cell antigen receptor pathway, whereas BET inhibitors downregulate some of these components (e.g., SYK, BLK, CD79A, and CD79B), thereby counteracting the adaptive response induced by idelalisib.276,277 Additionally, NF-κB activity is critical for the viability of some malignant cells and is maintained by the constitutive activity of IKK in the cytoplasm.278 By suppressing the expression of BRD2 and BRD4, BET inhibitors reduce IKK activity and block the downstream NF-κB-driven transcriptional program, which may strengthen the combined effect.97,101
Combination with mTOR signaling pathway inhibitors
Everolimus is an effective and safe oral inhibitor of mTOR, utilized in the treatment of breast carcinoma patients.279 This occurrence of resistance to everolimus has been linked to MYC upregulation, which is associated with higher BRD4 recruitment to MYC in everolimus-resistant cell lines.280,281 BET inhibition has been found to restore sensitivity in cells that are resistant to mTOR inhibitors.281 in vitro and in vivo studies have revealed that the combined therapy of everolimus and JQ1 exhibits higher effectiveness compared to the use of either drug alone.281 in vitro and xenograft models exhibited that OTX015, an innovative BET inhibitor, had synergistic antiproliferative effects when combined with everolimus.282 Additionally, the mTOR inhibitors rapamycin and Torin were previously found to enhance the susceptibility of JQ1-resistant cells to BET inhibition.280
mTOR has become a crucial therapeutic target in certain hematolymphoid tumors.283 BET inhibitors, when combined with mTOR inhibitors, have also shown strong synergistic effects against a variety of lymphomas and solid tumors.44,97,101,271,275,276,284,285 One possible mechanism is the inhibition of BET proteins, which leads to the restoration of sensitivity in drug-resistant cells towards mTOR inhibitors. However, since thrombocytopenia is a common toxicity of these two types of drugs, it is necessary to pay attention to drug side effects when using them in combination.44
A study using various molecular subtypes of patient-derived SCLC xenograft models demonstrated that, without appreciably raising toxicity, mTOR inhibition enhances BET inhibition’s anticancer activity in vivo.286 Moreover, BET inhibition induces apoptosis in SCLC models both in vitro and in vivo; the anticancer effect is further enhanced by cotreatment with mTOR inhibition.286 Activation of the intrinsic apoptotic pathway is the mechanism through which BET inhibition induces apoptosis in SCLC cells.287 BET inhibition upregulates RSK3, which activates the TSC2/mTOR/p70S6K1/BAD cascade to promote cell survival; however, this protective signaling can be blocked by mTOR inhibition to enhance BET inhibition-induced apoptosis (Fig. 5b).286
Combination with MEK inhibitors
Research has demonstrated evidence for synergistic benefits when treating ovarian cancer in vivo and in vitro with BET and MEK inhibitors, such as JQ1 and PD0325901 or trametinib, combined.288 BET inhibitors have been shown to induce cell cycle arrest rather than apoptosis in ovarian cancer cells, which may impose limitations on the potential therapeutic application for individuals with advanced ovarian cancer.288 However, the combined use of BET and MEK inhibitors can circumvent this constraint since co-administration of MEK inhibitors can nullify the BET inhibitor-induced feedback activation of the MAPK signaling pathway.288 Research has shown that the co-administration of BET and MEK inhibitors can coordinate the regulation of apoptotic molecules, such as BIM and BAD, and induce tumor cell apoptosis in ovarian cancer cells.288 Moreover, combining BET inhibitors with MEK inhibitors can also overcome resistance resulting from kinome reprogramming in neurofibromatosis type 1 (NF1)-deficient ovarian cancer cells, where MEK inhibitors transcriptionally induce the reactivation of RTK and its downstream pathways.289 Large-scale genomic studies have revealed that NF1, a RAS guanosine triphosphatase (GTPase)-activating protein that negatively regulates RAS family proteins, is lost in 12% of epithelial ovarian tumors.290,291 The loss of NF1 leads to the reactivation of RAS effector pathways, including PI3K/mTOR/AKT, RAF/MEK/ERK, and RALGDS signaling, which contributes to neoplastic proliferation and survival.290,292,293 MEK inhibitors trigger the transcriptional reactivation of RTK and its downstream pathways by destabilizing FOSL1, encompassing the RAF/MEK/ERK, JAK/STAT, and PI3K/AKT signaling pathways.289 However, in epithelial ovarian cancer cells lacking NF1, MEK inhibitor-induced RTK reprogramming renders them resistant to trametinib treatment, thereby reducing the growth inhibition of tumor cells by MEK inhibitors.289 BRD2 and BRD4 have been discovered to be pivotal players in the transcriptional regulation of RTKs in NF1-deficient cells, and functional inhibition of the BET protein has demonstrated the ability to counteract MEK inhibitor-triggered elevation of RTK RNA and protein expression.289 Therefore, BET and MEK inhibitors combined might serve as a beneficial therapeutic approach for NF1-deficient ovarian carcinomas; however, it is crucial to recognize that this combination therapy may not be effective for epithelial ovarian cancer cells that are NF1-sufficient.289 Overall, the combined use of BET and MEK inhibitors suggests a potentially advantageous strategy for addressing ovarian cancer therapeutically.
In TNBC, trametinib inhibits MEK to block the MEK/ERK pathway, resulting in a robust initial growth suppression. However, suppression of MEK/ERK triggers the activation of an alternate kinase pathway that inhibits growth. This upregulation allows the growth arrest to be overcome by an adaptive bypass response.294 BET inhibitors make trametinib-resistant cells more sensitive to the drug by inhibiting BRD4 and effectively reverse trametinib resistance by inhibiting the adaptive upregulation of RTKs.295 BET inhibitor and trametinib combined therapy produced synergistic growth inhibitory effects in different TNBC mouse models in vitro and in vivo.295 The concurrent administration of BET and MEK inhibitors has a synergistic effect on reducing the viability of TNBC cell lines and patient-derived xenograft models, suggesting that this treatment strategy may be a promising therapeutic option.296 Of particular note, this study found that cells exhibiting elevated expression levels of MYCN demonstrate heightened sensitivity towards BET inhibitors,296,297 providing a basis for extending the use of BET and MEK inhibitors in individuals who suffer from late-stage TNBC expressing MYCN.296 The finding has the potential to open up novel avenues for the development of more precise therapeutic interventions.
Limited research supporting the synergistic antitumor effects of BET and MEK inhibition in melanoma exists.298,299 One possible mechanism is that the co-administration of BET and MEK inhibitors can alleviate the transcriptional characteristics of MAPK and checkpoint inhibitor resistance, downregulate the transcription factor TCF19, and induce cell apoptosis.298
Combination with tyrosine kinase inhibitors
Lapatinib is a small-molecule tyrosine kinase inhibitor that targets HER2 as well as the epidermal growth factor receptor.300,301 It was approved by the FDA in 2007 for the treatment of advanced or metastatic breast cancer that overexpresses HER2 and was previously treated with anthracyclines, taxanes, and trastuzumab.300 However, resistance to lapatinib significantly limits its effectiveness against breast cancer.302 FOXOs are tumor suppressors that upregulate the expression of cyclin-dependent kinase inhibitors and pro-apoptotic proteins.303,304 When the PI3K/AKT signaling pathway is activated, the protein kinase AKT directly phosphorylates FOXO at three conserved sites, leading to the translocation of FOXO from the nucleus and inhibition of transcriptional activity.305 Lapatinib treatment upregulates MYC by inhibiting AKT-mediated phosphorylation of FOXO, and FOXO-mediated overexpression of MYC reduces the susceptibility of breast carcinoma cells to lapatinib.306 The sensitivity of HER2-positive cell lines and HER2-positive xenograft models to lapatinib can be enhanced by BET inhibitors via the reduction of lapatinib-induced upregulation of MYC (Fig. 5c).306 Another adaptive response contributing to lapatinib resistance involves kinome reprogramming through the reactivation of ERBB2/ERBB3 signaling, transcriptional upregulation, and activation of multiple tyrosine kinases.307 BET inhibitors can re-sensitize breast cancer cells to lapatinib by dissociating BRD4 and RNA Pol II from lapatinib-induced kinase genes.307 In summary, the use of a combined therapeutic strategy with BET inhibitors and lapatinib has promise as a more effective therapy modality for individuals diagnosed with breast carcinoma.
Sunitinib serves as an innovative oral multi-targeted tyrosine kinase inhibitor that received approval from the FDA in 2006 for the treatment of individuals with clear cell renal cell carcinoma or gastrointestinal stromal tumors.308 Prior research has demonstrated the therapeutic effect of sunitinib on melanoma, which is further enhanced by propranolol.309 BET inhibitors can hinder the development of melanoma through the noncanonical NF-κB/SPP1 pathway.103 Research has shown that the co-administration of BET inhibitors with sunitinib elicits a synergistic inhibitory effect on melanoma in vitro and in vivo.310 The main mechanism is the sensitization of melanoma cells to sunitinib via the inhibition of BET proteins.310 This inhibition leads to the suppression of GDF15 expression, which is controlled either directly through BRD4 transcription or indirectly through the BRD4/IL-6/STAT3 axis (Fig. 6a).310 This provides a promising alternative treatment option for melanoma.

Mechanistic modeling of the synergistic effect of BET inhibitors with sunitinib or tamoxifen. a BET inhibitors sensitize melanoma cells to Sunitinib by inhibiting GDF15 expression, either directly through inhibition of BRD4 transcription or indirectly through the BRD4/IL-6/STAT3 axis. b BET inhibition re-sensitizes ER-positive breast cancer cells to tamoxifen by reducing LOL expression
Combination with histone deacetylase (HDAC) inhibitors
HDACs are of paramount importance in the context of malignancy because they deacetylate histones and non-histone proteins that regulate the cell cycle, apoptosis, DNA damage response, metastasis, angiogenesis, and autophagy.311 HDAC inhibitors can counteract the abnormal acetylation state in cancer cells and activate tumor suppressors.311 In comparison to therapy with JQ1, mocetinostat, or valproic acid alone, the combined use of HDAC and BET inhibitors demonstrated synergistic benefits in breast cancer cell lines and significantly reduced cell viability.312 It was discovered that TNBC cells were more sensitive to mocetinostat-JQ1 and valproic acid-JQ1 combinations than to ER-positive cells.312 The main mechanism underlying this combination treatment is the attenuation of the RAS/MAPK signaling pathway and the upregulation of ubiquitin-specific peptidase 17, leading to decreased cell viability.312
HDAC and BET inhibitors have been demonstrated to elicit growth arrest and apoptosis in DLBCL,97,285,313 MCL,272 and cutaneous T-cell lymphoma314 cell lines with good synergistic activity. One possible mechanism for this combination is the application of HDAC inhibitors in AML, which results in histone hyperacetylation.315 Simultaneous histone hyperacetylation increases cellular dependence on BET proteins, rendering them sensitive to BET inhibition.316 Additionally, BET and HDAC inhibitors combined significantly attenuated the expression of MYC and BCL2, thereby increasing apoptosis induction.314 Novel compounds that possess the ability to simultaneously target BRD4 and HDAC proteins have been synthesized, taking advantage of the synergistic effects seen when combining BET and HDAC inhibitors.317,318,319,320
Combination with BCL2 inhibitors
BET inhibitors and BCL2 inhibitors showed significant synergy in several cancer cell lines, including DLBCL,173,313,321 MCL,272 CLL,322 cutaneous T-cell lymphoma, acute leukemia,323 and multiple myeloma.173 Treatment with both drugs induces apoptosis more than treatment with either drug alone, and BET inhibitors increase cancer cell sensitivity to BCL2 inhibitors.173,313,314,321,323 These effects may occur because of BET inhibitors that enable the downregulation of different anti-apoptotic proteins, including BCL-XL, BCL2, and BCL2A1, and the upregulation of pro-apoptotic proteins like BIM and/or PUMA, in leukemia, lymphoma, and multiple myeloma cell lines (Fig. 4d).173,313,314,321,323 In a study of germinal center B-cell (GCB)-like DLBCL cell lines, researchers found an inverse relationship between sensitivity to the BET inhibitor ABBV-075 and sensitivity to the BCL2 inhibitor venetoclax.173 Further, the level of apoptosis induced by BET inhibitor treatment of GCB-DLBCL cells was negatively correlated with BCL2 expression.173 These findings suggest that BET and BCL2 inhibitors might exhibit complementary effects in the GCB-DLBCL individuals and that BCL2 expression levels or previous responses to BCL2 inhibitors can be used to identify GCB-DLBCL subpopulations that are more likely to undergo apoptosis when treated with BET inhibitors.173,324
Combination with ATR inhibitors
ATR inhibitors lead to enhanced DNA damage and the initiation of apoptosis, whereas BET inhibitors induce DNA damage as well as the upregulation of genes involved in DNA repair.275,276 In studies on mouse MYC-induced lymphoma BL275 and MCL276 cell lines, it was found that the synergistic effects of BET and ATR inhibitors combined. The primary mechanism is that combined treatment of B-cell lymphomas with ATR and BET inhibitors results in gene expression profiles similar to those of the senescence-associated secretory pathway and endoplasmic reticulum stress.275 BET inhibitors enhance ATR inhibitor-induced cellular stress and cell death.275 Two main ATR inhibitors are currently being investigated in clinical trials,325,326 and although some toxicities (e.g., thrombocytopenia and anemia) may overlap, the combination of BET and ATR inhibitors holds immense potential.
One study on melanoma found that both human and mouse melanoma cells were sensitive to combination therapy with ATR and BET inhibitors in vitro and in vivo, possibly due to their synergistic induction of cell apoptosis and senescence-associated secretory pathway.327 This provides novel insights and directions for the treatment of melanoma and may serve as a reference for the treatment of many additional tumor forms.
Combination with AKT inhibitors
AKT inhibitors induce the expression of CDK6 by binding to the forkhead box class O 3a (FOXO3a)/BRD4 combination located at the CDK6 promoter.328 CDK6 is an oncogenic kinase that controls G1/S phase transition and cell cycle progression.329,330 Cell lines that express high levels of CDK6 exhibit increased RB phosphorylation and disruption of cell cycle arrest, rendering them resistant to AKT inhibitors.328 Resistance to PI3K/AKT inhibition can be promoted by the FOXO3a/BRD4/CDK6 axis. However, the combined inhibition of AKT and BET, which blocks the simultaneous PI3K/AKT and FOXO3a/BRD4/CDK6 pathways, proves to be more efficacious compared to monotherapy (Fig. 5c).328 in vivo experiments in mice have also confirmed that BET inhibitors sensitize luminal breast cancer cells to AKT inhibitors.328 In breast cancer cell lines, on the other hand, overexpression of AKT3 is a common characteristic of acquired resistance to AKT inhibitors, and BET inhibitors can reverse this effect by inhibiting AKT3 expression.331
Mutations in the gene encoding the substrate-binding adapter protein POZ of the E3 ubiquitin ligase SPOP are the most common in primary prostate cancer.332 Prostate cancer-specific SPOP mutations impair BET degradation, making cells more resistant to cell growth inhibition and apoptosis induced by BET inhibitors.145,234 Research has shown that BRD4 accumulation triggers an increase in the expression of GTPase RAC1 and genes involved in the cholesterol biosynthetic pathway, leading to the activation of the AKT/mTOR complex 1 (mTORC1) signaling pathway and the development of resistance to BET inhibitors.333 However, resistance can be overcome by the synergistic use of BET inhibitors in conjunction with AKT inhibitors, and this strategy has been shown to be effective against SPOP-mutant prostate cancer.333
Combination with BTK inhibitors
Inhibition of BTK is a well-established treatment for B-cell malignancies. BET inhibitors exhibit strong synergy with ibrutinib in B-cell lymphomas.276 Such effects have been noted across a range of lymphoma types, including DLBCL,101,284,334,335,336,337 MCL,272,276,336,337 BL,271 CLL, and anaplastic large T-cell lymphoma models.274 One plausible explanation for this synergy lies in the on-target side effects associated with BET inhibitors, which may restrict the obtainable clinical dosages. Combining BET inhibitors with ibrutinib could enable achieving comparable antitumor responses at lower doses, thereby mitigating overall toxicity. Another possible explanation is that although ibrutinib showed effectiveness in B-cell lymphoma, most patients didn’t observe a sustained response. Therefore, the efficacy of ibrutinib could be improved by a combination of BET inhibitors. In anaplastic large T-cell lymphoma, combining ibrutinib with a BET inhibitor yielded enhanced interleukin-2-inducible T-cell kinase (ITK) downregulation, leading to a more potent ITK inhibition.334
Combination with endocrine therapy
Compared with parental cells, BET inhibitors selectively inhibit the expression of ER alpha by inhibiting BRD3/4 and attenuate the growth of tamoxifen-resistant, ER-positive cells to an even larger extent.89 In addition, transcriptome array analysis of paired luminal breast cancer tumors showed that ENST00000456526, a luminal long non-coding RNA (lncRNA), is overexpressed in luminal breast cancer.338 ER-positive tamoxifen-resistant cells exhibit a higher level of lncRNA of luminal (LOL) expression compared with parental cells, and downregulation of LOL enhances the sensitivity to tamoxifen treatment in tamoxifen-resistant breast cancer cells and xenograft models (Fig. 6b).338 BET inhibition reverses tamoxifen resistance in ER-positive cells by reducing LOL expression.338 In conclusion, BET inhibitor-induced LOL knockdown may represent a potentially effective strategy for hindering the progression of luminal breast cancer and overcoming tamoxifen resistance in patients exhibiting a high level of LOL expression. Furthermore, JQ1 and fulvestrant combined hindered the proliferation of tumors in a tamoxifen-resistant murine xenograft model.89
ESE-15-ol is an antimitotic compound that interferes with microtubule dynamics in cells during active division and induces mitotic cell cycle arrest followed by apoptosis.339 The combined use of BET inhibitors and ESE-15-ol exhibited considerable synergistic activity against TNBC cells.340
Combination with Aurora kinase inhibitors
High-throughput drug screening has revealed that the combination of BET and Aurora kinase inhibitors is potentially effective in the treatment of ovarian cancer.288 The combination of the BET inhibitor JQ1 and Aurora kinase inhibitors has shown promising synergistic effects in drug-resistant ovarian cancer cell lines.341 However, further investigation is necessary to elucidate the underlying mechanisms, and clinical trials are warranted to assess the efficacy of this combined therapy for ovarian cancer treatment.
Combination with EZH2 inhibitors
A study on aggressive B-cell lymphomas identified a feed-forward loop involving MYC, miRNA, and EZH2; MYC overexpression facilitated the recruitment of EZH2 to the miR-26a promoter, resulting in a synergistic suppression of miR-26a.342,343 BET and EZH2 inhibitors were found to disrupt MYC activation, leading to synergistic inhibition of lymphoma proliferation and clonogenicity in aggressive lymphoma cells.342 Another study found that BET inhibitors downregulated EZH2 and H3K27me3, a marker of EZH2 activity, in DLBCL cells. Stronger downregulation was observed when BET and EZH2 inhibitors were combined.284 This combination therapy may be promising for addressing therapies aggressive B-cell lymphoma.
Aberrant activity of EZH2 and BRD4, two H3K27 modifiers, is a key driving factor in atypical teratoid/rhabdoid tumors (AT/RT), and targeted inhibition of these modifiers represents a potential therapy approach for AT/RT treatment.344,345,346,347,348,349 In AT/RT cells, H3K27me3 and H3K27ac levels are elevated and distributed at different chromatin regions to regulate specific gene expression and promote tumor growth.346,350 The proliferation of AT/RT cells is suppressed, their invasiveness is reduced, and the levels of H3K27me3 and H3K27ac are decreased by the specific suppression of EZH2 and BRD4 activity.351 The concurrent inhibition of EZH2 and BRD4 shows superior in vitro and in vivo therapeutic benefits, surpassing the therapeutic outcomes seen with individual agent treatments.351
The combination of BET and EZH2 inhibitors demonstrated superior tumor growth inhibition in diffuse intrinsic pontine glioma via blocking proliferation and promoting apoptosis in vitro as well as in vivo.352
A recent study assessed the combination of EZH2 and BET inhibitors within metastatic prostate cancer cells and found superior reduced cell viability, proliferation, and clonogenicity compared with single-agent treatment, while c-MYC and NF-kB expression were reduced.353 While this finding provides a novel approach to treating metastatic prostate cancer, the efficacy and safety of this combination require further validation via in vivo experiments and preclinical studies.
Combination with CREBBP/EP300 inhibitors
BET and CREBBP/EP300 inhibitors combined also demonstrated significant synergistic activity and increased cell death rates.354 A recent study on multiple myeloma cell lines found that the novel dual BET and CREBBP/EP300 inhibitors, NEO2734 and NEO1132, exhibited stronger in vitro antitumor effects compared with individual inhibitors targeting either BET or CREBBP/EP300.355 In multiple myeloma cell lines, the dual inhibitors induce significant G1 cell cycle arrest and reduce c-MYC and IRF4 protein levels, indicating their potential as a therapeutic option for multiple myeloma.355 NEO2734, a novel antitumor compound, has demonstrated superior antitumor activity in lymphoma, leukemia, and prostate cancer.356,357
The newly developed inhibitor XP-524, which targets EP300/CBP of histone acetyltransferase and BRD4, demonstrated higher potency and superior antitumor activity compared with single-agent BET inhibition in PDAC.358 In addition to inhibiting KRAS/MAPK signaling, XP-524 enhances self-peptide presentation and tumor recruitment of cytotoxic T lymphocytes, despite their refractory nature, even after full activation.358 Notably, one study found that the combination of XP-524 with an anti-PD-1 antibody in vivo reactivated the cytotoxic immune program and significantly prolonged survival.358 This suggests that XP-524 has promise as a novel treatment alternative for PDAC, and its combination with immune checkpoint inhibitors warrants further investigation.
Combination with PRMT5 inhibitors
PRMT5 overexpression has been regularly documented in cell lines for lymphoma and leukemia, and its increased expression has been shown to induce the symmetrical methylation of H3R8 and H4R3. Furthermore, studies on MCL have found that PRMT5 overexpression correlates with decreased miR-92b and miR-96 expression.359 Research has shown that selective inhibition of PRMT5 expression produces anti-lymphoma activity.360 BRD4 binds to the 5’ regulatory region of PRMT5, and treatment with BET inhibitors mis localizes BRD4 from this region. This downregulates arginine methyltransferase expression and reduces PRMT5 occupancy at the cancer suppressor miR-96-5p promoter, leading to the inhibition of miR-96-5P.361 This suggests that combining BET and PRMT5 inhibitors could be a new strategy for lymphoma treatment.
Combination with PLK inhibitors
In a study on BET inhibitor-acquired resistance in TNBC cell models, BET inhibitor-resistant cells were found to be particularly sensitive to PLK1 inhibition.362 Inhibition of PLK1 exhibited an antiproliferative effect in resistant cells, causing G2/M arrest and upregulation of the cell cycle proteins Bcl-2, pH3, and cyclin B, along with the induction of caspase-dependent apoptosis.362 This suggests that BET and PLK1 inhibitors combined may provide a new treatment option for BET inhibitor-resistant patients.
BET proteins are central regulators of AR and MYC-mediated transcription in CRPC,88 whereas PLK1 inhibitors not only affect the cell cycle but can also downregulate AR and MYC.363,364 Hence, a novel dual inhibitor of BET and PLK1, WNY0824, was synthesized, exhibiting nanomolar-level and equipotent inhibition against both BRD4 and PLK1.365 WNY0824 has demonstrated superior antitumor activity both in vitro and in vivo, providing an innovative therapeutic option for the treatment of CRPC.365
Conclusion
BRDs constitute a dynamically evolving class of epigenetic targets, with aberrant BET protein activation strongly linked to various diseases, including cancer. The emergence of BET inhibitors has yielded significant insights into the pivotal role of BET proteins in governing proto-oncogene transcription, underscoring their therapeutic potential. In recent decades, dozens of clinical trials have been conducted to validate the potential advantages of BET inhibitors in refractory cancer and non-cancerous ailments, yielding initial efficacy. However, addressing drug resistance and adverse effects remains a priority for future drug development. Rational drug combinations have emerged as a promising strategy against drug resistance, validated through clinical trials. Thrombocytopenia stands as a principal safety concern in ongoing clinical trials, and optimizing dosing schedules could ameliorate patient tolerability. The development of BD1- or BD2-selective inhibitors might enhance tolerability, and the advent of BET PROTACs could potentially offer enduring antitumor efficacy.
Preliminary evidence from preclinical and clinical studies suggests that BET inhibitors do not provide durable cytotoxic effects in human cancers when used as single agents. However, the therapeutic potential associated with combining targeted small-molecule inhibitors, immune checkpoint inhibitors, and other epigenetic therapies is significant (Fig. 5). While the potential toxicity of BET inhibitors could restrict this combination, research indicates that employing combination therapies at lower doses might offer effectiveness while mitigating toxicity concerns. In conclusion, BET inhibitors hold significant clinical promise in combination regimens for the future. Enhanced binding approaches and the evolution of next-generation compounds, extending beyond bromodomain targeting, are poised to unlock fresh avenues for the prospective clinical advancement of BET inhibitors as anticancer agents.
An additional critical concern pertains to the absence of biomarkers for predicting BETi sensitivity and the utilization of non-clinically applicable dosages in preclinical investigations, thereby constraining the translation of these agents into clinical practice. Several new approaches are now available to aid in the screening of BET inhibitors for personalized therapy and combination regimens that may improve clinical outcomes.366,367 Further mechanistic studies can help identify these biomarkers, and the development of novel, highly selective BET inhibitors will help prevent toxicity.
References
Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010).
Dhalluin, C. et al. Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496 (1999).
Filippakopoulos, P. et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231 (2012).
Dawson, M. A. et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533 (2011).
French, C. A. Small-Molecule Targeting of BET Proteins in Cancer. Adv. Cancer Res 131, 21–58 (2016).
Bell, C. C. et al. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat Commun 10, (2019).
Haynes, S. R. et al. The bromodomain: a conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res 20, 2603 (1992).
Tamkun, J. W. et al. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 68, 561–572 (1992).
Kim, S. C. et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 (2006).
Choudhary, C. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009).
Choudhary, C., Weinert, B. T., Nishida, Y., Verdin, E. & Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550 (2014).
Verdin, E. & Ott, M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 16, 258–264 (2015).
Prinjha, R. K., Witherington, J. & Lee, K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharm. Sci. 33, 146–153 (2012).
French, C. A. et al. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 63, 304–307 (2003).
Miyoshi, I. et al. BRD4 bromodomain gene rearrangement in aggressive carcinoma with translocation t(15;19). Am. J. Pathol. 159, 1987–1992 (2001).
Yang, Z., He, N. & Zhou, Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell Biol. 28, 967–976 (2008).
Rahl, P. B. et al. c-Myc regulates transcriptional pause release. Cell 141, 432–445 (2010).
Yang, Z. et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19, 535–545 (2005).
Dey, A., Nishiyama, A., Karpova, T., McNally, J. & Ozato, K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 20, 4899–4909 (2009).
Nicodeme, E. et al. Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123 (2010).
Ferri, E., Petosa, C. & McKenna, C. E. Bromodomains: Structure, function and pharmacology of inhibition. Biochem. Pharm. 106, 1–18 (2016).
Wang, Q., Shao, X., Leung, E. L. H., Chen, Y. & Yao, X. Selectively targeting individual bromodomain: Drug discovery and molecular mechanisms. Pharmacol. Res. 172, (2021).
Cochran, A. G., Conery, A. R. & Sims, R. J. Bromodomains: a new target class for drug development. Nat. Rev. Drug Discov. 18, 609–628 (2019).
Okada, Y. et al. hDOT1L links histone methylation to leukemogenesis. Cell 121, 167–178 (2005).
Jeffers, V., Yang, C., Huang, S. & Sullivan, W. J. Bromodomains in protozoan parasites: evolution, function, and opportunities for drug development. Microbiol. Mol. Biol. Rev. 81, (2017).
Mujtaba, S., Zeng, L. & Zhou, M. M. Structure and acetyl-lysine recognition of the bromodomain. Oncogene 26, 5521–5527 (2007).
Lloyd, J. T. & Glass, K. C. Biological function and histone recognition of family IV bromodomain-containing proteins. J. Cell Physiol. 233, 1877–1886 (2018).
Shi, J. et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 27, 2648–2662 (2013).
Zeng, L. & Zhou, M. M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 513, 124–128 (2002).
Ortega, E. et al. Transcription factor dimerization activates the p300 acetyltransferase. Nature 562, 538–544 (2018).
Ullah, M. et al. Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol. Cell Biol. 28, 6828–6843 (2008).
Marmorstein, R. & Zhou, M. M. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 6, (2014).
Mashtalir, N. et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell 175, 1272–1288.e20 (2018).
Xiao, A. et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 457, 57–62 (2009).
Sanchez, R. & Zhou, M. M. The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discov. Devel 12, 659–665 (2009).
Fujisawa, T. & Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 18, 246–262 (2017).
Koo, S. J. et al. ATAD2 is an epigenetic reader of newly synthesized histone marks during DNA replication. Oncotarget 7, 70323–70335 (2016).
Zhang, Q. et al. Structural Mechanism of Transcriptional Regulator NSD3 Recognition by the ET Domain of BRD4. Structure 24, 1201–1208 (2016).
Welti, J. et al. Targeting bromodomain and extra-terminal (BET) family proteins in castration-resistant prostate cancer (CRPC). Clin. Cancer Res. 24, 3149–3162 (2018).
Zeng, L. et al. Structural insights into human KAP1 PHD finger-bromodomain and its role in gene silencing. Nat. Struct. Mol. Biol. 15, 626–633 (2008).
Xue, J. et al. Tumour suppressor TRIM33 targets nuclear β-catenin degradation. Nat Commun 6, (2015).
Seeler, J. S. et al. Common properties of nuclear body protein SP100 and TIF1alpha chromatin factor: role of SUMO modification. Mol. Cell Biol. 21, 3314–3324 (2001).
Bottomley, M. J. et al. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nat. Struct. Biol. 8, 626–633 (2001).
Stathis, A. & Bertoni, F. BET proteins as targets for anticancer treatment. Cancer Discov. 8, 24–36 (2018).
Savitsky, P. et al. Multivalent histone and DNA Engagement by a PHD/BRD/PWWP triple reader cassette recruits ZMYND8 to K14ac-Rich chromatin. Cell Rep. 17, 2724–2737 (2016).
Li, D. & Roberts, R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol. Life Sci. 58, 2085–2097 (2001).
Denis, G. V., Vaziri, C., Guo, N. & Faller, D. V. RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F. Cell Growth Differ. 11, 417–424 (2000).
Kharenko, O. A. et al. RVX-297- a novel BD2 selective inhibitor of BET bromodomains. Biochem Biophys. Res Commun. 477, 62–67 (2016).
Ali, H. A. et al. A comprehensive review of BET protein biochemistry, physiology, and pathological roles. Front. Pharmacol. 13, (2022).
Lee, J. S., Smith, E. & Shilatifard, A. The language of histone crosstalk. Cell 142, 682–685 (2010).
Kouzarides, T. Chromatin modifications and their function. Cell 128, 693–705 (2007).
Lovén, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013).
Gyuris, A. et al. The chromatin-targeting protein Brd2 is required for neural tube closure and embryogenesis. Biochim. Biophys. Acta 1789, 413–421 (2009).
Padmanabhan, B., Mathur, S., Manjula, R. & Tripathi, S. Bromodomain and extra-terminal (BET) family proteins: New therapeutic targets in major diseases. J. Biosci. 41, 295–311 (2016).
Sun, R. et al. Bromodomain-containing protein 2 induces insulin resistance via the mTOR/Akt signaling pathway and an inflammatory response in adipose tissue. Cell Signal 30, 92–103 (2017).
Lamonica, J. M. et al. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc. Natl. Acad. Sci. USA 108, (2011).
Stonestrom, A. J. et al. Functions of BET proteins in erythroid gene expression. Blood 125, 2825–2834 (2015).
Ishii, H., Mimori, K., Mori, M. & Vecchione, A. Differentially expressed genes in endothelial differentiation. DNA Cell Biol. 24, 432–437 (2005).
Klein, K. et al. The bromodomain protein inhibitor I-BET151 suppresses expression of inflammatory genes and matrix degrading enzymes in rheumatoid arthritis synovial fibroblasts. Ann. Rheum. Dis. 75, 422–429 (2016).
Wang, N., Wu, R., Tang, D. & Kang, R. The BET family in immunity and disease. Signal Transduct. Target Ther. 6, (2021).
Devaiah, B. N. et al. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 23, 540–548 (2016).
Dey, A. et al. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol. Cell Biol. 20, 6537–6549 (2000).
Mochizuki, K. et al. The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J. Biol. Chem. 283, 9040–9048 (2008).
Wu, S. Y. & Chiang, C. M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 282, 13141–13145 (2007).
Bisgrove, D. A., Mahmoudi, T., Henklein, P. & Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl Acad. Sci. USA 104, 13690–13695 (2007).
Donati, B., Lorenzini, E. & Ciarrocchi, A. BRD4 and Cancer: going beyond transcriptional regulation. Mol. Cancer 17, (2018).
Moon, K. J. et al. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19, 523–534 (2005).
Rahman, S. et al. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell Biol. 31, 2641–2652 (2011).
Li, X. et al. BRD4 promotes DNA repair and mediates the formation of TMPRSS2-ERG gene rearrangements in prostate cancer. Cell Rep. 22, 796–808 (2018).
Stanlie, A., Yousif, A. S., Akiyama, H., Honjo, T. & Begum, N. A. Chromatin reader Brd4 functions in Ig class switching as a repair complex adaptor of nonhomologous end-joining. Mol. Cell 55, 97–110 (2014).
Zhao, J. Q. et al. Activation of telomerase rna gene promoter activity by NF-Y, Sp1, and the retinoblastoma protein and repression by Sp3. Neoplasia 2, 531–539 (2000).
Zhang, A. et al. Deletion of the telomerase reverse transcriptase gene and haploinsufficiency of telomere maintenance in Cri du chat syndrome. Am. J. Hum. Genet 72, 940–948 (2003).
Gaucher, J. et al. Bromodomain-dependent stage-specific male genome programming by Brdt. EMBO J. 31, 3809–3820 (2012).
Shang, E. et al. Identification of unique, differentiation stage-specific patterns of expression of the bromodomain-containing genes Brd2, Brd3, Brd4, and Brdt in the mouse testis. Gene Expr. Patterns 4, 513–519 (2004).
Shang, E., Nickerson, H. D., Wen, D., Wang, X. & Wolgemuth, D. J. The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development 134, 3507–3515 (2007).
Liu, D. et al. Cyclin A1 is required for meiosis in the male mouse. Nat. Genet 20, 377–380 (1998).
Nickerson, H. D., Joshi, A. & Wolgemuth, D. J. Cyclin A1-deficient mice lack histone H3 serine 10 phosphorylation and exhibit altered aurora B dynamics in late prophase of male meiosis. Dev. Biol. 306, 725–735 (2007).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Pott, S. & Lieb, J. D. What are super-enhancers? Nat. Genet 47, 8–12 (2015).
Mertz, J. A. et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl Acad. Sci. USA 108, 16669–16674 (2011).
Delmore, J. E. et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 (2011).
Miller, D. M., Thomas, S. D., Islam, A., Muench, D. & Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 18, 5546–5553 (2012).
Li, C. et al. Mutational landscape of primary, metastatic, and recurrent ovarian cancer reveals c-MYC gains as potential target for BET inhibitors. Proc. Natl Acad. Sci. USA 116, 619–624 (2019).
Kurimchak, A. M. et al. Resistance to BET bromodomain inhibitors is mediated by kinome reprogramming in ovarian cancer. Cell Rep. 16, 1273–1286 (2016).
Segura, M. F. et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 73, 6264–6276 (2013).
Chaidos, A., Caputo, V. & Karadimitris, A. Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: emerging preclinical and clinical evidence. Ther. Adv. Hematol. 6, 128–141 (2015).
Zou, Z. et al. Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA. Oncogene 33, 2395–2404 (2014).
Asangani, I. A. et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 510, 278–282 (2014).
Feng, Q. et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 24, 809–819 (2014).
Yang, L. et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 9, (2017).
Sun, C. et al. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell 33, 401–416.e8 (2018).
Villar-Prados, A. et al. Predicting novel therapies and targets: regulation of Notch3 by the bromodomain protein BRD4. Mol. Cancer Ther. 18, 421–436 (2019).
Liu, A., Fan, D. & Wang, Y. The BET bromodomain inhibitor i-BET151 impairs ovarian cancer metastasis and improves antitumor immunity. Cell Tissue Res. 374, 577–585 (2018).
Yin, M. et al. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat. Commun. 11, (2020).
Hu, X., li, J., Fu, M., Zhao, X. & Wang, W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct. Target Ther. 6, (2021).
Ott, C. J. et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 120, 2843–2852 (2012).
Boi, M. et al. The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical b-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 21, 1628–1638 (2015).
Yu, H., Lin, L., Zhang, Z., Zhang, H. & Hu, H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther 5, (2020).
Hayden, M. S. & Ghosh, S. Shared principles in NF-kappaB signaling. Cell 132, 344–362 (2008).
Israël, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol 2, (2010).
Ceribelli, M. et al. Blockade of oncogenic IκB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc. Natl Acad. Sci. USA 111, 11365–11370 (2014).
Huang, B., Yang, X.-D., Zhou, M.-M., Ozato, K. & Chen, L.-F. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell Biol. 29, 1375–1387 (2009).
Deng, G. et al. BET inhibitor suppresses melanoma progression via the noncanonical NF-κB/SPP1 pathway. Theranostics 10, 11428–11443 (2020).
Chung, C. W. et al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med Chem. 54, 3827–3838 (2011).
Sanchez, R., Meslamani, J. & Zhou, M. M. The bromodomain: from epigenome reader to druggable target. Biochim Biophys. Acta 1839, 676–685 (2014).
Belkina, A. C. & Denis, G. V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 12, 465–477 (2012).
Filippakopoulos, P. & Knapp, S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 13, 337–356 (2014).
Shi, J. & Vakoc, C. R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 54, 728–736 (2014).
Stratikopoulos, E. E. & Parsons, R. E. Molecular pathways: targeting the PI3K pathway in cancer-BET inhibitors to the rescue. Clin. Cancer Res. 22, 2605–2610 (2016).
Theodoulou, N. H., Tomkinson, N. C. O., Prinjha, R. K. & Humphreys, P. G. Clinical progress and pharmacology of small molecule bromodomain inhibitors. Curr. Opin. Chem. Biol. 33, 58–66 (2016).
Shorstova, T., Foulkes, W. D. & Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 124, 1478–1490 (2021).
Liu, Z. et al. Drug discovery targeting bromodomain-containing protein 4. J. Med. Chem. 60, 4533–4558 (2017).
Ali, I., Choi, G. & Lee, K. BET Inhibitors as Anticancer Agents: A Patent Review. Recent. Pat. Anticancer Drug Discov. 12, (2017).
Raux, B. et al. Exploring selective inhibition of the first bromodomain of the human bromodomain and extra-terminal domain (BET) proteins. J. Med. Chem. 59, 1634–1641 (2016).
Seal, J. et al. Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I-BET151 (GSK1210151A). Bioorg. Med Chem. Lett. 22, 2968–2972 (2012).
Odore, E. et al. Phase I population pharmacokinetic assessment of the oral bromodomain inhibitor OTX015 in patients with haematologic malignancies. Clin. Pharmacokinet. 55, 397–405 (2016).
Berthon, C. et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 3, e186–e195 (2016).
Coudé, M. M. et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 6, 17698–17712 (2015).
Baratta, M. G. et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc. Natl. Acad. Sci. USA 112, 232–237 (2015).
Piha-Paul, S. A. et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. JNCI Cancer Spectr. 4, (2019).
Zhang, G., Smith, S. G. & Zhou, M. M. Discovery of chemical inhibitors of human bromodomains. Chem. Rev. 115, 11625–11668 (2015).
Hu, R. et al. BRD4 inhibitor suppresses melanoma metastasis via the SPINK6/EGFR-EphA2 pathway. Pharmacol. Res. 187, (2023).
Deng, G. et al. EEF2K silencing inhibits tumour progression through repressing SPP1 and synergises with BET inhibitors in melanoma. Clin. Transl. Med. 12, (2022).
Jiang, Y. et al. Bromodomain inhibition attenuates the progression and sensitizes the chemosensitivity of osteosarcoma by repressing GP130/STAT3 Signaling. Front. Oncol. 11, (2021).
Wu, S. et al. The novel BRDT inhibitor NHWD870 shows potential as a male contraceptive in mice. Acta Biochim. Biophys. Sin. (Shanghai) 54, 1789–1800 (2022).
Amorim, S. et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 3, e196–e204 (2016).
Borthakur, G. et al. A phase 1 study of the pan-bromodomain and extraterminal inhibitor mivebresib (ABBV-075) alone or in combination with venetoclax in patients with relapsed/refractory acute myeloid leukemia. Cancer 127, 2943–2953 (2021).
Stathis, A. et al. Clinical response of carcinomas harboring the BRD4–NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Discov. 6, 492–500 (2016).
Vangamudi, B. et al. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-mutant cancers: insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res. 75, 3865–3878 (2015).
Zhang, G. et al. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J. Med Chem. 56, 9251–9264 (2013).
Cheung, K. et al. BET N-terminal bromodomain inhibition selectively blocks Th17 cell differentiation and ameliorates colitis in mice. Proc. Natl Acad. Sci. USA 114, 2952–2957 (2017).
Gacias, M. et al. Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem. Biol. 21, 841–854 (2014).
Watson, R. J. et al. GSK789: a selective inhibitor of the first bromodomains (BD1) of the bromo and extra terminal domain (BET) proteins. J. Med Chem. 63, 9045–9069 (2020).
Picaud, S. et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 110, 19754–19759 (2013).
Gilham, D. et al. RVX-208, a BET-inhibitor for treating atherosclerotic cardiovascular disease, raises ApoA-I/HDL and represses pathways that contribute to cardiovascular disease. Atherosclerosis 247, 48–57 (2016).
Nicholls, S. J. et al. Efficacy and safety of a novel oral inducer of apolipoprotein a-I synthesis in statin-treated patients with stable coronary artery disease a randomized controlled trial. J. Am. Coll. Cardiol. 57, 1111–1119 (2011).
Nikolic, D., Rizzo, M., Mikhailidis, D. P., Wong, N. C. & Banach, M. An evaluation of RVX-208 for the treatment of atherosclerosis. Expert Opin. Investig. Drugs 24, 1389–1398 (2015).
Ray, K. K. et al. Effect of apabetalone added to standard therapy on major adverse cardiovascular events in patients with recent acute coronary syndrome and type 2 diabetes: a randomized clinical trial. JAMA 323, 1565–1573 (2020).
Faivre, E. J. et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 578, 306–310 (2020).
Sheppard, G. S. et al. Discovery of N-Ethyl-4-[2-(4-fluoro-2,6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]-6-methyl-7-oxo-1 H-pyrrolo[2,3- c]pyridine-2-carboxamide (ABBV-744), a BET Bromodomain Inhibitor with Selectivity for the Second Bromodomain. J. Med. Chem. 63, 5585–5623 (2020).
Gilan, O. et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 368, 387–394 (2020).
Jake Slavish, P. et al. Bromodomain-selective BET inhibitors are potent antitumor agents against MYC-driven pediatric cancer. Cancer Res. 80, 3507–3518 (2020).
Chen, D. et al. Discovery, structural insight, and bioactivities of BY27 as a selective inhibitor of the second bromodomains of BET proteins. Eur. J. Med. Chem. 182, (2019).
Carrasco, K. et al. CRCM5484: A BET-BDII selective compound with differential anti-leukemic drug modulation. J. Med. Chem. 65, 5660–5674 (2022).
Dai, X. et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 23, 1063–1071 (2017).
Lu, J. et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 22, 755–763 (2015).
Winter, G. E. et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015).
Li, H. et al. Protein degradation technology: a strategic paradigm shift in drug discovery. J. Hematol. Oncol. 14, 138 (2021).
Zou, Y., Ma, D. & Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 37, 21–30 (2019).
Dong, J., Cheng, X. D., Zhang, W. D. & Qin, J. J. Recent update on development of small-molecule STAT3 inhibitors for cancer therapy: from phosphorylation inhibition to protein degradation. J. Med. Chem. 64, 8884–8915 (2021).
Paiva, S. L. & Crews, C. M. Targeted protein degradation: elements of PROTAC design. Curr. Opin. Chem. Biol. 50, 111–119 (2019).
Wang, P. & Zhou, J. Proteolysis targeting chimera (PROTAC): a paradigm-shifting approach in small molecule drug discovery. Curr. Top. Med. Chem. 18, 1354–1356 (2018).
Tian, C. & Burgess, K. PROTAC compatibilities, degrading cell-surface receptors, and the sticky problem of finding a molecular glue. ChemMedChem. 16, 316–318 (2021).
Ottis, P. et al. Assessing different E3 ligases for small molecule induced protein ubiquitination and degradation. ACS Chem. Biol. 12, 2570–2578 (2017).
Sun, B. et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 32, 343–352 (2018).
Tarantelli, C. et al. Abstract A179: The BRD4 degrader MZ1 exhibits potent antitumoral activity in diffuse large B cell lymphoma of the activated B cell-like type. Mol. Cancer Ther. 17, A179–A179 (2018).
Winter, G. E. et al. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 67, 5–18.e19 (2017).
Zengerle, M., Chan, K. H. & Ciulli, A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 10, 1770–1777 (2015).
Gadd, M. S. et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 13, 514–521 (2017).
Raina, K. et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl Acad. Sci. USA 113, 7124–7129 (2016).
Saenz, D. T. et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia 31, 1951–1961 (2017).
Dombret, H. et al. A Phase 1 Study of the BET-Bromodomain Inhibitor OTX015 in patients with advanced acute leukemia. Blood 124, 117 (2014).
Lewin, J. et al. Phase Ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in Patients with selected advanced solid tumors. J. Clin. Oncol. 36, 3007–3014 (2018).
Dawson, M. A. et al. A Phase I/II open-label study of molibresib for the treatment of relapsed/refractory hematologic malignancies. Clin. Cancer Res. 29, (2023).
Piha-Paul, S. A. et al. First-in-human study of mivebresib (ABBV-075), an oral pan-inhibitor of bromodomain and extra terminal proteins, in patients with relapsed/refractory solid tumors. Clin. Cancer Res. 25, 6309–6319 (2019).
Hilton, J. et al. BMS-986158, a Small Molecule Inhibitor of the Bromodomain and Extraterminal Domain Proteins, in Patients with Selected Advanced Solid Tumors: Results from a Phase 1/2a Trial. Cancers (Basel) 14, (2022).
Blum, K. A. et al. A Phase I Study of Pelabresib (CPI-0610), a Small-Molecule Inhibitor of BET Proteins, in Patients with Relapsed or Refractory Lymphoma. Cancer Res. Commun. 2, 795–805 (2022).
Shapiro, G. I. et al. A Phase 1 study of RO6870810, a novel bromodomain and extra-terminal protein inhibitor, in patients with NUT carcinoma, other solid tumours, or diffuse large B-cell lymphoma. Br. J. Cancer 124, 744–753 (2021).
Aggarwal, R. R. et al. A Phase Ib/IIa Study of the Pan-BET Inhibitor ZEN-3694 in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer. Clin. Cancer Res. 26, 5338–5347 (2020).
Mascarenhas, J. et al. MANIFEST: Pelabresib in Combination With Ruxolitinib for Janus Kinase Inhibitor Treatment-Naïve Myelofibrosis. J. Clin. Oncol. https://doi.org/10.1200/JCO.22.01972 (2023).
Mascarenhas, J. et al. MPN-375 BET Inhibitor Pelabresib (CPI-0610) combined with ruxolitinib in patients with myelofibrosis - JAK Inhibitor-Naïe or with suboptimal response to ruxolitinib - preliminary data from the MANIFEST Study. Clin. Lymphoma Myeloma Leuk. 22, S335–S336 (2022).
Dickinson, M. et al. Phase 1b study of the BET protein inhibitor RO6870810 with venetoclax and rituximab in patients with diffuse large B-cell lymphoma. Blood Adv. 5, 4762–4770 (2021).
Bui, M. H. et al. Preclinical Characterization of BET Family Bromodomain Inhibitor ABBV-075 Suggests Combination Therapeutic Strategies. Cancer Res. 77, 2976–2989 (2017).
Fiskus, W. et al. Superior efficacy of cotreatment with BET protein inhibitor and BCL2 or MCL1 inhibitor against AML blast progenitor cells. Blood Cancer J. 9, (2019).
Huang, Y. et al. Synergistic effect of PARP inhibitor and BRD4 inhibitor in multiple models of ovarian cancer. J. Cell Mol. Med. 27, 634–649 (2023).
Oien, D. B. et al. BET inhibition targets ABC-DLBCL constitutive B-cell receptor signaling through PAX5. Blood Adv. https://doi.org/10.1182/BLOODADVANCES.2022009257 (2023).
Kharenko, O. A., Patel, R. G., Calosing, C. & van der Horst, E. H. Combination of ZEN-3694 with CDK4/6 inhibitors reverses acquired resistance to CDK4/6 inhibitors in ER-positive breast cancer. Cancer Gene Ther. 29, 859–869 (2022).
Falchook, G. et al. Development of 2 bromodomain and extraterminal inhibitors with distinct pharmacokinetic and pharmacodynamic profiles for the treatment of advanced malignancies. Clin. Cancer Res. 26, 1247–1257 (2020).
Moreno, V. et al. Phase I study of CC-90010, a reversible, oral BET inhibitor in patients with advanced solid tumors and relapsed/refractory non-Hodgkin’s lymphoma. Ann. Oncol. 31, 780–788 (2020).
Postel-Vinay, S. et al. First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur. J. Cancer 109, 103–110 (2019).
Balduini, C. L. et al. Effects of the R216Q mutation of GATA-1 on erythropoiesis and megakaryocytopoiesis. Thromb. Haemost. 91, 129–140 (2004).
Tubman, V. N. et al. X-linked gray platelet syndrome due to a GATA1 Arg216Gln mutation. Blood 109, 3297–3299 (2007).
Yu, C. et al. X-linked thrombocytopenia with thalassemia from a mutation in the amino finger of GATA-1 affecting DNA binding rather than FOG-1 interaction. Blood 100, 2040–2045 (2002).
Fujiwara, Y., Browne, C. P., Cunniff, K., Goff, S. C. & Orkin, S. H. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc. Natl Acad. Sci. USA 93, 12355–12358 (1996).
Nichols, K. E. et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat. Genet 24, 266–270 (2000).
Aiyer, S. et al. Altering murine leukemia virus integration through disruption of the integrase and BET protein family interaction. Nucleic Acids Res. 42, 5917–5928 (2014).
Hellert, J. et al. A structural basis for BRD2/4-mediated host chromatin interaction and oligomer assembly of Kaposi sarcoma-associated herpesvirus and murine gammaherpesvirus LANA proteins. PLoS Pathog. 9, (2013).
Liang, J. et al. Epstein-Barr virus super-enhancer eRNAs are essential for MYC oncogene expression and lymphoblast proliferation. Proc. Natl Acad. Sci. USA 113, 14121–14126 (2016).
Zhao, Y. et al. Inhibition of BET family proteins suppresses african swine fever virus infection. Microbiol. Spectr. 10, (2022).
Board, N. L., Moskovljevic, M., Wu, F., Siliciano, R. F. & Siliciano, J. D. Engaging innate immunity in HIV-1 cure strategies. Nat. Rev. Immunol. 22, 499–512 (2022).
Wei, P., Garber, M. E., Fang, S. M., Fischer, W. H. & Jones, K. A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92, 451–462 (1998).
Zhu, Y. et al. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 11, 2622–2632 (1997).
Zhou, M. et al. Bromodomain protein Brd4 regulates human immunodeficiency virus transcription through phosphorylation of CDK9 at threonine 29. J. Virol. 83, 1036–1044 (2009).
Karn, J. A new BET on the control of HIV latency. Cell Cycle 12, 545–546 (2013).
Hsu, S. C. et al. The BET Protein BRD2 cooperates with CTCF to enforce transcriptional and architectural boundaries. Mol. Cell 66, 102–116.e7 (2017).
Matzuk, M. M. et al. Small-molecule inhibition of BRDT for male contraception. Cell 150, 673–684 (2012).
Bolden, J. E. et al. Inducible in vivo silencing of Brd4 identifies potential toxicities of sustained BET protein inhibition. Cell Rep. 8, 1919–1929 (2014).
Spiltoir, J. I. et al. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 63, 175–179 (2013).
Mills, R. J. et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell 184, 2167–2182.e22 (2021).
Cully, M. Cardiovascular disease: BET inhibitor attenuates heart failure. Nat. Rev. Drug Discov. 16, 453 (2017).
Bhadury, J. et al. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc. Natl. Acad. Sci. USA 111, (2014).
Fong, C. Y. et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 525, 538–542 (2015).
Rathert, P. et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547 (2015).
Izaguirre-Carbonell, J. et al. Critical role of Jumonji domain of JMJD1C in MLL-rearranged leukemia. Blood Adv. 3, 1499–1511 (2019).
Guo, L. et al. A combination strategy targeting enhancer plasticity exerts synergistic lethality against BETi-resistant leukemia cells. Nat. Commun. 11, (2020).
Shu, S. et al. Synthetic lethal and resistance interactions with BET bromodomain inhibitors in triple-negative breast cancer. Mol. Cell 78, 1096–1113.e8 (2020).
Wright, S. et al. Interrogating bromodomain inhibitor resistance in KMT2A-rearranged leukemia through combinatorial CRISPR screens. Proc. Natl. Acad. Sci. USA 120, (2023).
Shu, S. et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529, 413–417 (2016).
Liu, B. et al. BRD4-directed super-enhancer organization of transcription repression programs links to chemotherapeutic efficacy in breast cancer. Proc. Natl. Acad. Sci. USA 119, (2022).
De Palma, M. & Lewis, C. E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 23, 277–286 (2013).
Dijkgraaf, E. M. et al. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. 73, 2480–2492 (2013).
Kozin, S. V. et al. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 70, 5679–5685 (2010).
Mazzieri, R. et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 19, 512–526 (2011).
Qiao, J. et al. Macrophages confer resistance to BET inhibition in triple-negative breast cancer by upregulating IKBKE. Biochem. Pharmacol. 180, (2020).
Wang, R. et al. Uncovering BRD4 hyperphosphorylation associated with cellular transformation in NUT midline carcinoma. Proc. Natl. Acad. Sci. USA 114, E5352–E5361 (2017).
Wilson, A. J., Stubbs, M., Liu, P., Ruggeri, B. & Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 149, 575–584 (2018).
Yokoyama, Y. et al. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. 76, 6320–6330 (2016).
Momeny, M. et al. Inhibition of bromodomain and extraterminal domain reduces growth and invasive characteristics of chemoresistant ovarian carcinoma cells. Anticancer Drugs 29, 1011–1020 (2018).
Bagratuni, T. et al. JQ1 inhibits tumour growth in combination with cisplatin and suppresses JAK/STAT signalling pathway in ovarian cancer. Eur. J. Cancer 126, 125–135 (2020).
Tomita, H., Tanaka, K., Tanaka, T. & Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 7 www.impactjournals.com/oncotarget/ (2016).
Landen, C. N. et al. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol. Cancer Ther. 9, 3186–3199 (2010).
Dai, Y., Jin, S., Li, X. & Wang, D. The involvement of Bcl-2 family proteins in AKT-regulated cell survival in cisplatin resistant epithelial ovarian cancer. Oncotarget 8 www.impactjournals.com/oncotarget/ (2017).
Dasari, S. & Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 740, 364–378 (2014).
Yang, L. et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. http://stm.sciencemag.org/ (2017).
Yardley, D. A. et al. nab-Paclitaxel Plus Carboplatin or Gemcitabine vs Gemcitabine Plus Carboplatin as First-Line Treatment for Patients With Triple-Negative Metastatic Breast Cancer: Results From the tnAcity Trial. https://doi.org/10.1093/annonc/mdy201/5033593. (2018)
von Minckwitz, G. et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): A randomised phase 2 trial. Lancet Oncol. 15, 747–756 (2014).
Tutt, A. et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 24, 628–637 (2018).
Gerratana, L., Fanotto, V., Pelizzari, G., Agostinetto, E. & Puglisi, F. Do platinum salts fit all triple negative breast cancers? Cancer Treat. Rev. 48, 34–41 (2016).
Mio, C. et al. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int J. Cancer 144, 755–766 (2019).
Pérez-Peña, J. et al. In silico analysis guides selection of BET Inhibitors for triple-negative breast cancer treatment. Mol. Cancer Ther. 15, 1823–1833 (2016).
Meyerhardt, J. A. & Mayer, R. J. Systemic therapy for colorectal cancer. N. Engl. J. Med 352, 476–487 (2005).
Tan, X. et al. BET inhibitors potentiate chemotherapy and killing of SPOP-mutant colon cancer cells via induction of DR5. Cancer Res. 79, 1191–1203 (2019).
Mert, U. & Sanlioglu, A. D. Intracellular localization of DR5 and related regulatory pathways as a mechanism of resistance to TRAIL in cancer. Cell Mol. Life Sci. 74, 245–255 (2017).
Janouskova, H. et al. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat. Med 23, 1046–1054 (2017).
Karakashev, S. et al. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep. 21, 3398–3405 (2017).
Matheson, C. J., Backos, D. S. & Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 37, 872–881 (2016).
Wardlaw, C. P., Carr, A. M. & Oliver, A. W. TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair 22, 165–174 (2014).
Li, L. et al. Targeting Poly(ADP-Ribose) Polymerase and the c-Myb-Regulated DNA Damage Response Pathway in Castration-Resistant Prostate Cancer. www.SCIENCESIGNALING.org (2014).
Moudry, P. et al. TOP BP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J. Cell Biol. 212, 281–288 (2016).
Miller, A. L. et al. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine 44, 419–430 (2019).
Fehling, S. C., Miller, A. L., Garcia, P. L., Vance, R. B. & Yoon, K. J. The combination of BET and PARP inhibitors is synergistic in models of cholangiocarcinoma. Cancer Lett. 468, 48–58 (2020).
Pawar, A., Gollavilli, P. N., Wang, S. & Asangani, I. A. Resistance to BET inhibitor leads to alternative therapeutic vulnerabilities in castration-resistant prostate cancer. Cell Rep. 22, 2236–2245 (2018).
Poggio, M. et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell 177, 414–427.e13 (2019).
Hogg, S. J. et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 18, 2162–2174 (2017).
Zhu, H. et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 16, 2829–2837 (2016).
Lai, X. et al. Modeling combination therapy for breast cancer with BET and immune checkpoint inhibitors. Proc. Natl Acad. Sci. USA 115, 5534–5539 (2018).
Gowrishankar, K. et al. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-κB. PLoS One 10, (2015).
Pan, Y. et al. Synergistic inhibition of pancreatic cancer with anti-PD-L1 and c-Myc inhibitor JQ1. Oncoimmunology 8, (2019).
Liu, C. et al. Bromo- and extraterminal domain protein inhibition improves immunotherapy efficacy in hepatocellular carcinoma. Cancer Sci. 111, 3503–3515 (2020).
Miao, X., Wu, Z., Jiang, Y., Liu, H. & Gong, W. An efficient combination immunotherapy for antitumor immunity without accelerating cardiac allograft rejection. Immunology 169, (2023).
Wang, H. et al. BET inhibitor JQ1 enhances anti-tumor immunity and synergizes with PD-1 blockade in CRC. J. Cancer 13, 2126–2137 (2022).
Wang, J. et al. BRD4-IRF1 axis regulates chemoradiotherapy-induced PD-L1 expression and immune evasion in non-small cell lung cancer. Clin. Transl. Med. 12, (2022).
Mao, W. et al. Immunogenicity of prostate cancer is augmented by BET bromodomain inhibition. J. Immunother. Cancer 7, (2019).
Stratikopoulos, E. E. et al. Kinase and BET inhibitors together clamp inhibition of PI3K signaling and overcome resistance to therapy. Cancer Cell 27, 837–851 (2015).
Karin Bauer, Daniela Berger, Christoph C. Zielinski, Peter Valent & Thomas W. Grunt. Hitting two oncogenic machineries in cancer cells: cooperative effects of the multi-kinase inhibitor ponatinib and the BET bromodomain blockers JQ1 or dBET1 on human carcinoma cells SUPPLEMENTARY MATERIALS. www.oncotarget.com (2018).
Shigeta, S. et al. Targeting BET Proteins BRD2 and BRD3 in Combination with PI3K-AKT Inhibition as a Therapeutic Strategy for Ovarian Clear Cell Carcinoma. Mol. Cancer Ther. 20, 691–703 (2021).
Elkabets, M. et al. MTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. 5, (2013).
Janku, F. et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J. Clin. Oncol. 30, 777–782 (2012).
Serra, V. et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 30, 2547–2557 (2011).
Rodrik-Outmezguine, V. S. et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 1, 248–259 (2011).
O’Reilly, K. E. et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508 (2006).
Chandarlapaty, S. et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19, 58–71 (2011).
Britschgi, A. et al. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell 22, 796–811 (2012).
Risom, T. et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat. Commun. 9, (2018).
Miao, X. et al. BET protein inhibition evidently enhances sensitivity to PI3K/mTOR dual inhibition in intrahepatic cholangiocarcinoma. Cell Death Dis. 12, (2021).
Lee, H. S., Lee, S. & Cho, K. H. Cotargeting BET proteins overcomes resistance arising from PI3K/mTOR blockade-induced protumorigenic senescence in colorectal cancer. Int J. Cancer 147, 2824–2837 (2020).
Wendel, H. G. et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 428, 332–337 (2004).
Sander, S. et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell 22, 167–179 (2012).
Pourdehnad, M. et al. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl Acad. Sci. USA 110, 11988–11993 (2013).
Derenzini, E. et al. BET inhibition-induced GSK3β feedback enhances lymphoma vulnerability to PI3K inhibitors. Cell Rep. 24, 2155–2166 (2018).
Tomska, K. et al. Drug-based perturbation screen uncovers synergistic drug combinations in Burkitt lymphoma. Sci. Rep. 8, (2018).
Sun, B. et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood 126, 1565–1574 (2015).
Tarantelli, C. et al. Copanlisib synergizes with conventional and targeted agents including venetoclax in B- And T-cell lymphoma models. Blood Adv. 4, 819–829 (2020).
Kim, E. et al. The BET inhibitor GS-5829 targets chronic lymphocytic leukemia cells and their supportive microenvironment. Leukemia 34, 1588–1598 (2020).
Muralidharan, S. V. et al. BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene 35, 4689–4697 (2016).
Tarantelli, C. et al. BET bromodomain inhibitor birabresib in mantle cell lymphoma: In vivo activity and identification of novel combinations to overcome adaptive resistance. ESMO Open 3, (2018).
Tarantelli, C. et al. PQR309 is a novel dual PI3K/mTOR inhibitor with preclinical antitumor activity in lymphomas as a single agent and in combination therapy. Clin. Cancer Res. 24, 120–129 (2018).
Eric Davis, R., Brown, K. D., Siebenlist, U. & Staudt, L. M. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 194, 1861–1874 (2001).
Lebwohl, D. et al. Development of everolimus, a novel oral mTOR inhibitor, across a spectrum of diseases. Ann. N. Y Acad. Sci. 1291, 14–32 (2013).
Marcotte, R. et al. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell 164, 293–309 (2016).
Bihani, T. et al. Resistance to everolimus driven by epigenetic regulation of MYC in ER+ breast cancers. Oncotarget. 6 www.impactjournals.com/oncotarget/ (2014).
Vázquez, R. et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget 8 www.impactjournals.com/oncotarget/www.impactjournals.com/oncotarget (2017).
Gupta, M. et al. Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood 114, 2926–2935 (2009).
Bernasconi, E. et al. Preclinical evaluation of the BET bromodomain inhibitor BAY 1238097 for the treatment of lymphoma. Br. J. Haematol. 178, 936–948 (2017).
Gaudio, E. et al. Bromodomain inhibitor OTX015 (MK-8628) combined with targeted agents shows strong in vivo antitumor activity in lymphoma. 7 www.impactjournals.com/oncotarget (2016).
Kumari, A. et al. mTOR inhibition overcomes RSK3-mediated resistance to BET inhibitors in small cell lung cancer. JCI Insight 8, (2023).
Christensen, C. L. et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 26, 909–922 (2014).
Jing, Y. et al. Concomitant BET and MAPK blockade for effective treatment of ovarian cancer. 7 www.oncotarget.com (2015).
Kurimchak, A. M. et al. Intrinsic resistance to MEK inhibition through BET protein–mediated kinome reprogramming in NF1-deficient ovarian cancer. Mol. Cancer Res. 17, 1721–1734 (2019).
Nissan, M. H. et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 74, 2340–2350 (2014).
Bell, D. et al. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011).
Johannessen, C. M. et al. The NF1 tumor suppressor critically regulates TSC2 and mTOR. https://doi.org/10.1073/pnas.0503224102 (2005).
Rajalingam, K., Schreck, R., Rapp, U. R. & Albert, Š. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta, Mol. Cell Res. 1773, 1177–1195 (2007).
Duncan, J. S. et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321 (2012).
Zawistowski, J. S. et al. Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacologic targeting of the P-TEFb complex. Cancer Discov. 7, 302–321 (2017).
Schafer, J. M. et al. Targeting MYCN-expressing triple-negative breast cancer with BET and MEK inhibitors. Sci. Transl. Med. 12, (2020).
Puissant, A. et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 3, 309–323 (2013).
Echevarría‐Vargas, I. M. et al. Co-targeting BET and MEK as salvage therapy for MAPK and checkpoint inhibitor-resistant melanoma. EMBO Mol. Med. 10, (2018).
Tiago, M. et al. Targeting BRD/BET proteins inhibits adaptive kinome upregulation and enhances the effects of BRAF/MEK inhibitors in melanoma. Br. J. Cancer 122, 789–800 (2020).
Moy, B., Kirkpatrick, P., Kar, S. & Goss, P. Lapatinib. Nat. Rev. Drug Discov. 6, 431–432 (2007).
Dhillon, S. & Wagstaff, A. J. Lapatinib. Drugs 67, 2101–2108 (2007).
D’Amato, V. et al. Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat. Rev. 41, 877–883 (2015).
Eijkelenboom, A. & Burgering, B. M. T. FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 14, 83–97 (2013).
Greer, E. L. & Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 24, 7410–7425 (2005).
Webb, A. E. & Brunet, A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem. Sci. 39, 159–169 (2014).
Matkar, S. et al. An epigenetic pathway regulates sensitivity of breast cancer cells to HER2 inhibition via FOXO/c-Myc Axis. Cancer Cell 28, 472–485 (2015).
Stuhlmiller, T. J. et al. Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep. 11, 390–404 (2015).
Li, H. et al. The beneficial role of sunitinib in tumor immune surveillance by regulating tumor PD-L1. Adv. Sci. (Weinh) 8, (2020).
Kuang, X. et al. Propranolol enhanced the anti-tumor effect of sunitinib by inhibiting proliferation and inducing G0/G1/S phase arrest in malignant melanoma. Oncotarget 9, 802–811 (2017).
Zeng, F. et al. BET inhibitors synergize with sunitinib in melanoma through GDF15 suppression. Exp. Mol. Med 55, 364–376 (2023).
Li, Y. & Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 6 (2016).
Borbely, G., Haldosen, L.-A., Dahlman-Wright, K. & Zhao, C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget 6 www.impactjournals.com/oncotarget/ (2015).
Li, W. et al. Targeting MYC activity in double-hit lymphoma with MYC and BCL2 and/or BCL6 rearrangements with epigenetic bromodomain inhibitors. J. Hematol. Oncol. 12, (2019).
Kim, S. R. et al. BET inhibition in advanced cutaneous T cell lymphoma is synergistically potentiated by BCL2 inhibition or HDAC inhibition. Oncotarget 9, 29193–29207 (2018).
George, P. et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood 105, 1768–1776 (2005).
Fiskus, W. et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol. Cancer Ther. 13, 1142–1154 (2014).
Amemiya, S., Yamaguchi, T., Hashimoto, Y. & Noguchi-Yachide, T. Synthesis and evaluation of novel dual BRD4/HDAC inhibitors. Bioorg. Med. Chem. 25, 3677–3684 (2017).
He, S. et al. Potent dual BET/HDAC inhibitors for efficient treatment of pancreatic cancer. Angew. Chem. - Int. Ed. 59, 3028–3032 (2020).
Huang, Y., Liu, N., Pan, Z., Li, Z. & Sheng, C. BET-HDAC dual inhibitors for combinational treatment of breast cancer and concurrent candidiasis. J. Med Chem. 66, 1239–1253 (2023).
Burmeister, A. et al. Establishment and Evaluation of Dual HDAC/BET Inhibitors as Therapeutic Options for Germ Cell Tumors and Other Urological Malignancies. Mol. Cancer Ther. 21, 1674–1688 (2022).
Esteve-Arenys, A. et al. The BET bromodomain inhibitor CPI203 overcomes resistance to ABT-199 (venetoclax) by downregulation of BFL-1/A1 in in vitro and in vivo models of MYC+/BCL2+ double hit lymphoma. Oncogene 37, 1830–1844 (2018).
Carrà, G. et al. Inhibition of bromodomain and extra-terminal proteins increases sensitivity to venetoclax in chronic lymphocytic leukaemia. J. Cell Mol. Med. 24, 1650–1657 (2020).
Peirs, S. et al. Targeting BET proteins improves the therapeutic efficacy of BCL-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia 31, 2037–2047 (2017).
Souers, A. J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 (2013).
Yap, T. A. et al. First-in-human trial of the oral ataxia telangiectasia and rad3-related (Atr) inhibitor bay 1895344 in patients with advanced solid tumors. Cancer Discov. 11, 80–91 (2021).
Dillon, M. et al. A phase I study of ATR inhibitor, AZD6738, as monotherapy in advanced solid tumours (PATRIOT part A, B). Ann. Oncol. 30, v165–v166 (2019).
Muralidharan, S. V. et al. BET bromodomain inhibitors synergize with ATR inhibitors in melanoma. Cell Death Dis. 8, (2017).
Liu, J. et al. Targeting the BRD4/FOXO3a/CDK6 axis sensitizes AKT inhibition in luminal breast cancer. Nat. Commun. 9, (2018).
Leung, B. S. & Potter, A. H. Mode of estrogen action on cell proliferative kinetics in CAMA-1 cells. I. Effect of serum and estrogen. Cancer Invest. 5, 187–194 (1987).
Sherr, C. J. Cancer cell cycles. Science 274, 1672–1674 (1996).
Stottrup, C., Tsang, T. & Chin, Y. R. Upregulation of AKT3 confers resistance to the AKT Inhibitor MK2206 in breast cancer. Mol. Cancer Ther. 15, 1964–1974 (2016).
Song, Y. et al. The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol. Cancer 19, (2020).
Zhang, P. et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat. Med. 23, 1055–1062 (2017).
Boi, M. et al. Therapeutic efficacy of the bromodomain inhibitor OTX015/ MK-8628 in ALK-positive anaplastic large cell lymphoma: an alternative modality to overcome resistant phenotypes The AIRC 5xMille Consortium ‘Genetics-Driven Targeted Management of Lymphoid Malignancies’. 7 www.impactjournals.com/oncotarget (2016).
Gaudio, E. et al. Combination of the MEK inhibitor pimasertib with BTK or PI3K-delta inhibitors is active in pre-clinical models of aggressive lymphomas Key-Message. http://annonc.oxfordjournals.org/ (2016).
Schaffer, M. et al. Identification of potential ibrutinib combinations in hematological malignancies using a combination high-throughput screen. Leuk. Lymphoma 59, 931–940 (2018).
Tarantelli, C. et al. The Bruton tyrosine kinase inhibitor zanubrutinib (BGB-3111) demonstrated synergies with other anti-lymphoma targeted agents. Haematologica 104, e307–e309 (2019).
Sun, W. et al. Transcriptome analysis of luminal breast cancer reveals a role for LOL in tumor progression and tamoxifen resistance. Int J. Cancer 145, 842–856 (2019).
Stander, B. A. et al. In Vitro Evaluation of ESE-15-ol, an Estradiol Analogue with Nanomolar Antimitotic and Carbonic Anhydrase Inhibitory Activity. PLoS One 7, (2012).
Mqoco, T., Stander, A., Engelbrecht, A. M. & Joubert, A. M. A Combination of an Antimitotic and a Bromodomain 4 Inhibitor Synergistically Inhibits the Metastatic MDA-MB-231 Breast Cancer Cell Line. Biomed. Res. Int. 2019 (2019).
Sun, Y. et al. Epigenetic heterogeneity promotes acquired resistance to BET bromodomain inhibition in ovarian cancer. Am. J. Cancer Res. 11, 3021–3038 (2021).
Zhao, X. et al. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia 27, 2341–2350 (2013).
Zhang, X. et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell 22, 506–523 (2012).
Kurmasheva, R. T. et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 64, (2017).
Torchia, J. et al. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell 30, 891–908 (2016).
Johann, P. D. et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 29, 379–393 (2016).
Erkek, S. et al. Comprehensive analysis of chromatin states in atypical teratoid/rhabdoid tumor identifies diverging roles for SWI/SNF and polycomb in gene regulation. Cancer Cell 35, 95–110.e8 (2019).
Tang, Y. et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 20, 732–740 (2014).
Alimova, I. et al. Inhibition of MYC attenuates tumor cell self-renewal and promotes senescence in SMARCB1-deficient Group 2 atypical teratoid rhabdoid tumors to suppress tumor growth in vivo. Int J. Cancer 144, 1983–1995 (2019).
Wang, X. et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet 49, 289–295 (2017).
Ishi, Y. et al. Therapeutic targeting of EZH2 and BET BRD4 in pediatric rhabdoid tumors. Mol. Cancer Ther. 21, 715–726 (2022).
Zhang, Y. et al. Combination of EZH2 inhibitor and BET inhibitor for treatment of diffuse intrinsic pontine glioma. Cell Biosci. 7, (2017).
Negri, A. et al. Simultaneous administration of EZH2 and BET inhibitors inhibits proliferation and clonogenic ability of metastatic prostate cancer cells. J. Enzyme Inhib. Med. Chem. 38 (2023).
Picaud, S. et al. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for Leukemia therapy. Cancer Res. 75, 5106–5119 (2015).
Ryan, K. R., Giles, F. & Morgan, G. J. Targeting both BET and CBP/EP300 proteins with the novel dual inhibitors NEO2734 and NEO1132 leads to anti-tumor activity in multiple myeloma. Eur. J. Haematol. 106, 90–99 (2021).
Spriano, F. et al. Antitumor activity of the dual BET and CBP/EP300 inhibitor NEO2734. Blood Adv. 4, 4124–4135 (2020).
Yan, Y. et al. The novel BET-CBP/p300 dual inhibitor NEO2734 is active in SPOP mutant and wild-type prostate cancer. EMBO Mol. Med. 11, (2019).
Principe, D. R. et al. XP-524 is a dual-BET/EP300 inhibitor that represses oncogenic KRAS and potentiates immune checkpoint inhibition in pancreatic cancer. Proc. Natl. Acad. Sci. USA 119, (2022).
Pal, S. et al. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 26, 3558–3569 (2007).
Alinari, L. et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 125, 2530–2543 (2015).
Mensah, A. A. et al. Bromodomain and extra-terminal domain inhibition modulates the expression of pathologically relevant microRNAs in diffuse large B-cell lymphoma. Haematologica 103, 2049–2058 (2018).
Nieto-Jimenez, C. et al. Inhibition of the mitotic kinase PLK1 overcomes therapeutic resistance to BET inhibitors in triple negative breast cancer. Cancer Lett. 491, 50–59 (2020).
Zhang, Z. et al. Plk1 inhibition enhances the efficacy of androgen signaling blockade in castration-resistant prostate cancer. Cancer Res. 74, 6635–6647 (2014).
Gutteridge, R. E. A., Ndiaye, M. A., Liu, X. & Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 15, 1427–1435 (2016).
Xu, Y. et al. Novel Dual BET and PLK1 Inhibitor WNY0824 Exerts Potent Antitumor Effects in CRPC by Inhibiting Transcription Factor Function and Inducing Mitotic Abnormality. Mol. Cancer Ther. 19, 1221–1231 (2020).
Tyler, D. S. et al. Click chemistry enables preclinical evaluation of targeted epigenetic therapies. Science 356, 1397–1401 (2017).
Arfaoui, A. et al. A genome-wide RNAi screen reveals essential therapeutic targets of breast cancer stem cells. EMBO Mol. Med. 11, (2019).
Zaware, N. & Zhou, M. M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 26, 870–879 (2019).
Acknowledgements
We thank BioRender (biorender.com) for providing technical support on drawing figures.
Funding
This work was supported by the National Natural Science Foundation of China (nos. 81872507 and 82173238), the Heilongjiang Provincial Natural Science Foundation Key Projects (ZD2020H007), the Harbin Medical University Cancer Hospital (CN) Nn10 Project (Nn10py2017-01), the China Postdoctoral Science Foundation (296266), the Heilongjiang Province Doctoral Post-doctoral Fund (LBH-Z21177), the Fundamental Research Funds for the Provincial Universities (2022-KYYWF-0291), and the Harbin Medical University Haiyan Youth Fund (JJQN2022–3).
Author information
Authors and Affiliations
Contributions
G.L., C.Y. and Z.-Q.W. designed the manuscript. Z.-Q.W., Z.-C.Z. and Y.-Y.W. wrote the manuscript. Z.-Q.W. and Y.-Y.W. drew the figures and tables. Z.-Q.W., C.Y., Y.-N.P. and S.-H.L. mainly revised the manuscript. C.Y. and T.-B.L. made some revisions of the review. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All authors have read and approved of its submission to this journal.
Additional information
Consent for publication
All authors have read and approved of its submission to this journal.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, ZQ., Zhang, ZC., Wu, YY. et al. Bromodomain and extraterminal (BET) proteins: biological functions, diseases, and targeted therapy. Sig Transduct Target Ther 8, 420 (2023). https://doi.org/10.1038/s41392-023-01647-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01647-6
