
Abstract
The proper transfer of genetic information from DNA to RNA to protein is essential for cell-fate control, development, and health. Methylation of DNA, RNAs, histones, and non-histone proteins is a reversible post-synthesis modification that finetunes gene expression and function in diverse physiological processes. Aberrant methylation caused by genetic mutations or environmental stimuli promotes various diseases and accelerates aging, necessitating the development of therapies to correct the disease-driver methylation imbalance. In this Review, we summarize the operating system of methylation across the central dogma, which includes writers, erasers, readers, and reader-independent outputs. We then discuss how dysregulation of the system contributes to neurological disorders, cancer, and aging. Current small-molecule compounds that target the modifiers show modest success in certain cancers. The methylome-wide action and lack of specificity lead to undesirable biological effects and cytotoxicity, limiting their therapeutic application, especially for diseases with a monogenic cause or different directions of methylation changes. Emerging tools capable of site-specific methylation manipulation hold great promise to solve this dilemma. With the refinement of delivery vehicles, these new tools are well positioned to advance the basic research and clinical translation of the methylation field.
Similar content being viewed by others
Introduction
The three protagonists of the central dogma of molecular biology, DNA, RNA, and protein, are subjected to various post-synthesis chemical modifications. The flow of genetic information from DNA to RNA to protein and consequent protein load and function are strictly regulated by post-synthesis modifications, which gives rise to phenotypic variations in cells/organisms with the same/similar genetic origins. One of the most prevalent modifications is methylation which uses S-adenosylmethionine (SAM) as the donor of a methyl group and replaces a hydrogen atom. As a result, the physicochemical properties of the methylated substrates are altered, including stability and affinity for binding partners or methyl-binding proteins (“readers”). The existence of demethylases (“erasers”) that remove methylation installed by methyltransferases (“writers”) at DNA, RNAs, and proteins indicates the dynamic property of the different methylation pathways. This dynamic nature is consistent with its key regulatory roles in health, and the disturbance of the dynamics is associated with various diseases and constitutes a rationale for therapeutic remedies.1
In mammalian genomes, DNA methylation has been identified at the carbon-5 position of cytosine and recently at the nitrogen-6 position of adenosine, generating C5-methylcytosine (5mC) and N6-methyladenosine (6 mA), respectively. 5mC is the predominant DNA modification and occurs almost exclusively at the symmetric CpG dinucleotides in most somatic cells or tissues; specifically, 60–80% of the 32.28 million CpG dinucleotides in the human genome are methylated.2,3 The majority of remaining unmethylated CpG dinucleotides are located near the transcription start sites in dense clusters known as CpG islands. Besides, non-CpG methylation, namely CpH methylation (where H = A, T or C), is prevalent in human embryonic stem cells and brain.2,4 Recently, A DNA cytosine methylation atlas of normal human cell types has been determined by deep whole-genome bisulfite sequencing, providing a key resource for the investigation of gene regulation and disease-associated variation, and abundant tissue-specific biomarkers for liquid biopsies.5 The effect of DNA cytosine methylation is context-dependent; for example, its presence on gene regulatory sequences (promoters or enhancers) usually causes transcriptional silence, whereas it is not associated with repression and may promote transcription when present on gene bodies.6 DNA adenine methylation, in contrast to DNA cytosine methylation, is a relatively new type of epigenetic modification. Although its existence, genomic distribution pattern, and biological functions in more recently evolved eukaryotes are still being debated mainly due to the low abundance of 6 mA, multiple studies have reported that 6 mA is implicated in regulating transcription, transposon activity, disease, and other functions.7,8,9
The six billion bases of the human genome are wrapped around ~30 million histone octamers (H2A, H2B, H3, and H4) termed chromatin. Histone methylation, primarily on the side chains of lysine (Lys) and arginine (Arg) residues, either upregulates or downregulates transcription depending on the location within histone proteins and the degree of methylation. For example, histone Lys residues can be mono-, di- or tri-methylated; mono-methylation at H3K27 and di- and tri-methylation at H3K4, H3K36, and H3K79 are generally associated with gene activation, while tri-methylation at H3K27 and H3K9 with gene repression.10,11 Histone Arg residues can be mono-, symmetrically, or asymmetrically di-methylated (me1, me2s, or me2as, respectively); H3R2me2s, H3R17me2as, and H4R3me2as generally act as activation marks, while H3R2me2as, H3R8me2s, and H4R3me2s are repressive marks.12,13,14,15 There is extensive crosstalk between histone methylation and DNA methylation. Together with histone acetylation which is often interdependent or mutually exclusive with certain types of histone methylation and DNA methylation, they form the fundamental mechanism of epigenetic regulation that assures the somatic inheritance of gene expression patterns.
Beyond epigenetic regulation, methylation of RNAs and non-histone proteins provides two additional layers for governing gene expression and function. All types of RNAs including messenger RNA (mRNA), ribosomal RNA (rRNA), transfer RNA (tRNA), micro RNA (miRNA), and long non-coding RNA (lncRNA) are substrates for methylation reaction. More than 70 types of RNA methylation have been identified, such as N7-methylguanosine (m7G), N6-methyladenosine (m6A), C5-methylcytosine (m5C; not to be confused with DNA N6-methyladenosine (6 mA) and C5-methylcytosine (5mC)), and 2′-O-methyl (Nm) (where N = A, U, G, or C). Broad interest in RNA methylation biology has been re-inspired by the discovery of the significant level and function of mRNA internal modifications, primarily m6A which is the most abundant one and regulates splicing, localization, translation, and stability of mRNAs. Nonhistone protein methylation also mainly occurs at Lys and Arg residues and shares the common set of catalytic enzymes with histone methylation to regulate the activity, stability, and subcellular localization of methylated proteins. As histones are just a subset of the thousands of proteins targeted for methylation, the interpretation of the mechanisms of protein methylation writers, erasers, and readers in health and diseases is challenging. Moreover, there is extensive crosstalk among protein, RNA, and DNA methylation in various biological processes, generating a sophisticated regulatory network. In this Review, we summarize the operating system of methylation across the central dogma, which involves writers, erasers, readers, and reader-independent outputs. We then discuss how dysregulation of the system contributes to neurological disorders, cancer, and aging, and the present and emerging therapeutic strategies.
Mechanism and function of DNA/RNA/protein methylation
Writers and erasers of methylation
The effectors in DNA, RNA, and protein methylation pathways are categorized into three groups: writers, erasers, and readers, which add, remove, and recognize methyl signals, respectively (Fig. 1). There are ~200 genes in the human genome encoding known or putative SAM-dependent methyltransferases, which have been grouped according to distinct conserved structures. The seven-β-strand domain (7βS) superfamily is the largest group with roughly 130 members, containing DNA methyltransferase (DNMT), Nol1/Nop2/Sun (NSUN), MT-A70, and protein Arg methyltransferase (PRMT) subfamilies, and catalyzes a wide range of substrates including nucleic acids, proteins, and metabolites. For instance, C5-cytosine methylation in DNA is catalyzed by three active writers of the DNMT family: DNMT3A, DNMT3B, and DNMT1. DNMT3A and DNMT3B are mainly responsible for de novo DNA methylation and DNMT1 for the maintenance of the established DNA methylation pattern during cell division. Two other members of the DNMT family, DNMT2 (also known as tRNA aspartic acid methyltransferase 1) and DNMT3L, are not catalytically active DNA methyltransferases. DNMT2 functions as a tRNA methyltransferase, and DNMT3L acts as a de novo DNA methyltransferase cofactor that stimulates their activity specifically in the germline.16 C5-cytosine methylation in mRNA is primarily catalyzed by NSUN2 and NSUN6 of the NSUN family which contains a conserved SUN domain with enzymatic activity. NSUN2 catalyzes the m5C sites that locate at the 5′ ends of hairpin structures and have a 3′ G-rich triplet motif, while NSUN6 acts on the m5C sites that locate at the loops of hairpin structures and have a 3′ UCCA motif.17,18,19,20,21 Another writer that installs m5C in mRNA is DNMT2, especially at the DNA damage sites.22,23 For DNA N6-adenosine methylation, three putative N6-adenosine methyltransferases have been reported, i.e., methyltransferase-like 4 (METTL4), METTL3- METTL14 complex of MT-A70 family, and N-6 adenine-specific DNA Methyltransferase 1 (N6AMT1) (also known as KMT9).24,25,26 Bewilderingly, METTL4 can catalyze N6-methylation of 2′-O-methyladenosine (Am) to generate N6,2′-O-dimethyladenosine (m6Am) in U2 small nuclear RNA (snRNA); METTL3- METTL14 complex is well established as RNA m6A writer; N6AMT1 has been reported to be involved in protein methylation at glutamine (Gln) and Lys residues; thus further study is needed to confirm the specificity and physiological relevance of these putative N6-adenosine methyltransferases.7,9 The majority of m6A in mRNAs are catalyzed by METTL3- METTL14 complex that prefers the sequence motif RRACH (R = A or G; H = A, C, or U), of which METTL3 is the catalytic subunit and METTL14 is an allosteric adaptor.27 Additional adaptors for this writer complex are Wilms’ tumor 1-associated protein (WTAP), Vir like m6A methyltransferase associated protein, zinc finger CCCH domain-containing protein 13 (ZC3H13), RNA binding motif protein 15/15B (RBM15/15B), and HAKAI (also known as CBLL1). The remaining small number of m6A in mRNAs are catalyzed by METTL16 which prefers the UAC(m6A)GAGAA sequence presented as a loop in a hairpin structure. Histone and nonhistone protein methylation at Arg residue is performed by the PRMT family, of which nine members have been identified in the human genome. They are categorized into three types: PRMT1,2,3,4 (also known as CARM1),6, and 8 are type I enzymes that perform mono- and asymmetric di-methylation; PRMT5 and 9 are type II enzymes that mediate mono- and symmetric di-methylation; PRMT7, the only type III PRMT, only be able to catalyze mono-methylation of Arg. Most PRMTs methylate Gly–Arg-rich motifs within their nonhistone substrates, while PRMT4 methylates Pro-Gly-Met-rich motifs and PRMT5 can di-methylate both motifs. Protein Lys methylation is primarily catalyzed by the SET (Su(var)3–9, Enhancer-of-zeste, Trithorax) domain family which is the second largest group with roughly 50 members. Based on sequence similarities surrounding the SET domain, these Lys methyltransferases (KMTs) are classified into seven main subfamilies, i.e., SUV39, SET1, SET2, EZ, SMYD, SUV4-20, and RIZ (PRDM).28 Several additional 7βS superfamily KMTs with no SET domain have been identified, including DOT1L,29 METTL13,30 VCPKMT,31 and the above-mentioned N6AMT1.32 KMTs exhibit high specificity with regard to the location within histone proteins and the degree of methylation. For example, SUV39H1 of SUV39 family, the first identified human KMT, catalyzes trimethylation of H3K9 (H3K9me3);33,34 mixed-lineage leukemia 3 (MLL3) and MLL4 of SET1 family catalyze monomethylation and dimethylation of H3K4 (H3K4me1 and H3K4me2);35 DOTL1 can mono-, di- or tri-methylate H3K79 in a non-processive manner to generate H3K79me1/2/3.36,37

Biochemical processes of reversible DNA/RNA/protein methylation. a C5-cytosine methylation and demethylation in DNA and RNAs. Blue fonts and arrows represent components of the DNA methylation pathway, purple fonts and arrows represent components of the RNA methylation pathway, black fonts and arrows represent common components, and dashed arrows indicate potential steps. Methyl groups and carbon atoms are highlighted in gold. b N6-adenosine methylation and demethylation in DNA and RNAs. The rule of color usage is the same as that of in (a). c Protein lysine methylation and demethylation. d Protein arginine methylation and demethylation. Dashed arrows indicate potential steps
The methyl groups on DNA, RNAs, and proteins can be removed by Fe(II) and α-ketoglutarate (α-KG) dependent dioxygenase superfamily, including ten-eleven translocation (TET), AlkB, and Jumonji C (JmjC) subfamilies. DNA 5mC demethylation is mediated by TET family members (TET1, 2, and 3), in which 5mC is iteratively oxidized to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). All these oxidized derivatives are unable to be recognized by DNMT1, are passively lost during DNA replication, and are replaced with unmethylated cytosine. Alternatively, 5fC and 5caC can be actively reverted to unmethylated cytosine in a DNA replication-independent manner by thymine DNA glycosylase (TDG)-mediated base excision repair. Interestingly, TET enzymes can also mediate the stepwise oxidation of m5C in mRNA, resulting in hm5C, f5C, and ca5C.38,39,40 Whether and how f5C and ca5C, like their DNA counterparts, contribute to methylation reversibility remains unknown. Although decarboxylation of ca5C in mRNA provides a possible pathway to restore unmethylated cytosine,41 evidence of these steps still lacks. Nine AlkB members are identified in the human genome, including ALKBH1-8 and FTO (fat mass and obesity-associated protein). It has been proposed ALKBH1 and 4 demethylate 6 mA of DNA, while ALKBH5 and FTO demethylate m6A of mRNAs. Oxidization of DNA 6 mA by ALKBH1 or 4 generates an unstable intermediate 6-hydroxymethyladenine (6hmA) that undergoes spontaneous loss of the methyl group as formaldehyde and regenerates an unmethylated adenine. Similarly, FTO can successively oxidize RNA m6A to N6-hydroxymethyladenosine(hm6A) and N6-formyladenosine (f6A) which undergo spontaneous hydrolyzation to generate unmethylated adenine and formaldehyde (from hm6A) or formic acid (from f6A). Unlike FTO, ALKBH1, and 4, ALKBH5 can efficiently catalyze the fragmentation of the hemiaminal intermediate to generate formaldehyde and unmethylated adenine directly.42 JmjC family with more than 30 members constitutes the largest class of demethylases, and roughly 20 members of the family have been assigned as lysine demethylases (KDMs) that can demethylate mono-, di-, and tri-methylated Lys using a strategy similar to the demethylation of N6-methyladenosine by AlkB family.43 Based on the domain architecture of the full-length proteins, the family members are classified into seven groups: JHDM1, JHDM2, PHF2/PHF8, JARID1/JARID2, JHDM3/JMJD2, UTX/UTY, and JmjC-domain-only groups. Like KMTs, KDMs exhibit specificity with regard to the site within histone proteins and the degree of methylation. For example, JHDM1A of the JHDM1 group, the first identified JmjC domain-containing demethylase, specifically demethylates H3K36me1/2.44 Besides Fe(II) and α-KG-dependent dioxygenases, two members of the superfamily of the flavin adenine dinucleotide (FAD)-dependent amine oxidases were characterized as KDMs, lysine-specific demethylase 1 (LSD1, also known as KDM1A) and LSD2 (also known as KDM1B). They oxidize the methylamine to generate a labile intermediate, imine, which is hydrolyzed to give formaldehyde and demethylated substrate by a non-enzymatic process. The LSD enzymes only demethylate mono- and di-methylated Lys residues, not tri-methylated ones, due to the limitations of the imine-forming catalytic mechanism.45 LSD1, the first histone demethylase identified, can catalyze the demethylation of H3K4me1/2, H3K9me1/2, and nonhistone substrates (e.g., DNMT1 and p53), while LSD2 has only been shown activity on H3K4me1/2.41,46 Although a dedicated methylarginine demethylases (RDMs) is yet to be identified, some members of the JmjC family have been proposed as RDM candidates, including JMJD6, JMJD1B, and JMJD2A.47,48,49 JMJD6 was the first reported RDM that specifically demethylates H3R2me2 and H4R3me1/2,48 however, multiple studies showed that JMJD6 functions as a lysyl hydroxylase rather than a RDM.50,51,52 JMJD1B, a KDM of the JmjC family for H3K9me2 demethylation, also catalyzes the demethylation of H4R3me2s and H4R3me1.47 Additional multiple KDMs of the JmjC family, including JMJD2A, have been reported to possess RDM activity in vitro, but their RDM activities and functions in vivo have not been reported.49 The biochemical processes of writing and erasing methyl signals at DNA/RNAs/proteins are summarized in Fig. 1.
Functional interpretation of methylation: readers and beyond
Readers
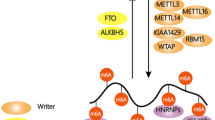
The functional consequences of DNA/RNA/protein methylation depend on the site and/or the degree of methylation. Methylation substantially alters the hydration, hydrophobicity, and hydrogen-bonding capacity of the methylated residues, which in turn directly or indirectly influence the local structure, interacting proteins, stability, localization, and activity of the methylated macromolecules. The most widely studied mechanism of functional interpretation of methylation is the recruitment of effector proteins (also termed “readers”) at the methylated sites, which triggers downstream cellular processes (Fig. 2). Canonical direct and robust methyl signal readers are those that contain conserved methyl-group binding domains.

Methylation of DNA/RNAs/proteins regulates the flow of genetic information. In the central dogma of molecular biology, genetic information is transmitted from DNA to RNA to protein. DNA and histone methylation has essential roles in regulating chromatin opening (involving activating histone methylation, e.g., H3K4me3, H3K36me3, and H3R2me2s) for gene transcription or compaction (involving DNA cytosine methylation and repressive histone methylation, e.g., H3K9me3, H3K27me3, and H3R8me2s) for gene silencing. The methylation of mRNAs (m6A and m5C) regulates the splicing, localization, translation, and stability of the mRNAs. Nonhistone protein methylation influences the activity, stability, and subcellular localization of translated proteins. Collectively, methylation of the different macromolecules constitutes a multilayer dynamic regulation of biological processes. A large array of readers that contain conserved methyl-group binding domains are involved in interpreting these post-synthesis chemical modifications
Three types of domains can bind 5mC of DNA, i.e., methyl-CpG-binding domain (MBD) (represented by MeCP2, MBD1, MBD2, and MBD4 proteins), Set and RING-associated (SRA) domain (including UHRF1 and UHRF2), and methyl-CpG binding Cys2His2 Zinc finger (C2H2 ZF) motifs (represented by Kaiso, ZBTB4, and ZFP57)53 (Fig. 2). Structural studies reveal that these domains use distinct physicochemical mechanisms to specifically recognize methylated CpG (mCpG) dinucleotides: the interaction between the MBD domain of MeCP2 and methylated CpG is driven by hydration of the major groove of methylated DNA rather than cytosine methylation itself;54 SRA domain of UHRF1 flips 5mC out of the DNA helix and accommodates it in a binding pocket with planar stacking, hydrogen bonding, and van der Waals interactions;55,56,57 ZF motifs of Kaiso recognize mCpG sites through hydrophobic and methyl CH···O hydrogen-bonding interactions.58,59 Functionally, MeCP2, MBD1, or MBD2 recognizes methylated CpG-island promoters and subsequently recruits histone deacetylases (HDACs) and histone H3K9 methyltransferases (SUV39H1 and SETDB1) through transcription repression domains (TRDs), resulting in transcriptional gene silence and heterochromatin formation.60 Additionally, MeCP2 is implicated in the translation of CpG methylation in gene-body regions into alternative splicing,61 and can specifically recognize hydroxymethylated CA repeats to prevent nucleosome deposition and regulate the transcription of CA repeat–enriched genes.62 MBD4 has a unique C-terminal glycosylase domain capable of correcting the mC→T mutation which is one of the primary sources of somatic mutation caused by spontaneous deamination of 5mC.63,64,65 UHRF1 (Ubiquitin-like with plant homeodomain and RING finger domains 1) recognizes hemimethylated DNA and catalyzes ubiquitylation of histone H3 lysine 18 (H3K18) and/or H3K23, providing a docking site for DNMT1 that faithfully propagates the DNA methylation patterns following replication.66,67 Intriguingly, UHRF2 preferentially binds to 5hmC via its SRA domain, and subsequent allosteric activation of its E3 ligase activity by 5hmC catalyzes K33-linked polyubiquitination of X-ray repair cross-complementing protein 1, which in turn instructs completion of DNA demethylation by TDG-mediated base excision repair.68,69 Kaiso and ZBTB4 are members of the BTB/POZ transcription factor family and can attract corepressor complexes, such as NCoR, SMRT, and Sin3/HDAC, via BTB/POZ domain to repress gene transcription.70,71,72 ZFP57 possesses a KRAB domain able to interact with KRAB-associated protein 1 (KAP1; also known as TRIM28) co-repressor complex and functions as a master regulator of genomic imprinting to regulate allelic expression of the imprinted genes.73,74
YT521-B homology (YTH) domain can read m6A of mRNA, including YTH domain family 1–3 (YTHDF1-3) and YTH domain-containing 1–2 (YTHDC1-2) proteins75 (Fig. 2). Biophysical studies of the YTH domains of YTHDF2 and YTHDC1 shows that aromatic cages (formed by Trp486, Trp432, and Trp491 in YTHDF2; Trp377 and Trp428 in YTHDC1) contribute to m6A recognition and binding through the cation–π interactions between the N6-methyl moiety and the side chains of the aromatic residues.76,77,78 All of the five YTH proteins except YTHDC2 contain intrinsically disordered regions (IDRs) and undergo liquid-liquid phase separation (LLPS) in the presence of mRNAs with multiple m6A signals, forming nuclear and cytoplasmic condensates (e.g., nuclear YTHDC1-m6A condensates (nYACs); cytosolic P-bodies, stress granules, and neuronal RNA granules), which is crucial in the control of fate and function of the m6A-modified mRNAs.79,80 YTHDC1 is a nuclear m6A reader that controls alternative splicing, alternative polyadenylation, nuclear export, and stability of m6A-modified mRNAs.79,81 In addition, YTHDC1 is implicated in the regulation of gene transcription and transposon silence by readout m6A signal of chromatin-associated noncoding regulatory RNAs (e.g., long non-coding RNA X-inactive specific transcript and enhancer RNAs) and transposon-derived RNAs (e.g., intracisternal A-type particle, ERVK and LINE1 RNAs).81,82,83,84 YTHDC2 possesses RNA helicase activity that can promote translation and degradation of m6A-modified mRNAs by resolving secondary structures and cooperating with the 5ʹ→3ʹ exoribonuclease XRN1, respectively.85,86,87 Unlike other members of the YTH family that preferentially bind to m6A sites, YTHDC2 weakly binds to m6A and possesses other RNA-binding domains besides the YTH domain,88 and recent studies argue that the role of YTHDC2 in germ cell development is independent of m6A recognition,89,90 thus raising doubt about the biological relevance of its m6A-reading activity. Earlier studies proposed that each YTHDF protein mediates different effects on m6A-modified mRNAs: YTHDF1 stimulates translation through interacting with the translation initiation factor eIF3;91 YTHDF2 promotes degradation by recruiting CCR4–NOT deadenylase complex and subsequent deadenylation,92 or by facilitating RNase P/MRP complex-mediated endoribonucleolytic cleavage when the m6A-modified mRNAs contain HRSP12-binding site (an adaptor) and RNase P/MRP cleavage site,93 or by interacting with UPF1 to promote decapping and subsequent 5ʹ→3ʹ exoribonucleolytic cleavage;94 YTHDF3 has both translation and decay effects via cooperating with YTHDF1 and YTHDF2.95 In addition, YTHDF1 and YTHDF3 (but not YTHDF2) promote m6A-mediated stress granule formation in osteosarcoma (U2OS) cells,96 while YTHDF2 and YTHDF3 (but not YTHDF1) mediate the localization of the m6A-modified mRNAs to neurites.97 However, earlier and especially two later studies challenged the view of distinct function, and proposed that all three YTHDF proteins function similarly and act redundantly to accelerate the decay of m6A-modified mRNAs, with no direct effect on translation.92,98,99,100 This is consistent with the fact that the three YTHDF paralogs show high sequence identity. The role of YTHDF1 in the regulation of mRNA stability is confirmed by multiple studies, and the effect of YTHDF3 is linked to the other two YTHDF proteins, therefore, one focus of the debate is the translation-stimulating function of YTHDF1. Further exploration and more data are required to clarify whether YTHDF proteins function in similar or distinct ways or a unified explanation will be found to reconcile the contrasting observations in the future.
Similar to the role of aromatic cages in YTH domains, variant aromatic cages consisting of two to four aromatic residues (Phe, Tyr, or Trp, and occasionally His) are involved in the specific interactions with methyl-lysine motifs of proteins through the cation-π interactions between the methylated ammonium group and the aromatic cage.101,102 There are nine types of aromatic-cage-containing domains capable to recognize methylated lysines, i.e., Tudor, chromo, malignant brain tumor (MBT), proline-tryptophan-tryptophan-proline, tryptophan-aspartate 40 (WD40), plant homeodomain (PHD), ankyrin repeats, bromo-adjacent homology, and cysteine-tryptophan103 (Fig. 2). Among these, Tudor, PHD, and WD40 domains are also capable of accommodating methyl-arginine motifs.104 The effects of histone and nonhistone protein methylations are versatile and context-dependent, and different readers with these domains mediate different biological outputs. For example, TAF3, a subunit of the basal transcription factor TFIID, utilizes PHD domain to bind H3K4me3 at gene promoters and stimulate RNA polymerase II-mediated transcription.105 Heterochromatin protein 1 (HP1) recognizes H3K9me2/3 via the chromo domain to instruct heterochromatin formation, contributing to gene transcriptional silence and stabilization of H3K9 methyltransferases and demethylases.106,107 Di-methylation of p53 at K370 (K370me2) can be recognized by the Tudor domains of 53BP1 or PHF20, promoting transcriptional activity and stability of p53, respectively;108,109 whereas mono-methylation at K382 (K382me1) is read by the triple MBT repeats of the chromatin compaction factor L3MBTL1, inhibiting p53-mediated transcriptional activation.110
Currently, such conserved methyl-group binding domains dedicatedly for 6 mA in DNA and m5C in mRNAs have yet to be identified. Interestingly, the YTH domain of YTHDC1 can efficiently bind to 6 mA in single-stranded and lesion-containing double-stranded DNAs in vitro.111,112 As YTHDC1 is recruited by m6A in RNA hybridized with DNA at DSB sites and stimulates homologous recombination-mediated repair of DSBs by stabilizing DNA:RNA hybrids, it is tempting to hypothesize that YTHDC1 may be recruited to DNA damage sites by 6 mA in DNA in vivo and play a role in the damage repair and maintenance of genome stability.111,113 However, no evidence was found that YTHDC1 could localize to ultraviolet-induced damage sites, making the hypothesis suspicious.114 Aly/REF export factor (ALYREF), a reader of m5C in mRNA, promotes nuclear export of the modified mRNAs.21 Although no apparent methyl-group binding domain was found in ALYREF, sequence alignment analysis using MBD and YTH family proteins as references along with experimental validation identified a conserved amino acid (K171) crucial for the specific binding,21 suggesting a potential conserved methyl-group binding domain might exist when more readers of m5C are available.
A different group of methyl signal readers uses common DNA or RNA binding domains to preferentially bind to methylated DNA or RNA in a sequence-dependent manner, such as Rel-homology domains (RHDs) and homeodomains for DNA, K homology (KH) domains, Arg-Gly-Gly repeat (RGG) domains, and cold shock domains (CSD) for RNA. NFAT (RHD) transcription factors and many members of the extended homeodomain (e.g., homeodomain, POU, and NKX) transcription factor family prefer to bind to CpG-methylated DNA sequences through direct hydrophobic interactions between the homeodomains and the C5-methyl group.115 Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs; including IGF2BP1-3) use KH domains to recognize m6A-modified mRNAs, which promotes mRNA stability by preventing degradation in the P-body or boosting storage in stress granules under stress conditions and facilitates mRNA translation by shuttling to ribosome fractions during recovery from stress.116 Proline-rich coiled-coil 2A (Prrc2a) utilizes the GRE domain (enriched in glycine, arginine, and glutamic acid) to compete for binding of m6A-modified mRNAs with YTHDF2 and stabilizes the Olig2 transcripts which are involved in oligodendrocyte specification and myelination.117 Fragile X mental retardation protein (FMRP) has three KH and one RGG domains and prefers m6A-modified mRNAs, which modulates the nuclear export, translation, and stability of the targets by interacting with CRM1, YTHDF1, and YTHDF2, respectively.118,119,120,121 FMRP can also act as an m5C reader that preferentially binds to DNA:RNA hybrids containing m5C-modified mRNAs at DSB sites, and the KH RNA binding domain of FMRP is required for the functional readout of the methyl signal, in which FMRP promotes completion of homologous recombination repair by facilitating TET1-mediated demethylation of m5C.22 Similarly, RAD52 recognizes m5C mRNA in DNA:RNA hybrids at DNA damage sites and promotes homologous recombination-mediated DSB repair by recruiting RAD51.23 The potential domain of RAD52 responsible for m5C recognition has yet to be identified. Y-box binding protein 1 (YBX1) uses a CSD domain to bind m5C-modified mRNAs through CH–π interactions between the indole ring of Trp65 and the methyl group of m5C, which stabilizes the mRNAs by recruiting an mRNA-stability maintainer ELAVL1.122 Interestingly, YBX1 plays a role in regulating the stability of m6A-modified mRNA targets via interaction between its CSD domain and IGF2BPs.123 These studies point out the dual roles of certain common RNA-binding proteins (e.g., FMRP and YBX1) in the functional interpretation of both m5C and m6A signals in mRNAs.
A distinct subgroup of readers (also called indirect readers) binds methylated substrates using common domains upon methylation-induced structural shift and exposure of the specific binding motifs, which is best demonstrated in the RNA field known as “m6A structural switch”. Several nuclear ribonucleoproteins (HNRNPs) including HNRNPC, HNRNPG, and HNRNPA2B1 belong to this subgroup, and function in transcript processing, including splicing.124,125,126 They use RNA recognition motifs or RGG domains to bind exposed recognition sites due to destabilized hairpin stem around the m6A:U pair or other unknown physicochemical mechanisms.125,126,127,128 Although IGF2BPs can directly recognize m6A via a GGAC motif, there is evidence that they can bind different RNA targets through the “m6A structural switch” mechanism.129 It is conceivable that any RNA-binding protein could benefit from an m6A structural switch when its binding motifs are near or overlapping with m6A sites. However, it is often difficult to clearly distinguish between direct binding and RNA-structure-dependent binding, as both mechanisms have been seen for proteins including HNRNPA2B1 and IGF2BPs.126,129,130 Since methylation can alter the local structure of DNA and proteins, such a structural switch mechanism might also be applied to potential indirect readers of DNA and protein methylation.
Beyond readers
Methylation of DNA, RNAs, or proteins can exert biological effects independent of readers. In contrast to readers attracted by methylation, methylation can directly repel binding proteins that prefer unmethylated targets. The most important protein repelled by DNA cytosine methylation is a C2H2 ZF protein, CCCTC-binding factor (CTCF). CTCF is implicated in a variety of regulatory processes, including chromatin architecture, transcriptional activity, alternative splicing, and alternative polyadenylation.131,132,133,134,135 The binding of most major classes of transcription factors, including bHLH-, bZIP-, and ETS-families, is inhibited by DNA methylation-mediated steric hindrance.115 ZF-CxxC domain-containing proteins, such as CXXC finger protein 1, histone lysine transferase MLLs, and histone lysine demethylase JHDM1A/B (KDM2A/B), recognize unmethylated CpG dinucleotides to regulate epigenetic modification, while methylation blocks their binding to DNA due to a steric clash between the methyl group and the protein backbones.136,137,138,139 The structural and binding analysis identified some RNA-binding proteins repelled by m6A in mRNAs, including stress granule proteins G3BP1/2, pluripotency regulator LIN-28 homolog A (LIN28A), and EW RNA binding protein 1 (EWSR1), and the further study confirmed that m6A can modulate mRNA stability and turnover by repelling G3BP1.118,129 m6A deposited by nematode METT-10 (the ortholog of mammalian METTL16) at the 3′ splice site represses proper splicing and protein production of the targeted mRNAs through physically blocking the binding of the essential splicing factor U2AF35, and this mechanism of splicing regulation is conserved in mammals.140 Although in vivo mRNA targets of mammalian METTL16 remain to be characterized, the finding highlights the biological significance of m6A-mediated direct inhibition of protein binding. Recognition and binding of unmodified lysine or arginine of histone proteins including H3K4 and H3R2 are performed by a separate group of PHD domains, including those of BRAF35–HDAC complex protein (BHC80), autoimmune regulator, tripartite motif-containing protein 24 (TRIM24) and DNMT3L, KDM5A, UHRF1, and DPF3b. These PHD domains replace the aromatic cages with a combination of acidic and hydrophobic residues, facilitating hydrogen-bonding interactions with the unmethylated H3K4 or H3R2.141 In contrast, methylation of these sites decreases hydrogen-bonding capacity and disrupts binding by the PHD domain-containing proteins. Along with proteins repelled by DNA methylation, factors repelled by histone methylation are important components of the chromatin-based regulation network of gene transcription. Methylation of nonhistone proteins can directly repress their interaction with other proteins, playing a role in the regulation of signaling transduction and gene expression. For example, methylation of MAPK kinase kinase 2 (MAP3K2) at Lys260 prevents the binding of protein phosphatase 2A complex (a key negative regulator of the MAPK pathway), resulting in elevated MAP3K2 signaling and promotion of Ras-driven cancer.142 Mono-methylation of a crucial lysine within the nuclear export signal sequence of YAP, a key effector of the Hippo pathway, blocks its interaction with the nuclear exporter CRM1, which results in the retention of YAP in the nucleus and stimulates YAP-mediated transcription activity and tumorigenesis143 (Fig. 4b). Methylation of transcriptional coactivator bromodomain-containing protein 4 (BRD4) at Lys99 compromises its interaction with transcription factor E2F1, leading to reduced expression of translation-related genes and decreased total mRNA translation.144
Furthermore, if the nonhistone proteins are nucleic acid-binding proteins, methylation can directly affect their DNA/RNA binding affinity positively or negatively. For instance, methylation of two lysines of p65 (a subunit of NF-κB) enhances the binding of p65 to targeted DNA sites by producing new hydrophobic contacts, resulting in the activation of downstream genes.145 Methylation of the RGG3 domain of Ewing’s sarcoma abolishes its interaction with the substrate DNA containing G-quadruplex structure, while retaining its ability to bind the mutant counterpart lacking the G-quadruplex structure.146 Methylation of the coiled-coil domain of p54nrb, a subunit of paraspeckle-associated protein complexes, prevents the binding of p54nrb to mRNAs with double-stranded RNA structure, which reduces paraspeckle-mediated nuclear retention of the mRNAs.147 The RNA binding activity of cellular nucleic acid binding protein, a zinc-finger protein that binds structured RNAs, is inhibited upon arginine methylation.148,149 These cases imply that protein methylation-mediated interfering binding of nucleic acids is associated with DNA/RNA higher structure, which may be due to interfered hydrogen bonding or introduced steric clashes.
Methylation can change the local or global structure of methylated DNA, RNAs, and proteins, which directly mediates the effects of methylation. DNA cytosine methylation stabilizes the double helix structure and in turn slows down the DNA unwinding, replication, and transcription.150 Furthermore, cytosine methylation causes profound alteration in the conformation of both nucleosomal and linker DNA, resulting in enhanced contacts between the 5mC-modified DNA and histone proteins.151,152 Such more stable and compact nucleosomes restrict DNA accessibility and facilitate the formation of repressive chromatin. DNA methylation also modulates the formation of certain non-canonical DNA (non-B DNA) structures including G-quadruplexes, which affects gene expression.153 Methylation in the mRNA coding sequence (CDS) including m5C and m6A can regulate codon-anticodon interactions, influencing translation efficiency in a codon-specific manner.85,154 Moreover, the three-dimensional (3D) structures of mRNAs can be altered or stabilized upon methylation, which regulates their stability, localization, splicing, and translation efficiency. The above-mentioned “m6A structural switch” is a good example in which methylation-induced structural alteration matters. A transcriptome-wide study showed m5C may commonly compromise the mRNA translation, which is likely associated with the stabilized secondary structures upon methylation by facilitating base stacking and enhancing the hydrogen-bonding strength with guanosine.17,155 Methylation of histone H3K79 and H4K20 alters nucleosomal surface and higher-order chromatin structure.156,157 Specifically, mono-methylation of H4K20 directly stimulates chromatin openness by interfering with chromatin folding, thereby promoting the transcription of housekeeping genes.156
Protein methylation, especially lysine methylation, competitively inhibits other post-translational modifications of the same residues, such as acetylation, ubiquitination, and crotonylation.158 There are at least 29 types of lysine modification across 219 species including humans, and their dysregulation is involved in abnormal biological processes and human diseases.159 For example, tri-methylation of H3K9 and H3K27 blocks the acetylation of the two sites and keeps the genes from activation.160,161 Mono-methylation of lysine 120 on histone H2B (H2BK120) in cancer cells prevents the ubiquitination of H2BK120 and down-regulates transcription of downstream tumor-suppressor genes.162 Nonhistone protein methylation (e.g., K372 of p53 and K302 of estrogen receptor α protein) can stabilize the methylated proteins by inhibiting polyubiquitination-dependent proteolysis.163,164 The biological significance of the switch between methylation and the other modifications (besides ubiquitination and acetylation) at specific lysine residues is poorly understood. In addition, a recent study showed that di-methylation of the autophagy initiation protein ULK1 at R170 directly stimulates its autophosphorylation of spatially closed T180, which activates ULK1-mediated hypoxic stress adaptation.165 These progresses indicate complex interactions between methylation and other post-translational modifications within proteins.
Methylation in neurological disorders, cancer, and aging
Neurological disorders
The development and function of the nervous system require cell-type-specific precise control of methylation pattern and readout at DNA, RNA, and protein dimensions, which is involved in the proliferation and differentiation of neural precursors, neuronal maturation, gliogenesis, synaptogenesis, and common brain physiology. Disruption of methylation patterns or factors has been linked to various human neurological disorders, such as Rett syndrome (RTT) and Fragile X syndrome (FXS).
Rett syndrome
Unlike symmetrical CpG methylation that is maintained by DNMT1 during genome replication, asymmetrical non-CpG methylation is lost in replicating cells.166 Since post-mitotic neurons do not undergo replication, they accumulate exceptionally high levels of non-CpG methylation alongside the CpG methylation, most prevalently in CpA dinucleotides (mCpA).4,167 The deposition of mCpA is performed by DNMT3A at gene bodies of lowly expressed genes during early life in the brain and recruits the reader MeCP2 to repress transcription of the targeted genes in the adult brain168 (Fig. 3b). Consistently, the evolutionary analysis revealed that non-CpG methylation is confined to vertebrates and enriched within a highly conserved set of developmental genes silenced in adult brains, and MeCP2 originated at the onset of vertebrates, suggesting the emergence of non-CpG methylation and its reader may facilitate the evolution of sophisticated cognitive abilities of vertebrate lineage.169 As a result, loss-of-function mutations in the MECP2 gene cause a severe neurological disorder known as RTT. The MeCP2 protein consists of an N-terminal domain, an MBD that recognizes methylated cytosine, an intervening domain (ID), a TRD able to recruit HDAC3-containing NCoR/SMRT co-repressor complex, and a C-terminal domain (Fig. 3a). RTT-causing mutations are largely confined to the MBD and TRD domains, supporting MeCP2 serves as a bridge between methylated DNA and the co-repressor complexes.170,171 RTT is characterized by an initial normal early development followed by progressive neurological dysfunction and developmental regression, which could be explained by the postnatal accumulation of non-CpG methylation and MeCP2.172 The binding of MeCP2 to DNA is correlated with the number of methylated cytosines, therefore, long genes with more methylcytosines, especially mCpA, are preferentially silenced by MeCP2 in neurons.173 Mice expressing a chimeric MeCP2 protein containing the DNA-binding domain of MBD2 that cannot bind mCpA develop severe RTT-like phenotypes, while mice with about half MeCP2 expression level show only very mild behavioral phenotypes, suggesting the irreplaceable functional significance of mCpA cannot be simply explained by doubling the abundance of methylcytosines in neurons; instead, the distribution of mCpA that shows more cell-type-specific than mCpG is crucial.174,175

Function of MeCP2 and FMR1 and their mutations in RTT and FXS respectively. a The majority of RTT-causing mutations are located in the methyl-CpG-binding domain (MBD) and transcriptional repression domain (TRD) of MeCP2. ID, intervening domain; NTD, N-terminal domain; CTD, C-terminal domain. b Molecular functions of MeCP2: MeCP2 recognizes mCpG and mCpA and recruits NCOR-SMRT co-repressor complex to compact chromatin and suppress transcription; MeCP2 binds the hydroxymethylated CA repeats and protects them from nucleosome invasion. Both functions are abolished upon RTT-causing mutations. c CGG trinucleotide repeat expansion (>200 repeats) in the 5′-untranslated region of the FMR1 gene causes DNA hypermethylation and histone methylation shift, which silences the FMR1 gene in FXS patients. d Multiple important domains of FMRP, including a tandem Agenet (Agn) domain that binds DNA and other proteins, a nuclear localization sequence (NLS), a nuclear export sequence (NES), and several RNA- binding domains (KH1, KH2, and RGG box). PRMT1 performs arginine methylation of RGG motifs within FMRP. e FMRP regulates the histone methylation states by modulating the translation of writers (MLL1 and SETD2), and binds a fraction of m6A-modified mRNAs (probably with a G-quadruplex structure) to modulate their nuclear export, stability, and translation, which is implicated in the regulation of neural differentiation, development, and function. f Methylation and phosphorylation of FMRP have opposing effects on the neuronal granule assembly and activity-dependent translation through modulating FMRP-mediated phase separation
In contrast, MeCP2 can also directly mediate gene activation through interacting with co-activator complexes at the promoters of targeted genes. Particularly, ~85% of thousands of genes can be positively regulated in the hypothalamus of mice by MeCP2.176 The exact mechanism of MeCP2-mediated gene activation remains unclear, and three models have been proposed. (1), cyclic AMP‑responsive element‑binding protein 1(CREB1) was identified as a MeCP2-interacting co-activator, and their co-occupancy at the promoter of an activated targeted gene, somatostatin (Sst), was confirmed.176 However, RTT-associated mutations have not been reported to impact the interaction between MeCP2 and CREB1. (2), HDAC3 recruited at promoters by MeCP2 can deacetylate the transcription factor forkhead box O3 (FOXO3) to stimulate the transcription of a subset of neuronal genes, including brain-derived neurotrophic factor (BDNF) gene.177 The RTT-causing mutation R306C in the TRD domain of MeCP2 inhibits the recruitment of HDAC3 and FOXO3, leading to the downregulation of targeted genes.177 (3), the transcription factor 20 (TCF20) complex interacts with the MBD-ID domain of MeCP2, which may regulate the transcription of downstream genes, such as BDNF.178 The interaction between MeCP2 and TCF20 complex is disrupted by the RTT-causing missense mutations in the MBD-ID region and by a missense mutation in a subunit of the TCF20 complex that was found in a patient with RTT-like syndrome.178 Although the latter two models seem to be compatible with mutation studies, all the current MeCP2-mediated gene activation models place MeCP2 at the transcription start sites or promoters where the level of both CG and non-CG methylation is low and cannot explain the essential role of the gene-body enrichment of non-CG methylation in the pathogenesis of RTT.174,179 More complex, MeCP2 mediates both negative and positive transcriptional regulation of targeted genes, such as one of the most and best-studied downstream genes, BDNF. Specifically, MeCP2 represses promoter III–dependent transcription of exon III–containing BDNF mRNA in the absence of neuronal activity, while the release of MeCP2 from promoter III upon phosphorylation is a prerequisite for de-repression.180 This differential regulation may be related to the location of the methylation and MeCP2 binding, resulting in different adjacent interacting proteins recruited by the surrounding DNA motifs. In addition to mCpG and mCpA, MeCP2 can recognize hydroxymethylated CpA (hmCpA) repeats through Arg133 and repel nucleosomes62 (Fig. 3b). The Arg133 is a potent RTT-causing mutation site, and loss-of-function mutation of MeCP2 alters chromatin architecture and genome-wide transcription of CpA repeat-enriched genes.62 The interaction of MeCP2 with hydroxymethylated DNA has a different thermodynamic signature, compared to that of methylated or unmethylated DNA.181 These studies show the complex regulatory role of MeCP2 in gene transcription through read mCpG, mCpA, and hmCpA signals.
Besides transcriptional control, it has been reported that MeCP2 binding of mCpG at the gene bodies can promote exon recognition in the alternative mRNA splicing process via recruitment of HDACs and subsequently altered DNA polymerase II elongation rate in two human non-neuron cell lines.61 Indeed, hundreds of aberrant splicing events occur in the cortex of Mecp2 knockout mice, including genes critical for synaptic plasticity (Gria2, Nrxns, and Nlgn1).182 However, the mechanism identified in the non-neuron cell lines is not responsible for the altered splicing in the cortex of the RTT mouse model. Instead, MeCP2 interacts with several regulators of RNA splicing, including Y box-binding protein 1 (YB-1), lens epithelium-derived growth factor p75 (LEDGF/p75), or RNA-binding fox-2 (RBFOX2).182,183,184 The TRD domain of MeCP2 is involved in the interaction with YB-1 and LEDGF, and binding of methylated DNA is not required for their interaction, leaving the role of DNA methylation in the MeCP2-mediated alternative splicing unresolved.183,184 In contrast, the MBD and ID domains of MeCP2 are implicated in association with RBFOX2, which promotes the formation of large assemblies of splicing regulator (LASR) condensates in a DNA methylation-dependent manner through the LLPS property of MeCP2.182,185 Furthermore, RTT-causing missense mutations within MBD compromise the formation of MeCP2/RBFOX/LASR condensates.182 These results indicate MeCP2 can function as a bridge to link methylated DNA and mRNA splicing modulators. Future study using Dnmt3a conditional knockout model or the chimeric MeCP2 protein that can distinguish non-CG methylation from CpG methylation in neurons is promising for comprehensively understanding the mechanism of methylation-mediated splicing regulation in regards to specific methylation types, which may be implicated in the pathogenesis of RTT.168,174
Fragile X syndrome
FXS, an X-linked neurodevelopmental disorder, is a leading inherited form of intellectual disability and autism spectrum disorder (ASD), afflicting ~1 in 4000 males and 7000 females. Nearly all cases of FXS are caused by CGG trinucleotide repeat expansion (>200 repeats) in the 5′-untranslated region of the fragile X mental retardation 1 gene (FMR1), leading to transcriptional silence and loss of the gene product FMRP (Fig. 3c). The mechanism underlying FMR1 inactivation is of particular interest since FMR1 reactivation can serve as a therapeutic strategy for FXS.186,187 Despite intensive research, the exact mechanism of the repeat expansion-induced gene silence remains unresolved.188 One important factor is the DNA cytosine hypermethylation of the FMR1 promoter and the repeat region. Demethylation of the FMR1 gene by DNMT inhibitors (e.g., azacitidine and decitabine) or CRISPR-mediated DNA demethylation reactivates the FMR1 expression and rescues FXS neurons.186,189,190,191 Consistently, rare individuals with normal intelligence have a completely unmethylated or partially methylated mutated FMR1 gene capable of producing FMRP proteins.192,193,194 The presence of expanded CGG-repeats in the 5′-untranslated region of the FMR1 mRNA can stimulate the formation of DNA:RNA hybrid, which stalls RNA polymerase II (Pol II) transcription and causes gain of repressive histone marks including H3K9me2 and H3K27me3 and loss of activating histone mark H3K4me2195,196 (Fig. 3c). Combing the DNMT inhibitor (decitabine) with H3K9 or H3K27 HMT inhibitor (chaetocin or 3-deazaneplanocin A, respectively) potentiates the effect of reactivating treatment and prevents re-silencing, compared with decitabine treatment alone.186,197 This suggests a combination of DNA and repressive histone methylation mediates stable transcriptional silencing of the mutated FMR1 gene. Since both DNA and histone methylation is stable and inheritable during cell divisions, the methylation pattern can be maintained in the absence of the initial stimulus.198,199,200 Strikingly, removal of the CGG repeat from FXS patient-derived cells by genome editing can stimulate extensive demethylation of the upstream CpG island within the FMR1 promoter, shift repressive histone methylation to active modification, and initiate FMR1 transcription.201 This suggests CGG repeat expansion is not only required for the establishment of the silence state of FMR1 but also involved in the maintenance of the silence. A recent genome-wide loss-of-function genetic screening uncovered 155 candidate genes predicted to be involved in the maintaining silence of an FMR1 reporter in the haploid and FXS patient-derived pluripotent stem cells (PSCs), including transcriptional co-repressor ZNF217, chromatin remodeling factor SMARCD1, and succinate-metabolism factor C6orf57 (involved in the regulation of α-KG-dependent histone demethylation process).202 Among these, only DNMT1 disruption resulted in robust and partial expression of FMR1 mRNA, implying different repressive mechanisms exist to function redundantly for stable silence of the FMR1 gene.202
FMRP is a widely expressed RNA-binding protein and plays an important role in nearly all aspects of brain development and function, including neurogenesis, neuronal maturation, and excitability203 (Fig. 3d). Initially, it was revealed that FMRP function as a translation repressor by stalling ribosome translocation, and recent studies showed it also regulates alternative splicing, poly(A) tail length, localization, and stability of mRNAs.119,121,204,205,206 More than 1000 mRNAs in the brain are targets of FMRP, among these, ~20 of which are histone methylation modifiers, e.g., MLL3 (H3K4me1/2 writer) and SETD2 (H3K36me3 writer) that regulate transcription and alternative splicing of genes related to neural function, respectively207,208 (Fig. 3e). The principle of target mRNA selection is a focus of the field. m6A modification within the mRNAs contributes to the target specificity. FMRP preferentially binds m6A-modified mRNAs to maintain the mRNA stability in adult mouse cerebral cortex and to promote nuclear export of the mRNAs that regulate mouse neural differentiation.119,121 YTHDF, the unique cytoplasmic YTH protein in Drosophila, regulates FMR1(the Drosophila FMRP homolog) target selection in an m6A-dependent manner, which represses the translation of key mRNAs implicated in axonal growth.209 A similar mechanism of indirectly choosing m6A-modified mRNAs might work in mammals as YTHDF2 can interact with FMRP in an RNA-independent manner.119 Moreover, since the m6A modification is more prevalent in the human brain than the mouse, m6A might contribute to human-specific mRNA targeting by FMRP.210,211 It should be noted that only a fraction of m6A-modified mRNA is recognized by human FMRP, suggesting m6A cooperates with other factors to define a subset of FMRP targets.120 One possibility is the FMRP-interacting protein, like the mechanism in Drosophila.209 Alternatively, m6A may cooperate with secondary RNA structure, such as G-quadruplex, to restrict FMRP specificity, which is supported by the co-localization of m6A and G-quadruplex-forming sequences and preference of FMRP for RNA G-quadruplex structure212,213,214 (Fig. 3e).
The recognition of the G-quadruplex structure is executed by the RGG domain of FMRP.215,216 Methylation of this domain compromises the interaction of FMRP with G-quadruplex-containing mRNAs and polyribosomes, facilitating translation217,218,219 (Fig. 3d). Coupling with phosphorylation, methylation/demethylation of FMRP protein regulates reversible neuronal granule assembly for activity-dependent translation control at the synapse220 (Fig. 3f). Collectively, the biological function of FMR1 gene is associated with methylation status of chromatin, mRNA targets, and the protein per se. Deciphering the details of the underlying molecular mechanisms may provide the opportunity to develop new therapies for FXS.
Cancer
Cancer is a leading cause of death worldwide with almost 10.0 million deaths in 2020, and the global cancer burden is expected to increase by 47%, i.e., from 19.3 million cases in 2020 to 28.4 million cases in 2040.221 The increase in incidence is associated with an aging and growing population as well as changes in the distribution and prevalence of the cancer risk factors, such as excess body weight, physical inactivity, ionizing radiation, chronic infection, and certain environmental pollutants. The transformation of normal, healthy cells into lethal cancer cells with multiple hallmark capabilities, including sustaining proliferative signaling, resisting cell death, avoiding immune destruction, inducing/accessing vasculature, unlocking phenotypic plasticity, and activating invasion and metastasis, is enabled at least by genome instability/mutation and non-genetic alterations within the cells. Aberrant and adaptive DNA/RNA/protein methylation landscape contributes to every stage of cancer progression, from initiation to metastasis and treatment resistance (Fig. 4). The use of single-cell and spatial technologies has provided unprecedented insights into the mechanism of methylation in tumorigenesis.

Mutational cancer driver methylation modifier genes and the role of methylation dysregulation across the central dogma in tumor initiation and progression. a Distribution of the prevalence of methylation modifiers with cancer driver mutations across 66 cancer types. All data are retrieved from IntOGen database. AML acute myeloid leukemia, SBCC skin basal cell carcinoma, MDPS myelodysplastic syndrome neoplasm, HC hepatic cancer, VV vulval cancer, SSCC skin squamous cell carcinoma, ESCA esophageal carcinoma, RCCC renal clear cell carcinoma, HNSC head and neck squamous cell carcinoma, ALL acute lymphoblastic leukemia, DLBCL diffuse large B cell lymphoma, LY lymphoma, AN anus cancer, MBL medulloblastoma, CM cutaneous melanoma of the skin, BLCA bladder cancer. b Methylation remodeling of DNA, RNA, histone, and nonhistone proteins contributes to tumor initiation and progression. Aging, genetic mutations, or environmental stimuli induce methylation remodeling of DNA/RNAs/proteins and causes oncogene activation and TSG silence. A gear set is used as a metaphor for the link between different methylation pathways and their roles in tumorigenesis. The common mechanisms that cause oncogene/TSG disturbance by methylation remodeling at DNA, RNA, and protein levels are recapitulated in the boxes
Instability of genome and methylome in cancer
Cancer genomes accumulate different numbers and types of mutations (e.g., point mutations, copy number alterations, genomic rearrangements) in coding and non-coding sequences, due to various exogenous and endogenous DNA damaging agents, including ultraviolet, ionizing radiation, chemicals, replication errors, spontaneous hydrolytic reactions, and reactive oxygen intermediates.222,223,224 A relatively small proportion of mutations, known as drivers, contribute to tumorigenesis and are thus under positive selection with observed frequency and patterns of mutations deviating from that of expected neutral mutations (known as passengers).225 Dysregulation of DNA, RNA, histone, and nonhistone protein methylation contributes to genome instability/mutations and cancer progression. DNA cytosine methylation can directly promote genetic mutations by spontaneous hydrolytic deamination of 5mC, which leads to C→T transition mutations and is a common cause of somatic point mutations in tumor suppressor genes (TSGs) including TP53.223,226 Indirectly, promoter methylation-induced silence of DNA repair genes (e.g., MLH1, BRCA1, MGMT) compromises the ability to repair DNA damages and results in genome instability and hypermutation during tumor initiation and progression.227,228,229 Loss of DNA methylation at transposable elements reactivates their activity, and subsequent random insertion into the genome potentiates cancer-driver mutations.230 Dynamic mRNA modifications including m6A and m5C are involved in homologous-recombination-mediated DNA repair through the formation and resolution cycle of R-loops.22,23,113 Deficiency of the mRNA methylation or disruption of the dynamics compromises the repair process or induces new damages due to the persistence of R-loops, respectively.22,23,113,231 Elevation of m6A modification in human cancers promotes telomere shortening and genomic instability through degradation of m6A-modified HMBOX1 mRNAs.232 Methylated histones often provide docking sites for repairing proteins at damaged sites. For example, H3K9me3 and H3K4me3 recruit TIP60 and RIF1 for homology-dependent and non-homologous end-joining (NHEJ) repair respectively.233,234 An increasing number of DNA repair proteins or regulators have been identified to be substrates of PRMTs, especially PRMT1 and PRMT5. PRMT1 methylates USP11, MRE11, and 53BP1 while PRMT5 methylates RUVBL1, FEN1, RAD9, and TDP1 to facilitate DNA repair and maintain genome stability.235,236 In addition, PRMT5 methylates DDX5 and the carboxy-terminal domain of Pol II to resolve the R-loop and prevent DNA damage.235 Therefore, maintenance of proper methylome at DNA, RNA, and protein levels is required for genome integrity.
The pattern of DNA methylation in most cancers is characterized as genome-wide hypomethylation accompanied by focal hypermethylation at CpG islands in promoters.237 The exceptions, including follicular thyroid cancer, acute lymphoblastic leukemia, and acute myeloid leukemia, only show a hypermethylation phenotype without global hypomethylation.238,239 Genes susceptible to promoter CpG-island hypermethylation in adult human cancers are bivalently marked with H3K4me3 and H3K27me3 in embryonic stem cells. The bivalent marks let lineage-controlling developmental genes in a repressed and “transcription-ready” state, while DNA hypermethylation, probably a result of loss of H3K4me3/H3K27me3 bivalency in cancer, locks the silenced state and reduces regulatory plasticity, promoting the maintenance of stem cell features and carcinogenesis.240,241,242 In addition, DNA methylation-induced transcriptional silence frequently occurs in a number of TSGs involved in cancer-related cellular pathways, such as DNA repair (MLH1, BRCA1, MGMT), cell cycle (CDKN2A/B, Rb), p53 network (TP73, HIC1), Ras signaling (RASSF1A), and apoptosis (TMS1, DAPK1).243 Global hypomethylation occurs primarily in lamina-associated and late-replicating regions, maybe a result of inefficient methylation maintenance during excess mitotic cell division.244,245 The oncogenic role of global hypomethylation has been attributed to transcriptional activation of transposable elements and/or oncogenes involved in the regulation of cell proliferation, angiogenesis, immortality, metastasis, and tumor suppressor pathways.246,247 However, this classical oncogenic view has been challenged by a study suggesting that global hypomethylation and associated topological alterations have a tumor-suppressive role in colorectal cancer through inhibiting stemness and invasion programs and activating antitumor immunity genes.248 The existence of cancer types without global hypomethylation indicates that global hypomethylation maybe not a prerequisite for carcinogenesis.238,239 Moreover, DNA methylation is required for maintaining the integrity of higher-order genome architecture, and the hypomethylation treatment caused a similar tumor-suppressive topological genome reorganization in human colon cancer cells.248,249 It is important to learn more about how cancer cells balance the oncogenic and tumor-suppressive effects of global hypomethylation.
Methylome has high intrinsic plasticity that is needed for the differentiation of hundreds of cell types in our body with a unique same genome. Operating at the interface between the genome and the environment, it readily changes upon environmental stimuli, which can precede oncogenic mutations and predispose cells to driver mutations through above-mentioned mechanisms.250 Different cell types, either within the same tissue or between tissues, display strongly divergent methylome landscapes.251,252 This relates to different types and frequencies of mutations in tumors with different cell-of-origin.253 Strikingly, H3K9me3 alone can account for more than 40% of mutation rate variation in human cancer cells, and the numeral increases to 55% when combined with other types of histone methylation and chromatin organization features.254 Tumors with more DNA hypomethylation regions have higher frequencies of copy number variations, and regions suffering from differential methylation during cancer progression overlap with mutational hotspots.255
Mutually, genome instability/mutations lead to further disruption of the methylome. A comprehensive mutational cancer driver gene identifying pipeline called IntOGen integrates more than 28,000 tumors of 66 cancer types and identifies 568 driver genes, including 20 DNA and histone (de)methylation enzymes253 (Fig. 4a). Two genes (MLL3 and MLL4) are the extremely wide drivers that drive more than 30 malignancies through mutations, with maximum mutation frequencies in skin cancers, i.e., 55% for MLL4 in skin basal cell carcinoma (SBCC) and 37% for MLL3 in skin squamous cell carcinoma (Fig. 4a). Nine genes including NSD2, MLL2, and JARID1C act as drivers in only one to three tumor types, and the rest nine genes including DNMT3A, EZH2, and SET2 drive 5–18 malignancies (Fig. 4a). On the other hand, mutations at or near key sites of methylation can inhibit or ectopically enhance the modification activity. For example, H3K27M and H3K36M, two oncohistone mutations identified in 78% of diffuse intrinsic pontine gliomas and 95% of chondroblastomas, act as dominant-negative inhibitors of the H3K27 and H3K36 methyltransferases, leading to a global loss of H3K27 and H3K36 methylation, respectively.256,257 Mutations in DNA-binding motifs of CTCF and other regulatory factors significantly influence the methylation level of the CpGs in the neighboring regions, which is associated with cancer subtypes and patient survival.258 The deposition of m6A is regulated by cis-elements 50-nt downstream of the m6A sites, and mutations of these elements or the m6A site itself can influence the m6A deposition and the mRNA fate and subsequently the fitness of cancers.259,260 Therefore, the instability of genome and methylome across the central dogma and their interaction fuel tumor initiation and evolution.
Methylation and cancer initiation and progression
Recent sequencing studies indicate that cancer driver mutations are not rare in normal healthy tissues and can occur early in life.261,262 Only sporadic cells with these mutations transform into a malignant state. The oncogenic competence of a given mutation within the cells depends on the gene-expression programs which are associated with the microenvironment and the cell of origin and are shaped by methylation from DNA to proteins263,264 (Fig. 4b). For example, BRAFV600E mutation readily transforms neural crest and melanoblast lineages but less so in the melanocytes in human pluripotent stem cell-derived cancer model and transgenic zebrafish model.264 Multiple DNA and histone (de)methylases (e.g., EZH2, TET1, and SET2) are highly expressed in these progenitor cells, and gain-of-function EZH2 mutations sensitize melanocytes to BRAFV600E-mediated transformation by global redistribution of H3K27me3, silence of ciliary genes, and activation of WNT/β-catenin signaling in mouse models.264,265,266 WNT pathway is also activated by age-associated DNA methylation remodeling that facilitates oncogenic BRAF mutations to drive colon cancer initiation in mouse colon-derived age-mimic organoid and aged animal models, which may be related to the higher risk of BRAF-mediated transformation from sessile serrated lesions in older individuals, despite these pre-cancerous polyps are equally represented across the age spectrum.267,268 Unlike DNA and protein methylation modifiers that frequently mutate during tumorigenesis (Fig. 4a), RNA methyltransferases and demethylases are frequently ectopic expressed in cancer tissues.269 The role of mRNA m6A modification in tumorigenesis is complex with both tumor-promoting and suppressing effects, depending on both specific sites and alteration of the m6A level (Fig. 4b). A panel of oncogenes (e.g., ADAM19, mTORC2, SP1) or TSGs (e.g., FOXM1, ASB2, LATS1) are targets of m6A pathway, which is co-opted by oncogenic mutations (e.g., RAS, p53, MLL-fusion) to initiate tumors.270,271,272,273,274 Furthermore, fluctuation in the m6A reading process can independently cause amplification of oncogenic signals while shrinkage in TSG signals (Fig. 4b). Alteration of methylomes also bridges exogenous carcinogens and transformation by driver mutations; for example, chronic cigarette smoke replaces H3K4me3/H3K27me3 bivalent histone marks with 5mC at promoters of a set of low-expression genes and primes human bronchial epithelial cells for cancer initiation by a single KRAS mutation.275 In addition to potentiating the oncogenic competence of driver mutations, methylation alteration plays a leading role in the initiation of some cancer types, which has been best demonstrated in some pediatric tumors with very few or no recurrent somatic mutations. For example, childhood ependymomas lack recurrent single nucleotide and focal copy number variations, instead displaying a switch between H3K27me3 and 5mC marks and response to inhibitors that target either DNMTs or PRC2/EZH2.276,277 Strikingly, MLL1 (also known as KMT2A) genomic rearrangements that induce leukemia in an H3K79 methylation-dependent manner are sufficient to induce infant acute lymphoblastic leukemia in the background of fetal-specific gene expression programs.278,279 Although the crucial role of methylation reprogramming in cell transformation has been appreciated, unraveling the exact mechanism of how it drives cancer initiation remains a formidable challenge, due to the rarity and transient nature of these events. This is further aggravated by the complex reading systems of the methylation signals and the coexistence of driver and passenger methylation changes.280
After initiation, the tumor cells proliferate and progress toward aggressive cancers with increasing intratumoral heterogeneity (ITH) of cellular subpopulations that are associated with treatment resistance, metastasis, and relapse (Fig. 4b). ITH has traditionally been ascribed to genetic variation, recent studies indicate that epigenetic variation that is usually manifested as plastic DNA and histone methylomes is a major driver of phenotypic ITH with underlying transcriptomic heterogeneity. DNA methylation changes in human cancers are dominated by stochasticity and occur at different rates across the genome.280 An assay of CpG methylation changes in patient samples with chronic lymphocytic leukemia (CLL) showed that variation within DNA fragments (termed locally disordered methylation) rather than the variation between concordantly methylated fragments constitutes the basis of intratumor methylation heterogeneity.281 For instance, a fragment with 5 CpG sites has 25 possible patterns or 32 epialleles. Such methylation heterogeneity contributes to transcriptional variation by regulating the activity of promoter, enhancer, or CTCF-mediated insulation, which may promote cancer evolution and causes adverse clinical outcomes.281,282 Although DNA methylation modifiers are rarely mutated in CLL, such mutations in individual samples (DNMT3A-Q153* and TET1-N789I) further increase the methylation heterogeneity, implying the difference in expression level, activity, or recruitment of methylation modifiers in the cancer cells causes the DNA methylation variation.281 Several histone demethylases, such as KDM5B (catalyzes H3K4me3 demethylation), are overexpressed in human tumors, which is associated with higher transcriptomic heterogeneity.283 Unlike locally disordered manner in DNA methylation, the difference in methylation peak broadness correlates with stochastic gene transcription.284 Specifically, higher KDM5B activity decreases H3K4me3 peak broadness, which results in rare events of active transcription, larger gene expression fluctuations, and elevated cellular transcriptomic heterogeneity. Conversely, broad H3K4me3 domains are associated with rapid activation of transcription with smaller fluctuations, contributing to high transcriptional consistency. Phenotypic ITH may be further enhanced by the m6A modification of mRNAs since the installation and downstream effects of m6A are heterogeneous across individual cells.252,285 The resource of m6A heterogeneity and the mechanism of how it contributes to ITH remain unknown, and the development of new detecting methods at the single cell level (e.g., scDART-seq) will help to resolve the fundamental questions.252 Tumor evolution can be driven by post-translational modification of nonhistone proteins, resulting in a non-stochastic probability of cancer cells with higher fitness.286 EZH2-catalyzed methylation of β-catenin enhances its stability by inhibiting ubiquitination-mediated degradation and activates Wnt–β-catenin signaling, which sustains self-renewal of cancer stem cells and may contribute to heterogeneity and recurrence of hepatocellular carcinoma287 (Fig. 4b).
Methylation and metastasis
Metastasis is the major cause of cancer-related death. Although large-scale prospective clinical sequencing found an association between genomic alterations and metastatic patterns, there are few metastasis-specific driver gene alterations compared to primary lesions.288,289 Metastasis involves multiple and even opposite steps including detachment and intravasation from primary sites and extravasation and colonization at distal sites, which requires a high degree of gene expression plasticity and reversibility that are readily achieved by non-genetic rather than genetic alterations, therefore, non-genetic variations, such as methylation changes at DNA, RNA, and protein levels, could be the main drivers of primary-to-metastasis transition. For example, dynamic changes of methylation level in CDH1 (encodes a cell-cell adhesion glycoprotein E-cadherin) promoter contribute to the induction of epithelial-mesenchymal transition (EMT) in primary thyroid cancer and of the reverse process, mesenchymal-epithelial transition (MET), in lymph node metastases.290 Concerted histone and DNA hypomethylation, as a result of KMT2C deficiency and its link to DNMT3A, promotes metastasis of small cell lung cancer through activating metastasis-promoting MEIS/HOX genes.247 Inheritable metastasis-promoting gene expression signatures can be achieved by biotic and non-biotic factor-mediated methylome alteration, such as cell-cell contact and hypoxia which are unlikely to induce gene mutation. Specifically, in the circulatory system DNA methylation levels at binding sites of proliferation- and stemness-associated transcription factor genes, such as OCT4, NANOG, and SOX2, can be affected by the clustering state of circulating tumor cells, which is associated with the different metastatic capability of cluster and single circulating tumor cells of breast cancer patients and mouse models.291 Hypoxia promotes EMT and stem cell phenotypes through directly or indirectly suppressing oxygen-dependent H3K27me3 demethylases KDM6A/B, resulting in the persistence of H3K27me3, and subsequent silence of key genes (e.g., DICER)292,293 (Fig. 4b). Increasing evidence indicated that elevation in mRNA m6A level is involved in the EMT and cancer metastasis.294,295 Upregulation of METTL3 or downregulation of FTO promotes methylation of SNAIL or Wnt pathway transcripts, respectively, which enhances the translation or stability of target mRNAs that mediate the EMT process294,295 (Fig. 4b). Although RNA m6A modification itself is not considered to be inheritable, Genetic analyses supported the contribution of mRNA methylation to human disease heritability.285 The inheritance can be achieved by inheritable epigenetic changes in its writers (e.g., METTL3) and erasers (e.g., FTO) or alterations in H3K36me3 as it guides m6A deposition globally.296 Otherwise, a recent study showed RNA m6A modification regulates chromatin accessibility and gene transcription via recruitment DNA demethylase TET1 in normal and cancer cells,297 constituting a potential mechanism for the propagation of phenotypic changes during cell division. Beyond inheritance, plasticity, and reversibility, methylation change is a continuous variable that gives rise to continuous phenotypes of cancer cells, whereas genetic alterations usually produce discrete or binary phenotypic changes.286,298 Such continuous property greatly increases ITH within tumors and subsequent metastasis, which facilitates cancer cells to adapt to the dynamic environments during different stages of metastasis by achieving the fittest gene expression signatures.
In addition to methylation fluctuation, the activity of reader proteins influences the downstream events and promotes metastasis. For example, upregulation of the RNA m6A reader YTHDC1 facilitates TGFβ-mediated lung metastasis of triple-negative breast cancer through promoting nuclear export of SMAD3 transcripts and expression.299 Furthermore, YTHDF3 expression is specifically upregulated in brain metastasis but not in other metastases, which promotes the translation of brain metastasis-associated m6A-modified mRNAs including EGFR, GJA1, and ST6GALNAC5300 (Fig. 4b). These results suggest that different readout of methylation signals may correlate with organ-specific patterns of metastasis.
Methylation and tumor microenvironment
The initiation and progression of tumors are determined by both tumor cell-intrinsic properties and the surrounding tissue/organ environment. The tumor microenvironment (TME) is typically composed of immune cells (e.g., T lymphocytes, natural killer cells, macrophages, neutrophils, dendritic cells), stromal cells (e.g., fibroblasts and mesenchymal stromal cells), blood and lymphatic vessels, extracellular matrix (e.g., collagens, fibronectins, and elastin) and other secreted molecules by tumor and non-tumor cells (e.g., cytokines, growth factors, and chemokines) (Fig. 5). These TME components that usually suppress tumor in their naïve states are reshaped by tumor cells to support growth, angiogenesis, immune evasion, local invasion, metastasis, and therapy resistance of tumors, which involves gene expression reprogramming in non-tumor cells. Although a recent study observed increased somatic copy number alterations in tumor-associated fibroblasts of patients with colorectal cancer,301 generally these non-tumor cells are genetically stable and their tumor-supporting gene expression programs are established through non-genetic mechanisms, including methylation of DNA, RNAs, and proteins.302,303 Through direct cell-cell contact, secreted molecules, or tumor-induced extracellular physicochemical changes (e.g., hypoxia, elevated extracellular potassium), tumor cells induce alteration of methylome landscape and subsequent gene expression changes in TME cells, which can be fixed to robustly facilitate tumor progression due to the inheritability of chromatin methylation.304,305,306,307 In the following discussion, we focus on how methylation remodeling in the various TME cells regulates tumor development and progression (Fig. 5).

Methylation remodeling of non-tumor cells in the tumor microenvironment (TME). The TME is populated by various cell types including immune cells (e.g., T lymphocytes, natural killer cells, macrophages, neutrophils, and dendritic cells), stromal cells (e.g., fibroblasts and mesenchymal stromal cells), and blood and lymphatic vessels. To support tumor development and immune evasion, tumor cells induce alterations of the methylome landscape and subsequent gene expression changes in these TME cells. Examples of methylation remodeling of chromatin, mRNAs, and nonhistone proteins (represented by cartoons in the figure) are summarized
T cells
The immune landscape within TME can be classified into three main categories based on the spatial distribution of T cells: (1) immune-inflamed landscape in which T cells infiltrate and distribute throughout the tumor; (2) immune-excluded landscape in which T cells accumulate at the periphery of tumor; (3) immune-desert landscape in which T cells are completely lacking or at very low numbers.308 The expression of DNA/RNA methylation regulators or methylation profiling of tumors can be used to identify the immune landscape type of individual tumors with mathematical tools, which is consistent with the important roles of methylation in the regulation of tumor immune landscape.309,310,311 There are two major classes of T cells: CD4+ and CD8+ T cells. CD8+ T cells, also known as cytotoxic T cells, recognize antigens presented by MHC-I molecules on the tumor cell surface and kill the tumor cells. Accordingly, tumor cells downregulate the expression of MHC-I molecules or neoantigens to evade immune surveillance of the T cells through promoter hypermethylation of the encoding genes.312,313 Differentiation and activation of naïve CD8+ T cells, featured by the induction of a transcriptional program that facilitates rapid expansion, migrating into tumor tissue, and expression of key cytokines (e.g., IFNγ and IL-2) and effector proteins (e.g., granzymes and perforins), is associated with global DNA and histone methylome remodeling.314,315 For example, the DNA 5mC signal is erased from the promoters of Gzmb (encoding granzyme), Ifng (encoding IFNγ), and the inhibitory receptor Pdcd1 genes to derepress their expression in the activated CD8+ T cells. In the setting of acute infections, most activated CD8+ T cells die and a small portion survive and differentiate into memory CD8+ T cells after clearance of infectious agents, with much of the activated effector genes being silenced again by DNA methylation in the memory CD8+ T cells. These memory CD8+ T cells can provide long-term immunity against pathogens and cancers, as they can rapidly replenish effector CD8+ T cells upon restimulation.316 However, in the setting of cancer, chronic stimulation by antigens and/or inflammation causes their differentiation into a dysfunctional or exhausted phenotype, resulting in failure of cancer eradication by T cells in many patients. The differentiation to exhaustion phenotype is progressive, consistent with progressive alterations of the underlying transcriptome and epigenome.317 Terminally exhausted CD8+ T cells show poor proliferative and cytolytic capacity, low expression of effector cytokines (e.g., IFNγ, IL-2, and TNFα), and sustained upregulation of multiple inhibitory receptors (e.g., PD-1, CTLA-4, TIM3, and LAG3), which is associated with aberrant DNA and histone methylation, i.e., suppression of effector-related genes by DNA methylation, repressive histone methylation or histone bivalency (H3K4me3/H3K27me3), and activation of inhibitory genes by DNA demethylation and activating histone modification.305,318,319 Epigenetic stability of exhausted CD8+T cells prevents function recovery by simple immune-checkpoint blockade therapy.318,320 Not only endogenous CD8+ T cells, but also engineered adoptive CD8+ T cells undergo exhaustion-associated DNA methylation reprogramming that dampens chimeric antigen receptor (CAR) T-cell therapy, characterized by the methylation and silence of genes associated with memory potential (e.g., TCF7 and LEF1) and demethylation and activation of exhaustion-driver genes (e.g., TOX, CX3CR1, and BATF).321,322,323 Therefore, understanding the mechanisms of T cell-exhaustion induction is crucial yet challenging for cancer prevention and immunotherapy.
Persistent tumor antigen exposure, inhibitory receptors/ligands, suppressive cytokines, and harsh TME (e.g., hypoxia and nutrient levels) contribute to CD8+ T cell exhaustion.324 Methylome remodeling is involved in the integration of these signals within the T cells, as well as the upregulation of these signals in tumor and TME cells. Tumor antigen overstimulation is doubtlessly a crucial inducer for CD8+ T cell exhaustion, and multiple transcription factors or regulators are identified, such as HMG-box transcription factor TOX, T cell factor-1, and death-associated protein like 1 (Dapl1).325,326,327 However, the details of how they respond to upstream antigen overstimulation and orchestrate downstream methylome remodeling remain largely unknown. While Yin Yang-1 (YY1) transcription factor recruits EZH2 histone methyltransferase to the IL-2 locus and inhibits the production of the pivotal cytokine in an in vitro exhaustion model created by persistent antigen and co-stimulatory signal stimulation, whether YY1 regulates T cell exhaustion in vivo in a similar way remains to be investigated.328 Inhibitory receptors and their ligands are ectopically upregulated in tumor-infiltrating CD8+ T cells and tumor cells respectively, marking T cell exhaustion and immunosuppression. DNA hypomethylation, loss of repressive histone methylation (e.g., H3K9me3, H3K27me3), and/or mRNA methylation contribute to their upregulation in tumor tissue.319 It should be noted that the effect of mRNA methylation is dependent on the reader proteins in different cancer types, e.g., YTHDF2 promotes degradation of m6A-modified PD-L1 mRNA in intrahepatic cholangiocarcinoma while IGF2BP3 enhances the stability of the modified PD-L1mRNA in breast cancer, resulting in an opposite influence on T cell exhaustion and tumor immune escape.329,330 Suppressive cytokines, including IL-10 and TGFβ that are secreted by tumor cells and TME cells (including immune cells and stromal cells), promote T cell exhaustion and tend to be overproduced in different cancer types by epigenetic alterations. For example, both promoter and gene-body hypomethylation of IL-10 contribute to the upregulation of IL-10 by enabling access to activating transcription factors, such as STAT3 and Sp1.331,332 The development and differentiation of T cells require adequate oxygen and nutrient supplies which are undermined in TME by tumor cells. For nutrient supply, a prominent example is the tug-of-war between cancer and T cells for methionine uptake, an amino required for the synthesis of the common methyl donor SAM for DNA, RNA, and protein methylation in T cells.333 Cancer cells express high level of the methionine transporter SLC43A2 and outcompete for methionine, leading to lower intracellular level of SAM and loss of histone H3K79 methylation in CD8+ T cell, which in turn transcriptionally downregulates the expression of STAT5 transcription factor and causes T cell death and dysfunction.333 Exposure to hypoxia in TME directly inhibits the activity of oxygen-sensitive histone demethylases including KDM5A (H3K4me3 demethylase) and KDM6A (H3K27me3 demethylase), leading to increased histone bivalency and reduced transcription of genes required for robust effector response in terminally exhausted CD8+ T cells.305 Notably, the bivalent gene set was not significantly changed in the chronic viral infection-inducted exhaustion, suggesting bivalency is specific to TME-driven exhaustion of T cells.305
CD4+ T cells recognize antigens presented by MHC-II molecules that are mainly expressed in professional antigen-presenting cells (APCs), such as dendritic cells, B cells, and macrophages. Depending on their patterns of cytokine and function, CD4+ T cells are classified into multiple subtypes including T-helper 1(Th1), Th2, Th17, and regulatory T cells (Treg). These subtypes extensively regulate immune responses within TME, resulting in either tumor progression or regression. Th1 cells are characterized by the production of IL-2 and IFNγ, which suppresses tumor progression at least by supporting CD8+ T cell function, facilitating recruitment of antigen-presenting cells, and inhibiting tumor angiogenesis. In contrast, Treg cells, characterized by expression of transcription factor forkhead box P3 (FOXP3) and the high-affinity heterotrimeric IL-2 receptor (CD25), generally inhibit antitumor immunity through secreting immunosuppressive cytokines (e.g., TGFβ, IL-10, and IL-35), sequestrating IL-2, producing immunomodulatory metabolite (adenosine), expressing inhibitory receptors (e.g., CTLA-4 and LAG-3).334 The roles of Th2 and Th17 cells in tumor progression are ambivalent, depending on the tumor phenotypes.335 The differentiation and function of the different subtypes depend on a particular cytokine milieu upon activation by antigens, followed by the expression of master regulators and methylome remodeling.336,337 For example, deposition of activating histone mark H3K4me3 is associated with the expression of signature-cytokine genes (e.g., Ifng, Il4, and Il17) in their corresponding CD4+ T cell lineages (Th1, Th2, and Th17); active DNA demethylation at the Treg-specific demethylated region within the FOXP3 locus and maintenance of global DNA methylation is required for Treg differentiation and stability of suppressive function;338,339 METTL3-meditated mRNA methylation is implicated in the control of homeostatic proliferation and differentiation of CD4+ T cells and the maintenance of Treg suppressive functions by targeting IL-7/STAT5/SOCS and IL-2/STAT5/SOCS pathways, respectively.340,341 In addition, methylome remodeling mediates transdifferentiation between CD4+ T subtypes. The poised chromatin state, featured by the presence of histone bivalency and lack of DNA methylation, at lineage-specific loci in nonexpressing lineages contributes to this cellular plasticity. For example, TBX21 (a master regulator gene of Th1 differentiation) is marked by histone bivalency in nonexpressing lineages and involved in KDM6B(an H3K27me3 demethylase)-mediated transdifferentiation of Th2, Th17, and Treg cells to Th1 cells.342,343
Given the importance of the methylation program in the lineage commitment of CD4+ T cells, tumors often induce immunosuppression through methylation remodeling of key effector genes or lineage-specific master regulator genes, skewing toward protumor Treg rather than antitumor Th1 phenotype. Ifng promoter region is significantly more methylated in CD4+ T cells of TME than that of tumor-draining lymph nodes from patients with colon cancer, showing Th1 lineage restriction of tumor-infiltrating lymphocytes.344 Treg cells are epigenetically reprogrammed in the TME for complete activation with the help of several important transcription factors, such as BATF and IRF4.345,346,347 After activation, EZH2-mediated histone H3K27 methylation is involved in the maintenance of FOXP3 expression and Treg identity in non-lymphoid tissues, which contributes to cancer immune tolerance.348,349 Moreover, PRMT1- or PRMT5-mediated arginine methylation of FOXP3 is crucial for its transcriptional activity and the function of Treg, and interference with such arginine methylation improves antibody-mediated targeted therapy in a mouse model by inhibiting Treg function and inducing tumor immunity.350,351 Therefore, through these methylation reprogramming mechanisms Treg cells accumulate in TME and compromise antitumor immunity of infiltrating CD8+ T cells, contributing to tumor development and progression.
B cells
Like the T cells, B cells are heterogenous and contain multiple subsets, including antibody-secreting plasma cells and regulatory B (Breg) cells. They play different roles in the tumor immune response, resulting in either tumor regression or progression, depending on the phenotype of both tumor and B cells. The antitumor effects of B cells are achieved by producing antibodies that can mediate antibody-dependent cell cytotoxicity and phagocytosis of cancer cells by natural killer cells and macrophages respectively, presenting antigen to T cells, secreting proinflammatory cytokines (IFNγ and IL-12) that promote cytotoxic immune responses, and directly killing cancer cells with granzyme B and death ligand TRAIL.352 Such antitumor effects of B cells are augmented when they form tertiary lymphoid structures (TLSs) with other immune cells including T cells and dendritic cells.353,354 Consequently, the presence of B-cell-rich TLSs is often correlated with a positive prognosis and a strong indicator of better survival upon immune-checkpoint blockade treatment in various tumor types, such as sarcoma, melanoma, renal cell carcinoma, and lung cancer.355,356,357,358 Moreover, de novo induction of TLS in tumors improves immunotherapy in a mouse model of resistant insulinomas.359 Alternatively, B cells in patients often fail to perform an antitumor function but promote tumorigenesis through secreting immunosuppressive cytokines (e.g., IL-10, IL-35, and TGFβ) and small metabolite (GABA) and expressing inhibitory ligand (PD-L1), leading to inhibition of cytotoxic CD8+ T cells and natural killer cells.352,360 Immunosuppressive and tumor-promoting B cells are generally attributed to the presence of Breg subsets. A recent study showed that IL-35 is involved in Breg differentiation, implying a positive feedback loop exists to consolidate the immunosuppressive role of B cells in tumors.361 The balance between antitumor B cell lineages and Breg cells determines the immune response and is affected by cancers. Pancreatic cancer shifts the B cell differentiation away from plasma phenotype and toward Breg phenotype through IL-35/STAT3/PAX5 pathway, which supports tumor growth, suppresses CD8+ T cell-mediated immunity, and dampens anti-PD1 immunotherapy.361,362,363 Upregulation of several lysine demethylases (e.g., KDM6A/B) and removal of repressive H3K27me3 histone marks at downstream target genes contribute to this Breg differentiation.361 How methylation reprogramming of DNA, RNAs, and proteins orchestrates B-cell activation and differentiation in the peripheral lymphoid system is relatively well documented,364 while counterpart information in tumors is much less. Compared with T cells, B cell infiltration in TME is relatively few. However, as the importance of B cells in tumor development and treatment is emerging, especially recent pan-cancer studies showed that the presence of tumor-infiltrating B cells and/or activated natural killer cells is correlated with exceptional responses to treatment,365,366 it is expected that the gap will be filled soon in future.
Natural killer cells
Natural killer (NK) cells belong to the innate lymphoid cell family and possess remarkable cytotoxic potential. They constitute the first line of immunity against tumor initiation and progression by directly killing tumor cells or enhancing both innate and adaptive immune responses. The former is achieved by releasing granzymes and perforin or expressing death ligands (e.g., FasL and TRAIL), and the latter is mediated by secreting pro-inflammatory cytokines (e.g., IFNγ).367 NK cells do not express somatically rearranged T or B cell antigen receptors to specifically pick up tumor cells, instead, they target tumor cells with MHC class I-deficiency (missing-self signal) and/or stress-induced (e.g., DNA damage) activating ligand overexpression. The two signals correspond to inhibitory and activating receptors on the NK cell surface, the balance of which regulates whether NK cells initiate attack. Inhibitory receptors including the killer cell immunoglobulin (Ig)‑like receptors (KIRs) bind MHC-I molecules to protect healthy cells from NK cell attack, while tumor cells downregulate MHC-I expression in an attempt to evade CD8+ T cell response and activate NK cell response. Alternatively, the inhibitory signals are overridden by upregulated activating ligands in tumor cells with the full expression of MHC-I.368 Suppressing ligand-receptor interactions between tumor and NK cells constitutes an important mechanism to escape detection and eradication by NK cells, which is underpinned by methylation reprogramming. For instance, IDH-mutation mediated DNA hypermethylation and EZH2-catalyzed H3K27 methylation transcriptionally suppress the expression of NK group 2D (NKG2D) ligands (e.g., ULBP1 and ULBP3) in glioma and hepatocellular carcinoma, respectively.369,370 Furthermore, circular EZH2 RNA derived from circularization of exon-2 and -3 of the EZH2 gene encodes a functional shortened version protein called EZH2-92aa which represses the transcription of multiple NKG2D ligands both directly (by binding to the gene promoters) and indirectly (by stabilizing EZH2).371
NK cells show distinct DNA methylation and hydroxymethylation landscapes among innate lymphoid cell lineages,372 and their differentiation and activation are regulated epigenetically and epitranscriptionally.373,374 EZH2-catalyzed H3K27 methylation negatively regulates the differentiation and function of NK cells through downregulating NKG2D receptor and IL-15 receptor β chain (IL-15Rβ/CD122), a cytokine pathway crucial for the survival, proliferation, and activation of NK cells.375 Therefore, inhibition of EZH2 has the potential to kill two birds with one stone: upregulation of NKG2D ligands in tumor cells to expose them to NK cells and increasing NKG2D receptor and IL-15Rβ in NK cells to promote effector function, which ultimately mediates NK cell-based tumor eradication. METTL3-catalyzed m6A modification of SHP-2 mRNAs is required for SHP-2 expression and response to IL-15, whereas the expression of METTL3 is reduced in tumor-infiltrating NK cells from patients with hepatocellular carcinoma, compromising tumor immune-surveillance function of NK cells.374 The m6A reader YTHDF2 that works downstream of the IL-15 signaling pathway is required for the antitumor activity of NK cells by forming a STAT5-YTHDF2 positive feedback loop and targeting TARDBP mRNAs.376 Like CD8+ T cell exhaustion, chronic exposure to activating stimuli during tumorigenesis can cause NK cell exhaustion, accompanied by global DNA methylation changes.377,378 Other mechanisms contributing to tumor evasion of NK cells include expressing immune checkpoint ligands (PD-L1, LAG3, and TIM-3), releasing immunomodulatory molecules (IL-10, TGFβ, and prostaglandin E2), and expressing inhibitory NK cell ligands (HLA-E and HLA-G).379 However, whether and how methylation changes within NK cells affect antitumor activity upon these inhibitory interactions conducted by cancer cells remain unknown, and filling up the gap would help to develop more effective NK cell-based immunotherapy.
Macrophages
Macrophages are derived from myeloid cells in the bone marrow and abundantly populate the majority of solid tumors. They exhibit a high degree of phenotypic and functional plasticity, with pro-inflammatory (M1) and anti-inflammatory (M2) states representing the two ends of the phenotypic spectrum of macrophages.380 M1 macrophages coordinate innate and adaptive immune responses and restrain tumor development through phagocytosis, antigen presentation, and secretion of pro-inflammatory cytokines (e.g., TNFα and IL-6); while M2 macrophages that normally function in wound healing and tissue repair promote tumor progression and metastasis by secreting immunosuppressive factors (e.g., IL-10), pro-angiogenic cytokines (e.g., VEGF), growth factors (e.g., EGF), and extracellular matrix remodeling proteases (e.g., matrix metallopeptidases).381 The phenotypes and the underlying epigenetic identity and gene expression signature of macrophages are ready to be sculpted by the niche environments, which provides a rationale for the education of macrophages in the TME towards M2 state through hypoxia, tumor-derived metabolites (e.g., lactic acid), and secreted cytokines (e.g., IL-4).382,383,384 As a result, high macrophage infiltration often correlates with poor patient prognosis.
Multiple modifiers of DNA/RNA/protein methylation are implicated in regulating the balance between immunosuppressive and proinflammatory polarization of TAMs, and controversial results were reported for certain of them. IL-1R-MyD88 axis-induced TET2 expression in tumor-associated macrophages (TAMs) maintains the expression of immunosuppressive genes (Arg1, Mgl2, and Il4) and M2 macrophage function, which inhibits the antitumor T cell response and increases melanoma tumor burden.385 DNMT3B promotes macrophage polarization to classically activated M1 phenotype in murine adipose tissue macrophages and RAW264.7 macrophage cells by promoter methylation and silence of peroxisome proliferator activated receptor γ (Pparg) gene which encodes a key transcription factor promoting M2 polarization.386 Consistently, DNMT inhibitor (DNMTi) treatment in a mouse model of pancreatic ductal adenocarcinoma (PDAC) increases M2 polarization of tumor-infiltrating macrophages that express markers including Arg1, Chi3l3/4, and the resistin-like molecule (RELM)-alpha/FIZZ1.387 In contrast, GARP- and integrin αV/β8-mediated interaction between PDAC tumor cells and M1-like macrophages induces metabolic and phenotypic reprogramming to M2-like macrophages in a DNMT-dependent manner, in which some glucose metabolism and OXPHOS genes are methylated and downregulated.388 DNMTi treatment increases M1 macrophages while reducing M2 macrophages in the TME of murine models of ovarian cancer, through activating type I IFN signaling.389,390 The different roles of DNA methylation writers and erasers may be associated with tumor types, the specific TME, and upstream signaling pathways. Things are more complex for the modifiers of protein methylation in the regulation of macrophage polarization. Enzymatic activity-dependent and -independent mechanisms and opposite roles of the same modifier or the modifiers that catalyze the same methylation reaction are observed. For example, KDM6B facilitates macrophage M1 polarization through H3K27 demethylation-dependent and independent pathways and serves as a target of cancer-derived exosomal miR-138-5p which promotes the progression of breast cancer;391,392 while acting downstream of PERK (encoded by EIF2AK3) signaling, α-KG synthesis and KDM6B-catalyzed H3K27 demethylation at M2 genes (IRF4, MGL2, and PPARG) reinforces immunosuppressive activity of TME-educated macrophages.393 SETD4 positively regulates TLR-induced IL-6 and TNFα production in LPS-stimulated macrophages by directly methylating histone H3K4 of their promoters,394 while another H3K4 methyltransferase Ash1l negatively modulates this process by methylation and induction of the ubiquitin-editing enzyme A20 that mediates deubiquitination of NF-κB signal modulator NEMO and transducer TRAF6,395 indicating different target choices contribute to the divergent influences of these protein (de)methyltransferases on macrophage polarization. However, major information about the function of protein (de)methyltransferases in macrophage polarization, including SETD4, Ash1l, PRMT1, and G9a, is obtained using classical inflammatory macrophage models, and therefore the further study of TAM models is needed to get a precise conclusion of their roles in the tumor progression.396 The role of RNA m6A methylation in TAM polarization is emerging and also under debate.397 Initially, METTL3/METTL14-YTHDF1-3 axes were shown to drive the polarization bias of TAMs toward M1 phenotype, promote CD8+ T cell function, and inhibit colon cancer progression by targeting mRNAs of immunosuppressive factors for degradation, such as IL-1 receptor-associated kinase 3 (IRAK3, also known as IRAKM) and Epstein-Barr virus-induced protein 3 (Ebi3).398,399 In contrast, a recent study in colon cancer displayed that the METTL3-m6A-YTHDF1 axis fosters immunosuppression of tumor-infiltrating myeloid cells including M2 macrophages, through enhancing the translation of m6A-modified Jak1 mRNA.400 Furthermore, this study showed that lactylation modification of METTL3 is crucial for its target RNA capture. These results imply a sophisticated regulatory network of RNA methylation pathway that may involve post-translational modification of writers, target choice, and readout manner for modulating the macrophage phenotypes in the TME, which requires further exploration and clarification.
Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) are heterozygous populations of bone marrow-derived immature myeloid cells with an immunosuppressive function in TME. Phenotypically, MDSCs are characterized by the expression of myeloid markers (CD33 and CD11b) but lack MHC II (HLA-DR) expression, and they are generally classified into two main subgroups according to their morphological resemblance to monocytes and neutrophils, i.e., monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs) (also known as granulocyte-MDSCs (G-MDSCs). Multiple mechanisms are used by MDSCs to dampen cytotoxic T cells and NK cells, such as the production of nitric oxide (NO) and reactive oxygen species (ROS), secretion of immunosuppressive cytokines (IL-10 and TGFβ), expression of ARG1, indoleamine 2, 3-dioxygenase, and immune checkpoint molecules (PD-L1 and CTLA-4), differentiation into suppressive TAMs (mostly from M-MDSCs).401,402 The differentiation, activation, and survival of MDSCs are regulated by tumor-derived factors (e.g., GM-CSF, IL- 6, and IL-1β), followed by methylation remodeling of DNA/RNA/protein.
Distinct DNA methylation patterns are observed between antigen-presenting myeloid cells (macrophages and dendritic cells) and myeloid suppressive cells under normal physiological conditions and within different MDSC subsets in TME.403,404 Immunosuppressive molecules including TGFβ1, TIM-3, and ARG1 are highly expressed with their promoters highly demethylated in CD33+HLA-DR– cells compared with antigen-presenting myeloid cells,404 and genes related to DNA methylation are deregulated in tumor-infiltrating MDSCs.403 Tumor-derived factors, such as prostaglandin E2, shift differentiation from immune-promoting dendritic cells to MDSCs by upregulating DNMT3A and consequently methylating and inhibiting immunogenic-associated genes (e.g., S1PR4, RUNX1, and FAS).405 In addition to specific gains of DNA methylation during this differentiation, extensive demethylation also occurs, consistent with the fact that TET2 expression is upregulated and contributes to the immunosuppressive function of MDSCs during melanoma progression.385 IFN regulatory factor 8 (IRF8) is an important transcription factor for the differentiation and apoptosis of myeloid cells, and IRF8 deficiency facilitates MDSC differentiation and correlates with increased MDSC levels in cancer patients.406,407 Although IRF8 promoter is hypermethylated in tumor-induced MDSCs, DNA methylation regulates MDSC accumulation via an IRF8-independent mechanism in which IL6-STAT3-DNMT axis silences TNFα expression and suppresses downstream RIP1-dependent necroptosis to support MDSC survival.408 Similarly, SETD1B bypasses a normal role for IRF8 in stimulating iNOS expression in MDSCs and instead directly deposits H3K4me3 signals at the NOS2 promoter to activate the gene expression in tumor-induced MDSCs.409 Another H3K4 methyltransferase, MLL1, negatively regulates the expansion, activation, and differentiation of PMN-MDSCs, and tumor-secreted factors, as well as GM-CSF + IL-6, activate STAT3/CEBPβ to induce microRNAs targeting MLL1-complex (containing WDR5 and ASH2L) during accumulation and activation of PMN-MDSCs.410 Although the role of H3K27 acetylation in the regulation of MDSC-associated genes expression (e.g., NOS2, Arg1) in tumors was reported,411 whether and how methylation of H3K27, as well as other modification sites of histones, modulate the fate of tumor-infiltrating MDSCs need further investigation. Like the function of the METTL3-m6A-YTHDF1 axis in TAMs mentioned above, this axis strengthens immunosuppressive functions (e.g., generation of NO and ROS) of tumor-infiltrating MDSCs by upregulating JAK1-STAT3 signaling pathway.400 Moreover, intratumoral MDSC expansion is associated with high levels of METTL3 in tumor tissues of patients with cervical cancer, and the induction of CD33+CD11b+HLA-DR− MDSCs and tumor-derived MDSCs in vitro is weakened in METTL3-deficient CD33+ cells.412 The potential role of the RNA methylation pathway in regulating MDSC differentiation and homeostasis requires further confirmation and study in vivo, as well as the exact mechanism and downstream targets.
Dendritic cells
Dendritic cells (DCs) are heterogeneous and comprise four main subtypes: conventional DC 1 (cDC1), conventional DC 2 (cDC2), plasmacytoid DC (pDC), and monocyte-derived DC (moDC).413 DCs not only are professional APCs that present antigens on MHC II molecules to prime CD4+ T cells but also are the main APCs that crosspresent tumor-associated antigens on MHC I molecules to activate CD8+ T cells, which is essential for antitumor immunity. The activation and maturation of DCs are stimulated by damage-associated molecular patterns of necrotic cells and/or type I interferons within TME. On the other hand, DCs are implicated in the induction of central and peripheral immune tolerance, and they achieve tolerogenic/immunosuppressive functions under the stimulation of various factors, such as IL-10, TGFβ, and vitamin D3.414 This is often co-opted by tumors to shift the phenotypes of infiltrating DCs from immunogenic to tolerogenic, which dampens antitumor immunity and promotes tumor progression.415 The maturation and functional plasticity of DCs in TME involves methylation remodeling across DNA/RNA/protein and related writers, erasers, and readers.
Different DNA methylation levels and patterns are needed for the development of DC subsets, with pDC showing the highest DNA methylation levels across differentially methylated regions (DMRs) and most sensitive to hypomethylating perturbation.416,417 DNMT1 deficiency leads to hypomethylation and upregulation of suppressor of cytokine signaling (SOCS)1 and impairs the maturation of tumor-associated DCs.418 Meanwhile, TET2-mediated DNA demethylation along with the JAK3-STAT6 pathway is crucial for moDC differentiation upon IL-4 and GM-CSF stimulation, and elevating the enzymatic activity of TET2 by vitamin C treatment during the differentiation improves the immunogenic properties of moDCs, supporting the proliferation and function of T cells and the cancer immunotherapy.419,420,421,422 In contrast, TET2 along with the JAK2-STAT3 pathway is indispensable for the vitamin D-mediated establishment of tolerogenic/immunosuppressive moDCs.423 Similarly, histone methylation (H3K4me3 and H3K27me3) regulates the differentiation of moDCs into either immunogenic or tolerogenic phenotypes, depending on the environmental stimuli.424 Tumor-derived TGFβ induces alteration in H3K4me3 and H3K27me3 landscapes and represses the maturation and function of DCs, which involves downregulation of MHC class II, costimulatory molecules, the chemokine receptor CCR7, and type I IFN.425,426,427 CCR7 is a key determinant for the migration of mature DCs (primarily cDC1s) to the lymph nodes where T cell priming occurs, and distinct levels of H3K27me3 account for different migration abilities of DC subsets.413,428 Besides H3K4 and H3K27 methylation, DOT1L-catalyzed H3K79 methylation at the Forkhead box transcription factor M1 (FOXM1) promoter enhances FOXM1 expression that inhibits the DC maturation via the Wnt5a signaling pathway, which is associated with the poor survival in pancreatic cancer and colon cancer patients with high DC infiltration.429 The m6A-YTHDF1 axis promotes DC activation and function to augment CD4+ T cell proliferation by enhancing the translation of some immune-related transcripts, including Tirap, CD80, and CD40 mRNAs.430 However, this axis negatively regulates the antigen crosspresentation ability of cDCs by enhancing the translation of targeted lysosomal protease transcripts, which restrains the priming of CD8+ T cells and antitumor immunity.431 The underlying mechanism for the distinct roles of m6A in DC function modulation remains unknown. Nonetheless, these studies indicate a flexible role of RNA methylation, as well as DNA and protein methylation, in DC-mediated antitumor immunity.
Neutrophils
Neutrophils constitute 50–70% of circulating leukocytes in humans and are essential components of innate immunity against invading pathogens. Like macrophages, tumor-associated neutrophils polarize to either antitumor (N1) or protumor (N2) phenotype. The N1 neutrophils are short-lived and use tools of anti-infection to eliminate tumor cells, including performing antibody-dependent cell-mediated cytotoxicity (ADCC), releasing chromatin DNA filaments (known as neutrophil extracellular traps (NETs)), and generating superoxide (H2O2 and HOCl), and stimulating adaptive antitumor immune responses. In contrast, the N2 neutrophils are long-lived, low cytotoxic, and positively involved in tumor initiation, growth, metastasis, angiogenesis, and immune evasion.432 Notably, neutrophils participate in the entire metastatic process from primary tumors to the distant organs, which involves distinct subsets of neutrophils: MMP-9+TIMP- neutrophils promote angiogenesis and intravasation through releasing MMP-9 at the primary tumor sites;433 neutrophils cluster with circulating cancer cells (CTCs) to promote cell cycle progression within the bloodstream;434 NET DNA acts as both chemotactic factor and trap to facilitate CTC landing at distant organs;435,436 a subset of neutrophils featured by P2RX1 deficiency foster immunosuppressive metastatic TME through upregulating immune exhaustion-related genes (e.g., PD-L1).437 The protumor effect of neutrophils dominates in patients, and consequently, neutrophil-based assays, including the number of tumor-associated neutrophils, circulating neutrophil level, neutrophil to lymphocyte ratio, and CD8+ T cell to neutrophil ratio, are developed to predict patient prognosis and responsive to treatments.438 The phenotypes of neutrophils are shaped by tumor cells and other cells within TME through released cytokines, such as TGFβ, IL-6, and IFNβ. TGFβ439 and IL-6440 drive N2 phenotype, while IFNβ441 stimulates N1 phenotype. Furthermore, in tumor-bearing mice TGFβ can reverse mature antitumor neutrophils to pro-tumor immunosuppressive counterparts that accumulate in cancer patients, indicating phenotypic plasticity of mature neutrophils.442
Although DNA/RNA/protein methylation remodeling in tumor cells for motivating, recruiting, and polarization of neutrophils has been studied,443,444 the methylation mechanism for intrinsically regulating neutrophils is still largely unknown, due to technical difficulties in isolating tumor-associated neutrophils from human samples at high purity. Dynamic DNA methylation remodeling is involved in the neutrophil differentiation from the multipotent common myeloid progenitors,445 and dysregulation in DNA methylation contributes to neutrophil-based autoimmune vasculitis,446 suggesting the role of DNA methylation in modulating neutrophil fate and function, which is pertinent to diseases. Tumor-infiltrating neutrophils form a positive feedback loop with glioblastoma cells to facilitate further neutrophil infiltration and tumor progression through releasing High Mobility Group Box 1(HMGB1) and activating the NF-κB signaling pathway in glioblastoma.447 The exposure of nuclear-localized HMGB1 is a result of NET formation, and mono-methylation of HMGB1 at Lys42 changes the protein conformation and causes its cytoplasmic localization.448 The oncoprotein growth factor independent 1 (GFI-1) is essential for neutrophil differentiation and maturation after the myeloblast stage and acts as a transcriptional repressor that cooperates with suppressive histone modifiers, such as G9a (H3K9 methylation) and LSD1 (H3K4 demethylation).449 These results imply a potential role of histone and nonhistone protein methylation in regulating neutrophil recruitment and function in tumors. A recent study demonstrated the migration of neutrophils to infectious sites is empowered by the m6A demethylase ALKBH5 that changes the degradation of migration-related transcripts (CXCR2, NLRP12, PTGER4, WNK1, and TNC).450 Up-regulation of CXCR2 is also crucial for neutrophil recruitment in tumors,438 suggesting a potential role of neutrophil RNA methylation in regulating tumor progression.
Endothelial cells
When tumors reach about 2 mm3 in volume, passive diffusion cannot meet the further demand for oxygen and nutrients, and tumors have to establish their blood supply system.306 Endothelial cells (ECs) form tumor blood vessels to deliver nutrients and other essentials for tumor growth. In addition, tumor blood vessels enable the dissemination of tumor cells to distant organs. Stimulated by excessive proangiogenic factors (e.g., VEGF, PDGF, and EGF), the new tumor vessels are malformed and dysfunctional, characterized by irregular diameters, fragility, leakiness, tortuosity, and abnormal blood flow. These characteristics prevent drug delivery, cause hypoxia, foster an immunosuppressive environment, and facilitate intravasation and metastasis. Recent studies have proposed that tumor-associated ECs play a more active role in TME remodeling and tumor progression than merely providing a physical channel. EC-derived surface proteins (e.g., VCAM-1) and soluble factors (e.g., CXCL2, soluble Notch ligands, LAMA1, and INHBB) orchestrate tumor cell behavior, promote cancer stem cell phenotype and survival, and recruit and educate tumor-associated neutrophils and macrophages.451,452,453,454 To achieve these above-mentioned protumor function, tumor ECs alters their gene expression program upon crosstalk with the tumor cells and other TME cells.454,455
Genome-wide DNA methylation profiling uncovered thousands of differentially methylated loci that contain hundreds of differentially expressed genes in human prostate tumor-derived ECs versus normal ECs.456 DNA hypomethylation of some proangiogenic genes (e.g., VEGF) caused by intracellular adenosine accumulation within ECs in a hypoxic environment promotes EC proliferation, migration, and angiogenic sprouting.457 DNA demethylation at the promoter region upregulates biglycan (a small leucine-rich secretory proteoglycan) expression in TECs from high metastatic tumors, which promotes tumor cell migration and intravasation through activating TLRs/ NF-κB/ ERK signaling pathway.458 CYP24A1 is hypermethylated and silenced in the tumor ECs compared with that of normal ECs, which contributes to the selective calcitriol-mediated growth inhibition in ECs.459,460 However, the exact function of DNA methylation change in the CYP24A1 gene in tumor vasculature remains unknown. Using DNMT and HDAC inhibitors, 81 genes including functionally validated anti-angiogenesis genes (clusterin, fibrillin 1, and quiescin Q6) were identified specifically and epigenetically silenced in tumor-conditioned versus quiescent ECs.461 Although the mechanism of DNMT-mediated gene silence remains elusive as no significant DNA methylation alteration was detected in the promoter CpG islands of seven tested genes, histone modification change, i.e., loss of H3 acetylation and H3K4 methylation, was proposed to be associated with the gene silence. Similarly, histone H3 deacetylation and H3K4 demethylation are involved in the silence of intercellular adhesion molecule-1 (ICAM-1) in tumor ECs, leading to reduced leukocyte-endothelial cell adhesion and consequently less inflammatory infiltration.462 In contrast, the expression of cell adhesion molecules in the ECs of distant tissues is crucial for circulating tumor cell adhesion, extravasation, and metastasis. Tumor cells induce an inflammatory response and consequent upregulation of cell adhesion molecules (e.g., E-selectin and VCAM-1) in the ECs at premetastatic sites.463 PRMT5-mediated methylation of the homeobox transcription factor HOXA9 is essential for the expression of cell adhesion molecules during the inflammatory response of human umbilical vein ECs.464 In addition, several histone methyltransferases (EZH2, G9a, and DOT1L) were shown to regulate the inflammatory response, cell cycle progression, viability,465 and angiogenesis of normal ECs, suggesting protein methyltransferases may play a more important role than currently appreciated in the regulating tumor ECs and tumor progression, which deserves further exploration. Although RNA methylation is relatively well studied in other systems (e.g., cardiovascular system) where it affects angiogenesis and vascular function, information about the potential role of m6A modification in tumor ECs is scarce.466 VEGF which has been considered the most crucial angiogenic growth factor is positively regulated by METTL3-catalyzed m6A modification in bone marrow stem cells.467 In tumor but not normal ECs, the m6A reader Hu antigen R (HuR) specifically accumulates in the cytoplasm to enhance the mRNA stability of VEGF and cyclooxygenase-2, promoting the survival, migration, and tube formation of tumor ECs.468,469 These results indicate DNA/RNA/protein methylation mechanism plays a key role in regulating the gene expression reprogramming and phenotypic plasticity of tumor ECs. Further studying of the methylation remodeling mechanism in tumor ECs may aid in developing new strategies to normalize or target tumor vasculature.
Fibroblasts
Cancer-associated fibroblasts (CAFs) are a major tumor stroma component (can reach 90% of tumor mass in pancreatic cancer) that participate in the TME shaping. They are responsible for the production of the bulk of extracellular components, including growth factors, cytokines, and extracellular matrix, as well as for ECM structure remodeling.470 Through these, CAFs stimulate tumor proliferation and metastasis, enhance angiogenesis, drive the desmoplastic reaction, suppress the immune response, and foster therapeutic resistance, resulting in poor prognosis of many tumor types. There are distinct subtypes of CAFs, including at least three main subtypes: myofibroblastic, inflammatory, and antigen-presenting subtypes that express α-smooth-muscle-actin, IL-6/LIF, and MHC class II, respectively.471,472 Identification of sub-subsets with specific markers (e.g., LRRC15 and NetG1) further subdivides the three main subtypes into more subpopulations, underpinning the complexity and heterogeneity of CAFs.473,474 Different external cues from TME drive the heterogeneity of CAFs. For example, tumor-derived IL-1 promotes LIF expression and differentiation into inflammatory CAFs, while TGFβ antagonizes this process by suppressing the expression of IL-1 receptor and facilitates differentiation into myofibroblastic subtype.472 In addition, multiple cells of origin generating CAFs contribute to heterogeneity and functional divergence, including tissue-resident fibroblasts, pericytes, mesenchymal stromal cells, and adipocytes.475 The genome of CAFs is stable, but their gene expression programs show highly heterogeneous. The transition from normal and quiescent fibroblasts into activated CAFs in TME involves distinct methylation alteration in DNA, histones, and likely RNAs.476,477
Similar to tumor cells, global DNA hypomethylation accompanying focal hypermethylation was observed in CAFs compared with that in normal fibroblasts.478,479 Specific genes that are hypomethylated, upregulated, and functionally validated in CAFs include ADAM12, RUNX1, CXCR4, and glycolytic genes (PKM and LDHA). ADAM12 functions in tumor-stromal cell interactions;480,481 RUNX1 mediates the transition from normal fibroblasts to CAFs;482 CXCR4 promotes tumor cell invasion probably through regulating the production of IL-8 in CAFs;483 PKM and LDHA, along with hypomethylated HIF-1α, promote glucose metabolic rewiring in CAFs to produce metabolites (lactate and pyruvate) that support anabolism of tumor cells.484 Independence on DNA synthesis and an increase in 5hmC level suggest the demethylation process involves TET-mediated active DNA demethylation mechanism.485,486 Indeed, tumor-derived lactate promotes α-KG synthesis in mesenchymal stem cells, facilitating TET activation and global demethylation reaction during differentiation into CAFs.483 On the other hand, the silence of negative regulators of CAF activation signaling is a prerequisite. STAT3 and TGFβ signaling pathways are essential for differentiation into inflammatory and myofibroblastic CAFs, respectively.472 DNA methylation-mediated silence of the negative regulators, SHP-1 and SOCS1, is involved in the constitutive activation of STAT3 signaling, leading to the conversion of fibroblasts and sustained pro-invasive and pro-tumor phenotype of CAFs.487,488,489 SMAD3, an important transcription factor downstream of TGFβ signaling, is selectively hypermethylated in CAFs, which is linked with hyperresponsiveness to TGFβ signaling in terms of contractility and ECM deposition.478 Moreover, DNMT3B and TGFβ form a positive feedback loop through miR-200s/221, leading to a stably high level of DNMT3B expression and contributing to the maintenance of CAF phenotype.490 Methylation remodeling of histone constitutes another mechanism for maintaining the protumor capacity of CAFs. Loss of EZH2-mediated H3K27 methylation is implicated in the regulation of protumor secretome of CAFs, such as ADAMTS1, WNT5A, and IGF2 which facilitate cancer cell growth, migration, and invasion.307,491 Nicotinamide N-methyltransferase that consumes SAM to generate SAH cause histone hypomethylation and global gene expression alteration in CAFs, which is necessary and sufficient for the expression of CAF markers and secretion of protumor factors.492 The methylated product, 1-methylnicotinamide, can be taken up by infiltrating T cells and influences its tumor-killing function by inducing and reducing the expression of TNFα and IFNγ, respectively.493 Furthermore, the expression of lysine demethylases (e.g., KDM2A and LSD1) actively demethylates promoter-associated histones, rewiring the gene expression programs and promoting the conversion and function of CAFs.494,495 Although the current studies bias histone hypomethylation, histone methylation deserves further investigation as there is emerging evidence supporting the role of histone methylation gain (e.g., H3K4 methylation) in regulating CAF phenotype.494 Few studies focusing on the role of RNA methylation in regulating the differentiation and function of CAFs. METTL3-catalyzed m6A modification of COL10A1 transcripts in CAFs enhances the mRNA stability and protein level, which protects lung squamous cell carcinoma from apoptosis-induced oxidative stress and promotes cancer cell proliferation.496 Zfp217-FTO-YTHDF2 axis is involved in the adipogenic differentiation of mouse fibroblast cells,497 suggesting a potential role of m6A remodeling in regulating CAF fate.
Aging
Aging is characterized by functional decline with time at the molecular, cellular, tissue, and organismal levels, constituting a predominant risk factor for mortality and various diseases, such as cancer, diabetes, cardiovascular diseases, and neurodegenerative diseases. Twelve hallmarks of aging are recapitulated at the molecular and cellular levels: genomic instability, epigenetic alterations, telomere attrition, loss of proteostasis, mitochondrial dysfunction, deregulated nutrient sensing, stem cell exhaustion, altered intercellular communication, cellular senescence, disabled macroautophagy, chronic inflammation, and dysbiosis.498,499 Although these hallmarks are interconnected in a complex network, some degree of hierarchical relationship exists between them, with epigenetic changes at the top of the hierarchy.498,500 Studies in simple organisms (e.g., yeast, worms, and flies) lead to the proposition of the “information theory of aging”, which states that loss of epigenetic information and collapse of gene expression networks with time caused aging,501,502,503,504 and recently this theory has been experimentally validated in mammal.505
DNA methylation and aging
As cells aging, genome-wide DNA methylation alteration occurs, featuring both loss of and gain of methylation at different sites (Fig. 6). The methylation alteration is conserved among species including mice, monkeys, and humans, and its rate is associated with lifespan.506 Loss of DNA methylation occurs primarily at repetitive DNA sequences where transposable elements enrich. These mobile sequences are highly methylated and form heterochromatin to suppress their activity within young cells. Hypomethylation contributes to heterochromatin loss and activation of (retro)transposition, leading to genomic instability and an increase in disease risk. As transposition-induced double-strand breaks can further aggravate loss of DNA methylation and heterochromatin through re-localization of the methyltransferases,505,507,508 loss of DNA methylation and activation of transposable elements may form a positive feedback loop. Reduced expression and/or activity of DNMTs contribute to the global demethylation process during aging. The expression of DNMTs responds to age-related decline of growth hormone, and their enzymatic activities are diminished due to reduced SAM/SAH ratio.509 Outside the heterochromatin regions, selected hypomethylation occurs in regulatory elements of genes (e.g., enhancers and promoters), resulting in elevated expression of the functional genes. Lineage-specific transcription factor binding and chromatin states in young cells facilitate gene demethylation with aging in specific cell types, such as ITGAL in T cells and Nkx6-1 in pancreatic β cells.510,511 Hypermethylation and age-associated heterochromatin formation usually occur at CpG-island-containing genes, such as metabolism-associated genes and cell-cycle genes, contributing to metabolic dysfunction and permanent close of the proliferation program, respectively.510,512,513 Genome-scale studies of DNA methylation dynamics in stem cells revealed that Polycomb target genes are preferentially hypermethylated with age, contributing to stem cell exhaustion during aging.514,515 Two alternative theories have been proposed for the hypermethylation of Polycomb target genes: cooperation, or competition between de novo DNA methyltransferases and Polycomb complexes, which remains further clarification.509 Since these hyper- or hypomethylated CpG sites show predictable and consistent shifts in average methylation level as individuals age, they are termed age-associated differentially methylated positions (aDMPs) or differentially methylated regions (aDMRs) that involve multiple contiguous CpGs. aDMPs and aDMRs might link to genes and pathways (as mentioned above) involved in an intrinsic age-related functional decline process occurring over chronological time516 (Fig. 6).

DNA methylation changes during aging and methylation clocks. Both global loss of and local gain of DNA methylation occur with age. age-associated differentially methylated positions (aDMPs) reflect an intrinsic age-related functional decline process occurring over chronological time in a population, while age-associated variably methylated positions (aVMPs) recapitulate the inter-individual heterogeneity in health status. A variety of DNA methylation clocks have been developed, generally using ElasticNet regression, to assess chronological age (chronological clocks) or biological age (biological clocks). A chronological clock can precisely predict calendar age irrespective of health conditions, while a biological clock can distinguish between people with different health conditions
Unlike aDMPs, there are CpG sites that show an increase in methylation variance with age, while the mean methylation level does not necessarily shift. These sites are referred to as age-associated variably methylated positions (aVMPs) or variably methylated regions if implicate multiple contiguous CpGs.516 Such methylation drifts with aging was first reported in studies of monozygotic twins, in which inter-individual variation in both global and locus-specific methylation is larger in the elderly than in the young.517 aVMPs represent a stochastic alteration of methylation accumulated during age as a result of differential environmental stimulation, contributing to deviation of biological age from chronological age. That is, aVMPs reflect inter-individual heterogeneity in health status (Fig. 6). Along with hypermethylated or hypomethylated CpG sites that converge on the mean (i.e., methylation level transition from 0→50% or 100→50%), aVMPs increase epigenetic disorder as measured by Shannon entropy.516,518 The heterogeneous methylation landscape is correlated with increased gene expression variability in cells and tissues of elders, including stem cells, pancreas, heart, and immune cells.519,520,521,522 Notably, this increased gene expression noise is an intrinsic mechanism of cellular aging, independent of cellular composition.520,521 Furthermore, some aVMPs are associated with gene expression in trans, which may affect fundamental aging mechanisms including DNA repair, apoptosis, and cellular metabolism.518 These results underscore the close correlation between DNA methylation and age, and the deconvolution of individual CpG sites facilitates the development of age-tracking clocks (see below).
Not only correlation but also DNA methylation change is a causative factor for aging and rejuvenation. DNMT1 haploinsufficiency-mediated insufficient DNA methylation promotes age-related diseases in mouse model.523 Ectopic DNA methylation interferes with the expression and function of pro-longevity transcription factors, such as FOXO3A and NRF2,500,524,525 indicating the essential role of DNA methylation homeostasis for lifespan. In line with this, centenarians and supercentenarians show younger-than-expected DNA methylation states at hundreds of CpG sites enriched for neuropsychiatric- and cancer-related genes,526 and prevention of age-related DNA methylation alteration contributes to successful age-delaying interventions, such as caloric restriction.512,527 More direct evidence that supports DNA methylation-mediated rejuvenation comes from the cellular reprogramming experiment in aged mice, in which ectopic expression of Yamanaka factors (OCT4, SOX2, KLF4) recovers youthful DNA methylation pattern and improves tissue function in a TET1/2-dependent manner.528 The expression and activity of TETs are also induced in other rejuvenating strategies in human cell and mouse models, including caloric restriction, heterochronic parabiosis, and metformin treatment.512 Moreover, a recent study showed that TET3-catalyzed 5hmC functions to inhibit intragenic transcription and assure transcriptional fidelity in airway smooth muscle cells, which prevents chronic inflammation in the lung.529 As loss of faithful transcription and increase in inflammation are drivers of aging,498,530 the discovery implies the additional benefit of TET expression for age-delaying.
DNA methylation is the best characterized age-associated epigenetic biomarker, and the DNA methylation clock (also known as epigenetic clock) is currently the most promising biological age predictor.531 It is built upon a linear regression model, usually, ElasticNet regression (Fig. 6). In the principle of the minimal cost function, informative CpG sites are selected and endowed with positive or negative coefficients, while non-informative CpG sites get the coefficient value of 0. The sum of each product of individual methylation level with its coefficient gives the estimated age. There are a variety of DNA methylation clocks developed with different characteristics and purposes, which can be grouped into three categories: chronological clocks, biological clocks, and hybrid chronological-biological clocks532 (Fig. 6). Chronological clocks, the first-generation DNA methylation clocks, are trained on exclusively on calendar age, such as Horvath’s pan-tissue clock and Hannum’s blood-specific clock which adopt 353 and 71 CpG sites, respectively.533,534 Profiling DNA methylation age at the single-cell level leads to the development of a statistical framework for the single-cell epigenetic clock, namely scAge, which recapitulates the chronological age of tissues and exhibits heterogeneity among cells.535 These chronological clocks predict calendar age with high accuracy (Pearson correlation between predicted age and chronological age can be over 0.9 in test data of Horvath’s and Hannum’s studies), even near-perfectly when enough training samples are available.533,534,536 Since calendar age per se is a strong predictor of organismal functional state and has biological meaning, residual values of estimated DNA methylation age by these chronological clocks and chronological age can be used to predict all-cause mortality to a certain degree.537 Lower DNA methylation age than chronological age was observed in centenarians and supercentenarians.526,538 Nevertheless, the capacity of chronological clocks to distinguish between healthy versus unhealthy individuals of the same calendar age is still limited and unsatisfactory, and increased prediction of chronological age accompanies decreased association with mortality.532,536,539 To deal with the problem, biological clocks, such as GrimAge and DunedinPACE, focus on biological phenotypes rather than calendar age, which enables them to predict age-related morbidity and all-cause mortality better than chronological clocks.540,541 GrimAge integrates DNAm surrogates (containing 1030 CpG sites) of seven plasma proteins and smoking pack-year to predict lifespan,542 and DunedinPACE trains DNA methylation data (containing 173 CpG sites) on a composite estimator of aging that reflects 19 biomarkers of organ-system integrity, including cardiovascular, immune, metabolic, hepatic, renal, dental, and pulmonary systems.541 Hybrid clocks track both chronological and biological age, as exemplified by PhenoAge. To build PhenoAge, DNAm data were trained on a combined phenotypic age estimator consisting of chronological age and nine biomarkers related to lifespan and healthspan, and 513 CpG sites were selected.543 Compared with chronological and biological clocks, hybrid clocks display intermediate prediction power for age-related diseases and lifespan.540,541 The development of these DNA methylation clocks greatly facilitates the evaluation of the aging rate and screening of potential anti-aging treatments.
The selected CpG sites and epigenetic clock scores are mathematical products, while the biological meaning and mechanism of epigenetic clock ticks remain elusive. Indeed, many CpG sites of epigenetic clocks are not located within genes, and their methylation changes are not strongly associated with gene expression.544 In addition, the linear models of epigenetic clocks presume that DNA methylation changes with age at a constant rate. In contrast, there are at least two nonlinear patterns where the rate of age-related DNA methylation changes is variable and age-dependent. The first pattern describes that some CpG sites exhibit rapid DNA methylation changes in early life and the change rate is stabilized in later life, which may be associated with the rapid development in early life.533,545 The second pattern describes a reverse mode: stable DNA methylation level in early life followed by rapid change in late life.546 CpG sites with a second pattern may be more pertinent to senescence and age-related diseases as a result of biological roles performed by the resident genes. For example, two CpG sites whose DNA methylation levels increase with age at an increasing rate are identified in the promoter region of KLF14.546 Since KLF14 directly regulates Treg cell differentiation via suppressing FOXP3 expression,547 hypermethylation and silence of KLF14 may contribute to increased levels of Treg cells and immunosenescence.546,548 Understanding what drives DNA methylation clocks tick and reconciling the current linear model with the non-linear aging process may shed light on the aging mechanisms and encourage the design of new age-tracking clocks.
Histone and non-histone protein methylation in aging
Intertwined with DNA methylation, histone methylation, especially at H3 lysines 4, 9, 27, and 36, changes and play a role during aging. The sum of these methylation changes links to common alterations in 3D genome structure with age, featuring loss of heterochromatin in repressive compartments, weakened euchromatin in active compartments, interfacing topological compartment switch, and elevated epigenetic disorder549 (Fig. 7a). Both H3K9me2 and H3K9me3 are histone marks for constitutive heterochromatin, and their levels change during aging. In human fibroblasts, the expression of methyltransferases for H3K9me2, G9a/GLP heterodimer, is reduced in an age-dependent manner, resulting in global reduction of H3K9me2 in the physiologically aged cells550 (Fig. 7a). Tethering H3K9me2-marked heterochromatin to the nuclear lamina, a structure called lamina-associated domain (LAD), is essential for gene regulation and lifespan.549,551 In addition to histone substrate, G9a/GLP methylate Lamin B1 (LMNB1) at K417, a component of the nuclear lamina, to facilitate the LAD formation by modulating the stability and localization of LMNB1 in young human fibroblasts, whereas this process is dampened in the old cells due to G9a/GLP deficiency550 (Fig. 7a). Studies in worms demonstrate H3K9me2 is required for lifespan extension induced by mitochondrial stress or specific gene mutations in components of the H3K4 methyltransferase complex (i.e., set-2, ash-2, and wdr-5),552,553 supporting a positive role of H3K9me2 in longevity regulation. However, a recent study showed H3K9me2 and its methyltransferases restrict the lifespan of long-lived worms with a mutation in insulin-like receptor gene daf-2.554 It will be intriguing to see whether such genomic context-dependent regulation of longevity by H3K9 di-methylation works in mammals. A decline in the global level of H3K9me3 was observed during normal mammalian aging, as well as in patients with premature aging diseases, such as Hutchinson-Gilford progeria syndrome (HGPS) and Werner syndrome.500,555,556 The expression of SUV39H1, an H3K9me3 methyltransferase, decreases with age in hematopoietic stem cells (HSC), contributing to perturbed heterochromatin and decreased B lymphopoiesis in the elders.557 Paradoxically, SUV39H1 is stabilized by progerin in a progeria mouse model (Zmpste24 mutation) that mimics HGPS, leading to increased H3K9me3, which dampens DNA repair and shortens lifespan.558 A decrease in global H3K9me3 level is involved in the α-KG-mediated rejuvenation of bone marrow MSCs, which ameliorates age-related osteoporosis in mice.559 Region-specific conversion of H3K9me2 to H3K9me3 and loss of H3K9me3 occur in somatic tissues of aged worms, potentially contributing to aging phenotype.560 Considering the distinct role of H3K9me2 and H3K9me3 in genomic structural organization,561 these results suggest the different role of the two repressive histone marks in the regulation of lifespan, as well as the complex mechanism of action for H3K9me3. Consistently, the H3K9me3 level decreases in the aging intestine of flies,562 while its level increases in the aging brain, accompanied by changes in its distribution.563 Single-cell epigenome analysis of mouse brain shows age-related loss of H3K9me3 occurs in the excitatory neurons but not in inhibitory neurons or glial cells, that is, cell-type specific alteration of this mark during the organ aging.564 Both H3K9me2 and H3K9me3 are implicated in the repression of senescence-associated secretory phenotype (SASP) genes.565 Therefore, further studies are needed to untangle to what extent the function of H3K9me2 and H3K9me3 is overlapped and divergent, as well as to clarify their tissue-specific and context-dependent mechanism in longevity regulation.

Histone, nonhistone protein, and RNA methylation changes with age. a Alterations in histone and nonhistone protein methylation and 3D genome structure with age. G9a/GLP-mediated methylation of Lamin B1 (LMNB1) promotes heterochromatin assembly at the nuclear periphery and formation of lamina-associated domains (LADs) in young cells, which ensures transcriptional silence; whereas defects in both histone methylation (H3K9me2/3) and nonhistone protein methylation are associated with chromatin detachment from the nuclear lamina and a shift to a euchromatin state. b Overall changes of m6A modification with age in different tissues including brain, peripheral blood mononuclear cells, ovary, and intestine, and crucial age-related targets. Changes in the expression of writers or erasers account for the methylation fluctuation. Although the m6A level and expression of modifiers do not differ significantly between young and old mouse hearts, they do show aging-related differences in response to acute cardiac ischemia/reperfusion injury, which may be associated with reduced tolerance to ischemic injury with age
In line with the heterochromatin loss theory of aging, H3K27me3 which marks facultative heterochromatin acts as a positive regulator of lifespan. In worms, increasing the global H3K27me3 level by knockdown of H3K27me3 demethylase UTX-1 extends lifespan.566,567 A global higher level of H3K27me3 contributes to a stable epigenome and exceptional longevity in naked mole rats, compared with that in mice.568 The total H3K27me3 level is reduced with age in worms, senescent mammalian fibroblast cells, and progeroid mammalian cells (e.g., HGPS patients and engineered ICE mice).505,556,567,569 Down-regulation of the methyltransferase EZH2 is implicated in the decline of H3K27me3 level in these normal and pathological aging mammalian cells.556,569 Loss of H3K27me3 facilitates transcriptional activation of multiple pro-aging genes/pathways, such as SASP genes, insulin/IGF-1 signaling pathway, cell identity-related genes, and p16INK4A.505,566,569,570 In contrast, there’s plenty of evidence for a negative role of H3K27 tri-methylation in longevity regulation. H3K27me3 increases with age in flies, murine HSCs and quiescent satellite cells, and brain tissues of senescence-accelerated prone mouse 8.571,572,573,574 Deposition of H3K27me3 dampens a battery of stress responses (e.g., heat shock response, oxidative stress response, and mitochondrial unfolded protein response) and is detrimental to lifespan in worms and flies, which is rescued by overexpressing the H3K27me3 demethylases or downregulating PRC2 complex, leading to a longer lifespan.575,576,577 The correlation of the murine demethylase homologs (PHF8 and KDM6B) with longevity and mitochondrial stress response was also observed.575 Furthermore, enhancing glycolysis is helpful for longer and healthier lifespans in flies and humans, whereas the expression of glycolytic genes is suppressed with age by H3K27me3 deposition in Drosophila.571,578 Therefore, the positive or negative regulatory role of H3K27 tri-methylation in longevity is dependent on the specific loci and cell types. Since both genome-wide gain and loss of H3K27me3 can occur in aging mammalian cells,570,573 it is conceivable the net impact of the redistribution on aging relies on the specific context. In addition, lifespan extension in worms by overexpressing the H3K27 demethylase JMJD-3.2 does not require its catalytic activity.579 Similarly, upregulation of KDM6A ameliorates age-induced cognitive deficits in male mice independent of its demethylase activity,580 providing an additional regulatory layer in age and age-related diseases by H3K27 methylation modifiers.
Like H3K27me3, the role of H3K4me3 in lifespan regulation is highly context-dependent, and both positive and negative roles have been observed. In C. elegans, downregulation of H3K4me3 level, by disrupting the methyltransferase complex (e.g., SET-2), transient exposure to ROS during early development, or metformin treatment, extends lifespan, while upregulation of the mark via inactivating the demethylases (RBR-2 or SPR-5) shortens lifespan.581 The pro-longevity effect of H3K4me3 deficiency in worms is dependent on the presence of an intact adult germline, germline-intestine communication, accumulation of mono-unsaturated fatty acids, and increased stress resistance.581,582,583,584 Similarly, the global H3K4me3 level is sensitive to ROS and negatively regulates the stress resistance in mammalian cells.584 Elevation of H3K4me3 level with age is observed in murine HSCs and human immune cells.573,585 Furthermore, the H3K4me3 level and its methyltransferases are upregulated in the prefrontal cortex of patients and mouse models with Alzheimer’s disease (AD), and pharmacological targeting of the SET1/MLL HMTs ameliorates cognitive and synaptic deficits in AD mice.586 Specific gene targeted for H3K4 methylation during mammalian aging is exemplified by WTAP: downregulation of the demethylase KDM5A in senescent nucleus pulposus cells facilitates H3K4me3 deposition at the WTAP promoter and enhances the gene expression, promoting the progression of intervertebral disc degeneration (IVDD), one of the most strongly age-associated degenerative diseases.587 Paradoxically, knockdown of H3K4 demethylases (RBR-2 or LSD-1) is sufficient to extend longevity in worms,588,589 and a recent study showed a decrease in H3K4me3 level by mutation of the methyltransferase SET-2 shortens lifespan, accompanying with loss of fertility but normal intestinal fat stores.590 In humans, the level of H3K4me1 and H3K4me3 is decreased in aged HSCs,591 and the number of genes with H3K4me3 loss surpasses that of genes with H3K4me3 gain (in a 6:1 ratio) during postnatal development and aging of prefrontal neurons,592 implying a pro-aging effect of H3K4me3 deficiency. Multiple sources of variation, such as genetic backgrounds, uncontrollable daily fluctuations in the microenvironment, and quality and purity of reagents, can cause stochasticity in aging phenotype and potentially contributes to the different lifespan extension outcomes in the model organisms.590,593,594 In addition, studies in yeast revealed accumulation of H3K4me3 with age contributes to loss of rDNA heterochromatin and genome-wide increase in pervasive transcription that potentially impairs lifespan, meanwhile maintaining normal lifespan by facilitating the expression of many genes crucial for healthy aging, such as NAD+ biosynthesis genes and histone genes.595,596 Therefore, beneficial and detrimental effects of H3K4 methylation on aging phenotype are potentially influenced by the multiple variable sources and disproportionally manifested at cell, tissue, and organismal levels, the combination of which biases longevity outcomes.
H3K36me3 was initially reported to be a pro-longevity histone mark in yeast, worms, and flies. Genetic studies in these simple organisms showed that loss of H3K36me3 by inactivating the methyltransferase shortens lifespan, while gain of H3K36me3 by deleting the demethylase extends lifespan.597,598 Mechanically, H3K36me3 at gene bodies prevents cryptic transcription597 and/or limits transcriptional change with age.598 A similar mechanism was recently demonstrated in murine hematopoietic and neural stem cells and human mesenchymal stem cells, in which loss of H3K36me3 within gene bodies during aging compromises transcriptional fidelity.530 Specifically, the recruitment of H3K4me3 demethylase and de novo DNA methyltransferases is suppressed in the absence of H3K36me3, generating intragenic permissive chromatin state (i.e., accumulation of H3K4me3 and unmethylated CpGs) that supports cryptic transcription in the aged mammalian stem cells. The level of H3K36me3 also declines with age in brain tissues of a senescence-accelerated mouse model.574 SETD2-mediated H3K36 trimethylation promotes osteogenesis of bone marrow mesenchymal stem cells (MSCs) by facilitating the transcriptional initiation and elongation of the Lbp gene in mouse models, and such axis is decreased in aged bone marrow, which may contribute to age-related osteoporosis.599 The DNA methylation age is substantially accelerated in patients with loss-of-function mutations in the H3K36 methyltransferase NSD1.600 These results indicate H3K36 methylation-mediated transcriptional regulation is an important and conserved anti-aging mechanism from yeast to humans.
In addition to protein lysine methylation, protein arginine methylation and methyltransferases (PRMTs) are involved in lifespan regulation. PRMT1 is a positive regulator of stress tolerance and lifespan in worms and moths by asymmetrically methylating the crucial pro-longevity transcription factors, DAF16/FOXO and SKN-1/NRF.601,602,603 Asymmetric arginine methylation that is catalyzed by type I PRMTs is downregulated during replicative and H2O2-induced premature senescence in human fibroblasts.604 The expression of three PRMT family members (PRMT1, PRMT4, and PRMT5) in rats is tissue-specific and age-dependent, suggesting tissue-specific role of PRMTs in the aging process of different tissues.605 Consistently, tissue-specific expression of PRMT8 and proper accumulation of asymmetric dimethyl arginines in murine postmitotic neurons are required for protection against the age-related increase in DNA damage and cell death.606 As PRMT activity against histone mixtures changes when tissues age,605 future studies should determine whether PRMTs transcriptionally regulate age-related genes directly by histone arginine methylation.
RNA methylation and aging
The global m6A RNA modification is diminished in peripheral blood mononuclear cells of elders and in replicative senescent human fibroblasts, which is associated with changes in the expression of AGO2 and the abundance of miRNAs607 (Fig. 7b). The protein level of m6A methyltransferases (METTL3 or METTL14) is reduced in the fibroblasts and mesenchymal stem cells (MSCs) derived from HGPS patients, due to the loss of Lamin A-mediated protection from proteasomal degradation.608,609 Cell cycle-related genes, including MIS12, are enriched in the subset of demethylated mRNAs, contributing to premature aging.608 Depletion of METTL3 and RNA m6A was also observed in MSCs of individuals with Werner syndrome,608 suggesting dysfunction of the m6A modification pathway is common during normal and premature aging with different etiological factors. In line with this, de-repression of endogenous retroviruses (ERVs) is common during aging and contributes to cellular senescence, while the m6A-YTHDF pathway directly restricts mRNAs of ERVs (e.g., intracisternal A-particles) to maintain cellular integrity.610 Overexpression of METTL14 alleviates replicative senescence and premature senescence in human skin fibroblasts and Zmpste24−/− mouse embryonic fibroblasts, respectively,609 while upregulation of demethylase FTO is implicated in the IL-17A-induced cellular senescence of human endothelial cells.611 On the other hand, m6A modification is enhanced during senescence and contributes to senescence and age-related diseases. For example, METTL3 expression and m6A level are elevated in fibroblast-like synoviocytes (FLS) of patients with osteoarthritis, which inhibits autophagy and promotes senescence of the cells by targeting ATG7 mRNAs (an enzyme crucial for the formation of autophagosomes) for degradation.612 Inhibition of METTL3 ameliorates the FLS senescence and osteoarthritis progression in mouse models. WTAP- or METTL14-mediated m6A modification of non-coding RNAs, such as lncRNA NORAD and miRNA miR-34a-5p, promotes senescence of nucleus pulposus cells (NPCs) and development of IVDD.587,613 Things are more complex. Low fluid shear stress induces redistribution of more than one thousand m6A peaks, in which hyper-/hypomethylated genes are enriched in aging-related (e.g., mTOR, insulin, and ERRB) and oxidative stress-related (e.g., HIF1A, NFE2L2, and NFAT5) pathways, which mediates the cellular response to oxidative stress and senescence.614 ALKBH5-mediated demethylation of DNMT3B transcripts increases DNMT3B expression, which in turn downregulates E4F1 expression by DNA methylation, contributing to NPC senescence and IVDD degeneration.615 These results suggest that m6A RNA modification works in a cell-type-specific and context-dependent manner to promote or delay cellular senescence.
Unlike the relatively well-studied cellular senescence, the studies about the role of m6A RNA methylation, especially mRNA methylation, in the aging of organs and organisms are in their infancy. This is partly due to the absence of genes encoding mRNA m6A methyltransferases METTL3/METTL14 in C. elegans, the most used models for organismal aging. However, two rRNA m6A methyltransferase genes ZCCHC4 and METTL5 in their genome regulate lifespan.616,617 zcchc-4 mutation extends worm longevity under homeostatic conditions while mettl5 mutation does it under stress conditions, which is associated with translational adjustment to cope with endogenous and environmental stresses.616,617,618 METTL5-deficient mice show abnormal morphology and behavior,619 nevertheless, whether mammalian rRNA m6A methyltransferase homologs regulate aging and lifespan remains unknown.
The correlation of mRNA m6A modification pathway with the aging of certain mammalian organs, including intestine, ovary, heart, and brain, has been recorded (Fig. 7b). The levels of m6A modification decrease in mouse intestine as a consequence of aging.620 Further study demonstrated METTL14-mediated m6A methylation is required for intestinal integrity and normal lifespan of Drosophila melanogaster, partly through the downstream target, Lamin B receptor (LBR).620 Reduced expression of FTO and ensuing elevated m6A level occur in the aged mouse ovary, as well as in granulosa cells of the aged human ovary.621,622 Consequently, the degradation of a key downstream target mRNA, FOS, is interrupted with the help of reader protein IGF2BP2, contributing to ovarian aging.621 Although the m6A level and expression of modifiers do not show significant differences in young and old mouse hearts, they exhibit aging-related differences in response to acute cardiac ischemia/reperfusion injury, which might be associated with reduced tolerance to ischemic injury with age.623 The potential role of m6A in brain aging acquires the most attention, as m6A methylation strongly associates with brain development and function.624 The level of m6A methylation is increased with age in the human brain tissue region BA9.625 A similar trend occurs in the mouse cerebral cortex and hippocampus.625,626 In contrast, a recent study reported an opposite trend across mouse brain regions.627 Abnormal m6A methylation has been observed in neurodegenerative diseases, such as Parkinson’s disease (PD) and AD.625,628 The precise mechanisms, including directionality, magnitude, and functional consequences of m6A alterations, are controversial,625,627,629,630,631 with the latest studies supporting human AD is associated with loss of m6A methylation.627,632 Moreover, upregulation of METTL3 alleviates AD in both in vitro and in vivo models,632 indicating m6A methylation matters in brain aging and neurodegenerative diseases and serves as a potential therapeutic target. In addition, an intron sequence polymorphism of the YTHDF2 gene with enhanced transcription is associated with exceptionally long lifespan in humans, implying high expression of YTHDF2 is beneficial for longevity.633 Consistently, YTHDF2 is responsible for the degradation of m6A-modified mRNAs encoding inflammatory pathway genes and maintains HSC function upon aging in mice,634 and its upregulation underlies the anti-senescence effects of melatonin or senolytics cocktail (dasatinib and quercetin) in human cells by suppressing the MAPK-NF-κB pathway.611,635 The above preliminary results warrant future studies to clarify whether changes in the m6A methylation pathway have a causative relationship with mammalian aging.
The role of RNA m5C methylation in senescence and aging is emerging, implicating nearly all types of RNAs, i.e., rRNAs, tRNAs, miRNAs, mRNAs, and lncRNAs. Two rRNA m5C methyltransferases, NSUN-1 and -5 that each methylate one of the two known m5C sites in the large ribosomal subunit, play an important role in the lifespan regulation in simple organisms. Deficiency of NSUN5-mediated rRNA m5C modification extends lifespan in yeast, worms, and flies, which is associated with the enhanced translation of stress-responsive mRNAs as a result of altered local ribosome structure and translational fidelity.636 Soma-specific rather than whole-animal knockdown of NSUN-1 in worms prolongs lifespan, accompanied by reduced body size and impaired fecundity.637 While NSUN5 knockout mice show a decrease in body weight, lean mass, and protein synthesis in several tissues, whether NSUN-1 and −5 regulate the lifespan of mammals remains future clarification.638 The two tRNA methyltransferases, DNMT2 and NSUN2, stabilize m5C-modified tRNAs by protecting them from angiogenin-mediated cleavage and promoting stress tolerance.639 Overexpression of DNMT2 extends the lifespan of flies,640 while the silence of DNMT2 suppresses proliferation and induces senescence in human fibroblasts.641 Loss of NSUN2-mediated tRNA m5C modification contributes to neurodevelopmental disease as a result of an accumulation of tRNA-derived small RNA fragments.642 Beyond m5C methylation and tRNA substrates, NSUN2 plays protective roles in preventing neurodegeneration (e.g., AD) and delaying replicative senescence, through targeting miRNAs (miR-125b) and mRNAs (p27 and CDK1) for m6A and m5C modification, respectively.643,644 Unlike generally downregulated during replicative senescence, the NSUN2 expression is stimulated in the stress conditions (e.g., oxidative stress and high glucose level), leading to premature senescence of cells.645,646 Such processes involve downstream senescence-related mRNAs, including SHC, p21, and p16, and the m5C methylation promotes their stability and/or translation.645,646,647 Interestingly, NSUN2-mediated-m5C and METTL3/14-mediated m6A modifications mutually promote each other at the 3′UTR of p21, which synergistically enhances the translation of p21, promoting oxidative stress-induced cellular senescence.645 In addition, m5C modification at the lncRNA subunit TERC of telomerase holoenzyme is necessary for telomerase function,648 and interruption of the RNA methylation causes telomere attrition, driving replicative senescence of mouse and human cells.648,649 The RNA methyltransferase responsible for the methylation of lncRNA TERC is yet to be investigated.
Targeting methylation for therapy
Targeting aberrant DNA methylome
Two types of small molecular inhibitors are developed to inhibit DNMTs: nucleoside analogs and non-nucleoside molecules. The nucleoside DNMT inhibitors, including azacitidine (5′-azacytidine) and decitabine (5-aza-2′-deoxycytidine), are incorporated into replicating DNA, resulting in covalently sequestering DNMTs (DNMT1, DNMT3A, and DNMT3B), degradation of these enzymes by proteasome, and DNA damage. Azacytidine is also integrated into RNA, inhibiting protein synthesis. Therefore, these inhibitors cause direct cytotoxicity at high doses and are used at low concentrations to induce genome-wide DNA demethylation. Azacytidine and decitabine were approved in the early 2000s for the treatment of myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), and chronic myelomonocytic leukemia.650 The anti-cancer activities of the DNMT inhibitors involve both directly targeting tumor cells and enhancement of antitumor immunity. A direct consequence of DNA demethylation is the reactivation of silenced tumor suppressor genes, leading to cell-cycle arrest, differentiation, and apoptosis.651 De-repression of MHC-I genes and cancer/testis antigens promote T cells to recognize cancer cells, and de-repression of ERVs induces a state of viral mimicry in cancer cells, which induces type I interferon signaling and facilitates immune elimination. As mentioned above DNA methylation dynamics is important for the function of nontumor cells within TME, and the anti-cancer effects of DNMT inhibitors are associated with the modulation of these cells. For example, decitabine treatment promotes CD8+ T cell activation and effector function, which is due in part to the overexpression of NFATc1 short isoforms and reversal of exhaustion-associated DNA methylation program.318,652 Demethylation-mediated expression reprogramming rather than direct cytotoxicity is consistent with the prolonged time for initial response.653 Although a positive response to azacytidine treatment is well-documented in patients with MDS, the hypomethylating agent causes demethylation and upregulation of an oncogene (SALL4) in 30–40% of patients, associated with a worse outcome.653,654 That is, DNMT inhibition activates not only tumor suppressor genes but also oncogenes, discounting the therapeutic effect of global demethylation. Moreover, these nucleoside DNMT inhibitors show limited clinical efficacy as monotherapy in solid cancers. DNMT inhibition stimulates the recruitment of MDSCs to TME, dampening antitumor immunity.655 Besides the tumor-intrinsic mechanisms, low chemical stability and poor pharmacokinetic properties of the nucleoside DNMT inhibitors also account for the limited clinical efficacy.
To overcome the poor pharmacokinetic properties and dose-limiting toxicity of nucleoside analogs, non-nucleoside DNMT inhibitors that directly block the catalytic activity are pursued. RG-108, one of the first nonnucleoside inhibitors, can effectively inhibit bacterial M.SssI CpG methyltransferase (half maximal inhibitory concentration (IC50) = 0.012 μM),656 while was reported later with reduced potency against human DNMT1 (half maximal effective concentration (EC50) = 390 μM).657 Quinoline-based inhibitors, such as SGI-1027 and its analog MC3343, inhibit DNMT1 and DNMT3A far more potent than RG-108.657 The antitumor effect of MC3343 was confirmed in an osteosarcoma patient-derived xenograft.657 Intriguingly, there is an inverse relationship between MC3343 sensitivity and DNMT1/3A expression (r > 0.6), but not for the traditional inhibitor decitabine, nevertheless this relationship needs further validation.657 GSK3685032 which contains a dicyanopyridine moiety is a potent first-in-class selective inhibitor of DNMT1 (IC50 = 0.036 μM). It works by competing with the active-site loop of DNMT1 for penetration into hemimethylated DNA between two CpG base pairs.658 Compared with decitabine, GSK3685032 exhibits improved tolerability and efficacy in mouse models of AML and may provide more benefit in the clinic.658 Following studies show the demethylation and antitumor effect of GSK3685032 in peripheral nerve sheath tumor and colorectal cancer in vitro or in vivo,659,660 and identify several additional dicyanopyridine-containing DNMT1-selective, nonnucleoside inhibitors.661 Some old drugs, such as hydralazine, procainamide, and epigallocatechin gallate, are repurposed to inhibit DNMT activity and reactivate silenced genes in cancer or noncancer cells.662,663,664 Epigallocatechin gallate, a major polyphenol from green tea, competitively represses DNMT activity possibly by forming hydrogen bonds with the five key amino acids in the catalytic pocket.662 A different study shows epigallocatechin gallate possesses significant cytotoxicity and genotoxicity in human cancer cell lines, but fails to cause significant demethylation of genomic DNA.665 Epigallocatechin gallate also can inhibit HDACs and regulate noncoding RNA networks in different types of cancer.666 A similar pleiotropic effect likely applies to other repositioned old drugs.
While DNMT inhibitors have been used to target aberrant DNA methylation for the treatment of TET2, IDH1, and IDH2 mutant diseases,667,668 specific agonists or inhibitors are developed (Table 1). Vitamin C is crucial for the TET-mediated oxidation of 5mC via physical interaction with the TET catalytic domain and providing an electron to reduce Fe3+ to Fe2+.669 Vitamin C treatment mimics the effects of TET2 restoration in the Tet2-deficient mouse model of leukemia, which promotes DNA demethylation and suppresses leukemia progression.670 The majority of TET2 mutations in AML are heterozygous, making vitamin C a potent hypomethylating agent for therapy.671 In a 1-year clinical trial with oral 1 g/day vitamin C supplementation, the proportion of hypermethylated loci is reduced, accompanied by reduced gene expression divergence between lymphoma predisposition TET2+/− and control TET2+/+ individuals.672 Two clinical trials are engaged using vitamin C alone or in combination with azacytidine in MDS patients with TET2 mutations, i.e., NCT03433781 (Recruiting) and NCT03397173 (Completed, yet no result has been published). TET2 loss-of-function mutations and IDH1/2 neomorphic mutations are mutually exclusive in the AML cohort, and they show similar DNA methylation alteration.673 IDH mutants produce oncometabolite D-2-hydorxyglutarate (D2-HG) that competitively inhibits α-KG-dependent TET enzymes. In addition to disturbed DNA demethylation, JMJC domain demethylase-mediated histone demethylation and FTO-mediated RNA demethylation are also impaired.674,675,676 Therefore, it is not surprising that treatment of IDH mutant tumors with DNA demethylating agents works modestly,282,650,677 and inhibitors targeting mutant IDH proteins are developed. Enasidenib and ivosidenib were approved by FDA in 2017 and 2018 for adult patients with relapsed or refractory (R/R) AML with IDH2 and IDH1 mutations, respectively. The clinical trials showed the overall response rate is 41.6% for ivosidenib in IDH1-mutated patients with R/R AML (NCT02074839) and 38.8% for enasidenib in IDH2-mutated patients (NCT01915498).678,679 Clearance of IDH mutant clones is correlated with the complete remission.678,679 The frequency of IDH1 mutations is high in patients with glioma (~70% of lower-grade gliomas), therefore, the development of inhibitors capable to penetrate the blood-brain barrier is essential for therapy in these patients.680 Vorasidenib is the first-in-class, brain-penetrant dual inhibitor of mutant IDH1/2 for glioma therapy.681 In a first-in-human phase I trial (NCT02481154), vorasidenib is well tolerated, effectively reduces D2-HG level by about 93% in patients with recurrent or progressive IDH1-mutant glioma, and displays a preliminary antitumor effect in the subset of patients with non-enhancing glioma.682,683 DS-1001, another new brain-penetrant mutant IDH1 inhibitor, exhibits well distribution to the mouse brain and the ability to reduce D2-HG and suppress tumor growth in preclinical studies.684 The first-in-human phase I study of DS-1001 (NCT03030066) reported patients with recurrent or progressive IDH1-mutant gliomas respond to the treatment with well tolerance.680 A study of the inhibitor in patients with chemotherapy- and radiotherapy-naïve IDH1-mutated glioma is ongoing (NCT04458272).
Targeting aberrant histone methylome
EZH2 and H3K27me3 methylome
EZH2 overexpression or gain-of-function mutation is associated with cancer progression and poor prognoses in many solid cancers and hematologic malignancies, such as prostate cancer, breast cancer, lymphoma, and AML.685,686,687 Inhibition of EZH2 or other PRC2 components diminishes the H3K27me3 level and derepresses the expression of tumor-suppressive genes, impairing cell proliferation and tumor growth in vivo. Multiple inhibitors are developed to target the SET domain of EZH2 to inhibit the catalytic activity, including EPZ-6438 (tazemetostat),688 CPI-1205,689 GSK126,690 C24,691 and UNC1999.692 Tazemetostat, a first-in-class oral EZH2 inhibitor, was approved by FDA in 2020 for the treatment of relapsed follicular lymphoma (Table 1). Supporting the approval, tazemetostat monotherapy exhibited meaningful and durable responses in patients with R/R follicular lymphoma in a phase II trial (NCT01897571).693 Specifically, the objective response rate is 69 and 35% and includes 13 and 4% complete response in the EZH2 gain-of-function mutant and wild-type cohorts, respectively.693 The antagonistic relationship between PRC2 and SWI/SNF chromatin remodeling complex provides a rationale for the use of EZH2 inhibitors in cancers with loss-of-function mutations in SWI/SNF complex members where unopposed EZH2 activity increases H3K27me3 level and causes excessive gene silence.694 Preclinical studies indicate inhibition of EZH2 is synthetic lethal with inactivation of SWI/SNF in a range of cancers with mutations in SWI/SNF subunits, such as PBRM1, ARID1A, SMARCA2, and SMARCB1.695,696,697,698 An international, open-label, phase II basket study (NCT02601950) in advanced epithelioid sarcoma with loss of SMARCB1 showed tazemetostat is well tolerated with clinical activity (15% overall response);699 the results of this trial supported the FDA approval in 2020 for patients with metastatic or locally advanced epithelioid sarcoma not eligible for complete resection. Likewise, the pathognomonic SS18-SSX fusion protein in synovial sarcomas induces a state of SMARCB1-deficiency, conferring cancer cells with this chromosomal translocation sensitive to tazemetostat.700 Some H3K27me3-high malignancies are relatively tolerant to EZH2 inhibitors due to EZH1 compensation for EZH2 loss.701 This necessitates the development of EZH1/2 dual inhibitors, including valemetostat. Preclinical studies demonstrated that valemetostat is superior to an EZH2 selective inhibitor OR-S0 in terms of H3K27me3 depletion and antitumor efficacy.702 In a phase II trial of valemetostat in R/R adult T-cell leukemia/lymphoma (ATL) (NCT04102150), this inhibitor showed promising efficacy and tolerability, i.e., twenty-five patients have an overall response rate of 48%, including five complete remissions,703 which supports the drug approval in 2022 for treatment of aggressive ATL in Japan. Clinical trials of valemetostat in other cancer types (e.g., peripheral T-cell lymphoma, B-cell lymphoma, and tumors with SMARCB1 deficiency) and of two other dual inhibitors (HM97662 and HH2853) are underway704 (Table 1).
Despite these advances, the EZH2 enzymatic inhibitors cannot fully suppress the oncogenic function of EZH2, as non-catalytic or non-canonical activity of EZH2 also contributes to tumorigenesis.705 EZH2 acts as a coactivator for critical transcription factors to promote gene expression and oncogenesis, such as androgen receptor in prostate cancer,706 NF-κB in breast cancer,707 and MYC/p300 in MLL1-rearranged (MLL-r) leukemia.708 To repress the multifaceted activities of EZH2, an EZH2-targeting degrader MS177 was developed based on proteolysis-targeting chimera (PROTAC) technology.708 MS177 induces effective degradation of both canonical EZH2–PRC2 and noncanonical EZH2–MYC complexes, contributing to fast and more potent suppression of cancer growth compared with the enzymatic inhibitors.708
Careful monitoring of patients is required when targeting EZH2, as PRC2 can also function as a tumor suppressor. Loss-of-function mutations of EZH2 or other core PRC2 components (EED and SUZ12) are observed in multiple cancers, such as MDS,709,710 T cell acute lymphoblastic leukemia,711,712 malignant peripheral nerve sheath tumor (MPNST),713 and melanoma.714 Moreover, inactivating mutations appear to dominate all of the EZH2 mutations among 10,967 pan-cancer samples in The Cancer Genome Atlas.715,716 PRC2 inactivation leads to a global loss of H3K27me2/3 and aberrant transcriptional activation of carcinogenic signaling pathways (e.g., Ras signaling pathway) and developmentally silenced master regulators.713,714 Therapeutically targeting loss-of-function mutations of PRC2 members remains an unmet need. A recent RNAi screen targeting 565 known epigenetic/chromatin regulators identifies DNMT1 synthetic lethality with PRC2 inactivation in MPNST.659 Mechanistically, PRC2 inactivation enhances DNMT inhibition-mediated activation of retrotransposons and subsequent viral mimicry response.659 DNMT and EZH2 inhibitors synergize to amplify antitumor immune response in hepatocellular carcinoma with wild-type EZH2,717 consolidating DNMT1-targeted therapy can be used to treat PRC2-loss cancer. Additionally, PRC2 loss induces an epigenetic switch in NF1 mutant cancers, sensitizing these cancers to BRD4-based therapies.714 Collectively, different strategies are developed to target cancers with EZH2 hyperactivity (inhibition of EZH2 enzymatic activity alone, EZH1/2 dual inhibition, or suppression of both canonical and non-canonical activity of EZH2) or with PRC2 loss (synthetic lethality with other targeted therapy), which provides an excellent paradigm to target a histone modifier with context-dependent functions and multiple acting mechanisms.
Inhibitors of other histone methyltransferases and demethylases
High-quality inhibitors have been developed for other histone methyltransferases, including DOT1L, PRMT5, G9a, GLP, SMYD2, SMYD3, SETD7, SUV420H1/2, and PRDM9.651 These inhibitors enable convenient evaluation of cancer cell dependency on the methyltransferases in different contexts and exploration of potential clinical use. The advancement of the DOT1L inhibitor into clinical trials provides a paradigm (Table 1). DOTIL drives the progression of multiple cancers, such as MLL-r leukemia,718 renal cell carcinoma,719 ovarian cancer,720 and breast cancer.721 DOT1L-mediated H3K79 methylation promotes transcriptional elongation and/or enhancer-promoter interaction,722 which is hijacked by oncogenic transcription factors, such as MLL-fusion proteins in MLL-r leukemia718 and estrogen receptor α in breast cancer,721 to activate downstream target genes. For this reason, DOT1L inhibitors were tested to treat MLL-r leukemia and antiestrogen-resistant breast cancer. Pinometostat (EPZ-5676), a first-in-class SAM competitive inhibitor, showed potent and selective inhibition with a subnanomolar affinity for DOT1L and encouraging curative effect in preclinical models.723 In a phase I trial in adult patients with advanced acute leukemias, particularly those bearing MLL-r, pinometostat diminishes H3K9me2 level and shows modest clinical activity, i.e., 2 of 51 patients bearing MLL-r experience complete remission.724 To further improve DOT1L inhibition-mediated therapy for MLL-r leukemia, the combination of pinometostat with existing standard-of-care drugs for acute leukemias, including DNMT inhibitors, was suggested according to synergistic anti-cancer activity observed in preclinical studies.725 Moreover, inhibition of DOT1L shows potent anti-cancer activity in a nude rat xenograft model of Dnmt3a-mutant AML.726 These results fueled two trials assessing pinometostat in combination with either standard-of-care chemotherapy (NCT03724084) or with azacytidine (NCT03701295) to treat R/R MLL-r leukemia. Currently, the former trial is terminated and the latter is completed, yet no result has been published.
Chemical interference of histone demethylases for cancer therapy also acquires great attention, with inhibitors of LSD1 and KDM4C advancing into clinical trials (Table 1). The FAD-dependent H3K4/H3K9 demethylase LSD1 is aberrantly expressed in multiple types of cancer, promoting cancer progression through regulating chromatin accessibility. Targeting LSD1 is becoming an emerging option for cancer therapy. There are two types of LSD1 inhibitors: irreversible and reversible inhibitors.727 Tranylcypromine (TCP), an inhibitor of monoamine oxidases (MAOs), has been identified as able to irreversibly repress LSD1, due to the sequence similarity between LSD1 and MAOs.728 Preclinical studies showed TCP treatment represses clonogenic potential and induces the differentiation of MLL-r leukemia stem cells and similarly the differentiation of all-trans-retinoic acid (ATRA)-insensitive AML cells through selective increase in H3K4me2 at related genes,729,730 which supports initiation of a phase I clinical trial to assess the safety and activity of ATRA plus TCP in patients with R/R AML and MDS(NCT02273102). Encouraging results that recapitulate preclinical studies were recorded, i.e., LSD1 inhibition sensitizes AML cells to ATRA, with an acceptable safety profile.731 The selective increase rather than a large-scale genome-wide increase of H3K4me2 may contribute to the low toxicity of LSD1 inhibition.729,730 Potential side effects of TCP as an LSD1 inhibitor, including drowsiness, dizziness, and orthostatic hypotension, are mostly attributed to the concomitant inhibition of MAOs.732 To improve selectivity and potency, multiple new inhibitors are developed using TCP as the lead compound, including ORY-1001, ORY-2001, GSK2879552, IMG-7289, and INCB059872. Clinical trials of these compounds for a range of cancers are ongoing, as well as other diseases including Alzheimer’s disease, multiple sclerosis, and myelofibrosis. Unlike irreversible inhibitors that covalent bind to the cofactor FAD of LSD1, reversible inhibitors act through competing for substrate or FAD-binding site or allosteric regulating LSD1 activity.733 Reversible inhibitors are clinically preferred due to potentially safer metabolism and lower toxicity. A large number of reversible LSD1 inhibitors are developed;727 two of them, CC-90011 (substrate competitive inhibitor)734 and SP2577 (allosteric inhibitor)735 have entered clinical trials for therapy of cancers including sarcomas, non-Hodgkin’s lymphomas, and small cell lung cancer (Table 1). Preliminary results of the phase I trial (NCT03600649) of SP-2577 in patients with R/R Ewing sarcoma exhibit a manageable safety profile with proof-of-concept preliminary antitumor activity, which supports the planned phase II expansion.736 KDM4C is an α-KG-dependent histone H3K9 demethylase that is amplified in various types of cancers, such as esophageal squamous cell carcinoma (ESCC), breast cancer, and medulloblastoma.737,738 Our lab and others demonstrated that KDM4C promotes proliferation and stemness of cancer cells by removing H3K9me3 from serine pathway genes (PHGDH and PSAT1) and stem cell-related genes (NOTCH1, NANOG, and ALDH1A3), respectively, which is disrupted by inhibition of KDM4C.737,739,740,741 Caffeic acid (CA), a micromolar KDM4C inhibitor,742 suppresses the demethylation activity in ESCC cells, accompanied by reduced cancer cell stemness,739,743 and phase III trials of targeting KDM4C with CA in ESCC patients are ongoing744 (NCT04648917 and NCT03070262).
Targeting aberrant mRNA methylome
Targeting the mRNA methylation pathway presents a new direction for cancer therapy.745 METTL3 is the primary mRNA m6A methyltransferase, and the interest in the development of METTL3 inhibitors has been stimulated since the reported key oncogenic role of the gene in leukemia in 2017.746,747 STM2457 is a non-SAM analogous small molecule but competes for the SAM-binding site of METTL3 (IC50 = 16.9 nM), and is the first bioavailable METTL3 inhibitor that exhibits anti-cancer efficacy in a preclinical cancer model.748 Administration of tumors with STM2457 compromises AML growth and promotes differentiation and apoptosis, accompanied with selective depletion of m6A on known leukemogenic mRNAs and downregulation of their expression.748 Following, a derivative of STM2457, STC-15, was developed and showed activities in stimulating innate immune pathways, suppressing tumor growth, and augmenting the efficacy of anti-PD1 therapy with durable antitumor immunity in preclinical models of colorectal cancer and lymphoma.749 A phase I clinical trial of STC-15 in patients with advanced malignancies is ongoing (NCT05584111) (Table 1). Other SAM-competitive inhibitors of METTL3 include adenine derivatives,750 UZH1a,751 and UZH2.752 Among these, UZH2, a 1,4,9-triazaspiro[5.5]undecan-2-one derivative, shows the highest potency (IC50 = 5 nM) and favorable in vitro absorption–distribution–metabolism–excretion properties.752 Two allosteric inhibitors of METTL3, 43n (IC50 = 2.81 μM) and eltrombopag (IC50 = 3.65 μM), were recently discovered, and their effects on m6A level and cell proliferation were demonstrated in AML cells.753,754 Although current allosteric inhibitors have micromolar rather than nanomolar inhibitory efficacy of SAM competitive inhibitors, investment in the development of efficient allosteric inhibitors is warranted as most methyltransferases from DNA to proteins possess preserved SAM binding regions. Collectively, the development of these METTL3 inhibitors will greatly advance the functional study and therapeutic targeting of mRNA m6A methylation in cancer and other diseases.
Site-specific correction of methylation for precise therapy
Although knowledge about the molecular and cellular mechanisms of DNA/RNA/protein methylation actions in various diseases increases explosively over the past decades, the translation of it into clinical practice is still challenging with only modest success in the cancer field. The methylation modifiers generally have genome/transcriptome/proteome-wide effects, regulating both tumor suppressor genes and oncogenes. Targeting these modifiers with current small-molecule compounds leads to global loss or gain of cognate methylation marks and global changes in gene expression, as well as in nonhistone protein function. This will inevitably introduce undesirable biological effects and cause cytotoxicity, restricting the use of these compounds. For instance, in hematologic malignancies where hypomethylating therapy is relatively more successful, demethylation and upregulation of oncogene occur in patients after DNMT inhibitor treatment.654 The risk would be much greater when administration of these compounds aims to reactivate a specific gene for the treatment of monogenic disorders, such as FRM1 for FXS and MeCP2 for RTT.189,755,756 In addition to potential off-target toxicity, monotherapy with DNA hypomethylating agents showed limited efficacy in terms of reactivation of these stable silenced target genes.755,757
The development of tools (hereafter designated as methylation editors) capable of site-specific manipulating DNA and histone methylation status holds great promise to address the above challenges. Rather than methylome-wide alteration induced by directly targeting methylation machinery, chromatin methylation editors use sequence-specific DNA-binding domains (DBDs) to place the methylation machineries at the defined loci to perform DNA/histone methylation or demethylation, which in turn modulates the transcription of the targeted genes. Three generations of programmable DBD platforms are developed based on zinc fingers (ZFs), transcription activator-like effectors, and catalytically dead CRISPR-dCas system, with the CRISPR-based editors getting the most attention due to their easy reprogramming758,759 (Fig. 8a). Chromatin methylation editors are now available for the commonly studied methylation substrates, including CpG (by DNMT3A or TET1), H3K4 (by PRDM9, SMYD3, and LSD1), H3K9 (by KRAB, G9a, and HP1), H3K27 (by EZH2 and FOG1), and H3K79 (by DOT1L)760 (Fig. 8b, c).

Principle of site-specific manipulating DNA, histone, and RNA methylation. a Three generations of programmable DNA-binding domain (DBD) platforms for site-specific chromatin methylation manipulation: zinc fingers (ZFs), transcription activator-like effectors (TALEs), and catalytically dead CRISPR-dCas system. b Fusion of dCas9 to DNA methyltransferases (e.g., DNMT3A) or demethylases (e.g., TET1) allows for targeted DNA methylation or demethylation, respectively. c Fusion of dCas9 to protein methylation machinery (e.g., KRAB) or demethylases (e.g., LSD1) enables artificial histone methylation or demethylation at defined loci, respectively. d Fusion of dCas13 to RNA methyltransferases (e.g., METTL3) or demethylases (e.g., FTO) allows for selective deposition or removal of m6A signals at specific transcripts, respectively
The application of these tools for methylation and disease correction generates encouraging preclinical results. dCas9-TET1-mediated demethylation of the mutant FMR1 gene reactivates the gene expression and rescues behavioral defects in the FXS patient-derived cells.191 By coupling dCas9-TET1-mediated demethylation with dCpf1-CTCF-mediated insulation, the healthy copy of MECP2 on the inactive X chromosome is effectively activated in RTT human embryonic stem cells and derived neurons, which rescues RTT-related neuronal abnormality.757 In cancers, dCas9-TET1 has been adopted to unleash silenced tumor suppressor genes, e.g., BRCA1 (in breast and cervical cancer),761 SARI (in colon cancer),762 and NNT (in lung cancer),763 to combat tumor progression and chemoresistance. On the other hand, dCas9-based repressors that contain DNA methyltransferases (e.g., DNMT3A and DNMT3L) and repressive histone methyltransferases (e.g., KRAB and EZH2) alone or in combination have been used to silence oncogenes in cancers, such as GRN in liver cancer,764 BRAF and HER2 in colon cancer,765,766 FGFR4 in breast cancer,767 and KRAS in pancreatic cancer.765 Targeted methylation of the promoter of amyloid precursor protein gene APP by dCas9-DNMT3A rescues neuron cell pathology in vitro and in vivo of mouse AD model.768 Alternative to directly target promoters or transcription start sites in the above cases, methylation editing of CTCF-binding sites can alter 3D genome structure, affect the interaction between the distal enhancer and promoters, and in turn, control gene transcription of targeted genes.769,770 Multiple oncogenes including MYC, TERT, and CCND1, are influenced by recurrent changes of the 3D chromosome architecture in diverse cancer types.771,772,773 Artificial methylation of a CTCF-binding site located 2 kb upstream of the MYC promoter by dCas9-DNMT prevents the docking of cancer-specific super-enhancers, which compromises MYC expression and cancer cell proliferation.774 Recently, an epigenomic controller (called OTX-2002) developed by Omega Therapeutics to target the CTCF-insulated loop domain-containing MYC has advanced to a clinical trial for MYC-associated hepatocellular carcinoma and other solid tumors (NCT05497453)775 (Fig. 9 and Table 1). These chromatin methylation editors can also be used to treat various diseases without methylation-based etiology, through upregulating or downregulating the expression of protective or pathogenic factors, respectively. For instance, ZFN-KRAB repressors that recognize the amplified CAG repeats within the mutant HTT are designed to selectively silence the mutant allele for the treatment of Huntington’s disease, and a one-time striatal AAV-mediated delivery can correct molecular, histopathological, and behavioral defects in the mouse model.776 Besides therapeutic efforts, methylation editors are helping dissect the causal relationship between methylation alteration and tumorigenesis, which will identify faithful cancer driver methylation changes that serve as potential therapeutic or diagnostic targets.280,777

Paradigm of site-specific manipulating chromatin methylation for precise cancer therapy. mRNAs encoding a methylation editor are delivered to liver cancer cells by lipid nanoparticles (LNPs), which suppresses oncogene expression by catalyzing DNA and/or histone methylation at the promoters of oncogenes (e.g., MYC) or at the CTCF-binding sites to abolish their interactions with potential enhancers
With the help of PAMmer oligonucleotides, dCas9 can target mRNAs, and the first generation of site-specific m6A editors was developed in 2019 by fusing dCas9 with the methyltransferases (METTL3/METTL14) or demethylases (ALKBH5 or FTO).778 Subsequently, the m6A editors are optimized by substituting dCas9 with smaller Cas13 proteins (Fig. 8d), with lower off-target activity and PAMmer oligonucleotide-independence.779,780,781,782 Furthermore, chemically or light-inducible versions of m6A editors are developed to manipulate mRNA methylation spatiotemporally.783,784 Editing individual transcripts in cancer cells inhibits proliferation, migration, or therapy resistance, such as EGFR and MYC in cervical cancer,780 FOXM1 and MYC in glioblastoma,779 FGFR4 in HER2-positive breast cancer,767 ZNF677 and BTG2 in renal cell carcinoma (RCC),785,786 as well as lncRNA NEAT1 in RCC.787 Temporal removal of m6A from the SOX2 transcripts is sufficient to modulate the differentiation of human pluripotent stem cells.788 Therefore, the implementation of RNA methylation editors represents a potentially new and effective way to specifically alter the expression of targeted genes for cell fate and disease control.
Conclusion and outlook
Reversible tagging methyl groups on DNA, RNAs, histones, and nonhistone proteins finetunes gene expression and function. Dysregulation of the process caused by genetic mutations or environmental stimuli promotes various diseases and accelerates aging. This general outline is right but not nearly enough. We need to delve deeply into the details and deconvolute the complex networks to find faithful disease-driver methylation targets,280 which is a formidable task. Advances in the technologies, including CRISPR-assisted library screening,777,789 spatial transcriptomics,790,791 and DNA/RNA/protein methylation assays,792,793,794 will undoubtedly help, however, the field has its intractable problems. The function of methylation pathways is highly context-dependent, making annotation of their role in diseases complex and difficult. Limitation in animal models, such as failure to recapitulate intratumor heterogeneity seen in humans in genetically engineered mouse models, lack of effective immune system in patient-derived xenograft models, and/or shortage of models to study cancer and aging simultaneously,795 is a commonplace problem when translating the preclinical finding into the clinic, which is likely be more severe in the methylation field due to the plastic roles of methylation pathways. Consistently, predictive biomarkers for patient selection remain elusive when targeting methylation.
Another intractable problem is the global changes in methylomes caused by current inhibition strategies (e.g., chemical or genetic interference of methylation modifiers), which confounds the interpretation of results and compromises the therapeutic effects. The root cause of even monogenic diseases (e.g., FXS and RTT), let alone multigenic diseases involving different directions of methylation changes (e.g., cancer and aging-related diseases), can hardly be effectively and safely tackled by simple interference with the modifiers. While monotherapy targeting methylomes has not generated the expected clinical outcomes, combining the epigenetic drugs with other therapies, including chemotherapy, radiation therapy, molecular targeted therapy, and immunotherapy, shows encouraging anti-cancer synergistic effects,796 which is associated with the important role of methylation remodeling in the regulating tumor immunogenicity, DNA repair, therapy resistance, among others. A number of clinical trials are ongoing to test different combination strategies (Table 1), and whether the combination therapy represents the direction of the future yet has no conclusion. However, given the context-dependent role of methylation pathways, lack of predictive biomarkers, and unidirectional methylome perturbance, rational design of combination drug regimens for individual cancer patients remains challenging.
The development of precise methylation editing technologies raises boundless opportunities to meet the above challenges. One key opportunity for the future will be cross-disciplinary cooperation to find appropriate methods to perform in vivo methylation editing for therapy. The expression changes of some genes, especially those involving gain of DNA and repressive histone methylation, can be long-lasting and heritable, which is clinically preferred as a single dose of administration can induce durable therapeutic effects.200,797,798 Using mRNA-lipid nanoparticle (mRNA-LNP)-based therapeutics that has been adopted to combat the COVID-19 pandemic during past years and liver-accumulation feature of LNPs, an epigenome editor (OTX-2002) targeting MYC for the treatment of liver cancer recently received clearance of Investigational New Drug by US FDA to start a phase I/II clinical trial (Fig. 9 and Table 1). This marks an exciting beginning, however, to realize the therapeutic promise of methylation editors, several hurdles related to science and technology need to be addressed. First, many genes show modest and transient gene expression alteration using the current tools. Therefore, the efficacy of methylation editors needs to be improved and the contextual cues for persistent changes to gene regulation require to be clarified.799,800,801 Second, the efficacy, specificity, and safety of in vivo delivery remain to be a bottleneck in the treatment of diseases, and specific requirements for delivery vehicles vary with diseases.802,803,804,805 Third, immune response to methylation editors in humans.806
Another key opportunity is that the causal relationship between the methylation alteration of specific sites or genes with disease phenotypes and its underlying mechanism could be finally answered, which cannot be achieved by current gene-deletion/-overexpression mimic models. The future diagnosis and selection of the most promising therapies for individual patients will rely on this knowledge. Particularly, DNA methylation-based non-invasive liquid biopsy (e.g., plasma cell-free DNA) tests hold great promise in cancer early detection, prognosis, and therapy monitoring, which would transform cancer survival.807,808,809,810,811 The sensitivity of individual gene-based methylation models or the generalization ability of genome-wide methylation models needs to be further improved for clinical implementation as a standard of care.812 Finding and validating the most informative DNA methylation biomarkers is crucial for future success when they are used alone or combined with other cancer markers (e.g., cell-free DNA mutations).813,814 In addition, like we can’t merely focus on DNA methylation and ignore the important role of RNA and protein methylation in the progression of mIDH-driven cancers, RNA and protein methylation may serve as orthogonal predictors, and synchronous detection of methylation across the central dogma might improve diagnostic power (especially in the diseases with disordered metabolism) based on tissue biopsies and even liquid biopsies.815
References
Cheng, Y. et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Sig. Transduct. Target. Ther. 4, 62 (2019).
Lister, R. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 (2009).
Gershman, A. et al. Epigenetic patterns in a complete human genome. Science 376, eabj5089 (2022).
Guo, J. U. et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 17, 215–222 (2014).
Loyfer, N. et al. A DNA methylation atlas of normal human cell types. Nature 613, 355–364 (2023).
Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492 (2012).
Boulias, K. & Greer, E. L. Means, mechanisms and consequences of adenine methylation in DNA. Nat. Rev. Genet. 23, 411–428 (2022).
Kong, Y. et al. Critical assessment of DNA adenine methylation in eukaryotes using quantitative deconvolution. Science 375, 515–522 (2022).
Shen, C., Wang, K., Deng, X. & Chen, J. DNA N6-methyldeoxyadenosine in mammals and human disease. Trends Genet. 38, 454–467 (2022).
Jambhekar, A., Dhall, A. & Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 20, 625–641 (2019).
Wang, H. et al. H3K4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature 615, 339–348 (2023).
Migliori, V. et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol. 19, 136–144 (2012).
Guccione, E. et al. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 449, 933–937 (2007).
Fabbrizio, E. et al. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 3, 641–645 (2002).
Yang, Y. & Bedford, M. T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 13, 37–50 (2013).
Ooi, S. K. T. et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717 (2007).
Schumann, U. et al. Multiple links between 5-methylcytosine content of mRNA and translation. BMC Biol. 18, 40 (2020).
Huang, T., Chen, W., Liu, J., Gu, N. & Zhang, R. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat. Struct. Mol. Biol. 26, 380–388 (2019).
Liu, J. et al. Sequence- and structure-selective mRNA m5C methylation by NSUN6 in animals. Natl Sci. Rev. 8, nwaa273 (2020).
Selmi, T. et al. Sequence- and structure-specific cytosine-5 mRNA methylation by NSUN6. Nucleic Acids Res. 49, 1006–1022 (2021).
Yang, X. et al. 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 27, 606–625 (2017).
Yang, H. et al. FMRP promotes transcription-coupled homologous recombination via facilitating TET1-mediated m5C RNA modification demethylation. Proc. Natl Acad. Sci. 119, e2116251119 (2022).
Chen, H. et al. m5C modification of mRNA serves a DNA damage code to promote homologous recombination. Nat. Commun. 11, 2834 (2020).
Xiao, C.-L. et al. N6-methyladenine DNA modification in the human genome. Mol. Cell 71, 306–318.e307 (2018).
Kweon, S.-M. et al. An adversarial DNA N6-methyladenine-sensor network preserves polycomb silencing. Mol. Cell 74, 1138–1147.e1136 (2019).
Woodcock, C. B. et al. Human MettL3–MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discov. 5, 63 (2019).
Liu, J. et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95 (2014).
Dillon, S. C., Zhang, X., Trievel, R. C. & Cheng, X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 6, 227 (2005).
Feng, Q. et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 12, 1052–1058 (2002).
Liu, S. et al. METTL13 methylation of eEF1A increases translational output to promote tumorigenesis. Cell 176, 491–504.e421 (2019).
Kernstock, S. et al. Lysine methylation of VCP by a member of a novel human protein methyltransferase family. Nat. Commun. 3, 103 (2012).
Metzger, E. et al. KMT9 monomethylates histone H4 lysine 12 and controls proliferation of prostate cancer cells. Nat. Struct. Mol. Biol. 26, 361–371 (2019).
Rea, S. et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599 (2000).
Peters, A. H. F. M. et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell 12, 1577–1589 (2003).
Lee, J.-E. et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife 2, e01503 (2013).
Min, J., Feng, Q., Li, Z., Zhang, Y. & Xu, R.-M. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 112, 711–723 (2003).
Frederiks, F. et al. Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat. Struct. Mol. Biol. 15, 550–557 (2008).
Fu, L. et al. Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 136, 11582–11585 (2014).
Huber, S. M. et al. Formation and abundance of 5-hydroxymethylcytosine in RNA. ChemBioChem 16, 752–755 (2015).
Basanta-Sanchez, M. et al. TET1-mediated oxidation of 5-formylcytosine (5fC) to 5-carboxycytosine (5caC) in RNA. ChemBioChem 18, 72–76 (2017).
Walport, L. J., Hopkinson, R. J. & Schofield, C. J. Mechanisms of human histone and nucleic acid demethylases. Curr. Opin. Chem. Biol. 16, 525–534 (2012).
Kaur, S., Tam, N. Y., McDonough, M. A., Schofield, C. J. & Aik, W. S. Mechanisms of substrate recognition and N6-methyladenosine demethylation revealed by crystal structures of ALKBH5–RNA complexes. Nucleic Acids Res 50, 4148–4160 (2022).
Kooistra, S. M. & Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 13, 297–311 (2012).
Tsukada, Y.-i. et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816 (2006).
Hou, H. & Yu, H. Structural insights into histone lysine demethylation. Curr. Opin. Struct. Biol. 20, 739–748 (2010).
Cornett, E. M., Ferry, L., Defossez, P.-A. & Rothbart, S. B. Lysine methylation regulators moonlighting outside the epigenome. Mol. Cell 75, 1092–1101 (2019).
Li, S. et al. JMJD1B demethylates H4R3me2s and H3K9me2 to facilitate gene expression for development of hematopoietic stem and progenitor cells. Cell Rep. 23, 389–403 (2018).
Chang, B., Chen, Y., Zhao, Y. & Bruick, R. K. JMJD6 is a histone arginine demethylase. Science 318, 444–447 (2007).
Walport, L. J. et al. Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat. Commun. 7, 11974 (2016).
Mantri, M. et al. Crystal structure of the 2-oxoglutarate- and Fe(II)-dependent Lysyl hydroxylase JMJD6. J. Mol. Biol. 401, 211–222 (2010).
Islam, M. S. et al. Biochemical and structural investigations clarify the substrate selectivity of the 2-oxoglutarate oxygenase JMJD6. J. Biol. Chem. 294, 11637–11652 (2019).
Webby, C. J. et al. Jmjd6 catalyses Lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 325, 90–93 (2009).
Mahmood, N. & Rabbani, S. A. DNA methylation readers and cancer: mechanistic and therapeutic applications. Front. Oncol. 9, 489 (2019).
Ho, K. L. et al. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol. Cell 29, 525–531 (2008).
Hashimoto, H. et al. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature 455, 826–829 (2008).
Avvakumov, G. V. et al. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 455, 822–825 (2008).
Arita, K., Ariyoshi, M., Tochio, H., Nakamura, Y. & Shirakawa, M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature 455, 818–821 (2008).
Nikolova, E. N., Stanfield, R. L., Dyson, H. J. & Wright, P. E. CH···O hydrogen bonds mediate highly specific recognition of methylated CpG sites by the zinc finger protein Kaiso. Biochemistry 57, 2109–2120 (2018).
Buck-Koehntop, B. A. et al. Molecular basis for recognition of methylated and specific DNA sequences by the zinc finger protein Kaiso. Proc. Natl Acad. Sci. 109, 15229–15234 (2012).
Du, Q., Luu, P.-L., Stirzaker, C. & Clark, S. J. Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics 7, 1051–1073 (2015).
Maunakea, A. K., Chepelev, I., Cui, K. & Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269 (2013).
Ibrahim, A. et al. MeCP2 is a microsatellite binding protein that protects CA repeats from nucleosome invasion. Science 372, eabd5581 (2021).
Hendrich, B., Hardeland, U., Ng, H.-H., Jiricny, J. & Bird, A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature 401, 301–304 (1999).
Sanders, M. A. et al. MBD4 guards against methylation damage and germ line deficiency predisposes to clonal hematopoiesis and early-onset AML. Blood 132, 1526–1534 (2018).
Pidugu, L. S. et al. Structural insights into the mechanism of base excision by MBD4. J. Mol. Biol. 433, 167097 (2021).
Nishiyama, A. et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature 502, 249–253 (2013).
Qin, W. et al. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 25, 911–929 (2015).
Zhou, T. et al. Structural basis for hydroxymethylcytosine recognition by the SRA domain of UHRF2. Mol. Cell 54, 879–886 (2014).
Liu, X. et al. UHRF2 commissions the completion of DNA demethylation through allosteric activation by 5hmC and K33-linked ubiquitination of XRCC1. Mol. Cell 81, 2960–2974.e2967 (2021).
Yoon, H.-G., Chan, D. W., Reynolds, A. B., Qin, J. & Wong, J. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol. Cell 12, 723–734 (2003).
Raghav, S. K. et al. Integrative genomics identifies the corepressor SMRT as a gatekeeper of adipogenesis through the transcription factors C/EBPβ and KAISO. Mol. Cell 46, 335–350 (2012).
Hodges, A. J., Hudson, N. O. & Buck-Koehntop, B. A. Cys2His2 zinc finger methyl-CpG binding proteins: getting a handle on methylated DNA. J. Mol. Biol. 432, 1640–1660 (2020).
Mackay, D. J. G. et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat. Genet. 40, 949–951 (2008).
Jiang, W. et al. ZFP57 dictates allelic expression switch of target imprinted genes. Proc. Natl Acad. Sci. 118, e2005377118 (2021).
Shi, H., Wei, J. & He, C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 (2019).
Xu, C. et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat. Chem. Biol. 10, 927–929 (2014).
Li, Y. et al. Atomistic and thermodynamic analysis of N6-methyladenosine (m6A) recognition by the reader domain of YTHDC1. J. Chem. Theory Comput. 17, 1240–1249 (2021).
Li, F., Zhao, D., Wu, J. & Shi, Y. Structure of the YTH domain of human YTHDF2 in complex with an m6A mononucleotide reveals an aromatic cage for m6A recognition. Cell Res. 24, 1490–1492 (2014).
Cheng, Y. et al. N6-Methyladenosine on mRNA facilitates a phase-separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell 39, 958–972.e958 (2021).
Ries, R. J. et al. m6A enhances the phase separation potential of mRNA. Nature 571, 424–428 (2019).
Widagdo, J., Anggono, V. & Wong, J. J. L. The multifaceted effects of YTHDC1-mediated nuclear m6A recognition. Trends Genet. 38, 325–332 (2022).
Patil, D. P. et al. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373 (2016).
Liu, J. et al. The RNA m6A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature 591, 322–326 (2021).
Lee, J.-H. et al. Enhancer RNA m6A methylation facilitates transcriptional condensate formation and gene activation. Mol. Cell 81, 3368–3385.e3369 (2021).
Mao, Y. et al. m6A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat. Commun. 10, 5332 (2019).
Kretschmer, J. et al. The m6A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5′–3′ exoribonuclease XRN1. RNA 24, 1339–1350 (2018).
Wu, R. et al. m6A methylation promotes white-to-beige fat transition by facilitating Hif1a translation. EMBO Rep. 22, e52348 (2021).
Zaccara, S., Ries, R. J. & Jaffrey, S. R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 20, 608–624 (2019).
Saito, Y. et al. YTHDC2 control of gametogenesis requires helicase activity but not m6A binding. Genes Dev. 36, 180–194 (2022).
Li, L. et al. The XRN1-regulated RNA helicase activity of YTHDC2 ensures mouse fertility independently of m6A recognition. Mol. Cell 82, 1678–1690.e1612 (2022).
Wang, X. et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015).
Du, H. et al. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4–NOT deadenylase complex. Nat. Commun. 7, 12626 (2016).
Park, O. H. et al. Endoribonucleolytic cleavage of m6A-containing RNAs by RNase P/MRP complex. Mol. Cell 74, 494–507.e498 (2019).
Boo, S. H. et al. UPF1 promotes rapid degradation of m6A-containing RNAs. Cell Rep. 39, 110861 (2022).
Shi, H. et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 27, 315–328 (2017).
Fu, Y. & Zhuang, X. m6A-binding YTHDF proteins promote stress granule formation. Nat. Chem. Biol. 16, 955–963 (2020).
Flamand, M. N. & Meyer, K. D. m6A and YTHDF proteins contribute to the localization of select neuronal mRNAs. Nucleic Acids Res. 50, 4464–4483 (2022).
Zaccara, S. & Jaffrey, S. R. A unified model for the function of YTHDF proteins in regulating m6A-modified mRNA. Cell 181, 1582–1595.e1518 (2020).
Lasman, L. et al. Context-dependent functional compensation between Ythdf m6A reader proteins. Genes Dev. 34, 1373–1391 (2020).
Kennedy, E. M. et al. Posttranscriptional m6A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe 19, 675–685 (2016).
Nielsen, P. R. et al. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature 416, 103–107 (2002).
Hughes, R. M., Wiggins, K. R., Khorasanizadeh, S. & Waters, M. L. Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect. Proc. Natl Acad. Sci. 104, 11184–11188 (2007).
Musselman, C. A., Lalonde, M.-E., Côté, J. & Kutateladze, T. G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227 (2012).
Gayatri, S. & Bedford, M. T. Readers of histone methylarginine marks. Biochim. Biophys. Acta 1839, 702–710 (2014).
Vermeulen, M. et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58–69 (2007).
Maeda, R. & Tachibana, M. HP1 maintains protein stability of H3K9 methyltransferases and demethylases. EMBO Rep. 23, e53581 (2022).
Lachner, M., O’Carroll, D., Rea, S., Mechtler, K. & Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120 (2001).
Huang, J. et al. p53 is regulated by the lysine demethylase LSD1. Nature 449, 105–108 (2007).
Cui, G. et al. PHF20 is an effector protein of p53 double lysine methylation that stabilizes and activates p53. Nat. Struct. Mol. Biol. 19, 916–924 (2012).
West, L. E. et al. The MBT repeats of L3MBTL1 link SET8-mediated p53 methylation at lysine 382 to target gene repression. J. Biol. Chem. 285, 37725–37732 (2010).
Yu, D. et al. Human MettL3-MettL14 RNA adenine methyltransferase complex is active on double-stranded DNA containing lesions. Nucleic Acids Res. 49, 11629–11642 (2021).
Woodcock, C. B. et al. Biochemical and structural basis for YTH domain of human YTHDC1 binding to methylated adenine in DNA. Nucleic Acids Res. 48, 10329–10341 (2020).
Zhang, C. et al. METTL3 and N6-methyladenosine promote homologous recombination-mediated repair of DSBs by modulating DNA-RNA hybrid accumulation. Mol. Cell 79, 425–442 (2020).
Xiang, Y. et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576 (2017).
Yin, Y. et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356, eaaj2239 (2017).
Huang, H. et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295 (2018).
Wu, R. et al. A novel m6A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res 29, 23–41 (2019).
Edupuganti, R. R. et al. N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 24, 870–878 (2017).
Zhang, F. et al. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum. Mol. Genet. 27, 3936–3950 (2018).
Hsu, P. J. et al. The RNA-binding protein FMRP facilitates the nuclear export of N6-methyladenosine-containing mRNAs. J. Biol. Chem. 294, 19889–19895 (2019).
Edens, B. M. et al. FMRP modulates neural differentiation through m6A-dependent mRNA nuclear export. Cell Rep. 28, 845–854 (2019).
Chen, X. et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell Biol. 21, 978–990 (2019).
Feng, M. et al. YBX1 is required for maintaining myeloid leukemia cell survival by regulating BCL2 stability in an m6A-dependent manner. Blood 138, 71–85 (2021).
Liu, N. et al. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 518, 560–564 (2015).
Liu, N. et al. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 45, 6051–6063 (2017).
Wu, B. et al. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat. Commun. 9, 420 (2018).
Zhou, K. I. et al. Regulation of co-transcriptional pre-mRNA splicing by m6A through the low-complexity protein hnRNPG. Mol. Cell 76, 70–81.e79 (2019).
Cieniková, Z., Damberger, F. F., Hall, J., Allain, F. H. T. & Maris, C. Structural and mechanistic insights into poly(uridine) tract recognition by the hnRNP C RNA recognition motif. J. Am. Chem. Soc. 136, 14536–14544 (2014).
Sun, L. et al. RNA structure maps across mammalian cellular compartments. Nat. Struct. Mol. Biol. 26, 322–330 (2019).
Alarcón, C. R. et al. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell 162, 1299–1308 (2015).
Nanavaty, V. et al. DNA methylation regulates alternative polyadenylation via CTCF and the cohesin complex. Mol. Cell 78, 752–764.e756 (2020).
Phillips, J. E. & Corces, V. G. CTCF: master weaver of the genome. Cell 137, 1194–1211 (2009).
Renda, M. et al. Critical DNA binding interactions of the insulator protein CTCF: a small number of zinc fingers mediate strong binding, and a single finger-DNA interaction controls binding at imprinted loci. J. Biol. Chem. 282, 33336–33345 (2007).
Shukla, S. et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74–79 (2011).
Soochit, W. et al. CTCF chromatin residence time controls three-dimensional genome organization, gene expression and DNA methylation in pluripotent cells. Nat. Cell Biol. 23, 881–893 (2021).
Xu, C., Bian, C., Lam, R., Dong, A. & Min, J. The structural basis for selective binding of non-methylated CpG islands by the CFP1 CXXC domain. Nat. Commun. 2, 227 (2011).
Blackledge, N. P. et al. CpG islands recruit a histone H3 lysine 36 demethylase. Mol. Cell 38, 179–190 (2010).
Cierpicki, T. et al. Structure of the MLL CXXC domain–DNA complex and its functional role in MLL-AF9 leukemia. Nat. Struct. Mol. Biol. 17, 62–68 (2010).
Farcas, A. M. et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife 1, e00205 (2012).
Mendel, M. et al. Splice site m6A methylation prevents binding of U2AF35 to inhibit RNA splicing. Cell 184, 3125–3142 (2021).
Sanchez, R. & Zhou, M.-M. The PHD finger: a versatile epigenome reader. Trends Biochem. Sci. 36, 364–372 (2011).
Mazur, P. K. et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 510, 283–287 (2014).
Fang, L. et al. SET1A-mediated mono-methylation at K342 regulates YAP activation by blocking its nuclear export and promotes tumorigenesis. Cancer Cell 34, 103–118 (2018).
Vershinin, Z. et al. BRD4 methylation by the methyltransferase SETD6 regulates selective transcription to control mRNA translation. Sci. Adv. 7, eabf5374 (2021).
Lu, T. et al. Role of lysine methylation of NF-κB in differential gene regulation. Proc. Natl Acad. Sci. 110, 13510–13515 (2013).
Takahama, K., Kino, K., Arai, S., Kurokawa, R. & Oyoshi, T. Identification of Ewing’s sarcoma protein as a G-quadruplex DNA- and RNA-binding protein. FEBS J. 278, 988–998 (2011).
Hu, S.-B. et al. Protein arginine methyltransferase CARM1 attenuates the paraspeckle-mediated nuclear retention of mRNAs containing IRAlus. Genes Dev. 29, 630–645 (2015).
Wei, H.-M. et al. Arginine methylation of the cellular nucleic acid binding protein does not affect its subcellular localization but impedes RNA binding. FEBS Lett. 588, 1542–1548 (2014).
Kim, D. N. et al. Zinc-finger protein CNBP alters the 3-D structure of lncRNA Braveheart in solution. Nat. Commun. 11, 148 (2020).
Rausch, C. et al. Cytosine base modifications regulate DNA duplex stability and metabolism. Nucleic Acids Res. 49, 12870–12894 (2021).
Li, S., Peng, Y., Landsman, D. & Panchenko, A. R. DNA methylation cues in nucleosome geometry, stability and unwrapping. Nucleic Acids Res. 50, 1864–1874 (2022).
Choy, J. S. et al. DNA methylation increases nucleosome compaction and rigidity. J. Am. Chem. Soc. 132, 1782–1783 (2010).
Stevens, A. J., de Jong, L. & Kennedy, M. A. The dynamic regulation of G-quadruplex DNA structures by cytosine methylation. Int. J. Mol. Sci. 23, 2407 (2022).
Ranjan, N. & Leidel, S. A. The epitranscriptome in translation regulation: mRNA and tRNA modifications as the two sides of the same coin? FEBS Lett. 593, 1483–1493 (2019).
Banks, K. M. & Evans, T. In Epitranscriptomics (eds S. Jurga & J. Barciszewski) 423–433 (Springer International Publishing, 2021).
Shoaib, M. et al. Histone H4 lysine 20 mono-methylation directly facilitates chromatin openness and promotes transcription of housekeeping genes. Nat. Commun. 12, 4800 (2021).
Lu, X. et al. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat. Struct. Mol. Biol. 15, 1122–1124 (2008).
Yang, X.-J. & Seto, E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell 31, 449–461 (2008).
Zhang, W. et al. CPLM 4.0: an updated database with rich annotations for protein lysine modifications. Nucleic Acids Res. 50, D451–D459 (2022).
Pasini, D. et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 38, 4958–4969 (2010).
Ji, H. et al. HDAC3 deficiency promotes liver cancer through a defect in H3K9ac/H3K9me3 transition. Cancer Res. 79, 3676–3688 (2019).
Kogure, M. et al. The oncogenic polycomb histone methyltransferase EZH2 methylates lysine 120 on histone H2B and competes ubiquitination. Neoplasia 15, 1251–IN1210 (2013).
Chuikov, S. et al. Regulation of p53 activity through lysine methylation. Nature 432, 353–360 (2004).
Subramanian, K. et al. Regulation of estrogen receptor α by the SET7 lysine methyltransferase. Mol. Cell 30, 336–347 (2008).
Li, J. et al. Oxygen-sensitive methylation of ULK1 is required for hypoxia-induced autophagy. Nat. Commun. 13, 1172 (2022).
Law, J. A. & Jacobsen, S. E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220 (2010).
Xie, W. et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148, 816–831 (2012).
Stroud, H. et al. Early-life gene expression in neurons modulates lasting epigenetic states. Cell 171, 1151–1164 (2017).
de Mendoza, A. et al. The emergence of the brain non-CpG methylation system in vertebrates. Nat. Ecol. Evol. 5, 369–378 (2021).
You, S.-H. et al. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol. 20, 182–187 (2013).
Lyst, M. J. et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 16, 898–902 (2013).
Chen, L. et al. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl Acad. Sci. 112, 5509–5514 (2015).
Gabel, H. W. et al. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93 (2015).
Tillotson, R. et al. Neuronal non-CG methylation is an essential target for MeCP2 function. Mol. Cell 81, 1260–1275 (2021).
Samaco, R. C. et al. A partial loss of function allele of Methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum. Mol. Genet. 17, 1718–1727 (2008).
Chahrour, M. et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229 (2008).
Nott, A. et al. Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat. Neurosci. 19, 1497–1505 (2016).
Zhou, J. et al. Disruption of MeCP2-TCF20 complex underlies distinct neurodevelopmental disorders. Proc. Natl Acad. Sci. 119, e2119078119 (2022).
Ciernia, A. V. & LaSalle, J. The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nat. Rev. Neurosci. 17, 411–423 (2016).
Chen, W. G. et al. Derepression of BDNF tanscription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889 (2003).
Ortega-Alarcon, D. et al. Unexpected thermodynamic signature for the interaction of hydroxymethylated DNA with MeCP2. Int. J. Biol. Macromol. 232, 123373 (2023).
Jiang, Y. et al. Rett syndrome linked to defects in forming the MeCP2/Rbfox/LASR complex in mouse models. Nat. Commun. 12, 5767 (2021).
Young, J. I. et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl Acad. Sci. 102, 17551–17558 (2005).
Li, R. et al. Misregulation of alternative splicing in a mouse model of Rett syndrome. PLoS Genet. 12, e1006129–e1006129 (2016).
Zhang, H. et al. MeCP2-induced heterochromatin organization is driven by oligomerization-based liquid-liquid phase separation and restricted by DNA methylation. Nucleus 13, 1–34 (2022).
Vershkov, D. et al. FMR1 reactivating treatments in fragile X iPSC-derived neural progenitors in vitro and in vivo. Cell Rep. 26, 2531–2539 (2019).
Rais, M. et al. Functional consequences of postnatal interventions in a mouse model of fragile X syndrome. Neurobiol. Dis. 162, 105577 (2022).
Malik, I., Kelley, C. P., Wang, E. T. & Todd, P. K. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 22, 589–607 (2021).
Chiurazzi, P., Pomponi, M. G., Willemsen, R., Oostra, B. A. & Neri, G. In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum. Mol. Genet. 7, 109–113 (1998).
Sapozhnikov, D. M. & Szyf, M. Unraveling the functional role of DNA demethylation at specific promoters by targeted steric blockage of DNA methyltransferase with CRISPR/dCas9. Nat. Commun. 12, 5711 (2021).
Liu, X. S. et al. Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell 172, 979–992 (2018).
Smeets, H. J. M. et al. Normal phenotype in two brothers with a full FMR1 mutation. Hum. Mol. Genet. 4, 2103–2108 (1995).
Rousseau, F., Robb, L. J., Rouillard, P. & Kaloustian, V. M. D. No mental retardation in a man with 40% abnormal methylation at the FMR-1 locus and transmission of sperm cell mutations as premutations. Hum. Mol. Genet. 3, 927–930 (1994).
Hagerman, R. J. et al. High functioning fragile X males: Demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am. J. Med. Genet. 51, 298–308 (1994).
Colak, D. et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005 (2014).
Kumari, D. & Usdin, K. Polycomb group complexes are recruited to reactivated FMR1 alleles in Fragile X syndrome in response to FMR1 transcription. Hum. Mol. Genet. 23, 6575–6583 (2014).
Kumari, D., Sciascia, N. & Usdin, K. Small molecules targeting H3K9 methylation prevent silencing of reactivated FMR1 alleles in fragile X syndrome patient derived cells. Genes 11, 356 (2020).
Greer, E. L. & Shi, Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 (2012).
Petryk, N., Bultmann, S., Bartke, T. & Defossez, P.-A. Staying true to yourself: mechanisms of DNA methylation maintenance in mammals. Nucleic Acids Res. 49, 3020–3032 (2021).
Takahashi, Y. et al. Transgenerational inheritance of acquired epigenetic signatures at CpG islands in mice. Cell 186, 715–731 (2023).
Park, C.-Y. et al. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 13, 234–241 (2015).
Vershkov, D., Yilmaz, A., Yanuka, O., Nielsen, A. L. & Benvenisty, N. Genome-wide screening for genes involved in the epigenetic basis of fragile X syndrome. Stem Cell Rep. 17, 1048–1058 (2022).
Richter, J. D. & Zhao, X. The molecular biology of FMRP: new insights into fragile X syndrome. Nat. Rev. Neurosci. 22, 209–222 (2021).
Shin, J. et al. Oppositional poly(A) tail length regulation by FMRP and CPEB1. RNA 28, 756–765 (2022).
Zhou, L.-T. et al. A novel role of fragile X mental retardation protein in pre-mRNA alternative splicing through RNA-binding protein 14. Neuroscience 349, 64–75 (2017).
Darnell, J. C. et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011).
Shah, S. et al. FMRP control of ribosome translocation promotes chromatin modifications and alternative splicing of neuronal genes linked to autism. Cell Rep. 30, 4459–4472 (2020).
Korb, E. et al. Excess translation of epigenetic regulators contributes to fragile X syndrome and is alleviated by Brd4 inhibition. Cell 170, 1209–1223 (2017).
Worpenberg, L. et al. Ythdf is a N6-methyladenosine reader that modulates Fmr1 target mRNA selection and restricts axonal growth in Drosophila. EMBO J. 40, e104975 (2021).
Kang, Y. et al. A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat. Neurosci. 24, 1377–1391 (2021).
Yoon, K.-J. et al. Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell 171, 877–889 (2017).
Yoshida, A., Oyoshi, T., Suda, A., Futaki, S. & Imanishi, M. Recognition of G-quadruplex RNA by a crucial RNA methyltransferase component, METTL14. Nucleic Acids Res. 50, 449–457 (2022).
Jara-Espejo, M., Fleming, A. M. & Burrows, C. J. Potential G-quadruplex forming sequences and N6-methyladenosine colocalize at human pre-mRNA intron splice sites. ACS Chem. Biol. 15, 1292–1300 (2020).
Darnell, J. C. et al. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107, 489–499 (2001).
Vasilyev, N. et al. Crystal structure reveals specific recognition of a G-quadruplex RNA by a β-turn in the RGG motif of FMRP. Proc. Natl Acad. Sci. 112, E5391–E5400 (2015).
Athar, Y. M. & Joseph, S. RNA-binding specificity of the human fragile X mental retardation protein. J. Mol. Biol. 432, 3851–3868 (2020).
Blackwell, E., Zhang, X. & Ceman, S. Arginines of the RGG box regulate FMRP association with polyribosomes and mRNA. Hum. Mol. Genet. 19, 1314–1323 (2010).
Dolzhanskaya, N., Merz, G., Aletta, J. M. & Denman, R. B. Methylation regulates the intracellular protein-protein and protein-RNA interactions of FMRP. J. Cell Sci. 119, 1933–1946 (2006).
Stetler, A. et al. Identification and characterization of the methyl arginines in the fragile X mental retardation protein Fmrp. Hum. Mol. Genet. 15, 87–96 (2005).
Tsang, B. et al. Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc. Natl Acad. Sci. 116, 4218–4227 (2019).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Dietlein, F. et al. Genome-wide analysis of somatic noncoding mutation patterns in cancer. Science 376, eabg5601 (2022).
Degasperi, A. et al. Substitution mutational signatures in whole-genome-sequenced cancers in the UK population. Science 376, abl9283 (2022).
Tubbs, A. & Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656 (2017).
Sherman, M. A. et al. Genome-wide mapping of somatic mutation rates uncovers drivers of cancer. Nat. Biotechnol. 40, 1634–1643 (2022).
Rideout, W. M., Coetzee, G. A., Olumi, A. F. & Jones, P. A. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 249, 1288–1290 (1990).
Simpkins, S. B. et al. MLH1 promoter methylation and gene silencing is the primary cause of microsatellite instability in sporadic endometrial cancers. Hum. Mol. Genet. 8, 661–666 (1999).
Shen, L. et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J. Natl Cancer Inst. 97, 1330–1338 (2005).
Rice, J. C., Massey-Brown, K. S. & Futscher, B. W. Aberrant methylation of the BRCA1 CpG island promoter is associated with decreased BRCA1 mRNA in sporadic breast cancer cells. Oncogene 17, 1807–1812 (1998).
Burns, K. H. Transposable elements in cancer. Nat. Rev. Cancer 17, 415–424 (2017).
Costantino, L. & Koshland, D. Genome-wide map of R-loop-induced damage reveals how a subset of R-loops contributes to genomic instability. Mol. Cell 71, 487–497 (2018).
Lee, J. H. et al. Regulation of telomere homeostasis and genomic stability in cancer by N6-adenosine methylation (m6A). Sci. Adv. 7, eabg7073 (2021).
Bayley, R. et al. H3K4 methylation by SETD1A/BOD1L facilitates RIF1-dependent NHEJ. Mol. Cell 82, 1924–1939 (2022).
Sulkowski, P. L. et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 582, 586–591 (2020).
Guccione, E. & Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 20, 642–657 (2019).
Sanchez-Bailon, M. P. et al. Arginine methylation and ubiquitylation crosstalk controls DNA end-resection and homologous recombination repair. Nat. Commun. 12, 6313 (2021).
Feinberg, A. P. & Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 4, 143–153 (2004).
Hetzel, S. et al. Acute lymphoblastic leukemia displays a distinct highly methylated genome. Nat. Cancer 3, 768–782 (2022).
Lee, J.-J. et al. Gene-specific promoter hypermethylation without global hypomethylation in follicular thyroid cancer. Int. J. Oncol. 33, 861–869 (2008).
Ohm, J. E. et al. A stem cell–like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 39, 237–242 (2007).
Schlesinger, Y. et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 39, 232–236 (2007).
Kumar, D., Cinghu, S., Oldfield, A. J., Yang, P. & Jothi, R. Decoding the function of bivalent chromatin in development and cancer. Genome Res. 31, 2170–2184 (2021).
Esteller, M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum. Mol. Genet. 16, R50–R59 (2007).
Zhou, W. et al. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 50, 591–602 (2018).
Endicott, J. L., Nolte, P. A., Shen, H. & Laird, P. W. Cell division drives DNA methylation loss in late-replicating domains in primary human cells. Nat. Commun. 13, 6659 (2022).
Van Tongelen, A., Loriot, A. & De Smet, C. Oncogenic roles of DNA hypomethylation through the activation of cancer-germline genes. Cancer Lett. 396, 130–137 (2017).
Na, F. et al. KMT2C deficiency promotes small cell lung cancer metastasis through DNMT3A-mediated epigenetic reprogramming. Nat. Cancer 3, 753–767 (2022).
Johnstone, S. E. et al. Large-scale topological changes restrain malignant progression in colorectal cancer. Cell 182, 1474–1489 (2020).
Du, Q. et al. DNA methylation is required to maintain both DNA replication timing precision and 3D genome organization integrity. Cell Rep. 36, 109722 (2021).
Ushijima, T., Clark, S. J. & Tan, P. Mapping genomic and epigenomic evolution in cancer ecosystems. Science 373, 1474–1479 (2021).
Kelsey, G., Stegle, O. & Reik, W. Single-cell epigenomics: Recording the past and predicting the future. Science 358, 69–75 (2017).
Tegowski, M., Flamand, M. N. & Meyer, K. D. scDART-seq reveals distinct m6A signatures and mRNA methylation heterogeneity in single cells. Mol. Cell 82, 868–878 (2022).
Martínez-Jiménez, F. et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 20, 555–572 (2020).
Schuster-Böckler, B. & Lehner, B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488, 504–507 (2012).
Zhao, S. G. et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 52, 778–789 (2020).
Lu, C. et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849 (2016).
Lewis, P. W. et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013).
Wang, M. et al. Identification of DNA motifs that regulate DNA methylation. Nucleic Acids Res. 47, 6753–6768 (2019).
An, M., Wang, H. & Zhu, Y. Mutations in m6A consensus motifs are suppressed in the m6A modified genes in human cancer cells. PLoS One 15, e0236882 (2020).
Luo, Z., Zhang, J., Fei, J. & Ke, S. Deep learning modeling m6A deposition reveals the importance of downstream cis-element sequences. Nat. Commun. 13, 2720 (2022).
Moore, L. et al. The mutational landscape of normal human endometrial epithelium. Nature 580, 640–646 (2020).
Tang, J. et al. The genomic landscapes of individual melanocytes from human skin. Nature 586, 600–605 (2020).
Weiss, J. M. et al. Anatomic position determines oncogenic specificity in melanoma. Nature 604, 354–361 (2022).
Baggiolini, A. et al. Developmental chromatin programs determine oncogenic competence in melanoma. Science 373, eabc1048 (2021).
Souroullas, G. P. et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 22, 632–640 (2016).
Zingg, D. et al. EZH2-mediated primary cilium deconstruction drives metastatic melanoma formation. Cancer Cell 34, 69–84 (2018).
Tao, Y. et al. Aging-like spontaneous epigenetic silencing facilitates Wnt activation, stemness, and BrafV600E-induced tumorigenesis. Cancer Cell 35, 315–328 (2019).
Fennell, L. et al. Braf mutation induces rapid neoplastic transformation in the aged and aberrantly methylated intestinal epithelium. Gut 71, 1127–1140 (2022).
Lan, Q. et al. The critical role of RNA m6A methylation in cancer. Cancer Res 79, 1285–1292 (2019).
Raj, N. et al. The Mettl3 epitranscriptomic writer amplifies p53 stress responses. Mol. Cell 82, 2370–2384 (2022).
Dong, L. et al. Relaxed initiation pausing of ribosomes drives oncogenic translation. Sci. Adv. 7, eabd6927 (2021).
Huang, H., Weng, H. & Chen, J. m6A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell 37, 270–288 (2020).
Xu, Y. et al. The N6-methyladenosine METTL3 regulates tumorigenesis and glycolysis by mediating m6A methylation of the tumor suppressor LATS1 in breast cancer. J. Exp. Clin. Cancer Res. 42, 10 (2023).
Li, Z. et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell 31, 127–141 (2017).
Vaz, M. et al. Chronic cigarette smoke-induced epigenomic changes precede sensitization of bronchial epithelial cells to single-step transformation by KRAS mutations. Cancer Cell 32, 360–376 (2017).
Bayliss, J. et al. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci. Transl. Med. 8, 366ra161–366ra161 (2016).
Mack, S. C. et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450 (2014).
Rice, S. et al. A human fetal liver-derived infant MLL-AF4 acute lymphoblastic leukemia model reveals a distinct fetal gene expression program. Nat. Commun. 12, 6905 (2021).
Krivtsov, A. V. et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 14, 355–368 (2008).
Pan, H. et al. Discovery of candidate DNA methylation cancer driver genes. Cancer Discov. 11, 2266–2281 (2021).
Landau, D. A. et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell 26, 813–825 (2014).
Flavahan, W. A. et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016).
Hinohara, K. et al. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell 34, 939–953 (2018).
Benayoun, B. A. et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 158, 673–688 (2014).
Zhang, Z. et al. Genetic analyses support the contribution of mRNA N6-methyladenosine (m6A) modification to human disease heritability. Nat. Genet. 52, 939–949 (2020).
Jacob Berger, A. et al. IRS1 phosphorylation underlies the non-stochastic probability of cancer cells to persist during EGFR inhibition therapy. Nat. Cancer 2, 1055–1070 (2021).
Zhu, P. et al. lnc-β-Catm elicits EZH2-dependent β-catenin stabilization and sustains liver CSC self-renewal. Nat. Struct. Mol. Biol. 23, 631–639 (2016).
Nguyen, B. et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 185, 563–575 (2022).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Jensen, K. et al. Dynamic changes in E-cadherin gene promoter methylation during metastatic progression in papillary thyroid cancer. Exp. Ther. Med. 1, 457–462 (2010).
Gkountela, S. et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 176, 98–112 (2019).
van den Beucken, T. et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat. Commun. 5, 5203 (2014).
Chakraborty, A. A. et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 363, 1217–1222 (2019).
Jeschke, J. et al. Downregulation of the FTO m6A RNA demethylase promotes EMT-mediated progression of epithelial tumors and sensitivity to Wnt inhibitors. Nat. Cancer 2, 611–628 (2021).
Lin, X. et al. RNA m6A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat. Commun. 10, 2065 (2019).
Huang, H. et al. Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature 567, 414–419 (2019).
Deng, S. et al. RNA m6A regulates transcription via DNA demethylation and chromatin accessibility. Nat. Genet. 54, 1427–1437 (2022).
Oakes, C. C. et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat. Genet. 48, 253–264 (2016).
Tan, B. et al. RNA N(6) -methyladenosine reader YTHDC1 is essential for TGF-beta-mediated metastasis of triple negative breast cancer. Theranostics 12, 5727–5743 (2022).
Chang, G. et al. YTHDF3 induces the translation of m6A-enriched gene transcripts to promote breast cancer brain metastasis. Cancer Cell 38, 857–871 (2020).
Zhou, Y. et al. Single-cell multiomics sequencing reveals prevalent genomic alterations in tumor stromal cells of human colorectal cancer. Cancer Cell 38, 818–828 (2020).
Bejarano, L., Jordāo, M. J. C. & Joyce, J. A. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 11, 933–959 (2021).
Qiu, W. et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat. Genet. 40, 650–655 (2008).
Vodnala, S. K. et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 363, eaau0135 (2019).
Ford, B. R. et al. Tumor microenvironmental signals reshape chromatin landscapes to limit the functional potential of exhausted T cells. Sci. Immunol. 7, eabj9123 (2022).
Anderson, N. M. & Simon, M. C. The tumor microenvironment. Curr. Biol. 30, R921–R925 (2020).
Tyan, S.-W. et al. Breast cancer cells induce stromal fibroblasts to secrete ADAMTS1 for cancer invasion through an epigenetic change. PLoS One 7, e35128 (2012).
Hegde, P. S., Karanikas, V. & Evers, S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin. Cancer Res. 22, 1865–1874 (2016).
Zhang, B. et al. m6A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 19, 53 (2020).
Meng, Q. et al. DNA methylation regulator-mediated modification patterns and tumor microenvironment characterization in gastric cancer. Mol. Ther. Nucleic Acids 24, 695–710 (2021).
Guidry, K. et al. DNA methylation profiling identifies subgroups of lung adenocarcinoma with distinct immune cell composition, DNA methylation age, and clinical outcome. Clin. Cancer Res. 28, 3824–3835 (2022).
Nie, Y. et al. DNA hypermethylation is a mechanism for loss of expression of the HLA class I genes in human esophageal squamous cell carcinomas. Carcinogenesis 22, 1615–1623 (2001).
Rosenthal, R. et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485 (2019).
Scharer, C. D., Barwick, B. G., Youngblood, B. A., Ahmed, R. & Boss, J. M. Global DNA methylation remodeling accompanies CD8 T Cell effector function. J. Immunol. 191, 3419–3429 (2013).
Pace, L. et al. The epigenetic control of stemness in CD8 + T cell fate commitment. Science 359, 177–186 (2018).
Huang, Q. et al. The primordial differentiation of tumor-specific memory CD8 + T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell 185, 4049–4066 (2022).
Philip, M. et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017).
Ghoneim, H. E. et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 170, 142–157 (2017).
Sasidharan Nair, V., Toor, S. M., Taha, R. Z., Shaath, H. & Elkord, E. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin. Epigenet. 10, 104 (2018).
Pauken, K. E. et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165 (2016).
Delgoffe, G. M. et al. The role of exhaustion in CAR T cell therapy. Cancer Cell 39, 885–888 (2021).
Zebley, C. C. et al. CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep. 37, 110079 (2021).
Prinzing, B. et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci. Transl. Med. 13, eabh0272 (2021).
Wherry, E. J. & Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015).
Zhu, L. et al. Dapl1 controls NFATc2 activation to regulate CD8 + T cell exhaustion and responses in chronic infection and cancer. Nat. Cell Biol. 24, 1165–1176 (2022).
Khan, O. et al. TOX transcriptionally and epigenetically programs CD8 + T cell exhaustion. Nature 571, 211–218 (2019).
Zhao, X., Shan, Q. & Xue, H.-H. TCF1 in T cell immunity: a broadened frontier. Nat. Rev. Immunol. 22, 147–157 (2022).
Balkhi, M. Y., Wittmann, G., Xiong, F. & Junghans, R. P. YY1 upregulates checkpoint receptors and downregulates Type I cytokines in exhausted, chronically stimulated human T cells. iScience 2, 105–122 (2018).
Qiu, X. et al. M6A demethylase ALKBH5 regulates PD-L1 expression and tumor immunoenvironment in intrahepatic cholangiocarcinoma. Cancer Res 81, 4778–4793 (2021).
Wan, W. et al. METTL3/IGF2BP3 axis inhibits tumor immune surveillance by upregulating N6-methyladenosine modification of PD-L1 mRNA in breast cancer. Mol. Cancer 21, 60 (2022).
Drennan, S. et al. IL-10 production by CLL cells is enhanced in the anergic IGHV mutated subset and associates with reduced DNA methylation of the IL10 locus. Leukemia 31, 1686–1694 (2017).
Rivas, J. R. et al. Interleukin-10 suppression enhances T-cell antitumor immunity and responses to checkpoint blockade in chronic lymphocytic leukemia. Leukemia 35, 3188–3200 (2021).
Bian, Y. et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585, 277–282 (2020).
Renaude, E. et al. Epigenetic reprogramming of CD4+ helper T cells as a strategy to improve anticancer immunotherapy. Front. Immunol. 12, 669992 (2021).
Knochelmann, H. M. et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 15, 458–469 (2018).
Baessler, A. et al. Tet2 coordinates with Foxo1 and Runx1 to balance T follicular helper cell and T helper 1 cell differentiation. Sci. Adv. 8, eabm4982 (2022).
Luckheeram, R. V., Zhou, R., Verma, A. D. & Xia, B. CD4 + T cells: differentiation and functions. Clin. Dev. Immunol. 2012, 925135 (2012).
Toker, A. et al. Active demethylation of the Foxp3 locus leads to the generation of stable regulatory T cells within the thymus. J. Immunol. 190, 3180–3188 (2013).
Helmin, K. A. et al. Maintenance DNA methylation is essential for regulatory T cell development and stability of suppressive function. J. Clin. Investig. 130, 6571–6587 (2020).
Li, H.-B. et al. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 548, 338–342 (2017).
Tong, J. et al. m6A mRNA methylation sustains Treg suppressive functions. Cell Res. 28, 253–256 (2018).
Wei, G. et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4 + T Cells. Immunity 30, 155–167 (2009).
Li, Q. et al. Critical role of histone demethylase Jmjd3 in the regulation of CD4 + T-cell differentiation. Nat. Commun. 5, 5780 (2014).
Janson, P. C. J., Marits, P., Thörn, M., Ohlsson, R. & Winqvist, O. CpG methylation of the IFNG gene as a mechanism to induce immunosupression in tumor-infiltrating lymphocytes. J. Immunol. 181, 2878–2886 (2008).
Alvisi, G. et al. IRF4 instructs effector Treg differentiation and immune suppression in human cancer. J. Clin. Investig. 130, 3137–3150 (2020).
Itahashi, K. et al. BATF epigenetically and transcriptionally controls the activation program of regulatory T cells in human tumors. Sci. Immunol. 7, eabk0957 (2022).
Itahashi, K., Irie, T. & Nishikawa, H. Regulatory T-cell development in the tumor microenvironment. Eur. J. Immunol. 52, 1216–1227 (2022).
DuPage, M. et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity 42, 227–238 (2015).
Wang, D. et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep. 23, 3262–3274 (2018).
Nagai, Y. et al. PRMT5 associates with the FOXP3 homomer and when disabled enhances targeted p185erbB2/neu tumor immunotherapy. Front. Immunol. 10, 174 (2019).
Kagoya, Y. et al. Arginine methylation of FOXP3 is crucial for the suppressive function of regulatory T cells. J. Autoimmun. 97, 10–21 (2019).
Sharonov, G. V., Serebrovskaya, E. O., Yuzhakova, D. V., Britanova, O. V. & Chudakov, D. M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 20, 294–307 (2020).
Castino, G. F. et al. Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. OncoImmunology 5, e1085147 (2016).
Garaud, S., Dieu-Nosjean, M.-C. & Willard-Gallo, K. T follicular helper and B cell crosstalk in tertiary lymphoid structures and cancer immunotherapy. Nat. Commun. 13, 2259 (2022).
Petitprez, F. et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560 (2020).
Cabrita, R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020).
Cho, J.-W. et al. Dysregulation of TFH-B-TRM lymphocyte cooperation is associated with unfavorable anti-PD-1 responses in EGFR-mutant lung cancer. Nat. Commun. 12, 6068 (2021).
Helmink, B. A. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020).
Johansson-Percival, A. et al. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat. Immunol. 18, 1207–1217 (2017).
Zhang, B. et al. B cell-derived GABA elicits IL-10+ macrophages to limit anti-tumour immunity. Nature 599, 471–476 (2021).
Mirlekar, B. et al. Balance between immunoregulatory B cells and plasma cells drives pancreatic tumor immunity. Cell Rep. Med. 3, 100744 (2022).
Mirlekar, B. et al. B cell–derived IL35 drives STAT3-dependent CD8 + T-cell exclusion in pancreatic cancer. Cancer Immunol. Res. 8, 292–308 (2020).
Pylayeva-Gupta, Y. et al. IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Discov. 6, 247–255 (2016).
Xiao, F. et al. Epigenetic regulation of B cells and its role in autoimmune pathogenesis. Cell. Mol. Immunol. 19, 1215–1234 (2022).
Wheeler, D. A. et al. Molecular features of cancers exhibiting exceptional responses to treatment. Cancer Cell 39, 38–53 (2021).
Weiner, A. B. et al. Plasma cells are enriched in localized prostate cancer in Black men and are associated with improved outcomes. Nat. Commun. 12, 935 (2021).
Morvan, M. G. & Lanier, L. L. NK cells and cancer: you can teach innate cells new tricks. Nat. Rev. Cancer 16, 7–19 (2016).
Laskowski, T. J., Biederstädt, A. & Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 22, 557–575 (2022).
Zhang, X. et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol. 18, 1402–1412 (2016).
Bugide, S., Green, M. R. & Wajapeyee, N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc. Natl Acad. Sci. 115, E3509–E3518 (2018).
Zhong, J. et al. Circular EZH2-encoded EZH2-92aa mediates immune evasion in glioblastoma via inhibition of surface NKG2D ligands. Nat. Commun. 13, 4795 (2022).
Peng, V. et al. Whole-genome profiling of DNA methylation and hydroxymethylation identifies distinct regulatory programs among innate lymphocytes. Nat. Immunol. 23, 619–631 (2022).
Bugide, S., Janostiak, R. & Wajapeyee, N. Epigenetic mechanisms dictating eradication of cancer by natural killer cells. Trends Cancer 4, 553–566 (2018).
Song, H. et al. METTL3-mediated m6A RNA methylation promotes the anti-tumour immunity of natural killer cells. Nat. Commun. 12, 5522 (2021).
Yin, J. et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc. Natl Acad. Sci. 112, 15988–15993 (2015).
Ma, S. et al. The RNA m6A reader YTHDF2 controls NK cell antitumor and antiviral immunity. J. Exp. Med. 218, e20210279 (2021).
Myers, J. A. et al. Balanced engagement of activating and inhibitory receptors mitigates human NK cell exhaustion. JCI Insight 7, e150079 (2022).
Merino, A. et al. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J. Clin. Investig. 129, 3770–3785 (2019).
Avella Patino, D. M. et al. Epigenetic regulation of cancer immune cells. Semin. Cancer Biol. 83, 377–383 (2022).
Lawrence, T. & Natoli, G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761 (2011).
Vitale, I., Manic, G., Coussens, L. M., Kroemer, G. & Galluzzi, L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 30, 36–50 (2019).
Subramanian, S. et al. Long-term culture-expanded alveolar macrophages restore their full epigenetic identity after transfer in vivo. Nat. Immunol. 23, 458–468 (2022).
Colegio, O. R. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014).
DeNardo, D. G. & Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 19, 369–382 (2019).
Pan, W. et al. The DNA methylcytosine dioxygenase Tet2 sustains immunosuppressive function of tumor-infiltrating myeloid cells to promote melanoma progression. Immunity 47, 284–297 (2017).
Yang, X. et al. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 28, 565–574 (2014).
Gonda, T. A. et al. A DNA hypomethylating drug alters the tumor microenvironment and improves the effectiveness of immune checkpoint inhibitors in a mouse model of pancreatic cancer. Cancer Res. 80, 4754–4767 (2020).
Zhang, M. et al. Pancreatic cancer cells render tumor-associated macrophages metabolically reprogrammed by a GARP and DNA methylation-mediated mechanism. Sig. Transduct. Target. Ther. 6, 366 (2021).
Stone, M. L. et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl Acad. Sci. 114, E10981–E10990 (2017).
Travers, M. et al. DFMO and 5-azacytidine increase M1 macrophages in the tumor microenvironment of murine ovarian cancer. Cancer Res. 79, 3445–3454 (2019).
Xun, J. et al. Cancer-derived exosomal miR-138-5p modulates polarization of tumor-associated macrophages through inhibition of KDM6B. Theranostics 11, 6847–6859 (2021).
De Santa, F. et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. 28, 3341–3352 (2009).
Raines, L. N. et al. PERK is a critical metabolic hub for immunosuppressive function in macrophages. Nat. Immunol. 23, 431–445 (2022).
Zhong, Y. et al. The novel methyltransferase SETD4 regulates TLR agonist-induced expression of cytokines through methylation of lysine 4 at histone 3 in macrophages. Mol. Immunol. 114, 179–188 (2019).
Xia, M. et al. Histone methyltransferase Ash1l suppresses interleukin-6 production and inflammatory autoimmune diseases by inducing the ubiquitin-editing enzyme A20. Immunity 39, 470–481 (2013).
Niu, Y., Chen, J. & Qiao, Y. Epigenetic modifications in tumor-associated macrophages: a new perspective for an old foe. Front. Immunol. 13, 836223 (2022).
Yin, H. et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat. Commun. 12, 1394 (2021).
Dong, L. et al. The loss of RNA N6-adenosine methyltransferase Mettl14 in tumor-associated macrophages promotes CD8 + T cell dysfunction and tumor growth. Cancer Cell 39, 945–957 (2021).
Tong, J. et al. Pooled CRISPR screening identifies m6A as a positive regulator of macrophage activation. Sci. Adv. 7, eabd4742 (2021).
Xiong, J. et al. Lactylation-driven METTL3-mediated RNA m6A modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol. Cell 82, 1660–1677 (2022).
Yang, Y., Li, C., Liu, T., Dai, X. & Bazhin, A. V. Myeloid-derived suppressor cells in tumors: from mechanisms to antigen specificity and microenvironmental regulation. Front. Immunol. 11, 1371 (2020).
Grover, A., Sanseviero, E., Timosenko, E. & Gabrilovich, D. I. Myeloid-derived suppressor cells: a propitious road to clinic. Cancer Discov. 11, 2693–2706 (2021).
Sasidharan Nair, V. et al. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin. Epigenet. 12, 13 (2020).
Saleh, R., Toor, S. M., Taha, R. Z. & Al-Ali, D. DNA methylation in the promoters of PD-L1, MMP9, ARG1, galectin-9, TIM-3, VISTA and TGF-β genes in HLA-DR(-) myeloid cells, compared with HLA-DR(+) antigen-presenting cells. Epigenetics 15, 1275–1288 (2020).
Rodríguez-Ubreva, J. et al. Prostaglandin E2 leads to the acquisition of DNMT3A-dependent tolerogenic functions in human myeloid-derived suppressor cells. Cell Rep. 21, 154–167 (2017).
Waight, J. D. et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J. Clin. Investig. 123, 4464–4478 (2013).
Ibrahim, M. L. & Liu, K. IRF8 deficiency in colonic epithelial cells promotes inflammation-mediated colon tumorigenesis and myeloid-derived suppressor cell differentiation. J. Immunol. 198, 66.20 (2017).
Smith, A. D. et al. Autocrine IL6-mediated activation of the STAT3–DNMT axis silences the TNFα–RIP1 necroptosis pathway to sustain survival and accumulation of myeloid-derived suppressor cells. Cancer Res. 80, 3145–3156 (2020).
Redd, P. S. et al. SETD1B activates iNOS expression in myeloid-derived suppressor cells. Cancer Res. 77, 2834–2843 (2017).
Zhang, Z., Huang, X., Wang, E., Huang, Y. & Yang, R. Suppression of Mll1-complex by Stat3/Cebpβ-induced miR-21a/21b/181b maintains the accumulation, homeostasis, and immunosuppressive function of polymorphonuclear myeloid-derived suppressor cells. J. Immunol. 204, 3400–3415 (2020).
de Almeida Nagata, D. E. et al. Regulation of tumor-associated myeloid cell activity by CBP/EP300 bromodomain modulation of H3K27 acetylation. Cell Rep. 27, 269–281 (2019).
Ni, H.-h., Zhang, L., Huang, H., Dai, S.-q & Li, J. Connecting METTL3 and intratumoural CD33+ MDSCs in predicting clinical outcome in cervical cancer. J. Transl. Med. 18, 393 (2020).
Gardner, A. & Ruffell, B. Dendritic cells and cancer immunity. Trends Immunol. 37, 855–865 (2016).
Morante-Palacios, O., Fondelli, F., Ballestar, E. & Martínez-Cáceres, E. M. Tolerogenic dendritic cells in autoimmunity and inflammatory diseases. Trends Immunol. 42, 59–75 (2021).
Scarlett, U. K. et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 209, 495–506 (2012).
Singh Rawat, B., Venkataraman, R. & Budhwar, R. Methionine- and choline-deficient diet identifies an essential role for DNA methylation in plasmacytoid dendritic cell biology. J. Immunol. 208, 881–897 (2022).
Czeh, M. & Stäble, S. DNMT1 deficiency impacts on plasmacytoid dendritic cells in homeostasis and autoimmune disease. J. Immunol. 208, 358–370 (2022).
Liu, L. et al. Integrated nanovaccine with microRNA-148a inhibition reprograms tumor-associated dendritic cells by modulating miR-148a/DNMT1/SOCS1 axis. J. Immunol. 197, 1231–1241 (2016).
Vento-Tormo, R. et al. IL-4 orchestrates STAT6-mediated DNA demethylation leading to dendritic cell differentiation. Genome Biol. 17, 4 (2016).
Morante-Palacios, O. et al. Vitamin C enhances NF-κB-driven epigenomic reprogramming and boosts the immunogenic properties of dendritic cells. Nucleic Acids Res. 50, 10981–10994 (2022).
Magrì, A. et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 12, eaay8707 (2020).
Jeong, Y.-J. et al. Vitamin C treatment of mouse bone marrow-derived dendritic cells enhanced CD8+ memory T cell production capacity of these cells in vivo. Immunobiology 219, 554–564 (2014).
Català-Moll, F. et al. Vitamin D receptor, STAT3, and TET2 cooperate to establish tolerogenesis. Cell Rep. 38, 110244 (2022).
Huang, Y. et al. Global mapping of H3K4me3 and H3K27me3 reveals chromatin state-based regulation of human monocyte-derived dendritic cells in different environments. Genes Immun. 13, 311–320 (2012).
Kel, J. M., Girard-Madoux, M. J. H., Reizis, B. & Clausen, B. E. TGF-β Is required to maintain the pool of immature Langerhans cells in the epidermis. J. Immunol. 185, 3248–3255 (2010).
Melief, C. J. M. Cancer immunotherapy by dendritic cells. Immunity 29, 372–383 (2008).
Terra, M. et al. Tumor-derived TGFβ alters the ability of plasmacytoid dendritic cells to respond to innate immune signaling. Cancer Res. 78, 3014–3026 (2018).
Moran, T. P., Nakano, H., Kondilis-Mangum, H. D., Wade, P. A. & Cook, D. N. Epigenetic control of Ccr7 expression in distinct lineages of lung dendritic cells. J. Immunol. 193, 4904–4913 (2014).
Zhou, Z. et al. Epigenetically modulated FOXM1 suppresses dendritic cell maturation in pancreatic cancer and colon cancer. Mol. Oncol. 13, 873–893 (2019).
Wang, H. et al. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat. Commun. 10, 1898 (2019).
Han, D. et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature 566, 270–274 (2019).
Masucci, M. T., Minopoli, M. & Carriero, M. V. Tumor associated neutrophils. their role in tumorigenesis, metastasis, prognosis and therapy. Front. Oncol. 9, 1146 (2019).
Bekes, E. M. et al. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am. J. Pathol. 179, 1455–1470 (2011).
Szczerba, B. M. et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 566, 553–557 (2019).
Yang, L. et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 583, 133–138 (2020).
Liu, R., Zhao, E., Wang, F. & Cui, H. CCDC25: precise navigator for neutrophil extracellular traps on the prometastatic road. Sig. Transduct. Target. Ther. 5, 162 (2020).
Wang, X. et al. Identification of a subset of immunosuppressive P2RX1-negative neutrophils in pancreatic cancer liver metastasis. Nat. Commun. 12, 174 (2021).
Raftopoulou, S., Valadez-Cosmes, P., Mihalic, Z. N., Schicho, R. & Kargl, J. Tumor-mediated neutrophil polarization and therapeutic implications. Int. J. Mol. Sci. 23, 3218 (2022).
Fridlender, Z. G. et al. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 16, 183–194 (2009).
Zhu, Q. et al. The IL-6–STAT3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell Death Dis. 5, e1295–e1295 (2014).
Jablonska, J., Leschner, S., Westphal, K., Lienenklaus, S. & Weiss, S. Neutrophils responsive to endogenous IFN-β regulate tumor angiogenesis and growth in a mouse tumor model. J. Clin. Investig. 120, 1151–1164 (2010).
Sagiv, J. Y. et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 10, 562–573 (2015).
Nishida, J. et al. Epigenetic remodelling shapes inflammatory renal cancer and neutrophil-dependent metastasis. Nat. Cell Biol. 22, 465–475 (2020).
He, J. et al. METTL3 restrains papillary thyroid cancer progression via m6A/c-Rel/IL-8-mediated neutrophil infiltration. Mol. Ther. 29, 1821–1837 (2021).
Rönnerblad, M. et al. Analysis of the DNA methylome and transcriptome in granulopoiesis reveals timed changes and dynamic enhancer methylation. Blood 123, e79–e89 (2014).
Ciavatta, D. J. et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J. Clin. Investig. 120, 3209–3219 (2010).
Zha, C. et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol. Med 17, 154–168 (2020).
Ito, I., Fukazawa, J. & Yoshida, M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J. Biol. Chem. 282, 16336–16344 (2007).
Ostuni, R., Natoli, G., Cassatella, M. A. & Tamassia, N. Epigenetic regulation of neutrophil development and function. Semin. Immunol. 28, 83–93 (2016).
Liu, Y. et al. m6A demethylase ALKBH5 is required for antibacterial innate defense by intrinsic motivation of neutrophil migration. Sig. Transduct. Target. Ther. 7, 194 (2022).
Alsina-Sanchis, E. et al. Endothelial RBPJ is essential for the education of tumor-associated macrophages. Cancer Res. 82, 4414–4428 (2022).
Wieland, E. et al. Endothelial notch1 activity facilitates metastasis. Cancer Cell 31, 355–367 (2017).
Lu, J. et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 23, 171–185 (2013).
Hongu, T. et al. Perivascular tenascin C triggers sequential activation of macrophages and endothelial cells to generate a pro-metastatic vascular niche in the lungs. Nat. Cancer 3, 486–504 (2022).
Croix, B. S. et al. Genes expressed in human tumor endothelium. Science 289, 1197–1202 (2000).
Luo, W. et al. Isolation and genome-wide expression and methylation characterization of CD31+ cells from normal and malignant human prostate tissue. Oncotarget 4, 1472–1483 (2013).
Xu, Y. et al. Intracellular adenosine regulates epigenetic programming in endothelial cells to promote angiogenesis. EMBO Mol. Med. 9, 1263–1278 (2017).
Maishi, N. et al. Tumour endothelial cells in high metastatic tumours promote metastasis via epigenetic dysregulation of biglycan. Sci. Rep. 6, 28039 (2016).
Deeb, K. K. et al. Differential vitamin D 24-hydroxylase/CYP24A1 gene promoter methylation in endothelium from benign and malignant human prostate. Epigenetics 6, 994–1000 (2011).
Chung, I. et al. Epigenetic silencing of CYP24 in tumor-derived endothelial cells contributes to selective growth inhibition by calcitriol. J. Biol. Chem. 282, 8704–8714 (2007).
Hellebrekers, D. M. E. I. et al. Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res. 67, 4138–4148 (2007).
Hellebrekers, D. M. E. I. et al. Epigenetic regulation of tumor endothelial cell anergy: silencing of intercellular adhesion molecule-1 by histone modifications. Cancer Res. 66, 10770–10777 (2006).
Auguste, P. et al. The host inflammatory response promotes liver metastasis by increasing tumor cell arrest and extravasation. Am. J. Pathol. 170, 1781–1792 (2007).
Bandyopadhyay, S. et al. HOXA9 methylation by PRMT5 is essential for endothelial cell expression of leukocyte adhesion molecules. Mol. Cell. Biol. 32, 1202–1213 (2012).
Ciesielski, O. et al. The epigenetic profile of tumor endothelial cells. Effects of combined therapy with antiangiogenic and epigenetic drugs on cancer progression. Int. J. Mol. Sci. 21, 2606 (2020).
Li, M., Zha, X. & Wang, S. The role of N6-methyladenosine mRNA in the tumor microenvironment. Biochim. Biophys. Acta 1875, 188522 (2021).
Chen, X. et al. Regulatory role of RNA N6-methyladenosine modification in bone biology and osteoporosis. Front. Endocrinol. 10, 911 (2020).
Kurosu, T. et al. HuR keeps an angiogenic switch on by stabilising mRNA of VEGF and COX-2 in tumour endothelium. Br. J. Cancer 104, 819–829 (2011).
Yue, B. et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol. Cancer 18, 142 (2019).
Chen, X. & Song, E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 18, 99–115 (2019).
Huang, H. et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 40, 656–673 (2022).
Biffi, G. et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9, 282–301 (2019).
Francescone, R. et al. Netrin G1 promotes pancreatic tumorigenesis through cancer-associated fibroblast–driven nutritional support and immunosuppression. Cancer Discov. 11, 446–479 (2021).
Dominguez, C. X. et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 10, 232–253 (2020).
Lavie, D., Ben-Shmuel, A., Erez, N. & Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 3, 793–807 (2022).
Hu, M. et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet. 37, 899–905 (2005).
Marks, D. L., Olson, R. L. & Fernandez-Zapico, M. E. Epigenetic control of the tumor microenvironment. Epigenomics 8, 1671–1687 (2016).
Vizoso, M. et al. Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis 36, 1453–1463 (2015).
Jiang, L. et al. Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res. 68, 9900–9908 (2008).
Peduto, L. et al. ADAM12 is highly expressed in carcinoma-associated stroma and is required for mouse prostate tumor progression. Oncogene 25, 5462–5466 (2006).
Yu, J. et al. Unlike pancreatic cancer cells pancreatic cancer associated fibroblasts display minimal gene induction after 5-aza-2’-deoxycytidine. PLoS One 7, e43456 (2012).
Halperin, C. et al. Global DNA methylation analysis of cancer-associated fbroblasts reveals extensive epigenetic rewiring linked with RUNX1 upregulation in breast cancer stroma. Cancer Res. 82, 4139–4152 (2022).
Bhagat, T. D. et al. Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. eLife 8, e50663 (2019).
Becker, L. M. et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep. 31, 107701 (2020).
Shakya, R. et al. Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res. 73, 885–896 (2013).
Götze, S., Schumacher, E. C., Kordes, C. & Häussinger, D. Epigenetic changes during hepatic stellate cell activation. PLoS One 10, e0128745 (2015).
Albrengues, J. et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 6, 10204 (2015).
Xiao, Q. et al. Cancer-associated fibroblasts in pancreatic cancer are reprogrammed by tumor-induced alterations in genomic DNA methylation. Cancer Res. 76, 5395–5404 (2016).
Avalle, L. et al. STAT3 induces breast cancer growth via ANGPTL4, MMP13 and STC1 secretion by cancer associated fibroblasts. Oncogene 41, 1456–1467 (2022).
Tang, X. et al. Autocrine TGF-β1/miR-200s/miR-221/DNMT3B regulatory loop maintains CAF status to fuel breast cancer cell proliferation. Cancer Lett. 452, 79–89 (2019).
Maeda, M. et al. Cancer cell niche factors secreted from cancer-associated fibroblast by loss of H3K27me3. Gut 69, 243–251 (2020).
Eckert, M. A. et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 569, 723–728 (2019).
Kilgour, M. K. et al. 1-Methylnicotinamide is an immune regulatory metabolite in human ovarian cancer. Sci. Adv. 7, eabe1174 (2021).
Chen, J.-Y., Li, C.-F., Lai, Y.-S. & Hung, W.-C. Lysine demethylase 2A expression in cancer-associated fibroblasts promotes breast tumour growth. Br. J. Cancer 124, 484–493 (2021).
Liu, C. et al. LSD1 stimulates cancer-associated fibroblasts to drive Notch3-dependent self-renewal of liver cancer stem–like cells. Cancer Res. 78, 938–949 (2018).
Li, Y. et al. Cancer-associated fibroblasts hinder lung squamous cell carcinoma oxidative stress-induced apoptosis via METTL3 mediated m6A methylation of COL10A1. Oxid. Med. Cell. Longev. 2022, 4320809 (2022).
Song, T. et al. Zfp217 mediates m6A mRNA methylation to orchestrate transcriptional and post-transcriptional regulation to promote adipogenic differentiation. Nucleic Acids Res. 47, 6130–6144 (2019).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell 186, 243–278 (2023).
Booth, L. N. & Brunet, A. The aging epigenome. Mol. Cell 62, 728–744 (2016).
Oberdoerffer, P. et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918 (2008).
Sinclair, D. A. & LaPlante, M. D. Lifespan: Why We Age—And Why We Don’t Have To. (Atria books, 2019).
Jiang, N. et al. Dietary and genetic effects on age-related loss of gene silencing reveal epigenetic plasticity of chromatin repression during aging. Aging 5, 813–824 (2013).
Lu, T. et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454 (2014).
Yang, J.-H. et al. Loss of epigenetic information as a cause of mammalian aging. Cell 186, 305–326 (2023).
Maegawa, S. et al. Caloric restriction delays age-related methylation drift. Nat. Commun. 8, 539 (2017).
Gasior, S. L., Wakeman, T. P., Xu, B. & Deininger, P. L. The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 357, 1383–1393 (2006).
O’Hagan, H. M., Mohammad, H. P. & Baylin, S. B. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 4, e1000155 (2008).
Jung, M. & Pfeifer, G. P. Aging and DNA methylation. BMC Biol. 13, 7 (2015).
Avrahami, D. et al. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved β cell function. Cell Metab. 22, 619–632 (2015).
Zhang, Z., Deng, C., Lu, Q. & Richardson, B. Age-dependent DNA methylation changes in the ITGAL (CD11a) promoter. Mech. Ageing Dev. 123, 1257–1268 (2002).
Zhang, W., Qu, J., Liu, G.-H. & Belmonte, J. C. I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 21, 137–150 (2020).
Li, X. et al. Lipid metabolism dysfunction induced by age-dependent DNA methylation accelerates aging. Sig. Transduct. Target. Ther. 7, 162 (2022).
Beerman, I. et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 12, 413–425 (2013).
Teschendorff, A. E. et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 20, 440–446 (2010).
Seale, K., Horvath, S., Teschendorff, A., Eynon, N. & Voisin, S. Making sense of the ageing methylome. Nat. Rev. Genet. 23, 585–605 (2022).
Talens, R. P. et al. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell 11, 694–703 (2012).
Slieker, R. C. et al. Age-related accrual of methylomic variability is linked to fundamental ageing mechanisms. Genome Biol. 17, 191 (2016).
Martinez-Jimenez, C. P. et al. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 355, 1433–1436 (2017).
Enge, M. et al. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell 171, 321–330 (2017).
Hernando-Herraez, I. et al. Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nat. Commun. 10, 4361 (2019).
Bahar, R. et al. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441, 1011–1014 (2006).
Liu, L. et al. Insufficient DNA methylation affects healthy aging and promotes age-related health problems. Clin. Epigenet. 2, 349–360 (2011).
Gao, Q. et al. Inhibition of DNA methyltransferase aberrations reinstates antioxidant aging suppressors and ameliorates renal aging. Aging Cell 21, e13526 (2022).
Zha, S., Li, Z., Chen, S., Liu, F. & Wang, F. MeCP2 inhibits cell functionality through FoxO3a and autophagy in endothelial progenitor cells. Aging 11, 6714–6733 (2019).
Komaki, S. et al. Epigenetic profile of Japanese supercentenarians: a cross-sectional study. Lancet Healthy Longev. 4, e83–e90 (2023).
Wakeling, L. A. et al. SIRT1 affects DNA methylation of polycomb group protein target genes, a hotspot of the epigenetic shift observed in ageing. Hum. Genom. 9, 14 (2015).
Lu, Y. et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 588, 124–129 (2020).
Wu, F. et al. Spurious transcription causing innate immune responses is prevented by 5-hydroxymethylcytosine. Nat. Genet. 55, 100–111 (2023).
McCauley, B. S. et al. Altered chromatin states drive cryptic transcription in aging mammalian stem cells. Nat. Aging 1, 684–697 (2021).
Jylhävä, J., Pedersen, N. L. & Hägg, S. Biological age predictors. EBioMedicine 21, 29–36 (2017).
Field, A. E. et al. DNA methylation clocks in aging: categories, causes, and consequences. Mol. Cell 71, 882–895 (2018).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, 3156 (2013).
Hannum, G. et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367 (2013).
Trapp, A., Kerepesi, C. & Gladyshev, V. N. Profiling epigenetic age in single cells. Nat. Aging 1, 1189–1201 (2021).
Zhang, Q. et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 11, 54 (2019).
Marioni, R. E. et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 16, 25 (2015).
Horvath, S. et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging 7, 1159–1170 (2015).
Maddock, J. et al. DNA methylation age and physical and cognitive aging. J. Gerontol. A Biol. Sci. Med. Sci. 75, 504–511 (2020).
McCrory, C. et al. GrimAge outperforms other epigenetic clocks in the prediction of age-related clinical phenotypes and all-cause mortality. J. Gerontol. A Biol. Sci. Med. Sci. 76, 741–749 (2021).
Belsky, D. W. et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. eLife 11, e73420 (2022).
Lu, A. T. et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 11, 303–327 (2019).
Levine, M. E. et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 10, 573–591 (2018).
Horvath, S. & Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 19, 371–384 (2018).
Alisch, R. S. et al. Age-associated DNA methylation in pediatric populations. Genome Res. 22, 623–632 (2012).
Johnson, N. D. et al. Non-linear patterns in age-related DNA methylation may reflect CD4 + T cell differentiation. Epigenetics 12, 492–503 (2017).
Sarmento, O. F. et al. A novel role for Kruppel-like factor 14 (KLF14) in T-regulatory cell differentiation. Cell. Mol. Gastroenterol. Hepatol. 1, 188–202 (2015).
Garg, S. K. et al. Aging is associated with increased regulatory T-cell function. Aging Cell 13, 441–448 (2014).
Liu, Z. et al. Large-scale chromatin reorganization reactivates placenta-specific genes that drive cellular aging. Dev. Cell 57, 1347–1368 (2022).
Rao, R. A. et al. KMT1 family methyltransferases regulate heterochromatin–nuclear periphery tethering via histone and non-histone protein methylation. EMBO Rep. 20, e43260 (2019).
Kind, J. et al. Single-cell dynamics of genome-nuclear lamina interactions. Cell 153, 178–192 (2013).
Lee, T. W.-S., David, H. S., Engstrom, A. K., Carpenter, B. S. & Katz, D. J. Repressive H3K9me2 protects lifespan against the transgenerational burden of COMPASS activity in C. elegans. eLife 8, e48498 (2019).
Tian, Y. et al. Mitochondrial stress induces chromatin reorganization to promote longevity and UPRmt. Cell 165, 1197–1208 (2016).
Huang, M. et al. H3K9me1/2 methylation limits the lifespan of daf-2 mutants in C. elegans. eLife 11, e74812 (2022).
Zhang, W. et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348, 1160–1163 (2015).
Shumaker, D. K. et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl Acad. Sci. 103, 8703–8708 (2006).
Djeghloul, D. et al. Age-associated decrease of the histone methyltransferase SUV39H1 in HSC perturbs heterochromatin and B lymphoid differentiation. Stem Cell Rep. 6, 970–984 (2016).
Liu, B. et al. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat. Commun. 4, 1868 (2013).
Wang, Y. et al. Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat. Commun. 11, 5596 (2020).
Li, C. L. & Pu, M. Region-specific H3K9me3 gain in aged somatic tissues in Caenorhabditis elegans. PLoS Genet 17, e1009432 (2021).
Poleshko, A. et al. H3K9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis. eLife 8, e49278 (2019).
Jeon, H.-J. et al. Effect of heterochromatin stability on intestinal stem cell aging in Drosophila. Mech. Ageing Dev. 173, 50–60 (2018).
Wood, J. G. et al. Chromatin remodeling in the aging genome of Drosophila. Aging Cell 9, 971–978 (2010).
Zhang, Y. et al. Single-cell epigenome analysis reveals age-associated decay of heterochromatin domains in excitatory neurons in the mouse brain. Cell Res. 32, 1008–1021 (2022).
Soto-Palma, C., Niedernhofer, L. J., Faulk, C. D. & Dong, X. Epigenetics, DNA damage, and aging. J. Clin. Investig. 132, e158446 (2022).
Jin, C. et al. Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 14, 161–172 (2011).
Maures, T. J., Greer, E. L., Hauswirth, A. G. & Brunet, A. The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell 10, 980–990 (2011).
Tan, L. et al. Naked mole rat cells have a stable epigenome that resists iPSC reprogramming. Stem Cell Rep. 9, 1721–1734 (2017).
Bracken, A. P. et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 21, 525–530 (2007).
Shah, P. P. et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 27, 1787–1799 (2013).
Ma, Z. et al. Epigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in Drosophila. eLife 7, e35368 (2018).
Liu, L. et al. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 4, 189–204 (2013).
Sun, D. et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 14, 673–688 (2014).
Wang, C. M., Tsai, S. N., Yew, T. W., Kwan, Y. W. & Ngai, S. M. Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology 11, 87–102 (2010).
Merkwirth, C. et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223 (2016).
Labbadia, J. & Morimoto, R. I. Repression of the heat shock response is a programmed event at the onset of reproduction. Mol. Cell 59, 639–650 (2015).
Siebold, A. P. et al. Polycomb repressive complex 2 and trithorax modulate Drosophila longevity and stress resistance. Proc. Natl Acad. Sci. 107, 169–174 (2010).
Santo, E. E. et al. FOXO3A-short is a novel regulator of non-oxidative glucose metabolism associated with human longevity. Aging Cell 22, e13763 (2023).
Guillermo, A. R. R. et al. H3K27 modifiers regulate lifespan in C. elegans in a context-dependent manner. BMC Biol. 19, 59 (2021).
Shaw, C. K. et al. X chromosome factor Kdm6a enhances cognition independent of its demethylase function in the aging XY male brain. J. Gerontol. A Biol. Sci. Med. Sci. https://doi.org/10.1093/gerona/glad007 (2023).
Greer, E. L. et al. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466, 383–387 (2010).
Castillo-Quan, J. I. et al. An antisteatosis response regulated by oleic acid through lipid droplet–mediated ERAD enhancement. Sci. Adv. 9, eadc8917 (2023).
Han, S. et al. Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature 544, 185–190 (2017).
Bazopoulou, D. et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature 576, 301–305 (2019).
Cheung, P. et al. Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell 173, 1385–1397 (2018).
Cao, Q. et al. Targeting histone K4 trimethylation for treatment of cognitive and synaptic deficits in mouse models of Alzheimer’s disease. Sci. Adv. 6, eabc8096 (2020).
Li, G. et al. WTAP-mediated m6A modification of lncRNA NORAD promotes intervertebral disc degeneration. Nat. Commun. 13, 1469 (2022).
McColl, G. et al. Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J. Biol. Chem. 283, 350–357 (2008).
Ni, Z., Ebata, A., Alipanahiramandi, E. & Lee, S. S. Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 11, 315–325 (2012).
Caron, M. et al. Loss of SET1/COMPASS methyltransferase activity reduces lifespan and fertility in Caenorhabditis elegans. Life Sci. Alliance 5, e202101140 (2022).
Adelman, E. R. et al. Aging human hematopoietic stem cells manifest profound epigenetic reprogramming of enhancers that may predispose to leukemia. Cancer Discov. 9, 1080–1101 (2019).
Cheung, I. et al. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl Acad. Sci. 107, 8824–8829 (2010).
Onken, B. et al. Metformin treatment of diverse Caenorhabditis species reveals the importance of genetic background in longevity and healthspan extension outcomes. Aging Cell 21, e13488 (2022).
Lucanic, M. et al. Impact of genetic background and experimental reproducibility on identifying chemical compounds with robust longevity effects. Nat. Commun. 8, 14256 (2017).
Mei, Q. et al. Set1-catalyzed H3K4 trimethylation antagonizes the HIR/Asf1/Rtt106 repressor complex to promote histone gene expression and chronological life span. Nucleic Acids Res. 47, 3434–3449 (2019).
Cruz, C. et al. Tri-methylation of histone H3 lysine 4 facilitates gene expression in ageing cells. eLife 7, e34081 (2018).
Sen, P. et al. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 29, 1362–1376 (2015).
Pu, M. et al. Trimethylation of Lys36 on H3 restricts gene expression change during aging and impacts life span. Genes Dev. 29, 718–731 (2015).
Wang, L. et al. H3K36 trimethylation mediated by SETD2 regulates the fate of bone marrow mesenchymal stem cells. PLoS Biol. 16, e2006522 (2018).
Martin-Herranz, D. E. et al. Screening for genes that accelerate the epigenetic aging clock in humans reveals a role for the H3K36 methyltransferase NSD1. Genome Biol. 20, 146 (2019).
Li, H. et al. Arginine methylation of SKN-1 promotes oxidative stress resistance in Caenorhabditis elegans. Redox Biol. 21, 101111 (2019).
Takahashi, Y. et al. Asymmetric arginine dimethylation determines life span in C. elegans by regulating forkhead transcription factor DAF-16. Cell Metab. 13, 505–516 (2011).
Zhang, X.-S., Li, W.-S. & Xu, W.-H. Activation of protein arginine methyltransferase 1 and subsequent extension of moth lifespan is effected by the ROS/JNK/CREB signaling axis. J. Biol. Chem 299, 102950 (2023).
Lim, Y., Lee, E., Lee, J., Oh, S. & Kim, S. Down-regulation of asymmetric arginine methylation during replicative and H2O2-induced premature senescence in WI-38 human diploid fibroblasts. J. Biochem. 144, 523–529 (2008).
Hong, E., Lim, Y., Lee, E., Oh, M. & Kwon, D. Tissue-specific and age-dependent expression of protein arginine methyltransferases (PRMTs) in male rat tissues. Biogerontology 13, 329–336 (2012).
Simandi, Z. et al. Arginine methyltransferase PRMT8 provides cellular stress tolerance in aging motoneurons. J. Neurosci. 38, 7683–7700 (2018).
Min, K.-W. et al. Profiling of m6A RNA modifications identified an age-associated regulation of AGO2 mRNA stability. Aging Cell 17, e12753 (2018).
Wu, Z. et al. METTL3 counteracts premature aging via m6A-dependent stabilization of MIS12 mRNA. Nucleic Acids Res. 48, 11083–11096 (2020).
Zhang, J. et al. Lamin A safeguards the m6A methylase METTL14 nuclear speckle reservoir to prevent cellular senescence. Aging Cell 19, e13215 (2020).
Chelmicki, T. et al. m6A RNA methylation regulates the fate of endogenous retroviruses. Nature 591, 312–316 (2021).
Fan, T., Du, Y., Zhang, M., Zhu, A. R. & Zhang, J. Senolytics cocktail dasatinib and quercetin alleviate human umbilical vein endothelial cell senescence via the TRAF6-MAPK-NF-κB axis in a YTHDF2-dependent manner. Gerontology 68, 920–934 (2023).
Chen, X. et al. METTL3-mediated m6A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann. Rheum. Dis. 81, 85–97 (2022).
Zhu, H., Sun, B., Zhu, L., Zou, G. & Shen, Q. N6-methyladenosine induced miR-34a-5p promotes TNF-α-induced nucleus pulposus cell senescence by targeting SIRT1. Front. Cell Dev. Biol. 9, 642437 (2021).
Xu, Z. et al. m6A modification mediates endothelial cell responses to oxidative stress in vascular aging induced by low fluid shear stress. Oxid. Med. Cell. Longev. 2023, 8134027 (2023).
Li, G. et al. m6A hypomethylation of DNMT3B regulated by ALKBH5 promotes intervertebral disc degeneration via E4F1 deficiency. Clin. Transl. Med. 12, e765 (2022).
Sendinc, E., Valle-Garcia, D., Jiao, A. & Shi, Y. Analysis of m6A RNA methylation in Caenorhabditis elegans. Cell Discov. 6, 47 (2020).
Bhalla, S. et al. Patient similarity network of newly diagnosed multiple myeloma identifies patient subgroups with distinct genetic features and clinical implications. Sci. Adv. 7, eabg9551 (2021).
Hansen, M. et al. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 6, 95–110 (2007).
Ignatova, V. V. et al. The rRNA m6A methyltransferase METTL5 is involved in pluripotency and developmental programs. Genes Dev. 34, 715–729 (2020).
Zhang, Z. et al. METTL14 regulates intestine cellular senescence through m6A modification of lamin B receptor. Oxid. Med. Cell. Longev. 2022, 9096436 (2022).
Jiang, Z.-x. et al. The m6A mRNA demethylase FTO in granulosa cells retards FOS-dependent ovarian aging. Cell Death Dis. 12, 744 (2021).
Sun, X. et al. Decreased expression of m6A demethylase FTO in ovarian aging. Arch. Gynecol. Obstet. 303, 1363–1369 (2021).
Su, X. et al. Aging-associated differences in epitranscriptomic m6A regulation in response to acute cardiac ischemia/reperfusion injury in female mice. Front. Pharmacol. 12, 654316 (2021).
Livneh, I., Moshitch-Moshkovitz, S., Amariglio, N., Rechavi, G. & Dominissini, D. The m6A epitranscriptome: transcriptome plasticity in brain development and function. Nat. Rev. Neurosci. 21, 36–51 (2020).
Shafik, A. M. et al. N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer’s disease. Genome Biol. 22, 17 (2021).
Huang, H., Song, R., Wong, J. J.-L., Anggono, V. & Widagdo, J. The N6-methyladenosine RNA landscape in the aged mouse hippocampus. Aging Cell 22, e13755 (2023).
Castro-Hernández, R. et al. Conserved reduction of m6A marks during aging and neurodegeneration is linked to altered translation of synaptic transcripts. bioRxiv, 2022.2006.2008.495100 (2022).
Chen, X. et al. Down-regulation of m6A mRNA methylation is involved in dopaminergic neuronal death. ACS Chem. Neurosci. 10, 2355–2363 (2019).
Huang, H., Camats-Perna, J., Medeiros, R., Anggono, V. & Widagdo, J. Altered expression of the m6A methyltransferase METTL3 in Alzheimer’s disease. eneuro 7, ENEURO.0125-0120.2020 (2020).
Zhao, F. et al. METTL3-dependent RNA m6A dysregulation contributes to neurodegeneration in Alzheimer’s disease through aberrant cell cycle events. Mol. Neurodegener. 16, 70 (2021).
Han, M. et al. Abnormality of m6A mRNA methylation is involved in Alzheimer’s Disease. Front. Neurosci. 14, 98 (2020).
Tang, Z. et al. KDM1A-mediated upregulation of METTL3 ameliorates Alzheimer’s disease via enhancing autophagic clearance of p-Tau through m6A-dependent regulation of STUB1. Free Radic. Biol. Med. 195, 343–358 (2023).
Cardelli, M. et al. A polymorphism of the YTHDF2 gene (1p35) located in an Alu-rich genomic domain is associated with human longevity. J. Gerontol. A Biol. Sci. Med. Sci. 61, 547–556 (2006).
Mapperley, C. et al. The mRNA m6A reader YTHDF2 suppresses proinflammatory pathways and sustains hematopoietic stem cell function. J. Exp. Med. 218, e20200829 (2020).
Zhu, R. et al. Melatonin antagonizes ovarian aging via YTHDF2-MAPK-NF-κB pathway. Genes Dis. 9, 494–509 (2022).
Schosserer, M. et al. Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat. Commun. 6, 6158 (2015).
Heissenberger, C. et al. The ribosomal RNA m5C methyltransferase NSUN-1 modulates healthspan and oogenesis in Caenorhabditis elegans. eLife 9, e56205 (2020).
Heissenberger, C. et al. Loss of the ribosomal RNA methyltransferase NSUN5 impairs global protein synthesis and normal growth. Nucleic Acids Res. 47, 11807–11825 (2019).
Tuorto, F. et al. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 19, 900–905 (2012).
Lin, M.-J., Tang, L.-Y., Reddy, M. N. & Shen, C. K. J. DNA Methyltransferase gene dDnmt2 and longevity of drosophila*. J. Biol. Chem. 280, 861–864 (2005).
Lewinska, A. et al. Reduced levels of methyltransferase DNMT2 sensitize human fibroblasts to oxidative stress and DNA damage that is accompanied by changes in proliferation-related miRNA expression. Redox Biol. 14, 20–34 (2018).
Blanco, S. et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 33, 2020–2039 (2014).
Kim, Y. A. et al. RNA methyltransferase NSun2 deficiency promotes neurodegeneration through epitranscriptomic regulation of tau phosphorylation. Acta Neuropathol. 145, 29–48 (2023).
Tang, H. et al. NSun2 delays replicative senescence by repressing p27 (KIP1) translation and elevating CDK1 translation. Aging 7, 1143–1158 (2015).
Li, Q. et al. NSUN2-mediated m5C methylation and METTL3/METTL14-mediated m6A methylation cooperatively enhance p21 translation. J. Cell. Biochem. 118, 2587–2598 (2017).
Cai, X. et al. RNA methyltransferase NSUN2 promotes stress-induced HUVEC senescence. Oncotarget 7, 19099–19110 (2016).
Zhang, X. et al. The tRNA methyltransferase NSun2 stabilizes p16INK4 mRNA by methylating the 3′-untranslated region of p16. Nat. Commun. 3, 712 (2012).
Tang, H. et al. HuR regulates telomerase activity through TERC methylation. Nat. Commun. 9, 2213 (2018).
Cheng, X. et al. HuB and HuD repress telomerase activity by dissociating HuR from TERC. Nucleic Acids Res. 49, 2848–2858 (2021).
Bates, S. E. Epigenetic therapies for cancer. N. Engl. J. Med. 383, 650–663 (2020).
Conery, A. R., Rocnik, J. L. & Trojer, P. Small molecule targeting of chromatin writers in cancer. Nat. Chem. Biol. 18, 124–133 (2022).
Loo Yau, H. et al. DNA hypomethylating agents increase activation and cytolytic activity of CD8 + T cells. Mol. Cell 81, 1469–1483 (2021).
Silverman, L. R. et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J. Clin. Oncol. 20, 2429–2440 (2002).
Liu, Y.-C. et al. Demethylation and up-regulation of an oncogene after hypomethylating therapy. N. Engl. J. Med. 386, 1998–2010 (2022).
Traynor, S. et al. DNA methyltransferase inhibition promotes recruitment of myeloid-derived suppressor cells to the tumor microenvironment through induction of tumor cell-intrinsic interleukin-1. Cancer Lett. 552, 215982 (2023).
Brueckner, B. et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 65, 6305–6311 (2005).
Manara, M. C. et al. A quinoline-based DNA methyltransferase inhibitor as a possible adjuvant in osteosarcoma therapy. Mol. Cancer Ther. 17, 1881–1892 (2018).
Pappalardi, M. B. et al. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat. Cancer 2, 1002–1017 (2021).
Patel, A. J. et al. PRC2-inactivating mutations in cancer enhance cytotoxic response to DNMT1-targeted therapy via enhanced viral mimicry. Cancer Discov. 12, 2120–2139 (2022).
Wiseman, A. K. et al. Chromosome-specific retention of cancer-associated DNA hypermethylation following pharmacological inhibition of DNMT1. Commun. Biol. 5, 528 (2022).
Horton, J. R. et al. Structural characterization of dicyanopyridine containing DNMT1-selective, non-nucleoside inhibitors. Structure 30, 793–802 (2022).
Fang, M. Z. et al. Tea polyphenol (–)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 63, 7563–7570 (2003).
Cornacchia, E. et al. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J. Immunol. 140, 2197–2200 (1988).
Qadir Nanakali, N. M. et al. The role of dietary polyphenols in alternating DNA methylation in cancer. Crit. Rev. Food Sci. Nutr. https://doi.org/10.1080/10408398.2022.2100313 (2022).
Stresemann, C., Brueckner, B., Musch, T., Stopper, H. & Lyko, F. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res. 66, 2794–2800 (2006).
Li, F., Qasim, S., Li, D. & Dou, Q. P. Updated review on green tea polyphenol epigallocatechin-3-gallate as a cancer epigenetic regulator. Semin. Cancer Biol. 83, 335–352 (2022).
Bensberg, M. et al. TET2 as a tumor suppressor and therapeutic target in T-cell acute lymphoblastic leukemia. Proc. Natl Acad. Sci. 118, e2110758118 (2021).
Turcan, S. et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT inhibitor decitabine. Oncotarget 4, 1729–1736 (2013).
Yin, R. et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 135, 10396–10403 (2013).
Cimmino, L. et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079–1095 (2017).
Ngo, B., Van Riper, J. M., Cantley, L. C. & Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 19, 271–282 (2019).
Taira, A. et al. Vitamin C boosts DNA demethylation in TET2 germline mutation carriers. Clin. Epigenet. 15, 7 (2023).
Figueroa, M. E. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 (2010).
Xu, W. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011).
Lu, C. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012).
Elkashef, S. M. et al. IDH mutation, competitive inhibition of FTO, and RNA methylation. Cancer Cell 31, 619–620 (2017).
McMurry, H., Fletcher, L. & Traer, E. IDH inhibitors in AML—promise and pitfalls. Curr. Hematol. Malig. Rep. 16, 207–217 (2021).
Stein, E. M. et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 133, 676–687 (2019).
DiNardo, C. D. et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 378, 2386–2398 (2018).
Natsume, A. et al. The first-in-human phase I study of a brain-penetrant mutant IDH1 inhibitor DS-1001 in patients with recurrent or progressive IDH1-mutant gliomas. Neuro Oncol. 25, 326–336 (2022).
Konteatis, Z. et al. Vorasidenib (AG-881): a first-in-class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma. ACS Med. Chem. Lett. 11, 101–107 (2020).
Mellinghoff, I. K. et al. Vorasidenib, a dual inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first-in-human phase I trial. Clin. Cancer Res. 27, 4491–4499 (2021).
Mellinghoff, I. et al. ACTR-66. A phase 1, open-lable, perioperative study of ivosidenib (AG-120) and vorasidenib (AG-881) in recurrent IDH1 mutant, low-grade glioma: updated results. Neuro Oncol. 21, vi28–vi29 (2019).
Machida, Y. et al. A potent blood–brain barrier-permeable mutant IDH1 inhibitor suppresses the growth of glioblastoma with IDH1 mutation in a patient-derived orthotopic xenograft model. Mol. Cancer Ther. 19, 375–383 (2020).
Varambally, S. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629 (2002).
Morin, R. D. et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 42, 181–185 (2010).
Xu, B., Konze, K. D., Jin, J. & Wang, G. G. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp. Hematol. 43, 698–712 (2015).
Knutson, S. K. et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 8, 890–896 (2012).
Vaswani, R. G. et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J. Med. Chem. 59, 9928–9941 (2016).
McCabe, M. T. et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112 (2012).
Yang, X. et al. Structure–activity relationship studies for enhancer of Zeste Homologue 2 (EZH2) and enhancer of Zeste Homologue 1 (EZH1) inhibitors. J. Med. Chem. 59, 7617–7633 (2016).
Konze, K. D. et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 8, 1324–1334 (2013).
Morschhauser, F. et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 21, 1433–1442 (2020).
Wilson, B. G. et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18, 316–328 (2010).
Rehman, H. et al. ARID1A-deficient bladder cancer is dependent on PI3K signaling and sensitive to EZH2 and PI3K inhibitors. JCI Insight 7, e155899 (2022).
Chan-Penebre, E. et al. Selective killing of SMARCA2- and SMARCA4-deficient small cell carcinoma of the ovary, hypercalcemic type cells by inhibition of EZH2: In vitro and in vivo preclinical models. Mol. Cancer Ther. 16, 850–860 (2017).
Knutson, S. K. et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl Acad. Sci. 110, 7922–7927 (2013).
Bitler, B. G. et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 21, 231–238 (2015).
Gounder, M. et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol. 21, 1423–1432 (2020).
Kawano, S. et al. Preclinical evidence of anti-tumor activity induced by EZH2 inhibition in human models of synovial sarcoma. PLoS One 11, e0158888 (2016).
Yamagishi, M. et al. Targeting excessive EZH1 and EZH2 activities for abnormal histone methylation and transcription network in malignant lymphomas. Cell Rep. 29, 2321–2337 (2019).
Honma, D. et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 108, 2069–2078 (2017).
Izutsu, K. et al. An open-label, single-arm, phase 2 trial of valemetostat in relapsed or refractory adult T-cell leukemia/lymphoma. Blood 141, 1159–1168 (2022).
Dou, F. et al. Valemetostat: first approval as a dual inhibitor of EZH1/2 to treat adult T-cell leukemia/lymphoma. Drug Discov. Ther. 16, 297–299 (2022).
Kim, K. H. et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 21, 1491–1496 (2015).
Xu, K. et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science 338, 1465–1469 (2012).
Lee, S. T. et al. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol. Cell 43, 798–810 (2011).
Wang, J. et al. EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat. Cell Biol. 24, 384–399 (2022).
Ernst, T. et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 42, 722–726 (2010).
Nikoloski, G. et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 42, 665–667 (2010).
Ntziachristos, P. et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 18, 298–302 (2012).
Zhang, J. et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 (2012).
Lee, W. et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 46, 1227–1232 (2014).
De Raedt, T. et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514, 247–251 (2014).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1–pl1 (2013).
Zhang, L. et al. DNMT and EZH2 inhibitors synergize to activate therapeutic targets in hepatocellular carcinoma. Cancer Lett. 548, 215899 (2022).
Okada, Y. et al. hDOT1L links histone methylation to leukemogenesis. Cell 121, 167–178 (2005).
Hou, Y. et al. DOT1L promotes cell proliferation and invasion by epigenetically regulating STAT5B in renal cell carcinoma. Am. J. Cancer Res. 13, 276–292 (2023).
Zhang, Y., Wang, Y., Valdivia, A., Huang, H. & Matei, D. DOT1 L regulates ovarian cancer stem cells by activating β-catenin signaling. Mol. Cancer Res. 21, 140–154 (2023).
Nassa, G. et al. Inhibition of histone methyltransferase DOT1L silences ERα gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci. Adv. 5, eaav5590 (2019).
Godfrey, L. et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nat. Commun. 10, 2803 (2019).
Daigle, S. R. et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 122, 1017–1025 (2013).
Stein, E. M. et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 131, 2661–2669 (2018).
Klaus, C. R. et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J. Pharmacol. Exp. Ther. 350, 646–656 (2014).
Rau, R. E. et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 128, 971–981 (2016).
Dong, J. et al. A comprehensive comparative study on LSD1 in different cancers and tumor specific LSD1 inhibitors. Eur. J. Med. Chem. 240, 114564 (2022).
Schmidt, D. M. Z. & McCafferty, D. G. trans-2-phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 46, 4408–4416 (2007).
Schenk, T. et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 18, 605–611 (2012).
Harris, W. J. et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21, 473–487 (2012).
Tayari, M. M. et al. Clinical responsiveness to all-trans retinoic acid is potentiated by LSD1 inhibition and associated with a quiescent transcriptome in myeloid malignancies. Clin. Cancer Res. 27, 1893–1903 (2021).
Fiedorowicz, J. G. & Swartz, K. L. The role of monoamine oxidase inhibitors in current psychiatric practice. J. Psychiatr. Pract. 10, 239–248 (2004).
Fang, Y., Liao, G. & Yu, B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J. Hematol. Oncol. 12, 129 (2019).
Kanouni, T. et al. Discovery of CC-90011: A potent and selective reversible inhibitor of lysine specific demethylase 1 (LSD1). J. Med. Chem. 63, 14522–14529 (2020).
Sorna, V. et al. High-throughput virtual screening identifies novel N′-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J. Med. Chem. 56, 9496–9508 (2013).
Reed, D. R. et al. Phase 1 trial of seclidemstat (SP-2577) in patients with relapsed/refractory Ewing sarcoma. J. Clin. Oncol. 39, 11514–11514 (2021).
Zhao, E. et al. KDM4C and ATF4 cooperate in transcriptional control of amino acid metabolism. Cell Rep. 14, 506–519 (2016).
Manni, W., Jianxin, X., Weiqi, H., Siyuan, C. & Huashan, S. JMJD family proteins in cancer and inflammation. Sig. Transduct. Target. Ther. 7, 304 (2022).
Jia, R. et al. GASC1 promotes stemness of esophageal squamous cell carcinoma via NOTCH1 promoter demethylation. J. Oncol. 2019, 1621054 (2019).
Loh, Y.-H., Zhang, W., Chen, X., George, J. & Ng, H.-H. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 21, 2545–2557 (2007).
Lang, T. et al. Disruption of KDM4C-ALDH1A3 feed-forward loop inhibits stemness, tumorigenesis and chemoresistance of gastric cancer stem cells. Sig. Transduct. Target. Ther. 6, 336 (2021).
Nielsen, A. L. et al. Identification of catechols as histone–lysine demethylase inhibitors. FEBS Lett. 586, 1190–1194 (2012).
Yuan, X. et al. KDM4C, a H3K9me3 histone demethylase, is involved in the maintenance of human ESCC-initiating cells by epigenetically enhancing SOX2 expression. Neoplasia 18, 594–609 (2016).
Jia, R. et al. GASC1-adapted neoadjuvant chemotherapy for resectable esophageal squamous cell carcinoma: a prospective clinical biomarker trial. J. Oncol. 2020, 1607860 (2020).
Li, H., Zhang, Q., Feng, Q., You, Q. & Guo, X. The development of small molecules targeting methyltransferase-like 3. Drug Discov. Today 28, 103513 (2023).
Barbieri, I. et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 552, 126–131 (2017).
Vu, L. P. et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 23, 1369–1376 (2017).
Yankova, E. et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601 (2021).
Ofir-Rosenfeld, Y. et al. STC-15, an oral small molecule inhibitor of the RNA methyltransferase METTL3, inhibits tumour growth through activation of anti-cancer immune responses associated with increased interferon signalling, and synergises with T cell checkpoint blockade. Eur. J. Cancer 174, S123 (2022).
Bedi, R. K. et al. Small-molecule inhibitors of METTL3, the major human epitranscriptomic writer. ChemMedChem 15, 744–748 (2020).
Moroz-Omori, E. V. et al. METTL3 inhibitors for epitranscriptomic modulation of cellular processes. ChemMedChem 16, 3035–3043 (2021).
Dolbois, A. et al. 1,4,9-triazaspiro[5.5]undecan-2-one derivatives as potent and selective METTL3 inhibitors. J. Med. Chem. 64, 12738–12760 (2021).
Lee, J.-H. et al. Eltrombopag as an allosteric inhibitor of the METTL3-14 complex affecting the m6A methylation of RNA in acute myeloid leukemia cells. Pharmaceuticals 15, 440 (2022).
Lee, J.-H., Kim, S., Jin, M. S. & Kim, Y.-C. Discovery of substituted indole derivatives as allosteric inhibitors of m6A-RNA methyltransferase, METTL3-14 complex. Drug Dev. Res. 83, 783–799 (2022).
Kumari, D. & Usdin, K. Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27. Hum. Mol. Genet. 25, 3689–3698 (2016).
Grimm, N.-B. & Lee, J. T. Selective Xi reactivation and alternative methods to restore MECP2 function in Rett syndrome. Trends Genet. 38, 920–943 (2022).
Qian, J. et al. Multiplex epigenome editing of MECP2 to rescue Rett syndrome neurons. Sci. Transl. Med. 15, eadd4666 (2023).
Waryah, C. B., Moses, C., Arooj, M. & Blancafort, P. in Epigenome Editing: Methods and Protocols (eds A. Jeltsch & M. G. Rots) 19-63 (Springer New York, 2018).
Chavez, M., Chen, X., Finn, P. B. & Qi, L. S. Advances in CRISPR therapeutics. Nat. Rev. Nephrol. 19, 9–22 (2023).
Nakamura, M., Gao, Y., Dominguez, A. A. & Qi, L. S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 23, 11–22 (2021).
Choudhury, S. R., Cui, Y., Lubecka, K., Stefanska, B. & Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 7, 46545–46556 (2016).
Wang, Q. et al. Targeted demethylation of the SARI promotor impairs colon tumour growth. Cancer Lett. 448, 132–143 (2019).
Xu, C. et al. CRISPR-based DNA methylation editing of NNT rescues the cisplatin resistance of lung cancer cells by reducing autophagy. Arch. Toxicol. 97, 441–456 (2023).
Wang, H. et al. Epigenetic targeting of granulin in hepatoma cells by synthetic CRISPR dCas9 epi-suppressors. Mol. Ther. Nucleic Acids 11, 23–33 (2018).
Rosenbluh, J. et al. Complementary information derived from CRISPR Cas9 mediated gene deletion and suppression. Nat. Commun. 8, 15403 (2017).
O’Geen, H. et al. Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenet. Chromatin 12, 26 (2019).
Zou, Y. et al. N6-methyladenosine regulated FGFR4 attenuates ferroptotic cell death in recalcitrant HER2-positive breast cancer. Nat. Commun. 13, 2672 (2022).
Park, H. et al. CRISPR/dCas9-Dnmt3a-mediated targeted DNA methylation of APP rescues brain pathology in a mouse model of Alzheimer’s disease. Transl. Neurodegener. 11, 41 (2022).
Liu, X. S. et al. Editing DNA methylation in the mammalian genome. Cell 167, 233–247 (2016).
Fang, C. et al. Cancer-specific CTCF binding facilitates oncogenic transcriptional dysregulation. Genome Biol. 21, 247 (2020).
Xu, Z. et al. Structural variants drive context-dependent oncogene activation in cancer. Nature 612, 564–572 (2022).
Oh, S. et al. Enhancer release and retargeting activates disease-susceptibility genes. Nature 595, 735–740 (2021).
Chachoua, I. et al. Canonical WNT signaling-dependent gating of MYC requires a noncanonical CTCF function at a distal binding site. Nat. Commun. 13, 204 (2022).
Schuijers, J. et al. Transcriptional dysregulation of MYC reveals common enhancer-docking mechanism. Cell Rep. 23, 349–360 (2018).
Feehley, T., O’Donnell, C. W., Mendlein, J., Karande, M. & McCauley, T. Drugging the epigenome in the age of precision medicine. Clin. Epigenet. 15, 6 (2023).
Zeitler, B. et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 25, 1131–1142 (2019).
Ahmed, M. et al. CRISPRi screens reveal a DNA methylation-mediated 3D genome dependent causal mechanism in prostate cancer. Nat. Commun. 12, 1781 (2021).
Liu, X.-M., Zhou, J., Mao, Y., Ji, Q. & Qian, S.-B. Programmable RNA N6-methyladenosine editing by CRISPR-Cas9 conjugates. Nat. Chem. Biol. 15, 865–871 (2019).
Xia, Z. et al. Epitranscriptomic editing of the RNA N6-methyladenosine modification by dCasRx conjugated methyltransferase and demethylase. Nucleic Acids Res 49, 7361–7374 (2021).
Li, J. et al. Targeted mRNA demethylation using an engineered dCas13b-ALKBH5 fusion protein. Nucleic Acids Res 48, 5684–5694 (2020).
Wilson, C., Chen, P. J., Miao, Z. & Liu, D. R. Programmable m6A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat. Biotechnol. 38, 1431–1440 (2020).
Chang, C., Ma, G., Cheung, E. & Hutchins, A. P. A programmable system to methylate and demethylate N6-methyladenosine (m6A) on specific RNA transcripts in mammalian cells. J. Biol. Chem. 298, 102525 (2022).
Zhao, J., Li, B., Ma, J., Jin, W. & Ma, X. Photoactivatable RNA N6-methyladenosine editing with CRISPR-Cas13. Small 16, 1907301 (2020).
Shi, H., Xu, Y., Tian, N., Yang, M. & Liang, F.-S. Inducible and reversible RNA N6-methyladenosine editing. Nat. Commun. 13, 1958 (2022).
Li, A. et al. ZNF677 suppresses renal cell carcinoma progression through N6-methyladenosine and transcriptional repression of CDKN3. Clin. Transl. Med. 12, e906 (2022).
Qi, F. et al. BTG2 suppresses renal cell carcinoma progression through N6-methyladenosine. Front. Oncol. 12, 1049928 (2022).
Chen, J. et al. Targeted methylation of the LncRNA NEAT1 suppresses malignancy of renal cell carcinoma. Front. Cell Dev. Biol. 9, 777349 (2021).
Chen, X. et al. Targeted RNA N6-methyladenosine demethylation controls cell fate transition in human pluripotent stem cells. Adv. Sci. 8, 2003902 (2021).
Zhou, Y. et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509, 487–491 (2014).
Tian, L., Chen, F. & Macosko, E. Z. The expanding vistas of spatial transcriptomics. Nat. Biotechnol. https://doi.org/10.1038/s41587-022-01448-2 (2022).
Zhang, L. et al. Clinical and translational values of spatial transcriptomics. Sig. Transduct. Target. Ther. 7, 111 (2022).
Yue, X. et al. Simultaneous profiling of histone modifications and DNA methylation via nanopore sequencing. Nat. Commun. 13, 7939 (2022).
Liu, Y. et al. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 37, 424–429 (2019).
Wang, Y. & Jia, G. Detection methods of epitranscriptomic mark N6-methyladenosine. Essays Biochem 64, 967–979 (2020).
Anczuków, O. et al. Challenges and opportunities for modeling aging and cancer. Cancer Cell 41, 641–645 (2023).
Morel, D., Jeffery, D., Aspeslagh, S., Almouzni, G. & Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—past lessons and future promise. Nat. Rev. Clin. Oncol. 17, 91–107 (2020).
Escobar, T. M. et al. Active and repressed chromatin domains exhibit distinct nucleosome segregation during DNA replication. Cell 179, 953–963 (2019).
Amabile, A. et al. Inheritable silencing of endogenous genes by hit-and-run targeted epigenetic editing. Cell 167, 219–232 (2016).
Chan, W. F. et al. Activation of stably silenced genes by recruitment of a synthetic de-methylating module. Nat. Commun. 13, 5582 (2022).
O’Geen, H., Tomkova, M., Combs, J. A., Tilley, E. K. & Segal, D. J. Determinants of heritable gene silencing for KRAB-dCas9 + DNMT3 and Ezh2-dCas9 + DNMT3 hit-and-run epigenome editing. Nucleic Acids Res. 50, 3239–3253 (2022).
Nuñez, J. K. et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 184, 2503–2519 (2021).
Dilliard, S. A., Cheng, Q. & Siegwart, D. J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl Acad. Sci. 118, e2109256118 (2021).
Han, X. et al. Ligand-tethered lipid nanoparticles for targeted RNA delivery to treat liver fibrosis. Nat. Commun. 14, 75 (2023).
Khirallah, J., Eimbinder, M., Li, Y. & Xu, Q. Clinical progress in genome-editing technology and in vivo delivery techniques. Trends Genet 39, 208–216 (2023).
Wang, X. et al. Preparation of selective organ-targeting (SORT) lipid nanoparticles (LNPs) using multiple technical methods for tissue-specific mRNA delivery. Nat. Protoc. 18, 265–291 (2023).
Katti, A., Diaz, B. J., Caragine, C. M., Sanjana, N. E. & Dow, L. E. CRISPR in cancer biology and therapy. Nat. Rev. Cancer 22, 259–279 (2022).
Xi, Y. et al. Multi-omic characterization of genome-wide abnormal DNA methylation reveals diagnostic and prognostic markers for esophageal squamous-cell carcinoma. Sig. Transduct. Target. Ther. 7, 53 (2022).
Papanicolau-Sengos, A. & Aldape, K. DNA methylation profiling: an emerging paradigm for cancer diagnosis. Annu. Rev. Pathol. 17, 295–321 (2022).
Jiang, H. et al. DNA methylation markers in the diagnosis and prognosis of common leukemias. Sig. Transduct. Target. Ther. 5, 3 (2020).
Hao, X. et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proc. Natl Acad. Sci. 114, 7414–7419 (2017).
Chemi, F. et al. cfDNA methylome profiling for detection and subtyping of small cell lung cancers. Nat. Cancer 3, 1260–1270 (2022).
Koch, A. et al. Analysis of DNA methylation in cancer: location revisited. Nat. Rev. Clin. Oncol. 15, 459–466 (2018).
Crosby, D. et al. Early detection of cancer. Science 375, eaay9040 (2022).
Sturm, D. et al. Multiomic neuropathology improves diagnostic accuracy in pediatric neuro-oncology. Nat. Med. 29, 917–926 (2023).
Vorperian, S. K. et al. Cell types of origin of the cell-free transcriptome. Nat. Biotechnol. 40, 855–861 (2022).
Acknowledgements
This work was supported by the pilot program of Southwest University (SWU-XDZD22006), the Natural Science Foundation of Chongqing (cstc2022ycjh-bgzxm0145), and the Chongqing Postdoctoral Science Foundation (cstc2021jcyj-bsh0128). All figures were created with BioRender and Adobe Illustrator.
Author information
Authors and Affiliations
Contributions
R.L., E.Z., C.Y., and M.N.A. wrote the manuscript; H.Y. drew figures; H.C. provided the idea and revised the manuscript. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, R., Zhao, E., Yu, H. et al. Methylation across the central dogma in health and diseases: new therapeutic strategies. Sig Transduct Target Ther 8, 310 (2023). https://doi.org/10.1038/s41392-023-01528-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01528-y
This article is cited by
-
OTUD4 promotes the progression of glioblastoma by deubiquitinating CDK1 and activating MAPK signaling pathway
Cell Death & Disease (2024)
