
Abstract
Genome instability has been identified as one of the enabling hallmarks in cancer. DNA damage response (DDR) network is responsible for maintenance of genome integrity in cells. As cancer cells frequently carry DDR gene deficiencies or suffer from replicative stress, targeting DDR processes could induce excessive DNA damages (or unrepaired DNA) that eventually lead to cell death. Poly (ADP-ribose) polymerase (PARP) inhibitors have brought impressive benefit to patients with breast cancer gene (BRCA) mutation or homologous recombination deficiency (HRD), which proves the concept of synthetic lethality in cancer treatment. Moreover, the other two scenarios of DDR inhibitor application, replication stress and combination with chemo- or radio- therapy, are under active clinical exploration. In this review, we revisited the progress of DDR targeting therapy beyond the launched first-generation PARP inhibitors. Next generation PARP1 selective inhibitors, which could maintain the efficacy while mitigating side effects, may diversify the application scenarios of PARP inhibitor in clinic. Albeit with unavoidable on-mechanism toxicities, several small molecules targeting DNA damage checkpoints (gatekeepers) have shown great promise in preliminary clinical results, which may warrant further evaluations. In addition, inhibitors for other DNA repair pathways (caretakers) are also under active preclinical or clinical development. With these progresses and efforts, we envision that a new wave of innovations within DDR has come of age.
Similar content being viewed by others

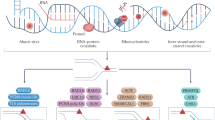
Introduction
Cells are constantly under DNA damage stress posed by endogenous or environmental agents.1,2 A complex DNA damage response (DDR) network has been evolved to maintain the integrity and fidelity of genomic DNA. These DDR networks include DNA repair pathways themselves and a repertoire of regulatory signaling events which are closely related to other cellular processes such as cell cycle, immunogenicity and apoptosis.3,4,5,6,7 Defects in DDR pathways or exposure to carcinogens can lead to accumulated DNA damage and genome instability, which could favor carcinogenesis.8,9 Disrupting DDR processes in cancer cells would aggregate genomic DNA damage and ultimately trigger senescence or programmed cell death.1,7 Now DNA repair defect has been validated as one of the targetable hallmarks in cancer.10
The scenarios for DDR inhibitors in clinic have been portrayed as: synthetic lethality, replication stress, and potentiation of chemo- or radio- therapy.11 Synthetic lethality is described as malfunction in one certain DDR mechanism renders cells more reliant on other somewhat redundant DDR pathways to survive.12,13 Hitherto synthetic lethality remains the only approved strategy in clinic for DDR targeting therapy, such as Poly (ADP-ribose) polymerase (PARP) inhibitors’ success in breast cancer gene (BRCA) mutation or homologous recombination deficient (HRD) solid tumors.14 Replication stress represents a phenomenon that DNA synthesis slows down or replication fork stalls in S phase, which is characterized by extended single strand DNA (ssDNA) exposure.15,16,17 In cancer cells, uncontrolled proliferation, deregulated cell cycle progression or exhausted dNTPs due to nucleotide analog chemotherapy treatment, would cause replication stress. To avoid more catastrophic genome instabilities due to replication stress, ssDNA-bound replication protein A (RPA) would activate the ataxia–telangiectasia and Rad3 related (ATR) - checkpoint kinase 1 (CHK1) - Wee1-like protein kinase (WEE1) - cyclin dependent kinase 1 or 2 (CDK1/2) axis to control the replication firing and arrest cell cycle progress.18 Albeit the intriguing potentiality to use replication stress as predictive biomarkers for ATR, CHK1 or WEE1 inhibitors, more indicative and predictive biomarkers are required to be verified for patient stratification in clinic.11,19 Combination with DNA damage inducing agents such as chemotherapy and radiation is the initial purpose of targeting DDR processes.20,21 However this strategy have been confounded for years because of overlapped toxicity, difficult to dosing, and intolerable damage to normal tissues.22,23
The first-ever DNA repair inhibitor, PARP inhibitor olaparib, was approved in 2014 for the late line treatment of BRCA deficient ovarian cancer24 (Fig. 1). Hitherto at least 6 PARP inhibitors have been launched worldwide, and the indications have been expanded to breast cancer, prostate cancer and pancreatic cancer25 (Table 1). Now the PARP inhibitor development strategy has moved to selectively inhibiting PARP1 which could maintain the efficacy while mitigating side effects.26,27 Beyond PARP, a subset of DNA damage checkpoints have emerged as antitumor targets in clinic, including WEE1, ATR, CHK1, ataxia–telangiectasia mutated (ATM), checkpoint kinase 2 (CHK2), protein kinase membrane associated tyrosine/threonine 1 (PKMYT1), polo-like kinase 1 (PLK1) and even tumor suppressor p53 (Table 2). Inhibitors of WEE1, ATR, CHK1 and PLK1 have also achieved preliminary response in certain types of cancer patients. Recently, small molecule inhibitors of Polymerase theta (Polθ), DNA repair protein RAD51 homolog 1 (RAD51), ubiquitin carboxyl-terminal hydrolase 1 (USP1), poly (ADP-Ribose) glycohydrolase (PARG) and werner syndrome helicase (WRN) were reported, some of which have moved into clinical investigations (Table 2). Concerning the DDR mechanisms and inhibitors have been widely reviewed elsewhere,10,28,29,30,31,32 we embark on the newly progress and recently identified DDR targets and inhibitors in this manuscript. Due to the span of our knowledge, we cannot cover all the progress of targets and inhibitors within DDR space. However inspired by these intriguing progresses and findings, we prospect a new wave of innovations within DDR targets in the near future.

Timeline to show the selected key milestones in DDR mechanism identification, DDR correlation with carcinogenesis and drug discovery. As early as 1775, the linkage between cancer predisposition and environmental insult was observed. However, until 1940s to 1960s, the correlations of carcinogenesis and DNA damage induced by chemicals or radiation became clear with the emergence of molecular biology. Since 1970s, DDR pathways have been depicted as a spectrum of catalytic processes, protein-protein interactions and protein-DNA interactions. Deficiencies in DDR pathways could facilitate carcinogenesis, and can be targeted by small molecule drugs, such as PARP inhibitors’ approval for the treatment of BRCA mutant ovarian cancer. All these efforts eventually led to the grant of Nobel Prize in Chemistry in DDR area in 2015. Now a great deal of interest has been evoked for the study of DDR mechanisms as well as antitumor drug discovery
A historical perspective about DDR and cancer
In 1944, DNA was first-time identified as genetic information carrier (Fig. 1).33 About 10 years later in 1953, Watson and Francis Crick resolved the double helix structure of DNA,34 which laid a foundation for molecular biology as well as DDR mechanistic studies. From 1940s to 1960s, one type of direct reversal repair mechanism, photoreactivation to resolve cyclobutane pyrimidine dimers induced by ultraviolet (UV) exposure, was discovered.35,36,37 Then in 1970s, Tomas Lindahl observed the spontaneous decay of DNA which evoked the ground breaking identification of the first DNA repair enzyme, a uracil DNA glycosylase.38,39,40 Over the following decades, hundreds of proteins involved in kinds of DDR pathways, such as PARP (1980),41 DNA-dependent protein kinase catalytic subunit (DNA-PKcs, 1985),42 ATM (1995),43 CHK1 (1996),44 etc, were identified. In 2015, the Nobel Prize in Chemistry was granted to Tomas Lindahl, Paul Modrich and Aziz Sancar for their seminal study in DNA repair mechanism. Now the underlying mechanisms of DDR including protein-protein interaction, protein-nucleic acids interactions, catalytic processes, are still rapidly evolving.
The first evidence of the correlation between environmental insult and cancer can be traced back to 1775, when Percival Pott linked the predisposition of scrotal cancer to exposure to soot45,46 (Fig. 1). It was widely accepted by 1955 that chemical mutagens could lead to cancer susceptibility by increasing gene mutation rates.47 With the understanding of DNA chemical structure, Phil Lawley and Peter Brooks demonstrated that mustard gas and alkylating agents could form covalent DNA adducts, which impaired normal template functions.48,49 Shortly after that, they further illustrated that polynuclear aromatic hydrocarbons (also a component of tobacco smoke) exposure could result in DNA adducts and facilitate cancer initiation.50 This finding provides strong evidence for the linkage between chemical alterations in DNA and carcinogenesis. The correlation between radiation and cancer was only observed decades after the discovery of X-ray in 1895. A report in 1958 from United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR) concluded that in atomic bomb survivors, radiation-induced mutations were responsible for carcinogenesis51 (Fig. 1).
Defects in DDR genes accounted for dozens of hereditary diseases as well as carcinogenesis (Table 3). In 1969, Jim Cleaver linked the skin cancer predisposition of xeroderma pigmentosum (XP) to unrepaired DNA damage.52 XP patients developed skin cancer at the median age of 8 years53 (Fig. 1 and Table 3). Subsequently, these unrepaired DNA damages were attributed to mutation in NER genes. In 1990s, a colorectal cancer risk factor, Lynch syndrome, was shown to be related to familial mutations in mismatch repair (MMR) proteins54,55,56,57 (Fig. 1 and Table 3). Colorectal cancer patients with defective MMR (dMMR) are characterized by instabilities of simple repeated sequences in their genomes. dMMR has been widely known as microsatellite instability and used as a predictive biomarker for immunotherapy.58,59 Also in 1990s, women with familial mutations in genes BRCA1 and BRCA2 were found to be prone to breast cancer or ovarian cancer.60,61,62 Interestingly, familial mutations in BRCA1 exhibit a different cancer spectrum from BRCA2 mutations.63 BRCA1 mutations are predominately implicated in breast and ovarian cancers,64 whereas BRCA2 mutations are predisposed to breast, prostate, pancreatic, melanoma and ovarian cancers (Table 3). Nowadays BRCA mutations have been validated as biomarkers for patient selection for PARP inhibitors in clinic.25 Hitherto dozens of DDR genes have been identified to be associated with cancer predisposition (Table 3). Their potential use as biomarkers and/or antitumor targets are still under active exploitation.
Caretakers and gatekeepers in DDR
DDR proteins can be roughly classified into caretakers and gatekeepers.65 Caretakers protect the genome DNA by directly repairing DNA damage, while gatekeepers render the DNA repair fine-tuned with cell cycle or cell death.66 Caretakers and gatekeepers cooperatively maintain the genome integrity. Different types of DNA damage activate corresponding repair pathways. Of note, these DDR pathways are partially redundant, which may explain why synthetic lethal interactions are common within DDR proteins.67
Caretakers in DDR include damage sensors, signaling/mediator proteins, and effectors.68 As aforementioned, one of the simplest DNA damage UV-induced cyclobutane pyrimidine dimers can be repaired by light stimulated photolyase proteins (photoreactivation)69,70 (Fig. 2e). Small base modifications such as methylation induced by alkylating agents, oxidants and UV could cause mismatch and mutagenesis.71 Direct reversal repair enzymes can remove base modifications without the help of other proteins (Fig. 2e). For instance, O6-methylguanine DNA methyltransferase (MGMT) demethylates O6-methylguanine lesions through a suicide mechanism, transferring the methyl group to MGMT itself which leads to degradation.72 AlkB human homolog 2 and 3 (ALKBH2 and ALKBH3) directly erase methylation on N1-adenosine and N3-cytosine in a process described as “flip-out”.73,74 Another mechanism to tackle with small base modifications is base excision repair (BER)75,76 (Fig. 2a). DNA glycosylases will sense and remove the damaged base such as 8‑oxoguanine (8‑oxoG) and 5‑hydroxycytosine, leaving abasic sites or known as apurinic or apyrimidinic (AP) sites. Then AP endonuclease 1 (APE1) produce a “nick,” that is a single strand break (SSB). So the downstream effector proteins are shared between BER and SSB repair (SSBR)77,78 (Fig. 2a). The main difference is the sensor protein, as PARP1 recognize SSB in other conditions (for instance, induced by topoisomerase I inhibitors). The remaining BER process can be either short patch (single nucleotide replacement; the predominant way) or long patch (2 to 13 nucleotides replacement), depending on the accessibility of SSB ends. For bulk DNA adducts or crosslinks that would distort helix, nucleotide excision repair (NER) will be activated79,80 (Fig. 2b). Global genome NER (GG‑NER) probes the genome helix distorting lesion and transcription-coupled NER (TC‑NER) removes the lesions blocking transcription. Mismatch repair (MMR) deals with replication errors,81,82 including single nucleotide mismatches as well as nucleotide insertions and deletions (Fig. 2c). Like BER, both NER and MMR are also multiwise ‘cut and patch’ type reactions. Another economic but error-prone way to deal with DNA lesion is translesion synthesis (TLS)83 (Fig. 2d). As high fidelity repair during replication would induce breaks and replication fork collapse, TLS may help restore to duplex DNA and avoid more catastrophic consequences. Fanconi anemia (FA)84,85 pathway is responsible for the repair of interstrand crosslinks (ICLs) (Fig. 2d). FA core complex recognize crosslinks and recruit nucleases to incise the damaged nucleotide. In turn the effector proteins of NER, TLS or HR complete the repair.

DNA repair mechanisms for damages on a single strand or interstrand crosslink. a BER and SSBR share the signaling/mediator proteins (such as XRCC1) and effectors (such as Polβ and TDP), while the major difference is the damage sensors. DNA glycosylases and APE1 deal with small base modifications and generate a nick (SSB). Other SSBs can be directly recognized by PARP1. PARP1 and PARG dynamically modulate PARylation level to regulate SSBR process. b NER deals with bulk damages that distort helix structure. These damages can be either sensed on genome by XPC-RAD23B-CETN2 complex (GG-NER), or during transcription (TC-NER) by CSA-CSB complex which bind to RNA pol II. The following processes of GG-NER and TC-NER are shared. TFIIA-XPA complex recruit endonucleases to remove distorted DNA. Then PCNA in complex with DNA polymerases are loaded to carry out gap-filling synthesis. c MMR corrects mismatches, insertions or deletions during replication. MSH2 heterodimerizes with MSH3 or MSH6 to form sensors of MMR. In turn MLH1-PMS2 and EXO1 cooperate to remove nucleotides including damages. Like NER, PCNA mediates the resting gap-filling synthesis. d ICLs could be recognized by FA core complex. The effectors of ICL repair are shared with TLS, HR or NER. USP1 mediated ubiquitination on FANCD2, FANCI or PCNA could regulate the recruitment of other repair proteins in FA or TLS, respectively. e Direct repair can effectively repair DNA damages by photoreactivation or MGMT, ALKBH2, and ALKBH3 mediated removal of methylated DNA damage without any help from other proteins. Ub ubiquitination; PAR poly (ADP-ribose)
Double strand break (DSB) is the most lethal type of DNA damage, as even one DSB could trigger cell death. 4 major DSB repair (DSBR)86,87 pathways have been identified (Fig. 3): homologous recombination (HR),88 nonhomologous end joining (NHEJ),89 single strand annealing (SSA)90 and Polθ-mediated end joining (TMEJ).91 NHEJ is the predominant but error-prone DSBR pathway, which could bridge DSB ends blunt or with very short overhangs. HR is error-free but only activated in G2 and M phase with the presence of homologous sister chromatin as template. As NHEJ sensor KU70/KU80 heterodimers are abundant in cells, HR sensor MRE11–RAD50–NBS1 (MRN) complex need to outcompete KU70/KU80 in the recognition of DSB ends (Fig. 3). The end resection is carried out bidirectionally from DSB ends. Eventually the long ssDNA overhangs could prevent NHEJ and facilitate HR. TMEJ recognize < 5 base pair (bp) microhomology in ssDNA overhangs after end resection91 (Fig. 3). Albeit error-prone, TMEJ can complete the repair when HR proteins are deficient. SSA can occur between two homologous 3′ ssDNA ends after extensive end resection (Fig. 3). In contrast, short-range end resection is sufficient to facilitate TMEJ.

Major double strand break repair pathways. In G1 phase, the DSBs are recognized by Ku70/80 heterodimers, which recruit DNA-PKcs to form an active DNA-PK. Then DNA-PK complexes with other effector proteins to carry out end processing and ligation. After DNA replication, MRN complex may compete with Ku70/80 in the recognition of DSB ends. Endonuclease activity of MRN form a nick distant away from break point. Then 3’-5’ exonuclease MRN and 5’-3’ exonuclease EXO1 or BLM-DNA2 heterodimer carry out end resection, leaving long ssDNA exposure. MRN can also activate ATM, which phosphorylates MDC1 or γH2AX to amplify the repair signaling. Exposed ssDNA can be coated and protected by RPA, RPA interacts with ATR-ATRIP heterodimer and subsequently ATR kinase activity could be activated. RAD51 in complex with BRCA2 replaces RPA and mediates homology search for HR. If the HR process is deficient, TMEJ and SSA could compensate after end resection. Even <5 bp microhomology is sufficient for activating TMEJ, but long-range homology is required for SSA. P phosphorylation
As with gatekeepers, 3 major DNA damage checkpoints have been depicted in cells: G1/S, intra S, and G2/M checkpoint92,93,94 (Fig. 4). The cell cycle will be arrested to allow DNA repair and avoid the presence of damaged DNA in replication or mitosis. ATR-CHK1-WEE1 axis, ATM-CHK2-p53 axis, PKMYT1, and DNA-PK are the best-known DNA damage checkpoint. PLK1 and aurora kinase A (Aurora-A) are also involved in damage checkpoint regulation. Of note, ATM and ATR orchestrate both DNA damage repair and checkpoint pathways.

DNA damage checkpoints would be activated to by the presence of DNA damage, leading to cell cycle arrest to allow for DNA repair. ATM-CHK2-p53 axis and ATR-CHK1-WEE1 axis will be activated in response to DSBs and ssDNA exposure, respectively. PKMYT1 behaves nonredundantly from WEE1 in regulation of CDK1 activity. Aurora-A and PLK1 are implicated in mitotic entry partially through phosphorylation on WEE1 and PKMYT1 that result in their degradation. Arrows in blue denote phosphatase activity. P phosphorylation
PARP inhibitors
PARP1 is the prominent sensor of SSB or DSB, mediates poly-ADP-ribosylation (PARylation) on PARP1 protein itself and a subset of other substrates95,96,97 (Fig. 2a). Auto-PARylated PARP1 mediates the recruitment of X-Ray repair cross complementing 1 (XRCC1), which orchestrates the following repair process via forming complexes with other proteins.77 Additionally, PARP1 also involves in other repair mechanisms of NER,98 HR,99 TMEJ100 and other physiological processes such as chromatin remodeling,101 transcription,102 DNA replication,103 inflammation,104 metabolism,105 and aging.106 In 2005, two seminal studies demonstrated the hypersensitivity of BRCA1 or BRCA2 mutant cells to PARP1 inhibition,107,108 which paved the way for the approval of PARP1 inhibitors in patients with BRCA mutation. Both BRCA1 and BRCA2 are indispensable components of effective HR, so BRCA1 or BRCA2 mutations are strong indicators of HRD. Now all the 6 approved PARP1 inhibitors (olaparib,24 rucaparib,109 niraparib,110 talazoparib,111 pamiparib,112 and fluzoparib113) have been reckoned as first-generation inhibitors (Fig. 5c), for their dual inhibition to both PARP1 and PARP2, and even off-target activity against other PARPs or other targets.28 Building on the experiences of first-generation inhibitors, PARP1 selective or specific inhibitors (next-generation PARP1 inhibitors), have emerged26 (Fig. 5d).

Paradigm shift in the development of PARP inhibitors. a Cytotoxic mechanisms of PARP inhibitors. PARP inhibitors could disrupt catalytic activity as well as cause PARP trapping on DNA, both leading to unrepaired cytotoxic DNA damage accumulation. b Higher therapeutic index of PARP1 selective inhibitors diversified combo opportunities. The reduced hematological toxicities of next-generation PARP1 selective inhibitors may warrant combinations with chemotherapy, while drugs with nonoverlapped toxicities may be better combo choice for first-generation PARP inhibitors. AUC: area under curve. c Chemical structures of 6 launched first-generation PARP inhibitors: olaparib, rucaparib, niraparib, talazoparib, fluzoparib and pamiparib. d PARP1 selective inhibitors AZD5305 and AZD9574
First-generation PARP inhibitors
All the approved first-generation PARP inhibitors are nicotinamide adenine dinucleotide (NAD+) competitive inhibitors of both PARP1 and PARP2,14,25 some of which even inhibit other PARP subtypes. Moreover, these inhibitors also trap PARP1/2 protein to genome DNA due to attenuated auto-PARylation of PARP1/2, which is reminiscent of topoisomerase inhibitors114,115,116 (Fig. 5a). To some extent the trapping capability of PARP inhibitors dominates over enzyme inhibition in the contribution to efficacy and toxicity117 (Table 4). Initially, it was hypothesized that SSBs induced by PARP inhibition would convert into DSBs that rely on BRCA1 and BRCA2 to repair. However several studies found that PARP inhibitors failed to accumulate SSBs even in BRCA mutant cells.118,119 This led to other 2 models to explain synthetic lethal mechanism between PARP1 and BRCA1/2. One is that trapped PARP would interfere with replication fork and elicit its collapse and DSBs in S phase, then HR repair is activated to resolve damages. Another model anticipates that PARP restart the stalled replication fork in a different way from HR. As with first-generation PARP inhibitors, although their enzymatic inhibition activities are comparable, the trapping activities and cytotoxic effects are significantly different.120,121 By mechanism, trapping activity may result from attenuated auto-PARyaltion, allosteric effect, and other reasons122 (Fig. 5a). Interestingly trapping abilities are inversely correlated with maximal tolerated dose (MTD) in clinic.123,124 For example, trapping activity of talazoparib is 100 fold more potent than olaparib (Table 4), the clinical monotherapy dose of talazoparib is 1 mg QD (once a day) whereas olaparib is 300 mg BID (twice a day).
PARP inhibitors have been approved for the treatment of ovarian cancer, HER2-negative breast cancer, pancreatic ductal adenocarcinoma (PDAC) and metastatic castration-resistant prostate cancer (mCRPC) (Table 1). Recently, olaparib, rucuparib and niraparib voluntarily withdraw the indication of late-line treatment therapy for ovarian cancer patients, due to the potential detrimental effect on patient overall survival. The approvals of PARP inhibitors for the treatment of late line ovarian cancer were largely based on objective response rate (ORR) and median duration of response (mDOR), and median overall survival (mOS) data have not yet matured at that time.125,126,127 Of note, median progression free survival (mPFS) but not mOS are primary endpoints for most clinical trials of PARP inhibitors, which have supported other approvals in ovarian cancer. However, the recent updates of PARP inhibitors as maintenance therapy seem more promising.128,129,130,131,132 For example, in BRCA mutant ovarian cancer patients responsive to first-line platinum-based chemotherapy,133 olaparib maintenance therapy prolonged both mPFS (56.0 vs 13.8 months, hazard ratio: 0.33) and mOS (not reached vs 75.2 months, hazard ratio: 0.55) than placebo control.
The biomarkers for patient selection in early approvals were predominately BRCA mutations (Table 1). Subsequently, HRD including not only BRCA but other HR repair gene deficiencies could benefit from PARP inhibitors in ovarian cancer or prostate cancer.130,134,135,136 Furthermore, in platinum sensitive ovarian cancer patients, olaparib, rucaparib and niraparib extended mPFS irrespective of BRCA or HR repair gene background. Maybe platinum-sensitive patients harbor other vulnerable gene signatures beyond BRCA mutations and HRD. Now first-generation PARP inhibitors are still widely exploited in clinic for more indications, novel combinations and biomarkers.
Next generation PARP1 selective inhibitors
Albeit with their impressive efficacies in clinic, hematological toxicities such as anemia, neutropenia, and thrombocytopenia are common adverse events (AEs) during first-generation PARP inhibitors treatment.137 These safety risks lead to dose discontinuation and reduce combination possibilities with chemotherapy or other kinds of therapies. The discovery of next generation PARP1 selective inhibitors can be attributed to fundamental mechanistic understandings of different PARP proteins. Several studies point out that PARP2 is linked with hematological toxicities,138,139 but PARP1, the primary responder in DDR, is predominately required for efficacy. Double knockout of both PARP1 and PARP2 would impair normal embryonic development.140 Moreover, PARP5A/B inhibition are believed to be responsible for gastrointestinal adverse effects.141 In this sense a PARP1 selective inhibitor may reduce the toxicity whilst maintain the antitumor activity, thus leaving a higher therapeutic index for more combination choice (Fig. 5b).
NMS-03305293 (also known as NMS-P293), the first claimed PARP1 selective inhibitor proceeded to clinical investigation, is developed as a potent PARP1 enzyme inhibitor but not a trapper (Table 4).142 NMS-03305293 selectively suppresses the HR deficient cell growth in vitro and in vivo, accompanied by the significantly PAR reduction. Strikingly, NMS-03305293 could penetrate the blood brain barrier and shows synergistic effect when combined with temozolomide (TMZ) in glioblastoma (GBM) tumor models.143 Now NMS-03305293 is under clinical investigation by Nerviano Medical Sciences in collaboration with Merck (Table 2).
The second PARP1 selective inhibitor in clinic, AZD5305, displays over 400 fold selectivity to PARP2 in a fluorescence polarization competition assay (Table 4 and Fig. 5d).117 Different than NMS-03305293, AZD5305 is also a strong PARP1 trapper.144 In a well-established cell-based trapping assay, AZD5305 selectively trap PARP1 at nanomolar range, which was much more potent than olaparib. Strikingly AZD5305 failed to trap PARP2 even at micromolar concentration. With optimized pharmacokinetics, AZD5305 caused tumor regression at 100 fold lower dosage than olaparib. In addition, AZD5305 retained the selective killing to BRCA deficient cell line both in vitro and in vivo, when compared to BRCA wild type isogenic cells. In rat toxicity studies, AZD5305 demonstrated minimal reduction with respect to reticulocytes, erythroids, neutrohils and platelets, which was ascribed to the avoidance of PARP2 inhibition and less promiscuity in secondary pharmacology.145 Preliminary data from first-in-human clinical trial (PETRA) showed that AZD5305 outcompeted first-generation PARP inhibitors in safety profile.146 As reported, AZD5305 dosage has been escalated to 140 mg daily, with only 3% patients require a dose reduction due to AEs, versus 25% - 53% in patients receiving a full dose of first-generation PARP inhibitors. In addition, AZD5305 has achieved higher steady state Ctrough/target effective concentration (TEC) fold than first-generation PARP inhibitors, even at the starting dose 10 mg daily (Ctrough /TEC: 7.12). Remarkably, patients resistant to prior PARP inhibitor treatment also responded to AZD5305. All these findings warrant a wide therapeutic index of AZD5305 and more combination opportunities in clinic (Table 2).
Recently, AZD9574, another PARP1 selective inhibitor with improved brain penetrant property, initiated first-in-human clinical trial. AZD9574 retained the selectivity and potency of AZD5305 (Table 4 and Fig. 5d), and dramatically regressed tumor growth in both subcutaneous and intracranial models.147 With low P-glycoprotein (P-gp)/breast cancer resistance protein (BCRP) driven efflux, AZD9574 displayed higher Kpuu in both rat (0.31) and monkey (0.79). In parallel, the rat Kpuu of first-generation PARP inhibitors were all < 0.1 and for AZD5305 was < 0.05.148,149 Hence AZD9574 can be explicitly differentiated from other PARP inhibitors as the first brain penetrant PARP1 selective inhibitor and trapper in clinic (Table 2).
DDR gatekeepers as antitumor targets
ATR inhibitors
In human, ATR gene is essential in development and its deficiency resulted in a rare autosomal recessive disorder called Seckel syndrome (Table 3), which is featured by intrauterine growth retardation, microcephaly, and developmental defects.150 ATR kinase belongs to the phosphatidylinositol 3-kinase-related kinase (PIKK) family and functions as the apical responder to ssDNA exposure.151 ssDNAs are abundant in numerous physiological processes including DNA replication, HR, NER, and cancer cells with replication stress. ssDNA-bound RPA recruits ATR in complex with ATR interaction protein (ATRIP) to the sites of replication stress or DNA damage. Upon the loading of Rad9–Rad1–Hus1 (9–1–1) complex to these sites, DNA topoisomerase 2-binding protein 1 (TOPBP1) will be recruited and serves as an allosteric activator of ATR/ATRIP complex (Fig. 3). The activated ATR/ATRIP mediates the phosphorylation of a broad range of substrates involved in DNA repair, control of replication firing, restart of stalled replication fork and cell cycle arrest.152 p53, CHK1, BRCA1, WRN and minichromosome maintenance 2 (MCM2) are among the best-known ATR substrates. The activated CHK1 catalyzes the inhibitory phosphorylation on CDC25 phosphatases and stimulatory phosphorylation on WEE1, which converge on the prevention of CDK1 activation and lead to cell cycle arrest152 (Fig. 4). MCM2 in complex with MCM7 forms a helicase that unwinds the DNA duplex during replication.16 It is suggested that ATR was indispensable for regulating replication in both normal tissues and cancer cells. Concerning cancer cells suffering from replication stress, it may confer a window for pharmacological ATR inhibition.153
Most clinical-stage ATR inhibitors, such as berzosertib, ceralasertib, elimusertib, gartisertib, and camonsertib are all ATP-competitive with highly selectivity over other PIKK members or other kinases, whereas their potencies on ATR are different (Table 5 and Fig. 6a).154 These ATR inhibitors accumulated DNA damage, demonstrated hypersensitivity in ATM mutant cancer cell lines and synergies with radiation, chemotherapy or PARP inhibitors in CDX (cell line derived xenograft) and PDX (patient-derived xenograft) models.155,156,157,158 These preclinical findings are consistent with ATR function in the maintenance of genome integrity, and overlapped downstream effectors of ATR and ATM may imply a synthetic lethal relationship. Furthermore, ATR inhibition can facilitate antitumor microenvironment by reducing PDL1 expression, promoting CD3+ or NK infiltration and activation of nucleic acid sensing pathway.159,160 Thus ATR inhibitors also showed synergistic effect with immune-oncology therapeutics such as anti-PD(L)1 antibodies.161 Interestingly given the differences in ATR potency and physicochemical property, the safety profile of ATR inhibitors as monotherapy in patients illustrated somewhat similarities. Hematological toxicities including anemia, neutropenia, and thrombocytopenia were common in ceralasertib, elimusertib, or camonsertib monotherapy.162,163,164 One exception is berzosertib, which is intravenously administrated once or twice a week.165 No dose-limiting toxicities (DLTs) were observed during berzosertib dose escalation, and the monotherapy recommended phase 2 dosage (RP2D) was determined at 240 mg/m2 due to the limit of infusion volumes. However, relatively lower patient compliance due to intravenous dosing route may impede the possibility for more intensive schedule for berzosertib.

Chemical structures of selected DNA damage checkpoint inhibitors. a ATR inhibitors berzosertib, M4344, elimusertib and ceralasertib. b WEE1 inhibitors adavosertib and azenosertib. c CHK1/2 inhibitor prexasertib. d PKMYT1 inhibitor RP6306
Berzosertib (previously known as M6620 or VX970 or VE822, developed by Vertex and Merck) was the first ATR inhibitor entering clinical investigations166 (Fig. 6a). The preliminary clinical data of berzosertib in combination with chemotherapy have been extensively reported. By combination with cisplatin,165 partial response was observed in 4 out of 31 patients who experienced disease progression following standard therapy. In later-line nonsmall cell lung cancer (NSCLC) patients,167 berzosertib combined with gemcitabine led to an ORR at 10.5% (90% confidence interval (CI), 3.7 – 22.5%) and DCR (disease control rate) at 68.4% (90% CI, 53.9 – 80.7%), respectively. Of note, patients with high tumor mutation burden (TMB) or loss of heterozygosity (LOH) score tended to be more responsive to berzosertib and gemcitabine co-treatment. In platinum-resistant high-grade serous ovarian cancer (HGSOC) (Table 6),168 berzosertib plus gemcitabine significantly prolonged mPFS compared to gemcitabine alone (22.9 vs 14.7 weeks, hazard ratio 0.57, one-sided log-rank test p=0·044). Of note, the safety profiles were comparable in combination group and gemcitabine monotherapy. However, the ORR was lower in combination group, which is uncommon. In the second line small cell lung cancer (SCLC) patients,169 the addition of berzosertib to standard chemotherapy topotecan achieved partial response in 9 out of 25 patients. 17/25 patients experienced tumor regressions. Strikingly, most major AEs can be attributed to topotecan, but not the combination. Now berzosertib is still under active clinical explorations by combination with chemotherapy, radiation, PARP inhibitor or anti-PD(L)1 antibodies (Table 2).
In addition to berzosertib, Vertex and Merck also developed an oral ATR inhibitor gartisertib (also known as M4344) in clinic (Fig. 6a). Gartisertib seemed to be more potent than berzosertib at cellular level.158 Interestingly, cancer cell lines with replication stress (RepStress) and neuroendocrine (NE) gene expression signatures were hypersensitive to gartisertib. RepStress and NE gene expression signatures are of candidate predictive biomarkers to stratify patients for ATR inhibitors.
Ceralasertib (also known as AZD6738, developed by AstraZeneca) is the first oral ATR inhibitor in clinic (Fig. 6a). Ceralasertib was optimized from a lead compound AZ20, with improved solubility and avoidance of CYP3A4 time dependent inhibition.156 Albeit ceralasertib demonstrates efficacy as single agent, the clinical development is centered on combination. For example, in melanoma patients resistant to prior anti-PD1 therapy,170 ceralasertib (dose escalation from 40 mg QD to 240 mg BID) in combination with paclitaxel delivered ORR and DCR at 33.3% (95% CI, 18.0–51.8) and 60.6% (95% CI, 42.1%–77.1%), mPFS and mOS at 3.6 (95% CI, 2.0–5.8) and 7.4 (95% CI, 5.7–11.9) months, respectively. The RP2D was determined at 240 mg BID days 1-14 every 28 days. Interestingly, in another trial treating melanoma patients resistant to prior anti-PD1 therapy,171 ceralasertib at fixed dosage (240 mg BID days 15-28 every 28 days) combined with anti-PDL1 antibody durvalumab generated ORR and DCR at 31.0% and 63.3%, mPFS and mOS at 7.1 (95% CI, 3.6-10.6) and 14.2 (95% CI, 9.3-19.1) months, respectively. Seemingly that the addition of durvalumab to ceralasertib improved the duration of clinical activity than paclitaxel. In advanced gastric cancer (AGS),172 co-treatment of ceralasertib and durvalumab also brought benefit. Of note, AGS patients with loss of ATM expression or HRD benefited more than those with intact ATM or low HR signature. By combination with olaparib, ceralasertib also showed preliminary response in HGSOC,173 SCLC174 and breast cancer.175 Impressively, 6 out of 13 PARP inhibitor resistant HGSOC patients demonstrated partial response upon co-treatment of olaparib and ceralasertib, indicating that ATR inhibition could circumvent PARP inhibitor resistance.173 Recently, ceralasertib in combination with durvalumab initiates a phase 3 study for the treatment of later line NSCLC patients (Table 2).
Elimusertib (also known as BAY1895344, developed by Bayer) was more potent than ceralasertib and berzosertib at cellular level, meanwhile was comparable to M4344158,176 (Fig. 6a). In a CDX model, elimusertib monotherapy outcompeted ceralasertib and berzosertib at their MTD dosages, due to its longer and sufficient exposure above antiproliferative IC50. At the MTD dosage in human (40 mg BID 3 days on/4 days off),163,177 elimusertib brought preliminary single agent benefit to patients in clinic, but only 5 out 143 patients achieved PR. In patients with ATM loss, the ORR was slightly increased to 9% and DCR was 65%. Of note, a less intensive dosing schedule 3 days on/11 days off may help mitigate toxicities. Interestingly, two intermittent strategy, 40 mg/kg, BID, 3 days on/4 days off and 60 mg/kg, BID, 3 days on/11 days off generated comparable efficacy in several CDX models. So a less intensive schedule with enhanced dosage may not sacrifice efficacy and can improve tolerability.178 Now elimusertib is under investigation by combination with chemotherapy, niraparib, or anti-PD1 antibody pembrolizumab (Table 2). And the dosing schedule of elimusertib in the combination scenario may need more explorations.
Likewise, another oral ATR inhibitor camonsertib (also known as RP3500, developed by Repare) reported single-agent activity in clinic.179 The RP2D of camonsertib monotherapy was determined at MTD of 160 mg QD, 3 days on/4 days off.164 In platinum drugs or PARP inhibitors pretreated ovarian cancer, 5 out of 20 benefited from camonsertib monotherapy, including 1 complete response, 3 PRs and 1 CA125 reduction (Table 6). The ORRs were modest in patients harboring ATM (12%) or BRCA (14%) deficiency. For the combination scenario, camonsertib and PARP inhibitors dosed concomitantly 3 days on/4 days off outperformed sequential (PARPi for 3 days followed by camonsertib for 3 days, then 1 day off) in preclinical evaluations. And shortened duration of drug exposure help ameliorate tolerability with minimal effect on red blood cell and reticulocyte.180 Now camonsertib is co-developed by Repare and Roche.
WEE1 inhibitors
WEE1 kinase catalyzes the inhibitory phosphorylation of CDK1 and CDK2 on conservative Tyr15, thereby acting as a G2/M checkpoint and a guardian for DNA replication.181,182 With the presence of DNA damage or uncompleted DNA replication, ATR-CHK1 axis phosphorylates and stimulates WEE1, which in turn inactivates CDK1 to avoid premature mitosis (Fig. 4). Otherwise to override G2/M checkpoint, PLK1-mediated WEE1 phosphorylation promotes WEE1 degradation via the ubiquitin ligase complex. During S phase, WEE1 is implicated in the maintenance of genome integrity through CDK2-regulated replication initiation and Mus81-Eme1 endonuclease mediated processing of stalled replication forks.183,184 In light of its sophisticated functions, WEE1 depletion or inhibition render cancer cells die of replicative or mitotic catastrophe, and hypersensitive to chemotherapy or radiation as expected.185,186
Adavosertib (also known as AZD1775 or MK1775) is the first ATP-competitive WEE1 inhibitor in clinic (Fig. 6b). In preclinical animal studies, adavosertib showed antitumor effect either as single agent or a sensitizer to chemotherapy such as gemcitabine, 5-fluorouracil and platinum drugs.186,187,188 In vitro, the combination with chemotherapy resulted in premature mitosis and mitotic catastrophe, whereas single agent activity of adavosertib was more related to DNA damage accumulation in S phase and replicative catastrophe. Albeit cancer cells with G1 checkpoint dysregulation are believed to be more reliant on G2/M checkpoint to maintain homeostasis, the correlation of adavosertib sensitivity and TP53 status appeared to be controversial.189 Of note, an unbiased mass spectrometry (MS)-based chemical proteomics survey uncovered a set of kinases hit by adavosertib, and adavosertib was equally potent against WEE1 and PLK1.190 Severe AEs especially gastrointestinal toxicities and myelosuppression were observed in patients receiving adavosertib.191,192 Although these AEs were broadly identified in DDR targeting agents, the contributions of off-target kinase inhibition cannot be neglected. Limited by therapeutic index in clinic, the optimal dosage and schedule for adavosertib as monotherapy or in combination with chemotherapy were both determined in unconventional intermittent manners. Even with these challenges, adavosertib achieved preliminary response in various cancer conditions. In patients with recurrent uterine serous carcinoma (USC),193 adavosertib monotherapy brought about an ORR of 29.4% (95%CI 15.1-47.5%), and mPFS and mDOR were determined at 6.1 and 9.0 months, respectively. In high-grade serous ovarian cancer patients that were refractory to or relapse after platinum drugs treatment,194 adavosertib plus gemecitabine extended both mPFS (4.6 vs 3.0 months, hazard ratio 0.55, log-rank p = 0.015) and mOS (11.4 vs 7.2 months, hazard ratio 0·56, log-rank p = 0.017) compared to gemcitabine alone (Table 6). Also in late line ovarian cancer patients who progressed on PARP inhibitor treatment,195 adavosertib monotherapy induced an ORR of 23% (90% CI, 12%-38%), a clinical benefit rate (CBR) of 63% (90% CI, 48%-76%), and mPFS of 5.5 (90% CI, 3.9–6.9) months (Table 6). In another noncomparative arm, adavosertib and olaparib co-treatment delivered an ORR of 29% (90% CI, 14%-44%), a CBR of 89% (90% CI, 76%-96%) and mPFS of 6.8 (90% CI, 4.3–8.3) months. The benefit was achieved irrespective of BRCA background, however, grade 3/4 adverse effects were common in both arms. In locally advanced pancreatic cancer patients,196 adavosertib in combination with gemcitabine and radiation extended mPFS and mOS to 9.4 and 21.4 months respectively, both longer than historical results. Recently, adavosertib was deprioritized by AstraZeneca.
In light of adavosertib experiences, two more selective ATP-competitive WEE1 inhibitors azenosertib (also known as ZnC3, developed by Zentalis)197 and Debio0123 (developed by Debio)198 were under clinical investigations. Although structurally analogous to adavosertib (Fig. 6b), azenosertib was obviously less promiscuous in a panel of kinases. Strikingly, the higher selectivity of azenosertib left a safer AE profile in clinic when compared to adavosertib at similar dosage of 300 mg QD.199,200 Moreover, azenosertib can be dosed continuously while adavosertib had to be intermitted due to safety issues. Likewise, azenosertib obtained preliminary response in USC patients as monotherapy or platinum-resistant ovarian cancer by combination with chemotherapy (Table 6). Recently, azenosertib was shown to be more sensitive in cyclin E1 overexpression ovarian cancer cell line in vitro and in vivo. Cyclin E1 overexpression via CCNE1 amplification or independent mechanisms is quite common in HGSOC patients, which may be employed for responder enrichment in clinic.201 Now Zentalis teams up with Pfizer and GlaxoSmithKline (GSK) for the development of azenosertib (Table 2).
The structure of Debio0123 remains undisclosed. Compared to adavosertib, Debio0123 curtailed the activity against PLK1,202 which may ameliorate tolerability. In a dose-escalation phase 1 trial, target engagement in patients has been confirmed by using skin tissue pCDK1 reduction as a surrogate.203 More patient data have not yet come with respect to safety profile and response of Debio0123. Recently, it is reported that Debio0123 can penetrant blood-brain barrier (BBB) with mean brain-to-plasma concentration ratios of ~0.6 and 1.52 and 4 in mice, rats, and monkeys, respectively.204 Of note, Debio0123 monotherapy or in combination with TMZ produced remarkable efficacy in orthotopic GBM models. A new clinical trial of Debio0123 in combination with TMZ and radiation has just initiated for the treatment of GBM (Table 2).
ATM inhibitors
ATM, another member of PIKK family, plays an integral role in DSB response.205 Mutations in ATM gene are associated with a hereditary genomic instability disorder called ataxia-telangiectasia (Table 3), which is featured by progressive ataxia, telangiectasias, weakened immune system, and hypersensitivity to ionizing radiation.206,207 ATM could be recruited to DSB sites by MRN complex, in turn mediates the phosphorylation of a subset of substrates such as Serine 139 on histone H2AX (referred as γH2AX) and mediator of DNA damage checkpoint 1 (MDC1) to orchestrate DSB response network208 (Fig. 3). Of note, ATM and its well-documented substrate CHK2 both phosphorylated p53, leads to p53 stabilization and G1/S checkpoint activation (Fig. 4). In S phase, activated ATM-CHK2 axis induces phosphorylation and degradation of phosphatase CDC25A. CDC25A is responsible for the removal of inhibitory phosphorylation of CDK2, which is required for DNA replication. Albeit regarded as the most lethal type of DNA damage, DSB is scarce in normal physiological conditions.209 This may explain why ATM was not as essential as ATR to normal cell.
Historically KuDOS Pharma (acquired by AstraZeneca) reported a series of small molecule ATM inhibitors.205 These ATM inhibitors failed to cause cytotoxic effect as single agent, but sensitized cancer cells to DSB inducers such as radiation and topoisomerase inhibitors. In this way, ATM inhibitors should be developed for combination scenarios. Consistent with ATM biological function, ATM inhibitors cannot potentiate DNA alkylating agents, platinum drugs and taxanes in vitro. AZD0156 (developed by AstraZeneca) is the first ATM inhibitor entering clinical evaluations. AZD0156 was of high potency and showed remarkable selectivity over other PIKK family kinases.210 AZD0156 abrogated the DSB repair signaling induced by IR in vitro and showed synergistic effect when combined with IR or isomerase inhibitors in vivo. Of note, AZD0156 also potentiate PARP inhibitor olaparib in PDX models.211 Combination of AZD0156 and olaparib led to enhanced accumulation of cells arrested in G2/M phase and triggered more apoptosis. However severe AEs especially hematological toxicities emerged in patients treated with AZD0156 and olaparib combination.212 We anticipated that systemic administration of AZD0156 and olaparib exacerbated on-target toxicities in blood. Now AZD0156 has been removed from AstraZeneca pipeline.
Other 2 potent and selective ATM inhibitors AZD1390 and M4076 are now under clinical investigations. Compared to AZD0156, AZD1390 demonstrated brain-penetrant capability in both cynomolgus monkey (Kpuu = 0.33)213 and healthy human (Kpuu = 0.24 mL*cm−3, determined by positron emission tomography using radiolabeled AZD1390).214 In mouse intracranial xenograft models, oral administrated AZD1390 dramatically extended survival by combination with radiation and temozolomide. Of interest, glioma cell line screen indicated that cells harboring TP53 mutation were more sensitive to AZD1390 and radiation combination compared to TP53 wildtype.213 This may be attributed to S phase accumulation of TP53 mutant glioma cell lines, which render cells more reliant on HR to repair radiation-induced DSBs. M4076 also displayed synergistic effect when combined with radiation, topoisomerase, and PARP inhibitors in preclinical models, albeit the BBB permeability of M4076 was not reported.215 A recent study illustrated that residual cancer cells which survive oncogene-targeted therapies developed synthetically dependency on ATM, and combination of AZD0156 and osimertinib (a 3rd generation EGFR inhibitor) generated synergistic effect and eradicate residual cancer cells in vivo.216 This may broaden the combination opportunities for ATM inhibitor in clinic.
DNA-PK inhibitors
DNA-PK is the major signaling/mediator protein in NHEJ, the error-prone but default DSBR pathway for cells outside S or G2 phase217,218 (Fig. 3). As a member of PIKK kinase family, DNA-PK enzyme consists of a catalytic subunit (DNA-PKcs) and a regulatory heterodimer Ku (Ku70/Ku80). Ku70/Ku80 heterodimers are abundant in cells, so as to instantly recognize and localize DSB ends which are blunt or with very short ssDNA overhangs. DNA-PKcs is then recruited to the heterodimer to form an active DNA-PK complex (Fig. 3). DNA-PK serves as a scaffold for loading other NHEJ effector proteins, which will complete end processing and ligation processes. Besides, DNA-PKcs involves in other cellular processes such as replication stress response,219 transcription,220 telomere maintenance & capping221,222 and innate immunity.223 Strikingly, deficiencies in DNA-PKcs encoding gene PRKDC dampen T and B cell development and lead to severe combined immunodeficiency (SCID) in mice.224 With its versatile roles in physiological processes, DNA-PKcs may be essential to certain normal tissues.
Targeting DNA-PKcs by siRNA or pharmacological inhibition leads to potentiation of cancer cells to radiation and chemotherapy.225,226 This finding evoked a great deal of interest in DNA-PKcs inhibitor development, some of which have advanced into clinical investigation.227 Unfortunately, most DNA-PKcs inhibitors have been deprioritized from clinical development, including DNA-PKcs selective inhibitors M3814228 and AZD7648.229 Both M3814 and AZD7648 are ATP-competitive inhibitors, and demonstrated selectivity over other PIKK kinases. As expected, M3814 or AZD7648 potentiated radiation and chemotherapy both in vitro and in vivo. AZD7648 was also explored in combination studies with olaparib in cells with ATM deficiency, as ATM deficiency may cause synthetic lethality with DNA-PKcs inhibition. Preliminary clinical data indicated that M3814 was well tolerated as monotherapy, accompanied by limited patient response.230 By combination with radiation, the tolerated dose was lowered for M3814, even though preliminary efficacy was observed.231 With limited information, we cannot precisely rule out the underlying reasons for the discontinuation of M3814 and AZD7648. But the unsatisfactory patient responses and potential competition with ATM inhibitors M4076 and AZD1390 when combined with radiation should be taken into consideration.
CHK1/2 inhibitors
Cell cycle checkpoint CHK1 and CHK2 are key downstream regulators of ATR and ATM, respectively232,233 (Fig. 4). Albeit ATR-CHK1 axis and ATM-CHK2 axis aforementioned are activated by different conditions, substrates and signaling circuities of CHK1 and CHK2 are partially overlapped. With respect to DDR-associated cell cycle regulation, CHK2 is in principle implicated in G1 checkpoint whereas CHK1 is mainly activated in intra S and G2/M checkpoint. CHK1 knockout in mice resulted in early embryonic lethality, in contrast CHK2 knockout mice developed normally, which implies that CHK1 is more essential than CHK2.232 Providing ATR inhibitors are hypersensitive in ATM-deficient conditions, yet the synthetic lethal relationship between CHK1 and CHK2 remain elusive. Based on the extent of CHK2 potency, most clinical-stage CHK1 inhibitors can be classified into CHK1-selective (for example rabusertib and SRA737) or CHK1/2 dual inhibitors (for example AZD7762, PF-477736 and prexasertib).234 Albeit entering clinical investigations for more than a decade, there has been a long track records of deprioritization in the development of CHK1 inhibitors, irrespective of their CHK1/2 selectivity. Notably, prexasertib (Fig. 6c) and LY2880070 are still under active development (Table 2).
As an ATP-competitive inhibitor, prexasertib (also known as ACR-368 or LY2606368) potently inhibited CHK1, and CHK2 to a lesser extent.235 Prexasertib treatment induced replication catastrophe, premature mitosis and apoptosis in cancer cells. In vivo prexasertib inhibited the growth of tumor models with various histological backgrounds and potentiate chemotherapy and PARP inhibitors.236,237,238,239 The intravenous dose of prexasertib in clinic was determined at MTD, 105 mg/m2 once every 14 days.240 The most common treatment-emergent adverse event (TEAE) was grade 4 neutropenia, typically lasting <5 days. In heavily pretreated platinum-resistant high-grade serous ovarian cancer (HGSOC) patients (Table 6),241 monotherapy of prexasertib brought an ORR of 30.7%, and the clinical benefit rate (PR+CR+SD >4 months) was determined at 84.6%. The mPFS and mDOR among PRs were 5.8 and 5.5 months, respectively. As an intravenous and less selective CHK1 inhibitor, only one dose in each cycle may considerably balance compliance, safety and efficacy for prexasertib. Providing that the T1/2 of prexasertib was around 11 - 12 hours, the duration of exposure at RP2D is shortened compared to other DNA damage associated cell cycle checkpoint inhibitors dosing more intensively. Now prexasertib is developed by Acrivon Therapeutics, that employed a diagnosis test for the stratification of patients sensitive to prexasertib.
LY2880070 (also known as ESP-001) is claimed as an oral and selective ATP-competitive CHK1 inhibitor, however to our knowledge preclinical data of LY2880070 is still unavailable. The dosing escalation study of LY2880070 monotherapy compared QD and BID dosing days 1 - 5 in every 21-day cycle in patients.242 Although the AUCs of 200 mg BID (MTD) and 400 QD were comparable, 400 mg QD was not tolerated, which may be ascribed to the enhanced Cmax. As the T1/2 of LY2880070 was as short as 5.35 ± 2.3 hours, the median steady state Cmin of 200 mg BID schedule was enhanced and remained above IC50 for 24 hours. However, the best response of LY2880070 monotherapy was stable disease in 16% patients. LY2880070 was also explored by combination with low dose gemcitabine in advanced/metastatic HGSOC patients243 (Table 6). The RP2D of LY2880070 in this scenario was 50 mg BID days 1 - 5 weekly, which is more intensive than the MTD as monotherapy. As of data reported, 59.3% patients achieved disease control but the ORR was only 7.3%. Now the clinical study of LY2880070 combined with low dose gemcitabine in genetically selected HGSOC subpopulation is conducted by Esperas Pharma. Thus for both prexasertib and LY288070, new biomarkers for patient selection is of extremely importance in future clinical trials.
Recently, a new oral and CHK1 selective inhibitor XS-02 was disclosed.244 In a cell-based CHK1 enzymatic activity analysis, XS-02 showed comparable potency with prexasertib but more potent than LY2880070 and SRA737. In vivo, XS-02 illustrated meaningful antitumor effect in several xenograft models either as single agent or by combination with a PARP inhibitor. XS-02 demonstrated favorable bioavailability and safety profile across species. All in all XS-02 is a new oral CHK1 selective inhibitor with improved potency than LY2880070 and SRA737. The IND filing of XS-02 is expected in second half of 2023.
PKMYT1 inhibitors
PKMYT1 also belongs to WEE1 kinase family that mediates the inhibitory phosphorylation of CDK1245 (Fig. 4). Albeit PKMYT1 and WEE1 are seemingly redundant in negative regulation of CDK1, there are several major discrepancies:246 (i) WEE1 phosphorylates both CDK1 and CDK2 at Tyr15 but PKMYT1 only phosphorylates CDK1 at Thr14; (ii) WEE1 is mainly nuclear-localized, while PKMYT1 is cytoplasmic via a membrane-tether to endoplasmic reticulum and Golgi complex; (iii) PKMYT1 could sequester CDK1 to prevent its entry into nucleus. Importantly, it seems that PKMYT1 was dispensable for normal cell cycle progression, whereas WEE1 was somehow broad essential, given that WEE1 knockout mice died of defective development.247
Recently, a genome-wide clustered regularly interspaced short palindromic repeat (CRISPR) knockout screen revealed that PKMYT1 was synthetic lethal with CCNE1 amplification in cancer cells.248 CCNE1 amplification is prevalent in uterine, ovarian, stomach and other cancer types, which represent an unmet clinical need. Mechanistically, CCNE1 amplification activated the transcription program MMB–FOXM1, which upregulated PKMYT1 substrate, cyclin B – CDK1 complexes. CCNE1 amplification engendered replication stress and extended S phase. In light of these interesting findings, RP6306, a clinical-stage selective PKMYT1 inhibitor was developed249 (Fig. 6d). RP6306 demonstrated biased cytotoxicity to CCNE1 amplification cancer cells, whereas adavosertib was both cytotoxic irrespective of CCNE1 background. RP6306 treatment resulted in activated CDK1, premature mitosis entry and DNA damage, which is reminiscent of WEE1 inhibition by adavosertib. In xenograft animal models harboring CCNE1 amplification or FBXW7 (encode the E3 ubiquitin ligase which degrades CCNE1) loss, RP6306 dramatically inhibited tumor growth either as monotherapy or in combination with gemcitabine. Now the ongoing clinical trials of RP6306 recruit patients with CCNE1 amplification or FBXW7 loss. We wonder whether the different characteristics of PKMYT1 and WEE1 could bring about a wider therapeutic index for RP6306 than adavosertib in clinic.
PLK1 inhibitors
PLK1 is the best studied member of human polo-like serine/threonine kinase family. Like other PLKs, PLK1 is comprised of a C-terminal polo-box domain (PBD) and an N-terminal kinase domain.250,251 PBD domains aid in the localization and substrate recognition of PLK1 within cell. To achieve full activation, PLK1 needs to be phosphorylated by upstream kinase Aurora-A and its cofactor Bora at threonine 210 within T-loop (Fig. 4). The best known physiological function of PLK1 is its role in G2/M phase, including timing of mitotic entry and exit, centrosome regulation, coordination of spindle assembly, correct chromosomal segregation and cytokinesis.252 PLK1 expression is exquisitely regulated throughout cell cycle: upregulated in G2/M phase while keep at low level in interphase.253 PLK1 function in the course of DNA replication,254 DDR255 and DNA damage associated cell cycle checkpoint256 has only been unveiled in last a few years. During replication and especially replicative stress, PLK1 phosphorylates a subset of substrates including origin recognition complex 2 (ORC2), minichromosome maintenance complex 2-7 (MCM2-7) and other components to regulate licensing and firing.257 At the end of replication, cyclin-B1/CDK1 complex facilitates the Aurora-A/Bora complex formation, which in turn activates PLK1. PLK1 then mediates inhibitory phosphorylation on WEE1 and PKMYT1 to promote their degradation and further activation of CDK1258 (Fig. 4). These intertwined feedback loops guarantee the smooth transition from DNA replication to mitosis. In the presence of DNA damages, ATM and ATR mediate phosphorylation and degradation of Bora, which will inhibit PLK1 activity.259 Moreover, PLK1 is also implicated in HR process,260,261 epithelial to mesenchymal transition (EMT),262 autophagy,263 apoptosis264 and even inflammatory response.265 These versatile functions are closely related to cancer initiation and progress, which make PLK1 an attractive target for cancer treatment.266 Two strategies have arisen for the development of PLK1 inhibitors, either targeting PBD domain or kinase domain. Right now, only ATP-competitive inhibitors are under active clinical development.
Volasertib (also known as BI6727, developed by Boehringer Ingelheim) is the most advanced ATP-competitive PLK1 inhibitor in clinic.267 Of note, volasertib potently inhibited PLK1 as well as PLK2 and PLK3, even though to a lesser extent. As PLK2 and PLK3 may function as tumor suppressors,268 this may conflict the antitumor effects of volasertib induced by PLK1 inhibition. In vitro, volasertib showed broad antiproliferation effect in cancer cell lines by inducing G2/M arrest and apoptosis. With favorable intravenous pharmacokinetic profile and high volume of distribution, volasertib demonstrated meaningful in vivo efficacy either as monotherapy or by combination with chemotherapy or radiation.269 The RP2D of volasertib monotherapy in patients was determined at 300 mg per administration every 3 weeks.270 As expected, the most frequent AEs were hematological toxicities including anemia, neutropenia, and thrombocytopenia. However, most reported clinical efficacies of volasertib monotherapy or in combination with other agents in solid tumors were less optimal.271 Though early results of volasertib in combination with low-dose cytarabine (LDAC) in acute myeloid leukemia (AML) patients seems intriguing,272 a following large phase 3 trial failed to reproduce the positive results.
Different from volasertib, onvansertib (also known as NMS1286937, developed by Cardiff Oncology) is an oral and potent ATP-competitive PLK1 inhibitor with high selectivity over PLK2 or PLK3.273,274 Onvansertib also demonstrated broad antiproliferation effects and produces remarkable in vivo efficacy either as single agent or in combination with chemotherapy. In patients, the MTD and RP2D of onvansertib was determined to be 24 mg/m2/day in 5 consecutive dosing followed by a 16-day holiday.275 The monotherapy DLTs were mainly thrombocytopenia and neutropenia, which are consistent with volasertib. Impressively, onvansertib adopted different strategies in the following clinical trials. Consistent with finding from a genome-wide RNA interference (RNAi) screen which identified that PLK1 inhibition is synthetic lethal with KRAS mutation,276 onvansertib showed a biased cytotoxicity to cells carrying KRAS mutation compared to wildtype isogenic.277 As a result, onvansertib is explored in a combination trial with folinic acid, 5-fluorouracil, and irinotecan (FOLFIRI) and bevacizumab (a VEGFR antibody) for treatment of 2nd line KRAS mutant colorectal cancer patients. According to a recent report, the ORR, DCR and mPFS were determined to be 35.4%, 91.7% and 9.3 months respectively, all remarkably better than historical data.278 Of note, the response rate in KRAS responders (≥ 90% decrease in KRAS mutant allele frequency in circulating tumor DNA (ctDNA) after 1 cycle of treatment) was considerably higher than that of KRAS nonresponders. As KRAS mutation hints replication stress, we anticipate that the role of PLK1 in replication may be the underlying mechanism of synthetic lethal relationship. Now onvansertib is also explored by combination with other agents in clinic (Table 2). We are looking forward to more mechanistic studies of PLK1 in disease condition to help patient selection in future.
Aurora-A inhibitors
As the upstream regulator of PLK1, Aurora-A is also an attractive antitumor target.279 Aurora-A, as well as Aurora-B and Aurora C, all belong to Aurora serine/threonine kinase family. These 3 paralogues share a conserved C-terminal kinase domain but the N-terminal domains are varied.280 Upon activation, Aurora kinases will auto-phosphorylate themselves on catalytic T-loop residues. Aurora kinases are all implicated in cell division: Aurora-A is responsible for centrosome maturation and segregation, and spindle assembly in mitosis;281 Aurora-B coordinates microtubule attachments to centrosome and phosphorylates histone H3 (pHH3) in mitosis;282 whereas Aurora-C is mainly expressed in testis and involves in meiosis and embryonic development.283 The different physiological functions of Aurora kinases suggest the necessity for developing selective Aurora-A inhibitors. As aforementioned, Aurora-A can also regulate mitotic entry (Fig. 4). In addition to activating PLK1, Aurora-A also mediates phosphorylation of BRCA1 at serine 308 to promote G2/M transition.284 Moreover, the inhibitory phosphorylation of p73 at serine 235 by Aurora-A leads to abrogation of DNA damage induced apoptotic response and mitotic spindle assembly checkpoint (SAC).285 Like PLK1, Aurora-A expression peaks in G2/M phase but decays in interphase in normal cells. However, Aurora-A overexpression is observed in numerous cancer types irrespective of cell cycle phases.286 In cancer cells, Aurora-A suppresses apoptosis and autophagy, activates the Wnt/β-catenin signaling pathway and promotes EMT.287 Of note, Aurora-A inhibition is synthetic lethal with tumor suppressor gene deficiencies such as RB1, SNF5, SMARCA4 or ARIDA1A.286 Interestingly, Aurora-A is also associated with resistance to EGFR288 or PI3K-mTOR-Akt289 pathway inhibitors, and the addition of Aurora-A inhibitors can circumvent the resistance in preclinical studies. In KRASG12C mutant tumor cells, Aurora-A facilitates the interaction between KRAS and C-RAF and is associated with adaptive reactivation of KRAS after KRASG12C inhibitor treatment.290 Combination of an Aurora-A inhibitor and a KRASG12C inhibitor shows synergistic effect in vitro and in vivo. All these evidence makes Aurora-A an attractive antitumor target.
Alisertib (also known as MLN8237, developed by Millennium) is the most advanced oral and ATP-competitive Aurora-A inhibitor in clinical-stage.291,292 Alisertib showed > 200 fold selectivity over Aurora-B either in both enzymatic and cell based assays. Treatment of alisertib resulted in delayed mitotic entry, accumulation of tetraploid (4N) cells and M phase cells with abnormal mitotic spindles and misaligned chromosomes, which were consistent with Aurora-A physiological functions. Alisertib moderately inhibited or even suppressed in vivo tumor growth in models covering solid tumors and lymphoma. Importantly, even at in vivo MTD dosage, Aurora-B was not inhibited at all as illustrated by no changes in pHH3 in vivo.291 The RP2D of alisertib as single agent in clinic was determined to be 50 mg BID 7 days on/14 days off.293 The main DLTs of alisertib were fatigue, nausea, neutropenia, and stomatitis. Stomatitis may be correlated with benzodiazepine-like structure of alisertib, but not Aurora-A inhibition itself.294 Although with some promising results in several phase1/2 studies, alisertib alone failed to show superiority with respect to efficacy in a large phase 3 clinical trial when compared to chemotherapy for the treatment of peripheral T-cell lymphoma (PTCL) as a single agent.295 Of note, the rates of severe adverse events appeared comparable in both arms. Alisertib was also explored by combination with chemotherapy or other targeting therapy in clinic. However, most of the combinations discontinued or failed due to limited efficacy or intolerability.294 Of note, preliminary results of osimertinib plus alisertib in osimertinib-resistant NSCLC patients was disclosed.296 The benefit in this arm was inferior to another combination of osimertinib and sapanisertib (an mTOR inhibitor), although the TEAEs were comparable for both arms. Overall, it seems difficult to balance risk and benefit for alisertib in patients. Concerning that another highly selective Aurora-A inhibitor LY3295668 has been discontinued,297 we suspect that future development of Aurora-A inhibitors requires more thorough understanding of the role of Aurora-A in tumors.
p53 Y220C reactivators
As the best known tumor suppressor, p53 (encoded by TP53) regulates transcription of a spectrum of genes involved in genome integrity maintenance, cell cycle checkpoint, apoptosis and other physiological processes.298,299 Upon DNA damage, p53 would be phosphorylated and activated by ATR, ATM, CHK1 or CHK2, leading to cell cycle arrest (Fig. 4), DDR gene expression or cell death. TP53 mutation are frequently found in almost 50% of tumor patients.300 These mutations either disrupt the binding to DNA or destabilize p53, and eventually attenuation of p53 function in transcription regulation.301,302 Generally loss of function of tumor suppressors is difficult to target directly, by alternative synthetic lethal strategies are readily employed in these conditions, which is exemplified by ATR or WEE1 inhibitors in TP53 deficient tumors. However in the cases of TP53 mutation, several small molecule reactivators which can restore p53 function have proceeded to clinical trials.303,304 In particular, hot spot mutation Y220C is amenable for selective reactivator development.
p53Y220C accounts for around 1.8% of all p53 mutations, and is broadly detected across various solid tumor types.300 Unlike other hot spot mutations, Y220C locates distant away from DNA binding interface of p53. Y220C inactivates p53 by destabilization of p53 DNA binding domain by around 4 kcal/mol.305 Of the most importance, Y220C left a cavity on p53 surface which can be bound by small molecules.306 A set of binders have been developed,307,308,309 which promoted p53Y220C stability, restored conformation and transcription regulation, and selectively led to TP53Y220C mutant tumor cell apoptosis. In this sense, p53Y220C may be a promising tumor agnostic target.
PC14586 is the first clinical-stage selective p53Y220C reactivator. In preclinical evaluations, PC14586 restored p53Y220C to wildtype conformation, induced p53-regulated gene expression such as p21 and MDM2, and regressed TP53Y220C mutant tumor in vivo.310 Remarkably, in an engineered mouse model carrying TP53Y220C mutation, PC14586 combined an anti-PD1 antibody led to 6 out of 10 complete response and dramatically extended median survival time.311 Recently, preliminary response was reported in patients harboring TP53Y220C mutation during PC14586 dose escalation.312 PC14586 reached maximal tolerated dose at 1500 mg BID with acceptable safety profile. Now the clinical study is still ongoing to determine RP2D for PC14586 (Table 2).
Other DDR targets on the rise
Polθ
Polθ, a 290 kDa protein which contains an N-terminal helicase-like domain (HelD) and a C-terminal polymerase domain (PolD), serves the predominate role in TMEJ91,100 (Fig. 3). Both TMEJ and HR require DSB end resection, whereas unlike HR was only active in the presence of homologous chromatins as template, Polθ can be facilitated by even < 5 bp microhomology in ssDNA overhangs.86 Polθ HelD is an ssDNA-activated ATPase and serves to remove ssDNA-bound RPA, while PolD was responsible for DNA synthesis from microhomology sites. Albeit TMEJ is intrinsically error-prone compared to HR, TMEJ rendered cell survive by avoiding more catastrophic genome aberrations.313 Recently, POLQ down regulation was shown to be synthetic lethal with a group of other DDR gene deficiencies, including those from HR and NHEJ.314 Hence TMEJ is reckoned as the salvage pathway to deficient HR or NHEJ, and Polθ inhibitors was exploited in these conditions, especially HRD.315
Novobiocin316 and ART558,317 which inhibit HelD and PolD, respectively, represent two strategies to disrupt Polθ function. Both novobiocin and ART558 phenocopied POLQ selective dependency in HRD cancer cells. Interestingly, loss of functional 53BP1/Shieldin complex in HRD cells conferred resistance to PARP inhibition, but hypersensitive to Polθ inhibitors.317 Concerning 53BP1 and Shieldin complex channeled NHEJ by preventing DSB end resection, these findings suggested that end resection is indispensable for DSBR choice towards TMEJ. Of note, novobiocin circumvent PARPi resistance in HRD PDX models by monotherapy or in combination with PARP inhibitors. RP6685 was another PolD inhibitor reported by Repare, which selectively killed BRCA2 knockout cancer cell line compared to wildtype isogenic.318 RP6685 enhanced micronuclei and DNA damage marker γH2AX in BRCA2 knockout tumor models. These studies validate the potential of Polθ inhibition in clinic, however targeting which domain will be better is not conclusive so far.
Recently, the first in class Polθ PolD inhibitor ART4215 initiated a phase 2 clinical trial in combination with talazoparib for the treatment of BRCA deficient breast cancer (Table 2). For Polθ HelD domain, Ideaya disclosed an inhibitor, which displayed synergistic effect with niraparib in BRCA deficient model. The first-in-human study of Ideaya’s compound is expected in 2023.
RAD51
RAD51 recombinase is an ATPase and functions as a critical effector in HR.319 After end resection at the DSB sites, the exposed ssDNAs are coated and protected by RPA. Subsequently BRCA2 in complex with RAD51 and other proteins displaces RPA, and RAD51 forms homopolymeric filaments with ssDNA (Fig. 3). Then RAD51 nucleoprotein filament conducts homology search and strand invasion to a sister chromatin, and use it as the template for DNA synthesis. Additionally in cells struggling with replication stress, RAD51 promotes replication fork reversal, inhibits fork degradation and orchestrates break induced replication (BIR).320 Of note, RAD51 strand exchange activity is required for HR and BIR but dispensable for replication fork reversal. Mutations in RAD51 as one type of HRD are related to cancer susceptibility and FA-like syndromes.321 In brief, RAD51 was essential for genome integrity. Different modes of inhibition have been reported for RAD51, including ssDNA binding disruption, oligomerization interference, and inhibition to D-loop formation.322,323,324 All these RAD51 inhibitors showed antiproliferation effect in a range of cell lines and potentiate other antitumor drugs such as cisplatin and topoisomerase inhibitors.
To our knowledge CYT0851 remains the only clinical-stage compound claimed as RAD51 inhibitor (Table 2), albeit the precise mode of inhibition was still undisclosed.325,326 Interestingly, CYT0851 was recently verified as an inhibitor of monocarboxylate transporter which medicates monocarboxylated biomolecules transportation. CYT0851 was optimized from hits identified through a phenotypic screen, which selectively inhibited high activation inducted cytidine deaminase (AID) expression cancer cell growth but spared normal cells with low AID expression. AID stochastically deaminates cytidines throughout genome, leading to point mutation, SSBs and DSBs.327 Ectopic AID is broadly expressed in multiple solid tumor types and nonhodgkin lymphoma (nHL), which confers dependency on HR and RAD51. In AID positive cells, CYT0851 reduced RAD51 foci formation and induced γH2AX expression, showed >30 fold selectivity over AID knockout cells. In vivo CYT0851 suppressed AID positive tumor growth and potentiate a PARP inhibitor. Recently, the clinical data of CYT0851 dose escalation study was reported.328 As a single agent, CYT0851 displayed favorable PK and safety profile, and preliminary response was observed in heavily pre-treated patients, especially nHL. Now the RP2D has been determined, meanwhile dose expansion and combination study is still ongoing (Table 2).
USP1
USP1 in complex with UAF1 is one type of deubiquitinases that regulates FA and TLS through deubiquitination of several platform proteins in these DDR processes329 (Fig. 2d). For instance during FA for the response to interstrand crosslinks induced by platinum drugs, USP1 deubiquitinates monoubiquitinated FANCD2 (ub-FANCD2) and FANCI (ub-FANCI), either of which acts as a scaffold to recruit other repair proteins. While in TLS and DNA replication, USP1 mediates deubiquitination of monoubiquitinated proliferating nuclear antigen (PCNA) which would disrupt unscheduled recruitment of error-prone TLS polymerases such as Polκ and REV1.330 Both Polκ and REV1 would introduce single nucleotide mutations and cause genome instability, in this sense USP1 serves a protective role for genome integrity.
Recently, a genome-wide CRISPR knockout screen identified USP1 to be selective essential in breast & ovarian cancer cell lines harboring HRD, especially BRAC1/2 deficiency.331 It is prospected that BRCA1 deficient cells was characterized by fork instability and cannot tolerate more instabilities by the absence of active USP1.330 By mechanism persistent monoubiquitinated PCNA was responsible for synthetic lethal relationship between USP1 and BRCA1. Thus USP1 inhibitor was anticipated to be effective either as monotherapy in BRCA deficient cells or by combination with other DNA damaging agents including platinum drugs. Interestingly, USP1 with defective autocleavage activity cannot recycle itself from DNA, a phenomenon called USP1 trapping.332 But to our knowledge there has been no USP1 inhibitor claimed USP1 trapping capability so far.
The first in class clinical-stage USP1 inhibitor, KSQ4279, is highly potent (Ki: 1.2 nM) and selective over other deubiquitinases. Interestingly, kinetic analysis indicated that KSQ4279 is allosteric and substrate uncompetitive. KSQ4279 demonstrated hyperactive in cancer cells with HRD or BRCA deficiencies in vitro and in vivo.331 Of note, KSQ4279 led to cell cycle arrest, accumulated DNA damage and replication fork degradation in BRCA1 deficient cells, which is consistent with USP1 biological functions. A CRISPR screen revealed that PCNA loss was associated with KSQ4279 resistance but loss of BER genes including PARP1 conferred hypersensitivity to KSQ4279. Particularly, KSQ4279 potentiate olaparib in PARPi insensitive or partially resistant PDX models. Now KSQ4279 was evaluated in a phase 1 clinical trial (Table 2).
PARG
In contrary to PARP, PARG is responsible for the catabolism of PAR333 and consequently the release of DNA repair complex from genome. Lines of evidence suggest protective roles of PARG in SSBR (Fig. 2a), DSBR and especially replication. As dynamic and stringent regulation of PARylation is indispensable for optimal DDR, PARG is also validated as a potential DDR target for cancer treatment.334 PARG inhibition was shown to be hypersensitive in cells with replication vulnerability, leading to failure to restart stalled replication fork and persistent replication stress.335 Although a set of PARG inhibitors have been reported, none of them reaches clinic to our knowledge. Recently, a clinical candidate PARG inhibitor IDE161, was disclosed by Ideaya. Accordingly, IDE161 behaved a different profile than PARP inhibitors in a panel of cancer cell lines irrespective of HRD, suggesting different dependency of PARG and PARP in cancers. In vivo IDE161 can impressively regress tumor growth even in PARPi-resistant PDX models. IDE161 is expected to enter clinic soon.
WRN
WRN helicase is recently identified as an intriguing synthetic lethal target in dMMR or microsatellite instability high (MSI-H) cancers.336,337,338 In dMMR/MSI-H cancer cells featured by (TA)n-dinucleotide repeat expansions, WRN could unwind non-B DNA cruciform-like structures formed by (TA)n repeats during replication. Otherwise without the presence of functional WRN, the replication fork will be stalled at cruciform-like structures and resulted in DSBs and apoptosis. Given that DNA cruciform-like structures were only detected in MSI-H tumors, WRN is presumed collateral essential in MSI-H rather than MSS (microsatellite stable) tumors. Meanwhile in BRCA2 deficient cells, WRN compensated BRCA2 function in safeguarding genome stability through rescuing the stalled replication forks and suppressing MRE11-mediated fork degradation.339 These cumulative evidence suggest the potential of WRN as a synthetic lethal target and prompt a certain of medicinal efforts for the development of WRN inhibitors.340 However to our knowledge, none of WRN inhibitors has proceeded into clinic hitherto.
Conclusion and future perspective
With decades of antitumor innovations, small molecule drugs such as chemotherapy and targeted therapy have dramatically changed cancer treatment paradigm.341,342,343 Chemotherapy drugs unbiasedly attack essential substances, leaving a narrow or even inverted therapeutic index (TI) in patients. The TIs of targeted therapy are generally high, due to selective targeting oncogenic gene aberrations within cancer cells (EGFR, KRAS, etc), or genes essential in restricted lineages (BTK, BCL2, etc). Another high-TI example is synthetic lethal, which is prevalent within DDR genes exemplified by selective essential of PARP1 in BRCA deficiency or HRD. However, ATR, CHK1 and WEE1 appear to be broad essential for cancer cells across various histological origins.344 For these targets, development strategies cannot simply copy conventional targeted therapy, somehow even akin to chemotherapy. Although the essentiality of DDR genes are different, there are several commonalities can be compiled to enhance the probability of success for DDR targeting therapy.
(i) Enhance selectivity to mitigate off-target toxicities. Dissecting that PARP1 and PARP2 behave differently in the contributions to efficacy and hematological toxicity, PARP1 selective inhibitors with reduced toxicity also achieved higher TEC in clinic.146 Likewise, azenosertib can be dosed continuously with more manageable safety profile compared to adavosetib partially in that azenosertib was more selective in the kinase selectivity profile.197,200 Hence selectivity enhancement can not only reduce off-target toxicity but deepen or prolong on-target inhibition.
(ii) Identify predictive biomarker. The success of PARP inhibitors showcased the power of predictive biomarker. New DDR targets USP1, PKMYT1, and WRN were identified in given gene aberrations. In preclinical evaluations, cell line panel, PDXs, and organoids enable the characterization of responders and nonresponders for a DDR inhibitor.345 For the existed clinical-stage CHK1/2 inhibitor prexasertib, a companion diagnostic test has been employed to select patients in a newly initiated clinical trial (NCT05548296) (Table 2). Concerning on-target toxicity is almost unavoidable, a new mechanism or drug with clear predictive biomarker may help stratify and enrich clinical trial population, so as to widen the therapeutic index.
(iii) Combination with more cancer hallmarks and modalities. Combinations seems to be a permanent topic for DDR targeting therapy. Typically DDR inhibitors were combined with chemotherapy or other DDR inhibitors, which would magnify genome instabilities, lead to enhanced efficacy as well as toxicity. Recently, olaparib plus bevacizumab significantly prolonged mPFS as first line maintenance therapy in ovarian cancer patients with HRD (PAOLA-1 trial).346 The addition of olaparib did not increase the known toxicity of bevacuzumab. In a similar vein, olaparib in combination with abiraterone (a CYP17 inhibitor) boosted benefit for first line metastatic castration-resistant prostate cancer, also with no additional toxicities compared to either drug alone (PROpel trial).347 These evidence hints that DDR targeting therapy can exploit combinations with drugs targeting different cancer hallmarks with nonoverlapped toxicities. Antibody drug conjugate (ADC) can be regarded as tumor-oriented delivery of chemotherapy. Trastuzumab deruxtecan (T-DXd), an ADC composed of anti-HER2 antibody and a cytotoxic topoisomerase I inhibitor, showed synergistic effect both in vitro and in vivo by combination with ceralasertib, adavosertib, AZD1390 or AZD5305.348,349 Of note, no synergistic interaction was observed in the in vitro human bone marrow assay treated with T-DXd combined with ceralasertib or adavosertib. The localized cytotoxic effect of ADC may warrant further investigation in combination with DDR targeting therapy (Table 2).
(iv) New target identification and evaluation. By using phenotypic genome-wide CRISPR knockout screen, new synthetic lethal gene PKMYT1 and USP1 were identified in the condition of CCNE1 amplification and HRD, respectively. It is notable that in recent years CRISPR knockout screen has been broadly applied in the search of synthetic lethal pair, sensitive or resistant biomarkers and new target opportunities.350 The gene essentiality analysis is necessary for the prediction of efficacy and toxicity. Nowadays Cancer Cell Line Encyclopedia (CCLE),351 The Cancer Genome Atlas (TCGA)352 and other databases have empowered the essentiality evaluation in normal tissues, restricted lineages and cancer cells. In particular, the DDR gene associated hereditary disease can also provide a path to delineate physiological function and clinical scenarios for DDR targets.353
As cancer remains as one of the top health threats to humanity, new MOAs (mechanism of actions) and drugs are still of great requirements to improve cancer prognosis. Inspired by the precedent success and thriving advancements, we believe the new wave of innovations targeting DDR network will open up new opportunity to expand the toolkit for antitumor treatment.
References
Roos, W. P., Thomas, A. D. & Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 16, 20–33 (2016).
Voulgaridou, G. P., Anestopoulos, I., Franco, R., Panayiotidis, M. I. & Pappa, A. DNA damage induced by endogenous aldehydes: current state of knowledge. Mutat. Res. 711, 13–27 (2011).
Huang, R. & Zhou, P. K. DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 6, 254 (2021).
Pearl, L. H., Schierz, A. C., Ward, S. E., Al-Lazikani, B. & Pearl, F. M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 15, 166–180 (2015).
Brown, J. S., Sundar, R. & Lopez, J. Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Br. J. Cancer 118, 312–324 (2018).
Chabanon, R. M. et al. Targeting the DNA damage response in immuno-oncology: developments and opportunities. Nat. Rev. Cancer 21, 701–717 (2021).
Surova, O. & Zhivotovsky, B. Various modes of cell death induced by DNA damage. Oncogene 32, 3789–3797 (2013).
Jeggo, P. A., Pearl, L. H. & Carr, A. M. DNA repair, genome stability and cancer: a historical perspective. Nat. Rev. Cancer 16, 35–42 (2016).
Larsen, B. D. et al. Cancer cells use self-inflicted DNA breaks to evade growth limits imposed by genotoxic stress. Science 376, 476–483 (2022).
Pilie, P. G., Tang, C., Mills, G. B. & Yap, T. A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 16, 81–104 (2019).
O’Connor, M. J. Targeting the DNA Damage Response in Cancer. Mol. Cell 60, 547–560 (2015).
Huang, A., Garraway, L. A., Ashworth, A. & Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 19, 23–38 (2020).
Ashworth, A. & Lord, C. J. Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 15, 564–576 (2018).
Lord, C. J. & Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158 (2017).
Gaillard, H., Garcia-Muse, T. & Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 15, 276–289 (2015).
Dobbelstein, M. & Sorensen, C. S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 14, 405–423 (2015).
Forment, J. V. & O’Connor, M. J. Targeting the replication stress response in cancer. Pharmacol. Ther. 188, 155–167 (2018).
Sorensen, C. S. & Syljuasen, R. G. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 40, 477–486 (2012).
Ngoi, N. Y. L., Pham, M. M., Tan, D. S. P. & Yap, T. A. Targeting the replication stress response through synthetic lethal strategies in cancer medicine. Trends Cancer 7, 930–957 (2021).
Begg, A. C., Stewart, F. A. & Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 11, 239–253 (2011).
Goldstein, M. & Kastan, M. B. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 66, 129–143 (2015).
Matulonis, U. A. & Monk, B. J. PARP inhibitor and chemotherapy combination trials for the treatment of advanced malignancies: does a development pathway forward exist? Ann. Oncol. 28, 443–447 (2017).
Martorana, F., Da Silva, L. A., Sessa, C. & Colombo, I. Everything Comes with a Price: The Toxicity Profile of DNA-Damage Response Targeting Agents. Cancers (Basel) 14, 953–974 (2022).
Deeks, E. D. Olaparib: first global approval. Drugs 75, 231–240 (2015).
Curtin, N. J. & Szabo, C. Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat. Rev. Drug Discov. 19, 711–736 (2020).
Ngoi, N. Y. L., Leo, E., O’Connor, M. J. & Yap, T. A. Development of Next-Generation Poly(ADP-Ribose) Polymerase 1-Selective Inhibitors. Cancer J. 27, 521–528 (2021).
Papeo, G. et al. Discovery of 2-[1-(4,4-Difluorocyclohexyl)piperidin-4-yl]-6-fluoro-3-oxo-2,3-dihydro-1H-isoind ole-4-carboxamide (NMS-P118): A Potent, Orally Available, and Highly Selective PARP-1 Inhibitor for Cancer Therapy. J. Med. Chem. 58, 6875–6898 (2015)..
Dias, M. P., Moser, S. C., Ganesan, S. & Jonkers, J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 18, 773–791 (2021).
Shah, S. M. et al. Therapeutic implications of germline vulnerabilities in DNA repair for precision oncology. Cancer Treat. Rev. 104, 102337 (2022).
Cleary, J. M., Aguirre, A. J., Shapiro, G. I. & D’Andrea, A. D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 78, 1070–1085 (2020).
Brandsma, I., Fleuren, E. D. G., Williamson, C. T. & Lord, C. J. Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin. Investig. Drugs 26, 1341–1355 (2017).
Lord, C. J. & Ashworth, A. The DNA damage response and cancer therapy. Nature 481, 287–294 (2012).
Portin, P. The birth and development of the DNA theory of inheritance: sixty years since the discovery of the structure of DNA. J. Genet. 93, 293–302 (2014).
Franklin, R. E. & Gosling, R. G. Evidence for 2-chain helix in crystalline structure of sodium deoxyribonucleate. Nature 172, 156–157 (1953).
Setlow, R. B. & Carrier, W. L. The Disappearance of Thymine Dimers from DNA: An Error-Correcting Mechanism. Proc. Natl Acad. Sci. USA 51, 226–231 (1964).
Cleaver, J. R. & Painter, R. B. Evidence for repair replication of HeLa cell DNA damaged by ultraviolet light. Biochim. Biophys. Acta 161, 552–554 (1968).
Schuster, R. C. Dark Repair of Ultraviolet Injury in E. Coli during Deprivation of Thymine. Nature 202, 614–615 (1964).
Lindahl, T. & Andersson, A. Rate of chain breakage at apurinic sites in double-stranded deoxyribonucleic acid. Biochemistry 11, 3618–3623 (1972).
Lindahl, T. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc. Natl Acad. Sci. USA 71, 3649–3653 (1974).
Lindahl, T. & Nyberg, B. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry 13, 3405–3410 (1974).
Durkacz, B. W., Omidiji, O., Gray, D. A. & Shall, S. (ADP-ribose)n participates in DNA excision repair. Nature 283, 593–596 (1980).
Walker, A. I., Hunt, T., Jackson, R. J. & Anderson, C. W. Double-stranded DNA induces the phosphorylation of several proteins including the 90 000 mol. wt. heat-shock protein in animal cell extracts. EMBO J. 4, 139–145 (1985).
Savitsky, K. et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749–1753 (1995).
Walworth, N. C. & Bernards, R. rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science 271, 353–356 (1996).
Hall, E. J. From chimney sweeps to oncogenes: the quest for the causes of cancer. Radiology 179, 297–306 (1991).
Waldron, H. A. A brief history of scrotal cancer. Br. J. Ind. Med. 40, 390–401 (1983).
Burdette, W. J. The significance of mutation in relation to the origin of tumors: a review. Cancer Res. 15, 201–226 (1955).
Brookes, P. & Lawley, P. D. The reaction of mustard gas with nucleic acids in vitro and in vivo. Biochem. J. 77, 478–484 (1960).
Brookes, P. & Lawley, P. D. The reaction of mono- and di-functional alkylating agents with nucleic acids. Biochem. J. 80, 496–503 (1961).
Brookes, P. & Lawley, P. D. Evidence for the Binding of Polynuclear Aromatic Hydrocarbons to the Nucleic Acids of Mouse Skin: Relation between Carcinogenic Power of Hydrocarbons and Their Binding to Deoxyribonucleic Acid. Nature 202, 781–784 (1964).
Lindell, B. & Sowby, D. The 1958 UNSCEAR report. J. Radiol. Prot. 28, 277–282 (2008).
Cleaver, J. E. Xeroderma pigmentosum: a human disease in which an initial stage of DNA repair is defective. Proc. Natl Acad. Sci. USA 63, 428–435 (1969).
Black, J. O. Xeroderma Pigmentosum. Head. Neck Pathol. 10, 139–144 (2016).
Leach, F. S. et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 75, 1215–1225 (1993).
Fishel, R. et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 75, 1027–1038 (1993).
Bronner, C. E. et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 368, 258–261 (1994).
Papadopoulos, N. et al. Mutation of a mutL homolog in hereditary colon cancer. Science 263, 1625–1629 (1994).
Marabelle, A. et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 38, 1–10 (2020).
Mandal, R. et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364, 485–491 (2019).
Castilla, L. H. et al. Mutations in the BRCA1 gene in families with early-onset breast and ovarian cancer. Nat. Genet. 8, 387–391 (1994).
Wooster, R. et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 378, 789–792 (1995).
Easton, D. F., Bishop, D. T., Ford, D. & Crockford, G. P. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 52, 678–701 (1993).
King, M. C., Marks, J. H. & Mandell, J. B. New York Breast Cancer Study, G. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302, 643–646 (2003).
Ford, D., Easton, D. F., Bishop, D. T., Narod, S. A. & Goldgar, D. E. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 343, 692–695 (1994).
Deininger, P. Genetic instability in cancer: caretaker and gatekeeper genes. Ochsner J. 1, 206–209 (1999).
Matthews, H. K., Bertoli, C. & de Bruin, R. A. M. Cell cycle control in cancer. Nat. Rev. Mol. Cell. Biol. 23, 74–88 (2022).
Setton, J. et al. Synthetic lethality in cancer therapeutics: the next generation. Cancer Discov. 11, 1626–1635 (2021).
Brown, J. S., O’Carrigan, B., Jackson, S. P. & Yap, T. A. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discov. 7, 20–37 (2017).
Sancar, A. Mechanisms of DNA Repair by Photolyase and Excision Nuclease (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 55, 8502–8527 (2016).
Ramirez-Gamboa, D. et al. Photolyase production and current applications: a review. Molecules 27, 5998–6014 (2022).
Mishina, Y., Duguid, E. M. & He, C. Direct reversal of DNA alkylation damage. Chem. Rev. 106, 215–232 (2006).
Bai, P. et al. The dual role of DNA repair protein MGMT in cancer prevention and treatment. DNA Repair (Amst.) 123, 103449 (2023).
Yang, C. G. et al. Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature 452, 961–965 (2008).
Li, Q. et al. Rhein Inhibits AlkB Repair Enzymes and Sensitizes Cells to Methylated DNA Damage. J. Biol. Chem. 291, 11083–11093 (2016).
Dianov, G. L. & Hubscher, U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 41, 3483–3490 (2013).
Almeida, K. H. & Sobol, R. W. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. DNA Repair (Amst.) 6, 695–711 (2007).
Caldecott, K. W. DNA single-strand break repair and human genetic disease. Trends Cell Biol. 32, 733–745 (2022).
Caldecott, K. W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631 (2008).
Scharer, O. D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 5, a012609 (2013).
Marteijn, J. A., Lans, H., Vermeulen, W. & Hoeijmakers, J. H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell. Biol. 15, 465–481 (2014).
Li, G. M. Mechanisms and functions of DNA mismatch repair. Cell Res. 18, 85–98 (2008).
Iyer, R. R., Pluciennik, A., Burdett, V. & Modrich, P. L. DNA mismatch repair: functions and mechanisms. Chem. Rev. 106, 302–323 (2006).
Sale, J. E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 5, a012708 (2013).
Nepal, M., Che, R., Zhang, J., Ma, C. & Fei, P. Fanconi anemia signaling and cancer. Trends Cancer 3, 840–856 (2017).
Clauson, C., Scharer, O. D. & Niedernhofer, L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol. 5, a012732 (2013).
Scully, R., Panday, A., Elango, R. & Willis, N. A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 20, 698–714 (2019).
Chapman, J. R., Taylor, M. R. & Boulton, S. J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47, 497–510 (2012).
Wright, W. D., Shah, S. S. & Heyer, W. D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 293, 10524–10535 (2018).
Zhao, B., Rothenberg, E., Ramsden, D. A. & Lieber, M. R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 21, 765–781 (2020).
Rossi, M. J., DiDomenico, S. F., Patel, M. & Mazin, A. V. RAD52: Paradigm of Synthetic Lethality and New Developments. Front. Genet. 12, 780293 (2021).
Ramsden, D. A., Carvajal-Garcia, J. & Gupta, G. P. Mechanism, cellular functions and cancer roles of polymerase-theta-mediated DNA end joining. Nat. Rev. Mol. Cell Biol. 23, 125–140 (2022).
Suski, J. M., Braun, M., Strmiska, V. & Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 39, 759–778 (2021).
Groelly, F. J., Fawkes, M., Dagg, R. A., Blackford, A. N. & Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 23, 78–94 (2023).
Branzei, D. & Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 9, 297–308 (2008).
Bai, P. Biology of Poly(ADP-Ribose) polymerases: the factotums of cell maintenance. Mol. Cell 58, 947–958 (2015).
Vyas, S. et al. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun. 5, 4426 (2014).
Ray Chaudhuri, A. & Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621 (2017).
Robu, M. et al. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc. Natl Acad. Sci. USA 110, 1658–1663 (2013).
Haince, J. F. et al. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 283, 1197–1208 (2008).
Mateos-Gomez, P. A. et al. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257 (2015).
Sinha, S., Molla, S. & Kundu, C. N. PARP1-modulated chromatin remodeling is a new target for cancer treatment. Med. Oncol. 38, 118 (2021).
Jubin, T. et al. Poly ADP-ribose polymerase-1: Beyond transcription and towards differentiation. Semin. Cell Dev. Biol. 63, 167–179 (2017).
Bryant, H. E. et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615 (2009).
Kunze, F. A. & Hottiger, M. O. Regulating Immunity via ADP-Ribosylation: Therapeutic Implications and Beyond. Trends Immunol. 40, 159–173 (2019).
Bai, P. & Canto, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 16, 290–295 (2012).
Vida, A., Abdul-Rahman, O., Miko, E., Brunyanszki, A. & Bai, P. Poly(ADP-Ribose) Polymerases in Aging - Friend or Foe? Curr. Protein Pept. Sci. 17, 705–712 (2016).
Farmer, H. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005).
Bryant, H. E. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005).
Syed, Y. Y. Rucaparib: First global approval. Drugs 77, 585–592 (2017).
Scott, L. J. Niraparib: First global approval. Drugs 77, 1029–1034 (2017).
Hoy, S. M. Talazoparib: First global approval. Drugs 78, 1939–1946 (2018).
Markham, A. Pamiparib: First approval. Drugs 81, 1343–1348 (2021).
Lee, A. Fuzuloparib: First approval. Drugs 81, 1221–1226 (2021).
Pommier, Y., O’Connor, M. J. & de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 8, 362ps317 (2016).
Murai, J. et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 72, 5588–5599 (2012).
Shen, Y., Aoyagi-Scharber, M. & Wang, B. Trapping Poly(ADP-Ribose) polymerase. J. Pharmacol. Exp. Ther. 353, 446–457 (2015).
Krastev, D. B., Wicks, A. J. & Lord, C. J. PARP inhibitors - Trapped in a toxic love affair. Cancer Res. 81, 5605–5607 (2021).
Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 5, 387–393 (2011).
Strom, C. E. et al. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 39, 3166–3175 (2011).
Murai, J. et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 13, 433–443 (2014).
Mariappan, L., Jiang, X. Y., Jackson, J. & Drew, Y. Emerging treatment options for ovarian cancer: focus on rucaparib. Int. J. Women’s Health 9, 913–924 (2017).
Zandarashvili, L. et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 368, eaax6367 (2020).
LaFargue, C. J., Dal Molin, G. Z., Sood, A. K. & Coleman, R. L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 20, e15–e28 (2019).
Brown, J. S., Kaye, S. B. & Yap, T. A. PARP inhibitors: the race is on. Br. J. Cancer 114, 713–715 (2016).
Kim, G. et al. FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 21, 4257–4261 (2015).
Balasubramaniam, S. et al. FDA approval summary: rucaparib for the treatment of patients with deleterious BRCA mutation-associated advanced ovarian cancer. Clin. Cancer Res. 23, 7165–7170 (2017).
Moore, K. N. et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 20, 636–648 (2019).
Poveda, A. et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 22, 620–631 (2021).
Mirza, M. R. et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 375, 2154–2164 (2016).
Coleman, R. L. et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 1949–1961 (2017).
Sabatier, R. et al. Efficacy and safety of maintenance olaparib and bevacizumab in ovarian cancer patients aged >/=65 years from the PAOLA-1/ENGOT-ov25 trial. Eur. J. Cancer 181, 42–52 (2023).
Moore, K. et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 379, 2495–2505 (2018).
Bellier, C. et al. Olaparib First-Line Maintenance Monotherapy in BRCA-Mutated Epithelial Ovarian Cancer: Descriptive Analysis of the First French Real-World Data Study. Drugs Real World Outcomes https://doi.org/10.1007/s40801-022-00349-9 (2023).
Hodgson, D. R. et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 119, 1401–1409 (2018).
Mateo, J. et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 373, 1697–1708 (2015).
Abida, W. et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase II TRITON2 Study. Clin. Cancer Res. 26, 2487–2496 (2020).
Leo, E. et al. A head-to-head comparison of the properties of five clinical PARP inhibitors identifies new insights that can explain both the observed clinical efficacy and safety profiles. Proc. Am. Assoc. Cancer Res. 78, LB-273 (2018).
Farres, J. et al. PARP-2 sustains erythropoiesis in mice by limiting replicative stress in erythroid progenitors. Cell Death Differ. 22, 1144–1157 (2015).
Farres, J. et al. Parp-2 is required to maintain hematopoiesis following sublethal gamma-irradiation in mice. Blood 122, 44–54 (2013).
Menissier de Murcia, J. et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 22, 2255–2263 (2003).
Zhong, Y. et al. Tankyrase inhibition causes reversible intestinal toxicity in mice with a therapeutic index < 1. Toxicol. Pathol. 44, 267–278 (2016).
Alessia, M. et al. NMS-P293, a novel potent and selective PARP-1 inhibitor with high antitumor efficacy and tolerability. Proc. Am. Assoc. Cancer Res. 76, 1223 (2016).
Montagnoli, A. et al. NMS-P293, a PARP-1 selective inhibitor with no trapping activity and high CNS penetration, possesses potent in vivo efficacy and represents a novel therapeutic option for brain localized metastases and glioblastoma. Proc. Am. Assoc. Cancer Res. 78, 4843 (2018).
Illuzzi, G. et al. Preclinical characterization of AZD5305, a next generation, highly selective PARP1 inhibitor and trapper. Clin. Cancer Res. 28, 4724–4736 (2022).
Gill, S. J. et al. The novel PARP1-selective inhibitor AZD5305 has reduced hematological toxicity when compared to PARP1/2 inhibitors in pre-clinical models. Proc. Am. Assoc. Cancer Res. 81, 1374 (2021).
Yap, T. A. et al. PETRA: First in class, first in human trial of the next generation PARP1-selective inhibitor AZD5305 in patients (pts) with BRCA1/2, PALB2 or RAD51C/D mutations. Proc. Am. Assoc. Cancer Res. 82, CT007 (2022).
Ghosh, A. et al. Structure-based and property-based drug design of AZD9574, a CNS penetrant PARP1 selective inhibitor and trapper. Proc. Am. Assoc. Cancer Res. 82, 6302 (2022).
Jamal, K. et al. AZD9574 is a novel, brain penetrant PARP-1 selective inhibitor with activity in an orthotopic, intracranial xenograft model with aberrant DNA repair. Proc. Am. Assoc. Cancer Res. 82, 2609 (2022).
Andy, P. et al. Evaluation of the CNS penetration of a next generation PARP inhibitor, AZD9574, in cynomolgus monkey using positron emission tomography. Proc. Am. Assoc. Cancer Res. 82, 5076 (2022).
Lecona, E. & Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 18, 586–595 (2018).
Saldivar, J. C., Cortez, D. & Cimprich, K. A. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 18, 622–636 (2017).
Cimprich, K. A. & Cortez, D. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 (2008).
Karnitz, L. M. & Zou, L. Molecular pathways: targeting ATR in cancer therapy. Clin. Cancer Res. 21, 4780–4785 (2015).
Barnieh, F. M., Loadman, P. M. & Falconer, R. A. Progress towards a clinically-successful ATR inhibitor for cancer therapy. Curr. Res. Pharmacol. Drug Discov. 2, 100017 (2021).
Knegtel, R. et al. Rational Design of 5-(4-(Isopropylsulfonyl)phenyl)-3-(3-(4-((methylamino)methyl)phenyl)isoxazol-5-yl)pyrazin-2-amine (VX-970, M6620): Optimization of Intra- and Intermolecular Polar Interactions of a New Ataxia Telangiectasia Mutated and Rad3-Related (ATR) Kinase Inhibitor. J. Med. Chem. 62, 5547–5561 (2019).
Foote, K. M. et al. Discovery and characterization of AZD6738, a potent inhibitor of ataxia telangiectasia mutated and rad3 related (ATR) kinase with application as an anticancer agent. J. Med. Chem. 61, 9889–9907 (2018).
Lucking, U. et al. Damage incorporated: discovery of the potent, highly selective, orally available ATR Inhibitor BAY 1895344 with favorable pharmacokinetic properties and promising efficacy in monotherapy and in combination treatments in preclinical tumor models. J. Med. Chem. 63, 7293–7325 (2020).
Jo, U. et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells With Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol. Cancer Ther. 20, 1431–1441 (2021).
Feng, X. et al. ATR inhibition potentiates ionizing radiation-induced interferon response via cytosolic nucleic acid-sensing pathways. EMBO J. 39, e104036 (2020).
Vendetti, F. P. et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J. Clin. Invest. 128, 3926–3940 (2018).
Wengner, A. M. et al. Synergistic activity of the ATR inhibitor BAY1895344 in combination with immune checkpoint inhibitors in preclinical tumor models. Proc. Am. Assoc. Cancer Res. 79, 272 (2019).
Dillon, M. T. et al. CT084: A Phase I dose-escalation study of ATR inhibitor monotherapy with AZD6738 in advanced solid tumors (PATRIOT Part A). Proc. Am. Assoc. Cancer Res. 77, CT084 (2017).
Yap, T. A. et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 11, 80–91 (2021).
Fontana, E. et al. 5MO Comprehensive dose-finding strategy for single-agent RP-3500, a highly selective inhibitor of ataxia telangiectasia and Rad3-related (ATR) kinase. Ann. Oncol. 33, 5MO (2022).
Shapiro, G. I. et al. Phase 1 study of the ATR inhibitor berzosertib in combination with cisplatin in patients with advanced solid tumours. Br. J. Cancer 125, 520–527 (2021).
Gorecki, L., Andrs, M., Rezacova, M. & Korabecny, J. Discovery of ATR kinase inhibitor berzosertib (VX-970, M6620): Clinical candidate for cancer therapy. Pharmacol. Ther. 210, 107518 (2020).
Plummer, R. et al. A phase 1b study evaluating the safety and preliminary efficacy of berzosertib in combination with gemcitabine in patients with advanced non-small cell lung cancer. Lung Cancer 163, 19–26 (2022).
Konstantinopoulos, P. A. et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 21, 957–968 (2020).
Thomas, A. et al. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell 39, 566–579.e567 (2021).
Kim, S. T. et al. Phase I Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination with Weekly Paclitaxel in Refractory Cancer. Clin. Cancer Res. 27, 4700–4709 (2021).
Kim, R. et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Ann. Oncol. 33, 193–203 (2022).
Kwon, M. et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced gastric cancer. J. Immunologist. Cancer 10, e005041 (2022).
Wethington, S. L. et al. Combination of PARP and ATR inhibitors (olaparib and ceralasertib) shows clinical activity in acquired PARP inhibitor-resistant recurrent ovarian cancer. J. Clin. Onco. 39, 5516 (2021).
Sehhoon, P. et al. The clinical efficacy of olaparib monotherapy or combination with ceralasertib (AZD6738) in relapsed small cell lung cancer. J. Clin. Onco. 39, 8562 (2021).
Dean, E. et al. Ceralasertib (cer) in combination with olaparib (ola) in patients (pts) with advanced breast cancer (BC): Results of phase I expansion cohorts. Proc. Am. Assoc. Cancer Res. 81, PS11-18 (2021).
Wengner, A. M. et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 19, 26–38 (2020).
Yap, T. A. et al. Phase Ib expansion trial of the safety and efficacy of the oral ataxia telangiectasia and Rad3-related (ATR) inhibitor elimusertib in advanced solid tumors with DNA damage response (DDR) defects. Proc. Am. Assoc. Cancer Res. 82, CT006 (2022).
Wengner, A. M. et al. Optimization of treatment schedule for the combination therapy of ATR inhibitor elimusertib and PARP inhibitor niraparib in preclinical tumor models. Proc. Am. Assoc. Cancer Res. 83, 311 (2023).
Yap, T. A. et al. Genomic and pathologic determinants of response to RP-3500, an ataxia telangiectasia and Rad3-related inhibitor (ATRi), in patients (pts) with DNA damage repair (DDR) loss-of-function (LOF) mutant tumors in the Phase 1/2 TRESR trial. Proc. Am. Assoc. Cancer Res. 82, CT030 (2022).
Roulston, A. et al. RP-3500: A Novel, Potent, and Selective ATR Inhibitor that is Effective in Preclinical Models as a Monotherapy and in Combination with PARP Inhibitors. Mol. Cancer Ther. 21, 245–256 (2022).
Matheson, C. J., Backos, D. S. & Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 37, 872–881 (2016).
Geenen, J. J. J. & Schellens, J. H. M. Molecular Pathways: Targeting the Protein Kinase Wee1 in Cancer. Clin. Cancer Res. 23, 4540–4544 (2017).
Dominguez-Kelly, R. et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 194, 567–579 (2011).
Beck, H. et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell Biol. 32, 4226–4236 (2012).
Wang, Y., Decker, S. J. & Sebolt-Leopold, J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer Biol. Ther. 3, 305–313 (2004).
Guertin, A. D. et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol. Cancer Ther. 12, 1442–1452 (2013).
Hirai, H. et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 8, 2992–3000 (2009).
Rajeshkumar, N. V. et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin. Cancer Res. 17, 2799–2806 (2011).
Kreahling, J. M. et al. MK1775, a selective Wee1 inhibitor, shows single-agent antitumor activity against sarcoma cells. Mol. Cancer Ther. 11, 174–182 (2012).
Wright, G. et al. Dual Targeting of WEE1 and PLK1 by AZD1775 Elicits Single Agent Cellular Anticancer Activity. ACS Chem. Biol. 12, 1883–1892 (2017).
Do, K. et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J. Clin. Oncol. 33, 3409–3415 (2015).
Leijen, S. et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 34, 4371–4380 (2016).
Liu, J. F. et al. Phase II Study of the WEE1 Inhibitor Adavosertib in Recurrent Uterine Serous Carcinoma. J. Clin. Oncol. 39, 1531–1539 (2021).
Lheureux, S. et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 397, 281–292 (2021).
Westin, S. N. et al. EFFORT: EFFicacy Of adavosertib in parp ResisTance: A randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J. Clin. Oncol. 39, 5505 (2021).
Cuneo, K. C. et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 37, 2643–2650 (2019).
Huang, P. Q. et al. Discovery of ZN-c3, a Highly Potent and Selective Wee1 Inhibitor Undergoing Evaluation in Clinical Trials for the Treatment of Cancer. J. Med. Chem. 64, 13004–13024 (2021).
O’Dowd, C. et al. Antitumor activity of the novel oral highly selective Wee1 inhibitor Debio 0123. Proc. Am. Assoc. Cancer Res. 79, 4423 (2019).
Meric-Bernstam, F. et al. Safety and clinical activity of single-agent ZN-c3, an oral WEE1 inhibitor, in a phase 1 trial in subjects with recurrent or advanced uterine serous carcinoma (USC). Proc. Am. Assoc. Cancer Res. 82, CT029 (2022).
Pasic, A. et al. A phase 1b dose-escalation study of ZN-c3, a WEE1 inhibitor, in combination with chemotherapy (CT) in subjects with platinum-resistant or refractory ovarian, peritoneal, or fallopian tube cancer. Proc. Am. Assoc. Cancer Res. 82, CT148 (2022).
Ma, J. et al. Cyclin E1 protein overexpression sensitizes ovarian cancer cells to ZN-c3, a novel, selective and oral bioavailable inhibitor of Wee1. Proc. Am. Assoc. Cancer Res. 83, 2153 (2023).
Piggott, L., Chessex, A. V., Luong, N., Tschumi, B. & Vuagniaux, G. The WEE1 inhibitor Debio 0123 enhances the efficacy of standard of care DNA damaging agents in lung cancer models. Proc. Am. Assoc. Cancer Res. 82, 2303 (2022).
Gelderblom, H. et al. 84P Pharmacodynamic marker modulation of the selective WEE1 inhibitor Debio 0123 in patient biopsies from phase I clinical trial. Ann. Oncol. 33, 84P (2022).
Piggott, L. et al. Debio 0123 is a selective WEE1 inhibitor that effectively penetrates the brain and demonstrates anti-tumor activity in preclinical models of glioblastoma. Proc. Am. Assoc. Cancer Res. 83, 6185 (2023).
Weber, A. M. & Ryan, A. J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 149, 124–138 (2015).
Lavin, M. F. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 9, 759–769 (2008).
Lee, J. H. & Paull, T. T. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat. Rev. Mol. Cell Biol. 22, 796–814 (2021).
Shiloh, Y. & Ziv, Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210 (2013).
Alvarez-Quilon, A. et al. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nat. Commun. 5, 3347 (2014).
Pike, K. G. The Identification of Potent, Selective, and Orally Available Inhibitors of Ataxia Telangiectasia Mutated (ATM) Kinase: The Discovery of AZD0156 (8-{6-[3-(Dimethylamino)propoxy]pyridin-3-yl}-3-methyl-1-(tetrahydro-2 H-pyran-4-yl)-1,3-dihydro-2 H-imidazo[4,5- c]quinolin-2-one. J. Med. Chem 61, 3823–3841 (2018).
Riches, L. C. et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer Ther. 19, 13–25 (2020).
Chen, Y. et al. Adaptive oncology phase 1 study of first-in-class inhibitor of ataxia telangiectasia mutated protein kinase (ATM), in combination with olaparib. Proc. Am. Assoc. Cancer Res. 78, 4909 (2018).
Durant, S. T. et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 4, eaat1719 (2018).
Jucaite, A. et al. Brain exposure of the ATM inhibitor AZD1390 in humans-a positron emission tomography study. Neuro Oncol. 23, 687–696 (2021).
Zimmermann, A. et al. A New Class of Selective ATM Inhibitors as Combination Partners of DNA Double-Strand Break Inducing Cancer Therapies. Mol. Cancer Ther. 21, 859–870 (2022).
Ali, M. et al. Small-molecule targeted therapies induce dependence on DNA double-strand break repair in residual tumor cells. Sci. Transl. Med. 14, eabc7480 (2022).
Yue, X., Bai, C., Xie, D., Ma, T. & Zhou, P. K. DNA-PKcs: A Multi-Faceted Player in DNA Damage Response. Front. Genet. 11, 607428 (2020).
Mohiuddin, I. S. & Kang, M. H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 9, 635 (2019).
Lin, Y. F., Shih, H. Y., Shang, Z., Matsunaga, S. & Chen, B. P. DNA-PKcs is required to maintain stability of Chk1 and Claspin for optimal replication stress response. Nucleic Acids Res. 42, 4463–4473 (2014).
Woodard, R. L., Anderson, M. G. & Dynan, W. S. Nuclear extracts lacking DNA-dependent protein kinase are deficient in multiple round transcription. J. Biol. Chem. 274, 478–485 (1999).
Fisher, T. S. & Zakian, V. A. Ku: a multifunctional protein involved in telomere maintenance. DNA Repair (Amst.) 4, 1215–1226 (2005).
Sui, J. et al. DNA-PKcs phosphorylates hnRNP-A1 to facilitate the RPA-to-POT1 switch and telomere capping after replication. Nucleic Acids Res. 43, 5971–5983 (2015).
Ferguson, B. J., Mansur, D. S., Peters, N. E., Ren, H. & Smith, G. L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 1, e00047 (2012).
Blunt, T. et al. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell 80, 813–823 (1995).
Schwartz, C., Rohr, O. & Wallet, C. Targeting the DNA-PK complex: Its rationale use in cancer and HIV-1 infection. Biochem. Pharmacol. 160, 80–91 (2019).
Damia, G. Targeting DNA-PK in cancer. Mutat. Res. 821, 111692 (2020).
Hu, S. et al. Small molecule DNA-PK inhibitors as potential cancer therapy: a patent review (2010-present). Expert Opin. Ther. Pat. 31, 435–452 (2021).
Zenke, F. T. et al. Pharmacologic Inhibitor of DNA-PK, M3814, Potentiates Radiotherapy and Regresses Human Tumors in Mouse Models. Mol. Cancer Ther. 19, 1091–1101 (2020).
Fok, J. H. L. et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 10, 5065 (2019).
van Bussel, M. T. J. et al. A first-in-man phase 1 study of the DNA-dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br. J. Cancer 124, 728–735 (2021).
Mau-Sorensen, M. et al. Safety, clinical activity and pharmacological biomarker evaluation of the DNA-dependent protein kinase (DNA-PK) inhibitor M3814: Results from two phase I trials. Ann. Oncol. 29, 5396 (2018).
Bartek, J. & Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3, 421–429 (2003).
Neizer-Ashun, F. & Bhattacharya, R. Reality CHEK: Understanding the biology and clinical potential of CHK1. Cancer Lett. 497, 202–211 (2021).
Dent, P. Investigational CHK1 inhibitors in early phase clinical trials for the treatment of cancer. Expert Opin. Investig. Drugs 28, 1095–1100 (2019).
King, C. et al. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 14, 2004–2013 (2015).
Parmar, K. et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 25, 6127–6140 (2019).
Sen, T. et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res. 77, 3870–3884 (2017).
Lowery, C. D. et al. Broad Spectrum Activity of the Checkpoint Kinase 1 Inhibitor Prexasertib as a Single Agent or Chemopotentiator Across a Range of Preclinical Pediatric Tumor Models. Clin. Cancer Res. 25, 2278–2289 (2019).
Lowery, C. D. et al. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin. Cancer Res. 23, 4354–4363 (2017).
Hong, D. et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J. Clin. Oncol. 34, 1764–1771 (2016).
Zurcher, G. et al. A phase II study of prexasertib, a cell cycle checkpoint kinase 1 (CHK1) inhibitor, in platinum-resistant recurrent high-grade serous ovarian cancer (HGSOC) with BRCA wild-type (BRCAwt). Proc. Am. Assoc. Cancer Res. 82, CT113 (2022).
Miller, W. H. et al. A phase Ib study of oral Chk1 inhibitor LY2880070 as monotherapy in patients with advanced or metastatic cancer. J. Clin. Oncol. 38, 3579 (2020).
Quincy, S. C. et al. A phase Ib study of oral Chk1 inhibitor LY2880070 in combination with gemcitabine in patients with advanced or metastatic cancer. J. Clin. Oncol. 38, 3581 (2020).
Xin, G. et al. Discovery of a novel and oral CHK1 inhibitor for the treatment of solid tumors. Proc. Am. Assoc. Cancer Res. 83, 482 (2023).
Schmidt, M. et al. Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules 22, 2045–2061 (2017).
Ghelli Luserna di Rora, A., Cerchione, C., Martinelli, G. & Simonetti, G. A WEE1 family business: regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 13, 126 (2020).
Tominaga, Y., Li, C., Wang, R. H. & Deng, C. X. Murine Wee1 plays a critical role in cell cycle regulation and pre-implantation stages of embryonic development. Int. J. Biol. Sci. 2, 161–170 (2006).
Gallo, D. et al. CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibition. Nature 604, 749–756 (2022).
Szychowski, J. et al. Discovery of an Orally Bioavailable and Selective PKMYT1 Inhibitor, RP-6306. J. Med. Chem. 65, 10251–10284 (2022).
Zhang, J., Zhang, L., Wang, J., Ouyang, L. & Wang, Y. Polo-like Kinase 1 Inhibitors in Human Cancer Therapy: Development and Therapeutic Potential. J. Med. Chem. 65, 10133–10160 (2022).
Chiappa, M. et al. Present and Future Perspective on PLK1 Inhibition in Cancer Treatment. Front. Oncol. 12, 903016 (2022).
Seki, A., Coppinger, J. A., Jang, C. Y., Yates, J. R. & Fang, G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 320, 1655–1658 (2008).
Schmucker, S. & Sumara, I. Molecular dynamics of PLK1 during mitosis. Mol. Cell Oncol. 1, e954507 (2014).
Lee, M., Daniels, M. J. & Venkitaraman, A. R. Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression. Oncogene 23, 865–872 (2004).
Chabalier-Taste, C. et al. Polo-like kinase 1 mediates BRCA1 phosphorylation and recruitment at DNA double-strand breaks. Oncotarget 7, 2269–2283 (2016).
Liu, Z., Sun, Q. & Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 10, 22–32 (2017).
Lemmens, B. et al. DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation. Mol. Cell 71, 117–128 e113 (2018).
Watanabe, N. et al. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc. Natl Acad. Sci. USA 102, 11663–11668 (2005).
Qin, B., Gao, B., Yu, J., Yuan, J. & Lou, Z. Ataxia telangiectasia-mutated- and Rad3-related protein regulates the DNA damage-induced G2/M checkpoint through the Aurora A cofactor Bora protein. J. Biol. Chem. 288, 16139–16144 (2013).
Yata, K. et al. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol. Cell 45, 371–383 (2012).
Peng, B. et al. PARP1 and CHK1 coordinate PLK1 enzymatic activity during the DNA damage response to promote homologous recombination-mediated repair. Nucleic Acids Res. 49, 7554–7570 (2021).
Fu, Z. & Wen, D. The Emerging Role of Polo-Like Kinase 1 in Epithelial-Mesenchymal Transition and Tumor Metastasis. Cancers (Basel) 9, 131–145 (2017).
Ruf, S. et al. PLK1 (polo like kinase 1) inhibits MTOR complex 1 and promotes autophagy. Autophagy 13, 486–505 (2017).
Matthess, Y., Raab, M., Knecht, R., Becker, S. & Strebhardt, K. Sequential Cdk1 and Plk1 phosphorylation of caspase-8 triggers apoptotic cell death during mitosis. Mol. Oncol. 8, 596–608 (2014).
Li, M., Liu, Z. & Wang, X. Exploration of the Combination of PLK1 Inhibition with Immunotherapy in Cancer Treatment. J. Oncol. 2018, 3979527 (2018).
Elsayed, I. & Wang, X. PLK1 inhibition in cancer therapy: potentials and challenges. Future Med. Chem. 11, 1383–1386 (2019).
Lansing, T. J. et al. In vitro biological activity of a novel small-molecule inhibitor of polo-like kinase 1. Mol. Cancer Ther. 6, 450–459 (2007).
Bahassi el, M. Polo-like kinases and DNA damage checkpoint: beyond the traditional mitotic functions. Exp. Biol. Med. (Maywood) 236, 648–657 (2011).
Rudolph, D. et al. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 15, 3094–3102 (2009).
Schoffski, P. et al. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur. J. Cancer 48, 179–186 (2012).
Van den Bossche, J. et al. Spotlight on Volasertib: Preclinical and Clinical Evaluation of a Promising Plk1 Inhibitor. Med. Res. Rev. 36, 749–786 (2016).
Dohner, H. et al. Randomized, phase 2 trial of low-dose cytarabine with or without volasertib in AML patients not suitable for induction therapy. Blood 124, 1426–1433 (2014).
Valsasina, B. et al. NMS-P937, an orally available, specific small-molecule polo-like kinase 1 inhibitor with antitumor activity in solid and hematologic malignancies. Mol. Cancer Ther. 11, 1006–1016 (2012).
Beria, I. et al. NMS-P937, a 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivative as potent and selective Polo-like kinase 1 inhibitor. Bioorg. Med. Chem. Lett. 21, 2969–2974 (2011).
Weiss, G. J. et al. Phase I dose escalation study of NMS-1286937, an orally available Polo-Like Kinase 1 inhibitor, in patients with advanced or metastatic solid tumors. Invest. N. Drugs 36, 85–95 (2018).
Luo, J. et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137, 835–848 (2009).
Ahn, D. H. et al. 436P Phase Ib/II study of the polo-like kinase 1 (PLK1) inhibitor, onvansertib, in combination with FOLFIRI and bevacizumab for second line treatment of KRAS-mutated metastatic colorectal cancer. Ann. Oncol. 31, 436P (2020).
Lenz, H. J., Ridinger, M., Samuelsz, E., Smeal, T. & Ahn, D. 397P Early decreases in KRAS mutant allele frequency (MAF) predicts clinical benefit to the PLK1 inhibitor onvansertib in combination with FOLFIRI/bev in 2L treatment of metastatic colorectal carcinoma (mCRC). Ann. Oncol. 33, 397P (2022).
Damodaran, A. P., Vaufrey, L., Gavard, O. & Prigent, C. Aurora A Kinase Is a Priority Pharmaceutical Target for the Treatment of Cancers. Trends Pharmacol. Sci. 38, 687–700 (2017).
Pradhan, T., Gupta, O., Singh, G. & Monga, V. Aurora kinase inhibitors as potential anticancer agents: Recent advances. Eur. J. Med. Chem. 221, 113495 (2021).
Magnaghi-Jaulin, L., Eot-Houllier, G., Gallaud, E. & Giet, R. Aurora A Protein Kinase: To the Centrosome and Beyond. Biomolecules 9, 28–46 (2019).
Borah, N. A. & Reddy, M. M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules 26, 1981–2010 (2021).
Yang, K. T., Tang, C. J. & Tang, T. K. Possible Role of Aurora-C in Meiosis. Front. Oncol. 5, 178 (2015).
Yang, G. et al. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin. Cancer Res. 16, 3171–3181 (2010).
Katayama, H. et al. Aurora kinase-A inactivates DNA damage-induced apoptosis and spindle assembly checkpoint response functions of p73. Cancer Cell 21, 196–211 (2012).
Mou, P. K. et al. Aurora kinase A, a synthetic lethal target for precision cancer medicine. Exp. Mol. Med. 53, 835–847 (2021).
Yan, M. et al. Aurora-A Kinase: A Potent Oncogene and Target for Cancer Therapy. Med. Res. Rev. 36, 1036–1079 (2016).
Shah, K. N. et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 25, 111–118 (2019).
Donnella, H. J. et al. Kinome rewiring reveals AURKA limits PI3K-pathway inhibitor efficacy in breast cancer. Nat. Chem. Biol. 14, 768–777 (2018).
Xue, J. Y. et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 577, 421–425 (2020).
Manfredi, M. G. et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin. Cancer Res. 17, 7614–7624 (2011).
Niu, H., Manfredi, M. & Ecsedy, J. A. Scientific Rationale Supporting the Clinical Development Strategy for the Investigational Aurora A Kinase Inhibitor Alisertib in Cancer. Front. Oncol. 5, 189 (2015).
Dees, E. C. et al. Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin. Cancer Res. 18, 4775–4784 (2012).
Liewer, S. & Huddleston, A. Alisertib: a review of pharmacokinetics, efficacy and toxicity in patients with hematologic malignancies and solid tumors. Expert Opin. Investig. Drugs 27, 105–112 (2018).
O’Connor, O. A. et al. Randomized Phase III Study of Alisertib or Investigator’s Choice (Selected Single Agent) in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma. J. Clin. Oncol. 37, 613–623 (2019).
Elamin, Y. Y. et al. Results of a phase 1b study of osimertinib plus sapanisertib or alisertib for osimertinib-resistant, EGFR-mutant non–small cell lung cancer (NSCLC). J. Clin. Oncol. 40, 9105 (2022).
Du, J. et al. Aurora A-Selective Inhibitor LY3295668 Leads to Dominant Mitotic Arrest, Apoptosis in Cancer Cells, and Shows Potent Preclinical Antitumor Efficacy. Mol. Cancer Ther. 18, 2207–2219 (2019).
Toufektchan, E. & Toledo, F. The Guardian of the Genome Revisited: p53 Downregulates Genes Required for Telomere Maintenance, DNA Repair, and Centromere Structure. Cancers (Basel) 10, 135–149 (2018).
Levine, A. J. The many faces of p53: something for everyone. J. Mol. Cell Biol. 11, 524–530 (2019).
Bouaoun, L. et al. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 37, 865–876 (2016).
Baugh, E. H., Ke, H., Levine, A. J., Bonneau, R. A. & Chan, C. S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 25, 154–160 (2018).
Joerger, A. C., Ang, H. C. & Fersht, A. R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl Acad. Sci. USA 103, 15056–15061 (2006).
Hu, J. et al. Targeting mutant p53 for cancer therapy: direct and indirect strategies. J. Hematol. Oncol. 14, 157 (2021).
Hassin, O. & Oren, M. Drugging p53 in cancer: one protein, many targets. Nat. Rev. Drug Discov. 22, 127–144 (2022).
Boeckler, F. M. et al. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl Acad. Sci. USA 105, 10360–10365 (2008).
Basse, N. et al. Toward the rational design of p53-stabilizing drugs: probing the surface of the oncogenic Y220C mutant. Chem. Biol. 17, 46–56 (2010).
Wilcken, R. et al. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J. Am. Chem. Soc. 134, 6810–6818 (2012).
Bauer, M. R. et al. Harnessing Fluorine-Sulfur Contacts and Multipolar Interactions for the Design of p53 Mutant Y220C Rescue Drugs. ACS Chem. Biol. 11, 2265–2274 (2016).
Bauer, M. R. et al. Targeting Cavity-Creating p53 Cancer Mutations with Small-Molecule Stabilizers: the Y220X Paradigm. ACS Chem. Biol. 15, 657–668 (2020).
Melissa, D. et al. PC14586: The first orally bioavailable small molecule reactivator of Y220C mutant p53 in clinical development. Proc. Am. Assoc. Cancer Res. 81, LB006 (2021).
Puzio-Kuter, A. M. et al. Small molecule reactivators of Y220C mutant p53 modulate tumor infiltrating leukocytes and synergize with immune checkpoint inhibitors. Proc. Am. Assoc. Cancer Res. 82, 1295 (2022).
Ecaterina Elena, D. et al. First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a TP53 Y220C mutation. J. Clin. Oncol. 40, 3003 (2022).
Ceccaldi, R. et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518, 258–262 (2015).
Feng, W. et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 10, 4286 (2019).
Higgins, G. S. & Boulton, S. J. Beyond PARP-POLtheta as an anticancer target. Science 359, 1217–1218 (2018).
Zhou, J. et al. A first-in-class Polymerase Theta Inhibitor selectively targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2, 598–610 (2021).
Zatreanu, D. et al. Poltheta inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 12, 3636 (2021).
Bubenik, M. et al. Identification of RP-6685, an Orally Bioavailable Compound that Inhibits the DNA Polymerase Activity of Poltheta. J. Med. Chem. 65, 13198–13215 (2022).
Bonilla, B., Hengel, S. R., Grundy, M. K. & Bernstein, K. A. RAD51 Gene Family Structure and Function. Annu. Rev. Genet. 54, 25–46 (2020).
Wassing, I. E. & Esashi, F. RAD51: Beyond the break. Semin. Cell Dev. Biol. 113, 38–46 (2021).
Nalepa, G. & Clapp, D. W. Fanconi anaemia and cancer: an intricate relationship. Nat. Rev. Cancer 18, 168–185 (2018).
Huang, F., Mazina, O. M., Zentner, I. J., Cocklin, S. & Mazin, A. V. Inhibition of homologous recombination in human cells by targeting RAD51 recombinase. J. Med. Chem. 55, 3011–3020 (2012).
Lv, W., Budke, B., Pawlowski, M., Connell, P. P. & Kozikowski, A. P. Development of Small Molecules that Specifically Inhibit the D-loop Activity of RAD51. J. Med. Chem. 59, 4511–4525 (2016).
Demeyer, A., Benhelli-Mokrani, H., Chenais, B., Weigel, P. & Fleury, F. Inhibiting homologous recombination by targeting RAD51 protein. Biochim. Biophys. Acta Rev. Cancer 1876, 188597 (2021).
Day, M., Lapierre, J.-M., O’Shea, T. & Mills, K. A novel RAD51 inhibitor, CYT-0851, shows anticancer activity in preclinical models of pancreatic cancer. Proc. Am. Assoc. Cancer Res. 79, C14 (2019).
Guy, J. L., Maclay, T., Day, M., Burness, M. L. & Mills, K. RAD51 inhibition using CYT-0851, shows anti-cancer activity in cellular models of breast cancer and acts synergistically with PARP inhibitors. Proc. Am. Assoc. Cancer Res. 80, P2-05-05 (2020).
Hasham, M. G. et al. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat. Immunol. 11, 820–826 (2010).
Lynch, R. C. et al. First-in-human phase I/II study of CYT-0851, a first-in-class inhibitor of RAD51-mediated homologous recombination in patients with advanced solid and hematologic cancers. J. Clin. Oncol. 39, 3006 (2021).
Garcia-Santisteban, I., Peters, G. J., Giovannetti, E. & Rodriguez, J. A. USP1 deubiquitinase: cellular functions, regulatory mechanisms and emerging potential as target in cancer therapy. Mol. Cancer 12, 91 (2013).
Lim, K. S. et al. USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol. Cell 72, 925–941.e924 (2018).
Cadzow, L. et al. KSQ-4279: A first-in-class USP1 inhibitor for the treatment of cancers with homologous recombination deficiencies. Proc. Am. Assoc. Cancer Res. 82, ND01 (2022).
Coleman, K. E. et al. USP1-trapping lesions as a source of DNA replication stress and genomic instability. Nat. Commun. 13, 1740 (2022).
Harrision, D., Gravells, P., Thompson, R. & Bryant, H. E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP) - Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front. Mol. Biosci. 7, 191 (2020).
Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 34, 360–394 (2020).
Pillay, N. et al. DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 35, 519–533.e518 (2019).
Chan, E. M. et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568, 551–556 (2019).
van Wietmarschen, N. et al. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 586, 292–298 (2020).
Behan, F. M. et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568, 511–516 (2019).
Datta, A. et al. WRN helicase safeguards deprotected replication forks in BRCA2-mutated cancer cells. Nat. Commun. 12, 6561 (2021).
Orlovetskie, N., Serruya, R., Abboud-Jarrous, G. & Jarrous, N. Targeted inhibition of WRN helicase, replication stress and cancer. Biochim. Biophys. Acta Rev. Cancer 1867, 42–48 (2017).
Peters, S., Mok, T., Passaro, A. & Janne, P. A. The Promising Evolution of Targeted Therapeutic Strategies in Cancer. Cancer Discov. 11, 810–814 (2021).
Hahn, W. C. et al. An expanded universe of cancer targets. Cell 184, 1142–1155 (2021).
Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 12, 31–46 (2022).
Chang, L., Ruiz, P., Ito, T. & Sellers, W. R. Targeting pan-essential genes in cancer: Challenges and opportunities. Cancer Cell 39, 466–479 (2021).
Tuveson, D. A. Fighting the Sixth Decade of the Cancer War with Better Cancer Models. Cancer Discov. 11, 801–804 (2021).
Ray-Coquard, I. et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 381, 2416–2428 (2019).
Saad, F. et al. PROpel: Phase III trial of olaparib (ola) and abiraterone (abi) versus placebo (pbo) and abi as first-line (1L) therapy for patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 40, 11 (2022).
Wallez, Y. et al. Activity and tolerability of combination of trastuzumab deruxtecan with the next generation PARP1-selective inhibitor AZD5305 in preclinical models. Proc. Am. Assoc. Cancer Res. 82, 1142 (2022).
Wallez, Y. et al. Activity and tolerability of combinations of trastuzumab deruxtecan (T-DXd) with inhibitors of the DNA damage response in preclinical models. Proc. Am. Assoc. Cancer Res. 82, 5298 (2022).
Fellmann, C., Gowen, B. G., Lin, P. C., Doudna, J. A. & Corn, J. E. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat. Rev. Drug Discov. 16, 89–100 (2017).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019).
Blum, A., Wang, P. & Zenklusen, J. C. SnapShot: TCGA-Analyzed Tumors. Cell 173, 530 (2018).
Bhattacharjee, S. & Nandi, S. Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy. Cancers (Basel) 10, 298–318 (2018).
Acknowledgements
We thank Drs Charles Ding, Kevin Chen, Chi-Chung Chan, Ling Yang, Yi-Wei Wang, Bao-Ming Ge and Xiao-Hong Yu for their kind help in this study.
Author information
Authors and Affiliations
Contributions
C.-S.H., X.-Y.F. and L.-Q. designed and organized the manuscript. L.-Q. and X.-Y.F. wrote and revised the manuscript. C.-S.H., Q.-W.Y., Z.-Y. and H.-L.H. contributed insights based on their experiences in DDR targeting small-molecule drug discovery. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Q., Qian, W., Zhang, Y. et al. A new wave of innovations within the DNA damage response. Sig Transduct Target Ther 8, 338 (2023). https://doi.org/10.1038/s41392-023-01548-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01548-8
This article is cited by
-
High-throughput screening assay for PARP-HPF1 interaction inhibitors to affect DNA damage repair
Scientific Reports (2024)
