
Abstract
Neuropsychiatric disorders are multifactorial disorders with diverse aetiological factors. Identifying treatment targets is challenging because the diseases are resulting from heterogeneous biological, genetic, and environmental factors. Nevertheless, the increasing understanding of G protein-coupled receptor (GPCR) opens a new possibility in drug discovery. Harnessing our knowledge of molecular mechanisms and structural information of GPCRs will be advantageous for developing effective drugs. This review provides an overview of the role of GPCRs in various neurodegenerative and psychiatric diseases. Besides, we highlight the emerging opportunities of novel GPCR targets and address recent progress in GPCR drug development.
Similar content being viewed by others


Introduction
The nervous system employs membrane receptors to detect extracellular stimuli and transmit signals across the cell membrane. As the largest membrane protein family, G protein-coupled receptors (GPCRs) allow the nervous system to respond accurately to external stimuli and internal states. GPCRs are structurally similar transmembrane proteins containing seven transmembrane (TM) α-helices linked by three extracellular loops and three intracellular loops.1 The unique ligand binding pockets formed by the 7TM regions allow the receptor to engage with various stimuli, including neurotransmitters, nucleotides, amines, peptides, cytokines, and hormones in the extracellular environment (Fig. 1).2 Through expressing GPCRs with different ligand-recognizing abilities, the nervous system could filter and select particular signals to respond.3 Furthermore, the intrinsic ligand selectivity of neuronal GPCRs allows crosstalk and proper integration between signal transduction pathways. GPCRs drive signal transduction via two major modulators: heterotrimeric G protein and arrestins. Characterizing the physiological functions of GPCRs in the nervous system and pathological mechanisms in disease models could accelerate GPCR-targeted drug development.

Structure features of active GPCR. a Orthosteric pocket forms by the helical core of 5-HT2A receptor (marine blue, PDB 6WHA). b Solvent-accessible surface. Hydrophobic surface (red); hydrophilic surface (white). c Activated GPCR opens cytosolic pocket for G protein coupling. d Heterotrimeric G protein, Monomeric Gαi, Gβγ. d Activated 5-HT2A receptor forms a cytoplasmic pocket which allows G-protein coupling
The progressive dysfunction of neural tissues in the central and peripheral nervous systems is the hallmark feature of neurodegenerative diseases. Neurodegenerative diseases are increasing in the elderly population.4 It is estimated that neurodegenerative diseases affect over 50 million people across the globe.5 Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, and Multiple sclerosis are representative examples. Currently, there is no effective cure. The pathogenesis and underlying mechanisms of neurodegenerative diseases remain poorly understood. At present, symptom control is the primary treatment objective.6 It is estimated that neurodegenerative diseases will become the second most common cause of death.7
Alzheimer’s disease and dementias are in the top-ten ranking leading cause of death globally.8 Deposition of the insoluble and phosphorylated β-amyloid peptide (derived from amyloid precursor protein) in the brain parenchyma of Alzheimer’s disease patients affects functions/regeneration of various forms of neurons.9 The resulting widespread neuron damage affects synaptic communication leading to cognitive deficits, regional brain shrinkage, and brain atrophy;10 Huntington’s disease could appear in childhood or adolescence. Aberrant expansion of DNA segment containing CAG trinucleotide repeats in the huntingtin gene is a hallmark feature.11 Large CAG repeat is associated with early symptoms manifestation.12 Symptoms include poor coordination, chorea (involuntary dance-like movements), slow movement, seizures, and slurred speech; Parkinson’s disease affects motion control. Rigidity, tremor, and slow movement (bradykinesia) are frequently observed. Risk factors include genetic polymorphism, chronic inflammation, and metabolic disorders.13 Multiple sclerosis is a relapsing-remitting disease caused by an autoimmune attack in the central nervous system. Damage of myelin sheath in multiple areas by immune cells causes cognitive impairment, fatigue, muscle weakness, tremor, and vision problems.14
Brain disorders are frequently associated with mental/psychiatric illnesses.15 Mental illness is burdening the healthcare system with enormous unmet medical needs.16,17 Serious mental illness is closely linked to reduced life expectancy due to a higher risk of cardiovascular morbidity and mortality.18 Common mental illnesses include anxiety, depression, bipolar disorder, attention deficit hyperactivity disorder, and schizophrenia. Both children and adolescents are vulnerable to mental illnesses. Mental health condition is interlinked with physical health. The generation of suicide ideation/attempts and self-destructive thoughts are closely related to psychiatric diseases.19 Patients with degenerative diseases could also present emotional symptoms adding complexity to disease diagnosis and management. Recent studies reveal that hospitalized patients with COVID-19 and survivors display different levels of neuropsychiatric complications and the underlying mechanisms remain to be explored.20
GPCRs are one of the most intensively exploited targets for drug development. Approximately 35% of the FDA-listed drugs act through GPCRs.21,22 With our increasing understanding of the neuronal relay functions of GPCRs in the nervous system, many GPCRs are perceived as promising druggable targets for neurodegenerative and psychiatric diseases. This review summarizes the multifaceted role of GPCRs in chronic neurodegenerative conditions exemplified by Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, and Multiple sclerosis. The emerging role of GPCRs on psychiatric illnesses, including Schizophrenia, Bipolar disorder, Depression, Attention deficit hyperactivity disorder, and Tourette’s disorder, are discussed. We also highlight the emerging opportunities for the previously unexplored GPCRs and provides examples of pharmaceutical development of GPCR-targeted therapeutics.
G protein-coupled receptors signaling
Synaptic transmission can be classified into two types: fast and slow synaptic transmission.23 In fast synapses, GPCRs such as glutamate and GABA (γ-aminobutyric acid) receptors generate membrane depolarizing signals in less than 1/1000 s. In slow synapses, biogenic amines, peptides, and amino acid receptors generate signals in hundreds of milliseconds to minutes.23 GPCRs are structurally similar membrane proteins (Fig. 1). They elicit different intracellular signal pathways by interacting with heterotrimeric G proteins (α, β, and γ). GPCRs can be stabilized by an array of neurotransmitters and neurological modulators, including ions, hormones (peptide or non-peptide), vitamins, metabolites (ATP, fatty acids, etc.), natural products, and pharmacological ligands.24 A plethora of GPCR signaling events are involved in developing neuropsychiatric disorders. Understanding the downstream signaling events of disease-associated GPCR is essential for designing efficacious therapy.
Human GPCR can be classified into five distinct subtypes: rhodopsin (class A), secretin (class B1), adhesion (class B2), glutamate (class C), and frizzled (class F).1 To date, over 750 ligand-bound or apo-GPCR structures (including 96 CNS-related GPCRs) have been reported (Table 1). For details: https://gpcrdb.org. The transmembrane helical core exhibits high similarity. The helical core forms the orthosteric binding pocket for cognate ligands. GPCR can be divided into three different functional regions: (1) extracellular region including N-terminus, extracellular loops (ECLs), and extracellular ends of the transmembrane helices are involved in ligand recognition and selectivity;25 (2) intracellular region consisting of C-terminus, intracellular loops (ICLs) and intracellular ends of the transmembrane helices provide docking cavity for G proteins/ arrestins and interacts with different regulatory proteins such as GPCR kinases;26 (3) helical core in-between extracellular and intracellular region deliver and covert ligand signals via unique conformational change (Fig. 1b).27,28
Activated receptors generate second messengers via the G protein. In heterotrimeric form, the G protein is inactive. After binding to the intracellular cavity formed by GPCR, the GDP-binding pocket on the Gα subunit of heterotrimeric G proteins is opened, facilitating subsequent exchange for GTP.29 GTP is physiologically more abundant as compared to GDP.30 The nucleotide exchange is a rate-limiting step in the G protein activation process.29 GTP binding prevents Gα protein from forming heteromer with Gβγ subunit.31 The free Gα and Gβγ subunits modulate different downstream effector pathways. By hydrolyzing GTP to GDP, the active GTP-bound Gα subunit returns to an inactive state and forms a complex with the Gβγ subunit again. G proteins are classified based on their Gα subunit. There are four different Gα protein families: Gαi/o, Gαs, Gαq/11, and Gα12/13. Each family regulates a specific set of downstream responses. Individual GPCR could mediate different functions in different cellular contexts via preferential G protein coupling (Figs. 1 and 2).

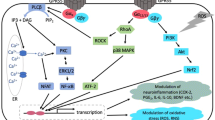
GPCR-regulated downstream signaling pathways in neurodegenerative and psychiatric disorders. CAMK calmodulin-dependent protein kinase, BACE1 β-site APP cleaving protein 1, TAK1 transforming growth factor-β-activated kinase, TAB1 TAK1 binding protein, PP2A protein-phosphatase 2A, PLC phospholipase C, PDK1 phosphoinositide-dependent kinase 1, diacylglycerol (DAG), IP3 inositol triphosphate, Akt protein kinase B, APP amyloid protein precursor, Bax B-cell lymphoma-2-associated X, Blk B lymphoid tyrosine kinase, cAMP cyclic adenosine monophosphate, Casp9 caspase 9, CREB cAMP response element binding protein, ERK1/2 extracellular signal-regulated-kinase, GSK3β glycogen synthase kinase 3β, Gβγ free heterotrimeric G protein beta/gamma subunits, IKBα inhibitory subunit of nuclear factor kappa-B alpha, IKKα/β inhibitor of kappa-B kinase, MEK mitogen-activated protein, NFT neurofibrillary tangles, NF-κB nuclear factor kappa-B, PI3K phosphoinositide 3-kinase, PKA protein kinase A, Raf1 Raf-1 proto-oncogene, serine/threonine kinase, Ras Ras Sarcoma oncoproteins, Rho Ras homologous proteins, ROCK Rho-associated coiled-coil containing kinases, SRF serum response factor
Gα proteins: Gαs and Gαi/o
Gαs (stimulatory regulator of adenylyl cyclase G protein activates adenylyl cyclase) promotes the generation of 3’-5’-cyclic adenosine monophosphate (cAMP) from ATP by adenylate cyclase. cAMP is essential for protein kinase A (PKA)-mediated signal transduction;32 In contrast, Gαi/o suppresses adenylyl cyclase activity, which prevents cAMP accumulation and reduces PKA activity. cAMP is a crucial regulator of the phosphoinositide 3-kinase/AKT murine thymoma viral oncogene homolog (PI3K/AKT) signaling pathway. It has been shown that PI3K/AKT is associated with the inflammatory response in multiple neurodegenerative diseases.33,34,35 cAMP is also linked to calcium dynamics in neuronal cells and neurodegenerative diseases. Details can be found in the comprehensive review by Sobolczyk and Boczek.36
Gα protein: Gαq/11
Gαq activates phospholipase C (PLC), which hydrolyzes phosphatidylinositol 4,5-biphosphate into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG activates protein kinase C, which phosphorylates various downstream signaling proteins. IP3 stimulates calcium efflux from the endoplasmic reticulum through specific IP3 receptors. Calcium signaling is essential for the release of neurotransmitters.37,38 For instance, dysregulation of the dopamine D1 receptor-mediated PLC/IP3/Ca2+ pathway in the anterior cortex of the brain is associated with mental illness in rats.39,40 PLC/IP3/Ca2+ pathway regulates the electrical response of the neuron.41 Impaired Ca2+ homeostasis by Aβ exposure is one of the underlying causes of amyloid toxicity in Alzheimer’s disease.42 In psychiatric disorders, Ca2+ signaling regulates neuronal connectivity, synaptic plasticity, and glial functions.43
Gα protein: Gα12/13
Gα12/13 binding can stimulate Rho family GTPases.44 Rho GTPases activate the cytosolic Rho protein by promoting GDP/GTP exchange.45 Activated Rho is released from inhibitory protein, migrates to the plasma membrane, and modulates multiple downstream effectors.46 One of which is ROCK1/2 (Rho kinase). The Rho-ROCK pathway is essential in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington’s disease.47 ROCK activity is closely associated with neuronal cell loss, impaired synaptic functions, and cytoskeleton modulation in central nervous system disorders.47 Rho/ROCK signaling modulates the activity of transcription regulators such as AP-1, MRTF-A, YAP, NF-κB, and serum response factor.47,48 Rho family GTPases are essential for axon guidance, cell polarity, and synapse formation.49 It has been shown that Rho GTPase regulates neuronal cell survival by inhibiting AKT signaling.50
GPCR kinases (GRKs)
Activated GPCR is subjected to desensitization to protect the cell from sustained stimulation.51 After peak response, ligand-bound receptor activity will return to basal level.52 Receptor phosphorylation by a family of GPCR kinases (GRKs), including GRK1/7, GRK2/3, and GRK4/5/6, is an essential first step to switch off sustained signaling.53,54 GRKs are second messenger-independent kinases (e.g., in contrast to PKA, which is dependent on cAMP levels). Serine/threonine residues on the GPCR carboxyl-terminal tail are common phosphorylation sites targeted by GRKs.55 GRKs translocate from cytoplasm to plasma membrane and initiate receptor phosphorylation by binding to Gβγ.56,57 GRK could also interfere with G protein binding through direct interaction.58 GRK level is affected by inflammatory responses in neonatal and adult neurons.59 GRK dysfunction is associated with cognitive impairment and tau hyperphosphorylation in Alzheimer-like pathology.60 Colocalization of GRK with amyloid plaques is observed in brain tissues of Alzheimer’s disease patients.61 Patients of Parkinson’s disease with dementia have increased GRK3/5 transcripts.62 GRK might promote the formation of pathological Lewy bodies in sporadic Parkinson’s disease, but the mechanism is yet to be defined.63 In psychiatric disorders, upregulating brain GRKs are observed in schizophrenia and major depression.64,65
Arrestins in GPCR desensitization
Active GPCR is ready for the arrestins (signal terminators) binding after GRK phosphorylation. Arrestins can be classified into visual arrestins (arrestin 1 and arrestin 4) and non-visual arrestins (β-arrestin 1/2 or arrestin 2/3). Visual arrestins express exclusively in retina photoreceptors. They regulate light-activated rhodopsin signaling.66,67 β-arrestin 1/2 are ubiquitously expressed cytoplasmic proteins (Fig. 3a, b).52 β-arrestins and G proteins compete for the receptors. They bind to the same inter-helical cavity on the intracellular region (Fig. 3c).68 β-arrestin reduce G protein singling by hindering interaction between receptor and heterotrimeric G proteins. Further, β-arrestins facilitate receptor recycling by promoting internalization and cellular trafficking.69,70 The C-edge of arrestin protein with proximity to the membrane surface functions membrane anchor to stabilize the arrestin-active receptor complex (Fig. 3a).71 Recent studies illustrate the association of β-arrestin in multiple physiological functions and neuropsychiatric disorders.72,73 Phosphorylation of PI3K/AKT is remarkably reduced in the β-arrestin 2-deficient adult neural stem cells, indicating the crucial role of β-arrestin 2-PI3K/Akt pathway in adult hippocampal neurogenesis.74,75

GPCR-G protein/arrestin complexes. a Crystal structure of arrestin 1 (PDB 1CF1) showing the membrane-anchoring c-loop. b Solvent-accessible surface. Hydrophobic surface (red); Hydrophilic surface (white). c Biased signaling of serotonin 5-HT2B receptors. Activated 5-HT2B receptor (PDB 7SRQ) is preferentially coupled to Gαs protein (PDB 7SRR). The receptor could also couple to β-arrestin 1 (PDB 7SRS). Gαs and β-arrestin 1 engaged on the same cavity formed by the cytoplasmic receptor interface
Biased signaling of GPCRs
G protein-biased signaling is regarded as the canonical signaling pathway employed by GPCRs.
β-arrestin can modulate GPCR signal transduction in G protein-independent mechanism. β-arrestin can use the receptor as a structural component to generate an intracellular signaling complex consisting of agonist-occupied receptor and nonreceptor tyrosine kinases (c-Src).76 β-arrestin can maintain ERK signaling by acting as a scaffold for ERK mitogen-activated protein kinase.77 Other downstream effectors of β-arrestins include phosphatases and transcription factors.78 β-arrestins can act as a scaffold protein for specific downstream effectors.79,80 In the mouse model, β-arrestin 2 exerts anti-inflammatory functions by inhibiting nuclear factor kappa-B.81 Maintaining the arrestin-dependent signaling of M1 muscarinic acetylcholine receptor can prevent the insoluble misfolded proteins accumulation in Alzheimer’s disease model which thereby slowing down neurodegenerative disease progression.82
β-arrestin is important for astrocyte-mediated pro-inflammatory cytokine production.83 In mouse Parkinson’s disease models, β-arrestin 2-biased ligands suppress glia-derived inflammation and prevent neuron loss.84 IL-1β produced by the inflammation site is suppressed by β-arrestin 2.84 As compared to agonists which facilitate G protein and β-arrestin signaling at the same time, a β-arrestin-biased agonist for δ-opioid receptor can effectively control anxiety-like behaviour by activating ERK1/2 in the limbic structures of the brain.85 Hence, identifying therapeutic modulators that could preferentially stabilize GPCR structure for G proteins or β-arrestins is important for developing effective treatments for neurodegenerative and psychiatric diseases.
Examples of GPCR-regulated modulators in disease development
β-site APP cleaving protein 1 (BACE1)
The proteolytic activity of BACE1 promotes the generation of β-amyloid (Aβ) peptides from amyloid precursor protein in Alzheimer’s disease.86 BACE1 expression can be activated by muscarinic acetylcholine receptor M1/M3 via PKC and MAP kinase signaling cascades.87 BACE1 activity is modulated by other GPCRs, such as the A2A and delta-opioid receptors.33 It has been shown that selective activation of the M2 receptor will suppress BACE1 expression via PKA-mediated signaling events.33
cAMP-response element binding protein (CREB)
GWAS analysis indicates that genes involved in the cAMP/PKA/CREB pathway are genetically associated with schizophrenia and bipolar disorder.88 CREB is a transcription factor activated by phosphorylation after GPCR activation. The binding of CREB to a specific cAMP response element (CRE) in the transcription regulatory region enhances particular gene transcription. For instance, neurotransmitter-activated dopamine D1 receptor on dopaminergic neurons can elicit transcription brain derived growth factor (BDNF) and other neurotrophins.89 In patients with bipolar disorder and schizophrenia, CREB expression is remarkably reduced in the dorsolateral prefrontal cortex and cingulate gyrus.90 While CREB protects neuronal cells in neurogenerative diseases, constitutively active CREB can reduce hippocampal neuron numbers and trigger sporadic epileptic seizures.91,92 It has been shown that the CREB modulator could enhance synaptic plasticity, which is beneficial for schizophrenia treatment.89
DARPP-32 and PP1
DARPP-32 (dopamine- and cyclic-AMP-regulated phosphoprotein of molecular weight 32,000) regulates neuronal excitability levels by prolonged depolarizations and voltage oscillations.93 DARPP-32 is the downstream target of Gi-coupling receptors such as the D2 dopamine receptor. DARPP-32 functions as a protein phosphatase-1 (PP1) inhibitor, a eukaryotic serine/threonine protein, upon phosphorylation at Thr-34 by PKA. PP1 is a phosphatase with multiple physiological functions. PP1 controls clock component PER2 accumulation in neurons, influencing circadian rhythm by light-mediated clock resetting.94 PP1 is an inducer of long-term synaptic depression in the hippocampus.95 Dysregulation of glutamate and dopamine signaling is common in neurodegenerative and neuropsychiatric disorders. Quantitative modeling results suggested that DARPP-32 could integrate dopamine and glutamate signals in striatal neurons.96 PP1 signaling reduces GABA(A) receptors in neostriatal medium spiny neurons depending on PKA and DARPP.97
GPCRs in neuropsychiatric diseases
Class A GPCR (rhodopsin)
Structural insights
Class A GPCR is the most heavily investigated GPCR family for drug development. Ligand binding to the unique pocket stabilizes GPCR in a particular conformation.98 Comparative analysis reveals that the outward bending/rotation of intracellular TM6 is a universal structure feature of receptor activation throughout the GPCR superfamily (Fig. 4a).98 Hydrophobic packing interactions between the transmembrane helices help to maintain the active conformation of TM6.99 Apart from TM6, rearrangements of other transmembrane helices, including TM3/5/7, open the intracellular milieu to facilitate recruitment of G protein.100 Class A GPCR has a consensus binding interface for G protein coupling.101 The receptors employ unique structure motifs as microswitches to transmit external stimuli (Fig. 4b). D3.49R3.50Y3.51 motif (Ballesteros–Weinstein number) at the intracellular region of TM3 forms the classic “ionic lock” with E6.30 on TM6 to constrain the receptor in the ground state.102,103 Disruption of the ionic lock is an activation feature of class A GPCRs.104 Side chains of Y7.53 (NPxxY motif) on TM7 and W6.48 (CWxP motif) on TM6 are subjected to orientation rearrangement during receptor activation.105,106 The P5.50I3.40F6.44 motif, formed by a group of hydrophobic residues on TM3/5/6, is also a crucial switch for receptor activation.107 Polar interactions and aromatic stacking interactions between the conserved aromatic residues are frequently observed in the ligand binding region of activated class A GPCRs.27

Class A GPCR activation. a Prominent outward bending of TM5 and TM6 opens the cytoplasmic pocket of inactive serotonin 5-HT2A receptor (orange, PDB 6A93) for the binding of G protein. Active 5-HT2A receptor (marine blue, PDB 6WHA). b Microswitches involved in 5-HT2A receptor activation. c Structural features of class C GPCRs. Inactive (PDB 7MTQ) and active metabotropic glutamate receptor 2 mGlu2R (PDB 7MTR). VFT extracellular venus flytrap domain, CRD cysteine-rich domain, TMD transmembrane domain, PAM positive allosteric modulator
Acetylcholine receptors (muscarinic)
Acetylcholine is a neurotransmitter employed by cholinergic neurons in the brain and spinal cord.108 Muscarinic acetylcholine receptors in the central and peripheral nervous systems have five distinct subtypes. M1, M3, and M5 receptors are excitatory M1-like receptors.109 In contrast, M2-like receptors (M2 and M4 receptors) inhibit adenylyl cyclase activity. All the subtypes are detected in the brain. M2 and M3 receptors are also found in peripheral tissues.110
Reduced acetylcholine signaling due to the loss of cholinergic neurons is common in Alzheimer’s disease.111 Amyloid-β proteins could interrupt the interaction between the M1 receptor and G protein.112 M1 receptor-knockout mice show Alzheimer’s disease-like pathology with age-dependent cognitive decline.113 M1 receptor function is impaired by the binding of tau protein, a microtubule-associated protein in the extracellular matrix, which is toxic in secreted form;114,115 Autoantibodies to recombinant human M1 receptors are detected in patients with schizophrenic disorders, mood disorders, and other psychiatric disorders.116,117
The M1 receptor is a promising target for schizophrenia treatment. Allosteric modulation of M1 receptor activity could improve cognitive performance with antipsychotic activity.118 However, substantial loss of cortical M1 receptor might affect the efficacy of positive allosteric modulator.119
M2 receptor reduction is noted in the frontal cortex of Alzheimer’s disease patients.120 Suppressing M2 receptor expression with siRNA alters the expression of β-site APP cleaving protein. This transmembrane aspartic endopeptidase is involved in beta-amyloid formation.121
M2 receptor is suspected to be related to the major depressive disorder and bipolar disorder development.122 M2-encoding gene is genetically associated with the cholinergic dysfunction seen in mood disorders.122
M3 receptor level is remarkably reduced in the post-mortem frontal cortex tissues of patients with bipolar disorder.123 However, conflicting results are observed in another study cohort.124 Genetic variants of the M3 receptor-encoding gene are associated with abnormal neural connectivity in schizophrenia and cannabis-induced hallucinations.125,126
Acetylcholine elevation is observed in Parkinson’s disease.127,128,129 Targeting the M4 receptor with various antagonists showed promising treatment results for Parkinson’s disease.130,131 M4 receptor is abundantly expressed in striatal neurons, which regulates the balance between acetylcholine and dopamine responses.132 M4 receptor promotes the development of the dopamine hypersensitivity phenotype of schizophrenia.133 It has been shown that the M5 receptor can potentiate drug addiction by reinforcing rewarded behavior.134
Adenosine receptor
Adenosine (A1A, A2A, A2B, A3A) receptors are synaptic modulators that transmit inhibitory signals from adenosine to excitatory synapses.135 Adenosine is also known as a “retaliatory metabolite” as it is produced exponentially from tissue under stress.136 Astrocytes release adenosine to modulate synaptic transmission during hypoxia.137 A1A and A2A receptors exhibit widespread expression in the brain.138 A1A and A3A receptors are Gi-coupling receptors. In contrast, A2A and A2B receptors prefer Gs for downstream signaling.
Although dopamine-replacement therapy is the mainstay treatment for Parkinson’s disease, it remains challenging to manage dyskinesia during replacement treatments.139 Animal study reveals that activating the A2A receptor will reduce the agonistic effects of dopaminergic D2 receptor-targeting drugs.140 As the A2A receptor is colocalized with D2 dopaminergic receptors, it is suggested that interactions between A2A and D2 receptors might be involved in the pathophysiology of Parkinson’s disease.141
Epidemiological data support that caffeine (a naturally occurring methylxanthine) consumption might reduce the risk of depression or depressive symptoms.142,143 The psychoactive function of caffeine is mediated via the non-selective antagonistic action on A1/A2A receptors.144 How A1/A2A receptors regulate depression-like behaviour remains unclear.145 It should be noted that caffeine at high doses might function other than adenosine receptor antagonists causing insomnia and anxiety.146,147
Activated A2A receptor suppresses nitric oxide (NO) production by inhibiting NO synthetase.12 NO signaling is associated with various neurodegenerative diseases, including Parkinson’s disease, amyotrophic lateral sclerosis, multiple sclerosis, amyotrophic lateral sclerosis, and Alzheimer’s disease.148 NO is a mediator of neuroinflammation, which triggers the microglial to release pro-inflammatory factors.149 NO induces protein S-nitrosylation (covalent addition of a NO group to a cysteine thiol/sulfhydryl), imposing endoplasmic reticulum stress in neurons.150,151 As A2A receptor activation affects synaptic plasticity and introduces memory deficits, antagonizing the A2A receptor might be helpful to control age-related cognitive impairments in Alzheimer’s disease.152
Adrenergic receptor
Brain adrenergic receptors on neurons and glia are activated by the monoamine neurotransmitter norepinephrine (produced primarily in the locus coeruleus of the brain stem) and epinephrine.153 Norepinephrine is produced from dopamine and converted into epinephrine. Norepinephrine and epinephrine released at synaptic junctions in the autonomic nervous system control classical fight-or-flight response.154 Norepinephrine controls response to environmental changes by regulating neuronal excitability.155 Epinephrine and norepinephrine also affect intelligence.156 Human has 2 adrenergic receptor subtypes: α-adrenergic (α1, α2A, α2B, α2C) receptors and β-adrenergic (β1, β2, β3) receptors. All the subtypes can be detected in the brain tissues.
Adrenergic receptor protects the central nervous system from uncontrolled inflammatory responses.157 In the neonatal Lewis rats model, norepinephrine protects neuronal damage from inflammation.158,159,160,161 Blocking β-adrenergic receptors signaling with beta-blockers (β-adrenergic antagonists) could exacerbate neuroinflammation in a mouse model of Alzheimer’s disease.162
Patients of Alzheimer’s disease and Parkinson’s disease show profound cell loss in locus coeruleus.163 Amyloid Aβ affects norepinephrine production and alters adrenergic receptor signaling in Alzheimer’s disease;164 Low norepinephrine level is linked to mood disorders such as anxiety, depression, and attention deficit hyperactivity disorder.156 α2-adrenergic receptors are established targets for antidepressant therapy.165 Depressed suicide victims showed high α2A-adrenergic receptor in the prefrontal cortex.166 Presynaptic α2-adrenergic receptor is an auto-receptor with the highest affinity to norepinephrine. Activated α2-adrenergic receptor inhibits norepinephrine synthesis and release.167 Thus, antagonizing presynaptic α2-adrenergic receptors could benefit depression treatment by enhancing norepinephrine release.168
Cannabinoid receptor
Cannabinoid signaling is involved in nociception, neurotransmission, and neuroprotection.169 It is also engaged in learning, memory, motor, food intake, anxiety, pain perception, and fear memories.170 Cannabinoid receptor type 1 (CB-1) is the primary subtype in the central nervous system. In comparison, the CB-2 receptor is mainly found in immune tissues.171 Cannabinoid receptors in the presynaptic nerve terminals can be activated by endogenous lipid endocannabinoids 2-arachidonoylglycerol (2-AG) and N-arachidonoyl-ethanolamine (AEA; anandamide).172 2-AG is a full agonist for cannabinoid receptors, while AEA is a weak partial agonist.173 Cannabinoid receptors can also be activated by phytocannabinoids such as Δ9-tetrahydrocannabinol and non-euphoric cannabidiol (CBD) extracted from cannabis.174,175
The CB-1 receptor is the dominant subtype in the brain.176,177 CB-1 receptor can be found in different neuronal types (e.g., GABAergic, glutamatergic, and serotonergic neurons) and controls cholinergic transmission.178,179 Exogenous administration of endocannabinoids protects neurons from β-amyloid (Aβ) neurodegeneration and apoptosis.180 Targeting cannabinoid receptors can improve spasticity (increase in muscle stiffness) and central neuropathic pain in patients with multiple sclerosis.181 Substantial reduction of CB-1 receptor in lateral globus pallidus and substantia nigra pars reticulata is associated with neurodegeneration in Huntington’s disease.182,183 Genetic polymorphisms on the CB-1 receptor are a risk factor for schizophrenia. CBD treatment is effective for neuroinflammatory-derived conditions such as epilepsy and anxiety.184
The pathological functions of the CB-2 receptor in inflammatory conditions (e.g., Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, stress response, and depression) are under active investigation.185,186 Inflammation is a driving factor of depression and could counter the effects of antidepressant therapies.187 CB-2 receptor-overexpressing mice showed a significant reduction in depressive-related behaviors.188 In contrast, pro-inflammatory chemokines and cytokines are markedly reduced in the brain of CB-2 receptor-deficient mice.189 CB-2 receptor can suppress microglial activation and prevent pro-inflammatory mediators release.190,191 In bipolar disorder, a neuropsychiatric disorder presenting with mood fluctuation, selective activation of the CB-2 receptor can stabilize mood and reduce mood swings.192
Other receptors for endogenous cannabinoids: GPR12, GPR18, and GPR55
GPR12 is phylogenetically related to the cannabinoid (CB-1 and CB-2) receptors.193 GPR12 is a constitutively active receptor.194 Apart from cannabidiol, lysophospholipid sphingosine 1-phosphate and phingosyl-phosphorylcholine are potential endogenous ligands for GPR12.193,195 GPR12 expressed mainly in the central nervous system (frontal cortex, piriform cortex, thalamus, hypothalamus, hippocampus, amygdala, and olfactory bulb).196 In mice, GPR12 expresses in the area controlling emotion and metabolism.195 GPR12 promotes neurite outgrowth by activating ERK1/2 signaling.197 Other functions include pain control, neurite outgrowth, and regeneration.193 SNP microarray-based genome-wide association study reveals a close association between GPR12 and antipsychotic response in schizophrenia treatment.198
GPR18 and GPR55 also act as receptors for endogenous cannabinoids 2-AG and AEA.199,200 GPR18 and GPR55 exhibit high structural similarity.201 GPR18 regulates polymorphonuclear cell infiltration and protects organs from acute immune responses.202 It has been shown that GPR18 could interact with the CB-2 receptor in activated microglia of Alzheimer’s disease model;203 GPR55 expresses predominantly in the brain.204 The receptor can be activated by endocannabinoids, phytocannabinoids, synthetic cannabinoid ligands, and lysophosphatidylinositol.205 GPR55 antagonist exhibits anti-inflammatory functions by modulating GPR55-expressing immune cells such as monocytes and microglia.206 Given the high expression of GPR55 in the striatum, GPR55 signaling is suspected to be involved in motor impairment in Parkinson’s disease.207
Dopamine receptor
Dopamine is a catecholamine neurotransmitter in the brain. Dopamine/ dopamine receptors are crucial for motor function, cognition, learning, and memory.208 There are two receptor subtypes: D1-like (D1 and D5) and D2-like (D2, D3, and D4).209 D1 and D2 receptors are the most abundantly expressed dopamine receptor subtypes in the brain.210
D1 and D2 receptors are significantly reduced in asymptomatic Huntington’s disease patients.12 In the early stage of Huntington’s disease, dopamine signaling is associated with the development of dance-like movements (chorea). Clinical studies show that dopamine receptor blockers or depleting agents control motor dysregulation, especially chorea.211 In the late stage, however, a remarkable reduction in dopamine/ dopamine metabolite level is observed.212 The D1 receptor is remarkably reduced in patients presenting mild to moderate functional impairment.213 It is noted that targeting the dopaminergic signaling cascade might lead to rapid cognitive decline in Huntington’s disease patients.214
Disturbances in the dopaminergic system are frequently observed in other neurodegeneration disorders, including Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis.215 Reduced dopamine receptors are correlated with the progression of Alzheimer’s disease;216 Loss of dopaminergic neurons is a hallmark feature of Parkinson’s disease. Activating D2-like receptors (D2/3 receptors) or increasing circulating dopamine are effective treatment strategies for symptomatic Parkinson’s disease;217 Dopamine dysregulation contributes to the demyelinating process (resulting from autoimmune attack) in multiple sclerosis.218 Dopamine can modulate pro-inflammatory cytokines secretion in T helper Th17 cells in uncontrolled neuroinflammatory responses.219,220
The development of β-arrestin-biased modulators might improve treatment outcomes and avoid side effects. Dopamine receptor agonist exhibits mild to serious side effects.221 This is partly caused by the activation of both G proteins and the β-arrestin signaling cascade.222,223 Many antipsychotics could interfere with dopamine-dependent β-arrestin 2 recruitment.83 Selective activating the D2 receptor-β-arrestin pathway with biased agonist is beneficial to correct dopamine signaling in schizophrenia.224
Histamine receptor
Histamine is an inflammatory biogenic amine synthesized from L-histidine. Histamine stimulates peripheral immune cells to release pro-inflammatory cytokines. In the central nervous system, histamine signaling in the tuberomammillary nucleus (TMN) controls sleep-wake, circadian and feeding rhythms.225 Elevated histamine increases blood-brain barrier permeability, allowing peripheral immune cells to enter and act on brain parenchyma.226
Four different histamine (H1–H4) receptors are reported.227 H1 and H2 receptors are expressed in the brain, central nervous system, and peripheral tissues.225 H1 receptor activation promotes neuron differentiation. In contrast, H2 receptor activation induces neural stem cell proliferation.228 H3 receptor is localized in the brain.229 H3 receptor is an important therapeutic target for cognitive disorders.230 The neurological function of the H4 receptor remains unclear.229 H4 receptor can be detected in the non-neuronal cells of the brain.229 H4 receptor activation is involved in the inflammatory responses regulated by mast cells, eosinophils, and T cells.229 Histamine acts on H1 and H3 receptors to control normal sleep/wake behavior.231
Alterations in histamine signaling are found in both neurodegenerative and psychiatric disorders.230 Due to structural similarity, H1 and H4 receptors are suggested to have cross-functional impacts on disease development. Positron emission tomography results show that reduced H1 or H4 receptor is present in a subgroup of Alzheimer’s disease, schizophrenic and depressed patients.232,233,234 The role of histamine signaling in Alzheimer’s disease remains controversial due to the conflicting results on histamine levels.232 H1 receptor upregulation is associated with myelin damage mediated by focal lymphocytes in multiple sclerosis.235 Targeting the H3 receptor with selective antagonists could stimulate the release of crucial neurotransmitters, including acetylcholine, dopamine, norepinephrine, and histamine.236 H4 receptor is involved in M1-activated microglia cells (primary inflammatory cells in the brain) driven neuroinflammation. Attenuating H4 receptor signaling is beneficial in controlling inflammation propagation in Parkinson’s disease.237
Melanin-concentrating hormone receptor
Melanin-concentrating hormone (MCH) is the pro-melanin expressed by the central nervous system.238 MCH is well documented for its function in controlling motivated behaviours, including feeding and drinking.239 Later studies suggest that MCH promotes non-REM sleep and modulates energy homeostasis.240,241 MCH receptor 1 is a stress modulator regulating fear and anxiety processes.242 MCH receptor 1-signaling is responsive to physiological- or neurochemical-controlling stress and affective states in genetically knockout models.243 MCH is associated with behavioural disorders and depressive symptoms observed in Huntington’s disease patients.244 Animals without MCH receptor expression exhibit schizophrenia-like phenotypes.245
Melatonin receptor
Melatonin (MT) or N-acetyl-5-methoxytryptamine is a neuroendocrine hormone produced by the pineal gland. MT is a regulator of the circadian rhythm (sleep-wake cycle). Melatonin is converted from tryptophan/ serotonin in the pinealocytes. Melatonin also functions as an antioxidant to protect tissues from free radical damage.246 The antioxidant activity of melatonin is essential in tissue (such as the brain) with high reactive oxygen species (ROS) resulting from oxygen consumption.247 Peripheral tissues, such as the gut and skin, could also secrete melatonin.248 Melatonin secretion is suppressed by daylight through the retino‐hypothalamic tract and reaches a peak at night. Rhythmic nocturnal secretion (secreted in the dark) allows melatonin to distributes throughout the body via circulation.
Circadian rhythm dysregulation is a common symptom presented by patients with neurodegenerative disease due to functional impairment of the retina-suprachiasmatic nucleus (SCN)-pineal axis.249 In Alzheimer’s disease, melatonin and MT1 receptor level in SCN and cortex diminishes remarkably.250,251 Pathological α-synuclein aggregation (a stepwise aggregation of presynaptic neuronal protein observed during Parkinson’s disease development) is reduced in animal models subjected to melatonin treatment.252,253 In multiple sclerosis and amyotrophic lateral sclerosis, melatonin demonstrates anti-apoptotic functions and offers neural protection from oxidative damage.254,255
Dysregulation in MT1/2 receptor signaling contributes to the pathological development of anxiety, sleep disorders (insomnia), and depression.256,257,258 In a post-mortem study on depressed patients, hypothalamic MT1 expression increased in the hypothalamic suprachiasmatic nucleus and is correlated with disease duration.259 Melatonin treatment can alleviate symptoms of psychiatric disorders with few side effects (even at high dosages).260 Exogenous melatonin may also be administered to control anxiety.261 Melatonin is an effective medication for sleep disturbances in depression.262 However, no solid empirical evidence supports melatonin or melatonin receptor agonists as the cure for depression. The use of melatonin to normalize the disrupted circadian cycle might not be sufficient to alleviate depression.
Sphingosine 1-phosphate (S1P) receptor
S1P is an active lysophospholipid. S1P exerts its biological functions through S1P receptor 1-5.263 S1P/S1P receptor controls angiogenesis, chemotaxis, and egress of lymphocytes (from bone marrow, thymus, and lymphoid tissues).263 S1P receptor-expressing immune cells in lymphoid tissues are attracted by the high S1P level in the bloodstream.264 S1P receptors on immune cells are inactivated in peripheral blood by receptor internalization.264 S1PR1 can be found in B, T, and dendritic cells.263 During inflammation, S1PR1 on immune cells is upregulated.265 S1PR1 enhances inflammation by activating neuroglia/microglia (immune cells orchestrating inflammatory response in the central nervous system).266 S1PR1 might contribute to the development of multiple sclerosis by promoting chronic and acute inflammation.263,267 Unlike S1PR1, S1PR5 is mainly detected in natural killer and dendritic cells.268 S1PR5 expression on natural killer cells is critical for its egress from lymph nodes and bone marrow.269 Hence, targeting S1P receptors might protect the brain from immune attacks by limiting lymphocytes from passing through the blood-brain barrier in multiple sclerosis.266
Opioid receptor
The opioid receptor family is composed of delta (δ)-opioid receptor (DOR), kappa (κ)-opioid receptor (KOR), mu (μ)-opioid receptor (MOR), and nociceptin receptor. Opioid receptor recognizes a variety of endogenous neuropeptides, including enkephalins, endorphins, and dynorphins.270 The endogenous opioids are one of the neuromodulators produced by the body to attenuate stressful states. Opioid receptor in the central and peripheral nervous system regulates stress and pain responses.271 locus coeruleus (LC) in the brain is the stress-integrating site. The opioid receptor can sensitize neurons in LC to corticotropin-releasing factor (CRF), a potent psychological mediator regulating stress-induced behaviors.272 Chronic or persistent acute stress can alter LC functions.273 Hyperactive LC is associated with psychiatric disorders.274 Dysregulation of the opioid receptors affects emotion processing in patients with major depressive disorders.275 Opioid receptor levels are related to neurocognitive deficits.276 Elevated opioid receptors level might elicit symptoms of schizophrenia resulting in treatment resistance.276
δ-opioid receptor and mu-opioid receptor exhibit opposite functions in the pathogenesis of Alzheimer’s disease. δ-opioid receptor agonist reduces expression of β-site APP cleaving enzyme 1 (BACE1), which cleaves amyloid precursor protein to initiate Aβ peptide production in PC12 cells (harbouring mimicked injury of Alzheimer’s disease).277,278 On the contrary, knocking down δ-opioid receptor increases BACE1 expression, leading to high production of Aβ42, the essential pathogenic Aβ peptides in Alzheimer’s disease with 42 amino acids.278 For the μ-opioid receptor, it is noted that agonist-induced receptor activation enhances BACE1 and Aβ42 expression.278 Hence, targeting δ-opioid/μ-opioid receptor signaling might benefit Alzheimer’s disease treatment; Parkinson’s disease patients have reduced brain kappa-opioid receptor levels.279 Activating κ-opioid receptor ameliorates Parkinsonian behaviours and restores locomotor in marmoset with Parkinsonism.280 In addition, κ-opioid receptor agonists can alleviate dyskinesia behaviour derived from L-DOPA in Parkinson’s disease rats.281
Serotonin receptor
Dysregulation of serotonin (5-hydroxytryptamine, 5-HT) receptors is observed in nearly all neurodegenerative and psychiatric disorders.282,283 5-HT receptors 1 and 2 are the most intensively studied drug targets. The receptors have various effects with multiple subtypes and alternative splice variants. 5-HT1 and 5-HT2 receptors have different expression patterns in the brain with similar or opposite functions.284 5-HT1 receptor has 5 subtypes: 5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E and 5-HT1F receptors. 5-HT1A receptor can be found in both serotonin neurons and non-serotonin neurons.285 5-HT1A receptor is associated with anxiety and mental traits in transgenic mice.285 Partial agonists targeting the 5-HT1A receptor are suggested to be useful in controlling alcohol abuse.286 Anterior cingulate cortex (ACC) is a brain region regulating emotion regulation, pain perception, and cognitive control.287 Patients with bipolar disorder, major depressive disorder, and schizophrenia have higher 5-HT1B receptor expression in the outer ACC layers compared to the inner ACC layers;288 5-HT2 receptor has 3 subtypes: 5-HT2A, 5-HT2B, and 5-HT2C receptors. 5-HT2 receptors are implicated in various neuropsychiatric phenotypes, including schizophrenia, attention deficit hyperactivity disorder, affective disorders, eating disorders, anxiety disorders, obsessive-compulsive disorder, suicide, and Alzheimer’s disease.289
Class C (glutamate)
Structural insights
Class C GPCRs are distinguished from other classes of GPCRs by two unique features. First, the orthosteric ligand binding pocket is located in the large extracellular venus flytrap domain (VFT). VFT is connected to the transmembrane helix via the cysteine-rich domain (CRD) (Fig. 4c). Among class C GPCRs, only the GABAB receptor lacks CRD; Second, class C GPCR forms hetero- or homo-dimers at physiological conditions.290,291,292,293,294 VFT domain forms an asymmetric dimer interface to facilitate dimer formation. Ligand engagement at either subunit is sufficient to activate the receptor.291,294,295 The surface interface between dimers is the potential binding site for the therapeutic modulator.292 The conformation rearrangement between ICL2 and ICL3, and C-terminus contributes to receptor activation.296,297,298
γ-aminobutyric acid B receptor
γ-aminobutyric acid B (GABAB) is an inhibitory neurotransmitter. GABAB receptor is a heterodimer consisting of two subunits, GABAB1 and GABAB2. GABAB1 expression is reduced in the brain of Alzheimer’s disease patients. The GABAB1 protein level is negatively associated with the neurofibrillary tangle.299 Results from a genome-wide association study (GWAS) show that GABAB1 SNPs are a risk factor for schizophrenia.300 GABAB2 SNPs are correlated with the development of Huntington’s disease.301 Activating GABAB receptor can ameliorate motor impairment and reduces inflammation/ oxidative damage in Parkinson’s disease models.302
Metabotropic glutamate receptors
The excitatory neurotransmitter glutamate mediates neuronal excitability via metabotropic glutamate receptors (mGluRs). Functional mGluR is a homodimeric receptor consisting of 8 members (mGluR1-8).303 Dysregulation of mGluR signaling pathways is observed in both neurodegenerative and psychiatric disorders.304
Group I mGluR composes of mGluR1 and mGluR5. mGluR1 localizes in the hippocampus, hypothalamus, periaqueductal gray, and amygdala, which are associated with anxiety.305 mGluR5 activity is linked to the cognitive symptoms of Alzheimer’s disease.306,307,308 Deleting mGluR5 improved spatial learning impairment and decreased Aβ oligomers in Alzheimer’s disease models.309 Interaction between mGluR5 and cellular prion protein could also play a part in the pathogenesis of Alzheimer’s disease.310,311 Activating mGluR5 promotes striatal neuron survival in Huntington’s disease models.312,313 mGluR5 knockout mice exhibit obvious schizophrenia symptoms, including reduced spatial memory and reduced sensorimotor gating.314
Group II mGluR consists of mGluR2 and mGluR3. Activating mGluR2 and mGluR3 can control panic-like behaviors and ameliorates acute stress responses in the anxiety model.315 Mutant huntingtin in Huntington’s disease is toxic to neurons.313 In the mouse model, activating mGluR2 and mGluR3 could enhance limb coordination by attenuating the generation of huntingtin aggregate.316 mGluR2 and mGluR3 demonstrate protective effects on the nigrostriatal system, which restores functional deficits in Parkinson’s disease rat model.317,318 Overexpression of mGluR2 in the neocortical layers, cerebellum, striatum, hippocampus, and thalamus/hypothalamus could build up glutamate-mediated excitotoxicity and promote Huntington’s disease progression.319,320,321,322
Group III mGluR includes mGluR4/6/7/8. mGluR4 activation ameliorates locomotion disorder in Parkinson’s disease rats.323 mGluR7/8 are associated with the anxiety-related phenotype.324,325 SNPs in mGluR7/8 are correlated to the susceptibility of schizophrenia.326,327,328,329
GPCR dimers
GPCRs can function in homodimeric or heterodimeric forms.330,331 The receptor complex consists of one GPCR dimer with two orthosteric binding sites and a heterotrimeric G protein.332 GPCR dimer exhibits different biochemical properties compared to the individual receptor. Activation of either one of the receptors is sufficient to promote dimer formation.333 Dimeric GPCR has a different ligand binding affinity as compared to the monomer.331 Receptor dimerization affects receptor trafficking in agonist-induced GPCR endocytosis.330 Closely related GPCR subtypes are more efficient in forming heteromers.334 Here, we focused on discussing two physiologically existing GPCR heterodimers (A2AR-D2R and mGluR2-5-HT2A).
Adenosine 2A receptor-dopamine D2 receptor (A2AR-D2R) heterodimer is located in the ventral striato-pallidal GABA neurons.335,336 A2AR-D2R heterodimer attracts attention in the field of Parkinson’s disease medication as ligands for A2AR can modulate dopamine signaling in Parkinson’s disease. Co-administration of dopamine precursor L-DOPA (L-3,4-dihydroxyphenylalanine) and dopamine receptor agonists could improve mobility in Parkinson’s disease.141 It has been shown that adenosine antagonists such as caffeine could enhance dopamine agonist action in Parkinson’s disease treatment.337 A2AR activation can suppress D2R-mediated Gi/o signaling.335 Stimulating A2AR with adenosine A2AR agonist in the nucleus accumbens produces behavioural effects similar to local dopamine depletion.338 Thus, the action of A2AR modulators should be considered in the drug design for Parkinson’s disease.
Serotonin type A 5-HT2A receptor and type C metabotropic glutamate 2 (mGlu2) receptor regulates psychoactive behavior in schizophrenia.339,340 5-HT2A receptor is a Gq-coupled receptor, while mGluR2 receptor signals through Gi.341 5-HT2A receptor is upregulated in the frontal cortex of schizophrenic subjects compared with normal subjects. In contrast, the expression level of mGluR2 is decreased.341 Balance between Gq and Gi is a predictive indicator of antipsychotic drug properties.342 5-HT2A receptor and mGluR2 can form stable complexes in physiological conditions which regulate Gq-Gi balance cooperatively.343 mGluR2 agonist reduces 5-HT2A receptor/Gq signaling in the frontal cortex of schizophrenic subjects.341 mGluR2 agonist can downregulate 5-HT2A receptor expression.344 On the contrary, it has been shown that the 5-HT2A receptor controls mGluR2 expression at the epigenetic level in the frontal cortex.342 Although the 5-HT2A receptor and mGluR2 regulate the activity of each other remains elusive, interrupting the functional crosstalk in the 5-HT2A receptor/ mGluR2 complex is a putative approach in schizophrenia treatment.345
Therapeutic development
The small molecules regulate GPCR activity by stabilizing receptors at unique conformational state (Fig. 5). To explore the GPCRs-based therapeutic strategies against neuropsychiatric disorders, we examined the clinically approved drugs (Fig. 5a) and compounds being tested in different stages of clinical trials (Fig. 5b) in the DrugBank database (https://go.drugbank.com/). In total, 92 drugs are being approved (Table 2). Forty-one candidates are undergoing clinical trials (Table 3). Selected receptors/drugs interaction are shown in Fig. 6.

GPCRs-targeting drugs for neurodegenerative diseases and psychiatric disorders. a Numbers of compounds approved for clinical use or under clinical trials. b Summary of the action modes of GPCR-targeted agents for treatment of neuropsychiatric diseases

Interactions between neuropsychiatric drugs with key residues in the orthosteric ligand binding pocket of GPCRs (e.g., adrenoceptors, dopamine receptors, histamine receptors, melatonin receptors, S1P1/5 receptors, and serotonin receptor). The small molecules regulate GPCR activity by stabilizing receptors at unique conformational state
Neurodegenerative diseases
Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disease. AD patients present with cognitive deficits, memory loss, and personality and behaviour changes. Currently, there is no curative treatment for AD. Reducing patients’ symptoms and delaying the disease’s progression is the primary objective of treatment. α1-adrenergic receptor, dopamine receptor, muscarinic acetylcholine receptor M3, histamine H1 receptor, and serotonin receptors are the primary therapeutic targets. Medication to control mental symptoms is another important objective, as patients manifest neuropsychiatric symptoms frequently.
Developing new drugs for AD is challenging, with high failure rates and long development periods. Several trials attempt to explore the use of GPCR agonism in AD treatment. SUVN-502 is in the Phase II trial (NCT02580305) to evaluate its safety and efficacy in moderate AD treatment.346 SUVN-502 is an orally active 5-HT6 receptor antagonist exhibiting effects by modulating cholinergic and glutamatergic neurotransmission.347 ∆9-tetrahydrocannabinol (THC) analog Nabilone (agonist targeting CB1/2 receptor) is under phase III investigation (NCT02351882) for its benefit on agitation, hyperactive behavioural symptoms of AD.348,349 Caffeine, the antagonist of adenosine receptor antagonist, could modify brain dysfunctions in various neurodegenerative diseases including AD, Parkinson’s disease, Huntington’s disease. The efficacy of caffeine on cognitive decline in AD dementia is undergoing examination in phase III clinical trial (NCT04570085).350
Guanfacine is an α2a-adrenergic agonist.351 Guanfacine could increase brain noradrenaline levels. Dual actions of Guanfacine on noradrenergic transmission and thalamocortical glutamatergic transmission have been reported.352 Guanfacine is a drug for treating children’s attention deficit/hyperactivity disorder (ADHD). The efficacy for improving cognition in AD is evaluated in the phase III trial (NCT03116126). The α1-adrenergic receptor antagonist, Prazosin, is being tested for its effectiveness on agitation in adults with AD in a phase III trial (NCT03710642). Prazosin is a drug for hypertension, benign prostatic hyperplasia, and post-traumatic stress disorder (PTSD) associated nightmares. Prazosin can cross the blood-brain barrier and act on the active α1-adrenoreceptor in the brain.
Brexpiprazole is classified as a novel class of antipsychotic with serotonin-dopamine modulating functions. It is an atypical antipsychotic that function as a partial agonist for serotonin and dopamine receptors. As a partial agonist, Brexpiprazole exerts smaller responses than the native ligands.353,354 The use of Brexpiprazole in AD agitation is now in phase III study (NCT03620981).
Parkinson’s disease
Parkinson’s disease (PD) is the second most prevalent age-related disorder. Early stage with mild symptoms did not require medication. Dopamine-like agonists, also known as dopamine-replacement therapy, are the primary treatment for symptomatic PD. As degeneration of the substantia nigra leading to striatal dopamine reduction is a leading cause of PD, re-introducing dopamine can improve motor problems dramatically and slow down PD progression.348,355
Levodopa is a dopamine precursor. It has long been used in controlling bradykinetic symptoms in PD. Levodopa can cross the blood-brain barrier and is known as a well-tolerated drug for dopamine-replacement therapy.356 However, Levodopa could lead to motor and psychiatric side effects.357 Amantadine could reduce dyskinesia (involuntary movements) in PD patients receiving Levodopa.358 Amantadine is an antiviral medicine with antiparkinsonian effects. Synergistic effects are observed when used in combination with Levodopa.359,360 Lisuride functions as a dopamine receptor agonist with 5-HT1A receptor agonist and 5-HT2B receptor antagonist for PD treatment.361 Piribedil is a dopamine agonist used with or without Levodopa in a phase III trial to treat idiopathic PD (NCT01519856).362 Bromocriptine is a dopamine D2 receptor agonist for early PD treatment. Bromocriptine works by activating post-synaptic dopamine receptors.363
Apomorphine is a morphine derivative. It functions as a D2 dopamine agonist for treating hypermobile “off” episodes of advanced PD, a stage in which PD symptoms get worse even with scheduled medication. It also prevents dyskinesia by functioning as a 5-HT1A receptor agonist.364 The A2A receptor in the basal ganglia is involved in the motor control of PD.365 At present, Istradefylline is the principal adenosine A2A receptor antagonist employed in adult PD patients presenting “off” episodes associated with Levodopa treatment.141
Pergolide is a long-acting dopamine receptor agonist approved in 1982 for treating PD. It functions on various GPCRs, including dopamine D2/3 receptor, α1/2-adrenergic receptor, and 5-HT receptors. It is used as adjunct therapy with Levodopa and carbidopa in the symptomatic treatment of PD.366 Ropinirole is a non-ergoline dopamine agonist, approved as monotherapy and as an adjunct to Levodopa in the treatment of PD.367
Benztropine is used to treat the molecular mechanism of anticholinergics PD.368 Benztropine inhibits dopamine uptake and exhibits varied binding affinities for muscarinic acetylcholine M1 and histamine H1 receptors.369 Biperiden, another anticholinergic drug launched in 1954, has an antagonistic effect on the muscarinic acetylcholine receptor.368
Pramipexole is a non-ergot-derived dopaminergic agonist for PD treatment. Pramipexole treatment enhances DA and 5-HT neurotransmission and increases tonic activation of post-synaptic D2 and 5-HT1A receptors in the forebrain.370 Apart from PD, Pramipexole can also be prescribed for psychiatric conditions such as treatment-resistant depression and bipolar disorder.371
Multiple sclerosis
Multiple sclerosis (MS) results from an immune attack by infiltrating inflammatory leukocytes in the central nervous system, causing hard, mottled pathologic changes and nerve conduction disorders.372,373 At present, medication aims to control GPCR-regulated immune cell function as one of the treatment regime for MS. In the database, 6 GPCR-related drugs are recorded. The drugs target multiple GPCRs, including adrenergic receptors, cannabinoid receptors, dopamine receptors, GABA receptors, opioid receptors, orphan GPCRs (GPR12/18/55), S1PR1/5, and chemokine receptors.
Baclofen is a derivative of the neurotransmitter γ-aminobutyric acid (GABA). Baclofen can help relax the stiff muscle (muscle spasticity) experienced by MS patients. Cannabidiol (CBD), one of the active components in cannabis, could improve mobility in MS by reducing depression, fatigue, inflammation, pain, and spasticity (stiff muscle with feelings of pain or tightness) in MS patients.374 Modafinil is a partial agonist for brain α1b-adrenoceptor. Pharmacological blockade of α1b-adrenoceptor shows benefit in controlling fatigue syndromes in MS. Modafinil exhibits clinical efficacy in psychiatric conditions, including treatment-resistant depression and attention deficit/hyperactivity disorder.375
Ozanimod, Siponimod, and Fingolimod are S1PR agonists that selectively bind to the S1PR1 and S1PR5 subtypes, inhibiting lymphocyte egress from lymph nodes.376 Ozanimod demonstrates a favourable safety profile in trials.377 Fingolimod may cause undesirable effects because of its interaction with other S1PR subtypes. Compared to Fingolimod, Siponimod has fewer off-target effects.
Ceralifimod is a selective S1PΡ1/5 agonist under investigation in phase II clinical trial NCT01226745 in patients with relapsing-remitting multiple sclerosis (a condition with relapses or exacerbations of old and new symptoms).267 Plozalizumab is another potential drug for MS treatment. It is a humanized anti-CCR2 monoclonal antibody targeting white blood cells.378 Plozalizumab may regulate inflammatory responses by targeting the CCL2‐CCR2 axis in MS.
Huntington’s disease
Huntington’s disease (HD) is a hereditary neurodegenerative disease. Symptoms include movement disorders and cognitive and psychiatric manifestations. Blocking and antagonizing dopamine are effective for HD treatment. Tetrabenazine is a reversible vesicular monoamine transporter 2 (VMAT) inhibitor that inhibits the reuptake of neurotransmitters in presynaptic neurons. VMAT helps to repackage the unbound dopamine taken up by the pre-synaptic terminal. Although it is first designed for schizophrenia treatment, clinical trials demonstrate efficacy in treating hyperkinetic movement disorders.379 Tetrabenazine also functions as a D2 post-synaptic receptor blocker at high doses and is used to treat uncontrolled muscle movement in HD.379 Haloperidol is a first-generation antipsychotic for schizophrenia and psychotic disorders.380 As a dopamine receptor antagonist, Haloperidol is used off-label for managing chorea associated with HD.381 For cognitive impairment, no effective targeted therapy is available at the present stage. Tiapride is in phase III for the treatment of HD (NCT00632645). Preclinical pharmacologic and behavioral research suggests that Tiapride is a selective blocker of dopamine D2 and D3 receptors in limbic brain regions.382
Psychiatric disorders
Schizophrenia
Schizophrenia is characterized by cognitive deficits and positive and negative symptoms with complex inheritance patterns.383 Patients may have positive, negative, cognitive, and general psychopathological disorders. According to the positive and negative syndrome scale (a psychiatric rating system), positive symptoms include delusions, hallucinations, conceptual disorganization, hallucinatory, excitement, grandiosity, suspiciousness, and hostility; Negative symptoms include blunted affect, emotional withdrawal, poor rapport, passive social withdrawal, difficulty in abstract thinking or stereotyped thinking and lack of spontaneity and flow of conversation. Schizophrenia patients could also present general cognitive disorders. Examples include anxiety, guilt feeling, tension, depression, poor attention or impulse control, and active social avoidance.384
Schizophrenia treatment is challenging because existing antipsychotics are antidopaminergic drugs that improve only positive symptoms such as agitation and aggression but have limited efficacy for negative and cognitive symptoms.385 Globally marketed antipsychotic drugs include typical antipsychotic drugs (mostly specific dopamine D2 receptor antagonists) and atypical antipsychotic drugs (such as dopamine D2 and 5-HT2A dual antagonists and D2/D3 partial agonists).
Aripiprazole, a blockbuster drug for controlling psychiatric symptoms, has high affinities for 5-HT1A, 5-HT2A, D2, and D3 receptors. It is a partial agonist of D2, D3, and 5-HT1A receptors and a 5-HT2A receptor antagonist.386 Aripiprazole is also a drug for bipolar disorders.387 Brexpiprazole, developed by Otsuka, is considered as the pharmacological successor to Aripiprazole. Brexpiprazole can also be used as an adjunct for major depressive disorder.388,389,390
Cariprazine is a D3/D2 partial agonist with moderate affinity for the 5-HT2A receptor.391 FDA approved it in 2016 for treating adult schizophrenia and bipolar disorder.
Lumateperone is an antipsychotic targeting multiple GPCRs. It is a post-synaptic dopamine D2 receptor antagonist, a presynaptic dopamine D2 receptor partial agonist, and a 5-HT2A receptor antagonist.392 Lumateperone can be used for positive & negative symptoms and cognitive dysfunction in schizophrenia.393 It can also be used in bipolar disorder treatment.393
Chlorpromazine blocks dopamine receptors, α-adrenergic receptors, and 5-HT receptors. It can quickly control the state of agitation and gradually eliminate hallucinations and delusions. Thus, it can apply as medication to control combativeness and aggressive behaviour in children.394
Risperidone can be used for various mental disorders, including schizophrenia and mood disorders. Risperidone has high affinities for 5-HT receptors and dopamine receptors and mildly inhibits α1-adrenergic receptors and histamine receptors.395
Olanzapine is developed based on clozapine with structural modification. It was approved to be marketed by FDA in 1996. Olanzapine not only inhibits dopamine receptors but also binds to serotonin receptors, and its affinity with serotonin receptors is far greater than its affinity with dopamine receptors.
Haloperidol is a widely used antipsychotic for positive symptoms of schizophrenia, Tourette syndrome, and behavioural disorders/hyperactivity in children.396 Haloperidol can block dopamine, α-adrenergic, and serotonin receptors. It is highly selective for dopamine receptors.
Spiperone is a potent dopamine D2 receptor antagonist bearing the butyrophenone scaffold. Although it displayed efficacy in treating drug-resistant schizophrenia, it is not yet approved by the FDA.397 Zotepine is an atypical antipsychotic drug for treating schizophrenia in Japan. It is a potent dopamine D1/D2 receptor and 5-HT2A receptor antagonist.398
Medication for schizophrenia is an active research area. Schizophrenia drugs generally target multiple GPCRs. For instance, Brilaroxazine, an investigational antipsychotic drug developed by Reviva, could stabilize the dopamine-serotonin system by partially activating D2, D3, D4, 5-HT1A, and 5-HT2A receptors. In addition, it antagonizes 5-HT6 and 5-HT7 receptors.399 A phase III clinical trial of Brilaroxazine for the safety and efficacy of the treatment of schizophrenia is now under recruitment (NCT05184335).
Zicronapine is a tetracyclic azepine developed by Lundbeck with affinities for 5-HT2A/2 C and D1/2 receptors.400 Phase III study of Zicronapine has been completed (NCT01295372).
Eltoprazine is a piperazine derivative that partially activates the 5-HT1A/2B receptor.401 It is tested in a phase II trial to investigate the treatment of schizophrenia and cognitive impairment (NCT01266174).
LuAF35700 is an antagonist targeting dopamine receptors, serotonin receptors, and α-adrenergic receptors.399 The efficacy and safety of the LuAF35700 have been examined in phase III randomized, double-blind trial (NCT02717195).
Roluperidone is a novel 5-HT2A and σ2 receptor antagonist developed by Minerva Neurosciences.402 Phase III studies have shown that Roperidone may treat negative symptoms in schizophrenia patients without causing post-synaptic dopaminergic blockade due to low or no affinity for dopamine and histamine receptors (NCT03397134).
Depression
The underlying mechanism of depression is not clear. According to the record in the DrugBank database, a total of 31 antidepressants target GPCRs. Examples include tricyclic antidepressants, bioamine neurotransmitters (serotonin, norepinephrine, and dopamine) reuptake blockers, and 5-HT2A receptor inhibitors.
Imipramine and Desipramine are examples of tricyclic drugs for major depressive disorders, anxiety, and ADHD.403 They have high affinities to 5-HT2C and 5-HT2A receptor subtypes. The pharmacological properties of Amitriptyline are similar to Imipramine. Amitriptyline can inhibit 5-HT reuptake with sedative, hypnotic and anticholinergic effects. A combination of Amitriptyline and Imipramine could block serotonin reuptake in the brain’s limbic (emotional) regions.
Currently, monoaminergic alterations involving serotonin receptors are a significant cause of depression.404 Selective or non-selective 5-HT reuptake inhibitors are the first-line treatment for depression. Representative drugs include Fluoxetine, Paroxetine, and Citalopram.405,406,407,408,409 Fluoxetine, a weak antagonist of 5-HT2C and 5-HT2A receptors, was approved for marketing in 1988 to treat major depressive disorder. Later, Paroxetine was approved in 1992. It is a highly selective reuptake inhibitor of 5-HT in neurons. Citalopram has a similar function in depression treatment. It is also a serotonin reuptake inhibitor. Nefazodone and Trazodone improve mood by antagonizing 5-HT2A/C receptors. They showed affinity to the 5-HT1A receptor.410,411 Pindolol can accelerate the effects of selective serotonin reuptake by antagonizing 5-HT1A and β-adrenergic receptors.405,412 Meanwhile, Mirtazapine and Mianserin have antagonistic properties on 5-HT2A/2C receptors. They exhibit inhibitory effects on presynaptic A2-adrenergic receptors. Both drugs improve sleep duration.408,413,414 Vortioxetine is a multi-mode antidepressant for major depressive disorder treatment in adults. Vortioxetine inhibits serotonin reuptake. It exerts different effects on different members of the 5-HT receptor. On one hand, Vortioxetine is an antagonist for 5-HT1D, 5-HT3, and 5-HT7 receptors. On the other hand, it is a partial agonist for the 5-HT1B receptor.415,416,417 Bupropion and its primary metabolite hydroxybupropion function by blocking 5-HT3A receptor.418 Agomelatine is an atypical antidepressant acting as a melatonin receptor (MT1/2) agonist and a 5-HT2C/2B receptor antagonist.419
Inhibitors of dopamine (DA) transporters are another class of antidepressants. Nortriptyline can bind directly to the DA transporter to inhibit dopamine uptake. It can be used in treatment-resistant depression.420,421,422 Brexpiprazole is a partial agonist on the 5-HT1A receptor and D2 receptor. Brexpiprazole can also be used in adult patients with schizophrenia.
Ansofaxine is a reuptake inhibitor for 5-HT, norepinephrine, and dopamine which is under clinical development for major depressive disorder (NCT04853407).423 5-methoxy-N, N-dimethyltryptamine (5-MEO-DMT) is a non-selective serotonin receptors agonist for depression (NCT04698603).
Anxiety disorders
Anxiety disorders are the most common psychiatric disorders. Anxiety is accompanied by other psychiatric disorders, including major depressive disorders, substance use disorders, and personality disorders.424
Partial agonists of the 5-HT1A receptor and selective 5-HT reuptake inhibitors are frequently used in anxiety treatment.425,426 Buspirone, the partial agonist for the 5-HT1A receptor, is approved for treating anxiety due to neurosis.427 Paroxetine428 and Escitalopram, the 5-HT reuptake inhibitors, can relieve anxiety symptoms and prevent recurrence in patients.409 Trazodone is used to treat anxiety disorders with depressive symptoms and is suitable for patients with significant psychomotor agitation, anxiety, and insomnia.429
Hydroxyzine is the most studied antihistamine for anxiety and the only FDA-approved antihistamine for treating anxiety. It is commonly used for anxiety, panic attacks, and insomnia in inpatients and outpatients.429,430
Drug targeting β-adrenoreceptor in the central nervous system can also relieve anxiety.431 Propranolol, the selective β1/2-adrenoceptor antagonist (β-blockers), is the first-line pharmacological treatment for anxiety disorders.432,433 Doxepin can be used for depression and anxiety. It is an antagonist of the histamine H1 and H2 receptors, 5-HT2A/2C receptors, and the muscarinic acetylcholine receptors (M1–M5).434
Naluzotan, the selective 5-HT1A receptor agonist, has been investigated for anxiety disorders and depression treatment (NCT00248183).435 Ansofaxine, a reuptake inhibitor of serotonin, norepinephrine, and dopamine, is a new-generation drug for anxiety management. The drug has completed phase III clinical trials in China to treat anxiety and depression (NCT04853407).
Bipolar disorder
Bipolar disorder (BD) is characterized by periodic mood disorders. Medication is the primary treatment to improve the psychosocial function and quality of life of patients with BD. Pharmacological management of acute depressive/manic episodes and prevention of recurrence is also essential. Atypical antipsychotics for bipolar disorder exhibit high affinities for multiple serotonergic receptors, including 5-HT1A, 5-HT2A-C, 5-HT6, and 5-HT7 receptors.
Quetiapine was approved by the FDA in 1997 for the symptomatic treatment of schizophrenia and is used as a first-line treatment to control depressive episodes of BD. It exerts therapeutic effects may by antagonizing 5-HT1A, 5-HT2A, D1, D2, and H1 receptors as well as α1/2- adrenergic receptors.436,437 Dexmedetomidine is an α2-adrenergic receptor agonist that can be used for the acute treatment of agitation associated with schizophrenia or bipolar I or II disorders.438 Risperidone, an atypical antipsychotic drug, is now used as maintenance therapy for patients with bipolar I disorder.439
Tianeptine is a novel antidepressant that stimulates serotonin, increases levels of 5-hydroxyindoleacetic acid in brain tissue and plasma, and decreases serotonin-induced behavior.440,441 Clinical trials are underway for the adjuvant treatment for BD with Tientidine (NCT00879372). Lumateperone, an antagonist with high binding affinity to the 5-HT2A receptor and moderate affinity to the post-synaptic D2 receptor, is being evaluated for treating BD, depression, and other neuropsychiatric and neurological disorders (NCT03249376, NCT02600507).
Tourette’s syndrome
Tourette’s syndrome (TS) is a neurodevelopmental disorder characterized by repetitive behaviours, including motor/phonic tics. TS is commonly coupled with obsessive-compulsive disorder (OCD) and ADHD.442 The underlying mechanism of TS remains poorly clarified.443,444,445 Abnormalities in synaptic neurotransmission involved in the cortico-striatal-thalamocortical circuitry are implicated in TS pathogenesis.446,447 Dopaminergic signaling in cortico-striatal-thalamocortical pathways might be associated with TS progression.444,448,449 α-adrenergic agonists are the first choice in TS treatment.450 Examples include Clonidine and Guanfacine.438,451 Aripiprazole is a partial agonist of dopamine D2 and 5-HT1A receptors. It can stabilize dopamine receptor and improves TS symptoms.452 In contrast, Pimozide exerts a therapeutic effect by inhibiting the dopamine D2 receptor in the central nervous system.453
Attention deficit hyperactivity disorder
Attention deficit hyperactivity disorder (ADHD) is a common psychiatric disorder affecting school-age children. It is a neurodevelopmental disorder with multifactorial etiological risk factors. ADHD is characterized by hyperactivity, impulsivity, and age-inappropriate symptoms of inattention.454 Irregularities in catecholamines circuits in the prefrontal cortex, such as dopamine and norepinephrine, are a leading cause of ADHD.455,456 Most ADHD drugs are designed to enhance catecholamine transmission in the prefrontal cortex.457
Methylphenidate can significantly reduce hyperactive behavior, increase attention concentration ability, and effectively improve the core symptoms of ADHD, so it is one of the most widely used first-line drugs approved by the FDA. Methylphenidate blocks dopamine D1 and D2 transporters, resulting in increased levels of synaptic dopamine, and also shows activity against serotonergic 5-HT1A receptors.351,458,459
Second-line drugs for ADHD include Atomoxetine, Guanfacine, and Clonidine.351,438,460 Atoroxetine is a non-stimulant medication that acts as a selective norepinephrine reuptake inhibitor in ADHD.440,461 Guanfacine is a phenylacetyl guanidine derivative, which is more selective than Clonidine in activating the α2-adrenergic receptor.351 Venlafaxine is a new type of selective serotonin and dopamine reuptake inhibitor. It is a dual-channel antidepressant. Venlafaxine inhibits the reuptake of serotonin by neuron endings at low doses and inhibits the reuptake function of neuron endings at a high dose to enhance attention. Amfetamine (AMF) acts on the cerebral cortex and reticular activation system. AMF stimulates adrenalin receptors and enhances neurotransmitter secretion, such as 5-HT and dopamine.462 Fluoxetine is a potent and selective serotonin reuptake inhibitor for ADHD treatment.463,464
Edivoxetine is an adrenergic absorption inhibitor. It is now in phase III development for ADHD with hyperactivity (NCT00922636, NCT00965419). Centanafadine is a triple-reuptake inhibitor for dopamine, norepinephrine, and serotonin reuptake. It is currently in phase III clinical trials (NCT03605849, NCT03605680, NCT03605836). SGS-742 has been investigated for ADHD treatment. It acts as a GABA-B receptor antagonist and could enhance the release of glutamate, aspartate, glycine, and somatostatin.
Example of emerging GPCR targets
Most of the GPCRs targeted by approved drugs for neuropsychiatric diseases belong to class A and C GPCRs. With the advance of biotechnology and increase in understanding of GPCR functions, new candidates are discovered in other GPCR families, including class A (orphan), class B1 (secretin), class B2 (adhesion), class C (calcium-sensing receptor), and class F.
Class A (orphan GPCR)
Orphan GPCRs are receptors whose cognate ligands are not discovered or validated in cellular/ animal models. Deorphanization with reverse pharmacology is currently an active area in GPCR research.
GPR17
GPR17 is activated by two different endogenous ligands: uracil nucleotides and cysteinyl-leukotrienes.465 Uracil nucleotides trigger astrocytic migration by upregulating membrane integrins.466 Cysteinyl-leukotrienes are lipid mediators secreted by inflammatory cells and nervous tissues.467 Cysteinyl‐leukotrienes can stimulate astrocyte proliferation via autocrine signaling.468 GPR17 is a sensor of local damage to the myelin sheath. GPR17 downregulation promotes the development of mature oligodendrocytes from myelin-producing oligodendrocyte precursors.469 GPR17 is involved in reconstructing and repairing demyelinating plaques formed by ongoing inflammatory processes.470 In a mouse model of multiple sclerosis, targeting GPR17 can delay the onset of autoimmune encephalomyelitis.471
GPR26
GPR26 is a brain-specific GPCR. GPR26 has high sequence homology with purinergic P2Y receptor and serotonin 5-HT5A receptor.472,473 GPR26 regulates emotion in animal models. GPR26 knockout mice exhibits anxiety- and depressive-like behaviors.474 Colocalization of GPR26 and neuronal nuclear inclusions is observed in brain tissues suggesting a potential link between GPR26 and neurodegenerative diseases.473
GPR37 and GPR37L1
GPR37 can be found in pre-myelinating/myelinating oligodendrocytes, dopaminergic neurons, and hippocampal neurons.475 GPR37 shares high sequence homology with peptide-activated GPCRs such as endothelin receptor B (ETB).475 In Parkinson’s disease, GPR37 acts as an adenosine A2A receptor inhibitor via receptor oligomerization;476 GPR37L1, in contrast, is found mainly in astrocytes and oligodendrocyte progenitor cells.475 GPR37L1 is involved in the adaptive myelination of oligodendrocytes which is critical for neural plasticity, learning, and memory in adults.477
GPR39
Zinc regulates behavior, cognition, and ability to learn.478 Dysregulation in zinc homeostasis is associated with progressive dementia and cognitive impairment. Zinc deficiency gives rise to various neuropsychiatric disorders, including epilepsy, seizures, and depression.479,480 Extracellular zinc can activate zinc-sensing receptor GPR39.481,482 Zinc stimulates GPR39-mediated signal transduction and induces calcium mobilization in HEK293 cells.483 Zinc-activated GPR39 increases expression of K+/Cl− cotransporter 2 (KCC2), the Cl- outward transporter in neurons.484 Further, GPR39 increases Na+/H+ exchanger activity in hippocampal neurons in a pH-dependent process.485
GPR40
GPR40 (also known as free fatty acid receptor 1) is the receptor for medium and long-chain unsaturated fatty acids. GPR40 activates the NOD-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome pathway by blocking the formation of apoptosis-associated speck-like protein containing a CARD (an inflammasome component).486 GPR40 promotes hypothalamic neurogenesis by enhancing cell proliferation and survival.487 GPR40 may associate with the development of epilepsy by altering N-methyl-d-aspartate receptor-mediated synaptic transmission.488 In Alzheimer’s disease model, activating the GPR40 receptor can reduce β-amyloid production and rescue cognitive deficits.489,490
GPR50
GPR50 exhibits high sequence homology with melatonin MT1/2 receptors. However, melatonin (the endogenous ligand for MT1/2 receptors) cannot bind to GPR50 directly.491 GPR50 can be detected in the pituitary, hypothalamus, and hippocampus intermedia.491,492 GPR50 enhances neuronal differentiation via notch and WNT/β-catenin.493 GPR50 might be involved in psychiatric illness by interacting with neurite outgrowth inhibitor NOGO-A.494 GPR50 is an X-linked gene (Xq28). It is suggested to be a sex-specific risk factor in bipolar affective disorder, major depressive disorder, and schizophrenia.495 GPR50 can antagonize the MT1 receptor by forming a heterodimer.496 The inhibitory effects are mediated via the large C-terminal tail, which blocks the β-arrestin recruitment and G protein coupling.495 MT2 receptor could also form a heterodimer with GPR50, but the functional consequence remains to be defined.496
GPR52
GPR52, a striatal-enriched orphan GPCR. GPR52 stabilizes HTT by cAMP-dependent but PKA-independent mechanisms.497 GPR52 antagonist can ameliorate Huntington disease-like phenotypes by diminishing mHTT protein levels.498 GPR52 is a potential target of antipsychotic drugs.499 GPR52 is associated with cognitive function, emotion, and psychosis-related/antipsychotic-like behaviors.204,499,500 GPR52 has high sequence homology with histamine H2 receptor and 5-HT4 receptor.204 GPR52 agonist treatment suppresses methamphetamine-induced hyperactivity suggesting that GPR52 might be involved in neurochemical sensitization.501 Recent study reveals that GPR52 is a self-activating receptor.502 The extracellular loop 2 is immersed deeply into the typical ligand binding pocket of GPR52, which maintains the constitutive active state at physiological conditions.503
Super-conserved receptors expressed in the brain
GPR27, GPR85, and GPR173 are super-conserved receptors expressed in the brain (SREB). GRR27 deletion is associated with speech delay, contractures, hypertonia, and blepharophimosis.504 GPR85 may function as a negative regulator in hippocampal adult neurogenesis and alters cognitive functions, including learning and memory.505 It has been reported that GPR85 is a risk factor for schizophrenia.505 GPR173 may function by interacting with phoenixin (a recently discovered peptide controlling reproductive hormone secretion, visceral pain, and pruritus) in hypothalamic neurons, which regulates memory and anxiety.506,507 In neuronal M17 cells, phoenixin promotes neuronal mitochondrial activity and biogenesis by activating the CREB pathway.508 Further, binding of gonadotropin-releasing hormone 1–5 (GnRH 1–5) to GPR173 could inhibit neuronal migration.509
GPR88
GPR88 expresses exclusively in the neuron of the rat brain throughout the striatum.510 In GABAergic medium spiny neurons (MSNs), GPR88 contributes to tonic GABAergic inhibition and responses to GABA release.511 GPR88 might play a part in prepulse inhibition of startle, apomorphine-induced climbing, and amphetamine-stimulated locomotor activity.512 Co-expression of GPR88 and D1 dopamine receptors is found in the brain.513 In Parkinson’s disease (unilateral 6-hydroxydopamine-lesioned rats), GPR88 expression is associated with L-DOPA-mediated behavioural changes.510 Antidepressant treatments can modulate GPR88 expression in rat brains.514 Morphine can regulate GPR88 expression in the amygdala via the mu-opioid receptor.515 GPR88 is genetically associated with various neuropsychiatric disorders, including schizophrenia, bipolar disorder, speech delay, and chorea.516,517
Class B1 (secretin)
Structural highlights
Class B1 GPCRs have a conserved extracellular N-terminal domain (ECD) with a three-layered α-β-β/α fold structure (100 to 160 residues) responsible for the binding of peptide hormones (Fig. 7).518,519,520 Peptide ligands stabilize receptors by interacting with both ECD and transmembrane core.521 N-terminus of the peptide interacts with the orthosteric pocket within the transmembrane domain.522,523 Class B1 GPCRs recognize peptide ligands with different C-terminus, ranging from disordered secondary structures to continuous α-helix.524,525 Like class A GPCRs, the cavity formed by the receptor cytoplasmic part allows anchoring of the α5 helix of G proteins.526,527 Among class B1 GPCRs, calcitonin and calcitonin gene-related peptide receptors, corticotropin-releasing factor receptors, and the glucagon receptor family are frequently reported to be involved in neurodegenerative diseases and psychiatric disorders.

Structural features of class B1 GPCR. Binding of peptide ligand activates calcitonin receptor-like receptor (PDB 6UVA)
Receptors for calcitonin and calcitonin gene-related peptides
Calcitonin (CT) and calcitonin gene-related peptides (CGRPs) are ligands of the CT receptor. CGRPs also exert their biological functions through CL (calcitonin receptor-like) receptors.528
The activity of CT and CL receptors is modulated by receptor activity-modifying protein (RAMP1-3).529 CT receptor-RAMP complexes can also interact with amylin. Therefore they are also known as amylin receptors (AMY1-3).529 CT receptors are implicated in neuroinflammation in Alzheimer’s disease.530 Antagonists targeting amylin receptors might be beneficial for Alzheimer’s disease treatment.531
Corticotropin-releasing factor receptor
Corticotropin-releasing hormone (CRF) regulates the neuroendocrine stress response.532 CRH exerts its biological function through two receptors: CRFR1 and CRFR2. Human corticotropin-releasing factor receptor 1 (CRFR1) exhibits widespread distribution in the central nervous system. In contrast, human CRFR2 is predominately expressed in peripheral tissues.532 CRFR1 signaling shows sex divergence in Alzheimer’s disease.533 CRFR1 antagonist treatment delays Alzheimer’s disease symptoms, including cognitive impairment and accumulation of Aβ amyloid plaques, by regulating oxidative stress in transgenic mice.534 CRF/CRFR1 signaling plays a crucial role in stress-induced behaviour.532 It has been shown that noise exposure can increase CRF/CRFR1 expression in the hippocampus.535 CRFR1 could sensitize 5-HT2 receptor signaling to modulate anxiety behavior.536 In addition, CRFR1 antagonist modulates gamma-aminobutyric acid (GABA)-ergic activity in the brain and controls fear response in rat anxiety models.537 Single-nucleotide polymorphisms of CRFR1/2 are positively associated with major depressive disorder.538,539,540
Glucagon receptor family
The glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1/2 (GLP-1/2) are gut peptide hormones.541 The hormones can pass through the blood-brain barrier.542 GIP and GIP receptors are expressed throughout the central nervous system.543,544 Protease-resistant analog of GIP is designed to treat type 2 diabetes mellitus by controlling weight and improving glycaemic control.545,546 Clinical trials indicate that GIP and GLP-1 analogs exhibit therapeutic effects for neurodegenerative diseases.547 GLP-1 enhances the supportive function of astrocytes to neurons.548 Activated GLP-2 receptor protects hippocampal cells from glutamate-induced cell death and increases the growth of astrocytes.549 GLP-1 mimetic reduces oxidative stress and inflammation and promotes neuron formation.550,551 GIP can alleviate amyloid beta-induced toxicity in Alzheimer’s disease and relieve symptoms of Parkinson’s disease.541,542
Class B2 (adhesion)
Structural highlights
Class B2 GPCR, also known as adhesion GPCR, has a large extracellular domain (ECD). ECD is responsible for the adhesive function exhibiting high structural diversity (Fig. 8a, b).552 Adhesion GPCRs are essential for the early development of the nervous system and the brain.553 The receptor allows neural cells to communicate with the surrounding environment and migrate to destinate sites to carry out specific functions.554 In mouse Purkinje neurons, adhesion GPCR is required to generate intricate dendritic structures for synaptic connections.554 Adhesion GPCRs are further classified into ADGRL, ADGRE, ADGRA, ADGRC, ADGRD, ADGRF, ADGRB, ADGRG, and ADGRV subfamilies.555

Structural features of class B2 adhesion GPCR ADGRL3 (PDB 7SF7). a Schematic representation of ADGRL3 showing characteristic large N-termini. Source: https://gpcrdb.org/protein/agrl3_human/. b GPCR of adhesion GPCR coupled to Gαs protein after activation by tethered agonist. c Tethered agonist (TA) indicated in spheres. TA occupies the orthosteric pocket of ADGRL3 which as self-agonist for receptor activation
Nearly all class B2 orthologs have the GPCR autoproteolysis inducing domain (GAIN). The GAIN domain is located at the juxtamembrane region.556 GAIN domain is crucial for the maturation and function of adhesion GPCR. GAIN possesses intrinsic autoproteolytic activity and cleaves at the integral cysteine-rich GPCR proteolysis site (GPS).556 Autoproteolysis give rise to two noncovalently associated fragments: N-terminal fragment (NTF) with most of the extracellular domain; and C-terminal fragment (CTF) consisting of a small proportion of the GAIN domain and most of the entire transmembrane domain (Fig. 8a).554,557,558
The activation mechanism of adhesion GPCR is the least understood among different GPCR classes. Most adhesion GPCRs are orphan GPCRs as their natural ligands remain poorly defined.552 Receptor activation may follow the tethered-peptide-agonist models.558 The stalk region bends approximately 180º downward into the core of the 7TM domain, which functions as tethered agonist to initiate G protein signaling (Fig. 8c).559,560 Cleavage-independent mechanisms may exist for receptor activation.560 Ligand binding at the GAIN domain might induce conformational changes, which initiate transient G protein signaling.561 Upon activation, the intracellular milieu is in the open conformation facilitating G protein coupling. Adhesion GPCRs could employ non–G protein such as PDZ/SH3 domain-proteins and arrestins for signal transduction.562
Examples of class B2 GPCR
Adhesion G protein-coupled receptor B1 (ADGRB1 or brain-specific angiogenesis inhibitor 1, BAI1) regulates synaptic plasticity in learning and memory processes in the hippocampus.563 ADGRB1 is a post-synaptic receptor controlling excitatory synapse development.564,565 Forced ADGRB1 attenuates toxin-induced neuronal cell death.566 ADGRB1 is associated with dopaminergic neuronal loss in Parkinson’s disease.566
Adhesion G protein-coupled receptor B3 (ADGRB3) is enriched in post-synaptic density and cerebellar Purkinje cells.563,567,568 ADGRB3 modulates synaptic connection in the cerebellum.568 SNPs and gene amplification in ADGRB3 are associated with familial schizophrenia.569 Other psychiatric conditions, such as bipolar disorder, are suggested to be linked with ADGRB3.570
Adhesion G protein-coupled receptor L3 (ADGRL3) is genetically associated with attention deficit/hyperactivity disorder (ADHD) in adults.571 Knockout mice models show enhanced locomotive activity, improved levels of impulsivity, and working memory deficits.572 Maternal smoking during pregnancy is an environmental risk factor for ADHD.573 In fibroblast cells, nicotine exposure could stimulate ADGRL3 expression.571 The downstream ADGRL3 signaling events leading to ADHD remains poorly defined.574 ADGRL3 might alter monoaminergic signaling by modulating the expression of dopamine and serotonin transporters.575
Class C (glutamate)
Calcium-sensing receptor
Calcium-sensing (CaS) receptor participates in the regulation of Ca2+ homeostasis. In Alzheimer’s disease model, elevated expression of CaS receptor is observed in the hippocampal CA1 area and dentate gyrus, which is in accord with the β-amyloid plaques increase.576 CaS receptor impeding amyloid-β42 oligomers (Aβ42-os) proteolysis via direct interaction, leading to Aβ42-os aggregation and oversecretion.577 CaS receptor inhibitor sustains mental competence by promoting Aβ42 proteolysis.577 Inhibiting the CaS receptor improves memory and cognitive defects caused by β-amyloid in mice.578 CaS receptor might induce cognitive defects via eliciting cytosolic phospholipase A2 and prostaglandin E2 signaling pathway.578
Class F
Structural highlights of Class F GPCR
Class F GPCR contains a large extracellular and cysteine-rich (CRD) domain (Fig. 9).579 CRD is essential for the stability and activity of class F GPCRs.580 FZD gene family is highly conserved in mammals with conserved structural features. FZD is a receptor for the WNT family of lipoglycoprotein, which mediates signal transduction via canonical WNT-β-catenin pathway and β-catenin-independent noncanonical pathways. The secretory WNT binds to the cysteine-rich domain at the extracellular side. The Lys-Thr-X-X-X-Trp (KTXXXW) motif located at the C-terminal is essential for activating the canonical WNT/ β-catenin pathway.581,582 WNT signaling regulates neuronal polarization and axon specification polarity by activating atypical protein kinase C in rat hippocampal neurons.583 Further, WNT signaling governs collateral or terminal branching of the axon, dendrite outgrowth and guidance, dendritic spine formation, synapse formation/plasticity, and elimination.584 WNT/FZD signaling alterations are observed in several neurological disorders, including Alzheimer’s disease and Huntington’s disease.585,586 The transmembrane region is compact and hydrophilic.580,587 Similar to class A GPCR, outward bending of TM6 and an inward shift of TM5 at the cytoplasmic side is observed in the active class F GPCR.580

Structural features of class F GPCR. Smoothened homolog SMO (PDB 5L7D) with large extracellular and cysteine-rich (CRD) domain. Solvent-accessible surface. Hydrophobic surface (red); hydrophilic surface (white)
Class F receptors frizzled (FZD1-10) and smoothened (SMO) are closely associated with embryonic development and tissue homeostasis.588 Reported FZD ligands include frizzled-related proteins (SFRPs) and R-spondin.589,590 FZD1 is found in dopamine-synthesizing neurons, which form an astrocyte-DA autoprotective loop via WNT1/FZD1/β-catenin signaling.591 FZD1 enhances myelin preservation and neuronal survival;592 FZD3 is genetically related to substance-induced psychosis and schizophrenia;593,594 Neuronal degeneration observed in amyotrophic lateral sclerosis is regulated by WNT5a/FZD4 signaling.595 WNT5a/FZD5 activity is associated with neuronal inflammatory signaling;596 Genetic FZD6 variants are associated with neural tube defects in the central nervous system;597 FZD9 deletion is noted in patients with Williams-Beuren syndrome, a rare genetic disorder with mild to moderate intellectual disability or learning difficulties598 FZD10 may play a role in brain vascular development;599 SMO is the receptor for hedgehog proteins involved in neuronal/ glial proliferation and tissue regeneration.600
Concluding remarks
GPCRs are cooperatively involved in the manifestations of neuropsychiatric disorders. Elucidating the intrinsic signaling preference of G proteins or arrestins helps to improve drug efficacy and side-effect profiles. GPCR can work in the dimeric form in disease development. Characterizing the allosteric interactions and the functional consequences of GPCR dimers might provide insights into the pathogenesis of neuropsychiatric disorders. Apart from acting directly in the nervous system, GPCRs might contribute to disease development via the immune system.220
Target identification is challenging as the clinical presentations are resulted from heterogeneous biological, genetic, and environmental factors. Nevertheless, the increasing understanding of GPCR functions opens a new possibility in drug discovery. Most of the drugs targeting GPCR lack subtype-selectivity.601 Local drug administration may require to avoid debilitating side effects.602 The development of psychiatric medications remains slow as the pharmaceutical industry pays more attention to antidepressants and antipsychotic drug development.603 Therefore, developing specific therapeutic modulators which could recognize subtypes with high specificity is crucial for effective drug development.602
Benefiting from the advances in crystallography and cryo-electron microscopy technology, the resolved GPCR structures increase our understanding of GPCR functions in pathological conditions. Detailed protein structures could reveal crucial ligand binding features in physiological conditions.215,547,604 Detailed receptor/ligand profile could facilitate lead compound identification and drug optimization. Hence, harnessing our knowledge of molecular mechanisms and structural information of GPCR will be advantageous for developing effective treatments against neuropsychiatric disorders.
References
Yang, D. et al. G protein-coupled receptors: structure- and function-based drug discovery. Signal Transduct. Target. Ther. 6, 7 (2021).
Shoichet, B. K. & Kobilka, B. K. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 33, 268–272 (2012).
Denis, C. et al. Probing heterotrimeric G protein activation: applications to biased ligands. Curr. Pharm. Des. 18, 128–144 (2012).
Heemels, M. T. Neurodegenerative diseases. Nature 539, 179 (2016).
Kumar, D., Md Ashraf, G., Bilgrami, A. L. & Imtaiyaz Hassan, M. Emerging therapeutic developments in neurodegenerative diseases: a clinical investigation. Drug Discov. Today. 27, 103305 (2022).
Armstrong, M. J. & Okun, M. S. Diagnosis and treatment of Parkinson disease: a review. J. Am. Med. Assoc. 323, 548–560 (2020).
Duraes, F., Pinto, M. & Sousa, E. Old drugs as new treatments for neurodegenerative diseases. Pharmaceuticals 11, 44 (2018).
Organization, W. H. The top 10 causes of death. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (2020).
Breijyeh, Z. & Karaman, R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules 25, 5789 (2020).
Roe, J. M. et al. Asymmetric thinning of the cerebral cortex across the adult lifespan is accelerated in Alzheimer’s disease. Nat. Commun. 12, 721 (2021).
McColgan, P. & Tabrizi, S. J. Huntington’s disease: a clinical review. Eur. J. Neurol. 25, 24–34 (2018).
Brodie, C., Blumberg, P. M. & Jacobson, K. A. Activation of the A2A adenosine receptor inhibits nitric oxide production in glial cells. FEBS Lett. 429, 139–142 (1998).
Brown, R. C., Lockwood, A. H. & Sonawane, B. R. Neurodegenerative diseases: an overview of environmental risk factors. Environ. Health Perspect. 113, 1250–1256 (2005).
Dobson, R. & Giovannoni, G. Multiple sclerosis—a review. Eur. J. Neurol. 26, 27–40 (2019).
Lyketsos, C. G. Lessons from neuropsychiatry. J. Neuropsychiatry Clin. Neurosci. 18, 445–449 (2006).
Chaudhury, P. K., Deka, K. & Chetia, D. Disability associated with mental disorders. Indian J. Psychiatry 48, 95–101 (2006).
Garcia-Gutierrez, M. S. et al. Biomarkers in psychiatry: concept, definition, types and relevance to the clinical reality. Front. Psychiatry 11, 432 (2020).
Ilyas, A., Chesney, E. & Patel, R. Improving life expectancy in people with serious mental illness: should we place more emphasis on primary prevention? Br. J. Psychiatry 211, 194–197 (2017).
Bradvik, L. Suicide risk and mental disorders. Int. J. Environ. Res. Public Health. 15, 2028 (2018).
Xie, Q., Liu, X. B., Xu, Y. M. & Zhong, B. L. Understanding the psychiatric symptoms of COVID-19: a meta-analysis of studies assessing psychiatric symptoms in Chinese patients with and survivors of COVID-19 and SARS by using the Symptom Checklist-90-Revised. Transl. Psychiatry 11, 290 (2021).
Hauser, A. S. et al. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 (2017).
Sriram, K. & Insel, P. A. G Protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol. 93, 251–258 (2018).
Greengard, P. The neurobiology of slow synaptic transmission. Science 294, 1024–1030 (2001).
Wacker, D., Stevens, R. C. & Roth, B. L. How ligands illuminate GPCR molecular pharmacology. Cell. 170, 414–427 (2017).
Wheatley, M. et al. Lifting the lid on GPCRs: the role of extracellular loops. Br. J. Pharmacol. 165, 1688–1703 (2012).
Kim, H. R. et al. Structural mechanism underlying primary and secondary coupling between GPCRs and the Gi/o family. Nat. Commun. 11, 3160 (2020).
Smith, S. O. Deconstructing the transmembrane core of class A G protein-coupled receptors. Trends Biochem. Sci. 46, 1017–1029 (2021).
Miyagi, H. et al. The discovery of a new antibody for BRIL-fused GPCR structure determination. Sci. Rep. 10, 11669 (2020).
Mahoney, J. P. & Sunahara, R. K. Mechanistic insights into GPCR-G protein interactions. Curr. Opin. Struct. Biol. 41, 247–254 (2016).
Traut, T. W. Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem. 140, 1–22 (1994).
Sprang, S. R. Activation of G proteins by GTP and the mechanism of Galpha-catalyzed GTP hydrolysis. Biopolymers. 105, 449–462 (2016).
Forn, J., Krueger, B. K. & Greengard, P. Adenosine 3’,5’-monophosphate content in rat caudate nucleus: demonstration of dopaminergic and adrenergic receptors. Science 186, 1118–1120 (1974).
Desale, S. E., Chidambaram, H. & Chinnathambi, S. G-protein coupled receptor, PI3K and Rho signaling pathways regulate the cascades of Tau and amyloid-beta in Alzheimer’s disease. Mol. Biomed. 2, 17 (2021).
Rai, S. N. et al. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox. Res. 35, 775–795 (2019).
Singh, S. & Singh, T. G. Role of nuclear factor Kappa B (NF-kappaB) signalling in neurodegenerative diseases: an mechanistic approach. Curr. Neuropharmacol. 18, 918–935 (2020).
Sobolczyk, M. & Boczek, T. Astrocytic calcium and cAMP in neurodegenerative diseases. Front. Cell Neurosci. 16, 889939 (2022).
Berridge, M. J. Inositol trisphosphate and calcium signalling. Nature 361, 315–325 (1993).
Durkee, C. A. et al. Gi/o protein-coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia 67, 1076–1093 (2019).
Berridge, M. J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta. 1793, 933–940 (2009).
Ponce, A. et al. G-protein-gated inward rectifier K+ channel proteins (GIRK1) are present in the soma and dendrites as well as in nerve terminals of specific neurons in the brain. J. Neurosci. 16, 1990–2001 (1996).
Luscher, C. et al. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19, 687–695 (1997).
Demuro, A., Parker, I. & Stutzmann, G. E. Calcium signaling and amyloid toxicity in Alzheimer disease. J. Biol. Chem. 285, 12463–12468 (2010).
Nakao, A., Matsunaga, Y., Hayashida, K. & Takahashi, N. Role of oxidative stress and Ca(2+) signaling in psychiatric disorders. Front. Cell Dev. Biol. 9, 615569 (2021).
Dutt, P., Nguyen, N. & Toksoz, D. Role of Lbc RhoGEF in Galpha12/13-induced signals to Rho GTPase. Cell. Signal. 16, 201–209 (2004).
Mosaddeghzadeh, N. & Ahmadian, M. R. The RHO family GTPases: mechanisms of regulation and signaling. Cells. 10, 1831 (2021).
Siehler, S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 158, 41–49 (2009).
Koch, J. C. et al. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol. Ther. 189, 1–21 (2018).
Miano, J. M. Role of serum response factor in the pathogenesis of disease. Lab. Invest. 90, 1274–1284 (2010).
Zou, Y. Breaking symmetry - cell polarity signaling pathways in growth cone guidance and synapse formation. Curr. Opin. Neurobiol. 63, 77–86 (2020).
Stankiewicz, T. R. & Linseman, D. A. Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell Neurosci. 8, 314 (2014).
Rajagopal, S. & Shenoy, S. K. GPCR desensitization: acute and prolonged phases. Cell. Signal. 41, 9–16 (2018).
Lefkowitz, R. J. & Shenoy, S. K. Transduction of receptor signals by beta-arrestins. Science 308, 512–517 (2005).
Stadel, J. M. et al. Catecholamine-induced desensitization of turkey erythrocyte adenylate cyclase is associated with phosphorylation of the beta-adrenergic receptor. Proc. Natl Acad. Sci. USA 80, 3173–3177 (1983).
Benovic, J. L. et al. Beta-adrenergic receptor kinase: primary structure delineates a multigene family. Science 246, 235–240 (1989).
Thomsen, A. R. B. et al. GPCR-G protein-beta-arrestin super-complex mediates sustained G protein signaling. Cell 166, 907–919 (2016).
Moore, C. A., Milano, S. K. & Benovic, J. L. Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 69, 451–482 (2007).
Pitcher, J. A. et al. The G protein-coupled receptor kinase 2 is a microtubule-associated protein kinase that phosphorylates tubulin. J. Biol. Chem. 273, 12316–12324 (1998).
Carman, C. V. et al. Selective regulation of Galpha(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. J. Biol. Chem. 274, 34483–34492 (1999).
Degos, V. et al. G protein-coupled receptor kinase 2 and group I metabotropic glutamate receptors mediate inflammation-induced sensitization to excitotoxic neurodegeneration. Ann. Neurol. 73, 667–678 (2013).
Zhao, J. et al. GRK5 influences the phosphorylation of Tau via GSK3β and contributes to Alzheimer’s disease. J. Cell Physiol. 234, 10411–10420 (2019).
Pennington, L. K. et al. How is adaptive potential distributed within species ranges? Evolution 75, 2152–2166 (2021).
Bychkov, E. R. et al. Arrestins and two receptor kinases are upregulated in Parkinson’s disease with dementia. Neurobiol. Aging. 29, 379–396 (2008).
Arawaka, S. et al. The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson’s disease. J. Neurosci. 26, 9227–9238 (2006).
Grange-Midroit, M. et al. Regulation of GRK 2 and 6, beta-arrestin-2 and associated proteins in the prefrontal cortex of drug-free and antidepressant drug-treated subjects with major depression. Brain Res. Mol. Brain Res. 111, 31–41 (2003).
Funk, A. J., Haroutunian, V., Meador-Woodruff, J. H. & McCullumsmith, R. E. Increased G protein-coupled receptor kinase (GRK) expression in the anterior cingulate cortex in schizophrenia. Schizophr. Res. 159, 130–135 (2014).
Kuhn, H. Light-regulated binding of rhodopsin kinase and other proteins to cattle photoreceptor membranes. Biochemistry 17, 4389–4395 (1978).
Haider, R. S. et al. Arrestin-1 engineering facilitates complex stabilization with native rhodopsin. Sci. Rep. 9, 439 (2019).
Zhou, X. E. et al. Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell 170, 457–469 e413 (2017).
Shenoy, S. K. & Lefkowitz, R. J. beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 (2011).
Oakley, R. H. et al. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275, 17201–17210 (2000).
Lally, C. C., Bauer, B., Selent, J. & Sommer, M. E. C-edge loops of arrestin function as a membrane anchor. Nat. Commun. 8, 14258 (2017).
Pottie, E., Dedecker, P. & Stove, C. P. Identification of psychedelic new psychoactive substances (NPS) showing biased agonism at the 5-HT(2A)R through simultaneous use of β-arrestin 2 and miniGα(q) bioassays. Biochem. Pharmacol. 182, 114251 (2020).
Liu, P. et al. Ligand-induced activation of ERK1/2 signaling by constitutively active G(s)-coupled 5-HT receptors. Acta Pharmacol. Sin. 40, 1157–1167 (2019).
Wang, Y. et al. β-arrestin 2 mediates cardiac ischemia-reperfusion injury via inhibiting GPCR-independent cell survival signalling. Cardiovasc. Res. 113, 1615–1626 (2017).
Eichel, K., Jullié, D. & von Zastrow, M. β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol. 18, 303–310 (2016).
Luttrell, L. M. et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283, 655–661 (1999).
Tohgo, A. et al. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J. Biol. Chem. 278, 6258–6267 (2003).
Xiao, K. et al. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc. Natl Acad. Sci. USA 104, 12011–12016 (2007).
DeFea, K. A. Beta-arrestins as regulators of signal termination and transduction: how do they determine what to scaffold? Cell. Signal. 23, 621–629 (2011).
Reiter, E., Ahn, S., Shukla, A. K. & Lefkowitz, R. J. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197 (2012).
Campo, G. M. et al. Beta-arrestin-2 negatively modulates inflammation response in mouse chondrocytes induced by 4-mer hyaluronan oligosaccharide. Mol. Cell. Biochem. 399, 201–208 (2015).
Scarpa, M. et al. Biased M1 muscarinic receptor mutant mice show accelerated progression of prion neurodegenerative disease. Proc. Natl Acad. Sci. USA 118, e2107389118 (2021).
Masri, B. et al. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl Acad. Sci. USA 105, 13656–13661 (2008).
Zhu, J. et al. Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of beta-arrestin2 and NLRP3. Cell Death Differ 25, 2037–2049 (2018).
Ko, M. J. et al. beta-Arrestin-dependent ERK signaling reduces anxiety-like and conditioned fear-related behaviors in mice. Sci. Signal. 14, eaba0245 (2021).
Yan, R. Physiological functions of the beta-site amyloid precursor protein cleaving enzyme 1 and 2. Front. Mol. Neurosci. 10, 97 (2017).
Zhao, J. et al. GRK5 influences the phosphorylation of tau via GSK3beta and contributes to Alzheimer’s disease. J. Cell Physiol. 234, 10411–10420 (2019).
Forero, D. A. et al. A network of synaptic genes associated with schizophrenia and bipolar disorder. Schizophr. Res. 172, 68–74 (2016).
Wang, H. et al. cAMP response element-binding protein (CREB): a possible signaling molecule link in the pathophysiology of schizophrenia. Front. Mol. Neurosci. 11, 255 (2018).
Ren, X. et al. Alteration of cyclic-AMP response element binding protein in the postmortem brain of subjects with bipolar disorder and schizophrenia. J. Affect. Disord. 152-154, 326–333 (2014).
Ao, H., Ko, S. W. & Zhuo, M. CREB activity maintains the survival of cingulate cortical pyramidal neurons in the adult mouse brain. Mol. Pain. 2, 15 (2006).
Lopez de Armentia, M. et al. cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J. Neurosci. 27, 13909–13918 (2007).
Vergara, R. et al. Spontaneous voltage oscillations in striatal projection neurons in a rat corticostriatal slice. J. Physiol. 553, 169–182 (2003).
Schmutz, I. et al. Protein phosphatase 1 (PP1) is a post-translational regulator of the mammalian circadian clock. PLoS ONE 6, e21325 (2011).
Mulkey, R. M., Herron, C. E. & Malenka, R. C. An essential role for protein phosphatases in hippocampal long-term depression. Science 261, 1051–1055 (1993).
Fernandez, E., Schiappa, R., Girault, J. A. & Le Novere, N. DARPP-32 is a robust integrator of dopamine and glutamate signals. PLoS Comput. Biol. 2, e176 (2006).
Flores-Hernandez, J. et al. D(1) dopamine receptor activation reduces GABA(A) receptor currents in neostriatal neurons through a PKA/DARPP-32/PP1 signaling cascade. J. Neurophysiol. 83, 2996–3004 (2000).
Hauser, A. S. et al. GPCR activation mechanisms across classes and macro/microscales. Nat. Struct. Mol. Biol. 28, 879–888 (2021).
Latorraca, N. R., Venkatakrishnan, A. J. & Dror, R. O. GPCR dynamics: structures in motion. Chem. Rev. 117, 139–155 (2017).
Hilger, D., Masureel, M. & Kobilka, B. K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 25, 4–12 (2018).
Wess, J. Molecular basis of receptor/G-protein-coupling selectivity. Pharmacol. Ther. 80, 231–264 (1998).
Manglik, A. & Kruse, A. C. Structural basis for G protein-coupled receptor activation. Biochemistry 56, 5628–5634 (2017).
Rovati, G. E., Capra, V. & Neubig, R. R. The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol. Pharmacol. 71, 959–964 (2007).
Unal, H. & Karnik, S. S. Domain coupling in GPCRs: the engine for induced conformational changes. Trends Pharmacol. Sci. 33, 79–88 (2012).
Zhou, Q. et al. Common activation mechanism of class A GPCRs. eLife 8, e50279 (2019).
Filipek, S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 55, 114–120 (2019).
Hilger, D. The role of structural dynamics in GPCR-mediated signaling. FEBS J. 288, 2461–2489 (2021).
Ferreira-Vieira, T. H., Guimaraes, I. M., Silva, F. R. & Ribeiro, F. M. Alzheimer’s disease: targeting the cholinergic system. Curr. Neuropharmacol. 14, 101–115 (2016).
Ishii, M. & Kurachi, Y. Muscarinic acetylcholine receptors. Curr. Pharm. Des. 12, 3573–3581 (2006).
Caulfield, M. P. & Birdsall, N. J. International union of pharmacology. XVII. classification of muscarinic acetylcholine receptors. Pharmacol. Rev. 50, 279–290 (1998).
Whitehouse, P. J. et al. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 10, 122–126 (1981).
Janickova, H. et al. Uncoupling of M1 muscarinic receptor/G-protein interaction by amyloid beta(1-42). Neuropharmacology 67, 272–283 (2013).
Medeiros, R. et al. Loss of muscarinic M1 receptor exacerbates Alzheimer’s disease-like pathology and cognitive decline. Am. J. Pathol. 179, 980–991 (2011).
Morozova, V. et al. Normal and pathological tau uptake mediated by M1/M3 muscarinic receptors promotes opposite neuronal changes. Front. Cell. Neurosci. 13, 403 (2019).
Gomez-Ramos, A. et al. Extracellular tau is toxic to neuronal cells. FEBS Lett. 580, 4842–4850 (2006).
Tanaka, S. et al. Autoantibodies against four kinds of neurotransmitter receptors in psychiatric disorders. J. Neuroimmunol. 141, 155–164 (2003).
Borda, T. et al. Antibodies against cerebral M1 cholinergic muscarinic receptor from schizophrenic patients: molecular interaction. J. Immunol. 168, 3667–3674 (2002).
Scarr, E. Muscarinic receptors: their roles in disorders of the central nervous system and potential as therapeutic targets. CNS Neurosci. Ther. 18, 369–379 (2012).
Hopper, S., Pavey, G. M., Gogos, A. & Dean, B. Widespread changes in positive allosteric modulation of the muscarinic m1 receptor in some participants with schizophrenia. Int. J. Neuropsychopharmacol. 22, 640–650 (2019).
Lai, M. K. et al. Psychosis of Alzheimer’s disease is associated with elevated muscarinic M2 binding in the cortex. Neurology 57, 805–811 (2001).
Zuchner, T., Schliebs, R. & Perez-Polo, J. R. Down-regulation of muscarinic acetylcholine receptor M2 adversely affects the expression of Alzheimer’s disease-relevant genes and proteins. J. Neurochem. 95, 20–32 (2005).
Jeon, W. J., Dean, B., Scarr, E. & Gibbons, A. The role of muscarinic receptors in the pathophysiology of mood disorders: a potential novel treatment? Curr. Neuropharmacol. 13, 739–749 (2015).
Gibbons, A. S. et al. Decreased muscarinic receptor binding in the frontal cortex of bipolar disorder and major depressive disorder subjects. J. Affect. Disord. 116, 184–191 (2009).
Jeon, W. J., Gibbons, A. S. & Dean, B. The use of a modified [3H]4-DAMP radioligand binding assay with increased selectivity for muscarinic M3 receptor shows that cortical CHRM3 levels are not altered in mood disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 47, 7–12 (2013).
Cheng, Z. et al. A regulatory variant of CHRM3 is associated with cannabis-induced hallucinations in European Americans. Transl. Psychiatry 9, 309 (2019).
Wang, Q. et al. The CHRM3 gene is implicated in abnormal thalamo-orbital frontal cortex functional connectivity in first-episode treatment-naive patients with schizophrenia. Psychol. Med. 46, 1523–1534 (2016).
Chambers, N. E. et al. Effects of muscarinic acetylcholine m1 and m4 receptor blockade on dyskinesia in the hemi-Parkinsonian rat. Neuroscience 409, 180–194 (2019).
Langmead, C. J., Watson, J. & Reavill, C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 117, 232–243 (2008).
Dencker, D. et al. Muscarinic acetylcholine receptor subtypes as potential drug targets for the treatment of schizophrenia, drug abuse and Parkinson’s disease. ACS Chem. Neurosci. 3, 80–89 (2012).
Mayorga, A. J. et al. Characterization of the muscarinic receptor subtype mediating pilocarpine-induced tremulous jaw movements in rats. Eur. J. Pharmacol. 364, 7–11 (1999).
Miller, N. R. et al. Synthesis and SAR of N-(4-(4-alklylpiperazin-1-yl)phenyl)benzamides as muscarinic acetylcholine receptor subtype 1 (M1) anatgonists. Bioorg. Med. Chem. Lett. 20, 2174–2177 (2010).
Costa, A. et al. Deletion of muscarinic acetylcholine receptor 3 in microglia impacts brain ischemic injury. Brain Behav. Immun. 91, 89–104 (2021).
Dean, B. & Scarr, E. Possible involvement of muscarinic receptors in psychiatric disorders: a focus on schizophrenia and mood disorders. Curr. Mol. Med. 15, 253–264 (2015).
Weiner, D. M., Levey, A. I. & Brann, M. R. Expression of muscarinic acetylcholine and dopamine receptor mRNAs in rat basal ganglia. Proc. Natl Acad. Sci. USA 87, 7050–7054 (1990).
Dunwiddie, T. V. & Masino, S. A. The role and regulation of adenosine in the central nervous system. Annu. Rev. Neurosci. 24, 31–55 (2001).
Perez-Pinzon, M. A., Lutz, P. L., Sick, T. J. & Rosenthal, M. Adenosine, a “retaliatory” metabolite, promotes anoxia tolerance in turtle brain. J. Cereb. Blood Flow Metab. 13, 728–732 (1993).
Martin, E. D. et al. Adenosine released by astrocytes contributes to hypoxia-induced modulation of synaptic transmission. Glia 55, 36–45 (2007).
Fredholm, B. B. et al. Adenosine and brain function. Int. Rev. Neurobiol. 63, 191–270 (2005).
Zheng, J., Zhang, X. & Zhen, X. Development of adenosine A2A receptor antagonists for the treatment of Parkinson’s disease: a recent update and challenge. ACS Chem. Neurosci. 10, 783–791 (2019).
Popoli, P., Pezzola, A. & de Carolis, A. S. Modulation of striatal adenosine A1 and A2 receptors induces rotational behaviour in response to dopaminergic stimulation in intact rats. Eur. J. Pharmacol. 257, 21–25 (1994).
Cieslak, M., Komoszynski, M. & Wojtczak, A. Adenosine A(2A) receptors in Parkinson’s disease treatment. Purinergic Signal 4, 305–312 (2008).
Smith, A. P. Caffeine, cognitive failures and health in a non-working community sample. Hum. Psychopharmacol. 24, 29–34 (2009).
Lucas, M. et al. Coffee, caffeine, and risk of depression among women. Arch. Intern. Med. 171, 1571–1578 (2011).
Ferre, S. An update on the mechanisms of the psychostimulant effects of caffeine. J. Neurochem. 105, 1067–1079 (2008).
Yamada, K., Kobayashi, M. & Kanda, T. Involvement of adenosine A2A receptors in depression and anxiety. Int. Rev. Neurobiol. 119, 373–393 (2014).
Fredholm, B. B., Yang, J. & Wang, Y. Low, but not high, dose caffeine is a readily available probe for adenosine actions. Mol. Aspects Med. 55, 20–25 (2017).
Temple, J. L. et al. The safety of ingested caffeine: a comprehensive review. Front. Psychiatry 8, 80 (2017).
Tewari, D. et al. Role of nitric oxide in neurodegeneration: function, regulation, and inhibition. Curr. Neuropharmacol. 19, 114–126 (2021).
Liy, P. M., Puzi, N. N. A., Jose, S. & Vidyadaran, S. Nitric oxide modulation in neuroinflammation and the role of mesenchymal stem cells. Exp. Biol. Med. 246, 2399–2406 (2021).
Sun, J., Steenbergen, C. & Murphy, E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid. Redox Signal. 8, 1693–1705 (2006).
Nakato, R. et al. Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci. Rep. 5, 14812 (2015).
Batalha, V. L. et al. The caffeine-binding adenosine A2A receptor induces age-like HPA-axis dysfunction by targeting glucocorticoid receptor function. Sci. Rep. 6, 31493 (2016).
Perez, D. M. alpha1-adrenergic receptors in neurotransmission, synaptic plasticity, and cognition. Front. Pharmacol. 11, 581098 (2020).
McCorry, L. K. Physiology of the autonomic nervous system. Am. J. Pharm. Educ. 71, 78 (2007).
O’Donnell, J. et al. Norepinephrine: a neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem. Res. 37, 2496–2512 (2012).
Jung, Y. H. et al. Relationships between catecholamine levels and stress or intelligence. Neurochem. Res. 44, 1192–1200 (2019).
Feinstein, D. L., Kalinin, S. & Braun, D. Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: noradrenergic signaling system. J. Neurochem. 139, 154–178 (2016).
Frohman, E. M., Vayuvegula, B., Gupta, S. & van den Noort, S. Norepinephrine inhibits gamma-interferon-induced major histocompatibility class II (Ia) antigen expression on cultured astrocytes via beta-2-adrenergic signal transduction mechanisms. Proc. Natl Acad. Sci. USA 85, 1292–1296 (1988).
Feinstein, D. L. Suppression of astroglial nitric oxide synthase expression by norepinephrine results from decreased NOS-2 promoter activity. J. Neurochem. 70, 1484–1496 (1998).
Ballestas, M. E. & Benveniste, E. N. Elevation of cyclic AMP levels in astrocytes antagonizes cytokine-induced adhesion molecule expression. J. Neurochem. 69, 1438–1448 (1997).
Etienne-Manneville, S., Chaverot, N., Strosberg, A. D. & Couraud, P. O. ICAM-1-coupled signaling pathways in astrocytes converge to cyclic AMP response element-binding protein phosphorylation and TNF-alpha secretion. J. Immunol. 163, 668–674 (1999).
Evans, A. K. et al. Beta-adrenergic receptor antagonism is proinflammatory and exacerbates neuroinflammation in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 146, 105089 (2020).
Marien, M. R., Colpaert, F. C. & Rosenquist, A. C. Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res. Brain Res. Rev. 45, 38–78 (2004).
Bekdash, R. A. The cholinergic system, the adrenergic system and the neuropathology of Alzheimer’s disease. Int. J. Mol. Sci. 22, 1273 (2021).
Cottingham, C. & Wang, Q. alpha2 adrenergic receptor dysregulation in depressive disorders: implications for the neurobiology of depression and antidepressant therapy. Neurosci. Biobehav. Rev. 36, 2214–2225 (2012).
Garcia-Sevilla, J. A. et al. Up-regulation of immunolabeled alpha2A-adrenoceptors, Gi coupling proteins, and regulatory receptor kinases in the prefrontal cortex of depressed suicides. J. Neurochem. 72, 282–291 (1999).
Wang, B. et al. Effects of alpha2A adrenoceptors on norepinephrine secretion from the locus coeruleus during chronic stress-induced depression. Front. Neurosci. 11, 243 (2017).
Zhang, H. T. et al. Postsynaptic alpha-2 adrenergic receptors are critical for the antidepressant-like effects of desipramine on behavior. Neuropsychopharmacology 34, 1067–1077 (2009).
An, D., Peigneur, S., Hendrickx, L. A. & Tytgat, J. Targeting cannabinoid receptors: current status and prospects of natural products. Int. J. Mol. Sci. 21, 5064 (2020).
Busquets Garcia, A., Soria-Gomez, E., Bellocchio, L. & Marsicano, G. Cannabinoid receptor type-1: breaking the dogmas. F1000Res. 5, F1000 (2016).
Turcotte, C., Blanchet, M. R., Laviolette, M. & Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell. Mol. Life Sci. 73, 4449–4470 (2016).
Lu, H. C. & Mackie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 79, 516–525 (2016).
Sugiura, T. et al. Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J. Biol. Chem. 275, 605–612 (2000).
Pertwee, R. G. Cannabinoid pharmacology: the first 66 years. Br. J. Pharmacol. 147, S163–S171 (2006).
Amin, M. R. & Ali, D. W. Pharmacology of medical cannabis. Adv. Exp. Med. Biol. 1162, 151–165 (2019).
Osei-Hyiaman, D. et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 115, 1298–1305 (2005).
Cavuoto, P. et al. The expression of receptors for endocannabinoids in human and rodent skeletal muscle. Biochem. Biophys. Res. Commun. 364, 105–110 (2007).
Pertwee, R. G. Endocannabinoids and their pharmacological actions. Handb. Exp. Pharmacol. 231, 1–37 (2015).
Soria-Gomez, E. et al. Habenular CB1 receptors control the expression of aversive memories. Neuron 88, 306–313 (2015).
Chen, X., Zhang, J. & Chen, C. Endocannabinoid 2-arachidonoylglycerol protects neurons against beta-amyloid insults. Neuroscience 178, 159–168 (2011).
Rice, J. & Cameron, M. Cannabinoids for treatment of MS symptoms: state of the evidence. Curr. Neurol. Neurosci. Rep. 18, 50 (2018).
Glass, M., Faull, R. L. & Dragunow, M. Loss of cannabinoid receptors in the substantia nigra in Huntington’s disease. Neuroscience 56, 523–527 (1993).
Richfield, E. K. & Herkenham, M. Selective vulnerability in Huntington’s disease: preferential loss of cannabinoid receptors in lateral globus pallidus. Ann. Neurol. 36, 577–584 (1994).
Fernandez-Ruiz, J. et al. Cannabidiol for neurodegenerative disorders: important new clinical applications for this phytocannabinoid? Br. J. Clin. Pharmacol. 75, 323–333 (2013).
Bie, B., Wu, J., Foss, J. F. & Naguib, M. An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr. Opin. Anaesthesiol. 31, 407–414 (2018).
Morcuende, A. et al. Immunomodulatory role of CB2 receptors in emotional and cognitive disorders. Front. Psychiatry 13, 866052 (2022).
Miller, A. H. & Raison, C. L. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 16, 22–34 (2016).
Garcia-Gutierrez, M. S., Perez-Ortiz, J. M., Gutierrez-Adan, A. & Manzanares, J. Depression-resistant endophenotype in mice overexpressing cannabinoid CB(2) receptors. Br. J. Pharmacol. 160, 1773–1784 (2010).
Schmole, A. C. et al. Cannabinoid receptor 2 deficiency results in reduced neuroinflammation in an Alzheimer’s disease mouse model. Neurobiol. Aging 36, 710–719 (2015).
Klegeris, A., Bissonnette, C. J. & McGeer, P. L. Reduction of human monocytic cell neurotoxicity and cytokine secretion by ligands of the cannabinoid-type CB2 receptor. Br. J. Pharmacol. 139, 775–786 (2003).
Ehrhart, J. et al. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflammation 2, 29 (2005).
Arjmand, S. et al. Bipolar disorder and the endocannabinoid system. Acta Neuropsychiatr. 31, 193–201 (2019).
Allende, G. et al. Advances in neurobiology and pharmacology of GPR12. Front. Pharmacol. 11, 628 (2020).
Martin, A. L., Steurer, M. A. & Aronstam, R. S. Constitutive activity among orphan class-A G protein coupled receptors. PLoS ONE 10, e0138463 (2015).
Ignatov, A. et al. Role of the G-protein-coupled receptor GPR12 as high-affinity receptor for sphingosylphosphorylcholine and its expression and function in brain development. J. Neurosci. 23, 907–914 (2003).
Laun, A. S., Shrader, S. H., Brown, K. J. & Song, Z. H. GPR3, GPR6, and GPR12 as novel molecular targets: their biological functions and interaction with cannabidiol. Acta Pharmacol. Sin. 40, 300–308 (2019).
Lu, X., Zhang, N., Dong, S. & Hu, Y. Involvement of GPR12 in the induction of neurite outgrowth in PC12 cells. Brain Res. Bull. 87, 30–36 (2012).
Zhao, M. et al. Different responses to risperidone treatment in Schizophrenia: a multicenter genome-wide association and whole exome sequencing joint study. Transl. Psychiatry 12, 173 (2022).
McHugh, D. GPR18 in microglia: implications for the CNS and endocannabinoid system signalling. Br. J. Pharmacol. 167, 1575–1582 (2012).
Ross, G. R., Lichtman, A., Dewey, W. L. & Akbarali, H. I. Evidence for the putative cannabinoid receptor (GPR55)-mediated inhibitory effects on intestinal contractility in mice. Pharmacology 90, 55–65 (2012).
Morales, P. et al. Therapeutic exploitation of GPR18: beyond the cannabinoids? J. Med. Chem. 63, 14216–14227 (2020).
Chiang, N., Dalli, J., Colas, R. A. & Serhan, C. N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 212, 1203–1217 (2015).
Reyes-Resina, I. et al. Molecular and functional interaction between GPR18 and cannabinoid CB2 G-protein-coupled receptors. Relevance in neurodegenerative diseases. Biochem. Pharmacol. 157, 169–179 (2018).
Sawzdargo, M. et al. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, PsiGPR53 and GPR55: GPR55 is extensively expressed in human brain. Brain Res. Mol. Brain Res. 64, 193–198 (1999).
Balenga, N. A., Henstridge, C. M., Kargl, J. & Waldhoer, M. Pharmacology, signaling and physiological relevance of the G protein-coupled receptor 55. Adv. Pharmacol. 62, 251–277 (2011).
Saliba, S. W. et al. Anti-neuroinflammatory effects of GPR55 antagonists in LPS-activated primary microglial cells. J.Neuroinflammation 15, 322 (2018).
Celorrio, M. et al. GPR55: a therapeutic target for Parkinson’s disease? Neuropharmacology 125, 319–332 (2017).
Arias-Carrion, O. & Poppel, E. Dopamine, learning, and reward-seeking behavior. Acta Neurobiol. Exp. 67, 481–488 (2007).
Missale, C. et al. Dopamine receptors: from structure to function. Physiol. Rev. 78, 189–225 (1998).
Baik, J. H. Dopamine signaling in food addiction: role of dopamine D2 receptors. BMB Rep. 46, 519–526 (2013).
Bachoud-Levi, A. C. et al. International guidelines for the treatment of Huntington’s disease. Front. Neurol. 10, 710 (2019).
Kish, S. J., Shannak, K. & Hornykiewicz, O. Elevated serotonin and reduced dopamine in subregionally divided Huntington’s disease striatum. Ann. Neurol. 22, 386–389 (1987).
Sedvall, G. et al. Dopamine D1 receptor number-a sensitive PET marker for early brain degeneration in Huntington’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 243, 249–255 (1994).
Harris, K. L., Kuan, W. L., Mason, S. L. & Barker, R. A. Antidopaminergic treatment is associated with reduced chorea and irritability but impaired cognition in Huntington’s disease (Enroll-HD). J. Neurol. Neurosurg. Psychiatry 91, 622–630 (2020).
Ranjbar-Slamloo, Y. & Fazlali, Z. Dopamine and noradrenaline in the brain; overlapping or dissociate functions? Front. Mol. Neurosci. 12, 334 (2019).
Pan, X. et al. Dopamine and dopamine receptors in Alzheimer’s disease: a systematic review and network meta-analysis. Front. Aging Neurosci. 11, 175 (2019).
Hisahara, S. & Shimohama, S. Dopamine receptors and Parkinson’s disease. Int. J. Med. Chem. 2011, 403039 (2011).
Melnikov, M., Pashenkov, M. & Boyko, A. Dopaminergic receptor targeting in multiple sclerosis: is there therapeutic potential? Int. J. Mol. Sci. 22, 5313 (2021).
Fu, J. et al. The role of Th17 cells/IL-17A in AD, PD, ALS and the strategic therapy targeting on IL-17A. J. Neuroinflammation 19, 98 (2022).
Melnikov, M. & Lopatina, A. Th17-cells in depression: Implication in multiple sclerosis. Front. Immunol. 13, 1010304 (2022).
Borovac, J. A. Side effects of a dopamine agonist therapy for Parkinson’s disease: a mini-review of clinical pharmacology. Yale J. Biol. Med. 89, 37–47 (2016).
Urs, N. M. et al. Deletion of GSK3beta in D2R-expressing neurons reveals distinct roles for beta-arrestin signaling in antipsychotic and lithium action. Proc. Natl Acad. Sci. USA 109, 20732–20737 (2012).
Beaulieu, J. M. & Gainetdinov, R. R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182–217 (2011).
Urs, N. M., Peterson, S. M. & Caron, M. G. New concepts in dopamine D2 receptor biased signaling and implications for schizophrenia therapy. Biol. Psychiatry 81, 78–85 (2017).
Haas, H. L., Sergeeva, O. A. & Selbach, O. Histamine in the nervous system. Physiol. Rev. 88, 1183–1241 (2008).
Abbott, N. J. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol. Neurobiol. 20, 131–147 (2000).
Rocha, S. M. et al. Histamine: a new immunomodulatory player in the neuron-glia crosstalk. Front. Cell Neurosci. 8, 120 (2014).
Burfoot, C. Chronic sport-induced hypohydration. Med. J. Aust. 158, 140 (1993).
Mehta, P. et al. Enigmatic histamine receptor H4 for potential treatment of multiple inflammatory, autoimmune, and related diseases. Life 10, 50 (2020).
Nuutinen, S. & Panula, P. Histamine in neurotransmission and brain diseases. Adv. Exp. Med. Biol. 709, 95–107 (2010).
John, J., Wu, M. F., Boehmer, L. N. & Siegel, J. M. Cataplexy-active neurons in the hypothalamus: implications for the role of histamine in sleep and waking behavior. Neuron 42, 619–634 (2004).
Higuchi, M. et al. Histamine H(1) receptors in patients with Alzheimer’s disease assessed by positron emission tomography. Neuroscience 99, 721–729 (2000).
Kano, M. et al. Decreased histamine H1 receptor binding in the brain of depressed patients. Eur. J. Neurosci. 20, 803–810 (2004).
Iwabuchi, K. et al. Histamine H1 receptors in schizophrenic patients measured by positron emission tomography. Eur. Neuropsychopharmacol. 15, 185–191 (2005).
Passani, M. B. & Ballerini, C. Histamine and neuroinflammation: insights from murine experimental autoimmune encephalomyelitis. Front. Syst. Neurosci. 6, 32 (2012).
Esbenshade, T. A. et al. The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br. J. Pharmacol. 154, 1166–1181 (2008).
Zhou, P. et al. Histamine-4 receptor antagonist JNJ7777120 inhibits pro-inflammatory microglia and prevents the progression of Parkinson-like pathology and behaviour in a rat model. Brain Behav. Immun. 76, 61–73 (2019).
Nahon, J. L. et al. The rat melanin-concentrating hormone messenger ribonucleic acid encodes multiple putative neuropeptides coexpressed in the dorsolateral hypothalamus. Endocrinology 125, 2056–2065 (1989).
Diniz, G. B. & Bittencourt, J. C. The melanin-concentrating hormone as an integrative peptide driving motivated behaviors. Front. Syst. Neurosci. 11, 32 (2017).
Benedetto, L. et al. Microinjection of melanin concentrating hormone into the lateral preoptic area promotes non-REM sleep in the rat. Peptides 39, 11–15 (2013).
Macneil, D. J. The role of melanin-concentrating hormone and its receptors in energy homeostasis. Front. Endocrinol. 4, 49 (2013).
Roy, M. et al. Genetic inactivation of melanin-concentrating hormone receptor subtype 1 (MCHR1) in mice exerts anxiolytic-like behavioral effects. Neuropsychopharmacology 31, 112–120 (2006).
Smith, D. G. et al. Melanin-concentrating hormone-1 receptor modulates neuroendocrine, behavioral, and corticolimbic neurochemical stress responses in mice. Neuropsychopharmacology 31, 1135–1145 (2006).
Aziz, A. et al. Hypocretin and melanin-concentrating hormone in patients with Huntington disease. Brain Pathol. 18, 474–483 (2008).
Vawter, M. P. et al. Melanin concentrating hormone signaling deficits in Schizophrenia: ssociation with memory and social impairments and abnormal sensorimotor gating. Int. J. Neuropsychopharmacol. 23, 53–65 (2020).
Tan, D. X. et al. The changing biological roles of melatonin during evolution: from an antioxidant to signals of darkness, sexual selection and fitness. Biol. Rev. Camb. Philos. Soc. 85, 607–623 (2010).
Gupta, Y. K., Gupta, M. & Kohli, K. Neuroprotective role of melatonin in oxidative stress vulnerable brain. Indian J. Physiol. Pharmacol. 47, 373–386 (2003).
Tan, D. X. et al. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal. Res. 42, 28–42 (2007).
Wu, Y. H. & Swaab, D. F. The human pineal gland and melatonin in aging and Alzheimer’s disease. J. Pineal. Res. 38, 145–152 (2005).
Pandi-Perumal, S. R. et al. Physiological effects of melatonin: role of melatonin receptors and signal transduction pathways. Prog. Neurobiol. 85, 335–353 (2008).
Wu, Y. H. et al. Decreased MT1 melatonin receptor expression in the suprachiasmatic nucleus in aging and Alzheimer’s disease. Neurobiol. Aging. 28, 1239–1247 (2007).
de Oliveira, G. A. P. & Silva, J. L. Alpha-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson’s disease. Commun. Biol. 2, 374 (2019).
Su, L. Y. et al. Melatonin attenuates MPTP-induced neurotoxicity via preventing CDK5-mediated autophagy and SNCA/alpha-synuclein aggregation. Autophagy 11, 1745–1759 (2015).
Vakilzadeh, G. et al. The effect of melatonin on behavioral, molecular, and histopathological changes in cuprizone model of demyelination. Mol. Neurobiol. 53, 4675–4684 (2016).
Rogerio, F. et al. Superoxide dismutase isoforms 1 and 2 in lumbar spinal cord of neonatal rats after sciatic nerve transection and melatonin treatment. Brain Res. Dev. Brain Res. 154, 217–225 (2005).
Alghamdi, B. S. The neuroprotective role of melatonin in neurological disorders. J. Neurosci. Res. 96, 1136–1149 (2018).
Boiko, D. I. et al. Melatonergic receptors (Mt1/Mt2) as a potential additional target of novel drugs for depression. Neurochem. Res. 47, 2909–2924 (2022).
Liu, J. et al. MT1 and MT2 melatonin receptors: a therapeutic perspective. Annu. Rev. Pharmacol. Toxicol. 56, 361–383 (2016).
Wu, Y. H. et al. Alterations of melatonin receptors MT1 and MT2 in the hypothalamic suprachiasmatic nucleus during depression. J. Affect. Disord. 148, 357–367 (2013).
Weishaupt, J. H. et al. Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J. Pineal Res. 41, 313–323 (2006).
Hansen, M. V. et al. Melatonin for pre- and postoperative anxiety in adults. Cochrane Database Syst. Rev. 2015, CD009861 (2015).
Germain, A. & Kupfer, D. J. Circadian rhythm disturbances in depression. Hum. Psychopharmacol. 23, 571–585 (2008).
Perez-Jeldres, T., Alvarez-Lobos, M. & Rivera-Nieves, J. Targeting sphingosine-1-phosphate signaling in immune-mediated diseases: beyond multiple sclerosis. Drugs 81, 985–1002 (2021).
Perez-Jeldres, T. et al. Targeting cytokine signaling and lymphocyte traffic via small molecules in inflammatory bowel disease: JAK inhibitors and S1PR agonists. Front. Pharmacol. 10, 212 (2019).
Aoki, M. et al. Sphingosine-1-phosphate signaling in immune cells and inflammation: roles and therapeutic potential. Mediat. Inflamm. 2016, 8606878 (2016).
Cohan, S. et al. Sphingosine-1-phosphate: its pharmacological regulation and the treatment of multiple sclerosis: a review article. Biomedicines 8, 227 (2020).
Roy, R., Alotaibi, A. A. & Freedman, M. S. Sphingosine 1-phosphate receptor modulators for multiple sclerosis. CNS Drugs 35, 385–402 (2021).
Rivera, J., Proia, R. L. & Olivera, A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat. Rev. Immunol. 8, 753–763 (2008).
Jaillard, C. et al. Edg8/S1P5: an oligodendroglial receptor with dual function on process retraction and cell survival. J. Neurosci. 25, 1459–1469 (2005).
Pradhan, A. A., Smith, M. L., Kieffer, B. L. & Evans, C. J. Ligand-directed signalling within the opioid receptor family. Br. J. Pharmacol. 167, 960–969 (2012).
Darcq, E. & Kieffer, B. L. Opioid receptors: drivers to addiction? Nat. Rev. Neurosci. 19, 499–514 (2018).
Weninger, S. C. et al. Stress-induced behaviors require the corticotropin-releasing hormone (CRH) receptor, but not CRH. Proc. Natl Acad. Sci. USA 96, 8283–8288 (1999).
Borodovitsyna, O., Flamini, M. D. & Chandler, D. J. Acute stress persistently alters locus coeruleus function and anxiety-like behavior in adolescent rats. Neuroscience 373, 7–19 (2018).
Wong, M. L. et al. Pronounced and sustained central hypernoradrenergic function in major depression with melancholic features: relation to hypercortisolism and corticotropin-releasing hormone. Proc. Natl Acad. Sci. USA 97, 325–330 (2000).
Jelen, L. A., Stone, J. M., Young, A. H. & Mehta, M. A. The opioid system in depression. Neurosci. Biobehav. Rev. 140, 104800 (2022).
Moustafa, S. R. et al. The endogenous opioid system in schizophrenia and treatment resistant schizophrenia: increased plasma endomorphin 2, and kappa and mu opioid receptors are associated with interleukin-6. Diagnostics 10, 633 (2020).
Tan, J. & Evin, G. Beta-site APP-cleaving enzyme 1 trafficking and Alzheimer’s disease pathogenesis. J. Neurochem. 120, 869–880 (2012).
Xu, Y. et al. Opposite roles of delta- and mu-opioid receptors in BACE1 regulation and Alzheimer’s Injury. Front. Cell Neurosci. 14, 88 (2020).
Yamada, M. et al. The expression of mRNA for a kappa opioid receptor in the substantia nigra of Parkinson’s disease brain. Brain Res. Mol. Brain Res. 44, 12–20 (1997).
Maneuf, Y. P. et al. Functional implications of kappa opioid receptor-mediated modulation of glutamate transmission in the output regions of the basal ganglia in rodent and primate models of Parkinson’s disease. Brain Res. 683, 102–108 (1995).
Ikeda, K. et al. TRK-820, a selective kappa opioid receptor agonist, could effectively ameliorate L-DOPA-induced dyskinesia symptoms in a rat model of Parkinson’s disease. Eur. J. Pharmacol. 620, 42–48 (2009).
Marazziti, D. Understanding the role of serotonin in psychiatric diseases. F1000Res 6, 180 (2017).
Svob Strac, D., Pivac, N. & Muck-Seler, D. The serotonergic system and cognitive function. Transl. Neurosci. 7, 35–49 (2016).
Nichols, D. E. & Nichols, C. D. Serotonin receptors. Chem. Rev. 108, 1614–1641 (2008).
Piszczek, L. et al. Modulation of anxiety by cortical serotonin 1A receptors. Front. Behav. Neurosci. 9, 48 (2015).
Belmer, A., Patkar, O. L., Lanoue, V. & Bartlett, S. E. 5-HT1A receptor-dependent modulation of emotional and neurogenic deficits elicited by prolonged consumption of alcohol. Sci. Rep. 8, 2099 (2018).
Braem, S. et al. The role of anterior cingulate cortex in the affective evaluation of conflict. J. Cogn. Neurosci. 29, 137–149 (2017).
Veldman, E. R., Svedberg, M. M., Svenningsson, P. & Lundberg, J. Distribution and levels of 5-HT1B receptors in anterior cingulate cortex of patients with bipolar disorder, major depressive disorder and schizophrenia—an autoradiography study. Eur. Neuropsychopharmacol. 27, 504–514 (2017).
Norton, N. & Owen, M. J. HTR2A: association and expression studies in neuropsychiatric genetics. Ann. Med. 37, 121–129 (2005).
Papasergi-Scott, M. M. et al. Structures of metabotropic GABAB receptor. Nature 584, 310–314 (2020).
Park, J. et al. Structure of human GABAB receptor in an inactive state. Nature 584, 304–309 (2020).
Shaye, H. et al. Structural basis of the activation of a metabotropic GABA receptor. Nature 584, 298–303 (2020).
Du, J. et al. Structures of human mGlu2 and mGlu7 homo- and heterodimers. Nature 594, 589–593 (2021).
Gao, Y. et al. Asymmetric activation of the calcium-sensing receptor homodimer. Nature 595, 455–459 (2021).
Kim, Y. et al. Structural basis for activation of the heterodimeric GABAB receptor. J. Mol. Biol. 432, 5966–5984 (2020).
Shen, C. et al. Structural basis of GABAB receptor-Gi protein coupling. Nature 594, 594–598 (2021).
Lin, S. et al. Structures of Gi-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature 594, 583–588 (2021).
Seven, A. B. et al. G-protein activation by a metabotropic glutamate receptor. Nature 595, 450–454 (2021).
Patel, H., Dobson, R. J. B. & Newhouse, S. J. A meta-analysis of Alzheimer’s disease brain transcriptomic data. J. Alzheimers Dis. 68, 1635–1656 (2019).
Niu, H. M. et al. Comprehensive functional annotation of susceptibility SNPs prioritized 10 genes for schizophrenia. Transl. Psychiatry 9, 56 (2019).
Philpott, A. L. et al. A GABBR2 gene variant modifies pathophysiology in Huntington’s disease. Neurosci. Lett. 620, 8–13 (2016).
Tyagi, R. K. et al. Possible role of GABA-B receptor modulation in MPTP induced Parkinson’s disease in rats. Exp. Toxicol. Pathol. 67, 211–217 (2015).
Hovelso, N. et al. Therapeutic potential of metabotropic glutamate receptor modulators. Curr. Neuropharmacol. 10, 12–48 (2012).
Crupi, R., Impellizzeri, D. & Cuzzocrea, S. Role of metabotropic glutamate receptors in neurological disorders. Front. Mol. Neurosci. 12, 20 (2019).
Mikulecka, A. & Mares, P. Effects of mGluR5 and mGluR1 antagonists on anxiety-like behavior and learning in developing rats. Behav. Brain Res. 204, 133–139 (2009).
Farlow, M. R., Graham, S. M. & Alva, G. Memantine for the treatment of Alzheimer’s disease: tolerability and safety data from clinical trials. Drug Saf. 31, 577–585 (2008).
Willard, S. S. & Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 9, 948–959 (2013).
Hamilton, A. et al. Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol. Brain. 7, 40 (2014).
Hamilton, A., Zamponi, G. W. & Ferguson, S. S. Glutamate receptors function as scaffolds for the regulation of beta-amyloid and cellular prion protein signaling complexes. Mol. Brain. 8, 18 (2015).
Haas, L. T. et al. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain 139, 526–546 (2016).
Abd-Elrahman, K. S. & Ferguson, S. S. G. Noncanonical metabotropic glutamate receptor 5 signaling in Alzheimer’s disease. Annu. Rev. Pharmacol. Toxicol. 62, 235–254 (2022).
Doria, J. G. et al. Metabotropic glutamate receptor 5 positive allosteric modulators are neuroprotective in a mouse model of Huntington’s disease. Br. J. Pharmacol. 169, 909–921 (2013).
Doria, J. G. et al. The mGluR5 positive allosteric modulator, CDPPB, ameliorates pathology and phenotypic signs of a mouse model of Huntington’s disease. Neurobiol. Dis. 73, 163–173 (2015).
Matosin, N. & Newell, K. A. Metabotropic glutamate receptor 5 in the pathology and treatment of schizophrenia. Neurosci. Biobehav. Rev. 37, 256–268 (2013).
Schoepp, D. D. et al. LY354740, an mGlu2/3 receptor agonist as a novel approach to treat anxiety/stress. Stress 6, 189–197 (2003).
Li, S. H., Colson, T. L., Abd-Elrahman, K. S. & Ferguson, S. S. G. Metabotropic glutamate receptor 2/3 activation improves motor performance and reduces pathology in heterozygous zQ175 Huntington disease mice. J. Pharmacol. Exp. Ther. 379, 74–84 (2021).
Chan, H. et al. Neuroprotection and functional recovery associated with decreased microglial activation following selective activation of mGluR2/3 receptors in a rodent model of Parkinson’s disease. Parkinsons Dis. 2010, 190450 (2010).
Murray, T. K. et al. Evaluation of the mGluR2/3 agonist LY379268 in rodent models of Parkinson’s disease. Pharmacol. Biochem. Behav. 73, 455–466 (2002).
Arnett, P. A. et al. Depression in multiple sclerosis: relationship to working memory capacity. Neuropsychology 13, 546–556 (1999).
Beal, M. F., Ferrante, R. J., Swartz, K. J. & Kowall, N. W. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. 11, 1649–1659 (1991).
Bruno, V. et al. Selective blockade of metabotropic glutamate receptor subtype 5 is neuroprotective. Neuropharmacology 39, 2223–2230 (2000).
Schiefer, J. et al. The metabotropic glutamate receptor 5 antagonist MPEP and the mGluR2 agonist LY379268 modify disease progression in a transgenic mouse model of Huntington’s disease. Brain Res. 1019, 246–254 (2004).
Le Poul, E. et al. A potent and selective metabotropic glutamate receptor 4 positive allosteric modulator improves movement in rodent models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 343, 167–177 (2012).
Cryan, J. F. et al. Antidepressant and anxiolytic-like effects in mice lacking the group III metabotropic glutamate receptor mGluR7. Eur. J. Neurosci. 17, 2409–2417 (2003).
Robbins, M. J. et al. Evaluation of the mGlu8 receptor as a putative therapeutic target in schizophrenia. Brain Res. 1152, 215–227 (2007).
Shibata, H. et al. Association study of polymorphisms in the group III metabotropic glutamate receptor genes, GRM4 and GRM7, with schizophrenia. Psychiatry Res. 167, 88–96 (2009).
Ohtsuki, T. et al. A polymorphism of the metabotropic glutamate receptor mGluR7 (GRM7) gene is associated with schizophrenia. Schizophr Res. 101, 9–16 (2008).
Niu, W. et al. Association study of GRM7 polymorphisms and schizophrenia in the Chinese Han population. Neurosci. Lett. 604, 109–112 (2015).
Takaki, H. et al. Positive associations of polymorphisms in the metabotropic glutamate receptor type 8 gene (GRM8) with schizophrenia. Am. J. Med. Genet. B128B, 6–14 (2004).
Gomes, I. et al. G protein-coupled receptor heteromers. Annu. Rev. Pharmacol. Toxicol. 56, 403–425 (2016).
Terrillon, S. & Bouvier, M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 5, 30–34 (2004).
Ferre, S. et al. Allosteric mechanisms within the adenosine A2A-dopamine D2 receptor heterotetramer. Neuropharmacology 104, 154–160 (2016).
Hillion, J. et al. Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J. Biol. Chem. 277, 18091–18097 (2002).
Ramsay, D. et al. Homo- and hetero-oligomeric interactions between G-protein-coupled receptors in living cells monitored by two variants of bioluminescence resonance energy transfer (BRET): hetero-oligomers between receptor subtypes form more efficiently than between less closely related sequences. Biochem. J. 365, 429–440 (2002).
Ferre, S. et al. An update on adenosine A2A-dopamine D2 receptor interactions: implications for the function of G protein-coupled receptors. Curr. Pharm. Des. 14, 1468–1474 (2008).
Borroto-Escuela, D. O. et al. Disruption of A2AR-D2R heteroreceptor complexes after A2AR transmembrane 5 peptide administration enhances cocaine self-administration in rats. Mol. Neurobiol. 55, 7038–7048 (2018).
Fuxe, K. & Ungerstedt, U. Action of caffeine and theophyllamine on supersensitive dopamine receptors: considerable enhancement of receptor response to treatment with DOPA and dopamine receptor agonists. Med. Biol. 52, 48–54 (1974).
Font, L. et al. Intra-accumbens injections of the adenosine A2A agonist CGS 21680 affect effort-related choice behavior in rats. Psychopharmacology 199, 515–526 (2008).
Muguruza, C. et al. Dysregulated 5-HT(2A) receptor binding in postmortem frontal cortex of schizophrenic subjects. Eur. Neuropsychopharmacol. 23, 852–864 (2013).
Moreno, J. L. et al. Identification of three residues essential for 5-hydroxytryptamine 2A-metabotropic glutamate 2 (5-HT2A·mGlu2) receptor heteromerization and its psychoactive behavioral function. J. Biol. Chem. 287, 44301–44319 (2012).
Moreno, J. L. et al. Allosteric signaling through an mGlu2 and 5-HT2A heteromeric receptor complex and its potential contribution to schizophrenia. Sci. Signal. 9, ra5 (2016).
Ibi, D. Role of interaction of mGlu2 and 5-HT2A receptors in antipsychotic effects. Pharmacol. Biochem. Behav. 221, 173474 (2022).
Fribourg, M. et al. Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell 147, 1011–1023 (2011).
Moreno, J. L. et al. Chronic treatment with LY341495 decreases 5-HT(2A) receptor binding and hallucinogenic effects of LSD in mice. Neurosci. Lett. 536, 69–73 (2013).
Poulie, C. B. M., Liu, N., Jensen, A. A. & Bunch, L. Design, synthesis, and pharmacological characterization of heterobivalent ligands for the putative 5-HT2A/mGlu2 receptor complex. J. Med. Chem. 63, 9928–9949 (2020).
Nirogi, R. et al. Discovery and development of 1-[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-1-piperazinyl)methyl]-1H-indole dimesylate monohydrate (SUVN-502): a novel, potent, selective and orally active serotonin 6 (5-HT(6)) receptor antagonist for potential treatment of Alzheimer’s disease. J. Med. Chem. 60, 1843–1859 (2017).
Nirogi, R. et al. SUVN-502, a novel, potent, pure, and orally active 5-HT6 receptor antagonist: pharmacological, behavioral, and neurochemical characterization. Behav. Pharmacol. 30, 16–35 (2019).
Koenig, A. M., Arnold, S. E. & Streim, J. E. Agitation and Irritability in Alzheimer’s disease: evidenced-based treatments and the black-box warning. Curr. Psychiatry Rep. 18, 3 (2016).
Ruthirakuhan, M. et al. Agitation, oxidative stress, and cytokines in Alzheimer disease: biomarker analyses from a clinical trial with nabilone for agitation. J. Geriatr. Psychiatry Neurol. 33, 175–184 (2020).
Londzin, P. et al. Potential of caffeine in Alzheimer’s disease-a review of experimental studies. Nutrients 13, 537 (2021).
Sharma, A. & Couture, J. A review of the pathophysiology, etiology, and treatment of attention-deficit hyperactivity disorder (ADHD). Ann. Pharmacother. 48, 209–225 (2014).
Fukuyama, K., Nakano, T., Shiroyama, T. & Okada, M. Chronic administrations of guanfacine on mesocortical catecholaminergic and thalamocortical glutamatergic transmissions. Int. J. Mol. Sci. 22, 4122 (2021).
Kikuchi, T. et al. Discovery research and development history of the dopamine D2 receptor partial agonists, aripiprazole and brexpiprazole. Neuropsychopharmacol. Rep. 41, 134–143 (2021).
Caraci, F. et al. New antipsychotic drugs for the treatment of agitation and psychosis in Alzheimer’s disease: focus on brexpiprazole and pimavanserin. F1000Res 9, F1000 (2020).
Aurora, R. N. et al. The treatment of restless legs syndrome and periodic limb movement disorder in adults-an update for 2012: practice parameters with an evidence-based systematic review and meta-analyses: an American academy of sleep medicine clinical practice guideline. Sleep 35, 1039–1062 (2012).
Salat, D. & Tolosa, E. Levodopa in the treatment of Parkinson’s disease: current status and new developments. J. Parkinsons Dis. 3, 255–269 (2013).
Crosby, N., Deane, K. H. & Clarke, C. E. Amantadine in Parkinson’s disease. Cochrane Database Syst. Rev. 2003, CD003468 (2003).
Marmol, S., Feldman, M., Singer, C. & Margolesky, J. Amantadine revisited: a contender for initial treatment in Parkinson’s disease? CNS Drugs 35, 1141–1152 (2021).
Dragasevic-Miskovic, N., Petrovic, I., Stankovic, I. & Kostic, V. S. Chemical management of levodopa-induced dyskinesia in Parkinson’s disease patients. Expert Opin. Pharmacother. 20, 219–230 (2019).
Machado-Alba, J. E., Calvo-Torres, L. F., Gaviria-Mendoza, A. & Castrillon-Spitia, J. D. [Prescribing patterns of antiparkinson drugs in a group of Colombian patients, 2015]. Biomedica 38, 417–426 (2018).
Cacabelos, R. Parkinson’s disease: from pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 18, 551 (2017).
Perez-Lloret, S. & Rascol, O. Piribedil for the treatment of motor and non-motor symptoms of Parkinson disease. CNS Drugs 30, 703–717 (2016).
Lieberman, A. N. & Goldstein, M. Bromocriptine in Parkinson disease. Pharmacol. Rev. 37, 217–227 (1985).
Kvernmo, T., Houben, J. & Sylte, I. Receptor-binding and pharmacokinetic properties of dopaminergic agonists. Curr. Top Med. Chem. 8, 1049–1067 (2008).
Dungo, R. & Deeks, E. D. Istradefylline: first global approval. Drugs 73, 875–882 (2013).
Pezzoli, G., Canesi, M., Pesenti, A. & Mariani, C. B. Pergolide mesylate in Parkinson’s disease treatment. J. Neural. Transm. Suppl. 45, 203–212 (1995).
Nashatizadeh, M. M., Lyons, K. E. & Pahwa, R. A review of ropinirole prolonged release in Parkinson’s disease. Clin. Interv. Aging. 4, 179–186 (2009).
LiverTox: Clinical and Research Information on Drug-induced Liver Injury https://pubmed.ncbi.nlm.nih.gov/31643176/ (National Institute of Diabetes and Digestive and Kidney Diseases, 2012).
Kulkarni, S. S., Kopajtic, T. A., Katz, J. L. & Newman, A. H. Comparative structure-activity relationships of benztropine analogues at the dopamine transporter and histamine H(1) receptors. Bioorg. Med. Chem. 14, 3625–3634 (2006).
Chernoloz, O., El Mansari, M. & Blier, P. Long-term administration of the dopamine D3/2 receptor agonist pramipexole increases dopamine and serotonin neurotransmission in the male rat forebrain. J. Psychiatry Neurosci. 37, 113–121 (2012).
Moirand, R., Galvao, F. & Donde, C. Pramipexole and selegiline combination therapy in a case of treatment-resistant depression. J. Clin. Psychopharmacol. 39, 684–685 (2019).
Alavi, M. S., Karimi, G. & Roohbakhsh, A. The role of orphan G protein-coupled receptors in the pathophysiology of multiple sclerosis: a review. Life Sci. 224, 33–40 (2019).
McGinley, M. P., Goldschmidt, C. H. & Rae-Grant, A. D. Diagnosis and treatment of multiple sclerosis: a review. J. Am. Med. Assoc. 325, 765–779 (2021).
Rudroff, T. & Sosnoff, J. Cannabidiol to improve mobility in people with multiple sclerosis. Front. Neurol. 9, 183 (2018).
Minzenberg, M. J. & Carter, C. S. Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacology 33, 1477–1502 (2008).
Callegari, I., Derfuss, T. & Galli, E. Update on treatment in multiple sclerosis. La Presse Médicale 50, 104068 (2021).
Comi, G. et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 18, 1009–1020 (2019).
Gilbert, J. et al. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 107, 906–911 (2011).
Paleacu, D. Tetrabenazine in the treatment of Huntington’s disease. Neuropsychiatr. Dis. Treat. 3, 545–551 (2007).
Dold, M. et al. Haloperidol versus first-generation antipsychotics for the treatment of schizophrenia and other psychotic disorders. Cochrane Database Syst. Rev. 1, CD009831 (2015).
Videnovic, A. Treatment of Huntington disease. Curr. Treat. Options Neurol. 15, 424–438 (2013).
Lissek, S. et al. Opposing effects of dopamine antagonism in a motor sequence task-tiapride increases cortical excitability and impairs motor learning. Front. Behav. Neurosci. 8, 201 (2014).
Tashiro, M. et al. Central effects of fexofenadine and cetirizine: measurement of psychomotor performance, subjective sleepiness, and brain histamine H-1-receptor occupancy using C-11-doxepin positron emission tomography. J. Clin. Pharmacol. 44, 890–900 (2004).
Shankar, G. & Nate, C. Positive and negative syndrome scale as a long-term outcome measurement tool in patients receiving clozapine ODT- a pilot study. Pharm. Pract. 5, 42–45 (2007).
Li, P., Snyder, G. L. & Vanover, K. E. Dopamine targeting drugs for the treatment of schizophrenia: past, present and future. Curr. Top Med. Chem. 16, 3385–3403 (2016).
de Bartolomeis, A., Tomasetti, C. & Iasevoli, F. Update on the mechanism of action of aripiprazole: translational insights into antipsychotic strategies beyond dopamine receptor antagonism. CNS Drugs 29, 773–799 (2015).
Fleischhacker, W. W. Aripiprazole. Expert. Opin. Pharmacother. 6, 2091–2101 (2005).
Citrome, L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int. J. Clin. Pract. 69, 978–997 (2015).
Maeda, K. et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J. Pharmacol. Exp. Ther. 350, 589–604 (2014).
Oosterhof, C. A., El Mansari, M. & Blier, P. Acute effects of brexpiprazole on serotonin, dopamine, and norepinephrine systems: an in vivo electrophysiologic characterization. J. Pharmacol. Exp. Ther. 351, 585–595 (2014).
Mahmood, D. et al. New paradigms of old psychedelics in schizophrenia. Pharmaceuticals 15, 640 (2022).
Kumar, B. & Kuhad, A. Lumateperone: a new treatment approach for neuropsychiatric disorders. Drugs Today 54, 713–719 (2018).
Edinoff, A. et al. Lumateperone for the treatment of Schizophrenia. Psychopharmacol. Bull. 50, 32–59 (2020).
Ahmed, R. et al. Evaluation of the use of chlorpromazine for agitation in pediatric patients. Ment. Health Clin. 11, 40–44 (2021).
Moller, H. J. Risperidone: a review. Expert Opin. Pharmacother. 6, 803–818 (2005).
Rahman, S. & Marwaha, R. Haloperidol (StatPearls Publishing, 2022).
Davis, C. in xPharm: The Comprehensive Pharmacology Reference (eds Enna, S. J. & Bylund, D. B.) 1–3 (Elsevier, 2007).
Green, B. Zotepine: a clinical review. Expert. Opin. Drug. Metab. Toxicol. 5, 181–186 (2009).
Capuzzi, E. et al. Experimental serotonergic agents for the treatment of schizophrenia. J. Exp. Pharmacol. 13, 49–67 (2021).
Citrome, L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs 27, 879–911 (2013).
Tiihonen, J., Hakola, P., Paanila, J. & Turtiainen, M. Eltoprazine for aggression in schizophrenia and mental retardation. Lancet 341, 307 (1993).
Davidson, M. et al. Efficacy and safety of roluperidone for the treatment of negative symptoms of schizophrenia. Schizophr. Bull. 48, 609–619 (2022).
Zhao, J. et al. A simple, rapid and reliable method to determine imipramine and desipramine in mouse serum using ultra-high-performance liquid chromatography-quadrupole-time-of-flight mass spectrometry. J. Chromatogr. Sci. 54, 561–568 (2016).
Jesulola, E., Micalos, P. & Baguley, I. J. Understanding the pathophysiology of depression: From monoamines to the neurogenesis hypothesis model - are we there yet? Behav. Brain Res. 341, 79–90 (2018).
Dawson, L. A. & Nguyen, H. Q. The role of 5-HT1A and 5-HT1B/1D receptors on the modulation of acute fluoxetine-induced changes in extracellular 5-HT: the mechanism of action of (+/-)pindolol. Neuropharmacology 39, 1044–1052 (2000).
Fernandez-Perez, S., Pache, D. M. & Sewell, R. D. Co-administration of fluoxetine and WAY100635 improves short-term memory function. Eur. J. Pharmacol. 522, 78–83 (2005).
Werling, L. L., Keller, A., Frank, J. G. & Nuwayhid, S. J. A comparison of the binding profiles of dextromethorphan, memantine, fluoxetine and amitriptyline: treatment of involuntary emotional expression disorder. Exp. Neurol. 207, 248–257 (2007).
Sato, H. et al. Histamine H-1 receptor occupancy by the new-generation antidepressants fluvoxamine and mirtazapine: a positron emission tomography study in healthy volunteers. Psychopharmacology 230, 227–234 (2013).
Bareggi, S. R., Mundo, E., Dell’Osso, B. & Altamura, A. C. The use of escitalopram beyond major depression: pharmacological aspects, efficacy and tolerability in anxiety disorders. Expert Opin. Drug. Metab. Toxicol. 3, 741–753 (2007).
Avila, A. et al. Does nefazodone improve both depression and Parkinson disease? A pilot randomized trial. J. Clin. Psychopharmacol. 23, 509–513 (2003).
Shin, D. S. et al. A novel assessment of nefazodone-induced hERG inhibition by electrophysiological and stereochemical method. Toxicol. Appl. Pharmacol. 274, 361–371 (2014).
Joseph, S. S. et al. Intrinsic sympathomimetic activity of (-)-pindolol mediated through a (-)-propranolol-resistant site of the beta1-adrenoceptor in human atrium and recombinant receptors. Naunyn-Schmiedebergs Arch. Pharmacol 368, 496–503 (2003).
Olianas, M. C., Dedoni, S. & Onali, P. The atypical antidepressant mianserin exhibits agonist activity at κ-opioid receptors. Br. J. Pharmacol. 167, 1329–1341 (2012).
Pawlyk, A. C. et al. Effects of the 5-HT2A antagonist mirtazapine in rat models of thermoregulation. Brain Res. 1123, 135–144 (2006).
Bang-Andersen, B. et al. Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder. J. Med. Chem. 54, 3206–3221 (2011).
Mørk, A. et al. Vortioxetine (Lu AA21004), a novel multimodal antidepressant, enhances memory in rats. Pharmacol. Biochem. Behav. 105, 41–50 (2013).
D’Agostino, A., English, C. D. & Rey, J. A. Vortioxetine (brintellix): a new serotonergic antidepressant. Pharmacy Ther. 40, 36–40 (2015).
Pandhare, A. et al. The antidepressant bupropion is a negative allosteric modulator of serotonin type 3A receptors. Neuropharmacology. 113, 89–99 (2017).
Smeraldi, E. & Delmonte, D. Agomelatine in depression. Expert Opin. Drug Saf. 12, 873–880 (2013).
Bravo, L., Llorca-Torralba, M., Berrocoso, E. & Micó, J. A. Monoamines as drug targets in chronic pain: focusing on neuropathic pain. Front. Neurosci. 13, 1268 (2019).
Nojimoto, F. D. et al. The tricyclic antidepressants amitriptyline, nortriptyline and imipramine are weak antagonists of human and rat alpha1B-adrenoceptors. Neuropharmacology 59, 49–57 (2010).
Nierenberg, A. A. et al. Nortriptyline for treatment-resistant depression. J. Clin. Psychiatry 64, 35–39 (2003).
Mi, W. et al. Efficacy, safety, and tolerability of ansofaxine (LY03005) extended-release tablet for major depressive disorder: a randomized, double-blind, placebo-controlled, dose-finding, phase 2 clinical trial. Int. J. Neuropsychopharmacol. 25, 252–260 (2022).
Craske, M. G. & Stein, M. B. Anxiety. Lancet 388, 3048–3059 (2016).
Bandelow, B., Michaelis, S. & Wedekind, D. Treatment of anxiety disorders. Dialogues Clin. Neurosci. 19, 93–107 (2017).
Garcia-Garcia, A. L., Newman-Tancredi, A. & Leonardo, E. D. 5-HT(1A) receptors in mood and anxiety: recent insights into autoreceptor versus heteroreceptor function. Psychopharmacology 231, 623–636 (2014).
de Boer, S. F., Lesourd, M., Mocaer, E. & Koolhaas, J. M. Selective antiaggressive effects of alnespirone in resident-intruder test are mediated via 5-hydroxytryptamine1A receptors: a comparative pharmacological study with 8-hydroxy-2-dipropylaminotetralin, ipsapirone, buspirone, eltoprazine, and WAY-100635. J. Pharmacol. Exp. Ther. 288, 1125–1133 (1999).
van Zeeland, Y. R. et al. Pharmacokinetics of paroxetine, a selective serotonin reuptake inhibitor, in Grey parrots (Psittacus erithacus erithacus): influence of pharmaceutical formulation and length of dosing. J. Vet. Pharmacol. Ther. 36, 51–58 (2013).
Khouzam, H. R. A review of trazodone use in psychiatric and medical conditions. Postgrad. Med. 129, 140–148 (2017).
Guaiana, G., Barbui, C. & Cipriani, A. Hydroxyzine for generalised anxiety disorder. Cochrane Database Syst. Rev. 8, Cd006815 (2010).
Noyes, R. Jr Beta-adrenergic blocking drugs in anxiety and stress. Psychiatr. Clin. North Am. 8, 119–132 (1985).
Srinivasan, A. V. Propranolol: A 50-year Historical Perspective. Ann. Indian Acad. Neurol. 22, 21–26 (2019).
Baker, J. G. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br. J. Pharmacol. 144, 317–322 (2005).
Almasi, A. & Meza, C. E. StatPearls (StatPearls Publishing LLC., 2022).
Rickels, K. et al. Effects of PRX-00023, a novel, selective serotonin 1A receptor agonist on measures of anxiety and depression in generalized anxiety disorder: results of a double-blind, placebo-controlled trial. J. Clin. Psychopharmacol. 28, 235–239 (2008).
Jain, M. Quetiapine associated central serous chorioretinopathy: implicit role of serotonin and dopamine pathways. Indian J. Ophthalmol. 67, 292–294 (2019).
Suttajit, S., Srisurapanont, M., Maneeton, N. & Maneeton, B. Quetiapine for acute bipolar depression: a systematic review and meta-analysis. Drug Des. Devel. Ther. 8, 827–838 (2014).
Millan, M. J. et al. S18616, a highly potent, spiroimidazoline agonist at alpha(2)-adrenoceptors: I. receptor profile, antinociceptive and hypothermic actions in comparison with dexmedetomidine and clonidine. J. Pharmacol. Exp. Ther. 295, 1192–1205 (2000).
Fenton, C. & Scott, L. J. Risperidone: a review of its use in the treatment of bipolar mania. CNS Drugs 19, 429–444 (2005).
Dziedzicka-Wasylewska, M. et al. Effect of repeated treatment with tianeptine and fluoxetine on central dopamine D(2) /D(3) receptors. Behav. Pharmacol. 13, 127–138 (2002).
Samuels, B. A. et al. The behavioral effects of the antidepressant tianeptine require the mu-opioid receptor. Neuropsychopharmacology 42, 2052–2063 (2017).
Kleimaker, A. et al. Networks in the field of Tourette syndrome. Front. Neurol. 12, 624858 (2021).
Ünal, D. & Akdemir, D. [Neurobiology of Tourette Syndrome]. Turk psikiyatri dergisi = Turkish journal of psychiatry 27, 275–285 (2016).
O’Rourke, J. A., Scharf, J. M., Yu, D. & Pauls, D. L. The genetics of Tourette syndrome: a review. J. Psychosom. Res. 67, 533–545 (2009).
Rose, O. et al. Tourette syndrome research highlights from 2018. F1000Res 8, 988 (2019).
Felling, R. J. & Singer, H. S. Neurobiology of Tourette syndrome: current status and need for further investigation. J. Neurosci. 31, 12387–12395 (2011).
Singer, H. S., Morris, C. & Grados, M. Glutamatergic modulatory therapy for Tourette syndrome. Med. Hypotheses 74, 862–867 (2010).
Paschou, P. The genetic basis of gilles de la Tourette syndrome. Neurosci. Biobehav. Rev. 37, 1026–1039 (2013).
Huertas-Fernández, I. et al. GDNF gene is associated with tourette syndrome in a family study. Mov. Disord. 30, 1115–1120 (2015).
Hallett, M. Tourette syndrome: update. Brain Dev. 37, 651–655 (2015).
Stahl, S. M. Mechanism of action of alpha 2A-adrenergic agonists in attention-deficit/hyperactivity disorder with or without oppositional symptoms. J. Clin. Psychiatry 71, 223–224 (2010).
Cox, J. H. & Cavanna, A. E. Aripiprazole for the treatment of Tourette syndrome. Expert Rev. Neurother. 21, 381–391 (2021).
Silva, M. R., Bernardi, M. M., Cruz-Casallas, P. E. & Felicio, L. F. Pimozide injections into the Nucleus accumbens disrupt maternal behaviour in lactating rats. Pharmacol. Toxicol. 93, 42–47 (2003).
Luo, Y., Weibman, D., Halperin, J. M. & Li, X. A review of heterogeneity in attention deficit/hyperactivity disorder (ADHD). Front. Hum. Neurosci. 13, 42 (2019).
Del Campo, N., Chamberlain, S. R., Sahakian, B. J. & Robbins, T. W. The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol. Psychiatry 69, e145–e157 (2011).
Wu, C. S., Shang, C. Y., Lin, H. Y. & Gau, S. S. Differential treatment effects of methylphenidate and atomoxetine on executive functions in children with attention-deficit/hyperactivity disorder. J. Child Adolesc. Psychopharmacol. 31, 187–196 (2021).
Arnsten, A. F. & Pliszka, S. R. Catecholamine influences on prefrontal cortical function: relevance to treatment of attention deficit/hyperactivity disorder and related disorders. Pharmacol. Biochem. Behav. 99, 211–216 (2011).
Faraone, S. V. The pharmacology of amphetamine and methylphenidate: relevance to the neurobiology of attention-deficit/hyperactivity disorder and other psychiatric comorbidities. Neurosci. Biobehav. Rev. 87, 255–270 (2018).
Markowitz, J. S., DeVane, C. L., Ramamoorthy, S. & Zhu, H. J. The psychostimulant d-threo-(R,R)-methylphenidate binds as an agonist to the 5HT(1A) receptor. Pharmazie 64, 123–125 (2009).
Creighton, C. J. et al. Synthesis and biological evaluation of the major metabolite of atomoxetine: elucidation of a partial kappa-opioid agonist effect. Bioorg. Med. Chem. Lett. 14, 4083–4085 (2004).
Fu, D. et al. The mechanism, clinical efficacy, safety, and dosage regimen of atomoxetine for ADHD therapy in children: a narrative review. Front. Psychiatry 12, 780921 (2021).
Reese, E. A. et al. Trace amine-associated receptor 1 displays species-dependent stereoselectivity for isomers of methamphetamine, amphetamine, and para-hydroxyamphetamine. J. Pharmacol. Exp. Ther. 321, 178–186 (2007).
Chanrion, B. et al. Inverse agonist and neutral antagonist actions of antidepressants at recombinant and native 5-hydroxytryptamine2C receptors: differential modulation of cell surface expression and signal transduction. Mol. Pharmacol. 73, 748–757 (2008).
Cryan, J. F. & Lucki, I. Antidepressant-like behavioral effects mediated by 5-Hydroxytryptamine(2C) receptors. J. Pharmacol. Exp. Ther. 295, 1120–1126 (2000).
Ciana, P. et al. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 25, 4615–4627 (2006).
Bagchi, S. et al. The P2Y2 nucleotide receptor interacts with alphav integrins to activate Go and induce cell migration. J. Biol. Chem. 280, 39050–39057 (2005).
Ghosh, A., Chen, F., Thakur, A. & Hong, H. Cysteinyl leukotrienes and their receptors: emerging therapeutic targets in central nervous system disorders. CNS Neurosci. Ther. 22, 943–951 (2016).
Ciccarelli, R. et al. Cysteinyl-leukotrienes are released from astrocytes and increase astrocyte proliferation and glial fibrillary acidic protein via cys-LT1 receptors and mitogen-activated protein kinase pathway. Eur. J. Neurosci. 20, 1514–1524 (2004).
Coppolino, G. T. et al. Differential local tissue permissiveness influences the final fate of GPR17-expressing oligodendrocyte precursors in two distinct models of demyelination. Glia 66, 1118–1130 (2018).
Dziedzic, A., Miller, E., Saluk-Bijak, J. & Bijak, M. The GPR17 receptor-a promising goal for therapy and a potential marker of the neurodegenerative process in multiple sclerosis. Int. J. Mol. Sci. 21, 1852 (2020).
Parravicini, C. et al. Development of the first in vivo GPR17 ligand through an iterative drug discovery pipeline: A novel disease-modifying strategy for multiple sclerosis. PLoS ONE 15, e0231483 (2020).
Lee, D. K. et al. Cloning and characterization of additional members of the G protein-coupled receptor family. Biochim. Biophys. Acta. 1490, 311–323 (2000).
Mori, F. et al. G protein-coupled receptor 26 immunoreactivity in intranuclear inclusions associated with polyglutamine and intranuclear inclusion body diseases. Neuropathology 36, 50–55 (2016).
Zhang, L. L. et al. GPR26-deficient mice display increased anxiety- and depression-like behaviors accompanied by reduced phosphorylated cyclic AMP responsive element-binding protein level in central amygdala. Neuroscience 196, 203–214 (2011).
Mouhi, S., Martin, B. & Owino, S. Emerging roles for the orphan GPCRs, GPR37 and GPR37 L1, in stroke pathophysiology. Int. J. Mol. Sci. 23, 4028 (2022).
Morato, X. et al. The Parkinson’s disease-associated GPR37 receptor interacts with striatal adenosine A2A receptor controlling its cell surface expression and function in vivo. Sci. Rep. 7, 9452 (2017).
An, J. et al. G protein-coupled receptor GPR37-like 1 regulates adult oligodendrocyte generation. Dev. Neurobiol. 81, 975–984 (2021).
Howland, J. G. & Wang, Y. T. Synaptic plasticity in learning and memory: stress effects in the hippocampus. Prog. Brain Res. 169, 145–158 (2008).
Portbury, S. D. & Adlard, P. A. Zinc signal in brain diseases. Int. J. Mol. Sci. 18, 2506 (2017).
Davis, C. M. et al. GPR39 localization in the aging human brain and correlation of expression and polymorphism with vascular cognitive impairment. Alzheimers Dement. 7, e12214 (2021).
Rychlik, M. & Mlyniec, K. Zinc-mediated neurotransmission in Alzheimer’s disease: a potential role of the GPR39 in dementia. Curr. Neuropharmacol. 18, 2–13 (2020).
Khan, M. Z. A possible significant role of zinc and GPR39 zinc sensing receptor in Alzheimer disease and epilepsy. Biomed. Pharmacother. 79, 263–272 (2016).
Hershfinkel, M., Moran, A., Grossman, N. & Sekler, I. A zinc-sensing receptor triggers the release of intracellular Ca2+ and regulates ion transport. Proc. Natl Acad. Sci. USA 98, 11749–11754 (2001).
Chorin, E. et al. Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J. Neurosci. 31, 12916–12926 (2011).
Ganay, T. et al. Regulation of neuronal pH by the metabotropic Zn(2+)-sensing Gq-coupled receptor, mZnR/GPR39. J. Neurochem. 135, 897–907 (2015).
Park, J. et al. GPR40 agonist inhibits NLRP3 inflammasome activation via modulation of nuclear factor-kappaB and sarco/endoplasmic reticulum Ca(2+)-ATPase. Life Sci. 287, 120127 (2021).
Engel, D. F. et al. Activation of GPR40 induces hypothalamic neurogenesis through p38- and BDNF-dependent mechanisms. Sci. Rep. 10, 11047 (2020).
Yang, Y. et al. GPR40 modulates epileptic seizure and NMDA receptor function. Sci. Adv. 4, eaau2357 (2018).
Liu, C. et al. GPR40 receptor agonist TAK-875 improves cognitive deficits and reduces beta-amyloid production in APPswe/PS1dE9 mice. Psychopharmacology 238, 2133–2146 (2021).
Chen, J. J., Gong, Y. H. & He, L. Role of GPR40 in pathogenesis and treatment of Alzheimer’s disease and type 2 diabetic dementia. J. Drug. Target. 27, 347–354 (2019).
Khan, M. Z., He, L. & Zhuang, X. The emerging role of GPR50 receptor in brain. Biomed. Pharmacother. 78, 121–128 (2016).
Hamouda, H. O. et al. Detection of the human GPR50 orphan seven transmembrane protein by polyclonal antibodies mapping different epitopes. J. Pineal. Res. 43, 10–15 (2007).
Ma, Y. X. et al. G protein coupled receptor 50 promotes self-renewal and neuronal differentiation of embryonic neural progenitor cells through regulation of notch and wnt/beta-catenin signalings. Biochem. Biophys. Res. Commun. 458, 836–842 (2015).
Grunewald, E., Kinnell, H. L., Porteous, D. J. & Thomson, P. A. GPR50 interacts with neuronal NOGO-A and affects neurite outgrowth. Mol. Cell Neurosci. 42, 363–371 (2009).
Thomson, P. A. et al. Sex-specific association between bipolar affective disorder in women and GPR50, an X-linked orphan G protein-coupled receptor. Mol. Psychiatry 10, 470–478 (2005).
Levoye, A. et al. The orphan GPR50 receptor specifically inhibits MT1 melatonin receptor function through heterodimerization. EMBO J. 25, 3012–3023 (2006).
Yao, Y. et al. A striatal-enriched intronic GPCR modulates huntingtin levels and toxicity. eLife 4, e05449 (2015).
Komatsu, H. Discovery of the first druggable GPR52 antagonist to treat Huntington’s disease. J. Med. Chem. 64, 938–940 (2021).
Komatsu, H. et al. Anatomical transcriptome of G protein-coupled receptors leads to the identification of a novel therapeutic candidate GPR52 for psychiatric disorders. PLoS ONE 9, e90134 (2014).
Komatsu, H. Novel therapeutic GPCRs for psychiatric disorders. Int. J. Mol. Sci. 16, 14109–14121 (2015).
Setoh, M. et al. Discovery of the first potent and orally available agonist of the orphan G-protein-coupled receptor 52. J. Med. Chem. 57, 5226–5237 (2014).
Watkins, L. R. & Orlandi, C. Orphan G protein coupled receptors in affective disorders. Genes 11, 694 (2020).
Lin, X. et al. Structural basis of ligand recognition and self-activation of orphan GPR52. Nature 579, 152–157 (2020).
Pariani, M. J., Spencer, A., Graham, J. M. Jr & Rimoin, D. L. A 785kb deletion of 3p14.1p13, including the FOXP1 gene, associated with speech delay, contractures, hypertonia and blepharophimosis. Eur. J. Med. Genet. 52, 123–127 (2009).
Chen, Q. et al. SREB2/GPR85, a schizophrenia risk factor, negatively regulates hippocampal adult neurogenesis and neurogenesis-dependent learning and memory. Eur. J. Neurosci. 36, 2597–2608 (2012).
Yuan, T. et al. Phoenixin: a newly discovered peptide with multi-functions. Protein Pept. Lett. 24, 472–475 (2017).
McIlwraith, E. K. & Belsham, D. D. Phoenixin: uncovering its receptor, signaling and functions. Acta Pharmacol. Sin. 39, 774–778 (2018).
Yang, Y. et al. Phoenixin 20 promotes neuronal mitochondrial biogenesis via CREB-PGC-1alpha pathway. J. Mol. Histol. 51, 173–181 (2020).
Larco, D. O. et al. GnRH-(1-5) Inhibits TGF-beta signaling to regulate the migration of immortalized gonadotropin-releasing hormone neurons. Front. Endocrinol. 9, 45 (2018).
Massart, R. et al. Striatal GPR88 expression is confined to the whole projection neuron population and is regulated by dopaminergic and glutamatergic afferents. Eur. J. Neurosci. 30, 397–414 (2009).
Lovinger, D. M. New twist on orphan receptor GPR88 function. Nat. Neurosci. 15, 1469–1470 (2012).
Logue, S. F. et al. The orphan GPCR, GPR88, modulates function of the striatal dopamine system: a possible therapeutic target for psychiatric disorders? Mol. Cell Neurosci. 42, 438–447 (2009).
Marley, A., Choy, R. W. & von Zastrow, M. GPR88 reveals a discrete function of primary cilia as selective insulators of GPCR cross-talk. PLoS ONE 8, e70857 (2013).
Conti, B. et al. Region-specific transcriptional changes following the three antidepressant treatments electro convulsive therapy, sleep deprivation and fluoxetine. Mol. Psychiatry 12, 167–189 (2007).
Befort, K. et al. Mu-opioid receptor activation induces transcriptional plasticity in the central extended amygdala. Eur. J. Neurosci. 27, 2973–2984 (2008).
Alkufri, F., Shaag, A., Abu-Libdeh, B. & Elpeleg, O. Deleterious mutation in GPR88 is associated with chorea, speech delay, and learning disabilities. Neurol. Genet. 2, e64 (2016).
Del Zompo, M. et al. Association study in three different populations between the GPR88 gene and major psychoses. Mol. Genet. Genomic Med. 2, 152–159 (2014).
de Graaf, C. et al. Extending the structural view of class B GPCRs. Trends Biochem. Sci. 42, 946–960 (2017).
Parthier, C., Reedtz-Runge, S., Rudolph, R. & Stubbs, M. T. Passing the baton in class B GPCRs: peptide hormone activation via helix induction? Trends Biochem. Sci. 34, 303–310 (2009).
Lagerstrom, M. C. & Schioth, H. B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 7, 339–357 (2008).
Pal, K., Melcher, K. & Xu, H. E. Structure and mechanism for recognition of peptide hormones by Class B G-protein-coupled receptors. Acta Pharmacol. Sin. 33, 300–311 (2012).
Alexander, S. P. et al. The concise guide to pharmacology 2015/16: G protein-coupled receptors. Br. J. Pharmacol. 172, 5744–5869 (2015).
Hoare, S. R. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov. Today 10, 417–427 (2005).
Liang, Y. L. et al. Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497 (2018).
Liang, Y. L. et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123 (2017).
Ma, S. et al. Molecular basis for hormone recognition and activation of corticotropin-releasing factor receptors. Mol. Cell 77, 669–680 e664 (2020).
Krumm, B. & Roth, B. L. A structural understanding of Class B GPCR selectivity and activation revealed. Structure 28, 277–279 (2020).
Edvinsson, L., Grell, A. S. & Warfvinge, K. Expression of the CGRP family of neuropeptides and their receptors in the trigeminal ganglion. J. Mol. Neurosci. 70, 930–944 (2020).
Benemei, S., Nicoletti, P., Capone, J. A. & Geppetti, P. Pain pharmacology in migraine: focus on CGRP and CGRP receptors. Neurol. Sci. 28, S89–S93 (2007).
Singh, Y. et al. Calcitonin gene-related peptide (CGRP): a novel target for Alzheimer’s disease. CNS Neurosci. Ther. 23, 457–461 (2017).
Fu, W. et al. Role of microglial amylin receptors in mediating beta amyloid (Abeta)-induced inflammation. J. Neuroinflammation 14, 199 (2017).
Dedic, N., Chen, A. & Deussing, J. M. The CRF family of neuropeptides and their receptors– mediators of the central stress response. Curr. Mol. Pharmacol. 11, 4–31 (2018).
Yan, Y., Dominguez, S., Fisher, D. W. & Dong, H. Sex differences in chronic stress responses and Alzheimer’s disease. Neurobiol. Stress 8, 120–126 (2018).
Zhang, C. & Rissman, R. A. Corticotropin-releasing factor receptor-1 modulates biomarkers of DNA oxidation in Alzheimer’s disease mice. PLoS ONE 12, e0181367 (2017).
Gai, Z. et al. Effects of chronic noise on the corticotropin-releasing factor system in the rat hippocampus: relevance to Alzheimer’s disease-like tau hyperphosphorylation. Environ. Health Prev. Med. 22, 79 (2017).
Magalhaes, A. C. et al. CRF receptor 1 regulates anxiety behavior via sensitization of 5-HT2 receptor signaling. Nat. Neurosci. 13, 622–629 (2010).
Skorzewska, A. et al. Corticotropin releasing factor receptor 1 antagonist differentially inhibits freezing behavior and changes gamma-aminobutyric acidergic activity in the amygdala in low- and high-anxiety rats. J. Physiol. Pharmacol. 68, 35–46 (2017).
Ishitobi, Y. et al. Association of CRHR1 and CRHR2 with major depressive disorder and panic disorder in a Japanese population. Am. J. Med Genet. B Neuropsychiatr. Genet. 159B, 429–436 (2012).
Liu, Z. et al. Association of corticotropin-releasing hormone receptor1 gene SNP and haplotype with major depression. Neurosci. Lett. 404, 358–362 (2006).
Xiao, Z. et al. Interaction between CRHR1 and BDNF genes increases the risk of recurrent major depressive disorder in Chinese population. PLoS ONE 6, e28733 (2011).
Verma, M. K., Goel, R., Krishnadas, N. & Nemmani, K. V. S. Targeting glucose-dependent insulinotropic polypeptide receptor for neurodegenerative disorders. Expert Opin. Ther. Targets. 22, 615–628 (2018).
Zhang, Z. Q. & Holscher, C. GIP has neuroprotective effects in Alzheimer and Parkinson’s disease models. Peptides 125, 170184 (2020).
Paratore, S. et al. Gastric inhibitory polypeptide and its receptor are expressed in the central nervous system and support neuronal survival. Cent. Nerv. Syst. Agents Med. Chem. 11, 210–222 (2011).
Kim, D. S. et al. A new treatment strategy for Parkinson’s disease through the gut-brain axis: the glucagon-like peptide-1 receptor pathway. Cell Transplant. 26, 1560–1571 (2017).
Holst, J. J. & Rosenkilde, M. M. GIP as a therapeutic target in diabetes and obesity: insight from incretin co-agonists. J. Clin. Endocrinol. Metab. 105, e2710–e2716 (2020).
Irwin, N. et al. GIP(Lys16PAL) and GIP(Lys37PAL): novel long-acting acylated analogues of glucose-dependent insulinotropic polypeptide with improved antidiabetic potential. J. Med. Chem. 49, 1047–1054 (2006).
Holscher, C. Incretin analogues that have been developed to treat type 2 diabetes hold promise as a novel treatment strategy for Alzheimer’s disease. Recent Pat. CNS Drug Discov. 5, 109–117 (2010).
Zheng, J. et al. GLP-1 improves the supportive ability of astrocytes to neurons by promoting aerobic glycolysis in Alzheimer’s disease. Mol. Metab. 47, 101180 (2021).
Amato, A. & Mule, F. Protective potential of glucagon like peptide 2 (GLP-2) against the neurodegeneration. Neural Regen. Res. 14, 1901–1902 (2019).
Duarte, A. I. et al. Liraglutide protects against brain amyloid-beta1-42 accumulation in female mice with early Alzheimer’s disease-like pathology by partially rescuing oxidative/nitrosative stress and inflammation. Int. J. Mol. Sci. 21, 1746 (2020).
Hansen, H. H. et al. The GLP-1 receptor agonist liraglutide improves memory function and increases hippocampal CA1 neuronal numbers in a senescence-accelerated mouse model of Alzheimer’s disease. J. Alzheimers Dis. 46, 877–888 (2015).
Hamann, J. et al. International union of basic and clinical pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol. Rev. 67, 338–367 (2015).
Ganesh, R. A., Venkataraman, K. & Sirdeshmukh, R. GPR56: An adhesion GPCR involved in brain development, neurological disorders and cancer. Brain Res. 1747, 147055 (2020).
Langenhan, T., Piao, X. & Monk, K. R. Adhesion G protein-coupled receptors in nervous system development and disease. Nat. Rev. Neurosci. 17, 550–561 (2016).
Lala, T. & Hall, R. A. Adhesion G protein-coupled receptors: structure, signaling, physiology, and pathophysiology. Physiol. Rev. 102, 1587–1624 (2022).
Promel, S., Langenhan, T. & Arac, D. Matching structure with function: the GAIN domain of adhesion-GPCR and PKD1-like proteins. Trends Pharmacol. Sci. 34, 470–478 (2013).
Liebscher, I. et al. A guide to adhesion GPCR research. FEBS J. 289, 7610–7630 (2021).
Barros-Alvarez, X. et al. The tethered peptide activation mechanism of adhesion GPCRs. Nature 604, 757–762 (2022).
Ping, Y. Q. et al. Structures of the glucocorticoid-bound adhesion receptor GPR97-Go complex. Nature 589, 620–626 (2021).
Qu, X. et al. Structural basis of tethered agonism of the adhesion GPCRs ADGRD1 and ADGRF1. Nature 604, 779–785 (2022).
Folts, C. J., Giera, S., Li, T. & Piao, X. Adhesion G protein-coupled receptors as drug targets for neurological diseases. Trends Pharmacol. Sci. 40, 278–293 (2019).
Magalhaes, A. C., Dunn, H. & Ferguson, S. S. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br. J. Pharmacol. 165, 1717–1736 (2012).
Zhu, D. et al. BAI1 regulates spatial learning and synaptic plasticity in the hippocampus. J. Clin. Investig. 125, 1497–1508 (2015).
Stephenson, J. R. et al. Brain-specific angiogenesis inhibitor-1 signaling, regulation, and enrichment in the postsynaptic density. J. Biol. Chem. 288, 22248–22256 (2013).
Tu, Y. K., Duman, J. G. & Tolias, K. F. The adhesion-GPCR BAI1 promotes excitatory synaptogenesis by coordinating bidirectional trans-synaptic signaling. J. Neurosci. 38, 8388–8406 (2018).
Choi, J. S., Bae, W. Y., Nam, S. & Jeong, J. W. New targets for Parkinson’s disease: adhesion G protein-coupled receptor B1 is downregulated by AMP-activated protein kinase activation. OMICS 22, 493–501 (2018).
Scuderi, C. et al. Biallelic intragenic duplication in ADGRB3 (BAI3) gene associated with intellectual disability, cerebellar atrophy, and behavioral disorder. Eur. J. Hum. Genet. 27, 594–602 (2019).
Duman, J. G., Tu, Y. K. & Tolias, K. F. Emerging roles of BAI adhesion-GPCRs in synapse development and plasticity. Neural Plast 2016, 8301737 (2016).
DeRosse, P. et al. The genetics of symptom-based phenotypes: toward a molecular classification of schizophrenia. Schizophr. Bull. 34, 1047–1053 (2008).
McCarthy, M. J., Nievergelt, C. M., Kelsoe, J. R. & Welsh, D. K. A survey of genomic studies supports association of circadian clock genes with bipolar disorder spectrum illnesses and lithium response. PLoS ONE 7, e32091 (2012).
McNeill, R. V. et al. Expression of the adult ADHD-associated gene ADGRL3 is dysregulated by risk variants and environmental risk factors. World J. Biol. Psychiatry 22, 335–349 (2021).
Mortimer, N. et al. Dissociation of impulsivity and aggression in mice deficient for the ADHD risk gene Adgrl3: evidence for dopamine transporter dysregulation. Neuropharmacology 156, 107557 (2019).
Langley, K., Holmans, P. A., van den Bree, M. B. & Thapar, A. Effects of low birth weight, maternal smoking in pregnancy and social class on the phenotypic manifestation of attention deficit hyperactivity disorder and associated antisocial behaviour: investigation in a clinical sample. BMC Psychiatry 7, 26 (2007).
Mathiasen, S. et al. G12/13 is activated by acute tethered agonist exposure in the adhesion GPCR ADGRL3. Nat. Chem. Biol. 16, 1343–1350 (2020).
Wallis, D. et al. Initial characterization of mice null for Lphn3, a gene implicated in ADHD and addiction. Brain Res. 1463, 85–92 (2012).
Gardenal, E. et al. Increased calcium-sensing receptor immunoreactivity in the hippocampus of a triple transgenic mouse model of Alzheimer’s disease. Front. Neurosci. 11, 81 (2017).
Chiarini, A., Armato, U. & Liu, D. & Dal Pra, I. Calcium-sensing receptors of human neural cells play crucial roles in Alzheimer’s disease. Front. Physiol. 7, 134 (2016).
Feng, C. et al. Calcium-sensing receptor mediates beta-amyloid-induced synaptic formation impairment and cognitive deficits via regulation of cytosolic phospholipase A2/prostaglandin E2 metabolic pathway. Front. Aging Neurosci. 12, 144 (2020).
Schulte, G. International union of basic and clinical pharmacology. LXXX. The class Frizzled receptors. Pharmacol. Rev. 62, 632–667 (2010).
Xu, L. et al. Cryo-EM structure of constitutively active human Frizzled 7 in complex with heterotrimeric Gs. Cell Res. 31, 1311–1314 (2021).
Umbhauer, M. et al. The C-terminal cytoplasmic Lys-thr-X-X-X-Trp motif in frizzled receptors mediates Wnt/beta-catenin signalling. EMBO J. 19, 4944–4954 (2000).
Schulte, G. & Kozielewicz, P. Structural insight into Class F receptors—what have we learnt regarding agonist-induced activation? Basic Clin. Pharmacol. Toxicol. 126, 17–24 (2020).
Zhang, X. et al. Dishevelled promotes axon differentiation by regulating atypical protein kinase C. Nat. Cell Biol. 9, 743–754 (2007).
He, C. W., Liao, C. P. & Pan, C. L. Wnt signalling in the development of axon, dendrites and synapses. Open Biol. 8, 180116 (2018).
Zeng, C. M., Chen, Z. & Fu, L. Frizzled receptors as potential therapeutic targets in human cancers. Int. J. Mol. Sci. 19, 1543 (2018).
Palomer, E. et al. Epigenetic repression of Wnt receptors in AD: a role for Sirtuin2-induced H4K16ac deacetylation of Frizzled1 and Frizzled7 promoters. Mol. Psychiatry27, 3024–3033 (2022).
Yang, S. et al. Crystal structure of the Frizzled 4 receptor in a ligand-free state. Nature 560, 666–670 (2018).
Turku, A. et al. Residue 6.43 defines receptor function in class F GPCRs. Nat. Commun. 12, 3919 (2021).
Bhat, R. A., Stauffer, B., Komm, B. S. & Bodine, P. V. Structure-function analysis of secreted frizzled-related protein-1 for its Wnt antagonist function. J. Cell Biochem. 102, 1519–1528 (2007).
de Lau, W. B., Snel, B. & Clevers, H. C. The R-spondin protein family. Genome Biol 13, 242 (2012).
L’Episcopo, F. et al. A Wnt1 regulated Frizzled-1/beta-Catenin signaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: therapeutical relevance for neuron survival and neuroprotection. Mol. Neurodegener. 6, 49 (2011).
Gonzalez, P. et al. Frizzled 1 and Wnt1 as new potential therapeutic targets in the traumatically injured spinal cord. Cell Mol. Life Sci. 77, 4631–4662 (2020).
Jeong, S. H. et al. Investigation of genetic association between human Frizzled homolog 3 gene (FZD3) and schizophrenia: results in a Korean population and evidence from meta-analysis. Psychiatry Res. 143, 1–11 (2006).
Kishimoto, M. et al. The Frizzled 3 gene is associated with methamphetamine psychosis in the Japanese population. Behav. Brain Funct. 4, 37 (2008).
Jiang, X. et al. The mechanism of the WNT5A and FZD4 receptor mediated WNT/beta-catenin pathway in the degeneration of ALS spinal cord motor neurons. Biochem. Biophys. Res. Commun. 609, 23–30 (2022).
Calandria, J. M. et al. cRel and Wnt5a/Frizzled 5 receptor-mediated inflammatory regulation reveal novel neuroprotectin D1 targets for neuroprotection. Cell Mol. Neurobiol. 43, 1077–1096 (2023).
De Marco, P. et al. FZD6 is a novel gene for human neural tube defects. Hum. Mutat. 33, 384–390 (2012).
Zhou, J. et al. Atypical deletion of Williams-Beuren syndrome reveals the mechanism of neurodevelopmental disorders. BMC Med. Genomics. 15, 79 (2022).
Hot, B. et al. FZD10-Galpha13 signalling axis points to a role of FZD10 in CNS angiogenesis. Cell. Signal. 32, 93–103 (2017).
Traiffort, E., Angot, E. & Ruat, M. Sonic Hedgehog signaling in the mammalian brain. J. Neurochem. 113, 576–590 (2010).
Kruse, A. C. et al. Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat. Rev. Drug Discov. 13, 549–560 (2014).
Liu, H. et al. Structure-guided development of selective M3 muscarinic acetylcholine receptor antagonists. Proc. Natl Acad. Sci. USA 115, 12046–12050 (2018).
Krystal, J. H. & State, M. W. Psychiatric disorders: diagnosis to therapy. Cell 157, 201–214 (2014).
Ho, B. C., Wassink, T. H., Ziebell, S. & Andreasen, N. C. Cannabinoid receptor 1 gene polymorphisms and marijuana misuse interactions on white matter and cognitive deficits in schizophrenia. Schizophr. Res. 128, 66–75 (2011).
Hayslett, R. L. & Tizabi, Y. Effects of donepezil, nicotine and haloperidol on the central serotonergic system in mice: implications for Tourette’s syndrome. Pharmacol. Biochem. Behav. 81, 879–886 (2005).
Nisijima, K. et al. Memantine, an NMDA antagonist, prevents the development of hyperthermia in an animal model for serotonin syndrome. Pharmacopsychiatry 37, 57–62 (2004).
Mancini, M. et al. Memantine alters striatal plasticity inducing a shift of synaptic responses toward long-term depression. Neuropharmacology 101, 341–350 (2016).
Richelson, E. & Souder, T. Binding of antipsychotic drugs to human brain receptors focus on newer generation compounds. Life Sci. 68, 29–39 (2000).
Bymaster, F. P. et al. Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology 14, 87–96 (1996).
Krogsgaard-Larsen, N., Jensen, A. A. & Kehler, J. Novel 7-phenylsulfanyl-1,2,3,4,10,10a-hexahydro-pyrazino[1,2-a]indoles as dual serotonin 5-HT2C and 5-HT6 receptor ligands. Booing. Med. Chem. Lett. 20, 5431–5433 (2010).
Rowley, M., Bristow, L. J. & Hutson, P. H. Current and novel approaches to the drug treatment of schizophrenia. J. Med. Chem. 44, 477–501 (2001).
Hertel, P. Comparing sertindole to other new generation antipsychotics on preferential dopamine output in limbic versus striatal projection regions: mechanism of action. Synapse 60, 543–552 (2006).
Hrib, N. J. et al. Structure-activity relationships of a series of novel (piperazinylbutyl)thiazolidinone antipsychotic agents related to 3-[4-[4-(6-fluorobenzo[b]thien-3-yl)-1-piperazinyl]butyl]-2,5,5- trimethyl-4-thiazolidinone maleate. J. Med. Chem. 39, 4044–4057 (1996).
Hutchison, K. E. et al. The effect of olanzapine on craving and alcohol consumption. Neuropsychopharmacology 31, 1310–1317 (2006).
Campiani, G. et al. Novel atypical antipsychotic agents: rational design, an efficient palladium-catalyzed route, and pharmacological studies. J. Med. Chem. 48, 1705–1708 (2005).
Kongsamut, S. et al. Iloperidone binding to human and rat dopamine and 5-HT receptors. Eur. J. Pharmacol. 317, 417–423 (1996).
Pettersson, F. et al. Synthesis and evaluation of a set of 4-phenylpiperidines and 4-phenylpiperazines as D2 receptor ligands and the discovery of the dopaminergic stabilizer 4-[3-(methylsulfonyl)phenyl]-1-propylpiperidine (huntexil, pridopidine, ACR16). J. Med. Chem. 53, 2510–2520 (2010).
Gao, M., Shi, Z., Wang, M. & Zheng, Q. H. [11C]olanzapine, radiosynthesis and lipophilicity of a new potential PET 5-HT2 and D2 receptor radioligand. Bioorg. Med. Chem. Lett. 23, 1953–1956 (2013).
Nasrallah, H. A. Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol. Psychiatry 13, 27–35 (2008).
Sunahara, R. K. et al. Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature 350, 614–619 (1991).
Pearlstein, R., Vaz, R. & Rampe, D. Understanding the structure-activity relationship of the human ether-a-go-go-related gene cardiac K+ channel. A model for bad behavior. J. Med. Chem. 46, 2017–2022 (2003).
Cahir, M. & King, D. J. Antipsychotics lack alpha 1A/B adrenoceptor subtype selectivity in the rat. Eur. Neuropsychopharmacol. 15, 231–234 (2005).
Kroeze, W. K. et al. HI-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology 28, 519–526 (2003).
Sultan, M. A. & Courtney, D. B. Adjunctive trazodone and depression outcome in adolescents treated with serotonin re-uptake inhibitors. J. Canad. Acad. Child Adolesc. Psychiatry 26, 233–240 (2017).
Balsara, J. J. et al. Effects of the antidepressant trazodone, a 5-HT2A/2C receptor antagonist, on dopamine-dependent behaviors in rats. Psychopharmacology 179, 597–605 (2005).
LeWitt, P. A. Subcutaneously administered apomorphine—pharmacokinetics and metabolism. Neurology 62, S8–S11 (2004).
Succu, S. et al. Stimulation of dopamine receptors in the paraventricular nucleus of the hypothalamus of male rats induces penile erection and increases extra-cellular dopamine in the nucleus accumbens: involvement of central oxytocin. Neuropharmacology 52, 1034–1043 (2007).
Imming, P., Sinning, C. & Meyer, A. Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discov. 5, 821–834 (2006).
Chen, X., Ji, Z. L. & Chen, Y. Z. TTD: therapeutic target database. Nucleic Acids Res. 30, 412–415 (2002).
Overington, J. P., Al-Lazikani, B. & Hopkins, A. L. How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 (2006).
de Leeuw van Weenen, J. E. et al. The dopamine receptor D2 agonist bromocriptine inhibits glucose-stimulated insulin secretion by direct activation of the alpha2-adrenergic receptors in beta cells. Biochem. Pharmacol. 79, 1827–1836 (2010).
Cussac, D. et al. Agonist-directed trafficking of signalling at serotonin 5-HT2A, 5-HT2B and 5-HT2C-VSV receptors mediated Gq/11 activation and calcium mobilisation in CHO cells. Eur. J. Pharmacol. 594, 32–38 (2008).
Knight, J. A. et al. Pharmacological analysis of the novel, rapid, and potent inactivation of the human 5-Hydroxytryptamine7 receptor by risperidone, 9-OH-Risperidone, and other inactivating antagonists. Mol. Pharmacol. 75, 374–380 (2009).
Davis, M. P. Recent advances in the treatment of pain. F1000 Med. Rep. 2, 63 (2010).
Du, L. & Li, M. Modeling the interactions between alpha(1)-adrenergic receptors and their antagonists. Curr. Comput. Aided Drug Des. 6, 165–178 (2010).
Mukai, M. et al. Effects of rifampin on the pharmacokinetics of a single dose of istradefylline in healthy subjects. J. Clin. Pharmacol. 58, 193–201 (2018).
Dupre, K. B., Eskow, K. L., Negron, G. & Bishop, C. The differential effects of 5-HT(1A) receptor stimulation on dopamine receptor-mediated abnormal involuntary movements and rotations in the primed hemiparkinsonian rat. Brain Res. 1158, 135–143 (2007).
Kovoor, A. et al. D2 dopamine receptors colocalize regulator of G-protein signaling 9-2 (RGS9-2) via the RGS9 DEP domain, and RGS9 knock-out mice develop dyskinesias associated with dopamine pathways. J. Neurosci. 25, 2157–2165 (2005).
Deleu, D., Northway, M. G. & Hanssens, Y. Clinical pharmacokinetic and pharmacodynamic properties of drugs used in the treatment of Parkinson’s disease. Clin. Pharmacokinet. 41, 261–309 (2002).
Onofrj, M., Bonanni, L. & Thomas, A. An expert opinion on safinamide in Parkinson’s disease. Expert Opin. Investig. Drugs 17, 1115–1125 (2008).
Perachon, S., Schwartz, J. C. & Sokoloff, P. Functional potencies of new antiparkinsonian drugs at recombinant human dopamine D1, D2 and D3 receptors. Eur. J. Pharmacol. 366, 293–300 (1999).
Gornemann, T. et al. Characterization of the molecular fragment that is responsible for agonism of pergolide at serotonin 5-Hydroxytryptamine2B and 5-Hydroxytryptamine2A receptors. J. Pharmacol. Exp. Ther. 324, 1136–1145 (2008).
Hutcheson, J. D., Setola, V., Roth, B. L. & Merryman, W. D. Serotonin receptors and heart valve disease-it was meant 2B. Pharmacol. Ther. 132, 146–157 (2011).
Gilliland, S. L. & Alper, R. H. Characterization of dopaminergic compounds at hD2short, hD4.2 and hD4.7 receptors in agonist-stimulated [35S]GTPgammaS binding assays. Naunyn Schmiedebergs Arch. Pharmacol. 361, 498–504 (2000).
Lam, Y. W. F. Clinical pharmacology of dopamine agonists. Pharmacotherapy 20, 17S–25S (2000).
Chernoloz, O., El Mansari, M. & Blier, P. Sustained administration of pramipexole modifies the spontaneous firing of dopamine, norepinephrine, and serotonin neurons in the rat brain. Neuropsychopharmacology 34, 651–661 (2009).
Piercey, M. F. Pharmacology of pramipexole, a dopamine D-3-preferring agonist useful in treating Parkinson’s disease. Clin. Neuropharmacol. 21, 141–151 (1998).
Wood, M. D. et al. Pharmacological profile of antipsychotics at monoamine receptors: atypicality beyond 5-HT2A receptor blockade. CNS Neurol. Disord. Drug Targets. 5, 445–452 (2006).
Moallem, N. & Ray, L. A. Quetiapine improves response inhibition in alcohol dependent patients: a placebo-controlled pilot study. Pharmacol. Biochem. Behav. 100, 490–493 (2012).
Uys, M. M., Shahid, M. & Harvey, B. H. Therapeutic potential of selectively targeting the alpha(2C)-adrenoceptor in cognition, depression, and schizophrenia-new developments and future perspective. Front. Psychiatry 8, 144 (2017).
Vehof, J. et al. Association of genetic variants of the histamine H1 and muscarinic M3 receptors with BMI and HbA1c values in patients on antipsychotic medication. Psychopharmacology 216, 257–265 (2011).
Shill, H. A. & Stacy, M. Update on ropinirole in the treatment of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 5, 33–36 (2009).
Scheller, D. et al. The in vitro receptor profile of rotigotine: a new agent for the treatment of Parkinson’s disease. Naunyn Schmiedebergs Arch. Pharmacol 379, 73–86 (2009).
Fu, Z. et al. The gamma-aminobutyric acid type B (GABAB) receptor agonist baclofen inhibits morphine sensitization by decreasing the dopamine level in rat nucleus accumbens. Behav. Brain Funct. 8, 20 (2012).
de Beaurepaire, R. A review of the potential mechanisms of action of baclofen in alcohol use disorder. Front. Psychiatry 9, 506 (2018).
Yang, K. H. et al. The nonpsychoactive cannabinoid cannabidiol inhibits 5-hydroxytryptamine3A receptor-mediated currents in Xenopus laevis oocytes. J. Pharmacol. Exp. Ther. 333, 547–554 (2010).
Bih, C. I. et al. Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics 12, 699–730 (2015).
Laprairie, R. B., Bagher, A. M., Kelly, M. E. & Denovan-Wright, E. M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 172, 4790–4805 (2015).
Chen, C. R. et al. Modafinil exerts a dose-dependent antiepileptic effect mediated by adrenergic alpha(1) and histaminergic H-1 receptors in mice. Neuropharmacology 53, 534–541 (2007).
Cohen, J. A. et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 18, 1021–1033 (2019).
O’Sullivan, C., Schubart, A., Mir, A. K. & Dev, K. K. The dual S1PR1/S1PR5 drug BAF312 (Siponimod) attenuates demyelination in organotypic slice cultures. J. Neuroinflammation. 13, 31 (2016).
Zu Heringdorf, D. M., Ihlefeld, K. & Pfeilschifter, J. Pharmacology of the sphingosine-1-phosphate signalling system. Handb. Exp. Pharmacol. 215, 239–253 (2013).
David, O. J., Kovarik, J. M. & Schmouder, R. L. Clinical pharmacokinetics of fingolimod. Clin. Pharmacokinet. 51, 15–28 (2012).
Seeman, P. Atypical antipsychotics: mechanism of action. Can. J. Psychiatry 47, 27–38 (2002).
Bisson, W. H. et al. Discovery of antiandrogen activity of nonsteroidal scaffolds of marketed drugs. Proc. Natl Acad. Sci. USA 104, 11927–11932 (2007).
Tuppurainen, H. et al. Dopamine D2/3 receptor binding potential and occupancy in midbrain and temporal cortex by haloperidol, olanzapine and clozapine. Psychiatry Clin. Neurosci. 63, 529–537 (2009).
Kulagowski, J. J. et al. 3-((4-(4-Chlorophenyl)piperazin-1-yl)-methyl)-1H-pyrrolo-2,3-b-pyridine: an antagonist with high affinity and selectivity for the human dopamine D4 receptor. J. Med. Chem. 39, 1941–1942 (1996).
Leopoldo, M. et al. Structure-affinity relationship study on N-[4-(4-arylpiperazin-1-yl)butyl]arylcarboxamides as potent and selective dopamine D(3) receptor ligands. J. Med. Chem. 45, 5727–5735 (2002).
Zhang, X. et al. trans-1-[(2-Phenylcyclopropyl)methyl]-4-arylpiperazines: mixed dopamine D(2)/D(4) receptor antagonists as potential antipsychotic agents. J. Med. Chem. 43, 3923–3932 (2000).
Pandey, D. K. et al. A novel 5-HT(2A) receptor antagonist exhibits antidepressant-like effects in a battery of rodent behavioural assays: approaching early-onset antidepressants. Pharmacol. Biochem. Behav. 94, 363–373 (2010).
Nojimoto, F. D. et al. The tricyclic antidepressants amitriptyline, nortriptyline and imipramine are weak antagonists of human and rat alpha(1B)-adrenoceptors. Neuropharmacology 59, 49–57 (2010).
Cottingham, C., Percival, S., Birky, T. & Wang, Q. Tricyclic antidepressants exhibit variable pharmacological profiles at the alpha(2A) adrenergic receptor. Biochem. Biophys. Res. Commun. 451, 461–466 (2014).
Ozdogan, U. K., Lahdesmaki, J., Mansikka, H. & Scheinin, M. Loss of amitriptyline analgesia in alpha 2A-adrenoceptor deficient mice. Eur. J. Pharmacol. 485, 193–196 (2004).
Benbouzid, M. et al. Delta-opioid receptors are critical for tricyclic antidepressant treatment of neuropathic allodynia. Biol. Psychiatry 63, 633–636 (2008).
Onali, P., Dedoni, S. & Olianas, M. C. Direct agonist activity of tricyclic antidepressants at distinct opioid receptor subtypes. J. Pharmacol. Exp. Ther. 332, 255–265 (2010).
Connelly, W. M. et al. The histamine H4 receptor is functionally expressed on neurons in the mammalian CNS. Br. J. Pharmacol. 157, 55–63 (2009).
Nguyen, T. et al. Discovery of a novel member of the histamine receptor family. Mol. Pharmacol. 59, 427–433 (2001).
Di Matteo, V., De Blasi, A., Di Giulio, C. & Esposito, E. Role of 5-HT(2C) receptors in the control of central dopamine function. Trends. Pharmacol. Sci. 22, 229–232 (2001).
Liu, J., Reid, A. R. & Sawynok, J. Spinal serotonin 5-HT7 and adenosine A1 receptors, as well as peripheral adenosine A1 receptors, are involved in antinociception by systemically administered amitriptyline. Eur. J. Pharmacol. 698, 213–219 (2013).
Lucchelli, A. et al. The interaction of antidepressant drugs with enteric 5-HT7 receptors. Naunyn. Schmiedebergs Arch. Pharmacol. 362, 284–289 (2000).
Pithadia, A. B. & Jain, S. M. 5-Hydroxytryptamine receptor subtypes and their modulators with therapeutic potentials. J. Clin. Med. Res. 1, 72–80 (2009).
Wood, M. & Reavill, C. Aripiprazole acts as a selective dopamine D2 receptor partial agonist. Expert Opin. Investig. Drugs 16, 771–775 (2007).
Cosi, C. et al. Clozapine, ziprasidone and aripiprazole but not haloperidol protect against kainic acid-induced lesion of the striatum in mice, in vivo: role of 5-HT1A receptor activation. Brain Res. 1043, 32–41 (2005).
Shahid, M., Walker, G. B., Zorn, S. H. & Wong, E. H. Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J. Psychopharmacol. 23, 65–73 (2009).
Stahl, S. M. Mechanism of action of cariprazine. CNS Spectr. 21, 123–127 (2016).
McCormack, P. L. Cariprazine: first global approval. Drugs 75, 2035–2043 (2015).
Davies, M. A. et al. The highly efficacious actions of N-desmethylclozapine at muscarinic receptors are unique and not a common property of either typical or atypical antipsychotic drugs: is M1 agonism a pre-requisite for mimicking clozapine’s actions? Psychopharmacology 178, 451–460 (2005).
Wu, S. N. et al. Association of DRD2 polymorphisms and chlorpromazine-induced extrapyramidal syndrome in Chinese schizophrenic patients. Acta Pharmacol. Sin. 27, 966–970 (2006).
Vasudevan, S. R., Moore, J. B., Schymura, Y. & Churchill, G. C. Shape-based reprofiling of FDA-approved drugs for the H(1) histamine receptor. J. Med. Chem. 55, 7054–7060 (2012).
von Coburg, Y. et al. Potential utility of histamine H3 receptor antagonist pharmacophore in antipsychotics. Bioorg. Med. Chem. Lett. 19, 538–542 (2009).
Bolden, C., Cusack, B. & Richelson, E. Antagonism by antimuscarinic and neuroleptic compounds at the 5 cloned human muscarinic cholinergic receptors expressed in chinese-hamster ovary cells. J. Pharmacol. Exp. Ther. 260, 576–580 (1992).
Cusack, B., Nelson, A. & Richelson, E. Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology 114, 559–565 (1994).
Wander, T. J., Nelson, A., Okazaki, H. & Richelson, E. Antagonism by neuroleptics of serotonin 5-HT1A and 5-HT2 receptors of normal human brain in vitro. Eur. J. Pharmacol. 143, 279–282 (1987).
Morphy, R. & Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 48, 6523–6543 (2005).
Fernandez, J. et al. Discovery of new tetracyclic tetrahydrofuran derivatives as potential broad-spectrum psychotropic agents. J. Med. Chem. 48, 1709–1712 (2005).
Navailles, S., De Deurwaerdere, P. & Spampinato, U. Clozapine and haloperidol differentially alter the constitutive activity of central serotonin2C receptors in vivo. Biol. Psychiatry 59, 568–575 (2006).
Glennon, R. A. et al. 2-Substituted tryptamines: agents with selectivity for 5-HT(6) serotonin receptors. J. Med. Chem. 43, 1011–1018 (2000).
Taverne, T. et al. Novel benzothiazolin-2-one and benzoxazin-3-one arylpiperazine derivatives with mixed 5HT1A/D2 affinity as potential atypical antipsychotics. J. Med. Chem. 41, 2010–2018 (1998).
Vanover, K. E. et al. Pharmacological characterization of AC-90179 [2-(4-methoxyphenyl)-N-(4-methyl-benzyl)-N-(1-methyl-piperidin-4-yl)-acetamide hydrochloride]: a selective serotonin 2A receptor inverse agonist. J. Pharmacol. Exp. Ther. 310, 943–951 (2004).
Schotte, A. et al. Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology 124, 57–73 (1996).
Hagino, Y. & Watanabe, M. Effects of clozapine on the efflux of serotonin and dopamine in the rat brain: the role of 5-HT1A receptors. Can. J. Physiol. Pharmacol. 80, 1158–1166 (2002).
Zhao, A. L. et al. Dopamine D4 receptor gene exon III polymorphism and interindividual variation in response to clozapine. Int. J. Neurosci. 115, 1539–1547 (2005).
Heiser, P. et al. Effects of clozapine and its metabolites on the 5-HT2 receptor system in cortical and hippocampal cells in vitro. Prog. Neuropsychopharmacol. Biol. Psychiatry 28, 297–302 (2004).
Haubmann, C., Hubner, H. & Gmeiner, P. 2,2-Dicyanovinyl as a nonaromatic aryl bioisostere: synthesis, binding experiments and SAR studies of highly selective dopamine D4 receptor ligands. Bioorg. Med. Chem. Lett. 9, 1969–1972 (1999).
Takano, A. et al. Time course of dopamine D2 receptor occupancy by clozapine with medium and high plasma concentrations. Prog. Neuropsychopharmacol. Biol. Psychiatry 30, 75–81 (2006).
Glennon, R. A. Higher-end serotonin receptors: 5-HT(5), 5-HT(6), and 5-HT(7). J. Med. Chem. 46, 2795–2812 (2003).
Menon, D. V. et al. Central sympatholysis as a novel countermeasure for cocaine-induced sympathetic activation and vasoconstriction in humans. J. Am. Coll. Cardiol. 50, 626–633 (2007).
Kalkman, H. O., Subramanian, N. & Hoyer, D. Extended radioligand binding profile of iloperidone: a broad spectrum dopamine/serotonin/norepinephrine receptor antagonist for the management of psychotic disorders. Neuropsychopharmacology 25, 904–914 (2001).
Bobo, W. V. Asenapine, iloperidone and lurasidone: critical appraisal of the most recently approved pharmacotherapies for schizophrenia in adults. Expert Rev. Clin. Pharmacol. 6, 61–91 (2013).
Roth, B. L., Lopez, E., Patel, S. & Kroeze, W. K. The multiplicity of serotonin receptors: uselessly diverse molecules or an embarrassment of riches? Neuroscientist 6, 252–262 (2000).
Liao, Y. et al. New (sulfonyloxy)piperazinyldibenzazepines as potential atypical antipsychotics: chemistry and pharmacological evaluation. J. Med. Chem. 42, 2235–2244 (1999).
Herrick-Davis, K., Grinde, E. & Teitler, M. Inverse agonist activity of atypical antipsychotic drugs at human 5-hydroxytryptamine2C receptors. J. Pharmacol. Exp. Ther. 295, 226–232 (2000).
Hjerde, E., Dahl, S. G. & Sylte, I. Atypical and typical antipsychotic drug interactions with the dopamine D2 receptor. Eur. J. Med. Chem. 40, 185–194 (2005).
Phillips, S. T. et al. Binding of 5H-dibenzo [a,d] cycloheptene and dibenz [b,f] oxepin analogs of clozapine to dopamine and serotonin receptors. J. Med. Chem. 38, 708–714 (1995).
Li, Z., Ichikawa, J. & Meltzer, H. Y. A comparison of the effects of loxapine with ziprasidone and thioridazine on the release of dopamine and acetylcholine in the prefrontal cortex and nucleus accumbens. Psychopharmacology 167, 315–323 (2003).
Seeman, P. & Tallerico, T. Antipsychotic drugs which elicit little or no parkinsonism bind more loosely than dopamine to brain D2 receptors, yet occupy high levels of these receptors. Mol. Psychiatry 3, 123–134 (1998).
Tarazi, F. I. & Stahl, S. M. Iloperidone, asenapine and lurasidone: a primer on their current status. Expert. Opin. Pharmacother. 13, 1911–1922 (2012).
George, M. et al. Newer antipsychotics and upcoming molecules for schizophrenia. Eur. J. Clin. Pharmacol. 69, 1497–1509 (2013).
Green, B., Pettit, T., Faith, L. & Seaton, K. Focus on levomepromazine. Curr. Med. Res. Opin. 20, 1877–1881 (2004).
Teitler, M. et al. Clozapine and other competitive antagonists reactivate risperidone-inactivated h5-HT7 receptors: radioligand binding and functional evidence for GPCR homodimer protomer interactions. Psychopharmacology 212, 687–697 (2010).
Leysen, J. E., Janssen, P. M. F., Megens, A. A. H. P. & Schotte, A. Risperidone—a novel antipsychotic with balanced serotonin-dopamine antagonism, receptor occupancy profile, and pharmacological activity. J. Clin. Psychiat. 55, 5–12 (1994).
Cicek, E., Cicek, I. E. & Uguz, F. Bilateral pretibial edema associated with paliperidone palmitate long-acting injectable: a case report. Clin. Psychopharmacol. Neurosci. 15, 184–186 (2017).
Narita, M. et al. Suppression of dopamine-related side effects of morphine by aripiprazole, a dopamine system stabilizer. Eur. J. Pharmacol. 600, 105–109 (2008).
Strange, P. G. Antipsychotic drug action: antagonism, inverse agonism or partial agonism. Trends Pharmacol. Sci. 29, 314–321 (2008).
Kim, D. H., Maneen, M. J. & Stahl, S. M. Building a better antipsychotic: receptor targets for the treatment of multiple symptom dimensions of schizophrenia. Neurotherapeutics 6, 78–85 (2009).
Leung, J. Y. et al. Cardiovascular side-effects of antipsychotic drugs: the role of the autonomic nervous system. Pharmacol. Ther. 135, 113–122 (2012).
Corena-McLeod, M. Comparative pharmacology of risperidone and paliperidone. Drugs. R&D 15, 163–174 (2015).
Cincotta, S. L. & Rodefer, J. S. Emerging role of sertindole in the management of schizophrenia. Neuropsychiatr. Dis. Treat. 6, 429–441 (2010).
Mork, A., Witten, L. M. & Arnt, J. Effect of sertindole on extracellular dopamine, acetylcholine, and glutamate in the medial prefrontal cortex of conscious rats: a comparison with risperidone and exploration of mechanisms involved. Psychopharmacology 206, 39–49 (2009).
Lindstrom, E. & Levander, S. Sertindole: efficacy and safety in schizophrenia. Expert. Opin. Pharmacother. 7, 1825–1834 (2006).
Cavallotti, C., Nuti, F., Bruzzone, P. & Mancone, M. Age-related changes in dopamine D2 receptors in rat heart and coronary vessels. Clin. Exp. Pharmacol. Physiol. 29, 412–418 (2002).
Lipina, T. V. & Vishnivetskaia, G. B. [Effect of sulpiride on immobility reflex and pinch-induced catalepsy in CBA/Lac male mice with various social status]. Zhurnal vysshei nervnoi deiatelnosti imeni I P Pavlova 59, 482–487 (2009).
Ciszowski, K., Szpak, D. & Wilimowska, J. [Toxicity of sulpiride]. Przegl. Lek. 67, 606–609 (2010).
Seeman, P. Dopamine D2 receptors as treatment targets in schizophrenia. Clin. Schizophr. Relat. Psychoses. 4, 56–73 (2010).
Bymaster, F. P. et al. Muscarinic mechanisms of antipsychotic atypicality. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 1125–1143 (2003).
Ablordeppey, S. Y. et al. Identification of a butyrophenone analog as a potential atypical antipsychotic agent: 4-[4-(4-chlorophenyl)-1,4-diazepan-1-yl]-1-(4-fluorophenyl)butan-1-one. Bioorg. Med. Chem. 16, 7291–7301 (2008).
Rollema, H. et al. 5-HT(1A) receptor activation contributes to ziprasidone-induced dopamine release in the rat prefrontal cortex. Biol. Psychiatry 48, 229–237 (2000).
Graham, J. M. et al. 1-Aminoindanes as novel motif with potential atypical antipsychotic properties. Bioorg. Med. Chem. Lett. 18, 489–493 (2008).
Khalifa, A. E. Zuclopenthixol facilitates memory retrieval in rats: possible involvement of noradrenergic and serotonergic mechanisms. Pharmacol. Biochem. Behav. 75, 755–762 (2003).
Lublin, H. et al. Zuclopenthixol, a combined dopamine D1/D2 antagonist, versus haloperidol, a dopamine D2 antagonist, in tardive dyskinesia. Eur. Neuropsychopharmacol. 1, 541–548 (1991).
Horacek, J. et al. Mechanism of action of atypical antipsychotic drugs and the neurobiology of schizophrenia. CNS Drugs 20, 389–409 (2006).
Leucht, S., Pitschel-Walz, G., Engel, R. R. & Kissling, W. Amisulpride, an unusual “atypical” antipsychotic: a meta-analysis of randomized controlled trials. Am. J. Psychiatry 159, 180–190 (2002).
Rosenzweig, P. et al. A review of the pharmacokinetics, tolerability and pharmacodynamics of amisulpride in healthy volunteers. Hum. Psychopharmacol. 17, 1–13 (2002).
Tyson, P. J., Roberts, K. H. & Mortimer, A. M. Are the cognitive effects of atypical antipsychotics influenced by their affinity to 5HT-2A receptors? Int. J. Neurosci. 114, 593–611 (2004).
Abbas, A. I. et al. Amisulpride is a potent 5-HT7 antagonist: relevance for antidepressant actions in vivo. Psychopharmacology 205, 119–128 (2009).
Rehni, A. K., Singh, T. G. & Chand, P. Amisulpride-induced seizurogenic effect: a potential role of opioid receptor-linked transduction systems. Basic Clin. Pharmacol. Toxicol. 108, 310–317 (2011).
Weizman, T. et al. The antinociceptive effect of amisulpride in mice is mediated through opioid mechanisms. Eur. J. Pharmacol. 478, 155–159 (2003).
Xiberas, X. et al. In vivo extrastriatal and striatal D2 dopamine receptor blockade by amisulpride in schizophrenia. J. Clin. Psychopharmacol. 21, 207–214 (2001).
Burstein, E. S. et al. Intrinsic efficacy of antipsychotics at human D2, D3, and D4 dopamine receptors: identification of the clozapine metabolite N-desmethylclozapine as a D2/D3 partial agonist. J. Pharmacol. Exp. Ther. 315, 1278–1287 (2005).
Lim, H. D. et al. Evaluation of histamine H1-, H2-, and H3-receptor ligands at the human histamine H4 receptor: identification of 4-methylhistamine as the first potent and selective H4 receptor agonist. J. Pharmacol. Exp. Ther. 314, 1310–1321 (2005).
Glusa, E. & Pertz, H. H. Further evidence that 5-HT-induced relaxation of pig pulmonary artery is mediated by endothelial 5-HT(2B) receptors. Br. J. Pharmacol. 130, 692–698 (2000).
Nasu, R. et al. Quantitative prediction of catalepsy induced by amoxapine, cinnarizine and cyclophosphamide in mice. Biopharm. Drug Dispos. 21, 129–138 (2000).
Miller, G. M. et al. Primate trace amine receptor 1 modulation by the dopamine transporter. J. Pharmacol. Exp. Ther. 313, 983–994 (2005).
Xie, Z. & Miller, G. M. Trace amine-associated receptor 1 as a monoaminergic modulator in brain. Biochem. Pharmacol. 78, 1095–1104 (2009).
Xie, Z. & Miller, G. M. Trace amine-associated receptor 1 is a modulator of the dopamine transporter. J. Pharmacol. Exp. Ther. 321, 128–136 (2007).
Vallender, E. J., Xie, Z., Westmoreland, S. V. & Miller, G. M. Functional evolution of the trace amine associated receptors in mammals and the loss of TAAR1 in dogs. BMC Evol. Biol. 10, 51 (2010).
Siemian, J. N., Zhang, Y. & Li, J. X. Trace amine-associated receptor 1 agonists RO5263397 and RO5166017 attenuate quinpirole-induced yawning but not hypothermia in rats. Behav. Pharmacol. 28, 590–593 (2017).
Revel, F. G. et al. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl Acad. Sci. USA 108, 8485–8490 (2011).
Luptak, M. et al. Novel approaches in schizophrenia-from risk factors and hypotheses to novel drug targets. World J. Psychiatry 11, 277–296 (2021).
Wolinsky, T. D. et al. The Trace Amine 1 receptor knockout mouse: an animal model with relevance to schizophrenia. Genes Brain Behav. 6, 628–639 (2007).
Boido, A., Boido, C. C. & Sparatore, F. Synthesis and pharmacological evaluation of aryl/heteroaryl piperazinyl alkyl benzotriazoles as ligands for some serotonin and dopamine receptor subtypes. II Farmaco 56, 263–275 (2001).
Malt, E. A. et al. Altered dopamine D2 receptor function in fibromyalgia patients: a neuroendocrine study with buspirone in women with fibromyalgia compared to female population based controls. J. Affect. Disord. 75, 77–82 (2003).
Osei-Owusu, P. & Scrogin, K. E. Buspirone raises blood pressure through activation of sympathetic nervous system and by direct activation of alpha1-adrenergic receptors after severe hemorrhage. J. Pharmacol. Exp. Ther. 309, 1132–1140 (2004).
Howland, R. H. Buspirone: Back to the Future. J. Psychosoc. Nurs. Ment. Health Serv. 53, 21–24 (2015).
Pache, D. M., Fernandez-Perez, S. & Sewell, R. D. Buspirone differentially modifies short-term memory function in a combined delayed matching/non-matching to position task. Eur. J. Pharmacol. 477, 205–211 (2003).
Pastoor, D. & Gobburu, J. Clinical pharmacology review of escitalopram for the treatment of depression. Expert Opin. Drug Metab. Toxicol. 10, 121–128 (2014).
Bartlett, D. Drug-induced serotonin syndrome. Crit. Care Nurse. 37, 49–54 (2017).
Fisar, Z. Drugs related to monoamine oxidase activity. Prog. Neuropsychopharmacol. Biol. Psychiatry 69, 112–124 (2016).
Moore, N., Verdoux, H. & Fantino, B. Prospective, multicentre, randomized, double-blind study of the efficacy of escitalopram versus citalopram in outpatient treatment of major depressive disorder. Int. Clin. Psychopharmacol. 20, 131–137 (2005).
Baldwin, D. S. et al. Evidence-based guidelines for the pharmacological treatment of anxiety disorders: recommendations from the British Association for Psychopharmacology. J. Psychopharmacol. 19, 567–596 (2005).
Nevels, R. M., Gontkovsky, S. T. & Williams, B. E. Paroxetine-the antidepressant from hell? probably not, but caution required. Psychopharmacol. Bull. 46, 77–104 (2016).
Zhang, D. et al. Paroxetine in the treatment of premature ejaculation: a systematic review and meta-analysis. BMC Urol. 19, 2 (2019).
Altamura, A. C. et al. Understanding the pharmacokinetics of anxiolytic drugs. Expert Opin. Drug Metab. Toxicol. 9, 423–440 (2013).
Spahr, L. et al. Histamine H1 blocker hydroxyzine improves sleep in patients with cirrhosis and minimal hepatic encephalopathy: a randomized controlled pilot trial. Am. J. Gastroenterol. 102, 744–753 (2007).
Sugimoto, Y., Inoue, K. & Yamada, J. The tricyclic antidepressant clomipramine increases plasma glucose levels of mice. J. Pharmacol. Sci. 93, 74–79 (2003).
Hentall, I. D., Kurle, P. J. & White, T. R. Correlations between serotonin level and single-cell firing ln the rat’s nucleus raphe magnus. Neuroscience 95, 1081–1088 (2000).
Burgi, S., Baltensperger, K. & Honegger, U. E. Antidepressant-induced switch of beta 1-adrenoceptor trafficking as a mechanism for drug action. J. Biol. Chem. 278, 1044–1052 (2003).
Samnick, S. et al. Technetium-99m labeled 1-(4-fluorobenzyl)-4-(2-mercapto-2-methyl-4-azapentyl)-4-(2-mercapto-2-methylpropylamino)-piperidine and iodine-123 metaiodobenzylguanidine for studying cardiac adrenergic function: a comparison of the uptake characteristics in vascular smooth muscle cells and neonatal cardiac myocytes, and an investigation in rats. Nucl. Med. Biol. 31, 511–522 (2004).
Osadchii, O. E., Woodiwiss, A. J., Deftereos, D. & Norton, G. R. Temporal changes in myocardial adrenergic regulation with the progression to pump dysfunction after chronic beta-adrenoreceptor activation in rats. Pflugers Arch. 455, 251–260 (2007).
Prenner, L. et al. Reduction of high-affinity beta2-adrenergic receptor binding by hyperforin and hyperoside on rat C6 glioblastoma cells measured by fluorescence correlation spectroscopy. Biochemistry 46, 5106–5113 (2007).
Gillman, P. K. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br. J. Pharmacol. 151, 737–748 (2007).
Stahl, S. M. Selective histamine H1 antagonism: novel hypnotic and pharmacologic actions challenge classical notions of antihistamines. CNS Spectr. 13, 1027–1038 (2008).
Singh, H. & Becker, P. M. Novel therapeutic usage of low-dose doxepin hydrochloride. Expert Opin. Investig. Drugs 16, 1295–1305 (2007).
Wellman, P. J., Miller, D. K. & Ho, D. H. Noradrenergic modulation of ephedrine-induced hypophagia. Synapse 48, 18–24 (2003).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
Ma, G. et al. Pharmacological effects of ephedrine alkaloids on human alpha(1)- and alpha(2)-adrenergic receptor subtypes. J. Pharmacol. Exp. Ther. 322, 214–221 (2007).
Reimold, M. et al. Occupancy of dopamine D(1), D (2) and serotonin (2A) receptors in schizophrenic patients treated with flupentixol in comparison with risperidone and haloperidol. Psychopharmacology 190, 241–249 (2007).
Mahapatra, J. et al. Flupenthixol decanoate (depot) for schizophrenia or other similar psychotic disorders. Cochrane Database Syst. Rev 2014, CD001470 (2014).
Li, X., Murray, W. V., Jolliffe, L. & Pulito, V. Novel arylpiperazines as selective alpha1-adrenergic receptor antagonists. Bioorg. Med. Chem. Lett. 10, 1093–1096 (2000).
Peddi, S., Roth, B. L., Glennon, R. A. & Westkaemper, R. B. Structural determinants for high 5-HT(2A) receptor affinity of spiro[9,10-dihydroanthracene]-9,3(‘)-pyrrolidine (SpAMDA). Bioorg. Med. Chem. Lett. 14, 2279–2283 (2004).
Zanoveli, J. M., Nogueira, R. L. & Zangrossi, H. Jr Chronic imipramine treatment sensitizes 5-HT1A and 5-HT 2 A receptors in the dorsal periaqueductal gray matter: evidence from the elevated T-maze test of anxiety. Behav. Pharmacol. 16, 543–552 (2005).
Csaba, G., Kovacs, P. & Pallinger, E. Prolonged effect of the tricyclic antidepressant, mianserin on the serotonin and histamine content of young rats’ white blood cells and mast cells. A case of late-imprinting. Pharmacol. Res. 48, 457–460 (2003).
Grinshpoon, A., Valevski, A., Moskowitz, M. & Weizman, A. Beneficial effect of the addition of the 5-HT 2A/2C and alpha2 antagonist mianserin to ongoing haloperidol treatment in drug-resistant chronically hospitalized schizophrenic patients. Eur. Psychiatry 15, 388–390 (2000).
Dekeyne, A., Iob, L. & Millan, M. J. Discriminative stimulus properties of the selective and highly potent alpha(2)-adrenoceptor agonist, S18616, in rats. Behav. Pharmacol. 14, S41–S41 (2003).
Millan, M. J. Serotonin 5-HT2C receptors as a target for the treatment of depressive and anxious states: focus on novel therapeutic strategies. Therapie 60, 441–460 (2005).
Lee, H. Y. et al. Association of the adrenergic alpha 2a receptor-1291C/G polymorphism with weight change and treatment response to mirtazapine in patients with major depressive disorder. Brain Res. 1262, 1–6 (2009).
Waldinger, M. D., Berendsen, H. H. & Schweitzer, D. H. Treatment of hot flushes with mirtazapine: four case reports. Maturitas 36, 165–168 (2000).
Peckham, A. M., De La Cruz, A. & Dufresne, R. L. Kappa opioid receptor antagonism: Are opioids the answer for treatment resistant depression? Ment. Health Clin. 8, 175–183 (2018).
Salvi, V., Mencacci, C. & Barone-Adesi, F. H1-histamine receptor affinity predicts weight gain with antidepressants. Eur. Neuropsychopharmacol. 26, 1673–1677 (2016).
Talvik, M. et al. A cross-validation study on the relationship between central D2 receptor occupancy and serum perphenazine concentration. Psychopharmacology175, 148–153 (2004).
Dolzan, V. et al. Polymorphisms in dopamine receptor DRD1 and DRD2 genes and psychopathological and extrapyramidal symptoms in patients on long-term antipsychotic treatment. Am. J. Med. Genet. B 144B, 809–815 (2007).
Hoffmann, C. et al. Comparative pharmacology of human beta-adrenergic receptor subtypes-characterization of stably transfected receptors in CHO cells. Naunyn. Schmiedebergs Arch. Pharmacol. 369, 151–159 (2004).
Rubenstein, L. A., Zauhar, R. J. & Lanzara, R. G. Molecular dynamics of a biophysical model for beta2-adrenergic and G protein-coupled receptor activation. J. Mol. Graph. Model. 25, 396–409 (2006).
Ozdogan, U. K., Lahdesmaki, J., Hakala, K. & Scheinin, M. The involvement of alpha 2A-adrenoceptors in morphine analgesia, tolerance and withdrawal in mice. Eur. J. Pharmacol. 497, 161–171 (2004).
Xie, Z. et al. Rhesus monkey trace amine-associated receptor 1 signaling: enhancement by monoamine transporters and attenuation by the D2 autoreceptor in vitro. J. Pharmacol. Exp. Ther. 321, 116–127 (2007).
Liu, J. F. & Li, J. X. TAAR1 in addiction: looking beyond the tip of the iceberg. Front. Pharmacol. 9, 279 (2018).
Pei, Y., Asif-Malik, A. & Canales, J. J. Trace amines and the trace amine-associated receptor 1: pharmacology, neurochemistry, and clinical implications. Front. Neurosci. 10, 148 (2016).
Ferry, G. et al. Old and new inhibitors of quinone reductase 2. Chem. Biol. Interact. 186, 103–109 (2010).
Davies, S. J. et al. Sequential drug treatment algorithm for agitation and aggression in Alzheimer’s and mixed dementia. J. Psychopharmacol. 32, 509–523 (2018).
Millan, M. J., Girardon, S., Monneyron, S. & Dekeyne, A. Discriminative stimulus properties of the dopamine D3 receptor agonists, PD128,907 and 7-OH-DPAT: a comparative characterization with novel ligands at D3 versus D2 receptors. Neuropharmacology 39, 586–598 (2000).
Martinez, D. et al. Cocaine dependence and d2 receptor availability in the functional subdivisions of the striatum: relationship with cocaine-seeking behavior. Neuropsychopharmacology 29, 1190–1202 (2004).
Lehmann, A. GABAB receptors as drug targets to treat gastroesophageal reflux disease. Pharmacol. Ther. 122, 239–245 (2009).
Chaudhry, B. Z., Cohen, J. A. & Conway, D. S. Sphingosine 1-phosphate receptor modulators for the treatment of multiple sclerosis. Neurotherapeutics 14, 859–873 (2017).
Scatton, B. et al. The preclinical pharmacologic profile of tiapride. Eur. Psychiatry 16, 29s–34s (2001).
Dawson, L. A. et al. In vitro and in vivo characterization of the non-peptide NK3 receptor antagonist SB-223412 (talnetant): potential therapeutic utility in the treatment of schizophrenia. Neuropsychopharmacology 33, 1642–1652 (2008).
Deeks, E. D. & Keating, G. M. Blonanserin: a review of its use in the management of schizophrenia. CNS Drugs 24, 65–84 (2010).
Litman, R. E. et al. The selective neurokinin 3 antagonist AZD2624 does not improve symptoms or cognition in schizophrenia: a proof-of-principle study. J. Clin. Psychopharmacol. 34, 199–204 (2014).
Morales, P., Hurst, D. P. & Reggio, P. H. in Phytocannabinoids: Unraveling the Complex Chemistry and Pharmacology of Cannabis Sativa, Vol 103 Progress in the Chemistry of Organic Natural Products-Series (eds Kinghorn, A. D. et al.) 103–131 (Springer, 2017).
Langlois, X. et al. Pharmacology of JNJ-37822681, a specific and fast-dissociating D2 antagonist for the treatment of schizophrenia. Eur. Neuropsychopharmacol. 20, S502–S502 (2010).
Dedic, N. et al. SEP-363856, a novel psychotropic agent with a unique, non-D(2) receptor mechanism of action. J. Pharmacol. Exp. Ther. 371, 1–14 (2019).
Nyandege, A., Kolanos, R., Roth, B. L. & Glennon, R. A. Further studies on the binding of N1-substituted tryptamines at h5-HT6 receptors. Bioorg. Med. Chem. Lett. 17, 1691–1694 (2007).
Su, T. P., Hayashi, T. & Vaupel, D. B. When the endogenous hallucinogenic trace amine N,N-dimethyltryptamine meets the sigma-1 receptor. Sci. Signal. 2, pe12 (2009).
Rusch, D. et al. Subunit-dependent modulation of the 5-hydroxytryptamine type 3 receptor open-close equilibrium by n-alcohols. J. Pharmacol. Exp. Ther. 321, 1069–1074 (2007).
Riga, M. S. et al. The serotonergic hallucinogen 5-methoxy-N,N-dimethyltryptamine disrupts cortical activity in a regionally-selective manner via 5-HT(1A) and 5-HT(2A) receptors. Neuropharmacology 101, 370–378 (2016).
Di Fabio, R. et al. Discovery and biological characterization of (2R,4S)-1’-acetyl-N-(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl-2-(4-fluoro-2-methylphenyl)-N-methyl-4,4’-bipiperidine-1-carboxamide as a new potent and selective neurokinin 1 (NK1) receptor antagonist clinical candidate. J. Med. Chem. 54, 1071–1079 (2011).
Acknowledgements
The project is supported by grants from Science, Technology and Innovation Commission of Shenzhen Municipality (Project code JCYJ20200109150019113, GXWD20201231105722002); the Kobilka Institute of Innovative Drug Discovery and Presidential Fellowship at the Chinese University of Hong Kong, Shenzhen, China; National Natural Science Foundation of China (Project code 32271263 to Y.D., Project code 82173690 to S.L., 81825020 and 82150208 to H.L.); and the Lingang Laboratory (Project code LG-QS-202206-02 to S.L.)
Author information
Authors and Affiliations
Contributions
T.S.W., G.L., and S.L. prepared and revised the manuscript. T.S.W., W.G., G.C., S.G., and M.Z. contributed to the writing and figures/tables preparation. T.S.W., S.W., and H.L. reviewed and revised the manuscript. Y.D. supervised the project, designed the content and revised the manuscript. All authors contributed to the article. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wong, TS., Li, G., Li, S. et al. G protein-coupled receptors in neurodegenerative diseases and psychiatric disorders. Sig Transduct Target Ther 8, 177 (2023). https://doi.org/10.1038/s41392-023-01427-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01427-2
This article is cited by
-
Microbiota–gut–brain axis and its therapeutic applications in neurodegenerative diseases
Signal Transduction and Targeted Therapy (2024)
-
GPR37 processing in neurodegeneration: a potential marker for Parkinson’s Disease progression rate
npj Parkinson's Disease (2024)
-
Pathology of pain and its implications for therapeutic interventions
Signal Transduction and Targeted Therapy (2024)
-
First report on chemometrics-driven multilayered lead prioritization in addressing oxysterol-mediated overexpression of G protein-coupled receptor 183
Molecular Diversity (2024)
-
Structure, function and drug discovery of GPCR signaling
Molecular Biomedicine (2023)
