
Abstract
Aging is characterized by systemic chronic inflammation, which is accompanied by cellular senescence, immunosenescence, organ dysfunction, and age-related diseases. Given the multidimensional complexity of aging, there is an urgent need for a systematic organization of inflammaging through dimensionality reduction. Factors secreted by senescent cells, known as the senescence-associated secretory phenotype (SASP), promote chronic inflammation and can induce senescence in normal cells. At the same time, chronic inflammation accelerates the senescence of immune cells, resulting in weakened immune function and an inability to clear senescent cells and inflammatory factors, which creates a vicious cycle of inflammation and senescence. Persistently elevated inflammation levels in organs such as the bone marrow, liver, and lungs cannot be eliminated in time, leading to organ damage and aging-related diseases. Therefore, inflammation has been recognized as an endogenous factor in aging, and the elimination of inflammation could be a potential strategy for anti-aging. Here we discuss inflammaging at the molecular, cellular, organ, and disease levels, and review current aging models, the implications of cutting-edge single cell technologies, as well as anti-aging strategies. Since preventing and alleviating aging-related diseases and improving the overall quality of life are the ultimate goals of aging research, our review highlights the critical features and potential mechanisms of inflammation and aging, along with the latest developments and future directions in aging research, providing a theoretical foundation for novel and practical anti-aging strategies.
Similar content being viewed by others

Introduction
Aging is a common, complex, and natural phenomenon. Aging research began in 1939 with the observation that restricting calorie intake could prolong life both in mice and rats.1 To further explain aging from the perspective of harmful inflammation and weakened immunity, inflammaging was introduced as an evolutionary perspective on immunosenescence, referring to the phenomenon of low-grade, chronic damage resulting from increased inflammation levels within the body.2 Later, inflammaging has been considered a hallmark of aging.3 Meanwhile, it is worth mentioning that can also damage the immune system, leading to immunosenescence during aging. For example, studies have shown that women living longer than men,4 in which older men showed higher activity of inflammation-related modules, with a more dramatic decrease in the ratio of naive T and B cells compared to older women.4,5 In addition, centenarians have been found to possess stronger anti-inflammatory abilities, suggesting that inflammation and immunity may a significant impact on the process of aging.6,7
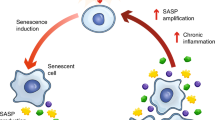
Considering the complexity of aging, multi-modal and multi-perspective studies are important. The process and accumulation of cellular senescence contribute significantly to the development of organ damage and diseases in organisms. Organ and organismal aging are often accompanied by the generation of inflammatory responses, and inflammation-associated molecular patterns promote cellular senescence, which in turn can lead to further inflammation, creating a vicious cycle (Fig. 1). In this review, we have discussed the concept of inflammaging across spatial and temporal scales, and complex factors leading to aging. We have also reviewed aging models, cutting-edge technologies in aging studies, and anti-aging strategies. Considering that preventing and alleviating the aging diseases and improving quality of life are the ultimate goals of aging research, our review shows current progress and directions in aging studies and provides a theoretical basis for new and feasible anti-aging strategies.

Inflammaging at the molecular, cellular, and organ levels. During the aging process, almost all cells in the body undergo senescence, a state characterized by a dysfunctional state and senescence-associated secretory phenotype (SASP). While immune cells play a crucial role in recognizing and eliminating these senescent cells, they are also affected by SASP, leading to a phenomenon called immunosenescence. Immunosenescence can impair the immunity to respond to infections and diseases, making the organism more vulnerable to illnesses. Moreover, the accumulation of senescent cells can trigger inflammation in organs, leading to organ damage and an increased risk of age-related diseases. This process is exacerbated by positive feedback loops that drive the accumulation of inflammation and organ damage, leading to further inflammation and an even higher risk of aging-related diseases
Inflammaging at the cellular level
As the basic unit of the body, cellular senescence and the accompanying low-energy effects drive organismal aging. Recent studies have systematically summarized the biomarkers of cellular aging.8,9 Immunocytes, as key regulators of aging cells, have always been a focus of research due to their dysfunctional changes during aging.10,11 As early as 1969, Walford proposed “the immunologic theory of aging”,12 which further developed into the concept of immunosenescence, which is mainly manifested by a decrease of the body’s immune response to endogenous and exogenous antigens, leading to a decrease of the individual’s anti-tumor capacity and the ability to clear senescent cells (Fig. 1).13 Immunosenescence is a multifactorial cascade of events with different types of immune cells exhibiting different sensitivities.14,15 However, due to the inherent complexity of the mechanisms of immunosenescence, it is imperative to conduct research on immune cellular changes in multi-modal and systematic ways.
Hematopoietic stem cells (HSCs)
Senescence of HSCs is the basis of immunosenescence. Senescent HSC differentiate into various types of dysfunctional immune cells, driving immunosenescence.16 Inflammation drives impaired self-renewal activity and accelerates aging of HSCs. Exposure to inflammatory stimuli during the early to mid-life stages in mice can lead to the eventual development of peripheral blood hemocytopenia, bone marrow (BM) cytopenia, and BM adipocyte accumulation, features that together constitute typical features of hematopoiesis in the elderly.17 The primary features of senescent HSC are characterized by changes in their self-renewal, differentiation bias, and energy metabolism (Fig. 2).

Characterization of HSC differentiation into immune cells during aging. Inflammation in senescent bone marrow impairs the function of HSCs. HSCs differentiate into various immune cells, and their senescence leads to changes in the number and functions of immune cells. Common features of immune cell senescence include a decline in performing immune functions and an increase in the release of inflammatory factors
The increased myeloid/megakaryocytic differentiation bias is a major feature of senescent HSC.18 Numerous pro-inflammatory cytokines and growth factors, including granulocyte colony-stimulating factor (G-CSF), macrophage colony-stimulating factor (M-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-1, IL-3, IL-6, TNF-α, IFNs, and Flt3 ligands, have been found to promote the differentiation of hematopoietic stem/progenitor cells (HSPCs) towards myeloid cells over lymphoid cells, leading to imbalanced myelopoiesis and lymphopoiesis.19,20,21 For example, plasma cells that have accumulated in the bone marrow of aged mice can create a feedback loop of pro-inflammatory cytokines, such as IL-1 and TNF-α, which promote HSC myeloid differentiation bias.22 According to recent studies, bone marrow cells in senescent mice secrete more IL-1α/β,23 while damaged endosteum produces IL-1β to drive inflammation in the central bone marrow in trans to impede hematopoietic regeneration.24 When faced with in vivo stimulation by lipopolysaccharide (mimicking external microbial stimuli), senescent mice exhibited increased and prolonged IL-1α/β reactions.23 These illustrate IL1 as a key mediator of niche inflammatory damage to HSC. Conversely, neutralizing transforming growth factor (TGF) -β was found to reverse the age-related bias of HSCs towards megakaryocytic differentiation, leading to a greater generation of lymphoid progenitors and a more balanced lineage output of HSCs in transplantation experiments. In addition, inhibiting IL-6 improved the function of erythroid progenitors in aged mice.25 The results indicate that inflammaging is a key mediator of age-related HSC myeloid/ megakaryocyte differentiation biases.
HSC aging leads to a diminished capacity for self-renewal (Fig. 2), as shown by a significant increase in the number of mouse HSCs with age, but not a corresponding increase in the capacity of HSCs to undergo self-renewal. Studies in aged mouse HSCs have shown that older HSCs have overall reduced cell cycle activity.26,27 Notably, IFN-γ, a crucial pro-inflammatory cytokine, appears to have a dual role in regulating HSC proliferation. On the one hand, IFN-γ has been observed to stimulate HSC proliferation during infections.28,29,30 On the other hand, conflicting evidence has suggested that IFN-γ can hinder HSC regeneration by restricting self-renewal, rather than impacting quiescence or cell cycle progression.30,31,32,33
During inflammation, HSCs shift their energy metabolism from relying on anaerobic glycolysis to oxidative respiration.34,35 Accumulated reactive oxygen species (ROS) stress may trigger excessive DNA damage and HSC senescence and/or apoptosis.36 Binding to C-X-C motif chemokine ligand (CXCL) 12, C-X-C motif chemokine receptor (CXCR) 4 serves as a crucial mediator in numerous physiological and pathological processes, including inflammatory responses of the immune systems, regulation of hematopoiesis, induction of angiogenesis, as well as tumor invasion and metastasis.37 Mice with disrupted CXCR4 receptors experience a rise in endogenous production of ROS, which activates p38 mitogen-activated protein kinases (MAPK), triggers an increase in DNA double-strand breaks, and leads to apoptosis. As a result, there is a notable decline in the HSC repopulation potential.37,38 This depletion of HSC pools can be attributed to the elevated ROS levels, which are not related to the loss of quiescence in CXCR4-deficient HSCs. The CXCR4/CXCL12 axis has been found to limit apoptosis, DNA damage, and ROS elevation in HSCs by reducing mitochondrial oxidative stress.38 These findings suggest that inflammation could hasten the aging of HSCs and accelerate HSC functional decline (Fig. 2).
Neutrophils
The role of neutrophils throughout the inflammatory response involves activation, migration, and clearance of pathogens and damaged tissues. The age-related decline in neutrophil function has a substantial influence on the development and advancement of various age-related diseases. Neutrophil development and numbers do not appear to be systematically altered with advancing age (Fig. 2).
Immunosenescence of neutrophils occurs in a low-grade inflammatory environment, with specific abnormalities in their metabolism and function, including decreased phagocytic capacity,39 abnormalities in adhesion and chemotaxis,40,41 increased apoptosis,40,42 abnormal neutrophil trap network release,43 and abnormal toll-like receptor function.44 In addition, as the organism ages, the transcriptomic and epigenomic profiles of neutrophils undergo significant remodeling.45
Past studies have focused on changes in neutrophils maintained in culture for a few hours in vitro, as they defined neutrophil senescence as its phenotypic change from release into the bloodstream to disappearance in the absence of inflammation. Another phenotypic change observed in neutrophils during in vitro culture is the downregulation of CXCR2 (CXCL1 receptor), a potent neutrophil chelator that has been shown to promote the release of neutrophils into the circulation and migration to sites of inflammation.46,47 In mice, senescence can disrupt the normal movement of neutrophils across epithelial layers in injured tissues through a CXCL1-mediated mechanism, resulting in abnormal neutrophil trafficking and consequential damage to distant organs.48 CXCL1 can also attract neutrophils to the liver of older mice, where they generate ROS and trigger tissue senescence and inflammation.49 Therefore, the role of neutrophils in defending against inflammation and pathogens is greatly weakened during aging (Fig. 2).
Monocytes/macrophages
Apart from neutrophils, macrophages act as the initial responders to infections and participate in identifying, engulfing, and breaking down cellular debris and pathogens. The deterioration of macrophage function is a critical contributor to immunosenescence, where the capability of macrophages to effectively clear senescent cells from tissues reduces with aging (Fig. 2). Aged macrophages exhibit changes like reduced autophagy50 and a defect in their ability to fight viral infections.51
Aged macrophages display a noteworthy increase in SASP components, such as TNF-α, IL-6, and IL-1β. Furthermore, the ERCC1 gene deletion, which accelerates immune aging, was found to be responsible for the failure to excise the coding sequence for the DNA repair protein ERCC1 (ERCC1 gene deletion accelerates immune deficiency).10 Of particular importance, the anti-inflammatory cytokine IL-10 exhibited a decrease.52 This may lead to a tissue environment more prone to fibrosis, as IL-10 has been found to possess anti-fibrotic properties by suppressing pro-fibrotic molecules, including TGF-β.53,54 Besides, senescent macrophages show significant upregulation of both cell-cycle checkpoint inhibitors p16INK4a and p21CIP1 in a mouse model with deficiency in repairing DNA damage10 and downregulate both glycolysis and mitochondrial oxidative phosphorylation, which leads to an energy-depleted state that impairs the functioning of macrophages (Fig. 2).55
Natural killer (NK) cells
NK cells are fundamental cells of the innate immune system and are regarded as the primary defense mechanism for human health. Recent findings indicate that NK cells play a central role in the immune surveillance of aging cells, and that dysfunctional NK cell activity is associated with infections, malignant tumors, inflammatory diseases, and an increased burden of aging cells with advancing age.56 While age does not seem to affect the number of NK progenitors in the peripheral blood or bone marrow,57 most studies suggest that the aging process causes an elevation of the overall number of NK cells in older adults.58,59 However, this increase in NK cell number is accompanied by a decline in their ability to proliferate and kill targets (Fig. 2).60,61,62 Specifically, there tends to be a decrease in the proportion of immature CD56 bright NK cells and an increase in the percentage of CD56 dim NK cells.61,63 CD56 dim cells produce many cytokines and mainly play an immunomodulatory role. They also account for more than 90% of NK cells, the majority of which are cytotoxic and have strong killing activity. Moreover, changes in the expression of NKp30, NKp46, and DNAM1 (NK activation receptors) in the elderly can impair the immune surveillance function of NK cells.64,65,66 Due to age-related functional decline, NK cells from younger donors exhibit a greater potential for expansion than those from older donors when subjected to in vitro stimulation with IL-2, underscoring the susceptibility of NK cells to age-related dysfunction.61 Also, the signs of reduced NK cell effector functions, such as decreased cytotoxicity, as well as lower expression of perforin and granzyme and reduced secretion of IFN-α and IFN-γ but more IL-1, IL-4, IL-6, IL-8, IL-10, and TNF-α are identified.67,68 Besides, with increasing donor age, the frequency of T cell precursors in CD34+Lin- cells tends to decrease, while the frequency of NK/T cell precursors tends to increase.69 This suggests that the lymphoid differentiation potential of peripheral blood precursor cells shifts from T cells to NK/T cells with age, meaning that more HSCs differentiate into NK/T cells. Meanwhile, a notable rise in the quantity of both NK and NKT cells occurs after the age of 60 (Fig. 2).70
B cells
B cells always work as antibody producers have an essential role in immunity.71 Age-related changes in B cell composition are the main reason for decreased antibody response to vaccination and infection in older adults (Fig. 2). Lymphopoiesis of B cells continues during the life cycle. The output of B cells is severely affected by changes in the microecology of the bone marrow, such as decreased pro-B cell-survival cytokine IL-7 level.72 The number of B-cell precursors and antibody-producing plasma cells in mouse and human bone marrow decreases with age.73 Further, the proliferative potency of lymphoid progenitor is also impaired by ageing, while that of myeloid progenitor does not changes.74 Different from mice, as individuals age, there is a decline in the proportion and absolute number of B cells in the peripheral blood.75,76 Especially, the aging process is associated with a rise in the proportion of late-stage exhausted memory B cells,77 while the percentage of memory B cells that exhibit a positive correlation with influenza vaccine responses significantly decreases with age.70,78 Furthermore, the number of B cells mobilized after antigenic stimulation is only 1/10 to 1/50 that of normal adult animals in the elderly. Similarly, the seropositive protection rate in those aged 60-74 years after influenza vaccination was 41% to 58%, decreasing to 29% to 46% for those 75 years or older. Meanwhile, a collapse in B cell diversity has been discovered.79 However, the elderly tend to exhibit an increase in autoantibodies, which can elevate the risk of developing autoimmune diseases.80
B cells not only produce antibodies, but also play regulatory effector functions in the development of memory T-cells (Fig. 2). Memory B cells are more prevalent in older adults and can produce various pro-inflammatory cytokines and chemokines such as IL-1α, IL-1β, IL-6, and TNF-α, suggesting their potential involvement in inflammatory disorders during inflammaging.81 Moreover, aging mice exhibit increased frequencies of age-associated B cells (ABCs) in their bone marrow, which secrete higher levels of TNF-α, a cytokine that impairs the generation of young pro-B cells.82 This observation suggests that bone marrow-resident ABCs may contribute to altered B cell development with age.
T cells
As fighters of pathogens, their dysfunction makes the mice less resistant to infection and get muscle atrophy. These dysregulated T cells even release many inflammatory molecules to accelerate aging,83 which emphasizes the role of T cells in aging. As a crucial immune cell type, T cell replenishment is achieved by the export from the thymus and self-renewal of peripheral naive T cells. In general, CD4 T cells are adaptable to the challenges of aging and keep naive-memory imbalance to a minor level. Compared with CD4 naive cells, the naive-memory imbalance in CD8 T cells is considerable. A decline in the number of circulating naive CD8 T cells is the most significant and consistently observed marker of immunosenescence in healthy older adults. Like CD4 T cells, BATF/IRF4 also promotes the transformation of naive CD8 T cells to effector CD8 T cells, which upregulates transcription factors related to effector functions, including T-bet, Runx3, and Blimp-1.84
With aging, the number of T helper cells (Th) and T regulatory cells (Treg) increases. The levels of cytokines secreted by Th1 and Th2 cells diminishes with age, making the body less able to defend itself against external pathogens. Elderly individuals exhibit increased expression of TGF-β receptor 3 (TGFβR3) on naive CD4 cells. This leads to the activation of a transcription factor network that includes PU.1, BATF, and IRF4, ultimately resulting in a preference for Th9 differentiation.85,86 The increased Th9 leads to the increased secretion of the signature cytokine IL-9, which mediates various inflammatory responses and is involved in the differentiation of autoimmune diseases and inflammatory diseases.85 Although Treg cells increase in number with age, their suppressing capability declines significantly, which may contribute to inflammation in the elderly.87,88 At the molecular level, the damage to signal transduction, such as decreased CD28-mediated JNK kinase and Raf-1/MEK/ERK kinase activation, results in a hypo-responsiveness of T cell receptor (TCR) signal transduction.87 Meanwhile, effector memory CD45RA (EMRA) CD8 T cells show significant SASP, including high levels of IL-18 and disintegrin and metalloproteinase 28 (ADAM28, a proteinase involved in the cleavage of membrane-bound TNF-α).89,90 However, it is noteworthy that the cell cycle of EMRA CD8 T cells is partially reversible, which is different to senescent T cells.91
In old people, highly differentiated T cells, especially memory T cells display the loss of co-stimulatory molecules such as CD27/28, representing an earlier stage of senescence or exhaustion.92,93 Exhausted T cells display several hallmarks similar to aging ones, such as mitochondrial dysfunction94 and epigenetic dysregulation.95 Traditionally, it is believed that Exhausted T cells lack the function of secreting inflammatory, anti-inflammatory, and cytotoxic effector molecules.96 However, Denis et al. have recently substantiated that exhausted GZMK-expressing CD8 T cells can accelerate the inflammatory phenotypes.97
In the peripheral blood lymphocyte subsets of healthy adults in different ages, it was found that the decreased naive CD4 and CD8 T cell number, increased memory CD4 or CD8 T cell number, and decreased CD28 expression on T cells.70 Numerous studies have shown a close association between increased stimulation by various antigens in vitro, especially cytomegalovirus (CMV) infection, and an increase in effector memory T cells,98 resulting in the activation of naive lymphocytes into memory lymphocytes and their long-term presence in vivo.13,99 This process leads to an increased number of memory CD4 and CD8 T lymphocytes with age, and a decrease of TCR diversity in naive T cells, which suppresses the responsiveness of T cells to neoantigens (Fig. 2).
In summary, as the body ages, most immune cells exhibit senescent characteristics, which manifests internally as difficulty in clearing senescent/damaged cells and externally as weakening of the body’s resistance.
Inflammaging at the organ level
As a result of the effects of cellular senescence, chronic inflammation, and immunosenescence, the pathological aging of organs increases the level of inflammation and makes repair difficult, ultimately leading to diseases.10
Lymphoid organs
The primary lymphoid organs, including the bone marrow and thymus, are responsible for immune cell development. However, with advancing age, these organs undergo a functional decline, which results in compromised capability of replenishing the immune cell reservoir. Senescence of the lymphatic organs promotes immunosenescence and plays a key role in organ inflammaging.
Aged bone marrow promotes HSC-related immunosenescence
The bone marrow, which serves as the site of hematopoiesis, is a complex environment where bone cells and hematopoietic cells interact with each other. Recent studies have highlighted the importance of the aging bone marrow microenvironment as a key contributor to the aging process. One significant finding is that a higher percentage of senescent bone marrow mesenchymal stem cells (MSCs) have been observed in older individuals compared to younger individuals. This was determined by DNA damage, elevated ROS, and accumulation of SASP-expressing cells. The SASP-generated inflammatory environment can change the expression profile of healthy MSCs and disrupt the expression of factors indispensable for lymphocyte survival (Table 1).100,101,102 Senescent MSCs generating inflammatory factors further impair the function and clonogenicity of young HSCs. Aging has been linked to several hematopoietic system-related issues, such as an increased occurrence of anemias, compromised adaptive immune responses,103 and a higher susceptibility to myelodysplastic and myeloproliferative disorders (Fig. 3).104

Aging-organ atlas. Aging manifests as a decline in organ function and an increased susceptibility to diseases. Organs are mainly divided into immune organs, sterile organ, and others. Functional changes in cells are shown in each organ
The aging of bone tissue inevitably affects HSCs. With age, red bone marrow is gradually replaced by fat cells, leading to yellow bone marrow formation that inhibits hematopoietic function.105 The decreased secretion of nutrient factors by bone marrow stromal cells can result in an enhanced differentiation of HSCs into myeloid cells and a reduced differentiation into lymphocytes. This imbalance in myeloid/lymphoid differentiation is one of the manifestations of HSC aging.105,106,107 Importantly, aged HSCs tend to differentiate more towards myeloid cells, while their ability to support lymphoid cell maturation decreases. This leads to a reduction in the number of precursors for T and B cells with increasing age.108,109 Taken together, with aging, the number of HSCs increases, while their function including self-renewal and clonogenicity, decreases. In addition to the HSC changes mentioned above, aging marrow also has decreased Wnt signaling and the accumulation of senescent cells and inflammatory cytokines.110
Aged thymus promotes T cell-related immunosenescence
The thymus is a central T-lymphatic organ that produces functional initial T-lymphocytes and immune tolerance. In most mammals, aging is accompanied by degeneration of the thymus gland. In humans, thymocyte numbers and hormone secretion levels typically increase during early development and then decrease over time. In addition, the majority of functional cells are substituted with senescent fibroblasts and adipocytes, and stromal cells during thymus aging.88,111,112 In the aged mouse thymus, elevated levels of phosphorylated histone H2AX and the p53 binding protein suggest heightened oxidative stress and DNA damage, consequently leading to cellular senescence,113 providing support for the notion that the aging thymus exhibits a greater proportion of senescent cells.
Thymic degeneration results in reduced generation of new T-cells, an accumulation of memory T-cells, and a decline in the diversity of T-cell receptors. As a consequence, this leads to a weakened immune response and decreased overall immunity. It has been observed that apparent aging-associated alteration, especially a progressively reduced population of naive T cells, in murine T cell compartment during thymic involution.114 However, in human, there is a progressive loss of CD8+ naive T cells while, notably, a relatively stable naive CD4+ compartment is efficiently maintained via homeostatic proliferation.115,116,117
Consequently, both in mice and humans, age-related variations in the production of naive T cells from the thymus result in qualitative disparities in the overall T cell repertoire.118,119,120,121 In addition, it has been observed that naive T cells from older individuals exhibit reduced responsiveness to the superantigen toxic shock syndrome toxin-1 compared to younger individuals,122,123 which may be caused by low dual specificity phosphatase 6 levels in naive T cells, resulting in a rise in the threshold of TCR activation.
Aged spleen promotes T and B cell-related immunosenescence
The spleen acts as a secondary lymphoid organ promoting immune defense and is the main pivotal organ for initiating the activities required for the adaptive immune responses. During the aging process, significant alterations occur in the cellular composition and microarchitecture of the spleen. The clear distinction between T-cell and B-cell areas within the white pulp becomes less defined, and there are noticeable changes in the organization and functionality of marginal zone macrophages, stromal cells, and marginal metallophilic macrophages.124,125
Furthermore, recent advancements in single-cell RNA sequencing studies have revealed that the proportion of T cells in the spleen decreases with age, while the relative abundance of plasma cells increases.126 Impaired migration of B cells and the phagocytic capacity of macrophages in the marginal zone can also be seen in aged spleens.127 Also, impaired function of microenvironment-mediated antigen-presenting cells was observed, which may provide an explanation for the observed delayed responses to stimulation even in T cells derived from young HSCs (Fig. 3).102,128,129
Aged lymph nodes promote immunosenescence
Lymph nodes serve as crucial sites where T cells and B cells reside and where immune responses are initiated, playing a vital role in establishing an effective immune response. However, the number, integrity, and functionality of lymph nodes undergo significant declines with age, as evidenced by previous studies.102,125 The exact cause of lymph node atrophy remains unknown; however, it can result in the deterioration of the microenvironment where immune cells reside, thereby negatively impacting immune function. Age-related alterations in cellularity and the functionality of different cell types within the lymph nodes have been extensively documented.130 Specifically, the number of fibroblastic reticular cells in lymph nodes diminishes, resulting in a compressed and less reticular stromal network.131 In addition, older individuals, aged 60 years and above, exhibit increased fat deposition and fibrosis in their lymph nodes.124,125 Moreover, stromal cells within aged lymph nodes exhibit reduced replication potential when stimulated and are unable to maintain a balance of naive T cells.126,127,131 The accumulation of senescent cells in lymph nodes, along with heightened inflammation, may negatively impact the migration and recruitment of immune cells, thereby serving as detrimental factors (Fig. 3).130
Sterile organs
Brain
The main cause of brain aging appears to be neuroinflammation132 via aged brain cells and a weakened immune system. The process of brain aging significantly contributes to the decline of various cognitive functions, encompassing decreased speed of information processing, reduced capacity of working memory, impaired spatial memory, and diminished plasticity (Fig. 3).133
Aging of brain cells including neurons and glial cells (i.e., microglia and astrocytes) leads to the upregulation of inflammatory-related pathways, causing brain function weakness and increased inflammation damage. During the aging process, microglia gradually lose their ability to efficiently clear misfolded proteins that are linked to neurodegeneration. This impairment in protein clearance significantly contributes to the neuroinflammatory response observed in the brain, with microglia playing a central role in this process. Except for the supporting role, Shao et al. found that astrocytes can also be a mastermind of neuroinflammation, depending on the Dopamine D2 receptor (Drd2), normally an important brake on it.134 During aging, the level of Drd2 and its ligand dopamine both decline with high neuroinflammation in the brain. Subsequently, activated astrocytes produce SASP factors, such as IL-1β, IL-6, TNF-α, IFN-γ, COX-2, and other inflammatory factors (Table 1). In turn, these factors further promote astrocyte activation. The excessive production of pro-inflammatory mediators disrupts the intricate equilibrium necessary for the induction of long-term potentiation, leading to a decrease in the production of brain plasticity-related molecules such as BDNF and IGF-1, consequently impairing synaptic plasticity.135 Remarkably, even older adults without neurological impairments demonstrate a gradual escalation in neuroinflammation, characterized by elevated homeostatic levels of inflammatory cytokines and reduced production of anti-inflammatory molecules (Fig. 3).136
Immunosenescence and inflammaging can both contribute to neuroinflammation, resulting in impaired neuronal function and the accumulation of brain tissue damage.137,138 Consequently, various central nervous system disorders, including Alzheimer’s disease, Parkinson’s disease, and stroke, are characterized by degenerative neurological conditions.139
Heart
Most cardiac tissue is composed of cardiomyocytes, cardiac fibroblasts, and macrophages. The aging process in the heart is characterized by the gradual occurrence of several hallmarks, including progressive cardiomyocyte hypertrophy, the gradual onset of cardiac fibrosis, and the presence of inflammation (Fig. 3).140
Hypertrophic cardiomyocytes, characterized by heightened oxygen and energy requirements, create a hypoxic environment of low oxygen levels. This imbalance in oxygen levels leads to the generation of excessive free radicals, which can potentially damage cellular components. In response to hypoxia, cardiomyocytes release pro-inflammatory cytokines and chemokines. These molecules stimulate an immune response and contribute to an increase in the number of macrophages within the left ventricle.140 In addition, because mature cardiomyocytes have a low rate of proliferation, the injured area in the aging heart is replaced by fibrotic scar tissue, resulting in organ failure.141
The main effector cells in cardiac fibrosis are activated myofibroblasts. Long-term inflammation promotes cardiac and vascular fibrosis. The cardioprotective effects of AMPK and GDF11 on cardiomyocytes have been extensively documented, and the decline in AMPK and GDF11 expression associated with aging is likely a contributing factor to the heightened cardiac fibrosis observed during the aging process.142
In the steady-state heart, macrophages play a crucial role in eliminating senescent and dying cells, contributing to the normal homeostatic maintenance of the myocardium and facilitating tissue repair following injury. However, in the aging heart, macrophages recruited to the site of infarction exhibit a pro-inflammatory M1 phenotype initially, but subsequently transition to an anti-inflammatory M2 phenotype after myocardial infarction (MI). This phenotypic switch promotes angiogenesis and scar formation, aiding in the recovery process.143
In addition, vascular smooth muscle cells (VSMCs) play a crucial role in coordinating vascular function alongside endothelial cells, regulating blood pressure, vascular tone, and blood flow. However, during the aging process, the dysfunction and decline of VSMCs have a detrimental impact on the structural integrity of the aorta, ultimately leading to the development of transthoracic aortic aneurysms.142 Classical molecular IGF-1 signaling causes cardiac hypertrophy and heart failure.142 IGF-1 increases cellular senescence in VSMCs by inducing DNA damage and increasing ROS production via the p53 pathway.
Kidney
As individuals age, the kidneys undergo various structural impairments, such as fibrosis, and experience functional issues, including mitochondrial dysfunction (Fig. 3).144,145,146 Moreover, older individuals become more susceptible to acute kidney injury (AKI) and chronic kidney disease (CKD).123,147 Age-related alterations in multiple cell types, such as tubular epithelial cells, resident and circulating leukocytes, contribute to kidney injury. Proximal tubular cells depend on autophagy to effectively eliminate defective mitochondria and other organelles under both normal and pathological conditions.148,149 However, during aging, their diminished proliferative capacity leads to impaired clearance abilities.150
Aging in the kidneys is associated with various physiological changes, including chronic low-grade inflammation. This inflammatory state, often referred to as inflammaging, has been observed to have detrimental effects on the kidneys. Chronic low-grade inflammation in the kidneys can impair the normal repair mechanisms that occur following injury. This inflammation hampers intrinsic cellular repair mechanisms following injury and promotes immunosenescence and organ damage.151,152 Elderly individuals exhibit immunological phenotypes characterized by reduced numbers of naive lymphocytes, increased pro-inflammatory T cells, and diminished phagocytic activity in monocyte lineage cells, similar to CKD patients. These alterations in the kidneys form the basis for prevalent pathological conditions that are commonly observed in both elderly individuals and patients with CKD.123
Liver
Aging raises the risk of chronic liver disease and liver fibrosis, which is highly related to hepatic stellate cells, hepatocytes, and macrophages. Liver cells initially activate compensatory mechanisms in response to time-dependent damage caused by aging, which can lead to the development of pathologies of the liver if overstimulated.153 Activated hepatic stellate cells are the major functional population during liver fibrogenesis.154 During senescence, their replication, immune-recruiting signals, and clearance are important for the regulation of liver fibrogenesis.155 An illustration of the contribution of senescent hepatocytes to hepatic stellate cell activation and liver fibrogenesis is evident in p53-deficient mice with nutrition-induced steatohepatitis. It was discovered that these mice displayed reduced levels of hepatocyte p21, as well as decreased activation of hepatic stellate cells and expression of fibrotic markers such as SMA and collagen.156 This finding supports the involvement of senescent hepatocytes in the activation of hepatic stellate cells and the development of liver fibrosis. Furthermore, M2 macrophages secrete pro-fibrogenic mediators, including TGF-β1, which promote the progression of liver fibrosis.157 In brief, the recruitment and mobilization of immune cells, the accumulation of inflammation, and the activation of hepatic stellate cells and hepatocytes contributes to the development of liver fibrosis and the aging process (Fig. 3).
Other organs
Skin
During the aging process, the skin accumulates senescent cells that, despite their inability to divide, remain metabolically active. These senescent cells exhibit an altered secretome known as SASP, which significantly disrupts the skin microenvironment.158 For instance, senescent dermal fibroblasts secrete a higher amount of extracellular vesicles (EV) compared to their non-senescent counterparts. This increased EV secretion hampers the normal differentiation of keratin-forming cells and compromises the skin’s barrier function. In addition, it triggers the elevated production of the pro-inflammatory cytokine IL-6.159
Furthermore, skin aging can occur due to age-related factors or exposure to environmental stressors like ultraviolet radiation.
The process of skin aging can also have systemic effects on the overall aging process of the body, primarily through the activation of SASP.160 The presence of p16-positive cells in the skin, which is a marker of cellular senescence, has been found to be associated with markers of CD4+ T-cell senescence and biological age.161,162 Notably, the microbiome of skin has been found to predict a person’s actual age accurately.160,163 While the numbers of CD4 T cells remain consistent with age, the levels of CD8 T cells are higher in older skin compared to younger skin.164 The ratio of cutaneous CD4 T cells to CD8 T cells is greater in aged individuals, but the number of CD4 T cells is not elevated in aged skin.165 Moreover, aged skin exhibits increased numbers of regulatory T cells (Tregs)166 and elevated expression of the immunosuppressive receptor PD-1,165,167 which may contribute to weakened adaptive immunity. These changes could be a response to an inflammatory state exacerbated by impaired epidermal barrier function or fibroblast senescence, further promoting an inflammatory microenvironment (Fig. 3).
Lung
The aging process brings about notable transformations in the structure and function of the lungs, including a decline in mucociliary clearance and heightened vulnerability to pulmonary infections.168,169 These alterations contribute to the onset and progression of various lung diseases like idiopathic pulmonary fibrosis (IPF) and chronic obstructive pulmonary disease (COPD).169 Several cell types within the lungs undergo modifications during aging, including respiratory epithelial cells, lung progenitor cells, lung immune cells, and lung interstitial cells.169 Among these cell types, alveolar epithelial type II cells (AT2) are a significant population responsible for regenerating the alveolar parenchyma. However, as these cells age, the airway epithelium experiences quantitative and qualitative defects. The number of basal and spherical cells decreases, while the count of AT2 cells remains unchanged but exhibits impairments in self-renewal and differentiation capacity.170,171,172 Moreover, age-related changes in the lung environment, such as alterations in extracellular matrix (ECM) components, tissue and circulating cytokines, SASP, and structural abnormalities, can lead to abnormal intercellular communication mechanisms. This is evident through distorted interactions with microbial pathogens and a shift in innate and adaptive immunity towards increased inflammation, disrupted adaptive immune responses, and impaired immune surveillance (Fig. 3).173
The increased susceptibility of elderly individuals to lung diseases can be ascribed to age-related changes in immunity and anti-infection responses. The phagocytic capacity of pulmonary and alveolar macrophages diminishes with age, impairing the clearance of pathogens from the lungs.51,174,175 Dendritic cells, neutrophils, and NK cells also experience age-related alterations in their numbers and functionality.169 In addition, the aging process is linked with a decrease in CD4, and CD8 T cell populations. The decline in naive T cell numbers is accompanied by an increase in the number of memory T cells. The CD4 to CD8 lymphocyte ratio in bronchoalveolar lavage fluid tends to rise with age, indicating a reduction in the pool of naive T cells available for conversion into memory cells in response to new antigens. Furthermore, aging is associated with reduced CD4 and CD8 T cell responses, diminished TCR repertoire diversity, impaired Th cell differentiation, and reduced Th cell activity.176 These age-related changes in T cell number and function can compromise influenza vaccination immunity and cytotoxicity against the virus. The adaptive immune response to antigens also declines with age, which explains why older individuals are more susceptible to environmental stimuli. Notably, immune cell disorganization associated with aging may contribute to the heightened severity of COVID-19 and chronic obstructive pulmonary disease (COPD) in the elderly.169,177
To recapitulate, age-related changes in T cell-mediated adaptive immune responses enhance vulnerability to infectious agents and result in severe diseases.
Gut
Age-related perturbations in the gut microbiome have emerged as crucial factors contributing to age-related pathological conditions, including chronic inflammation,178 neurodegeneration,179 cognitive decline,180 and type 1 and type 2 diabetes.181 The gut microbiota comprises probiotic, commensal, and pathogenic bacteria, and the imbalance between intestinal flora and aging mutually influences and exacerbates each other. Older adults (>65 years) exhibit reduced microbiota diversity compared to adults, along with greater inter-individual variation in microbiota composition.182 This is characterized by diminished populations of beneficial bacteria such as Bifidobacterium, Bacillus, E. coli, Clostridium XIV, Blautia coccoides-Eubacterium rectal, and Bacteroidetes, and increased presence of Enterobacteriaceae.183 However, it’s important to note that data regarding age-related changes in microbiome composition can vary among populations.
Age-related alterations in the intestinal microbiota, particularly due to prolonged immune system stimulation, can contribute to the accumulation of inflammation and a decline in immune system function known as immunosenescence.184 Interestingly, two previous studies have indicated that changes in the relative abundance of gut microbiota are more likely to be influenced by inflammation rather than age, with TNF playing a significant role.185,186 This suggests that the changes in gut microbiota are more closely related with inflammatory processes rather than solely being a consequence of aging (Fig. 3).187,188
Mechanisms of inflammaging
Consensus features of inflammaging
While the precise interpretation of senescent cell markers remains incomplete and requires further investigation, there is a consensus regarding certain essential characteristics of senescent cells, primarily focusing on the SASP (Table 1). The SASP, considered to be molecular inflammation, is a universal, dynamic, and complex phenomenon arising with cellular senescence. It is the phenomenon of senescent cells secreting pro-inflammatory cytokines.189 The SASP possesses the capacity to perpetuate senescence itself or influence the surrounding tissue microenvironment, consequently affecting the entire organism. Classic SASP factors contain pro-inflammatory and immune-modulatory cytokines, chemokines, proteases, and growth factors (Table 1).190,191,192,193 According to its complex composition, the SASP has been implicated in the majority of the nonautonomous effects observed in senescent cells, including inflammation, immune evasion, tumor promotion, senescence reinforcement, paracrine senescence, and so on.194 Recent analysis has identified a set of shared components within the SASP that are consistent across various inducers of senescence and different cell types. Interestingly, some of these components overlap with aging markers observed in human plasma, including serine protease inhibitors, stanniocalcin 1, and growth differentiation factor 15.190 Furthermore, dysfunctional mitochondria,195 persistent DNA damage response,196 CCAAT/enhancer-binding protein β (C/EBPβ) and NF-κB,197 mTOR, and other factors are involved in regulating SASP.194
In addition, SASP encompasses additional characteristics such as lipofuscin accumulation within lysosomes, increased cytoplasmic DNA, activation of anti-apoptotic pathways, and alterations in the nucleus, including the loss of Lamin B1, telomere shortening, senescence-associated heterogeneous chromatin aggregation, and the presence of telomerase-associated foci.198 At the transcriptional level, p16 and p21 are the most commonly used markers to identify senescent cells. These features and markers have been extensively employed to detect senescent cells in various tissues, both in the context of individual senescence and other pathological conditions.199
Triggers of inflammaging
Inflammaging develops from cold-inflammaging with a less than 2-fold increase of pro-inflammatory mediators in plasma, compared to healthy adults.200 This slightly altered level is a positive response for maintaining homeostatic stability. However, during aging, the homeostasis imbalance arises and progresses, leading to increased cytokine response (2- to 4-fold increase) mediated by the chronic activated innate immune system. The transition is highly influenced by several vital triggers, including cellular senescence with the secretion of SASP, which have been already mentioned above, dysbiosis caused by microbiome and their metabolites, and endogenous molecular garbage caused by abnormal cell death.
Oxidative stress
Oxidative stress leads to oxidative damage to biomolecules (especially DNA),201 causing endogenous damage-associated molecular patterns (DAMPs) production and cytokine release in the organism.202,203 Cytokines activate downstream signaling pathways of pattern recognition receptors,203 causing systemic chronic inflammatory responses in the body.204 Consequently, oxidative stress is recognized as a concurrent occurrence within the inflammatory process, amplifying the inflammatory response through oxidation. At the same time, inflammation promotes oxidation through inflammatory mediators.205,206 Based on the close relationship among oxidative stress, inflammation, and aging, Dela Fuente et al. proposed the theory of aging by oxidation-inflammation (oxi-inflamm-aging)207,208 and concluded that oxidative stress leads to inflammatory aging. Oxidative stress has been ensured as a crucial factor for cellular senescence through shortening telomere and causing DNA double-strand breaks.209,210 Moreover, infections,211 environmental pollution,212,213 and adverse lifestyle habits214 can increase oxidative stress.
Microbiome
Recently, there has been an increasing focus on studying age-specific changes in the intestinal microbiome and its role in regulating inflammation. A healthy gut microbiome is essential for body metabolism, infection resistance, inflammation regulation, prevention of autoimmunity and cancer, and brain-gut axis regulation.215 However, with age, there is a decrease in beneficial microorganisms216,217 and an accumulation of potentially pro-inflammatory microorganisms in the gut,218 leading to a change in microbial composition and a decrease in microbial diversity. Moreover, this phenomenon exists simultaneously in species such as Drosophila,219 fish,220 mice,221 rats,222 and humans.223 The detailed gut microbiota changes with aging have been collected and discussed by Du et al.224 Recent studies have revealed that the transplantation of fecal matter from young donors into the gastrointestinal tract of middle-aged fish can effectively prolong lifespan and delay the onset of behavioral decline,220 and that fecal transplantation from young mice slows HSC senescence in the bone marrow.225 Lachnospiraceae and tryptophan-associated metabolites have emerged as key players in important biological processes, but the exact mechanism and involvement of other factors are not yet clear. Nonetheless, it is evident that the senescence-related remodeling of microorganisms mediates the accumulation of chronic inflammation, which is highly correlated with their metabolites and their induced immune responses.
Inflammatory cell death
Eukaryotic cells possess the ability to activate various self-destructive mechanisms, but the type of cell death can be classified as either inflammatory or non-inflammatory. In normal tissues, cell death serves as a highly conserved process that promotes a stable cell population through the elimination of surplus, impaired, or aged cells. Consequently, the human body generates over 150 billion deceased cells on a daily basis.226 A newly developed conception, garb-aging, reveals that the production of inflammatory cell death modalities, endogenous molecular garbage (e.g., mitochondrial RNA, misplaced molecules, and cell debris), is a causal inflammatory stimuli that can accelerate inflammaging.227,228 Certain cytokines, such as IL-1β and IL-6, have clearly emerged as key to promoting inflammaging.83,229,230,231,232 Moreover, inflammatory death of internal cells due to exogenous factors such as infection also promotes the progression of inflammaging.17,233 During aging, the imbalance between the increased production and decreased disposal via autophagy, mitophagy, and proteasome, stimulus the innate immune system and thereby triggers the body from a pre-inflammatory state towards a pro-inflammatory state.234
Necrosis
As the body ages, tissues and cells gradually experience damage, leading to a decline in their abilities and functions. Aging cells may be more susceptible to damage from external stimuli, increasing the risk of necrosis. In addition, certain age-related diseases such as cardiovascular diseases and neurodegenerative disorders may be accompanied by cellular necrosis.
Necrosis is traditionally considered an unprogrammed and unregulated form of cell death that occurs due to overwhelming external stimuli.235 It is characterized by cellular swelling, loss of membrane integrity, release of intracellular contents (DAMPs and pathogen-associated molecular pattern, PAMPs) into the extracellular environment, an increase in intracellular calcium concentration, and the generation of ROS. These events ultimately lead to irreversible cellular damage. DAMPs, such as HMGB1, uric acid, nucleosomes, and members of the heat shock protein family (HSP 70, HSP 60, and GP96), can directly or indirectly activate and recruit immune cells, thereby triggering inflammation or immunosuppression.236 It is important to note that these factors can be released during the entire process of cell death, even when cells are still metabolically active. Consequently, cells in the process of dying may contribute to carcinogenesis even before the appearance of obvious necrotic changes (Fig. 4).236

Schematic diagram of six inflammatory cell death molecular patterns. The source of inflammation comes from cell death in addition to the release of SASP from senescent cells. Both immune response-mediated and damage signaling-mediated cell death promote inflammation to some extent. Various modes of cell death (in addition to apoptosis) release large amounts of inflammatory factors
Necroptosis
Necroptosis (previously named programmed necrosis) is a regulated form of inflammatory necrosis that occurs when apoptosis (the programmed cell death process) fails. Its identification questioned the conventional notion that necrosis is exclusively an inert process induced by overwhelming stress. Necroptosis is distinguished by the early disruption of plasma membrane integrity, release of intracellular contents, and enlargement of organelles. Apoptosis is generally regarded as non-immunogenic since the regulated dismantling of apoptotic cells restricts the liberation of DAMPs. However, necroptosis triggers inflammation through the massive release of DAMPs from the disintegrating cell.237
The contribution of DAMPs from dying cells in the RIPK1-RIPK3 inflammasome-dependent pathway of cytokine production varies. Upregulation of RIPK3 has been observed in hepatocytes, suggesting that RIPK3-dependent necroptosis may play a role in inflammation and hepatocyte death. Immunostaining with antibodies recognizing phosphorylated mixed lineage kinase domain-like protein (MLKL) serves as a specific marker of necroptosis.238,239 In addition, immunostaining using antibodies that target phosphorylated MLKL has been identified as a specific marker for necroptosis.240
Focus on liver, its aging has been linked to an increase in necroptosis, and this process has been found to contribute to chronic liver inflammation, which in turn appears to be involved in the development of liver fibrosis.241 On the other hand, in the livers of old mice (specifically, those aged 18 months and older), there was a significant upregulation of phosphorylated MLKL and MLKL oligomers, which are markers associated with necroptosis. In addition, the phosphorylation of RIPK3 and RIPK1, two key proteins involved in necroptosis signaling, was also significantly increased in the livers of old mice compared to young mice. In comparison to young mice, hepatocytes and liver macrophages from old mice had higher levels of necroptosis markers and higher expression of pro-inflammatory cytokines M1 macrophage markers, pro-inflammatory cytokines (TNF-α, IL-1, and IL-6), and fibrosis markers. In the livers of old mice, short-term treatment with the necroptosis inhibitor necrostatin-1s (Nec-1s) reduced necroptosis, M1 macrophage markers, cellular senescence, fibrosis, and pro-inflammatory cytokines.241 Importantly, nerve injury-induced protein 1 (Ninjurin1/Ninj 1) plays a crucial role in facilitating the ultimate breach of the plasma membrane that takes place in necroptosis, pyroptosis, and secondary necrosis. Secondary necrosis refers to the phenomenon where cells undergoing apoptosis fail to be engulfed by adjacent phagocytes (Fig. 4).242 These findings suggest an age-associated dysregulation of necroptosis signaling, indicating a potential role for necroptosis in aging and related pathologies.
Pyroptosis
As the body ages, tissues and cells gradually experience damage, leading to a decline in their abilities and functions. Similar to other forms of inflammatory cell death, aging cells may also be more susceptible to damage from external stimuli that can trigger pyroptosis, increasing the risk of pyroptosis occurrence. In addition, age-related diseases such as neurodegenerative disorders and cardiovascular diseases may be accompanied by cellular pyroptosis. Specifically, pyroptosis may play a specific role in aging. Inflammation and cell death have important regulatory roles in aging, and pyroptosis, as an inflammatory form of cell death, may contribute to the inflammatory response and cellular dysregulation in the aging process.243 Moreover, pyroptosis may be involved in the development and progression of age-related diseases.
In the non-classical pathway, human-derived caspase-4, 5, and murine-derived caspase-11 can be activated upon direct contact with bacterial lipopolysaccharide (LPS). LPS cleaves gasdermin D (GSDMD), which indirectly activates caspase-1, leading to pyroptosis. Alternatively, caspase-1 can be recruited and activated by inflammatory vesicles that detect danger signals. Activated caspase-1 cleaves and activates inflammatory factors, which in turn cleave the N-terminal sequence of GSDMD. This results in the binding of GSDMD to the membrane and the generation of membrane pores, ultimately leading to pyroptosis (Fig. 4). Evidence of pyroptosis, coincided with elevated levels of IL-1 and IL-18, and inflammasome activation, has been illustrated in various conditions such as atherosclerosis, neurodegenerative diseases, cancer, and chimeric antigen receptor (CAR)-T therapy.244,245,246,247
Ferroptosis
As the body ages, changes in iron levels and iron metabolism may occur. The accumulation of iron in cells during the aging process may be associated with the development of age-related diseases such as neurodegenerative diseases248 and cardiovascular diseases.249 This iron accumulation can lead to increased generation of ROS within cells, thereby triggering iron-dependent cell death known as ferroptosis. The main drivers of ferroptosis are the inactivation of the lipid repair enzyme glutathione peroxidase 4 (GPX4) and the induction of ROS, particularly lipid ROS. GPX4 plays a cytoprotective role by reducing cellular lipid hydroperoxide levels, which are associated with inflammation. In cancer cells, certain inflammatory cytokines such as TNF, PGE2, IL-1, and IL-6 have been shown to directly affect GPX4 levels and activity. Treatment with TNF, for example, downregulates GPX4, leading to ferroptosis.
Ferroptosis is an inflammatory form of cell death that is distinct from apoptosis. It is characterized by iron-dependent lipid peroxidation and can contribute to various pathological processes, including neurodegenerative diseases, inflammatory diseases, autoimmune diseases, and cancer. The inactivation of the lipid repair enzyme glutathione peroxidase 4 (GPX4) and the induction of ROS, particularly lipid ROS, are the main causes of iron death. GPX4 has been shown to have a cytoprotective effect by lowering the levels of cellular lipid hydroperoxides.250 Several pro-inflammatory cytokines, including TNF, PGE2, IL-1, and IL-6, have been demonstrated to exert a direct influence on the levels and function of GPX4 within cancer cells;251 for example, TNF treatment causes GPX4 downregulation that can lead to ferroptosis.252 High mobility group box 1 (HMGB1), a DAMP, has been implicated in inflammation and its pathogenesis.253,254 In the context of ferroptosis, inhibiting HMGB1 release has been shown to limit the inflammatory response during cell death. Anti-HMGB1 antibodies have demonstrated their ability to reduce the inflammatory response in macrophages induced by ferroptotic cells.252 Ferroptosis inhibitors have shown promise in the treatment of certain diseases due to their anti-inflammatory properties. In an oxalate-induced mouse model of AKI, evidence of inflammation was observed, and the ferroptosis inhibitor Ferrostatin-1 successfully inhibited neutrophil infiltration and the expression of pro-inflammatory cytokines such as CXCL-2 and IL-6.255,256 Conversely, the ferroptosis inducer RSL-3 significantly increased the protein levels of pro-inflammatory cytokines like TNF, IL-1, and IL-6, exacerbating hepatosteatosis, lobular inflammation, and apoptosis (Fig. 4).257 The results indicate a possible interplay between ferroptosis and inflammation.
In addition, certain biological processes and molecular mechanisms associated with aging may be related to ferroptosis. During the aging process, alterations in cellular function and metabolism can increase the sensitivity of cells to external stimuli, including sensitivity to ferroptosis. Recent studies have reported the involvement of ferroptosis as a mechanism that promotes skeletal muscle aging.258 With skeletal muscle aging, there is a decreased expression of Tfr1 and an increased expression of Slc39a14, which is enriched on the cell membrane surface of aging mouse skeletal muscle cells. This increase in Slc39a14 leads to enhanced non-transferrin-bound iron uptake, resulting in the accumulation of free iron ions within skeletal muscle and the occurrence of ferroptosis.258
Lastly, the interaction between ferroptosis and aging may be bidirectional. On one hand, ferroptosis may play a role in the development of certain age-related diseases, accelerating tissue and cellular aging processes. On the other hand, the cellular functional and metabolic changes that occur during the aging process may increase the sensitivity of cells to ferroptosis, further promoting disease progression.
NETosis
NETosis is a special form of cell death closely associated with inflammation and immune response. It is a cell death program executed by neutrophils and is characterized by the release of net-like structures (neutrophil extracellular traps/ NETs). These structures are composed of DNA, histones, and microbial toxins, among other components, and serve the purpose of capturing and killing microorganisms.228 NETosis plays a significant role in various pathologies, including COVID-19, Kawasaki syndrome, and rheumatoid arthritis (RA). Excessive NETosis has been implicated in the development of cytokine storms and thrombosis.259 In COVID-19, NETosis can be caused by virus-infected epithelial and endothelial cells, thereby activating inflammatory cytokines and platelets. Excessive NETosis, accompanied by increased circulating free DNA and Neutrophil Extracellular (NE)-DNA complexes, is also found in acute Kawasaki syndrome, a vasculitis occurring in children.260
In RA, the disease pathology is characterized by the accumulation of DNA-MPO complexes and the presence of antibodies targeting guanylated histones (NETosis markers).261,262 In myocarditis, NETosis probably promotes PMN trafficking via MK and substantially contributes to cardiac inflammation.263 In systemic lupus erythematosus, NETosis activates the plasmacytoid and induces the production of IFN-α and ROS, which contribute the following further inflammation.264,265,266,267 On the other hand, the anti-microbial effects of NETosis have been observed to slow down the spread of pathogens in infected lesions. NETs in staphylococcal skin infections inhibit the penetration of pathogens into the bloodstream.268 Knockout of the PAD4 gene in mice prevents NET formation and leads to more severe necrotizing fasciitis caused by streptococcus pyogenes. In summary, NETosis could cause inflammation or conversely slow the onset of age-related diseases (Fig. 4).
The ability of neutrophils to undergo NETosis may be affected by aging.269 Senescent neutrophils exhibit several distinct characteristics during NETosis, including a reduced capacity to release NETs, instability in the quality of formed NETs, and decreased activity of DNA degrading enzymes (DNases) within NETs. These age-related changes can result in impaired functionality of aging neutrophils, leading to deficiencies in their ability to effectively combat microbial infections and regulate inflammatory responses.
Moreover, aging is often accompanied by a phenomenon called inflammaging, which refers to a chronic low-grade inflammatory state. Inflammaging can further contribute to the occurrence of NETosis. This persistent inflammatory condition enhances the activity of inflammatory cells, including neutrophils, thereby increasing the likelihood of NETosis. A previous study found that aged mice exhibited an increased propensity for NETosis compared to younger mice. This heightened NETosis activity was associated with the activation of peptidylarginine deiminase 4 (PAD4), an enzyme involved in the formation of NETs. The excessive formation of NETs, in turn, was implicated in the development of age-related organ fibrosis.270
In conclusion, the process of aging can adversely affect the ability of neutrophils to undergo NETosis. Senescent neutrophils may experience limitations in NET release, compromised stability of formed NETs, and reduced DNase activity within NETs. In addition, the presence of inflammaging, the age-associated inflammatory state, can intensify the occurrence of NETosis by stimulating inflammatory cell activity. However, further research is necessary to fully comprehend the intricate mechanisms and interactions between aging and NETosis.
PANoptosis
PANoptosis, is a united modality of inflammatory programmed cell death, accompanied by markers of apoptosis, necrosis, and pyroptosis pathways.271,272,273,274,275 Influenza A virus was first discovered to cause PANoptosis, followed by many other infections, of bacterial, fungal, and viral origin.275
PANoptosis is involved in the occurrence of cytokine storms (CS) characterized by excessive cytokine production.276 The combination of TNF-α and IFN-γ activates the JAK/STAT1/IRF1 signaling pathway, leading to the production of nitric oxide (NO). This NO release triggers PANoptosis through the involvement of GSDME (pyroptotic), CASP8/3/7 (apoptotic), and pMLKL (necroptotic) pathways. In vivo, blocking CS by giving mice anti-TNF-α and anti-IFN-γ antibodies prevents death from SARS-CoV-2 infection, hemophagocytic lymph histiocytosis, and LPS shock (sepsis).276 This highlights the critical role of TNF-α and IFN-γ released from PANoptosis in driving cytokine storms during infections and inflammatory conditions.275 Cytokines or PAMPs trigger the assembly of a multiprotein complex called the PANoptosome. This complex includes various molecules necessary for the activation of downstream programmed cell death (PCD) effectors such as GSDMD, GSDME, CASP3/7, and MLKL (Fig. 4). No direct evidence yet links PANoptosis to aging. However, aging can affect cell responses to inflammation and cell death. Further research is needed to explore the potential connection between aging and PANoptosis, shedding light on its impact on immune and cell death mechanisms.
In summary, DAMPs from senescent, damaged, and dying cells trigger various cell death modalities, including necrosis, pyroptosis, necroptosis, PANoptosis, NETosis, and ferroptosis (Fig. 4). DAMPs bind to specific receptors, initiating inflammation and orchestrating a coordinated response involving immune cells. This response includes the recruitment of neutrophils and monocytes, which play crucial roles in tissue repair and healing processes. When leukocytes fail to clear immunostimulatory molecules, inflammation persists, which further causes cancer and aging. Endogenous DAMPs can activate PRRs and non-PRR transmembrane proteins, resulting in massive inflammation, cellular senescence, diseases of the organs, and aging.277,278,279 The shift of understanding in aging mechanisms, from the cellular and organ level to the molecular level, aid in identifying novel targets for anti-inflammatory therapies and effective anti-aging interventions.
Classical models to study aging
Aging model systems can simulate human physiological and pathological processes to reveal aging mechanisms and guide anti-aging research. To date, aging models consist of in vitro models (e.g., physical, chemical, and biological induced models) and in vivo models (e.g., animal models, premature aging models, and centenarian).
In vitro models
Here, we describe the main in vitro models of senescence used in research, classified according to different stimuli: replicative senescence (RS), oncogene-induced senescence (OIS), and chemotherapy-induced senescence (CIS).
Replicative senescence (RS) models
Replicative senescence is closely associated with the shortening of telomeres. In the laboratory aging of human diploid fibroblasts (HDFs), as the cells undergo a certain number of population doublings, telomeres become shorter, leading to cell cycle arrest, reduced cell saturation density, and increased cell surface and volume.280 Hydrogen peroxide is commonly used to induce stress-induced premature senescence (SIPS), which shares similarities with replicative senescence. When young HDFs are exposed to prolonged low doses of hydrogen peroxide, they enter irreversible G1 cell cycle arrest and exhibit senescence-associated beta-galactosidase activity. These cellular senescence markers are accompanied by increased expression of p21, gadd45, and enhanced p53 binding activity.281 In addition, DNA repair capability decreases, and telomere shortening accelerates. Hydrogen peroxide-induced senescence also triggers inflammation, characterized by the upregulation of pro-inflammatory cytokines such as IL-6, TNF-α, and MCP-1.282,283
Oncogene-induced senescence (OIS)
Oncogene-induced senescence (OIS) is observed following the activation of various oncogenes such as B-RAFV600E or H-RAS G12V, as well as the loss of tumor suppressor proteins like PTEN or NF-1, in different cell types.284 OIS is often associated with DNA replication stress and hyper-replication. It is characterized by the upregulation of CDK inhibitors, including p15INK4B, p16INK4A, p21CIP1, and an increased senescence-associated β-galactosidase (SA-β-Gal) activity.285,286
Kuilman et al. discovered that OIS is specifically associated with the induction of an inflammatory gene expression profile, which includes the expression of various genes such as the pleiotropic cytokines IL-6, IL-1α, IL-1β, and IL-8. In addition, the transcription factor C/EBPbeta collaborates with IL-6 to enhance initiation of the pro-inflammatory cascade, as demonstrated in cells carrying B-RAFV600E and H-RAS G12V mutations.287
Chemotherapy-induced senescence (CIS)
Chemotherapy-induced cellular senescence is a commonly used cellular model. Drugs like doxorubicin can induce cells to enter a senescent state.288 In this model, cells treated with doxorubicin display characteristic features of senescence. For instance, the expression of 4-HNE and GPX4 increases, while SIRT1 expression decreases. Furthermore, these senescent cells exhibit elevated levels of pro-inflammatory cytokines like IL-6, IL-17, and TNF-α, along with reduced levels of the anti-inflammatory cytokine IL-4, indicating the presence of inflammation.289 Similarly, treatment of melanoma cells with Palbociclib leads to cell cycle arrest at the G0/G1 phase, accompanied by SA-βgal and SASP that includes factors such as IL-6, IL-8, and CXCL1.290 In addition, doxorubicin-induced senescence in H9c2 myocardial cells results in increased expression of 4-HNE and GPX4, decreased SIRT1 expression, and heightened levels of pro-inflammatory cytokines (IL-6, IL-17, and TNF-α), while the anti-inflammatory cytokine IL-4 is reduced.291,292,293 Furthermore, primary human astrocytes exposed to X-rays exhibit increased expression of senescence-associated proteins (p16INK4a and Hp1γ) and cytokines associated with SASP, such as IL-1β and IL-6.294,295,296 Further details of other in vitro models are shown in Table 2.
In vivo models
The mouse has quickly emerged as the preferred mammalian model organism in aging research. This is primarily attributed to several factors, including its relatively short lifespan compared to humans, the close similarity of its genome and physiology to humans, and the ease with which its genetics can be manipulated, including the availability of various mutant strains. These advantages make mice an excellent model for studying the aging process and investigating potential interventions and treatments for age-related conditions.297 Mouse models of accelerated aging involve physically induced models (e.g., radiation and O3), chemically induced models (e.g., D-galactose and D-galactose-combined therapy), “senescence-prone” mice (e.g., SAMP), and premature aging models (e.g., HGPS) (Table 2).
Induced or genetic aging models
In physically induced models, the inhalation of ozone is a frequently employed technique for inducing premature senescence. When male BALB/c mice are exposed to ozone at a concentration of 1.2 mg/m3 for 10 h per day, they exhibit thymic atrophy and an elevated level of oxidative damage. Subsequently, there is a decline in the immune function of the mice, which is closely associated with oxidative stress. This decline is characterized by an increase in IL-6 levels, reduced splenocyte proliferation, decreased production of IL-2, diminished natural killer (NK) cell activity, and a weakened antigen-specific response.298
For chemical induction models, the D-galactose-induced aging model is widely preferred in chemical induction studies due to its convenience, higher survival rate, and minimal side effects. This model effectively mimics aging in vivo by inducing changes in various tissues and organs. When mice are treated with certain concentrations of D-galactose, they exhibit increased levels of ROS and inflammatory cytokines such as NOS-2, IL-1β, IL-6, TNF-α, and NF-κB. These alterations contribute to the aging-like phenotype observed in this model. Moreover, researchers have developed combined methods involving D-galactose to induce premature aging. For instance, the D-galactose and AlCl3 model and the D-galactose and NaNO2 model are commonly used. These combinations enhance the aging effects and provide additional insights into the mechanisms underlying accelerated aging.299
Senescence-accelerated mouse/prone (SAMP) strains, specifically the SAMP1/Yit substrain, have been recognized as valuable models for studying the genetic aspects of aging. In particular, the SAMP1/Yit mice have been used as a model for Crohn’s disease, which is a chronic and recurring inflammatory bowel disease. The mice exhibit both acute and chronic inflammation in the ileum and cecum, displaying a non-continuous pattern of inflammation.300 On the other hand, the SAMP8 mouse strain is considered an excellent model for investigating Alzheimer’s disease (AD), a cognitive decline disorder that predominantly affects the elderly. The SAMP8 mice exhibit reduced expression and lower activity of anti-aging factors including silent information regulator type (sirtuin/Sirt), Forkhead box class O (FoxOs), and Klothos. These factors play crucial roles in the aging process.301
Aging models with apparent inflammation phenotypes include Nfkb1 deficient mice (Nfkb1−/−). Nfkb1−/− mice have shortened lifespan, kyphosis, osteoporosis, tissue inflammation, and gliosis of the central nervous system.302 Interestingly, the accumulation of senescent cells with telomere-dysfunction in Nfkb1−/− tissues can be effectively hindered through the implementation of anti-inflammatory or antioxidant treatment in mice.303 This noteworthy observation underscores the promising utilization of these mice as valuable models for investigating age-related interventions. Details of other in vivo aging models are also shown in Table 2.
Premature aging models
Premature aging models can be categorized into two main types: progeroid syndrome models and other models exhibiting premature aging phenotypes. Progeroid syndromes are exceptionally uncommon human disorders characterized by early onset aging and a reduced lifespan. These syndromes include laminopathies, such as Hutchinson-Gilford progeria syndrome (HGPS), which disrupt the balance of the nuclear envelope, as well as conditions that affect telomere length and DNA repair mechanisms, like Werner syndrome and Cockayne syndrome.
The identification of specific mutations causing these syndromes has enabled the development of mouse models that simulate premature aging.304 For instance, mouse models of HGPS have been created by modifying the Lmna gene or its processing enzyme. Among these models, the LmnaG609G mouse model (featuring the mutation 1827C > T; Gly609Gly) closely resembles human phenotypes.305 This includes heightened inflammation markers like IL-6, caspase 1, and Nlrp3, increased oxidative stress, persistent DNA damage, and cell cycle arrest.306,307 In addition, two independently generated mouse models deficient in the Zmpste24 gene (Zmpste24 − /− mice) also exhibit elevated expression of caspase 1 and Nlrp3, severe growth retardation, dilated cardiomyopathy, muscular dystrophy, lipodystrophy and premature death.307,308
Classic longevity animal model
Many animals naturally have long lifespans. Decoding the underlying mechanisms for provide insights for developing anti-aging strategies. One such remarkable example is the naked mole rat, a socially oriented mammal that dwells in subterranean burrows. This extraordinary creature holds the esteemed distinction of being the longest-living rodent, with a maximum lifespan that surpasses an astonishing 30 years.309 With aging, naked mole rats will not lose their physiological functions, and their mortality will not increase significantly.310 As a successful aging specifications, nude mole also achieved resistance to tumor through a variety of methods, such as efficient DNA damage repair, synthesis of unique anti-inflammatory high molecular weight hyaluronan.311 The special longevity mechanism of naked mole makes it a good animal model for longevity research and provides a blueprint for exploring the strategies of delaying human aging.
Planaria is a type of flatworms. For planarians, small fragments of almost any tissue can be regenerated into an entire individual. This has led it to be considered immortal. The shortening of telomeres during cell division is a major obstacle to infinite cell division, and planarians overcome this problem by upregulating telomerase expression during regeneration.312 On the other hand, planarians also achieve resistance to tumors through a variety of means, such as efficient DNA repair mechanisms.313 All of these mechanisms provide enlightenment for us to study human aging.
As an amphibian, the salamander is also very long-lived. Their limbs and many of their organs are capable of regenerating. Axolotls age very slowly, and their phenotypes are less pronounced. Its physiological mechanism to clear senescent cells in time prevents senescent cells from accumulating in its body, which may explain its slow aging.314 The salamander’s large genome may have provided lines of defense for potentially harmful mutations, such as regulators. This also laid the foundation for the prevention of tumors.315
Turtles are a typical group of long-lived animals. The protection of the carapace keeps them from predators. During diving, turtles are chronically deprived of oxygen, which allows them to upregulate glutathione-related enzymes to clear away ROS.316 Inhibition of ROS and unique telomerase and perhaps DNA repair mechanisms make turtles’ longevity possible.317
Centenarian human models
Unlike artificial models, the centenarian model, as a naturally occurring model, has become an indispensable tool for human beings to decipher longevity. Unlike other people, centenarians have significantly different levels of hormones, cholesterol, etc. in their bodies. Inflammation levels in centenarians show a better balance compared to others, and therefore inflammation levels have been used to predict healthy lifespan. The offspring of centenarians also typically maintain lower levels of chronic inflammation in their bodies, and these lower levels can increase as the centenarian ages, but ultimately those who can maintain lower levels of inflammation may have the best chance of maintaining their bodies in good health. This suggests that genes may be a key factor in maintaining low levels of inflammation in centenarians.6 In addition, the low susceptibility of centenarians to diseases raises the question of whether their immune system is stronger. Studies have shown lower levels of B cells, similar numbers of T cells, and a higher percentage of cytotoxic T cells in the peripheral blood of supercentenarians.318 Compared with animal models, the centenarian model can more realistically reflect the changes in human organs as well as peripheral blood components and has indispensable reference value for human anti-aging research. In conclusion, the long-lived elders are the result of “natural experiments”. They show us that it is possible for individuals to live longer and healthier lives, even if they are influenced by risky genes or if they choose to ignore health information on their own.
Cutting-edge single-cell technology
Single-cell technology reveals organismal activity at the level of the genome, epigenome, transcriptome, proteome, and metabolome. A recent achievement of single-cell technology in the aging field is an aging atlas in different species at a multi-omics level, which allows us to understand aging with a more systemic view.
Aging is extremely heterogeneous, especially from a transcriptome perspective.198 The aging transcriptome landscape in mouse, rat, and cynomolgus monkey is presented in Table 3, including samples, time, platform, cell numbers, and main conclusions. In the future, spatial single-cell technologies (spatial transcriptome and spatial metabolome) make it possible to construct three-dimensional aging atlases at the organ level.319
Intervention strategies in aging
Lifestyle interventions
A healthy lifestyle has long been recognized as the most effective way to maintain health and fight aging.320 More and more research has proven that maintaining a healthy lifestyle, such as adequate nutrition,321 moderate exercise,322 and good mental state323 can effectively delay aging. Balanced and adequate nutrition intake has a positive effect on aging. Many of the nutrients that people take in, such as minerals, probiotics, etc., play an important role in alleviating inflammation and regulating immunity. Long-term polyphenyl-rich dietetic pattern has been proved to improve intestinal permeability and the level of inflammatory markers.324 Previous studies have also proved that the intake of probiotics, such as Lactobacillus pentosus var. plantarum C29, has been proved to significantly reduce the level of systemic inflammatory factors and the expression of aging markers p16 and p53.325 Similarly, consumption of polyunsaturated fatty acids has been shown to significantly reduce levels of inflammatory cytokines throughout the body326 The continuous intake of vitamins, such as vitamin C and vitamin E, can effectively improve the function of immune cells in the elderly, Such as the chemotaxis and phagocytosis of neutrophils.327 The supplementation of minerals, such as zinc, can increase the naive T cell subgroup328 and improve the homeostasis of Th1 and Th2 cells.329 These all emphasize the importance of maintaining balanced nutrition intake.
Exercise is an efficient strategy for delaying aging330 through various mechanisms, such as DNA damage331 and oxidative stress.332 Recent study found that middle-aged marathon/triathletes had higher telomerase activity and longer telomere length in circulating white blood cells compared to the control group.333 Correspondingly, resistance training for five months in older overweight or obese women reduced the number of P16-expressing cells in their thigh fat tissue.334 These evidence suggest that exercise can effectively reduce the appearance of age-related markers, such as p16, so as to achieve the effect of delaying aging.
Keeping a good mental state can also delay aging. Psychological stress affects neuroendocrine function through hypothalamus-pituitary-adrenal axis.335 The continuous activation of this circuit leads to the continuous increase of glucocorticoid level, which will lead to hippocampal atrophy, a phenomenon closely related to aging.323 In addition. A longitudinal study found that the elevation of inflammatory markers increased in elderly people with high self-reported stress levels during follow-up. This may reflect the internal relationship between psychological stress and inflammation.336 These findings suggest that improving mental health and alleviating psychological stress have a positive effect on aging. Lifestyle choices are closely associated with ageing. Keeping a healthy life sometimes means a longer life.
Anti-inflammation strategies
Recent studies have demonstrated that the pro-inflammatory cytokine network is one potential anti-aging target, using anti-inflammation drugs such as metformin, aspirin, rapamycin, and ibuprofen. For example, metformin can reduce chronic inflammation and improve healthy mid-life aging by acting on possible targets such as IKK/NF-κB in patients with Type 2 diabetes,337 as well as GPX7/NRF2338 and a recently found target, PEN2.339 Aspirin can postpone the occurrence of replicative senescence by decreasing oxidative stress. The latest studies have identified CD36 as key to SASP-related mechanisms and reduced SASP secretion in senescent cells by silencing CD36 in senescent muscle tissue cells using CD36-specific short interfering RNA.340 In general, current strategies of anti-inflammation and immune enhancement, including potential targets and main adaptation diseases are summarized in Table 4.
Senolytic drug for eliminating senescent cells
Of all the anti-ageing cell therapies, Senolytics (removal of senescent cells) are the most well-developed and specific but also the most controversial. Since 2015, several Senolytics have gone from identification to clinical trials. The first Senolytics-like drug combination was Dasatinib and Quercetin. Dasatinib removes senescent human adipocyte progenitor cells, while Quercetin is more potent in killing senescent human endothelial cells and bone marrow stem cells in mice.341 The most potent removal of senescent cells was achieved when these two compounds were combined. In several mouse experiments, the treatment alleviated inflammation342 and age-related diseases of the intestines343 and bone.344 In the first human trial, ‘Dasatinib + Quercetin’ treatment increased patients’ 6-minute walking distance by an average of 21.5 meters. However, other indicators including Pulmonary function, clinical chemistries, frailty index (FI-LAB), and reported health did not change significantly.345 Further research is needed on the core indicators such as SASP levels which will also predict efficacy and side effects over time. In clinical trials in patients with diabetic nephropathy, Dasatinib, and Quercetin significantly reduced senescent cell levels as well as circulating SASP levels over 11 days.346 Considering the potential toxicity of the inflammation produced by cell death and grab, there is a lack of long-term follow-up studies for this therapy and the current evidence is insufficient to support its long-term anti-aging effectiveness. In addition, another Senolytic called Fenofibrate can induce selective elimination of senescence cells through upregulating PPARα expression.347 Excessive clearance of senescent cells (especially for senescent liver sinusoidal endothelial cells or adipocytes) has also been reported to accompany the dilemma faced by cells that cannot be replenished in time, although Quercetin + Dasatinib clears mainly p16 macrophages.348
(CAR-)T/NK for eliminating senescent cells
On the other hand, immune cell-mediated clearance of senescent cells is emerging as a promising strategy to fight multiple chronic diseases and aging.349 It was found that anti-uPAR-CAR-T cells were effective in removing senescent cells in vitro and pre-cancerous and malignant cells in mouse models of liver and lung in the presence of potentially toxic.350 Previously, CAR-T therapy targeting FAP was found to significantly reduce cardiac fibrosis and improve heart function. Surprisingly, FAP-CAR-T cells did not target other normal cells in the body and did not cause recruitment and infiltration of immune cells and increased levels of inflammatory factors, such as IL-1 and IL-6.351 In addition, clearance of senescent cells by NK cell-based immune cells prolongs the lifespan of mice and appeared to be safer.352 In the aged mice and healthy/obese volunteers, adoptive NK cell infusion significantly reduced senescence markers and SASP levels without significant toxic side effects.353,354 In addition, their combination with the immunomodulatory factor Acein significantly decreased the expression of tissue senescence markers and age-related genes in a mouse model of senescence.353
Stem cell therapy
Stem cell therapy has become an effective strategy for the treatment of aging-related diseases. Many studies have shown that the number and function of different somatic cell populations declines with age, which diminishes the regenerative potential of tissues and organs. However, there is limited evidence that loss of stem cell function is a major driver of age-related pathology and shortened lifespan.355,356,357,358 Trials have found that after stem cell injections in aging debilitated patients, many symptoms improve, and inflammatory marker levels decrease.359 MSC transplantation for acute stroke improves patient symptoms and promotes neurological recovery in ischemic stroke patients without adverse effects.360,361,362 Meanwhile, mesenchymal stem cell transplantation for Parkinson’s disease can significantly improve the daily activities and motor functions of Parkinson’s disease patients without side effects, as well as being safe and reliable.363,364 In addition, HSC transplantation is a very rapidly and effective clinical treatment for age-related diseases such as AML.
Organ regeneration and transplantation
Similar to stem cell transplantation, organ transplantation is a very effective anti-aging modality because it can “repair” the damage caused by aging in the most simple and brutal way. For example, many risk indices for age-related diseases were improved after thymic regeneration in humans (taking three commonly used drugs: growth hormone, dehydroepiandrosterone, and metformin),365 and biological age was reversed.366 However, in order to use this technology in the field of anti-aging, two problems must first be solved: the shortage of spare organ stock and rejection after transplantation. Currently, most donated organs come from relatives, cerebrally dead donors, or even animals. For organs from old donors, Senolytics are used to rejuvenate aging organs.367 Meanwhile, the world’s first “transplantation” of a porcine heart into a human was initially successful and has not caused hyperacute rejection in humans. If animal organs are successfully transplanted and the cost is controlled, human disease treatment and anti-aging will take a step forward.
Resistance and reversal of aging is the ultimate goal of aging research. Anti-inflammation, removal/improvement of senescent cells, stem cell therapy and organ transplantation have become crucial ways to reverse aging in humans.
Discussion
The global population has been experiencing an aging trend, and the elderly population is more susceptible to infections, increased mortality, and morbidity.368,369 Chronic inflammation appears to be closely linked to aging, and this review focuses on inflammaging, providing an overview of the molecular, cellular, and organ levels of the human body. At the cellular level, external DAMPs activate different immune cells, promoting inflammation and leading to immunosenescence. Dysfunctional immune cells cannot clear senescent cells in a timely manner, leading to inflammation and the development of aging-related diseases and/or normal aging in different organs (Fig. 1). In addition to the separate analysis of molecules, cells, and organs, we also hope to closely link the cells, organs and molecule with single-cell multi-omics (including genome, transcriptome, epigenome, proteome and metabolome) and spatial omics.
The construction of animal models is an effective way to study aging (Table 2). The present review systematically summarizes the characteristics of different aging models, both in mice and in vitro cell lines. To date, an aging atlas of the whole organ has been profiled in mice. Considering the differences between different species,370 it will be important to construct non-human primate and human aging models in the future. In addition, several resource tools have been developed to assist in aging research, such as SeneQuest to promote the discovery of genes associated with senescence (available at http://Senequest.net).371 On the other hand, other technologies, such as machine learning which integrates multi-modal and multidimensional big data, are also important to address the complexity of aging problems (Table 4). A recent paper summarizes the development and application of aging clocks with the help of machine learning analysis of histological data. This illustrates the ability of machine learning to identify novel biomarkers of biological aging and provides a boost to early warning and intervention in aging and precision medicine strategies.372 In 2022, Li et al. developed Nvwa, a deep-learning-based strategy, which predicts gene expression and identifies conserved regulatory programs underlying cell types at the cross-species single-cell level.373 In the future, we also hope to find similar cross-species conservative regulatory elements in the aging process through development of machine learning algorithms.
The immune system has a remarkable ability to remember and respond to different stimuli and experiences, leading to heterogeneity in immunosenescence among individuals. This heterogeneity can result from differences in the type, dose, intensity, and temporal sequence of antigenic stimuli to which each person is exposed. To address this issue, Franceschi et al. proposed the concept of “immunobiography,” which considers the unique history of antigenic exposure that shapes an individual’s immune system over time.374 While immunobiography provides a comprehensive framework for understanding immune aging, it does not take into account other factors that can influence the accumulation of inflammation and the aging process, such as genetics and social factors. Therefore, a more holistic approach that considers multiple factors may be necessary to fully understand the complexities of immunosenescence and inflammaging.
Preventing and alleviating the diseases of aging and improving quality of life are the ultimate goals of aging research. Current anti-aging strategies include eliminating senescent cells, stem cell therapy, and organ transplantation, whose essence is anti-inflammatory. However, what came first, the aging (chicken) or the inflammation (egg)? The exact causality between inflammation and aging deserves further studies for efficient intervention of aging-associated diseases and enhancing well-being.
References
McCay, C. M., Maynard, L. A., Sperling, G. & Barnes, L. L. Retarded growth, life span, ultimate body size and age changes in the albino rat after feeding diets restricted in calories. Nutr. Rev. 33, 241–243 (1975).
Fulop, T. et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front. Immunol. 8, 1960 (2017).
Campisi, J. et al. From discoveries in ageing research to therapeutics for healthy ageing. Nature 571, 183–192 (2019).
Takahashi, T. & Iwasaki, A. Sex differences in immune responses. Science 371, 347–348 (2021).
Marquez, E. J. et al. Sexual-dimorphism in human immune system aging. Nat. Commun. 11, 751 (2020).
Arai, Y. et al. Inflammation, but not telomere length, predicts successful ageing at extreme old age: a longitudinal study of semi-supercentenarians. EBioMedicine 2, 1549–1558 (2015).
Sayed, N. et al. An inflammatory aging clock (iAge) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nat. Aging 1, 598–615 (2021).
Hernandez-Segura, A., Nehme, J. & Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453 (2018).
Aging Biomarker, C. et al. Biomarkers of aging. Sci. China Life Sci. 66, 893–1066 (2023).
Yousefzadeh, M. J. et al. An aged immune system drives senescence and ageing of solid organs. Nature 594, 100–105 (2021).
Carrasco, E. et al. The role of T cells in age-related diseases. Nat. Rev. Immunol. 22, 97–111 (2022).
Walford, R. L. The immunologic theory of aging. Gerontologist 4, 195–197 (1964).
Pawelec, G. Age and immunity: What is “immunosenescence”? Exp. Gerontol. 105, 4–9 (2018).
Kennedy, B. K. et al. Geroscience: linking aging to chronic disease. Cell 159, 709–713 (2014).
St Sauver, J. L. et al. Risk of developing multimorbidity across all ages in an historical cohort study: differences by sex and ethnicity. BMJ Open 5, e006413 (2015).
Liggett, L. A. & Sankaran, V. G. Unraveling Hematopoiesis through the Lens of Genomics. Cell 182, 1384–1400 (2020).
Bogeska, R. et al. Inflammatory exposure drives long-lived impairment of hematopoietic stem cell self-renewal activity and accelerated aging. Cell Stem Cell 29, 1273–1284.e1278 (2022).
Rundberg Nilsson, A. et al. Human and murine hematopoietic stem cell aging is associated with functional impairments and intrinsic megakaryocytic/erythroid bias. PLoS ONE 11, e0158369 (2016).
Jahandideh, B. et al. The pro-Inflammatory cytokines effects on mobilization, self-renewal and differentiation of hematopoietic stem cells. Hum. Immunol. 81, 206–217 (2020).
Yamashita, M. & Passegue, E. TNF-alpha coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell 25, 357–372.e357 (2019).
Ho, N. P. & Takizawa, H. Inflammation regulates haematopoietic stem cells and their niche. Int. J. Mol. Sci. 23, 1125 (2022).
Pioli, P. D. et al. Plasma cells are obligate effectors of enhanced myelopoiesis in aging bone marrow. Immunity 51, 351–366.e356 (2019).
Kovtonyuk, L. V. et al. IL-1 mediates microbiome-induced inflammaging of hematopoietic stem cells in mice. Blood 139, 44–58 (2022).
Mitchell, C. A. et al. Stromal niche inflammation mediated by IL-1 signalling is a targetable driver of haematopoietic ageing. Nat. Cell Biol. 25, 30–41 (2023).
Valletta, S. et al. Micro-environmental sensing by bone marrow stroma identifies IL-6 and TGFbeta1 as regulators of hematopoietic ageing. Nat. Commun. 11, 4075 (2020).
Li, X. et al. Mechanisms and rejuvenation strategies for aged hematopoietic stem cells. J. Hematol. Oncol. 13, 31 (2020).
Janzen, V. et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443, 421–426 (2006).
Baldridge, M. T. et al. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797 (2010).
Florez, M. A. et al. Interferon gamma mediates hematopoietic stem cell activation and niche relocalization through BST2. Cell Rep. 33, 108530 (2020).
Qin, Y. & Zhang, C. The regulatory role of ifn-gamma on the proliferation and differentiation of hematopoietic stem and progenitor cells. Stem Cell Rev. Rep. 13, 705–712 (2017).
de Bruin, A. M. et al. Interferon-gamma impairs proliferation of hematopoietic stem cells in mice. Blood 121, 3578–3585 (2013).
Yang, L. et al. IFN-gamma negatively modulates self-renewal of repopulating human hemopoietic stem cells. J. Immunol. 174, 752–757 (2005).
Qin, Y. et al. Interferon gamma inhibits the differentiation of mouse adult liver and bone marrow hematopoietic stem cells by inhibiting the activation of notch signaling. Stem Cell Res. Ther. 10, 210 (2019).
Mistry, J. J. et al. ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection. Proc. Natl Acad. Sci. USA 116, 24610–24619 (2019).
Mistry, J. J. et al. Free fatty-acid transport via CD36 drives beta-oxidation-mediated hematopoietic stem cell response to infection. Nat. Commun. 12, 7130 (2021).
Shao, L. et al. Reactive oxygen species and hematopoietic stem cell senescence. Int. J. Hematol. 94, 24–32 (2011).
Shi, Y., Riese, D. J. 2nd & Shen, J. The role of the CXCL12/CXCR4/CXCR7 chemokine axis in cancer. Front. Pharmacol. 11, 574667 (2020).
Zhang, Y. et al. CXCR4/CXCL12 axis counteracts hematopoietic stem cell exhaustion through selective protection against oxidative stress. Sci. Rep. 6, 37827 (2016).
Butcher, S., Chahel, H. & Lord, J. M. Review article: ageing and the neutrophil: no appetite for killing? Immunology 100, 411–416 (2000).
Dubey, M. et al. Nitric oxide-mediated apoptosis of neutrophils through caspase-8 and caspase-3-dependent mechanism. Cell Death Dis. 7, e2348 (2016).
Niwa, Y., Kasama, T., Miyachi, Y. & Kanoh, T. Neutrophil chemotaxis, phagocytosis and parameters of reactive oxygen species in human aging: Cross-sectional and longitudinal studies. Life Sci. 44, 1655–1664 (1989).
Liles, W. C. et al. Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J. Exp. Med. 184, 429–440 (1996).
Hazeldine, J. et al. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell 13, 690–698 (2014).
Qian, F. et al. Reduced bioenergetics and toll-like receptor 1 function in human polymorphonuclear leukocytes in aging. Aging 6, 131–139 (2014).
Lu, R. J. et al. Multi-omic profiling of primary mouse neutrophils predicts a pattern of sex and age-related functional regulation. Nat. Aging 1, 715–733 (2021).
Eash, K. J., Greenbaum, A. M., Gopalan, P. K. & Link, D. C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Invest. 120, 2423–2431 (2010).
Adrover, J. M., Nicolas-Avila, J. A. & Hidalgo, A. Aging: a temporal dimension for neutrophils. Trends Immunol. 37, 334–345 (2016).
Barkaway, A. et al. Age-related changes in the local milieu of inflamed tissues cause aberrant neutrophil trafficking and subsequent remote organ damage. Immunity 54, 1494–1510.e1497 (2021).
Lagnado, A. et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 40, e106048 (2021).
Stranks, A. J. et al. Autophagy controls acquisition of aging features in macrophages. J. Innate Immun. 7, 375–391 (2015).
Wong, C. K. et al. Aging impairs alveolar macrophage phagocytosis and increases influenza-induced mortality in mice. J. Immunol. 199, 1060–1068 (2017).
Zhang, B., Bailey, W. M., Braun, K. J. & Gensel, J. C. Age decreases macrophage IL-10 expression: implications for functional recovery and tissue repair in spinal cord injury. Exp. Neurol. 273, 83–91 (2015).
Shamskhou, E. A. et al. Hydrogel-based delivery of Il-10 improves treatment of bleomycin-induced lung fibrosis in mice. Biomaterials 203, 52–62 (2019).
Campbell, R. A., Docherty, M. H., Ferenbach, D. A. & Mylonas, K. J. The role of ageing and parenchymal senescence on macrophage function and fibrosis. Front. Immunol. 12, 700790 (2021).
Minhas, P. S. et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature 590, 122–128 (2021).
Brauning, A. et al. Aging of the immune system: focus on natural killer cells phenotype and functions. Cells 11, 1017 (2022).
Muzzioli, M., Stecconi, R., Moresi, R. & Provinciali, M. Zinc improves the development of human CD34+ cell progenitors towards NK cells and increases the expression of GATA-3 transcription factor in young and old ages. Biogerontology 10, 593–604 (2009).
Sansoni, P. et al. Lymphocyte subsets and natural killer cell activity in healthy old people and centenarians. Blood 82, 2767–2773 (1993).
Le Garff-Tavernier, M. et al. Human NK cells display major phenotypic and functional changes over the life span. Aging Cell 9, 527–535 (2010).
Facchini, A. et al. Increased number of circulating Leu 11+ (CD 16) large granular lymphocytes and decreased NK activity during human ageing. Clin. Exp. Immunol. 68, 340–347 (1987).
Gounder, S. S. et al. Effect of aging on NK cell population and their proliferation at ex vivo culture condition. Anal. Cell Pathol. 2018, 7871814 (2018).
Witkowski, J. M., Larbi, A., Le Page, A. & Fulop, T. Natural killer cells, aging, and vaccination. Interdiscip. Top. Gerontol. Geriatr. 43, 18–35 (2020).
Solana, R., Campos, C., Pera, A. & Tarazona, R. Shaping of NK cell subsets by aging. Curr. Opin. Immunol. 29, 56–61 (2014).
Campos, C. et al. Expression of NKp30, NKp46 and DNAM-1 activating receptors on resting and IL-2 activated NK cells from healthy donors according to CMV-serostatus and age. Biogerontology 16, 671–683 (2015).
Campos, C. et al. Effect of age and CMV on NK cell subpopulations. Exp. Gerontol. 54, 130–137 (2014).
Lian, J., Yue, Y., Yu, W. & Zhang, Y. Immunosenescence: a key player in cancer development. J. Hematol. Oncol. 13, 151 (2020).
Wolf, N. K., Kissiov, D. U. & Raulet, D. H. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol. 23, 90–105 (2023).
Martin-Fontecha, A. et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat. Immunol. 5, 1260–1265 (2004).
Kyoizumi, S. et al. Age-associated changes in the differentiation potentials of human circulating hematopoietic progenitors to T- or NK-lineage cells. J. Immunol. 190, 6164–6172 (2013).
Qin, L. et al. Aging of immune system: Immune signature from peripheral blood lymphocyte subsets in 1068 healthy adults. Aging 8, 848–859 (2016).
Wang, S. S. et al. Tumor-infiltrating B cells: their role and application in anti-tumor immunity in lung cancer. Cell. Mol. Immunol. 16, 6–18 (2019).
Labrie, J. E. 3rd et al. Bone marrow microenvironmental changes underlie reduced RAG-mediated recombination and B cell generation in aged mice. J. Exp. Med. 200, 411–423 (2004).
Caraux, A. et al. Circulating human B and plasma cells. Age-associated changes in counts and detailed characterization of circulating normal CD138- and CD138+ plasma cells. Haematologica 95, 1016–1020 (2010).
Min, H., Montecino-Rodriguez, E. & Dorshkind, K. Effects of aging on the common lymphoid progenitor to pro-B cell transition. J. Immunol. 176, 1007–1012 (2006).
Kogut, I., Scholz, J. L., Cancro, M. P. & Cambier, J. C. B cell maintenance and function in aging. Semin. Immunol. 24, 342–349 (2012).
Cepeda, S. et al. Age-associated decline in thymic B cell expression of aire and aire-dependent self-antigens. Cell Rep. 22, 1276–1287 (2018).
Colonna-Romano, G. et al. A double-negative (IgD-CD27-) B cell population is increased in the peripheral blood of elderly people. Mech. Ageing Dev. 130, 681–690 (2009).
Frasca, D. et al. Unique biomarkers for B-cell function predict the serum response to pandemic H1N1 influenza vaccine. Int. Immunol. 24, 175–182 (2012).
Gibson, K. L. et al. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell 8, 18–25 (2009).
Ma, S., Wang, C., Mao, X. & Hao, Y. B cell dysfunction associated with aging and autoimmune diseases. Front. Immunol. 10, 318 (2019).
Bulati, M. et al. B cells and immunosenescence: a focus on IgG+IgD-CD27- (DN) B cells in aged humans. Ageing Res. Rev. 10, 274–284 (2011).
Ratliff, M. et al. In senescence, age-associated B cells secrete TNFalpha and inhibit survival of B-cell precursors. Aging Cell 12, 303–311 (2013).
Desdín-Micó, G. et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 368, 1371–1376 (2020).
Kurachi, M. et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol. 15, 373–383 (2014).
Mittelbrunn, M. & Kroemer, G. Hallmarks of T cell aging. Nat. Immunol. 22, 687–698 (2021).
Hu, B. et al. Transcription factor networks in aged naive CD4 T cells bias lineage differentiation. Aging Cell 18, e12957 (2019).
Kovaiou, R. D. & Grubeck-Loebenstein, B. Age-associated changes within CD4+ T cells. Immunol. Lett. 107, 8–14 (2006).
Shanley, D. P., Aw, D., Manley, N. R. & Palmer, D. B. An evolutionary perspective on the mechanisms of immunosenescence. Trends Immunol. 30, 374–381 (2009).
Jowett, J. B. et al. ADAM28 is elevated in humans with the metabolic syndrome and is a novel sheddase of human tumour necrosis factor-alpha. Immunol. Cell Biol. 90, 966–973 (2012).
Callender, L. A. et al. Human CD8(+) EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell 17, e12675 (2018).
Di Mitri, D. et al. Reversible senescence in human CD4+CD45RA+CD27- memory T cells. J. Immunol. 187, 2093–2100 (2011).
Elyahu, Y. et al. Aging promotes reorganization of the CD4 T cell landscape toward extreme regulatory and effector phenotypes. Sci. Adv. 5, eaaw8330 (2019).
Fletcher, J. M. et al. Cytomegalovirus-specific CD4+ T cells in healthy carriers are continuously driven to replicative exhaustion. J. Immunol. 175, 8218–8225 (2005).
Callender, L. A. et al. Mitochondrial mass governs the extent of human T cell senescence. Aging Cell 19, e13067 (2020).
Ucar, D. et al. The chromatin accessibility signature of human immune aging stems from CD8(+) T cells. J. Exp. Med 214, 3123–3144 (2017).
Goronzy, J. J. & Weyand, C. M. Mechanisms underlying T cell ageing. Nat. Rev. Immunol. 19, 573–583 (2019).
Mogilenko, D. A. et al. Comprehensive profiling of an aging immune system reveals clonal GZMK(+) CD8(+) T cells as conserved hallmark of inflammaging. Immunity 54, 99–115.e112 (2021).
Aravinthan, A. et al. Hepatocyte expression of the senescence marker p21 is linked to fibrosis and an adverse liver-related outcome in alcohol-related liver disease. PLoS ONE 8, e72904 (2013).
Tarazona, R. et al. Immunosenescence: limitations of natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 66, 233–245 (2017).
Hagen, M. & Derudder, E. Inflammation and the alteration of B-cell physiology in aging. Gerontology 66, 105–113 (2020).
Day, R. B., Bhattacharya, D., Nagasawa, T. & Link, D. C. Granulocyte colony-stimulating factor reprograms bone marrow stromal cells to actively suppress B lymphopoiesis in mice. Blood 125, 3114–3117 (2015).
Budamagunta, V., Foster, T. C. & Zhou, D. Cellular senescence in lymphoid organs and immunosenescence. Aging 13, 19920–19941 (2021).
Pritz, T., Weinberger, B. & Grubeck-Loebenstein, B. The aging bone marrow and its impact on immune responses in old age. Immunol. Lett. 162, 310–315 (2014).
Chung, S. S. & Park, C. Y. Aging, hematopoiesis, and the myelodysplastic syndromes. Blood Adv. 1, 2572–2578 (2017).
Zhang, H. et al. The roles of bone remodeling in normal hematopoiesis and age-related hematological malignancies. Bone Res 11, 15 (2023).
Ambrosi, T. H. et al. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature 597, 256–262 (2021).
Zhao, E. et al. Bone marrow and the control of immunity. Cell. Mol. Immunol. 9, 11–19 (2012).
Beerman, I. et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl Acad. Sci. USA 107, 5465–5470 (2010).
Pang, W. W. et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl Acad. Sci. USA 108, 20012–20017 (2011).
Reya, T. et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 423, 409–414 (2003).
Aw, D., Taylor-Brown, F., Cooper, K. & Palmer, D. B. Phenotypical and morphological changes in the thymic microenvironment from ageing mice. Biogerontology 10, 311–322 (2009).
Kvell, K. et al. Wnt4 and LAP2alpha as pacemakers of thymic epithelial senescence. PLoS ONE 5, e10701 (2010).
Aw, D. et al. Architectural changes in the thymus of aging mice. Aging Cell 7, 158–167 (2008).
Hale, J. S., Boursalian, T. E., Turk, G. L. & Fink, P. J. Thymic output in aged mice. Proc. Natl Acad. Sci. USA 103, 8447–8452 (2006).
Wertheimer, A. M. et al. Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J. Immunol. 192, 2143–2155 (2014).
Czesnikiewicz-Guzik, M. et al. T cell subset-specific susceptibility to aging. Clin. Immunol. 127, 107–118 (2008).
den Braber, I. et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity 36, 288–297 (2012).
Goldrath, A. W., Bogatzki, L. Y. & Bevan, M. J. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J. Exp. Med. 192, 557–564 (2000).
Renkema, K. R. et al. Two separate defects affecting true naive or virtual memory T cell precursors combine to reduce naive T cell responses with aging. J. Immunol. 192, 151–159 (2014).
Rudd, B. D. et al. Nonrandom attrition of the naive CD8+ T-cell pool with aging governed by T-cell receptor:pMHC interactions. Proc. Natl Acad. Sci. USA 108, 13694–13699 (2011).
Kato, A., Takaori-Kondo, A., Minato, N. & Hamazaki, Y. CXCR3(high) CD8(+) T cells with naive phenotype and high capacity for IFN-gamma production are generated during homeostatic T-cell proliferation. Eur. J. Immunol. 48, 1663–1678 (2018).
Li, G. et al. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat. Med. 18, 1518–1524 (2012).
Sato, Y. & Yanagita, M. Immunology of the ageing kidney. Nat. Rev. Nephrol. 15, 625–640 (2019).
Aw, D. et al. Disorganization of the splenic microanatomy in ageing mice. Immunology 148, 92–101 (2016).
Turner, V. M. & Mabbott, N. A. Influence of ageing on the microarchitecture of the spleen and lymph nodes. Biogerontology 18, 723–738 (2017).
Tabula Muris, C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595 (2020).
Turner, V. M. & Mabbott, N. A. Ageing adversely affects the migration and function of marginal zone B cells. Immunology 151, 349–362 (2017).
Lefebvre, J. S. et al. The aged microenvironment contributes to the age-related functional defects of CD4 T cells in mice. Aging Cell 11, 732–740 (2012).
Li, G., Smithey, M. J., Rudd, B. D. & Nikolich-Zugich, J. Age-associated alterations in CD8alpha+ dendritic cells impair CD8 T-cell expansion in response to an intracellular bacterium. Aging Cell 11, 968–977 (2012).
Thompson, H. L., Smithey, M. J., Surh, C. D. & Nikolich-Zugich, J. Functional and homeostatic impact of age-related changes in lymph node stroma. Front. Immunol. 8, 706 (2017).
Becklund, B. R. et al. The aged lymphoid tissue environment fails to support naive T cell homeostasis. Sci. Rep. 6, 30842 (2016).
Ownby, R. L. Neuroinflammation and cognitive aging. Curr. Psychiatry Rep. 12, 39–45 (2010).
Ron-Harel, N. & Schwartz, M. Immune senescence and brain aging: can rejuvenation of immunity reverse memory loss? Trends Neurosci. 32, 367–375 (2009).
Shao, W. et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via alphaB-crystallin. Nature 494, 90–94 (2013).
Di Benedetto, S. et al. Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci. Biobehav. Rev. 75, 114–128 (2017).
Barrientos, R. M., Kitt, M. M., Watkins, L. R. & Maier, S. F. Neuroinflammation in the normal aging hippocampus. Neuroscience 309, 84–99 (2015).
Giunta, B. et al. Inflammaging as a prodrome to Alzheimer’s disease. J. Neuroinflammation 5, 51 (2008).
von Bernhardi, R., Tichauer, J. E. & Eugenin, J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem. 112, 1099–1114 (2010).
Sikora, E. et al. Cellular senescence in brain aging. Front. Aging Neurosci. 13, 646924 (2021).
Meschiari, C. A. et al. The impact of aging on cardiac extracellular matrix. Geroscience 39, 7–18 (2017).
Ruiz-Meana, M. et al. Cardiomyocyte ageing and cardioprotection: consensus document from the ESC working groups cell biology of the heart and myocardial function. Cardiovasc. Res. 116, 1835–1849 (2020).
Obas, V. & Vasan, R. S. The aging heart. Clin. Sci. 132, 1367–1382 (2018).
Ma, Y., Mouton, A. J. & Lindsey, M. L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 191, 15–28 (2018).
O’Sullivan, E. D., Hughes, J. & Ferenbach, D. A. Renal aging: causes and consequences. J. Am. Soc. Nephrol. 28, 407–420 (2017).
Sato, Y. & Yanagita, M. Immune cells and inflammation in AKI to CKD progression. Am. J. Physiol. Ren. Physiol. 315, F1501–F1512 (2018).
Schmitt, R. & Melk, A. Molecular mechanisms of renal aging. Kidney Int. 92, 569–579 (2017).
Sato, Y. et al. Heterogeneous fibroblasts underlie age-dependent tertiary lymphoid tissues in the kidney. JCI Insight 1, e87680 (2016).
Kimura, T. et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 22, 902–913 (2011).
Yamamoto, T. et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 12, 801–813 (2016).
Schmitt, R., Marlier, A. & Cantley, L. G. Zag expression during aging suppresses proliferation after kidney injury. J. Am. Soc. Nephrol. 19, 2375–2383 (2008).
Kotas, M. E. & Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827 (2015).
Nathan, C. & Ding, A. Nonresolving inflammation. Cell 140, 871–882 (2010).
Pinto, C. et al. Aging and the biological response to liver injury. Semin Liver Dis. 40, 225–232 (2020).
Ezhilarasan, D., Sokal, E. & Najimi, M. Hepatic fibrosis: it is time to go with hepatic stellate cell-specific therapeutic targets. Hepatobiliary Pancreat. Dis. Int. 17, 192–197 (2018).
Udomsinprasert, W. et al. Cellular senescence in liver fibrosis: implications for age-related chronic liver diseases. Expert Opin. Ther. Targets 25, 799–813 (2021).
Tomita, K. et al. p53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 57, 837–843 (2012).
Wen, Y., Lambrecht, J., Ju, C. & Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 18, 45–56 (2021).
Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705 (2013).
Choi, E. J., Kil, I. S. & Cho, E. G. Extracellular vesicles derived from senescent fibroblasts attenuate the dermal effect on keratinocyte differentiation. Int. J. Mol. Sci. 21, 1022 (2020).
Franco, A. C., Aveleira, C. & Cavadas, C. Skin senescence: mechanisms and impact on whole-body aging. Trends Mol. Med. 28, 97–109 (2022).
Waaijer, M. E. et al. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 11, 722–725 (2012).
Waaijer, M. E. C. et al. Are skin senescence and immunosenescence linked within individuals? Aging Cell 18, e12956 (2019).
Huang, S. et al. Human skin, oral, and gut microbiomes predict chronological age. mSystems 5, e00630–19 (2020).
Pilkington, S. M. et al. Aged human skin accumulates mast cells with altered functionality that localize to macrophages and vasoactive intestinal peptide-positive nerve fibres. Br. J. Dermatol. 180, 849–858 (2019).
Zuelgaray, E. et al. Increased expression of PD1 and CD39 on CD3(+) CD4(+) skin T cells in the elderly. Exp. Dermatol. 28, 80–82 (2019).
Lages, C. S. et al. Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J. Immunol. 181, 1835–1848 (2008).
Vukmanovic-Stejic, M. et al. The characterization of varicella zoster virus-specific T cells in skin and blood during aging. J. Invest. Dermatol. 135, 1752–1762 (2015).
Kovacs, E. J., Boe, D. M., Boule, L. A. & Curtis, B. J. Inflammaging and the Lung. Clin. Geriatr. Med. 33, 459–471 (2017).
Cho, S. J. & Stout-Delgado, H. W. Aging and lung disease. Annu. Rev. Physiol. 82, 433–459 (2020).
Ortega-Martinez, M. et al. Analysis of cell turnover in the bronchiolar epithelium through the normal aging process. Lung 194, 581–587 (2016).
Wansleeben, C. et al. Age-related changes in the cellular composition and epithelial organization of the mouse trachea. PLoS ONE 9, e93496 (2014).
Watson, J. K. et al. Distal lung epithelial progenitor cell function declines with age. Sci. Rep. 10, 10490 (2020).
Schneider, J. L. et al. The aging lung: Physiology, disease, and immunity. Cell 184, 1990–2019 (2021).
Hearps, A. C. et al. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 11, 867–875 (2012).
Li, Z. et al. Aging-impaired filamentous actin polymerization signaling reduces alveolar macrophage phagocytosis of bacteria. J. Immunol. 199, 3176–3186 (2017).
Zhou, X. & McElhaney, J. E. Age-related changes in memory and effector T cells responding to influenza A/H3N2 and pandemic A/H1N1 strains in humans. Vaccine 29, 2169–2177 (2011).
Zheng, Y. et al. A human circulating immune cell landscape in aging and COVID-19. Protein Cell 11, 740–770 (2020).
Rehman, T. Role of the gut microbiota in age-related chronic inflammation. Endocr. Metab. Immune Disord. Drug Targets 12, 361–367 (2012).
Friedland, R. P. Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J. Alzheimers Dis. 45, 349–362 (2015).
Magnusson, K. R. et al. Relationships between diet-related changes in the gut microbiome and cognitive flexibility. Neuroscience 300, 128–140 (2015).
Paun, A. & Danska, J. S. Modulation of type 1 and type 2 diabetes risk by the intestinal microbiome. Pediatr. Diabetes 17, 469–477 (2016).
Claesson, M. J. et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184 (2012).
Rondanelli, M. et al. Review on microbiota and effectiveness of probiotics use in older. World J. Clin. Cases 3, 156–162 (2015).
DeJong, E. N., Surette, M. G. & Bowdish, D. M. E. The gut microbiota and unhealthy aging: disentangling cause from consequence. Cell Host Microbe 28, 180–189 (2020).
Chen, L. et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat. Immunol. 18, 541–551 (2017).
Thevaranjan, N. et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 21, 455–466 e454 (2017).
Valentini, L. et al. Small intestinal permeability in older adults. Physiol. Rep. 2, e00281 (2014).
Wilms, E. et al. Intestinal barrier function is maintained with aging—a comprehensive study in healthy subjects and irritable bowel syndrome patients. Sci. Rep. 10, 475 (2020).
Olivieri, F. et al. DNA damage response (DDR) and senescence: shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget 6, 35509–35521 (2015).
Basisty, N. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599 (2020).
Coppe, J. P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 (2008).
Wiley, C. D. et al. SILAC analysis reveals increased secretion of hemostasis-related factors by senescent cells. Cell Rep. 28, 3329–3337.e3325 (2019).
Davalos, A. R. et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 201, 613–629 (2013).
Birch, J. & Gil, J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 34, 1565–1576 (2020).
Correia-Melo, C. et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742 (2016).
Rodier, F. et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 (2009).
Kulkarni, A. S., Gubbi, S. & Barzilai, N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 32, 15–30 (2020).
Cohn, R. L., Gasek, N. S., Kuchel, G. A. & Xu, M. The heterogeneity of cellular senescence: insights at the single-cell level. Trends Cell Biol. 33, 9–17 (2023).
Casella, G. et al. Transcriptome signature of cellular senescence. Nucleic Acids Res. 47, 11476 (2019).
Giunta, S., Wei, Y., Xu, K. & Xia, S. Cold-inflammaging: When a state of homeostatic-imbalance associated with aging precedes the low-grade pro-inflammatory-state (inflammaging): meaning, evolution, inflammaging phenotypes. Clin. Exp. Pharmacol. Physiol. 49, 925–934 (2022).
von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 27, 339–344 (2002).
Sies, H., Berndt, C. & Jones, D. P. Oxidative Stress. Annu. Rev. Biochem. 86, 715–748 (2017).
Adebayo, M., Singh, S., Singh, A. P. & Dasgupta, S. Mitochondrial fusion and fission: the fine-tune balance for cellular homeostasis. FASEB J. 35, e21620 (2021).
Lin, J. B. et al. Macrophage microRNA-150 promotes pathological angiogenesis as seen in age-related macular degeneration. JCI Insight 3, e120157 (2018).
Furman, D. et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat. Med. 23, 174–184 (2017).
Furman, D. et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 25, 1822–1832 (2019).
De la Fuente, M. & Miquel, J. An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 15, 3003–3026 (2009).
Martinez de Toda, I., Ceprian, N., Diaz-Del Cerro, E. & De la Fuente, M. The role of immune cells in oxi-inflamm-aging. Cells 10, 2974 (2021).
Barascu, A. et al. Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J. 31, 1080–1094 (2012).
Di Leonardo, A., Linke, S. P., Clarkin, K. & Wahl, G. M. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 8, 2540–2551 (1994).
Wieczfinska, J., Kleniewska, P. & Pawliczak, R. Oxidative Stress-Related Mechanisms in SARS-CoV-2 Infections. Oxid. Med Cell Longev. 2022, 5589089 (2022).
Renz, H. et al. An exposome perspective: early-life events and immune development in a changing world. J. Allergy Clin. Immunol. 140, 24–40 (2017).
Yuan, J. et al. Long-term persistent organic pollutants exposure induced telomere dysfunction and senescence-associated secretary phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 73, 1027–1035 (2018).
Carroll, J. E. et al. Partial sleep deprivation activates the DNA damage response (DDR) and the senescence-associated secretory phenotype (SASP) in aged adult humans. Brain Behav. Immun. 51, 223–229 (2016).
Biagi, E. et al. Through ageing, and beyond: gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 5, e10667 (2010).
Claesson, M. J. et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl Acad. Sci. USA 108, 4586–4591 (2011).
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005).
Allen, J. M. et al. Exercise training-induced modification of the gut microbiota persists after microbiota colonization and attenuates the response to chemically-induced colitis in gnotobiotic mice. Gut Microbes 9, 115–130 (2018).
Clark, R. I. et al. Distinct shifts in microbiota composition during Drosophila aging impair intestinal function and drive mortality. Cell Rep. 12, 1656–1667 (2015).
Smith, P. et al. Regulation of life span by the gut microbiota in the short-lived African turquoise killifish. eLife 6, e27014 (2017).
Langille, M. G. et al. Microbial shifts in the aging mouse gut. Microbiome 2, 50 (2014).
Flemer, B. et al. Fecal microbiota variation across the lifespan of the healthy laboratory rat. Gut Microbes 8, 428–439 (2017).
van Soest, A. P. M. et al. Associations between pro- and anti-inflammatory gastro-intestinal microbiota, diet, and cognitive functioning in dutch healthy older adults: the NU-AGE study. Nutrients 12, 3471 (2020).
Du, Y. et al. Effects of anti-aging interventions on intestinal microbiota. Gut Microbes 13, 1994835 (2021).
Zeng, X. et al. Fecal microbiota transplantation from young mice rejuvenates aged hematopoietic stem cells by suppressing inflammation. Blood 141, 1691–1707 (2023).
Bianconi, E. et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 40, 463–471 (2013).
Franceschi, C. et al. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 28, 199–212 (2017).
Franceschi, C. et al. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590 (2018).
Li, X. et al. Cell deaths: Involvement in the pathogenesis and intervention therapy of COVID-19. Signal Transduct. Target. Ther. 7, 186 (2022).
Binet, F. et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 369, eaay5356 (2020).
Del Valle, D. M. et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 26, 1636–1643 (2020).
Laing, A. G. et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 26, 1623–1635 (2020).
Lee, S. et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 599, 283–289 (2021).
Santoro, A., Bientinesi, E. & Monti, D. Immunosenescence and inflammaging in the aging process: age-related diseases or longevity? Ageing Res. Rev. 71, 101422 (2021).
Vanlangenakker, N., Vanden Berghe, T. & Vandenabeele, P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 19, 75–86 (2012).
Yee, P. P. & Li, W. Tumor necrosis: A synergistic consequence of metabolic stress and inflammation. Bioessays 43, e2100029 (2021).
Kaczmarek, A., Vandenabeele, P. & Krysko, D. V. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223 (2013).
Gautheron, J. et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 6, 1062–1074 (2014).
Roychowdhury, S. et al. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 57, 1773–1783 (2013).
Wang, H. et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell. 54, 133–146 (2014).
Mohammed, S. et al. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell 20, e13512 (2021).
Kayagaki, N. et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591, 131–136 (2021).
Miao, E. A. et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142 (2010).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010).
Voet, S., Srinivasan, S., Lamkanfi, M. & van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 11, e10248 (2019).
Xia, X. et al. The role of pyroptosis in cancer: pro-cancer or pro-“host”? Cell Death Dis. 10, 650 (2019).
Liu, X., Belmonte, J. C. I., Zhang, W. & Liu, G. H. A beta-galactosidase kiss of death for senescent cells. Cell Res. 30, 556–557 (2020).
Ward, R. J. et al. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060 (2014).
Fang, X., Ardehali, H., Min, J. & Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 20, 7–23 (2023).
Li, C. et al. Activation of glutathione peroxidase 4 as a novel anti-inflammatory strategy. Front. Pharmacol. 9, 1120 (2018).
Kim, S. et al. Circulating levels of inflammatory cytokines and risk of colorectal adenomas. Cancer Res. 68, 323–328 (2008).
Wen, Q. et al. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 510, 278–283 (2019).
Son, G. H. et al. MicroRNA-548 regulates high mobility group box 1 expression in patients with preterm birth and chorioamnionitis. Sci. Rep. 9, 19746 (2019).
Splichal, I. et al. High mobility group box 1 and TLR4 signaling pathway in gnotobiotic piglets colonized/infected with L. amylovorus, L. mucosae, E. coli Nissle 1917 and S. Typhimurium. Int. J. Mol. Sci. 20, 6294 (2019).
Linkermann, A. et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl Acad. Sci. USA 111, 16836–16841 (2014).
Sun, Y. et al. The emerging role of ferroptosis in inflammation. Biomed. Pharmacother. 127, 110108 (2020).
Qi, J. et al. Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am. J. Pathol. 190, 68–81 (2020).
Ding, H. et al. Transferrin receptor 1 ablation in satellite cells impedes skeletal muscle regeneration through activation of ferroptosis. J. Cachexia Sarcopenia Muscle 12, 746–768 (2021).
Barnes, B. J. et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J. Exp. Med. 217, e20200652 (2020).
Yoshida, Y. et al. Enhanced formation of neutrophil extracellular traps in Kawasaki disease. Pediatr. Res. 87, 998–1004 (2020).
Khandpur, R. et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 5, 178ra140 (2013).
Wang, W., Peng, W. & Ning, X. Increased levels of neutrophil extracellular trap remnants in the serum of patients with rheumatoid arthritis. Int. J. Rheum. Dis. 21, 415–421 (2018).
Weckbach, L. T. et al. Midkine drives cardiac inflammation by promoting neutrophil trafficking and NETosis in myocarditis. J. Exp. Med 216, 350–368 (2019).
Lood, C. et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 22, 146–153 (2016).
Garcia-Romo, G. S. et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra20 (2011).
Thiam, H. R., Wong, S. L., Wagner, D. D. & Waterman, C. M. Cellular mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 36, 191–218 (2020).
Nguyen, G. T., Green, E. R. & Mecsas, J. Neutrophils to the ROScue: mechanisms of NADPH oxidase activation and bacterial resistance. Front. Cell. Infect. Microbiol. 7, 373 (2017).
Yipp, B. G. et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 18, 1386–1393 (2012).
Sabbatini, M. et al. Aging hampers neutrophil extracellular traps (NETs) efficacy. Aging Clin. Exp. Res. 34, 2345–2353 (2022).
Martinod, K. et al. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J. Exp. Med. 214, 439–458 (2017).
Banoth, B. et al. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J. Biol. Chem. 295, 18276–18283 (2020).
Christgen, S. et al. Identification of the PANoptosome: a molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 10, 237 (2020).
Malireddi, R. K. S. et al. RIPK1 distinctly regulates yersinia-induced inflammatory cell death, PANoptosis. Immunohorizons 4, 789–796 (2020).
Samir, P., Malireddi, R. K. S. & Kanneganti, T. D. The PANoptosome: a deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 10, 238 (2020).
Wang, Y. & Kanneganti, T. D. From pyroptosis, apoptosis and necroptosis to PANoptosis: a mechanistic compendium of programmed cell death pathways. Comput. Struct. Biotechnol. J. 19, 4641–4657 (2021).
Karki, R. et al. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184, 149–168.e117 (2021).
Li, T. & Chen, Z. J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299 (2018).
Mangan, M. S. J. et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 17, 688 (2018).
Roers, A., Hiller, B. & Hornung, V. Recognition of endogenous nucleic acids by the innate immune system. Immunity 44, 739–754 (2016).
Toussaint, O., Medrano, E. E. & von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 35, 927–945 (2000).
Duan, J., Duan, J., Zhang, Z. & Tong, T. Irreversible cellular senescence induced by prolonged exposure to H2O2 involves DNA-damage-and-repair genes and telomere shortening. Int. J. Biochem. Cell Biol. 37, 1407–1420 (2005).
Xu, M. et al. Hydrogen peroxide-induced senescence reduces the wound healing-promoting effects of mesenchymal stem cell-derived exosomes partially via miR-146a. Aging Dis. 12, 102–115 (2021).
Zoico, E. et al. Senolytic effects of quercetin in an in vitro model of pre-adipocytes and adipocytes induced senescence. Sci. Rep. 11, 23237 (2021).
Zhu, H. et al. Oncogene-induced senescence: from biology to therapy. Mech. Ageing Dev. 187, 111229 (2020).
Ribeiro, J. D. et al. ZRF1 controls oncogene-induced senescence through the INK4-ARF locus. Oncogene 32, 2161–2168 (2013).
Innes, A. J. et al. XPO7 is a tumor suppressor regulating p21(CIP1)-dependent senescence. Genes Dev. 35, 379–391 (2021).
Kuilman, T. et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 (2008).
Sun, T. et al. Characterization of cellular senescence in doxorubicin-induced aging mice. Exp. Gerontol. 163, 111800 (2022).
Kirsch, V. et al. In vitro characterization of doxorubicin-mediated stress-induced premature senescence in human chondrocytes. Cells 11, 1106 (2022).
Wang, T. H. et al. Palbociclib induces DNA damage and inhibits DNA repair to induce cellular senescence and apoptosis in oral squamous cell carcinoma. J. Formos. Med. Assoc. 120, 1695–1705 (2021).
Zhang, S. et al. Roflumilast attenuates doxorubicin-induced cardiotoxicity by targeting inflammation and cellular senescence in cardiomyocytes mediated by SIRT1. Drug Des. Devel. Ther. 15, 87–97 (2021).
Zhang, Y. et al. Apocynum venetum leaf extract alleviated doxorubicin-induced cardiotoxicity through the AKT/Bcl-2 signaling pathway. Phytomedicine 94, 153815 (2022).
Yoshida, A., Lee, E. K. & Diehl, J. A. Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended inhibition of CDK4/6. Cancer Res. 76, 2990–3002 (2016).
Tian, Y. T. et al. Autophagy regulates X-ray radiation-induced premature senescence through STAT3-Beclin1-p62 pathway in lung adenocarcinoma cells. Int. J. Radiat. Biol. 98, 1432–1441 (2022).
Limbad, C. et al. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 15, e0227887 (2020).
Turnquist, C. et al. Radiation-induced astrocyte senescence is rescued by Delta133p53. Neuro-oncology 21, 474–485 (2019).
Azzu, V. & Valencak, T. G. Energy metabolism and ageing in the mouse: a mini-review. Gerontology 63, 327–336 (2017).
Feng, R., He, W. & Ochi, H. A new murine oxidative stress model associated with senescence. Mech. Ageing Dev. 122, 547–559 (2001).
Azman, K. F. & Zakaria, R. D-Galactose-induced accelerated aging model: an overview. Biogerontology 20, 763–782 (2019).
Pizarro, T. T. et al. SAMP1/YitFc mouse strain: a spontaneous model of Crohn’s disease-like ileitis. Inflamm. Bowel Dis. 17, 2566–2584 (2011).
Liu, B., Liu, J. & Shi, J. S. SAMP8 mice as a model of age-related cognition decline with underlying mechanisms in Alzheimer’s disease. J. Alzheimers Dis. 75, 385–395 (2020).
Bernal, G. M. et al. Loss of Nfkb1 leads to early onset aging. Aging 6, 931–943 (2014).
Jurk, D. et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2, 4172 (2014).
Koks, S. et al. Mouse models of ageing and their relevance to disease. Mech. Ageing Dev. 160, 41–53 (2016).
Kreienkamp, R. & Gonzalo, S. Metabolic dysfunction in Hutchinson-Gilford progeria syndrome. Cells 9, 395 (2020).
Squarzoni, S. et al. Interleukin-6 neutralization ameliorates symptoms in prematurely aged mice. Aging Cell 20, e13285 (2021).
Gonzalez-Dominguez, A. et al. Inhibition of the NLRP3 inflammasome improves lifespan in animal murine model of Hutchinson-Gilford Progeria. EMBO Mol. Med. 13, e14012 (2021).
Pendas, A. M. et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 31, 94–99 (2002).
Valenzano, D. R., Aboobaker, A., Seluanov, A. & Gorbunova, V. Non-canonical aging model systems and why we need them. EMBO J. 36, 959–963 (2017).
Buffenstein, R. Negligible senescence in the longest living rodent, the naked mole-rat: insights from a successfully aging species. J. Comp. Physiol. B 178, 439–445 (2008).
Yamamura, Y., Kawamura, Y., Oka, K. & Miura, K. Carcinogenesis resistance in the longest-lived rodent, the naked mole-rat. Cancer Sci. 113, 4030–4036 (2022).
Tan, T. C. et al. Telomere maintenance and telomerase activity are differentially regulated in asexual and sexual worms. Proc. Natl Acad. Sci. USA 109, 4209–4214 (2012).
Barghouth, P. G., Thiruvalluvan, M., LeGro, M. & Oviedo, N. J. DNA damage and tissue repair: What we can learn from planaria. Semin. Cell Dev. Biol. 87, 145–159 (2019).
Yun, M. H., Davaapil, H. & Brockes, J. P. Recurrent turnover of senescent cells during regeneration of a complex structure. eLife 4, e05505 (2015).
Poetsch, A. R., Boulton, S. J. & Luscombe, N. M. Genomic landscape of oxidative DNA damage and repair reveals regioselective protection from mutagenesis. Genome Biol. 19, 215 (2018).
Willmore, W. G. & Storey, K. B. Glutathione systems and anoxia tolerance in turtles. Am. J. Physiol. 273, R219–R225 (1997).
Plot, V., Criscuolo, F., Zahn, S. & Georges, J. Y. Telomeres, age and reproduction in a long-lived reptile. PLoS ONE 7, e40855 (2012).
Hashimoto, K. et al. Single-cell transcriptomics reveals expansion of cytotoxic CD4 T cells in supercentenarians. Proc. Natl Acad. Sci. USA 116, 24242–24251 (2019).
Liao, J. et al. Uncovering an organ’s molecular architecture at single-cell resolution by spatially resolved transcriptomics. Trends Biotechnol. 39, 43–58 (2021).
Sharma, R., Diwan, B., Sharma, A. & Witkowski, J. M. Emerging cellular senescence-centric understanding of immunological aging and its potential modulation through dietary bioactive components. Biogerontology 23, 699–729 (2022).
Dasgupta, M., Sharkey, J. R. & Wu, G. Inadequate intakes of indispensable amino acids among homebound older adults. J. Nutr. Elder. 24, 85–99 (2005).
Li, Y. et al. Impact of healthy lifestyle factors on life expectancies in the US population. Circulation 138, 345–355 (2018).
O’Hara, R. et al. Serotonin transporter polymorphism, memory and hippocampal volume in the elderly: association and interaction with cortisol. Mol. Psychiatry 12, 544–555 (2007).
Del Bo, C. et al. A polyphenol-rich dietary pattern improves intestinal permeability, evaluated as serum zonulin levels, in older subjects: The MaPLE randomised controlled trial. Clin. Nutr. 40, 3006–3018 (2021).
Jeong, J. J. et al. Orally administrated Lactobacillus pentosus var. plantarum C29 ameliorates age-dependent colitis by inhibiting the nuclear factor-kappa B signaling pathway via the regulation of lipopolysaccharide production by gut microbiota. PLoS ONE 10, e0116533 (2015).
Kiecolt-Glaser, J. K. et al. Omega-3 supplementation lowers inflammation in healthy middle-aged and older adults: a randomized controlled trial. Brain Behav. Immun. 26, 988–995 (2012).
De la Fuente, M. et al. Vitamin C and vitamin C plus E improve the immune function in the elderly. Exp. Gerontol. 142, 111118 (2020).
Wong, C. P., Magnusson, K. R., Sharpton, T. J. & Ho, E. Effects of zinc status on age-related T cell dysfunction and chronic inflammation. Biometals 34, 291–301 (2021).
Uciechowski, P. et al. TH1 and TH2 cell polarization increases with aging and is modulated by zinc supplementation. Exp. Gerontol. 43, 493–498 (2008).
Zhang, X. et al. Exercise counters the age-related accumulation of senescent cells. Exerc. Sport Sci. Rev. 50, 213–221 (2022).
Radak, Z. et al. Exercise training decreases DNA damage and increases DNA repair and resistance against oxidative stress of proteins in aged rat skeletal muscle. Pflug. Arch. 445, 273–278 (2002).
Gomez-Cabrera, M. C., Domenech, E. & Vina, J. Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radic. Biol. Med. 44, 126–131 (2008).
Werner, C. et al. Physical exercise prevents cellular senescence in circulating leukocytes and in the vessel wall. Circulation 120, 2438–2447 (2009).
Justice, J. N. et al. Cellular senescence biomarker p16INK4a+ cell burden in thigh adipose is associated with poor physical function in older women. J. Gerontol. A Biol. Sci. Med. Sci. 73, 939–945 (2018).
Moore, R. C., Straus, E. & Campbell, L. M. in Handbook of Mental Health and Aging (eds Hantke, N., Etkin, A. & O’Hara, R.) 37–58 (Academic Press, 2020).
Casaletto, K. B. et al. Perceived stress is associated with accelerated monocyte/macrophage aging trajectories in clinically normal adults. Am. J. Geriatr. Psychiatry 26, 952–963 (2018).
Moiseeva, O. et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 12, 489–498 (2013).
Fang, J. et al. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 17, e12765 (2018).
Ma, T. et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature 603, 159–165 (2022).
Moiseeva, V. et al. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature 613, 169–178 (2023).
Zhu, Y. et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Islam, M. T. et al. Senolytic drugs, dasatinib and quercetin, attenuate adipose tissue inflammation, and ameliorate metabolic function in old age. Aging Cell 22, e13767 (2023).
Saccon, T. D. et al. Senolytic combination of dasatinib and quercetin alleviates intestinal senescence and inflammation and modulates the gut microbiome in aged mice. J. Gerontol. A Biol. Sci. Med. Sci. 76, 1895–1905 (2021).
Novais, E. J. et al. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 12, 5213 (2021).
Justice, J. N. et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40, 554–563 (2019).
Hickson, L. J. et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446–456 (2019).
Nogueira-Recalde, U. et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 45, 588–605 (2019).
Grosse, L. et al. Defined p16(High) senescent cell types are indispensable for mouse healthspan. Cell Metab. 32, 87–99.e86 (2020).
Song, P., An, J. & Zou, M. H. Immune clearance of senescent cells to combat ageing and chronic diseases. Cells 9, 671 (2020).
Amor, C. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020).
Aghajanian, H. et al. Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433 (2019).
Baker, D. J. et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Bai, Z. et al. Combining adoptive NK cell infusion with a dopamine-releasing peptide reduces senescent cells in aged mice. Cell Death Dis. 13, 305 (2022).
Tang, X. et al. Characterization of age-related immune features after autologous NK cell infusion: Protocol for an open-label and randomized controlled trial. Front. Immunol. 13, 940577 (2022).
Dorronsoro, A. et al. Mesenchymal stem cell-derived extracellular vesicles reduce senescence and extend health span in mouse models of aging. Aging Cell 20, e13337 (2021).
Kretlow, J. D. et al. Donor age and cell passage affects differentiation potential of murine bone marrow-derived stem cells. BMC Cell Biol. 9, 60 (2008).
Paul, C. & Robaire, B. Ageing of the male germ line. Nat. Rev. Urol. 10, 227–234 (2013).
Shen, J. et al. Transplantation of mesenchymal stem cells from young donors delays aging in mice. Sci. Rep. 1, 67 (2011).
Tompkins, B. A. et al. Allogeneic mesenchymal stem cells ameliorate aging frailty: a Phase II randomized, double-blind, placebo-controlled clinical trial. J. Gerontol. A Biol. Sci. Med. Sci. 72, 1513–1522 (2017).
Prasad, K. et al. Intravenous autologous bone marrow mononuclear stem cell therapy for ischemic stroke: a multicentric, randomized trial. Stroke 45, 3618–3624 (2014).
Tsang, K. S. et al. Phase I/II randomized controlled trial of autologous bone marrow-derived mesenchymal stem cell therapy for chronic stroke. World J. Stem Cells 9, 133–143 (2017).
Vu, Q. et al. Meta-analysis of preclinical studies of mesenchymal stromal cells for ischemic stroke. Neurology 82, 1277–1286 (2014).
Berrio Sanchez, J., Cucarian Hurtado, J., Barcos Nunes, R. & de Oliveira, A. A. Mesenchymal stem cell transplantation and aerobic exercise for Parkinson’s disease: therapeutic assets beyond the motor domain. Rev. Neurosci. 30, 165–178 (2019).
Staff, N. P., Jones, D. T. & Singer, W. Mesenchymal stromal cell therapies for neurodegenerative diseases. Mayo Clin. Proc. 94, 892–905 (2019).
Mohammed, I., Hollenberg, M. D., Ding, H. & Triggle, C. R. A critical review of the evidence that metformin is a putative anti-aging drug that enhances healthspan and extends lifespan. Front. Endocrinol. 12, 718942 (2021).
Fahy, G. M. et al. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell 18, e13028 (2019).
Iske, J. et al. Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nat. Commun. 11, 4289 (2020).
Thompson, W. W. et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289, 179–186 (2003).
Bulut, O., Kilic, G., Dominguez-Andres, J. & Netea, M. G. Overcoming immune dysfunction in the elderly: trained immunity as a novel approach. Int. Immunol. 32, 741–753 (2020).
Medetgul-Ernar, K. & Davis, M. M. Standing on the shoulders of mice. Immunity 55, 1343–1353 (2022).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019).
Rutledge, J., Oh, H. & Wyss-Coray, T. Measuring biological age using omics data. Nat. Rev. Genet. 23, 715–727 (2022).
Li, J. et al. Deep learning of cross-species single-cell landscapes identifies conserved regulatory programs underlying cell types. Nat. Genet. 54, 1711–1720 (2022).
Franceschi, C. et al. Immunobiography and the heterogeneity of immune responses in the elderly: a focus on inflammaging and trained immunity. Front. Immunol. 8, 982 (2017).
Schmid, N. et al. Insights into replicative senescence of human testicular peritubular cells. Sci. Rep. 9, 15052 (2019).
Lawless, C. et al. A stochastic step model of replicative senescence explains ROS production rate in ageing cell populations. PLoS One 7, e32117 (2012).
Dong, C. M. et al. A stress-induced cellular aging model with postnatal neural stem cells. Cell Death Dis. 5, e1116 (2014).
Li, N. et al. Exosomes derived from M2 microglia cells attenuates neuronal impairment and mitochondrial dysfunction in Alzheimer’s disease through the PINK1/Parkin pathway. Front. Cell. Neurosci. 16, 874102 (2022).
Wang, J. et al. The protective effects of Agomelatine against Abeta1-42 oligomers-induced cellular senescence mediated by SIRT6 and Agomelatine’s potential in AD treatment. Hum. Cell. 34, 1734–1743 (2021).
Callies, S. et al. A population pharmacokinetic model for doxorubicin and doxorubicinol in the presence of a novel MDR modulator, zosuquidar trihydrochloride (LY335979). Cancer Chemother. Pharm. 51, 107–118 (2003).
Salinger-Martinovic, S. et al. Impact of ellagic acid application on doxorubicin-induced cardiovascular toxicity model. Can. J. Physiol. Pharm. 99, 185–191 (2021).
Marouille, A. L. et al. Pharmacokinetic/pharmacodynamic model of neutropenia in real-life palbociclib-treated patients. Pharmaceutics 13, 1708 (2021).
Pandey, P. K. et al. Model-based X-ray-induced acoustic computed tomography. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 68, 3560–3569 (2021).
Close, D. M. & Bernhard, W. A. Comprehensive model for X-ray-induced damage in protein crystallography. J. Synchrotron Radiat. 26, 945–957 (2019).
Martic, I., Wedel, S., Jansen-Durr, P. & Cavinato, M. A new model to investigate UVB-induced cellular senescence and pigmentation in melanocytes. Mech. Ageing Dev. 190, 111322 (2020).
Shi, C., Li, Q. & Zhang, X. Platycodin D protects human fibroblast cells from premature senescence induced by H2O2 through improving mitochondrial biogenesis. Pharmacology 105, 598–608 (2020).
Zhou, Y. et al. Mussel oligopeptides protect human fibroblasts from hydrogen peroxide (H2O2)-induced premature senescence. Arch. Gerontol. Geriatr. 58, 293–299 (2014).
Yao, Y. et al. Osthole delays tert-butyl hydroperoxide-induced premature senescence in neural stem cells. Cell. Reprogram. 20, 268–274 (2018).
Ma, W. et al. Psoralen plus UVA (PUVA) induced premature senescence as a model for stress-induced premature senescence. Exp. Gerontol. 37, 1197–1201 (2002).
Debacq-Chainiaux, F. et al. Screening of senescence-associated genes with specific DNA array reveals the role of IGFBP-3 in premature senescence of human diploid fibroblasts. Free Radic. Biol. Med. 44, 1817–1832 (2008).
Giusto, K., Wanczyk, H., Jensen, T. & Finck, C. Hyperoxia-induced bronchopulmonary dysplasia: better models for better therapies. Dis. Model Mech. 14, dmm047753 (2021).
Tedeschi, T., Lee, K., Zhu, W. & Fawzi, A. A. Limited hyperoxia-induced proliferative retinopathy: a model of persistent retinal vascular dysfunction, preretinal fibrosis and hyaloidal vascular reprogramming for retinal rescue. PLoS ONE 17, e0267576 (2022).
Fukasawa, K. & Vande Woude, G. F. Mos overexpression in Swiss 3T3 cells induces meiotic-like alterations of the mitotic spindle. Proc. Natl Acad. Sci. USA 92, 3430–3434 (1995).
Tsai, J. et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl Acad. Sci. USA 105, 3041–3046 (2008).
Prahallad, A. et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103 (2012).
Drosten, M. et al. H-Ras and K-Ras oncoproteins induce different tumor spectra when driven by the same regulatory sequences. Cancer Res. 77, 707–718 (2017).
Viosca, J., Schuhmacher, A. J., Guerra, C. & Barco, A. Germline expression of H-Ras(G12V) causes neurological deficits associated to Costello syndrome. Genes Brain Behav. 8, 60–71 (2009).
Venturelli, S. et al. Differential induction of apoptosis and senescence by the DNA methyltransferase inhibitors 5-azacytidine and 5-aza-2’-deoxycytidine in solid tumor cells. Mol. Cancer Ther. 12, 2226–2236 (2013).
Vogt, M. et al. Independent induction of senescence by p16INK4a and p21CIP1 in spontaneously immortalized human fibroblasts. Cell Growth Differ. 9, 139–146 (1998).
Zhou, J. M. et al. Senescence and telomere shortening induced by novel potent G-quadruplex interactive agents, quindoline derivatives, in human cancer cell lines. Oncogene 25, 503–511 (2006).
Chen, X. et al. Cyclin E overexpression sensitizes triple-negative breast cancer to Wee1 kinase inhibition. Clin. Cancer Res. 24, 6594–6610 (2018).
Scaltriti, M. et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc. Natl Acad. Sci. USA 108, 3761–3766 (2011).
Innes, A. J. & Gil, J. IMR90 ER:RAS: a cell model of oncogene-induced senescence. Methods Mol. Biol. 1896, 83–92 (2019).
Mertens, J. et al. Aging in a dish: iPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu. Rev. Genet. 52, 271–293 (2018).
Chang, C. Y. et al. Induced pluripotent stem cell (iPSC)-based neurodegenerative disease models for phenotype recapitulation and drug screening. Molecules 25, 2000 (2020).
Mertens, J. et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17, 705–718 (2015).
Sunderland, P. et al. ATM-deficient neural precursors develop senescence phenotype with disturbances in autophagy. Mech. Ageing Dev. 190, 111296 (2020).
Silva, M. J., Brodt, M. D. & Uthgenannt, B. A. Morphological and mechanical properties of caudal vertebrae in the SAMP6 mouse model of senile osteoporosis. Bone 35, 425–431 (2004).
Chen, H., Zhou, X., Emura, S. & Shoumura, S. Site-specific bone loss in senescence-accelerated mouse (SAMP6): a murine model for senile osteoporosis. Exp. Gerontol. 44, 792–798 (2009).
Chen, H. & Kubo, K. Y. Segmental variations in trabecular bone density and microstructure of the spine in senescence-accelerated mouse (SAMP6): a murine model for senile osteoporosis. Exp. Gerontol. 47, 317–322 (2012).
Zheng, Q., Schaefer, A. M. & Nonet, M. L. Regulation of C. elegans presynaptic differentiation and neurite branching via a novel signaling pathway initiated by SAM-10. Development 138, 87–96 (2011).
de Almeida Rezende, M. S. et al. D-(+)-Galactose-induced aging: a novel experimental model of erectile dysfunction. PLoS ONE 16, e0249487 (2021).
Haider, S. et al. Naringenin protects AlCl3/D-galactose induced neurotoxicity in rat model of AD via attenuation of acetylcholinesterase levels and inhibition of oxidative stress. PLoS ONE 15, e0227631 (2020).
Bagheri, S. M. & Dashti, R. M. Influence of asafoetida on prevention and treatment of memory impairment induced by d-galactose and NaNO2 in mice. Am. J. Alzheimers Dis. Other Demen. 30, 607–612 (2015).
Peng, X. M. et al. The mechanism of memory enhancement of acteoside (verbascoside) in the senescent mouse model induced by a combination of D-gal and AlCl3. Phytother. Res. 29, 1137–1144 (2015).
Chan, L. S. A., Gu, L. C. & Wells, R. A. The effects of secondary iron overload and iron chelation on a radiation-induced acute myeloid leukemia mouse model. BMC Cancer 21, 509 (2021).
Brennan, T. A. et al. Mouse models of telomere dysfunction phenocopy skeletal changes found in human age-related osteoporosis. Dis. Model Mech. 7, 583–592 (2014).
Hale, C. M. et al. Dysfunctional connections between the nucleus and the actin and microtubule networks in laminopathic models. Biophys. J. 95, 5462–5475 (2008).
Yang, S. H. et al. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Invest. 116, 2115–2121 (2006).
Macias, A. et al. Paclitaxel mitigates structural alterations and cardiac conduction system defects in a mouse model of Hutchinson-Gilford progeria syndrome. Cardiovasc. Res. 118, 503–516 (2022).
Butala, P. et al. Zmpste24-/- mouse model for senescent wound healing research. Plast. Reconstr. Surg. 130, 788e–798e (2012).
Corujo-Ramirez, A. M. et al. Genetic inhibition of sFRP3 prevents glial reactivity in a mouse model of accelerated aging. Int. Neurourol. J. 24, 72–78 (2020).
van de Wal, M. A. E. et al. Ndufs4 knockout mouse models of Leigh syndrome: pathophysiology and intervention. Brain 145, 45–63 (2022).
Schermer, B. et al. Transcriptional profiling reveals progeroid Ercc1(-/Delta) mice as a model system for glomerular aging. BMC Genom. 14, 559 (2013).
Wong, A., Kieu, T. & Robbins, P. D. The Ercc1(-/Delta) mouse model of accelerated senescence and aging for identification and testing of novel senotherapeutic interventions. Aging 12, 24481–24483 (2020).
Lim, C. K. W. et al. Treatment of a mouse model of ALS by in vivo base editing. Mol. Ther. 28, 1177–1189 (2020).
Nabeshima, Y.-I. Klotho deficient mouse: an in vivo model for human aging. Drug Discov. Today.: Dis. Models 1, 223–227 (2004).
Yamashita, K., Yotsuyanagi, T., Yamauchi, M. & Young, D. M. Klotho mice: a novel wound model of aged skin. Plast. Reconstr. Surg. Glob. Open 2, e101 (2014).
Park, J. Y. et al. Homeostatic imbalance between apoptosis and cell renewal in the liver of premature aging Xpd mice. PLoS ONE 3, e2346 (2008).
Verma, M. et al. iPSC-derived neurons from patients with POLG mutations exhibit decreased mitochondrial content and dendrite simplification. Am. J. Pathol. 193, 201–212 (2023).
Woodbridge, P. et al. POLG mutations in Australian patients with mitochondrial disease. Intern. Med. J. 43, 150–156 (2013).
Smith, A. L. et al. Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis. Nat. Cancer 1, 976–989 (2020).
Goytisolo, F. A. & Blasco, M. A. Many ways to telomere dysfunction: in vivo studies using mouse models. Oncogene 21, 584–591 (2002).
Nugent, C. I. & Lundblad, V. The telomerase reverse transcriptase: components and regulation. Genes Dev. 12, 1073–1085 (1998).
Hoffman, J. L. et al. Alcohol drinking exacerbates neural and behavioral pathology in the 3xTg-AD mouse model of Alzheimer’s disease. Int. Rev. Neurobiol. 148, 169–230 (2019).
Stover, K. R., Campbell, M. A., Van Winssen, C. M. & Brown, R. E. Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer’s disease. Behav. Brain Res. 289, 29–38 (2015).
Wolf, A. et al. A comprehensive behavioral test battery to assess learning and memory in 129S6/Tg2576 mice. PLoS ONE 11, e0147733 (2016).
Stewart, S., Cacucci, F. & Lever, C. Which memory task for my mouse? A systematic review of spatial memory performance in the Tg2576 Alzheimer’s mouse model. J. Alzheimers Dis. 26, 105–126 (2011).
Oka, K. et al. The naked mole-rat as a model for healthy aging. Annu. Rev. Anim. Biosci. 11, 207–226 (2023).
Savina, A. et al. Single-cell transcriptomics reveals age-resistant maintenance of cell identities, stem cell compartments and differentiation trajectories in long-lived naked mole-rats skin. Aging 14, 3728–3756 (2022).
Sharp, Z. D. & Bartke, A. Evidence for down-regulation of phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR)-dependent translation regulatory signaling pathways in Ames dwarf mice. J. Gerontol. A Biol. Sci. Med. Sci. 60, 293–300 (2005).
Flurkey, K., Papaconstantinou, J., Miller, R. A. & Harrison, D. E. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc. Natl Acad. Sci. USA 98, 6736–6741 (2001).
Tabula Muris, C. et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018).
Schaum, N. et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583, 596–602 (2020).
Ma, S. et al. Caloric restriction reprograms the single-cell transcriptional landscape of rattus norvegicus aging. Cell 180, 984–1001.e1022 (2020).
Wang, S. et al. Single-cell transcriptomic atlas of primate ovarian aging. Cell 180, 585–600.e519 (2020).
Ma, S. et al. Single-cell transcriptomic atlas of primate cardiopulmonary aging. Cell Res. 31, 415–432 (2021).
Zhang, W. et al. A single-cell transcriptomic landscape of primate arterial aging. Nat. Commun. 11, 2202 (2020).
Wang, S. et al. Deciphering primate retinal aging at single-cell resolution. Protein Cell 12, 889–898 (2021).
Zhu, Y. et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435 (2016).
Roos, C. M. et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15, 973–977 (2016).
Soukas, A. A., Hao, H. & Wu, L. Metformin as anti-aging therapy: is it for everyone? Trends Endocrinol. Metab. 30, 745–755 (2019).
Cameron, A. R. et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ. Res. 119, 652–665 (2016).
Knowler, W. C. et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 346, 393–403 (2002).
Wu, H. et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 23, 850–858 (2017).
Zhao, P. et al. Anti-aging pharmacology in cutaneous wound healing: effects of metformin, resveratrol, and rapamycin by local application. Aging Cell 16, 1083–1093 (2017).
Bissler, J. J. et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 381, 817–824 (2013).
McCormack, F. X. et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N. Engl. J. Med. 364, 1595–1606 (2011).
Tang, Y. et al. Alterations in polyamine metabolism in patients with lymphangioleiomyomatosis and tuberous sclerosis complex 2-deficient cells. Chest 156, 1137–1148 (2019).
Park, S., Kim, B. K. & Park, S. K. Effects of fisetin, a plant-derived flavonoid, on response to oxidative stress, aging, and age-related diseases in Caenorhabditis elegans. Pharmaceuticals 15, 1528 (2022).
Yousefzadeh, M. J. et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28 (2018).
Zia, A., Farkhondeh, T., Pourbagher-Shahri, A. M. & Samarghandian, S. The role of curcumin in aging and senescence: Molecular mechanisms. Biomed. Pharmacother. 134, 111119 (2021).
Yan, Z. et al. Curcumin exerts a protective effect against premature ovarian failure in mice. J. Mol. Endocrinol. 60, 261–271 (2018).
Liu, J. et al. Piperlongumine restores the balance of autophagy and apoptosis by increasing BCL2 phosphorylation in rotenone-induced Parkinson disease models. Autophagy 14, 845–861 (2018).
Shi, W. et al. Piperlongumine attenuates high calcium/phosphate-induced arterial calcification by preserving P53/PTEN signaling. Front. Cardiovasc. Med 7, 625215 (2020).
Liu, X. et al. Senolytic activity of piperlongumine analogues: Synthesis and biological evaluation. Bioorg. Med. Chem. 26, 3925–3938 (2018).
Go, J. et al. Piperlongumine decreases cognitive impairment and improves hippocampal function in aged mice. Int. J. Mol. Med. 42, 1875–1884 (2018).
Berk, M. et al. Effect of aspirin vs placebo on the prevention of depression in older people: a randomized clinical trial. JAMA Psychiatry 77, 1012–1020 (2020).
McNeil, J. J. et al. Effect of aspirin on all-cause mortality in the healthy elderly. N. Engl. J. Med. 379, 1519–1528 (2018).
Gaziano, J. M. et al. Use of aspirin to reduce risk of initial vascular events in patients at moderate risk of cardiovascular disease (ARRIVE): a randomised, double-blind, placebo-controlled trial. Lancet 392, 1036–1046 (2018).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81900176, 82130003, 82222003, 92268117, 82161138028), the National Key Research and Development Program of China (2022YFA1103500), Zhejiang Provincial Key Research and Development Program (2021C03010), the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (2020R01006). We thank Francis Kaming Chan, Yufei Li, Yufei Wang, and Yifei Shang for help with data collection and manuscript revision. Figures 2 and 3 were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
X.L., C.L., W.Z., Y.W., P.Q., and H.H. wrote the manuscript. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Li, C., Zhang, W. et al. Inflammation and aging: signaling pathways and intervention therapies. Sig Transduct Target Ther 8, 239 (2023). https://doi.org/10.1038/s41392-023-01502-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01502-8
This article is cited by
-
Bazi Bushen ameliorates age-related energy metabolism dysregulation by targeting the IL-17/TNF inflammatory pathway associated with SASP
Chinese Medicine (2024)
-
Insights into vaccines for elderly individuals: from the impacts of immunosenescence to delivery strategies
npj Vaccines (2024)
-
Decoding regulators of gut aging in nonhuman primates
Nature Aging (2024)
-
Intestinal stem cells: guardians of homeostasis in health and aging amid environmental challenges
Experimental & Molecular Medicine (2024)
-
Kdm6a-CNN1 axis orchestrates epigenetic control of trauma-induced spinal cord microvascular endothelial cell senescence to balance neuroinflammation for improved neurological repair
Bone Research (2024)
