
Abstract
Immune cell infiltration in response to myocyte death regulates extracellular matrix remodeling and scar formation after myocardial infarction (MI). Caspase-recruitment domain family member 9 (CARD9) acts as an adapter that mediates the transduction of pro-inflammatory signaling cascades in innate immunity; however, its role in cardiac injury and repair post-MI remains unclear. We found that Card9 was one of the most upregulated Card genes in the ischemic myocardium of mice. CARD9 expression increased considerably 1 day post-MI and declined by day 7 post-MI. Moreover, CARD9 was mainly expressed in F4/80-positive macrophages. Card9 knockout (KO) led to left ventricular function improvement and infarct scar size reduction in mice 28 days post-MI. Additionally, Card9 KO suppressed cardiomyocyte apoptosis in the border region and attenuated matrix metalloproteinase (MMP) expression. RNA sequencing revealed that Card9 KO significantly suppressed lipocalin 2 (Lcn2) expression post-MI. Both LCN2 and the receptor solute carrier family 22 member 17 (SL22A17) were detected in macrophages. Subsequently, we demonstrated that Card9 overexpression increased LCN2 expression, while Card9 KO inhibited necrotic cell-induced LCN2 upregulation in macrophages, likely through NF-κB. Lcn2 KO showed beneficial effects post-MI, and recombinant LCN2 diminished the protective effects of Card9 KO in vivo. Lcn2 KO reduced MMP9 post-MI, and Lcn2 overexpression increased Mmp9 expression in macrophages. Slc22a17 knockdown in macrophages reduced MMP9 release with recombinant LCN2 treatment. In conclusion, our results demonstrate that macrophage CARD9 mediates the deterioration of cardiac function and adverse remodeling post-MI via LCN2.
Similar content being viewed by others


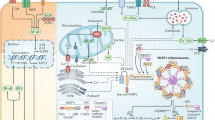
Introduction
Heart disease is the main cause of death worldwide,1 and acute myocardial infarction (MI) is a common cardiovascular disorder leading to necrosis of cardiomyocytes. Owing to lack of regenerative potential of the adult heart, damaged myocardium is replaced by scar, which adversely affects cardiac function. Consequently, patients who survive an MI often experience heart failure. Efforts to reduce cardiac injury or facilitate repair post-MI may help to prevent heart failure. Innate immunity is elicited and plays an essential regulatory role post-MI in response to danger-associated molecular patterns (DAMPs) released by dead myocytes.2 Immune cells are recruited to amplify inflammation. Monocyte infiltration into the infarcted heart starts soon after MI via C-C motif chemokine ligand2 / C-C motif chemokine receptor (CCR2) signaling,3 and begins to differentiate into macrophages, which replaced the resident macrophages in the ischemic area within 24 hours post-MI.4 Macrophages are highly plastic and can adopt various activation states.5,6 At 1 to 3 days post-MI, monocyte-derived macrophages exhibit pro-inflammatory characteristics, secreting inflammatory cytokines and chemokines. On the one hand, they contribute to the clearance of necrotic cells, facilitating the repair of infarcted heart. On the other hand, excessive inflammatory reaction may extend ischemic injury by damaging surviving cardiomyocytes of the border zone and promoting protease activity.7 Increased interleukin 6 are associated with poor outcomes in MI patients.8 Pro-inflammatory macrophages impair healing9; whereas suppressing pro-inflammatory macrophage polarization reduces necrosis of cardiomyocytes10 and cardiac rupture11 post-MI. Targeting CCR2 using siRNA decreases monocyte infiltration, leading to a decrease in infarct size and improvement of cardiac function.12,13
The extracellular matrix (ECM) which is a dynamic structure is critical in cardiac injury, repair, and remodeling post-MI. Macrophages and inflammation also regulate ECM. Infiltrated macrophage is one of the primary sources of metalloproteinases (MMPs), which mediate the degradation of ECM.5 ECM degradation facilitates cardiac healing in the early stages. Optimal scar formation requires proper synthesis, cross-linking, and ECM alignment, which helps to maintain cardiac function post-MI. However, excessive ECM degradation may result in insufficient collagen deposition, leading to left ventricle (LV) thinning and dilation. Additionally, plasma MMP9 levels are correlated with LV dysfunction post-MI.14 Conversely, knockout (KO) of Mmp9 or selective MMP inhibition can decrease MI-induced cardiac rupture and LV dilation in mice.15,16 Moreover, overexpression of tissue inhibitor of metalloproteinase 1 (Timp1) preserves cardiac function in MI rats;17 whereas, Timp1 KO exacerbates LV remodeling post-MI.18 Thus, modulation of the macrophage function to reduce MMP expression or activation may decrease the risk of post-MI cardiac injury.
Pattern recognition receptors (PRRs) recognize DAMPs and induce activation of downstream inflammatory pathways. The proteins of the intracellular caspase recruitment domain (CARD) family act as linkers between PPRs and downstream signaling molecules.19,20,21 In particular, CARD9 is a critical adapter of activated PRRs, such as C-type lectin receptors (CLRs), intracellular nucleotide oligomerization domain (NOD)-like receptors, toll-like receptors (TLRs), and some other intracellular nucleic acid-sensing PPRs in response to fungal, bacterial, viral, and parasitic infections.22,23,24,25,26 CARD9 is mainly expressed by myeloid cells. With microorganism stimulation, PPRs recruit spleen tyrosine kinase (Syk) or receptor interacting protein 2 (RIP2), which further activates CARD9. After activation, CARD9 binds to the CARD domain of BCL10 immune signaling adaptor, which interacts with MALT1 paracaspase to form a CARD9/BCL10/MALT1complex, activating downstream nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and mitogen-activated protein kinases (MAPKs) signaling.23,27,28 Mice with Card9 knockout (KO) are susceptible to certain microbial infections.28,29 CARD9 also participates in noninfectious inflammation regulation.30,31,32 Furthermore, Card9 KO improves insulin resistance by blocking MAPK pathways in high-fat diet treated mice,31 and knockdown of Card9 in acute pancreatitis rats inhibits NF-κB and p38 MAPK signaling and attenuates pancreatic, lung, and liver tissue injury.32 However, whether CARD9 is a key regulator between innate immunity and cardiac injury in MI remains unclear. In a previous study, we showed that CARD9 mediated necrotic cell-induced NF-κB signaling activation in macrophages and promoted inflammatory cytokine expression and neointima formation in vein grafts.33 The role of CARD9 has also been studied in several cardiac disease models. Card9 depletion decreases angiotensin II-induced cardiac fibrosis,34 prevents pressure overload-induced heart failure and fibrosis,35 and alleviates cardiac injury in obese mice.36 Exploring the function of CARD9 and underlying mechanisms in cardiac injury and remodeling post-MI will provide us the new targets or strategies for MI therapy.
In the present study, we investigated the expression of CARD9 in ischemic myocardium and revealed its role in cardiac injury and remodeling post-MI using Card9 KO mice. We found that CARD9 mainly located in macrophages and its regulation of lipocalin 2 (LCN2) through NF-κB activation may contribute to its adverse effects post-MI.
Results
CARD9 expression is increased in infarcted heart
MI was induced in mice. Three days after infarction, the expression of Card family genes was examined, and the mRNA levels of Card9 and Card11 were found to be increased the most among all the Card family genes (Fig. 1a). Subsequently, we assayed the protein levels of CARD9 in myocardium at different time following infarction and found that CARD9 expression was induced as early as 1 day, peaked on 3 days, and descended on 7 days post-MI (Fig. 1b). To determine the cellular localization of CARD9, immunofluorescent staining was performed on infarcted heart sections. We observed that CARD9 co-stained with F4/80, a macrophage marker, indicating that CARD9 was expressed in macrophages (Fig. 1c). To explore CARD9 expression levels in various heart cells, we isolated CD45-, CD45+ F4/80-, and CD45+ F4/80+ cells from infarcted mouse hearts using enzyme digestion and magnetic- or fluorescence-activated cell sorting. We found that Card9 mRNA expression was highest in CD45+ F4/80+ macrophages, lower in CD45+ F4/80- bone marrow (BM)-derived cells, and lowest in CD45- cells (Fig. 1d). Furthermore, a large amount of CARD9 protein was observed in CD45+ F4/80+ macrophages, a small amount of CARD9 in CD45+ F4/80- cells, and virtually no expression of CARD9 in CD45- cells (Fig. 1e), which was consistent with the mRNA data. Because CARD9 expression was mainly detected in CD45+ cells, we investigated CARD9 expression in distinct populations of CD45+ immune cells. We obtained a published single-cell RNA sequencing (scRNA-seq) dataset (GSE163465)37 of cardiac CD45+ cells in mice after MI from the Gene Expression Omnibus (GEO) database and observed that macrophages contributed the most to Card9 expression post-MI (Supplementary Fig. 1). These results confirmed Card9 expression in macrophages post-MI.

CARD9 is increased in mouse hearts post-MI and mainly expressed in macrophages. a The mRNA levels of Card family genes in below-ligature cardiac tissues at 3 days post-MI, quantified as fold induction with respect to the sham group after normalization to Gapdh (n = 7). b CARD9 protein levels in cardiac tissues of sham and MI model mice at the indicated time points post-MI, quantified as fold induction with respect to the sham group after normalization to GAPDH (n = 6). c Representative co-staining of CARD9 and F4/80 on cardiac sections at 3 days post-MI (n = 4). Nuclei were stained with DAPI. CD45-, CD45+ F4/80- and CD45+ F4/80+ cells sorted from hearts of mice at 3 days post-MI. d Card9 mRNA expression in the three population of MACS-sorted cells, quantified as fold change with respect to CD45- group after normalization to Gapdh. Hearts from 4-5 mice were pooled as one sample; n = 4 samples. e CARD9 protein levels in three population of FACS-sorted cells. Hearts from three mice were used for sorting. Scale bar: 50 µm. Data represent the mean ± SEM. Two-tailed unpaired t-test (a) and one-way ANOVA followed by Tukey’s test for post hoc comparison (b and d) were used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001
Card9 KO enhances LV function and attenuates reversed remodeling post-MI
To determine the involvement of CARD9 in MI, we used Card9 KO mice. The survival of the mice was analyzed and cardiac function was determined at 28 days post-MI. Card9 KO increased the survival rate of MI mice from 70% to 82% (Fig. 2a), significantly decreased the LV end-systolic internal diameter, and increased the LV ejection fraction (Supplementary Table 1). Collectively, our results demonstrated that Card9 deficiency restores cardiac function post-MI.

Card9 knockout attenuates infarct size and adverse left ventricular remodeling post-MI. a Survival analysis of mice post-MI (n = 8, 8, 30, 41). b Ratios of heart weight to body weight at 28 days post-MI (n = 6). c H&E and Masson’s trichrome staining of cardiac sections at 28 days after MI. Infarct size is presented as the ratio of infarcted area length to left ventricular circumference (n = 6). d H&E and wheat germ agglutinin (WGA) staining of non-infarcted area. The bar graft shows cross-sectional area of myocytes (n = 6). e Sirus Red staining. Fibrotic area in non-infarcted region was measured and quantified as fold induction with respect to WT group (n = 6). f Flow cytometric analysis of CD45.1+ and CD45.2+ cells in the peripheral blood of CD45.2 mice transplanted with BM cells from CD45.1 mice (n = 3). g Detection of donor BM cell infiltration into the heart at 3 days post-MI. h Bar grafts show the percentage of CD45.1+ or CD45.2+ cells in g (n = 4). i Masson’s trichrome staining on cardiac sections from mice with BMT at 28 days post-MI and measurement of infarct size (n = 4-5). Scale bar: 1 mm (c, e top and i), 50 μm (d and e bottom). Data represent the mean ± SEM. One-way ANOVA followed by Tukey’s test (b and i) and two-tailed unpaired t-test (c, d, and e) were used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001
We further investigated whether CARD9 affects pathological remodeling. Card9 KO attenuated heart weight/body weight ratio (Fig. 2b). Moreover, Card9 KO reduced the infarct size compared with wild-type (WT) mice at 28 days post-MI (Fig. 2c) and suppressed the enlargement of cardiomyocytes (Fig. 2d) and interstitial fibrosis in non-ischemic regions (Fig. 2e). Collectively, these data provide the first evidence that Card9 KO attenuates adverse cardiac remodeling following MI.
To exclude the influence of Card9 KO on other Card family member expression, we examined the mRNA levels of these genes in BM-derived macrophages (BMDMs), BM neutrophils, heart, liver, lung, and kidney tissues, and found Card9 KO did not affect Card family expression in these cells and tissues (Supplementary Fig. 2). Moreover, Card9 KO mice exhibited no changes in body weight (Supplementary Fig. 3a) and the morphology of vital organs, such as the liver, lungs, and kidneys (Supplementary Fig. 3b).
As the expression of Card11 was also significantly increased post-MI, we evaluated its role. We found that Card11 KO did not change infarct size or cardiac function at 28 days post-MI (Supplementary Fig. 4a, b).
Card9 KO in BM cells results in protective effects post-MI
In the early post-MI stages, most macrophages are derived from infiltrated monocytes,4,38,39 exhibiting pro-inflammatory and pro-injury effects. CCR2 is a commonly used marker to distinguish the origin of cardiac macrophages. CCR2- macrophages are embryonically derived, and CCR2+ macrophages are derived from circulating monocytes.4,40 We analyzed Card9 expression in CCR2+ and CCR2- macrophages using the cardiac CD45+ scRNA-seq dataset (GSE163465)37 and found the expression of Card9 seemed marginally higher in CCR2+ macrophages, although the difference was not significant (p = 0.0556, Supplementary Fig. 5). To further explore the importance of CARD9 expression in infiltrating myeloid cells in MI, BM transplantation (BMT) experiments were performed in WT and Card9 KO mice. We used mice expressing the CD45.1 allele as donors and CD45.2 mice with the same background as recipients to estimate BM reconstitution. Flow cytometric analysis confirmed that over 95% of the blood cells (with erythrocytes removed) from the CD45.2 recipients expressed the CD45.1 allele at 8 weeks after BMT (Fig. 2f), indicating successful BM reconstitution. These CD45.2 recipient mice were subjected to MI, and we observed that more than 10% of cells in the heart expressed CD45.1, whereas less than 0.5% of cells were CD45.2+ at 3 days post-MI (Fig. 2g, h), suggesting that many donor BM-derived cells had infiltrated into the heart.
WT and Card9 KO recipient mice were induced MI after BM reconstitution. We found that transplantation of Card9 KO BM cells into WT mice reduced the infarct size at 28 days post-MI compared to that in WT mice transplanted with WT BM cells. In contrast, transplantation of WT BM cells into Card9 KO mice increased infarct size compared to that in Card9 KO mice transplanted with Card9 KO BM cells (Fig. 2i). These results confirmed that CARD9 from BM-derived cells contributed to cardiac injury and remodeling post-MI.
Card9 KO has no significant effect on angiogenesis but inhibits cardiomyocyte apoptosis and MMP expression
To explore the mechanism for CARD9 regulating MI, we first evaluated the angiogenic effect of Card9 KO by staining endothelial cell marker at 7 days post-MI and observed that Card9 KO did not significantly increase the number of microvessels in the border regions (Supplementary Fig. 6a). Moreover, we isolated BM-derived macrophages from WT and Card9 KO mice (Supplementary Fig. 6b) and treated them with necrotic cells. We found that the levels of vascular endothelial growth factor A and fibroblast growth factor 2 were lower in Card9-deficient cells (Supplementary Fig. 6c), suggesting that mechanisms other than angiogenesis are responsible for the protective role of Card9 KO.
Subsequently, we examined immune cell infiltration and found reduced macrophage and neutrophil recruitment in the border region of Card9 KO mice at 3 days post-MI (Supplementary Fig. 7a, b) and fewer apoptotic cells (Fig. 3a), which were identified as cardiomyocytes (Fig. 3b). Moreover, cleaved caspase 3 in Card9 KO hearts were reduced (Fig. 3c). Thus, Card9 KO protects cardiomyocytes from secondary apoptosis after MI.

Card9 knockout attenuates apoptosis of cardiomyocytes and Mmp expression post-MI. a TUNEL assay in border region of hearts at 3 days after MI. b TUNEL and α-actinin co-staining in the border region of WT hearts. Nuclei were stained with DAPI. c Cleaved caspase 3 protein levels in WT and Card9 KO hearts at 3 days after MI, quantified as fold induction with respect to WT after normalization to Caspase 3 (n = 4). d qPCR analysis of Card9 expression in Card9-overexpressing (OE) and Ctrl RAW264.7 cells, quantified as fold change with respect to Ctrl after normalizing to Gapdh (n = 3). e TUNEL assay in H9C2 cells treated with conditioned medium from Ctrl and Card9 OE RAW264.7 cells. The percentage of apoptotic cells was calculated (n = 3). f The mRNA expression of Mmp2, Mmp3 and Mmp9 in hearts at 3 days after MI (n = 7), quantified as fold induction with respect to WT Sham after normalization to Gapdh. g MMP2, MMP3 and MMP9 protein levels in hearts, quantified as fold change with respect to WT after normalization to GAPDH (n = 4). Scale bar: 50 μm. Data represent the mean ± SEM. Two-tailed unpaired t-test (c, d, e and g) and one-way ANOVA followed by Tukey’s test (f) were used for statistical analysis. *p < 0.05, **p < 0.01
To further evaluate the function of macrophage Card9 on cardiomyocyte apoptosis, Card9 was overexpressed in RAW264.7 macrophages (Fig. 3d). H9C2 cardiomyocytes were treated with conditioned medium from Card9-overexpressing (OE) and control RAW264.7 cells, and it was observed that Card9 OE conditioned medium increased the apoptosis of H9C2 cells (Fig. 3e).
Excessive expression of ECM-degrading proteases can cause cardiac injury following MI. We then assessed the expression of Mmp genes and found that Card9 KO decreased Mmp2, Mmp3, and Mmp9 mRNA levels (Fig. 3f) as well as the protein levels (Fig. 3g). Additionally, MMP2 and MMP9 staining were reduced in Card9 KO hearts (Supplementary Fig. 7c, d). Regulation of MMPs may contribute to the protective effects in Card9 KO mice post-MI. Moreover, we examined ECM-related gene expression 7 days post-MI and found that Card9 KO increased Col1a1, Col3a1, and Fn1 mRNA expression (Supplementary Fig. 8).
Card9 KO attenuates LCN2 expression in macrophages via NF-κB post-MI
To investigate the molecular mechanism of CARD9 regulating the extent of cardiac injury after MI, we conducted RNA sequencing using infarcted heart tissue from WT and Card9 KO mice. Gene ontology (GO) enrichment analysis on differentially expressed genes (DEGs) between WT MI and Card9 KO MI groups showed that the downregulated genes were associated with secreted proteins, proteins involved in inflammation, and MMP-related proteins; whereas, the upregulated genes were ECM remodeling-related (Supplementary Fig. 9a, b). Among the downregulated genes in Card9 KO hearts, the change in Lcn2 expression compared to that in WT hearts was the most significant (Fig. 4a and supplementary Fig. 9c). In contrast, its expression was increased in MI hearts compared to that in sham hearts. The regulation of Lcn2 mRNA expression by CARD9 was confirmed in the heart post-MI (Fig. 4b). Moreover, we found the mRNA levels of Lcn2 were elevated at 1 day and 3 days after MI and returned to the baseline level at 7 days (Fig. 4b). The change in plasma LCN2 levels also showed a similar pattern (Fig. 4c); however, Card9 KO inhibited the increase in LCN2 levels at 3 days post-MI (Fig. 4b, c). These results suggest that LCN2 may be produced by inflammatory cells. Therefore, we co-stained LCN2 with macrophage marker F4/80 and found that LCN2 was expressed in macrophages (Fig. 4d). Subsequently, we stimulated BMDMs from WT and Card9 KO mice with necrotic cells, and found that Card9 KO counteracted the increase in Lcn2 mRNA expression (Fig. 4e) and protein release into the cell culture supernatant (Fig. 4f). In Card9 OE RAW264.7 cells, the mRNA expression (Fig. 4g) and protein release (Fig. 4h) of LCN2 also increased.

Card9 knockout attenuates Lcn2 expression in macrophages post-MI. a Heat map of DEGs between WT-MI and Card9 KO-MI groups (n = 2). b Lcn2 mRNA expression in WT and Card9 KO hearts post-MI (n = 6–8). c Detection of LCN2 plasma levels by ELISA (n = 6–9). d Co-staining of LCN2 and F4/80 in the border region of WT cardiac tissues at 3 days after MI. Nuclei were stained with DAPI. e Lcn2 mRNA expression in necrotic cell-stimulated WT and Card9 KO BMDMs, quantified as fold induction with respect to WT Ctrl after normalization to Gapdh. f ELISA analysis of LCN2 levels in cell culture supernatant (n = 3). g qPCR analysis of Lcn2 expression in Card9 OE RAW264.7 cells, quantified as fold change with respect to Ctrl after normalizing to Gapdh. h ELISA analysis of LCN2 levels in cell culture supernatant (n = 3). Scale bar: 50 μm. Data represent the mean ± SEM. Two-way ANOVA followed by Bonferroni’s test (b, c), one-way ANOVA followed by Tukey’s test (e and f), and two-tailed unpaired t-test (g and h) were used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001
LCN2 is reported to be regulated by NF-κB,41,42 and we have previously observed that Card9 KO attenuates necrotic smooth muscle cells-induced NF-κB activation.33 Thus, we hypothesized that CARD9 regulated LCN2 via NF-κB. To test this hypothesis, we first examined NF-κB activation in cardiac tissue after MI, and found that Card9 KO attenuated the phosphorylation of its p65 subunit (Fig. 5a). Card9 KO also abolished necrotic cell-induced phosphorylation of p65 in BMDMs (Fig. 5b). When Card9 OE RAW264.7 macrophages were treated with NF-κB inhibitor, the mRNA expression and protein release of LCN2 were inhibited (Fig. 5c, d). These data indicated that CARD9 may regulate LCN2 expression through NF-κB.

CARD9 regulates Lcn2 expression via NF-κB activation. a Protein levels of phosphorylated p65 in WT and Card9 KO hearts at 3 days after MI, quantified as fold induction with respect to WT Sham after normalization to total p65 (n = 4). b Protein level of phosphorylated p65 in necrotic cell-treated WT and Card9 KO macrophages, quantified as fold induction with respect to WT Ctrl after normalization to total p65 (n = 4). Card9 OE RAW264.7 cells were treated with ammonium pyrrolidine dithiocarbamate (PDTC). c Lcn2 mRNA expression was analyzed by qPCR, quantified as fold change with respect to Ctrl vehicle after normalizing to Gapdh (n = 3). d LCN2 protein in cell culture supernatant detected by ELISA (n = 3). Data represent the mean ± SEM. One-way ANOVA followed by Tukey’s test were used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001
LCN2 mediates the effect of CARD9 post-MI
To determine the function of LCN2 in MI, we ligated the left coronary artery of Lcn2 KO mice. We observed that Lcn2 KO considerably increased the survival (Fig. 6a), diminished infarct size (Fig. 6b), and ameliorated cardiac function (Fig. 6c) post-MI in these mice. However, the administration of recombinant mouse LCN2 to Card9 KO mice after MI increased death, although it was not statistically significant owing to the limited number of mice (Supplementary Fig. 10a), enlarged infarct size (Supplementary Fig. 10b), and worsened cardiac function (Supplementary Fig. 10c).

LCN2 mediates cardiac injury in MI and enhanced MMP9 expression in macrophages. a Survival curve of WT and Lcn2 KO mice post-MI (n = 18,13). b Masson’s trichrome staining at 28 days post-MI. Infarct size is presented as the ratio of infarcted area length to left ventricular circumference (n = 6). c Echocardiographic analysis of left ventricular end-systolic internal diameter (LVIDs), left ventricular end-diastolic internal diameter (LVIDd), ejection fraction (EF) and fractional shortening (FS) (n = 8, 6). d Slc22a17 mRNA expression in cardiac tissues of sham and 3 days post-MI groups, quantified as fold induction with respect to Sham after normalization to Gapdh (n = 6). Co-staining of SLC22A17 and Mac 3 (e) or α-actinin (f) in the border region of WT hearts from sham and 3 days post-MI mice. g MMP9 staining in WT and Lcn2 KO mouse hearts at 3 days post-MI. Nuclei were stained with DAPI. h Mmp9 mRNA expression in BMDMs from WT and Lcn2 KO mice at 3 days post-MI, quantified as fold induction with respect to WT after normalization to Gapdh (n = 3). i qPCR analysis of Lcn2 and Mmp9 expression in Lcn2-overexpressed RAW264.7 cells, quantified as fold induction with respect to Ctrl after normalizing to Gapdh (n = 3). j BMDMs were transfected with Slc22a17 siRNA and Slc22a17 and Mmp9 expression was assayed by qPCR, quantified as fold induction with respect to si-negative ctrl (NC) after normalization to Gapdh (n = 3). k ELISA analysis of MMP9 protein in cell culture supernatant (n = 3). Scale bar: 1 mm (b) and 50 μm (e, f and g). Data represent the mean ± SEM. Two-tailed unpaired t-test was used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001
LCN2 induces Mmp9 expression in macrophages
To explore the mechanism by which LCN2 regulates the extent of cardiac injury after MI, we first identified the expression and cellular localization of the LCN2 receptor solute carrier family 22 member 17 (SLC22A17) in the cardiac tissue. We found Slc22a17 mRNA expression elevated after MI (Fig. 6d), and immunofluorescence staining revealed SLC22A17 expression in both cardiomyocytes and macrophages (Supplementary Fig. 11a, b). Moreover, the intensity of SLC22A17 staining was enhanced in the MI hearts (Fig. 6e, f).
LCN2 stimulation induces cardiomyocyte apoptosis,43 possibly contributing to the effect of LCN2 on cardiac injury post-MI. Additionally, we assessed MMP9 expression in MI hearts and observed less MMP9 staining in Lcn2 KO cardiac sections (Fig. 6g). We isolated BMDMs from WT and Lcn2 KO MI mice and detected decreased Mmp9 expression in Lcn2 KO cells (Fig. 6h). Furthermore, Lcn2-overexpression in RAW264.7 resulted in increased Mmp9 expression (Fig. 6i). These results suggested that LCN2 regulates MMP9 expression after MI. Subsequently, we interrupted SLC22A17 expression in WT BMDMs to identify the involvement of SLC22A17 in LCN2-mediated regulation of MMP9 and found that SLC22A17 knockdown reduced Mmp9 mRNA expression (Fig. 6j) and MMP9 release into the supernatant with recombinant LCN2 treatment (Fig. 6k).
Discussion
Our study revealed a critical role for CARD9 in macrophage in regulating cardiac injury post-MI. CARD9 is an adapter molecule that mediates pathogen-associated molecular pattern (PAMP)-activated downstream signaling of PRRs. However, whether CARD9 is involved in DAMP-induced innate immunity remains unclear. Our previous study showed that necrotic smooth muscle cells in vein grafts activated NF-κB in macrophages through CARD9, revealing the function of CARD9 in mediating necrotic cell-induced inflammation for the first time.33 MI also activates innate immunity. DAMPs released by necrotic cells activate inflammatory cells that infiltrate the border region. Dectin-1 and Dectin-2, which belong to the CLR family of PRRs, are involved in cardiac ischemia.44,45 We detected an increase in Card9 expression in the myocardium, beginning as early as 1 day after MI, peaking at 3 days, and decreasing thereafter, suggesting that CARD9 is involved in myocardial ischemic injury. Furthermore, using Card9 KO mice, we identified CARD9 as a key mediator of injury and adverse remodeling after MI.
Our results demonstrated that CARD9 was mainly expressed in CD45+ F4/80+ macrophages. Furthermore, a BMT experiment confirmed that CARD9 in BM-derived cells mediated myocardial ischemic injury. Alteration of the ECM is vital for the repair post-MI, particularly for collagen-based scar formation. Macrophages, particularly classically activated macrophages, are an essential source of MMPs that degrade ECM following MI.46 Strategies that alter macrophage polarization and decrease MMP expression or activation may help to preserve the structural integrity and function of the heart post-MI. We found that Card9 KO attenuated Mmp9 expression following MI, which may have contributed to the protective effect of Card9 KO. In exploring the downstream consequences of CARD9 activation, we identified LCN2 using RNA sequencing. LCN2 is a glycoprotein first identified in neutrophils. However, further studies have also found that LCN2 may be released by various cells, such as epithelial cells, macrophages, and hepatocytes, during inflammation or injury.47,48,49 LCN2 binds directly to MMP9, thereby protecting it from degradation and enhancing its proteolytic activity.50 LCN2 serum levels are increased in both patients with MI and animal models48,51 and are associated with adverse outcomes.51,52 Here, we demonstrated that Lcn2 deletion enhanced cardiac function and limited infarct scar size post-MI, which is consistent with a previous study.53 The direct interaction between LCN2 and MMP9 may be one of the mechanisms underlying the post-MI regulatory role of LCN2. Moreover, LCN2 has been reported to induce apoptosis in cultured cardiomyocytes by inhibiting autophagy and increasing intracellular iron accumulation,43 and LCN2-siderophore-iron complexes increase ROS generation and decrease oxidative phosphorylation in mitochondria, which can also cause injury to cardiomyocytes.54 We found that Lcn2 overexpression increased Mmp9 expression in macrophages, and Lcn2 KO reduced MMP9 post-MI. Thus, LCN2 may mediate post-MI cardiac injury by regulating cardiomyocyte apoptosis and MMP9 expression.
CARD9 is located in the cytoplasm and, therefore, cannot directly regulate the expression of LCN2. However, the presence of an NF-κB response element in the LCN2 promoter has been reported, and NF-κB is a positive regulator of LCN2 expression.41,42,55 Our study showed that CARD9 mediated NF-κB activation in the heart following MI. When NF-κB activation was inhibited, Card9 overexpression-induced Lcn2 expression in macrophages was diminished, which is consistent with the results of Card9 KO. Our results suggest that CARD9 might regulate Lcn2 expression in macrophages via NF-κB activation.
CARD9 also expresses in neutrophils and dendritic cells in addition to macrophages. CARD9 deficiency impairs neutrophil function in fungal infections and autoimmune diseases.30,56,57 Neutrophil-specific deletion of CARD9 inhibits inflammatory reactions in mice with autoantibody-induced arthritis and dermatitis.30 Neutrophils infiltrate immediately after the onset of MI and are involved in injury and repair processes. We detected much lower CARD9 expression in CD45+ F4/80- cells than in macrophages, indicating that CARD9 mainly regulates macrophage function; however, we cannot exclude the effect of CARD9 on other myeloid cells. CARD9 has been reported to be increased in H9C2 cells and neonatal rat cardiomyocytes treated with hypoxia/reoxygenation or hydrogen peroxide, inhibiting apoptosis and promoting autophagy of cardiomyocytes during myocardial ischemia/reperfusion (I/R).58,59 The differences with our findings may be attributed to the distinct mechanisms of I/R and MI. Indeed, in I/R, oxidative stress owing to consistent ROS production during reperfusion is the primary cause of cardiomyocyte injury; whereas in MI, ischemia induces necrosis and apoptosis of cardiomyocytes, and infiltration of immune cells in the border region contributes to healing or further injury.
Revealing the role of CARD9 and LCN2 in MI provides us potential molecular targets for cardiac ischemic diseases. Because LCN2 is a secreted protein, blocking its effect using antibodies may be a new therapeutic strategy for MI. Several other Card genes have been detected increased post-MI in addition to Card9. A previous study shows that Card3 KO in mice alleviates LV remodeling and dysfunction post-MI and Card3 overexpression in cardiomyocytes has the opposite effects.60 CARD6 and CARD15 protect against pressure overload-induced cardiac hypertrophy.61,62 The effects of other CARD family members in cardiac ischemia can be explored in the future.
In conclusion, the present study demonstrated the vital role of the innate immune protein CARD9 in cardiac injury after MI. CARD9 upregulated Lcn2 expression via NF-κB activation in macrophages, which in turn increased MMP9 expression, contributing to the enlargement of the injured area and adverse remodeling (Supplementary Fig. 12).
Materials and methods
Animals and MI model
Card9 KO mice were constructed as previously described.28 Lcn2 KO mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). CD45.1 strain mice were obtained from Cyagen (Suzhou, Jiangsu, China). The above strains are all on a C57BL/6 genetic background. Ten- to fourteen-week -old Card9 or Lcn2 KO male mice were used, and Wild-type (WT) littermates and C57BL/6 mice were used as controls. All mice could freely get food and water. The experiments were conformed to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Capital Medical University. Experimental MI in mice was induced by ligation of the left anterior descending coronary artery as previously described.63 Anesthesia of mice was achieved with 1.5- 3% isoflurane in 100% oxygen. The same surgery was performed on sham group mice without ligation. Recombinant mouse LCN2 (Sino Biological, Beijing, China) was given to Card9 KO mice via angular vein injection on day -0, -1, and -3 post-MI at a dose of 30 µg per mouse. Mice were sacrificed by carbon dioxide (CO2) at different time points post-MI, and their hearts were harvested for further experiments.
Cell isolation and culture
BMDMs were isolated following protocols we previously described.33 Obtained cells were cultured in RPMI-1640 medium (Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco), penicillin-streptomycin and 50 ng/mL of macrophage colony-stimulating factor (PeproTech, Rocky Hill, NJ). Three days later, the BMDMs were harvested with trypsin and reseeded in culture plates for further use.
The RAW264.7 cells and H9C2 cardiomyocytes were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) with 10% FBS and penicillin-streptomycin. Confluent monolayers of H9C2 cells were harvested by trypsinization and resuspended in the BMDM growth medium at the density of 1×106 cells/mL. Necrotic cardiomyocytes used for macrophage stimulation in vitro were produced by subjecting the cell suspensions to five freeze - thaw cycles in liquid nitrogen and 37 °C.64,65
Necrotic cells were given to WT and Card9 KO BMDMs at a 3:1 ratio (necrotic cells: macrophages), and an equal volume of medium with five freeze - thaw cycles was used for control experiments. Four hours after stimulation, the medium was removed and fresh medium was added. After an additional 20 h of culture, the supernatants of cells were collected for LCN2 detection.
Bone marrow transplantation
BMT was carried out as previously described.66 Briefly, eight- to ten-week-old male recipient mice received total body irradiation at a dose of 10 Gy from a cobalt-60 source. BM cells were isolated from age-matched male donors and suspended in a serum-free RPMI-1640 medium. Four hours after irradiation, a 200 µL aliquot of BM cell suspension containing 1 × 107 cells was injected into recipient mice through the angular vein. The mice were kept with acidified water containing 100 mg/L fluconazole and 100 mg/L levofloxacin after irradiation for 2 weeks and switched to acidified water without antibiotics for an additional 6 weeks. Eight weeks after BMT, the mice were given a MI surgery. CD45.2 strain mice received BM cells from CD45.1 strain were used for BM reconstitution evaluation. Cells with CD45.1 or CD45.2 expression in the blood were analyzed by flow cytometry.
Echocardiographic imaging
Transthoracic echocardiography of mice was carried out with a Vevo 770 system66 (Fujifilm VisualSonics, Ontario, Canada) during anesthesia with 1.5–3% isoflurane in 100% oxygen at 28 days post-MI. Two-dimensional images of the short axis of LV were taken. LV wall thickness and internal diameters were measured for five consecutive beats and averaged in M-mode. Ejection fraction and fractional shortening were calculated.
Gene overexpression and knockdown
A plasmid expressing mouse Card9 with a C-terminal FLAG-tag and a neomycin resistance gene was constructed by GeneChem (Shanghai, China). A plasmid expressing mouse Lcn2 with a C-terminal FLAG-tag was purchased from Sino Biological. siRNA against mouse Slc22a17 (target sequence: 5’-CCATCACAACCTTCTCTGT-3’) was synthesized by Rui Biotech (Guangzhou, China).
For Card9 overexpression, Card9 plasmid transfection into RAW264.7 cells were carried out using Lipofectamine 3000 (Invitrogen), and the same vector expressing GFP was used as a control. The culture medium was discarded and a fresh selective medium containing G418 (Geneticin, Gibco) was added to cells after 24 hours. The selective medium was refreshed every 3 days for a period of 14 days, after which Card9-overexpressing cells were obtained. Equivalent numbers of Card9-overexpressing and control RAW264.7 cells were plated, and the cell culture supernatant was collected after 48 hours for detection of LCN2 levels and treatment of H9C2 cells as conditioned medium. Cells were treated with ammonium pyrrolidinedithiocarbamate (PDTC; Sigma-Aldrich, St. Louis, MO, USA), the NF-κB inhibitor, at 10 μM for 24 hours, and then RNA was isolated and the supernatant was collected for LCN2 detection. The same volume of vehicle was used as control.
For Lcn2 overexpression, RAW264.7 cells were transfected with Lcn2 plasmid using the 4D-Nucleofector X-unit (Lonza, Basel, Switzerland). An empty plasmid was used as a control. Forty-eight hours after transfection, RNA was isolated and overexpression of Lcn2 was confirmed by quantitative PCR (qPCR) analysis.
For Slc22a17 knockdown, WT BMDMs were transfected with si-Slc22a17 and si-negative control using Lipofectamine RNAiMAX Reagent (Invitrogen) and cultured for 24 h. The medium was then refreshed, and recombinant mouse LCN2 was added to the cells at 2 μg/mL 48 h after transfection. After incubation for an additional 24 hours, RNA was isolated and the cell culture supernatant was collected to examine MMP9 protein levels.
Assessment of cardiomyocyte apoptosis
Death of cardiomyocytes at the border region of the infarcted heart was detected using the DeadEndTM Fluorometric TUNEL kit (Promega, Madison, WI, USA).
H9C2 cells were treated with conditioned medium from Card9-overexpressing and control RAW264.7 cells for 48 h, and then their apoptosis was evaluated using TUNEL staining.
Enzyme-linked immunosorbent assay (ELISA)
Plasma was separated from ethylenediaminetetraacetic acid (EDTA) anti-coagulated blood samples of mice by centrifugation at 3000 rpm/min for 10 min at 4 °C. Cell culture medium was subjected to a centrifugation at 2000 ×g for 10 min at 4 °C in a refrigerated centrifuge to collect supernatant. LCN2 levels were determined using mouse LCN2 ELISA kits (LCN2 in plasma: R&D Systems, Minneapolis, MN, USA; LCN2 in cell culture supernatant: Abcam, Cambridge, UK). MMP9 levels in the cell culture supernatants were detected using a mouse MMP9 ELISA kit (RayBiotech, Norcross, GA, USA).
Cardiac cell sorting
Hearts from WT mice 3 days post-MI were perfused with cold PBS, cut into small pieces, and dissociated with collagenase Ia (Sigma-Aldrich) at 37 °C for 30 min to obtain single-cell suspensions, which were then used for fluorescence-activated cell sorting (FACS) and magnetic-activated cell sorting (MACS). For FACS, the cells were incubated with PerCP-Cy5.5-labeled anti-CD45 (550994, BD Biosciences, San Jose, CA, USA) and PE anti-F4/80 (12480182, eBioscience, San Diego, CA, USA) at 4 °C for 30 min in the dark, and the expression of the two molecules was analyzed on FACS Aria III (BD Bioscience). CD45-, CD45+F4/80-, and CD45+F4/80+ cell populations were collected, and CARD9 protein expression in these populations was assayed with western blotting. For MACS, the cells were first incubated with biotin anti-CD45 (13045182, eBioscience) and sorted using the Release Mouse Biotin Positive Selection Kit (STEMCELL, Seattle, WA, USA) to obtain CD45- and CD45+ cells. Subsequently, CD45+ cells were incubated with PE anti- F4/80 and sorted using a mouse PE-positive Selection Kit (STEMCELL) to obtain CD45+F4/80- and CD45+F4/80+ cells. Card9 mRNA level in these cells was evaluated by qPCR.
Flow cytometric analysis
EDTA anti-coagulated whole blood and cardiac single-cell samples were incubated with PE-CF594 anti-CD45.1 (562452) and V500 anti-CD45.2 (562129), which were purchased from BD Biosciences, for 30 min in the dark. Red blood cells were removed with exposure to the lysis buffer for 1 min. Data were collected with an LSR Fortessa cell analyzer (BD Biosciences).
Quantitative PCR
Total RNA isolation, complementary DNA synthesis, and qPCR analysis were performed following protocols we previously described.67 The expression of genes was normalized to Gapdh. The primers are shown in Supplementary Table 2.
Histopathology
Heart tissues were fixed, embedded and cut into sections as described previously.33 Subsequently, the cardiac sections were subjected to various staining procedures: hematoxylin and eosin (H&E) staining for morphological observation, Sirius red and Masson’s trichrome staining for fibrosis and infarct area analysis, and wheat germ agglutinin (Sigma-Aldrich) staining for cellular hypertrophy evaluation. For immunohistochemical analysis, sections were labeled with primary antibodies followed by HRP-linked secondary antibodies. Diaminobenzidine tetrahydrochloride was used for the colorimetric reaction. For immunofluorescence, frozen sections were labeled with primary antibodies followed by Alexa Flour 488- or Alexa Flour 555-conjugated secondary antibodies (Invitrogen). Antibodies against Mac3 (sc-19991), MMP2 (sc-10736), and CARD9 (sc-49408) were purchased from Santa Cruz Biotechnology; anti-CD31 (DIA-310) from Dianova; anti-MMP9 (ab38898 and ab283575), neutrophils (ab2557), F4/80 (ab6640), and SLC22A17 (ab124506) from Abcam; anti-α-actinin (A7811) from Sigma-Aldrich; anti-LCN2 (AF1857) from R&D System. Anti-CD31 was diluted to 1:50, and all other primary antibodies were diluted to 1:100. A Nikon ECLIPSE Ni microscope (Nikon, Japan) or a laser confocal microscope (Leica, Buffalo Grove, IL, USA) were used for image capture. ImagePro Plus 3.0 was used for analysis.
Western blotting
Protein from tissues and cells were isolated using RIPA buffer. The protein levels of targets were detected using western blotting method as described in our previous study.33 Primary antibodies against CARD9 (sc-99054) and MMP2 (sc-10736) were obtained from Santa Cruz Biotechnology; MMP3 (ab52915) and MMP9 (ab38898) from Abcam; cleaved caspase 3 (9661 S), caspase 3 (9662 S), phosphorylated NF-κB p65 (3033 S), and NF-κB p65 (8242 S) from Cell Signaling Technology; GAPDH (TA-08) from ZSGB-Bio. The primary antibodies were all diluted to 1:1000. Infrared fluorescence-labeled secondary antibodies were used with a dilution of 1:10000. The membranes were scanned using an Odyssey Infrared Imaging System (Li-Cor Bioscience, Lincoln, NE, USA).
Statistics
All data are presented as the mean ± SEM. Two-tailed unpaired t-test was performed to compare two groups, and one-way or two-way ANOVA were used for multiple group comparisons; the difference was statistically significant if p < 0.05.
Data availability
The raw data of RNA-Seq have been deposited in NCBI GEO under the accession number GSE235257. The data that support the findings of this study is available from the corresponding author upon reasonable request.
References
Virani, S. S. et al. Heart disease and stroke statistics-2021 update: a report from the American heart association. Circulation 143, e254–e743 (2021).
Frangogiannis, N. G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 11, 255–265 (2014).
Jung, K. et al. Endoscopic time-lapse imaging of immune cells in infarcted mouse hearts. Circ. Res. 112, 891–899 (2013).
Heidt, T. et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 115, 284–295 (2014).
Kubota, A. & Frangogiannis, N. G. Macrophages in myocardial infarction. Am. J. Physiol. Cell. Physiol. 323, C1304–C1324 (2022).
Lavine, K. J. et al. The macrophage in cardiac homeostasis and disease: JACC macrophage in CVD series (Part 4). J. Am. Coll. Cardiol. 72, 2213–2230 (2018).
Saxena, A., Russo, I. & Frangogiannis, N. G. Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl. Res. 167, 152–166 (2016).
Gruzdeva, O. et al. Relationship key factor of inflammation and the development of complications in the late period of myocardial infarction in patients with visceral obesity. BMC Cardiovasc. Disor. 17, 36 (2017).
Panizzi, P. et al. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J. Am. Coll. Cardiol. 55, 1629–1638 (2010).
Hu, Y. et al. Class A scavenger receptor attenuates myocardial infarction-induced cardiomyocyte necrosis through suppressing M1 macrophage subset polarization. Basic Res. Cardiol. 106, 1311–1328 (2011).
Ishikawa, S. et al. Apoptosis inhibitor of macrophage depletion decreased M1 macrophage accumulation and the incidence of cardiac rupture after myocardial infarction in mice. PLoS One 12, e0187894 (2017).
Leuschner, F. et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 29, 1005–1010 (2011).
Majmudar, M. D. et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 127, 2038–2046 (2013).
Kelly, D. et al. Plasma tissue inhibitor of metalloproteinase-1 and matrix metalloproteinase-9: novel indicators of left ventricular remodelling and prognosis after acute myocardial infarction. Eur. Heart J. 29, 2116–2124 (2008).
Ducharme, A. et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Invest. 106, 55–62 (2000).
Lindsey, M. L. et al. Selective matrix metalloproteinase inhibition reduces left ventricular remodeling but does not inhibit angiogenesis after myocardial infarction. Circulation 105, 753–758 (2002).
Jayasankar, V. et al. Inhibition of matrix metalloproteinase activity by TIMP-1 gene transfer effectively treats ischemic cardiomyopathy. Circulation 110, II180–II186 (2004).
Creemers, E. E. et al. Deficiency of TIMP-1 exacerbates LV remodeling after myocardial infarction in mice. Am. J. Physiol. Heart. Circ. Physiol. 284, H364–H371 (2003).
Hruz, P. & Eckmann, L. Caspase recruitment domain-containing sensors and adaptors in intestinal innate immunity. Curr. Opin. Gastroenterol. 24, 108–114 (2008).
Jia, X. M. et al. CARD9 mediates Dectin-1-induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity. J. Exp. Med. 211, 2307–2321 (2014).
Blonska, M. et al. The CARMA1-Bcl10 signaling complex selectively regulates JNK2 kinase in the T cell receptor-signaling pathway. Immunity 26, 55–66 (2007).
Hu, A. et al. CARD9 in host immunity to fungal, bacterial, viral, and parasitic infections: An update. Front. Microbiol. 13, 1021837 (2022).
Colonna, M. All roads lead to CARD9. Nat. Immunol. 8, 554–555 (2007).
Hara, H. et al. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol. 8, 619–629 (2007).
Gross, O. et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442, 651–656 (2006).
Roth, S. et al. Rad50-CARD9 interactions link cytosolic DNA sensing to IL-1beta production. Nat. Immunol. 15, 538–545 (2014).
Roth, S. & Ruland, J. Caspase recruitment domain-containing protein 9 signaling in innate immunity and inflammation. Trends Immunol 34, 243–250 (2013).
Hsu, Y. M. et al. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol. 8, 198–205 (2007).
Cao, Z. F. et al. Ubiquitin ligase TRIM62 regulates CARD9-mediated anti-fungal immunity and intestinal inflammation. Immunity 43, 715–726 (2015).
Nemeth, T., Futosi, K., Sitaru, C., Ruland, J. & Mocsai, A. Neutrophil-specific deletion of the CARD9 gene expression regulator suppresses autoantibody-induced inflammation in vivo. Nat. Commun. 7, 11004 (2016).
Zeng, X. et al. The essential function of CARD9 in diet-induced inflammation and metabolic disorders in mice. J. Cell. Mol. Med. 22, 2993–3004 (2018).
Yang, Z. W., Meng, X. X., Zhang, C. & Xu, P. CARD9 gene silencing with siRNA protects rats against severe acute pancreatitis: CARD9-dependent NF-B and P38MAPKs pathway. J. Cell. Mol. Med. 21, 1085–1093 (2017).
Liu, Y. et al. CARD9 mediates necrotic smooth muscle cell-induced inflammation in macrophages contributing to neointima formation of vein grafts. Cardiovasc. Res. 108, 148–158 (2015).
Ren, J. et al. Proinflammatory protein CARD9 is essential for infiltration of monocytic fibroblast precursors and cardiac fibrosis caused by Angiotensin II infusion. Am. J. Hypertens. 24, 701–707 (2011).
Peterson, M. R. et al. A potential role of caspase recruitment domain family member 9 (Card9) in transverse aortic constriction-induced cardiac dysfunction, fibrosis, and hypertrophy. Hypertens. Res. 43, 1375–1384 (2020).
Cao, L. et al. CARD9 knockout ameliorates myocardial dysfunction associated with high fat diet-induced obesity. J. Mol. Cell. Cardiol. 92, 185–195 (2016).
Jin, K. et al. Single-cell RNA sequencing reveals the temporal diversity and dynamics of cardiac immunity after myocardial infarction. Small Methods 6, e2100752 (2022).
Dick, S. A. et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 20, 29–39 (2019).
Bajpai, G. et al. Tissue Resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res. 124, 263–278 (2019).
Epelman, S. et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104 (2014).
Zhao, P., Elks, C. M. & Stephens, J. M. The induction of lipocalin-2 protein expression in vivo and in vitro. J. Biol. Chem. 289, 5960–5969 (2014).
Ghosh, S. et al. Activating the AKT2-nuclear factor-kappaB-lipocalin-2 axis elicits an inflammatory response in age-related macular degeneration. J. Pathol. 241, 583–588 (2017).
Xu, G. et al. Lipocalin-2 induces cardiomyocyte apoptosis by increasing intracellular iron accumulation. J. Biol. Chem. 287, 4808–4817 (2012).
Fan, Q. et al. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation 139, 663–678 (2019).
Yan, X. et al. Dectin-2 deficiency modulates Th1 differentiation and improves wound healing after myocardial infarction. Circ. Res. 120, 1116–1129 (2017).
Raeeszadeh-Sarmazdeh, M., Do, L. D. & Hritz, B. G. Metalloproteinases and their inhibitors: potential for the development of new therapeutics. Cells 9, 1313 (2020).
Jaberi, S. A. et al. Lipocalin-2: Structure, function, distribution and role in metabolic disorders. Biomed. Pharmacother. 142, 112002 (2021).
Hemdahl, A. L. et al. Expression of neutrophil gelatinase-associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 26, 136–142 (2006).
Yan, Q. W. et al. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes 56, 2533–2540 (2007).
Yan, L., Borregaard, N., Kjeldsen, L. & Moses, M. A. The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL). Modulation of MMP-9 activity by NGAL. J. Biol. Chem. 276, 37258–37265 (2001).
Yndestad, A. et al. Increased systemic and myocardial expression of neutrophil gelatinase-associated lipocalin in clinical and experimental heart failure. Eur. Heart J. 30, 1229–1236 (2009).
Nymo, S. H. et al. Serum neutrophil gelatinase-associated lipocalin (NGAL) concentration is independently associated with mortality in patients with acute coronary syndrome. Int. J. Cardiol. 262, 79–84 (2018).
Martinez-Martinez, E. et al. Aldosterone Target NGAL (Neutrophil Gelatinase-Associated Lipocalin) Is Involved in Cardiac Remodeling After Myocardial Infarction Through NFkappaB Pathway. Hypertension 70, 1148–1156 (2017).
Song, E. et al. Holo-lipocalin-2-derived siderophores increase mitochondrial ROS and impair oxidative phosphorylation in rat cardiomyocytes. Proc. Natl. Acad. Sci. USA 115, 1576–1581 (2018).
Iannetti, A. et al. The neutrophil gelatinase-associated lipocalin (NGAL), a NF-kappaB-regulated gene, is a survival factor for thyroid neoplastic cells. Proc. Natl. Acad. Sci. USA 105, 14058–14063 (2008).
Drewniak, A. et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood 121, 2385–2392 (2013).
Drummond, R. A. et al. CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog 11, e1005293 (2015).
Li, Y. et al. CARD9 inhibits mitochondria-dependent apoptosis of cardiomyocytes under oxidative stress via interacting with Apaf-1. Free. Radic. Biol. Med. 141, 172–181 (2019).
Li, Y. et al. CARD9 promotes autophagy in cardiomyocytes in myocardial ischemia/reperfusion injury via interacting with Rubicon directly. Basic. Res. Cardiol. 115, 29 (2020).
Li, L. et al. Regulatory role of CARD3 in left ventricular remodelling and dysfunction after myocardial infarction. Basic. Res. Cardiol. 110, 56 (2015).
Li, L. et al. Caspase recruitment domain 6 protects against cardiac hypertrophy in response to pressure overload. Hypertension 64, 94–102 (2014).
Zong, J. et al. NOD2 deletion promotes cardiac hypertrophy and fibrosis induced by pressure overload. Lab. Invest. 93, 1128–1136 (2013).
Gao, E. et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 107, 1445–1453 (2010).
Sauter, B. et al. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 191, 423–433 (2000).
Lim, D. M. & Wang, M. L. Toll-like receptor 3 signaling enables human esophageal epithelial cells to sense endogenous danger signals released by necrotic Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G91–G99 (2011).
Li, Y. et al. Cardiac fibroblast-specific activating transcription factor 3 protects against heart failure by suppressing MAP2K3-p38 signaling. Circulation 135, 2041–2057 (2017).
Ren, W. et al. The complement C3a-C3aR axis promotes development of thoracic aortic dissection via regulation of MMP2 expression. J. Immunol. 200, 1829–1838 (2018).
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (grant numbers 81300120, 81770250 to Y.L.; 81790622 and 81430050 to J.D.) and Beijing Natural Science Foundation (grant number Z200026 to C.Z.). Card9 KO mice were kindly given by Prof. Xin Lin from Tsinghua University.
Author information
Authors and Affiliations
Contributions
Ya.L., Yu.L., and J.D. conceived and designed the research; Ya.L. carried out and analyzed most of the experiments; Y.S. and J.Z. conducted mouse myocardial infarction operation and some echocardiography; YW helped with research design and animal experiments; M.Z., Y.S., H.L., X.W., and S.G. participates in the pathologic analyses, W.B. and cell culture; M.Z. conducted some sequencing data analysis; C.Z. and P.Y. participates in bone marrow transplantation and flow cytometric assay; C.P. and L.J. helped with data analysis; Ya.L., Yu.L., and J.D. wrote and edited the manuscript. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Shao, Yh., Zhang, Jm. et al. Macrophage CARD9 mediates cardiac injury following myocardial infarction through regulation of lipocalin 2 expression. Sig Transduct Target Ther 8, 394 (2023). https://doi.org/10.1038/s41392-023-01635-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01635-w
