
Abstract
The ever-increasing prevalence of noncommunicable diseases (NCDs) represents a major public health burden worldwide. The most common form of NCD is metabolic diseases, which affect people of all ages and usually manifest their pathobiology through life-threatening cardiovascular complications. A comprehensive understanding of the pathobiology of metabolic diseases will generate novel targets for improved therapies across the common metabolic spectrum. Protein posttranslational modification (PTM) is an important term that refers to biochemical modification of specific amino acid residues in target proteins, which immensely increases the functional diversity of the proteome. The range of PTMs includes phosphorylation, acetylation, methylation, ubiquitination, SUMOylation, neddylation, glycosylation, palmitoylation, myristoylation, prenylation, cholesterylation, glutathionylation, S-nitrosylation, sulfhydration, citrullination, ADP ribosylation, and several novel PTMs. Here, we offer a comprehensive review of PTMs and their roles in common metabolic diseases and pathological consequences, including diabetes, obesity, fatty liver diseases, hyperlipidemia, and atherosclerosis. Building upon this framework, we afford a through description of proteins and pathways involved in metabolic diseases by focusing on PTM-based protein modifications, showcase the pharmaceutical intervention of PTMs in preclinical studies and clinical trials, and offer future perspectives. Fundamental research defining the mechanisms whereby PTMs of proteins regulate metabolic diseases will open new avenues for therapeutic intervention.
Similar content being viewed by others
Introduction
Rapid economic development, ageing of the population, and evolved lifestyles have the outcome of creating a dramatic worldwide growth in chronic metabolic disorders. These preventable lifestyle-related diseases include hyperglycemia, hyperlipidemia, hypertension, obesity and its consequence, nonalcoholic fatty liver disease (NAFLD).
The 10th International Diabetes Federation (IDF) indicates a global diabetes prevalence of nearly 10% (537 million), and the cases are predicted to reach 783 million by 2045; 90 percent of these cases are type 2 diabetes.1 The global prevalence of NAFLD is 38%, growing to nonalcoholic steatohepatitis (NASH) and hepatocellular carcinoma.2 As of 2015, nearly 712 million individuals (604 million adults, 108 million children) were obese worldwide, and the prevalence of obesity more than doubled from 1.1% in 1980 to 3.85% in 2015. Childhood obesity had an even higher rate of increase.3 The seminal issue is that metabolic disorders are closely related to the consequences of cardiovascular diseases (CVDs) and all-cause mortality.4 Over the past decades, CVD cases have increased from 271 million in 1990, reaching 523 million in 2019. The total cardiovascular deaths rose to 18.6 million in 2019, up from 12.1 million in 1990.5 CVD has become the predominant contributor to global mortality and disability.5,6 Alarmingly, the occurrence and hospitalization for metabolic disorders and CVD in young adults are increasing.5,6
With the rising incidence of metabolic diseases and CVD, attention has been focused on the global cardiometabolic disease epidemic, with negative impacts on lifespan and socioeconomic burden. In America, 90% of the annual healthcare expenditures (3.7 trillion dollars) are directed to the population with chronic diseases and mental health issues.7 Metabolic diseases represent both a huge social burden but also provide an opportunity for high cost-effectiveness for efficacious interventions. Since most metabolic diseases are preventable and treatable, their prevention and control will yield great societal and economic benefits.
The Human Genome Project has revealed the human genome includes about 20,000 to 25,000 genes, whereas, due to alternative splicing, metabolism and PTMs, the human proteome includes over 1 million proteins. Posttranslational modifications (PTMs) are central to the complexity and diverse functional roles of the proteome. PTMs are the biochemical modifications of proteins after protein biosynthesis. PTMs dynamically regulate protein activity, location and molecular interactions by modifying or introducing functional groups such as phosphoryl, methyl, acetyl and glycosyl groups. PTMs generally occur in proteins serving as important structures or exhibiting crucial functions, such as histones, membrane proteins and secretory proteins. PTMs are usually reversible, and the irreversible alterations arise from proteolytic modifications. PTMs take place in various cellular compartments, such as nucleus, cytosol, endoplasmic reticulum (ER) and the Golgi complex.8
Protein phosphorylation was the first discovered PTM and this phenomena was first identified in 1906.9 Another 50 years passed before the specific observation of protein kinase activity in 1954.10 In the 1960s, the general importance of PTMs was appreciated, as the discovery of histone acetylation governing the transcription of genes was put forward in 196411 and protein phosphorylation participating in cell metabolism was reported in 1969.12 The biological relevance of the newborn field of PTM sparked much excitement across scientific communities. However, the investigation of PTMs had decades of stagnation because of poor PTM detection technology and a lack description of functional activity and consequences. Fortunately, the enhanced accessibility of genomic sequencing data and the rapid development of detection approaches such as mass spectrometry (MS)-based proteomics, radioactive isotope labeling, peptide/protein array, immunoprecipitation and proximity ligation assay (PLA) have ended the long struggle and led to a golden age of PTM research.13
To date, owing to advanced detection technologies over 600 types of PTMs have been identified.13 These PTMs affect enzyme function and assembly, receptor activation, protein interactions, cell interactions, protein solubility, molecular trafficking, protein stability, protein folding, protein turnover, protein localization, cell metabolism, and signaling pathways.14 The most general PTMs include protein phosphorylation, methylation, acetylation, SUMOylation, neddylation, ubiquitination, glycosylation, palmitoylation, glutathionylation, S-nitrosylation, and ADP ribosylation. Aberrant PTMs are implicated in diverse human diseases, including metabolic disorders and CVDs.
Due to the aberrant regulatory role of PTMs in diseases, multiple important therapeutic agents regulating PTMs, such as kinase agonists/inhibitors, histone deacetylase inhibitors, and histone methyltransferase inhibitors, are discovered to treat a variety of illnesses. The c-Abl tyrosine kinase inhibitor, imatinib (Glevec®), which received Food and Drug Administration (FDA) approval in 2001, was the first “smart” kinase inhibitor developed with a specific target identified to treat chronic myeloid leukemia.15 This remarkable progress arouses awareness of the significance of protein phosphorylation, allowing protein kinases to serve as the second most prominent drug target category, following G-protein-coupled receptors.16 Drugs targeting PTMs have thus provided potential therapeutic strategies in the study of diverse diseases, including metabolic disorders.17,18,19
In summary, as shown in Fig. 1, an increasing number of human subjects acquire metabolic diseases which occurred in the liver (fatty acid liver), pancreas (diabetes), adipose tissue (obesity), blood fat (hyperlipidemia) and heart (atherosclerosis). Numerous proteins and PTMs are implicated in the progression of these metabolic diseases. To put into practice the preventive and treatment options for metabolic diseases, both medical and lifestyle, a thorough understanding of PTMs and metabolic disorders is needed. Here, we systematically review and profile the most recent advances in the roles of PTMs in metabolic diseases.

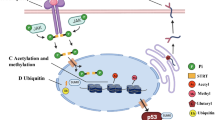
PTMs in metabolic diseases. An increasing number of people are suffering from metabolic diseases, such as fatty liver, diabetes, obesity, hyperlipidemia and atherosclerosis. The liver, pancreas, adipose tissue, blood vessels, and heart are the main affected organs. Numerous proteins and PTMs (such as phosphorylation, acetylation, methylation, ubiquitination, etc.) are involved in the normal biology of these organs and the whole body. Abnormal PTMs thus are involved in the progression of these metabolic diseases and can be therapeutically targeted. The figure is generated with BioRender (https://biorender.com)
Principles and mechanisms of protein posttranslational modifications (PTMs)
Transcription, translation, and PTMs provide a multilayer dynamic network for biochemical and physiological diversity and complexity. Dynamic reversibility enables PTMs to regulate cellular processes and signal transduction most efficiently. One protein can be modified by various PTMs at a time or modified by one specific PTM at different stages. Figure 2 illustrates the historical milestones of PTMs research. It can be seen that since the discovery of first type of identified PTM (phosphorylation in 19069) it takes more than one hundred years to the latest discovery of lactylation in 2019.20 Advances in detection technology will provide researchers with more opportunities to explore the biological functions of novel PTMs and accelerate the research of the roles of PTMs in human diseases, which could hold the promise of novel therapies. Below, we will describe the principles and mechanisms of the most common PTMs.

The historical milestones and schematic illustration of different PTMs. Protein phosphorylation was the first discovered PTM and this phenomenon was first identified in 1906. Since then, other common PTMs such as disulfide formation, palmitoylation, citrullination, methylation, ADP ribosylation, etc were being discovered. Recently, a novel PTM, lactylation, was reported in 2019. Advanced availability of genomic sequence information and the rapid development of detection approaches will lead to a golden age of PTM research and afford abundant novel therapeutic targets for human diseases. The figure is generated with BioRender (https://biorender.com). ADPr adenosine diphosphate (ADP)-ribose, Ac the acetyl group, β-OHB β-hydroxybutyrate, CHO cholesterol, GSH glutathione, H2S hydrogen sulfide, Me the methyl group, NEDD8 neuronal precursor cell-expressed developmentally downregulated protein 8, NO nitric oxide, P the phosphate group, SOH sulfenic acid, SSG glutathione disulfide, SSH the persulfide group, SSR disulfide formation, Ub ubiquitin, PTMs post translational modifications
Phosphorylation
Protein phosphorylation was first identified in 1906 in the egg-yolk protein, phosvitin, by Phoebus A. Levene.9 The enzymatic process of protein phosphorylation was first explained half a century later in 1954 when protein kinase activity was observed for the first time.10 Phosphorylation is an enzymatic reaction of protein kinases catalyzing the linkage between amino acid residues of the protein and phosphate groups in adenosine triphosphate (ATP). The reversibility arises from the actions of protein phosphatases which catalyze dephosphorylation by removing phosphate groups.
Protein kinases and phosphatases dynamically regulate the state of protein phosphorylation. According to the target phosphorylated amino acid residues, protein kinases can be characterized into three groups: serine protein kinases, threonine protein kinases, and tyrosine protein kinases. Protein phosphatases can also be clustered according to substrate specificity. Phosphorylation sites can be serine, threonine, tyrosine, cysteine, arginine, proline, aspartic acid, and histidine residues, but the most common target sites are serine (Ser), threonine (Thr), and tyrosine (Tyr). Protein phosphorylation receives the most attention and is the most intensively studied PTM. Phosphorylation usually occurs in the cytosol or nucleus and is considered a fundamental, universal and essential mechanism regulating protein activity and functions. Phosphorylation can rapidly control the function of proteins through two mechanisms.21 One is by allostery to activate enzyme activity (typically Ser/Thr, Tyr residues), as in the example of glycogen synthase kinase-3 via serine/threonine kinase. Another is by combining interaction domains to activate signal transduction (usually Tyr residues), as the instance of Src homology 2 domain-mediated autoinhibited conformation of the tyrosine kinase domain.
There are many research approaches for studying protein phosphorylation, such as kinase activity assays, phosphatase treatment, in vitro phosphorylation assays using γ-32P-ATP autoradiography, phosphor-specific antibody development, phospho-tag SDS-PAGE, ELISA (cell-based and enzyme-linked), immunohistochemistry (IHC), immunocytochemistry (ICC), flow cytometry, MS, and phosphoproteomics.22
Nearly 30% of human genome-coded proteins contain covalently bound phosphate. Reversible protein phosphorylation modulates almost every aspect of cellular processes relevant to replication, transcription, apoptosis, the immune response, environmental responsiveness, and cell metabolism.14 Abnormal phosphorylation has been recognized as the cause or consequence of diverse diseases, including tumors, CVDs, and metabolic disorders. Thus, drugs targeting protein kinases provide attractive therapeutic strategies against several diseases.17,18,19
Acetylation
In 1964, protein acetylation was first identified on histone proteins isolated from calf thymus nuclei in vitro by V.G. Allfrey.11 In the 1990s, the first histone acetyltransferases (HATs) and histone deacetylases (HDACs) in mammals were discovered, which were recognized as transcriptional regulatory factors owing to the observation of p53 acetylation.23 Acetyltransferases add acetyl coenzyme A (acetyl-CoA)-derived acetyl groups (COCH3) to the ε-amino group in the lysine. Conversely, deacetylases catalyze the removal of the acetyl group from the side chains in lysine. Acetylated modifications include one irreversible type (Nα-acetylation) and two reversible types (Nε-acetylation and O-acetylation). These three types of acetylation can occur on diverse amino acids with different frequencies, and lysine Nε-acetylation is more commonly reported.
At present, there are three types of human deacetylases: HDAC1, HDAC2, HDAC3, and HDAC8 make up the class I HDACs; class II HDACs are grouped into class IIa (HDAC4, HDAC5, HDAC7, and HDAC9) and class IIb (HDAC6 and HDAC10); and class III HDACs are nicotinamide adenine dinucleotide (NAD+)-dependent sirtuins (SIRTs) containing SIRT1 to SIRT7. Acetylation is directly connected to acetyl-CoA levels. Mitochondrial and nonmitochondrial acetyl-CoA are independently engendered and can locally trigger acetylation. Recent research has shown that acetyl-CoA can regulate acetylation in a nonenzymatic manner.24
Several acetyl-lysine research tools include convenient acetylation detection kits, mass spectrometry with acetylated affinity beads, and immunofluorescence by specific acetyl-lysine antibodies.25
The dynamic balance between histone acetylation and deacetylation in cell nucleus changes chromatin structure and regulates gene expression. Acetylation stimulates chromatin de-condensation and promotes gene expression, whereas deacetylation induces suppression of gene expression. In the past decade, increasingly advanced proteomic information has vastly expanded the number of known acetylated nonhistone proteins. A considerable amount of nonhistone protein acetylation has been identified and found to be associated with vital cellular biology, including gene expression, DNA damage repair, cell cycle control, cell fate, protein folding, protein–protein interaction, autophagy, signal transduction, and cell metabolism.23 Therefore, disruption in acetylation is implicated in diverse conditions and diseases, including immune disorders, ageing, tumors, neurological conditions, metabolic disorders, and heart diseases.26,27,28
Methylation
In 1959, protein methylation was initially reported in bacterial flagellar proteins by Richard Ambler and Maurice Rees.29 Methylation is the catalytic process of transferring methyl groups from active methyl compounds to amino acid residues. After decades of inactivity, due to advances in biology in the late 20th century, the investigation of protein methylation flourished leading to discoveries of extensive protein methylation and its potential functions. Methylation occurs mainly in nucleus and usually modifies nuclear proteins (for instance, histones). Protein methylation can occur on several amino acid residues. Methylation mainly modifies lysine and arginine residues.
There are expected to be more than 100 lysine methyltransferases (KMTs) in humans,30 such as SUV39H1 and enhancer of zeste homolog 2 (EZH2). Also, there are nine protein arginine methyltransferases (PRMTs) in mammals. PRMT1 primarily catalyzes asymmetric di-methylation, and PRMT5 is mainly responsible for symmetric demethylation.31 The arginine residue can be methylated up to two times and the lysine residue up to three times. One proton will be removed from the ɛ-amino group at each methylation, but these do not influence the total charge and will subsequently reduce the hydrogen-bonding capacity and increase the hydrophobicity. Methylation is an epigenetic regulatory process that mediates the transcriptional availability of DNA. Histone methylation occurs much slower than other histone PTMs (for example, phosphorylation and acetylation), indicating epigenetic stability.
Protein methylation can be investigated by methylation-site specific antibodies, mapping with mass spectrometry, protein or peptide immunoprecipitation (IP) with isotopic labeling or methionine labeling, and novel proteomic strategies to identify methylated substrates.32
Methylation is associated with various cell activities, including transcriptional regulation, epigenetic silencing, RNA processing and export, and signal transduction.33 Dysregulation in protein methylation results in multiple diseases, including cancer, mental abnormalities, metabolic disorders, and CVDs.34,35,36
Ubiquitination
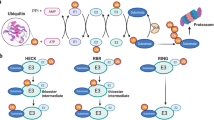
In 1975, ubiquitination was first discovered by Gideon Goldstein.37 Ubiquitin (Ub, 8.6 kDa) is a polypeptide of 76 amino acids that is highly conserved and widely expressed in eukaryotes. During ubiquitination, activated ubiquitin proteins are attached to Nε of the lysine residue of proteins and the subsequent modifications occur via a series of enzymes. Three steps are involved in ubiquitination: ubiquitin activation, conjugation and ligation. The ubiquitin-activating E1 enzyme activates ubiquitin in an ATP-dependent manner. Then, the ubiquitin-conjugating E2 enzyme is bound to the activated Ub-E1 complex, transferring ubiquitin from E1 to E2. At last, the ubiquitin E 3 ligase enzyme attaches to the lysine residues on the target protein and the C-terminal glycine on ubiquitin, leading to the subsequent modification and related effects.38
Most species contain only one E1 enzyme, ubiquitin-like modifier activating enzyme 1 (UBA1).39 E2 enzymes are a polygenic family, and their members vary in different species. Approximately 40 E2 enzymes have been discovered in humans, such as UBE2A, UBE2B, and UBE2C.40 Since E3 ligases link to the substrates and govern the peculiarity of ubiquitination, humans have over 600 E3 ligases. Based on different structures and functions, E3 ligases are classified into four types: HECT, RING-finger, U-box, and RBR types.41
Ubiquitination can modify all 20 amino acids, but lysine ubiquitination is the predominant form of ubiquitination. Ubiquitination is a well-recognized mechanism in endogenous protein degradation through the ubiquitin-proteasome system (UPS). This milestone discovery of UPS won the 2004 Nobel Prize in Chemistry. Functionally, the tag of one single ubiquitination drives the subsequent addition of ubiquitin and the growth of a polyubiquitin chain. The 26S proteasome will finally identify and degrade the polyubiquitinated protein, recycling the amino acids and ubiquitin.38 Once polyubiquitination occurs, the process becomes irreversible and the protein is destined for degradation. However, not all ubiquitination contributes to protein degradation.
Ubiquitin binding is reversible and can activate or inactivate proteins and regulate interactions among different proteins. The deubiquitinating enzyme (DUB) is a large family, including over 100 enzymes involved in removing a single ubiquitin and cleaving polyubiquitin chains. The regulation of ubiquitination depends on the conjugation of ubiquitin by ubiquitin ligases, in which deubiquitinating enzymes remove ubiquitin and counter the process.42
Current methodologies to study ubiquitination include activity-based probes (ABPs) targeting enzymes or the 26S proteasome, Ub tagging-based experiments, MS-based ubiquitination omics, ubiquitination site profiling with anti-diGly antibodies, ubiquitination-site specific antibodies or Ub COmbined FRActional DIagonal Chromatography (COFRADIC) and computational prediction.43
Ubiquitination is of great importance in the preservation and differentiation of stem cells and various cellular processes, such as cell proliferation, DNA repair, replication, transcription, protein degradation, autophagy and apoptosis, innate immunity and signal transduction.44 Dysfunction of ubiquitination is closely involved in various diseases, such as tumors, metabolic diseases, inflammatory diseases, and neurodegenerative diseases.45,46,47
SUMOylation
Small ubiquitin-related modifier (SUMO)-related PTMs were primarily found in yeast with the discovery of the yeast orthologue SMT3 in a genetic inhibition screening for the Mif2 protein by Meluh and Koshland in 1995.48 The SUMO protein is a 10-kDa polypeptide that links to the ɛ-amino groups of lysine residues via isopeptide bonds, and this is termed SUMOylation. SUMO proteins carry similarity (less than 20%) with ubiquitin in amino acid sequence. The N-terminus of all SUMO proteins share a formless sequence of 10–25 amino acids that is absent from ubiquitinated proteins. The SUMO family varies in diverse species, and there are two isoforms in yeasts, eight in plants, three in mammals, and four in humans.49
SUMOylation is a highly dynamic enzymatic cascade requiring SUMO-activating E1 enzyme (SAE1/UBA2), SUMO-conjugating E2 enzyme (UBC9), and SUMO E3 ligase, similar to but distinct from ubiquitination. In contrast to hundreds of identified ubiquitin E3 ligases, only a tiny number of SUMO E3s have been recognized, including nuclear pore protein RanBP2, tripartite motif-containing (TRIM) families, protein inhibitor of activated STAT (PIAS) and polycomb group protein Pc2.50 SUMOylation can occur in cell nuclei, cytoplasm, plasma membrane, ER, and mitochondria and is accordingly widespread in eukaryotic organisms. The acceptor lysine motif in target proteins is commonly found as ΨKxE/E (where Ψ refers to the hydrophobic residue such as valine, isoleucine, or phenylalanine; K is the SUMO-conjugated lysine; x represents any amino acid; D or E is an acidic residue),51 although increasingly non-consensus acceptor sites are being identified. SUMOylated modification alters protein localization, activity, and stability by covering or appending interaction surfaces. Some specific short SUMO-interaction motifs (SIMs) have been recognized noncovalently in target proteins, SUMO enzymes, and downstream effectors.52
SUMOylation is a reversible modification. Sentrin/SUMO-specific protease (SENP) mediates deSUMOylation, where SUMO is deconjugated to the target amino acids. The human SENP proteins include seven members: SENP1-3 and SENP5-8, but SENP8 shows no action on SUMO and has specificity for the NEDD8 protein.53
Methodologies to investigate SUMOylation include purifying SUMOylated protein, SUMO-fluorescent conjugation analysis, surface plasmon resonance-based SUMO-SIM interactions, PLA, biotin or histidine-tag assay, polymeric protein scaffold-based assay, and MS-based proteomics.54
SUMOylation contributes to many biological processes, including chromatin organization, DNA repair, transcription control, accumulation of macromolecules, cell cycle progression, trafficking, gene expression,55 and cell signaling pathways. There are many reports of abnormal SUMOylation in diseases, including tumors, Alzheimer’s disease, Parkinson’s disease, CVD, and metabolic disorders.56,57
Neddylation
Neddylation is a PTM akin to ubiquitination and was first reported in 1997 by Tetsu Kamitani.58 Neddylation attaches the ubiquitin-like protein neuronal precursor cell-expressed developmentally downregulated protein 8 (NEDD8) to the lysine residues of proteins. The ubiquitin superfamily includes 17 members, including ubiquitin, SUMO, and NEDD8. NEDD8, an 81-amino acid polypeptide, has remarkable similarity with ubiquitin, which shares 80% homogeneity and 60% identity with ubiquitin.58 NEDD8 is a conserved and predominantly nuclear protein.
Analogous to ubiquitination, protein neddylation is a highly dynamic enzymatic cascade that requires NEDD8-E1-activating enzyme (NAE), NEDD8-E2-conjugating enzyme (UBE2F, UBE2M), and specific NEDD8-E3 ligases. The most distinctive substrate of NEDDylated modification is the cullin subunit of Cullin-RING ligase (CRL). NEDD8 attaches to the lysine group at the C-terminus of cullins, spacing the CRL negative factor CAND1 and promoting CRL assembly for activation.59 All presently known NEDD8 E3 ligases could serve as E3 ubiquitin enzymes, and the majority of E3 NEDD8 ligases have the RING domain. CRLs are the principal family belonging to multiunit E3 ligases, regulating the breakdown of ~20% of proteasome-controlled proteins. The most common and studied NEDD8 E3 enzymes include RXB1 (CRL1, CRL2, CRL3, CRL4 complexes) and the homolog RXB2 (CRL5).60 De-neddylation enzymes detach NEDD8 from target proteins, including NEDD8-specific protease 1 (NEDP1), CNS5-derived eight-subunit COP9 signalosome (CSN), spinal-cerebral-ataxia related protein 3, and ubiquitin-specific peptidase 21 (USP21).61
The neddylation detection assay includes coincubation experiments, cellular thermal shift assays, isothermal titration calorimetry (ITC), biolayer interferometry (BLI), and high-throughput screening (HTS) combined with molecular docking, facilitating the confirmation of potential targets and the development of novel regulators.62
Overall, protein neddylation affects protein localization, stability and function. Neddylation participates in diverse cell processes, including DNA damage, cell apoptosis, immune regulation, and signaling pathways.59,60 Abnormal neddylation is involved in various diseases, including tumors,59 neurodegenerative disease, metabolic diseases and CVD.63,64
Glycosylation
In 1981, N-Glycosylation was the first type of glycosylated modification studied by E Bause and G Legler.65 Glycosylation is thought to be the most abundant and complex PTM, and accordingly, it vastly expands the diversity of the proteome. Glycosylation describes a series of reversible enzymatic processes of the glycoconjugates (composed of glycans or carbohydrate chains) covalently linked to the protein or lipid via glycosyltransferase or glycosidase. Glycoconjugates differ in their glycan composition, linkage, structure, and length, thus facilitating diversity. Glycosylation modifies approximately one-half of the plasma proteins, while membranes and secreted proteins are commonly glycosylated. Glycosylation can occur in cytosol, sarcolemma membrane, endoplasmic reticulum and the Golgi complex. According to the linked residues, glycopeptide bonds, and attached oligosaccharides, glycosylation can be categorized as N-glycosylation (linked to asparagine residues), O-glycosylation (attached to serines and threonines), C-glycosylation, S-glycosylation, glypiation, and phosphoglycosylation.66 N-glycosylation and O-glycosylation are two key types of protein glycosylation.
N-glycosylation represents the most common glycosylation, attaching N-acetylglucosamine (GlcNAc) to the conserved motif Asn-X-Ser/Thr by a β1-glycosidic linkage. N-glycosylation includes three processes: N-glycan synthesis, transfer, and modification. N-glycanidine biosynthesis and transfer only occur in ER, but the modification can occur in both ER and Golgi complex. Glycosyltransferases such as ALG7 and ALG13/14 produce N glycans. Oligosaccharyltransferase (OST) transfers the oligosaccharide chain to asparagine. Glycosyltransferases and glycosidases-mediated shearing and processing culminate in the formation of complex, heterogeneous N-glycan chains.65 N-glycanase (PNGase) specifically hydrolyzes asparagine (Asn)-linked oligosaccharides, mediating deglycosylation.67 N-glycosylation usually takes place cotranslationally, in which glycoconjugates are bound to the target protein during translation and transport into the endoplasmic reticulum.65 N-glycosylation thus regulates the functions of a majority of glycoproteins. The approaches to identify site-specific glycosylation are specific enzymatic proteolysis, fractionation of glycopeptides (liquid or affinity chromatography), and glycopeptide analysis by MS. Specifically, different specific endoglycosidases combined with isotope dimethyl labeling could quantitatively investigate the N-glycoproteome.68
O-glycosylation links GlcNAc and N-acetylgalactosamine (GalNAc) to serines or threonines from the oxygen atom in hydroxyl groups. O-glycosylation often occurs posttranslationally in the Golgi apparatus. O-glycans are formed by conserved O-GlcNAc transferase and reversibly broken down by the highly conserved O-glcNAcase. O-linked glycosylation is vital in the synthesis of mucins.65 No generic enzymes can directly deglycosylate O-glycans, making O-glycan release difficult and making analysis challenging. O-glycosidase fails to completely cleave complex O-glycans, and chemical methods (β-elimination, peeling reaction, and end-capping strategies) must be applied for intact glycan release.69
Approaches to study protein glycosylation include glycogene-chip; glycosyltransferase and glycosidase activity detection with radiochemistry, chromatography, spectrophotometry, and bioorthogonal chemical reporters; glycan analysis by lectin binding assays, chromatography, mass spectrometry, and novel fragmenting technologies (electron capture and transfer dissociation); and glycopeptide enrichment.70
Glycosylation is crucial in regulating cellular processes, including protein folding, degradation, secretion, molecular trafficking and clearance, cell adhesion, cell-cell interactions, signal transduction, receptor activation, and endocytosis.66,71 Dysregulation in glycosylation affects the development of human diseases, including tumors, atherosclerosis, diabetes, liver cirrhosis, and Alzheimer’s disease.72,73,74
Palmitoylation
Palmitoylation was first reported by J Folch in 1951.75 Myristoylation, and prenylation are the three major types of lipidation, describing the covalent binding of lipids to proteins, palmitoylation. These PTMs occur by thioester linkages of various fatty acids, including palmitate, myristic acid, stearic acid, octanoic acid, and cholesterol. Palmitate, a sixteen-carbon saturated fatty acid, can attach to cysteine residues by a thioester bond. This is considered a palmitoylated modification, which can increase the hydrophobicity of proteins and promote protein-lipid bilayer interactions.76 The labile and reversible thioester linkage of palmitate dynamically changes protein-palmitoylation levels in response to physiological stimulation, providing a critical potentiality to regulate cell development and signaling. The initial discovery of palmitoyltransferases was in yeast by Bartels in 1999.77 Palmitoyl-CoA is linked to target proteins by palmitoyltransferases and detached by thioesterases.
Chemical approaches to investigate protein palmitoylation include radio-labeled fatty acid reporters with autoradiographic detection or biorthogonal fatty acid reporters with bioorthogonal reactions.78 Protein palmitoylation plays critical roles in protein sorting, protein functions, protein-protein interactions, apoptosis, neuronal development, and signal transduction.76 Several pieces of evidence have indicated the crucial roles of palmitoylation in neurological diseases, cancers, and metabolic disorders.79,80,81
Myristoylation
Myristoylation was first reported in the bovine brain by Alastair Aitken in 1982.82 During myristoylation, the fourteen-carbon saturated fatty acid, myristic acid, attaches to the N-terminal of glycine by a covalent bond after cleavage of the initiator methionine. Myristoylation usually takes place co-translationally and irreversibly on cytoplasmic eukaryotic proteins. However, posttranslational myristoylation also occurs in cell apoptosis.83 Myristoylated proteins are frequently transported to the membrane according to the orientation of the myristoyl group, which usually promotes membrane binding. N-myristoyl transferases (NMTs) mediate attachment using myristoyl-coenzyme A as a substrate. The existence of NMT has been identified in most eukaryotes, but not in prokaryotes.84 Lower eukaryotes only express a single type of NMT, while mammals have two isozymes, NMT1 and NMT2. A few reports indicate the existence of demyristoylation, but the evidence is scarce, and the mechanism is still less understood.85
Myristoylation is vital in protein stability, protein localization, protein structure maturation, extracellular communication, immune response, cell metabolism, and signal transduction.86 Dysregulation in protein myristoylation has been reported in the development of cancer, neurological diseases,87 viral and bacterial infections, and metabolic disorders.84,88,89
Prenylation
Prenylation was first discovered in yeast by KamiIya in 1978.75 Prenylation is an irreversible modification ubiquitously occurring in all eukaryotic cells. Prenylation describes the covalent addition of isoprenoids to the carboxyl-terminal or cysteine residues.
Prenylation includes two major forms90: farnesylation (linkage of a fifteen-carbon farnesyl pyrophosphate) and geranylation (attachment of a twenty-carbon geranylgeranyl pyrophosphate). Three isoprenyl transferases catalyze these modifications. Farnesyltransferase (FTase) modulates the combination of a single geranylgeranyl group, whereas geranylgeranyltransferase type-1 (GGTase-I) adds a single geranylgeranyl group. The common sequence in C-terminal of the target cysteine is the “CaaX” box, in which “C” represents a cysteine, “a” represents the aliphatic amino acid, “X” could be any amino acid responsible for the attached isoprenoid.91 GGTase-II catalyzes dual geranylgeranyl groups attaching to double cysteine residues in motifs like “CCXX” or “CXC“.91 Prenylated proteins experience farnesylation, proteolytic removal of the “aaX” sequences, carboxymethylation and finally become oriented and the plasma membrane. Well-known prenylated proteins include Ras superfamily proteins, Ras-related small guanosine triphosphate-binding proteins (G proteins), and trimeric G proteins.91
Several tools to study protein prenylation include chemical proteomic analysis with alkyne-containing probes.92 Prenylation is the first necessary process in membrane targeting and binding and involves subsequent protein-protein interactions, protein trafficking, cell movement, cell growth, differentiation, and proliferation.93 Disruption in prenylated modification is observed in the pathogenesis of tumors, neurodegenerative diseases, bone diseases and cardiometabolic diseases.94,95
Cholesterylation
Cholesterol modification (cholesterylation) was first found in hedgehog (Hh) proteins by Porter in 1996.96 For the next 20 years, hedgehog was considered the only cholesterylated protein until another cholesterol-modified protein, smoothened (SMO), was reported by Song and colleagues in 2017.97 Cholesterol modifies SMO at the Asp95 residue, which is necessary for the Hh protein pathway and embryonic development. Song and colleagues subsequently revealed that cholesterylation of SMO is an autocholesterylation process promoted by calcium.98 Biorthogonal labeling can be applied to analyse and identify novel cholesteryled proteins.99 Several approaches to detecting protein cholesterylation exist, such as biorthogonal labeling with azido-conjugated cholesterol analogs and alkynyl sterol probes.99
Glutathionylation
The primary report of protein glutathionylation dated back to 1985 by Ziegler.100 However, the comprehensive understanding of protein glutathionylation associating reactive oxygen/nitrogen species (ROS/RNS) did not emerge until the last decade. S-glutathionylation describes the bond formed between glutathione (GSH) and the thiol group (-SH) of cysteines via a mixed disulfide linkage. This reversibly adds a negative charge and a tripeptide to alter the protein structure, charge, and functions. Glutathione S-transferase (GST) can catalyze S-glutathionylation, or S-glutathionylation can occur spontaneously.101 Thioredoxin, glutaredoxin (Grx), and sulfiredoxin can regulate the reversal of S-glutathionylation.101 The rate of GSH and glutathione disulfide (GSSG) mainly serves as the sensor of the intracellular redox state and can be reduced by oxidative or nitrosative stress under physiological or pathological conditions.102 Many proteins undergo S-glutathionylation, covering the cytoskeleton, cell metabolism, kinase, calcium pathway, antioxidant homeostasis, protein folding, and signal transduction.103
Given the potential significance of glutathionylation, numerous developing techniques could identify protein glutathionylation. The basis for the current proteomic investigation is labeling glutathione with 35S radiolabelling and biotinylation.104 There is also novel computational prediction by the position-specific matrix.105
Due to the abundance and significance of glutathione, the S-glutathionylation cycle plays vital roles in cell fate, cell proliferation, differentiation, apoptosis, antioxidant homeostasis, cell metabolism, immune response, inflammation and signaling pathways.102,106 An imbalance in S-glutathionylation results in a series of diseases, such as infection, tumors, neurodegenerative diseases,107 CVDs, and metabolic diseases.106,108,109,110
S-nitrosylation
Although S-nitrosylation was primarily investigated by Shigeru Oae and Koichi Shinhama in 1983,111 it took another 30 years before S-nitrosylation was recognized as nitric oxide (NO)-dependent PTM.112 S-nitrosylation is the covalent incorporation between the nitrosyl moiety of NO and target molecules. S-nitrosylation occurs at the cysteine thiol group, producing protein S-nitrosothiols (SNOs). If nitrosylation occurs at a transition metal, this is termed metal nitrosylation. Awarded as “the Molecule of Year” in 1992, the dissolved gas, NO, is of great significance in biology and has been associated with extensive research and numerous awards, including the Nobel Prize in 1998. In the classical NO pathway, NO induces the generation of cyclic guanosine monophosphate (cGMP) and activation of cGMP-dependent protein kinase (PKG) signaling through attachment to guanylyl cyclase (GC).113 In the nonclassical NO pathway, S-nitrosylation mediates the major mechanism.114 S-nitrosylation can consume NO to prevent the reaction between NO and ROS and guard cysteine thiols against ROS-induced oxidation at a low ROS level. Most proteins act as substrates for S-nitrosylation. Some enzymes are involved in S-nitrosylation and de-nitrosylation, but the mechanism of dynamic regulation has remained less explored.114 There are precise space and time mechanisms regulating S-nitrosylation and denitrosylation. For instance, the location of the target cysteine, the specific motif sequence “I/LXC-X2-D/E” in cysteine, a highly hydrophobic region, and the suitable environment jointly confirm the specificity of S-nitrosylation.115
Tools to detect protein S-nitrosylation include biotin-switch-based mass spectrometry, immunochemistry with specific anti-SNO antibodies, chemical strategies by gold nanoparticles, organomercury resin capture, organophosphine-related biotin labeling, and labeling based on one-step disulfide production.116
S-nitrosylation regulates various cellular mechanisms, including transcription, protein stability, localization, trafficking and interaction, cell growth and apoptosis, cell metabolism, signaling pathways, and further protein modification phosphorylation, acetylation, and ubiquitination.114 An imbalance in S-nitrosylation is implicated in the occurrence of various human diseases, such as cancer,117 neurodegenerative diseases, respiratory diseases, cardiovascular diseases, and metabolic disorders.118,119,120
Sulfhydration
First identified in mouse liver lysates by protein analysis in 2009, sulfhydration describes the PTM involving the alteration of the thiol group (-SH) in reactive cysteine residues to a persulfide (-SSH) group, leading to the enhanced reactivity of the cysteine residue, akin to nitrosylation.121 Hydrogen sulfide (H2S) functions as an imperative gasotransmitter/signaling molecule and is crucial in physiological processes analogous to NO. Mechanically, H2S facilitates its role through protein sulfhydration. H2S physiologically modifies nearly 10%-25% of hepatic proteins by S-sulfhydration, including tubulin, actin, and glyceraldehyde-3-phosphate dehydrogenase.122 Sulfhydration regulates protein function and mostly depends on the structure and spatial arrangement of sulfhydrated residues. Sulfhydration protects cysteine residues against oxidative damage, leading to remission of permanent injury and amelioration of protein function. Sulfhydration is similar to nitrosylation, by which both are reversible and occur on the cysteine residue, but they are differentiated from each other. Sulfhydration is more common than nitrosylation, as 25–50% sulfhydrated proteins are detected in murine liver.122 Sulfhydration generally activates enzyme activity, whereas nitrosylation usually suppresses protein function.123
Approaches to exploring protein sulfhydration include biotin-switch analysis, cysteinyl labeling examination, tag-switch assessment, protein persulfide detection, and mass spectrometry analysis.124 Sulfhydration orchestrates various processes, including inflammation, endoplasmic reticulum stress, signal transduction, blood pressure, and vascular tension.125 Disruption in sulfhydration mediates abundant diseases, such as Alzheimer’s disease, Parkinson’s disease, CVDs and metabolic disorders.126,127,128,129
Citrullination
The citrullinated modification was first reported in detail by Rogers in 1958.130 Citrullination, also known as deimination, is an irreversible chemical process converting arginine to citrulline, during which positively charged arginine is chemically hydrolyzed to uncharged citrulline and neutral urea. Citrulline is a nongenetically coded type of amino acid, and citrullination only takes place posttranslationally. This charge conversation will affect protein structure, charge, hydrogen bond generation, protein-protein interactions, and even protein denaturation.
Citrullinated modifications can involve numerous cellular proteins, including those in the nucleus, cytoplasm, mitochondria, and cell membrane. This modification is catalyzed by peptidylarginine deiminases (PADs), enzymes that appear to be activated by high calcium concentrations. The catalytic process of PAD enzymes was initially described in 1977.131 Five calcium-dependent isozymes (PAD1, PAD2, PAD3, PAD4, PAD6) are identified in humans, which share a 50% similar sequence.132 Diverse PAD enzymes are distributed widely in cells and tissues. Especially, PAD4 is found only in the nucleus and is essential in histone deamination, whereas the other four isozymes are located in the cytoplasm.133
Current strategies to study protein citrullination include COLDER assessment, immunochemistry with specific anti-citrullination antibodies, mass spectrometry, chemical derivatization targeting citrulline, and phenylglyoxal probe-based assays.134
The activity and balance of PADs play a role in citrullination and cellular processes, including protein stability and structure, protein-protein interactions, cell apoptosis, and cell death.135 Abnormalities in protein citrullination lead to multiple sclerosis, cancer, rheumatoid arthritis, systemic lupus erythematosus,136 Alzheimer’s disease and metabolic disorders.137,138,139
ADP ribosylation
Protein adenosine diphosphate (ADP)-ribosylation was primarily defined by Chambon in the early 1960s.140 ADP-ribosylation transfers ADP-ribose (ADPr) from NAD+ to the target protein and releases nicotinamide (Nam). This modification includes mono-ADP-ribosylation (MARylation) and poly-ADP-ribosylation (PARylation). PARylation possesses specific characteristics due to the synthesis and nature of ADP-ribose chains. ADP-ribosylation takes place in the side chains with sulfur, nitrogen, or nucleophilic oxygen, leading to S-, N-, or O-glycosidic attachment to the ribose. ADPr carries an adenine ring, two ribose moieties, and two negative charges, enabling hydrophobic interactions and hydrogen linkage. In this manner, ADP ribosylation offers diverse modalities to change protein structure and functions. ADP-ribosylation is a reversible event where ADP-ribosyltransferases (“writers”) covalently add ADPr, whereas ADP-ribosylglycohydrolases (“erasers”) remove ADPr. “Readers” describes the interaction with ADPr.141
Based on structural homology, the ADP-ribosyltransferase (ART) superfamily is characterized as ART diphtheria toxin like (ARTD) and ART cholera toxin like (ARTC). ARTDs include the majority of poly (ADP-ribose) polymerases (PARPs) and tankyrases (TNKS). The PARP family includes two tankyrases: tankyrase 1 (TNKS1, also termed PARP5A or ARTD5) and tankyrase 2 (TANK2, also named PARP5B or ARTD6).142 Viruses, prokaryotes, and eukaryotes all share conserved ART domains.
Hydrolases remove ADPr, which varies in structure and function, including MacroD1, MacroD2, terminal ADP-ribose protein glycohydrolase 1 (TARG1), poly-ADP-ribose glycohydrolase (PARG), and ADP-ribosyl-acceptor hydrolases (ARHs).143
Approaches to exploring protein ADP-ribosylation include chemical tools (such as α-ribosyl amino acids, (pyro)phosphate, ADP-ribosylated peptides, and analogues, polyADPr chains), macroGreen, fluorescence-related assessment, and molecular toolbox.144
ADP ribosylation regulates major cellular processes, including DNA repair, cell growth and differentiation, cell metabolism, stress responses, and immunity.145 Dysregulation of ADP-ribosylation can lead to human diseases, including cancer, ischaemia-reperfusion-like tissue injury, heart disease, neurological disorders,146 and metabolic disorders.147,148,149
Carbonylation
The introduction of carbonyl groups into protein was first reported during studies of glutamine synthesis in 1983.150 Protein carbonylation (PCO) is a type of protein oxidation that produces carbonyl groups such as reactive ketones, aldehydes, or lactam, facilitated by reactive oxygen species (ROS). PCO is a nonenzymatic and deleterious PTM, as the introduction of carbonyl groups into target proteins marks oxidative damage and destroys protein structure and function.
PCO is divided into four groups: the breakage of protein and polypeptide main chains; the oxidation of amino acid side chains; lipid peroxide addition to active site; and glycation oxidation products.151 The technology for determining carbonyl content is based on the formation of 2,4-dinitrophenylhydrazone. Spectrophotometry and chromatography can measure the total protein carbonyl content.
Valuable tools to study protein carbonylation include measuring the carbonyl level by the Levine spectrophotometric assay based on the chromogenic reaction with 2,4-dinitrophenylhydrazine (DNPH), ELISA, western blot with anti-DNPH antibodies, and in-gel detection assay by fluorescence.152
PCO leads to irreversible damage. PCO acts as the hallmark of oxidative stress and is closely implicated in regulating protein function and cell senescence.153 Dysregulation of PCO is seen in skeletal muscle dysfunction, Alzheimer’s disease, and metabolic disorders.154,155,156 The physiological roles of PCO in oxidant signaling indicate that drugs controlling carbonyl content might possess clinical value.
Other oxidative modifications
Protein oxidative modifications are an appreciable group of protein PTMs, which are induced by ROS, reactive sulfur species (RSS) or reactive nitrogen species (RNS). Cysteine is the molecular basis for thiol-mediated redox control. Common oxidative reversible alterations of cysteine thiols include S-nitrosylation (-SNO), S-sulfenylation (-SOH), glutathionylation (-SSG), disulfide formation (-SSR) and S-sulfhydration (-SSH). Furthermore, the biologically stable modifications mainly cover S-sulfinylation (-SO2H) and S-sulfonylation (-SO3H). We have described some oxidative modifications such as S-nitrosylation, glutathionylation, and sulfhydration, above, so here we will give a brief introduction to other protein oxidative modifications.
S-sulfenylation (-SOH)
The study of S-sulfenylation commenced in 1976.157 S-Sulfenylation is a process where hydrogen peroxide (H2O2) converts oxidized specific cysteine thiols to sulfenic acid (-SOH). Most interaction between cysteine thiol groups and H2O2 is slow, which depends on the protein microenvironment, pH, pKa(-SH), and the presence of bulky groups around the thiol groups.158S-Sulfenylation serves as an intermediate redox sensor leading toward other oxidative modifications, including S-glutathionylation and disulfide formation. This reversible modification can control molecular thiol switches to modulate protein stability, activity, interactions, conformational alteration, and cellular location.159 Approaches to identifying SOH are usually indirect, including protein engineering techniques, chemical labeling, single-molecule force-clamp spectroscopy, and mass spectrometry.160 Aberrant sulfenylation contributes to numerous diseases, including tumors, senility, CVDs, obesity, diabetes and neurodegenerative diseases.158
S-sulfinylation (-SO2H)
Protein thiol oxidation yields SOH, which is oxidized further to form S-sulfinic acid (-SO2H). This reversible process is sulfinylation (-SO2H). Hyperoxidation of SOH to SO2H relies on SOH ionization and the nucleophilic assault of H2O2. The generation of SO2H is commonly related to oxidative stress. Sulfiredoxin decreases S-sulfinic acid (-SO2H) back to the thiol in an ATP-dependent manner.161 The reversibility of SO2H indicates that sulfinic acid formation plays a role in redox regulation, which enables H2O2 signals to exert regulatory effects. It was reported that 5% of the hepatic cysteines in the rat are present as SO2H.162 S-sulfinylation is an elusive modification, and the identification depends on chemical probes, electrophilic probes and mass spectrum.163
S-sulfonylation (-SO3H)
Protein thiol oxidation can yield SOH, further generate SO2H, and finally, produce S-sulfonic acid (-SO3H). This is the process of sulfonylation (-SO3H), which irreversibly deactivates proteins.164 GSH could bind to SOH and form the protein glutathione mixed disulfide (PSSG), fundamentally avoiding the further oxidation of lipoate and blocking the irreversible alteration of SOH to SO3H.165
Disulfide formation (-SSR)
Disulfide bonds were found in coagulated egg albumin in 1907 by Heffter and in 1911 by Arnold.166 This two-electron reaction requires an electron acceptor or oxidant. Disulfide-bond formation in cellular proteins occurs as a series of catalyzed processes that are essential to the function of membrane and secreted proteins. Disulfide bonds primarily take place in the periplasmic space for prokaryotic cells or ER for eukaryotic cells.167 Disulfide bonds appear either intramolecularly (on two cysteines in the same polypeptide chain) or intermolecularly (between two proteins). The mixed disulfide is the disulfide bond linking the cysteine and a thiol-containing redox reagent (like dithiothreitol or glutathione). Intramolecular disulfide bonds attribute to stabilizing the tertiary structures of proteins, while intermolecular disulfide bonds contribute to stabilizing the quarternary structure.
The reversibility of disulfide formation allows its regulatory effects on protein folding, assembly, structure, stability, and function. Two redox-sensitive cysteines and the related disulfide bonds can serve as redox-sensitive switches. Redox-sensing switches can be present in abundant proteins, including enzymes, receptor proteins, sensor proteins, and transcriptional factors.168 Disulfide bonds can be detected by nuclear magnetic resonance, X-ray crystallography, LC-Fourier transform tandem mass spectrometry (FT MS/MS), and MassMatrix MS/MS search engine.169
Intramolecular disulfide bonds have been reported to be involved in G protein signaling,170 antioxidant enzyme Prdx1, thiol peroxidase, cholesterol metabolism, multiple myeloma, Alzheimer’s disease171 and amyotrophic lateral sclerosis. Intermolecular disulfide bonds play a role in innate immunity, prion diseases, Alzheimer’s disease, and vascular diseases. Mixed disulfide bonds are involved in immune response and celiac disease.172
Novel types of PTMs
Recently some novel PTMs have emerged. Here, we will briefly introduce these novel types of PTMs.
Succinylation
Succinylation is a unique, recently discovered, and less understood PTM. Succinylation was first identified in Escherichia coli in the context of the catalytic activation of homoserine by Ran Rosen in 2004.173 Protein succinylation is conserved in prokaryotes and eukaryotes, describing the process of the covalent attachment of the succinyl group derived from succinyl-CoA to the lysine residue. Since the succinyl group is large (100 Da), the succinylated PTM results in a significant mass change and alters the physiological charge from −1 to +1; accordingly, succinylation possesses a significant effect on protein structure and function compared to acetylation (40 Da) or methylation (14 Da).174 Succinylated modification can occur in the nuclei, cytoplasm, and mitochondria.
As the principal regulator of succinylation, succinyl-CoA is positively associated with nonenzymatic succinylation. In addition, the α-ketoglutarate dehydrogenase complex (α-KGDHC) controls succinylation either by direct enzymatic succinylation or by regulating the levels of succinyl-CoA.175 Carnitine palmitoyltransferase 1A (CPT1A), another lysine succinyltransferase in mammalian cells, promotes succinylation without changing succinyl-CoA levels.176 NAD+-dependent SIRT5 is a desuccinylase that functions in all cell compartments.175
Because of the low content and a broad dynamic range of succinylated proteins in cells, enrichment of succinylated peptides is required to increase their abundance before mass spectrometroscopic analysis, and then quantitative analysis of the enriched succinylated peptide samples is performed using traditional quantitative proteomic analysis tools. Moreover, several computational predictions based on websites and deep learning methods are becoming increasingly common.177
Succinyl-CoA serves as a crucial metabolic intermediate in tricarboxylic acid (TCA) cycle and a vital donor of succinylation at the same time, allowing succinylation to govern cell metabolism and signal transduction.178 Accumulating evidence indicates that protein succinylation is involved in transcription modification, immune response, and cell metabolism covering the TCA cycle, urea cycle and fatty acid metabolism with altered metabolism.179,180,181 Current data has shown that dysfunction of succinylation leads to many diseases, such as inflammatory diseases, tuberculosis, ischaemia-reperfusion-like tissue injury, and metabolic diseases.182,183,184
Crotonylation
Lysine crotonylation (Kcr) was first reported in male germinal cells and human somatic cells by Zhao and colleagues in 2011 and was recognized as an epigenetic research highlight of 2011 by the journal, Cell, in the same year.185 The crotonyl group has an exclusive C-C π‐bond, leading to a rigid configuration. Kcr is usually found on histones in transcriptionally active chromatin regions and is closely associated with reproductive regulation. Kcr can be regulated reversibly by crotonyltransferases and decrotonylases. Kcr can be controlled by a nonenzymatic mechanism, and crotonyl-CoA profusion is one modulating factor of Kcr.186
Kcr “writer” refers to enzymes that catalyze histone crotonylation, which is influenced by intracellular crotonyl-CoA. Both genetic and environmental factors can regulate Kcr. Three main HAT families exhibit extended histone crotonyltransferase (HCT) activities, including p300/CREB-binding protein (p300/CBP), MYST, and GNAT (Gcn5-related N-acetyltransferase).187
The group of Li and colleagues identified a novel crotonylation “reader” (the AF9 YEATS structural domain) in 2016.188 AF9 YEATS structural domain can directly link Kcr to transcriptional activity. Double plant homeodomain finger proteins, YEATS domain proteins, and bromodomain proteins have been identified as three major families of readers.
In 2017, a crotonylation “eraser” appeared with the discovery that HDACs, but not the sirtuin family, are the main histone decarboxylases.186 Histone crotonylation is dynamically regulated in mammalian cells in the same way as histone acetylation. CDYL can negatively regulate histone crotonylation by serving as a crotonyl-CoA hydratase to change crotonyl-CoA to β-hydroxybutyric-CoA. Moreover, crotonylation can also occur on nonhistones.189
Current tools for the study of crotonylation are water-soluble phosphine warhead-based probes and bioinformatics detection by deep learning.190
Lysine crotonylation is associated with numerous cellular processes including DNA damage and repair, differentiation of stem cells, spermatogenesis, and inflammation.186,187,191 Dysregulation of lysine crotonylation is involved in diverse human diseases, including tumors, neuropsychiatric disease, and cardiovascular diseases.192,193
Beta-hydroxybutyryration
Beta-hydroxybutyryration (Kbhb) is a novel acylation modification mediated by β-hydroxybutyrate (β-OHB), first proposed by Zhao and colleagues in 2016.194 The acyltransferase p300 catalyzes the attachment of β-OHB to lysine, whereas HDAC1 and HDAC2 reversibly eliminate Kbhb. By this, β-OHB has been simply considered a functional carrier transferring energy from the liver to peripheral tissues upon starvation stress. β-OHB is also an important signaling and epigenetic regulatory molecule that regulates all aspects of life functions in vivo. β-OHB can mediate lysine Kbhb on histones of several hunger-associated genes, assisting the body to quickly adapt to metabolic shifts caused by energy shortage.195 Subsequent studies have revealed that in addition to histones, β-OHB can modify nonhistone proteins and participate in regulating diseases such as cancer and cardiometabolic diseases.196,197,198,199
Many key metabolic enzymes have multiple Kbhb sites, such as the urea cycle rate-limiting enzyme CPS1, the ketogenic rate-limiting enzyme HMGCS2, and S-adenosyl-L-homocysteine hydrolase (AHCY) in the methionine cycle.200 β-OHB can regulate cellular functions by directly affecting intracellular acetyl-CoA, succinyl-CoA, and NAD+ levels or by inhibiting histone deacetylase (HDAC) activity, thereby altering protein acetylation, succinylation, and other downstream molecular events. Kbhb-altered proteins are broadly distributed in the cytoplasm, mitochondria, and nucleus, suggesting a broad impact of β-hydroxybutyrylation modifications on cellular functions.194,201 Immunochemistry with crotonylation-modified pan antibodies, site-specific antibodies, and mass spectrometric techniques are the major detection methods for Kbhb.
Lactylation
Zhao and colleagues identified the novel histone acylation code, lactylation, in 2019.20 The researchers determined that lactylation occurred on histone lysine in human and mouse cells, which could trigger gene transcription directly. Enzymatic lysine lactylation transfers the lactyl group from lactyl coenzyme A (lactyl-CoA) to lysine, catalyzed by lysine acetyltransferase (KAT) enzymatic P300 and regulated by lactyl-CoA.20 Nonenzymatic lysine lactylation is derived from methylglyoxal, a glycolytic by-product, producing lactoylglutathione (LGSH).202 Lactylation results from lactic acid produced by cellular glucose metabolism and is regulated by lactic acid, glycolysis, and mitochondrial oxidative metabolism. Histone lactylation is abundant in late M1 macrophage polarization and shows diverse temporal dynamics compared with histone acetylation.20 Immunochemistry with specific antibodies and mass spectrometric techniques are the primary detection methods for lactylation. Lactylation is involved in gene expression, cell differentiation,203 and inflammation. Aberrant lactylation has been proposed in cancer, fibrosis, and cardiometabolic diseases.204,205,206,207
PTMs in metabolic diseases
PTMs in diabetes
Diabetes occurs in several major forms, including type 1 diabetes mellitus (T1DM), type 2 diabetes mellitus (T2DM), and gestational diabetes mellitus (GDM) and others, each of which is diagnosed and characterised by hyperglycemia. Diabetes has a strong association with the development and progression of life-threatening CVD. The processes of diabetes are intricate and interactive, including various cellular responses and signaling cascades regulated by PTMs. For example, various kinases and phosphatases regulate glucose-stimulated insulin secretion in pancreatic beta cells. PTMs can establish the link between gluconeogenesis, the TCA cycle and glycolysis to affect beta cell viability and function. In this section, we discuss the roles of various PTMs in diabetes (Fig. 3).

A holistic summary and illustration of the crosstalk between PTMs and diabetes. The pathogenesis of diabetes is complex and interactive, involving various cellular responses and signaling cascades regulated by PTMs. (1) Balance the actions of kinases and phosphatases in regulating glucose-stimulated insulin secretion from pancreatic beta cells. (2) Establish the link between gluconeogenesis, the TCA cycle and glycolysis. (3) Directly cause modification of certain proteins or induce PTMs secondary to various cellular processes to maintain beta cell function and viability. Different colors and shapes represent different types of PTMs. Activation and inhibition effects are displayed in “arrows” and “inhibition” symbols, respectively. The figure is generated with BioRender (https://biorender.com). CUL4A cullin 4A, ERK extracellular regulated protein kinases, FOXO1 forkhead box O1, GCK glucokinase, GLUT glucose transporter, GSK-3 glycogen synthase kinase 3, IRS insulin receptor substrate, HDAC histone deacetylase inhibitor, JNK c-Jun N-terminal kinase, MKP1 mitogen-activated protein kinase phosphatase, MEKK mitogen-activated extracellular signal-regulated kinase kinase, NF-κB nuclear factor-k-gene binding, PTP1B protein tyrosine phosphatase 1B, PTEN phosphatase and tensin homolog, PPAR peroxisome proliferators-activated receptors, PDK1 3-phosphoinositide-dependent protein kinase 1, PP2A proteinphosphatase2A, SENP2 sentrin-specific protease 2, SREBP1 sterol-regulatory element binding protein 1, SIRT sirtuins, TRIB3 tribbles pseudokinase 3, TXNIP thioredoxin interacting protein
Phosphorylation in diabetes
Protein phosphorylation is an important PTM that balances the actions of kinases and phosphatases in regulating glucose-stimulated insulin secretion from pancreatic beta cells. Glucose homeostasis is mainly dependent on signaling cascades mediated by protein kinases and phosphatases which determine the output of metabolic processes by controlling PTMs of different substrates. The insulin receptor (INSR, IR) activates various downstream targets, such as PI3K/AKT (PKB), mitogen-activated protein kinases 3/1 (MAPK3/1), extracellular signal-regulating kinase 1/2 (ERK1/2) and AMP-activated protein kinase (AMPK), to control energy homeostasis and stimulate energy catabolic processes. Thus, the PI3K/AKT, MAPK and AMPK pathways are required for insulin-dependent regulation of metabolic activity. As exemplified above, the insulin-PI3K/AKT pathway is negatively regulated by PTPN1 (PTP1B), PTEN and PP2A, which can dephosphorylate and inhibit IR, IRS1/2, PIP3, and AKT. Thus, PTMs of proteins of the insulin signaling pathway can impair or improve metabolic pathways.
Phosphorylation events and kinases in islets are associated with insulin secretion. Based on the SILAC proteomics, 8539 phosphosites derived from 2487 proteins were identified in the islets, and 170 phosphosites (98 were upregulated and 72 were downregulated) are differentially expressed in response to a short-term high glucose challenge.
IR is essential for insulin action and plays an important role in pancreatic cells. Deletion of IR reduced β and α cell mass and induced hyperglycaemia in mice. Ins1-/-:Ins2f/f mouse β cells lose about 50% of insulin production, resulting in robust hyperglycemia, β cell proliferation, hormone expression disorders and alleviation of ER stress. This is associated with hyperphosphorylation of AKT, leading to reduced DNA damage inducible transcript 3 (DDIT3), tribbles pseudokinase 3 (TTIB3), activating transcription factor 4 (ATF4) and phospho-eIF2α expression.208 Overexpression and activation of AKT in pancreatic cells regulate the phosphorylation/dephosphorylation of signaling factors such as forkhead box O1 (FOXO1), glycogen synthase kinase 3β (GSK3β) and mammalian target of rapamycin 1 (mTORC1) and its downstream target to regulate β cell mass and proliferation.209 Previous studies have established that the overactivity of AKT is sufficient to increase the proliferation of β-cells via cyclin D1.209,210
AMPK is the most intensively studied protein kinase in the treatment of metabolic syndrome. Activated AMPK phosphorylates substrates and can stimulate glucose uptake and inhibit glycogen synthesis.
PTP1B and PTEN antagonize insulin signaling by dephosphorylating the IR and IRS1/2. PTP1B deficiency and the partial reduction of Pten results in improved glucose tolerance and protects against insulin resistance in mice.211
A study found that protein phosphatase-2C alpha (PP2C alpha) directly dephosphorylated the p85 subunit of PI3K to stimulate its catalytic activity and enhance insulin sensitivity. Heart- and liver-specific knockout of Ppp2r2a increases the phosphorylation of important insulin signaling molecules, such as AKT, GSK-3α/β, FOXO1 and GS, resulting in increased insulin sensitivity and improved glucose tolerance in the heart and liver.212
In addition, the data also indicated that all PKA and PKC substrates in the db/db mouse islets were dephosphorylated and that a hyperglycemic environment can increase the phosphorylation of the β cell-specific transcription factor PDX1 through GSK3 kinase, leading to β cell failure.19
Serine/Threonine Kinase 25 (STK25) and CK2 are both serine/threonine kinases. Overexpression of STK25 is known to aggravate muscle insulin resistance and increase intramyocellular lipid accumulation.213 Inhibition of CK2 reduced the phosphorylation of class I HDACs to activate adipocyte thermogenesis and protected mice from diet-induced obesity and insulin resistance.214
Acetylation in diabetes
The acetylation of proteins is a pathway that is a reversible PTM regulated through the function of specific types of enzymes and this process functions as a main regulator of human metabolism. Acetylation is a PTM dependent on acetylases and deacetylases for catalyzing acetylation and deacetylation processes, respectively. Acetyl-CoA provides acetyl groups for acetylation and acts as an essential constituent of gluconeogenesis, the TCA cycle and glycolysis. As a consequence, there is an established link between acetylation and hyperglycemia and the insulin resistance of metabolic syndrome.
The differential expression of NAD-dependent deacetylase SIRT3 between Goto-Kakizaki (GK) rats and nondiabetic Brown Norway (BN) rats can support the causality between protein acetylation and impaired glucose homeostasis.
The spectrum study of lysine acetylation in the diabetic kidney identified 39 differentially expressed proteins, most of which were intermediate metabolic enzymes.215 Hyperglycemia-induced acetylation of retinal histones H3 and H4 regulates the activities of several proinflammatory proteins that participate in the pathogenesis of diabetic retinopathy (DR). HFD feeding can enhance acetylation of the fatty acid β-oxidation enzymes β-hydroxylacyl coenzyme A dehydrogenase (β-HAD) and long-chain acyl-CoA dehydrogenase (LCAD); it can also lead to the dysregulation of the insulin signaling pathway. In a mutant mouse model of CREB-binding protein (CBP), increased insulin sensitivity and glucose tolerance were demonstrated. A monogenic autosomal form of T2DM,MODY (Maturity Onset Diabetes of the Young), was determined to be associated with histone acetyltransferase (HATs) and HDACs. Some HDAC inhibitors can improve insulin resistance to ameliorate inflammation. Some HDAC3 inhibitors can improve glycemia, promote insulin secretion and protect β cell function in the prediabetic stage.26,216 HDAC4, HDAC5, and HDAC9 are key regulators that promote the development of the β/δ-cell lineage. Moreover, inhibition of HDAC6 in pancreatic islets downregulates insulin signaling. Increased HDAC7 levels impair insulin secretion and contribute to β cell dysfunction in type 2 diabetic islets. SIRT6 mediates the deacetylation of histone H3 to restrain Txnip expression in beta cells, thereby maintaining beta cell function and viability.
Notably, compounds that modify lysine acetylation, such as resveratrol, are known to inhibit early diabetic retinopathy in diabetes.217
Methylation in diabetes
Histone methylation and nonhistone protein methylation are all types of PTMs termed methylation. Protein methylation mainly occurs at lysine or arginine residues and is appended with either one to three methyl groups by N-methyltransferase in the cytosol. Protein methylation is often associated with gene repression or activation depending on the degree and position of the modifications.
Some work has been done on diabetes-associated biochemical modification of metabolic enzymes via methylation. The expression of nicotinamide N-methyltransferase (NNMT) is increased in the white adipose tissue (WAT) and liver tissue of patients with insulin resistance or T2DM. Deletion of NNMT in the livers of C57BL6/J mice lowers fasting plasma glucose levels.218 The histone methyltransferase SETDB2-associated pathway IFN-β-SETDB2-H3K9me3 is dysfunctional in diabetes and induces nuclear factor kappa B (NF-κB)-mediated inflammation.219 The lack of histone methyltransferase G9a suppresses CD36 and M1 macrophage genes in type 2 diabetic patients.220 PRMT1 plays an essential role in maintaining mature β-cell identity.35 Deficiency of PRMT5 impairs glucose tolerance and glucose-stimulated insulin secretion in a mouse model. However, the compensatory increase in H3R8me2 can accelerate the binding of the brahma-related gene-1 (BRG1) chromatin remodeling enzyme to the insulin gene promoter.221
Patients with T1DM have an increased demethylation level of H3K9me2 in blood lymphocytes.222 Methylation of H3K4me1 was increased in patients with T2DM in the transcription factor NF-κB promoter region.223
T1DM and T2DM show increased H3K9me3 of the Slc2a4 promoter and decrease glucose transporter type 4 (GLUT4) expression, thus contributing to glycemic impairment.224 KDM6A, one of the known H3K27me2/3 demethylases, has higher protein levels in the kidneys of diabetic OVE26 mice.224 Combination therapy with telmisartan and esculetin, attenuates increased levels of histone PTMs such as H3K9me2, H3K9Ac, H2AK119Ub, and H2BK120Ub in type 2 diabetic cardiomyopathy.225
Ubiquitination in diabetes
Ubiquitination is a PTM resulting from the covalent linking of each successive ubiquitin to the previous ubiquitin at lysine by polyubiquitination or mono-ubiquitination. Ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2) and ubiquitin ligase enzyme (E3) coordinate the action of ubiquitin-proteasome system homeostasis and degradation.
Inhibition of ubiquitin-activating enzyme E1 blocks ubiquitination of the key molecules of insulin signaling and prevents palmitate-inducible insulin resistance.226 UBC9 protein expression is decreased in muscle tissues from T2DM patients.227 Haploinsufficiency of UBC13 can ameliorate HFD-induced insulin resistance.228 Ubiquitin-conjugating enzyme E2O (UBE2O) is significantly upregulated in obese individuals with T2DM.229
The Really Interesting New Gene (RING) family is emerging as the most important ubiquitin ligase. RING ligases play a crucial part in PI3K/AKT-mediated glucose metabolism. Cullin-RING ligase complexes (CRLs) are the most abundant RING E3 ligases. SKP2 (substrate of CRL1) can ubiquitinate and promote the degradation of FOXO1. Cul4A-RING E3 ubiquitin ligase suppresses PTP1B activity and suppresses the expression of genes associated with gluconeogenesis.230 TRIM family proteins are involved in the progression of diabetes and the development of diabetic complications. For instance, TRIM13 attenuates DN-induced collagen synthesis by promoting the ubiquitination of C/EBP homologous protein (CHOP).231 TRIM32 inhibition increased PI3K-AKT-FOXO signaling in the liver and skeletal muscle and enhanced glucose uptake.232 Mitsugumin 53/ TRIM72 promotes ubiquitin-dependent degradation of the insulin receptor and insulin receptor substrate-1, resulting in T2DM.46 E3 ubiquitin ligase F-box and WD repeat domain-containing 7 (FBW7) boosted EZH2 ubiquitination and proteasome degradation to inhibit tumor necrosis factor-α (TNF-α)-induced pancreatic β-cell apoptosis. FBW7 inhibits T1DM via the EZH2/ZBTB16 axis in vivo and in vitro.233
In addition, chronic hyperglycemia-induced oxidative stress can lead to ER stress and defective insulin secretion by disturbing the ubiquitin-proteasome system. Sections of pancreatic tissues from Zucker diabetic fatty rats show that a large number of Ub-proteins formed in the cytosol of pancreatic cells and β-cells. This response may promote autophagy to protect β-cells from cellular damage during hyperglycemia. Genetic deletion of thioredoxin-interacting protein (Txnip) in cells can increase protein ubiquitination of Xbp1, decrease gluconeogenesis and increase insulin sensitivity.234 An increased level of UBE2v1- and Lys63-ubiquitinated proteins was found in patients with T2DM, and the latter is involved in the pathological process of tubular damage in diabetic nephropathy.235
Several drugs can ameliorate diabetes by modulating protein ubiquitination. For example, inhibition of progestin and adipoQ receptor 3 (PAQR3) mediates STUB1-peroxisome-proliferator-activated receptor γ (PPARγ) protein ubiquitination and degradation to accelerate diabetic wound healing.236
Sumoylation in diabetes
SUMOylation is an evolutionarily conserved PTM in which a SUMO is covalently attached to the lysine (K) residue of target proteins.237 The SUMOylation process involves an activating enzyme 1 (E1, Uba2/Aos1), conjugating enzyme 2 (E2, UBC9), SUMO ligation enzyme 3 (E3, such as PIAS, RanBP2, and Pc2), and SENPs responsible for deSUMOylation. Sumoylation regulates diverse biological processes.
Type 2 diabetic patients with severe insulin resistance have lower UBC9 protein expression in skeletal muscle.227 Mice depleted of the unique SUMO conjugation E2 enzyme UBC9 in pancreatic beta cells spontaneously develop diabetes because of β cell death occurring as a result of the accumulation of reactive oxygen species.238 Gli-similar 3 (Glis3) is an insulin-regulated-associated transcription factor. Interestingly, PIAS-family proteins and UBC9 can sumoylate Glis3 to downregulate insulin transcription.239
The SUMO deconjugation enzyme SENP1 is involved in insulin secretion in T2DM and adipocyte inflammation in T1DM. SENP1 is localized with insulin granules in β cells, and deletion of SENP1 in β cells of mice impaired glucose tolerance.240 Adipocyte-specific deletion of SENP1 aggravated the SUMOylation of the NF-κB essential molecule (NEMO) and symptoms of T1DM.241
SUMOylation is associated with the incidence of T1DM in Asian populations.242 SUMOylation can also regulate β cell function to prevent the development of diabetes. Beta cells cultured in low glucose (2 mM) media show increased SUMOylation of MafA and interference with the transcription of the insulin gene. A high glucose environment increases the SUMOylation of PDX-1 to enhance insulin gene expression.243
SUMOylation affects insulin exocytosis. SUMO1 inhibits the voltage-dependent K(+) (Kv) channel Kv2.1, leading to decreased β-cell excitability and insulin exocytosis.244 SUMO1 blunts the exocytotic response of β-cells to Ca2+ to decrease glucose-stimulated insulin secretion.245
Based on this evidence, regulators of SUMOylation deserve additional study in the context of PTM and metabolic disease.
Neddylation in diabetes
Cullin neddylation is a process mediated by NEDD8-E1, E2, and E3 enzymes that sequentially transfer NEDD8 to a cullin protein. Inhibition of cullin neddylation rapidly decreases hepatic glucose generation, attenuates hyperglycemia and improves hepatic insulin signaling in mice. Dysfunction of Cullin 3 RING E3 ubiquitin ligase causes vasoconstriction and increased sodium reabsorption in diabetes64
Glycosylation in diabetes
Glycosylation includes glycosyltransferases and glycosidases. N-glycosylation is a subtype of glycosylation where polynucleotides and polypeptides are linked with asparagine by an N-glycosidic bond. Increased levels of highly branched N-glycans in plasma indicate an increased risk of diabetes.246
Reduced Glut-2 murine N-glycosylation and GlcNAcT-IV expression are associated with diabetes induced by a high-fat diet.247 GlycA is identified as a marker of systemic inflammation that originates from N-acetylglucosamine, and systemic inflammation may likely contribute to T2DM occurrence by causing insulin resistance and β cell dysfunction.248 Supplementation with sialic acid or the sialic acid precursor N-acetyl-D-mannosamine may restore anti-inflammatory properties and preserve insulin sensitivity.249
In other types of diabetes, fucosylated N-glycans are a novel biomarker of HNF1A-MODY, and N-glycans of human milk lactoferrin and secretory immunoglobulin A have been altered in gestational diabetes mellitus.250 Unlike the traditional forms of glycosylation, O-GlcNAcylation is an O-linked β-N-acetylglucosamine (O-GlcNAc) group covalently bound to threonine and/or serine residues of proteins. O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) control the dynamic cycling of O-GlcNAcylation. An early study found that the incidence and age of incidence of T2DM were linked with a region on chromosome 10q in the Mexican American population.251 The SNP of the enzyme O-GlcNAcase encoded by MGEA5 on 10q24.1-q24.3 may increase diabetes risk in Mexican Americans.252
OGT is highly expressed in islets. O-GlcNAcylation is essential for the function and survival of β cells. β-cell specific OGT-KO mice cannot maintain glucose homeostasis and regulate pancreatic β-cell function. OGA overexpression in β cells decreases insulin secretion and impairs glucose tolerance.253 Notably, the knockout phenotype of oga-1 (Oga-/-) is similar to human T2DM.254
Glucose increases the O-GlcNAcylation of Pdx-1 to increase its DNA binding to the A-box in the HR2 region of the GPR40 promoter to stimulate insulin secretion.255
Elevated O-GlcNAc-modified protein levels not only affect pancreatic islets but also affect kidney cells and cardiac, liver, muscle and fat tissues. The increased O-GlcNAcylation extent of cytoskeletal proteins (α-actin, α-tubulin, actinin 4, myosin) is associated with morphological changes in the diabetic kidney. Elevated O-GlcNAcylation in the liver can respond to hyperglycemia by accelerating gluconeogenesis/de novo lipogenesis through FoxO1, PGC-1α, CRTC2, carbohydrate-responsive element-binding protein (ChREBP) and liver X receptor (LXR).
GlcNAcylation can reflect the glycemic status at individual sites based on solid-phase chemical derivatization and chemoenzymatic tagging. O-GlcNAc modification can be a potential biomarker and assist in the identification of prediabetic patients.
Palmitoylation in diabetes
Protein palmitoylation is defined as the process by which palmitic acid molecules reversibly attach to cysteine residues via thioester bonds. Palmitoylation has been implicated in the metabolic dysregulation of islet β-cells. Previously, loss of the small GTPase ARF family member ARL15 gene reduced insulin secretion in a human β-cell line. ARL15 is located in the Golgi network, indicating a palmitoylation-dependent Golgi-based role.81 Phosphatidylinositol 4-kinase II-alpha (PI4KIIα) palmitoylation positively contributes to enhancing insulin signaling.256
Myristoylation in diabetes
Myristoylation is a crucial fatty acid acylation catalyzed by NMTs, which can add a myristoyl group to an amino-terminal glycine residue of a protein.84 STZ-induced diabetic animal results in a two-fold increase in liver NMT activity in animals and sodium orthovanadate can normalize liver NMT activity in STZ-induced diabetic rats. Furthermore, liver NMT activity is inversely proportional to plasma insulin levels. The effects of diabetes on NMT remain unclear.
Prenylation in diabetes
Prenylation includes protein farnesylation and geranylgeranylation, which are catalyzed by farnesyl transferase (FTase) and geranylgeranyl transferase (GGTase), respectively. Several pharmacological and molecular biological experiments have indicated that protein prenylation represents a committed step in glucose-stimulated insulin secretion. Mevalonic acid (MVA) is a precursor for the biosynthesis of FPP and GGPP. HMG-CoA reductase inhibitors (statins) inhibit the synthesis of MVA and sequentially inhibit GSIS. Knockdown of the FTase-β subunit suppresses insulin release.95
S-glutathionylation in diabetes
S-glutathionylation is a modification of cysteine and the disulfide bond formed only between the cysteine of protein. Protein S-glutathionylation mediates thiol redox signaling and likely plays a significant role in the pathogenesis of diabetes.106,108 S-glutathionylation at residue C215 of PTP1B can modify insulin signaling, leading to decreased activity. In addition to PTP1B, PTEN also undergoes S-glutathionylation and influences the PI3K-Akt pathway in hepatocytes from rats fed an HFD. S-glutathionylation, which is involved in the deactivation of Akt and downstream of Akt, is an inhibitor of IKKβ that can regulate insulin resistance in diabetes. Furthermore, S-glutathionylation of hemoglobin of diabetic patients was found to be increased in blood samples.257
S-nitrosylation in diabetes
Protein S-nitrosylation (SNO) is a reversible modification of cysteine thiols mediated by NO. S-nitrosylation plays an important role in the pathogenesis of insulin resistance. S-nitrosylation of insulin signaling molecules is elevated in patients with T2DM. Protein-nitrosylation was observed in diabetic rats and led to mitochondrial dysfunction. S-nitrosylation is involved in insulin resistance by activating and inactivating Akt. Nitric oxide (NO) inhibits the Abeta-degrading activities of insulin-degrading enzyme (IDE) through S-nitrosylation.258 Aspirin treatment reduces iNOS protein levels and S-nitrosylation of IRbeta, IRS-1 and Akt to improve insulin resistance and signaling.118
To date, multiple functions of protein S-nitrosylation have been associated with the fate of insulin. S-nitrosylation of Ryanodine Receptor 2 (RyR2) promotes calcium release and insulin secretion. Hyper-nitrosylation of RyR2 in β-cells impairs GSIS and blood glucose clearance.259 Sulfonylurea receptor (SUR) is a component of KATP channels, and its S-nitrosylation inhibits ATP binding. Gain-of-function SUR1 mutations lead to neonatal diabetes.260
Sulfhydration in diabetes
S-sulfhydration is similar to S-nitrosylation in some chemical features. H2S, a novel gasotransmitter, acts as a major donor for protein S-sulfhydration. S-sulfhydration modifies cysteines in proteins to mediate most cellular responses. H2S increased S-sulfhydration of kelch like ech associated protein 1 (KEAP1) and nuclear erythroid 2-related factor 2 (NRF2) nuclear translocation to attenuate diabetes-accelerated atherosclerosis.126
Citrullination in diabetes
Citrullination is catalyzed by the PAD enzyme family. A recent study has shown that daily hypodermic injections of BB-Cl-amidine (a pan-PAD inhibitor) can protect against the onset of diabetes in NOD mice.261 Moreover, citrullination caused by inflammation occurs almost exclusively in the pancreas and can be considered a marker of beta cell dysfunction or T1D.137
ADP-ribosylation in diabetes
ADP-ribosylation is a PTM catalyzed by the enzymatic transfer of ADP-ribose from NAD+ onto target proteins. This PTM usually occurs on cysteine, arginine and asparagine residues. PARP was demonstrated to be a pathogenic marker of diabetes and diabetic complications in vitro and in vivo. Hyperglycemia increases PARP activation in diabetic patients and decreases GAPDH and α-enolase enzymatic activity. NAD+ is used as a substrate for ADP ribosylation. Activated PARP can cause the depletion of NAD+ to destroy islet cells. The destruction of the PARP gene completely protected mice from diabetes.262
Carbonylation in diabetes
Carbonylation is induced by the oxidative stress generated by activated platelets. T2DM additionally enhances carbonylation of human platelet proteins. Rat model of T1DM loss the activity of SERCA2a and diastolic dysfunction occurs by carbonylation.154
PTMs in obesity
Obesity has much to do with excess calorie intake leading to excessive accumulation of adipose tissues. PTMs can regulate the activity of enzymes or cytokines connected with obesity, thereby engaging the treatment of obesity-related metabolic diseases. High fat and glucose levels can trigger the secretion of insulin from pancreatic β cells and PTMs of a series of kinases, generate fatty acids that are taken up by adipose tissue and induce obesity. (Fig. 4).

Overview of the roles of PTM in obesity. Obesity is associated with excess calorie intake leading to excessive accumulation of adipose tissues. PTMs can regulate the activity of enzymes or cytokines associated with obesity, thereby engaging the occurrence and treatment of obesity-related metabolic diseases. (1) High glucose levels, for example after ingestion of carbohydrates, trigger the secretion of insulin from pancreatic β cells and the activation of a series of kinases downstream of PI3K, such as Akt, PKC, and mTORC1, which stimulates glucose uptake and utilization, and generate FAs that are taken up by adipose tissue. (2) Activated transcription factors, including SREBP-1, PPARγ and PGC-1α, for transcriptional activation to promote de novo lipogenesis. (3) Some HDAC family members inhibit the thermogenic program in BAT to regulate HFD-induced leptin resistance and obesity. Activation and inhibition effects are displayed in “arrows” and “inhibitors”, respectively. The figure is generated with BioRender (https://biorender.com). AKT protein kinase B, ACL ATP citrate lyase, ACC acetyl CoA carboxylase, FFAs free fatty acids, FOXO1 forkhead box O1, G3P glyceraldehyde 3-phosphate, JNK c-Jun N-terminal kinase, mTOR mechanistic target of rapamycin, PIP2 phosphati-dylinositol-4,5-bisphosphate, PDK1 3-phosphoinositide-dependent protein kinase 1, PTEN phosphatase and tensin homolog, PGC1 peroxisome proliferators-activated receptorγcoactivator 1, PPARγ peroxisome proliferator-activated receptor
Phosphorylation in obesity
Obesity is a metabolic disease induced in its simplest manifestation by an imbalance between energy intake and expenditure. Chronic nutritional excess leads to adipocyte hypertrophy, which further promotes obesity-associated diseases.
Protein kinases regulate a number of biological processes by phosphorylation. Human GSK3β-overexpressing mice had greater body weight due to an increase in fat mass.263 Inactivation of GSK3β by Dyrk1A phosphorylation suppresses the expression of adipogenic proteins, potentially playing a part in the pathological process of obesity. Deficiency of MAP kinase interacting serine/threonine kinase 1 (MNK1) or MNK2 can protect against HFD-induced weight gain.264
Phosphatidylinositol-3,4,5-triphosphate [PtdIns(3,4,5)P3] is one of the most important phosphoinositides (PIs), and Akt is the most well-known target. The phosphorylation of AKT is catalyzed by PtdIns(3,4,5)P3. AKT-dependent FOXO1 phosphorylation decreased in DIO mice. Inhibition of hepatic atypical protein kinase C (aPKC) decreased excessive expression of lipogenic enzymes and improved weight gain.265 Activation of AKT2 mediates the stimulation of de novo lipogenesis. The phosphorylation of p66Shc is associated with obesity induced by excess nutrients.266
The deficiency of MARK4, an AMPK-related family member, enhances insulin-stimulated AKT phosphorylation to activate brown fat to diminish diet-induced obesity.267 Leptin, insulin and glucose adjust food intake by (de)phosphorylation of hypothalamic AMPK. Leptin regulation has been linked to PTP1B. Neuronal PTP1B knockout mice have lower body and adiposity weight, but adipose PTP1B deficiency increases body weight.268 JAK2 is a downstream effector of the leptin receptor, and its dephosphorylation depends on PTP1B to stimulate appetite-associated hormones. The biological activity of leptin can also be regulated by STAT3. STAT3 phosphorylation deficiency in the hypothalamus results in central leptin-induced resistance and obesity.269
The underlying causes of obesity may be cellular lipid and glucose imbalance or dysregulation. Further research on protein phosphorylation is required to determine its role as a target for treating obesity-associated metabolic diseases.
Acetylation in obesity
Protein acetylation is especially relevant to obesity. Protein acetylation is a component of a variety of metabolic reactions, such as glucose metabolism, the TCA cycle and fatty acid pathway. The dynamic regulation of white adipose tissue (WAT), brown adipose tissue (BAT) and beige adipose tissue can affect body obesity to a great extent. The levels of histone H3 lysine 9 and 18 acetylation at the Tnfα and Ccl2 genes are upregulated in obese mouse livers.270
Type I HDACs have deacetylation domains. Some studies demonstrated that HDAC1 inhibits the thermogenic program in BAT through the deacetylation of H3K27.271 Acute strenuous exercise can induce hyperacetylation of H4 and decrease HDAC2 activity in LPS-stimulated peripheral blood mononuclear cells (PBMCs) of obese males.272HDAC3 acts as a coactivator of oestrogen-related receptor α (ERRα) to maintain the capacity for thermogenesis in BAT by deacetylating PGC-1α, ERRα and UCP1.273,274 HDAC5 regulates HFD-induced leptin resistance and obesity via STAT3 deacetylation at Lys685 to improve the effect of leptin in the hypothalamus.275 HDAC6 and acetylated α-tubulin also control adipogenesis.276 In diet-induced obesity mice, the lipogenic transcription factor sterol regulatory element-binding protein-1 (SREBP-1) directly upregulated HDAC8 to promote insulin resistance.277 HDAC9 is associated with adipocyte differentiation and obesity. HDAC11 inhibits the expression of UCP1 in BAT to be a novel regulator of obesity.278
Type III HDACs include SIRT1-7. SIRT1 can accelerate the deacetylation of PPARγ to induce the browning of WAT.279 SIRT2 deacetylates the p65 subunit of NF-κB and RIP-1.280 SIRT3 and SIRT4 are located in mitochondria and regulate energy expenditure.281 SIRT5 and related acylation can reduce liver steatosis in ob/ob mice.282 SIRT6 is a FOXO1 deacetylase that drives lipid catabolism.283
As mentioned above, protein acetylation, energy metabolism and adiposity go hand in hand.
Methylation in obesity
An obesity study quantified histone methylation in diet-induced obesity mice. The study identified 4 glutamate methylation sites and 1 histidine methylation site with statistical significance. Among them, H2A E67me1 and H4 E74me1 might be associated with the pathological process of obesity.
Methylations of H3K4, H3K36, and H3K79 can activate transcription, while H3K9, H3K27, and H4K20 methylation can suppress transcription. Reversible histone methylation is catalyzed by histone methyltransferases (HMTs) and histone demethylases (HDMs), which have been shown to regulate energy metabolism.284
Lysine-specific demethylase 1 (LSD1) is the first identified HDM that demethylates H3K4 monomethylation/dimethylation (H3K4me1/me2) and can also reverse methylation of H3K9me1/me2.285
EHMT1 and EHMT2 are H3K9 methyltransferases. EHMT1 expression positively regulates brown adipose energy homeostasis by stabilizing the PRDM16 protein and depositing the suppressive H3K9me2 and H3K9me3.286 Specific deletion of adipose EHMT1 leads to obesity, systemic insulin resistance and adaptive thermogenesis.286 Lacking muscle-specific EHMT2 are resistant to high-fat diet (HFD)-induced obesity and hepatic steatosis in female mice.287 JMJD1A is another H3K9 demethylase that binds to the Ucp1 gene and decreases levels of H3K9me2 to regulate metabolic gene expression and obesity resistance.288
KMT5c catalyze the methylation of H4K20. Kmt5c knockout mice with decreased repressive marker H4K20me3 are obese when fed with an HFD and develop glucose intolerance.289
In the methylation of H3K27, enhancer of zeste homolog 2 (EZH2) promotes adipogenic differentiation, body weight and adipose tissue mass by catalyzing trimethylation of H3K27.290
Disruption of telomeric silencing-1 like (DOT-1L) regulates the BAT-selective gene program by promoting H3K79 methylation, especially H3K79me2 modification. Deletion of DOT-1L in thermogenic adipocytes can protect mice from diet-induced obesity.291
Ubiquitination in obesity
In obesity, adipocyte differentiation is controlled by numerous transcriptional cascades. PPARγ is a nuclear receptor that is associated with obesity and metabolic diseases by converting adipocytes from their precursors. E3 ubiquitin ligase-mediated protein ubiquitination and proteasome-dependent degradation of PPARγ gradually exhibited clear mechanisms in the development of obesity.
Ubiquitin-conjugating enzymes act as important factors affecting the process of ubiquitination. UBE2O is expressed preferentially in metabolic tissues. Ubc2o-/-mice showed a distinct reduction in overall fat mass and showed a reduction in body weight relative to their WT counterparts.229 Another conjugating enzyme simultaneously catalyzed ISGylation and ubiquitination reactions called UBE2L6, which were upregulated in WAT from obese humans and mice. Deficiency of adipose-specific Ube2l6 stabilizes ATGL protein and reduces HFD-induced obesity and insulin resistance.292
Knockdown of the E3 ubiquitin ligase IDOL in mice decreased circulating levels of cholesterol, triglycerides, hepatosteatosis and fat mass.293 Deletion of TRIM28 induces obesity via epigenetic mechanisms in embryonic development.294 TRIM72/MG53 deficiency targets the insulin receptor and IRS1 protein to attenuate HFD-induced obesity.295
PPARγ-mediated ubiquitination and degradation of lysine 150 of selenoprotein S (SelS) and lysine 47-48 of selenoprotein K (SelK) is required for adipocyte differentiation. CUL2-APPBP2 is the ubiquitin E3 ligase of the cullin–RING member family. CUL2 stabilized PRDM16 protein to repress adipocyte thermogenesis and counteracted diet-induced obesity by catalyzing its polyubiquitination.153 A study showed that fatty acid binding protein 4 (FABP4) was higher in the adipose tissues of obese diabetic patients. FABP4 regulates adipogenesis by downregulating PPARγ and attenuates the development of diet-induced obesity in mice.296
SUMOylation in obesity
The SUMOylation modification is reversible, in which the modified proteins can be deSUMOylated by SENPs. SUMO regulation is strongly associated with various diseases.
Depletion of UBC9 induces the expression of brown fat genes in human subcutaneous adipocytes.297
SUMO-specific protease SENP1 deficiency leads to hyper-SUMOylation of SIRT3, which can protect mice from HFD-induced obesity by increasing oxidative phosphorylation and energy expenditure.298 SENP2 deSUMOylated PPARα and promoted its ubiquitylation, which in turn inhibited FGF21 expression and fatty acid oxidation.299 Meanwhile, FGF21 null mice are lipodystrophy and have less body fat, which is associated with PPARγ SUMOylation at lysine 107.
Krüppel-like transcription factor 5 (KLF5) is also an essential regulator of lipid metabolism and is controlled by SUMOylation.300
Neddylation in obesity
NEDD8-based neddylation of PPARγ is crucial to conjugating and stabilizing PPARγ during adipogenesis and provides a potential anti-obesity therapeutic strategy that targets the neddylation of PPARγ. It remains to be investigated if the Neddylation signature in obesity is pathophysiologically relevant in obese animal models and human patients.
Glycosylation in obesity
N-glycosylation affects obesity-associated protein structure and function. Studies have shown that central obesity is involved in changes in IgG N-glycosylation. A low-calorie diet induced a marked effect on IgG N-glycosylation.301
O-GlcNAcylation of protein is a nutrient-sensing and cellular stress response. Excessive nutritional intake leads to metabolic disorders, including obesity and diabetes. Chronic ingestion of a high-fat diet can increase O-GlcNAc levels in cerebral arteries and the heart. It follows that there is a role for O-GlcNAc signaling in DIO and metabolic dysfunction. The levels of O-GlcNAcylation are determined by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). OGT targets adipose lipid desaturation to drive obesity, and the deletion of adipocyte OGT abolishes HFD-induced hyperphagia and obesity in mice.302 Loss of OGT can also decrease O-GlcNAcylation of lipid droplet-associated perilipin 1 (PLIN1), elevate PLIN1 phosphorylation and promote lipolysis in visceral fat to relieve diet-induced obesity.303
Palmitoylation in obesity
A palm oil-rich diet (HPD) causes obesity by inducing dynamic protein S-palmitoylation. It remains to be investigated if the palmitoylation signature is relevant to obesity in animal models and human subjects.
Myristoylation in obesity
Saturated FA activates Jun N-terminal kinase (JNK) by altering the membrane distribution of c-Src (a myristoylated protein) to affect obesity in mice and men.304
Prenylation in obesity
Mammalian geranylgeranyl diphosphate (GGPP) synthase catalyzed the synthesis of isoprenoid moieties for protein isoprenylation, whose expression is regulated in obesity and adipogenesis.305 Statins are a useful tool to investigate the role of prenylation in diabetes.
S-glutathionylation in obesity
S-glutathionylation, in which reactive oxygen species (ROS) react with cysteine residues of proteins to form glutathione (GSH), is removed by glutaredoxin-1 (Glrx). A previous study indicated that Glrx-lacking mice had increased protein S-glutathionylation and developed obesity; however, the mechanism is unknown.306 Another study indicated that deficiency of Glrx stabilized and increased C/EBPβ protein levels to stimulate 3T3L1 cell differentiation and adipogenesis.307
S-nitrosylation in obesity
In obese humans and DIO mice or ob/ob mice, total protein S-nitrosylation is increased. For example, the increased S-nitrosylation of PDE3B was detected in adipose tissue of DIO mice, which means PDE3B may be a specific target in adipocytes. S-Nitrosoglutathione reductase (GSNOR; alcohol dehydrogenase 5 [ADH5]) controls BAT homeostasis to regulate adipose thermogenesis.308
On the other hand, obesity-associated inflammation relates to endoplasmic reticulum dysfunction by S-nitrosylation. Obesity-increased inflammatory-associated iNOS activity causes S-nitrosylation of IRE1α (a key UPR regulator) and defective activity.309
Sulfhydration in obesity
Sulfhydration is a PTM of cysteine residues (RSH) to persulfides (RSSH) and is involved in H2S-based signal transduction.
H2S is a novel factor mediating obesity and associated metabolic diseases. Plasma H2S levels were proved to be reduced in overweight participants.310 Cystathionine g-lyase (CSE), cystathionine b-synthase (CBS), 3-mercaptopyruvate sulfurtransferase (3-MST) and selenium-binding-protein 1 (SELENBP1) are four principal mammalian H2S-generating enzymes. CSE is the most critical H2S-generating enzyme. The deletion of CSE in adipocytes can reduce adipose accumulation and enhance lipolysis by abolishing sulfhydration of plin-1.311 CSE, CBS, and 3-MST mRNA were reduced in the WAT of db/db mice, while only CSE was reduced in BAT.312
In differentiated human adipocytes, sulfhydration was increased in proteins participating in fatty acid metabolism and other metabolic signaling pathways.
ADP-ribosylation in obesity
ADP ribosylation was mediated by PARP family of enzymes. Physiological ADP ribosylation of histone H2B-Glu inhibits AMPK-mediated phosphorylation of adjacent H2B-Ser36, which is required for proadipogenic gene expression and fat metabolism programs.313
Carbonylation in obesity
During the development of obesity, levels of ROS in adipocytes are increased. These ROS products covalently modify the histidine, cysteine and lysine residues via protein carbonylation. Protein carbonylation levels are elevated threefold in the adipose tissue of diet-induced obese mice and obese human individuals.
Increased protein carbonylation has been linked to the antioxidant enzyme GSTA4 and the fatty acid-binding proteins. GSTA4 is decreased in obese mice and humans. Carbonylation of FABP4 on Cys-117 results in loss of fatty acid binding activity in mice. Treatment of TNF-α in 3T3-L1 adipocytes can decrease GSTA4 expression and increase protein carbonylation similar to obese states.314 GSTA4-null or accumulated ROS in obese C57BL/6J mice results in impaired glucose and lipid homeostasis.315
The accumulation of ROS is linked to adipose oxidative stress in the obese state. Extensive evidence has demonstrated that the accumulation of 4-hydroxynonenal (HNE) acts as a symbol of oxidative stress, which usually occurs in the blood and tissue of obese/diabetic patients.
S-sulfenylation in obesity
Brown and beige adipose tissues need uncoupling protein 1 (UCP1) to execute thermogenesis and Cys253 of UCP1 is sulfenylated during thermogenesis, which provides the strategy to improve therapeutic strategies for combating metabolic disorders.316,317
PTMs in fatty liver diseases
Various novel signaling pathways in fatty liver have been identified. Many proteins have multiple modification sites to modulate lipid synthesis, lipolysis, and fatty acid β-oxidation by phosphorylation, acetylation or SUMOylation of key substrates, such as ACC, SREBPs, GPAT, and farnesoid X receptor (FXR). Current efforts are concentrated on exploring the molecular mechanism of the interrelationship between fatty liver disease and PTMs of important factors. Here, we summarize the different effects of PTMs in the pathogenesis of NAFLD (Fig. 5).

Overview of the roles of PTM in fatty liver diseases. Non-alcoholic fatty liver disease (NAFLD) is characterized by fat accumulation in the liver. We summarize the diverse roles of PTMs in the pathogenesis of NAFLD to enrich the understanding of the molecular mechanisms of the intricate interrelationship between post-translational modification (PTM) of important factors and fatty liver disease. (1) Multiple endogenous and exogenous stimuli can lead to aberrant dynamic proteins posttranslational modifications to result in chronic liver disease. (2) Many proteins have multiple modification sites to modulate lipid synthesis, lipolysis, and fatty acid oxidation by phosphorylation, acetylation or SUMOylation of key substrates, such as ACC, SREBPs, GPAT and FXR. Activation and inhibition effects are displayed in “arrows” and “inhibitors”, respectively. The figure is generated with BioRender (https://biorender.com). AKT protein kinase B, ACLY ATP citrate lyase, ACC acetyl CoA carboxylase, ChREBP carbohydrate response element binding protein, FFAs free fatty acids, FASN fatty acid synthase, FXR farnesoid X receptor, FGF19 fibroblast growth factor 19, FATP fatty acid transport protein, GSK-3 glycogen synthase kinase 3, JNK c-Jun N-terminal kinase, MEKK mitogen-activated extracellular signal-regulated kinase kinase, Nck non-catalytic region of tyrosine kinase adaptor protein, NEFA non-esterified fatty acids, PAK p21-activated kinase, SREBP1 sterol-regulatory element binding protein 1, STAT5 signal transducer and activator of transcription 5, SCD1 stearoyl-CoA desaturase 1, TRAF TNF receptor associated factors
Phosphorylation in fatty liver disease
Protein kinases and phosphatases dynamically regulate protein phosphorylation. Protein kinases (PKs) families contain MAPK, ErbB, PKA-PKD, PI3K/Akt, and mTOR, acting on multiple downstream key protein targets in NAFLD and regulating hepatic gluconeogenesis, lipogenesis and inflammation. AMPK has multiple phosphorylation sites to modulate lipid synthesis, lipolysis, and fatty acid oxidation by phosphorylation of ACC, SREBPs, GPAT and so on. ACC can catalyze the carboxylation of acetyl-CoA to malonyl-CoA, which acts as an important regulatory site of fatty acid synthesis and oxidation pathways. The Ser79, Ser1200 and Ser1215 sites of ACC can be phosphorylated by AMPK. SREBPs directly promote genes involved in fatty acid uptake and triglyceride (TG) synthesis by phosphorylation. Sn-Glycerol-3-phosphate acyltransferase (GPAT) is the rate-determining enzyme that catalyzes TG synthesis. HSL catalyzes the rate-limiting step in TG hydrolysis. AMPK inhibits HSL by phosphorylating its Ser660 and Ser563 sites, thus suppressing lipolysis in adipocytes.318
Mitogen-activated protein kinases (MAPKs) include stress-responsive MAPKs, JNK, and p38 MAPK. and ERK1/2. JNK can be phosphorylated by MAPKK and MAPKKK. Lipid accumulation can enhance hepatic JNK, which contributes to liver injury.319 JNK-1 knockout mice showed reduced liver steatosis and TG accumulation.320 The activation of p38 MAPK promotes the expression of PPARα, CPT1A, and PGC-1α to suppress hepatic fat accumulation. PKC is increased accompanied by high hepatic lipid content in obese mouse models. PKC can directly phosphorylate IRS to reduce downstream insulin signaling.321 Akt signaling is triggered by activated-PI3K and requires phosphorylation of Thr308 and Ser473 via PDK-1 and mTORC2. Activated AKT-1 promotes the levels of transcription factors involved in DNL and increases the intracellular lipid content.322 High-fat diets can decrease the ratio of p-AKT/AKTt but increase the expression of SREBP1, LXR, ChREBP, ACC1, and fatty acid synthase (FASN).323
Acetylation in fatty liver disease
Proteomic analyses have identified a large number of acetylated proteins involved in intermediate metabolism. Reversible acetylation is controlled by acetyltransferases (KATs) and deacetylases (HDACs and SIRTs). Protein acetylation can regulate metabolism in chronic liver diseases. Hyperacetylated-LDHB has been detected in NAFL and NASH human samples.324
The zinc finger protein Snail1 recruits HDAC1/2 to induce the deacetylation of H3K9 and H3K27 to repress lipogenesis.325 HDAC3 controls the circadian rhythm of hepatic lipogenesis.326
SIRT1, an NAD+-dependent protein deacetylase, regulates lipid homeostasis by positively regulation of peroxisome proliferators-activated receptor α (PPARα). Exenatide (exendin-4) can improve hepatic steatosis via the SIRT1/heat shock factor 1/HSP pathway.327 SIRT2 regulated hepatic steatosis by HNF4α deacetylation.328 A natural chemical compound 2,3,5,4’-tetrahydroxy-stilbene-2-O-β-d-glucoside (TSG) reduces ROS formation and increases SIRT5 expression in mitochondria to ameliorate NAFLD.329 USP10 inhibits hepatic steatosis and inflammation by interacting with SIRT6.329 In mitochondria, MRG15 interacts with and deacetylates TUFM, which accelerates effects altogether and drives the progression from NAFLD to NASH with inflammation and fibrosis.330
Methylation in fatty liver disease
Aberrant histone methylation also takes participate in the process of CLD. Downregulation of glycine N-methyltransferase (Gnmt) occurred in the early stage of pathogenesis of NAFLD and promotes the development of NAFLD-derived HCC. Histone 3 lysine 9 methyltransferase enzyme (G9a) is downregulated in diet-induced animal models of obesity. Transgenerational HFD feeding reduces the accumulation of H3K9 histone methyltransferase in the LXRα and ERO1-α gene promoters and activates ChREBP-mediated both glycolytic and fatty acid synthesis.331
Histone demethylase Jumonji domain-containing protein 2B (JMJD2B) removes histone marks (H3K9me2 and H3K9me3) near the LXR response elements (LXREs) to play a role in liver X receptor α (LXRα)-mediated lipogenesis and contribute to hepatic steatosis. In addition, histone demethylase plant homeodomain finger 2 (Phf2) regulates H3K9me2 demethylation at carbohydrate-responsive element binding protein (ChREBP) to prevent NAFLD progression.332
Ubiquitination in fatty liver disease
Ubiquitination is an important PTM to cope with abnormally folded or damaged proteins which exert diverse functions in chronic liver disease.
E3 Ub ligases and DUBs play important roles in protein ubiquitination and deubiquitination. The activation of E3 ubiquitin ligase Ubr1, inducing polyubiquitination of Plin2 to prevent steatosis in mouse livers.333 E3 ubiquitin ligase-tripartite motif-containing protein 31 (TRIM31) promotes degradation of Rhbdf2 by K48-linked polyubiquitination to alleviate NAFLD in mouse hepatocytes.334 Knockout of Kindlin-2 destroys the stability of Foxo1 by promoting its ubiquitination and degradation through Skp2 E3 ligase-dependent ubiquitination, which can protect against fatty liver.335 Ring finger protein 5 (RNF5) can directly combine with HRD1 and promote ubiquitination and degradation to inhibit NASH progression.336 Moreover, sorting nexin 8 (SNX8) promoted FASN protein proteasomal degradation and protected NAFLD by recruiting the E3 ligase tripartite motif containing 28 (TRIM28).337 The E3 Ub ligases TRIM8 and TRIM16 can directly bind to TAK1 to promote its phosphorylation and activate JNK/p53 and NF-κB signaling. TRIM8 and TRIM16 can mitigate hepatic steatosis and fibrogenesis in NASH.338 The E3 ligase FBXW5 mediates ASK1 ubiquitination and exacerbates NASH.339 Liver and adipocytic MKRN1 is an E3 ubiquitin ligase for AMPK. MKRN1-null mice can suppress diet-induced metabolic syndrome.340
DUBs catalyze the removal of ubiquitin from protein substrates in the process of ubiquitination. Hepatic USP4 is directly bound to deubiquitinated TAK1, leading to amelioration of metabolic dysfunction.341 The USP7/ZNF638 axis mediates de novo lipogenesis.342 Ubiquitin-specific peptidase 10 (USP10) decreases over time in patients with NAFLD and in HFD-fed mice. USP10 can interact with Sirt6 and inhibit its ubiquitination and degradation to inhibit hepatic steatosis and inflammation.343 USP14 expression has been revised upwards in the livers of HFD, db/db mice and NAFLD patients and plays an indispensable role in hepatosteatosis via stabilization of FASN.344 Moreover, USP14 deubiquitinates HIF1-α to maintain its stability in hepatocellular carcinoma.345 The deubiquitinase cylindromatosis (CYLD) interacts with TAK1 and removes its K63-linked polyubiquitin chain to mitigate NASH.346
SUMOylation in fatty liver disease
An increasing body of evidence suggests that SUMOylation is closely associated with the progression of liver diseases. For example, UBC9 is the only known E2-conjugating enzyme involved in SUMOylation to regulate hepatic fibrosis. The SUMO-1-conjugating enzyme UBC9 reduces the transcriptional activity of two sumoylation sites in SREBP-1a to inhibit lipid production.347 In addition, small ubiquitin-related modifier (SUMO) E3 ligase sumoylates SREBP1c at Lys98, reinforcing the interaction between SREBP1c and PIASγ, which can regulate hepatic lipid metabolism during nutritional deprivation.348
The de-SUMOylation enzyme SENP2 decreased CCl4-induced mouse fibrosis in liver tissues. Last but not least, liver receptor homolog 1 (LRH-1) is an important regulator of hepatic metabolism. The SUMOylation-defective mutant of LRH-1 mice developed NAFLD and early symptoms of NASH under the regulation of OSBPL3 when fed a high-fat and high-sucrose diet.349
Neddylation in fatty liver disease
Recently, numerous mechanistic studies have been performed to elucidate the crucial role of neddylation in lipid metabolism. SRSF3 degradation via lysin11 neddylation partially protects mice from NAFLD and deletion of SRSF3 predisposes to hepatocellular carcinoma in mice.350 In addition, dysregulation of NRF2 by neddylation of cullin 3 was connected with AGER1 downregulation and NASH aggravation.351 It has been demonstrated that neddylation inhibition in vivo in NAFLD pre-clinical models inhibits mTOR activation and induced protein DEP-domain containing mTOR-interacting protein (DEPTOR), thus mediating anti-steatotic effects as well as boosting hepatic fatty acid oxidation.352
More recently, neddylation was described for the first time in liver fibrosis and is found to be deregulated in patients with liver fibrosis and CCl4-induced fibrosis mice.353 Preventing the neddylation-dependent degradation of serine-rich splicing factor 3 (SRSF3) protected mice from hepatic steatosis, fibrosis and inflammation.350 The neddylation modification of TGFβ-RII plays a critical role in HSC activation.353,354
The components of the neddylation pathway may become novel biomarkers for CLD diagnosis.
Glycosylation in fatty liver disease
Recent studies have indicated that many proteins involved in glycosylation play a part in the pathogenesis of NAFLD.
Recently, glycosyltransferase 8 domain containing 2 (Glt8D2) expression was found to be increased in patients with severe NAFLD.355
O-GlcNAcylation involving in liver metabolism by suppressing insulin signaling and activating lipogenic pathways. Protein O-GlcNAcylation was increased in NAFLD and NASH mice during lipid accumulation. Furthermore, O-GlcNAcylation can also modify the ChREBP and FXR in the liver.356 ChREBP is a key regulator of glycolysis and lipogenesis. O-GlcNAcylation can increase the ChREBP protein level.357
Palmitoylation in fatty liver disease
The latest research detected the upregulated palmitoylation of FAT/CD36 in NAFLD. Suppressed FAT/CD36 palmitoylation promotes FAT/CD36 localization change and avoids lipid accumulation in NAFLD.358
Prenylation in fatty liver disease
Protein prenylation includes protein farnesylation and geranylgeranylation. Abnormal expression of geranylgeranyl diphosphate synthase (GGPPS) breaking the balance of protein farnesylation and geranylgeranylation. The high expression of GGPPS was detected in the livers of NAFLD patients.359
Glutathionylation in fatty liver disease
Glutathione S-transferase π (GSTπ) is shown to promote S-glutathionylation. The decreased expression of GSTπ reduces protein S-glutathionylation and prevents hepatic lipid accumulation during liver development.360
S-nitrosylation in fatty liver disease
S-nitrosylation (SNO) can promote the conversion of NAFLD to NASH via the peroxisome PPARγ/SFRP5 pathway.361
Sulfhydration in fatty liver disease
S-sulfhydration forms a hydro persulfide moiety (-SSH) or polysulfide in the active cysteine residues. According to a prediction, one-third of proteins could be modified forming S-sulfhydration products. In a previous study, H2S showed a protective effect in HFD-induced NAFLD or CDA-induced NASH. CSE plays an important role in the methionine trans-sulfuration pathway. CSE/H2S promoted a protein sulfhydration of FXR at Cys138/141 sites to promote FXR activity and attenuate NAFLD. Deficiency of CSE in the liver promotes the pathological process of nonalcoholic steatohepatitis.362 Cystathionase (CTH) is another metabolic enzyme to mediate the synthesis of H2S in the liver. SREBF1/SREBP-1c was activated under an HFD-induced liver steatosis model. The upregulated Mir216a transcription directly decreased CTH-H2S sulfhydration signaling and ULK1-stimulated autophagy to promote hepatic steatosis.363
Strikingly, S-sulfhydration of Keap1 was decreased in the liver of NAFLD patients. H2S plays a protective role depending on the S-sulfhydration of Keap1 to alleviate liver damage through enhanced Nrf2-mediated antioxidant responses.364
ADP ribosylation in fatty liver disease
PARP1 is activated in the liver of HFD-fed mice and suppressed PPARα signaling.365 A PARP inhibitor olaparib reversed NAFLD by NAD+ elevation, increasing mitochondrial biogenesis and β-oxidation in liver under HFHS diet.366
Carbonylation in fatty liver disease
Protein carbonylation is associated with metabolic effects. Examining fatty nonalcoholic steatohepatitis carbonylated proteins by functional enrichment analysis, increased carbonylation was evident in proteins regulating Rho cytoskeletal pathways, nicotinic acetylcholine receptor signaling and chemokine/cytokine inflammatory pathways.367 Disruption of PTEN resulted in steatohepatitis and fibrosis in mice, but elevated Nrf-2 responses are not enough to relieve protein carbonylation in hepatocyte-specific PTEN deficient mice.368
PTMs and their roles in other metabolic conditions and diseases
PTMs in hyperlipidemia
Hyperlipidemia is a state of elevated levels of fats and lipids in the blood (TGs, cholesterol, or both), encompassing numerous genetic and acquired disorders. TG is the most popular and effective type of energy storage in animal tissues and originates from both dietary intake and endogenous (liver) generation.369 Cholesterol is the principal sterol in mammals and is mainly derived from dietary sources and partly endogenous synthesis by the liver and other tissues.369 Briefly, a range of apolipoproteins package TGs and cholesterol into lipoproteins, which are transported in vessels. Plasma lipoproteins are grouped into chylomicrons (CM), very low-density lipoprotein (VLDL), low-density lipoprotein (LDL), and high-density lipoproteins (HDL) based on size, density and apolipoprotein content.370
Exogenous TGs are converted to glycerol and fatty acids; the fatty acids are employed for energy metabolism (β oxidation of fatty acids) or storage. Extra energy is stored in the liver and adipose tissues as triglycerides via the fatty acid pathway.370 Fatty acids can also be produced from carbon sources undergoing several enzymatic processes known as de novo lipogenesis (DNL). Surplus cellular energy induces an increase in mitochondrial citrate, initiating DNL. Mitochondrial citrate travels across the plasma membrane with citrate/isocitrate carrier (CIC) and triggers synthesis through ATP-citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), FASN, and downstream lipid processing with stearoyl-CoA desaturase 1 (SCD1) and DGAT.371 SREBPs, LXRs, and ChREBPs mediate the transcription of CIC, ACLY, ACC, and FASN.372 Liver-derived lipoproteins travel through the bloodstream carrying endogenous triglycerides for uptake by peripheral tissues.
Dietary cholesterol is absorbed in the intestine, packaged and released as chylomicrons, and finally returns to the liver. Cholesterol biosynthesis originates from acetyl-CoA and requires more than 20 enzymes. Cholesterol biosynthesis mainly localizes in ER, where the liver is the leading site of biosynthesis. 3-Hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) and squalene monooxygenase (SM) are two rate-limiting enzymes, and SREBP2 is the master transcriptional regulator. Livers deliver endogenously produced and exogenously absorbed cholesterol to the blood with VLDL, after which VLDL gradually changes into circulating LDL for intake by peripheral tissues. HDL transports peripheral cholesterol back to the liver and intestine during the reverse cholesterol transport (RCT) process. Cholesterol participates in bile acids synthesis in the liver or contributes to steroid hormone synthesis. Excess cholesterol is esterified through acyl-coenzyme A:cholesterol acyltransferase (ACAT) for storage or release with lipoproteins.373
Excess intake and synthesis or insufficient catabolism of lipids will lead to hyperlipidemia. The metabolism of lipids is intricately regulated by various molecules and enzymes, involving abundant posttranslational modifications. As shown in Fig. 6, de novo lipogenesis, β-oxidation, cholesterol reverse transport, bile acid synthesis and lipoprotein transport are involved in lipid metabolism. Various PTMs play vital roles in regulating lipid homeostasis through diverse mechanisms. There is an incomplete understanding of PTMs in regulating lipid metabolism, and we thus will highlight the roles of common PTMs in lipid homeostasis and hyperlipidemia.

Diverse roles of PTMs in hyperlipidemia. Excessive intake and synthesis or insufficient catabolism of lipids lead to hyperlipidemia. De novo lipogenesis (DNL), reverse cholesterol transport (RCT), fatty acid β-oxidation, lipolysis, bile acid synthesis, and lipoprotein-related-regulation contribute to lipid homeostasis at multiple levels. We summarise the role of crosstalk among PTMs in regulating lipid metabolism. (1) During hepatic DNL, abundant PTMs such as phosphorylation (AMPK), (de)acetylation (SIRT1/2/6), methylation (EZH2, PRMT5), ubiquitination (FBW7, ASGR1), SUMOylation (SUMO1, SENP1, UBC9, PIAS1), sulfhydration (NsHS/H2S), citrullination (L-citrulline-AMPK) and ADP ribosylation (PARP2) can regulate the expression of ACLY, ACC, FAS, SCD via transcription factor mTOR, LXR, SREBP and CHREBP. (2) Fatty acid beta-oxidation. ADP ribosylation (PARP1) inhibits beta-oxidation via PPARα. (3) Lipolysis. LDL transports fatty acid from the liver to peripheral tissues. In adipose tissues, acetylation (SETDB1), ADP ribosylation (PARP1), sulfhydration (H2S) and phosphorylation (AMPK) modulate the lipolysis by regulating key enzyme HSL or transcription factor PPARγ. (4) Reverse cholesterol transport (RCT). SUMOylation (SUMO-LRH1), S-nitrosylation (NO-PON1-HDL), citrullination and ADP ribosylation regulate RCT by modifying HDL or relevant enzymes. (5) Bile acid transport. Ubiquitination (ASGR1) and glutathionylation (GSH) govern cholesterol excretion by promoting bile acid synthesis. (6) LDLR. Hepatic LDLR mediates LDL uptake and the subsequent degradation, abolishing excess serum LDL, which is inhibited by PCSK9-mediated LDLR degradation. Several PTMs such as phosphorylation acetylation, methylation, S-nitrosylation, ubiquitination, SUMOylation and glutathionylation could regulate LDLR functioning. Acetylation and ubiquitination modulate LDLR function via the SIRT6-PCSK9-LDLR axis and LXR-IDOL-LDLR axis. The red arrows indicate the triglyceride (TG) pathway, the blue arrows indicate the cholesterol (CHO) pathway, and the black arrows indicate the roles of PTM-related molecules in hyperlipidemia. The figure is generated with BioRender (https://biorender.com). ABCA1 ATP binding cassette transporter, ABCG1 ATP binding cassette transporter, ACC acetyl-CoA carboxylase, ACLY ATP-citrate lyase, AMPK AMP-activated protein kinase, ASGR1 asialoglycoprotein receptor 1, Apo apolipoprotein, CM chylomicrons, CYP7A1 cholesterol 7α-hydroxylase 1, ChREBP carbohydrate-responsive element-binding protein, EZH2 enhancer of zeste homolog 2, FAS fatty acid synthase, FFA fatty acid, GSH glutathione, H2S hydrogen sulfide, HDL high-density lipoprotein, HMGCR 3-Hydroxy-3-methylglutaryl coenzyme A reductase, HSL hormone-sensitive lipase, IDOL inducible degrader of LDLR, LDL low-density lipoprotein, LRH-1 liver receptor homolog-1, LXR liver X receptor, PARP poly (ADP-ribose) polymerase, PGC1α PPARγ coactivator 1α, PIAS1 protein inhibitor of activated STAT 1, PPAR peroxisome proliferator-activated receptor, PRMT protein arginine methyltransferase, SCD stearoyl-CoA desaturase, SENP1 sentrin/SUMO-specific protease 1, SETDB1 SUMOylated SET domain bifurcated 1, SIRT sirtuin, SR-BI scavenger receptor class B member 1, SREBP sterol regulatory element-binding protein, VLDL very low-density lipoprotein, mTOR mammalian target of rapamycin complex, PTMs post translational modifications
Phosphorylation in hyperlipidemia
Protein phosphorylation contributes to lipid metabolism by regulating critical substrates in lipolysis, lipid biosynthesis, and fatty acid β oxidation. AMPK, an energy-regulating kinase, regulates several critical molecules involved in lipid metabolism, such as ACC, SREBP, and some key enzymes.
ACC is a vital site in fatty acid biosynthesis and oxidation pathways with ACC1 and ACC2 isoforms. ACC1 mediates the process through which acetyl-CoA converses into malonyl-CoA in the synthesis pathway, and ACC2 suppresses carnitine palmitoyltransferase 1 (CPT1) to decrease β-oxidation. AMPK phosphorylates ACC1 at Ser79, leading to the inactivation of ACC1 and reduction in fatty acid synthesis.18 AMPK phosphorylates ACC2 at Ser219, resulting in the inhibition of ACC2 and increased fatty acid oxidation.374
SREBPs include three isoforms, SREBP1a, SREBP1c, and SREBP2, promoting related gene expression in lipid synthesis. AMPK phosphorylates SREBP1c at Thr426, Ser410, Ser430, or Ser372, inhibiting the expression of SREBP1c and suppressing TG synthesis.375 AMPK can also inhibit SREBP1c by reducing the transcription of the mTORC376 and the subsequent expression of FASN. AMPK can directly restrain SREBP2 by phosphorylation, repress HMGCR and cholesterol synthesis, and ameliorate dyslipidemia.375
Hormone-sensitive lipase (HSL) is the key rate-limiting enzyme catalyzing TG hydrolysis. AMPK agonist AICAR can suppress HSL activity by phosphorylation at Ser565 and inhibit lipolysis in adipocytes.318
HMGCR is a crucial rate-limiting enzyme in cholesterol synthesis. The balance between phosphorylation and dephosphorylation is suggested to influence HMGCR activity. AMPK inactivates HMGCR by phosphorylating Thr172, but AMPK activates HMGCR by phosphorylating Ser872 and promotes hypercholesterolemia.377,378
The role of protein kinases in lipid metabolism is widely known. Further exploration of novel therapeutic strategies targeting protein phosphorylation and protein kinases is essential for designing novel lipid-lowering therapies.
Acetylation in hyperlipidemia
Acetylation can markedly alter protein function through changes in hydrophobicity, solubility, and surface properties. Sirtuins (class III HDACs) are a group of remarkably conserved NAD+-dependent deacetylases catalyzing deacetylating reactions in crucial proteins related to lipid metabolism. Sirtuins are suggested to restrain lipogenesis and prevent lipid accumulation by promoting fatty acid beta-oxidation or exporting surplus lipids.
SIRT1 can deacetylate SREBP1c and cause SREBP1c ubiquitination and degradation, suppressing fatty acid and cholesterol synthesis.379 SIRT1 induces PPARα activation and enhances fatty acid beta-oxidation.380 SIRT2 is suggested to deacetylate ACLY in the liver381 and suppress PPARγ coactivator 1α (Pgc1α) in adipose tissue382 to inhibit lipid accumulation. SIRT6, regulated by SIRT1, promotes the acetylation of histone H3 lysine 9 to inhibit TG synthesis.27 Hepatic SIRT6 can inhibit PCSK9 transcription, preventing LDLR breakdown and thereby lowering plasma LDL levels in mice.383 Large yellow tea extract lowers blood lipids in leptin receptor knockout mice by inhibiting lipogenesis via activating the SIRT/SREBP pathway.384
The discovery of novel mechanisms of deacetylases regulating lipid homeostasis is an active field of exploration. Further identification of novel agents targeting protein acetylation and the related enzymes is warranted.
Methylation in hyperlipidemia
Protein methylation is a contributor to numerous human diseases. Methyltransferase (lysine methyltransferase, arginine methyltransferase) and demethyltransferase are the enzymes that regulate protein methylation.
Lysine methyltransferase is involved in lipid metabolism. LncRNA PU.1 AS can reduce plasma TG and hepatic TG by interacting with EZH2 (a histone-lysine N-methyltransferase) and decreasing lipogenesis via the EZH2/SIRT6/SREBP1c pathway.385
Arginine methyltransferase can also regulate lipid metabolism. Butyrylcholinesterase (BChE) interacts and colocalizes with PRMT5, regulating LDLR-mediated LDL intake via the MEK-ERK pathway. BChE-deficient mice exhibit PRMT5 degradation and are susceptible to hypercholesterolemia.34 PRMT5 can mediate symmetric arginine demethylation. PRMT5 has been demonstrated to promote SREBP1 SDM, activating enzymes regulating cholesterol synthesis386 and fatty acid synthesis.387
A better understanding of protein methylation in lipid metabolism is required to support the development of medicines for regulating lipid profiles.
Ubiquitination in hyperlipidemia
Multiple factors, such as transcription, translation and enzymatic activity, are involved in lipid homeostasis. Recently, evidence of the role of ubiquitin ligases in lipid metabolism has been provided. Here, we will describe the role of E3 ubiquitin ligases.
IDOL, the individual E3 ubiquitin ligase, regulates LDLR degradation to induce hypercholesterolemia through the LXR-IDOL-LDLR axis.388,389 IDOL regulates circulating lipid metabolism and the development of atherosclerosis independent of LDLR function.389 A few E3 ligases regulate SREBP. ITCH can degrade SIRT6 via ubiquitination to promote fatty acid beta-oxidation. ITCH deficiency can disturb nuclear SREBP clearance and decrease circulating cholesterol levels.390 FBW7, a member of the SKP1-cullin-1-F-box (SCF) complex ubiquitin ligases, regulates SREBP degradation, which is negatively mediated by microRNA-182.391 E3 ligase TRIM72/MG53 regulates the ubiquitin-dependent degradation of IRS1 and insulin receptors. TRIM72/MG53 deficiency would alleviate insulin resistance and hyperlipidemia in HFD-mice, while TRIM72/MG53 overexpression would aggravate insulin resistance and hyperlipidemia.295
Some E3 ligases play a role in cholesterol metabolism. Human TEB4, an E3 ligase resident in the ER membrane, degrades squalene monooxygenase (SM) to inhibit cholesterol synthesis.392 Glycoprotein 78 (gp78) can enhance the ubiquitinated degradation of HMGCR,393 insulin-induced gene 1 protein (INSIG1)394 and ApoB-100.395 Hepatic gp78 deficiency alleviates hyperlipidemia and insulin resistance through reducing lipid biosynthesis.
Recently, Song and colleagues found that inhibition of asialoglycoprotein receptor 1 (ASGR1) would upregulate ABCA1, ABCG5/ABCG8, LXRα, suppress SREBP and lipogenesis, subsequently facilitate cholesterol excretion and alleviate hyperlipidemia.45 Mechanistically, ASGR1 deficiency decreased LXRα ubiquitination, whereas overexpression of ASGR1 increased its ubiquitination.45
The role of protein ubiquitination of the UPS system in lipid metabolism is recognized. Further investigation of novel therapeutic approaches regulating E3 ligases and the UPS system may hold promise for the treatment of hyperlipidemia.
SUMOylation in hyperlipidemia
SUMOylation, a SUMO-mediated protein modification, influences many cellular processes by affecting protein stability, activity, interactions and cellular localization; accordingly, SUMOylation has been identified to participate in lipid homeostasis.
SUMO regulates cholesterol homeostasis involving cholesterol synthesis, ingestion, transport and bile acid metabolism. SUMOylated LRH-1 recruits corepressor prospero-related homeobox protein 1 (PROX1) and suppresses LRH-1-dependent genes associated with RCT.396 FXR, a bile-acid-stimulated nuclear receptor, restrains the biosynthesis and transport of bile acid through small heterodimer partner (SHP) and LRH-1. SUMO1 decreases the attachment of FXR to the promoter of SHP, inhibiting bile acid synthesis and transport.397 UBC9, the E2 SUMO-conjugating enzyme, can interact with SUMOylate SREBP2 at the lysine463 site, suppressing SREBP2 transcriptional activity and repressing cholesterol synthesis.347 Protein inhibitor of activated STAT 1 (PIAS1), with SUMO E3 ligase activity, inhibits LXR-dependent fatty acid synthesis and lipogenic genes, including Srebp1 and Fasn.398
SENP, mediating deSUMOylation, can also regulate lipid metabolism. SENP1 can raise LDLR expression through deSUMOylation.56 SUMOylated SET domain bifurcated 1 (SETDB1) inhibits the expression of Pparg and Cebpa (CCAAT Enhancer Binding Protein Alpha). SENP2 deficiency induces the accumulation of SUMOylated SETDB1, repressing the expression of Pparg and Cebpa, thus decreasing lipid storage in adipocytes.399
Further elucidation of protein SUMOylation may yield novel therapeutic targets for the treatment of hyperlipidemia.
Glycosylation in hyperlipidemia
Glycosylation of human lipoproteins exhibits high diversity, playing crucial roles in regulating lipoprotein metabolism.
N-glycosylation regulates proteins involved in lipid synthesis, package, and the abolition of lipoproteins. N-glycosylation in humans reduces LDL through enhanced LDLR expression.72 Glucose-induced N-glycosylation maintains SREBP cleavage-activating protein (SCAP) stability and decreases its interaction with INSIG1, permitting SREBP activation and downstream gene transcription.400 SCARB1 codes scavenger receptor class B member 1 (SR-BI), the primary receptor in selective HDL uptake into the liver. A loss-of-function genetic variant in SCARB1 (leucine replaces proline 376) induces altered N-glycosylation, increasing HDL-cholesteryl ester and preventing its uptake into hepatocytes due to decreased SR-BI.401
O-glycosylation links GlcNAc and GalNAc to target proteins. Site-specific glycan profiles of HDL-related ApoE are closely involved in HDL activity and function.402 The galNAc-T2 enzyme (encoded by GALNT2) catalyzes the GalNAc linkage in O-glycosylation. The GALNT2 variant has been shown to affect HDL and triglyceride levels in human genetics studies.403 However, reduced GalNAc-T2 function was related to increased HDL.404 Others demonstrated that GalNAc-T2 function defect reduced plasma HDL,405 which made the role of O-glycosylation in HDL metabolism somewhat mysterious. However, GALNT2 has been shown by glycoproteomics to serve as the inducer of phospholipid transfer protein (PLTP) and is required in maintaining HDL levels in mammals.405
A clearer understanding of how lipoproteins are regulated by protein glycosylation might provide new therapeutic insights into the treatment of dyslipidemia and its clinical consequences.
Lipidation (palmitoylation and myristoylation) in hyperlipidemia
Protein lipidation is an essential mechanism allowing proteins to shuttle between organelles and the membrane to alter structure, activity and function.
Palmitate, a sixteen-carbon saturated fatty acid, and can be linked to cysteine residues by thioester bonds. The rapid shift of palmitoylation and depalmitoylation reversibly regulates protein trafficking and function. ELMO domain containing 2 (ELMOD2), a nonclassical ADP-ribosylation factor (Arf)-GTPase activating protein, can handle the transport of adipocyte triglyceride lipase (ATGL) to lipid droplets. Through palmitoylation, ELMOD2 is bound to lipid droplets and regulates ATGL recruitment.80
Myristic acid, a fourteen-carbon saturated fatty acid, typically and irreversibly binds to the glycine residue by a covalent bond through myristoylation. In a study, adenovirus-mediated overexpression of hepatic NH(2)-terminal myristoylated signal-attached Akt (myr-Akt) leads to hypertriglyceridemia, hypoglycemia and hypoinsulinemia, which is regulated by Akt-mediated SREBP1 expression and fatty acid biosynthesis in an SREBP1-independent manner.88
Evidence of lipidation in hyperlipidemia is sparse, thus further exploration of lipidation in lipid metabolism might provide us with new perspectives on regulating dyslipidemia.
Glutathionylation in hyperlipidemia
S-glutathionylation is a vital redox regulatory mechanism involving the attachment of oxidized glutathione and protein thiol through a mixed disulfide linkage. In redox regulation, GSH undergoes S-glutathionylation and is then reversed via enzymatic or chemical reduction. Glutathione S-transferase and peroxiredoxins promote protein S-glutathionylation, while glutaredoxins (Glrx) mainly reverse it. S-glutathionylation and related enzymes play crucial roles in dyslipidemia.
S-glutathionylation can modify lipid metabolism-related enzymes and factors. Paraoxonase 1 (PON1) is an esterase related to HDL in serum that mediates macrophage cholesterol efflux. Oxidative stress can induce S-glutathionylation in PON1 and subsequent reversible inactivation.406 Elevated glutathionyl haemoglobin level is found in patients with hyperlipidemia, suggesting the role of protein glutathionylation and oxidative stress.110
Glutaredoxins are widely involved in lipid metabolism. Glrx knockout mice on a chow diet exhibit hyperlipidemia, obesity, and fatty liver. Glrx supplementation inhibits hepatic lipid levels in a short time, suggesting that upregulation of Glrx could potentially improve lipid metabolism.407 One possible explanation might be that SITR1 inactivation induces S-glutathionylation and promotes hyperacetylation and hyperactivation of SREBP.
The normal functioning of protein glutathionylation is crucial, and it may be a potential target for lipid hemostasis regulation.
S-nitrosylation in hyperlipidemia
Nitrosylation is the covalent incorporation between the nitrosyl moiety of NO and target molecules. Nitrosylation occurs at the thiol group of cysteine, which is known as S-nitrosylation. There is only scarce evidence of protein S-nitrosylation in hyperlipidemia, mainly regarding lipoprotein regulation. S-nitrosylation leads to an interaction between NO and HDL-related PON1 at cysteine 284, abolishing PON1 enzymatic activity and resulting in the inactivation of lipid peroxides and cholesterol efflux disorders.119 The phosphotyrosine binding domain protein ARH is an adaptor protein interacting with LDLR and is required for well-organized LDLR activity. ARH requires S-nitrosylation via NO to allow LDL uptake by LDLR.408
Evidence of S-nitrosylation in hyperlipidemia is sparse, further exploration and a better understanding of S-nitrosylation and lipid homeostasis may offer new opportunities in dyslipidemia research.
Sulfhydration in hyperlipidemia
Sulfhydration alters the thiol group of cysteine residues to a persulfide (-SSH) group, leading to the enhanced reactivity of the cysteine residue. H2S is essential in lipid homeostasis through protein sulfhydration.
H2S treatment decreases plasma triglycerides by stimulating hepatic autophagy via AMPK-mTOR signaling.409 H2S donors (NaHS or GYY4137) directly activate SIRT1 by sulfhydration, restrain cholesterol uptake in macrophages and inhibit cholesterol biosynthesis in the liver, thereby reducing plasma lipid levels in ApoE-/- mice.410 NaHS-derived H2S promotes HRD1 sulfhydration, thereby increasing the interaction between DGAT1, DGAT2 and HRD1, repressing lipid droplet development in the heart of db/db mice.129
Therefore, targeting H2S and related protein sulfhydration may contribute to novel strategies and targets for treating hyperlipidemia and its consequences.
Citrullination in hyperlipidemia
Citrullination is an irreversible chemical process converting arginine to citrulline; citrullination has an impact on protein denaturation, hydrogen bond generation, protein structure, charge and protein–protein interactions. To date, only limited studies have indicated that protein citrullination participates in lipid metabolism.
Citrulline could increase the expression of ATP-binding cassette transporter (ABCA1) and ATP-binding cassette subfamily G member 1 (ABCG1) in macrophages, thereby promoting reverse cholesterol transport.411 Treatment with L-citrulline promotes fatty acid β-oxidation and restrains hepatic fat accumulation by increasing AMPK phosphorylation.138
It is necessary to conduct more research to determine how citrullination regulates lipid metabolism.
ADP ribosylation in hyperlipidemia
ADP-ribosylation can be divided into MARylation and PARylation. PARP activity is modulated by a group of cholesterol-related compounds, including cholesterol derivatives, bile acids and steroid hormones. In turn, PARPs play a vital role in lipid homeostasis.
PARPs affect abundant lipid-activated nuclear receptors, including PPARγ, PPARα, and LXR. PARP1 inhibitors can reduce the expression of PPARγ-dependent molecules (including adiponectin and CD36) in the late adipogenesis stage in adipocytes.412 PARP1 could restrain PPARα transactivation directly via ADP-ribosylation, thereby reducing gene expression involving fatty acid β-oxidation in the liver.380 PARP1 can ADP-ribosylate LXR, regulating ABCA1-modulated cholesterol efflux in macrophages. PARP1 inhibitors increase cholesterol efflux and regulate lipid homeostasis via the LXR-ABCA1 axis.413 PARP7 is a mono-ADP-ribosylation polymerase that can also ADP-ribosylate LXR.414
PARPs can also influence lipid-related pathways such as fatty acid metabolism, cholesterol homeostasis and the regulation of lipoprotein. PARPs facilitate fatty acid biosynthesis and transport by stimulating the expression of related genes, including FASN, CD36, FABP3, FABP4, and FABP7.415,416 PRAP (PARP1, PARP2, PARP10) deficiency or inhibition promotes fatty acid β-oxidation, as indicated by elevated expression of related genes and an increased respiratory quotient.148,366,415 PARP2 is closely involved in cholesterol biosynthesis, and loss of PARP2 mediates cholesterol synthesis by stimulating SREBP1 and SREBP2 in skeletal muscle and liver.415,417 PARP2 deletion weakens the expression of hepatic ABCA1, thereby decreasing cholesterol flux.417
Multiple investigations have demonstrated that lower PARP activity is related to optimized HDL/LDL ratios and normal triglyceride levels. Pharmacological inhibition of tankyrase could lower serum triglyceride, cholesterol, nonesterified fatty acids, and glycerol in db/db mice.418 Suppression of PARP1 reduces plasma triglyceride, cholesterol, and LDL levels and enhances HDL levels in HFD-fed mice.419
ADP-ribosylation is closely involved in lipid homeostasis. Advances in identifying novel therapeutic targets will provide novel perspectives into targeted drug discovery.
Carbonylation in hyperlipidemia
Protein carbonylation induces oxidative stress-related damage. Protein carbonyl level can serve as an indicator of oxidative injury in individuals with familial hypercholesterolemia. Cyanate, a uremic toxin, works via protein carbonylation. Cyanate induces hyperlipidemia by suppressing the Nrf2/HO-1 pathway and mediates oxidative stress via the AMPK-mTOR pathway in mice.155
Inhibition of carbonylation is potentially beneficial to reducing oxidative stress and improving lipid metabolism. Annona crassiflora crude extract (CEAc) can improve hyperlipidemia with reduced triglycerides, lower cholesterol and higher HDL in mice by decreasing protein carbonylation and lipid peroxidation.420 Low molecular weight galactomannan-based standardized fenugreek seed extract (LMWGAL-TF) could also lower plasma lipids in HFD-fed mice by reducing hepatic FASN and leptin and reducing mitochondrial oxidative stress via downregulated protein carbonylation.421 Fish oils, abundant with ω-3 polyunsaturated fatty acids (ω-3 PUFAs), improve hyperlipidemia in high-caloric diet-fed rats through reducing oxidative stress and hepatic protein carbonylation.422
Protein carbonylation is a deleterious PTM that disrupts lipid homeostasis. Strategies to restrain protein carbonylation hold the promise of regulating lipid metabolism and improving dyslipidemia.
PTMs in atherosclerosis
Atherosclerosis occurs as a result of multiple factors, including matrix changes, lipid metabolism (hyperlipidemia), vascular changes, chronic unresolved inflammation, endothelial dysfunction, foam cell formation, hemodynamic stress, and blood coagulation. As shown in Fig. 7, dyslipidemia induces endothelial dysfunction, which promotes macrophage and vascular smooth muscle cell (VSMC)-derived foam cell formation, finally leading to the formation of atherosclerotic plaques. PTMs affect protein structures and biological properties, which are closely involved in these pathological processes. Aiming to understand and exploit the molecular mechanisms of atherosclerosis via proteomic investigations, a thorough comprehension of the role of PTMs is essential to gain insights into the potential regulation of the relevant cellular pathophysiology. Here, we will examine various PTMs in the context of atherosclerosis.

Multiple roles of PTMs in atherosclerosis. Atherosclerosis occurs as a result of multiple risk factors, including abnormal lipid metabolism (hyperlipidemia), endothelial dysfunction, foam cell formation, VSMC proliferation and migration, cell apoptosis and necrosis. a The holistic illustration of atherosclerotic progression: monocyte activation, rolling and adhesion; macrophage-derived foam cell formation; VSMC proliferation and migration; atherosclerotic plaque formation. Lipotoxic ox-LDL and inflammation injure endothelial cells, activate the main regulator NF-κB, promote expression of pro-inflammatory genes and induce inflammation. Ox-LDL induces macrophage inflammation and lipid uptake, which will facilitate foam cell formation. ABCA1 and ABCG1-mediated reverse cholesterol transport could alleviate lipid overload and inhibit foam cell generation. Ox-LDL also induces VSMC phenotypic transformation, and promote VSMC proliferation and migration, contributing to atherosclerotic plaque. To understand the comprehensive role of PTMs in atherogenesis, we summarise several pathways of PTMs in regulating the physiology and pathology of endothelial cells, macrophage and VSMC. b Endothelial dysfunction. Abundant PTMs regulate endothelial inflammation, such as phosphorylation (oxLDL-MAPK, AMPK-p300, JACD-PI3K/Akt), acetylation (HDAC9-IKK-NFκb, HDAC5-KLF2), methylation (ADMA-iNOS), ubiquitination (UPS-eNOS), SUMOylation (SUMO-IKK, Disturbed flow-SENP2-p53/ERK5-eNOS), glutathionylation (GPX1-oxLDL, GSH-NRF2/HIF1α-NFκB), S-nitrosylation (S-nitrosylated-NO-NFκB), sulfhydration (H2S-KEAP1-NRF2, oxLDL-H2S-CSE-NF-κB), ADP ribosylation (PARP1-NF-κB/NFAT/AP1, PARP1-eNOS, PARP10-NFκB, PARP12-TRIF-NFκB). c Macrophage inflammation and cholesterol efflux. Phosphorylation (PKCθ-ATF2-CD36), acetylation (HDAC1/2/3/6/8/9-ABCA1/ABCG1), ubiquitination (UBA1-oxLDL-NADPH, FBXW2-KSRP), Neddylation (CSN5-NFκB), glycosylation (ST3Gal-IV-CCR5) and ADP ribosylation (PARP1-LXR-ABCA1) regulating macrophage roles in atherosclerotic progression. d VSMC proliferation and migration. Phosphorylation (oxLDL-MAPK, AMPK-LDLR-ER stress), acetylation (HDAC2-KLF4/5, SIRT6- telomeres), ubiquitination (Peli1), and SUMOylation (SENP3-ROS, AMPKα2-UBC9/SUMO2/3-GPR120, SUMO-LRH1-PROX1-RCT, UBC9/PIASγ-PPARα, SENP-PPARα/PPARδ, AngII-ATF3-eNOS) mediate the VSMC function in atherogenesis. The blue and red boxes indicate the major pathways in atherosclerosis, and the black boxes indicate the roles of PTM-related molecules and enzymes in atherosclerotic regulation. The figure is generated with BioRender (https://biorender.com). Column1,Column2; ABCA1 ATP binding cassette transporter, ABCG1 ATP binding cassette transporter, ADMA asymmetric dimethylarginine, AMPK AMP-activated protein kinase, AngII angiotensin II, AP-1 activator protein-1, ATF activating transcription factor, CCL5 C-C motif chemokine ligand 5, CCR C-C motif chemokine receptor, CSE cystathionine gamma-lyase, CSN5 COP9 signalosome 5, ER endoplasmic reticulum, eNOS endothelial nitric oxide synthase, ERK5 extracellular signal-regulating kinase 5, GPX1 glutathione peroxidase 1, GSH glutathione, H2S hydrogen sulfide, HDAC histone deacetylase, HDL high-density lipoprotein, HIF-1α hypoxia-inducible factor 1α, HRD1 promote degradation protein 1, ICAM-1 intercellular adhesion molecule-1, KEAP1 kelch like ech associated protein 1, KLF kruppel-like factor, LDL low-density lipoprotein, LRH-1 liver receptor homolog-1, LXR liver X receptor, MAPK mitogen-activated protein kinase, MCP-1 monocyte chemoattractant protein 1, NF-κB nuclear factor kappa B, NRF2 nuclear erythroid 2-related factor 2, oxLDL oxidative LDL, PARP poly (ADP-ribose) polymerase, Peli1 Pellino1, PIAS1 protein inhibitor of activated STAT 1, PKC protein kinase C, PROX ROS, reactive oxygen species, SENP1 sentrin/SUMO-specific protease 1, SIRT sirtuin, TRIF toll-interleukin-1 receptor containing adapter-inducing interferon-β, UBA1 ubiquitin-like modifier activating enzyme 1, VCAM-1 vascular cell adhesion molecule 1
Phosphorylation in atherosclerosis
Protein phosphorylation is the most widely investigated PTM, and it is closely related to atherosclerosis. Abundant protein kinases have been reported to regulate the progress of atherosclerosis.
MAPKs, a Ser/Thr kinase family, are vital signal-transducing enzymes that are involved in cellular regulation. MAPK family signaling cascades mainly include p38α MAPK, ERK, and JNK.423 MAPKs are of relevance to atherosclerosis via matrix production, endothelial cell activation, macrophage inflammation and foam cell formation, VSMC proliferation and migration. MAPK activation mediates monocyte adhesion to activated endothelium mediated by oxidized LDL (ox-LDL).424 Apolipoprotein E (ApoE-/-) mice with p38α MAPK deficiency in macrophages show enhanced macrophage apoptosis in atherosclerotic plaques.425 JNK2 decreases the phosphorylation of scavenger receptor-A (Sr-A) cytoplasmic tail serine to fewer foam cells, which finally contributes to smaller plaques in ApoE-/-Jnk-/- mice.425 Oxidized LDL induces VSMC proliferation through MAPK activation.426 Knockout of MAPK phosphatase-1 (MKP-1) in ApoE-/- mice reduces atherosclerosis via reducing the contents of TNFα and interleukin-1α.427 In addition, other MAPK members or pathway-related molecules, such as activator protein-1 (AP-1)428 and MK2,429 are implicated in atherosclerosis via phosphorylation.
AMPK is a conserved Ser/Thr kinase responsible for energy homeostasis and the regulation of cell metabolism. AMPK kinases (including LBK1, CAMKKβ, and TAK1) activate AMPK via phosphorylation at Thr172, coping with depleted cellular ATP levels. AMPK then phosphorylates target proteins to activate catabolic pathways or suppress anabolic pathways to conserve and produce ATP. AMPK has been reported to possess vasoprotective and antiatherosclerotic effects by inhibiting oxidative stress, inflammation, and VSMC proliferation. Specifically, the double knockout of LDLR and AMPKα2 in mice promotes ER stress and accelerates the development of atherosclerosis.430 AMPK inhibits endothelial inflammation by phosphorylating the transcriptional coactivator p300 in human umbilical vein endothelial cells (HUVECs).431 Sterol SREBP-1c is phosphorylated by AMPK at Ser372, which reduces lipogenic transcription in hepatic cells.375
Furthermore, other related kinases and their substrates, including Rho-associated coiled-coil-containing kinases (ROCK),432 Akt kinase17 and protein kinase C (PKC) are also involved in the development of atherosclerosis.433 Ablation of ROCK1 in macrophage results in enhanced cholesterol efflux and thus reduced atherosclerosis.432 Junctional cadherin 5 associated (JCAD) aggravates arterial thrombosis and atherosclerosis via PI3K/Akt pathway.17 PKCθ enhances CD36 expression through triggering transcription factor 2 (ATF2) and promotes macrophage-derived foam cell formation, leading to atherosclerosis in ApoE-/- mice.433
The role of protein phosphorylation in the progression of atherosclerosis progression has been well recognized and it is a potential target for atherosclerosis prevention. Further research of novel therapeutic targets in regulating protein phosphorylation is critical for designing novel anti-atherosclerosis approaches.
Acetylation in atherosclerosis
Lysine acetylation can affect the process of atherosclerosis mainly via regulating acetylation and deacetylation of histones and nuclear proteins. Plasma interleukin-35 (IL-35) is increased in ApoE-/- mice and patients with hypercholesterolemia, and IL-35 suppresses mitochondrial ROS-induced H3K14 acetylation, which inhibits endothelial activation and alleviates the development of atherosclerosis.434
Increasing evidence has indicated the essential roles of HDACs in endothelial cell homeostasis and the development of atherosclerosis. The HDAC I, II and IV types are the typical HDACs; class III consists of sirtuin proteins. Among them, HDAC9 is well known because of its essential role in regulating human and mouse atherosclerosis. A genome-wide association meta-analysis (GWAS) reveals that HDAC9 is implicated in atherosclerotic aortic calcification at a genome-wide level and affects VSMC contractility phenotype through regulating calcification.435 HDAC9 deficiency in Ldlr-/- mice reduces aortic lesion size and protects against atherosclerosis through suppressing cholesterol efflux by inhibiting ABCA1/ABCG1 and PPARγ.436 HDAC9 can deacetylate IKK-α/β to activate NF-κB, leading to inflammation in macrophage437 and promoting endothelial-mesenchymal transition,28 both of which contribute to regulating atherosclerotic vulnerability.
Most typical HDACs are involved in endothelial cell biology, VSMC phenotype determination and macrophage-derived foam cell formation. Metformin-mediated anti-inflammatory role in the endothelium is caused by phosphorylation of HDAC5 and the subsequent activation of KLF2.438 Phosphorylated HDAC2 regulates the acetylation of KLF4 and KLF5 in VSMCs, mediating retinoic acid receptor agonist-induced VSMC proliferation.439 The VSMC proliferation stimulator Ang-II can activate HDAC5 via G protein-coupled receptor (GPCR)-kinase2 interacting protein 1 (GIT1).440 HDAC inhibitors such as TSA and ITF2357 (pan-HDACi) or genetic silencing of HDAC1/2/3/6/8 genes enhances histone acetylation and ABCA1/ABCG1 levels, thus restraining cholesterol accumulation and development of foam cells in macrophages.440
Furthermore, sirtuins also play a vital role in the progression of atherosclerosis. SRT3025, a SIRT1 activator, inhibits atherosclerosis in ApoE-/- mice through reducing hepatic PCSK9 expression and increasing LDLR expression.441 Human and mouse plaque VSMCs exhibit reduced SIRT6 expression. SIRT6 bounds to telomeres, affects H3K9 deacetylation and 53BP1 (p53 binding protein 1) binding, which preserves telomere integrity and extends VSMC lifespan and thus inhibits atherosclerosis.442
The balance of protein acetylation and deacetylation is tuned to govern cell biology and thus regulate atherosclerosis. Dysregulation of protein acetylation represents a cause of cardiovascular comorbidities. Advances in deciphering the role and mechanism of protein acetylation in atherosclerosis will potentially provide novel therapeutic targets in treating atherosclerosis.
Methylation in atherosclerosis
Protein methylation has been well recognized for regulating the function of nuclear and nucleic acid-binding proteins and to have an essential role in CVDs.
Histone methylation is of great importance in atherosclerosis. The histone methylation profile in human atherosclerotic lesions reveales that global H3K27me2 and H3K9me2 are reduced in atherosclerotic plaques, whereas H3K4me2 shows comparable levels in both atherosclerotic and normal carotid arteries.443
Furthermore, histone methyltransferases and a few intermediate products during the enzymatic process are implicated in atherosclerotic progression. Myocardin-related transcription factor A (MRTF-A) recruits the H3K27 methyltransferase ASH2, transactivates inducible nitric oxide synthase (iNOS), inducing endothelial inflammation.36 The Set7/9 lysine methyltransferase directly methylates the FXR at lysine 206 to regulate a series of target genes of bile acid homeostasis. These genes encode estrogen receptor, androgen receptor, p53, TAF10, p300/CBP-associated factor and RelA subunit of NF-κB, which are associated with atherosclerosis.444 PRMT and ensuing proteolysis of proteins generate asymmetric dimethylarginine (ADMA). ADMA regulates ROS by inhibiting NOS and reducing NO production. Plasma ADMA level is shown to increase in individuals with coronary artery disease,445 and ADMA is suggested to act as a risk marker.
Much more work is necessary to better understand the role of protein methylation in atherosclerosis. Advances in this area could afford new perspectives and guide treatments for atherosclerosis.
Ubiquitination in atherosclerosis
The ubiquitin-proteasome cascade pathway is crucial to protein metabolism and endogenous protein degradation in eukaryotic cells. The UPS is implicated in a wide range of physiological processes, including endothelial dysfunction, apoptosis, oxidative stress and foam cell formation, which are relevant to atherosclerosis. E1, E2, and E3 ligases are implicated in atherosclerotic progression.
UBA1 is a major E1-activating enzyme in the UPS cascade. UBA1 inhibitor PYR-41 can inhibit ox-LDL-induced proinflammatory cytokine expression, NADPH oxidases and lipid deposition in macrophage, thereby suppressing atherosclerosis in ApoE-/- mice with blunted proinflammatory responses in macrophage.446
Aggregated low-density lipoprotein (agLDL)-induced ubiquitin-conjugating E2 enzyme E2-25K facilitates lipid-bearing macrophages apoptosis via ubiquitination and degradation of p53, leading to foam cell formation.447
E3 ubiquitin ligases are recognised to play diverse and multifaceted roles in atherosclerosis-related inflammatory and metabolic processes. TRIM21 deficiency facilitates Th17 differentiation and promotes IL-17 expression, thereby enhancing collagen content in plaques and improving atherosclerotic plaque stability in Ldlr-/- mice.47 The E3 ligase IDOL triggers the degradation of LDLR. Transgenic expression of hepatic IDOL results in reduced LDLR and aggravated atherosclerosis in western diet-fed C57Bl/6J mice.448 Deleting the E3 ligase Pellino1 (Peli1) facilitates VSMC foam cell formation, induces proinflammatory cytokines and promotes atherosclerosis.449 E3 ubiquitin ligase FBXW2 can promote macrophage inflammation and atherosclerosis by ubiquitinating KH‐type splicing regulatory protein (KSRP) and reducing proinflammatory cytokines. Inhibition of FBXW2 might serve as a potential approach to treating atherosclerosis.450
Since the critical role of protein ubiquitination and the UPS system in atherosclerosis has gained widespread recognition, further exploration of new therapeutic approaches regulating ubiquitin ligases could contribute to the treatment of atherosclerosis.
SUMOylation in atherosclerosis
Recent studies have indicated that SUMOylation is implicated in dyslipidaemia, endothelial dysfunction, and VSMC proliferation, all processes which trigger the initiation and development of atherosclerosis. Next, we will elaborate on the role of SUMOylation in atherosclerosis from these three aspects.
Dyslipidemia in atherosclerosis is a crucial metabolic risk factor that includes high serum TG levels, increased LDL levels, and decreased HDL levels. SUMOylated LRH-1 interacts with PROX1 to inhibit RCT and thereby promotes atherosclerosis396. SUMO E2 enzyme UBC9 and SUMO E3 ligase PIASγ induce dyslipidaemia by SUMOylating PPARα at the K185 site and suppressing PPARα transcriptional activity.451 Correspondingly, SENPs promote fatty acid β-oxidation by eliminating SUMOylation in PPARα and PPARδ.452
During endothelial cell dysregulation, deficient nitric oxide (NO), enhanced adhesion molecules, endothelial apoptosis and senescence contribute to atherosclerotic pathology. SENP1 deficiency directly increases SUMOylation of GATA binding factor 2 (GATA2) and nuclear factor of kappa light polypeptide gene enhancer in B-cell inhibitor alpha (IκBα), leading to decreased GATA2 stability and NF-κB activity, thus reducing adhesion molecules expression.453 Disturbed flow causes phosphorylation of SENP2 via p90RSK kinase, which promotes SENP2 activity and downregulates SUMOylation of p53 and ERK5 under disturbed flow, leading to endothelial dysfunction evidenced by reduced eNOS and atherosclerotic plaque formation in Ldlr-/- mice.454 The SUMOylation of activation of transcription factor 3 (ATF3) could aggravate endothelial dysfunction induced by angiotensin II (Ang-II) by decreasing NO generation.455
It has been proposed that SUMOylation is essential to promote VSMC proliferation. Ox-LDL and Ang-II can increase SENP3 expression via a ROS-dependent way in VSMC. SENP3 overexpression facilitates VSMC proliferation, migration and vascular remodeling through suppressing de-SUMOylation of β-catenin.456 AMP-activated protein kinase α2 (AMPKα2) activation inhibits GPR120 SUMOylation via suppressing SUMO2/3 and Ubc9 in VSMC, mediating the anti-inflammatory and atheroprotective impacts of fish oil.57
From this perspective, protein SUMOylation plays a diverse and critical role in atherosclerosis. Further elucidation of protein SUMOylation in atherosclerosis could yield novel therapeutic targets of atheroprotection.
Neddylation in atherosclerosis
Studies have shown that protein neddylation is associated with dysfunction of the endothelium and macrophage activation.
Ox-LDL induces global neddylation, and overexpression of NEDD8 can reduce HDAC2 levels in human aortic endothelial cells (HAECs). Suppressing neddylation with the neddylation activating enzyme (NAE) inhibitor MLN4924 relieves ox-LDL-mediated endothelial dysfunction.457 MLN4924 can decrease inflammation in macrophages and endothelial cells. MLN4924 also increases HDAC6 activity, reducing endothelial dysfunction and atherosclerosis in vivo.458
The de-neddylation enzyme COP9 signalosome 5 (CSN5) was proposed to inhibit atherosclerosis by regulating macrophage activation through inhibiting NF-κB.63 Therefore, de-neddylation approaches may serve as a candidate strategy to decrease atherogenesis.
Much more exploration is required for a better understanding of the relationship between protein neddylation and atherosclerosis. Discoveries in this field might potentially identify novel therapeutic targets in treating atherosclerosis.
Glycosylation in atherosclerosis
Atherosclerosis is a complex and chronic inflammatory disease in which, in one early aspect, leukocytes are recruited to the arterial intima from the blood through a series of steps, including tethering, rolling, adhesion and transmigration, processes driven by adhesion molecules and chemokines. Glycosylation and various glycosyltransferases are contributors to atherosclerosis, as chemokine receptors and adhesion molecules are usually glycosylated proteins.
The lack of several glycosyltransferases inhibits atherosclerosis by decreasing leukocyte recruitment. α2,3-Sialyltransferase IV (ST3Gal-IV) deficiency in mice protects against atherosclerosis, as the glycosylation of C-C motif chemokine receptor 5 (CCR5) is impaired, leading to decreased leukocyte rolling.74 Loss of α(1,3) fucosyltransferases-IV (FucT-IV) and FucT-VII reduces the glycosylation of selectin ligands and leukocyte recruitment, decreasing atherosclerotic lesions.459,460 Core 2 β1,6 galactosyltransferase I deletion prevents atherosclerosis by reducing glycoproteins in selection ligands such as CD43, CD44, and CD162 and inhibiting their activity.461
Elevated levels of the protein glucose adducts and advanced glycosylation end products (AGEs) are found in individuals with diabetes. Increased AGEs promote oxidative stress and contribute to cardiovascular diseases. AGEs are produced mainly by protein and lipid nonenzymatic glycosylation. AGEs and receptor for AGE (RAGE) initiate signaling pathways, such as promoting adhesion molecule expression through the NF-κB pathway and activating NADPH and NOS.462
Further elucidation is necessary to better clarify the role of glycosylation in atherosclerosis, which might hold the potential to explore novel therapeutic drugs in CVD and its clinical sequelae.
Palmitoylation in atherosclerosis
Palmitoylation affects membrane fusion and cellular trafficking of proteins and is reversibly controled by palmitoyltransferases and acyl protein thioesterase (APT).
Palmitoylation is needed to target and localize endothelial nitric oxide synthase (eNOS) to the caveolae for optimal NO release.463 In endothelial cells with FASN deficiency, reduced eNOS palmitoylation causes impaired angiogenesis and endothelial dysfunction.464 ATP-1 insufficiency leads to prolonged palmitoylation of Ras-related protein (R-Ras), causing impaired trafficking of eNOS synthase and thus reduction of NO and endothelial dysfunction.463
Palmitoylation dysregulation could also cause abnormal foam cell formation. Oxidized high-density lipoprotein (ox-HDL) promotes CD36 palmitoylation, causes CD36 localization to lipid rafts and activates downstream pathway, which finally increases ox-HDL uptake in macrophages and foam cell development. Bromopalmitate-mediated suppression of CD36 palmitoylation reduces cell surface translocation and lightens oxHDL uptake.79
Limited knowledge of the function of protein palmitoylation in atherosclerosis hindered the research to explore novel strategies targeting palmitoylation, thereby more research is warranted to elaborate on the relationship between palmitoylation and atherosclerosis.
Myristoylation in atherosclerosis
Since eNOS dysfunction produces superoxide instead of NO, leading to endothelial dysfunction and atherosclerosis, eNOS is a potential therapeutic target in atherosclerosis. eNOS is acylated by palmitoylation and myristoylation and resides in caveolae and Golgi. N-myristoylation is required for eNOS acylation in the Golgi complex and is of great significance in determining eNOS activity.
Myristoylated pseudosubstrate of PKCζ (mPS), a myristoylated peptide inhibiting PKC activity, activates eNOS in endothelial cells. Myristoylation-induced eNOS activation depends on PI3K/Akt signaling and elevated cellular calcium. Other myristoylated peptides can also activate eNOS in a non-PKC-dependent manner.465
Moreover, LIM and cysteine-rich domains 1 (LMCD1) were recently reported to govern the proliferation and migration of human and mouse smooth muscle cells (SMCs) through controling NFATC1-mediated IL33 and E2F1-mediated CDC6 expression. LMCD1 myristoylation inhibits this regulation.89
Since very little information about myristoylation and atherosclerosis is available, much more work is required to offer a comprehensive understanding of myristoylation in atherosclerosis.
Prenylation in atherosclerosis
Protein prenylation is implicated in atherosclerosis through regulating lipid metabolism, smooth muscle cell proliferation and macrophage inflammation.
Prenylcysteine oxidase (PCYOX1) was recently found to be restricted to the metabolism of protein prenylation, which was related to lipoprotein oxidation and the atherogenic ApoB100 lipoproteins.94 PCYOX1 insufficiency in ApoE-/- mice attenuates the consequences of atherosclerosis by reducing lesion vulnerability, decreasing lipid peroxidation, and lowering serum lipids and inflammation.94 The farnesylation of small G-proteins is essential for cell growth and differentiation. Blocking the enzyme protein farnesyltransferase (PFT) suppresses the proliferation of human SMCs by suppressing the farnesylation of Ras protein.466 Inhibition of RAC1 farnesylation by geranylgeranyltransferase type I (GGTase-I) causes enhanced proinflammatory signaling in macrophages.94
The normal functioning of protein prenylation is of importance, and targeting protein prenylation might provide a novel opening in anti-atherosclerosis therapy.
Glutathionylation in atherosclerosis
Glutathione is a plentiful intracellular small-molecule antioxidant that is implicated in numerous cellular redox processes.
The pathological process of CVD is considered to involve changes in GSH concentration and oxidation state. Serum S-glutathionylated protein levels are elevated in patients with atherosclerosis. S-glutathionylated ApoB100 (an atherogenic lipoprotein) is positively related to peripheral vascular damage.467 The synthesis of GSH in macrophages was found to be negatively related to the pathogenesis of atherosclerosis. Ribo-cysteine-treated mice show an increase in the level of GSH and a significant reduction in circulating apoB, lipoprotein(a) and oxidized lipid.468
Glutathionylation-related key enzymes contribute to the regulation of atherosclerosis. In a study investigating the glutathione peroxidase 1 (GPX1) polymorphism in individuals with T2DM, GPX1 was found to possess a protective role in human endothelial cell dysfunction and atherosclerosis.469 GPX1 deficiency in diabetic ApoE-/- mice increases LDL oxidation and accelerates atherosclerosis.470 Ox-LDL restrains glutathione reductase (GR) activity and facilitates accumulating protein S-glutathionylation, ROS generation, and cell death in macrophages, promoting the development of atherosclerosis.471
Glutathionylation serves as an endothelial cell redox switch. SIRT1 glutathionylation restrains enzymatic activity through changing the protein structure and attaching to NAD+, causing endothelial cell apoptosis and senescence.472 S-glutathionylation inhibits p65 and p50 subnit of NF-κB, leading to angiogenesis and cell survival.473 S-glutathionylation also promotes anti-oxidation, anti-inflammation, and angiogenesis targeting HIF-a109 and KEAP1 (NRF2 inhibitor).474
Protein S-glutathionylation mediates oxidant-induced PTMs and has emerged as a vital redox regulator in macrophage and endothelial cell dysfunction. Advances in deciphering the underlying mechanism of how protein prenylation regulates atherosclerosis could facilitate novel perspectives in preventing atherosclerosis.
S-nitrosylation in atherosclerosis
NO acts as an endogenous mediator of cell respiration and regulator of cardiovascular physiology. NO exerts its influence via cyclic guanosine monophosphate (cGMP) or S-nitrosylated modification. Almost all primary cardiovascular functions of NO are implicated in S-nitrosylation.
S-nitrosylation regulates endothelial inflammation. S-nitrosylation inhibits proinflammatory cytokines and adhesion molecules by blocking the NF-κB pathway.475 S-nitrosylation regulates platelet activation in the development of atherosclerosis. NO suppresses platelet aggregation via S-nitrosylation.476 eNOS knock-out mice exhibit upregulated leukocyte rolling, increased exocytosis, and enhanced arteriolar thrombosis.477 Heat shock protein 90 (HSP90) S-nitrosylation inhibits the HSP90-ATPase activity 1 (AHA1) interaction but stimulates the HSP90-cell division cycle 37 (CDC37) association, which modulates endothelial dysfunction and exacerbates atherosclerosis.478 Guanine nucleotide-binding protein G(i) subunit alpha-2 (SNO-GNAI2) S-nitrosylation increases in individuals with diabetes and atherosclerosis. S-nitrosylation mediates the attachment of GNAI1 and C-X-C chemokine receptor type 5 (CXC-R5), inducing Hippo/YAP and accelerating atherosclerosis in diabetic mice.120
S-nitrosylated modifications play a role in cardiovascular disease and atherosclerotic progression via regulating NO activity. Further investigation of S-nitrosylation in cardiovascular function might offer us novel perspectives and opportunities in cardiovascular disease prevention research.
Sulfhydration in atherosclerosis
Hydrogen sulfide (H2S) as a dissolved gas is recognized as a member of the family of gasotransmitters. A considerable amount of evidence has suggested that H2S can protect against atherosclerosis. The atherosclerotic protective mechanisms of H2S include endothelium preservation, anti-inflammation, antioxidative responses, and vasorelaxation.128
S-sulfhydration has been recognized as one of the main mechanisms determining the physiological effects of H2S. A few studies have demonstrated H2S signaling via S-sulfhydration in attenuating atherosclerosis. KEAP1 protein is sulfhydrated at cysteine151, which enhanced KEAP1 thiolation, promoted NRF2 translocation and inhibited superoxide generation in endothelial cells. These increased NRF2-related antioxidant actions ameliorated the subsequent diabetes-accelerated atherosclerosis.479 Cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS) are two enzymes associated with the biosynthesis of H2S. Ox-LDL downregulates CSE/ H2S signaling and activates inflammation via the NF-κB signaling pathway in macrophages.480 Treatment with an H2S donor was found to cause CSE thiolation (C252, C255, C307, C310), boost its linkage to L-homocysteine, and alleviate hyperhomocysteinemia-related atherosclerosis in mice.127
The ordinary functioning of H2S regulated by the S-sulfhydration modification is pivotal in cellular biology. More research targeting the comprehensive role of S-sulfhydration in diverse physiologic and pathologic scenarios could potentially provide us with a new guide in anti-atherosclerosis therapies.
Citrullination in atherosclerosis
PADs catalyze the transition of arginine to citrulline, accordingly modifying protein charge and structure. Protein citrullination is associated with rheumatoid arthritis but there is only sparse evidence of its role in atherosclerosis.
Clinical evidence has revealed the relationship between citrullinated proteins and coronary heart disease. Several citrullinated proteins and PAD4 were identified in human coronary artery plaques. Anti-citrullinated protein antibodies (ACPAs) are related to the aortic plaque burden.481 High concentrations of serum ACPAs targeting Cit-histone H2B are also associated with elevated coronary artery calcium scores.139 These data suggest the possible role of citrullinated proteins as biomarkers for atherosclerosis.
Evidence of citrullination in regulating atherosclerosis is sparse, further exploration is needed to afford a comprehensive understanding of the effect of S-nitrosylation in atherosclerosis.
ADP ribosylation in atherosclerosis
PARP1 is the first identified and the classical PARP member that is activated by abnormal DNA or RNA breaks and mediates approximately 90% of total cellular PARP responses.482 PARP1 participates in atherosclerosis by regulating cholesterol efflux, inflammation, endothelial cell dysfunction, foam cell death, and cellular energy crisis. PARP1 can PARylate LXR and inhibit LXR-induced ABCA1 expression, thus reducing cholesterol efflux in macrophages.413 Proatherogenic factors (such as proinflammatory cytokines) cause PPAR1 hyperactivity, inducing overactivation of NF-κB, activated T cells (NF-AT), and the AP-1 pathway.483,484 Blocking PPAR1 improves endothelial dysfunction by boosting eNOS activity,485 reducing adhesion molecules,486 and advancing endothelium-dependent relaxation.149 PPAR1 inhibition decreases macrophage recruitment,487 and acetyl-coenzyme A acetyltransferase-1 (ACAT-1) mediates foam cell death and atherosclerosis in ApoE-/- mice.488 Hyperactivation of PPAR1 also leads to exhaustion of NAD+ and ATP, resulting in a cellular energy crisis.489
In addition to PPAR1, increasingly more other PPARs have been identified to participate in tissue inflammation. PPAR9 and PPAR14 together modulate the proinflammatory activation of macrophages.490 PPAR14-mediated SIRT1 ADP ribosylation appears to decrease the phosphorylation of SIRT1.490 PPAR10 ADP-ribosylates NF-κB essential modulator (NEMO) and suppresses its polyubiquitination, inhibiting NF-κB translocation.491 PARP12 binds to toll-interleukin-1 receptor containing adapter-inducing interferon-β (TRIF) and increases NF-κB-dependent gene expression.492
The diverse role of ADP ribosylation in endothelial dysfunction and atherosclerosis has gained widespread explanations. Further investigation of new therapeutic targets in regulating ADP ribosylation is crucial for developing novel anti-atherosclerosis therapies.
Carbonylation in atherosclerosis
Protein carbonylation is usually a deleterious PTM, and inhibition of carbonylation will potentially alleviate oxidative stress and modulate atherosclerosis.
In vitro, ox-LDL induces carbonylation of p65 of NF-κB, potentially worsening LPS-induced macrophage inflammation and related atherosclerosis.493
In vivo, D-carnosine octyl ester alleviates atherosclerosis in ApoE-/- mice by decreasing general serum carbonylated protein levels.494 Oolong tea derived-oolonghomobisflavan A (OFA) prevents atherosclerosis by reducing oxidative stress and carbonylation of ApoB100.156
Protein carbonylation is usually a deleterious PTM that disrupts normal cellular functions. Strategies to restrain protein carbonylation could benefit regulating cell homeostasis and prevent atherosclerosis.
Pharmaceutical intervention of PTMs in preclinical studies
The preclinical research of metabolic diseases which target protein modifications is an interesting field of study. We next discuss some representative compounds in preclinical studies and elucidate how these compounds impact each PTM in metabolic diseases. Detailed information can be seen in supplementary Table 1 which summarizes the pharmaceutical interventions of protein modifications in preclinical studies of metabolic diseases.
AMPK activators
Metformin is an AMPK activator that improves glucose control and insulin sensitivity, thus decreasing intestinal glucose absorption. Metformin promotes phosphorylation of AMPK and glucose production in primary rat hepatocytes.495
Ampkinone plays an indirect action in the phosphorylation of AMPK. DIO mice treated with ampkinone show reduced body weight, decreased fat mass weight and improved metabolic characteristics.496
In addition, the AMPK activator AICAR also inhibits the phosphorylation of IRS-1 by suppressing ERK phosphorylation. In db/db mice, intraperitoneal administration (0.25 g/kg) of AICAR can improve blood glucose levels by inhibiting the phosphorylation of ERK in adipose tissue.497
MAP kinase inhibitors
SD-169 is a selective ATP-competitive inhibitor of MAP kinases. SD-169 treatment provides a hypoglycemic effect and preserves beta cell mass in NOD mice.498
U0126 inhibits the MAPK/ERK pathway to ameliorate diabetic cardiomyopathy in STZ-induced diabetic mice. U0126 also reduces de novo fatty acid synthesis by inhibiting liver FASN expression. U0126 inhibits atherosclerosis in LDLR-deficient mice without adipogenesis side effects.499
PTP1B inhibitor
KY-226 is an inhibitor of PTP1B. Oral administration of KY-226 can increase the insulin-induced phosphorylation of insulin receptors, and reduce plasma glucose and triglyceride levels in db/db mice. In high-fat-diet-induced obese mice, KY-226 reduces body weight gain and increases phosphorylated STAT3 in the hypothalamus.500
JTT-551 has been used in the research of T2DM. In db/db mice, chronic administration of JTT-551 shows an anti-hyperglycemic effect. A single dose of JTT-551 in ob/ob mice enhanced the IR phosphorylation of the liver.501
MSI-1436 selectively inhibits PTP1B and enhances insulin-stimulated tyrosine phosphorylation of insulin receptors, causing fat-specific weight loss in DIO mice.502 DPM-1001 is an analogue of MSI-1436, which can enhance β-subunit phosphorylation and reduce diet-induced obesity.503
JAK1/2 inhibitor
Ruxolitinib is a selective JAK1/2 inhibitor. Inhibiting phosphorylation of the JAK2/STAT3/SOCS3 pathway significantly decreases the area of atherosclerotic plaques and balloon injury of the aorta in rabbits fed with HFD.504
Fedratinib (TG101348) is an ATP-competitive inhibitor of JAK2. TG101348 selectively decreases cellular phosphorylated STAT5 and substantially reduces aortic atherosclerosis in ApoE-/- mice.505
EGFR inhibitor
Erlotinib is a direct-acting inhibitor of EGFR tyrosine kinase and reduces EGFR autophosphorylation. Erlotinib can block EGFR-mediated STAT3 phosphorylation, significantly reducing myocardial structural and functional deficits in diabetic mice.506
Compound Y396 could inhibit the phosphorylation of EGFR. Y396 inhibits endothelial cell dysfunction induced by oxidative stress in diabetes.507
Histone deacetylase inhibition
Valproate improves kidney function in DN rat models by modulating acetylation of histone H4 in ERS-associated protein promoter.508
MGCD0103, an HDAC inhibitor, protects the pancreas from STZ-induced hyperglycemia, macrophage infiltration and oxidative stress.509
Vorinostat (SAHA) treatment ameliorates HFD-induced reduction of histone H3 acetylation and reverses memory impairment in insulin-resistant cognitively deficient mice by reducing BDNF levels.510
Tubastatin A is a specific HDAC6 inhibitor. Tubastatin A treatment reduces food intake, fat mass, hepatic steatosis and improves systemic glucose homeostasis by restoring leptin sensitivity.511
H3K27me3 demethylase inhibitor
GSK-J4 is the inhibitor of H3K27me3 demethylase towards KDM6B and KDM6A. It can reduce obesity-related properties in diet-induced obesity mice via the sensitised leptin signaling.512
Histone methyltransferase inhibitor
BIX01294 is an inhibitor of G9a histone methyltransferase. G9a histone methyltransferase mediates the phosphorylation of FOXO1 both in vivo and in vitro, regulating obesity and associated diseases.287
GSK126 is a specific EZH2 methyltransferase inhibitor. GSK126 alleviates obese phenotypes by promoting diet-induced differentiation of thermogenic beige adipocytes in obese mice and decreasing EZH2 enzymatic activity.513
USP30 inhibitor
MF-094 is a selective inhibitor of USP30. MF-094 can accelerate diabetic wound healing by deubiquitinating NLRP3 to inhibit the NLRP3 inflammasome.514
Ubiquitin-activating enzyme E1
PYR-41 is the first cell-permeable inhibitor of ubiquitin-activating enzyme E1, but invalid to E2. PYR-41 inhibits the ubiquitin-activating enzyme UBA1 to alleviate atherosclerosis and suppress the inflammatory response of macrophage in ApoE-/- mice.446
DUB inhibitor
GSK2643943A is a DUB inhibitor. GSK2643943A decreases the USP20 hydrolyzed K48- and K63-linkages and reduces HMGCR ubiquitination to inhibit diet-induced cholesterol biosynthesis.515
NEDD8-activating enzyme inhibitor
MLN4924 is a kind of small molecule NEDD8-activating enzyme (NAE) inhibitor. It has been demonstrated that MLN4924 effectively prevents diet-induced obesity and the disorder of glucose metabolism in mice.516 Similarly, MLN4924 causes pharmacological inhibition of cullin neddylation, which prolongs insulin action in hepatocytes and decreases hepatic glucose production in mice.
PARP inhibitors
PJ34 is a potent specific inhibitor of PARP1/2, that can reduce PARP activity, GAPDH ribosylation, and GAPDH translocation in the mouse model of T2DM.517
Clinical trials of metabolic diseases by targeting protein modifications
Growing evidence corroborates the essential role of PTMs in metabolic diseases. Families of drugs targeting protein modification enzymes include kinase agonists and inhibitors, HDAC inhibitors, histone methyltransferase inhibitors, and others. Here, we focus on clinical trials of metabolic diseases by agents that target protein modification. Table 1 shows the current clinical trials of metabolic diseases by targeting protein modification.
HMG-CoA reductase inhibitors (Statins)
The 3-hydroxymethylglutaryl coenzyme A reductase inhibitors (statins) is an effective prevention strategy for vascular diseases such as cardiovascular diseases and stroke for more than 20 years. Statins are used as hypolipidemic drugs which act as a primary response by inhibiting cholesterol synthesis. Statins reduce cholesterol biosynthesis, mainly in the liver, and consequently modulate endogenous lipid metabolism. The anti-atherosclerotic effects of statins are correlated with reduced LDL cholesterol levels. In addition, statins exert a cardiovascular protective impact by non-lipid dependent actions referred to as “pleiotropic” effects. Because statins lower the intermediates of the mevalonate pathway beyond cholesterol, they inhibit the posttranslational prenylation of small guanosine triphosphate-binding proteins and their downstream effectors.518 To date, statins also confer protective effects against NAFLD in addition to cardiovascular protection. In a clinical research using atorvastatin, the severity of NAFLD activity ratings were dramatically reduced in individuals with dyslipidemia (NCT02633956).519 Statins were also discovered to lower the risk of NAFLD in a 6-year follow-up cohort of more than 11 million individuals.520
ACC inhibitors
ACC is a rate-limiting molecule in DNL that converts acetyl-CoA to malonyl-CoA. ACC1 and ACC2 are two isoforms of ACC in mammals. AMPK phosphorylates ACC1 Ser79Ala and ACC2 Ser212Ala, regulating the fatty acid synthesis and oxidation.374,521,522 ACC inhibitors fall into two main classes: natural and non-natural (synthetic) compounds. These compounds inhibit ACC activity in three ways: they phosphorylate and inactivate ACC by activating AMPK, inhibiting carboxyltransferase (CT) activity, and acting as biotin carboxylase (BC) inhibitors. Curcumin, a natural compound found in turmeric, influences metabolism through AMPK/ACC pathway as mentioned previously. Curcumin also inhibits the O-GlcNAcylation pathway to restore aggravated liver metabolic damage and improve lipid accumulation.523 In a randomized, double-blind, placebo-controlled trial with 240 participants, researchers looked at the effectiveness of curcumin in preventing the onset of T2DM in a group of prediabetics. The data demonstrated that it can effectively prevent prediabetes from developing T2DM (NCT01052025).524 According to research by Lu et al., giving mice with gestational diabetes a high dosage of curcumin (100 mg/kg) might reduce their glucose and insulin intolerance. In a group of 118 T2DM patients, Panahi et al. conducted an RCT with a focus on the effects of curcuminoids on ghrelin, adiponectin, leptin, and TNF. When the treatment (1000 mg of curcumin plus 10 mg of piperine per day) was administered to patients with T2DM for 12 weeks, patients showed an inrecese in adiponectin and a decrease in the leptin: adiponectin ratio, leptin levels and TNF levels, independently of weight changes.525 A meta-analysis of nine trials with a combined total of 262 animals revealed that curcumin had a significant impact on blood vessel density and the pace of wound healing, suggesting that it could be a likely candidate for the treatment of diabetic foot ulcers.526
MK-4074 is a type of pan-ACC inhibitor and a liver-selective medication that dose-dependently reduces liver lipids in humans. A phase I, randomized, double-blind, and placebo-controlled clinical study that included 31 adults with fatty liver disease investigated the effect of MK-4074; however, this research was unfinished (NCT01431521).
The liver receives a preferential distribution of the ACC inhibitor PF-05221304, which is selective, orally accessible, and reversible. It has been tested in phase I and II clinical trials, which showed that with PF-05221304 monotherapy, liver fat reductions reached 50–65% (NCT03248882). When the drugs PF-05221304 and PF-06865571 were administered together, liver fat was reduced in contrast to the placebo; after 6 weeks with either drug alone, the placebo-adjusted LSM decrease in liver fat was -44.5% or -35.4%, respectively (NCT03776175). The findings of two trials show that raising the dose of PF-05221304 did not result in an overall rise in the frequency of adverse events, and that co-administration of PF-05221304 and PF-06865571 may be able to circumvent some of the drawbacks of ACC inhibition alone.527
Firsocostat (GS-0976), also known as ND-630, has been assessed in clinical trials. 20 mg GS-0976 treatment is capable of lowering hepatic steatosis, according to a phase II clinical research involving 126 patients with at least 18% hepatic steatosis (NCT02856555). In another open-label, proof-of-concept, phase II trial, fisocostat plus semaglutide, fisocostat plus cilofexor, or semaglutide monotherapy, could result in the greater capability of preventing hepatic steatosis and biochemical markers528(NCT03987074).
Resveratrol (RES)
Resveratrol (RES) is an important phenolic phytochemical that may improve mitochondrial function and prevent metabolic diseases. PGC-1 activity and acetylation are reduced by RES, respectively.529 A phase III study in patients with diabetic nephropathy indicated that with the treatment of RES, the urine albumin/creatinine ratio was markedly decreased and serum creatinine was unchanged, while serum antioxidant enzymes were significantly increased (NCT02704494).530 Patients with T2DM and CHD who received resveratrol treatment for 4 weeks saw improvements in their glycemic control, HDL cholesterol levels, total cholesterol to HDL cholesterol ratio, TAC, and MDA levels.531, Trans-resveratrol and hesperidin improved insulin resistance, dyslipidemia, hyperglycemia, hypertension, and low-grade inflammation in obese subjects.532 Kantartzis et al. conducted the largest randomized, placebo-controlled clinical trial to investigate the 3-month impact of 150 mg/d resveratrol in 112 overweight or NAFLD patients. They found that resveratrol treatment improved the liver status and did not affect fat content or cardiometabolic parameters in humans.533 However, Fogacci et al. suggested that the earlier study was designed without addressing the dose-dependent effects and oral administration of resveratrol might lead to poor bioavailability.533 Furthermore, a trial demonstrated that patients with resveratrol (≥300 mg/day) uptake had a significant decrease in total cholesterol, blood pressure, and blood glucose.534 In a comprehensive review and meta-analysis, resveratrol’s effectiveness and safety in controlling lipid and glucose levels in people with T2DM were shown.533
ASK1 inhibitor
Upstream control of p38/JNK signaling is provided by the enzyme apoptosis signal-regulating kinase 1 (ASK1). Oxidative stress, ER stress, and inflammatory signals can primarily activate ASK1. P38/JNK pathway is associated with numerous diseases; however, the application of P38/JNK inhibitors is limited due to their essential roles in cell survival and homeostasis.535 As a result, ASK1 seems to be a different therapeutic target for conditions and cell death caused by p38/JNK. The role of ASK1 in metabolic disorders has been demonstrated in mice, supporting clinical trials of ASK1 inhibitory compounds.
In 2016, a phase II clinical trial involving 333 diabetic nephropathy patients(NCT02177786) suggested that 18 mg selonsertib (GS-4997, an ASK1 inhibitor) treatment for 48 weeks showed a 71% reduction in eGFR.536 Another phase IIb clinical trial assessing the effect of selonsertib on diabetic kidney disease is still ongoing (NCT04026165).
In 2016, a phase II clinical trial involving 72 patients with NASH and stage 2-3 liver fibrosis (NCT02466516) showed that, 18 mg and 6 mg selonsertib-treated groups had a 43% and 30% reduction in fibrosis, respectively.537 In another IIb trial (NCT03449446), selonsertib combing firsocostat and cilofexor could effectively improve NASH activity and fibrosis.538
PARP inhibitors
It has been discovered that the hydroximic acid derivative BGP-15 inhibits PARP1. BGP-15 can also prevent p38 MAPK/JNK activation and induce phosphorylation of Akt and GSK-3,539 increasing insulin sensitivity. In a phase II clinical trial including 47 non-diabetic patients with impaired glucose tolerance, the BGP-15 treatment group demonstrated enhanced insulin sensitivity, and the drug was safe and well-tolerated.540 However, a phase II experiment that examined the safety and effectiveness of BGP-15 in T2DM patients was stopped early for lack of findings (NCT01069965).
FXR agonists
The Farnesoid X Receptor (FXR) is a member of the nuclear receptor family is a ligand-stimulated transcription factor. Many genes involved in the metabolism of bile acids, lipids, glucose, and amino acids are controlled by FXR. FXR may be phosphorylated, SUMOylated, and acetylated, and these modifications have an impact on the receptor functions.
The safety, tolerability, pharmacodynamics, and pharmacokinetics of tropifexor (LJN452) were studied in a clinical trial funded by Novartis Institutes for Biomedical Research. The results showed that in healthy subjects, tropifexor was well tolerated without altering plasma lipid levels, and had a pharmacokinetic profile suitable for once-daily administration.541 A phase II clinical trial enrolling 99 NASH patients showed that the decrease in total liver fibrosis was associated with the tropifexor therapy.542(NCT02855164).
In a phase II, double bind, placebo-control clinical trial with 140 noncirrhotic NASH patients, the 100 mg cilofexor (GS-9647) treatment group achieved a better result in NAFLD Activity Score (NAS), hepatic histology, and hepatic biochemistry543(NCT02854605). In two phase II co-medication clinical trials, cilofexor with semaglutide or firsocostat may be a better treatment strategy for NASH528,538 (NCT03449446, NCT03987074).
Caspase inhibitors
Caspases, a class of cysteine-dependent aspartate-specific proteases, are crucial for maintaining cellular and organismal homeostasis by mediating inflammatory response and cell death. PTMs that control caspase modification, including phosphorylation, nitrosylation, ubiquitination, SUMOylation, glutathionylation, and acetylation, are essential for both cell survival and death.544
To date, several clinical trials of emricasan (an irreversible pan-caspase inhibitor) have shown improved biomarkers.545 However, in a randomized, placebo-controlled phase II trial, 318 NASH patients were administrated with 5 mg emricasan, 50 mg emricasan or placebo; emricasan did not improve liver ballooning and NASH fibrosis546 (NCT02686762).546 In another phase II double-blind, placebo-controlled clinical trial with 217 NASH patients, emricasan is safe but has a poor therapeutic effect in patients with cirrhosis547(NCT03205345).
Glucokinase activators
Glucokinase (GCK) is one member of the hexokinase enzymes family and plays a critical role in glucose metabolism as a glucose-sensing. GCK regulates glucose homeostasis by catalyzing the phosphorylation of glucose into glucose-6 phosphate. GCK activity and cellular stability in beta cells are controlled by SUMOylation. S-nitrosylation of GCK at cysteine-371 promotes GCK enzymatic activity and increases insulin secretion. Glucokinase activators (GKAs), a particular class of T2DM medication, are created to target GCK. Several GKAs have recently been tested in clinical trials.
Dorzagliatin was the first GKA approved for T2DM in China. In a trial, 24 patients with T2DM were randomly assigned to receive dorzagliatin 75 mg once or twice daily for 28 days. Dorzagliatin displayed the capacity to improve β-cell function and regulate blood glucose. In a phase II, multicenter, randomized, double-blinded, and placebo-controlled clinical study enrolling 258 Chinese individuals with T2DM, dorzagliatin was safe and well tolerated, which finally improved glycemic control throughout 12 weeks548(NCT02561338). In another phase III clinical trial, dorzagliatin combined with metformin exhibited good glycemic management ability with safety549(NCT03141073).
In a phase III clinical study with 587 T2DM patients, MK-0941 (a new GKA) significantly reduced the levels of HbA1c and postmeal glucose in individuals that received conventional treatment of insulin glargine (metformin 1500 mg/day).550
AZD1656, another GKA, demonstrated good safety and tolerability in numerous ascending dose trials, and patients experienced lower fasting blood glucose levels.551 Moreover, the HbA1c and blood glucose levels were lowered by AZD1656 treatment in two different dose-range investigations552,553(NCT01152385; NCT01020123).
PTP1B inhibitors
The first member of the protein tyrosine phosphatase (PTP) family and a suppressor of the leptin and insulin signaling pathways is PTP1B. PTP1B is abundantly expressed in tissues that respond to insulin, negatively regulating both integrin and insulin by dephosphorylation. PTP1B enzyme activity is inhibited by S-nitrosylation of Cys215. In diverse animal models, several PTP1B inhibitors have demonstrated their potential for treating numerous diseases, such as diabetes, obesity, and cancer. However, few PTP1B inhibitors are suitable for clinical application and the trials about PTP1B inhibitors went to termination because of their poor efficacy and safety. For example, trodusquemine (MSI-1436) was studied in 4 phase I clinical trials which were completed without results from 2007 to 2018 (NCT00509132; NCT00806338; NCT00606112; NCT02524951).
HDAC inhibitors
HDAC, which removes the acetyl group from histone and non-histone proteins, is a novel molecular target in the treatment of type 2 diabetes and obesity. Clinical trials have looked into HDAC inhibitors.
In a clinical study with a double-blinded, randomized, placebo-controlled, crossover design, 18 T2DM patients accepted sodium phenylbutyrate (NaPB, an HDAC inhibitor) or a placebo. However, no difference was observed in the levels of insulin, triglycerides, FFA, or the amount of fat accumulating in muscle and the liver. The experiment indicated that NaPB increased peripheral insulin sensitivity when compared to placebo554(NTR7426). Another clinical trial involving 8 overweight or obese nondiabetic male subjects treated with phenylbutyrate (PBA) or placebo demonstrated that PBA prevented lipid-induced β-cell dysfunction.555
Concluding remarks and future perspectives
Novel PTMs in cardiometabolic diseases
In addition to the classic PTMs addressed in detail above, some novel PTMs emerge, such as succinylation, lactylation, beta-hydroxybutyryration, and lysine crotonylation. Next, we will describe the roles of some representative novel PTMs in metabolic diseases.
Succinylation and cardiometabolic disease
SIRT7 desuccinylates PRMT5, enhances PRMT5 activity, and induces SREBP1a arginine methylation. Arginine di-methylation of SREBP1a promotes the biogenesis of fatty acids, TGs and cholesterol.556 The data suggest that succinylation is a negative feedback regulator in cholesterol metabolism.557 Serum succinate levels were elevated in hyperlipidemia individuals and atherosclerotic mouse models,558 and succinate activates the succinate receptor 1 (SUCNR1)/renin-angiotensin system (RAS) pathway to increase Ang-II secretion, leading to endothelial cell damage.559 Succinate promotes VSMC proliferation and SMC migration. By the production of pro-inflammatory cytokines, succinate stimulates M1 polarization and causes atherosclerosis to form.184 Nine succinylation sites were identified in DIO mice.560 Increased protein succinylation has been identified in the muscle of type 1 diabetic rats and adipose tissue in type 2 diabetic mice.561 SIRT5-mediated desuccinylation of the K108 site of optineurin protects retinal ganglion cells in diabetic retinopathy.562 Succinated protein has been found in adipose tissue of DIO, db/db, and ob/ob mice.561 Supplementation with succinic acid protects mouse female offspring from HFD-induced obesity.563 Bioinformatics analysis showed that succinylation was the target of SIRT5, and liver overexpression of SIRT5 improved metabolic disorders in ob/ob mice. An earlier study using a mouse model of NAFLD caused by a high-fat, low-protein diet and carbon tetrachloride injection discovered substantial alterations in the liver’s succinylation levels.183
Lysine crotonylation and cardiometabolic disease
Lysine crotonylation is involved in cellular metabolism, cell cycle and tissue processes.564 Short-chain enyl-CoA hydrase (ECHS1) favors cardiac homeostasis by mediating crotonylation of histone proteins. Downregulation of ECHS1 increases histone crotonylation levels at H3K18 and H2BK12 and enhances the expression of genes associated with cardiac hypertrophy. The role of lysine crotonylation in metabolic diseases remains to be investigated. Improving crotonylation levels of oocytes can combat a decrease in oocyte quality in diabetics.193 Like acetylation, protein crotonylation is essential for controlling the browning of WAT. Acox2 is a lysine crotonylation regulator that controls the mice’s hepatic metabolic balance.192 It is still unknown how lysine crotonylation affects metabolic disorders.
Beta-hydroxybutyryration and cardiometabolic disease
Histone lysine β-hydroxybutyrylation (KbHb) is connected to upregulated genes in metabolic response pathways to starvation. Stimulation of ketone body production after starvation increases KbHb and lysine butyrylation.565 KbHb’s macronutrient pathway is enriched, including fatty acid β-oxidation, TCA cycle, glycolysis, amino acid metabolism, urea cycle, and ketogenesis.566 Elevated β-hydroxybutyric acid has been found to antagonize aortic endothelial damage and has a protective effect on diabetic vascular disease;197 at the same time, KbHb can inhibit the occurrence of kidney disease caused by diabetes.567 KbHb has the effect of improving obesity.568 Oral β-hydroxybutyric acid has been reported to improve blood lipid status by increasing HDL levels and reducing LDL/HDL ratios in Wistar rats.198 There is growing evidence that β-OHB is beneficial in vascular function and metabolism. In the Chinese sample, serum β-OHB levels were discovered to be related to coronary artery disease.569 Oral administration of ketone body 3-hydroxybutyrate (3-HB) can improve atherosclerosis in ApoE-/- mice by inhibiting macrophage autophagy.199
Lactylation and cardiometabolic disease
Histone lactylation mediates repolarization of macrophages from M1 to M2 by transitioning from glycolysis to oxidative phosphorylation, thereby providing an anti-atherosclerotic effect.204 Elevated serum lactate levels were found to be significantly associated with plaque load and atherosclerosis in the Community Atherosclerosis Risk (ARIC) study cohort.206 In human skeletal muscle, lactic acid-induced lactation is correlated with insulin intolerance.207
The association of novel protein modification sites suggested by high-sensitivity mass spectrometry
Mass spectrometry (MS) is a cutting-edge research tool for PTM analysis due to its advantages of excellent specificity and sensitivity. Numerous protein changes can be found using MS. With the improvement of analytical methods and mass spectrometry instruments, the methods for detecting PTMs are becoming increasingly comprehensive. Fourier transform (FT)-based mass spectrometers are characterized by high resolution and high sensitivity for PTM analysis. Discovery of structural mechanisms involving histone activity depends on crosslinked mass spectrometry (XL-MS).570 Using the LC-MS/MS method to assess the efficacy of the discovered modification sites, an MS/MS spectrogram can be produced. The isolation of target chromatin regions for the purpose of identifying histone posttranslational modifications is made easier by chromatin affinity purification using MS (ChAP-MS).571 The recently developed electron-transfer/higher-energy collision dissociation (ETHcD) has allowed for appreciable strides in addressing the uncertain locations of traditional fragmentation techniques in PTM omics.572 Affi-BAMS’ novel analytical immunoassay platform provides fast, highly specific quantitative data for protein and PTM targets. Accurate analysis of specific targets in complex proteomes is also possible.573 LC-FAIMS-MS/MS plays an important role in characterizing PTM crosstalk.574 Data-independent mass spectrometry techniques can identify precursor ion compliance with the modification, determine the quality and localization of peptide sequence modifications, and also record the peptide content in the sample, including the modified peptide. PTM site deletions can be detected in combination with LC-MS, isotope and antipeptide antibodies. LERLIC-MS/MS is used to study PTM modifications of complex biological samples in shotgun proteomics, which is directly coupled to tandem mass spectrometry (MS/MS). Broadband collision-induced dissociation (bbCID) mass spectrometry guide targeted proteomics, especially in the identification of glycopeptides and phosphopeptides.575 Low-intensity precursor ions can be used in liquid chromatography-electrospray ionization-quadrupole-time-of-flight mass spectrometry (LC-ESI-q-TOF MS/MS) to identify changed peptides.576
High-sensitivity mass spectrometry can efficiently and accurately identify metabolic disease-related proteins, and analyze the type and abundance of PTMs. For example, high-sensitivity mass spectrometry can aid diabetes risk assessment by analyzing amino acids, amines, and other polar metabolites, and the binding to tagging sites allows specific quantification of PTM sites.577 T2DM is linked with the type and site of PTM of hemoglobin, which may offer useful noninvasive biomarkers for protein damage in T2DM. The research of T2DM and other metabolic diseases is made possible by the detailed analysis of various sites in advanced glycosylation end products (AGEs).578 Mass spectrometry-based proteomics has discovered tens of thousands of PTM sites in the proteome. Proteomics techniques can measure and quantify PTMs that co-occur on the proteome or the same protein. At the same time, examining the steady-state levels of histone PTM in cells under various perturbations makes it possible to investigate the crosstalk between PTMs and its functional significance. Research on histone and nucleoproteins has recently benefited from the integration of proteomics based on high-resolution mass spectrometry and isolation of acetylated peptides.579 The latest PTM detection methods above have opened up the area of PTMs and their role in metabolic and cardiovascular diseases.
The crosstalk of different PTM omics
The interaction between multiple PTMs in a protein to jointly regulate the biochemical function of a protein through positive and negative regulatory interactions is called PTM crosstalk. Positive and negative crosstalk are two categories of crosstalk, depending on how the PTMs interact with each other. When multiple modifications of the same or distinct kinds are present in the same sequence area (within five amino acids), but do not impact the same residues, this is referred to as positive crosstalk. These changes could take place concurrently, or they could have a causal or chronological connection. Negative crosstalk occurs when two PTMs directly fight for the same amino acid, when one PTM obscures the recognition site of another PTM, or when PTMs “compete” for the same residue in a causal or temporal way.580 In important protein domains like histones, protein kinases, and RNA recognition motifs, which are involved in a variety of biological processes like RNA processing, DNA damage response, signal transduction, and cell cycle regulation, crosstalk between multiple PTMs happens more frequently.581
There is increasing evidence that PTM crosstalk is associated with the occurrence and development of various diseases, including metabolic diseases. Studies have suggested that interactions between O-GlcNAcylation and other PTMs may be involved in obesity and diabetes, and glucose toxicity may promote metabolic syndrome through abnormal O-GlcNAcylation and ubiquitination of these proteins in various tissues. Crosstalk between O-GlcNAcylation and phosphorylation initiated by insulin signaling is associated with cytoplasmic enzyme activity that regulates eNOS during vasodilation, and kinetic dysregulation between O-GlcNAcylation and eNOS phosphorylation leads to early endothelial dysfunction in diabetes-accelerated atherosclerosis.582 O-GlcNAc acylation in the insulin signaling pathway, which is the changed central pathway in diabetes, can prevent phosphorylation of the site and impair this signaling pathway.583 PTM crosstalk shows different mechanisms in different types of diseases, whereas in situ crosstalk may drive differential interactions in CVDs. p53 is a classic example of non-histone PTM crosstalk. By directly competing with the ubiquitination of the same residue, which is carried out by MDM2, p53’s CTD lysine acetylation inhibits ubiquitin-proteasome-dependent breakdown. We can better comprehend the etiology of diseases and identify novel targets for drug treatment with a thorough knowledge of PTM crosstalk.584
The superiority of targeting PTMs
Various PTMs play a role in almost every part of biological processes, and abnormal PTM states are frequently linked to human illnesses. A method for natural protein breakdown known as proteolysis-targeting chimera (PROTAC) successfully ubiquitinates target proteins using the in vivo UPS.585 A new medicinal approach to drug finding is using PROTAC-targeted protein degradation. The PROTAC complex consists of the E3 ligase and the target protein. In addition to offering novel chemical knockdown instruments for biological study, PROTACs have the potential to be used as therapeutic therapies for illnesses like obesity, hyperlipidemia, cancer, immune disorders, virus infections, and neurodegenerative diseases. PROTACs are divided into PROTACs that target protein kinases,586 nuclear receptors, and transcriptional regulators. In traditional cancer treatment, gene editing technology is used to remove cancer-related pathogenic proteins, such as CRISPR-Cas9 technology, genetic change through RNA interference, transcription activator-like effector nucleases, and recombination-based gene deletion. The expensive and time-consuming nature of conventional genome editing methods, especially in large nonhuman primates, has caused many inconveniences to researchers. In addition, some uncertainties, such as spontaneous mutations in knockout models, potential genetic compensation, and embryonic lethality, may lead to failure. PROTAC offers a potential strategy to knock down proteins of interest rapidly and reversibly, which is not possible with genome editing strategies.587 The advantages of PROTACs include event-driven activity, targeting undruggable proteins, overcoming traditional drug resistance, and other effects. Statistics show that more than 80% of human proteins are unreachable targets.588 Since POI ligands, E3 ubiquitin ligases, and linkers work together synergistically, and the ternary complex has a rigid shape, PROTAC molecules show the advantage of high selectivity for target proteins, which can convert non-druggable targets into druggable targets. PROTAC has the advantage of targeting non-druggable proteins.589 Current research on PROTACs is more focused on the treatment of cancer.
A method to achieve extensive substrate dephosphorylation has been developed, called phosphorylation targetan mosaicism (PhosTACs). Unlike PROTACs, PhosTAC focuses on recruiting Ser/Thr phosphatase into phosphate matrices to mediate its dephosphorylation, which can uniquely provide targeted function gain opportunities to manipulate protein activity; this mode of action provides PROTAC with greater selectivity, extended period of effect, and the ability to overcome drug resistance. PhosTAC works similarly to PROTAC and has great potential both in terms of biological tools and clinical treatment.590
In recent years, studies have found that PNPLA3 levels are lowered by PROTAC-mediated degradation, reducing liver TG content in PNPLA3-overexpressing (148 M) mice.591 An oral active VHL recruitment PROTAC (21c) can lead to HMGCR degradation as well as cholesterol reduction, which offers fresh methods for treating hyperlipidemia and associated metabolic illnesses.592 Lycoline binds directly to SCAP to inhibit SREBF, thereby improving HFD-induced obesity, hyperlipidemia and insulin resistance.593
Development of PTM database and research tools
In numerous biological functions, protein PTMs are crucial.594 PTM is closely linked to the onset and progression of many illnesses, including metabolic illnesses like obesity, diabetes, hypertension, and hyperlipidemia. Understanding basic biological processes and the frequency of diseases is significantly affected by PTM analysis. The PTM database is a database for researchers to study the protein structure of multiple species, their association with diseases, PTM sites, the relationship between enzymes and substrates, the relationship between substrates and PTM sites, protein interactions, cell signaling, and transcription. Information on regulation and PTM site prediction provides important information for the majority of researchers. Table 2 lists the main PTM databases.
Classical methods for the detection of protein PTMs include specific antibody development, radioisotope-labeled substrates, western blot analysis, chemically tagged peptides,595 and PTM-specific enzymes by peptides. The advancement of new biotechnologies to examine the landscape of different PTMs in various metabolic diseases will lead to the identification of new drug candidates. Figure 8 describes the crosstalk of PTMs, MS detection methods, PROATC, and PTM database.

Future outlook of PTMs research. To date, there are several PTM databases, and each one contains thousands of proteins and overlapping parts. PTM crosstalk regulates multiple PTMs on the same or different protein substrates. Mass spectrometry (MS) is a commonly used tool to study PTMs, and MS-based different PTM omics indicate the potential crosstalk. Recently, the proteolysis-targeted chimerism (PROTAC) technology for targeted protein degradation is an innovative strategy to treat various diseases. PhosTAC focuses on recruiting Ser/Thr phosphatase into phosphate matrices to mediate, which can uniquely provide targeted function gain opportunities to manipulate protein activity. PhosTAC works similarly to PROTAC and has great potential as biological and pharmacological tools. The figure is generated with BioRender (https://biorender.com)
In summary, PTMs control target protein function and stability, protein-protein interactions, and subcellular localization, contributing to almost all biological processes, thus providing novel and diverse mechanisms for the regulation of systematic biology. Targeting PTMs presents an opportunity for a novel or refined drug development that could provide a novel and more precise approach for the management of metabolic diseases.
References
International Diabetes Federation. IDF Diabetes Atlas, 10th edn. Brussels, Belgium: 2021. https://www.diabetesatlas.org.
Wai-Sun Wong, V., Ekstedt, M., Lai-Hung Wong, G. & Hagström, H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J. Hepatol. https://doi.org/10.1016/j.jhep.2023.04.036 (2023).
Afshin, A. et al. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 377, 13–27 (2017).
Malik, S. et al. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation 110, 1245–1250 (2004).
Roth, G. A. et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J. Am. Coll. Cardiol. 76, 2982–3021 (2020).
Dagenais, G. R. et al. Variations in common diseases, hospital admissions, and deaths in middle-aged adults in 21 countries from five continents (PURE): a prospective cohort study. Lancet 395, 785–794 (2020).
National Health Expenditure Data: Historical. Center for Medicare & Medicaid Services. December 15, 2021. Accessed May 5, 2022. https://www.cms.gov/Research-Statistics-Data-and-Systems/Statistics-Trends-and-Reports/NationalHealthExpendData/NationalHealthAccountsHistorical.
Blom, N., Sicheritz-Pontén, T., Gupta, R., Gammeltoft, S. & Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 (2004).
Levene, P. A. & Alsberg, C. L. The cleavage products of Vitellin. J. Biol. Chem. 2, 127–133 (1906).
Burnett, G. & Kennedy, E. P. The enzymatic phosphorylation of proteins. J. Biol. Chem. 211, 969–980 (1954).
Allfrey, V. G., Faulkner, R. & Mirsky, A. E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl Acad. Sci. USA 51, 786–794 (1964).
Linn, T. C., Pettit, F. H. & Reed, L. J. Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc. Natl Acad. Sci. USA 62, 234–241 (1969).
Tunyasuvunakool, K. et al. Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 (2021).
Ramazi, S. & Zahiri, J. Posttranslational modifications in proteins: resources, tools and prediction methods. Database (Oxford) 2021, baab012. https://doi.org/10.1093/database/baab012 (2021).
Cohen, P., Cross, D. & Jänne, P. A. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat. Rev. Drug Discov. 20, 551–569 (2021).
Cohen, P. Protein kinases-the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1, 309–315 (2002).
Liberale, L. et al. JCAD promotes arterial thrombosis through PI3K/Akt modulation: a translational study. Eur. Heart J. https://doi.org/10.1093/eurheartj/ehac641 (2022).
Lally, J. S. V. et al. Inhibition of acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab. 29, 174–182.e175 (2019).
Sacco, F. et al. Phosphoproteomics reveals the GSK3-PDX1 axis as a key pathogenic signaling node in diabetic islets. Cell Metab. 29, 1422–1432.e1423 (2019).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019).
Jin, J. & Pawson, T. Modular evolution of phosphorylation-based signalling systems. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2540–2555 (2012).
Yin, C. et al. Phosphorylation-mediated assembly of a semisynthetic fluorescent protein for label-free detection of protein kinase activity. Anal. Chem. 87, 6311–6318 (2015).
Verdin, E. & Ott, M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 16, 258–264 (2015).
Davies, M. N. et al. The acetyl group buffering action of carnitine acetyltransferase offsets macronutrient-induced lysine acetylation of mitochondrial proteins. Cell Rep. 14, 243–254 (2016).
Christensen, D. G. et al. Mechanisms, detection, and relevance of protein acetylation in prokaryotes. mBio 10, e02708–18 (2019).
Lei, L. et al. Histone deacetylase 3-selective inhibitor RGFP966 ameliorates impaired glucose tolerance through beta-cell protection. Toxicol. Appl. Pharm. 406, 115189 (2020).
Kim, H. S. et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 12, 224–236 (2010).
Lecce, L. et al. Histone deacetylase 9 promotes endothelial-mesenchymal transition and an unfavorable atherosclerotic plaque phenotype. J. Clin. Invest 131, e131178 (2021).
Schoenheimer, R., Ratner, S. & Rittenberg, D. Studies in protein metabolism: VII. The metabolism of tyrosine. J. Biol. Chem. 127, 333–344 (1939).
Husmann, D. & Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 26, 880–889 (2019).
Yang, Y. & Bedford, M. T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 13, 37–50 (2013).
Carlson, S. M. & Gozani, O. Emerging technologies to map the protein methylome. J. Mol. Biol. 426, 3350–3362 (2014).
Lee, D. Y., Teyssier, C., Strahl, B. D. & Stallcup, M. R. Role of protein methylation in regulation of transcription. Endocr. Rev. 26, 147–170 (2005).
Wang, D. et al. Hepatocellular BChE as a therapeutic target to ameliorate hypercholesterolemia through PRMT5 selective degradation to restore LDL receptor transcription. Life Sci. 293, 120336 (2022).
Kim, H. et al. PRMT1 is required for the maintenance of mature beta-cell identity. Diabetes 69, 355–368 (2020).
Lin, L., Zhang, Q., Fan, H., Zhao, H. & Yang, Y. Myocardin-related transcription factor A mediates LPS-induced iNOS transactivation. Inflammation 43, 1351–1361 (2020).
Goldstein, G. et al. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl Acad. Sci. USA 72, 11–15 (1975).
Callis, J. The ubiquitination machinery of the ubiquitin system. Arabidopsis Book 12, e0174 (2014).
Schulman, B. A. & Harper, J. W. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 10, 319–331 (2009).
Stewart, M. D., Ritterhoff, T., Klevit, R. E. & Brzovic, P. S. E2 enzymes: more than just middle men. Cell Res. 26, 423–440 (2016).
Yang, Q., Zhao, J., Chen, D. & Wang, Y. E3 ubiquitin ligases: styles, structures and functions. Mol. Biomed. 2, 23 (2021).
Reyes-Turcu, F. E., Ventii, K. H. & Wilkinson, K. D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 78, 363–397 (2009).
Sun, M. & Zhang, X. Current methodologies in protein ubiquitination characterization: from ubiquitinated protein to ubiquitin chain architecture. Cell Biosci. 12, 126 (2022).
Micel, L. N., Tentler, J. J., Smith, P. G. & Eckhardt, G. S. Role of ubiquitin ligases and the proteasome in oncogenesis: novel targets for anticancer therapies. J. Clin. Oncol. 31, 1231–1238 (2013).
Wang, J. Q. et al. Inhibition of ASGR1 decreases lipid levels by promoting cholesterol excretion. Nature 608, 413–420 (2022).
Wu, H. K. et al. Glucose-sensitive myokine/cardiokine MG53 regulates systemic insulin response and metabolic homeostasis. Circulation 139, 901–914 (2019).
Brauner, S. et al. Augmented Th17 differentiation in Trim21 deficiency promotes a stable phenotype of atherosclerotic plaques with high collagen content. Cardiovasc. Res. 114, 158–167 (2018).
Meluh, P. B. & Koshland, D. Evidence that the MIF2 gene of Saccharomyces cerevisiae encodes a centromere protein with homology to the mammalian centromere protein CENP-C. Mol. Biol. Cell 6, 793–807 (1995).
Ramazi, S., Allahverdi, A. & Zahiri, J. Evaluation of post-translational modifications in histone proteins: a review on histone modification defects in developmental and neurological disorders. J. Biosci 45, 135 (2020).
Chu, Y. & Yang, X. SUMO E3 ligase activity of TRIM proteins. Oncogene 30, 1108–1116 (2011).
Omenn, G. S. et al. Progress on Identifying and Characterizing the Human Proteome: 2018 Metrics from the HUPO Human Proteome Project. J. Proteome Res. 17, 4031–4041 (2018).
Sedek, M. & Strous, G. J. SUMOylation is a regulator of the translocation of Jak2 between nucleus and cytosol. Biochem. J. 453, 231–239 (2013).
Kunz, K., Piller, T. & Müller, S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci 131, jcs211904 (2018).
Matunis, M. J. & Rodriguez, M. S. Concepts and methodologies to study protein SUMOylation: an overview. Methods Mol. Biol. 1475, 3–22 (2016).
Celen, A. B. & Sahin, U. Sumoylation on its 25th anniversary: mechanisms, pathology, and emerging concepts. FEBS J. 287, 3110–3140 (2020).
Wang, J. Q. et al. SUMOylation of the ubiquitin ligase IDOL decreases LDL receptor levels and is reversed by SENP1. J. Biol. Chem. 296, 100032 (2021).
Yan, C. H. et al. AMPKα2 controls the anti-atherosclerotic effects of fish oils by modulating the SUMOylation of GPR120. Nat. Commun. 13, 7721 (2022).
Kamitani, T., Kito, K., Nguyen, H. P. & Yeh, E. T. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J. Biol. Chem. 272, 28557–28562 (1997).
Zhou, L., Jiang, Y., Luo, Q., Li, L. & Jia, L. Neddylation: a novel modulator of the tumor microenvironment. Mol. Cancer 18, 77 (2019).
Enchev, R. I., Schulman, B. A. & Peter, M. Protein neddylation: beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 16, 30–44 (2015).
Wu, J. T., Lin, H. C., Hu, Y. C. & Chien, C. T. Neddylation and deneddylation regulate Cul1 and Cul3 protein accumulation. Nat. Cell Biol. 7, 1014–1020 (2005).
Wang, S. Y. et al. Review of NEDDylation inhibition activity detection methods. Bioorg. Med. Chem. 29, 115875 (2021).
Asare, Y. et al. Inhibition of atherogenesis by the COP9 signalosome subunit 5 in vivo. Proc. Natl Acad. Sci. USA 114, E2766–E2775 (2017).
Zhang, Y., Guo, Q., Jiang, G. & Zhang, C. Dysfunction of Cullin 3 RING E3 ubiquitin ligase causes vasoconstriction and increased sodium reabsorption in diabetes. Arch. Biochem. Biophys. 710, 109000 (2021).
Bause, E. & Legler, G. The role of the hydroxy amino acid in the triplet sequence Asn-Xaa-Thr(Ser) for the N-glycosylation step during glycoprotein biosynthesis. Biochem. J. 195, 639–644 (1981).
Reily, C., Stewart, T. J., Renfrow, M. B. & Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 15, 346–366 (2019).
Hirsch, C., Blom, D. & Ploegh, H. L. A role for N-glycanase in the cytosolic turnover of glycoproteins. EMBO J. 22, 1036–1046 (2003).
Li, H. et al. Chemoenzymatic method for glycoproteomic N-glycan type quantitation. Anal. Chem. 92, 1618–1627 (2020).
Wilkinson, H. & Saldova, R. Current methods for the characterization of O-glycans. J. Proteome Res. 19, 3890–3905 (2020).
van Kooyk, Y., Kalay, H. & Garcia-Vallejo, J. J. Analytical tools for the study of cellular glycosylation in the immune system. Front. Immunol. 4, 451 (2013).
Schjoldager, K. T., Narimatsu, Y., Joshi, H. J. & Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 21, 729–749 (2020).
van den Boogert, M. A. W. et al. N-glycosylation defects in humans lower low-density lipoprotein cholesterol through increased low-density lipoprotein receptor expression. Circulation 140, 280–292 (2019).
Essawy, A. et al. O-linked N-acetylglucosamine transferase (OGT) regulates pancreatic alpha-cell function in mice. J. Biol. Chem. 296, 100297 (2021).
Döring, Y. et al. Deficiency of the sialyltransferase St3Gal4 reduces Ccl5-mediated myeloid cell recruitment and arrest: short communication. Circ. Res. 114, 976–981 (2014).
Kamiya, Y., Sakurai, A., Tamura, S. & Takahashi, N. Structure of rhodotorucine A, a novel lipopeptide, inducing mating tube formation in Rhodosporidium toruloides. Biochem. Biophys. Res. Commun. 83, 1077–1083 (1978).
el-Husseini Ael, D. & Bredt, D. S. Protein palmitoylation: a regulator of neuronal development and function. Nat. Rev. Neurosci. 3, 791–802 (2002).
Bartels, D. J., Mitchell, D. A., Dong, X. & Deschenes, R. J. Erf2, a novel gene product that affects the localization and palmitoylation of Ras2 in Saccharomyces cerevisiae. Mol. Cell. Biol. 19, 6775–6787 (1999).
Zhang, M. M. & Hang, H. C. Protein S-palmitoylation in cellular differentiation. Biochem. Soc. Trans. 45, 275–285 (2017).
Zhang, Y. et al. Oxidized high-density lipoprotein promotes CD36 palmitoylation and increases lipid uptake in macrophages. J. Biol. Chem. 298, 102000 (2022).
Suzuki, M. et al. ELMOD2 is anchored to lipid droplets by palmitoylation and regulates adipocyte triglyceride lipase recruitment. Mol. Biol. Cell 26, 2333–2342 (2015).
Wu, Y. et al. Palmitoylated small GTPase ARL15 is translocated within Golgi network during adipogenesis. Biol. Open 10, bio058420 (2021).
Aitken, A. et al. Identification of the NH2-terminal blocking group of calcineurin B as myristic acid. FEBS Lett. 150, 314–318 (1982).
Zha, J., Weiler, S., Oh, K. J., Wei, M. C. & Korsmeyer, S. J. Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 290, 1761–1765 (2000).
Meinnel, T., Dian, C. & Giglione, C. Myristoylation, an ancient protein modification mirroring eukaryogenesis and evolution. Trends Biochem. Sci. 45, 619–632 (2020).
Raju, R. V. & Sharma, R. K. Coenzyme A dependent myristoylation and demyristoylation in the regulation of bovine spleen N-myristoyltransferase. Mol. Cell. Biochem. 158, 107–113 (1996).
Adam, R. M. et al. Cholesterol sensitivity of endogenous and myristoylated Akt. Cancer Res. 67, 6238–6246 (2007).
Thinon, E. et al. Global profiling of co- and post-translationally N-myristoylated proteomes in human cells. Nat. Commun. 5, 4919 (2014).
Ono, H. et al. Hepatic Akt activation induces marked hypoglycemia, hepatomegaly, and hypertriglyceridemia with sterol regulatory element binding protein involvement. Diabetes 52, 2905–2913 (2003).
Govatati, S. et al. Myristoylation of LMCD1 leads to its species-specific derepression of E2F1 and NFATc1 in the modulation of CDC6 and IL-33 expression during development of vascular lesions. Arterioscler. Thromb. Vasc. Biol. 40, 1256–1274 (2020).
Palsuledesai, C. C. & Distefano, M. D. Protein prenylation: enzymes, therapeutics, and biotechnology applications. ACS Chem. Biol. 10, 51–62 (2015).
Zhang, F. L. & Casey, P. J. Protein prenylation: molecular mechanisms and functional consequences. Annu. Rev. Biochem. 65, 241–269 (1996).
Doerr, A. Probes for protein prenylation. Nat. Methods 16, 459 (2019).
Wang, M. & Casey, P. J. Protein prenylation: unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 17, 110–122 (2016).
Akula, M. K. et al. Protein prenylation restrains innate immunity by inhibiting Rac1 effector interactions. Nat. Commun. 10, 3975 (2019).
Matti, A., Kyathanahalli, C. & Kowluru, A. Protein farnesylation is requisite for mitochondrial fuel-induced insulin release: further evidence to link reactive oxygen species generation to insulin secretion in pancreatic beta-cells. Islets 4, 74–77 (2012).
Porter, J. A., Young, K. E. & Beachy, P. A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 274, 255–259 (1996).
Xiao, X. et al. Cholesterol modification of smoothened is required for Hedgehog signaling. Mol. Cell 66, 154–162.e110 (2017).
Hu, A. et al. Cholesterylation of Smoothened is a calcium-accelerated autoreaction involving an intramolecular ester intermediate. Cell Res. 32, 288–301 (2022).
Hu, A., Zhou, M. & Song, B. L. Analysis of protein cholesterylation by biorthogonal labeling. Methods Mol. Biol. 2374, 27–36 (2022).
Ziegler, D. M. Role of reversible oxidation-reduction of enzyme thiols-disulfides in metabolic regulation. Annu. Rev. Biochem. 54, 305–329 (1985).
Xiong, Y., Uys, J. D., Tew, K. D. & Townsend, D. M. S-glutathionylation: from molecular mechanisms to health outcomes. Antioxid. Redox Signal 15, 233–270 (2011).
Grek, C. L., Zhang, J., Manevich, Y., Townsend, D. M. & Tew, K. D. Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 288, 26497–26504 (2013).
Townsend, D. M. S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response. Mol. Inter. 7, 313–324 (2007).
Gao, X. H., Bedhomme, M., Veyel, D., Zaffagnini, M. & Lemaire, S. D. Methods for analysis of protein glutathionylation and their application to photosynthetic organisms. Mol. Plant 2, 218–235 (2009).
Anashkina, A. A. et al. A novel approach for predicting protein S-glutathionylation. BMC Bioinforma. 21, 282 (2020).
Mieyal, J. J., Gallogly, M. M., Qanungo, S., Sabens, E. A. & Shelton, M. D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal 10, 1941–1988 (2008).
Di Domenico, F. et al. Glutathionylation of the pro-apoptotic protein p53 in Alzheimer’s disease brain: implications for AD pathogenesis. Neurochem. Res. 34, 727–733 (2009).
Sanchez-Gomez, F. J., Espinosa-Diez, C., Dubey, M., Dikshit, M. & Lamas, S. S-glutathionylation: relevance in diabetes and potential role as a biomarker. Biol. Chem. 394, 1263–1280 (2013).
Jeon, D., Park, H. J. & Kim, H. S. Protein S-glutathionylation induced by hypoxia increases hypoxia-inducible factor-1α in human colon cancer cells. Biochem. Biophys. Res. Commun. 495, 212–216 (2018).
Niwa, T., Naito, C., Mawjood, A. H. & Imai, K. Increased glutathionyl hemoglobin in diabetes mellitus and hyperlipidemia demonstrated by liquid chromatography/electrospray ionization-mass spectrometry. Clin. Chem. 46, 82–88 (2000).
Oae, S. & Shinhama, K. Organic thionitrites and related substances. a review. Org. Preparations Proced. Int. 15, 165–198 (1983).
Stamler, J. S. et al. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc. Natl Acad. Sci. USA 89, 7674–7677 (1992).
Francis, S. H., Busch, J. L., Corbin, J. D. & Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 62, 525–563 (2010).
Hess, D. T., Matsumoto, A., Kim, S. O., Marshall, H. E. & Stamler, J. S. Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166 (2005).
Fernando, V. et al. S-nitrosylation: an emerging paradigm of redox signaling. Antioxidants 8, 404 (2019).
Devarie-Baez, N. O., Zhang, D., Li, S., Whorton, A. R. & Xian, M. Direct methods for detection of protein S-nitrosylation. Methods 62, 171–176 (2013).
Wang, Z. Protein S-nitrosylation and cancer. Cancer Lett. 320, 123–129 (2012).
Carvalho-Filho, M. A. et al. Expression of Concern: Aspirin attenuates insulin resistance in muscle of diet-induced obese rats by inhibiting inducible nitric oxide synthase production and S-nitrosylation of IRbeta/IRS-1 and Akt. Diabetologia https://doi.org/10.1007/s00125-017-4292-5 (2017).
Khatib, S., Artoul, F., Gershko, M., Markman, G. & Vaya, J. The synthesis and analysis of S-nitorsylated paraoxonase 1. Biochem. Biophys. Res. Commun. 444, 354–359 (2014).
Chao, M. L. et al. S-nitrosylation-mediated coupling of G-protein alpha-2 with CXCR5 induces Hippo/YAP-dependent diabetes-accelerated atherosclerosis. Nat. Commun. 12, 4452 (2021).
Foster, M. W., Hess, D. T. & Stamler, J. S. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol. Med. 15, 391–404 (2009).
Mustafa, A. K. et al. H2S signals through protein S-sulfhydration. Sci. Signal 2, ra72 (2009).
Sen, N. et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 10, 866–873 (2008).
Zhang, D., Du, J., Tang, C., Huang, Y. & Jin, H. H(2)S-induced sulfhydration: biological function and detection methodology. Front. Pharmacol. 8, 608 (2017).
Sen, N. et al. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 45, 13–24 (2012).
Xie, L. et al. Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes 65, 3171–3184 (2016).
Fan, J. et al. Hydrogen sulfide lowers hyperhomocysteinemia dependent on cystathionine γ lyase S-sulfhydration in ApoE-knockout atherosclerotic mice. Br. J. Pharmacol. 176, 3180–3192 (2019).
Wang, Z. J., Wu, J., Guo, W. & Zhu, Y. Z. Atherosclerosis and the hydrogen sulfide signaling pathway - therapeutic approaches to disease prevention. Cell. Physiol. Biochem. 42, 859–875 (2017).
Sun, Y. et al. Hydrogen sulphide reduced the accumulation of lipid droplets in cardiac tissues of db/db mice via Hrd1 S-sulfhydration. J. Cell. Mol. Med. 25, 9154–9167 (2021).
Rogers, G. E. & Simmonds, D. H. Content of citrulline and other amino-acids in a protein of hair follicles. Nature 182, 186–187 (1958).
Rogers, G. E., Harding, H. W. & Llewellyn-Smith, I. J. The origin of citrulline-containing proteins in the hair follicle and the chemical nature of trichohyalin, an intracellular precursor. Biochim. Biophys. Acta 495, 159–175 (1977).
Arita, K. et al. Structural basis for Ca(2+)-induced activation of human PAD4. Nat. Struct. Mol. Biol. 11, 777–783 (2004).
Vossenaar, E. R., Zendman, A. J., van Venrooij, W. J. & Pruijn, G. J. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays 25, 1106–1118 (2003).
Clancy, K. W., Weerapana, E. & Thompson, P. R. Detection and identification of protein citrullination in complex biological systems. Curr. Opin. Chem. Biol. 30, 1–6 (2016).
Tarcsa, E. et al. Protein unfolding by peptidylarginine deiminase. Substrate specificity and structural relationships of the natural substrates trichohyalin and filaggrin. J. Biol. Chem. 271, 30709–30716 (1996).
Alghamdi, M. et al. An overview of the intrinsic role of citrullination in autoimmune disorders. J. Immunol. Res. 2019, 7592851 (2019).
Yang, M. L. et al. Citrullination of glucokinase is linked to autoimmune diabetes. Nat. Commun. 13, 1870 (2022).
Kudo, M. et al. L-citrulline inhibits body weight gain and hepatic fat accumulation by improving lipid metabolism in a rat nonalcoholic fatty liver disease model. Food Sci. Nutr. 9, 4893–4904 (2021).
Geraldino-Pardilla, L. et al. Association of anti-citrullinated peptide antibodies with coronary artery calcification in rheumatoid arthritis. Arthritis Care Res. 69, 1276–1281 (2017).
Chambon, P., Weill, J. D. & Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 11, 39–43 (1963).
Lüscher, B. et al. ADP-ribosylation, a multifaceted posttranslational modification involved in the control of cell physiology in health and disease. Chem. Rev. 118, 1092–1136 (2018).
Kim, M. K. Novel insight into the function of tankyrase. Oncol. Lett. 16, 6895–6902 (2018).
Bu, X., Kato, J. & Moss, J. Emerging roles of ADP-ribosyl-acceptor hydrolases (ARHs) in tumorigenesis and cell death pathways. Biochem. Pharmacol. 167, 44–49 (2019).
García-Saura, A. G., Herzog, L. K., Dantuma, N. P. & Schüler, H. MacroGreen, a simple tool for detection of ADP-ribosylated proteins. Commun. Biol. 4, 919 (2021).
Palazzo, L., Mikolčević, P., Mikoč, A. & Ahel, I. ADP-ribosylation signalling and human disease. Open Biol. 9, 190041 (2019).
Martire, S., Mosca, L. & d’Erme, M. PARP-1 involvement in neurodegeneration: a focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev. 146-148, 53–64 (2015).
Zampieri, M. et al. Increased PARylation impacts the DNA methylation process in type 2 diabetes mellitus. Clin. Epigenet. 13, 114 (2021).
Huang, K. et al. PARP1-mediated PPARα poly(ADP-ribosyl)ation suppresses fatty acid oxidation in non-alcoholic fatty liver disease. J. Hepatol. 66, 962–977 (2017).
Tarantini, S. et al. Treatment with the poly(ADP-ribose) polymerase inhibitor PJ-34 improves cerebromicrovascular endothelial function, neurovascular coupling responses and cognitive performance in aged mice, supporting the NAD + depletion hypothesis of neurovascular aging. Geroscience 41, 533–542 (2019).
Levine, R. L. Oxidative modification of glutamine synthetase. I. Inactivation is due to loss of one histidine residue. J. Biol. Chem. 258, 11823–11827 (1983).
Madian, A. G., Regnier, F. E. & Zeng, A. In Protein Carbonylation 24–47 (2017).
Alomari, E. et al. Protein carbonylation detection methods: a comparison. Data Brief. 19, 2215–2220 (2018).
Wang, Q. et al. Post-translational control of beige fat biogenesis by PRDM16 stabilization. Nature 609, 151–158 (2022).
Shao, C. H. et al. Carbonylation contributes to SERCA2a activity loss and diastolic dysfunction in a rat model of type 1 diabetes. Diabetes 60, 947–959 (2011).
Hu, L. et al. Cyanate induces oxidative stress injury and abnormal lipid metabolism in liver through Nrf2/HO-1. Molecules 24, 3231 (2019).
Sukhbold, E. et al. Effects of oolonghomobisflavan A on oxidation of low-density lipoprotein. Biosci. Biotechnol. Biochem 81, 1569–1575 (2017).
Allison, W. S. Formation and reactions of sulfenic acids in proteins. Acc. Chem. Res. 9, 293–299 (1976).
Mailloux, R. J., Jin, X. & Willmore, W. G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2, 123–139 (2014).
Tian, Y. et al. Hydrogen peroxide positively regulates brassinosteroid signaling through oxidation of the BRASSINAZOLE-RESISTANT1 transcription factor. Nat. Commun. 9, 1063 (2018).
Beedle, A. E., Lynham, S. & Garcia-Manyes, S. Protein S-sulfenylation is a fleeting molecular switch that regulates non-enzymatic oxidative folding. Nat. Commun. 7, 12490 (2016).
Biteau, B., Labarre, J. & Toledano, M. B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425, 980–984 (2003).
Hamann, M., Zhang, T., Hendrich, S. & Thomas, J. A. Quantitation of protein sulfinic and sulfonic acid, irreversibly oxidized protein cysteine sites in cellular proteins. Methods Enzymol. 348, 146–156 (2002).
Doerr, A. Finding S-sulfinylated proteins. Nat. Methods 15, 859 (2018).
Lo Conte, M. & Carroll, K. S. The redox biochemistry of protein sulfenylation and sulfinylation. J. Biol. Chem. 288, 26480–26488 (2013).
Applegate, M. A., Humphries, K. M. & Szweda, L. I. Reversible inhibition of alpha-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry 47, 473–478 (2008).
Mirsky, A. E. & Anson, M. L. Sulfhydryl and disulfide groups of proteins: I. Methods of estimation. J. Gen. Physiol. 18, 307–323 (1935).
Sevier, C. S. & Kaiser, C. A. Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell Biol. 3, 836–847 (2002).
Nagahara, N. Intermolecular disulfide bond to modulate protein function as a redox-sensing switch. Amino Acids 41, 59–72 (2011).
Xu, H., Zhang, L. & Freitas, M. A. Identification and characterization of disulfide bonds in proteins and peptides from tandem MS data by use of the MassMatrix MS/MS search engine. J. Proteome Res. 7, 138–144 (2008).
Mishra, P. et al. Dynamic scaffolding in a G protein-coupled signaling system. Cell 131, 80–92 (2007).
Matsushima, Y., Yanagita, R. C. & Irie, K. Control of the toxic conformation of amyloid β42 by intramolecular disulfide bond formation. Chem. Commun. 56, 4118–4121 (2020).
Plugis, N. M., Palanski, B. A., Weng, C. H., Albertelli, M. & Khosla, C. Thioredoxin-1 selectively activates transglutaminase 2 in the extracellular matrix of the small intestine: implications for celiac disease. J. Biol. Chem. 292, 2000–2008 (2017).
Rosen, R. et al. Probing the active site of homoserine trans-succinylase. FEBS Lett. 577, 386–392 (2004).
Rardin, M. J. et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 18, 920–933 (2013).
Yang, Y. & Gibson, G. E. Succinylation links metabolism to protein functions. Neurochem. Res. 44, 2346–2359 (2019).
Wang, C. et al. CPT1A-mediated succinylation of S100A10 increases human gastric cancer invasion. J. Cell. Mol. Med. 23, 293–305 (2019).
Zhang, L., Liu, M., Qin, X. & Liu, G. Succinylation site prediction based on protein sequences using the IFS-lightGBM (BO) model. Comput. Math. Methods Med. 2020, 8858489 (2020).
Wellen, K. E. & Thompson, C. B. A two-way street: reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 13, 270–276 (2012).
Park, J. et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 50, 919–930 (2013).
Du, J. et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 (2011).
Zhang, Z. et al. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63 (2011).
Xu, Y. et al. iSuc-PseAAC: predicting lysine succinylation in proteins by incorporating peptide position-specific propensity. Sci. Rep. 5, 10184 (2015).
Cheng, Y., Hou, T., Ping, J., Chen, G. & Chen, J. Quantitative succinylome analysis in the liver of non-alcoholic fatty liver disease rat model. Proteome Sci. 14, 3 (2016).
Bories, G. F. P. & Leitinger, N. Macrophage metabolism in atherosclerosis. FEBS Lett. 591, 3042–3060 (2017).
Tan, M. et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 (2011).
Wei, W. et al. Class I histone deacetylases are major histone decrotonylases: evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 27, 898–915 (2017).
Sabari, B. R. et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 58, 203–215 (2015).
Li, Y. et al. Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Mol. Cell 62, 181–193 (2016).
Xu, W. et al. Global profiling of crotonylation on non-histone proteins. Cell Res 27, 946–949 (2017).
Bos, J. & Muir, T. W. A chemical probe for protein crotonylation. J. Am. Chem. Soc. 140, 4757–4760 (2018).
Liu, S. et al. Chromodomain protein CDYL acts as a crotonyl-CoA hydratase to regulate histone crotonylation and spermatogenesis. Mol. Cell 67, 853–866.e855 (2017).
Zhang, Y. et al. Acox2 is a regulator of lysine crotonylation that mediates hepatic metabolic homeostasis in mice. Cell Death Dis. 13, 279 (2022).
Luo, Y. et al. Procyanidin B2 improves oocyte maturation and subsequent development in type 1 diabetic mice by promoting mitochondrial function. Reprod. Sci. 27, 2211–2222 (2020).
Sabari, B. R., Zhang, D., Allis, C. D. & Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 18, 90–101 (2017).
Xie, Z. et al. Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol. Cell 62, 194–206 (2016).
Liu, K. et al. p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 10, 243 (2019).
Wu, X. et al. beta-hydroxybutyrate antagonizes aortic endothelial injury by promoting generation of VEGF in diabetic rats. Tissue Cell 64, 101345 (2020).
Caminhotto, R. O. et al. Oral β-hydroxybutyrate increases ketonemia, decreases visceral adipocyte volume and improves serum lipid profile in Wistar rats. Nutr. Metab. 14, 31 (2017).
Xu, Z., Zhang, M., Li, X., Wang, Y. & Du, R. Exercise ameliorates atherosclerosis via up-regulating serum β-hydroxybutyrate levels. Int. J. Mol. Sci. 23, 3788 (2022).
Koronowski, K. B. et al. Ketogenesis impact on liver metabolism revealed by proteomics of lysine β-hydroxybutyrylation. Cell Rep. 36, 109487 (2021).
Huang, H. et al. The regulatory enzymes and protein substrates for the lysine β-hydroxybutyrylation pathway. Sci. Adv. 7, eabe2771 (2021).
Gaffney, D. O. et al. Non-enzymatic lysine lactoylation of glycolytic enzymes. Cell Chem. Biol. 27, 206–213.e206 (2020).
Chen, A. N. et al. Lactylation, a novel metabolic reprogramming code: current status and prospects. Front. Immunol. 12, 688910 (2021).
Izzo, L. T. & Wellen, K. E. Histone lactylation links metabolism and gene regulation. Nature 574, 492–493 (2019).
Zhang, N. et al. Protein lactylation critically regulates energy metabolism in the protozoan parasite Trypanosoma brucei. Front. Cell Dev. Biol. 9, 719720 (2021).
Shantha, G. P. et al. Association of blood lactate with carotid atherosclerosis: the Atherosclerosis Risk in Communities (ARIC) Carotid MRI Study. Atherosclerosis 228, 249–255 (2013).
Maschari, D. et al. Lactate-induced lactylation in skeletal muscle is associated with insulin resistance in humans. Front. Physiol. 13, 951390 (2022).
Szabat, M. et al. Reduced insulin production relieves endoplasmic reticulum stress and induces beta cell proliferation. Cell Metab. 23, 179–193 (2016).
Bernal-Mizrachi, E., Wen, W., Stahlhut, S., Welling, C. M. & Permutt, M. A. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J. Clin. Invest. 108, 1631–1638 (2001).
Fatrai, S. et al. Akt induces beta-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes 55, 318–325 (2006).
Wong, J. T. et al. Pten (phosphatase and tensin homologue gene) haploinsufficiency promotes insulin hypersensitivity. Diabetologia 50, 395–403 (2007).
Goldsworthy, M. et al. Haploinsufficiency of the insulin receptor in the presence of a splice-site mutation in Ppp2r2a results in a novel digenic mouse model of type 2 diabetes. Diabetes 65, 1434–1446 (2016).
Chursa, U. et al. Overexpression of protein kinase STK25 in mice exacerbates ectopic lipid accumulation, mitochondrial dysfunction and insulin resistance in skeletal muscle. Diabetologia 60, 553–567 (2017).
Shinoda, K. et al. Phosphoproteomics identifies CK2 as a negative regulator of beige adipocyte thermogenesis and energy expenditure. Cell Metab. 22, 997–1008 (2015).
Kosanam, H. et al. Diabetes induces lysine acetylation of intermediary metabolism enzymes in the kidney. Diabetes 63, 2432–2439 (2014).
Lundh, M., Galbo, T., Poulsen, S. S. & Mandrup-Poulsen, T. Histone deacetylase 3 inhibition improves glycaemia and insulin secretion in obese diabetic rats. Diabetes Obes. Metab. 17, 703–707 (2015).
Kitada, M., Kume, S., Imaizumi, N. & Koya, D. Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1-independent pathway. Diabetes 60, 634–643 (2011).
Hong, S. et al. Nicotinamide N-methyltransferase regulates hepatic nutrient metabolism through Sirt1 protein stabilization. Nat. Med. 21, 887–894 (2015).
Kimball, A. S. et al. The histone methyltransferase Setdb2 modulates macrophage phenotype and uric acid production in diabetic wound repair. Immunity 51, 258–271.e255 (2019).
Wang, X. et al. Histone methyltransferases G9a mediated lipid-induced M1 macrophage polarization through negatively regulating CD36. Metabolism 114, 154404 (2021).
Motolani, A., Martin, M., Sun, M. & Lu, T. The structure and functions of PRMT5 in human diseases. Life 11, 1074 (2021).
Miao, F. et al. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes 57, 3189–3198 (2008).
Paneni, F. et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ. Cardiovasc. Genet. 8, 150–158 (2015).
Yonamine, C. Y. et al. Diabetes induces tri-methylation at lysine 9 of histone 3at Slc2a4 gene in skeletal muscle: a new target to improve glycemic control. Mol. Cell Endocrinol. 481, 26–34 (2019).
Kadakol, A., Malek, V., Goru, S. K., Pandey, A. & Gaikwad, A. B. Telmisartan and esculetin combination ameliorates type 2 diabetic cardiomyopathy by reversal of H3, H2A, and H2B histone modifications. Indian J. Pharm. 49, 348–356 (2017).
Ishii, M., Maeda, A., Tani, S. & Akagawa, M. Palmitate induces insulin resistance in human HepG2 hepatocytes by enhancing ubiquitination and proteasomal degradation of key insulin signaling molecules. Arch. Biochem. Biophys. 566, 26–35 (2015).
Kampmann, U. et al. GLUT4 and UBC9 protein expression is reduced in muscle from type 2 diabetic patients with severe insulin resistance. PLoS ONE 6, e27854 (2011).
Joo, E. et al. Ubc13 haploinsufficiency protects against age-related insulin resistance and high-fat diet-induced obesity. Sci. Rep. 6, 35983 (2016).
Vila, I. K. et al. A muscle-specific UBE2O/AMPKalpha2 axis promotes insulin resistance and metabolic syndrome in obesity. JCI Insight 4, e128269 (2019).
Kato, S., Ding, J., Pisck, E., Jhala, U. S. & Du, K. COP1 functions as a FoxO1 ubiquitin E3 ligase to regulate FoxO1-mediated gene expression. J. Biol. Chem. 283, 35464–35473 (2008).
Li, Y., Ren, D., Shen, Y., Zheng, X. & Xu, G. Altered DNA methylation of TRIM13 in diabetic nephropathy suppresses mesangial collagen synthesis by promoting ubiquitination of CHOP. EBioMedicine 51, 102582 (2020).
Cohen, S., Lee, D., Zhai, B., Gygi, S. P. & Goldberg, A. L. Trim32 reduces PI3K-Akt-FoxO signaling in muscle atrophy by promoting plakoglobin-PI3K dissociation. J. Cell Biol. 204, 747–758 (2014).
Guo, Y., Li, J., Fan, S. & Hu, Q. Suppressive role of E3 ubiquitin ligase FBW7 in type I diabetes in non-obese diabetic mice through mediation of ubiquitination of EZH2. Cell Death Discov. 7, 361 (2021).
Lee, S. et al. Thioredoxin-interacting protein regulates protein disulfide isomerases and endoplasmic reticulum stress. EMBO Mol. Med. 6, 732–743 (2014).
Pontrelli, P. et al. Deregulation of autophagy under hyperglycemic conditions is dependent on increased lysine 63 ubiquitination: a candidate mechanism in the progression of diabetic nephropathy. J. Mol. Med. 96, 645–659 (2018).
Qiu, J., Shu, C., Li, X. & Zhang, W. C. PAQR3 depletion accelerates diabetic wound healing by promoting angiogenesis through inhibiting STUB1-mediated PPARgamma degradation. Lab Invest. 102, 1121–1131 (2022).
Yang, Y. et al. Protein SUMOylation modification and its associations with disease. Open Biol. 7, 170167 (2017).
He, X. et al. Both conditional ablation and overexpression of E2 SUMO-conjugating enzyme (UBC9) in mouse pancreatic beta cells result in impaired beta cell function. Diabetologia 61, 881–895 (2018).
Hoard, T. M., Yang, X. P., Jetten, A. M. & ZeRuth, G. T. PIAS-family proteins negatively regulate Glis3 transactivation function through SUMO modification in pancreatic beta cells. Heliyon 4, e00709 (2018).
Attie, A. D. How do reducing equivalents increase insulin secretion? J. Clin. Invest. 125, 3754–3756 (2015).
Shao, L. et al. SENP1-mediated NEMO deSUMOylation in adipocytes limits inflammatory responses and type-1 diabetes progression. Nat. Commun. 6, 8917 (2015).
Wang, C. Y. & She, J. X. SUMO4 and its role in type 1 diabetes pathogenesis. Diabetes Metab. Res. Rev. 24, 93–102 (2008).
Shao, C. & Cobb, M. H. Sumoylation regulates the transcriptional activity of MafA in pancreatic beta cells. J. Biol. Chem. 284, 3117–3124 (2009).
Dai, X. Q., Kolic, J., Marchi, P., Sipione, S. & Macdonald, P. E. SUMOylation regulates Kv2.1 and modulates pancreatic beta-cell excitability. J. Cell Sci. 122, 775–779 (2009).
Dai, X. Q. et al. SUMOylation regulates insulin exocytosis downstream of secretory granule docking in rodents and humans. Diabetes 60, 838–847 (2011).
Bermingham, M. L. et al. N-glycan profile and kidney disease in type 1 diabetes. Diabetes Care 41, 79–87 (2018).
Ohtsubo, K. et al. Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell 123, 1307–1321 (2005).
Connelly, M. A., Otvos, J. D., Shalaurova, I., Playford, M. P. & Mehta, N. N. GlycA, a novel biomarker of systemic inflammation and cardiovascular disease risk. J. Transl. Med. 15, 219 (2017).
Tanigaki, K. et al. Hyposialylated IgG activates endothelial IgG receptor FcgammaRIIB to promote obesity-induced insulin resistance. J. Clin. Invest. 128, 309–322 (2018).
Smilowitz, J. T. et al. Human milk secretory immunoglobulin a and lactoferrin N-glycans are altered in women with gestational diabetes mellitus. J. Nutr. 143, 1906–1912 (2013).
Duggirala, R. et al. Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am. J. Hum. Genet. 64, 1127–1140 (1999).
Lehman, D. M. et al. A single nucleotide polymorphism in MGEA5 encoding O-GlcNAc-selective N-acetyl-beta-D glucosaminidase is associated with type 2 diabetes in Mexican Americans. Diabetes 54, 1214–1221 (2005).
Soesanto, Y. et al. Pleiotropic and age-dependent effects of decreased protein modification by O-linked N-acetylglucosamine on pancreatic beta-cell function and vascularization. J. Biol. Chem. 286, 26118–26126 (2011).
Forsythe, M. E. et al. Caenorhabditis elegans ortholog of a diabetes susceptibility locus: oga-1 (O-GlcNAcase) knockout impacts O-GlcNAc cycling, metabolism, and dauer. Proc. Natl Acad. Sci. USA 103, 11952–11957 (2006).
Kebede, M. et al. Glucose activates free fatty acid receptor 1 gene transcription via phosphatidylinositol-3-kinase-dependent O-GlcNAcylation of pancreas-duodenum homeobox-1. Proc. Natl Acad. Sci. USA 109, 2376–2381 (2012).
Schianchi, F. et al. Putative role of protein palmitoylation in cardiac lipid-induced insulin resistance. Int. J. Mol. Sci. 21, 9438 (2020).
Naito, C., Kajita, M. & Niwa, T. Determination of glutathionyl hemoglobin in hemodialysis patients using electrospray ionization liquid chromatography-mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 731, 121–124 (1999).
Cordes, C. M., Bennett, R. G., Siford, G. L. & Hamel, F. G. Nitric oxide inhibits insulin-degrading enzyme activity and function through S-nitrosylation. Biochem. Pharm. 77, 1064–1073 (2009).
Santulli, G. et al. Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J. Clin. Invest. 125, 1968–1978 (2015).
Nichols, C. G. KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476 (2006).
Sodre, F. M. C. et al. Peptidylarginine deiminase inhibition prevents diabetes development in NOD mice. Diabetes 70, 516–528 (2021).
Burkart, V. et al. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat. Med. 5, 314–319 (1999).
Pearce, N. J. et al. Development of glucose intolerance in male transgenic mice overexpressing human glycogen synthase kinase-3beta on a muscle-specific promoter. Metabolism 53, 1322–1330 (2004).
Sandeman, L. Y. et al. Disabling MNK protein kinases promotes oxidative metabolism and protects against diet-induced obesity. Mol. Metab. 42, 101054 (2020).
Sajan, M. P. et al. Akt-dependent phosphorylation of hepatic FoxO1 is compartmentalized on a WD40/ProF scaffold and is selectively inhibited by aPKC in early phases of diet-induced obesity. Diabetes 63, 2690–2701 (2014).
Ranieri, S. C. et al. Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proc. Natl Acad. Sci. USA 107, 13420–13425 (2010).
Sun, C. et al. Inactivation of MARK4, an AMP-activated protein kinase (AMPK)-related kinase, leads to insulin hypersensitivity and resistance to diet-induced obesity. J. Biol. Chem. 287, 38305–38315 (2012).
Bence, K. K. et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 12, 917–924 (2006).
Liu, H., Du, T., Li, C. & Yang, G. STAT3 phosphorylation in central leptin resistance. Nutr. Metab. 18, 39 (2021).
Mikula, M., Majewska, A., Ledwon, J. K., Dzwonek, A. & Ostrowski, J. Obesity increases histone H3 lysine 9 and 18 acetylation at Tnfa and Ccl2 genes in mouse liver. Int J. Mol. Med. 34, 1647–1654 (2014).
Liu, Y. et al. Protein acetylation: a novel modus of obesity regulation. J. Mol. Med. 99, 1221–1235 (2021).
Dorneles, G. P. et al. Acute strenuous exercise induces an imbalance on histone H4 acetylation/histone deacetylase 2 and increases the proinflammatory profile of PBMC of obese individuals. Oxid. Med. Cell Longev. 2017, 1530230 (2017).
Emmett, M. J. & Lazar, M. A. Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell Biol. 20, 102–115 (2019).
Kuang, Z. et al. The intestinal microbiota programs diurnal rhythms in host metabolism through histone deacetylase 3. Science 365, 1428–1434 (2019).
Kabra, D. G. et al. Hypothalamic leptin action is mediated by histone deacetylase 5. Nat. Commun. 7, 10782 (2016).
Forcioli-Conti, N., Esteve, D., Bouloumie, A., Dani, C. & Peraldi, P. The size of the primary cilium and acetylated tubulin are modulated during adipocyte differentiation: Analysis of HDAC6 functions in these processes. Biochimie 124, 112–123 (2016).
Tian, Y. et al. Histone deacetylase HDAC8 promotes insulin resistance and beta-catenin activation in NAFLD-associated hepatocellular carcinoma. Cancer Res. 75, 4803–4816 (2015).
Sun, L. et al. Programming and regulation of metabolic homeostasis by HDAC11. EBioMedicine 33, 157–168 (2018).
Qiang, L. et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell 150, 620–632 (2012).
Mendes, K. L., Lelis, D. F. & Santos, S. H. S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 38, 98–105 (2017).
Wei, T. et al. Sirtuin 3-mediated pyruvate dehydrogenase activity determines brown adipocytes phenotype under high-salt conditions. Cell Death Dis. 10, 614 (2019).
Du, Y. et al. SIRT5 deacylates metabolism-related proteins and attenuates hepatic steatosis in ob/ob mice. EBioMedicine 36, 347–357 (2018).
Jung, S. M. et al. Non-canonical mTORC2 signaling regulates brown adipocyte lipid catabolism through SIRT6-FoxO1. Mol. Cell 75, 807–822 e808 (2019).
Han, D. et al. Lysine methylation of transcription factors in cancer. Cell Death Dis. 10, 290 (2019).
Shi, Y. et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004).
Ohno, H., Shinoda, K., Ohyama, K., Sharp, L. Z. & Kajimura, S. EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature 504, 163–167 (2013).
Zhang, W. et al. Muscular G9a regulates muscle-liver-fat axis by musclin under overnutrition in female mice. Diabetes 69, 2642–2654 (2020).
Abe, Y. et al. Histone demethylase JMJD1A coordinates acute and chronic adaptation to cold stress via thermogenic phospho-switch. Nat. Commun. 9, 1566 (2018).
Zhao, Q. et al. KMT5c modulates adipocyte thermogenesis by regulating Trp53 expression. Proc. Natl Acad. Sci. USA 117, 22413–22422 (2020).
Yiew, N. K. H. et al. Enhancer of zeste homolog 2 (EZH2) regulates adipocyte lipid metabolism independent of adipogenic differentiation: role of apolipoprotein E. J. Biol. Chem. 294, 8577–8591 (2019).
Shuai, L. et al. DOT1L regulates thermogenic adipocyte differentiation and function via modulating H3K79 methylation. Diabetes 70, 1317–1333 (2021).
Wei, W., Li, Y., Li, Y. & Li, D. Adipose-specific knockout of ubiquitin-conjugating enzyme E2L6 (Ube2l6) reduces diet-induced obesity, insulin resistance, and hepatic steatosis. J. Pharm. Sci. 145, 327–334 (2021).
van Loon, N. M. et al. Inactivation of the E3 ubiquitin ligase IDOL attenuates diet-induced obesity and metabolic dysfunction in mice. Arterioscler. Thromb. Vasc. Biol. 38, 1785–1795 (2018).
Dalgaard, K. et al. Trim28 haploinsufficiency triggers bi-stable epigenetic obesity. Cell 164, 353–364 (2016).
Song, R. et al. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature 494, 375–379 (2013).
Odegaard, J. I. et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 (2007).
Koh, E. H. et al. Mitochondrial activity in human white adipocytes is regulated by the ubiquitin carrier protein 9/microRNA-30a axis. J. Biol. Chem. 291, 24747–24755 (2016).
Wang, T. et al. SENP1-Sirt3 signaling controls mitochondrial protein acetylation and metabolism. Mol. Cell 75, 823–834.e825 (2019).
Liu, Y. et al. Hepatic small ubiquitin-related modifier (SUMO)-specific protease 2 controls systemic metabolism through SUMOylation-dependent regulation of liver-adipose tissue crosstalk. Hepatology 74, 1864–1883 (2021).
Oishi, Y. et al. SUMOylation of Kruppel-like transcription factor 5 acts as a molecular switch in transcriptional programs of lipid metabolism involving PPAR-delta. Nat. Med. 14, 656–666 (2008).
Greto, V. L. et al. Extensive weight loss reduces glycan age by altering IgG N-glycosylation. Int J. Obes. 45, 1521–1531 (2021).
Li, M. D. et al. Adipocyte OGT governs diet-induced hyperphagia and obesity. Nat. Commun. 9, 5103 (2018).
Yang, Y. et al. O-GlcNAc transferase inhibits visceral fat lipolysis and promotes diet-induced obesity. Nat. Commun. 11, 181 (2020).
Holzer, R. G. et al. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 147, 173–184 (2011).
Vicent, D., Maratos-Flier, E. & Kahn, C. R. The branch point enzyme of the mevalonate pathway for protein prenylation is overexpressed in the ob/ob mouse and induced by adipogenesis. Mol. Cell Biol. 20, 2158–2166 (2000).
Kobayashi, T. et al. Mice lacking the glutamate-cysteine ligase modifier subunit are susceptible to myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 85, 785–795 (2010).
Watanabe, Y. et al. Protein S-glutathionylation stimulate adipogenesis by stabilizing C/EBPbeta in 3T3L1 cells. FASEB J. 34, 5827–5837 (2020).
Sebag, S. C. et al. ADH5-mediated NO bioactivity maintains metabolic homeostasis in brown adipose tissue. Cell Rep. 37, 110003 (2021).
Yang, L. et al. METABOLISM. S-nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 349, 500–506 (2015).
Whiteman, M. et al. Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia 53, 1722–1726 (2010).
Ding, Y., Wang, H., Geng, B. & Xu, G. Sulfhydration of perilipin 1 is involved in the inhibitory effects of cystathionine gamma lyase/hydrogen sulfide on adipocyte lipolysis. Biochem. Biophys. Res. Commun. 521, 786–790 (2020).
Katsouda, A., Szabo, C. & Papapetropoulos, A. Reduced adipose tissue H2S in obesity. Pharm. Res. 128, 190–199 (2018).
Margulies, C. E. & Ladurner, A. G. PARP-1 flips the epigenetic switch on obesity. Mol. Cell 79, 874–875 (2020).
Valerio, A. et al. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J. Clin. Invest. 116, 2791–2798 (2006).
Curtis, J. M. et al. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes 59, 1132–1142 (2010).
Oo, S. M. et al. Selenoprotein P-mediated reductive stress impairs cold-induced thermogenesis in brown fat. Cell Rep. 38, 110566 (2022).
Chouchani, E. T. et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 532, 112–116 (2016).
Anthony, N. M., Gaidhu, M. P. & Ceddia, R. B. Regulation of visceral and subcutaneous adipocyte lipolysis by acute AICAR-induced AMPK activation. Obesity 17, 1312–1317 (2009).
Schattenberg, J. M. et al. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology 43, 163–172 (2006).
Rinella, M. E. & Green, R. M. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 40, 47–51 (2004).
Boden, G. et al. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes 54, 3458–3465 (2005).
Calvisi, D. F. et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 140, 1071–1083 (2011).
Liu, X., Chen, S. & Zhang, L. Downregulated microRNA-130b-5p prevents lipid accumulation and insulin resistance in a murine model of nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab. 319, E34–E42 (2020).
Wang, T. et al. Acetylation of lactate dehydrogenase B drives NAFLD progression by impairing lactate clearance. J. Hepatol. 74, 1038–1052 (2021).
Liu, Y. et al. Insulin/Snail1 axis ameliorates fatty liver disease by epigenetically suppressing lipogenesis. Nat. Commun. 9, 2751 (2018).
Sun, Z. et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med. 18, 934–942 (2012).
Zheng, X. et al. SIRT1/HSF1/HSP pathway is essential for exenatide-alleviated, lipid-induced hepatic endoplasmic reticulum stress. Hepatology 66, 809–824 (2017).
Ren, H. et al. Sirtuin 2 prevents liver steatosis and metabolic disorders by deacetylation of hepatocyte nuclear factor 4alpha. Hepatology 74, 723–740 (2021).
Zhang, S. et al. 2,3,5,4’-tetrahydroxy-stilbene-2-O-beta-D-glucoside ameliorates NAFLD via attenuating hepatic steatosis through inhibiting mitochondrial dysfunction dependent on SIRT5. Phytomedicine 99, 153994 (2022).
Tian, C. et al. MRG15 aggravates non-alcoholic steatohepatitis progression by regulating the mitochondrial proteolytic degradation of TUFM. J. Hepatol. 77, 1491–1503 (2022).
Li, J. et al. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J. Hepatol. 56, 900–907 (2012).
Bricambert, J. et al. The histone demethylase Phf2 acts as a molecular checkpoint to prevent NAFLD progression during obesity. Nat. Commun. 9, 2092 (2018).
Zhang, Y. et al. Amelioration of hepatic steatosis by dietary essential amino acid-induced ubiquitination. Mol. Cell 82, 1528–1542.e1510 (2022).
Xu, M. et al. The E3 ubiquitin-protein ligase Trim31 alleviates non-alcoholic fatty liver disease by targeting Rhbdf2 in mouse hepatocytes. Nat. Commun. 13, 1052 (2022).
Gao, H. et al. Kindlin-2 haploinsufficiency protects against fatty liver by targeting Foxo1 in mice. Nat. Commun. 13, 1025 (2022).
Yang, Q. et al. The E3 ubiquitin ligase ring finger protein 5 ameliorates NASH through ubiquitin-mediated degradation of 3-hydroxy-3-methylglutaryl CoA reductase degradation protein 1. Hepatology 74, 3018–3036 (2021).
Hu, Y. et al. Fatty acid synthase-suppressor screening identifies sorting nexin 8 as a therapeutic target for NAFLD. Hepatology 74, 2508–2525 (2021).
Yan, F. J. et al. The E3 ligase tripartite motif 8 targets TAK1 to promote insulin resistance and steatohepatitis. Hepatology 65, 1492–1511 (2017).
Bai, L. et al. F-box/WD repeat-containing protein 5 mediates the ubiquitination of apoptosis signal-regulating kinase 1 and exacerbates nonalcoholic steatohepatitis in mice. Hepatology 70, 1942–1957 (2019).
Lee, M. S. et al. Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation. Nat. Commun. 9, 3404 (2018).
Zhao, Y. et al. Ubiquitin-specific protease 4 is an endogenous negative regulator of metabolic dysfunctions in nonalcoholic fatty liver disease in mice. Hepatology 68, 897–917 (2018).
Ni, W. et al. USP7 mediates pathological hepatic de novo lipogenesis through promoting stabilization and transcription of ZNF638. Cell Death Dis. 11, 843 (2020).
Luo, P. et al. Ubiquitin-specific peptidase 10 (USP10) inhibits hepatic steatosis, insulin resistance, and inflammation through Sirt6. Hepatology 68, 1786–1803 (2018).
Liu, B. et al. Proteome-wide analysis of USP14 substrates revealed its role in hepatosteatosis via stabilization of FASN. Nat. Commun. 9, 4770 (2018).
Lv, C. et al. USP14 maintains HIF1-alpha stabilization via its deubiquitination activity in hepatocellular carcinoma. Cell Death Dis. 12, 803 (2021).
Ji, Y. X. et al. The deubiquitinating enzyme cylindromatosis mitigates nonalcoholic steatohepatitis. Nat. Med. 24, 213–223 (2018).
Hirano, Y., Murata, S., Tanaka, K., Shimizu, M. & Sato, R. Sterol regulatory element-binding proteins are negatively regulated through SUMO-1 modification independent of the ubiquitin/26 S proteasome pathway. J. Biol. Chem. 278, 16809–16819 (2003).
Lee, G. Y. et al. PIASy-mediated sumoylation of SREBP1c regulates hepatic lipid metabolism upon fasting signaling. Mol. Cell Biol. 34, 926–938 (2014).
Stein, S. et al. Impaired SUMOylation of nuclear receptor LRH-1 promotes nonalcoholic fatty liver disease. J. Clin. Invest. 127, 583–592 (2017).
Kumar, D. et al. Degradation of splicing factor SRSF3 contributes to progressive liver disease. J. Clin. Invest. 129, 4477–4491 (2019).
Dehnad, A. et al. AGER1 downregulation associates with fibrosis in nonalcoholic steatohepatitis and type 2 diabetes. J. Clin. Invest. 130, 4320–4330 (2020).
Serrano-Macia, M. et al. Neddylation inhibition ameliorates steatosis in NAFLD by boosting hepatic fatty acid oxidation via the DEPTOR-mTOR axis. Mol. Metab. 53, 101275 (2021).
Zubiete-Franco, I. et al. Deregulated neddylation in liver fibrosis. Hepatology 65, 694–709 (2017).
Troeger, J. S. et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 143, 1073–1083 e1022 (2012).
Moylan, C. A. et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 59, 471–482 (2014).
Benhamed, F. et al. O-GlcNAcylation links ChREBP and FXR to glucose-sensing. Front. Endocrinol. 5, 230 (2014).
Guinez, C. et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 60, 1399–1413 (2011).
Zeng, S. et al. Inhibition of fatty acid translocase (FAT/CD36) palmitoylation enhances hepatic fatty acid beta-oxidation by increasing its localization to mitochondria and interaction with long-chain Acyl-CoA synthetase 1. Antioxid. Redox Signal 36, 1081–1100 (2022).
Zhao, Y., Wu, T. Y., Zhao, M. F. & Li, C. J. The balance of protein farnesylation and geranylgeranylation during the progression of nonalcoholic fatty liver disease. J. Biol. Chem. 295, 5152–5162 (2020).
Kong, A. N. et al. Signal transduction events elicited by cancer prevention compounds. Mutat. Res. 480-481, 231–241 (2001).
Wang, H., Li, F., Feng, J., Wang, J. & Liu, X. The effects of S-nitrosylation-induced PPARgamma/SFRP5 pathway inhibition on the conversion of non-alcoholic fatty liver to non-alcoholic steatohepatitis. Ann. Transl. Med. 9, 684 (2021).
Xu, W. et al. Hepatocellular cystathionine gamma lyase/hydrogen sulfide attenuates nonalcoholic fatty liver disease by activating farnesoid X receptor. Hepatology 76, 1794–1810 (2022).
Nguyen, T. T. P., Kim, D. Y., Im, S. S. & Jeon, T. I. Impairment of ULK1 sulfhydration-mediated lipophagy by SREBF1/SREBP-1c in hepatic steatosis. Autophagy 17, 4489–4490 (2021).
Zhao, S. et al. Hydrogen sulfide alleviates liver injury through the S-sulfhydrated-Kelch-like ECH-associated protein 1/nuclear erythroid 2-related factor 2/low-density lipoprotein receptor-related protein 1 pathway. Hepatology 73, 282–302 (2021).
Huang, K. et al. PARP1-mediated PPARalpha poly(ADP-ribosyl)ation suppresses fatty acid oxidation in non-alcoholic fatty liver disease. J. Hepatol. 66, 962–977 (2017).
Gariani, K. et al. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J. Hepatol. 66, 132–141 (2017).
Shearn, C. T. et al. Differential carbonylation of proteins in end-stage human fatty and nonfatty NASH. Free Radic. Biol. Med. 113, 280–290 (2017).
Petersen, D. R. et al. Elevated Nrf-2 responses are insufficient to mitigate protein carbonylation in hepatospecific PTEN deletion mice. PLoS ONE 13, e0198139 (2018).
Ginsberg, H. N. Lipoprotein physiology. Endocrinol. Metab. Clin. North Am. 27, 503–519 (1998).
Maldonado, E. N., Romero, J. R., Ochoa, B. & Aveldaño, M. I. Lipid and fatty acid composition of canine lipoproteins. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 128, 719–729 (2001).
Batchuluun, B., Pinkosky, S. L. & Steinberg, G. R. Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat. Rev. Drug Discov. 21, 283–305 (2022).
Viscarra, J. & Sul, H. S. Epigenetic regulation of hepatic lipogenesis: role in hepatosteatosis and diabetes. Diabetes 69, 525–531 (2020).
Luo, J., Yang, H. & Song, B. L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 21, 225–245 (2020).
O’Neill, H. M. et al. AMPK phosphorylation of ACC2 is required for skeletal muscle fatty acid oxidation and insulin sensitivity in mice. Diabetologia 57, 1693–1702 (2014).
Li, Y. et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388 (2011).
Davie, E., Forte, G. M. & Petersen, J. Nitrogen regulates AMPK to control TORC1 signaling. Curr. Biol. 25, 445–454 (2015).
Steinberg, G. R. & Carling, D. AMP-activated protein kinase: the current landscape for drug development. Nat. Rev. Drug Discov. 18, 527–551 (2019).
Loh, K. et al. Inhibition of adenosine monophosphate-activated protein kinase-3-hydroxy-3-methylglutaryl coenzyme A reductase signaling leads to hypercholesterolemia and promotes hepatic steatosis and insulin resistance. Hepatol. Commun. 3, 84–98 (2019).
Ponugoti, B. et al. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 285, 33959–33970 (2010).
Purushotham, A. et al. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9, 327–338 (2009).
Guo, L. et al. Enhanced acetylation of ATP-citrate lyase promotes the progression of nonalcoholic fatty liver disease. J. Biol. Chem. 294, 11805–11816 (2019).
Krishnan, J. et al. Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD + system. Genes Dev. 26, 259–270 (2012).
Tao, R., Xiong, X., DePinho, R. A., Deng, C. X. & Dong, X. C. FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J. Biol. Chem. 288, 29252–29259 (2013).
Wu, G. et al. Large yellow tea extract ameliorates metabolic syndrome by suppressing lipogenesis through SIRT6/SREBP1 pathway and modulating microbiota in leptin receptor knockout rats. Foods 11, 1638 (2022).
Dong, Z. et al. LncRNA PU.1 AS regulates arsenic-induced lipid metabolism through EZH2/Sirt6/SREBP-1c pathway. J. Environ. Sci. 85, 138–146 (2019).
Webb, L. M. et al. Protein arginine methyltransferase 5 promotes cholesterol biosynthesis-mediated Th17 responses and autoimmunity. J. Clin. Invest 130, 1683–1698 (2020).
Jia, Z. et al. Protein arginine methyltransferase PRMT5 regulates fatty acid metabolism and lipid droplet biogenesis in white adipose tissues. Adv. Sci. 7, 2002602 (2020).
Zelcer, N., Hong, C., Boyadjian, R. & Tontonoz, P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 325, 100–104 (2009).
Liang, C. et al. Idol depletion protects against spontaneous atherosclerosis in a hamster model of familial hypercholesterolemia. Oxid. Med. Cell. Longev. 2022, 1889632 (2022).
Stöhr, R. et al. ITCH modulates SIRT6 and SREBP2 to influence lipid metabolism and atherosclerosis in ApoE null mice. Sci. Rep. 5, 9023 (2015).
Jeon, T. I. et al. An SREBP-responsive microRNA operon contributes to a regulatory loop for intracellular lipid homeostasis. Cell Metab. 18, 51–61 (2013).
Foresti, O., Ruggiano, A., Hannibal-Bach, H. K., Ejsing, C. S. & Carvalho, P. Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. Elife 2, e00953 (2013).
Song, B. L., Sever, N. & DeBose-Boyd, R. A. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell 19, 829–840 (2005).
Liu, T. F. et al. Ablation of gp78 in liver improves hyperlipidemia and insulin resistance by inhibiting SREBP to decrease lipid biosynthesis. Cell Metab. 16, 213–225 (2012).
Liang, J. S. et al. Overexpression of the tumor autocrine motility factor receptor Gp78, a ubiquitin protein ligase, results in increased ubiquitinylation and decreased secretion of apolipoprotein B100 in HepG2 cells. J. Biol. Chem. 278, 23984–23988 (2003).
Stein, S. et al. SUMOylation-dependent LRH-1/PROX1 interaction promotes atherosclerosis by decreasing hepatic reverse cholesterol transport. Cell Metab. 20, 603–613 (2014).
Kim, D. H. et al. Critical role of RanBP2-mediated SUMOylation of Small Heterodimer Partner in maintaining bile acid homeostasis. Nat. Commun. 7, 12179 (2016).
Zhang, Y. et al. A role for protein inhibitor of activated STAT1 (PIAS1) in lipogenic regulation through SUMOylation-independent suppression of liver X receptors. J. Biol. Chem. 287, 37973–37985 (2012).
Zheng, Q. et al. Senp2 regulates adipose lipid storage by de-SUMOylation of Setdb1. J. Mol. Cell. Biol. 10, 258–266 (2018).
Cheng, C. et al. Glucose-mediated N-glycosylation of SCAP is essential for SREBP-1 activation and tumor growth. Cancer Cell 28, 569–581 (2015).
Zanoni, P. et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 351, 1166–1171 (2016).
Zhu, C. et al. Site-specific glycoprofiles of HDL-associated ApoE are correlated with HDL functional capacity and unaffected by short-term diet. J. Proteome Res. 18, 3977–3984 (2019).
Kathiresan, S. et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 41, 56–65 (2009).
Teslovich, T. M. et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 (2010).
Khetarpal, S. A. et al. Loss of function of GALNT2 lowers high-density lipoproteins in humans, nonhuman primates, and rodents. Cell Metab. 24, 234–245 (2016).
Rozenberg, O. & Aviram, M. S-Glutathionylation regulates HDL-associated paraoxonase 1 (PON1) activity. Biochem. Biophys. Res. Commun. 351, 492–498 (2006).
Shao, D. et al. Glutaredoxin-1 deficiency causes fatty liver and dyslipidemia by inhibiting sirtuin-1. Antioxid. Redox Signal 27, 313–327 (2017).
Zhao, Z. et al. S-nitrosylation of ARH is required for LDL uptake by the LDL receptor. J. Lipid Res. 54, 1550–1559 (2013).
Sun, L. et al. Hydrogen sulfide reduces serum triglyceride by activating liver autophagy via the AMPK-mTOR pathway. Am. J. Physiol. Endocrinol. Metab. 309, E925–E935 (2015).
Du, C. et al. Sulfhydrated sirtuin-1 increasing its deacetylation activity is an essential epigenetics mechanism of anti-atherogenesis by hydrogen sulfide. Antioxid. Redox Signal 30, 184–197 (2019).
Uto-Kondo, H. et al. Citrulline increases cholesterol efflux from macrophages in vitro and ex vivo via ATP-binding cassette transporters. J. Clin. Biochem. Nutr. 55, 32–39 (2014).
Lehmann, M. et al. ARTD1-induced poly-ADP-ribose formation enhances PPARγ ligand binding and co-factor exchange. Nucleic Acids Res. 43, 129–142 (2015).
Shrestha, E. et al. Poly(ADP-ribose) polymerase 1 represses liver X receptor-mediated ABCA1 expression and cholesterol efflux in macrophages. J. Biol. Chem. 291, 11172–11184 (2016).
Bindesbøll, C. et al. TCDD-inducible poly-ADP-ribose polymerase (TIPARP/PARP7) mono-ADP-ribosylates and co-activates liver X receptors. Biochem. J. 473, 899–910 (2016).
Bai, P. et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 13, 450–460 (2011).
Yeh, T. Y. et al. Hypermetabolism, hyperphagia, and reduced adiposity in tankyrase-deficient mice. Diabetes 58, 2476–2485 (2009).
Szántó, M. et al. Deletion of PARP-2 induces hepatic cholesterol accumulation and decrease in HDL levels. Biochim. Biophys. Acta 1842, 594–602 (2014).
Wang, H. et al. Tankyrase inhibition ameliorates lipid disorder via suppression of PGC-1α PARylation in db/db mice. Int J. Obes. 44, 1691–1702 (2020).
Liu, S. et al. Traditional Chinese medicine for bradyarrhythmia: evidence and potential mechanisms. Front. Pharmacol. 9, 324 (2018).
Ramos, L. P. A. et al. Antioxidant compounds from Annona crassiflora fruit peel reduce lipid levels and oxidative damage and maintain the glutathione defense in hepatic tissue of Triton WR-1339-induced hyperlipidemic mice. Biomed. Pharmacother. 142, 112049 (2021).
Kandhare, A. D., Bandyopadhyay, D. & Thakurdesai, P. A. Low molecular weight galactomannans-based standardized fenugreek seed extract ameliorates high-fat diet-induced obesity in mice via modulation of FASn, IL-6, leptin, and TRIP-Br2. RSC Adv. 8, 32401–32416 (2018).
Muñoz, S. et al. Targeting hepatic protein carbonylation and oxidative stress occurring on diet-induced metabolic diseases through the supplementation with fish oils. Mar. Drugs 16, 353 (2018).
Chang, L. & Karin, M. Mammalian MAP kinase signalling cascades. Nature 410, 37–40 (2001).
Li, D. & Mehta, J. L. Antisense to LOX-1 inhibits oxidized LDL-mediated upregulation of monocyte chemoattractant protein-1 and monocyte adhesion to human coronary artery endothelial cells. Circulation 101, 2889–2895 (2000).
Seimon, T. A. et al. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J. Clin. Invest. 119, 886–898 (2009).
Watanabe, T., Pakala, R., Katagiri, T. & Benedict, C. R. Synergistic effect of urotensin II with mildly oxidized LDL on DNA synthesis in vascular smooth muscle cells. Circulation 104, 16–18 (2001).
Shen, J. et al. Lack of mitogen-activated protein kinase phosphatase-1 protects ApoE-null mice against atherosclerosis. Circ. Res. 106, 902–910 (2010).
Meijer, C. A. et al. Activator protein-1 (AP-1) signalling in human atherosclerosis: results of a systematic evaluation and intervention study. Clin. Sci. 122, 421–428 (2012).
Jagavelu, K. et al. Systemic deficiency of the MAP kinase-activated protein kinase 2 reduces atherosclerosis in hypercholesterolemic mice. Circ. Res. 101, 1104–1112 (2007).
Dong, Y. et al. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 121, 792–803 (2010).
Zhang, Y., Qiu, J., Wang, X., Zhang, Y. & Xia, M. AMP-activated protein kinase suppresses endothelial cell inflammation through phosphorylation of transcriptional coactivator p300. Arterioscler. Thromb. Vasc. Biol. 31, 2897–2908 (2011).
Zhou, Q. et al. Rho-associated coiled-coil-containing kinase 2 deficiency in bone marrow-derived cells leads to increased cholesterol efflux and decreased atherosclerosis. Circulation 126, 2236–2247 (2012).
Raghavan, S., Singh, N. K., Gali, S., Mani, A. M. & Rao, G. N. Protein kinase Cθ via activating transcription factor 2-mediated CD36 expression and foam cell formation of Ly6C(hi) cells contributes to atherosclerosis. Circulation 138, 2395–2412 (2018).
Li, X. et al. IL-35 (Interleukin-35) suppresses endothelial cell activation by inhibiting mitochondrial reactive oxygen species-mediated site-specific acetylation of H3K14 (Histone 3 Lysine 14). Arterioscler. Thromb. Vasc. Biol. 38, 599–609 (2018).
Malhotra, R. et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat. Genet. 51, 1580–1587 (2019).
Cao, Q. et al. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler. Thromb. Vasc. Biol. 34, 1871–1879 (2014).
Asare, Y. et al. Histone deacetylase 9 activates IKK to regulate atherosclerotic plaque vulnerability. Circ. Res. 127, 811–823 (2020).
Tian, R. et al. Metformin ameliorates endotoxemia-induced endothelial pro-inflammatory responses via AMPK-dependent mediation of HDAC5 and KLF2. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 1701–1712 (2019).
Zheng, B. et al. HDAC2 phosphorylation-dependent Klf5 deacetylation and RARα acetylation induced by RAR agonist switch the transcription regulatory programs of p21 in VSMCs. Cell Res. 21, 1487–1508 (2011).
Pang, J. et al. GIT1 mediates HDAC5 activation by angiotensin II in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 28, 892–898 (2008).
Miranda, M. X. et al. The Sirt1 activator SRT3025 provides atheroprotection in Apoe-/- mice by reducing hepatic Pcsk9 secretion and enhancing Ldlr expression. Eur. Heart J. 36, 51–59 (2015).
Grootaert, M. O. J., Finigan, A., Figg, N. L., Uryga, A. K. & Bennett, M. R. SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ. Res. 128, 474–491 (2021).
Greißel, A. et al. Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thromb. Haemost. 114, 390–402 (2015).
Balasubramaniyan, N., Ananthanarayanan, M. & Suchy, F. J. Direct methylation of FXR by Set7/9, a lysine methyltransferase, regulates the expression of FXR target genes. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G937–G947 (2012).
Shivkar, R. R. & Abhang, S. A. Ratio of serum asymmetric dimethyl arginine (ADMA)/ nitric oxide in coronary artery disease patients. J. Clin. Diagn. Res. 8, Cc04–Cc06 (2014).
Liao, J. et al. Inhibition of the ubiquitin-activating enzyme UBA1 suppresses diet-induced atherosclerosis in apolipoprotein E-knockout mice. J. Immunol. Res. 2020, 7812709 (2020).
Kikuchi, J. et al. Induction of ubiquitin-conjugating enzyme by aggregated low density lipoprotein in human macrophages and its implications for atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 20, 128–134 (2000).
Calkin, A. C. et al. Transgenic expression of dominant-active IDOL in liver causes diet-induced hypercholesterolemia and atherosclerosis in mice. Circ. Res. 115, 442–449 (2014).
Burger, F., Baptista, D., Roth, A., Brandt, K. J. & Miteva, K. The E3 ubiquitin ligase Peli1 deficiency promotes atherosclerosis progression. Cells 11, 2014 (2022).
Wang, C. et al. E3 ligase FBXW2 is a new therapeutic target in obesity and atherosclerosis. Adv. Sci. 7, 2001800 (2020).
Wadosky, K. M. & Willis, M. S. The story so far: post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and SUMOylation. Am. J. Physiol. Heart Circ. Physiol. 302, H515–H526 (2012).
Koo, Y. D. et al. SUMO-specific protease 2 (SENP2) is an important regulator of fatty acid metabolism in skeletal muscle. Diabetes 64, 2420–2431 (2015).
Qiu, C. et al. The critical role of SENP1-mediated GATA2 deSUMOylation in promoting endothelial activation in graft arteriosclerosis. Nat. Commun. 8, 15426 (2017).
Heo, K. S. et al. Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J. Clin. Invest. 125, 1299–1310 (2015).
Zhang, Z. B. et al. Activating transcription factor 3 SUMOylation is involved in angiotensin II-induced endothelial cell inflammation and dysfunction. J. Mol. Cell. Cardiol. 92, 149–157 (2016).
Cai, Z. et al. Redox-sensitive enzyme SENP3 mediates vascular remodeling via de-SUMOylation of β-catenin and regulation of its stability. EBioMedicine 67, 103386 (2021).
Pandey, D. et al. NEDDylation promotes endothelial dysfunction: a role for HDAC2. J. Mol. Cell. Cardiol. 81, 18–22 (2015).
Nomura, Y., Nakano, M., Woo Sung, H., Han, M. & Pandey, D. Inhibition of HDAC6 activity protects against endothelial dysfunction and atherogenesis in vivo: a role for HDAC6 neddylation. Front. Physiol. 12, 675724 (2021).
Homeister, J. W., Daugherty, A. & Lowe, J. B. Alpha(1,3)fucosyltransferases FucT-IV and FucT-VII control susceptibility to atherosclerosis in apolipoprotein E-/- mice. Arterioscler. Thromb. Vasc. Biol. 24, 1897–1903 (2004).
Homeister, J. W. et al. The alpha(1,3)fucosyltransferases FucT-IV and FucT-VII exert collaborative control over selectin-dependent leukocyte recruitment and lymphocyte homing. Immunity 15, 115–126 (2001).
Wang, H. et al. Core2 1-6-N-glucosaminyltransferase-I is crucial for the formation of atherosclerotic lesions in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 29, 180–187 (2009).
Ott, C. et al. Role of advanced glycation end products in cellular signaling. Redox Biol. 2, 411–429 (2014).
García-Cardeña, G., Oh, P., Liu, J., Schnitzer, J. E. & Sessa, W. C. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc. Natl Acad. Sci. USA 93, 6448–6453 (1996).
Wei, X. et al. De novo lipogenesis maintains vascular homeostasis through endothelial nitric-oxide synthase (eNOS) palmitoylation. J. Biol. Chem. 286, 2933–2945 (2011).
Krotova, K. et al. Peptides modified by myristoylation activate eNOS in endothelial cells through Akt phosphorylation. Br. J. Pharm. 148, 732–740 (2006).
Cohen, L. H. et al. Inhibitors of prenylation of Ras and other G-proteins and their application as therapeutics. Biochem. Pharmacol. 60, 1061–1068 (2000).
Nonaka, K. et al. Serum levels of S-glutathionylated proteins as a risk-marker for arteriosclerosis obliterans. Circ. J. 71, 100–105 (2007).
Kader, T., Porteous, C. M., Williams, M. J., Gieseg, S. P. & McCormick, S. P. Ribose-cysteine increases glutathione-based antioxidant status and reduces LDL in human lipoprotein(a) mice. Atherosclerosis 237, 725–733 (2014).
Hamanishi, T. et al. Functional variants in the glutathione peroxidase-1 (GPx-1) gene are associated with increased intima-media thickness of carotid arteries and risk of macrovascular diseases in japanese type 2 diabetic patients. Diabetes 53, 2455–2460 (2004).
Lewis, P. et al. Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation 115, 2178–2187 (2007).
Wang, Y., Qiao, M., Mieyal, J. J., Asmis, L. M. & Asmis, R. Molecular mechanism of glutathione-mediated protection from oxidized low-density lipoprotein-induced cell injury in human macrophages: role of glutathione reductase and glutaredoxin. Free Radic. Biol. Med. 41, 775–785 (2006).
Shao, D. et al. A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress. J. Biol. Chem. 289, 7293–7306 (2014).
Liao, B. C., Hsieh, C. W., Lin, Y. C. & Wung, B. S. The glutaredoxin/glutathione system modulates NF-kappaB activity by glutathionylation of p65 in cinnamaldehyde-treated endothelial cells. Toxicol. Sci. 116, 151–163 (2010).
Carvalho, A. N. et al. S-Glutathionylation of Keap1: a new role for glutathione S-transferase pi in neuronal protection. FEBS Lett. 590, 1455–1466 (2016).
Lowenstein, C. J. Nitric oxide regulation of protein trafficking in the cardiovascular system. Cardiovasc. Res. 75, 240–246 (2007).
Pawloski, J. R., Swaminathan, R. V. & Stamler, J. S. Cell-free and erythrocytic S-nitrosohemoglobin inhibits human platelet aggregation. Circulation 97, 263–267 (1998).
Morrell, C. N. et al. Regulation of platelet granule exocytosis by S-nitrosylation. Proc. Natl Acad. Sci. USA 102, 3782–3787 (2005).
Zhao, S. et al. Hsp90 S-nitrosylation at Cys521, as a conformational switch, modulates cycling of Hsp90-AHA1-CDC37 chaperone machine to aggravate atherosclerosis. Redox Biol. 52, 102290 (2022).
Yang, G. et al. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal 18, 1906–1919 (2013).
Wang, X. H. et al. Dysregulation of cystathionine γ-lyase (CSE)/hydrogen sulfide pathway contributes to ox-LDL-induced inflammation in macrophage. Cell. Signal. 25, 2255–2262 (2013).
Sokolove, J. et al. Brief report: citrullination within the atherosclerotic plaque: a potential target for the anti-citrullinated protein antibody response in rheumatoid arthritis. Arthritis Rheum. 65, 1719–1724 (2013).
Ray Chaudhuri, A. & Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621 (2017).
Olabisi, O. A. et al. Regulation of transcription factor NFAT by ADP-ribosylation. Mol. Cell. Biol. 28, 2860–2871 (2008).
Zingarelli, B. et al. Activator protein-1 signalling pathway and apoptosis are modulated by poly(ADP-ribose) polymerase-1 in experimental colitis. Immunology 113, 509–517 (2004).
Garcia Soriano, F. et al. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat. Med. 7, 108–113 (2001).
von Lukowicz, T. et al. PARP1 is required for adhesion molecule expression in atherogenesis. Cardiovasc. Res. 78, 158–166 (2008).
Oumouna-Benachour, K. et al. Poly(ADP-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein E-deficient mice: effects on macrophage recruitment, nuclear factor-kappaB nuclear translocation, and foam cell death. Circulation 115, 2442–2450 (2007).
Hans, C. P. et al. Thieno[2,3-c]isoquinolin-5-one, a potent poly(ADP-ribose) polymerase inhibitor, promotes atherosclerotic plaque regression in high-fat diet-fed apolipoprotein E-deficient mice: effects on inflammatory markers and lipid content. J. Pharmacol. Exp. Ther. 329, 150–158 (2009).
Mathews, M. T. & Berk, B. C. PARP-1 inhibition prevents oxidative and nitrosative stress-induced endothelial cell death via transactivation of the VEGF receptor 2. Arterioscler. Thromb. Vasc. Biol. 28, 711–717 (2008).
Iwata, H. et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat. Commun. 7, 12849 (2016).
Verheugd, P. et al. Regulation of NF-κB signalling by the mono-ADP-ribosyltransferase ARTD10. Nat. Commun. 4, 1683 (2013).
Welsby, I. et al. PARP12, an interferon-stimulated gene involved in the control of protein translation and inflammation. J. Biol. Chem. 289, 26642–26657 (2014).
Muroya, T. et al. Oxidative modulation of NF-kappaB signaling by oxidized low-density lipoprotein. Biochem. Biophys. Res. Commun. 309, 900–905 (2003).
Menini, S. et al. D-Carnosine octylester attenuates atherosclerosis and renal disease in ApoE null mice fed a Western diet through reduction of carbonyl stress and inflammation. Br. J. Pharm. 166, 1344–1356 (2012).
Zhou, G. et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 (2001).
Oh, S. et al. Antidiabetic and antiobesity effects of Ampkinone (6 f), a novel small molecule activator of AMP-activated protein kinase. J. Med. Chem. 53, 7405–7413 (2010).
Shibata, T., Takaguri, A., Ichihara, K. & Satoh, K. Inhibition of the TNF-alpha-induced serine phosphorylation of IRS-1 at 636/639 by AICAR. J. Pharm. Sci. 122, 93–102 (2013).
Medicherla, S. et al. Preventive and therapeutic potential of p38 alpha-selective mitogen-activated protein kinase inhibitor in nonobese diabetic mice with type 1 diabetes. J. Pharm. Exp. Ther. 318, 99–107 (2006).
Wang, T. et al. The MEK inhibitor U0126 ameliorates diabetic cardiomyopathy by restricting XBP1’s phosphorylation dependent SUMOylation. Int J. Biol. Sci. 17, 2984–2999 (2021).
Ito, Y. et al. Therapeutic effects of the allosteric protein tyrosine phosphatase 1B inhibitor KY-226 on experimental diabetes and obesity via enhancements in insulin and leptin signaling in mice. J. Pharm. Sci. 137, 38–46 (2018).
Fukuda, S. et al. Pharmacological profiles of a novel protein tyrosine phosphatase 1B inhibitor, JTT-551. Diabetes Obes. Metab. 12, 299–306 (2010).
Lantz, K. A. et al. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity 18, 1516–1523 (2010).
Krishnan, N., Konidaris, K. F., Gasser, G. & Tonks, N. K. A potent, selective, and orally bioavailable inhibitor of the protein-tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models. J. Biol. Chem. 293, 1517–1525 (2018).
Yang, X. et al. Inhibition of JAK2/STAT3/SOCS3 signaling attenuates atherosclerosis in rabbit. BMC Cardiovasc. Disord. 20, 133 (2020).
Tang, Y. et al. Inhibition of JAK2 suppresses myelopoiesis and atherosclerosis in Apoe(-/-) mice. Cardiovasc. Drugs Ther. 34, 145–152 (2020).
Luo, W. et al. Inhibition of EGFR-STAT3 attenuates cardiomyopathy in streptozotocin-induced type 1 diabetes. J. Endocrinol. 242, 199–210 (2019).
Wang, Z. J. et al. A novel rhynchophylline analog, Y396, inhibits endothelial dysfunction induced by oxidative stress in diabetes through epidermal growth factor receptor. Antioxid. Redox Signal 32, 743–765 (2020).
Sun, X. Y. et al. Valproate attenuates diabetic nephropathy through inhibition of endoplasmic reticulum stress‑induced apoptosis. Mol. Med. Rep. 13, 661–668 (2016).
Lee, H. A. et al. Histone deacetylase inhibitor MGCD0103 protects the pancreas from streptozotocin-induced oxidative stress and beta-cell death. Biomed. Pharmacother. 109, 921–929 (2019).
Sharma, S. & Taliyan, R. Epigenetic modifications by inhibiting histone deacetylases reverse memory impairment in insulin resistance induced cognitive deficit in mice. Neuropharmacology 105, 285–297 (2016).
Cakir, I. et al. Histone deacetylase 6 inhibition restores leptin sensitivity and reduces obesity. Nat. Metab. 4, 44–59 (2022).
Wei, Y. et al. Restoration of H3k27me3 modification epigenetically silences Cry1 expression and sensitizes leptin signaling to reduce obesity-related properties. Adv. Sci. 8, 2004319 (2021).
Wu, X. et al. GSK126 alleviates the obesity phenotype by promoting the differentiation of thermogenic beige adipocytes in diet-induced obese mice. Biochem. Biophys. Res. Commun. 501, 9–15 (2018).
Li, X. et al. MF-094, a potent and selective USP30 inhibitor, accelerates diabetic wound healing by inhibiting the NLRP3 inflammasome. Exp. Cell Res. 410, 112967 (2022).
Lu, X. Y. et al. Feeding induces cholesterol biosynthesis via the mTORC1-USP20-HMGCR axis. Nature 588, 479–484 (2020).
Park, H. S. et al. PPARgamma neddylation essential for adipogenesis is a potential target for treating obesity. Cell Death Differ. 23, 1296–1311 (2016).
Long, C. A. et al. Poly-ADP-ribose-polymerase inhibition ameliorates hind limb ischemia reperfusion injury in a murine model of type 2 diabetes. Ann. Surg. 258, 1087–1095 (2013).
Oesterle, A., Laufs, U. & Liao, J. K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 120, 229–243 (2017).
Kimura, Y. et al. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J. Gastroenterol. 45, 750–757 (2010).
Lee, J. I., Lee, H. W., Lee, K. S., Lee, H. S. & Park, J. Y. Effects of statin use on the development and progression of nonalcoholic fatty liver disease: a nationwide nested case-control study. Am. J. Gastroenterol. 116, 116–124 (2021).
Munday, M. R. Regulation of mammalian acetyl-CoA carboxylase. Biochem. Soc. Trans. 30, 1059–1064 (2002).
Lee, M. S., Kim, K. J., Kim, D., Lee, K. E. & Hwang, J. K. meso-Dihydroguaiaretic acid inhibits hepatic lipid accumulation by activating AMP-activated protein kinase in human HepG2 cells. Biol. Pharm. Bull. 34, 1628–1630 (2011).
Lee, D. E., Lee, S. J., Kim, S. J., Lee, H. S. & Kwon, O. S. Curcumin ameliorates nonalcoholic fatty liver disease through inhibition of O-GlcNAcylation. Nutrients 11, 2702 (2019).
Chuengsamarn, S., Rattanamongkolgul, S., Luechapudiporn, R., Phisalaphong, C. & Jirawatnotai, S. Curcumin extract for prevention of type 2 diabetes. Diabetes Care 35, 2121–2127 (2012).
Panahi, Y. et al. Curcuminoids plus piperine modulate adipokines in type 2 diabetes mellitus. Curr. Clin. Pharm. 12, 253–258 (2017).
Dehghani, S. et al. Topical application of curcumin regulates the angiogenesis in diabetic-impaired cutaneous wound. Cell Biochem. Funct. 38, 558–566 (2020).
Calle, R. A. et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat. Med. 27, 1836–1848 (2021).
Alkhouri, N. et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: A randomised, open-label phase II trial. J. Hepatol. 77, 607–618 (2022).
Lagouge, M. et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127, 1109–1122 (2006).
Sattarinezhad, A., Roozbeh, J., Shirazi Yeganeh, B., Omrani, G. R. & Shams, M. Resveratrol reduces albuminuria in diabetic nephropathy: a randomized double-blind placebo-controlled clinical trial. Diabetes Metab. 45, 53–59 (2019).
Hoseini, A. et al. The effects of resveratrol on metabolic status in patients with type 2 diabetes mellitus and coronary heart disease. Food Funct. 10, 6042–6051 (2019).
Rabbani, N., Xue, M., Weickert, M. O. & Thornalley, P. J. Reversal of Insulin Resistance in overweight and obese subjects by trans-resveratrol and hesperetin combination-link to dysglycemia, blood pressure, dyslipidemia, and low-grade inflammation. Nutrients 13, 2374 (2021).
Zhang, T., He, Q., Liu, Y., Chen, Z. & Hu, H. Efficacy and safety of resveratrol supplements on blood lipid and blood glucose control in patients with type 2 diabetes: a systematic review and meta-analysis of randomized controlled trials. Evid. Based Complement Altern. Med. 2021, 5644171 (2021).
Huang, H. et al. The effects of resveratrol intervention on risk markers of cardiovascular health in overweight and obese subjects: a pooled analysis of randomized controlled trials. Obes. Rev. 17, 1329–1340 (2016).
Ogier, J. M., Nayagam, B. A. & Lockhart, P. J. ASK1 inhibition: a therapeutic strategy with multi-system benefits. J. Mol. Med 98, 335–348 (2020).
Chertow, G. M. et al. Effects of Selonsertib in Patients with Diabetic Kidney Disease. J. Am. Soc. Nephrol. 30, 1980–1990 (2019).
Loomba, R. et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology 67, 549–559 (2018).
Loomba, R. et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology 73, 625–643 (2021).
Sarszegi, Z. et al. BGP-15, a PARP-inhibitor, prevents imatinib-induced cardiotoxicity by activating Akt and suppressing JNK and p38 MAP kinases. Mol. Cell Biochem. 365, 129–137 (2012).
Literati-Nagy, B. et al. Improvement of insulin sensitivity by a novel drug, BGP-15, in insulin-resistant patients: a proof of concept randomized double-blind clinical trial. Horm. Metab. Res. 41, 374–380 (2009).
Badman, M. K. et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel non-bile acid FXR agonist tropifexor (LJN452) in healthy volunteers. Clin. Pharm. Drug Dev. 9, 395–410 (2020).
Naoumov, N. V. et al. Digital pathology with artificial intelligence analyses provides greater insights into treatment-induced fibrosis regression in NASH. J. Hepatol. 77, 1399–1409 (2022).
Patel, K. et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients With Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 72, 58–71 (2020).
Zamaraev, A. V., Kopeina, G. S., Prokhorova, E. A., Zhivotovsky, B. & Lavrik, I. N. Post-translational modification of caspases: the other side of apoptosis regulation. Trends Cell Biol. 27, 322–339 (2017).
Garcia-Tsao, G., Fuchs, M. & Shiffman, M. Emricasan (IDN-6556) lowers portal pressure in patients with compensated cirrhosis and severe portal hypertension. Hepatology 69, 717–28 (2019).
Harrison, S. A. et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J. Hepatol. 72, 816–827 (2020).
Frenette, C. et al. Emricasan to prevent new decompensation in patients with NASH-related decompensated cirrhosis. J. Hepatol. 74, 274–282 (2021).
Zhu, D. et al. Dorzagliatin monotherapy in Chinese patients with type 2 diabetes: a dose-ranging, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Diabetes Endocrinol. 6, 627–636 (2018).
Yang, W. et al. Dorzagliatin add-on therapy to metformin in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial. Nat. Med. 28, 974–981 (2022).
Meininger, G. E. et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care 34, 2560–2566 (2011).
Morrow, L. A. et al. Safety, pharmacokinetics and pharmacodynamics of multiple-ascending doses of the novel glucokinase activator AZD1656 in patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 14, 1114–1122 (2012).
Kiyosue, A., Hayashi, N., Komori, H., Leonsson-Zachrisson, M. & Johnsson, E. Dose-ranging study with the glucokinase activator AZD1656 as monotherapy in Japanese patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 15, 923–930 (2013).
Wilding, J. P., Leonsson-Zachrisson, M., Wessman, C. & Johnsson, E. Dose-ranging study with the glucokinase activator AZD1656 in patients with type 2 diabetes mellitus on metformin. Diabetes Obes. Metab. 15, 750–759 (2013).
Vanweert, F. et al. A randomized placebo-controlled clinical trial for pharmacological activation of BCAA catabolism in patients with type 2 diabetes. Nat. Commun. 13, 3508 (2022).
Xiao, C., Giacca, A. & Lewis, G. F. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes 60, 918–924 (2011).
Yuan, H. F. et al. PRMT5 confers lipid metabolism reprogramming, tumour growth and metastasis depending on the SIRT7-mediated desuccinylation of PRMT5 K387 in tumours. Acta Pharmacol. Sin. 43, 2373–2385 (2022).
Bonds, A. C. et al. Post-translational succinylation of Mycobacterium tuberculosis Enoyl-CoA hydratase EchA19 slows catalytic hydration of cholesterol catabolite 3-Oxo-chol-4,22-diene-24-oyl-CoA. ACS Infect. Dis. 6, 2214–2224 (2020).
Yang, L. et al. The Succinate receptor GPR91 is involved in pressure overload-induced ventricular hypertrophy. PLoS ONE 11, e0147597 (2016).
Amraei, R. & Rahimi, N. COVID-19, Renin-angiotensin system and endothelial dysfunction. Cells 9, 1652 (2020).
Nie, L. et al. The Landscape of histone modifications in a high-fat diet-induced obese (DIO) mouse model. Mol. Cell Proteom. 16, 1324–1334 (2017).
Thomas, S. A., Storey, K. B., Baynes, J. W. & Frizzell, N. Tissue distribution of S-(2-succino)cysteine (2SC), a biomarker of mitochondrial stress in obesity and diabetes. Obesity 20, 263–269 (2012).
Zhang, Y. et al. Sirt5-mediated desuccinylation of OPTN protects retinal ganglion cells from autophagic flux blockade in diabetic retinopathy. Cell Death Discov. 8, 63 (2022).
Liu, X. et al. Dietary succinate supplementation to maternal mice improves fetal brown adipose tissue development and thermogenesis of female offspring. J. Nutr. Biochem. 100, 108908 (2022).
Wan, J., Liu, H., Chu, J. & Zhang, H. Functions and mechanisms of lysine crotonylation. J. Cell Mol. Med. 23, 7163–7169 (2019).
Xu, H., Wu, M., Ma, X., Huang, W. & Xu, Y. Function and mechanism of novel histone posttranslational modifications in health and disease. Biomed. Res. Int. 2021, 6635225 (2021).
Koronowski, K. B. et al. Ketogenesis impact on liver metabolism revealed by proteomics of lysine beta-hydroxybutyrylation. Cell Rep. 36, 109487 (2021).
Martinez-Moreno, J. M. et al. Epigenetic modifiers as potential therapeutic targets in diabetic kidney disease. Int. J. Mol. Sci. 21, 4113 (2020).
Nasser, S. et al. Ketogenic diet administration to mice after a high-fat-diet regimen promotes weight loss, glycemic normalization and induces adaptations of ketogenic pathways in liver and kidney. Mol. Metab. 65, 101578 (2022).
Mu, H. et al. Association of serum β-hydroxybutyrate and coronary artery disease in an urban Chinese population. Front. Nutr. 9, 828824 (2022).
Gallego, L. D. et al. Structural mechanism for the recognition and ubiquitination of a single nucleosome residue by Rad6-Bre1. Proc. Natl Acad. Sci. USA 113, 10553–10558 (2016).
Byrum, S. D., Taverna, S. D. & Tackett, A. J. Purification of specific chromatin loci for proteomic analysis. Methods Mol. Biol. 1228, 83–92 (2015).
Yu, Q. et al. Electron-transfer/higher-energy collision dissociation (EThcD)-enabled intact glycopeptide/glycoproteome characterization. J. Am. Soc. Mass Spectrom. 28, 1751–1764 (2017).
Hamza, G. M. et al. Affinity-bead assisted mass spectrometry (affi-BAMS): a multiplexed microarray platform for targeted proteomics. Int. J. Mol. Sci. 21, 2016 (2020).
Adoni, K. R., Cunningham, D. L., Heath, J. K. & Leney, A. C. FAIMS enhances the detection of PTM crosstalk sites. J. Proteome Res. 21, 930–939 (2022).
Couto, N., Davlyatova, L., Evans, C. A. & Wright, P. C. Application of the broadband collision-induced dissociation (bbCID) mass spectrometry approach for protein glycosylation and phosphorylation analysis. Rapid Commun. Mass Spectrom. 32, 75–85 (2018).
Seo, J. et al. Strategy for comprehensive identification of post-translational modifications in cellular proteins, including low abundant modifications: application to glyceraldehyde-3-phosphate dehydrogenase. J. Proteome Res. 7, 587–602 (2008).
Mao, Y., Kleinberg, A. & Li, N. Isobaric tandem mass tag multiplexed post-translational modification quantitation of biopharmaceuticals by targeted high-resolution mass spectrometry. Anal. Chem. 92, 9682–9690 (2020).
Romanick, S. S. et al. Obesity-mediated regulation of cardiac protein acetylation: parallel analysis of total and acetylated proteins via TMT-tagged mass spectrometry. Biosci. Rep. 38, BSR20180721 (2018).
Morales-Tarre, O., Alonso-Bastida, R., Arcos-Encarnacion, B., Perez-Martinez, L. & Encarnacion-Guevara, S. Protein lysine acetylation and its role in different human pathologies: a proteomic approach. Expert Rev. Proteom. 18, 949–975 (2021).
Shi, X. et al. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 27, 636–646 (2007).
Li, G. X. H., Vogel, C. & Choi, H. PTMscape: an open source tool to predict generic post-translational modifications and map modification crosstalk in protein domains and biological processes. Mol. Omics 14, 197–209 (2018).
Zeidan, Q. & Hart, G. W. The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J. Cell Sci. 123, 13–22 (2010).
Zhu, Y. & Hart, G. W. Targeting O-GlcNAcylation to develop novel therapeutics. Mol. Asp. Med. 79, 100885 (2021).
Wu, Z., Huang, R. & Yuan, L. Crosstalk of intracellular post-translational modifications in cancer. Arch. Biochem. Biophys. 676, 108138 (2019).
Qi, S. M. et al. PROTAC: an effective targeted protein degradation strategy for cancer therapy. Front. Pharm. 12, 692574 (2021).
Zeng, S. et al. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: recent progress and future challenges. Eur. J. Med. Chem. 210, 112981 (2021).
Guo, J., Liu, J. & Wei, W. Degrading proteins in animals: “PROTAC“tion goes in vivo. Cell Res 29, 179–180 (2019).
Dang, C. V., Reddy, E. P., Shokat, K. M. & Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 17, 502–508 (2017).
Bondeson, D. P. et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 25, 78–87 e75 (2018).
Chen, P. H. et al. Modulation of phosphoprotein activity by phosphorylation targeting chimeras (PhosTACs). ACS Chem. Biol. 16, 2808–2815 (2021).
BasuRay, S., Wang, Y., Smagris, E., Cohen, J. C. & Hobbs, H. H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl Acad. Sci. USA 116, 9521–9526 (2019).
Luo, G. et al. Discovery of an orally active VHL-recruiting PROTAC that achieves robust HMGCR degradation and potent hypolipidemic activity in vivo. Acta Pharm. Sin. B 11, 1300–1314 (2021).
Zheng, Z. G. et al. Discovery of a potent SCAP degrader that ameliorates HFD-induced obesity, hyperlipidemia and insulin resistance via an autophagy-independent lysosomal pathway. Autophagy 17, 1592–1613 (2021).
Hendriks, I. A. et al. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 24, 325–336 (2017).
Greer, E. L. & Shi, Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 (2012).
Acknowledgements
This study was supported by grants from the National Key R&D Program of China (Grant No. 2021YFC2500500), the National Natural Science Foundation of China (Grant Nos. 82070464, 81941022, 81530025) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB38010100). This work was also supported by Program for Innovative Research Team of The First Affiliated Hospital of USTC (CXGG02), Anhui Provincial Key Research and Development Program (Grant No. 202104j07020051), Anhui Provincial Natural Science Foundation (Grant No. 2208085J08) and Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (Grant No. 2017BT01S131).
Author information
Authors and Affiliations
Contributions
Conceptualization: J.W. and S.X. Writing: X.W., M.X., S.C., M.G., S.X. Proofreading and revising: P.J.L. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
41392_2023_1439_MOESM1_ESM.docx
Supplementary Materials for Targeting protein modifications in metabolic diseases: molecular mechanisms and targeted therapies
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, X., Xu, M., Geng, M. et al. Targeting protein modifications in metabolic diseases: molecular mechanisms and targeted therapies. Sig Transduct Target Ther 8, 220 (2023). https://doi.org/10.1038/s41392-023-01439-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01439-y
This article is cited by
-
Protein lipidation in health and disease: molecular basis, physiological function and pathological implication
Signal Transduction and Targeted Therapy (2024)
-
Mechanisms and functions of protein S-acylation
Nature Reviews Molecular Cell Biology (2024)
-
Inter-organ crosstalk during development and progression of type 2 diabetes mellitus
Nature Reviews Endocrinology (2024)
-
Epigenetic meets metabolism: novel vulnerabilities to fight cancer
Cell Communication and Signaling (2023)
-
Glucose fluctuations aggravate myocardial fibrosis via activating the CaMKII/Stat3 signaling in type 2 diabtetes
Diabetology & Metabolic Syndrome (2023)
