
Abstract
Sirtuins (SIRTs) are nicotine adenine dinucleotide(+)-dependent histone deacetylases regulating critical signaling pathways in prokaryotes and eukaryotes, and are involved in numerous biological processes. Currently, seven mammalian homologs of yeast Sir2 named SIRT1 to SIRT7 have been identified. Increasing evidence has suggested the vital roles of seven members of the SIRT family in health and disease conditions. Notably, this protein family plays a variety of important roles in cellular biology such as inflammation, metabolism, oxidative stress, and apoptosis, etc., thus, it is considered a potential therapeutic target for different kinds of pathologies including cancer, cardiovascular disease, respiratory disease, and other conditions. Moreover, identification of SIRT modulators and exploring the functions of these different modulators have prompted increased efforts to discover new small molecules, which can modify SIRT activity. Furthermore, several randomized controlled trials have indicated that different interventions might affect the expression of SIRT protein in human samples, and supplementation of SIRT modulators might have diverse impact on physiological function in different participants. In this review, we introduce the history and structure of the SIRT protein family, discuss the molecular mechanisms and biological functions of seven members of the SIRT protein family, elaborate on the regulatory roles of SIRTs in human disease, summarize SIRT inhibitors and activators, and review related clinical studies.
Similar content being viewed by others
Introduction
The sirtuin (SIRT) protein family, which are conserved proteins belonging to class III histone deacetylases, comprises seven members.1 Notably, SIRTs share a nicotine adenine dinucleotide + (NAD) + -binding catalytic domain and may act specifically on different substrates depending on the biological processes in which they are involved.2 The sequence and length of SIRTs are different in both their N- and C-terminal domains, partially explaining their different localization and functions.2 Recently, more and more studies have shown their association with and involvement in different pathologies, such as (but not restricted to) cancer and cardiovascular diseases (CVDs).3,4,5,6 Additionally, increasing evidence supported the potential use of SIRT modulators for the treatment of different kinds of diseases,7,8,9,10,11 suggesting the critical roles of SIRTs in the diseases. Herein, to enhance our understanding of SIRTs, we provide a comprehensive summary of the roles of SIRTs in health and various diseases.
Historical review and structure of SIRT proteins
The history of SIRTs can be traced to founding member Sir2 nearly 40 years ago, which was first discovered in the budding Saccharomyces cerevisiae, and was originally known as mating-type regulator 1 protein.12 Subsequently, Sir2 has been found to function in transcriptional repression at ribosomal DNA loci,13 at silent mating-type loci14 and in telomeres,15 and this increasing knowledge has greatly improved exploration of its function. In the late 1990s, a study confirmed that Sir2 prolonged the lifespan of yeast by inhibiting genomic instability. Loss of Sir2 significantly shortened the lifespan of yeast, while an additional copy of Sir2 prolonged it by about 40%.16 Later evidence showed that Sir2 had NAD + -dependent HDAC enzymatic activity, which provided a molecular framework in which NAD-dependent histone deacetylation could be connected to genomic silencing and ageing in yeast, and possibly to higher eukaryotic metabolism as well, opening a new chapter of Sir2 enzymology.17 Sir2’s key role in the molecular mechanism of senescence in Caenorhabditis elegans was also later demonstrated.18 As Sir2 homologous genes have been successively isolated in bacteria, plants and mammals, the Sir2 homologous proteins in all species have been collectively referred to as SIRTs.19,20
Currently, seven mammalian homologs of yeast Sir2 named SIRT1 to SIRT7 have been identified, which are well-known as the β-NAD + or NAD + -dependent enzymes.21,22,23 Figure 1 shows a historical timeline summarizing studies on milestones in SIRT family members. Regarding to the molecular structures, SIRT1-7 share a chemically and structurally conserved catalytic core in general and there may be subtle differences in the infrastructure of active site.24 In detail, X-ray crystalline diffraction reveals that the catalytic core includes two bilobed globular domains consisting of approximately 275 amino acids residues, characterized by their necessity for NAD as a cofactor. The different N- and C-terminals of SIRT proteins are fairly variable in length, chemical composition, susceptibility to post-translational modifications (PTMs) (typically phosphorylation), and enable them to bind substrates.2,25,26 The large structural domain is composed of an inverted classical open α/β Rossmann-fold structure, which is a parallel β-sheet nucleotide-binding fold typical of many NAD-utilizing enzymes such as dehydrogenases; in addition, a smaller domain contains a zinc ribbon motif. These two domains form a pocket in the middle where NAD and acetylated peptides bind.2,27

The historical timeline on milestones in SIRT family members
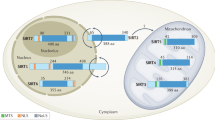
Differences among members of the SIRT protein family were initially attributed to their discrete pattern of subcellular localization.28 As far as we know, SIRT1 is mainly localized in the nucleus and shuttles to the cytosol under specific circumstances.29,30 SIRT2 is predominantly cytosolic but also exists in the nucleus in the G2 to M phase transition of the cell cycle.31 SIRT3-5 localize primarily to mitochondria, and have a mitochondrial targeting sequence.32,33,34 Additionally, SIRT6 and SIRT7 are nuclear proteins. Of them, SIRT6 is principally located in the chromatin and SIRT7 is mostly found in the nucleolus.35,36 Additionally, the localization and subcellular shuttling of SIRTs depend on different kinds of cell types and cell cycle oscillation.37 For example, SIRT1 could be primarily located in the cytosol in some subsets of neurons, as well as expressed in both nucleus and cytosol in ependymal cells.30 Moreover, SIRT2 is in the cytosol during most phases of cell cycle, while SIRT2 is expressed in nucleus and associates with chromatin and deacetylates the histone H4K16 during G2/M transition and mitosis.31
The catalytic activity level of SIRT protein family members is thought to be their second most significant difference. Of note, the regulation of catalytic activity of SIRTs involves multiple steps: (a) NAD + and acetyl lysine substrates binding; (b) the glycosidic bond cleavage; (c) acetyl transfer; and (d) O-acetyl-ADPR, nicotinamide, and deacetylated lysine products formation. Concretely, the initial reaction of NAD + glycosidic bond cleavage is proceeded through either an SN1-like mechanism, as supported by the structure of Hst2 bound to carba-NAD + ,38 or an SN2-like mechanism, as supported by the structure of Sir2Tm bound to NAD+ and an acetyl lysine-containing peptide.39 Furthermore, available studies suggested a complex array of PTMs regulated by SIRTs. Initially, Sir2 was considered solely as a deacetylase enzyme.17 However, the functional range of enzymatic activities of SIRTs has been greatly expanded in mammals. SIRT1-3 sustain strong deacetylase activities. SIRT4 has ADP-ribose transferase activity and can down-regulate glutamate dehydrogenase activity in β cells, thereby reducing insulin secretion response.33 SIRT5 is involved in regulating protein post translational modifications such as lysine succinylation, malonylation, and glutarylation, etc.40,41 Moreover, SIRT6 can function as NAD + -dependent monoADP-ribosyl transferase and long-chain fatty acyl deacetylases.42,43 Meanwhile, SIRT7, the latest discovered SIRT family protein, has been relatively less studied, which was first found to be a β-NAD + -dependent deacetylase enzyme and is localized in nucleoli that govern the transcription of RNA polymerase I.44,45 Numerous target proteins, including histone and non-histone, have been shown to be modified by SIRTs, and participates in the regulation of multiple fundamental cellular functions including glucose, and lipid metabolism, mitochondrial biogenesis, DNA repair, oxidative stress, apoptosis, and inflammation.46 Hence, SIRTs are now recognized as a major regulator of cellular physiology. Nevertheless, the SIRT protein family still has multiple proven and unproven catalytic modification activities. Given our current limited understanding of the SIRT protein family, more investigation is warranted in this area.
The regulatory role of SIRTs in cellular biology
The role of SIRTs in inflammation
Inflammation is an essential immune response that enables survival during infection or injury and maintains tissue homeostasis under a variety of noxious conditions.47 It comes at the cost of a transient decline in tissue function, which can in turn contribute to the pathogenesis of diseases involving altered homeostasis and a variety of physiological and pathological processes.48 The molecular process of inflammation is varied and depends on the type of inflamed cells and organs. The inflammatory response is composed of several inseparable pathways involving inflammatory cells, inflammatory mediators induced by sensor cells, inflammatory pathway components, and the target tissues that are affected by the inflammatory mediators.47 Recently, with greater in-depth understanding of the process of inflammation, numerous studies have successfully illustrated how the SIRT protein family has a close association with inflammation. In this section, we summarize the role of the SIRT family in the inflammatory response and the major signaling pathways (Fig. 2).

Overview of the roles of SIRTs in inflammation. a SIRTs mainly play an anti-inflammatory effect by regulating inflammatory mediators, however, early inhibition of SIRT2 may prevent neuroinflammation evidenced by reduced levels of GFAP, IL-β, IL-6, and TNF-α; (b) SIRTs could negatively regulate several pro-inflammatory cytokines; (c) SIRTs are involved in the regulation of NF-κB signaling pathway. https://biorender.com. ABCA1 ATP‑binding cassette A1, ABCG1 ATP‑binding cassette G1, Arf alternative reading frame, CaMKKβ Ca(2 + )/calmodulin-dependent protein kinase kinase β, CCR7 C‑C chemokine receptor type 7, CRIF1 CR6-interacting factor1, CTLA4 cytotoxic T lymphocyte–associated antigen 4, CTRP1 C1q/tumor necrosis factor-related protein 1, DBC1 deleted in breast cancer 1, DEPTOR DEP-domain containing mTOR-interacting protein, DMP1 dentin matrix protein-1, Ebi3 Epstein-Barr virus–induced gene 3, FGF21 fibroblast growth factor 21, FXR farnesoid X receptor, GFAP glial fibrillary acidic protein, HIF-α hypoxia-inducible factor-alpha, HMGB1 high-mobility group box 1, HNF4α hepatocyte nuclear factor 4α, HO1 heme oxygenase-1, ICOS inducible T cell co-stimulator, IFN-γ interferon-γ, IKKβ inhibitor kappa B kinaseβ, IRAK interleukin-1 receptor-associated kinase, IRF9 interferon regulatory factor 9, LXR liver X receptor, MCP monocyte chemotactic protein, MCPIP1 MCP-1 induced protein, MIP-2 macrophage inflammatory protein-2, MKP-1 mitogen-activated protein kinase phosphatase-1, NT5C3A pyrimidine 5'-nucleotidase, PAI-1 plasminogen activator inhibitor-1, PARP-1 peroxisome proliferator-activated receptor 1, PGRN progranulin, RORγt RAR-related orphan receptor γ-t, TAK1 transforming growth factor β activated kinase-1, TM thrombomodulin, VCAM-1 vascular cell adhesion molecule-1, XBP1 X-box binding protein 1
The effect of SIRTs in inflammatory cells
The cells involved in the inflammatory response include inflammatory cells such as macrophages, mast cells and endothelial cells. SIRTs, especially SIRT1 and SIRT6, can affect the secretion of inflammatory mediators and play a central role in regulating the differentiation of dendritic cells (DCs) and the activation of macrophages.49,50 For example, SIRT1 participates in mediating inflammatory signaling in DCs, consequentially modulating the balance of proinflammatory T helper type 1 cells and anti-inflammatory Foxp3(+) regulatory T cells. SIRT1 knockout (KO) in DCs restrained the generation of regulatory T cells while driving T helper 1 cell development, resulting in enhanced T-cell-mediated inflammation against microbial responses.49 Moreover, SIRT6 deficiency in macrophages resulted in inflammation with increases in acetylation and greater stability of the forkhead box protein O1 (FoxO1). Conversely, the ectopic overexpression of SIRT6 in KO cells reduced the inflammatory response.50 Moreover, results from in vivo experiments demonstrated that SIRT3 overexpression in transfused macrophages not only induced M2 macrophage polarization, but also alleviated inflammation.51 Based on these current studies, the SIRT family may regulate the activation or differentiation of inflammatory cells, such as DCs and macrophages in the immune system.
The effect of SIRTs on inflammatory mediators
Inflammatory mediators are chemicals produced during inflammation that cause an inflammatory response. In response to the inflammatory process, inflammatory cells release specialized substances, including vasoactive amines and peptides, eicosanoids, proinflammatory cytokines and acute-phase proteins, which mediate the inflammatory process by preventing further tissue damage and ultimately resulting in healing and restoration of tissue function.52 Overexpressed or activated SIRTs, mainly SIRT1–3, can reduce the inflammatory response through anti-inflammatory effects, such as tumor necrosis factor-alpha (TNF-α), a multifunctional pro-inflammatory cytokine, which is produced by macrophages/monocytes during acute inflammation, and plays a critical role with orchestrating the cytokine cascade in various inflammatory diseases.53 For instance, increased SIRT1 protein expression can reduce acetylation of the nuclear factor kappa-B (NF-κB) p65 subunit, which results in the suppression of TNF-α-induced NF-κB transcriptional activation and reduction of TNF-α secretion in a SIRT1-dependent manner.54,55 In addition, SIRT1 knockdown increased, while SIRT1 activator treatment decreased TNF-α secretion from macrophages.55 One recent study verified that SIRT6 suppressed inflammatory responses and downregulated the expression of inflammatory factors interleukin (IL)-6 and TNF-α via the NF-κB pathway.56 For example, both SIRT1 and SIRT6 inhibited TNF-α-induced inflammation of vascular adventitial fibroblasts through reactive oxygen species (ROS) and the protein kinase B (Akt) signaling pathway.57 SIRT1 exerted anti-inflammatory effects against IL-1β-mediated pro-inflammatory stress through the Toll-like receptor 2 (TLR2)/SIRT1/NF-κB pathway.58 SIRT1 deficiency increased microvascular inflammation in obese septic mice, while resveratrol treatment decreased leukocyte/platelet adhesion and E-selectin/intercellular adhesion molecule (ICAM-1) expression accompanied by increased SIRT1 expression and improved survival.59 In addition, SIRT1 and SIRT6 inhibited inflammation by decreasing pro-inflammatory cytokines such as IL-6, IL-β, cytochrome oxidase subunit 2 and ICAM-1.60 Moreover, SIRT1 exerted anti-inflammatory effects against IL-1β-mediated pro-inflammatory stress through the TLR2/SIRT1/NF-κB pathway.58 SIRT1 deficiency increased microvascular inflammation in obese septic mice, while resveratrol treatment decreased leukocyte/platelet adhesion and E-selectin/ICAM-1 expression accompanied by increased SIRT1 expression and improved survival.59 Recently, SIRT2 as modulators have been shown to be effective in inhibiting lipopolysaccharide-stimulated production of TNF-α to suppress neuroinflammation.61,62 Moreover, Kurundkar et al. have determined that SIRT3 deficiency altered the proinflammatory responses of macrophages to lipopolysaccharides, with a greater increase in TNF-α production.63 Several studies have also shown an anti-inflammatory effect of SIRT3, which downregulates IL-1β and IL-18, inhibits inflammasomes and attenuates oxidative stress.64,65 SIRT3 KO mice have significantly increased inflammatory cell infiltration.66 These studies highlight the critical role of SIRT3 in the process of inflammation. In conclusion, then, as one of the most important pro-inflammatory cytokines, inflammatory mediators are closely regulated by the SIRT protein family and is widely involved in inflammation.
Currently, the SIRT family mainly exerts an anti-inflammatory effect in response to tissue stress or disease development, but there are exceptions. For example, early SIRT2 inhibition prevented neuroinflammation evidenced by reduced levels of glial fibrillary acidic protein, IL-1β, IL-6 and TNF-α and by increased levels of glutamate receptor subunits GluN2A, GluN2B and GluA1; however, SIRT2 inhibition was unable to reverse cognitive decline or neuroinflammation.67 In this case, SIRT2 exhibited a temporary proinflammatory effect. Furthermore, both pro- and anti-inflammatory effects have been attributed to SIRT2 and SIRT3.68 Single deficiency of SIRT2 or SIRT3 had minor or no impact on the antimicrobial innate immune responses, while SIRT2/3−/− macrophages secreted increased levels of both proinflammatory and anti-inflammatory cytokines.68 From these results, then, most SIRT proteins appear to play anti-inflammatory roles, but limited reports have found the opposite effect, as just described for SIRT2. These inconsistent results might be due to the specificity of SIRT2 mechanisms in the SIRT family, or may be temporary effects manifested at different stages of the disease process. Therefore, more research is needed to explore the reasons for these discrepancies.
Overall, SIRTs can act in concert or compensate each other for certain immune functions.68 It is also worth noting that the effects of various SIRTs may differ between diseases, or even have opposite effects. Therefore, research on SIRTs has left a number of gaps which require further exploration to pinpoint the role of the SIRT family in inflammatory responses and the underlying mechanisms of action, which may account for the different results.
The effect of SIRTs on inflammatory pathway components
The signaling pathway of inflammation is complex, but inflammatory pathway components have begun to be elucidated over the past several years. Currently, there are many studies on the mechanisms by which the SIRT family participates in inflammation, especially pathways involving NF-κB, TNF-α, and the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome.
NF-κB is considered to be the central regulator of inflammation, which drives the expression of cytokines, chemokines, inflammasome components and adhesion molecules.69 It is mainly involved in immune and inflammatory responses and can induce the expression of downstream inflammatory cytokines.70,71 TNF-α is a pro-inflammatory cytokine mainly produced by macrophages and monocytes and is involved in normal inflammatory and immune responses.72 As an important component of innate immunity, the NLRP3 inflammasome plays an important role in the body’s immune response and inflammatory cell death (pyroptosis).73 In the following sections, we detail the role of the SIRT family as it affects three key inflammatory pathway components.
-
(1)
Majority of SIRTs exert anti-inflammatory effects by inhibiting the NF-κB pathway
NF-κB exists in multiple forms, with the heterodimer of p65 (RelA, Rel associated protein) and p50 subunits (p65/p50) being the most prevalent species.74 In the absence of stimulation, NF-κB is normally present in the cytoplasm in an inactive form. Upon stimulation by various pro-inflammatory cytokines (such as IL-1β, IL-6 and TNF-α), NF-κB rapidly translocates to the nucleus and regulates the transcription or expression of target genes.75,76 In addition, NF-κB activity can be modulated by PTMs of proteins, such as acetylation.77 Most members of the SIRT family are involved in regulation of the NF-κB pathway, primarily including SIRT1, SIRT2, SIRT6, and SIRT7.
Growing evidence suggests the significant role of SIRTs in the regulation of inflammation. SIRT1 has anti-inflammatory effects mediated by the deacetylation and inactivation of the p65 subunit of NF-κB.78 SIRT1 inhibits the transcriptional activity of NF-κB via deacetylation of the p65 (RelA) subunit at Ac-Lys310.78 Furthermore, the finding that lower SIRT1 activity levels may increase the expression of NF-κB, thus driving inflammation,79 also highlight the important role of SIRT1 during inflammation.
Repression of NF-κB activity is responsible for the anti-inflammatory effect of SIRT6.80 For instance, SIRT6 attenuated NF-κB expression by deacetylating histone H3K9 in the promoters of NF-κB target genes, hence decreasing inflammation.80 Additionally, SIRT6 overexpression suppressed NF-κB-mediated inflammatory responses in OA development.81 Since nuclear SIRT1 and SIRT6 deacetylate RelA/p65 and support its degradation by the proteasome, decreases in both SIRT1 or SIRT6 levels/activity increase NF-κB activity and amplify pro-inflammatory gene expression during chronic inflammation.82
Evidence concerning the role of SIRT7 in inflammatory processes has been somewhat inconsistent. In terms of mediating an anti-inflammatory response, knockdown of SIRT7 promoted the translocation of NF-κB p-p65 to the nucleus and subsequently increased the secretion of downstream inflammatory cytokines, while SIRT7 overexpression had the opposite effect.83,84 However, evidence also suggested that loss of SIRT7 promoted the translocation of NF-κB p65 to the cytoplasm.85 Thus, the roles of SIRT7 in p65 translocation is controversial. In addition, the decline of SIRT7 upregulated the levels of pro-inflammatory cytokines including IL-1β and IL-6 in human umbilical vein endothelial cells, while overexpression of SIRT7 effectively alleviated the inflammatory response.86 However, several studies have also revealed a pro-inflammatory role for SIRT7. For example, SIRT7-kidney-specific KO mice exhibited diminished inflammation with a reduction in the level of multiple inflammatory factors such as TNF-α, IL-1β and IL-6, and suppression of nuclear NF-κB p65 accumulation.87 These contradictory results imply that the regulatory effects of SIRT7 on the inflammatory process may be variable under specific pathologies, which will need further study.84
SIRT2 also participates in inflammatory responses. Inhibition of SIRT2 enhanced microglial activation and the release of pro-inflammatory cytokines via acetylation-dependent upregulation of NF-κB transcriptional activity.88 SIRT2 reduced the levels of pro-inflammatory cytokines and ameliorated the severity of arthritis by deacetylating the p65 subunit of NF-κB,89 further demonstrating the role of SIRT2 activation in suppression of the inflammatory response.
In summary, SIRTs are found to interfere with the NF-κB signaling pathway by preventing NF-κB translocation, influencing its expression and regulating its interactions, thereby having an anti-inflammatory function. Understanding the underlying molecular mechanisms of NF-κB pathway activation and its effects on inflammation may guide an approach to designing better pharmacological targets for alleviating inflammation and related therapies.
-
(2)
The activation of NLRP3 aggravates inflammation
NLRP3 is an important component of the NLRP3 inflammasome complex involved in inflammation.90,91 It is believed that activation of the NLRP3 inflammasome occurs in two sequential steps — first, it must be primed, and then it can be activated.71 When the body suffers from inflammatory disease, damage-associated molecules directly engage TLR4 and then quickly activate the NF-κB signaling pathway, resulting in augmented expression of NLRP3;92,93,94 this in turn generates inflammatory cytokines such as IL-1β, IL-18, TNF-α and transforming growth factor-beta (TGF-β) which aggravate inflammation.95 Some studies have found that SIRTs, especially SIRT1 and SIRT3, act on NLRP3 to exert anti-inflammatory functions. For example, SIRT1 plays an important protective role in the inflammation mediated by the attenuation of NLRP3 activity, which is the best characterized inflammasome.96,97 Mechanistic studies of acute liver injury98 demonstrated activation of a pathway involving SIRT1 and multipotent mesenchymal stromal/stem cell-mediated AMP-activated protein kinase (AMPK) α in macrophages, resulting in deacetylation of spliced X-box-binding protein 1 and subsequent inhibition of the NLRP3 inflammasome.
It was reported that mitophagy/autophagy blockade leads to the accumulation of damaged mitochondria generating ROS, and this in turn activates the NLRP3 inflammasome.99 For instance, a study carried out by Zhao et al. suggested that the mechanism of action by which SIRT3 protects against tissue damage involved the attenuation of ROS production and reduction of NLRP3 activity, resulting in the inhibition of oxidative stress and the downregulation of proinflammatory cytokines.64 However, little information is available on the relationship between SIRT3 and NLRP3; thus, further research is necessary to determine whether SIRT3 has a direct effect on the NLRP3 inflammasome.
-
(3)
The effect of SIRTs targeting noncoding RNAs on the inflammatory pathway
Current studies have mainly elucidated the role of the SIRT family in the inflammatory response. However, exploration of the molecular mechanism underlying how SIRTs affect inflammation is still limited, especially studies examining the interaction of SIRT1 with noncoding RNAs. For example, microRNAs (miRNAs) can negatively regulate inflammation by repressing SIRT1. Downregulation of miRNAs such as miR-217 and miR-543 mitigated the inflammatory response by regulating the SIRT1/AMPK/NF-κB signaling pathway.100 In the same way, miR-378 reduced SIRT1 activity and facilitated the inflammatory pathway involving NF-κB-TNFα by targeting 5'-AMPK subunit gamma-2.101 In addition, the RNase monocyte chemoattractant protein-induced protein 1 alleviated inflammatory responses by promoting the expression of SIRT1 mediated via miR-9.102 Furthermore, SIRT1 targets the p53/miR-22 axis to suppress inflammation, cyclooxygenase (COX)-2 and inducible nitric oxide synthase (iNOS) expression.103 These studies suggest that the regulation of SIRTs by noncoding RNAs may be a promising therapeutic strategy for inflammation-related diseases.
Conclusion
In summary, the SIRT family is involved in inflammation via various mechanisms. Although the details of SIRT-dependent regulation of inflammation are becoming clear, many unanswered questions remain. For example, further studies are needed to explore whether depletion of SIRTs is a common pathological change in the occurrence and development of inflammation-related diseases. Further attention is also needed to resolve some of the conflicting data and better understand the critical role of the SIRT family in the inflammatory response. The contradictory roles of the SIRT family in inflammation may result from their regulation of common signaling pathways under specific pathologic conditions. While determining what role the SIRT family plays in inflammation, researchers should also target its mechanism of action in order to lay the foundation for subsequent clinical translational studies. To summarize, we have focused on introducing relevant studies and the beneficial effects of the SIRT family through its regulation of inflammatory pathways, providing an important reference point for future studies.
The role of SIRTs in metabolism
Metabolism is the general term for a series of ordered chemical reactions that take place in the body to sustain life.104,105 These processes allow organisms to grow and reproduce, maintain their structure and respond to the external environment.106,107,108 Metabolism mainly includes glucose metabolism and lipid metabolism.104,109 Many metabolic processes occur in the mitochondria where SIRT3–5 proteins are located. In addition, SIRT proteins located in the nucleus may participate in regulating several metabolism-related genes.109,110 In this section, we focus on the SIRT proteins and their roles in maintaining metabolic homeostasis by participating in the regulation of glucose, glutamine, and lipid metabolism (Fig. 3).

Overview of the roles of SIRTs in cell metabolism. SIRTs participate in glucose metabolism, lipid metabolism, and other metabolisms via interacting with metabolism-related genes and enzymes. (i) In the nuclear, SIRT1 and SIRT6 activate the transcription factors HIF2α and HIF1α respectively through different manners and subsequently improve glycolysis. Besides, SIRT1 regulates gluconeogenesis by activating PGC1α and inhabiting FOXO1, thereby affecting the transcriptional activation of their target genes. SIRT1 also promotes fatty acid oxidation by activating PGC1α and promoting the expression of target genes. Besides the positive regulation, SIRT1 and SIRT6 suppress SREBP1 and transcriptionally represses lipogenesis. (ii) In cytoplasm, SIRT2 deacetylates and activates the rate-limiting enzyme PEPCK and promotes gluconeogenesis during low nutrient condition. Moreover, SIRT2 inhabits ACLY and deters lipid synthesis. (iii) Regarding SIRTs in mitochondria, SIRT4 and SIRT5 reduces PDH activity which converts pyruvate to acetyl CoA. Both SIRT3 and SIRT4 target GDH, but their enzymatic activities are opposite. Besides GDH, SIRT3 also improves IDH2 and LCAD activity, thus enhancing cellular respiration and stimulating β-oxidation of fatty acids. Moreover, SIRT5 represses IDH2 activity and may disrupt glutamine metabolism through GLS. Activation and inhibition effects are displayed in “arrows” and “inhibitors”, respectively. https://biorender.com. ACC acetyl-CoA carboxylase, ACLY ATP citrate lyase, ANT2 adenine nucleotide translocase 2, Bmal1 brain-muscle-Arnt-like protein-1, CDK2 cyclin-dependent kinase 2, ChREBP carbohydrate response element-binding protein, CPS1 carbamoyl phosphate synthetase 1, CPT1 carnitine palmitoyl transferase 1 A, eIF5A eukaryotic initiation factor 5A, GDH glutamate dehydrogenase, GLUT glucose transporter, HIF1/2α hypoxia-Inducible Factor-1/2α, HK2 hexokinase 2, HSF1 heat shock factor 1, IDH2 isocitrate dehydrogenase 2, LCAD long chain acyl CoA dehydrogenase, MCD malonyl CoA decarboxylase, MBD1 methyl-CpG-binding domain protein 1, MDH1 malate dehydrogenases 1, m-TORc1/2 mTOR complex 1/2, MyoD myogenic differentiation factor, NNMT nicotinamide N-methyl transferase, PARP poly (ADP-ribose) polymerase, PDH pyruvate dehydrogenase, PEPCK1 phosphoenolpyruvate carboxykinase, PFK phosphofructokinase-1, PK pyruvate kinase, PTP1B protein-tyrosine phosphatase 1B, RIPK1/3 receptor interacting protein kinases 1/3, SLC1A5 solute carrier family 1 member 5, SREBP1 sterol regulatory element binding protein 1, TRAP1 tumor necrosis factor receptor-associated protein 1, Tsc2 tuberous sclerosis complex 2, ZEB1 zinc finger E-box binding homeobox 1
The effect of SIRTs on glucose metabolism
Glucose metabolism refers to a series of complex chemical reactions after glucose, glycogen and other substances enter the body, including anaerobic glycolysis of glucose, aerobic oxidation, synthesis and decomposition of glycogen, and gluconeogenesis.111,112 Abnormal glucose metabolism and insulin resistance might cause metabolic diseases such as diabetes.113,114,115 The roles of SIRTs in glucose metabolism have been established. For example, SIRT1 is a key positive regulator of systemic insulin sensitivity and regulates pancreatic insulin secretion, thus contributing to increased systemic insulin sensitivity, which triggers glucose uptake and utilization.116,117,118 Mechanistically, SIRT1 participates in the regulation of glucose metabolism by upregulating AMPK, and activation of AMPK can ameliorate the glucose metabolic imbalance.116,119 Upregulated SIRT1 may reverse the development of diabetes by targeting the AMPK/acetyl CoA carboxylase signaling pathway.117 Similarly, decreased levels of SIRT1 may lead to AMPK deficiency, thereby impairing the improvement in glucose tolerance.119 Meanwhile, there are an interdependent relationship between AMPK and SIRT1,120,121 and activation of SIRT1 and its downstream signaling pathways could also be improperly triggered in AMPK-deficient states.121 Additionally, SIRT1 increases insulin sensitivity and lowers blood sugar by downregulating protein tyrosine phosphatase 1B, a key negative regulatory protein in the insulin signal transduction pathway.118 Thus, high expression of SIRT1 is benefit for maintaining blood sugar stability via the regulatory proteins of insulin signaling. However, the relationship between SIRT1 and other molecules (e.g., AMPK and protein tyrosine phosphatase 1B) that are closely associated with blood glucose regulation is still worth further exploration.
SIRT1, SIRT3, and SIRT6 also participate in glucose metabolism. The limited whole-body benefit of increasing hepatic SIRT3 during the development of diet-induced insulin resistance, which can be considered a pre-diabetic state, has also been demonstrated.122 Mechanistically, SIRT3 negatively regulates aerobic glycolysis by inhibiting hypoxia-inducible factor 1α (HIF-1α).123 SIRT6 takes part in the maintenance of glucose metabolic homeostasis in the whole body and in local tissues such as liver and skeletal muscle.124,125 For instance, SIRT6 in pancreatic β cells deacetylated FoxO1 and subsequently increased the expression of glucose-dependent transporter 2 to maintain the glucose-sensing ability of pancreatic β cells and systemic glucose tolerance.126 Improvement in SIRT6-mediated insulin signaling transduction has been reported in the liver of obese rats after exercise.127 Also, enhancement of insulin sensitivity in skeletal muscle and liver by physiological overexpression of SIRT6 has been described,128 suggesting potential functions of SIRT6 in glucose metabolism.
Finally, direct and indirect involvement of SIRTs in glucose metabolism may provide new insights into therapeutic targets for the treatment of abnormal glucose metabolism in the future. This may help reduce the human disease burden related to glucose metabolism, where SIRT proteins may play an important role in overcoming glucose metabolic diseases at an earlier time point.
The effect of SIRTs on lipid metabolism
Lipid metabolism means that most of the fat ingested by the human body is emulsified into small particles by bile, and the lipase secreted in the pancreas and small intestine hydrolyzes the fatty acids in the fat into free fatty acids, after which hydrolyzed small molecules are absorbed by the small intestine into the bloodstream.104,105,129 Notably, the SIRT protein family is involved in lipid metabolism.129,130 For SIRT1, Qiang et al. found that SIRT1-dependent cAMP Response Element Binding protein (Creb) deacetylation regulates lipid metabolism.131 Mechanistically, Lys136 is a substrate for SIRT1-dependent deacetylation that affects Creb activity by preventing cyclic adenosine monophosphate (cAMP)-dependent phosphorylation, leading to the promotion of hepatic lipid accumulation and secretion. Moreover, SIRT1 activates AMPK, which leads to lipid-lowering effects in vitro and in vivo.132 SIRT2 prevents liver steatosis and lipid metabolic disorders by deacetylation of hepatocyte nuclear factor 4α.133 Additionally, SIRT3 acts as a bridge in the lipid metabolism pathway. For example, pancreatic SIRT3 deficiency promoted hepatic steatosis by enhancing 5-hydroxytryptamine synthesis in mice with diet-induced obesity.134 In addition, roles for SIRT5 and SIRT6 were identified in lipid metabolism.135,136,137,138 For instance, SIRT5 inhibited preadipocyte differentiation and lipid deposition by activating AMPK and repressing mitogen-activated protein kinase (MAPK) signaling pathways, which has been verified in obese mice.135 Compared with control wild-type mice, SIRT6-KO mice had a significant increase in both body weight and fat mass and exhibited glucose intolerance and insulin resistance.138 Mechanistically, SIRT6-KO decreased expression of the adiponectin gene and Akt in white adipose tissue, while expression of the thermogenic gene UCP1 was diminished in brown adipose tissue.138
The effect of SIRTs on other metabolism
SIRT3 and SIRT4 have been found to play roles in regulating glutamine metabolism. In detail, Gonzalez-Herrera et al. reported that loss of SIRT3 promoted glutamine use in nucleotide biosynthesis.139 Conversely, SIRT4 inhibited glutamine metabolism in colorectal cancer cells, thereby acting as a tumor suppressor.140 In addition, SIRT3 affected mitochondrial metabolic reprogramming by activating the AMPK/peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) pathway, thereby maintaining the stability of mitochondrial membrane potential as well as mitochondrial structure.141 Moreover, silencing SIRT6 influenced collagen metabolism in human dermal fibroblasts by affecting the synthesis and degradation of collagen.142
Conclusion
As shown in the previous findings, SIRT1, SIRT3, and SIRT6 have been more frequently studied than other SIRTs in regulating human body metabolism, mainly through their effect on glucose and lipid metabolism. However, only a few studies have focused on the roles of other SIRT proteins, in particular SIRT2 and SIRT7. In the future, research should be focused on the role of these other SIRTs in regulating different metabolism subtypes. Overall, clarifying the various participating mechanisms of SIRTs in metabolism might provide future new ideas for research and novel therapeutic targets for the treatment of abnormal metabolism, thereby lessening the burden imposed on society by human lipid metabolism-related diseases.
The role of SIRTs in oxidative stress
Oxidative stress is considered to be an important factor in cell damage and is usually caused by the overproduction of ROS. Under physiological conditions, ROS are produced at low levels and are scavenged by the endogenous antioxidant system. When ROS exceed the scavenging capacity, however, cellular oxidative stress damage occurs.143 Oxidative stress plays an important role in the pathological process of various diseases.144 Recently, accumulating studies have shown that the SIRT protein family participates in the process of oxidative stress. Notably, SIRT proteins contribute to cellular tolerance to oxidative stress by regulating many genes and their related signaling pathways (as shown in Fig. 4). Herein, we review the regulation of different target genes or proteins by SIRTs, with the aim of understanding their mechanistic effects in the process of antioxidant stress damage.

Overview of the roles of SIRTs in oxidative stress. a The overall roles of SIRTs in regulating cellular oxidative stress. The effect of SIRTs on oxidative stress is mainly via affecting the following proteins, mainly including Nrf2, FOXOs and SOD. SIRT1 and SIRT6 could indirectly affecting Nrf2 signaling, thereby regulating oxidative stress. SIRT3 activates FOXO3, which leads to increasement of MnSOD, allowing for the elimination of ROS. In addition, SIRT1, SIRT2, and SIRT6 could upregulate the expression of SOD, then reducing the ROS and inhibiting the oxidative stress; (b) The regulatory effects of SIRTs on main proteins in oxidative stress. SIRT1 downregulation by NF-κB leads to oxidative stress. Moreover, SIRT3 regulates ROS generation, causing suppression of NF-κB activation, and SIRT6 reduces NF-kB activation and represses oxidative stress. c The roles of SIRTs in regulation of transcription factors. SIRT1 increases the expression of FOXO1, reducing the production of ROS and oxidative stress. SIRT1 inhibits oxidative stress by deacetylating P53 protein. Besides, SIRT1 could activate PGC-1α and alleviate oxidative stress injury. d The proteins less studied that activate or inhibit SIRT1. Activation and inhibition effects are displayed in green and red arrows, respectively. https://biorender.com. AT1 angiotensin type 1, ATF6 activating transcription factor 6, Bach1 BTB domain and CNC homolog 1, BIP binding immunoglobulin protein, CD36 cluster of differentiation 36, CHOP C/EBP-homologous protein, CoQ10 coenzyme Q10, COX2 cyclooxygenase-2, CPEB-1 cytoplasmic polyadenylation element binding protein 1, DPP4 dipeptidyl peptidase-4, DRG2 GTP-binding protein 2, FASTK Fas-activated serine/threonine kinase, FNDC5 fibronectin type III domain-containing 5, GCN5 general control non-repressed protein 5, GDF11 Growth differentiation factor 11, Hcy homocysteine, hnRNP heterogeneous nuclear ribonucleoprotein F, HO-1 heme oxygenase 1, Keap-1 kelch-like ECH-associated protein 1, LDH lactate dehydrogenase, LOX-1 lectin-like oxidized low-density lipoprotein receptor-1, Lsd lysine-specific demethylase 1, MIF migration inhibitory factor, MPO myeloperoxidase, NEU1 neuraminidase 1, NRLP3 NOD-like receptor thermal protein domain associated protein 3, OGG-1 BER enzyme 8oxoG DNA glycosylase I, PDGFR-α platelet derived growth factor receptor α, PGAM2 glycolytic enzyme phosphoglycerate mutase 2, PRMT protein arginine methyltransferase, α-SMA smooth muscle alpha actin, TIGAR TP53-induced glycolysis and apoptosis regulator, timp-1 tissue inhibitor of metalloproteinase 1, TOPK T‑lymphokine‑activated killer cell‑originated protein kinase, UCP2 uncoupling protein 2, Wt1 Wilms' tumor 1, Wt2 Wilms' tumor 2
The interaction between SIRT1, SIRT3, SIRT6 and AMPK
AMPK, is a major regulator of metabolic homeostasis and is often activated under oxidative stress conditions such as ischemia and hypoxia.145 SIRT1 participates in regulating AMPK and its related pathways. For example, AMPK can be activated by liver kinase B1 (LKB1), the upstream regulator of AMPK, while activated AMPK reduces oxidative stress injury by promoting insulin sensitivity, fatty acid oxidation and mitochondrial biosynthesis to generate ATP.146 SIRT1 overexpression leads to the deacetylation of LKB1, the translocation of LKB1 from the nucleus to the cytoplasm, and the activation of AMPK to alleviate oxidative stress.147 Additionally, SIRT1 lowers LKB1 activation in the liver, which subsequently abrogates Thr172-AMPKα phosphorylation, thereby increasing oxidative stress in severe acute hypoxia.148 It can be seen that SIRT1 may activate AMPK by regulating LKB1, thereby resisting oxidative stress damage and promoting cell survival.
In addition to the role of SIRT1 on AMPK, SIRT3 and SIRT6 can also interact with AMPK to exert an anti-oxidative effect on stress injury. Deficiency of AMPKα resulted in elevated expression of SIRT3, which modulated oxidative stress in heart tissue both in vitro and in vivo .149 It has also been shown that the AMPK activated SIRT3, limited oxidative stress and improved mitochondrial DNA integrity and function.150 In addition, SIRT3 reduced ROS and lipid peroxidation by improving mitochondrial function via deacetylation of LKB1 and activation of AMPK.151 As previously mentioned, a feedback loop may exist between AMPK and SIRT3. SIRT6 also promoted AMPK expression, thus upregulating antioxidant-encoding gene expression of manganese superoxide dismutase (MnSOD) and catalase (CAT), thereby suppressing oxidative stress.152 In brief, SIRT1, SIRT3 and SIRT6 act to counter oxidative stress by directly or indirectly interacting with AMPK. However, additional studies are required to clarify the relationship between other SIRT proteins and AMPK pathway under oxidative stress.
The effect of SIRT1, SIRT 2, and SIRT6 on Nuclear erythroid 2-related factor 2 (Nrf2)
Nrf2 is a leucine transcription factor that plays extremely important roles in antioxidant response element (ARE)-dependent transcriptional regulation of defense genes. When stimulated, Nrf2 dissociates from suppressor protein Keap1 in the nucleus and interacts with AREs to regulate the expression of antioxidant genes, suggesting a close association between Nrf2 and oxidative stress.153 Notably, SIRTs including SIRT1, SIRT2 and SIRT6 can activate Nrf2, regulate antioxidant gene expression, and thus fight oxidative stress damage. For example, SIRT1 activated Nrf2 by changing the structure of Keap1, leading to Nrf2 nuclear transfer and promoting the expression of antioxidant genes, such as glutathione S transferase and glucuronyl transferase.154,155 In addition, SIRT2 was downregulated in the spinal cord after peripheral nerve injury, which subsequently inhibited Nrf2 activity, leading to increased oxidative stress.156 The overexpression of SIRT6 in the brain through in vivo gene transfer enhanced Nrf2 signaling and reduced oxidative stress.157,158 SIRT6 protected human lens epithelial cells from oxidative damage via activation of Nrf2 signaling.159 Furthermore, SIRT6 protects cells against hydrogen peroxide-induced oxidative stress by promoting Nrf2/ARE signaling.160 Therefore, SIRTs can activate Nrf2, regulate antioxidant gene expression, and thus fight oxidative stress damage.
The effect of SIRT1 and SIRT3 on FoxOs
A family of SIRT targets are class O mammalian forkhead transcription factors (FoxO1, FoxO3, FoxO4 and FoxO6) which participate in regulating oxidative stress. FoxO1 can scavenge excessive ROS through the regulation of downstream target genes such as MnSOD and CAT, and thus reduce cellular oxidative stress damage. SIRT1 alleviates oxidative stress by controlling nuclear shuttling and transcriptional activity of FoxO1 and FoxO3a. For instance, SIRT1 induced the transfer of FoxO1 to the nucleus and increased the level of FoxO1 protein in adipocytes, reducing the production of ROS and oxidative stress.161 Moreover, SIRT1 promoted early-onset age-related hearing loss by suppressing FoxO3a-mediated oxidative stress resistance in vivo.162 Apart from SIRT1, SIRT3 has also been shown to participate in the regulation of oxidative stress via FoxO3.163,164 Mechanistically, SIRT3 activated FoxO3 gene expression, which increased transcription of MnSOD and CAT, enabling the elimination of ROS.165,166 The aforementioned studies show that SIRT1 and SIRT3 can interact with FoxOs to counteract oxidative stress.
The effect of SIRT1 and SIRT3 on PGC-1α
PGC-1α is a coactivator of peroxisome proliferator-activated receptor-γ, which can act to block oxidative stress damage by scavenging excess ROS, inducing antioxidant enzyme expression and maintaining mitochondrial function.167 SIRT1 can activate PGC-1α through deacetylation, scavenge ROS caused by oxidative stress, and alleviate oxidative stress injury. Activation of the SIRT1-PGC-1α axis implies activation of antioxidant defense mechanisms, alleviating mitochondrial oxidative stress.168,169,170 Additionally, PGC-1α and SIRT3 can interact directly. PGC-1α increased respiratory capacity and reduced oxidative stress through SIRT3-mediated reduction of mitochondrial ROS.171,172 Furthermore, loss of SIRT3 resulted in the expression of PGC-1α, which produced a decrease in mitochondrial respiration. Inhibition of SIRT3 reduced PGC-1α expression and mitochondrial function, thereby lowering oxidative stress resistance.173,174 Thus, both SIRT1 and SIRT3 may interact with PGC-1α in order to resist oxidative stress damage.
The effect of SIRT1 and SIRT6 on p53
p53 is a stress response transcription factor and was the earliest discovered physiological substrate of SIRT1. p53 can promote oxidative stress injury by regulating different target proteins and further induce cellular responses.175 p53 exerted pro-oxidant activity and promoted oxidative damage by regulating its transcriptional targets, including p53-inducible gene 3, glutathione/NADH, p-FoxO3a and B-cell lymphoma -2-associated-X-protein (Bax).176 In contrast, p53 can act as an antioxidant factor to suppress oxidative stress by regulating several redox-related proteins, such as MnSOD, glutathione peroxidase 1, and Jun N-terminal kinase (JNK).176 When cells are under oxidative stress, multiple sites in the N-terminal of p53 are phosphorylated and multiple lysine sites in the C-terminal are acetylated.177 SIRT1 has a negative regulatory effect on p53; for example, depletion of SIRT1 abolished the increase in oxidative stress induced by p53 acetylation in THP-1 cells.178 SIRT1 activation also reversed p53 expression and accumulation brought on by H2O2-induced oxidative stress.179 The small molecule activator SRT2104 enhanced renal SIRT1 expression and activity and deacetylated p53, resulting in activation of antioxidant signaling.180 As for the role of SIRT6 in oxidative stress, relevant studies have been limited. For instance, SIRT6 protected cardiomyocytes by inhibiting p53/Fas-dependent cell death and augmenting endogenous antioxidant defense mechanisms.181 Hence, SIRT1 and SIRT6 can inhibit p53 activity through deacetylation and reduce oxidative factor expression, promoting resistance to oxidative stress injury.
The effect of SIRT1, SIRT3, and SIRT6 on NF-κB
NF-κB is a nuclear transcription factor. Activated NF-κB factors promote the production of ROS that damage tissues and organs.182 When oxidative stress occurs, enhanced ROS activity can stimulate the activation of NF-κB and induce the expression of ICAM-1 and monocyte chemotactic factor 1, which further activate NF-κB and lead to oxidative stress.183 SIRTs inhibited transcription by deacetylating the NF-κB subunit Rel/p65, reducing the production of oxygen radicals.79 SIRT1, SIRT3 and SIRT6 inhibited the transcriptional activity of NF-κB through deacetylation, thereby resisting oxidative stress injury. For example, downregulation of SIRT1 protein levels by NF-κB led to oxidative stress.184 In addition, SIRT3 regulated ROS generation, causing suppression of NF-κB activation and oxygen radicals.185 Moreover, loss of SIRT6 in cutaneous wounds aggravated the proinflammatory response by increasing NF-κB activation and promoting oxidative stress.186 Therefore, SIRT1, SIRT3, and SIRT6 can block oxidative stress damage by inhibiting NF-κB activity.
The effect of SIRTs on oxidative stress through other pathways
Many molecules are upstream regulators of SIRTs and have a regulatory effect on them under oxidative stress. For example, the expression of SIRT1 and SIRT6 was decreased by oxidative stress-dependent miR-34a activation in epithelial cells.187 SIRT5 was upregulated by Krüppel-like factor (KLF) 6 silencing, thereby reducing oxidative stress.188 Meanwhile, SIRTs target many downstream factors, such as HIF-1α and endothelial nitric oxide synthase (eNOS), and then participate in regulating oxidative stress. Activation of HIF-1α is associated with oxidative stress and can regulate ROS formation through direct or indirect effects.189 For example, SIRT4 reduced the accumulation of ROS by inhibiting HIF-1α, which is also an important mechanism underlying SIRT4 activity in oxidative stress.190,191 In addition, eNOS dysfunction in an oxidative stress environment led to increased generation of ROS. SIRTs play important roles in regulating the activity of eNOS as well. For instance, upregulation of SIRT1 reduced eNOS acetylation (inactive state) and enhanced eNOS phosphorylation (active state).192 Activation of the SIRT1/eNOS pathway has been found to reduce ROS production by inhibiting NF-κB expression.193 In brief, the mechanisms by which SIRTs regulate oxidative stress are diverse, and there are many more regulatory pathways that need to be verified.
Conclusion
Together, these aforementioned studies reflect the importance of the SIRT protein family in oxidative stress and can be expected to stimulate future research in order to decipher the SIRT protein mechanisms. As summarized in Fig. 4, SIRTs are involved in the regulation of redox homeostasis and oxidative stress involving many key genes and molecules. Indeed, SIRTs play important roles in maintaining intracellular homeostasis which keeps cells healthy, making them ideal for redox regulation studies. Additionally, SIRTs enhance intracellular homeostasis by acting synergistically through different mechanisms.
Further in-depth studies are needed to identify and elucidate the exact role of each SIRT and to determine whether different SIRTs have functional redundancy or overlapping roles in homeostasis, which may be important for regulating oxidative stress in cells and important pathological manifestations. SIRTs should be developed as modulators of redox-related diseases, and may also provide a mechanistic basis for the development and discovery of antioxidants. Given the interest in SIRTs as drug targets and their redox importance, studies addressing these questions may also provide therapeutic opportunities for the treatment of metabolic, age-related and other redox-related diseases.
The role of SIRTs in cell apoptosis
Cell apoptosis is an active form of cell death that involves programmed cellular machineries leading to progressive self-destruction of the cell.194 As a type of programmed cell death, apoptosis is a basic cellular mechanism and may occur in numerous diseases. Notably, one of the most extensive biological functions regarding the SIRT protein family is participation in the process of cell apoptosis. The SIRT protein family has functions in both physiological conditions and diseases by regulating the acetylation modification and/or influencing various apoptosis-related proteins by pathway crosstalk, and thus takes part in the pathogenesis of many diseases including cancer, CVDs and others (Fig. 5).195,196

Overview of the roles of SIRTs in apoptosis. SIRT protein family has functions in both physiological conditions and diseases by regulating the acetylation modification and/or influencing various apoptosis-related proteins by crosstalk of pathways. Meanwhile, they can also be regulated by the molecules in the aforementioned process, such as microRNA, FoxO1, FoxO3a, TNF-α and NF-κB. a The roles of SIRT1 in regulating apoptosis by targeting apoptosis-related proteins and pathways; (b) The roles of SIRT3 in regulating apoptosis by targeting apoptosis-related proteins and pathways; (c) The roles of SIRT2, SIRT4 and SIRT5 in regulating apoptosis by targeting apoptosis-related proteins and pathways; (d) The roles of SIRT6 in regulating apoptosis by targeting apoptosis-related proteins and pathways; (e) The roles of SIRT7 in regulating apoptosis by targeting apoptosis-related proteins and pathways. https://biorender.com. ATM ataxia telangiectasia mutated, Cyt C cytochrome c, ELA elabela, GAPDH glyceraldehyde 3-phosphate dehydrogenase, HIC1 hypermethylated in cancer-1, HIPK2 homeodomain-interacting protein kinase-2, INZ inauhzin, JAK2 janus kinase 2, MALAT1 metastasis-associated lung adenocarcinoma transcript 1, Mcl-1 myeloid cell leukemia 1, MicRNA microRNA, MST1 mammalian sterile 20-like kinase 1, PLD2 phospholipase D2, RORA retinoid-related orphan receptor α, TSPYL2 testis-specific protein y-encoded-like 2, Yap yes-associated protein, ZMAT1 zinc finger matrin-type 1
The effect of SIRTs as histone deacetylases on apoptosis
Histones are the major protein components of chromatin, serve as spools around which DNA is wound, and play roles in gene regulation. The SIRT family-dependent epigenetic regulation of histone acetylation is an important link in the regulation of apoptosis.197 For example, SIRT1 can reduce the acetylation levels of histones in the promoters of genes, e.g., AR, BReast-CAncer susceptibility gene 1(BRCA1), ERS1, ERS2, EZH2 and EP300, which ultimately affected cancer cell apoptosis.197 Additionally, SIRT6 links histone H3K9 deacetylation to NF-κB-dependent gene expression and organismal life span.80 At the molecular level, SIRT6 binds to the promoters of extracellular signal-regulated kinase (ERK) 1 and ERK2 genes, and deacetylates histone H3K9, thereby inhibiting ERK1/2 expression.198 Moreover, SIRT6 induced the expression of GATA binding protein 5 (GATA5) through inhibition of Nkx3.2 transcription by deacetylating histone H3K9, thereby regulating GATA5-mediated signaling pathways to prevent endothelial injury.199 These studies have demonstrated the critical role of the SIRT protein family in regulating apoptosis. However, additional studies have found that the SIRT protein family regulates other novel modification types of histones, for example, sumoylation200 and ubiquitination.201 Whether these new types of histone modification participate in cell apoptosis remains largely unknown, which may be a new direction for further research.
The effect of SIRT1 on apoptosis by targeting apoptosis-related proteins and pathways
Among the SIRT protein family, SIRT1 is the most widely studied protein, especially in regulating cell apoptosis. A variety of transcription factors, including p53, NF-κB and FoxO, which act downstream of SIRT1, are closely related to cell apoptosis.103,202,203,204 Therefore, we focus here on how SIRT1 participates in regulating these three proteins and their related pathways.
-
(1)
SIRT1 mediates p53-dependent apoptosis by suppressing acetylated p53
As first discovered with non-histone targets of SIRT1, p53 plays a central role in the prevalence of diseases related to apoptosis.205,206,207 SIRT1 regulates p53 deacetylation, which is associated with the apoptosis-inhibiting signaling pathway, mainly including the p53-induced death domain protein Pidd,208 p21, Bax/Bad and caspases.209 For example, Zeng et al. reported that an extract of Anoectochilus roxburghii flavonoids reduced neuron apoptosis by positively regulating SIRT1 expression, thereby reducing expression of the apoptosis-related molecules p53, p21 and caspase-3, while increasing the ratio of B-cell lymphoma (Bcl)-2/Bax.210 SIRT1 also participated in the regulation of p53 protein through direct deacetylation. For example, SIRT1 deacetylating p53 at Lys379 inhibited p53-dependent apoptosis.211 In addition, SIRT1 can regulate the p53 signaling pathway by targeting proteins. The overexpression of SIRT1 resulted in markedly reduced mRNA and protein expression levels of p53 signaling pathway-related molecules (including p53 and Bax) in vitro, but increased Bcl mRNA and protein expression.212 p53 expression gradually decreased with increasing SIRT1 levels, thus indicating a gradual decrease in apoptosis.213 These findings thus show that SIRT1 inhibits apoptosis via inactivation of p53, suggesting a critical role for SIRT1 in regulating the p53 signaling pathway.
-
(2)
The SIRT1/NF-κB pathway is mainly involved in inflammation-induced apoptosis
Regarding the mechanism underlying SIRT1 involvement in apoptosis, NF-κB (p65) acetylation was significantly increased after inhibition/deletion of SIRT1.214 A large number of studies have shown that SIRT1 mediates NF-κB pathway modulation to mitigate inflammasome signaling and cellular apoptosis.203,214,215 For example, SIRT1 overexpression promoted mouse B lymphocytes cell proliferation, inhibited apoptosis, and upregulated pro-inflammatory cytokines by inhibiting the NF-κB pathway.216 Additionally, activating the NF-κB signaling pathway could ultimately induce apoptosis through regulation of the inflammatory process.217 Silencing interferon regulatory factor 9 curbed activity of the NF-κB signaling pathway by upregulating SIRT1, which further inhibited TNF-α induced changes in inflammatory cytokine secretion and promoted apoptosis.218 Therefore, it appears to be a double-edged sword that SIRT1 regulates NF-κB signaling to affect cellular inflammatory activation and apoptosis in different spatiotemporal dependencies.
-
(3)
SIRT1 regulates apoptosis by the regulation of FoxOs
FoxO transcription factors can control cell survival by regulating the expression of genes involved in cell-cycle progression and promoting apoptosis.219 SIRT1 is a key regulator of cell defenses and survival in response to stress, which deacetylates and represses FoxO-dependent apoptosis.219,220 SIRT1 mediates cell apoptosis through the deacetylation of FoxO proteins including FoxO1,221 and upregulation of SIRT1 can inhibit apoptosis via the FoxO1/β-catenin pathway.222 Moreover, SIRT1, FoxO1, and sterol regulatory element binding protein-1 (SREBP-1) may act as a pathway and play crucial roles in apoptosis. At both the protein and mRNA levels, SIRT1 and SREBP-1 were upregulated in progestin-resistant cells, while FoxO1 was downregulated.223 Interestingly, SIRT1 may be a potential target for cross-regulation of post-transcriptional modifications. For example, acetylation was required for FoxO3-induced apoptosis through phosphorylated-FoxO3 (p-FoxO3) formation, while expression or activation of SIRT1 blocked p-FoxO3 formation and apoptosis.224 Deacetylation of FoxO3 by SIRT1 resulted in S-phase kinase-associated protein 2-mediated FoxO3 ubiquitination and degradation.225 These fine-tuning mechanisms of FoxO3 regulation modulated by PTMs may be a new method to regulate apoptosis in a coordinated manner. In summary, then, SIRT1 can regulate the activity of FoxO, thereby modulating the balance between anti-apoptotic and apoptotic genes.226
-
(4)
miRNAs play important roles in the regulation of SIRT1
miRNAs, a subtype of non-coding RNAs, are small endogenous RNAs which can inhibit protein translation in apoptosis.227 Moreover, SIRT1 has been revealed to be targeted by miRNAs such as miR-34a, miR-181, miR-128, miR-449 and miR-30a-5p. For example, Yamakuchi et al. demonstrated a negative correlation between the expression of miR-34a and SIRT1, suggesting SIRT1 was a target of miR-34a.228 In addition, SIRT1 is a key player in the protection provided by miR-34a-5p inhibition during apoptosis.229 The overexpression of miR-181d-5p inhibited cell apoptosis and renal fibrosis in a mouse model by downregulating the SIRT1/p53 pathway.230 Furthermore, miR-181a increased FoxO1 acetylation and promoted granulosa cell apoptosis via SIRT1 downregulation.231 The previous study also suggested that miR-128 promoted apoptosis in human cancers via the p53/Bak axis.232 Upregulation of miR-128 promoted apoptosis in an epilepsy model in vivo and in vitro through the SIRT1/p53/Bax/cytochrome c/caspase signaling pathway.233 Other miRNAs, such as miR-449, have been investigated in a model of acute kidney injury model by detecting expression of its target SIRT1 and downstream factors p53/Bax.234 Inhibition of miR-449 elevated SIRT1 expression and inhibited acetylated p53 and Bax protein levels.234 Finally, miR-30a-5p targeted SIRT1 to activate the NF-κB/NLRP3 signaling pathway, resulting in cardiomyocyte apoptosis.227 These studies all demonstrate how miRNAs play important roles in the regulation of SIRT1, which should be further studied in various diseases in the future.
-
(5)
Other regulatory molecules or factors acting on SIRT1
Upstream of SIRT1, in addition to miRNAs, a novel fibroblast growth factor 1 variant could counteract adriamycin-induced apoptosis by decreasing p53 activity via upregulation of SIRT1-mediated p53 deacetylation.235 There have also been a series of studies on the anti-apoptotic effect of melatonin which regulates SIRT1 in various physiological processes.236,237,238,239 Additionally, some chemicals or drugs, like cambinol and ginsenoside Rc, have been shown to inhibit or activate SIRT1 to regulate the apoptotic process.240,241 Given that the above-mentioned molecules can regulate SIRT1-related signaling pathways, SIRT1 may be a potential therapeutic target in the apoptotic response.
The effect of SIRT2 on apoptosis
Several previous studies have suggested that SIRT2 has complex regulating mechanisms promoting or inhibiting apoptosis.242 In contrast to SIRT1, SIRT2 is predominantly a cytoplasmic protein and is able to deacetylate several cytoplasmic substrates,243 including p53,244 NF-κB,245 and FoxO3.246 In terms of its anti-apoptotic effects, SIRT2 downregulation alone is sufficient to cause apoptosis, and SIRT2 depletion leads to p53 accumulation causing activation of the p38 MAPK in cancer cell lines such as HeLa, but not in normal cells.247
On the other hand, SIRT2 can promote apoptosis mediated by the caspase, Bcl2/Bax and FoxO pathways. For example, She et al. demonstrated that the SIRT2 inhibitor AGK2 effectively reduced the levels of phospho-JNK and FoxO3a.248 As JNK is a well-known regulator of apoptosis, protein downregulation will lead to attenuation of the subsequent signaling cascade involving Bim, and eventually leads to suppression of the caspase cascade.248 In addition, SIRT2 overexpression induces cellular apoptosis via upregulating cleaved caspase 3 and Bax and downregulating anti-apoptotic protein Bcl-2,245 suggesting the important role of SIRT2 in apoptosis. As for the FoxO-related pathway, FoxO3a, which is the immediate downstream target for SIRT2-driven deacetylation, is a promoter of apoptotic pathways in many diseases.246,249 SIRT2 activates FoxO3a by deacetylating it, which promotes the activation of the pro-apoptotic pathways Akt/FoxO3a and JNK, and thus increases apoptosis. Additionally, the administration of specific inhibitors of SIRT2 attenuates neuronal cell death under ischemic conditions in vitro and in vivo.248 The confusing role of SIRT2 in the process of apoptosis might thus be attributed to regulation of different pathways affected by different conditions, but more studies verifying SIRT2 functions in apoptosis will be needed in the future.
The effect of SIRT3-7 on apoptosis
-
(1)
The critical roles of SIRT3-5 in regulating cell apoptosis
Three SIRT proteins, namely SIRT3–5, are localized to the mitochondrion, a dynamic organelle that functions as the primary site of endogenous apoptosis. Although mitochondrial SIRT proteins have not been as extensively studied as SIRT1, a growing body of studies have illustrated their importance in basic mitochondrial biology and apoptosis.
SIRT3 plays a pro-apoptotic role in that glycogen synthase kinase-3 β (GSK-3β)/Bax, Bax/Bcl-2 and bad/Bcl-x/L ratios regulate apoptosis.250,251 SIRT3 overexpression promoted apoptosis by enhancing caspase 9 cleavage in hepatocellular carcinoma (HCC) cells,252 and SIRT3 depletion downregulated cleaved caspase 3 levels in lung cancer (LC) cells.253 In contrast, several studies have found that SIRT3 has an anti-apoptotic effect. SIRT3 deficiency resulted in significantly increased apoptosis, increased Bax and caspase 3 mRNA levels, and decreased Bcl-2 mRNA levels in septic mice,254 and also significantly increased caspase 3 expression in SIRT3-KO mice. Thus, SIRT3 plays different roles in different diseases, both pro- and anti-apoptotic. A typical example is when SIRT3 expression inhibited the growth of cancer cells by promoting apoptosis and necroptosis. In a stress injury disease model, SIRT3 inhibited apoptosis and exerted a protective effect against various stressors. For example, SIRT3 deficiency produced more melanocyte apoptosis by inducing severe mitochondrial dysfunction and cytochrome c release into the cytoplasm.255 However, more research is needed in the future to determine whether SIRT3 promotes or inhibits apoptosis of the caspase 3 pathway in different types of diseases.
FoxO transcription factors are downstream targets of the serine/threonine protein kinase B/Akt, which promotes apoptosis signaling by affecting multiple mitochondria-targeting proteins.256 SIRT3 acetylation modulated FoxO1 and exerted apoptotic effects.51 In addition, SIRT3 post-translationally upregulated FoxO3a activity through deacetylation, dephosphorylation and deubiquitination to regulate apoptosis.257 Meanwhile, non-coding RNAs act as upstream regulators of SIRT3 to regulate apoptosis. For example, the miR-297 antagomir affected apoptosis by targeting SIRT3 to reduce the extent of IκBα and NF-κB phosphorylation and prevent activation of NLRP3.258 A similar study confirmed that SIRT3 was also a target of miR-421.259 Studies of the upstream and downstream regulatory mechanisms of SIRT3 regulating apoptosis are few and more research will be required in this area.
There are only limited studies on SIRT4 and cell apoptosis, but these few have indicated that SIRT4 prevents apoptosis by affecting the ratio of pro-caspase 9/caspase 9 or pro-caspase 3/caspase 3, and by altering Bax translocation.191,260 In addition, SIRT5 participates in the regulation of apoptosis as a deacetylated protein and may have an effect on apoptosis-related proteins. For example, SIRT5 deacetylated cytochrome c, a protein of the mitochondrial intermembrane space with a central function in oxidative metabolism as well as in apoptosis initiation.261 SIRT5 overexpression ameliorated cytochrome c leakage and activation of caspase 3 to alleviate apoptosis.262,263 Thus, these data implicate mitochondrial SIRTs as effective in protecting against pathological injury and apoptosis by inhibiting the cytochrome c/caspase 3 apoptosis pathway. Such research may form the basis for future treatment for apoptosis. However, the number of related studies on SIRT4 and SIRT5 is still limited and need to be expanded.
-
(2)
The role of SIRT6 and SIRT7 during apoptosis
At present, only a few studies have explored the role of SIRT6 and SIRT7, which could be a new research direction for the SIRT protein family. Both SIRT6 and SIRT7 mediate apoptosis by regulating p53.264,265 Furthermore, SIRT7 promoted cellular survival following genomic stress by attenuation of DNA damage and the p53 response.266 However, current studies on SIRT6 and SIRT7 are still in their infancy, and more research is needed in the future to explore their role in apoptosis.
Conclusion
In conclusion, one of the most extensive biological functions of the SIRT protein family is to participate in the process of apoptosis. As a family of bidirectional regulatory proteins, the function of SIRTs appears to be reversible depending on the cellular state. However, our current knowledge of SIRTs in apoptosis and its regulation is far from complete. More studies are needed in the future to explore the underlying molecular mechanisms of how the SIRT protein family is regulated in pathophysiological processes.
The role of SIRTs in autophagy
Autophagy is a cell self-digestion process via lysosomes that clears cellular waste, including aberrantly modified proteins or protein aggregates and damaged organelles.267 Recent studies have illustrated the important roles of the SIRT protein family in the autophagic process. Therefore, in this section, we aim to review recent research on the relationship between the SIRT protein family and autophagy, and discuss possible regulatory roles of SIRT proteins in autophagy, as well as the conditions under which they participate in autophagy in a positive or negative manner (Fig. 6).

Overview of the roles of SIRTs in autophagy. SIRTs can regulate a series of substrates involved in the process of macroautophagy and mitophagy. Meanwhile, they can also be regulated by a series of molecules in the aforementioned process. SIRTs are all involved in the regulation of macroautophagy, of which AMPK/mTOR signaling is the most common pathway. In addition, SIRT1, SIRT3, SIRT4, and SIRT5 are also involved in PINK1/Parkin-mediated mitophagy or Bnip3-mediated mitophagy. https://biorender.com. ACE2 angiotensin-converting enzyme 2, ATGL adipose triglyceride lipase, Bnip3 BCL2 interacting protein 3, CERKL ceramide kinase-like protein; circ, circular RNA; CUL4B, cullin 4B, eEF2 eukaryotic elongation factor-2, eEF2K eukaryotic elongation factor-2 kinase, EGFR epidermal growth factor receptor, ESRRA estrogen-related receptor α, FBXW7 F-box and WD repeat domain-containing 7, FoxM1 forkhead box M1, G6Pase-α glucose-6-phosphatase-α, GAS5 growth arrest specific 5, Hes‑1 hairy and enhancer of split‑1, HIF1α hypoxia-inducible factor 1 α, HIST1H1C histone cluster 1 H1 family member c, IPMK inositol polyphosphate multikinase, LDHB lactate dehydrogenase B, lncR long non-coding RNA, miR miRNA, NAT10 nucleolar protein N-acetyltransferase 10, NMNAT1 nicotinamide mononucleotide adenylyltransferase 1, Notch‑1 Notch homolog 1, OPA1 optic atrophy 1, p53 tumor protein p53, PINK PTEN induced putative kinase, PLIN5 perilipin 5, PTEN phosphatase and tensin homolog, SQSTM1/p62 sequestosome 1, TFEB transcription factor EB, TUG1 taurine-upregulated gene 1, TyrRS tyrosyl transfer-RNA synthetase, Ube2v1 ubiquitin-conjugating E2 enzyme variant 1
The effect of SIRT1 on autophagy
-
(1)
SIRT1 regulates autophagy through deacetylation
SIRT proteins affect protein acetylation level, and this modification is closely involved in autophagy. There are complex roles for SIRT1-related deacetylation in the regulation of autophagy.268,269 For example, SIRT1 deacetylates autophagy-related proteins (such as Beclin-1 and microtubule-associated protein light chain 3 (LC3)) to promote autophagy. Deacetylation of Beclin-1 lysine residue by SIRT1 impairs autophagic flux; thus, autophagosome fusion with lysosomes is compromised.270,271 SIRT1 promotes autophagy of cancer cells by reducing acetylation of LC3.272 LC3 and autophagy related (Atg)7 deacetylation is disrupted in germ-cell-specific SIRT1 KO mice, which affects the redistribution of LC3 from the nucleus to the cytoplasm and activation of autophagy.273 Suppression of SIRT1 enhances acetylation level of unc-51 like kinase 1 (ULK1) and induces ROS-dependent autophagy.274 Therefore, SIRT1 could directly regulate autophagy through deacetylation of autophagic proteins.
SIRT1 regulates autophagy via deacetylation of autophagy-related proteins as well as through deacetylation of mitochondrial proteins.275 Mitochondrial proteins participate in the process of mitophagy; a selective autophagic process that is critical for cellular homeostasis and eliminates dysfunctional mitochondria.276 For example, induction of autophagy by SIRT1/HIF-1α activation is a novel therapeutic option for peripheral nerve injury.277 SIRT1 activity is involved in mitochondrial biogenesis through PGC-1α and participates in the balance of autophagy regulatory proteins.278 Mitofusins2 (MFN2) is a mitochondrial fusion factor and increasing evidence has shown that it is involved in the regulation of autophagy.279 For example, MFN2 is deacetylated by SIRT1, and loss of SIRT1 causes a sequential chain of defective autophagy in an MFN2-dependent manner.280 Mechanistically, SIRT1 deacetylates K655 and K662 residues at the C terminus of MFN2, leading to autophagy activation.281
In conclusion, SIRT1 acts on autophagy-related proteins and transcriptional factors mainly through modification of acetylation, and affects the occurrence or degradation of autophagosomes. However, there have been limited studies on other PTMs of SIRT1, and more research is needed to explore the regulatory mechanism of SIRT1 in the future.
-
(2)
Upstream and downstream signaling pathway of SIRT1 in autophagy
AMPK is an evolutionarily conserved serine/threonine-protein kinase. Under various physiological and pathological conditions, AMPK acts as an activator of SIRT1 and is involved in the regulation of autophagy. For example, inositol polyphosphate multi kinase enhances autophagy-related transcription by stimulating AMPK-dependent SIRT1 activation.282 AMPK can also be activated as a downstream molecule of SIRT1. SIRT1 promoted autophagy via AMPK activation.283 Autophagy impairment is mediated by downregulation of SIRT1/FoxO3a/AMPK/ peroxisome proliferators-activated receptors (PPAR)-α signaling.284 The SIRT1 activator resveratrol increases cAMP content, expression of protein kinase A, as well as the activity of AMPK. Besides, resveratrol pretreatment reduces tumor necrosis factor α-induced inflammation and increases LC3B expression and sequestosome 1(SQSTM1)/p62 degradation in a concentration-dependent manner.285 Activation of the AMPK/SIRT1 pathway alleviates cell damage and promotes autophagic flux via downregulation of p62.286 Therefore, SIRT1 recognizes resveratrol-induced autophagy in vitro and in vivo via the cAMP/phosphorylated protein kinase A (PRKA)/AMPK/SIRT1 signaling pathway.287,288 AMPK acts as an upstream molecule to regulate expression of SIRT1 active agent. SIRT1 affects autophagy by binding to molecules directly. SIRT1 forms a molecular complex with Atg5, Atg7 and Atg8, and transiently increased expression of SIRT1 is sufficient to stimulate basal rates of autophagy.289 SIRT1 interacts with the Cullin 4B-Ring E3 ligase complex, which promotes autophagy of cancer cells.290 In conclusion, these molecules play important roles as the upstream or downstream of SIRT1 in the process of autophagy, and affect the occurrence and development of diseases.
-
(3)
Noncoding RNAs in SIRT1-regulated autophagy
A variety of miRNAs have been found to affect autophagy by directly regulating expression of SIRT1. For example, miR-124 and miR-142 represses autophagy via targeting SIRT1 in cancer cells.291 Silencing of miR-150-5p increases autophagy by targeting the SIRT1/p53/AMPK pathway.292 miR-138-5p affects insulin resistance through inducing autophagy in HepG2 cells by regulating SIRT1, and overexpression of SIRT1 increases Beclin-1 and LC3 II/I levels, and the number of green fluorescent protein-LC3 dots, and decreases p62 level.293 miR-145 inhibition upregulates SIRT1 and attenuates autophagy via NF-κB-dependent Beclin-1.294
Both long noncoding RNAs (lncRNAs) and circular RNAs (circRNAs) modulate autophagy associated with SIRT1. For instance, lncRNA metastasis-associated lung adenocarcinoma transcript 1 enhances ox- low-density lipoprotein (LDL)-induced autophagy through the SIRT1/MAPK/NF-κB pathway.295 lncRNA growth arrest specific 5 inhibits macroautophagy and forms a negative feedback regulatory loop with the miR-34a/SIRT1/mammalian target of rapamycin (mTOR) pathway.296
In conclusion, SIRT1 is a key regulator of the autophagic process. Through its deacetylase activity, SIRT1 is involved in the regulation of different autophagic proteins from initiation to degradation. The level and function of SIRT1 are also regulated by many signaling pathways, such as AMPK. Some studies have shown the regulation of SIRT1 by ncRNAs. SIRT1-mediated autophagic dysregulation leads to progression of various diseases. In the future, we need more research evidence to improve and supplement the mechanism of SIRT1.
Effect of SIRT2 on autophagy
It has been indicated that SIRT2 controls the functional ability of the autophagic system through acetylation.297 Genetic manipulation of SIRT2 levels in vitro and in vivo modulates the levels of α-synuclein acetylation, its aggregation, and autophagy.298 SIRT2 loss of function either with AK1 (a specific SIRT2 inhibitor) or by SIRT2 KO recovers microtubule stabilization and improves autophagy.299 Additionally, SIRT2 directly binds to the 3'UTR of transcription factor EB and facilitates its mRNA stability. Transcription factor EB is a key transcription factor involved in the regulation of many lysosome-related genes and plays a critical role in the fusion of autophagosomes and lysosomes, indicating that SIRT2 modulates autophagic components.300 Although the precise mechanism is unresolved, SIRT2 plays a key role in regulating autophagy in certain diseases, and more research is needed.
Effect of SIRT3–5 on autophagy
As mitochondrial SIRTs (mtSIRTs) members, SIRT3–5 are all involved in regulating energy metabolism and metabolic homeostasis through regulation of mitophagy.301,302 SIRT3 regulates autophagy by activating different downstream signaling pathways. For example, overexpression of SIRT3 activates macroautophagy through activating the AMPK/ULK1 pathway.301 SIRT3 promotes expression of autophagic proteins Beclin-1 and LC3II via downregulation of the Notch-1/Hes-1 pathway.303 Functional studies showed that SIRT3 reversed Bnip3 expression and promoted Bnip3-required mitophagy activity via the ERK-CREB signaling pathway.304 SIRT3 is involved in the regulation of autophagy; however, its role as an autophagy regulator, particularly the molecular mechanism, remains poorly understood. One recent study found that SIRT3 was directly inhibited by miR-874-5p and promoted autophagy, while depletion of miR-874-5p inhibited autophagy.305 A related study indicated that SIRT3 regulated the LKB1/AMPK/mTOR autophagic signaling pathway through the lncRNA DYNLRB2-2/miR-298/SIRT3 axis.306 Compared with SIRT1, the studies related to autophagy in SIRT3 are still lacking.
Mitochondria represent a major source of ROS that affect mitochondrial function, resulting in autophagic clearance of damaged mitochondria.183 Localized in the mitochondria, SIRT4 regulates proteins involved in metabolic reactions, antioxidant pathways and autophagy, thus maintaining mitochondrial homeostasis.307 Overexpression of SIRT4 inhibits ROS production and autophagy by activating the Akt/mTOR signaling pathway.308 Furthermore, the SIRT4/optic atrophy 1 axis is causally linked to mitochondrial dysfunction and altered mitochondrial dynamics that translates into aging-associated decreased mitophagy.301 So far, there are few relevant studies on SIRT4 regulation of autophagy. Further studies need to explore the role of SIRT4 as an mtSIRT in mitochondrial processes, such as autophagy (mitophagy).
Unlike SIRT4, which inhibits autophagy, the role of SIRT5 in regulating autophagy is contradictory. In the case of inhibition of autophagy by SIRT5, mitochondrial size is increased and mitophagy decreased upon SIRT5 overexpression, whereas the opposite effect is observed in SIRT5-silenced cells or upon treatment with the SIRT5 inhibitor MC3482.302 However, SIRT5 could enhance autophagy in gastric cancer (GC) cells via the AMPK/mTOR pathway.309 Additionally, SIRT5-induced deacetylation of lactate dehydrogenase B triggers hyperactivation of autophagy; a key event in tumorigenesis.310 Succinyl-proteomics in brown adipose tissue of normal and SIRT5 KO mice. Overacylation due to SIRT5 deficiency leads to defective autophagy/mitophagy.311 Besides their functions in energy metabolism and mitochondrial respiratory chain complexes, all three mtSIRTs participate in the regulation of mitochondrial morphology/dynamics. They seem to promote mitochondrial fusion and/or inhibit fission, and thus might attenuate mitophagic clearance of dysfunctional mitochondria.302 At present, the mechanism of action of mtSIRTs on autophagy is still unclear.
Effect of SIRT6 on autophagy is mainly through inhibition of Akt-related pathway
SIRT6 is essential for the regulation of autophagy in cells. For example, overexpression of the SIRT6 gene could inhibit apoptosis and induce autophagy, which might be involved in repairing kidney damage caused by lipopolysaccharide (LPS).312 Autophagy controls cellular senescence by eliminating damaged cellular components and is negatively regulated by Akt signaling through mTOR. SIRT6 overexpression induces autophagy via attenuation of insulin-like growth factor (IGF)/Akt/mTOR signaling.313 Lu et al. revealed that SIRT6 positively regulates autophagy in cardiomyocytes. Mechanistically, SIRT6 promotes nuclear retention of FoxO3 transcription factor via attenuating Akt signaling, which is responsible for autophagic activation.314 SIRT6 can be inhibited by upstream miR-122, resulting in a significant reduction in the levels of elabela, thereby preventing angiotensin II (Ang II)-mediated loss of autophagy.315 However, the mechanism of SIRT6 promotion of autophagy needs further study.
Effect of SIRT7 on autophagy needs further investigation
There are few studies about the effects of SIRT7 in autophagy. For example, silencing forkhead box M1 promotes apoptosis and autophagy through the SIRT7/mTOR/IGF2 pathway in GC cells.316 SIRT7 protects against chondrocyte degeneration in OA via autophagic activation.317 SIRT7 depletion significantly inhibits androgen-induced autophagy in LNCap and 22Rv1 cells (in vitro). SIRT7 plays an important role in tumor growth and metastases and immunohistochemical analysis of 93 specimens and bioinformatic analysis revealed that SIRT7 expression was positively associated with androgen receptor (AR) (in vivo).318 SIRT7 promotes prostate cancer autophagy indirectly via the AR signaling pathway.318 These results suggest that SIRT7 plays a positive role in promoting apoptosis. However, the number of studies on SIRT7 is still limited and further research is needed.
Conclusion
Autophagy is a highly conserved catabolic process and a major cellular pathway for the degradation of long-lived proteins and cytoplasmic organelles. Growing evidence has suggested that the SIRT protein family plays an important role in pathophysiology by mediating autophagy, maintaining cellular homeostasis, integrating cellular energy metabolism, and clearing damaged and waste cells. Although there is still a lot of work to be done, based on the current research, it is confident that the SIRT family might become a target for future research on autophagy. Investigating the exact mechanism of SIRT-mediated autophagy in different diseases is a new field to be explored in the future. Further studies should focus on the biological mechanism of SIRT co-regulating autophagy with various molecular signals and its role in different subcellular localization. Moreover, autophagy modulators of SIRTs may also provide new pharmacological targets.
Role of SIRTs in cell proliferation
Cell proliferation is the process by which a cell grows and divides to produce two daughter cells.319,320,321 Cell proliferation leads to an exponential increase in cell number and is, therefore, a rapid mechanism of tissue growth.321,322 Cell proliferation requires both cell growth and division to occur at the same time, which is the basis of organismal growth, development, reproduction and inheritance (Fig. 7).322,323,324

Overview of the roles of SIRTs in cell proliferation. (i) SIRTs participate in regulating cell proliferation by affecting a group of downstream proteins, including p53, p65, STAT3, FOXO1, AMPK, etc. (ii) SIRTs are also regulated by a series of ncRNAs and proteins, such as lncRNA PVT1, miR-34a, IFN-γ, MDM2, PRARα, eNOS, TCF3, etc, and subsequently promote or inhibit cell proliferation directly. (iii) In addition, SIRTs could activate or inhibit several signaling pathways, which perform important roles in cell proliferation, including JAK2/STAT3 signaling pathway, Wnt/β-catenin signaling pathway, PI3K/AKT signaling pathway, Notch signaling pathway, and ERK/STAT3 signaling pathway. Activation and inhibition effects are displayed in green and red arrows, respectively. https://biorender.com. ACAT1 acetyl coenzyme A acyltransferase1, Bmi-1 B-cell-specific Moloney murine leukemia virus integrationsite-1, CCAR2 cell cycle and apoptosis regulator protein 2, CDK9 cyclin-dependent kinase9, Drp1 dynamin-related protein 1, Erα estrogen receptor α, FASN fatty acid synthase, GLP-1 glucagon-like peptide-1, H1 histone1, HIF-2α hypoxia inducible factor-2α, K-Ras p21, MEF2D myocyte enhancer factor 2D, mitoCOX-2 mitochondria cyclooxygenase-2, MRP migration inhibitory-factor related protein, mTORC1 mTOR complex 1, Pcsk9 proprotein convertase subtilisin/kexin type 9, PD-L1 programmed death 1-ligand 1, POLD1 DNA polymerase delta 1, Pol-I DNA polymerase I, BBC3 Bcl-2 binding component 3, Rb retinoblastoma protein, SPEBP1 phosphatidylethanolamine binding protein 1, STAT1 signal transducer and activator of transcription 1, TCF3 transcription factor 3, Twist1 twist family bHLH transcription factor 1, ZEB2 zinc finger E-box binding homeobox 1
Effect of SIRT1 on cell proliferation
SIRT1 is involved in regulating cell proliferation in a bilateral way by regulating protein expression and acetylation.272,325 The opposite effects of SIRT1 on cell proliferation have been observed among different cell types or the regulation of different downstream molecules. For example, SIRT1 promotes cell proliferation by regulating LC3 and retinoblastoma (Rb) acetylation. At the molecular level, SIRT1 promotes the proliferation of endometrial cancer (EC) cells by reducing acetylation of LC3.272 SIRT1 deacetylates Rb protein in the Rb/ E2F transcription factor 1 (E2F1) complex, leading to dissociation of E2F1 and enhanced oligodendrocyte progenitor cell proliferation.326 SIRT1 directly regulates expression of transcription factor proteins, such as E2F1 and p53, subsequently promoting macrophage and HCC cell proliferation, respectively.327,328
However, SIRT1 can have an antiproliferative role via regulating expression of key proteins related to cell proliferation, such as AMPK and signal transducer and activator of transcription 3 (STAT3). For instance, SIRT1 exerts antiproliferative effects via the AMPK/mTOR pathway in the context of mutant p53 in HCC cells.329 SIRT1 overexpression inhibits the proliferation of renal cancer cells, while inhibition of SIRT1 expression has the opposite effects.325 SIRT1 might serve an anticancer role in cancer cells by upregulating expression of downstream AMPK.330 SIRT1 also inhibits GC cell proliferation via the STAT3/matrix metalloproteinase (MMP)-13 signaling pathway.331 SIRT1 has both promotive and inhibitory effects on proliferation in different cells. However, more studies are still needed to elucidate the mechanisms and establish under which conditions SIRT1 promotes or inhibits cell proliferation.
Effect of SIRT2 on cell proliferation
Participation of SIRT2 in cell proliferation was identified by a series of studies.332,333,334,335 At the molecular level, SIRT2 regulates Myc and results in promotion of cell proliferation. For example, SIRT2 enhances N-Myc and c-Myc protein stability and promotes cancer cell proliferation.332 On the contrary, SIRT2 functions as an HDAC and inhibits proliferation of neuroblastoma cells, renal podocytes, and neuroblastoma cells.336 SIRT2 upregulation reduces cell proliferation in renal podocytes under high-glucose conditions.337 The opposite effect of SIRT2 on cell proliferation might be due to the different cell types, which might be the direction for future studies.
Effect of SIRT3 on cell proliferation
SIRT3, the major deacetylase in mitochondria, also plays a bilateral role in regulating cell proliferation. For instance, SIRT3 is responsible for hydroxymethyl-transferase 2 (SHMT2) deacetylation, and the conversion of serine and glycine accomplished by SHMT2 deacetylation in mitochondria is significantly upregulated to support cell proliferation.338 Chen et al. found that increased activity of SIRT3 contributed to decreased ROS levels and increased cell proliferation.339 Conversely, the expression of SIRT3 is upregulated by Profilin-1, and subsequently negatively regulates HIF-1α protein levels and suppresses cell proliferation.340
Effect of SIRT4 on cell proliferation
SIRT4 inhibits proliferation of several types of cancer cells. For example, SIRT4 inhibits the proliferation of cancer cells by inhibiting glutamine metabolism.341,342 In addition, cell proliferation due to repression of SIRT4 by the mTORC1 pathway has been identified.343 Moreover, SIRT4 is the molecular switch mediating cellular proliferation through glutaminase (GLS)-mediated activation of the Akt/GSK3β/CyclinD1 pathway; mechanically, SIRT4 suppression activates glutaminase, thereby initiating Akt activation.344
Effect of SIRT5 on cell proliferation
SIRT5 promotes cell proliferation in most conditions by regulating activity of signaling proteins or protein PTM. For instance, SIRT5 promotes cell proliferation by increasing activity of the MAPK pathway through acetyl-CoA acetyltransferase 1.345,346 Moreover, citrate synthase desuccinylation by SIRT5 promotes cancer cell proliferation.347 Similarly, SHMT2 desuccinylation by SIRT5 drives cell proliferation.348 In addition, SIRT5 regulates cell proliferation directly or indirectly by influencing expression of transcription factors, such as E2F1 and pancreatic and duodenal homeobox 1 (PDX1).349 However, SIRT5 suppresses the proliferation of pancreatic β-cells in vitro by downregulating transcription of PDX1 by deacetylating H4K16.350 In conclusion, SIRT5 has dual functions in regulating proliferation of different cell types. However, the distinct mechanism for the bilateral roles of SIRT5 is worth further exploration.
Effect of SIRT6 on cell proliferation
SIRT6 is also reported to regulate cell proliferation in a bilateral manner via influencing downstream molecules, such as AMPK, ERK, Wnt signaling and the MAPK pathway. SIRT6 promotes expression of COX-2 by repressing AMPK signaling, thereby increasing cell proliferation.351 Moreover, overexpression of SIRT6 promotes cell proliferation via upregulating he phosphorylation of ERK.352 In addition, SIRT6 deletion promotes hematopoietic stem cell proliferation through aberrant activation of Wnt signaling.353 Using genetic and biochemical studies in vitro and in human multiple myeloma xenograft models, Cea et al. found that SIRT6 depletion enhanced cell proliferation via upregulating expression of MAPK.354 In conclusion, SIRT6 has both promotive and inhibitory effects on cell proliferation. The different results of SIRT6 in regulating cell proliferation need further study.
Effect of SIRT7 on cell proliferation
Previous studies have shown that SIRT7 has a positive role in regulating cell proliferation.355 Upregulation of SIRT7 protects against the proliferation of vascular smooth muscle cells (VSMCs) in atherosclerosis.355 Similarly, SIRT7 deficiency attenuates VSMC proliferation, thus attenuating neointimal formation following vascular injury.356 Moreover, SIRT7 depletion inhibits cancer cell proliferation by suppressing AR signaling and activating p38MAPK.318,357
Conclusion
The direct and indirect involvement of SIRTs in proliferation could provide new ideas and evidence in support of potential research and as therapeutic targets. This might be meaningful for the treatment of abnormal proliferation in the future, thereby reducing the human disease burden related to proliferation. However, at present, research is still focused on the effect of SIRTs on carcinoma, and other molecular mechanisms of proliferation is less researched. Therefore, research on other molecular mechanisms of proliferation should be increased in the future. More evidence from in vitro and in vivo models for different kinds of diseases to confirm undefined molecular mechanisms of proliferation as yet is awaited.
Roles of SIRTs in cell migration and invasion
Migration and invasion are vital phenotypes both in physiological and pathological status. They allow normal cells to change position within tissues during embryonic morphogenesis, wound healing, and immune-cell trafficking.358,359 Specifically, in human cancers, they allow neoplastic cells to enter lymphatic and blood vessels for undergoing metastatic growth in distant organs.360,361 An increasing number of studies have shown that SIRTs play important roles in the molecular mechanisms of cell migration and invasion, such as regulation of TGF-β signaling and epithelial-to-mesenchymal transition (EMT).362,363 Since these two phenotypes are hallmarks during tumor progression, we introduced the potential roles of SIRT protein family in cell migration and invasion, mainly depending on cancers (Fig. 8).

Overview of the roles of SIRTs in cell migration and invasion. SIRTs coordinate a multi-faceted regimen to control cell migration and invasion. In the nucleus, SIRT1, SIRT6, and SIRT7 may affect many key proteins, which also contain transcription factors, mainly involved in EMT process, TGF-β signaling, PI3K/Akt signaling, MMPs signaling, and AMPK signaling pathways, etc, thereby regulating cell migration and invasion. In the cytosol, SIRT2 could suppress cell migration and invasion by deacetylating target proteins such as AKR1C1 and IDH1. In mitochondria, SIRT3, SIRT4, and SIRT5 could participate in regulating cell migration and invasion via influencing various molecular mechanisms such as integrin adhesion and EMT. Activation and inhibition effects are displayed in green and red arrows, respectively. https://biorender.com. α7nAChR alpha7 subtype of nicotinic acetylcholine receptors, ISRE IFN-stimulated response element
Effect of SIRT1 on cell migration and invasion
SIRT1 deacetylates many key proteins, which also contain transcription factors, mainly involved in EMT and integrin adhesion, thereby regulating cell migration and invasion.271,363 EMT is the most well-established example of changes in cell–cell adhesion, which refers to nonepithelial cells that are loosely embedded in an extracellular matrix (ECM).364 Integrin adhesion activates pathways including TGF-β, phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/Akt, and AMPK signaling pathways.
Several studies have shown that SIRT1 protein levels are lower in lesion tissues than in adjacent tumor tissues or normal tissues of patients with cancer.365,366,367 This phenomenon is also observed in autoimmune disorders, which indicates that SIRT1 plays important roles in regulating cell migration and invasion.368 SIRT1, as a deacetylase, influences the biological functions of proteins via regulating protein deacetylation, such as deacetylation of Beclin-1. In melanoma cells, SIRT1 deacetylates Beclin-1 and then accelerates autophagic degradation of the epithelial marker E-cadherin, finally promoting EMT.271 Additionally, SIRT1 could regulate the expression levels of several proteins that participate in cell migration and invasion, resulting in promotion of EMT. Both in vivo and in vitro studies have shown that expression of SIRT1 in chondrosarcoma cells could effectively take part in the metastatic plasticity of the cells by inducing EMT, via enhancing expression of Twist protein, which is a critical transcriptional factor of EMT.363 Zinc finger E-box binding homeobox 1 is an E-cadherin‐related transcription factor. Yu et al. have reported that there is positive feedback between SIRT1 and Zinc finger E-box binding homeobox 1, which enhances EMT of osteosarcoma.369 SIRT1 induces deacetylation of Beclin-1 and then accelerates autophagic degradation of the epithelial marker E-cadherin, further promoting EMT in melanoma cells.271 Epidermal SIRT1 plays a role in wound repair. SIRT1 knockdown inhibits EMT, cell migration, and TGF-β signaling in keratinocytes.370 Furthermore, SIRT1 activates downstream PI3K/Akt and Notch signaling pathways, which alleviates H9c2 cell injury induced by hypoxia, via promoting cell proliferation, migration and invasion, and by inhibiting apoptosis.371 In non-small cell LC (NSCLC), the SIRT1-mediated AMPK/mTOR signaling pathway could promote A549 and H1299 cell proliferation, invasion and apoptosis.372
Expression of SIRT1 can be regulated by ncRNAs, which further influence its effects in regulating cell migration and invasion. For instance, in colorectal cancer (CRC) cells, downregulation of SIRT1, by miR-34a transfection, increases the level of acetylated-p53 and inhibits cell migration and invasion.373 This situation is also found in HCC.374 Expression of SIRT1 can also be regulated by lncRNAs or circRNAs in a ceRNA-dependent manner. For example, SIRT1 promotes cell migration and invasion in HCC. Expression of SIRT1 is upregulated by lncRNA MALAT1 via sponging miR-204, which might have a pivotal role in treatment and prognosis.375 Furthermore, SIRT1 promotes the migration of fibroblast-like synoviocytes in rheumatoid arthritis, which providing new insight into SIRT1 during RA progression. Mechanistically, SIRT1 is positively regulated by circ0088036 via sponging miR-140−3p.368
Effect of SIRT2 on cell migration and invasion
SIRT2 participates in regulating cell migration and invasion through deacetylating target proteins. STAT3 is an important protein for regulating cell invasion and migration.376,377 STAT3 has been shown to affect EMT in several cancers.378 Previous studies have shown that SIRT2 can deacetylate Aldo-keto reductase family 1 member C1 (AKR1C1), which is a member of the human aldo-keto reductase protein family that catalyzes NADP + -dependent reduction. AKR1C1 deacetylation further inhibits the transactivation of STAT3 target genes, thus suppressing migration in NSCLC cells and xenograft models.379 It has been reported that isocitrate dehydrogenase 1 (IDH1) affects cell migration in malignant tumors, such as glioblastoma.380 In human CRC, SIRT2-dependent IDH1 deacetylation represses CRC cell migration and invasion both in vitro and in vivo.381
Effect of SIRT3-5 on cell migration and invasion
SIRT3-5 are three main deacetylases that are located in mitochondria, which appear to be suppressors of cell migration and invasion. Previous studies have demonstrated that SIRT3 and SIRT4 negatively regulate EMT. For instance, transplantation of sh-SIRT3 cells in nude mice resulted in rapid tumor growth and larger tumors. At the molecular level, SIRT3 depletion inhibits EMT by lower E-cadherin expression, leading to tumor suppression.382 Sun et al. suggested that SIRT4 suppressed EMT through promoting E-cadherin expression in GC cells.383 Li et al. reported that SIRT3 was involved in the inhibitory effect of nicotinic alpha7 subtype of nicotinic acetylcholine receptors on platelet-derived growth factor-BB, an angiogenic factor, induced VSMC migration. Activation of alpha7 subtype of nicotinic acetylcholine receptors attenuates migration in platelet-derived growth factor-BB-treated VSMCs via a mitochondrial SIRT3-dependent manner.384
SIRT5 regulates cell migration and invasion in several cancer cells. For example, Dang et al. found that SIRT5 promoted migration and invasion of HCC cells.385 The opposite findings were reported by Yao et al. in that the inhibition of SIRT5 increased migration and invasion of HCC in hypoxic microenvironments.386 This inconsistent phenomenon might be attributed to the hypoxic status of tumor microenvironments. However, further studies are required for illustrating its deeper regulatory mechanisms.
Effect of SIRT6 and SIRT7 on cell migration and invasion
SIRT6 and SIRT7 are the least studied of the seven SIRTs to date, which are both located in the nucleus. Both of them have been found to play a role in cell migration and invasion via regulating EMT and/or MMP expression. For example, in human HCC, SIRT6 promotes N-cadherin and vimentin expression via deacetylating FoxO3a in HCC cells.387 SIRT6 upregulates expression of MMP9 probably through the MAPK/ERK1/2 pathway, with increased migration and invasion of OS cells.388 Liu et al. found that forced expression of SIRT6 attenuated EMT by suppressing the TGF-β1/ small mothers against decapentaplegic protein (Smad)3 pathway and N-terminal kinase (c-Jun) in rat models of asthma.362 A recent study has shown that SIRT7 promotes CRC cell invasion through the inhibition of E‐cadherin, which is the most important protein in EMT.389 Furthermore, SIRT7 is overexpressed in EC cells compared with normal endometrial cells. SIRT7 downregulation inhibits the invasiveness of EC cells.390
Conclusion
Taken together, the above-discussed findings suggest that SIRT proteins are involved in regulating cell migration and invasion during physiological processes and the development of human cancers. However, current research mainly focuses on the function of SIRT1 in regulating cell migration and invasion. Much work is still needed to pinpoint the precise molecular mechanisms governing the functions of other SIRTs, especially SIRT6 and SIRT7, under those conditions. It is meaningful to continue to explore the role of SIRT proteins in other diseases, which might provide future beneficial alternatives against those devastating diseases.
Regulatory role of SIRTs in human diseases
SIRTs and cancer
Cancer is currently the second most common contributor to premature mortality worldwide.391 Since an early diagnosis and effective treatment for patients with cancer are critical, the identification and application of effective biomarkers and novel drug targets are urgently required. Recent evidence reveals that aberrant expression of SIRTs occurs in almost all cancer types with different mechanisms, including those involved in cancer metabolism, genome stability, and the tumor microenvironment.3 The functions of SIRTs in the tumor process are characterized as tumor suppressor and/or oncogene, depending on genetic context and tumor type and stage.392 Moreover, SIRTs could exert regulatory roles in the response of the tumor to chemotherapy.393 These unique features suggest that SIRTs serve as potentially targetable markers and play important roles in cancer therapy. In this section, we summarize the recent studies of SIRTs in diverse cancers, which is shown in Fig. 9.

The roles of SIRTs in cancers. SIRTs are involved in a series of malignancies, including BC, LC, liver cancer, GC, PC, colorectal cancer, OC, EC, CC, malignant glioma, and leukemia. SIRTs act as tumor promoters (marked in red color), tumor suppressors (marked in green color), or both suppressor and promoter (marked in blue color). Major events in solid tumor development consist of tumor initiation, tumor proliferation, and tumor metastasis. Between these events, processes including cell proliferation, oxidative stress, apoptosis, angiogenesis, EMT, migration and invasion are promoted or inhibited. Depending on the tumor location, the metastasis site also varies, including lymph nodes, distant organs, liver, adjacent organs, etc. https://biorender.com
Breast cancer (BC)
BC is the most common malignancy throughout the world and is the fifth leading cause of cancer-related deaths.394 SIRT2 and SIRT4 are downregulated,395,396 while SIRT1 and SIRT7 are upregulated in BC tissues compared to adjacent tissues or normal tissues.357,397 Besides, increased SIRT2 and SIRT4 expression is correlated with longer overall survival,395,396 whereas increased SIRT1 and SIRT7 expression predicts a poor prognosis in patients with BC.398,399 These disparities might be in respect to the different roles of them in BC progression.400
Regarding BC development, SIRTs are generally considered as tumor suppressors but might act as tumor promoters as well. SIRT1 has been extensively explored in comparison to other SIRTs for their roles in BC, and may influence BC progression by regulating many processes, especially EMT. SIRT1 plays a critical role in regulating EMT-associated programming and thus, consequently, eliciting BC invasion and metastasis in patients with triple-negative BC.401 SIRT1 expression suppresses BC metastasis by reducing EMT, and invasiveness in nude mice.402 The effect of SIRT1 modulation on EMT in breast cancer-related cancer stem cells has also been observed. This study indicates that loss of SIRT1 destabilizes EMT inducer paired related homeobox 1, disinhibits KLF4, and activates transcription of aldehyde dehydrogenase 1, which encourages cancer stem cells, resulting in metastatic reversion.403 In addition to its tumor suppressive roles in BC, SIRT1 overexpression, altered EMT programming, and a decrease in tumor-suppressive miR-200a may be consistently involved in BC development and subsequent distant metastasis.404 The plausible explanation of the contradictory functions of SIRT1-mediated BC regulation might be due to tumor grade, tumor stage of BC, and the use of animal or human samples with a different pathological subtype.
Additionally, many other non-EMT factors that are known to function in other cellular processes in BC development could also be regulated by SIRT1. For example, the estrogen receptor (ER) and AR could mediate induction of estrogen- and androgen-responsive genes respectively and stimulate cell proliferation, and SIRT1 represses the transcriptional and proliferative response of BC cells to estrogens via an Erα-dependent mechanism.405 DNA polymerase delta1, the gene coding for DNA polymerase δ catalytic subunit p125, is upregulated by SIRT1, thus promoting proliferation and migration of BC cell line MCF-7.397 Metadherin, an oncogenic protein, has been implicated in promoting cancer progression, metastasis, and chemoresistance in BC.406 Activation of AMPK has been reported to reduce the expression of metadherin through enhanced SIRT1 activity along with GSK-3β in an independent manner in triple-negative BC.406
In addition to SIRT1, SIRT2 functions in a binary manner, as a tumor suppressor or promoter. SIRT2 acts as a tumor suppressor in BC by regulating mitosis and genome integrity. Evidence has shown that SIRT2 promotes BRCA1-BRCA1-associated RING domain protein 1 (BARD1) heterodimerization through deacetylation, thereby facilitating homologous recombination and tumor suppression.407 Additionally, in cancer biology, Slug, an EMT transcription factor, promotes tumor progression and metastasis.408 In basal-like BC, SIRT2 maintains Slug protein stability by deacetylation, which contributes to basal-like BC’s robust tumorigenic activity, along with enhanced invasive and metastatic capabilities.409 SIRT3, SIRT4 and SIRT7 illustrate different functions in BC progression. SIRT3 has been found to affect p53 by disruption of the ERα–p53 interaction, and decrease proliferation, colony formation, and migration in BC cells.410 Notably, SIRT4 could exert its tumor-suppressive activity in BC though negatively regulating SIRT1 expression via repressing glutamine metabolism, which suggests a novel crosstalk between mitochondrial and nuclear SIRT proteins in BC progression.411 SIRT7 depletion inhibits tumor growth via activating p38/MAPK signaling.357
Additionally, SIRT proteins can affect the sensitivity of BC cells to several drugs, including tamoxifen, paclitaxel and doxorubicin. For example, SIRT1 causes tamoxifen resistance in ER-α-positive BC cells through upregulation of multidrug resistance protein 2 by mediating deacetylation of FoxO1 protein.412 Subsequently, SIRT1 inhibition impairs nuclear FoxO1 and multidrug resistance protein 2 expression and augments the cytotoxic effect of paclitaxel and doxorubicin in tamoxifen-resistant BC cells.412 SIRT3 overexpression in BC cell line MTR-3 reduces the sensitivity of the resistant cells to tamoxifen.357,413 On the contrary, SIRT4 enhances the tamoxifen sensitivity of BC cells via inhibiting the STAT3 signaling pathway.
These findings indicate unique mechanisms of SIRT1 mediate BC regulation and its contribution to tumor development and resistance, which suggests that SIRTs are promising therapeutic targets in BC, and provides clinical strategy for overcoming drug resistance. However, the exact molecular mechanism is still uncertain and needs further investigation.
LC
LC is the leading cause of cancer-related deaths and the second most diagnosed cancer worldwide, with NSCLC being the most common type.394 Significant differences in SIRT expression between NSCLC tissues and nontumor lung tissue or adjacent tissue have been observed, which indicates that SIRTs are promising biomarkers in the diagnosis of LC.414,415,416 Notably, serum SIRT3 distinguished LC patients from healthy individuals with an area under the curve of 0.918 and optimal cutoff value of 3.12, reaching sensitivity of 86.4% and specificity of 94%.416 SIRTs could be potential prognostic factors for NSCLC.414,415,417 For example, high SIRT1-3 expression is associated with poor survival in patients with NSCLC.414,415
Evidence has suggested that SIRTs are key factors involved in tumor development and treatment in LC.418,419 Regarding LC progression, the SIRTs play conflicting roles. SIRT1 upregulated by SNHG10 suppresses NSCLC cell proliferation, as a tumor suppressor.420 Overexpression of SIRT1 protects NSCLC cells against osteopontin-induced NF-κB p65 acetylation and EMT, thus attenuating OPN-induced cell proliferation, migration and invasion.421 However, SIRT1 upregulated by circ_0001946, could promote cell growth in lung adenocarcinoma by activating the Wnt/β-catenin signaling pathway.422 SIRT2 and SIRT6 have been shown to exert both pro- and anticarcinogenic effects in the process of LC. For example, SIRT2 suppressed the migration of NSCLC cells by deacetylating AKR1C1, and inhibiting transactivation of STAT3 target genes.379 In addition, SIRT2 deacetylates the K100 residue of glycolytic enzyme phosphoglycerate mutase and facilitates its activation, resulting in enhanced NADPH production and accelerated tumor growth in NSCLC cells.423 Similarly, SIRT6 illustrates opposite functions in the promotion of LC development, as tumor suppressor and promoter.424,425 For instance, SIRT6 can coordinate with chromatin remodeler chromodomain-helicase-DNA-binding 4 to promote chromatin relaxation and DNA repair, thereby exerting an anticarcinogenic role in LC.424 In contrast, SIRT6 can also drive EMT and metastasis in NSCLC via snail-dependent transrepression of KLF4.425 This dual action of these SIRTs might depend upon the cellular context, tumor types, cancer stage, and their involvement in various cellular pathways,392,418 and further studies are needed to explore the exact mechanisms underlying their dual roles in LC.
Expression of SIRTs could have an influence on the chemoresistance and radioresistance of LC. SIRT1 promotes cisplatin resistance of NSCLC cells by elevating vascular endothelial growth factor A expression.426 SIRT1 is upregulated in cisplatin-resistant NSCLC tissues and cells compared to cisplatin-sensitive groups.291 SIRT1 silencing enhances the cisplatin sensitivity of H1299/cisplatin cells via suppressing autophagy. Upregulation of SIRT2 in NSCLC cells increases the sensitivity to cisplatin treatment while SIRT3 promotion reduces cisplatin resistance in LC by modulating the FoxO3/Cdc10-dependent transcript 1 protein axis.253,427 In relation to LC radioresistance, SIRT3 promotes DNA damage repair and radioresistance through ataxia telangiectasia mutated–Chk2 in NSCLC cells.428 SIRTs can affect radioresistance in LC through the regulation of tumor metabolism. Overexpression of SIRT6 inhibits key-enzyme generation in A549 cells to inhibit glycolysis and enhance radiosensitivity.428
In LC, SIRTs are involved in tumor development, chemoresistance and radioresistance, and exert regulatory functions by targeting different target proteins. Thus, SIRTs represent promising therapeutic targets in the perspective of precision medicine and provide new insights into therapeutic strategies for LC.
Gastrointestinal cancer
-
(1)
HCC
HCC, the most common type of primary liver cancer with a relatively high mortality, is the sixth most common cancer and the third-leading cause of cancer-related mortality worldwide.394 Several bioinformatics studies have reinforced that nonmitochondrial and mitochondrial SIRTs are differentially expressed in HCC. For example, nonmitochondrial SIRT1, SIRT2 and SIRT6 are expressed at higher levels,429,430,431 while mitochondrial SIRT3-5 are expressed at lower levels in HCC tissues compared with normal liver or surrounding tumor tissue.432,433,434 SIRTs could be prognostic markers for patients with HCC. For instance, high expression of SIRT1 and SIRT7 is highly associated with poor survival, whereas low tumor levels of SIRT4 predicts a decreased survival time in HCC patients.434,435,436
Recent studies have suggested that SIRTs could play regulatory roles in HCC development by regulating the metabolic state of the cancer cells.437 Referring to mitochondrial SIRTs, SIRT4 exerts a tumor-suppressive function in HCC by inhibiting glutamine metabolism.434 SIRT5 prevents tumor immune evasion and suppresses HCC development by orchestrating bile acid metabolism.438 However, SIRT5 also exerts a tumor-promoting function as a metabolic regulator. The activation of mitochondrial SIRT5 contributes to the promotion of growth and metastasis of HCC cells via glucose metabolism reprogramming from oxidative phosphorylation to glycolysis.439 The possible explanation for the dual role of SIRT5 in HCC could be related to its involvement in different metabolic processes, including glucose and lipid metabolism, which might result in opposite effects on tumor progression.3 In addition to mitochondrial SIRTs, the nonmitochondrial SIRTs can influence HCC by regulating cancer-related metabolism, especially glucose metabolism. SIRT1 and SIRT6 deacetylate hnRNP A1 to suppress glycolysis and growth in HCC.440 SIRT6, stabilized by ubiquitin-specific peptidase 48, attenuates HCC glycolysis and impedes metabolic reprogramming, thereby hampering HCC malignancy.441 SIRTs can also play roles in modulation of the cell cycle in HCC, which are essential for tumor development. Evidence has shown that SIRT4 upregulates cell-cycle governing genes p16 and p21 expression, suppresses CyclinB1/Cdc2 and Cdc25c, which normally induce cell-cycle progression, and suppresses survival to induce apoptosis in HCC cells.442
Therapeutic advances targeting SIRTs are currently being explored as it is suggested that modulating SIRT3 abundance via cyclin-dependent kinase (CDK) 4/6 inhibition might enhance HCC therapy when combined with sorafenib.443 SIRT3 downregulates the mRNA and protein levels of glutathione S-transferase π1, a phase II detoxification enzyme involved in metabolism of chemotherapeutic agents, and SIRT3 overexpression promotes chemotherapeutic-agent-induced or sorafenib-induced apoptosis, thereby enhancing the drug sensitivity of HCC cells.252 Aside from mitochondrially directed deacetylase activity, SIRT6 depletion is reported to downregulate multidrug resistance protein 1 expression through the suppression of CCAAT/enhancer-binding protein, promoting enhanced HCC chemosensitivity.444
-
(2)
Colorectal cancer
CRC ranks third in terms of cancer incidence worldwide and is the second most common cause of cancer deaths.394 Previous studies have shown that SIRT1 and SIRT7 are increased,389,445 whereas SIRT2, SIRT4 and SIRT6 are decreased in human CRC tissues compared to normal tissue, which suggests that SIRTs are potential diagnostic biomarkers for CRC.446,447,448 SIRTs are potential prognostic factors for CRC. For instance, overexpression of SIRT5 is correlated with poor prognosis in patients with CRC, while SIRT6 expression is related to improved survival.448,449 However, there still a need for further studies that make more clear analyses to verify the roles of SIRTs as biomarkers of CRC, such as receiver operating characteristic curve, sensitivity and specificity analyses.
The pleiotropic roles of SIRTs in the regulation of tumor cell metabolism and cell death are strongly linked to the progression of CRC. SIRT1 has been found to affect CRC in a dose-dependent manner by regulating glutamine metabolism and apoptotic pathways. Heterozygous deletion of SIRT1 induces c-Myc expression, enhancing glutamine metabolism and subsequent proliferation, autophagy and cancer formation. In contrast, homozygous deletion of SIRT1 triggers apoptotic pathways, increases cell death, diminishes autophagy, and reduces cancer formation.450 The dose-dependent regulation of cellular metabolism and apoptosis by SIRT1 mechanistically contributes to the observed dual roles of SIRT1 in tumorigenesis. SIRTs have an anticarcinogenic action via modulation of CRC-related metabolism. SIRT2-dependent IDH1 deacetylation regulates cellular metabolism and inhibits liver metastasis of CRC.381 SIRT4 upregulates E-cadherin expression and suppresses proliferation, migration and invasion through inhibition of glutamine metabolism in CRC cells.140 In addition to the anticarcinogenic effects of SIRTs, SIRT5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner.449 SIRTs exert their regulatory function in CRC development through the modulation of several autophagy-related pathways. In particular, SIRT5 can deacetylate lactate dehydrogenase B, thus promoting hyperactivation of autophagy and tumorigenesis in CRC.310
Recent evidence highlights that SIRTs are involved in various tumor processes related to chemoresistance and radioresistance in CRC. For example, overexpression of SIRT3 improves anticancer drug resistance of CRC cells through superoxide dismutase (SOD) 2 and PGC-1α regulation.451 In addition, SIRT4 increases the sensitivity of CRC cells to chemotherapeutic drug 5-fluorouracil by inhibiting the cell cycle.447 Regarding radioresistance of CRC, FoxQ1-mediated SIRT1 upregulation augments expression and nuclear translocation of β-catenin and benefits CRC-related intestinal pathological bacteria, thereby enhancing the radioresistance of CRC cells.452
SIRTs play a role in regulating CRC progression, which indicates that SIRT-small-molecule-activator/inhibitor-based therapy strategies is a rescue strategy for patients with CRC. However, there have been a limited number of studies of the molecular mechanisms of SIRTs in regulating CRC progression.
-
(3)
Gastric cancer
GC is the fifth most frequently diagnosed cancer with an incidence rate of 5.6%, and the fourth most common cause of cancer death with a mortality rate of 7.7% worldwide.394 During the past decades, SIRTs have been considered as potential druggable targets in the clinical treatment of GC. SIRT1 is upregulated in GC tissues and SIRT1 depletion promotes GC progression through activation of STAT3/MMP-13 signaling.331 SIRT4 and SIRT6 are downregulated in GC tissues, and their low expression is negatively correlated with tumor size and pathological grade, which predicts poor prognosis.383,453 Mechanistically, SIRT4 inhibits cell proliferation, migration, and invasion in GC via regulating EMT. SIRT6 inhibits the Janus kinase 2/STAT3 pathway, thereby suppressing the growth of GC. Regarding tumor resistance, SIRT6 silencing can overcome sorafenib resistance by promoting ferroptosis.454 Thus, SIRTs could act as novel biomarkers and therapeutic targets of GC.
-
(4)
Pancreatic cancer (PC)
PC has high mortality and ranks as the seventh leading cause of cancer-related deaths worldwide.394 PC is also affected by SIRT activity and expression. SIRT5 expression is directly correlated with favorable prognosis, as its loss promotes glutamic-oxaloacetic transaminase 1 acetylation, thus promoting cell proliferation by enhancing glutamine and glutathione metabolism.455 Upregulation of SIRT6 by tumor suppressor KLF10 activity influences glycolysis, EMT, and distant metastasis of PC.456 SIRTs are associated with drug resistance of PC. SIRT1 can facilitate chemoresistance of PC cells by regulating adaptive response to chemotherapy-induced stress.457
Collectively, SIRTs play important roles in tumor progression, chemoresistance and radioresistance in gastrointestinal cancer by regulating multiple cellular and physiological processes, including metabolism, cell cycle, cell death, and tumor microenvironment. Therefore, the potential selective modulation of SIRT protein family members represents a promising area in gastrointestinal cancer treatment. However, given the contribution of gastrointestinal cancer to worldwide morbidity and mortality, further research is needed to understand the exact mechanisms underlying the involvement of SIRTs in these cancers, especially for GC and PC.
Gynecological cancer
-
(1)
Ovarian cancer (OC)
OC is one of the most aggressive female malignancies, with poor prognosis.394 SIRT1-3 and SIRT6 are significantly decreased, while SIRT5 is significantly increased in OC tissues compared to normal or adjacent tissues.458,459,460 High expression of SIRT2 and SIRT5-7 is correlated with favorable survival, while high expression of SIRT1 and SIRT4 is associated with poor survival,458 suggesting that SIRTs could serve as novel prognostic biomarkers. SIRTs are implicated in the development and treatment of OC. For example, SIRT1 expression suppresses high motility group box-1 protein expression and acetylation, thus inhibiting OC migration, invasion and angiogenesis.461 However, MHY2245, a new SIRT1 inhibitor, exert antitumor activity against OC cells by blocking the pyruvate kinase M2/mTOR pathway.462 In addition to SIRT1, overexpression of SIRT3 dramatically suppresses OC cell metastatic capability by inhibiting EMT via downregulation of Twist.463 Regarding OC treatment, cisplatin has been a pivotal drug, however, cisplatin resistance hinders the prognosis of patients.464 Overexpression of SIRT2 significantly enhances the sensitivity of cisplatin-resistant counterpart cells to cisplatin in OC.465 In addition, SIRT5 can promote cisplatin resistance in OC by suppressing DNA damage in a ROS-dependent manner via regulation of the Nrf2/Heme Oxgenase-1 pathway.460
-
(2)
EC
EC is the most common gynecological cancer in high-income countries and its incidence is rising globally.466 Recent studies have shown that SIRTs participate in the development and progression of EC. For example, SIRT1 is elevated in EC cell lines and tissues and SIRT1 promotes autophagy and proliferation of EC cells by reducing acetylation of LC3.272 The expression of SIRT2 is increased in most human EC cell lines and SIRT2 overexpression promotes EC cell proliferation but inhibits apoptosis.467 In contrast to SIRT1 and SIRT2, SIRT6 might function as a tumor suppressor of EC cells. SIRT6 negatively affects the proliferation of AN3CA and KLE EC cells by repressing expression of the antiapoptotic protein surviving.468 Chemotherapy is crucial for postoperative adjuvant therapy of EC. SIRT1 promotes the growth and cisplatin resistance of EC cells.469 SIRT2 has been shown to promote cell stemness and activate the MEK/ERK signaling pathway while repressing chemosensitivity in EC.470
-
(3)
Cervical cancer (CC)
CC is one of the most severe and prevalent female malignancies and a global health issue.394 Abnormal expression of SIRTs in CC tissue may be related to disease progression. For instance, the expression of SIRT2 is decreased in CC tissue compared with paired adjacent tissue, and SIRT2 expression in tumor tissue is negatively correlated with tumor size, and lymph node metastasis, which predicts favorable survival.471 For mechanistic studies, SIRT1 has been found to be overexpressed in HPV-infected CC cells and SIRT1 expression is correlated with poor clinical outcomes in CC.472 SIRT1 enables HPV-infected CC cells to continue growing by nullifying absent in melanoma 2 inflammasome-mediated immunity. Moreover, SIRT3 contributes to the reprogramming of fatty acid synthesis by upregulating acetyl-coA carboxylase 1 to promote de novo lipogenesis by SIRT3 deacetylation, thereby promoting the invasion and metastasis of CC cells.473
SIRTs are implicated in tumor development and chemotherapy resistance in gynecological cancer including OC, EC, and CC, thus SIRTs might serve as indicators of prognosis and as promising therapeutic targets for gynecological cancer. However, evidence about the roles of SIRTs in gynecological cancer is still limited, so more studies are needed to further explore the underlying molecular mechanism by which SIRTs regulate tumor processes in these cancers.
Glioma
Glioma is the most common and malignant primary tumor of the central nervous system, with a poor prognosis, especially glioblastoma.474 SIRT1 and SIRT7 are upregulated,475,476 while SIRT3 and SIRT6 are downregulated in glioma tissues compared with normal or adjacent brain tissues.477,478 Glioma patients with higher SIRT1 or SIRT3 expression exhibit worse prognosis, whereas downregulation of SIRT5 is significantly correlated with shorter survival time in glioblastoma. These situations have suggested that SIRTs are promising prognostic biomarkers of glioma and might be involved in tumor progression.475,477,479
SIRT1 and SIRT6 exert a tumor suppressor effect in glioma. SIRT1-mediated p21-Activated kinase 1-deacetylation at K420 hinders autophagy and glioblastoma growth.480 Besides, SIRT6 suppresses glioma cell growth via induction of apoptosis, inhibition of oxidative stress, and inhibition of the activation of the Janus kinase 2/STAT3 signaling pathway.478 On the contrary, SIRT3 and SIRT7 are reported to play positive roles in the development of glioma. SIRT3 can stabilize Ku70–Bax interaction to enhance glioma cell viability.477 Moreover, SIRT7 affects the malignancy of glioma cells mainly by promoting glioma proliferation and invasion through ERK and STAT3 signaling.476 Evidence also suggests that SIRTs participate in the transformation of chemoresistance and radioresistance in glioma. For instance, SIRT1 inhibition increases the sensitivity of glioma cells for temozolomide via facilitation of intracellular ROS generation.475 In addition, CDK1-mediated SIRT3 activation could enhance mitochondrial function and contribute to adaptive radioresistance in glioma cells.481 Therefore, SIRTs are potential biomarkers for the prognosis and diagnosis of glioma and promising therapeutic targets.
Leukemia
Leukemia is a malignant clonal disease of hematopoietic stem cells, and most leukemias are sporadic and their specific etiology remains elusive.482 SIRTs participate in the development and therapeutic resistance of leukemia. SIRT1 promotes T-cell acute lymphoblastic leukemia progression by regulating the phosphorylation and degradation of p27 through deacetylating cyclin-dependent kinase 2.483 SIRT2 is overexpressed in primary acute myeloid leukemia blasts, and SIRT2 activation by nicotinamide phosphoribosyltransferase (NAMPT) reduces proliferation and induces apoptosis in human acute myeloid leukemia, possibly via the Akt/GSK-3β/β-catenin pathway.335 Inhibition of SIRT2 suppresses the in vitro growth and in vivo engraftment of T-cell acute lymphoblastic leukemia cells via diminished LIM domain only 2 (LMO2) deacetylation.484 This dual action in tumor development of SIRT2 might be due to different types of leukemia.
Regarding leukemia treatment, the combination of chemotherapeutics with SIRT modulators could provide a novel therapeutic strategy. For example, pharmacological targeting or RNAi-mediated knockdown of SIRT1 inhibits cell growth and sensitizes AML cells to tyrosine kinase inhibitor treatment.485 Moreover, shSIRT6-induced DNA repair deficiencies are potently synergistic with NAMPT targeting in acute myeloid leukemia treatment, which shows promising in vivo efficacy compared with monotherapy.486 SIRT7 expression increases with the positive response to treatment, but shows reduction when patients progress or relapse, which suggests that SIRT7 potentially serves as a general biomarker for monitoring treatment response in myeloid stem cell disorders.487 Accordingly, these results suggest that targeting SIRTs represents an attractive therapeutic strategy and provides a rationale for the novel combination-based treatments for leukemia.
Conclusion
We have reviewed the role of different SIRTs in diverse cancers, focusing on them as new anticancer therapeutic targets. Various investigations have indicated that different SIRTs show differential patterns of expression depending upon the pathological subtype, tumor grade and stage. It could be concluded that SIRTs serve as prognostic factors/biomarkers in patients with cancer. The discrepancy in the role of SIRTs in tumor progression and tumor chemoresistance or radioresistance might be attributed to various tumor types, stages, microenvironment, and their involvement in various tumor processes, such as cellular metabolism, cell death, cell cycle, and DNA damage/repair. Notably, several SIRTs exert a dual action in cancer. Thus, figuring out the underlying mechanisms and conditions that enable their opposing roles in cancer might be one of the main challenges and of great therapeutic significance. Collectively, SIRTs could be utilized as promising target molecules to be used as potential biomarkers for diagnosis and prognosis in patients with cancer. A variety of available SIRT modulators could be developed and further utilized to promote treatment efficacy of various cancers by themselves or, more likely, in combination with different anticancer drugs.
SIRTs and CVDs
Over the past decades, the incidence of CVDs, such as heart failure, atherosclerosis, and hypertension, has been increasing.488 CVDs are the major cause of mortality worldwide.489,490 According to the Global Burden of Disease Study 2019, prevalent cases of total CVDs have increased from 271 million to 523 million in 204 countries and territories between 1990 and 2019. The number of CVD deaths has also increased from 12.1 million to 18.6 million.491 Epigenetic modification plays a critical role in the occurrence and development of CVD488 and regulates the function and expression level of CVD-related genes through DNA methylation, histone modification, and non-coding RNA mechanism.492 Therefore, SIRT protein family has received much attention in CVD research due to its crucial role in regulating histone deacetylation.488 In addition to HDAC function, SIRTs also have multiple non-histone deacetylase and mono-ADP-ribosyl transferase activities.493 These functions also play an important role in CVDs (Fig. 10). SIRTs regulate crucial pathological processes, such as cell proliferation, cell senescence, DNA damage, oxidative stress, inflammation, and cell metabolism, thereby influencing the occurrence and development of CVDs.493,494

The roles of SIRTs in circulatory system. SIRT1, SIRT3 and SIRT6 play protective roles in CVDs, such as cardiac fibrosis, heart failure, atherosclerosis, and MI/R injury. In addition, the protective effect of SIRT2 is observed in cardiac hypertrophy, cardiac fibrosis, as well as atherosclerosis. Furthermore, SIRT4 has a protective effect on atherosclerosis and MI/R injury. However, SIRT4 may have an adverse effect on cardiac hypertrophy and fibrosis. In contrast, SIRT5 plays protective role in cardiac hypertrophy and fibrosis, and similar protective effect is also observed in MI/R injury. Finally, the protective effect of SIRT7 is observed in cardiac hypertrophy and atherosclerosis. https://biorender.com
Cardiac hypertrophy and fibrosis
Cardiac hypertrophy is an adaptive and compensatory mechanism for maintaining cardiac output during physiological and pathological stimuli.495 However, some detrimental processes, such as pressure or volume overload, can lead to pathological cardiac hypertrophy.495 Cardiac fibrosis induces fibroblast proliferation and excessive deposition of extracellular proteins.496 Pathological cardiac hypertrophy and fibrosis are the main characteristics of cardiac remodeling.497 It is crucial to reveal the molecular mechanisms associated with cardiac hypertrophy and fibrosis, as there are currently no effective treatments for cardiac remodeling.497 Therefore, SIRT proteins, which have been reported to play important roles in the occurrence and development of cardiac hypertrophy and fibrosis, have received extensive attention, especially SIRT1, SIRT3, and SIRT6.
-
(1)
Cardiac hypertrophy
The impact of SIRT1 on cardiac hypertrophy is inconsistent, with both alleviating and exacerbating effects having been reported.4 Some in vivo studies have suggested that SIRT1 overexpression can alleviate Ang II-induced cardiac hypertrophy by reducing cardiomyocyte apoptosis and promoting autophagy.498,499 In addition, SIRT1 overexpression can ameliorate cardiac hypertrophy induced by phenylephrine by inhibiting protein kinase C (PKC)‐ζ activation.500 However, some studies have shown the opposite effect. For example, SIRT1 exacerbated cardiac hypertrophy by promoting membrane localization and activation of Akt and phosphoinositide-dependent protein kinase 1, while impaired Akt activation in the hearts of SIRT1-deficient mice was related to decreased cardiac hypertrophy in response to physical exercise and Ang II.501 These opposite effects might be dependent on the degree of SIRT1 expression.502 For instance, the low (2.5-fold) or moderate (7.5-fold) overexpression of SIRT1 in the hearts of transgenic mice attenuated cardiac hypertrophy. However, a high overexpression (12.5-fold) level of SIRT1 increased cardiac hypertrophy.502 These conflicting effects imply that SIRT1 has different effects on cardiac hypertrophy in different contexts and models.503 Therefore, more studies are needed to further explore the complex effects of SIRT1 on cardiac hypertrophy.503
SIRT3 has a protective role in cardiac hypertrophy.504 Its expression was reduced in the hearts of Ang II-induced cardiac hypertrophic mice and in Ang II-treated cardiomyocytes.505 In addition, SIRT3 overexpression protects myocytes from hypertrophy, whereas SIRT3 silencing exacerbates Ang II-induced cardiomyocyte hypertrophy.505 Resveratrol can be used to activate SIRT3 with protective effects on hypertrophy through activation of SIRT3 and subsequent autophagy.506 However, the protective effects of resveratrol have not been observed after the addition of siRNA-SIRT3.506 SIRT3 promotes autophagy in Ang II-induced myocardial hypertrophy via deacetylation of FoxO1,507 blocks the cardiac hypertrophic response by augmenting FoxO3a-dependent antioxidant defense mechanisms in mice,164 and exerts protective effects against cardiac hypertrophy by reducing the level of acetylation and activity of poly (ADP-ribose) polymerase-1.508
SIRT6 also protects against cardiac hypertrophy.509 Both in vivo and in vitro studies have revealed that SIRT6 inhibits isoproterenol-induced cardiac hypertrophy via activation of autophagy.314 Specifically, SIRT6 promotes nuclear retention of FoxO3 transcription factor, possibly by attenuating Akt signaling, which is responsible for autophagy activation.314 In addition, SIRT6 protects cardiomyocytes from hypertrophy by decreasing the protein level of p300 and subsequently the acetylation and transcriptional activity of NF-κB p65 subunit.510 It also blocks IGF-Akt signaling and cardiac hypertrophy development by targeting c-Jun.511 Moreover, STAT3 suppression has been reported to be involved in the protective effect of SIRT6 against cardiomyocyte hypertrophy.512
In addition, SIRT2, SIRT5, and SIRT7 have protective effects on cardiac hypertrophy, while SIRT4 appears to have the opposite effect. The protein level of SIRT2 is reduced in cardiac hypertrophy, and SIRT2 overexpression attenuates agonist-induced cardiac hypertrophy in a cell-autonomous manner.513 On a molecular level, SIRT2 binds to and deacetylates the nuclear factor of activated T-cell c2 transcription factor, thereby regulating nuclear factor of activated T-cell c2 transcription activity and exerting protective effects on cardiac hypertrophy.513 In contrast, loss of SIRT2 has been reported to reduce AMPK activation, thereby promoting aging-related and Ang II-induced cardiac hypertrophy and blunting metformin-mediated cardioprotective effects.514 These findings have suggested that SIRT2 might be a potential target for the treatment of cardiac hypertrophy. In addition, SIRT5 prevents age-related cardiac hypertrophy,515 while SIRT7 also ameliorates stress-induced cardiac hypertrophy by interacting with and deacetylating GATA4.516 Interestingly, SIRT4 seems to have an adverse effect on cardiac hypertrophy. For instance, an in vivo study has revealed that SIRT4 overexpression aggravates Ang II-induced cardiac hypertrophy by inhibiting MnSOD activity.517 However, further studies are needed to confirm this result.
-
(2)
Cardiac fibrosis
In cardiac fibrosis, TGF-β is a key profibrotic cytokine that exerts profibrotic effects.518 TGF-β is involved in the protective effect of SIRT1, SIRT3, and SIRT6 on cardiac fibrosis by regulating the activity of Smad family transcription factors.519,520,521 For instance, activation of both SIRT1 and SIRT3 by resveratrol attenuates cardiac fibrosis in mice by inhibiting the TGF-β/Smad3 pathway519,520 and systematic SIRT6 KO induces cardiac fibrosis in mice by activating the TGF-β/Smad3 pathway.521 On a molecular level, the study has also shown that SIRT3 overexpression partially prevents the inflammatory and profibrotic effects by modulating the FOS/activator protein-1 pathway in human and rat cardiomyocytes.65 SIRT6 prevents Ang II-mediated cardiac fibrosis and injury by targeting AMPK-Angiotensin-converting enzyme 2 signaling.522
In addition, other SIRTs affect cardiac fibrosis. For example, SIRT2 overexpression protects against Ang II-induced cardiac fibrosis and rescues cardiac function.514 This protective effect of SIRT2 is associated with the promotion of AMPK activation by deacetylating the kinase LKB1.514 SIRT5 KO mice have shown increased fibrosis compared to age-matched wild-type mice,515 although relevant mechanisms need to be further explored. However, SIRT4 appears to contribute to cardiac fibrosis, and global SIRT4 KO in mice confers resistance to Ang II infusion by significantly suppressing fibrosis deposition.517 Similarly, enhanced expression and phosphorylation of SIRT7 plays a role in promoting cardiac fibrosis via activation of Smad2 and ERK signaling pathways.523 However, SIRT7 KO in mice has been reported to result in cardiac fibrosis.265
Overall, SIRTs play an important role in cardiac hypertrophy and fibrosis. SIRT1, SIRT3, and SIRT6 might protect against cardiac hypertrophy and fibrosis by affecting important biological processes and regulating downstream signaling pathways, such as autophagy and TGF-β/Smad3 pathways. Of note, SIRT1 might have bidirectional effects on cardiac hypertrophy, which might be dependent on the degree of SIRT1 expression. Furthermore, SIRT2 and SIRT5 might also have protective effects on cardiac hypertrophy and fibrosis. In contrast, SIRT4 might exacerbate cardiac hypertrophy and fibrosis. Evidence has suggested that SIRT7 has a protective effect on cardiac hypertrophy, but its effect on cardiac fibrosis is inconsistent. Considering that there are few studies on SIRT7 in cardiac hypertrophy and fibrosis, further research is needed in the future.
Heart failure
Heart failure is the most common endpoint of most CVDs,524 affecting an estimated 64.3 million people worldwide.391,525 It is a complex disease and involves various molecular and cellular alterations that affect the cardiac structure and impair the contractile function.526 However, the underlying mechanisms of heart failure remain not fully understood.527 Recently, growing evidence has suggested that SIRTs play key roles during the process of heart failure. The following section summarizes this evidence.
SIRT1 has beneficial effects on the development of heart failure. The expression of SIRT1 is decreased in the hearts of advanced heart failure patients and rat models.528,529 Heart failure is closely related to some biological processes, such as oxidative stress and cell apoptosis.530,531 SIRT1 might attenuate oxidative stress and protect cells from oxidative damage and apoptosis through several mechanisms.531 For example, levels of MnSOD, thioredoxin1, and Bcl-xL (an anti-apoptotic molecule) are significantly decreased in cardiomyocytes from individuals with advanced heart failure.528 The low expression of SIRT1 might downregulate antioxidants and upregulate pro-apoptotic molecules by increasing p53 acetylation and decreasing FoxO1 translocation in the nucleus.528 In addition, an in vivo study has suggested that SIRT1 overexpression reduces cardiomyocyte apoptosis through the NF-κB p65/miR-155/brain-derived neurotrophic factor (BDNF) signaling pathway, thereby alleviating heart failure in rats.532 Furthermore, reduced level and activity of sarco-endoplasmic reticulum Ca2+-ATPase (SERCA2a) are major features of heart failure, and SIRT1 KO elevated the acetylation of SERCA2a, which in turn leads to SERCA2a dysfunction and cardiac defects in a failing heart.533 In contrast, the pharmacological activation of SIRT1 restores SERCA2a activity via deacetylation at K492.533 Overall, the above evidence has indicated that SIRT1 is involved in the occurrence and development of heart failure and might be a promising therapeutic target for heart failure treatment.
In addition, mitochondrial energy metabolism disorder contributes to the progression of heart failure.534 Myocardial acetylproteomics demonstrates that there is extensive mitochondrial protein lysine hyperacetylation in mouse models of early-stage heart failure and in end-stage failing human hearts.534 As a mitochondrial deacetylase, SIRT3 plays an important role in maintaining the mitochondrial function,535 and provides a protective effect during heart failure.536 SIRT3 deficiency might impair cardiac mitochondrial function and aggravate heart failure during aging.537 In addition, SIRT3 is involved in the regulation of endothelial metabolism and angiogenesis, thereby affecting the occurrence and development of heart failure.538,539 For instance, an in vivo study has suggested that the endothelial‐specific SIRT3 KO disrupts glucose transport from endothelial cells to cardiomyocytes, decreases cardiomyocyte glucose utilization via apelin in a paracrine manner, and sensitizes pressure overload-induced heart failure.538
Similar protective effects during heart failure have also been observed in SIRT6.511 SIRT6 expression is significantly decreased in the hearts of patients with chronic heart failure as well as animal models of heart failure.527 SIRT6 overexpression increases the survival of transverse aortic constriction-induced heart failure mice, which might be associated with telomerase upregulation, such as telomerase reverse transcriptase and telomeric repeat binding factor 1.540
Compared to SIRT1, SIRT3, and SIRT6, studies on SIRT2,541 SIRT4, SIRT5,542 and SIRT7 in heart failure are limited. SIRTs might play important roles in the occurrence and development of heart failure and their further exploration is needed in the future. Studies on SIRT2, SIRT4, SIRT5, and SIRT7 might reveal promising research directions for the treatment of heart failure.
Atherosclerosis
Atherosclerosis is a chronic inflammatory disease4 that results from a series of events, including increased levels of LDL cholesterol in the plasma, dysfunctional endothelial cells, inflammation with immune cell infiltration, and ultimately plaque formation.494,543 SIRTs have been reported to directly affect atherogenesis and plaque stability by preventing endothelial cell dysfunction, VSMC senescence, and macrophage foam cell formation via regulation of key biological processes, such as DNA damage repair and anti-apoptosis and anti-inflammatory pathways.493
SIRT1 has a protective effect on atherosclerosis.544 A prior in vivo study has shown that endothelial cell-specific overexpression of SIRT1 protects against atherosclerosis in apolipoprotein E KO mice,545 which was associated with inhibited endothelial cell apoptosis via eNOS expression activation.545 In addition, SIRT1 activation by SRT1720 in aging mice ameliorates endothelial dysfunction by increasing COX-2 signaling and reducing oxidative stress and inflammation.546 On VSMC level, SIRT1 protects against DNA damage and inhibits atherosclerosis partly by activating the repair protein Nijmegen breakage syndrome-1.544 Moreover, macrophage foam cell formation is a key initiation event in the pathogenesis of atherosclerosis.547 SIRT1 activation reduces Lox-1-mediated foam cell formation via suppression of the NF-κB signaling pathway.548 In contrast, suppression of the SIRT1 signaling pathway by mTOR signaling promotes foam cell formation and inhibits foam cell egress.549 Several miRNAs have been revealed to have a key role in atherosclerosis by regulating the expression of SIRT1.550,551 For example, miR-217 downregulation might alleviate atherosclerosis via inhibition of macrophage apoptosis and inflammatory response.550 SIRT1 is a direct target of miR-217. SIRT1 silencing can eliminate the effects of miR-217 downregulation.550 The above evidence has suggested that SIRT1 is associated with the occurrence and development of atherosclerosis and might be a promising therapeutic target for atherosclerosis treatment.
Compared to SIRT1, a relatively limited number of studies have explored the roles of other SIRTs in atherosclerosis. SIRT2 decreases atherosclerotic plaque formation in LDL receptor-deficient mice by regulating macrophage polarization.552 SIRT3 gene expression is associated with endothelial cell apoptosis in atherosclerosis rats,553 and SIRT3/SOD2 signaling can be activated by circ_0,003,423, thereby protecting human umbilical vein endothelial cells from oxLDL-induced dysfunction.554 SIRT4 suppresses the PI3K/Akt/NF-κB signaling pathway and relieves oxLDL-induced human umbilical vein endothelial cells injury.555 SIRT6 protects against endothelial dysfunction, VSMC senescence, and atherosclerosis in mice.201,556,557 In addition, SIRT6 overexpression reduces oxLDL uptake in RAW macrophages, and SIRT6 knockdown enhances it and increases the expression of macrophage scavenger receptor 1.558 Finally, SIRT7 has been reported to regulate the VSMC proliferation and migration via the Wnt/β-catenin signaling pathway, which provides a promising therapeutic strategy for anti-atherosclerosis.559
In conclusion, the role of SIRT1 in atherosclerosis has received extensive attention. SIRT1 deficiency in endothelial cells, VSMCs, and monocytes/macrophages promotes atherosclerosis.560 Therefore, SIRT1 might be a potential therapeutic target for the treatment of atherosclerosis. Other SIRTs might also have protective effects on atherosclerosis. However, due to a relatively low number of studies, the relevant mechanisms need to be further explored in the future.
Coronary artery disease (CAD)
CAD is the result of atherosclerotic plaque development in the walls of coronary arteries.561 It is one of the most common causes of death in the developed countries and is responsible for about one in every five deaths.562 Current studies on the role of SIRTs in CAD mainly focus on SIRT1, which has a protective effect on CAD by regulating some crucial biological processes, such as oxidative stress, inflammation, cell apoptosis, and cell proliferation.
Epidemiological studies have suggested that genetic SIRT1 polymorphisms are associated with the risk of CAD,563 while the expression level of SIRT1 is reduced in CAD patients.564 SIRT1 inhibition causes oxidative stress and inflammation in CAD patients.565 On a molecular level, expression of downregulated SIRT1 in human CAD monocytes is related to the enhanced acetylated p53 expression levels.565 In contrast, SIRT1 overexpression in human CAD monocytes mitigates pro-apoptotic events and attenuates some proinflammatory events, such as upregulating expression of NF-κB and iNOS and NO concentrations.565 SIRT1 has been reported to be involved in the regulation of CAD via noncoding RNAs.566,567 For example, promoted expression of SIRT1 by elevated expression of lncRNA C2dat1 and subsequent suppressed miR-34a expression increases VSMC proliferation and migration in CAD.567
Except for SIRT1, epidemiological studies also suggest that genetic polymorphisms of SIRT3 and SIRT6 are associated with the risk of CAD,563 but the related mechanism needs to be further explored. Given the protective role of SIRT1 in CAD, other SIRTs might also be potential therapeutic targets for CAD. Therefore, exploring the roles of other SIRTs in CAD might be a promising research direction in the future.
Myocardial ischemia/reperfusion (MI/R) injury
In recent years, the morbidity and mortality of ischemic cardiac diseases, such as myocardial infarction, have shown an upward trend.568 With the development of recanalization technology, the treatment of myocardial infarction has made remarkable progress.568 However, MI/R injury can be induced as the treatments progress.569 MI/R injury is closely related to oxidative stress and apoptosis,4 and SIRTs play crucial roles in MI/R by controlling the above biological processes.
SIRT1 has a protective effect on MI/R injury and reduces the infarct area of the heart.570,571 Cardiac-specific SIRT1 KO mice have shown a significantly increased myocardial infarction area size.572 In contrast, cardiac-specific SIRT1 overexpression was significantly reduced in the myocardial infarction area.572 As for its potential mechanism, overexpression of SIRT1 leads to upregulation of antioxidant pathways mediated by FoxO1 and MnSOD and downregulation of pro-apoptotic pathways mediated by caspase-3 and Bax, thereby protecting the heart from MI/R injury.572 In addition, SIRT1 overexpression has been shown to be involved in ameliorating miRNA inhibition associated with MI/R injury.573,574 For example, upregulated SIRT1 expression resulting from miR-132 inhibition might ameliorate MI/R injury by inhibiting oxidative stress and pyroptosis through activation of PGC-1α/Nrf2 signaling.573 The SIRT1/AMPK/PGC-1α pathway is involved in the process by which lncRNA Oip5-as1 attenuates MI/R injury by sponging miR-29a.574 Like SIRT1, nuclear deacetylase SIRT6 also has a protective effect on MI/R injury. On a molecular level, SIRT6 protects against MI/R injury by increasing FoxO3α-dependent antioxidant defense mechanisms575 and attenuating aging-related charged multivesicular body protein 2B accumulation.576
In addition, the protective effects of mitochondrial SIRT3-5 have been observed in MI/R injury. An in vivo study has revealed that SIRT3 deficiency exacerbates MI/R injury.577 Both in vitro and in vivo models have shown that SIRT4 is downregulated in cardiomyocytes after MI/R injury, and that SIRT4 overexpression decreases myocardial infarct size.578 This protective effect of SIRT4 against MI/R injury has been reported to be associated with preserved mitochondrial function and reduced myocardial apoptosis.578 Similarly, a prior in vivo study has demonstrated that SIRT5 loss increased myocardial infarct size and MI/R injury, which might be associated with the effect of SIRT5 on modulating protein succinylation in the heart.579
This evidence suggests that SIRT1-6 might play critical roles in alleviating myocardial infarction and M/IR by regulating some important biological processes, such as oxidative stress and apoptosis. However, relevant molecular mechanisms behind these processes need to be further explored. Moreover, few studies have focused on the roles of SIRT2 and SIRT7 in M/IR injury, and further research is needed in the future.
Hypertension
Hypertension, defined as systolic blood pressure of ≥ 140 mmHg and/or diastolic blood pressure of ≥ 90 mmHg, is the risk factor for other CVDs,580 affecting an estimated 1.39 billion people worldwide in 2010. Its prevalence is still rising globally.581 In recent years, increasing studies have focused on the protective effects of SIRT1 and SIRT3 on hypertension.582,583 In vivo studies have shown that SIRT1 overexpression in VSMCs attenuates Ang II-induced hypertension in mice.582 Similarly, SIRT3 overexpression attenuates Ang II and deoxycorticosterone acetate salt-induced hypertension in transgenic mice.583 Both SIRT1 and SIRT3 have been reported to be involved in the regulation of oxidative stress in hypertension.584,585,586 For example, SIRT1 activation attenuates Klotho deficiency-induced arterial stiffness and hypertension by increasing AMPKα and eNOS activity.584 SIRT1 overexpression mediated by NAMPT alleviates Ang II-mediated ROS production.585 In addition, diminished SIRT3 expression and redox inactivation of SIRT3 leads to SOD2 inactivation and contributes to the pathogenesis of hypertension.586
SIRTs also play important roles in the complications of hypertension. For example, decreased urinary levels of SIRT1 can be seen as a non-invasive biomarker of early renal damage in hypertension.587 SIRT3 alleviates the development of hypertensive renal injury by suppressing EMT.588 Endothelial-specific deletion of SIRT6 significantly enhances blood pressure and exacerbates endothelial dysfunction and cardiorenal injury in experimental hypertension by targeting Nkx3.2-GATA5 signaling.589
These findings indicate that SIRTs have protective effects on the occurrence and development of hypertension and might be valuable predictive biomarkers as well as promising therapeutic targets for hypertension complications. However, relevant mechanisms still need to be further explored, especially for SIRT2, SIRT4, SIRT5, and SIRT7, which have not been extensively investigated.
Conclusion
This section summarized the effects of SIRTs on CVDs. The effects of SIRT1, SIRT3, and SIRT6 have received extensive attention. Most studies have shown that they have a protective effect on CVDs, such as cardiac fibrosis, heart failure, atherosclerosis, and M/IR injury. Compared to SIRT1, SIRT3, and SIRT6, studies on SIRT2, SIRT4, SIRT5, and SIRT7 are relatively limited, even though they play important roles in CVDs. The protective effects of SIRT2, SIRT5, and SIRT7 in several CVDs (e.g., hypertrophy) have been observed. Of note, SIRT4 might aggravate cardiac hypertrophy and fibrosis. Overall, SIRTs are promising therapeutic targets, and the pharmacological modulation of SIRTs can be used in the prevention and treatment of CVDs.
SIRTs and respiratory system diseases
Respiratory diseases are one of the biggest threats to human health.590 Common respiratory diseases, including asthma, chronic obstructive pulmonary disease (COPD), lung fibrosis (LF), coronavirus disease 2019 (COVID-19), and other lung injures, seriously affect physical and mental health.590 SIRTs have received considerable attention due to their important effects on respiratory diseases.591 Herein, we summarize the related studies in several common respiratory system diseases (Fig. 11).

The roles of SIRTs in respiratory system. SIRTs are involved in common respiratory diseases including COPD, asthma, lung fibrosis, COVID-19, and other lung injured diseases. SIRT1, SIRT3 and SIRT6 play protective effects in COPD, and these three members also have a positive effect on asthma. However, SIRT2 and SIRT7 could aggravate the occurrence of asthma. In lung fibrosis, the positive effects of SIRT1, SIRT3, SIRT6 and SIRT7 have been demonstrated. Besides, SIRT3 and SIRT6 contribute to the remission of lung injury, whereas SIRT1 play dual effects on the disease. Moreover, the activation of SIRT1 can effectively alleviate ventilator or paraquat-induced lung injury. Finally, SIRTs are also associated with COVID-19. https://biorender.com
COPD
COPD is a common disease characterized by persistent respiratory symptoms and progressive airflow obstruction.592 Most chronic respiratory disease-attributable deaths are due to COPD, which is the fourth leading cause of death worldwide and considered to be a global public health challenge.593,594,595 Oxidative stress, inflammation, and apoptosis are the most important influencing factors for COPD occurrence596 and are closely related to SIRT family.593 Cigarette smoking (CS) is a causative factor for COPD. The level of SIRT1 is substantially decreased in lungs of patients with COPD/emphysema, as well as in lungs of rodents exposed to CS.597 Moreover, SIRT1 has been found to have anti-inflammatory, anti-apoptotic, and antioxidant roles in the pathogenesis of COPD.598 For example, SIRT1 plays a pivotal role in regulating NF-κB-dependent proinflammatory mediators in lungs of smokers and patients with COPD.599 Apart from NF-κB regulation, SIRT1 also mediates COPD via deacetylation of the FoxO3 transcription factor and tumor suppressor p53 involved in lung cell senescence and oxidative stress-induced cellular apoptosis.597,599 Moreover, the SIRT1 activator SRT1720 might be able to inhibit LPS-induced cytokine release from cultured peripheral blood mononuclear cells in patients with COPD. Thus, pharmacological activation of SIRT1 might have considerable potential as a novel form of chronopharmacology in COPD.600
SIRT6 plays an important role in the regulation of autophagy in COPD.591 For example, reduced SIRT6 expression level is associated with COPD development through enhancement of cellular senescence created by insufficient autophagy during CS exposure.601 SIRT6 overexpression weakens autophagy via IGF–Akt–mTOR signaling.601 Similar to SIRT1, reduced SIRT6 level is also implicated in COPD.602 Therefore, SIRT6 deficiency might contribute to the development of COPD.
SIRT3 is a mitochondrial deacetylase regulating mitochondrial function, and its role in the pathogenesis of COPD has also been mentioned. For instance, SIRT3 inhibits airway epithelial mitochondrial oxidative stress, thereby contributing to attenuating the progression of COPD.603 Therefore, activating the SIRT3 signaling pathway might present a novel therapeutic target to slow or prevent the pathogenesis of COPD.
With the understanding of the positive roles of SIRT1, SIRT3, and SIRT6 in COPD, their pharmacological activation by specific agents might be a promising strategy against COPD. However, other SIRT family members have not yet been studied in the respiratory system. SIRTs mainly mediate this disease via inflammation- or autophagy-related pathways.604,605 In addition, COPD is commonly thought to be associated with other chronic diseases, especially those where accelerated aging is involved. Therefore, the defection of anti-aging molecules, such as SIRTs, has been proposed as a mechanism for accelerated lung aging in COPD.606 Given the severity and complexity of COPD, further studies are necessary to validate the exact roles of SIRTs.
LF
LF is a leading cause of death in the industrialized world, which significantly increases with age.607 An epidemiological study has shown that approximately 45% of global deaths have been attributed to fibrosis.607 The pathogenesis of LF is complex and involves environmental influences and microorganisms.608 Recent developments in the field of LF have pointed towards the pivotal role of SIRTs in regulating disease progression, thereby qualifying as potential anti-fibrotic drug targets.607 Four of the seven SIRTs (SIRT1, SIRT3, SIRT6, and SIRT7) have been investigated in LF, while the functional roles of the remaining SIRTs (SIRT2, SIRT4, and SIRT5) remain elusive.
SIRT1 loss might be involved in the pathogenesis of LF. Thus, its activation might be an effective treatment for LF. SIRT1 plays an important role in regulating alveolar epithelial cell 2 progenitor renewal and LF.609 Mechanistically, SIRT1 activation promotes self-renewal and differentiation of alveolar epithelial cell 2 in lung tissues of idiopathic pulmonary fibrosis (IPF) patients and aged mice.609 However, the opposite results have been reported for SIRT1 changes in LF. According to the study performed by Zeng et al., SIRT1 expression was significantly increased in lungs from patients with IPF, as well as in lungs from bleomycin-induced LF mouse models.610 Nevertheless, SIRT1 activation or overexpression attenuates LF through regulation of canonical TGF-β1/p300 signaling. In addition, SIRT1 activation has been used in aging-related LF prevention and therapy.611 As the expression of SIRT1 in LF is controversial, more studies are needed to explore this notion in the future.
Due to the preferential mitochondrial association with extended life span in humans, SIRT3 is a protein of particular interest in age-related diseases, including LF.612 For example, there is a SIRT3 deficiency within the murine aging lung, which promotes the fibrotic response mediated by TGF-β1.612 TGF-β1 is a major multifunctional cytokine that is known as a mediator implicated in LF pathogenesis.613 In addition, SIRT3 deficiency promotes LF by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis.614 Cheresh et al. have suggested that SIRT3 overexpression can ameliorate asbestos-induced pulmonary fibrosis.615 Thus, improvement in SIRT3 expression might be a novel therapeutic focus for managing patients with IF.
It is possible that SIRT6 participates in the inhibition of cellular fibrosis by regulating the TGF-β1 signaling pathway. SIRT6 can also be an ambitious target molecule for understanding the pathogenesis of IPF through the inhibitory role in TGF-β-induced cellular senescence.616 Additionally, Chen et al. have shown that targeting SIRT6 is a potential novel therapeutic strategy for pulmonary fibrosis that involves inactivating the TGF-β1/Smad2 signaling pathway.617 Furthermore, SIRT6 prevents TGF-β1-induced lung myofibroblast differentiation by inhibiting the TGF-β1/Smad2 and NF-κB signaling pathways.618 SIRT6 also inhibits EMT during IPF by inactivating TGF-β1/Smad3 signaling,619 highlighting the critical role of SIRT6 in LF. Moreover, all SIRTs show a tendency to be expressed at lower levels in fibroblasts from patients compared to controls, but the greatest decrease is observed with SIRT7.620 Furthermore, the decline in SIRT7 in LF has a profibrotic effect, which is mediated by changes in Smad3 levels.620
The above evidence shows that SIRT1, SIRT3, SIRT6, and SIRT7 are beneficial for preventing and improving the pathogenesis of LF. However, the modulatory roles of other SIRT members remain unclear. Mazumder et al. have reviewed the regulatory roles of under-reported SIRTs (mainly SIRT2, SIRT4, and SIRT5, which lack direct reported associations with LF) in basic cellular and mitochondrial metabolic pathways critical to LF.607 Overall, they have suggested that SIRTs appear to exert a protective action in LF, except SIRT2, which might have a pro-fibrotic action given its proinflammatory effects observed in asthma.607 In summary, studies on the function of SIRTs in regulating LF have potential.
Asthma
Asthma is a chronic inflammatory disease that is characterized by cough, breathlessness, and episodic wheezing caused by airway inflammation and hyperresponsiveness.621 It is estimated to influence about 300 million people all over the world, with a significantly increasing prevalence.621,622 Asthma affects all age groups, but particularly children623 SIRT1-targeting approach has been shown to be a potentially effective new strategy for the treatment of asthma.621,624 For example, SIRT1 protein levels are decreased in patients with severe asthma.625 SIRT1 exerts an anti-inflammatory effect on airway diseases, including asthma. Tang et al. have investigated the potential role of SIRT1 in regulating inflammation through modulation of IL-6 expression in an Akt-dependent manner during allergic asthma.626 Similarly, SIRT1 regulates IL-6 level via the Akt pathway, thereby affecting pulmonary function in asthma patients.627 In addition, SIRT1 inhibits the differentiation of IL-9-producing CD4+ T cells that are associated with allergic airway inflammation via mTOR-HIF-1α-dependent signaling coupled with glycolytic pathway.628 In addition, a study performed by Liu et al. has shown that anthocyanin inhibits airway inflammation by blocking the NF-κB pathway via the miR-138-5p/SIRT1 axis in asthmatic mice.621 All of these studies have demonstrated that SIRT1 suppresses the allergic airway inflammation that occurs in asthma and suggested that SIRT1 activation might represent a therapeutic strategy for asthma.
In addition to SIRT1, SIRT2, SIRT3, SIRT6, and SIRT7 have also been reported to be involved in asthma. For example, SIRT2 enhances allergic asthmatic inflammation, while pharmacologic SIRT2 ablation attenuates and genetic SIRT2 overexpression exaggerates the allergic asthmatic phenotype.629 Moreover, SIRT2 aggravates asthmatic inflammation by upregulating T-helper type 2 responses and macrophage polarization.630 In contrast, upregulation of SIRT3 expression reduces apoptosis in the bronchial epithelium and airway inflammation in asthma.631 Allergic asthma is a chronic inflammatory airway disease involving airway remodeling that severely limits airflow in the lungs.632 SIRT6 and SIRT7 expression levels have been found to be increased in human bronchial epithelial cells isolated from patients with asthma.633 Upregulated SIRT6 ameliorates airway remodeling through regulation of EMT in asthma.634 In contrast, upregulated SIRT7 promotes airway remodeling in asthma by regulating TGF-β1-induced airway smooth muscle cell proliferation and migration,635 indicating a different SIRT6 role during airway remodeling.
Asthma is a complex respiratory disease with an increasing incidence worldwide. Individuals with asthma need to receive emergency treatment if their symptoms become severe. However, there is currently no effective cure. Therefore, the pathological mechanism of asthma needs to be investigated further. Accumulating evidence shows that asthma is caused by chronic inflammation and that SIRTs have important effects on regulating chronic inflammatory responses. Future exploration of the molecular mechanisms of SIRT-mediated inflammatory response will provide more information for the development of novel therapeutic targets in asthma.
Lung injury
Acute lung injury (ALI) is a potentially life threatening and devastating disease with an extremely high rate of mortality.636 It is a clinical syndrome associated with respiratory dysfunction and is often a complication of sepsis.637 Additionally, ALI can develop into acute respiratory distress syndrome in more serious injuries, which lacks novel and efficient therapies.638,639 Inflammation and oxidative stresses are essential for the progression of ALI.640 However, the molecular mechanisms of sepsis-induced lung inflammatory injury are yet to be determined. SIRT1 has been widely reported to exert its anti-inflammatory function by regulating the production of proinflammatory cytokines.637,641 For instance, overall SIRT1 KO mice are highly susceptible to sepsis-induced inflammatory lung injury due to activation of proinflammatory transcription factor NF-κB.641 In addition, resveratrol is a potent SIRT1 activator that reduces ALI in an LPS-induced sepsis mouse model via activation of SIRT1.637 On the contrary, SIRT1 inhibitor EX-527 suppresses mTOR activation and alleviates ALI in mice with endotoxemia.642 This finding suggests that SIRT1 might be a detrimental factor under certain pathological conditions.
Evidence also shows that SIRT3 and SIRT6 have positive effects on ALI. SIRT3 promotes the expression of MnSOD, and this regulation is crucial for the protective effect of SIRT3 on hyperoxia-induced ALI.643 SIRT3 can also diminish inflammation and mitigate endotoxin-induced ALI.63 Kurundkar et al. have shown that SIRT3-deficient mice (SIRT3−/−) develop more severe ALI compared to wild-type controls (SIRT3+/+). Macrophages obtained from SIRT3−/− mice show significant alterations in mitochondrial bioenergetic and redox homeostasis in association with proinflammatory phenotype characterized by NLRP3 inflammasome activation.63 Similarly, SIRT6 regulates macrophage polarization to alleviate sepsis-induced acute respiratory distress syndrome via dual mechanisms both dependent on and independent of autophagy.644
Apart from the above-mentioned LPS and endotoxin, other external factors, such as ventilator and paraquat, also cause lung damage.645,646 Mechanical ventilation contributes to excessive mechanical stress and impaired physiological and structural lung integrity. HDAC inhibited by SIRT1-silencing RNA attenuates NAMPT expression in ventilator-induced lung injury.645 Paraquat, which is a highly toxic herbicide and primary lung attacker, results in severe ALI.647 A recent study has demonstrated that resveratrol reduces paraquat-induced lung injury by upregulating SIRT1 mRNA and protein expression in combination with the Nrf2 antioxidant pathway.646 Therefore, SIRT1 activation can effectively alleviate lung injury.
These results suggest that SIRT protein family plays an important role in maintaining normal homeostasis and protective mechanisms in the lung. SIRTs regulate lung injury by inhibiting the expression of inflammatory factors, although specific mechanisms require further investigation.
Coronavirus disease 2019
In December of 2019, a new strain of coronavirus, severe acute respiratory syndrome–coronavirus 2, was first identified and called COVID-19.648 The disease has been recognized as pandemic by the World Health Organization. A dysregulated inflammatory profile plays an important role in COVID-19 pathogenesis.649 It has been reported that the SIRT family has a part in this mechanism. The unbalanced p53/SIRT1 axis might impact lymphocyte homeostasis in COVID-19 patients.649 COVID-19 can be characterized not only by an increase in p53 transcription in circulating lymphocytes, but also by a persistently activated p53 form, possibly due to the low level of SIRT1.649 Therefore, increased SIRT1 expression might help to alleviate the pathogenesis of COVID-19. Additionally, serum SIRT3 levels are associated with the clinical outcome and prognosis of COVID-19 patients.650 SIRT3 levels are markedly lower in severe patients compared to those in the mild/moderate patients, indicating a positive role of SIRT3 in alleviating COVID-19.
In conclusion, SIRTs are involved in COVID-19 and may provide a new therapeutic strategy. However, the impact of SIRTs on regulation of inflammatory homeostasis in severe and mild cases of COVID-19 remains to be determined.
Conclusion
This section discusses how the SIRT family plays a vital role in various molecular pathways in the respiratory system. SIRTs might be targets for respiratory system-related adverse health events. Increased activity of individual SIRTs often has beneficial effects in pathophysiological conditions, whereas reduced activity is usually associated with disease conditions.651 This also seems to apply to the respiratory system diseases. In addition, epigenetic alteration is implied in the occurrence and development of various respiratory diseases.652 Since SIRTs are NAD-dependent deacetylases, their deacetylation activity via epigenetics might be a new research strategy. However, detailed epigenetic roles of SIRTs in the respiratory system still need to be further explored.
SIRTs and digestive system diseases
Digestive system diseases, including fatty liver diseases (FLDs), liver and intestinal ischemia-reperfusion injury (IRI), hepatitis B virus (HBV), pancreas diseases, and inflammatory bowel diseases (IBDs), are the most common clinical diseases.653,654 Increasing evidence has suggested that changes in SIRT activity and expression are associated with etiology of various digestive diseases.280,655 As discussed below, SIRTs play an important role in maintaining the homeostasis of digestive system function and participating in the occurrence and development of digestive diseases (Fig. 12).

The roles of SIRTs in digestive system, mainly including FLDs, liver and intestinal ischemia-reperfusion injury, HBV infection, pancreas diseases, and IBDs. In FLDs, SIRT1, SIRT2, SIRT3, and SIRT4 could provide protective effects, while the role of SIRT7 may be harmful. Notably, the effects of SIRT1, SIRT2, SIRT3, and SIRT4 may be dual effects; in different causes of liver injury, SIRT3 and SIRT6 are beneficial, while SIRT1 plays dual roles; in HBV infection, SIRT3, SIRT4, and SIRT6 can block viral replication, SIRT1, SIRT2, and SIRT5 may contribute to the HBV-induced pathomechanism in nontransformed hepatocytes, while the effect of SIRT7 could be dual; in liver ischemia-reperfusion injury and intestinal ischemia-reperfusion injury, SIRT1 and SIRT3 could reduce tissue damage, and SIRT6 could also protect intestinal ischemia/reperfusion injury, while SIRT2 augments liver ischemia-reperfusion injury; SIRT2, SIRT3, SIRT5, and SIRT7 have a protective role in inflammatory bowel diseases, however, SIRT1 may play opposite role; in other digestive diseases, SIRT1 acts inconsistently. https://biorender.com
Liver diseases
-
(1)
FLDs
The disease spectrum of FLDs, with high-fat/high-calorie diets, heavy alcohol consumption, and/or other causes of metabolic disorders, ranges from simple steatosis to steatohepatitis, fibrosis, and, ultimately, cirrhosis and carcinoma.656,657 Notably, nonalcoholic fatty liver disease (NAFLD) is the most common liver disease, with a worldwide prevalence of 25%. About 2 billion people consume alcohol worldwide and upwards of 75 million are at risk of alcohol-associated liver diseases.658,659 The accumulated evidence has shown that SIRTs have complex effects on the FLD. The dual effects of SIRT1-4 have been explored, while SIRT6 might only have a protective role. However, limited studies have investigated the roles of SIRT5 and SIRT7.
SIRTs might play a protective role in FLD, except for SIRT7. Decreased expression of SIRT1-3, SIRT5, and SIRT6 in patients with NAFLD or fibrosis have been observed in several studies.660,661,662 Molecular mechanism studies have also demonstrated this protective effect. For example, SIRT1 and SIRT6 deacetylate the carbohydrate response element-binding protein and sterol regulatory element-binding protein-1c, which are two major transcription factors responsible for the coordinated induction of glycolytic and lipogenic genes, thereby maintaining lipid homeostasis in the liver.5,136,663,664 In addition, SIRT1 and SIRT6 antagonize liver fibrosis by blocking the activation of hepatic stellate cells via the deacetylation function in a mouse model.655,665 Notably, SIRT1 might act as a key metabolic/energy sensor, which directly regulates transcriptional activity and/or gene expression of several crucial transcription factors and transcription co-activators that are involved in lipid metabolic homeostasis to play a protective role in FLD.666,667,668 These include PPAR-α, PPAR-γ co-activator 1 alpha, and NF-κB.666,667,668 Moreover, liver fibrosis, oxidative stress, and related gene expression are significantly elevated in hepatocyte-specific SIRT6-KO mice with nonalcoholic steatohepatitis.158,669
The beneficial effects of SIRT2-5 on FLD might also be mediated by various biological mechanisms. Although related studies on SIRT2 are limited, it has been demonstrated that SIRT2 prevented NAFLD by deacetylation of hepatocyte nuclear factor 4α, a master regulator of gene expression for bile acid, lipids, and glucose metabolism.670 SIRT3 and SIRT5 improve mitochondrial function and increase mitochondrial fatty acid oxidation to relieve hepatic steatosis.671,672 For example, hepatic overexpression of SIRT3 improves mitochondrial function by deacetylation of mitochondrial trifunctional proteins and long-chain acyl-coenzyme A dehydrogenase.671,672 In systematic SIRT5 KO mice, impaired mitochondrial medium-chain fatty acid oxidation drove periportal macrovascular steatosis.673 Results from in vitro models have shown that SIRT4 upregulation might inhibit high fat diet-induced lipid accumulation, inflammation, and fibrogenesis through the SIRT4/Smad4 axis. It can also inhibit hepatic stellate cell activation.662,674
Although SIRTs are involved in FLD as protective factors in most studies, the high expression of SIRT1, SIRT2, SIRT4, and SIRT7 in patients with alcoholic hepatitis or NAFLD and upregulation of SIRT3 after chronic alcohol exposure in mouse liver might highlight their harmful effects.224,661,675,676 Results from in vitro studies have suggested that elevated monocyte SIRT1 and SIRT7 levels can prevent p-FoxO3 formation and cause a defect in apoptosis in alcoholic hepatitis.224 Although hepatocyte apoptosis is related to disease severity, proinflammatory hepatic macrophages also undergo apoptosis in response to alcohol. Therefore, apoptosis serves as a mechanism that suppresses the inflammatory response in alcoholic liver disease.677 These results suggest that high SIRT1 and SIRT7 levels in myeloid cells could be a primary event leading to enhanced inflammation, possibly owing to the deleterious consequence of apoptosis.677 Intestinal SIRT1 also exerted a partially harmful effect on alcoholic liver disease by intensifying hepatic ferroptosis and inflammation due to the imbalance of gut microbiota.678 Thus, it is logical to speculate that intestinal SIRT1 might act as a proinflammatory factor. SIRT2 appears to have a deleterious effect on hepatic fibrosis via the SIRT2/ERK/c-Myc axis.675 Moreover, in both NAFLD and alcoholic fatty liver disease mouse models, liver-specific SIRT3 knockdown alleviated alcoholic feeding-induced liver injury and lipid accumulation, which was associated with improved autophagy induction.676,679 In addition, SIRT4 might have harmful effects on NAFLD, and its molecular mechanism may be partly associated with deacetylating and destabilizing mitochondrial trifunctional protein-α.661
Overall, the above findings demonstrate the complex role of SIRTs in FLD. Choosing to develop different SIRT agonists or inhibitors might be a new target for the control of FLD occurrence and development in a clinical setting.
-
(2)
Liver IRI
IRI is a major complication of hemorrhagic shock, resection, and transplantation. It is characterized by aseptic inflammation and liver cell death and acts as a risk factor involved in acute and chronic rejection.680 Current studies have mostly focused on the effect of SIRT1-3, which plays different roles via multiple molecular pathways in liver IRI.
SIRT1 and SIRT3 might have a beneficial effect against damage. SIRT1 was markedly decreased after IRI in human and mouse livers.280 High SIRT1 levels improved hepatocellular function and resulted in superior survival in human liver transplants.681 The results from in vivo studies showed that SIRT1 suppressed mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2-dependent manner.280 Moreover, overexpression of SIRT1 alleviated autophagy depletion and inflammation to partially mitigate hepatocellular injury during IRI.281,682,683 In addition, SIRT3 expression was suppressed in systematic KO of Takeda G protein-coupled receptor 5 in mice, thus leading to a proinflammatory response in macrophages, which significantly exacerbated liver injury and inflammatory response.684
Unlike SIRT1 and SIRT3, with an increasing expression in IRI liver tissues, SIRT2 potentially has a detrimental effect on liver IRI.685 SIRT2 deacetylates MAPK phosphatase‐1 and activates the MAPK signaling pathways during liver IRI, thereby augmenting inflammatory responses and enhancing cell death in a mouse model. Pharmacologic and genetic suppression of SIRT2 also provided additional evidence supporting this observation.685
Overall, SIRT1 and SIRT3 have protective effects on liver IRI, while SIRT2 might be harmful. Studies on the role of SIRT4-7 in this disease are lacking. Future research should clarify the role of SIRTs in liver IRI, including the exact molecular mechanisms.
-
(3)
HBV infection
HBV infection affects over 250 million chronic carriers, causing more than 800,000 deaths annually, although a safe and effective vaccine is available.686 Notably, current evidence shows that the expression of SIRTs might make a difference during HBV infection.
SIRT3, SIRT4, and SIRT6 are downregulated in patients who tested positive for HBV antigens or the cell for HBV replication.442,687,688 SIRT3 and SIRT6 inhibit HBV replication via epigenetic regulation.687,688 SIRT3 and SIRT6 induce a decrease in H3K9 acetylation on viral covalently closed circular DNA (cccDNA), serving as a template for all viral transcripts.687,688 Specifically, stable HBV X protein transfection suppresses SIRT4 expression, which demonstrates the interaction between HBV and SIRT4 in the context of HCC.442
However, slightly elevated mRNA levels of SIRT1, SIRT2, SIRT5, and SIRT7 in HBV-infected hepatocytes lead to global histone hypoacetylation signatures, which contribute to HBV-induced pathomechanism in non-transformed hepatocytes.689 Pharmaceutical agonists of SIRT1, such as resveratrol, activated HBV transcription, while small-molecule inhibitors of SIRT1, including sirtinol and Ex527, exhibited anti-HBV activity, showing that SIRTs might be an anti-HBV target.690 SIRT7 also has a protective function in cccDNA via desuccinylation. SIRT7 restricts HBV transcription and replication by catalyzing desuccinylation of histone H3 associated with cccDNA minichromosome.691
The exact molecular mechanisms underlying the alteration in SIRT expression are still not fully elucidated due to limited research, especially the conflicting roles of SIRT7 in HBV infection. Therefore, it is crucial to further examine the functions and molecular mechanisms of SIRTs in regulating the development of HBV infection and HBV-induced diseases.
-
(4)
Other liver diseases
SIRT1, SIRT3, and SIRT6 might have effects on other liver diseases, including acute liver failure, hepatitis C virus (HCV) infection, autoimmune liver diseases, cholestasis diseases, and liver injury induced by a variety of causes.692,693,694
In the acute liver failure induced by D-galactosamine/LPS, a dramatic decrease in SIRT1 levels has been documented in a rat model.695 SIRT1 might have a protective effect by inducing HIF-1α deacetylation to reduce the ROS levels in mice.692 The treatment by SITR1-activating compounds, including quercetin (natural polyphenol) and SRT1720 (synthetic SIRT1 activator), might also support the beneficial role of SIRT1 in acute liver failure.696
SIRT1 plays an important role in the process of HCV infection. For example, the related study showed that elevated SIRT1 at protein level had an anti-aging effect on senescent CD4+ T cells during HCV infection.697 In addition, HCV core protein could induce dysfunction of liver sinusoidal endothelial cell by down-regulation of SIRT1.698 Interesting, an in vivo study showed that HCV core protein 1b-induced hepatic steatosis could be alleviated in liver-specific SIRT1 KO mice by downregulation of PPAR-γ2 expression.699 Therefore, the role of SIRT1 in the process of HCV infection should be further studied.
Additionally, SIRT1 may have beneficial effects on autoimmune hepatitis. For instance, an in vivo study showed that SIRT1-null mice developed an autoimmune-like disease related with the accumulation of immune complexes in the liver.700 Meanwhile, evidence also suggested that the activation of SIRT1 by resveratrol could protected against concanavalin A-induced autoimmune hepatitis in aged mice by repressing the expression of p66shc.701 Interestingly, pregnancy induces a state of immune tolerance, which can lead to spontaneous improvement of clinical symptoms of autoimmune hepatitis.702 As for mechanism, this may be associated with the activation of SIRT1 by chorionic gonadotropin signaling.702
Moreover, SIRT1, SIRT3, and SIRT6 may play important roles in cirrhosis.665,669,703 The study suggested that SIRT1 and SIRT6 were decreased at protein level in the livers of patients with cirrhosis.665,669 Besides, an in vivo study also suggested that enhanced expression of SIRT3 by curcumin had protective effects on cirrhosis.703 However, the exact molecular mechanism regarding cirrhosis should be further explored.
SIRT1 and SIRT6 show protective effects in drug-induced liver injury. Suppressing SIRT1 by miR-128-3p aggravated doxorubicin-induced liver injury by promoting oxidative stress.693 Upregulated SIRT1 pathway by quercetin attenuated NLRP3 inflammasome activation and apoptosis to protect isoniazid-induced liver injury, while SIRT1 inhibitor EX527 reversed the protective effect.704 Moreover, overexpression or pharmacological SIRT6 activation enhanced glutathione and decreased N-acetyl-p-benzo-quinoneimine, thus alleviating acetaminophen-induced hepatotoxicity via normalization of liver damage, inflammatory infiltration, and oxidative stress in a mouse model.705
Interestingly, hepatocyte SIRT1 might be a detrimental rather than protective factor in the setting of endotoxemic liver injury. Mechanistically, SIRT1-deacetylated p65 and compromised NF-κB activity in hepatocytes leads to increased susceptibility to endotoxemic injury when confronted with LPS/TNF-α stimulation.706 However, the evidence points to a dual role by which SIRT1 overexpression might contribute to cholestasis disease progression. Based on a mouse model of cholestatic liver disease, intestine-specific deletion of SIRT1 impaired systemic bile acid homeostasis.707,708 In an in vivo model of cholestatic disease, SIRT1-overexpressing myeloid cells with macrophage activation contributed to liver injury and fibrosis by activating the inflammasome and attenuating autophagy.709 Therefore, the role of SIRTs might be varied in different liver diseases. In addition, a loss or decrease in levels of SIRT3 could be an underlying factor and contributor to a damage-permissive phenotype in radiation-induced long-term persistent liver injury.694
Collectively, SIRTs play complex roles in liver disease. SIRTs might act as a double-edged sword and might be related to a specific disease mechanism and cellular type of action. More studies are needed to explore the role of proinflammatory SIRT effects in liver disease.
Pancreatic diseases
The incidence of acute pancreatitis (AP) has increased globally to approximately 34 cases per 100,000 persons annually with an increased risk of death.710 Currently, few studies have explored the effects of SIRTs on AP. SIRT1 has been reported to have a low expression in a rat model and has shown a protective effect.711 Resveratrol protects against acute necrotizing pancreatitis in mice by enhancing SIRT1-mediated deacetylation of p53 and heat shock factor 1.711,712
However, SIRT1 has an opposing effect on chronic pancreatitis compared to AP. A related study has shown that SIRT1 was significantly upregulated in chronic pancreatitis and PC and in the absence of SIRT1 expression inhibition by miR-278 contributed to inflammation-induced EMT.713 The conflicting roles of SIRT1 in these studies imply a potentially different effect of SIRT1 on pancreatitis. Overall, further studies are needed to verify the exact role of SIRTs in this disease.
Intestinal diseases
-
(1)
IBDs
IBDs are lifelong and incurable chronic inflammatory diseases affecting 6.8 million people worldwide.714 Most SIRTs can alleviate IBD.
SIRT2 and SIRT6 are downregulated in IBD patients and their deletion promotes inflammatory responses by regulating the NF-κB pathway activation, which highlights their protective roles.715,716,717 In addition, SIRT2 inhibits Wnt/β-catenin signaling to maintain gut homeostasis.333 Evidence also supports the role of SIRT6 in the resistance of intestinal epithelium to injury, at least in part by preserving the expression of Rspo1, a critical growth factor in intestinal epithelial cells.715 Limited research has shown that systematic SIRT3, SIRT5, and SIRT7 KO mice were susceptible to colitis.718,719,720
However, the dual role of SIRT1 in IBD has been observed in several studies. There is a significant downregulation in mRNA and protein expression of SIRT1 in patients suffering from IBD.721 A decrease in SIRT1 with an increase in age has been shown to aggravate colitis and cause other impairments in a mouse model.722 SIRT1 deficiency induced the activation of paneth and goblet cells, increased NF-κB activity, and elevated the levels of proinflammatory genes and antimicrobial proteins in the small intestine.722 In addition, SIRT1 activation reduced apoptosis of intestinal epithelial cells via suppression of endoplasmic reticulum stress-mediated apoptosis-associated molecules CCAAT/enhancer-binding protein homologous protein and caspase-12 in both in vivo and in vitro models.723 Compared to the studies showing that a decreased expression of SIRT1 might be important in the pathogenesis of IBD, a decrease in SIRT1 might be protective in IBD. SIRT1 deletion might be useful in the improvement of disease conditions in colitis via induction of Foxp3 + T-regulatory cells, which are important for intestinal homeostasis.724,725 The loss of SIRT1 in thymic–derived natural Tregs did not affect rescue from autoimmune colitis, although it promoted Foxp3+ development from conventional T cell formation and attenuated autoimmune colitis.724 These conclusions suggest that the deleterious role of SIRT1 in immune-related diseases might be related to different T cell sources. Therefore, further studies are needed to verify the association between SIRT1 and more immune cells and to explore the role of SIRT4, which has not been well characterized based on current studies.
-
(2)
Intestinal ischemia/reperfusion injury
Studies have demonstrated the beneficial effects of SIRT1, SIRT3, and SIRT6 on intestinal IRI exerted by adjusting ROS generation and massive epithelial apoptosis in a mouse model, which are critical in the pathogenesis of intestinal IRI injury.229,726,727 For instance, SIRT1 suppressed epithelial ROS accumulation and apoptosis to attenuate intestinal IRI after miR-34a-5p systematic knockdown.229 Resveratrol protected intestinal subacute IRI via the SIRT1-NF-κB pathway in an iNOS-NO-inhibited manner. This might represent a novel prophylactic approach to intestinal IRI.728 SIRT3 alleviated intestinal IRI-induced mitochondrial oxidative damage and apoptosis through peroxiredoxin 3 deacetylation, a key protective factor in intestinal IRI.727 Moreover, based on both mouse and rat models, downregulating SIRT6 by miR-351-5p aggravated intestinal IRI by promoting oxidative stress, inflammation, and apoptosis.726 There have been limited research studies exploring the effect of other SIRTs on intestinal IRI.
-
(3)
Other intestinal diseases
In other intestinal diseases, including hirschsprung-associated enterocolitis and necrotizing enterocolitis, SIRT1 was downregulated and involved in inflammation.729,730 SIRT1 can be suppressed via miR-132 and miR-212 or downregulated retinoid-related orphan receptor α by exosomal miR-18a-5p and then activate the NF-κB signaling pathway, NLRP3 inflammasome, and caspase-1-mediated pyroptosis, thereby encouraging the inflammatory response in Hirschsprung-associated enterocolitis mice.729,731 Similarly, SIRT1 activation might decrease the damage caused by necrotizing enterocolitis by decreasing proinflammatory cytokines and oxidative stress proteins and by increasing the anti‑inflammatory cytokine pathway.730,732 SIRT1 alleviated the inflammatory response and intestinal epithelial barrier dysfunction by regulating the expression and inactivation of HIF-1α.730
In conclusion, current studies on intestinal system diseases have mostly focused on IBD. There are limited studies on other SIRTs, except SIRT1, which is a field worthy of further development. In addition, both protective and deleterious effects of SIRT1 have been explored in intestinal diseases. The proinflammatory effects of SIRT1 and regulation of different immune cells might play an important role in aggravating intestinal diseases. As SIRTs have various biological functions in intestinal diseases, they and their underlying mechanisms are promising novel targets for studying the development of intestinal diseases.
Conclusions
Most studies on the association between SIRTs and digestive system diseases have been completed in animal models, while the numbers of human studies are increasing. The current evidence demonstrates the role for SIRTs in digestive system diseases and identifies exciting opportunities to adjust SIRT activity to treat or prevent these diseases. Many questions remain unanswered, however, and more research needs to be done, especially on SIRT2, SIRT4, SIRT5, and SIRT7. The potentially divergent roles of different SIRTs in these diseases are not well verified, especially the conflicting roles.
Additionally, SIRT1, the most widely studied SIRT, might have opposing roles in different diseases, particularly in the inflammatory response. The proinflammatory effect of SIRT1 on digestive diseases contrasts with anti-inflammatory effects reported by most studies. These findings are essential because unraveling the less common negative effects of SIRT1 might contribute to a more comprehensive understanding of its generally accepted positive function.
Thus, in the future, more studies on a molecular level and in clinical populations are needed to confirm the role of SIRTs in digestive diseases.
SIRTs and nervous system diseases
Nervous system diseases directly affect the lives of hundreds of millions of people worldwide,733 and one in every nine people dies due to a disorder of the nervous system.734 Recently, there has been a gradual increase in research on the role of SIRTs in neurological diseases. An understanding of the latest progress and potential molecular mechanisms of SIRTs in neurological diseases will benefit further studies on the clinical diagnosis and treatment of these diseases. Therefore, this review mainly summarizes the current research progress on the role of SIRTs in neurological diseases (Fig. 13).

The role of SIRTs in nervous system, mainly including AD, HD, PD, brain injury, epilepsy, neuroinflammation, SCI, multiple sclerosis and ALS. SIRT1, SIRT3, and SIRT6 have protective effects on AD, multiple scierosis and amyotrophic lateral scierosis. SIRT1 plays a major protective role in HD. In PD, SIRT1 and SIRT3 provide protective effects. In addition, SIRT1, SIRT3, and SIRT5 are beneficial in both brain injury and epilepsy. SIRT1, SIRT2, and SIRT3 have roles in protecting against neuroinflammation. SIRT1 and SIRT6 could also protect SCI. Notably, the effects of SIRT2 may be harmful on AD, HD, PD, and brain injury. https://biorender.com
Alzheimer’s disease (AD)
AD is the most common neurodegenerative disorder that is associated with memory deficit and global cognitive decline.735,736 It is a brain disorder associated with gradual weakening of neurocognitive functions, neuroinflammation, and impaired signaling pathways.737 The SIRT proteins associated with AD mainly include SIRT1, although mitochondrial SIRTs represented by SIRT3 have also been the focus of research, as well as SIRT6 and SIRT2 that are located in the nucleus and cytosol, respectively.
Amyloid beta (Aβ) is a normal and soluble product of neuronal metabolism,738 and Aβ-mediated extracellular senile plaque is regarded as one of the major pathological lesions of AD.739 Previous evidence has suggested that SIRTs played important roles in the regulation of Aβ.739 For example, in vitro study has shown that overexpression of SIRT1 could reduce Aβ-induced senescence and mitochondrial dysfunctions,740 and related mechanism studies have suggested that SIRT1 could regulate Rho-associated kinase 1 or inflammation to attenuate the accumulation of Aβ.740,741,742 Similarly, SIRT3 also protects neurons against Aβ pathology and excitotoxicity.743 In contrast, SIRT2 may have adverse effect on Aβ pathology, and in vivo study has revealed that the suppression of SIRT2 deacetylase activity could alleviate Aβ pathology and cognitive deficits in the AD mouse model.744 As for the molecular mechanism, SIRT2 could influence the β‐secretase 1 by directly deacetylates reticulon 4B protein, thereby affecting the production of Aβ and ultimately promoting the development of AD.744
Tau is the major microtubule-associated protein of a mature neuron, and it is a central molecule in the pathogenesis of AD.745 Previous studies have highlighted the importance of PTMs (e.g., O-GlcNAcylation, phosphorylation, and acetylation) of Tau in AD.746,747,748 For example, the level of O-GlcNAcylation of tau in AD brain is reduced, and SIRT1 reduces O-GlcNAcylation of tau through CREB.749 Moreover, SIRT2 affects tau phosphorylation and autophagic flux in AD.750 There is evidence that tau acetylation occurs in AD brain at early stages of the disorder and that this phenomenon is involved in regulating the early accumulation of tau in AD brain. SIRT3 might play a role in tau acetylation and could be a potential target for developing novel therapies to alleviate tau accumulation.751 Collectively, these studies suggest that SIRT1-3 might play a role in PTMs of tau and could be potential targets for designing novel therapies to alleviate tau accumulation in AD brain.
In the healthy brain, high levels of H4K16ac and low levels of SIRT2 coexist with Fzd1 and Fzd7 promoters. A recent study reported a novel role of nuclear SIRT2 in regulating Fzd receptors in AD, wherein nuclear SIRT2 was hyperactivated in AD and FoxO1 recruited SIRT2 to Fzd1 and Fzd7 promoters, leading to a reduction in H4K16ac deacetylation.752 These findings suggest that SIRT2 inhibition is an attractive target for ameliorating the pathological effects of AD.
Several studies have shown that autophagy deficits occur in the early stage of AD, which contribute to the process of AD.753 SIRT-regulated autophagy impairment plays a key role in the neurodegenerative process of AD. Beclin-1 acetylation impairs the autophagic flux, which contributes to neurodegeneration in AD. Another study showed that SIRT1 enhances the deacetylation of Beclin-1, thus suggesting the beneficial role of SIRT1 in promoting autophagy in AD neurodegeneration.270 The loss of function of SIRT2 either through AK1 (a specific SIRT2 inhibitor) or through SIRT2 KO recovers microtubule stabilization and improves autophagy.299
AD is manifested through regional cerebral hypometabolism. SIRT3 has emerged as a key regulator of mammalian transcription in response to cellular metabolic state and stress.754 Recent studies have shown that SIRT3 dysfunction leads to mitochondrial and neuronal damage in AD, suggesting that SIRT3 has a protective role in hippocampal neurons.755 Intermittent food deprivation also reduces neuronal network hyperexcitability and ameliorates deficits in hippocampal synaptic plasticity in a SIRT3-dependent manner in animal models of AD.756 Pituitary adenylate cyclase activating polypeptide, a neurotrophin, stimulates mitochondrial SIRT3 production. Knockdown of SIRT3 compromises the neuroprotective effects of pituitary adenylate cyclase activating polypeptide in AD, and this effect was reversed by overexpression of SIRT3.757 SIRT3 expression mirrors the spatiotemporal deposition of Aβ in an AD mouse model and is also upregulated in the temporal neocortex of patients with AD.758
In clinical research, an inverse relationship was observed between serum levels of SIRT1, SIRT3, and SIRT6 and AD.759 Measurement of SIRT1, SIRT3, and SIRT6 levels in saliva could be used as an additional method for intravital noninvasive diagnosis of AD in advanced age patients.760 The SIRT protein family constitutes a unique molecular link between aging and human neurodegenerative diseases and offers a promising avenue for therapeutic intervention. However, the mechanisms of action of SIRTs in chronic neurodegenerative diseases in vivo remain unclear. Hence, further studies on the role and mechanism of the SIRT family in AD are required, which could provide promising avenues for therapeutic intervention.
Parkinson’s disease (PD)
PD is the most common movement disorder associated with older adults, and currently, there is no effective treatment or prevention methods other than symptomatic treatment. A previous study investigated the possible association of nine SIRT1 and SIRT2 SNPs with the risk of PD through a clinical case-control investigation in Chinese Han population. Further functional assays suggested that rs2015 might influence the expression of SIRT2 by affecting the binding of miR-8061 to the 3ʹ-UTR of SIRT2, eventually contributing to the risk of PD.761 Therefore, the SIRT family is involved in the pathology of PD. However, considering that an epidemiological investigation showed that variations in the SIRT genes do not affect the risk for PD,762 the association between SIRT gene polymorphisms and PD risk remains elusive and needs further studies to clarify.
The accumulation of misfolded α-synuclein in dopaminergic neurons is the leading cause of PD.763 Activated SIRT1 ameliorated LC3 deacetylation-mediated autophagic degradation of α-synuclein and improved motor defects and pathological changes in PD mice.763 Moreover, pharmacologically increased levels of SIRT3 could counteract αsyn-induced mitochondrial dysfunction by reducing αsyn oligomers and normalizing mitochondrial bioenergetics, thus supporting a protective role of SIRT3 in PD-associated pathways.764 Mitochondrial dysfunction is the main cause of dopaminergic (DAergic) neuronal loss in PD, and SIRT3 plays a key role in regulating mitochondrial function.765 The age-dependent elevation of mitochondrial oxidative stress is widely recognized as a major factor in the loss of dopaminergic neurons in the substantia nigra pars compacta in PD, and this process is associated with a decrease in SIRT3 protective function.766
SIRT2 also appears to play a different role in PD from other SIRT family members. In vitro and in vivo studies have shown that SIRT2 mediates exacerbation of alpha-synuclein toxicity in models of PD.298 NAD + metabolism is altered in sporadic PD patient-derived cells, which contributes to SIRT2 activation and subsequent decrease in the levels of acetylated α-tubulin.297 These results suggest that SIRT2 deletion was protective in PD models.
Collectively, these data support a protective role of SIRT1 and SIRT3 in PD-associated pathways, while SIRT2 might show different functions from the former two. Thus, further studies are required to investigate the role of SIRT2 in PD.
Huntington’s disease (HD)
HD is an incurable neurodegenerative disorder characterized by movement disorder, psychiatric symptoms, and cognitive decline. Brain-specific KO of SIRT1 results in exacerbation of brain pathology in a mouse model of HD, whereas overexpression of SIRT1 improves survival, neuropathology, and expression of BDNF in HD mice. Mechanistically, mutant huntingtin protein interferes with the CREB-regulated transcription coactivator 1-CREB interaction to repress BDNF transcription, and SIRT1 rescues this defect in vitro and in vivo; this finding suggests a key role of SIRT1 in transcriptional networks in HD brain and offers an opportunity for therapeutic development.767
HD has a complex pathogenesis mechanism, including protein aggregation and metabolic dysfunction. SIRT1 expression is increased in HD-affected brain regions, and metabolic pathways are altered in the hypothalamus of individuals with HD.768 An important finding is that the manipulation of sterol biosynthesis at the transcriptional level mimics SIRT2 inhibition, which demonstrates that the metabolic effects of SIRT2 inhibition are sufficient to diminish mutant huntingtin toxicity.769 This study demonstrated that inhibition of SIRT2 achieves neuroprotection in cellular and invertebrate models of HD. Therefore, both SIRT1 and SIRT2 play an important role in HD, and hence, the effect of SIRTs on HD needs to be further investigated.
Brain injury such as IRI and stroke
Brain injury, such as IRI and stroke, is a neurological disorder with high morbidity, high probability of mortality, and poor neurological outcome.770 The SIRT family is a highly potent therapeutic target to decrease IRI.771 SIRT1 plays an important role in neuroprotection against brain injury through oxidative, inflammatory, autophagy and apoptotic pathways.214,772,773 The regulation of autophagy proteins LC3-II and Beclin-1 by NAMPT was abolished in cultured SIRT1-KO neurons, thus suggesting that NAMPT promotes neuronal survival by inducing autophagy in a SIRT1-dependent manner during cerebral ischemia.774 SIRT1 deacetylates the RNA-binding protein quaking 6 and activates the transcription factor PGC-1α through post-transcriptional regulation of PPAR-γ expression, which significantly affects the synthesis of triglycerides in neurons of the cerebral IRI rat model, thereby inhibiting neuronal apoptosis.
Several studies have also shown the protective effects of mitochondrial SIRT3 and SIRT5 in IRI. Mitochondrial SIRT3 acts as a prosurvival factor to protect neurons from excitotoxic injury and exerts a protective role in ischemic stroke by regulating the HIF-1α/ vascular endothelial growth factor signaling pathway in astrocytes.775,776 Additionally, SIRT3 was found to be downregulated in response to cerebral IRI; therefore, strategies to enhance SIRT3 activity and activate the Wnt/β-catenin pathway could be therapeutic targets for treating cerebral IRI.777 SIRT5 has also been shown to mediate IR-induced brain damage by increasing the permeability of blood-brain barrier through degradation of the tight junction protein occluding.778
Notably, SIRT2 appears to have detrimental roles in an array of neurological disorders such as PD and HD. The current study demonstrated the neuroprotective effects of SIRT2 inhibition in ischemic stroke and identified the downregulation of the Akt/FoxO3a and MAPK pathways as intermediary mechanisms that might contribute to the reduction in apoptotic cell death by SIRT2 inhibition.248 In clinical practice, SIRT2 might serve as a marker of acute ischemic stroke (AIS) risk and prognosis. Serum SIRT2 expression was increased in patients with AIS as compared to that in non-AIS patients with high stroke risk factors. This finding supports the role of SIRT2 in facilitating disease monitoring and prognosis in patients with AIS.779
In conclusion, previous studies report the protective roles of SIRT1, SIRT3, and SIRT5 in IRI, while there is also evidence that SIRT2 appears to play a different role in IRI. More studies are required to elucidate the regulatory mechanisms and functional implications of the SIRT family in brain injury.
Other diseases
-
(1)
Multiple sclerosis and amyotrophic lateral sclerosis (ALS)
Multiple sclerosis is an autoimmune-mediated neurodegenerative disease with characteristic foci of inflammatory demyelination in the brain, spinal cord, and optic nerves.780 ALS is also a neurodegenerative disease characterized by degeneration of upper and lower motor neurons, which results in muscle weakness and eventual paralysis, and it is also known as motor neuron disease.781,782 To date, ALS remains as an incurable and devastating disease. Drug development efforts are mostly based on SOD1 gene -G93A mice that present a very strong and early phenotype, allowing only a short time window for intervention.783 An increased expression of SIRT1 was observed in the cerebral cortex, hippocampal formation, thalamus, and spinal cord of symptomatic SOD1 (G93A) transgenic mice, but the mechanisms and functional implications of increased SIRT1 expression require elucidation.784 In human postmortem tissue, increased mRNA and protein levels of SIRT3 were found in the spinal cord in patients with ALS.785 Moreover, enhanced SIRT6 activity abrogates the neurotoxic phenotype of astrocytes expressing ALS-linked mutant SOD1, thus indicating that SIRT6 could serve as a potential therapeutic target to prevent astrocyte-mediated motor neuron death in ALS.786 These studies illustrated the potential beneficial role of SIRT1, SIRT3, and SIRT6 in ALS.
-
(2)
Epilepsy
Epilepsy is a neurological disorder characterized by brain hyperexcitability and manifests as seizure.787 SIRT1 might represent a useful therapeutic target to rescue the expression of circadian rhythm genes and sleep patterns in patients with epilepsy.788 SIRT5 deficiency strikingly increased the mortality rate and severity of response to epileptic seizures, thus indicating that SIRT5 has a neuroprotective role in epileptic seizures and neurodegeneration.789 Mechanistically, SIRT1 protein expression could be inhibited by miR-128, and treatment with the SIRT1 agonist CAY10602 exerts neuroprotective effects on epilepsy.233 Similarly, SIRT3 could also protect neurons from kainic acid-induced excitotoxicity by mediating mitochondrial function with enhanced expression by inhibiting miR-134-5p.790 Overall, SIRT1, SIRT3, and SIRT5 appear to have a neuroprotective role in epilepsy. More molecular and clinical studies are required in the future to verify the effects of SIRTs on epilepsy.
-
(3)
Cognitive deficits
Cognitive deficits are common in patients with conditions such as PD, epilepsy, and psychotic depression.791,792,793 SIRT1 is a recognized longevity gene and has been shown to be associated with aging and its related diseases. SIRT1 is an important protective gene against hippocampal atrophy and its induced cognitive impairment during aging.794 Surgery-induced downregulation of hippocampal SIRT1 participates in cognitive impairment after surgery by inhibiting the autophagy process and activating apoptosis.795 Additionally, exposure to fluoride could lead to cognitive impairment, and the underlying mechanisms might be related to oxidative stress and mitochondrial dysfunction. Chronic long-term exposure to fluoride causes neural/synaptic damage and cognitive impairment through mitochondrial dysfunction and its associated oxidative stress, which is mediated at least in part by SIRT3 inhibition in mouse brain.796 The natural bisphenol compound honokiol upregulated the expression of SIRT3 protein in vivo and in vitro, and its protective effect against oxidative stress and mitochondrial dysfunction could be abrogated by SIRT3 shRNA.797 To date, few studies have been conducted on the relationship between SIRTs and cognitive deficits, and more research is required to explore this association.
-
(4)
Spinal cord injury (SCI)
SCI is a devastating condition with few effective treatments. Because posttraumatic inflammation contributes to the progression of neuronal degeneration, attenuating inflammation is important for reducing neural degeneration. The anti-inflammatory effect of SIRT1 has been reported to be involved in SCI.798 SIRT1 might have a neuroprotective effect by suppressing microglial activation and increasing the secretion of proinflammatory cytokines following SCI.799 After the trauma, spinal cord neurons were apparently damaged. Regulation of autophagy by the AMPK/SIRT1 pathway could restrain the damage of spinal cord neurons, which might be a potential intervention for SCI.286 SIRT6 might also play a vital role in the pathogenesis of SCI. Mechanistically, the upregulation of SIRT6 alleviated inflammation and oxidative stress and inhibited cell apoptosis in SCI.800 In terms of mechanism, multiple miRNAs such as miR-138-5p, miR-324-5p, and miR-30c have been reported to be involved in SCI by targeting SIRT1.801,802 These studies provide a promising biomarker of prognosis and therapy for spinal cord diseases.
-
(5)
Neuroinflammation and neuropathic pain
Diverse causes of neuropathic pain are associated with excessive inflammation in both the peripheral and central nervous system, which might contribute to the initiation and maintenance of persistent pain.803 SIRTs might serve as a potential therapeutic target for treating neuropathic pain. SIRT1 and SIRT2 deacetylases are reported to exert neuroprotective effects on neuroinflammation.61 SIRT1 activation attenuated Mn-induced oxidative stress and neuroinflammation in adult mice.804 Overexpression of SIRT2 alleviates neuropathic pain and neuroinflammation.805 The SIRT2 inhibitor AK-7 exacerbates traumatic brain injury through a potential mechanism involving increased acetylation and nuclear translocation of NF-κB p65, resulting in the upregulation of NF-κB target genes and proinflammatory cytokines.88 Another study suggested a key protective role of microglial SIRT2 in amnesic deficits associated with neuroinflammation.806 SIRT2-deficient mice (SIRT2(−/−)) showed morphological changes in microglia and an increase in proinflammatory cytokines upon intracortical injection of LPS.807 SIRT3 also regulates mitochondrial oxidative stress response and neuroinflammation. SIRT3-induced Mst1-JNK-SRV2 signaling pathway protected against neuroinflammation-mediated cell damage in BV-2 microglia.808 LPS induces oxidative stress and neuroinflammation in BV2 cells, which might be mediated in part by the downregulation of triggering receptor expressed on myeloid cells 2 and SIRT3. Triggering receptor expressed on myeloid cells 2 overexpression ameliorates LPS-induced oxidative stress and neuroinflammation by enhancing SIRT3 function through NAD + .809 Here, SIRT1-3 show anti-neuroinflammatory effects. More research is required to elucidate whether these SIRTs affect neuroinflammation and neuropathic pain through the same or different mechanisms.
Conclusion
An increasing number of studies predict that the effects of various SIRTs on neurological diseases might be different or even contrasting. However, it is worth noting that positive intervention of SIRT activity, such as through upregulation of SIRTs-activating molecules, might have profound therapeutic benefits on various nervous system diseases. The long-term effects of decreased SIRT levels per se or in chronic neurodegenerative conditions is an important question for future studies. Therefore, in the research process, we should discover new mechanisms of action to elucidate the different results. Additionally, research should be based not only on cellular and animal models but also on the relationship between inflammatory SIRTs and diseases from human epidemiology.
SIRTs and endocrine system diseases
Endocrine system regulation is important for the maintenance of homeostasis, and it controls hormonal functions under physiological conditions and behaviors as well as adaptations to social environments.810 Endocrine system disorders lead to various diseases such as diabetes mellitus (DM), obesity, and metabolic syndrome, which causes heavy disease burden worldwide.811 The activation of SIRT proteins enhances metabolic efficiency and upregulates mitochondrial oxidative metabolism, which are important for metabolic balance of human body.46 A growing number of studies have shown that SIRTs exert vital effects on maintaining metabolic health and controlling the occurrence and development of endocrine system diseases such as DM (Fig. 14),128,812 diabetic complication (Fig. 14),813,814,815 obesity,816 and metabolic syndrome.817

The roles of SIRTs in diabetes and related target organs injury. a SIRT1, SIRT2, SIRT3, SIRT4, and SIRT6 are associated with pathological processes in the occurrence and development of DM. SIRT1 and SIRT2 have dual functions, including both improving insulin sensitivity and reducing insulin responsiveness. SIRT3, SIRT4, and SIRT6 mainly exert protective effect on DM. b SIRT1, SIRT3, SIRT4, and SIRT7 play protective roles in diabetic nephropathy. Low levels of SIRT1 and SIRT3 are associated with renal fibrosis and reduced expressions of SIRT1, SIRT4, and SIRT7 are related to podocyte apoptosis. c Increasing SIRT1 expression can exert protective effect during the development of neuropathy in sensory neuron of spinal cord. Moreover, SIRT1 could also reverse neuron damage in hippocampus. In addition, SIRT3 may inhibit neuropathy in sciatic nerve. d SIRT1, SIRT3, and SIRT6 are reduced in the pathological process of diabetic retinopathy. Additionally, SIRT1 is reduced during the damage of blood-retinal barrier. e SIRT1, SIRT3, and SIRT6 act protective roles in the development of diabetic cardiomyopathy, which consists of heart failure, cardiac fibrosis, cardiac hypertrophy, myocardial infarction, vascular injury and atherosclerosis. https://biorender.com. HOTAIR HOX transcript antisense RNA, Mff mitochondrial fission factor, SGLT2 sodium-dependent glucose transporters 2
DM
Globally, more than 425 million people are living with DM, and its prevalence is expected to increase at least 50% by 2045.818 Worldwide, DM is the leading cause of blindness, nontraumatic lower extremity amputations, peripheral neuropathy, and end-stage kidney diseas.819,820,821,822 Numerous reports have suggested that SIRTs, especially SIRT1-3, SIRT5, and SIRT6, are associated with biological processes that participate in the development and progression of diabetes, such as glucose metabolism, mitochondrial function, and resistance against cellular stress.26,823,824 The expression of SIRTs in patients with DM has been reported inconsistently. The expression of SIRT1-3 is reduced in patients with DM,825,826,827 while the expression of SIRT5 and SIRT6 is elevated.350,828 Thus, the altered expression of SIRT proteins might affect the progression of DM.
SIRT proteins play important roles in the occurrence and development of DM by regulating glucose metabolism and maintaining insulin homeostasis.6 SIRT1 and SIRT2 have been found to have dual function in the development of DM, which might be due to the biological process occurring in the cells from different types of tissues or organs. For instance, SIRT1 overexpression could improve insulin sensitivity and reduce insulin resistance,829,830 while the downregulation of SIRT1 inhibits insulin-stimulated glucose transport in adipocytes in particular by inhibiting insulin signaling.831 Conversely, hepatic SIRT1 knockdown prevented fasting hyperglycemia by decreasing hepatic glucose production and increasing hepatic insulin responsiveness.832 SIRT2 could also promote glucose-dependent hepatic glucose uptake by deacetylating K126 of glucokinase regulatory protein.833 In contrast, the downregulation of SIRT2 ameliorated the reduced activity of Akt and increased insulin-stimulated glucose uptake in insulin-resistant neuro-2a cells.834 However, the detailed molecular mechanisms of these bilateral roles remain unclear and need further investigation.
SIRT3, SIRT4, and SIRT6 have been proven to exert a protective effect on DM. For example, SIRT3 KO severely impaired insulin-stimulated muscle glucose uptake, which further aggravated insulin resistance.812 Likewise, SIRT4 overexpression led to dyslipidemia, lipogenesis, and decreased fatty acid oxidation; this might be because SIRT4 can deactivate AMPK as well as directly inhibit insulin secretion at the cellular level.835 Moreover, SIRT6 induced PGC-1α acetylation and suppressed hepatic glucose production,836 and SIRT6 cooperated with p53 to deacetylate FoxO1 and transport FoxO1 from the nucleus to the cytosol, and suppressed the expression of gluconeogenic genes,837 all of which could alleviate diabetic hyperglycemia. Conversely, SIRT5 can promote the progression of DM. Experiments in two pancreatic β-cell lines (MIN6 and INS-1) suggest that SIRT5 inhibition facilitated pancreatic β-cell proliferation and insulin secretion.350 Moreover, SIRT5 negatively regulates the transcription of PDX1 through its deacetylase activity,350 and subsequently, the downregulation of PDX1 expression aggravates DM.838,839 These studies suggest that high expression of SIRT3, SIRT4, and SIRT6 and low expression of SIRT5 might exert protective effects on the development of DM.
In conclusion, there is limited research on the relationship between SIRT proteins and DM; most studies have shown that SIRT1-4 and SIRT6 exert protective effects on the development of DM, while SIRT5 promotes the progression of DM. However, other studies have found the downregulation of SIRT1 and SIRT2 contributes to improve DM. The difference in the effect of SIRT1 and SIRT2 on DM might be attributed to cells from different types of tissues or organs and required to be further clarified. Additionally, future studies could pay more attention to the role of SIRT proteins, especially SIRT7, in the development and progression of DM.
DM-related organ damage
The global epidemic of DM has led to a corresponding epidemic of complications of these disorders.840 Devastating macrovascular complications CVD and microvascular complications [such as diabetic kidney disease (DKD), diabetic retinopathy (DR), and diabetic neuropathy (DN)] lead to increased mortality, blindness, kidney failure, and an overall decreased quality of life in individuals with DM.841 SIRTs have been shown to have protective effects on the target organ damage caused by DM, such as diabetic cardiomyopathy (DCM),842 DKD,814,815 DR,129 and DN.843
-
(1)
DKD
DKD is recognized as a severe complication of DM and a dominant pathogeny of end-stage kidney disease, which causes severe health problems and large financial burden worldwide.844 During the past two decades, the morbidity and mortality of DKD have been rising rapidly worldwide,845 and the age-standardized prevalence of DKD in men and women was 15.48/1000 and 16.50/1000, respectively, in 2017.846 SIRT1 shows a protective role in the development of DKD. In detail, high expression of SIRT1 effectively protects the kidney and slows down the progression of DKD.814,815 On the one hand, increased SIRT1 activity protects against DM-induced podocyte injury and effectively mitigates the progression of DKD.814 On the other hand, stimulation of SIRT1 expression and signaling in DM protects the kidney against oxidative stress and nephropathy.815 Mechanistically, SIRT1 exhibited its renal protective effects through deacetylation of the transcription factor p53815 and activation of the transcription factors FoxO3a and Nrf2.815,847 For example, SIRT1 attenuated nephropathy progression in diabetic mice by downregulating acetylated p53 expression and upregulating FoxO3a expression.815 Moreover, increasing SIRT1 activation by resveratrol in both in vivo and in vitro studies promoted resistance to diabetic renal fibrosis by activating Nrf2, a leucine transcription factor.847 SIRT1 can effectively reduce the damage caused by DKD and slow down the progression of DKD.814,815 Therefore, SIRT1 might become a potential target for the clinical treatment of DKD.
-
(2)
DN
DN is the most prevalent diabetic complication, and at least 50% of individuals with diabetes develop DN over time.840 It substantially affects patients by increasing falls, thereby causing pain and reducing the quality of life.848 Accumulating evidence has demonstrated that SIRT1 modulates neuronal viability,849 neuronal differentiation,849 and synaptic plasticity,850 all of which are key factors largely linked to cognitive improvement. SIRT1 has also been proved to alleviate symptoms related to DN, including cognitive decline,843 neuropathic pain,851 and peripheral neuropathy.852 For instance, SIRT1 expression was decreased in the hippocampus of diabetic rats, which reduced dendritic length and spine densities and decreased TORC1, p-CREB, and BDNF protein levels, resulting in diabetes-related cognitive decline.843 Moreover, the upregulation of spinal SIRT1 relieved pain behavior, inhibited enhanced structural synaptic plasticity in diabetic rats and mice with diabetic neuropathic pain, and decreased the levels of synapse-associated proteins in diabetic neuropathic pain rats, diabetic mice, and high glucose-cultured spinal neurons.851 SIRT1 also regulated mitochondrial function in the peripheral nerve through PGC-1α, and the failure of the SIRT1-PGC-1α-mitochondrial transcription factor A (TFAM) signaling axis might result in the suppression of mitochondrial oxidative phosphorylation and development of peripheral neuropathy.852 Collectively, an understanding of the regulatory roles of SIRT1 proteins might help to develop them as promising therapeutic targets in DN treatment. However, recent studies mainly focus on SIRT1, and the molecular mechanisms of other SIRT proteins in regulating DN are still unclear and need further investigation.
-
(3)
DR and DCM
DR is a common and specific microvascular complication of DM and remains the leading cause of preventable blindness in working-aged people.853 It is identified in one third of patients with DM and is associated with increased risk of life-threatening systemic vascular complications, including stroke, coronary heart disease, and heart failure.853 Current studies have shown that SIRT1 can alleviate DR;129 however, related studies are still limited. Previous studies have revealed that overexpression of SIRT1 prevents the increase in capillary cell apoptosis and formation of degenerative capillaries,854 reduces DM-induced inflammation in the retina, and improves DM-induced visual function impairment.129
DCM is also a distinct form of heart disease that represents a major cause of death and disability in patients with diabetes, particularly in the more prevalent type 2 diabetes patient population.855 The activation of SIRT1 and SIRT3 contributes to inhibit the development of DCM. For example, SIRT1 activation inhibits ROS generation-induced oxidative stress and fibrosis, thereby attenuating DCM.856 The activation of SIRT3 also regulates fibrosis, inflammation, apoptosis, and oxidative stress in diabetic myocardial tissue149 and attenuates DCM through the reduction in p53 acetylation and TP53-induced glycolysis and apoptosis regulator expression together with upregulation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 3, which are the key regulators of phosphofructokinase and glycolysis.842 In contrast, SIRT3 deficiency aggravated hyperglycemic mitochondrial damage, increased ROS accumulation, promoted necroptosis, possibly activated the NLRP3 inflammasome, and finally exacerbated DCM in mice.857
Therefore, SIRT1 and SIRT3 show positive effects in a variety of diabetic complications, including DKD, DN, DR, and DCM, which indicated that these two SIRTs could serve as promising therapeutic targets in the clinical treatment of DM-related target organ damage. However, the molecular mechanisms of other SIRTs in regulating diabetic complications are not fully understood and require further studies.
Obesity
The Global Burden of Disease Obesity Collaborators have estimated that more than 603.7 million adult individuals are obese.858 Elevated body mass index values were responsible for 4 million deaths in 2015.858 Severe obesity is associated with a state of chronic inflammation,859 which results in an increase in the incidence of type 2 diabetes, CVD, hepatic steatosis, airway disease, neurodegeneration, biliary disease, and certain cancers.860 These obesity-associated disorders are subsequently linked to reduced life expectancy and premature death.861 SIRTs act as deacetylases that could affect a variety of metabolic and inflammatory pathways, potentially improving health and extending lifespan.816 Therefore, SIRT proteins might play an important role in controlling obesity and reducing other diseases caused by obesity.
Accumulated evidence suggests that SIRT1 and SIRT3 could suppress obesity by inhibiting adipogenesis and stimulating energy expenditure.862,863,864 The 3ʹ-UTR of SIRT1 mRNA binds directly to miR-146b and promotes adipogenesis through SIRT1 downregulation,862 while inhibition of hypothalamic SIRT1 enhanced the activity of the hypothalamic-pituitary-thyroid axis, which stimulated energy expenditure.863 Moreover, high expression of SIRT1 and PGC-1α activated by AMPK subsequently increased citrate synthase activity and improved muscle mitochondrial respiration on a fatty acid-derived substrate.865 The increased expression of SIRT1 similarly reduced acetylation of PGC-1α and FoxO1, which was associated with attenuation of high fat diet-induced mitochondrial dysfunction, insulin resistance, and obesity.866 Additionally, overexpression of SIRT3 activated macroautophagy by activating the AMPK-ULK1 pathway, leading to smaller lipid droplet size and reduced lipid accumulation. Similarly, SIRT3 overexpression induced the formation of perilipin-1-heat shock cognate 71-kDa protein-lysosome-associated membrane protein 2 complex to activate chaperone-mediated autophagy and cause instability of lipid droplets in adipocytes.864
In contrast, SIRT2 and SIRT6 promote the occurrence and development of obesity. The SIRT2- PGC-1α regulatory axis is negatively regulated by HIF-1α, which negates the intrinsic pathways of fatty acid catabolism in adipocytes and creates a metabolic state that supports the development of obesity.867 SIRT6 overexpression was found to exacerbate diet-induced obesity by decreasing STAT3 acetylation and lowering pro-opiomelanocortin expression in the hypothalamus.868
Overall, these findings suggest that high expression of SIRT1 and SIRT3 and low expression of SIRT2 and SIRT6 produced a metabolic state that inhibited the development of obesity, thereby reducing the occurrence of obesity. Therefore, the strategy of developing SIRT activators/inhibitors has important clinical significance to prevent obesity and control the occurrence and development of obesity and related diseases.
Other metabolic disorders
SIRT proteins are correlated with the occurrence and development of other metabolic diseases. The expression of SIRT1 and SIRT6 is downregulated in lipid metabolism-related diseases,869,870 and the expression of SIRT1 is downregulated in metabolic syndrome,178 which exerts an adverse effect on metabolic health.
SIRT1 and SIRT6 exert a crucial effect on lipid metabolism and are involved in the improvement of hepatic steatosis and hypercholesterolemia by inhibiting inflammation and promoting histone deacetylation.869,870 For instance, modest overexpression of SIRT1 shows lower lipid-induced inflammation and almost entirely protects from hepatic steatosis by induction of antioxidant proteins MnSOD and Nrf1, possibly through stimulation of PGC-1α and lower activation of proinflammatory cytokines such as TNF-α and IL-6 through downregulation of NF-κB activity.869 SIRT6 overexpression improves hypercholesterolemia in diet-induced or genetically obese mice, and the underlying biological mechanism might be due to the recruitment of SIRT6 by FoxO3 to the SREBP2 gene promoter where SIRT6 deacetylates histone H3 at lysine 9 and 56, thereby promoting a repressive chromatin state.870
Moreover, SIRT1 could confront metabolic syndrome by inhibiting inflammation. Mechanistically, post-transcriptional stabilization of SIRT1 by HuR repressed inflammation and hyperglycemia and induced E-selectin release and endothelial cell activation to counter metabolic syndrome.817 These findings show the protective roles of SIRT1 and SIRT6 in the development of various metabolic disorders. Although limited studies have been conducted on this topic, the modulation of SIRT proteins is thought to play a crucial role in the development and progression of metabolic disorders and is expected to be a therapeutic strategy of metabolic disorders.
Conclusion
In this section, we have reviewed the role of different SIRT proteins in diverse endocrine system diseases, and current studies are mainly focused on SIRT1-3 and SIRT6. Generally, SIRTs play protective roles in the occurrence and progression of a variety of endocrine system diseases. Of note, SIRT1 and SIRT2 exert a dual effect on the progression of DM, while SIRT6 overexpression exacerbates diet-induced obesity. Therefore, clarifying the specific mechanism of SIRT1 and SIRT2 in DM or revealing the mechanisms underlying their different effects might be of great significance for the clinical treatment of DM. Overall, SIRT proteins are promising therapeutic targets, and the pharmacological modulation of SIRTs could be used to prevent and treat endocrine system diseases.
SIRTs and urogenital system diseases
Urogenital system diseases include both urinary system diseases and genital system diseases which can contribute to the loss of some physiological functions, including reabsorption of nutrients, regulation of the balance of electrolytes and fluid, maintenance of acid–base homeostasis, and sexual reproduction.871,872 Thus, urogenital system diseases impose a serious economic and health burden on human development. Increasing evidence suggests that SIRT protein family activity and expression are associated with the occurrence and progression of various urogenital system diseases.873,874,875,876 Kidney disease is the most common urinary system disease, and can be divided into acute kidney disease and chronic kidney disease (CKD) according to the disease state.877 Therefore, in this section, we focus on the associations between the SIRT protein family and AKI, CKD, and genital system diseases (Fig. 15).

The roles of SIRTs in genitourinary system. SIRT protein family is involved in common of urogenital system including acute kidney disease, CKD (such as kidney fibrosis, kidney stone, aging-induced kidney injury, and vascular calcification in kidney), and genital system disease (mainly including erectile function, reproductive damage, male infertility, PCOS and endometriosis). SIRT1 play a protective effect in aforementioned disease. Moreover, the positive effects of SIRT3 and SIRT6 have been demonstrated in acute kidney disease, kidney fibrosis, vascular calcification in kidney, and male infertility. However, SIRT3 also play a protective role in kidney stone and PCOS, and SIRT6 is protectively associated aging-induced kidney injury. Besides, SIRT4-5 contribute to the remission of male infertility. Additionally, SIRT2 and SIRT7 can aggravate the occurrence of acute kidney disease, and SIRT2 also can aggravate the occurrence of kidney fibrosis. https://biorender.com
Acute kidney injury (AKI)
AKI is defined by a sudden loss of excretory function, in which slow deterioration of kidney function or persistent kidney dysfunction is associated with an irreversible loss of kidney cells and nephrons, which could lead to CKD.878,879 AKI mostly occurs as a complication of a single disease with a pooled incidence and mortality rate of 21%, respectively, and the incidence of AKI in intensive care units has increased in world regions over the past decades due to aging populations.880,881,882 Therefore, it is important to determine the molecular biological mechanisms of AKI.
Growing evidence has suggested that mitochondrial dysfunction is a major contributor to AKI.151,254,883,884,885 SIRT3-5 proteins, which are expressed in mitochondria, seem to play an important protective role in AKI.886 Among these, the protective role of SIRT3 has been reported to be related to improving mitochondrial function and ultimately improving apoptosis and eliminating ROS.151,254,883,884,885 For example, a sepsis-induced AKI model was constructed in wild-type and SIRT3 systematic KO mice. The results suggest that SIRT3 deficiency exacerbated histopathological and mitochondrial damage to the proximal tubules of the kidney. In addition, systematic KO of SIRT3 resulted in a significant increase in the apoptosis of kidney tubular epithelial cells, increased mRNA levels of Bax and Caspase-3, and decreased mRNA levels of Bcl-2.254 A previous study also demonstrated similar results, as SIRT3 deletion aggravated fatty acid oxidation dysfunction, resulting in increased apoptosis of kidney tissues and aggravated renal injury. Also, the activation of SIRT3 by honokiol increased ATP production, and reduced ROS and lipid peroxidation by improving mitochondrial function.151 Moreover, the overexpression of SIRT3 improved kidney function, modulated oxidative injury, repressed inflammatory damage, and reduced tubular epithelial cell apoptosis. SIRT3 overexpression attenuated ischemia-reperfusion-induced mitochondrial damage in renal tubular epithelial cells, as evidenced by decreased ROS production, increased antioxidant-sustained mitochondrial membrane potential, and inactivated mitochondria-initiated death signaling.885
Compared with the effects of SIRT3, although several studies have suggested the protective effect of SIRT1,887,888,889 the potential molecular mechanism regarding SIRT1 was inconsistent. More studies focused on different signaling pathways related to SIRT1 (such as the JNK signaling pathway and SIRT1/p53 up-regulated modulator of apoptosis/FoxO3a) rather than specific pathways. For example, a study suggested that, in vitro, SIRT1 attenuated the stress response by modulating the JNK signaling pathway, probably via deacetylation of the JNK phosphatase, DUSP16 of AKI.887 A previous study found that SIRT1/p53 up-regulated modulator of apoptosis/FoxO3a deacetylation by depleting miR-183-3p could improve renal tubulointerstitial fibrosis after AKI.873 Furthermore, kidney ischemia/reperfusion injury, which is a major cause of AKI, is associated with decreased AMPK phosphorylation and a five-fold increase in kidney SIRT1 expression. Activators of kidney AMPK might thus represent a novel therapeutic approach in patients susceptible to AKI.889 Moreover, the protection of NAD + in AKI is associated with SIRT1 expression and acts in a SIRT1-dependent manner. The NAD + /SIRT1/glycogen synthase kinase-3β/Nrf2 axis is an important mechanism that could protect against AKI and might be a potential therapeutic target in the treatment of AKI.890
There have been limited studies carried out on the associations between SIRT2, SIRT5-7 and AKI. Acetylation of MAPK phosphatase-1 was significantly increased in SIRT2-knockdown cells and decreased in SIRT2-overexpressed cells after cisplatin stimulation. SIRT2 systematic KO mice and SIRT2 transgenic mice showed amelioration and aggravation of renal injury, apoptosis, necroptosis, and inflammation induced by cisplatin.891 In addition, overexpression of SIRT5 and SIRT6 can repair kidney damage. For example, SIRT5 regulates the balance of mitochondrial versus peroxisomal fatty acid oxidation in proximal tubular epithelial cells to protect against AKI,892 and SIRT6 overexpression inhibited apoptosis induced by LPS and promoted autophagy in HK-2 cells.312 Previous studies found that SIRT7 deficient mice were protected against AKI, suggesting that this HDAC promotes tubular damage and kidney inflammation.893
In conclusion, the SIRT protein family could play an important role in AKI by regulating multiple cellular and physiologic processes including, apoptosis, oxidative stress, and mitochondrial function. Therefore, exploring treatment strategies using the SIRT protein family in AKI is a promising area.
Chronic kidney disease
CKD is characterized by progressive kidney dysfunction of at least three months duration, it affects about 10% of adults worldwide, and is ranked fourteenth in the list of leading causes of death.894,895,896 According to the World Health Organization estimates, 864,226 deaths (or 1.5% of deaths worldwide) were attributable to CKD in 2012.896 CKD arises from many heterogeneous disease pathways that alter the function and structure of the kidney irreversibly, over months or years. Diabetes and hypertension are the main causes of CKD in all high-income and middle-income countries, and many low-income countries.896 We describe details of associations between the SIRT protein family and both diabetic nephropathy and hypertensive nephropathy in the sections on endocrine system disease and cardiovascular system disease, respectively. In this section, we introduce the effects of the SIRT protein family on other types of CKD, including kidney fibrosis, kidney stones, aging-induced kidney injury, and vascular calcification (VC) in the kidney.
-
(1)
Kidney fibrosis
Regardless of the initial cause of disease, kidney fibrosis is the final common pathway in the evolution of virtually all types of CKD, which could contribute to loss of kidney functions (such as filtering and a reabsorbing).874,897,898 Thus, kidney fibrosis remains an important clinical problem in both developed countries and developing nations.899 In 2001, more than 400,000 patients were receiving treatment for chronic kidney failure in the United States, and the cost of treating this problem was approximately $22.8 billion.900 Therefore, we discuss the functions and molecular mechanisms of the SIRT protein family in kidney fibrosis to prevent and reduce the disease burden.
Among the mechanisms responsible for kidney fibrogenesis, the TGF-β signaling pathway is known to play a pivotal role in kidney tubulointerstitial fibrosis, which stimulates autocrine and paracrine released connective tissue growth factor (CTGF).901,902 Previous studies reported the effects of the SIRT protein family on TGF-β signaling pathway in kidney fibrosis.903,904,905,906,907 For example, overexpression of SIRT1 abolished TGF-β1-induced cell apoptosis and fibrosis, and suppressed CTGF expression via stimulation by TGF-β1 in mouse kidneys with unilateral ureteral obstruction (UUO).903 Similarly, a previous study also investigated the role of the SIRT1 activator, SRT1720, in UUO-induced tubulointerstitial fibrosis. The administration of SRT1720 increased SIRT1 levels and partially attenuated UUO-induced kidney fibrosis and apoptosis, and inhibited the levels of TGF-β1/CTGF.904 Moreover, genetic knockdown and chemical inhibition of SIRT2 attenuated TGF-β1-induced fibroblast activation and mouse double minute 2 protein expression.905 Furthermore, SIRT3 KO mice were susceptible to hyper-acetylated mitochondrial proteins and to severe kidney fibrosis. Pyruvate dehydrogenase E1α, which is the primary link between glycolysis and the tricarboxylic acid cycle, is hyper-acetylated at lysine 385 in tubular epithelial cells after TGF-β1 stimulation and is regulated by SIRT3.906 With regard to SIRT6, a study investigated the effect of proximal tubule-specific SIRT6 KO on UUO-induced kidney tubulointerstitial inflammation and fibrosis which suggested that the SIRT6 activator MDL-800 mitigated UUO-induced kidney tubulointerstitial inflammation and fibrosis. In an in vitro experiment, MDL-800 decreased the TGF-β1-induced activation of myofibroblasts and ECM production by regulating SIRT6-dependent β-catenin acetylation and the TGF-β1/Smad signaling pathway.907 The identification of strategies to prevent and/or treat fibrotic CKD is a daunting challenge, and no treatment is specifically targeted at kidney fibrosis.908 The effects of the SIRT protein family on the TGF-β signaling pathway may identify new targets for therapeutic intervention in kidney fibrosis.
In addition, apart from the TGF-β signaling pathway, other molecular mechanisms of kidney fibrosis regarding the effects of the SIRT protein family were also investigated. For example, SIRT1 attenuated kidney fibrosis by repressing HIF-2α;813 Endothelial SIRT1 deficiency induced nephrosclerosis through downregulation of matrix metalloproteinase-14, and restoration of matrix metalloproteinase-14 expression in SIRT1-depeleted mice improved the angiogenic and matrilytic functions of the endothelium, prevented kidney dysfunction, and attenuated nephrosclerosis;909 SIRT1 inhibited Ang II type 1 receptor and NF-κB expression in kidney fibroblasts and these mechanisms might play roles in alleviating UUO-induced damage.910
It is worth noting that downregulation of SIRT1 and SIRT2 might inhibit kidney interstitial fibroblast activation and attenuate kidney interstitial fibrosis in obstructive nephropathy. SIRT1/2 activity may contribute to kidney fibroblast activation and proliferation as well as kidney fibrogenesis through activation of epidermal growth factor receptor and platelet-derived growth factor receptor-β signaling. Blocking SIRT1/2 activation might have therapeutic potential for the treatment of CKD.911
Collectively, most studies showed that SIRT1-3 and SIRT6 were protective in the development of kidney fibrosis. However, one study showed that the downregulation of SIRT1 and SIRT2 contributed to improving this disease. The specific mechanism of these different effects of SIRT1 and SIRT2 on kidney fibrosis requires further clarification by more intensive studies.
-
(2)
Other chronic kidney injuries
With regard to other chronic kidney injuries, a previous study mainly focused on the effects of the SIRT protein family on kidney stones, aging-induced kidney injury, and VC in the kidney.
Limited studies have indicated that kidney stones showed downregulated expression of SIRT3 and SIRT1. Human peripheral blood monocytes from patients with kidney stones showed decreased SIRT3 expression, but increased FoxO1 acetylation compared with the normal controls,51 and the protective effect of SIRT3 could be mediated by activation of the nuclear factor erythroid 2-related factor/heme oxygenase-1 pathway.912 A previous study suggested that suppressing SIRT1 expression promoted calcium oxalate monohydrate-induced crystal-cell adhesion and exacerbated cell injury.913
Evidence of the effect SIRT1 and SIRT6 on aging-induced kidney injury is limited. SIRT1-induced deacetylation of HIF-1α might have protective effects against tubulointerstitial damage in aged kidney.914 A previous study found that reduction of podocyte SIRT1 led to aggravated aging-induced glomerulosclerosis and albuminuria. At the molecular level, knockdown of SIRT1 in podocytes was associated with reduced activation of the transcription factors PGC-1α/PPARγ, FoxO3, FoxO4, and p65 NF-κB, through SIRT1-mediated deacetylation.915 Moreover, SIRT6-deficient mice exhibited kidney hypertrophy with glomerular enlargement and proteinuria. In vitro, knockdown of SIRT6 in cultured primary murine podocytes induced shape changes with loss of process formation and cell apoptosis.916
VC is common in CKD and contributes to CVD. At the molecular level, soluble epoxide hydrolase interacted with SIRT3, which might destabilize SIRT3 and accelerate the degradation of SIRT3. Deletion of soluble epoxide hydrolase might preserve the expression of SIRT3, and thus maintain mitochondrial ATP synthesis and morphology, significantly suppressing calcification of VSMCs.875 In addition, SIRT6 is markedly downregulated in patients with CKD and VC. At the molecular level, SIRT6 suppressed the osteogenic transdifferentiation of VSMCs via regulation of runt-related transcription factor 2.917 A previous study also indicated that bone marrow mesenchymal stem cell-derived exosomes inhibited high phosphate-induced aortic calcification and ameliorated renal function via the SIRT6-high mobility group box 1 deacetylation pathway.918
The pathophysiology of CKD is complex and the etiologies diverse. There are still various unexplored associations between the SIRT protein family (such as SIRT2, SIRT4, SIRT5, and SIRT7) and different CKDs. Thus, these associations require more in-depth investigation. It could be implied that SIRT1 is an important survival factor and a potential therapeutic target in CKD.
Genital system diseases
To date, only a few studies have explored the SIRT protein family and genital system diseases. Two studies have emphasized the protective effect of resveratrol in erectile function and reproductive damage (caused by nicotine), which could positively modulate SIRT1.919,920 Moreover, through an improved level of SIRT1, polyunsaturated fatty acids supplementation attenuates oxidative damage in testis by reinforcing the antioxidant defense system.876 In addition, to investigate SIRT1 regarding adjuvant strategies in the treatment of male infertility, dysregulation of SIRT1 and mitochondrial SIRT (SIRT3-5) genes were associated with human male infertility.921,922 In female genital system disease, a limited number of studies have paid attention to polycystic ovary syndrome (PCOS) and endometriosis. PCOS patients had higher SIRT1 levels than healthy controls923 and involvement of the SIRT1/AMPK axis in autophagy activation in PCOS.924 SIRT3 deficiency in granulosa cells of PCOS patients might potentially induce impaired oocytes in PCOS.925 Furthermore, previous studies only explored the association between SIRT1 and endometriosis. For example, one study suggested that SIRT1 was over-expressed in eutopic endometrium of women with endometriosis and likely participates in the pathogenesis of endometriosis.926 Another two studies demonstrated that resveratrol has therapeutic potential601 and miRNA-34a927 might provide a potential biomarker for endometriosis therapeutics.
In summary, current studies on the SIRT protein family and genital system disease are still in their infancy, and more research is needed in the future to explore these associations.
Conclusion
In conclusion, current studies have successfully highlighted the critical role of SIRT1 in urogenital and genital system diseases. However, at the molecular level, previous studies did not concentrate on certain pathways; thus, the mechanism of the effect of SIRT1 was inconsistent between different studies. Furthermore, although other SIRT proteins have not been as extensively studied as SIRT1, the important effect of these proteins in urogenital disease should not be ignored. The association between the SIRT protein family and urogenital disease could still be a new direction for further research.
SIRTs and motor system diseases
Diseases of the motor system focus on abnormal bone metabolism and diseases resulting from skeletal muscle dysfunction, mainly including OA, osteoporosis, intervertebral disc degeneration (IDD) and skeletal muscle atrophy. In addition to the body’s own self-regulatory mechanisms, exogenous factors such as aging, mechanical stimulation, estrogen, and obesity are involved in the process of bone metabolism and skeletal muscle function.928 SIRTs are considered promising regulatory genes for bone and skeletal muscle metabolism, involved in processes such as differentiation of bone marrow MSCs, osteoblast viability, skeletal muscle fiber type conversion, endoplasmic reticulum stress and atrophy.929 Therefore, in this section, we focus on the functions of SIRTs in diseases of the locomotor system and the regulatory roles.
OA
OA is the most common joint disease, and is a type of degenerative disease.930 Chondrocyte senescence and apoptosis, ECM degradation with synovial inflammation, and dysfunction of the subchondral bone are the core pathological changes in OA.931 SIRTs may have different roles in influencing chondrocyte activity. Notably, SIRT1 is the best studied SIRT in OA, and negatively regulates important cellular biological processes impairing chondrocytes activity, including apoptosis and ECM degradation. For example, SIRT1 may reduce apoptosis and ECM degradation in OA chondrocytes via the Wnt/β-catenin signaling pathway to counteract aging-induced OA.932 Furthermore, SIRT1 is regulated by the circ0001103/miR-375 axis, which attenuates IL-1β-induced chondrocyte apoptosis and ECM degradation.933 In addition, SIRT1 can influence mitochondrial function, defense oxidative stress and inhibit senescence of chondrocytes. SIRT1 can reverse homocysteine-induced deleterious changes in chondrocytes that lead to OA via the SIRT1/PGC-1α/PPAR-γ cascade, including mitochondrial dysfunction and accumulation of oxidative stress.934,935 SIRT1 also improves the resistance of cartilage to oxidative stress by inhibiting epidermal growth factor receptor ubiquitination, thereby alleviating OA.936 Moreover, although there are many mechanisms affecting cellular senescence, SIRT1 can inhibit chondrocyte senescence and OA by negatively regulating the Wnt/β-catenin signaling pathway.937
SIRT2, SIRT3, and SIRT6 are also involved in the development of OA. Similar to SIRT1, SIRT2 and SIRT6 also play protective roles in disease progression. For instance, SIRT2 protects against the progression of OA by inhibiting degradation of the ECM by preventing the acetylation of p65.89 Moreover, SIRT6 can inhibit the senescence of chondrocytes by negatively regulating the NF-κB-mediated inflammatory response.81 However, SIRT3 was shown to have dual roles in disease progression. In detail, SIRT3 inhibited chondrocyte degeneration by maintaining mitochondrial homeostasis.938 SIRT3 alleviated OA by improving the resistance of cartilage to oxidative stress. Mechanistically, SIRT3 restored acetylation-dependent SOD2 activity in human OA cartilage.939 On the contrary, SIRT3 overexpression promoted OA chondrocyte apoptosis and reduced cell proliferation, finally resulting in OA progression.940 The molecular mechanism of the opposite effect of SIRT3 on OA is unclear, and is worth further study.
Overall, SIRT1-3, and SIRT6 have different effects on the viability and function of chondrocytes, and they play important roles in the occurrence and development of OA. Given that their complex mechanisms are not fully understood, more in-depth studies are needed on the interaction of SIRTs with cartilage, synovium, bones and joints. In particular, SIRT1 has an important role in the development of OA and is expected to be a therapeutic target for the treatment of OA in the future.
Osteoporosis
Osteoporosis is defined as a systemic skeletal disease characterized by low bone mass and deterioration of bone tissue microarchitecture, which increases bone fragility and fracture susceptibility.941 Bone exhibits continuous self-renewal, with replacement of old bone by new bone through osteoclast-mediated bone resorption and osteoblast-mediated bone formation, thereby repairing microstructural damage to bone, a process called bone reconstruction.942 In dynamic bone reconstruction, SIRTs not only promote osteoblast differentiation and inhibit osteoclast differentiation, but also inhibit osteoclast bone resorption, ensuring a positive balance between bone metabolism and increased bone mass through multiple pathways.943 SIRT1 KO mice have a low bone mass phenotype.944 Therefore, due to the confluence of cellular aging, energy metabolism and bone metabolism, SIRTs are of great significance in the study of osteoporosis pathogenesis.
Oxidative stress and aging are important factors that regulate the osteogenic differentiation process, and these can also be regulated by SIRT1 and thus are anti-osteoporosis.945 For instance, SIRT1 overexpression increased osteoblast osteogenesis through FoxO3a deacetylation and oxidative stress inhibition.946 Overexpression of SIRT1 might also reduce oxidative stress through the FoxO1 and β-catenin signaling pathways.222 In addition, SIRT1 plays a protective role in osteoporosis by regulating bone metabolism. For example, SIRT1 is regulated by the HIF-1α signaling pathway, which deacetylates sclerostin and activates the Wnt/β-catenin signaling pathway, leading to increased bone anabolism in osteoporosis.947 In postmenopausal osteoporosis, SIRT6 has been found to inhibit age-related bone loss by stabilizing ER alpha in preosteoblastic cells.948 Moreover, SIRTs not only regulate oxidative stress and aging signaling pathways to resist osteoporosis, but can also be activated by small molecule drugs such as resveratrol to affect bone metabolism. In osteoporosis, SIRT1 is activated by resveratrol and subsequently restores the levels of serum markers alkaline phosphatase and osteocalcin by inhibiting the NF-κB signaling pathway, which has a protective effect against osteoporosis.949,950
In brief, SIRT1 and SIRT6 can inhibit the development and progression of osteoporosis by resisting oxidative stress, aging and regulating bone metabolism. However, more in-depth and detailed studies are still needed to elucidate the regulatory mechanisms of other SIRTs on osteoporosis and to explore their clinical application value in the future.
IDD
IDD is an important pathological basis for degenerative spinal diseases, which manifests as increased degradation of the central nucleus pulposus matrix, thickening of the peripheral annulus fibrosus, and thinning and calcification of the cartilage endplates.951 SIRTs can inhibit the pathological process of IDD by inhibiting inflammation, cellular senescence, oxidative stress, and maintaining mitochondrial function. mRNA and protein expression levels of SIRT1 in degenerative nucleus pulposus tissues of intervertebral discs were reduced compared with control tissues and decreased with increasing disease severity.952 This suggests that there might be a protective effect of SIRT1 on IDD progression. Mechanistically, SIRT1 can resist the inflammatory response during IDD by inhibiting the transcriptional activity of NF-κB.58 Moreover, SIRT1 might inhibit disc degeneration by suppressing phosphorylation of activin 1 subunits c-Fos and c-Jun.953 It seems that SIRT1 might become a biological target for the treatment of IDD.
Furthermore, SIRT2, SIRT3, and SIRT6 have protective roles in the development of IDD. For example, SIRT2 reversed the action of IL-1β by inhibiting the p53/p21 pathway, inhibited oxidative stress and cellular senescence, and thus prevented the degradation of nuclear myeloid cells in IDD.954 It was also found that SIRT3 maintains nucleus pulposus cell homeostasis to prevent IDD mainly by regulating mitochondrial oxidative stress levels.955,956 In addition, SIRT6 mainly inhibits the inflammatory response and cellular senescence during IDD by inhibiting the transcriptional activity of NF-κB pathways.957
In conclusion, SIRTs, including SIRT1-3, and SIRT6, are involved in negative regulation of disease progression in IDD; however, the number of studies are limited. With the continuous discovery of acting molecules and the identification of deep molecular mechanisms, SIRTs are expected to become important targets in the prevention and treatment of IDD.
Skeletal muscle atrophy
Skeletal muscle atrophy, which is the accelerated degradation of skeletal muscle proteins, mainly involves a variety of chronic diseases, aging, and long-term muscle inactivity.958 SIRTs inhibit skeletal muscle atrophy and are associated with mechanisms such as mitochondrial dysfunction, autophagy and metabolism. For example, SIRT1 inhibited drug-induced mitochondrial dysfunction and thus alleviated skeletal muscle atrophy by activating its downstream signaling PGC-1α.959 Moreover, SIRT2 effectively inhibited the autophagic flux, thus maintaining protein metabolism homeostasis in skeletal muscle.960 SIRT3-mediated cellular metabolism has an inhibitory effect in skeletal muscle atrophy. Ang II caused skeletal muscle atrophy, and SIRT3 deficiency enhanced Ang II-induced fiber type transformation and mitochondrial metabolic reprogramming, exacerbating skeletal muscle atrophy.961 The incidence of skeletal muscle atrophy and sarcopenia is increasing year by year.962 As the research on SIRTs in skeletal muscle physiological and pathological processes continues to advance, SIRTs could be used as targets for the prevention and treatment of skeletal muscle-related diseases.
Conclusion
SIRTs are key nodes in several degenerative diseases of aging, including OA, osteoporosis, and IDD. SIRTs play a key role in bone homeostasis and can maintain the balance between bone formation and resorption by regulating the ratio of osteoblasts to osteoclasts. SIRTs enhance the viability of osteoblasts under unfavorable conditions by resisting senescence, inhibiting apoptosis and promoting autophagy. Therefore, given the critical role of the SIRTs pathway in bone homeostasis, it is likely to be a potential therapeutic target, laying a solid foundation for further studies in the future.
SIRTs and aging
Aging is associated with impaired adaptive and homeostatic mechanisms, leading to susceptibility to environmental or internal stresses with degeneration of multiple organ systems.963 Extensive studies have clearly revealed that SIRTs are important regulators of aging, which involves several biological processes, such as cellular senescence, metabolic regulation, genome fidelity, nutrient sensing, and circadian rhythms.964
It has long been known that mammalian aging is associated with cellular senescence, and SIRTs could play key roles in antagonism of aging and cellular senescence.965 For example, the activation of SIRT1 by La Ribonucleoprotein 7, a 7SK RNA binding protein, could ameliorate cellular senescence and aging through dampening p53 and NF-κB (p65) transcriptional activity.966 Besides, SIRT6 inhibition shortened human VSMC lifespan and induced senescence, associated with telomeric histone H3K9 hyperacetylation and p53 binding protein 1 binding, while SIRT6 overexpression preserved telomere integrity, delayed cellular senescence.201 Additionally, SIRTs could exert regulatory effects on aging by regulating cellular homeostasis in fundamental pathways such as genomic stability, nutrient sensing, and protein homeostasis.964 For instance, SIRT1 redistribution on chromatin induced by DNA damage, could promote DNA repair, enhance genomic stability, and suppress age-dependent transcriptional changes.967 SIRT3 deficiency resulted in the detachment of genomic lamina-associated domains from the nuclear lamina, increased chromatin accessibility and aberrant repetitive sequence transcription, and thereby leading to senescence phenotypes of human mesenchymal stem cells.968 Moreover, SIRT6 promoted resistance to DNA damage, suppressed genomic instability in mouse cells via deacetylation of Polβ, a base excision repair factor, and prevent the development of several progeroid pathologies.35 In addition to genomic stability, nutrient sensing is also known to be an important factor affecting aging.969 Evidence suggested that SIRT1 could control the gluconeogenic/glycolytic pathways in liver in response to fasting signals and modulate aging.970 The SIRTs-related roles in aging are also associated with the regulation of protein homeostasis. The related studies showed that the SIRT-activating compounds-resveratrol could attenuate copper-induced senescence by improving cellular proteostasis.971 Furthermore, the involvement of SIRTs in aging could be mediated by their roles in circadian rhythms.972 For instance, SIRT1 constituted a reciprocal negative regulation loop with the periodic gene, Period 2, and thus modulated circadian-clock maintenance and aging gene expression.973 SIRT6 deacetylated Period 2 and thus regulated circadian rhythms which were associated with aging and age-related diseases.974
Therefore, SIRTs have very important regulatory roles in aging through participating in diverse biological processes. Moreover, growing evidence has shown that SIRTs might be attractive anti-aging molecules involved in improving health, although it is still under debate and has not been fully defined.975 From this perspective, further studies are needed to uncover the exact roles and mechanisms of SIRTs in the aging process.
SIRT modulators
In view of the dual involvement of SIRTs in many biological processes, many laboratories have developed both SIRT inhibitors and activators, which might act as tools for studying SIRT function and potentially as treatments for different conditions and diseases. Generally, activators have better therapeutic potential than inhibitors. This might partly be attributed to higher target specificity in the enzyme family and fewer side effects.976 However, compared to inhibitors, the number of activators is small. The following sections describe in detail the most relevant SIRT inhibitors and activators identified so far.
SIRT activators
SIRT activators are mainly classified into natural polyphenols and nonrelated synthetic SIRT activators (Supplementary Table 1). The structures of these activators are shown in Fig. 16. Among them, resveratrol, a polyphenol commonly found in grapes and red wine, was the first SIRT1 activator identified in 2003.977 Resveratrol as an allosteric activator of SIRT1 can increase its activity by 50% (EC1.5) at 46.2 μM and extend the lifespan of many organisms, ranging from yeast to mammals.977 Evidence showed that resveratrol supplementation could help to reduce fasting glucose, insulin, and insulin resistance, increase high-density lipoprotein-cholesterol levels and total antioxidant capacity, and upregulate PPAR-γ and SIRT1 in the peripheral blood mononuclear cells of type 2 DM patients with coronary heart disease.978 Several other polyphenols, structurally related to resveratrol, were also found to activate SIRTs, including the chalcones butein and isoliquiritigenin, the flavones fisetin and quercetin, and the stilbene piceatannol.977 The compounds fistein and butein increased lifespan length in the yeast Saccharomyces cerevisiae by 33% and 5%, respectively,977 whereas quercetin increased the lifespan of the nematode Caenorhabditis elegans grown in 200 mM by approximately 20%.979 In addition, butein was reported to attenuate sepsis-induced brain injury through alleviation of cerebral inflammation, oxidative stress and apoptosis by SIRT1 signaling activation.980 Although piceatannol and isoliquiritigenin did not produce significant effects on lifespan, they were believed to activate prolonged survival.981 Isoliquiritigenin repressed the proliferation, migration, and invasion of NSCLC cells in vitro.825 Furthermore, in experimental diabetic neuropathy, isoliquiritigenin could reduce oxidative damage and alleviate mitochondrial impairment by SIRT1 activation.982

Structures of most relevant SIRT activators
As natural compounds did not show high activity on SIRT1, more potent compounds with a greater substrate-binding affinity for SIRT1 have been synthesized. SRT compounds, such as SRT1460 (EC1.5 = 2.9 μM),983 SRT1720 (EC1.5 = 0.16 μM),983 SRT2104 (EC1.5 = 0.43 μM),984 SRT2183 (EC1.5 = 0.36 μM),983 and SRT3025 (EC1.5 < 1 μM),985,986 were identified in 2007 as selective SIRT1 activators, which were more potent than resveratrol.983 They played important roles in the treatment of physiological and pathological conditions.983,987,988 For example, employment of SRT compounds in diet-induced and genetically obese mice improved insulin sensitivity and glucose tolerance, stimulated mitochondrial biogenesis, and regulated lipid metabolism, thus had beneficial effects on weight loss.983 Due to these promising activities, some SRT compounds have been evaluated in various clinical trials for the treatment of different conditions and diseases.989 SRT2104 was the most common intervention for healthy participants and type 2 diabetes patients in randomized controlled trials (RCTs).8,990,991
The development of SIRT6 activators was initially stimulated by early studies showing that free fatty acids containing 14–18 carbons acted as weak SIRT6 activators.992 UBCS039 is a pyrrolo[1,2-a]quinoxaline reported as the first synthetic activator of SIRT6 deacetylase activity (EC50 = 38 μM).993 Evidence shows that UBCS039 induced a time-dependent activation of autophagy and induced deacetylation of SIRT6-targeted histone in several human tumor cell lines.994 The bis benzenesulfonamide-based prodrug MDL-800 is also reported to be a potent and selective SIRT6 activator with an EC50 value of 10.3 μM.995 MDL-800 decreased both H3K9ac and H3K56ac at a concentration of 10 µM and showed a dose-dependent effect in Bel7405, PLC/PRF/5, and Bel7402 cell lines at 24 h and 48 h.995 Additionally, MDL-800 decreased TGF-β1-induced activation of myofibroblast and ECM production by regulating SIRT6-dependent β-catenin acetylation and the TGF-β1/Smad signaling pathway.907
SIRT inhibitors
Compared to SIRT activators, more studies have been conducted on SIRT inhibitors. A range of potent inhibitors were identified through a variety of development strategies, such as mechanism/structure based, or simply by virtual screening.976 Most studies focused on the inhibition of human SIRT1 and/or SIRT2. These available inhibitors are divided into several structural groups based on their mechanism of action and structural features (Supplementary Table 2). Figure 17 shows the structures of these inhibitors.

Structures of most relevant SIRT inhibitors
Nicotinamide and its analogs
Nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) are important precursors of NAD, in that NAD biosynthesis involves the conversion of nicotinamide to NMN and subsequent conversion of NMN to NAD.996 And the production of NMN is the key rate-limiting factor in mammalian NAD biosynthesis.996 Thus, NR and NMN might affect SIRT activity mainly by affecting the synthesis of NAD. Nicotinamide and its analog, AK-7, are reported to be SIRT inhibitors. Of these, nicotinamide is the endogenous inhibitor of SIRTs, which is formed from NAD + during catalysis. Nicotinamide inhibits SIRT1 and SIRT2 with IC50 values of approximately 120 μM and 100 μM for SIRT1 and SIRT2, respectively.997,998 Nicotinamide can inhibit the growth and viability of human prostate cancer cells through inhibition of SIRT1.999 In addition, it blocks proliferation and induces apoptosis of chronic lymphocytic leukemia cells.1000 AK-7, a benzamide (a nicotinamide mimic)-containing compound, shows selective SIRT2 inhibition.1001 An in vivo study showed that AK-7 improved behavioral and neuropathological phenotypes, prolonged survival, and improved HD neuropathology in R6/2 HD mice.1002 Furthermore, AK-7 limited the ability of adoptively transferred antigen-specific CD4 + T cells to cause autoimmune encephalomyelitis in mice and limited disease in lupus-prone MRL/lpr mice.1003 This might support the development of SIRT2 inhibitors as potential therapeutics for these diseases.
β-naphthol-containing inhibitors
β-naphthol acts as a key group for several SIRT inhibitors, including splitomicin, sirtinol, salermide, HR-73, and cambinol. Both sirtinol and splitomicin were identified through cell-based screens of more than 1000 compounds in yeast.1004,1005 Splitomicin inhibits human SIRT1 and SIRT2 with an IC50 value of 96 μM and 113 μM in vitro, respectively.9 Treatment with this molecule reduces deacetylase activity, enhances tissue factor mRNA expression in stimulated endothelial cells, and enhances NF-kB/p65 nuclear translocation.1006 In addition, evidence showed that splitomicin could reversed both ischemic preconditioning-mediated lysine deacetylation and ischemic preconditioning-induced cardioprotection.1007 Sirtinol (SIRT1 IC50 = 131 μM;1008 SIRT2 IC50 = 38-58 μM1005,1008) has been shown to induce apoptotic and autophagic cell death in MCF-7 human breast cancer cells.1009 Sirtinol induced senescence-like growth arrest in human LC H1299 cells and induced senescence-like growth arrest as well as apoptotic and autophagic cell death breast cancer MCF-7 cells.1009,1010 Structure-activity relationship studies on sirtinol resulted in improved analogs such as salermide, which has a greater inhibitory effect on SIRT1 and SIRT2 than sirtinol.1011 Salermide was reported to induce the reactivation of proapoptotic genes that were aberrantly repressed in cancer cells by SIRT1-mediated H4K16 deacetylation.1011 Also, salermide had potent antiproliferative on human leukemia MOLT4 cell lines, human breast MDA-MB-231, and colon RKO cancer cell lines and played an inhibitory role in colorectal carcinoma cancer stem cells.1012 HR-73 was identified as a splitomicin derivative, which inhibits the activity of SIRT1 in vitro with an IC50 lower than 5 μM.1013 It can decrease human immunodeficiency virus transcription through Tat acetylation.1013 Cambinol, a β-naphthol derivative, is the most promising SIRT inhibitor in this class of compounds.1014 It inhibits human SIRT1 and SIRT2 with IC50 values of 56 and 59 μM in vitro, respectively,240 and reduces the expression of poorly differentiated markers α-fetoprotein and glypican, and impairs cell migration in a dose-dependent manner.1015 It was also reported to reduce the expression of N-Myc protein and up-regulate the expression of the other SIRT1 target genes including early growth response 1, Kv channel interacting protein 4, and phospholipase C beta 1.1016
Indole derivatives
Large-scale fluorescence screening led to the emergence of pure indole Sir2 inhibitors in 2005, which are the only series of compounds with a simple indole as the scaffold identified to date.1017 These compounds include EX-527, AC-93253, inauhzin, and Ro31–8220. They act as inhibitors of SIRT1, which enhance cell survival and p53 acetylation.1018 EX-527, also called Selisistat, is the first known selective (over SIRT2/3) and cell-permeable SIRT1 (IC50 = 0.098 μM) inhibitor.1019 Evidence showed that EX-527 decreased tumor growth in endometrial and LC cells xenografted mice.469,1020 Additionally, EX-527 could decrease the viability of control HHUA cells and the survival of HEC151 cells and reduce cisplatin resistance in HEC1B cells with mutated and non-functional p53.469 AC-93253, a compound containing a modified indole ring, preferentially inhibits SIRT2 (IC50 = 6 μM)1021 and triggers the downregulation of melanoma progression markers and the inhibition of melanoma cell proliferation.1022 Another indole derivative, inauhzin, inhibits the deacetylase activity of SIRT1 with an IC50 value of 0.7-2 μM.1023 Inauhzin has potent anticancer activity and represses the growth of xenograft tumors derived from human LC H460 and colon cancer HCT116 cells harboring p53.1023 Also, inauhzin was found to induce ribosomal stress and the RPL11/RPL5- murine double minute 2 (MDM2) interaction, activating p53, and suppress cancer cell growth by dually targeting SIRT1 and inosine monophosphate dehydrogenase 2.1024 SIRTs use NAD as a co-substrate, whereas kinases use ATP as a co-substrate. Given that both NAD and ATP contain an adenosine moiety, kinase inhibitors might inhibit SIRTs. For example, a nM PKC inhibitor, Ro31–8220, shows inhibitory activity against SIRT1 and SIRT2, with IC50 values of 3.5 μM against SIRT1 and 0.8 μM against SIRT2.1025 In a human neuroblastoma cell line, Ro31–8220 was found to reduce PKC activity and the tau phosphorylation pattern.1026
SIRT-rearranging ligands (SirReals)
A family of aminothiazoles, basically known as SirReals, was found to act as a potent selective inhibitor of SIRT2. Of these, SirReal2 is a potent SIRT2 inhibitor (IC50 = 0.14 μM) with minimal effects on SIRT1 and SIRT3.1027 According to X-ray crystallography, SirReal2 exerts its potency and selectivity based on a ligand-induced structural rearrangement of the SIRT2 active site and interacts with residues in an unknown binding pocket located near the zinc-binding domain, known as the “selective pocket”.1027 The SIRT2 inhibition capability of SirReal2 has been confirmed in chondrocytes by the induction of several acetylations of H3.1028 SirReal2 was reported to increase the levels of phosphorylated Cx43 on S368 and the levels of acetylated MEK1/2, decrease the membrane localization of Cx43 between cumulus cells, and increase the Cx43 acetylation levels of cumulus-oocyte complexes.1029
Tenovins
Through phenotypic screening of 30,000 drug-like small molecules able to activate p53 and decrease tumor growth, Lain et al. discovered two compounds that were SIRT1 inhibitors: tenovin-1 and its more water soluble analog tenovin-6.1030 The poor water solubility of tenovin-1 prevents the accurate determination of an IC50 value, whereas IC50 values of tenovin-6 with better water solubility have been reported as follows: SIRT1 IC50 = 21 μM; SIRT2 IC50 = 10 μM;1030 SIRT3 IC50 = 67 μM.1030 Both compounds decrease tumor growth in vitro at one-digit micromolar concentrations, and delay tumor growth in vivo without significant general toxicity.1030 In the BL2 and ARN8 mouse xenograft model, tenovin-1 could reduce tumor growth,1030 while tenovin-6 was found to delay the growth of xenograft tumors derived from ARN8 cells.1030
Other SIRT inhibitors
Many other types of compounds have been reported as SIRT inhibitors. Some of them are worth mentioning. Suramin, a polyanionic urea derivative, was originally used as an adenosine receptor antagonist for the treatment of trypanosomiasis and has antiviral and anticancer activity.1031 It was later found to be a potent SIRT inhibitor with an IC50 of 297 nM, 1150 nM, and 22 μM for SIRT1, SIRT2, and SIRT5, respectively.1032,1033 Suramin has multiple biological effects, such as protection against disc degeneration, perturbation of mitochondrial membrane potential and ATP levels.10,1034 Aristoforin, a phloroglucinol derivative, was shown to inhibit SIRT1 (IC50 = 7 μM) and SIRT2 (IC50 = 21 μM).1035 AGK2 is a selective SIRT2 inhibitor (IC50 = 3.5 μM) identified from a concentrated compound library.1036 The design of MC2494 is inspired by AGK2, in which the 2,5-dichlorophenyl-substituted furan ring is replaced by a pyrrole bearing a 2-chlorobenzoyl moiety at the C4 position. MC2494 has been reported as a micromolar pan-SIRT inhibitor and regulated mitochondrial function in a leukemia cell line.1037 As the result of cambinol manipulation, MC2141 was identified in 2010 and was the prototype of a class of benzodeazaoxaflavins that inhibited SIRT1/2 in the low micromolar range.1038 Tripos 360702 showed SIRT2 inhibitory activity with IC50 values of 51 μM in a test, and could be considered a novel inhibitor of SIRT2.1039
Conclusion
After summarizing the activators and inhibitors of the SIRT protein family described above, we find that a substantial amount of progress has been made in past decades. However, as research on different types of SIRT modulators has been unbalanced and the clinical potential of these modulators in treating different diseases has been insufficient, there is still progress to be made. Currently, inhibitors of SIRT1/2 are relatively adequate, whereas there are no good inhibitors of SIRT3-7 to date, especially SIRT4 and SIRT7. Additionally, with regard to SIRT activators, a great deal of work has been conducted in the identification of molecules targeting SIRT1. Thus, further studies are needed to investigate activators and inhibitors of other SIRTs rather than SIRT1, which will eventually unlock the full therapeutic potential of SIRT molecules. We believe that SIRT modulators are a field worthy of research, the SIRT protein family will eventually pay off.
RCTs of SIRT proteins
We conducted an electronic search of relevant RCTs in PubMed (up to June 23, 2022) without restrictions. Additionally, relevant clinical trial registration sites were comprehensively examined, such as ClinicalTrials.gov, ISRCTN registry, EU Clinical Trials Register, and Iranian Registry of Clinical Trials. Literature retrieval was performed in duplicate by two independent reviewers. A total of 63 published RCTs were included, of which 43 studies mainly examined the effects of different interventions on SIRT protein expression in human samples, and 20 studies focused on the impact of SIRT activators (resveratrol and SRT2104) and SIRT inhibitor (nicotinamide) on physiological function in different participants. The characteristics of these studies are shown in Fig. 18 and listed in detail in Tables 1–2.

Characteristics of included randomized controlled trials by (a) regions; (b) condition of subject; (c) examination of tissue and samples; (d) years of recruitment and publication; (e) interventions
RCTs have been conducted on all continents, with Asia having the largest number of studies (n = 23), followed by Europe (n = 20) and North America (n = 12). Iran (n = 18), Italy (n = 5), and the United States (n = 12) ranked first in Asia, Europe, and North America, respectively. However, only one RCT was conducted in Oceania (Australia) and Africa (Egypt), respectively. These studies recruited from 2005 to 2019 and were published between 2012 and 2022. The peak years of study recruitment ranged from 2015 to 2016, whereas the majority of the studies were published between 2015 and 2020. Most studies examined SIRT protein expression in human samples, where blood samples (n = 40, 74.1%) were mostly used. Fat (n = 6, 11.1%) and muscle (n = 8, 14.8%) tissues were also used in several studies. Regardless of the tissue type, SIRT1 was the protein most focused on among the SIRT protein family. Some researchers evaluated SIRT3 protein expression in blood samples and adipose tissue.
Sixty-three RCTs included participants with more than 10 different diseases and conditions. Among them, the largest number of studies included participants with metabolic diseases (n = 30, 42.9%), including type 2 diabetes, obesity, and metabolic syndrome, followed by studies that recruited healthy participants (n = 17, 24.3%) such as healthy elderly participants, healthy employees, and healthy volunteers. A limited number of studies recruited participants with other diseases. For example, only three studies investigated a gynecological disease (polycystic ovarian syndrome), skin disease (systemic lupus erythematosus), and cancer (lymphoma), respectively.
With regard to the intervention/comparison in the study, about half of the studies (n = 34, 47.9%) focused on dietary interventions such as vitamin D and caloric restriction, with a few studies focusing on exercise interventions (n = 11, 15.5%) as well as drug and surgical treatments (n = 6, 8.5%). For example, supplementation of crocin or crocetin effectively improved gene expression of SIRT1 in coronary artery disease patients compared with the placebo.1040,1041 However, curcumin, administered to 67 overweight or obese patients with polycystic ovarian syndrome, led to a non-significant increase in SIRT1 expression, after 12 weeks compared to placebo.1042 With regard to exercise intervention, three RCTs in India highlighted that a yoga-based lifestyle intervention led to a significant increase in SIRT1.1043,1044,1045 On the contrary, among 70 rheumatoid arthritis patients, the mRNA expression levels of SIRT1 were not found to be statistically different in the yoga vs. non-yoga group.1046 As for medical treatment, three RCTs demonstrated that treatment with sildenafil, metformin, fenofibrate alone or in combination with pioglitazone up-regulated SIRT1 gene expression.1047,1048,1049 In addition, after Roux-en-Y gastric bypass, the expression of SIRT1 and SIRT3 increased compared to the baseline in 13 obese, non-diabetic patients.1050
Only 23.9% (n = 17) of published RCTs explored the effects of SIRT activators on physiological function. As a well-known SIRT activator,1051 resveratrol (n = 10, 14.1%) received more attention compared with SRT2104 (n = 7, 9.9%) in these studies. Several RCTs showed that resveratrol supplementation could effectively increase the expression or concentration of SIRT1.978,1052,1053,1054 Moreover, resveratrol performed important physiological functions by activating SIRT1, such as beneficial effects on neuro-inflammation and adaptive immunity.7 Similarly, SRT2014 played an important role by activating SIRT1, such as reduction in endotoxin-induced cytokine release and coagulation activation.1055 However, some intervention studies reported that resveratrol did not affect its putative molecular target.1056,1057,1058 For example, a double-blind randomized placebo-controlled proof-of-concept study conducted in the Netherlands suggested that the muscle mitochondrial biogenesis regulator SIRT1 was not improved by resveratrol.1056 A few studies suggested that significant clinical activities were not observed after supplementation with resveratrol and SRT2014.1059,1060,1061
As shown in Table 2, three RCTs (4.2%) focused on the impact of nicotinamide, a known SIRT inhibitor,976 on physiological function in different patients.1062,1063,1064 However, all three studies suggested that nicotinamide might not act through its putative molecular target. For example, findings from a long-term human clinical trial reported that NR supplementation did not affect SIRT activity in human skeletal muscle.1064 In addition, a clinical study evaluating the pharmacodynamics efficacy of nicotinamide as an inhibitor of SIRT revealed that over 12 months of nicotinamide treatment, no sustained inhibitions of SIRT activity were detected.1063 This might be attributed to the small sample size and short intervention duration in these three RCTs. Thus, further studies are needed to explore nicotinamide as a clinical therapeutic method by inhibiting SIRT activity.
Although several published RCTs have shown inconsistent findings, most studies have suggested that dietary, exercise, and drug interventions can enhance SIRT signaling, and SIRT activators played an important role in physiological functions by activating SIRTs. Given the important impact of the SIRT protein family on health and disease, the relatively small number of trials, study limitations and single study sites, further larger sample, longer intervention period, and multicenter RCTs are needed.
Conclusion
Since the discovery of the SIRT family members, the understanding of this protein family has become increasingly comprehensive and profound. The studies summarized herein provide strong evidence that SIRTs play important roles in the body. Considering that the roles of SIRTs vary in different types of biological processes and human diseases, it will be of great significance to focus attention on the mechanisms regarding the seven SIRTs under different conditions and the specific function of each SIRT. Recent advances in technology (e.g., development of omics, gene KO and knockin) may facilitate elucidation of the specific molecular mechanisms of SIRTs, providing new perspectives for the pathogenesis of human diseases and targets for treatments. Clinical trials to verify the biomarkers and therapeutic potential of SIRTs are still lacking and are warranted in the future. Thus, this review has systematically highlighted the recent advances with respect to the role of SIRTs, which may aid the design of future research, and thereby reveal the diagnostic and therapeutic potential of SIRTs.
Future directions
-
1.
To further clarify the biological regulation mechanism of SIRTs in different kinds of diseases and health conditions, and the interaction relationship between different kinds of SIRTs.
-
2.
To validate SIRTs as potential diagnostic and prognostic biomarkers for specific diseases at a large population level.
-
3.
To develop and validate more specific agonists and inhibitors of different kinds of SIRTs, and to explore and confirm their efficacy and safety in disease prevention and treatment in basic and clinical studies.
-
4.
SIRTs, such as SIRT7, which has been studied finitely, should be the focus of future research.
-
5.
Studies incorporating a multidisciplinary perspective provide a more comprehensive understanding of the roles of SIRTs.
References
North, B. J. & Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 5, 224 (2004).
Min, J., Landry, J., Sternglanz, R. & Xu, R. M. Crystal structure of a SIR2 homolog-NAD complex. Cell 105, 269–279 (2001).
Chalkiadaki, A. & Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 15, 608–624 (2015).
Kane, A. E. & Sinclair, D. A. Sirtuins and NAD(+) in the development and treatment of metabolic and cardiovascular diseases. Circ. Res. 123, 868–885 (2018).
Wang, R. H., Li, C. & Deng, C. X. Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int. J. Biol. Sci. 6, 682–690 (2010).
Yang, T., Fu, M., Pestell, R. & Sauve, A. A. SIRT1 and endocrine signaling. Trends Endocrinol. Metab. 17, 186–191 (2006).
Moussa, C. et al. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 14, 1 (2017).
Venkatasubramanian, S. et al. Cardiovascular effects of a novel SIRT1 activator, SRT2104, in otherwise healthy cigarette smokers. J. Am. Heart Assoc. 2, e000042 (2013).
Pasco, M. Y. et al. Characterization of sirtuin inhibitors in nematodes expressing a muscular dystrophy protein reveals muscle cell and behavioral protection by specific sirtinol analogues. J. Med. Chem. 53, 1407–1411 (2010).
Liu, Z. M. et al. Suramin attenuates intervertebral disc degeneration by inhibiting NF-kappaB signalling pathway. Bone Jt. Res 10, 498–513 (2021).
Outeiro, T. F. et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317, 516–519 (2007).
Rine, J., Strathern, J. N., Hicks, J. B. & Herskowitz, I. A suppressor of mating-type locus mutations in Saccharomyces cerevisiae: evidence for and identification of cryptic mating-type loci. Genetics 93, 877–901 (1979).
Gottlieb, S. & Esposito, R. E. A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell 56, 771–776 (1989).
Braunstein, M., Rose, A. B., Holmes, S. G., Allis, C. D. & Broach, J. R. Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes Dev. 7, 592–604 (1993).
Gottschling, D. E., Aparicio, O. M., Billington, B. L. & Zakian, V. A. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell 63, 751–762 (1990).
Kaeberlein, M., McVey, M. & Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580 (1999).
Imai, S., Armstrong, C. M., Kaeberlein, M. & Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 (2000).
Tissenbaum, H. A. & Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410, 227–230 (2001).
Brachmann, C. B. et al. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 9, 2888–2902 (1995).
Afshar, G. & Murnane, J. P. Characterization of a human gene with sequence homology to Saccharomyces cerevisiae SIR2. Gene 234, 161–168 (1999).
Carafa, V. et al. Sirtuin functions and modulation: from chemistry to the clinic. Clin. Epigenetics 8, 61 (2016).
Frye, R. A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 260, 273–279 (1999).
Frye, R. A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 273, 793–798 (2000).
Yuan, H. & Marmorstein, R. Structural basis for sirtuin activity and inhibition. J. Biol. Chem. 287, 42428–42435 (2012).
Chen, B. et al. The chemical biology of sirtuins. Chem. Soc. Rev. 44, 5246–5264 (2015).
Michan, S. & Sinclair, D. Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1–13 (2007).
Finnin, M. S., Donigian, J. R. & Pavletich, N. P. Structure of the histone deacetylase SIRT2. Nat. Struct. Biol. 8, 621–625 (2001).
Canto, C., Sauve, A. A. & Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Asp. Med. 34, 1168–1201 (2013).
Michishita, E., Park, J. Y., Burneskis, J. M., Barrett, J. C. & Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16, 4623–4635 (2005).
Tanno, M., Sakamoto, J., Miura, T., Shimamoto, K. & Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 282, 6823–6832 (2007).
Vaquero, A. et al. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 20, 1256–1261 (2006).
Schwer, B., North, B. J., Frye, R. A., Ott, M. & Verdin, E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 158, 647–657 (2002).
Haigis, M. C. et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126, 941–954 (2006).
Nakagawa, T., Lomb, D. J., Haigis, M. C. & Guarente, L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137, 560–570 (2009).
Mostoslavsky, R. et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329 (2006).
Emmons, M., Boulware, D., Sullivan, D. M. & Hazlehurst, L. A. Topoisomerase II beta levels are a determinant of melphalan-induced DNA crosslinks and sensitivity to cell death. Biochem. Pharmacol. 72, 11–18 (2006).
Buler, M., Andersson, U. & Hakkola, J. Who watches the watchmen? Regulation of the expression and activity of sirtuins. FASEB J. 30, 3942–3960 (2016).
Zhao, K., Harshaw, R., Chai, X. & Marmorstein, R. Structural basis for nicotinamide cleavage and ADP-ribose transfer by NAD(+)-dependent Sir2 histone/protein deacetylases. Proc. Natl Acad. Sci. USA 101, 8563–8568 (2004).
Hoff, K. G., Avalos, J. L., Sens, K. & Wolberger, C. Insights into the sirtuin mechanism from ternary complexes containing NAD+ and acetylated peptide. Structure 14, 1231–1240 (2006).
Rardin, M. J. et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 18, 920–933 (2013).
Du, J. et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 (2011).
Jiang, H. et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496, 110–113 (2013).
Chang, A. R., Ferrer, C. M. & Mostoslavsky, R. SIRT6, a Mammalian deacylase with multitasking abilities. Phys. Rev. 100, 145–169 (2020).
Barber, M. F. et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 487, 114–118 (2012).
Pande, S. & Raisuddin, S. Molecular and cellular regulatory roles of sirtuin protein. Crit. Rev. Food Sci. Nutr. 1-19 (2022) https://doi.org/10.1080/10408398.2022.2070722.
Haigis, M. C. & Sinclair, D. A. Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295 (2010).
Medzhitov, R. Inflammation 2010: new adventures of an old flame. Cell 140, 771–776 (2010).
Medzhitov, R. Origin and physiological roles of inflammation. Nature 454, 428–435 (2008).
Liu, G. et al. Dendritic cell SIRT1-HIF1alpha axis programs the differentiation of CD4+ T cells through IL-12 and TGF-beta1. Proc. Natl Acad. Sci. USA 112, E957–E965 (2015).
Woo, S. J. et al. Myeloid sirtuin 6 deficiency accelerates experimental rheumatoid arthritis by enhancing macrophage activation and infiltration into synovium. EBioMedicine 38, 228–237 (2018).
Xi, J. et al. Sirtuin 3 suppresses the formation of renal calcium oxalate crystals through promoting M2 polarization of macrophages. J. Cell Phys. 234, 11463–11473 (2019).
Abdulkhaleq, L. A. et al. The crucial roles of inflammatory mediators in inflammation: A review. Vet. World 11, 627–635 (2018).
Wu, L. et al. MiR-128-3p mediates TNF-alpha-induced inflammatory responses by regulating Sirt1 expression in bone marrow mesenchymal stem cells. Biochem. Biophys. Res. Commun. 521, 98–105 (2020).
Yang, H. et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-kappaB activity. PLoS One 7, e46364 (2012).
Jung, Y. J. et al. SIRT1 overexpression decreases cisplatin-induced acetylation of NF-kappaB p65 subunit and cytotoxicity in renal proximal tubule cells. Biochem. Biophys. Res. Commun. 419, 206–210 (2012).
Jiang, X. et al. MDL-800, the SIRT6 activator, suppresses inflammation via the NF-kappaB Pathway and Promotes Angiogenesis to Accelerate Cutaneous Wound Healing in Mice. Oxid. Med. Cell Longev. 2022, 1619651 (2022).
He, Y. et al. SIRT6 inhibits TNF-alpha-induced inflammation of vascular adventitial fibroblasts through ROS and Akt signaling pathway. Exp. Cell Res. 357, 88–97 (2017).
Shen, J. et al. SIRT1 Inhibits the Catabolic Effect of IL-1beta Through TLR2/SIRT1/NF-kappaB Pathway in Human Degenerative Nucleus Pulposus Cells. Pain. Phys. 19, E215–E226 (2016).
Wang, X., Buechler, N. L., Yoza, B. K., McCall, C. E. & Vachharajani, V. T. Resveratrol attenuates microvascular inflammation in sepsis via SIRT-1-Induced modulation of adhesion molecules in ob/ob mice. Obes. (Silver Spring) 23, 1209–1217 (2015).
Lappas, M. Anti-inflammatory properties of sirtuin 6 in human umbilical vein endothelial cells. Mediators Inflamm. 2012, 597514 (2012).
Zhang, Y., Anoopkumar-Dukie, S., Mallik, S. B. & Davey, A. K. SIRT1 and SIRT2 modulators reduce LPS-induced inflammation in HAPI microglial cells and protect SH-SY5Y neuronal cells in vitro. J. Neural Transm. (Vienna) 128, 631–644 (2021).
Wang, B. et al. SIRT2 plays significant roles in lipopolysaccharides-induced neuroinflammation and brain injury in mice. Neurochem Res 41, 2490–2500 (2016).
Kurundkar, D. et al. SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI Insight 4, e120722 (2019).
Zhao, W. Y., Zhang, L., Sui, M. X., Zhu, Y. H. & Zeng, L. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Sci. Rep. 6, 33201 (2016).
Palomer, X. et al. SIRT3-mediated inhibition of FOS through histone H3 deacetylation prevents cardiac fibrosis and inflammation. Signal Transduct. Target Ther. 5, 14 (2020).
Kim, D. et al. Absence of Sirt3 aggravates cisplatin nephrotoxicity via enhanced renal tubular apoptosis and inflammation. Mol. Med. Rep. 18, 3665–3672 (2018).
Diaz-Perdigon, T. et al. Early sirtuin 2 inhibition prevents age-related cognitive decline in a senescence-accelerated mouse model. Neuropsychopharmacology 45, 347–357 (2020).
Heinonen, T. et al. Dual deletion of the sirtuins SIRT2 and SIRT3 impacts on metabolism and inflammatory responses of macrophages and protects from endotoxemia. Front Immunol. 10, 2713 (2019).
Liu, T., Zhang, L., Joo, D. & Sun, S. C. NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2, 17023 (2017).
Koch, L. et al. LPS- and LTA-induced expression of IL-6 and TNF-alpha in neonatal and adult blood: role of MAPKs and NF-kappaB. Med. Inflamm. 2014, 283126 (2014).
Swanson, K. V., Deng, M. & Ting, J. P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019).
Chen, G. & Goeddel, D. V. TNF-R1 signaling: a beautiful pathway. Science 296, 1634–1635 (2002).
Sharma, B. R. & Kanneganti, T. D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 22, 550–559 (2021).
Fernando, K. K. M. & Wijayasinghe, Y. S. Sirtuins as potential therapeutic targets for mitigating neuroinflammation associated with Alzheimer’s disease. Front Cell Neurosci. 15, 746631 (2021).
He, L., Wang, J., Yang, Y., Li, J. & Tu, H. Mitochondrial sirtuins in Parkinson’s disease. Neurochem. Res. 47, 1491–1502 (2022).
Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1, a001651 (2009).
Huang, B., Yang, X. D., Lamb, A. & Chen, L. F. Posttranslational modifications of NF-kappaB: another layer of regulation for NF-kappaB signaling pathway. Cell Signal 22, 1282–1290 (2010).
Yeung, F. et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380 (2004).
Li, G. et al. SIRT1 inhibits rheumatoid arthritis fibroblast-like synoviocyte aggressiveness and inflammatory response via suppressing NF-kappaB pathway. Biosci. Rep. 38, BSR20180541 (2018).
Kawahara, T. L. et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136, 62–74 (2009).
Wu, Y. et al. Overexpression of Sirtuin 6 suppresses cellular senescence and NF-kappaB mediated inflammatory responses in osteoarthritis development. Sci. Rep. 5, 17602 (2015).
Vachharajani, V. T. et al. Sirtuins link inflammation and metabolism. J. Immunol. Res. 2016, 8167273 (2016).
Chen, K. L. et al. SIRT7 regulates lipopolysaccharide-induced inflammatory injury by suppressing the NF-kappaB signaling pathway. Oxid. Med. Cell Longev. 2019, 3187972 (2019).
Li, X. T., Zhang, Y. P., Zhang, M. W., Zhang, Z. Z. & Zhong, J. C. Sirtuin 7 serves as a promising therapeutic target for cardiorenal diseases. Eur. J. Pharm. 925, 174977 (2022).
Sobuz, S. U. et al. SIRT7 regulates the nuclear export of NF-kappaB p65 by deacetylating Ran. Biochim. Biophys. Acta Mol. Cell Res. 1866, 1355–1367 (2019).
Sun, S. et al. Vascular endothelium-targeted Sirt7 gene therapy rejuvenates blood vessels and extends life span in a Hutchinson-Gilford progeria model. Sci. Adv. 6, eaay5556 (2020).
Miyasato, Y. et al. Sirtuin 7 deficiency ameliorates cisplatin-induced acute kidney injury through regulation of the inflammatory response. Sci. Rep. 8, 5927 (2018).
Yuan, F. et al. SIRT2 inhibition exacerbates neuroinflammation and blood-brain barrier disruption in experimental traumatic brain injury by enhancing NF-kappaB p65 acetylation and activation. J. Neurochem. 136, 581–593 (2016).
Lin, J., Sun, B., Jiang, C., Hong, H. & Zheng, Y. Sirt2 suppresses inflammatory responses in collagen-induced arthritis. Biochem. Biophys. Res. Commun. 441, 897–903 (2013).
Meng, X. F. et al. Inhibition of the NLRP3 inflammasome provides neuroprotection in rats following amygdala kindling-induced status epilepticus. J. Neuroinflamm. 11, 212 (2014).
Cassel, S. L. & Sutterwala, F. S. Sterile inflammatory responses mediated by the NLRP3 inflammasome. Eur. J. Immunol. 40, 607–611 (2010).
Jin, X. et al. Dietary fatty acid regulation of the NLRP3 inflammasome via the TLR4/NF-kappaB signaling pathway affects chondrocyte pyroptosis. Oxid. Med. Cell Longev. 2022, 3711371 (2022).
Bauernfeind, F. G. et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 (2009).
Xie, N. et al. Rhodiola crenulate alleviates hypobaric hypoxia-induced brain injury via adjusting NF-kappaB/NLRP3-mediated inflammation. Phytomedicine 103, 154240 (2022).
Yamaguchi, K. et al. Indoxyl sulfate activates NLRP3 inflammasome to induce cardiac contractile dysfunction accompanied by myocardial fibrosis and hypertrophy. Cardiovasc. Toxicol. 22, 365–377 (2022).
Zou, P., Liu, X., Li, G. & Wang, Y. Resveratrol pretreatment attenuates traumatic brain injury in rats by suppressing NLRP3 inflammasome activation via SIRT1. Mol. Med. Rep. 17, 3212–3217 (2018).
Anders, H. J. & Muruve, D. A. The inflammasomes in kidney disease. J. Am. Soc. Nephrol. 22, 1007–1018 (2011).
Yu, M. et al. Notch-activated mesenchymal stromal/stem cells enhance the protective effect against acetaminophen-induced acute liver injury by activating AMPK/SIRT1 pathway. Stem Cell Res. Ther. 13, 318 (2022).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Xia, K., Zhang, Y. & Sun, D. miR217 and miR543 downregulation mitigates inflammatory response and myocardial injury in children with viral myocarditis by regulating the SIRT1/AMPK/NFkappaB signaling pathway. Int. J. Mol. Med. 45, 634–646 (2020).
Zhang, T. et al. MicroRNA-378 promotes hepatic inflammation and fibrosis via modulation of the NF-kappaB-TNFalpha pathway. J. Hepatol. 70, 87–96 (2019).
Han, S. et al. MCPIP1 alleviated lipopolysaccharide-induced liver injury by regulating SIRT1 via modulation of microRNA-9. J. Cell Physiol. 234, 22450–22462 (2019).
Lu, H. & Wang, B. SIRT1 exerts neuroprotective effects by attenuating cerebral ischemia/reperfusion-induced injury via targeting p53/microRNA-22. Int. J. Mol. Med. 39, 208–216 (2017).
DeBerardinis, R. J. & Keshari, K. R. Metabolic analysis as a driver for discovery, diagnosis, and therapy. Cell 185, 2678–2689 (2022).
Noda-Garcia, L., Liebermeister, W. & Tawfik, D. S. Metabolite-enzyme coevolution: from single enzymes to metabolic pathways and networks. Annu. Rev. Biochem. 87, 187–216 (2018).
Li, X. et al. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 16, 425–441 (2019).
Martinez-Reyes, I. & Chandel, N. S. Cancer metabolism: looking forward. Nat. Rev. Cancer 21, 669–680 (2021).
Meacham, C. E., DeVilbiss, A. W. & Morrison, S. J. Metabolic regulation of somatic stem cells in vivo. Nat. Rev. Mol. Cell Biol. 23, 428–443 (2022).
Brown, K. A. Metabolic pathways in obesity-related breast cancer. Nat. Rev. Endocrinol. 17, 350–363 (2021).
Menezes, L. F. & Germino, G. G. The pathobiology of polycystic kidney disease from a metabolic viewpoint. Nat. Rev. Nephrol. 15, 735–749 (2019).
Mulukutla, B. C., Yongky, A., Le, T., Mashek, D. G. & Hu, W. S. Regulation of glucose metabolism - a perspective from cell bioprocessing. Trends Biotechnol. 34, 638–651 (2016).
Lunt, S. Y. & Vander Heiden, M. G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464 (2011).
Dey, P., Kimmelman, A. C. & DePinho, R. A. Metabolic codependencies in the tumor microenvironment. Cancer Disco. 11, 1067–1081 (2021).
Han, X. Lipidomics for studying metabolism. Nat. Rev. Endocrinol. 12, 668–679 (2016).
Cappel, D. A. et al. Pyruvate-carboxylase-mediated anaplerosis promotes antioxidant capacity by sustaining TCA cycle and redox metabolism in liver. Cell Metab. 29, 1291–1305 (2019). e1298.
Peng, Y. et al. Does LKB1 mediate activation of hepatic AMP-protein kinase (AMPK) and sirtuin1 (SIRT1) after Roux-en-Y gastric bypass in obese rats? J. Gastrointest. Surg. 14, 221–228 (2010).
Zhang, Y., Ma, Y., Gu, M. & Peng, Y. lncRNA TUG1 promotes the brown remodeling of white adipose tissue by regulating miR204targeted SIRT1 in diabetic mice. Int. J. Mol. Med. 46, 2225–2234 (2020).
Gouranton, E. et al. Visfatin is involved in TNFalpha-mediated insulin resistance via an NAD(+)/Sirt1/PTP1B pathway in 3T3-L1 adipocytes. Adipocyte 3, 180–189 (2014).
Silvestre, M. F. et al. The AMPK-SIRT signaling network regulates glucose tolerance under calorie restriction conditions. Life Sci. 100, 55–60 (2014).
Canto, C. et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 (2009).
Canto, C. et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219 (2010).
Osborne, B. et al. Liver-specific overexpression of SIRT3 enhances oxidative metabolism, but does not impact metabolic defects induced by high fat feeding in mice. Biochem. Biophys. Res. Commun. 607, 131–137 (2022).
Chang, H. et al. TFB2M activates aerobic glycolysis in hepatocellular carcinoma cells through the NAD(+) /SIRT3/HIF-1alpha signaling. J. Gastroenterol. Hepatol. 36, 2978–2988 (2021).
Khan, D. et al. SIRT6 deacetylase transcriptionally regulates glucose metabolism in heart. J. Cell Physiol. 233, 5478–5489 (2018).
Cui, X. et al. SIRT6 regulates metabolic homeostasis in skeletal muscle through activation of AMPK. Am. J. Physiol. Endocrinol. Metab. 313, E493–E505 (2017).
Song, M. Y., Wang, J., Ka, S. O., Bae, E. J. & Park, B. H. Insulin secretion impairment in Sirt6 knockout pancreatic beta cells is mediated by suppression of the FoxO1-Pdx1-Glut2 pathway. Sci. Rep. 6, 30321 (2016).
Zhang, Y. et al. Exercise ameliorates insulin resistance and improves SIRT6-mediated insulin signaling transduction in liver of obese rats. Can. J. Physiol. Pharmacol. 99, 506–511 (2021).
Anderson, J. G. et al. Enhanced insulin sensitivity in skeletal muscle and liver by physiological overexpression of SIRT6. Mol. Metab. 4, 846–856 (2015).
Hammer, S. S. et al. Fasting and fasting-mimicking treatment activate SIRT1/LXRalpha and alleviate diabetes-induced systemic and microvascular dysfunction. Diabetologia 64, 1674–1689 (2021).
Jiang, Z. et al. The adiponectin-SIRT1-AMPK pathway in alcoholic fatty liver disease in the rat. Alcohol Clin. Exp. Res. 39, 424–433 (2015).
Qiang, L. et al. Proatherogenic abnormalities of lipid metabolism in SirT1 transgenic mice are mediated through Creb deacetylation. Cell Metab. 14, 758–767 (2011).
Chen, W. L., Kang, C. H., Wang, S. G. & Lee, H. M. alpha-Lipoic acid regulates lipid metabolism through induction of sirtuin 1 (SIRT1) and activation of AMP-activated protein kinase. Diabetologia 55, 1824–1835 (2012).
Ren, H. et al. Sirtuin 2 prevents liver steatosis and metabolic disorders by deacetylation of hepatocyte nuclear factor 4alpha. Hepatology 74, 723–740 (2021).
Ming, X. et al. Pancreatic sirtuin 3 deficiency promotes hepatic steatosis by enhancing 5-hydroxytryptamine synthesis in mice with diet-induced obesity. Diabetes 70, 119–131 (2021).
Hong, J. et al. SIRT5 inhibits bovine preadipocyte differentiation and lipid deposition by activating AMPK and repressing MAPK signal pathways. Genomics 112, 1065–1076 (2020).
Zhu, C. et al. SIRT6 controls hepatic lipogenesis by suppressing LXR, ChREBP, and SREBP1. Biochim. Biophys. Acta Mol. Basis Dis. 1867, 166249 (2021).
Masri, S. et al. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell 158, 659–672 (2014).
Xiong, X. et al. Fabp4-Cre-mediated Sirt6 deletion impairs adipose tissue function and metabolic homeostasis in mice. J. Endocrinol. 233, 307–314 (2017).
Gonzalez Herrera, K. N. et al. Small-molecule screen identifies de novo nucleotide synthesis as a vulnerability of cells lacking SIRT3. Cell Rep. 22, 1945–1955 (2018).
Miyo, M. et al. Tumour-suppressive function of SIRT4 in human colorectal cancer. Br. J. Cancer 113, 492–499 (2015).
Li, Y. et al. SIRT3 affects mitochondrial metabolic reprogramming via the AMPK-PGC-1alpha axis in the development of benign prostatic hyperplasia. Prostate 81, 1135–1148 (2021).
Baohua, Y. & Li, L. Effects of SIRT6 silencing on collagen metabolism in human dermal fibroblasts. Cell Biol. Int. 36, 105–108 (2012).
Maiese, K., Chong, Z. Z., Hou, J. & Shang, Y. C. The vitamin nicotinamide: translating nutrition into clinical care. Molecules 14, 3446–3485 (2009).
Maiese, K., Chong, Z. Z., Li, F. & Shang, Y. C. Erythropoietin: elucidating new cellular targets that broaden therapeutic strategies. Prog. Neurobiol. 85, 194–213 (2008).
Lan, F., Cacicedo, J. M., Ruderman, N. & Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 283, 27628–27635 (2008).
Galdieri, L., Gatla, H., Vancurova, I. & Vancura, A. Activation of AMP-activated protein kinase by metformin induces protein acetylation in prostate and ovarian cancer cells. J. Biol. Chem. 291, 25154–25166 (2016).
Vancura, A. et al. Reciprocal regulation of AMPK/SNF1 and protein acetylation. Int. J. Mol. Sci. 19, 3314 (2018).
Morales-Alamo, D. et al. Increased oxidative stress and anaerobic energy release, but blunted Thr172-AMPKalpha phosphorylation, in response to sprint exercise in severe acute hypoxia in humans. J. Appl. Physiol. (1985) 113, 917–928 (2012).
Guo, Z. et al. Neuraminidase 1 deficiency attenuates cardiac dysfunction, oxidative stress, fibrosis, inflammatory via AMPK-SIRT3 pathway in diabetic cardiomyopathy mice. Int. J. Biol. Sci. 18, 826–840 (2022).
Chen, L. Y., Wang, Y., Terkeltaub, R. & Liu-Bryan, R. Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthr. Cartil. 26, 1539–1550 (2018).
Li, M. et al. Sirt3 modulates fatty acid oxidation and attenuates cisplatin-induced AKI in mice. J. Cell Mol. Med. 24, 5109–5121 (2020).
Wang, X. X. et al. SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3alpha-dependent antioxidant defense mechanisms. Basic Res. Cardiol. 111, 13 (2016).
Tang, W., Jiang, Y. F., Ponnusamy, M. & Diallo, M. Role of Nrf2 in chronic liver disease. World J. Gastroenterol. 20, 13079–13087 (2014).
Zhang, Y. K., Wu, K. C. & Klaassen, C. D. Genetic activation of Nrf2 protects against fasting-induced oxidative stress in livers of mice. PLoS One 8, e59122 (2013).
Chen, Z. et al. Dipeptidyl peptidase-4 inhibition improves endothelial senescence by activating AMPK/SIRT1/Nrf2 signaling pathway. Biochem. Pharm. 177, 113951 (2020).
Zhao, M. et al. Sirt2 in the spinal cord regulates chronic neuropathic pain through Nrf2-mediated oxidative stress pathway in rats. Front Pharm. 12, 646477 (2021).
Zhang, W., Wei, R., Zhang, L., Tan, Y. & Qian, C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience 366, 95–104 (2017).
Ka, S. O., Bang, I. H., Bae, E. J. & Park, B. H. Hepatocyte-specific sirtuin 6 deletion predisposes to nonalcoholic steatohepatitis by up-regulation of Bach1, an Nrf2 repressor. FASEB J. 31, 3999–4010 (2017).
Sun, G. L., Huang, D., Li, K. R. & Jiang, Q. microRNA-4532 inhibition protects human lens epithelial cells from ultra-violet-induced oxidative injury via activating SIRT6-Nrf2 signaling. Biochem. Biophys. Res. Commun. 514, 777–784 (2019).
Yu, J. et al. SIRT6 protects retinal ganglion cells against hydrogen peroxide-induced apoptosis and oxidative stress by promoting Nrf2/ARE signaling via inhibition of Bach1. Chem. Biol. Interact. 300, 151–158 (2019).
Subauste, A. R. & Burant, C. F. Role of FoxO1 in FFA-induced oxidative stress in adipocytes. Am. J. Physiol. Endocrinol. Metab. 293, E159–E164 (2007).
Han, C. et al. Sirt1 deficiency protects cochlear cells and delays the early onset of age-related hearing loss in C57BL/6 mice. Neurobiol. Aging 43, 58–71 (2016).
Rangarajan, P., Karthikeyan, A., Lu, J., Ling, E. A. & Dheen, S. T. Sirtuin 3 regulates Foxo3a-mediated antioxidant pathway in microglia. Neuroscience 311, 398–414 (2015).
Sundaresan, N. R. et al. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest 119, 2758–2771 (2009).
Kwon, D. N., Park, W. J., Choi, Y. J., Gurunathan, S. & Kim, J. H. Oxidative stress and ROS metabolism via down-regulation of sirtuin 3 expression in Cmah-null mice affect hearing loss. Aging (Albany NY) 7, 579–594 (2015).
Yang, Y. et al. Activation of SIRT3 attenuates triptolide-induced toxicity through closing mitochondrial permeability transition pore in cardiomyocytes. Toxicol. Vitr. 34, 128–137 (2016).
Rius-Perez, S., Torres-Cuevas, I., Millan, I., Ortega, A. L. & Perez, S. PGC-1alpha, inflammation, and oxidative stress: an integrative view in metabolism. Oxid. Med. Cell Longev. 2020, 1452696 (2020).
Waldman, M. et al. PARP-1 inhibition protects the diabetic heart through activation of SIRT1-PGC-1alpha axis. Exp. Cell Res. 373, 112–118 (2018).
Zhu, H. et al. PARP-1 and SIRT-1 are interacted in diabetic nephropathy by activating AMPK/PGC-1alpha signaling pathway. Diabetes Metab. Syndr. Obes. 14, 355–366 (2021).
Wang, S. J. et al. Sirtuin 1 activation enhances the PGC-1alpha/mitochondrial antioxidant system pathway in status epilepticus. Mol. Med. Rep. 11, 521–526 (2015).
Rato, L. et al. Pre-diabetes alters testicular PGC1-alpha/SIRT3 axis modulating mitochondrial bioenergetics and oxidative stress. Biochim. Biophys. Acta 1837, 335–344 (2014).
Zhang, K., Cheng, H., Song, L. & Wei, W. Inhibition of the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1alpha)/Sirtuin 3 (SIRT3) pathway aggravates oxidative stress after experimental subarachnoid hemorrhage. Med. Sci. Monit. 26, e923688 (2020).
Chen, J. et al. Sirtuin 3 deficiency exacerbates age-related periodontal disease. J. Periodontal Res. 56, 1163–1173 (2021).
Paku, M. et al. SIRT3-mediated SOD2 and PGC-1alpha contribute to chemoresistance in colorectal cancer cells. Ann. Surg. Oncol. 28, 4720–4732 (2021).
Yuan, Y. et al. P53 contributes to cisplatin induced renal oxidative damage via regulating P66shc and MnSOD. Cell Physiol. Biochem. 37, 1240–1256 (2015).
Liu, X., Fan, L., Lu, C., Yin, S. & Hu, H. Functional role of p53 in the regulation of chemical-induced oxidative stress. Oxid. Med. Cell Longev. 2020, 6039769 (2020).
Gu, W. & Roeder, R. G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606 (1997).
de Kreutzenberg, S. V. et al. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes 59, 1006–1015 (2010).
Kim, H. J. et al. Carbon monoxide protects against hepatic ischemia/reperfusion injury by modulating the miR-34a/SIRT1 pathway. Biochim. Biophys. Acta 1852, 1550–1559 (2015).
Ma, F. et al. P53/NRF2 mediates SIRT1’s protective effect on diabetic nephropathy. Biochim. Biophys. Acta Mol. Cell Res. 1866, 1272–1281 (2019).
Wu, S. et al. Sirt6 protects cardiomyocytes against doxorubicin-induced cardiotoxicity by inhibiting P53/Fas-dependent cell death and augmenting endogenous antioxidant defense mechanisms. Cell Biol. Toxicol (2021).
Mulero, M. C. et al. DNA-binding affinity and transcriptional activity of the RelA homodimer of nuclear factor kappaB are not correlated. J. Biol. Chem. 292, 18821–18830 (2017).
Sies, H., Berndt, C. & Jones, D. P. Oxidative stress. Annu. Rev. Biochem. 86, 715–748 (2017).
Rada, P. et al. SIRT1 controls acetaminophen hepatotoxicity by modulating inflammation and oxidative stress. Antioxid. Redox Signal 28, 1187–1208 (2018).
Chen, I. C., Huang, H. H., Chen, P. F. & Chiang, H. C. Sirtuin 3 protects against urban particulate matter-induced autophagy in human bronchial epithelial cells. Toxicol. Sci. 152, 113–127 (2016).
Thandavarayan, R. A. et al. Sirtuin-6 deficiency exacerbates diabetes-induced impairment of wound healing. Exp. Dermatol 24, 773–778 (2015).
Baker, J. R. et al. Oxidative stress dependent microRNA-34a activation via PI3Kalpha reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci. Rep. 6, 35871 (2016).
Sun, J. et al. Kruppel-like factor 6 silencing prevents oxidative stress and neurological dysfunction following intracerebral hemorrhage via sirtuin 5/Nrf2/HO-1 Axis. Front Aging Neurosci. 13, 646729 (2021).
Stuart, J. A., Aibueku, O., Bagshaw, O. & Moradi, F. Hypoxia inducible factors as mediators of reactive oxygen/nitrogen species homeostasis in physiological normoxia. Med. Hypotheses 129, 109249 (2019).
Tong, Y. et al. VHL regulates the sensitivity of clear cell renal cell carcinoma to SIRT4-mediated metabolic stress via HIF-1alpha/HO-1 pathway. Cell Death Dis. 12, 621 (2021).
Shi, J. X., Wang, Q. J., Li, H. & Huang, Q. SIRT4 overexpression protects against diabetic nephropathy by inhibiting podocyte apoptosis. Exp. Ther. Med. 13, 342–348 (2017).
Ding, M. et al. SIRT1 protects against myocardial ischemia-reperfusion injury via activating eNOS in diabetic rats. Cardiovasc Diabetol. 14, 143 (2015).
Li, D. et al. Cardioprotection of CAPE-oNO2 against myocardial ischemia/reperfusion induced ROS generation via regulating the SIRT1/eNOS/NF-kappaB pathway in vivo and in vitro. Redox Biol. 15, 62–73 (2018).
Pistritto, G., Trisciuoglio, D., Ceci, C., Garufi, A. & D’Orazi, G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY) 8, 603–619 (2016).
Verdin, E., Hirschey, M. D., Finley, L. W. & Haigis, M. C. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem. Sci. 35, 669–675 (2010).
Nikoletopoulou, V., Markaki, M., Palikaras, K. & Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 1833, 3448–3459 (2013).
Rifai, K. et al. SIRT1-dependent epigenetic regulation of H3 and H4 histone acetylation in human breast cancer. Oncotarget 9, 30661–30678 (2018).
Li, Z. et al. Overexpressed SIRT6 attenuates cisplatin-induced acute kidney injury by inhibiting ERK1/2 signaling. Kidney Int 93, 881–892 (2018).
Yu, W. et al. Effects of resveratrol on H(2)O(2)-induced apoptosis and expression of SIRTs in H9c2 cells. J. Cell Biochem. 107, 741–747 (2009).
Han, X. et al. HDAC4 stabilizes SIRT1 via sumoylation SIRT1 to delay cellular senescence. Clin. Exp. Pharmacol. Physiol. 43, 41–46 (2016).
Grootaert, M. O. J., Finigan, A., Figg, N. L., Uryga, A. K. & Bennett, M. R. SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ. Res. 128, 474–491 (2021).
Sauve, A. A., Wolberger, C., Schramm, V. L. & Boeke, J. D. The biochemistry of sirtuins. Annu. Rev. Biochem. 75, 435–465 (2006).
Sarmah, D. et al. Sirtuin-1 - mediated NF-kappaB pathway modulation to mitigate inflammasome signaling and cellular apoptosis is one of the neuroprotective effects of intra-arterial mesenchymal stem cell therapy following ischemic stroke. Stem Cell Rev. Rep. 18, 821–838 (2022).
Brunet, A. et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015 (2004).
Yi, J. & Luo, J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta 1804, 1684–1689 (2010).
Sun, P., Yu, H., Zhang, W. Q., Hu, M. & Lv, R. Lentivirus-mediated siRNA targeting VEGF inhibits gastric cancer growth in vivo. Oncol. Rep. 28, 1687–1692 (2012).
Yarahmadi, S. et al. Inhibition of sirtuin 1 deacetylase by miR-211-5p provides a mechanism for the induction of cell death in breast cancer cells. Gene 711, 143939 (2019).
Lin, Y., Ma, W. & Benchimol, S. Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat. Genet 26, 122–127 (2000).
Ghosh, A. et al. A novel SIRT1 inhibitor, 4bb induces apoptosis in HCT116 human colon carcinoma cells partially by activating p53. Biochem. Biophys. Res. Commun. 488, 562–p569 (2017).
Zeng, Z. et al. Anoectochilus roxburghii flavonoids extract ameliorated the memory decline and reduced neuron apoptosis via modulating SIRT1 signaling pathway in senescent mice. J. Ethnopharmacol. 296, 115361 (2022).
Vaziri, H. et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 (2001).
Wei, X., Tan, J. & Gao, H. Role of sirtuin 1 in the brain development in congenital hypothyroidism rats via the regulation of p53 signaling pathway. Bioengineered 13, 9455–9466 (2022).
Bi, X. et al. The miRNA-34a/Sirt1/p53 pathway in a rat model of lens regeneration. Ann. Transl. Med. 10, 636 (2022).
Hernandez-Jimenez, M. et al. Silent information regulator 1 protects the brain against cerebral ischemic damage. Stroke 44, 2333–2337 (2013).
Lin, B. et al. Sirt1 improves heart failure through modulating the NF-kappaB p65/microRNA-155/BNDF signaling cascade. Aging (Albany NY) 13, 14482–14498 (2020).
Wang, Q. et al. Sirtuin 1 (Sirt1) overexpression in BaF3 cells contributes to cell proliferation promotion, apoptosis resistance and pro-inflammatory cytokine production. Med. Sci. Monit. 23, 1477–1482 (2017).
Harari, O. A. & Liao, J. K. NF-kappaB and innate immunity in ischemic stroke. Ann. NY Acad. Sci. 1207, 32–40 (2010).
Jiang, F., Zhou, H. Y., Zhou, L. F., Zeng, W. & Zhao, L. H. IRF9 affects the TNF-induced phenotype of rheumatoid-arthritis fibroblast-like synoviocytes via regulation of the SIRT-1/NF-kappaB signaling pathway. Cells Tissues Organs 209, 110–119 (2020).
Marfe, G. et al. Involvement of FOXO transcription factors, TRAIL-FasL/Fas, and sirtuin proteins family in canine coronavirus type II-induced apoptosis. PLoS One 6, e27313 (2011).
Motta, M. C. et al. Mammalian SIRT1 represses forkhead transcription factors. Cell 116, 551–563 (2004).
Yu, S. L. et al. SIRT1 suppresses in vitro decidualization of human endometrial stromal cells through the downregulation of forkhead box O1 expression. Reprod. Biol. 22, 100672 (2022).
Yao, H., Yao, Z., Zhang, S., Zhang, W. & Zhou, W. Upregulation of SIRT1 inhibits H2O2induced osteoblast apoptosis via FoxO1/betacatenin pathway. Mol. Med. Rep. 17, 6681–6690 (2018).
Wang, Y. et al. Roles of SIRT1/FoxO1/SREBP-1 in the development of progestin resistance in endometrial cancer. Arch. Gynecol. Obstet. 298, 961–969 (2018).
Li, Z., Bridges, B., Olson, J. & Weinman, S. A. The interaction between acetylation and serine-574 phosphorylation regulates the apoptotic function of FOXO3. Oncogene 36, 1887–1898 (2017).
Wang, F. et al. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene 31, 1546–1557 (2012).
Frampton, G. et al. The novel growth factor, progranulin, stimulates mouse cholangiocyte proliferation via sirtuin-1-mediated inactivation of FOXO1. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G1202–G1211 (2012).
Wu, Y. X., Xu, R. Y., Jiang, L., Chen, X. Y. & Xiao, X. J. MicroRNA-30a-5p promotes chronic heart failure in rats by targeting sirtuin-1 to activate the nuclear factor-kappaB/NOD-like receptor 3 signaling pathway. Cardiovasc Drugs Ther (2022).
Yamakuchi, M., Ferlito, M. & Lowenstein, C. J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl Acad. Sci. USA 105, 13421–13426 (2008).
Wang, G. et al. miR-34a-5p inhibition alleviates intestinal ischemia/reperfusion-induced reactive oxygen species accumulation and apoptosis via activation of SIRT1 signaling. Antioxid. Redox Signal 24, 961–973 (2016).
Fan, X., Yin, X., Zhao, Q. & Yang, Y. Hsa_circRNA_0045861 promotes renal injury in ureteropelvic junction obstruction via the microRNA-181d-5p/sirtuin 1 signaling axis. Ann. Transl. Med. 9, 1571 (2021).
Zhang, M. et al. miR-181a increases FoxO1 acetylation and promotes granulosa cell apoptosis via SIRT1 downregulation. Cell Death Dis. 8, e3088 (2017).
Adlakha, Y. K. & Saini, N. miR-128 exerts pro-apoptotic effect in a p53 transcription-dependent and -independent manner via PUMA-Bak axis. Cell Death Dis. 4, e542 (2013).
Chen, D. Z. et al. miR128 is upregulated in epilepsy and promotes apoptosis through the SIRT1 cascade. Int. J. Mol. Med. 44, 694–704 (2019).
Qin, W., Xie, W., Yang, X., Xia, N. & Yang, K. Inhibiting microRNA-449 attenuates cisplatin-induced injury in NRK-52E cells possibly via regulating the SIRT1/P53/BAX pathway. Med. Sci. Monit. 22, 818–823 (2016).
Xiao, M. et al. A new FGF1 variant protects against adriamycin-induced cardiotoxicity via modulating p53 activity. Redox Biol. 49, 102219 (2022).
Zhao, L. et al. Melatonin alleviates brain injury in mice subjected to cecal ligation and puncture via attenuating inflammation, apoptosis, and oxidative stress: the role of SIRT1 signaling. J. Pineal Res. 59, 230–239 (2015).
Hu, J. et al. Melatonin alleviates postinfarction cardiac remodeling and dysfunction by inhibiting Mst1. J. Pineal Res. 62 (2017).
Han, D. et al. Melatonin facilitates adipose-derived mesenchymal stem cells to repair the murine infarcted heart via the SIRT1 signaling pathway. J. Pineal Res. 60, 178–192 (2016).
Yang, Y. et al. Melatonin prevents cell death and mitochondrial dysfunction via a SIRT1-dependent mechanism during ischemic-stroke in mice. J. Pineal Res. 58, 61–70 (2015).
Heltweg, B. et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res 66, 4368–4377 (2006).
Huang, Q. et al. A SIRT1 activator, ginsenoside Rc, promotes energy metabolism in cardiomyocytes and neurons. J. Am. Chem. Soc. 143, 1416–1427 (2021).
Chen, X., Lu, W. & Wu, D. Sirtuin 2 (SIRT2): confusing roles in the pathophysiology of neurological disorders. Front Neurosci. 15, 614107 (2021).
Peck, B. et al. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer Ther. 9, 844–855 (2010).
Hoffmann, G., Breitenbucher, F., Schuler, M. & Ehrenhofer-Murray, A. E. A novel sirtuin 2 (SIRT2) inhibitor with p53-dependent pro-apoptotic activity in non-small cell lung cancer. J. Biol. Chem. 289, 5208–5216 (2014).
Li, Y. et al. Sirt2 suppresses glioma cell growth through targeting NF-kappaB-miR-21 axis. Biochem. Biophys. Res. Commun. 441, 661–667 (2013).
Keskin-Aktan, A., Akbulut, K. G., Abdi, S. & Akbulut, H. SIRT2 and FOXO3a expressions in the cerebral cortex and hippocampus of young and aged male rats: antioxidant and anti-apoptotic effects of melatonin. Biol. Futur. 73, 71–85 (2022).
Li, Y. et al. SIRT2 down-regulation in HeLa can induce p53 accumulation via p38 MAPK activation-dependent p300 decrease, eventually leading to apoptosis. Genes Cells 16, 34–45 (2011).
She, D. T., Wong, L. J., Baik, S. H. & Arumugam, T. V. SIRT2 inhibition confers neuroprotection by downregulation of FOXO3a and MAPK signaling pathways in ischemic stroke. Mol. Neurobiol. 55, 9188–9203 (2018).
Wang, Y. et al. SIRT2-mediated FOXO3a deacetylation drives its nuclear translocation triggering FasL-induced cell apoptosis during renal ischemia reperfusion. Apoptosis 22, 519–530 (2017).
Song, C. L. et al. Sirtuin 3 inhibits hepatocellular carcinoma growth through the glycogen synthase kinase-3beta/BCL2-associated X protein-dependent apoptotic pathway. Oncogene 35, 631–641 (2016).
Xiao, K. et al. Sirt3 is a tumor suppressor in lung adenocarcinoma cells. Oncol. Rep. 30, 1323–1328 (2013).
Tao, N. N. et al. Sirtuin 3 enhanced drug sensitivity of human hepatoma cells through glutathione S-transferase pi 1/JNK signaling pathway. Oncotarget 7, 50117–50130 (2016).
Cao, Y., Li, P., Wang, H., Li, L. & Li, Q. SIRT3 promotion reduces resistance to cisplatin in lung cancer by modulating the FOXO3/CDT1 axis. Cancer Med 10, 1394–1404 (2021).
Fan, H., Le, J. W., Sun, M. & Zhu, J. H. Sirtuin 3 deficiency promotes acute kidney injury induced by sepsis via mitochondrial dysfunction and apoptosis. Iran. J. Basic Med. Sci. 24, 675–681 (2021).
Yi, X. et al. SIRT3-dependent mitochondrial dynamics remodeling contributes to oxidative stress-induced melanocyte degeneration in vitiligo. Theranostics 9, 1614–1633 (2019).
Zhang, X., Tang, N., Hadden, T. J. & Rishi, A. K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 1978–1986, 2011 (1813).
Peng, Y. et al. Sirt3 suppresses calcium oxalate-induced renal tubular epithelial cell injury via modification of FoxO3a-mediated autophagy. Cell Death Dis. 10, 34 (2019).
Yue, Y., Du, Z., Tao, J. & Shi, L. Inhibition of microRNA-297 alleviates THLE-2 cell injury induced by hypoxia/reoxygenation by inhibiting NLRP3 inflammasome activation via sirtuin 3. Can. J. Physiol. Pharmacol. 100, 125–133 (2022).
Liu, Y., Qian, X. M., He, Q. C. & Weng, J. K. MiR-421 inhibition protects H9c2 cells against hypoxia/reoxygenation-induced oxidative stress and apoptosis by targeting Sirt3. Perfusion 35, 255–262 (2020).
Liu, B. et al. SIRT4 prevents hypoxia-induced apoptosis in H9c2 cardiomyoblast cells. Cell Physiol. Biochem 32, 655–662 (2013).
Schlicker, C. et al. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol. 382, 790–801 (2008).
Yang, C. N. et al. The possible role of sirtuin 5 in the pathogenesis of apical periodontitis. Oral. Dis. 27, 1766–1774 (2021).
Li, W., Yang, Y., Li, Y., Zhao, Y. & Jiang, H. Sirt5 attenuates cisplatin-induced acute kidney injury through regulation of Nrf2/HO-1 and Bcl-2. Biomed. Res. Int. 2019, 4745132 (2019).
Wood, M., Rymarchyk, S., Zheng, S. & Cen, Y. Trichostatin A inhibits deacetylation of histone H3 and p53 by SIRT6. Arch. Biochem. Biophys. 638, 8–17 (2018).
Vakhrusheva, O. et al. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 102, 703–710 (2008).
Kiran, S., Oddi, V. & Ramakrishna, G. Sirtuin 7 promotes cellular survival following genomic stress by attenuation of DNA damage, SAPK activation and p53 response. Exp. Cell Res. 331, 123–141 (2015).
Glick, D., Barth, S. & Macleod, K. F. Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12 (2010).
Hamai, A. & Codogno, P. New targets for acetylation in autophagy. Sci. Signal 5, pe29 (2012).
Yi, C. & Yu, L. How does acetylation regulate autophagy? Autophagy 8, 1529–1530 (2012).
Esteves, A. R., Filipe, F., Magalhaes, J. D., Silva, D. F. & Cardoso, S. M. The role of beclin-1 acetylation on autophagic flux in Alzheimer’s disease. Mol. Neurobiol. 56, 5654–5670 (2019).
Sun, T., Jiao, L., Wang, Y., Yu, Y. & Ming, L. SIRT1 induces epithelial-mesenchymal transition by promoting autophagic degradation of E-cadherin in melanoma cells. Cell Death Dis. 9, 136 (2018).
Huang, S. et al. Sirtuin 1 promotes autophagy and proliferation of endometrial cancer cells by reducing acetylation level of LC3. Cell Biol. Int. 45, 1050–1059 (2021).
Liu, C. et al. Sirt1 regulates acrosome biogenesis by modulating autophagic flux during spermiogenesis in mice. Development 144, 441–451 (2017).
Islam, A. et al. Capsaicin exerts therapeutic effects by targeting tNOX-SIRT1 axis and augmenting ROS-dependent autophagy in melanoma cancer cells. Am. J. Cancer Res. 11, 4199–4219 (2021).
Cho, J. H. et al. Downregulation of SIRT1 signaling underlies hepatic autophagy impairment in glycogen storage disease type Ia. PLoS Genet 13, e1006819 (2017).
Doblado, L. et al. Mitophagy in human diseases. Int. J. Mol. Sci. 22, 3903 (2021).
Romeo-Guitart, D., Leiva-Rodriguez, T., Fores, J. & Casas, C. Improved motor nerve regeneration by SIRT1/Hif1a-mediated autophagy. Cells 8, 1354 (2019).
Jiang, Y. et al. Elucidation of SIRT-1/PGC-1alpha-associated mitochondrial dysfunction and autophagy in nonalcoholic fatty liver disease. Lipids Health Dis. 20, 40 (2021).
Schrepfer, E. & Scorrano, L. Mitofusins, from mitochondria to metabolism. Mol. Cell 61, 683–694 (2016).
Biel, T. G. et al. Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2-dependent manner. Cell Death Differ. 23, 279–290 (2016).
Chun, S. K. et al. Loss of sirtuin 1 and mitofusin 2 contributes to enhanced ischemia/reperfusion injury in aged livers. Aging Cell 17, e12761 (2018).
Guha, P. et al. IPMK mediates activation of ULK signaling and transcriptional regulation of autophagy linked to liver inflammation and regeneration. Cell Rep. 26, 2692–2703 (2019). e2697.
Luo, G. et al. Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress. Int. J. Mol. Med. 43, 2033–2043 (2019).
Gautam, S. et al. The signaling pathways implicated in impairment of hepatic autophagy in glycogen storage disease type Ia. Hum. Mol. Genet. 29, 834–844 (2020).
Chen, M. L. et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 9, 2033–2045 (2013).
Yan, P. et al. Regulation of autophagy by AMP-activated protein kinase/sirtuin 1 pathway reduces spinal cord neurons damage. Iran. J. Basic Med. Sci. 20, 1029–1036 (2017).
Zhang, Y. et al. Resveratrol improves hepatic steatosis by inducing autophagy through the cAMP signaling pathway. Mol. Nutr. Food Res. 59, 1443–1457 (2015).
Guo, H., Chen, Y., Liao, L. & Wu, W. Resveratrol protects HUVECs from oxidized-LDL induced oxidative damage by autophagy upregulation via the AMPK/SIRT1 pathway. Cardiovasc Drugs Ther. 27, 189–198 (2013).
Lee, I. H. et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl Acad. Sci. USA 105, 3374–3379 (2008).
Leng, S. et al. SIRT1 coordinates with the CRL4B complex to regulate pancreatic cancer stem cells to promote tumorigenesis. Cell Death Differ. 28, 3329–3343 (2021).
Song, X. et al. miR-124 and miR-142 enhance cisplatin sensitivity of non-small cell lung cancer cells through repressing autophagy via directly targeting SIRT1. RSC Adv. 9, 5234–5243 (2019).
Dong, W., Zhang, H., Zhao, C., Luo, Y. & Chen, Y. Silencing of miR-150-5p Ameliorates Diabetic Nephropathy by Targeting SIRT1/p53/AMPK Pathway. Front Physiol. 12, 624989 (2021).
Luan, B. & Sun, C. MiR-138-5p affects insulin resistance to regulate type 2 diabetes progression through inducing autophagy in HepG2 cells by regulating SIRT1. Nutr. Res. 59, 90–98 (2018).
Dai, S. H. et al. microRNA-145 Inhibition Upregulates SIRT1 and Attenuates Autophagy in a Mouse Model of Lung Ischemia/Reperfusion Injury via NF-kappaB-dependent Beclin 1. Transplantation 105, 529–539 (2021).
Yang, J. et al. LncRNA MALAT1 enhances ox-LDL-induced autophagy through the SIRT1/MAPK/NF-kappaB pathway in macrophages. Curr. Vasc. Pharmacol. 18, 652–662 (2020).
Zhang, H. G., Wang, F. J., Wang, Y., Zhao, Z. X. & Qiao, P. F. lncRNA GAS5 inhibits malignant progression by regulating macroautophagy and forms a negative feedback regulatory loop with the miR34a/mTOR/SIRT1 pathway in colorectal cancer. Oncol. Rep. 45, 202–216 (2021).
Esteves, A. R. et al. Mitochondrial metabolism regulates microtubule acetylome and autophagy trough sirtuin-2: impact for Parkinson’s disease. Mol. Neurobiol. 55, 1440–1462 (2018).
de Oliveira, R. M. et al. The mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol. 15, e2000374 (2017).
Silva, D. F., Esteves, A. R., Oliveira, C. R. & Cardoso, S. M. Mitochondrial metabolism power SIRT2-dependent deficient traffic causing Alzheimer’s-disease related pathology. Mol. Neurobiol. 54, 4021–4040 (2017).
Wang, L. et al. Down regulation of SIRT2 reduced ASS induced NSCLC apoptosis through the release of autophagy components via exosomes. Front Cell Dev. Biol. 8, 601953 (2020).
Lang, A. et al. SIRT4 interacts with OPA1 and regulates mitochondrial quality control and mitophagy. Aging (Albany NY) 9, 2163–2189 (2017).
Polletta, L. et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 11, 253–270 (2015).
Wang, Y., Chang, J., Wang, Z. Q. & Li, Y. Sirt3 promotes the autophagy of HK2 human proximal tubular epithelial cells via the inhibition of Notch1/Hes1 signaling. Mol. Med. Rep. 24, 634 (2021).
Li, R. et al. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 18, 229–243 (2018).
Zhang, L. et al. MicroRNA-874-5p regulates autophagy and proliferation in pulmonary artery smooth muscle cells by targeting Sirtuin 3. Eur. J. Pharm. 888, 173485 (2020).
Li, Y., Sun, T., Shen, S., Wang, L. & Yan, J. LncRNA DYNLRB2-2 inhibits THP-1 macrophage foam cell formation by enhancing autophagy. Biol Chem (2019).
Zullo, A., Guida, R., Sciarrillo, R. & Mancini, F. P. Redox homeostasis in cardiovascular disease: the role of mitochondrial sirtuins. Front Endocrinol. (Lausanne) 13, 858330 (2022).
He, L. et al. SIRT4 suppresses doxorubicin-induced cardiotoxicity by regulating the AKT/mTOR/autophagy pathway. Toxicology 469, 153119 (2022).
Gu, W. et al. SIRT5 regulates autophagy and apoptosis in gastric cancer cells. J. Int. Med. Res. 49, 300060520986355 (2021).
Shi, L. et al. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol. Oncol. 13, 358–375 (2019).
Wang, G. et al. Regulation of UCP1 and mitochondrial metabolism in brown adipose tissue by reversible succinylation. Mol. Cell 74, 844–857.e847 (2019).
Zhang, Y., Wang, L., Meng, L., Cao, G. & Wu, Y. Sirtuin 6 overexpression relieves sepsis-induced acute kidney injury by promoting autophagy. Cell Cycle 18, 425–436 (2019).
Takasaka, N. et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 192, 958–968 (2014).
Lu, J. et al. SIRT6 suppresses isoproterenol-induced cardiac hypertrophy through activation of autophagy. Transl. Res 172, 96–112.e116 (2016).
Song, J. J. et al. MicroRNA-122 aggravates angiotensin II-mediated apoptosis and autophagy imbalance in rat aortic adventitial fibroblasts via the modulation of SIRT6-elabela-ACE2 signaling. Eur. J. Pharm. 883, 173374 (2020).
Yu, W. et al. Silencing forkhead box M1 promotes apoptosis and autophagy through SIRT7/mTOR/IGF2 pathway in gastric cancer cells. J. Cell Biochem. 119, 9090–9098 (2018).
Wu, S. Y., Du, Y. C. & Yue, C. F. Sirt7 protects chondrocytes degeneration in osteoarthritis via autophagy activation. Eur. Rev. Med. Pharmacol. Sci. 24, 9246–9255 (2020).
Ding, M. et al. SIRT7 depletion inhibits cell proliferation and androgen-induced autophagy by suppressing the AR signaling in prostate cancer. J. Exp. Clin. Cancer Res. 39, 28 (2020).
Conlon, I. & Raff, M. Size control in animal development. Cell 96, 235–244 (1999).
Neufeld, T. P., de la Cruz, A. F., Johnston, L. A. & Edgar, B. A. Coordination of growth and cell division in the Drosophila wing. Cell 93, 1183–1193 (1998).
Thompson, B. J. Developmental control of cell growth and division in Drosophila. Curr. Opin. Cell Biol. 22, 788–794 (2010).
Gaglia, G. et al. Temporal and spatial topography of cell proliferation in cancer. Nat. Cell Biol. 24, 316–326 (2022).
Cordero-Espinoza, L. et al. Dynamic cell contacts between periportal mesenchyme and ductal epithelium act as a rheostat for liver cell proliferation. Cell Stem Cell 28, 1907–1921.e1908 (2021).
Wang, J. et al. Tumor-derived adenosine promotes macrophage proliferation in human hepatocellular carcinoma. J. Hepatol. 74, 627–637 (2021).
Wang, X. et al. Role of SIRT1/AMPK signaling in the proliferation, migration and invasion of renal cell carcinoma cells. Oncol. Rep. 45, 109 (2021).
Jablonska, B. et al. Sirt1 regulates glial progenitor proliferation and regeneration in white matter after neonatal brain injury. Nat. Commun. 7, 13866 (2016).
Imperatore, F. et al. SIRT1 regulates macrophage self-renewal. EMBO J. 36, 2353–2372 (2017).
Lin, X. L., Li, K., Yang, Z., Chen, B. & Zhang, T. Dulcitol suppresses proliferation and migration of hepatocellular carcinoma via regulating SIRT1/p53 pathway. Phytomedicine 66, 153112 (2020).
Zhang, Z. Y. et al. SIRT1 regulates oncogenesis via a mutant p53-dependent pathway in hepatocellular carcinoma. J. Hepatol. 62, 121–130 (2015).
Valente, S. et al. 1,4-dihydropyridines active on the SIRT1/AMPK pathway ameliorate skin repair and mitochondrial function and exhibit inhibition of proliferation in cancer cells. J. Med Chem. 59, 1471–1491 (2016).
Zhang, S. et al. SIRT1 inhibits gastric cancer proliferation and metastasis via STAT3/MMP-13 signaling. J. Cell Physiol. 234, 15395–15406 (2019).
Liu, P. Y. et al. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ. 20, 503–514 (2013).
Li, C. et al. SIRT2 contributes to the regulation of intestinal cell proliferation and differentiation. Cell Mol. Gastroenterol. Hepatol. 10, 43–57 (2020).
Hao, C. et al. SIRT2 regulates proliferation and chemotherapy response of MLL-ENL-driven acute myeloid leukemia. Biochem. Biophys. Res. Commun. 596, 36–42 (2022).
Dan, L. et al. The role of sirtuin 2 activation by nicotinamide phosphoribosyltransferase in the aberrant proliferation and survival of myeloid leukemia cells. Haematologica 97, 551–559 (2012).
Zu, H. et al. SIRT2 functions as a histone delactylase and inhibits the proliferation and migration of neuroblastoma cells. Cell Disco. 8, 54 (2022).
Liu, S., Gao, X., Fan, Z. & Wang, Q. SIRT2 affects cell proliferation and apoptosis by suppressing the level of autophagy in renal podocytes. Dis. Markers 2022, 4586198 (2022).
Wei, Z. et al. Deacetylation of serine hydroxymethyl-transferase 2 by SIRT3 promotes colorectal carcinogenesis. Nat. Commun. 9, 4468 (2018).
Chen, I. C., Chiang, W. F., Liu, S. Y., Chen, P. F. & Chiang, H. C. Role of SIRT3 in the regulation of redox balance during oral carcinogenesis. Mol. Cancer 12, 68 (2013).
Yao, W. et al. Profilin-1 suppresses tumorigenicity in pancreatic cancer through regulation of the SIRT3-HIF1alpha axis. Mol. Cancer 13, 187 (2014).
Chen, Z. et al. SIRT4 inhibits the proliferation, migration, and invasion abilities of thyroid cancer cells by inhibiting glutamine metabolism. Onco. Targets Ther. 12, 2397–2408 (2019).
Wang, C. et al. Mammalian SIRT4 is a tumor suppressor of clear cell renal cell carcinoma by inhibiting cancer proliferation, migration and invasion. Cancer Biomark. 29, 453–462 (2020).
Csibi, A. et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 184, 2256 (2021).
Cui, Y. et al. SIRT4 is the molecular switch mediating cellular proliferation in colorectal cancer through GLS mediated activation of AKT/GSK3beta/CyclinD1 pathway. Carcinogenesis 42, 481–492 (2021).
He, S., Jia, Q., Zhou, L., Wang, Z. & Li, M. SIRT5 is involved in the proliferation and metastasis of breast cancer by promoting aerobic glycolysis. Pathol. Res. Pract. 235, 153943 (2022).
Guan, J. et al. Sirtuin 5 regulates the proliferation, invasion and migration of prostate cancer cells through acetyl-CoA acetyltransferase 1. J. Cell Mol. Med. 24, 14039–14049 (2020).
Ren, M. et al. Citrate synthase desuccinylation by SIRT5 promotes colon cancer cell proliferation and migration. Biol. Chem. 401, 1031–1039 (2020).
Yang, X. et al. SHMT2 desuccinylation by SIRT5 drives cancer cell proliferation. Cancer Res 78, 372–386 (2018).
Chang, L. et al. SIRT5 promotes cell proliferation and invasion in hepatocellular carcinoma by targeting E2F1. Mol. Med. Rep. 17, 342–349 (2018).
Ma, Y. & Fei, X. SIRT5 regulates pancreatic beta-cell proliferation and insulin secretion in type 2 diabetes. Exp. Ther. Med. 16, 1417–1425 (2018).
Ming, M. et al. SIRT6 promotes COX-2 expression and acts as an oncogene in skin cancer. Cancer Res 74, 5925–5933 (2014).
Zhang, C., Yu, Y., Huang, Q. & Tang, K. SIRT6 regulates the proliferation and apoptosis of hepatocellular carcinoma via the ERK1/2 signaling pathway. Mol. Med. Rep. 20, 1575–1582 (2019).
Wang, H. et al. SIRT6 controls hematopoietic stem cell homeostasis through epigenetic regulation of Wnt signaling. Cell Stem Cell 18, 495–507 (2016).
Cea, M. et al. Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood 127, 1138–1150 (2016).
Wang, H. et al. p53-dependent LincRNA-p21 protects against proliferation and anti-apoptosis of vascular smooth muscle cells in atherosclerosis by upregulating SIRT7 via MicroRNA-17-5p. J. Cardiovasc. Transl. Res. 14, 426–440 (2021).
Kimura, Y. et al. Sirt7 deficiency attenuates neointimal formation following vascular injury by modulating vascular smooth muscle cell proliferation. Circ. J. 85, 2232–2240 (2021).
Chen, K. L. et al. SIRT7 depletion inhibits cell proliferation, migration, and increases drug sensitivity by activating p38MAPK in breast cancer cells. J. Cell Physiol. 233, 6767–6778 (2018).
Friedl, P. & Brocker, E. B. The biology of cell locomotion within three-dimensional extracellular matrix. Cell Mol. Life Sci. 57, 41–64 (2000).
Trepat, X., Chen, Z. & Jacobson, K. Cell migration. Compr. Physiol. 2, 2369–2392 (2012).
Chambers, A. F., Groom, A. C. & MacDonald, I. C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2, 563–572 (2002).
Hamidi, H. & Ivaska, J. Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer 18, 533–548 (2018).
Liu, F. & Shang, Y. X. Sirtuin 6 attenuates epithelial-mesenchymal transition by suppressing the TGF-beta1/Smad3 pathway and c-Jun in asthma models. Int. Immunopharmacol. 82, 106333 (2020).
Feng, H. et al. The expression of SIRT1 regulates the metastaticplasticity of chondrosarcoma cells by inducing epithelial-mesenchymal transition. Sci. Rep. 7, 41203 (2017).
Thiery, J. P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442–454 (2002).
Yuan, Z. et al. Suppressive effect of microRNA-138 on the proliferation and invasion of osteosarcoma cells via targeting SIRT1. Exp. Ther. Med. 13, 3417–3423 (2017).
Miranda-Goncalves, V. et al. Lactate increases renal cell carcinoma aggressiveness through sirtuin 1-dependent epithelial mesenchymal transition axis regulation. Cells 9, 1053 (2020).
Latifkar, A. et al. Loss of sirtuin 1 alters the secretome of breast cancer cells by impairing lysosomal integrity. Dev. Cell 49, 393–408.e397 (2019).
Zhong, S. et al. Hsa_circ_0088036 promotes the proliferation and migration of fibroblast-like synoviocytes by sponging miR-140-3p and upregulating SIRT 1 expression in rheumatoid arthritis. Mol. Immunol. 125, 131–139 (2020).
Yu, X. J. et al. SIRT1-ZEB1-positive feedback promotes epithelial-mesenchymal transition process and metastasis of osteosarcoma. J. Cell Biochem. 120, 3727–3735 (2019).
Qiang, L., Sample, A., Liu, H., Wu, X. & He, Y. Y. Epidermal SIRT1 regulates inflammation, cell migration, and wound healing. Sci. Rep. 7, 14110 (2017).
Yao, Y., Fan, X., Yu, B., Li, T. & Zhang, Y. Knockdown of long noncoding RNA Malat1 aggravates hypoxia-induced cardiomyocyte injury by targeting miR-217. Adv. Clin. Exp. Med. 28, 719–728 (2019).
Li, G. & Zhong, S. MicroRNA-217 inhibits the proliferation and invasion, and promotes apoptosis of non-small cell lung cancer cells by targeting sirtuin 1. Oncol. Lett. 21, 386 (2021).
Lai, M. et al. MiR-34a inhibits migration and invasion by regulating the SIRT1/p53 pathway in human SW480 cells. Mol. Med. Rep. 11, 3301–3307 (2015).
Zhou, J. et al. microRNA-34a overexpression inhibits cell migration and invasion via regulating SIRT1 in hepatocellular carcinoma. Oncol. Lett. 14, 6950–6954 (2017).
Liang, Y. et al. A novel long non-coding RNA-PRLB acts as a tumor promoter through regulating miR-4766-5p/SIRT1 axis in breast cancer. Cell Death Dis. 9, 563 (2018).
Zhong, Z., Wen, Z. & Darnell, J. E. Jr Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98 (1994).
Stark, G. R. & Darnell, J. E. Jr. The JAK-STAT pathway at twenty. Immunity 36, 503–514 (2012).
Wang, B. et al. STAT3 aggravates TGF-beta1-induced hepatic epithelial-to-mesenchymal transition and migration. Biomed. Pharmacother. 98, 214–221 (2018).
Zhu, H. et al. The SIRT2-mediated deacetylation of AKR1C1 is required for suppressing its pro-metastasis function in non-small cell lung cancer. Theranostics 10, 2188–2200 (2020).
Shen, X. et al. Wildtype IDH1 affects cell migration by modulating the PI3K/AKT/mTOR pathway in primary glioblastoma cells. Mol. Med. Rep. 22, 1949–1957 (2020).
Wang, B. et al. SIRT2-dependent IDH1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Rep. 21, e48183 (2020).
Liu, L. et al. SIRT3 inhibits gallbladder cancer by induction of AKT-dependent ferroptosis and blockade of epithelial-mesenchymal transition. Cancer Lett. 510, 93–104 (2021).
Sun, H. et al. SIRT4 acts as a tumor suppressor in gastric cancer by inhibiting cell proliferation, migration, and invasion. Onco. Targets Ther. 11, 3959–3968 (2018).
Li, D. J. et al. Nicotinic ACh receptor alpha7 inhibits PDGF-induced migration of vascular smooth muscle cells by activating mitochondrial deacetylase sirtuin 3. Br. J. Pharmacol. 176, 4388–4401 (2019).
Dang, S. et al. MiR-299-3p functions as a tumor suppressor via targeting Sirtuin 5 in hepatocellular carcinoma. Biomed. Pharmacother. 106, 966–975 (2018).
Yao, B. et al. Hypoxia-induced miR-3677-3p promotes the proliferation, migration and invasion of hepatocellular carcinoma cells by suppressing SIRT5. J. Cell Mol. Med 24, 8718–8731 (2020).
Han, L. L., Jia, L., Wu, F. & Huang, C. Sirtuin6 (SIRT6) promotes the EMT of hepatocellular carcinoma by stimulating autophagic degradation of E-cadherin. Mol. Cancer Res. 17, 2267–2280 (2019).
Lin, H., Hao, Y., Zhao, Z. & Tong, Y. Sirtuin 6 contributes to migration and invasion of osteosarcoma cells via the ERK1/2/MMP9 pathway. FEBS Open Bio 7, 1291–1301 (2017).
Deng, Z. et al. Sirtuin 7 promotes colorectal carcinoma proliferation and invasion through the inhibition of E-cadherin. Exp. Ther. Med. 15, 2333–2342 (2018).
Mao, S., Ma, J. & Yu, H. Sirtuin-7 knockdown inhibits the growth of endometrial cancer cells by inducing apoptosis via the NF-kappaB signaling pathway. Oncol. Lett. 17, 937–943 (2019).
Collaborators, G. B. D. C. O. D. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1736–1788 (2018).
Mei, Z. et al. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 35, 182 (2016).
Olmos, Y., Brosens, J. J. & Lam, E. W. Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist Updat 14, 35–44 (2011).
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 71, 209–249 (2021).
Shi, P., Zhou, M. & Yang, Y. Upregulated tumor sirtuin 2 expression correlates with reduced TNM stage and better overall survival in surgical breast cancer patients. Ir. J. Med. Sci. 189, 83–89 (2020).
Shi, Q. et al. Decreased sirtuin 4 expression is associated with poor prognosis in patients with invasive breast cancer. Oncol. Lett. 12, 2606–2612 (2016).
Xu, Y., Qin, Q., Chen, R., Wei, C. & Mo, Q. SIRT1 promotes proliferation, migration, and invasion of breast cancer cell line MCF-7 by upregulating DNA polymerase delta1 (POLD1). Biochem. Biophys. Res. Commun. 502, 351–357 (2018).
Geng, Q., Peng, H., Chen, F., Luo, R. & Li, R. High expression of Sirt7 served as a predictor of adverse outcome in breast cancer. Int. J. Clin. Exp. Pathol. 8, 1938–1945 (2015).
Wu, M. et al. Expression of SIRT1 is associated with lymph node metastasis and poor prognosis in both operable triple-negative and non-triple-negative breast cancer. Med. Oncol. 29, 3240–3249 (2012).
Sinha, S., Sharma, S., Vora, J. & Shrivastava, N. Emerging role of sirtuins in breast cancer metastasis and multidrug resistance: Implication for novel therapeutic strategies targeting sirtuins. Pharm. Res 158, 104880 (2020).
Jin, M. S. et al. SIRT1 induces tumor invasion by targeting epithelial mesenchymal transition-related pathway and is a prognostic marker in triple negative breast cancer. Tumour Biol. 37, 4743–4753 (2016).
Simic, P. et al. SIRT1 suppresses the epithelial-to-mesenchymal transition in cancer metastasis and organ fibrosis. Cell Rep. 3, 1175–1186 (2013).
Shi, L. et al. A SIRT1-centered circuitry regulates breast cancer stemness and metastasis. Oncogene 37, 6299–6315 (2018).
Eades, G. et al. miR-200a regulates SIRT1 expression and epithelial to mesenchymal transition (EMT)-like transformation in mammary epithelial cells. J. Biol. Chem. 286, 25992–26002 (2011).
Moore, R. L. & Faller, D. V. SIRT1 represses estrogen-signaling, ligand-independent ERα-mediated transcription, and cell proliferation in estrogen-responsive breast cells. J. Endocrinol. 216, 273–285 (2013).
Gollavilli, P. N. et al. AMPK inhibits MTDH expression via GSK3β and SIRT1 activation: potential role in triple negative breast cancer cell proliferation. Febs J. 282, 3971–3985 (2015).
Minten, E. V. et al. SIRT2 promotes BRCA1-BARD1 heterodimerization through deacetylation. Cell Rep. 34, 108921 (2021).
Sterneck, E., Poria, D. K. & Balamurugan, K. Slug and E-cadherin: stealth accomplices? Front Mol. Biosci. 7, 138 (2020).
Zhou, W. et al. The SIRT2 deacetylase stabilizes slug to control malignancy of basal-like breast cancer. Cell Rep. 17, 1302–1317 (2016).
Pinterić, M. et al. Sirt3 exerts its tumor-suppressive role by increasing p53 and attenuating response to estrogen in MCF-7 Cells. Antioxid. (Basel) 9, 294 (2020).
Du, L. et al. Loss of SIRT4 promotes the self-renewal of breast cancer stem cells. Theranostics 10, 9458–9476 (2020).
Choi, H. K. et al. SIRT1-mediated FoxO1 deacetylation is essential for multidrug resistance-associated protein 2 expression in tamoxifen-resistant breast cancer cells. Mol. Pharm. 10, 2517–2527 (2013).
Xing, J. et al. SIRT4 enhances the sensitivity of ER-positive breast cancer to tamoxifen by inhibiting the IL-6/STAT3 signal pathway. Cancer Med 8, 7086–7097 (2019).
Grbesa, I. et al. Expression of sirtuin 1 and 2 is associated with poor prognosis in non-small cell lung cancer patients. PLoS One 10, e0124670 (2015).
Yang, G. C. et al. The expression and related clinical significance of SIRT3 in non-small-cell lung cancer. Dis. Markers 2017, 8241953 (2017).
Tao, F. et al. SIRT3 acts as a novel biomarker for the diagnosis of lung cancer: A retrospective study. Med. (Baltim.) 100, e26580 (2021).
Azuma, Y. et al. SIRT6 expression is associated with poor prognosis and chemosensitivity in patients with non-small cell lung cancer. J. Surg. Oncol. 112, 231–237 (2015).
Palmirotta, R. et al. Sirtuins and cancer: role in the epithelial-mesenchymal transition. Oxid. Med. Cell Longev. 2016, 3031459 (2016).
Yuan, H., Su, L. & Chen, W. Y. The emerging and diverse roles of sirtuins in cancer: a clinical perspective. Onco Targets Ther. 6, 1399–1416 (2013).
Zhang, Z. et al. Long noncoding RNA SNHG10 sponges miR-543 to upregulate tumor suppressive SIRT1 in nonsmall cell lung cancer. Cancer Biother. Radiopharm. 35, 771–775 (2020).
Li, X., Jiang, Z., Li, X. & Zhang, X. SIRT1 overexpression protects non-small cell lung cancer cells against osteopontin-induced epithelial-mesenchymal transition by suppressing NF-κB signaling. Onco Targets Ther. 11, 1157–1171 (2018).
Yao, Y., Hua, Q., Zhou, Y. & Shen, H. CircRNA has_circ_0001946 promotes cell growth in lung adenocarcinoma by regulating miR-135a-5p/SIRT1 axis and activating Wnt/β-catenin signaling pathway. Biomed. Pharmacother. 111, 1367–1375 (2019).
Xu, Y. et al. Oxidative stress activates SIRT2 to deacetylate and stimulate phosphoglycerate mutase. Cancer Res 74, 3630–3642 (2014).
Hou, T. et al. SIRT6 coordinates with CHD4 to promote chromatin relaxation and DNA repair. Nucleic Acids Res 48, 2982–3000 (2020).
Li, Z. et al. SIRT6 drives epithelial-to-mesenchymal transition and metastasis in non-small cell lung cancer via snail-dependent transrepression of KLF4. J. Exp. Clin. Cancer Res. 37, 323 (2018).
Wei, J. et al. MicroRNA-326 impairs chemotherapy resistance in non small cell lung cancer by suppressing histone deacetylase SIRT1-mediated HIF1α and elevating VEGFA. Bioengineered 13, 5685–5699 (2022).
Li, Z., Xie, Q. R., Chen, Z., Lu, S. & Xia, W. Regulation of SIRT2 levels for human non-small cell lung cancer therapy. Lung Cancer 82, 9–15 (2013).
Cao, K. et al. Sirt3 promoted DNA damage repair and radioresistance through ATM-Chk2 in non-small cell lung cancer cells. J. Cancer 12, 5464–5472 (2021).
Wen, X. et al. Ubiquitin-specific protease 22/silent information regulator 1 axis plays a pivotal role in the prognosis and 5-fluorouracil resistance in hepatocellular carcinoma. Dig. Dis. Sci. 65, 1064–1073 (2020).
Xu, J. et al. In-depth profiling and quantification of the lysine acetylome in hepatocellular carcinoma with a trapped ion mobility mass spectrometer. Mol. Cell Proteom. 21, 100255 (2022).
Lee, N. et al. SIRT6 depletion suppresses tumor growth by promoting cellular senescence induced by DNA damage in HCC. PLoS One 11, e0165835 (2016).
Wang, J. X. et al. Down-regulation of sirtuin 3 is associated with poor prognosis in hepatocellular carcinoma after resection. BMC Cancer 14, 297 (2014).
Guo, D. et al. Vimentin acetylation is involved in SIRT5-mediated hepatocellular carcinoma migration. Am. J. Cancer Res. 8, 2453–2466.
Wang, Y. S. et al. Sirtuin 4 depletion promotes hepatocellular carcinoma tumorigenesis through regulating adenosine-monophosphate-activated protein kinase alpha/mammalian target of rapamycin axis in mice. Hepatology 69, 1614–1631 (2019).
Liu, L. et al. SIRT1-mediated transcriptional regulation of SOX2 is important for self-renewal of liver cancer stem cells. Hepatology 64, 814–827 (2016).
Zhao, J. et al. SIRT7 regulates hepatocellular carcinoma response to therapy by altering the p53-dependent cell death pathway. J. Exp. Clin. Cancer Res. 38, 252 (2019).
McGinnis, C. D., Jennings, E. Q., Harris, P. S., Galligan, J. J. & Fritz, K. S. Biochemical mechanisms of sirtuin-directed protein acylation in hepatic pathologies of mitochondrial dysfunction. Cells 11, 2045 (2022).
Sun, R. et al. Loss of SIRT5 promotes bile acid-induced immunosuppressive microenvironment and hepatocarcinogenesis. J. Hepatol. 77, 453–466 (2022).
Bai, L. et al. Overexpression of SLC25A51 promotes hepatocellular carcinoma progression by driving aerobic glycolysis through activation of SIRT5. Free Radic. Biol. Med 182, 11–22 (2022).
Yang, H. et al. Sirtuin-mediated deacetylation of hnRNP A1 suppresses glycolysis and growth in hepatocellular carcinoma. Oncogene 38, 4915–4931 (2019).
Du, L. et al. USP48 Is Upregulated by Mettl14 to Attenuate Hepatocellular Carcinoma via Regulating SIRT6 Stabilization. Cancer Res 81, 3822–3834 (2021).
Huang, F. Y. et al. Tumor suppressive role of mitochondrial sirtuin 4 in induction of G2/M cell cycle arrest and apoptosis in hepatitis B virus-related hepatocellular carcinoma. Cell Death Disco. 7, 88 (2021).
Jo, H. et al. Modulation of SIRT3 expression through CDK4/6 enhances the anti-cancer effect of sorafenib in hepatocellular carcinoma cells. BMC Cancer 20, 332 (2020).
Xia, Y. Q. et al. SIRT6 depletion sensitizes human hepatoma cells to chemotherapeutics by downregulating MDR1 expression. Front Pharm. 9, 194 (2018).
Jiang, K. et al. Overexpression of SIRT1 is a poor prognostic factor for advanced colorectal cancer. Chin. Med. J. (Engl.). 127, 2021–2024 (2014).
Du, F. et al. SIRT2, a direct target of miR-212-5p, suppresses the proliferation and metastasis of colorectal cancer cells. J. Cell Mol. Med. 24, 9985–9998 (2020).
Huang, G. et al. Clinical and therapeutic significance of sirtuin-4 expression in colorectal cancer. Oncol. Rep. 35, 2801–2810 (2016).
Zhang, Y. et al. SIRT6, a novel direct transcriptional target of FoxO3a, mediates colon cancer therapy. Theranostics 9, 2380–2394 (2019).
Wang, Y. Q. et al. Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nat. Commun. 9, 545 (2018).
Ren, N. S. X. et al. Haploinsufficiency of SIRT1 enhances glutamine metabolism and promotes cancer development. Curr. Biol. 27, 483–494 (2017).
Paku, M. et al. SIRT3-mediated SOD2 and PGC-1α contribute to chemoresistance in colorectal cancer cells. Ann. Surg. Oncol. 28, 4720–4732 (2021).
Yang, M. et al. FOXQ1-mediated SIRT1 upregulation enhances stemness and radio-resistance of colorectal cancer cells and restores intestinal microbiota function by promoting β-catenin nuclear translocation. J. Exp. Clin. Cancer Res. 41, 70 (2022).
Zhou, J., Wu, A., Yu, X., Zhu, J. & Dai, H. SIRT6 inhibits growth of gastric cancer by inhibiting JAK2/STAT3 pathway. Oncol. Rep. 38, 1059–1066 (2017).
Cai, S. et al. SIRT6 silencing overcomes resistance to sorafenib by promoting ferroptosis in gastric cancer. Biochem. Biophys. Res. Commun. 577, 158–164 (2021).
Hu, T. et al. Metabolic rewiring by loss of sirt5 promotes kras-induced pancreatic cancer progression. Gastroenterology 161, 1584–1600 (2021).
Tsai, Y. C. et al. Upregulating sirtuin 6 ameliorates glycolysis, EMT and distant metastasis of pancreatic adenocarcinoma with krüppel-like factor 10 deficiency. Exp. Mol. Med. 53, 1623–1635 (2021).
Zhang, J. G. et al. Sirtuin 1 facilitates chemoresistance of pancreatic cancer cells by regulating adaptive response to chemotherapy-induced stress. Cancer Sci. 105, 445–454 (2014).
Zeng, Z. et al. Gene expression and prognosis of sirtuin family members in ovarian cancer. Med. (Baltim.) 99, e20685 (2020).
Zhang, J. et al. The histone deacetylase SIRT6 inhibits ovarian cancer cell proliferation via down-regulation of Notch 3 expression. Eur. Rev. Med. Pharm. Sci. 19, 818–824 (2015).
Sun, X. et al. SIRT5 promotes cisplatin resistance in ovarian cancer by suppressing DNA damage in a ROS-dependent manner via regulation of the Nrf2/HO-1 pathway. Front Oncol. 9, 754 (2019).
Jiang, W., Jiang, P., Yang, R. & Liu, D. F. Functional role of SIRT1-induced HMGB1 expression and acetylation in migration, invasion and angiogenesis of ovarian cancer. Eur. Rev. Med. Pharmacol. Sci. 22, 4431–4439 (2018).
Tae, I. H. et al. A new SIRT1 inhibitor, MHY2245, induces autophagy and inhibits energy metabolism via PKM2/mTOR pathway in human ovarian cancer cells. Int. J. Biol. Sci. 16, 1901–1916 (2020).
Dong, X. C., Jing, L. M., Wang, W. X. & Gao, Y. X. Down-regulation of SIRT3 promotes ovarian carcinoma metastasis. Biochem. Biophys. Res. Commun. 475, 245–250 (2016).
McGuire, W. P. 3rd & Markman, M. Primary ovarian cancer chemotherapy: current standards of care. Br. J. Cancer 89, S3–S8 (2003). Suppl 3.
Wang, W. et al. ROS-induced SIRT2 upregulation contributes to cisplatin sensitivity in ovarian cancer. Antioxid. (Basel) 9, 1137 (2020).
Crosbie, E. J. et al. Endometrial cancer. Lancet 399, 1412–1428 (2022).
Guo, Y., Zhao, N., Zhou, J., Dong, J. & Wang, X. Sirtuin 2 in endometrial cancer: a potential regulator for cell proliferation, apoptosis and RAS/ERK pathway. Technol. Cancer Res. Treat. 19, 1533033820980781 (2020).
Fukuda, T. et al. Putative tumor suppression function of SIRT6 in endometrial cancer. FEBS Lett. 589, 2274–2281 (2015).
Asaka, R. et al. Sirtuin 1 promotes the growth and cisplatin resistance of endometrial carcinoma cells: a novel therapeutic target. Lab. Invest. 95, 1363–1373 (2015).
Zhao, N., Guo, Y., Liu, P., Chen, Y. & Wang, Y. Sirtuin 2 promotes cell stemness and MEK/ERK signaling pathway while reduces chemosensitivity in endometrial cancer. Arch. Gynecol. Obstet. 305, 693–701 (2022).
Yang, L. P. et al. SIRT2 expression exhibits potential to serve as a biomarker for disease surveillance and prognosis in the management of cervical cancer patients. Med. (Baltim.) 99, e18668 (2020).
So, D. et al. Cervical cancer is addicted to SIRT1 disarming the AIM2 antiviral defense. Oncogene 37, 5191–5204 (2018).
Xu, L. X., Hao, L. J., Ma, J. Q., Liu, J. K. & Hasim, A. SIRT3 promotes the invasion and metastasis of cervical cancer cells by regulating fatty acid synthase. Mol. Cell Biochem. 464, 11–20 (2020).
Cheng, J., Meng, J., Zhu, L. & Peng, Y. Exosomal noncoding RNAs in Glioma: biological functions and potential clinical applications. Mol. Cancer 19, 66 (2020).
Chen, H. et al. Sirtuin 1 knockdown inhibits glioma cell proliferation and potentiates temozolomide toxicity via facilitation of reactive oxygen species generation. Oncol. Lett. 17, 5343–5350 (2019).
Mu, P. et al. Sirtuin 7 promotes glioma proliferation and invasion through activation of the ERK/STAT3 signaling pathway. Oncol. Lett. 17, 1445–1452 (2019).
Luo, K., Huang, W. & Tang, S. Sirt3 enhances glioma cell viability by stabilizing Ku70-BAX interaction. Onco. Targets Ther. 11, 7559–7567 (2018).
Feng, J. et al. SIRT6 suppresses glioma cell growth via induction of apoptosis, inhibition of oxidative stress and suppression of JAK2/STAT3 signaling pathway activation. Oncol. Rep. 35, 1395–1402 (2016).
Chen, X. et al. SIRT5 downregulation is associated with poor prognosis in glioblastoma. Cancer Biomark. 24, 449–459 (2019).
Feng, X. et al. Hypoxia-induced acetylation of PAK1 enhances autophagy and promotes brain tumorigenesis via phosphorylating ATG5. Autophagy 17, 723–742 (2021).
Liu, R. et al. CDK1-mediated SIRT3 activation enhances mitochondrial function and tumor radioresistance. Mol. Cancer Ther. 14, 2090–2102 (2015).
Bispo, J. A. B., Pinheiro, P. S. & Kobetz, E. K. Epidemiology and etiology of leukemia and lymphoma. Cold Spring Harb. Perspect. Med 10, a034819 (2020).
Wang, F. et al. SIRT1 regulates the phosphorylation and degradation of P27 by deacetylating CDK2 to promote T-cell acute lymphoblastic leukemia progression. J. Exp. Clin. Cancer Res. 40, 259 (2021).
Morishima, T. et al. LMO2 activation by deacetylation is indispensable for hematopoiesis and T-ALL leukemogenesis. Blood 134, 1159–1175 (2019).
Sasca, D. et al. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood 124, 121–133 (2014).
Zhang, P. et al. Targeting DNA damage repair functions of two histone deacetylases, HDAC8 and SIRT6, sensitizes acute myeloid leukemia to NAMPT inhibition. Clin. Cancer Res. 27, 2352–2366 (2021).
Kaiser, A. et al. SIRT7: an influence factor in healthy aging and the development of age-dependent myeloid stem-cell disorders. Leukemia 34, 2206–2216 (2020).
Shi, Y. et al. Epigenetic regulation in cardiovascular disease: mechanisms and advances in clinical trials. Signal Transduct. Target Ther. 7, 200 (2022).
Abdelsayed, M., Kort, E. J., Jovinge, S. & Mercola, M. Repurposing drugs to treat cardiovascular disease in the era of precision medicine. Nat. Rev. Cardiol. 19, 751–764 (2022).
Heimlich, J. B. & Bick, A. G. Somatic mutations in cardiovascular disease. Circ. Res 130, 149–161 (2022).
Roth, G. A. et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J. Am. Coll. Cardiol. 76, 2982–3021 (2020).
Xu, S. et al. Targeting epigenetics and non-coding RNAs in atherosclerosis: from mechanisms to therapeutics. Pharm. Ther. 196, 15–43 (2019).
Grootaert, M. O. J. & Bennett, M. R. Sirtuins in atherosclerosis: guardians of healthspan and therapeutic targets. Nat. Rev. Cardiol. 19, 668–683 (2022).
Winnik, S., Auwerx, J., Sinclair, D. A. & Matter, C. M. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur. Heart J. 36, 3404–3412 (2015).
Gallo, S., Vitacolonna, A., Bonzano, A., Comoglio, P. & Crepaldi, T. ERK: A key player in the pathophysiology of cardiac hypertrophy. Int. J. Mol. Sci. 20, 2164 (2019).
Papait, R., Serio, S. & Condorelli, G. Role of the epigenome in heart failure. Physiol. Rev. 100, 1753–1777 (2020).
Zeng, N. et al. Diverging targets mediate the pathological roleof miR-199a-5p and miR-199a-3p by promoting cardiac hypertrophy and fibrosis. Mol. Ther. Nucleic Acids 26, 1035–1050 (2021).
Wang, W. et al. Circ-SIRT1 inhibits cardiac hypertrophy via activating SIRT1 to promote autophagy. Cell Death Dis. 12, 1069 (2021).
Shen, T. et al. SIRT1 functions as an important regulator of estrogen-mediated cardiomyocyte protection in angiotensin II-induced heart hypertrophy. Oxid. Med. Cell Longev. 2014, 713894 (2014).
Li, J. et al. Sirtuin 1 represses PKC-ζ activity through regulating interplay of acetylation and phosphorylation in cardiac hypertrophy. Br. J. Pharm. 176, 416–435 (2019).
Sundaresan, N. R. et al. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal 4, ra46 (2011).
Alcendor, R. R. et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 100, 1512–1521 (2007).
Sadria, M. & Layton, A. T. Interactions among mTORC, AMPK and SIRT: a computational model for cell energy balance and metabolism. Cell Commun. Signal 19, 57 (2021).
Ding, Y. Q. et al. MicroRNA-214 contributes to Ang II-induced cardiac hypertrophy by targeting SIRT3 to provoke mitochondrial malfunction. Acta Pharm. Sin. 42, 1422–1436 (2021).
Yue, Z. et al. NMNAT3 is involved in the protective effect of SIRT3 in Ang II-induced cardiac hypertrophy. Exp. Cell Res. 347, 261–273 (2016).
Wang, H. N. et al. Effects of Sirt3-autophagy and resveratrol activation on myocardial hypertrophy and energy metabolism. Mol. Med. Rep. 22, 1342–1350 (2020).
Li, J. et al. Mouse Sirt3 promotes autophagy in AngII-induced myocardial hypertrophy through the deacetylation of FoxO1. Oncotarget 7, 86648–86659 (2016).
Feng, X. et al. SIRT3 inhibits cardiac hypertrophy by regulating PARP-1 activity. Aging (Albany NY) 12, 4178–4192 (2020).
Pillai, V. B., Samant, S., Hund, S., Gupta, M. & Gupta, M. P. The nuclear sirtuin SIRT6 protects the heart from developing aging-associated myocyte senescence and cardiac hypertrophy. Aging (Albany NY) 13, 12334–12358 (2021).
Shen, P. et al. SIRT6 suppresses phenylephrine-induced cardiomyocyte hypertrophy though inhibiting p300. J. Pharm. Sci. 132, 31–40 (2016).
Sundaresan, N. R. et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat. Med. 18, 1643–1650 (2012).
Zhang, X. et al. STAT3 suppression is involved in the protective effect of SIRT6 against cardiomyocyte hypertrophy. J. Cardiovasc. Pharm. 68, 204–214 (2016).
Sarikhani, M. et al. SIRT2 deacetylase represses NFAT transcription factor to maintain cardiac homeostasis. J. Biol. Chem. 293, 5281–5294 (2018).
Tang, X. et al. SIRT2 acts as a cardioprotective deacetylase in pathological cardiac hypertrophy. Circulation 136, 2051–2067 (2017).
Sadhukhan, S. et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl Acad. Sci. USA 113, 4320–4325 (2016).
Yamamura, S. et al. Cardiomyocyte sirt (sirtuin) 7 ameliorates stress-induced cardiac hypertrophy by interacting with and deacetylating GATA4. Hypertension 75, 98–108 (2020).
Luo, Y. X. et al. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur. Heart J. 38, 1389–1398 (2017).
Edgley, A. J., Krum, H. & Kelly, D. J. Targeting fibrosis for the treatment of heart failure: a role for transforming growth factor-β. Cardiovasc. Ther. 30, e30–e40 (2012).
Liu, Z. H. et al. SIRT1 activation attenuates cardiac fibrosis by endothelial-to-mesenchymal transition. Biomed. Pharmacother. 118, 109227 (2019).
Chen, T. et al. Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF-β/Smad3 pathway. Am. J. Physiol. Heart Circ. Physiol. 308, H424–H434 (2015).
Maity, S. et al. Sirtuin 6 deficiency transcriptionally up-regulates TGF-β signaling and induces fibrosis in mice. J. Biol. Chem. 295, 415–434 (2020).
Zhang, Z. Z. et al. The sirtuin 6 prevents angiotensin II-mediated myocardial fibrosis and injury by targeting AMPK-ACE2 signaling. Oncotarget 8, 72302–72314 (2017).
Wang, H. et al. Enhanced expression and phosphorylation of Sirt7 activates smad2 and ERK signaling and promotes the cardiac fibrosis differentiation upon angiotensin-II stimulation. PLoS One 12, e0178530 (2017).
Lüscher, T. F. Risk factors for and management of heart failure. Eur. Heart J. 36, 2267–2269 (2015).
Groenewegen, A., Rutten, F. H., Mosterd, A. & Hoes, A. W. Epidemiology of heart failure. Eur. J. Heart Fail 22, 1342–1356 (2020).
Chen, J., Chen, S., Zhang, B. & Liu, J. SIRT3 as a potential therapeutic target for heart failure. Pharm. Res 165, 105432 (2021).
Saiyang, X., Deng, W. & Qizhu, T. Sirtuin 6: A potential therapeutic target for cardiovascular diseases. Pharm. Res 163, 105214 (2021).
Lu, T. M. et al. Downregulation of Sirt1 as aging change in advanced heart failure. J. Biomed. Sci. 21, 57 (2014).
Wu, Y. X., Xu, R. Y., Jiang, L., Chen, X. Y. & Xiao, X. J. MicroRNA-30a-5p promotes chronic heart failure in rats by targeting sirtuin-1 to activate the nuclear factor-κB/NOD-like receptor 3 signaling pathway. Cardiovasc Drugs Ther (2022).
Akkafa, F. et al. Reduced SIRT1 expression correlates with enhanced oxidative stress in compensated and decompensated heart failure. Redox Biol. 6, 169–173 (2015).
Sun, S., Wang, C. & Weng, J. MicroRNA-138-5p drives the progression of heart failure via inhibiting sirtuin 1 signaling. Mol. Med. Rep. 23, 276 (2021).
Lin, B. et al. Sirt1 improves heart failure through modulating the NF-κB p65/microRNA-155/BNDF signaling cascade. Aging (Albany NY) 13, 14482–14498 (2020).
Gorski, P. A. et al. Role of SIRT1 in modulating acetylation of the sarco-endoplasmic reticulum Ca(2+)-ATPase in heart failure. Circ. Res. 124, e63–e80 (2019).
Horton, J. L. et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2, e84897 (2016).
Zhang, X. et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation 137, 2052–2067 (2018).
Tong, D. et al. NAD(+) repletion reverses heart failure with preserved ejection fraction. Circ. Res 128, 1629–1641 (2021).
Benigni, A. et al. Sirt3 deficiency shortens life span and impairs cardiac mitochondrial function rescued by Opa1 gene transfer. Antioxid. Redox Signal 31, 1255–1271 (2019).
Zeng, H., He, X. & Chen, J. X. Endothelial sirtuin 3 dictates glucose transport to cardiomyocyte and sensitizes pressure overload-induced heart failure. J. Am. Heart Assoc. 9, e015895 (2020).
He, X., Zeng, H. & Chen, J. X. Emerging role of SIRT3 in endothelial metabolism, angiogenesis, and cardiovascular disease. J. Cell Physiol. 234, 2252–2265 (2019).
Li, Y. et al. Cardioprotective effects of SIRT6 in a mouse model of transverse aortic constriction-induced heart failure. Front Physiol. 8, 394 (2017).
Walker, M. A. et al. Acetylation of muscle creatine kinase negatively impacts high-energy phosphotransfer in heart failure. JCI Insight 6, e144301 (2021).
Chang, X. et al. SIRT5-related desuccinylation modification contributes to quercetin-induced protection against heart failure and high-glucose-prompted cardiomyocytes injured through regulation of mitochondrial quality surveillance. Oxid. Med. Cell Longev. 2021, 5876841 (2021).
Hansson, G. K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352, 1685–1695 (2005).
Gorenne, I. et al. Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation 127, 386–396 (2013).
Zhang, Q. J. et al. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res 80, 191–199 (2008).
Gano, L. B. et al. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am. J. Physiol. Heart Circ. Physiol. 307, H1754–H1763 (2014).
Miao, H., Zeng, H. & Gong, H. microRNA-212 promotes lipid accumulation and attenuates cholesterol efflux in THP-1 human macrophages by targeting SIRT1. Gene 643, 55–60 (2018).
Stein, S. et al. SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur. Heart J. 31, 2301–2309 (2010).
Zheng, H. et al. mTOR signaling promotes foam cell formation and inhibits foam cell egress through suppressing the SIRT1 signaling pathway. Mol. Med. Rep. 16, 3315–3323 (2017).
Zhang, L. et al. MicroRNA-217 is involved in the progression of atherosclerosis through regulating inflammatory responses by targeting sirtuin 1. Mol. Med. Rep. 20, 3182–3190 (2019).
Yang, J. et al. LncRNA MALAT1 enhances ox-LDL-induced autophagy through the SIRT1/MAPK/NF-κB pathway in macrophages. Curr. Vasc. Pharm. 18, 652–662 (2020).
Zhang, B., Ma, Y. & Xiang, C. SIRT2 decreases atherosclerotic plaque formation in low-density lipoprotein receptor-deficient mice by modulating macrophage polarization. Biomed. Pharmacother. 97, 1238–1242 (2018).
Jing, S. H., Yu, B. & Qiao, H. Correlation between endothelial cell apoptosis and SIRT3 gene expression in atherosclerosis rats. Eur. Rev. Med. Pharm. Sci. 23, 9033–9040 (2019).
Wang, P., Zhang, H. & Wang, Y. Circ_0003423 alleviates oxidized low-density lipoprotein-induced endothelial cell injury by sponging miR-142-3p and activating sirtuin 3/Superoxide dismutase 2 pathway. J. Surg. Res. 277, 384–397 (2022).
Tao, Y. et al. SIRT4 suppresses the PI3K/Akt/NF-κB signaling pathway and attenuates HUVEC injury induced by oxLDL. Mol. Med. Rep. 19, 4973–4979 (2019).
Xu, S. et al. SIRT6 protects against endothelial dysfunction and atherosclerosis in mice. Aging (Albany NY) 8, 1064–1082 (2016).
Liu, Z., Wang, J., Huang, X., Li, Z. & Liu, P. Deletion of sirtuin 6 accelerates endothelial dysfunction and atherosclerosis in apolipoprotein E-deficient mice. Transl. Res. 172, 18–29.e12 (2016).
Arsiwala, T. et al. Sirt6 deletion in bone marrow-derived cells increases atherosclerosis - Central role of macrophage scavenger receptor 1. J. Mol. Cell Cardiol. 139, 24–32 (2020).
Zheng, J. et al. SIRT7 regulates the vascular smooth muscle cells proliferation and migration via Wnt/β-catenin signaling pathway. Biomed. Res. Int. 2018, 4769596 (2018).
Kitada, M., Ogura, Y. & Koya, D. The protective role of Sirt1 in vascular tissue: its relationship to vascular aging and atherosclerosis. Aging (Albany NY) 8, 2290–2307 (2016).
Zhang, Y. et al. MicroRNAs or long noncoding RNAs in diagnosis and prognosis of coronary artery disease. Aging Dis. 10, 353–366 (2019).
Cassar, A., Holmes, D. R. Jr, Rihal, C. S. & Gersh, B. J. Chronic coronary artery disease: diagnosis and management. Mayo Clin. Proc. 84, 1130–1146 (2009).
Song, X. et al. Association of sirtuin gene polymorphisms with susceptibility to coronary artery disease in a North Chinese population. Biomed. Res. Int. 2022, 4294008 (2022).
Heidari, L. et al. Reverse expression pattern of sirtuin-1 and histone deacetylase-9 in coronary artery disease. Arch. Physiol. Biochem., 1-8 (2020).
Chan, S. H. et al. SIRT1 inhibition causes oxidative stress and inflammation in patients with coronary artery disease. Redox Biol. 13, 301–309 (2017).
Du, S., Shen, S., Ding, S. & Wang, L. Suppression of microRNA-323-3p restrains vascular endothelial cell apoptosis via promoting sirtuin-1 expression in coronary heart disease. Life Sci. 270, 119065 (2021).
Wang, H. et al. Long noncoding RNAs C2dat1 enhances vascular smooth muscle cell proliferation and migration by targeting MiR-34a-5p. J. Cell Biochem. 120, 3001–3008 (2019).
Yang, M., Xi, N., Gao, M. & Yu, Y. Sitagliptin mitigates hypoxia/reoxygenation (H/R)-induced injury in cardiomyocytes by mediating sirtuin 3 (SIRT3) and autophagy. Bioengineered 13, 13162–13173 (2022).
Herr, D. J., Singh, T., Dhammu, T. & Menick, D. R. Regulation of metabolism by mitochondrial enzyme acetylation in cardiac ischemia-reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165728 (2020).
Zhao, S. & Yu, L. Sirtuin 1 activated by SRT1460 protects against myocardial ischemia/reperfusion injury. Clin. Hemorheol. Microcirc. 78, 271–281 (2021).
Du, J. K. et al. Upregulation of microRNA-22 contributes to myocardial ischemia-reperfusion injury by interfering with the mitochondrial function. Free Radic. Biol. Med 96, 406–417 (2016).
Hsu, C. P. et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 122, 2170–2182 (2010).
Zhou, Y., Li, K. S., Liu, L. & Li, S. L. MicroRNA-132 promotes oxidative stress-induced pyroptosis by targeting sirtuin 1 in myocardial ischaemia-reperfusion injury. Int. J. Mol. Med. 45, 1942–1950 (2020).
Niu, X. et al. lncRNA Oip5-as1 attenuates myocardial ischaemia/reperfusion injury by sponging miR-29a to activate the SIRT1/AMPK/PGC1α pathway. Cell Prolif. 53, e12818 (2020).
Wang, X. X. et al. SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3α-dependent antioxidant defense mechanisms. Basic Res. Cardiol. 111, 13 (2016).
Li, X. et al. SIRT6 Protects against myocardial ischemia-reperfusion injury by attenuating aging-related CHMP2B accumulation. J. Cardiovasc. Transl. Res 15, 740–753 (2022).
Porter, G. A., Urciuoli, W. R., Brookes, P. S. & Nadtochiy, S. M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts. Am. J. Physiol. Heart Circ. Physiol. 306, H1602–H1609 (2014).
Zeng, G., Liu, H. & Wang, H. Amelioration of myocardial ischemia-reperfusion injury by SIRT4 involves mitochondrial protection and reduced apoptosis. Biochem. Biophys. Res. Commun. 502, 15–21 (2018).
Boylston, J. A. et al. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J. Mol. Cell Cardiol. 88, 73–81 (2015).
Zhou, B., Perel, P., Mensah, G. A. & Ezzati, M. Global epidemiology, health burden and effective interventions for elevated blood pressure and hypertension. Nat. Rev. Cardiol. 18, 785–802 (2021).
Mills, K. T. et al. Global disparities of hypertension prevalence and control: a systematic analysis of population-based studies from 90 countries. Circulation 134, 441–450 (2016).
Gao, P. et al. Overexpression of SIRT1 in vascular smooth muscle cells attenuates angiotensin II-induced vascular remodeling and hypertension in mice. J. Mol. Med. (Berl.) 92, 347–357 (2014).
Dikalova, A. E. et al. Mitochondrial deacetylase sirt3 reduces vascular dysfunction and hypertension while sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress. Circ. Res. 126, 439–452 (2020).
Gao, D. et al. Activation of SIRT1 attenuates klotho deficiency-induced arterial stiffness and hypertension by enhancing AMP-activated protein kinase activity. Hypertension 68, 1191–1199 (2016).
Zhou, L. et al. NAMPT/SIRT1 attenuate Ang II-induced vascular remodeling and vulnerability to hypertension by inhibiting the ROS/MAPK pathway. Oxid. Med. Cell Longev. 2020, 1974265 (2020).
Dikalova, A. E. et al. Sirt3 Impairment and SOD2 hyperacetylation in vascular oxidative stress and hypertension. Circ. Res 121, 564–574 (2017).
Martinez-Arroyo, O. et al. Decreased urinary levels of SIRT1 as non-invasive biomarker of early renal damage in hypertension. Int. J. Mol. Sci. 21, 6390 (2020).
Lin, J. R. et al. Suppression of endothelial-to-mesenchymal transition by sirt (sirtuin) 3 alleviated the development of hypertensive renal injury. Hypertension 72, 350–360 (2018).
Guo, J. et al. Endothelial SIRT6 is vital to prevent hypertension and associated cardiorenal injury through targeting Nkx3.2-GATA5 signaling. Circ. Res. 124, 1448–1461 (2019).
Zhou, Y., Zhang, F. & Ding, J. As a modulator, multitasking roles of SIRT1 in respiratory diseases. Immune Netw. 22, e21 (2022).
Chun, P. Role of sirtuins in chronic obstructive pulmonary disease. Arch. Pharm. Res. 38, 1–10 (2015).
Labaki, W. W. & Rosenberg, S. R. Chronic obstructive pulmonary disease. Ann. Intern. Med. 173, Itc17–itc32 (2020).
Zhang, X. Y. et al. Roles of sirtuin family members in chronic obstructive pulmonary disease. Respir. Res 23, 66 (2022).
Soriano, J. B. et al. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Respiratory Med. 8, 585–596 (2020).
Gu, C. et al. Therapeutic effects of SRT2104 on lung injury in rats with emphysema via reduction of type II alveolar epithelial cell senescence. Copd 17, 444–451 (2020).
Barnes, P. J. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin. Chest. Med. 35, 71–86 (2014).
Yao, H. et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J. Clin. Invest. 122, 2032–2045 (2012).
Zhang, L. et al. SIRT1 attenuates endoplasmic reticulum stress and apoptosis in rat models of COPD. Growth Factors 38, 94–104 (2020).
Rajendrasozhan, S., Yang, S. R., Kinnula, V. L. & Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 177, 861–870 (2008).
Yao, H. et al. Disruption of sirtuin 1-mediated control of circadian molecular clock and inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 53, 782–792 (2015).
Taguchi, A. et al. Resveratrol suppresses inflammatory responses in endometrial stromal cells derived from endometriosis: a possible role of the sirtuin 1 pathway. J. Obstet. Gynaecol. Res. 40, 770–778 (2014).
Barnes, P. J. Mechanisms of development of multimorbidity in the elderly. Eur. Respir. J. 45, 790–806 (2015).
Zhang, M. et al. Sirtuin 3 inhibits airway epithelial mitochondrial oxidative stress in cigarette smoke-induced COPD. Oxid. Med. Cell Longev. 2020, 7582980 (2020).
Hwang, J. W. et al. Cigarette smoke-induced autophagy is regulated by SIRT1-PARP-1-dependent mechanism: implication in pathogenesis of COPD. Arch. Biochem. Biophys. 500, 203–209 (2010).
Jin, Y. et al. Double deletion of tetraspanins CD9 and CD81 in mice leads to a syndrome resembling accelerated aging. Sci. Rep. 8, 5145 (2018).
Barnes, P. J. Senescence in COPD and its comorbidities. Annu. Rev. Physiol. 79, 517–539 (2017).
Mazumder, S., Barman, M., Bandyopadhyay, U. & Bindu, S. Sirtuins as endogenous regulators of lung fibrosis: A current perspective. Life Sci. 258, 118201 (2020).
Varone, F. et al. Fibrotic hypersensitivity pneumonitis: diagnosis and management. Lung 198, 429–440 (2020).
Liang, J. et al. The ZIP8/SIRT1 axis regulates alveolar progenitor cell renewal in aging and idiopathic pulmonary fibrosis. J. Clin. Invest. 132, e157338 (2022).
Zeng, Z. et al. Activation and overexpression of Sirt1 attenuates lung fibrosis via P300. Biochem. Biophys. Res. Commun. 486, 1021–P1026 (2017).
Han, X. et al. Targeting Sirtuin1 to treat aging-related tissue fibrosis: From prevention to therapy. Pharm. Ther. 229, 107983 (2022).
Sosulski, M. L., Gongora, R., Feghali-Bostwick, C., Lasky, J. A. & Sanchez, C. G. Sirtuin 3 deregulation promotes pulmonary fibrosis. J. Gerontol. A Biol. Sci. Med. Sci. 72, 595–602 (2017).
Sime, P. J., Xing, Z., Graham, F. L., Csaky, K. G. & Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 100, 768–776 (1997).
Jablonski, R. P. et al. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. Faseb J. 31, 2520–2532 (2017).
Cheresh, P. et al. SIRT3 overexpression ameliorates asbestos-induced pulmonary fibrosis, mt-DNA damage, and lung fibrogenic monocyte recruitment. Int. J. Mol. Sci. 22, 6856 (2021).
Minagawa, S. et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 300, L391–L401 (2011).
Chen, P. et al. Sirtuin 6 inhibits MWCNTs-induced epithelial-mesenchymal transition in human bronchial epithelial cells via inactivating TGF-β1/Smad2 signaling pathway. Toxicol. Appl. Pharm. 374, 1–10 (2019).
Zhang, Q. et al. Sirtuin 6 inhibits myofibroblast differentiation via inactivating transforming growth factor-β1/Smad2 and nuclear factor-κB signaling pathways in human fetal lung fibroblasts. J. Cell Biochem. 120, 93–104 (2019).
Tian, K. et al. Sirtuin 6 inhibits epithelial to mesenchymal transition during idiopathic pulmonary fibrosis via inactivating TGF-β1/Smad3 signaling. Oncotarget 8, 61011–61024 (2017).
Wyman, A. E. et al. Sirtuin 7 is decreased in pulmonary fibrosis and regulates the fibrotic phenotype of lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 312, L945–l958 (2017).
Liu, Y. et al. Anthocyanins inhibit airway inflammation by downregulating the NF-κB pathway via the miR-138-5p/SIRT1 axis in asthmatic mice. Int. Arch. Allergy Immunol. 183, 539–551 (2022).
Kaur, R. & Chupp, G. Phenotypes and endotypes of adult asthma: Moving toward precision medicine. J. Allergy Clin. Immunol. 144, 1–12 (2019).
Luo, Y., Wang, M. & Tian, Y. Trends and age-period-cohort effects on incidence and mortality of asthma in Sichuan Province, China, 1990-2019. BMC Pulm. Med 22, 298 (2022).
Fukuda, Y. et al. Virus-induced asthma exacerbations: SIRT1 targeted approach. J. Clin. Med. 9, 2623 (2020).
Colley, T. et al. Defective sirtuin-1 increases IL-4 expression through acetylation of GATA-3 in patients with severe asthma. J. Allergy Clin. Immunol. 137, 1595–1597.e1597 (2016).
Tang, L. et al. Suppression of sirtuin-1 increases IL-6 expression by activation of the Akt pathway during allergic asthma. Cell Physiol. Biochem 43, 1950–1960 (2017).
Zhang, Y. Z. et al. Effects of SIRT1/Akt pathway on chronic inflammatory response and lung function in patients with asthma. Eur. Rev. Med. Pharm. Sci. 23, 4948–4953 (2019).
Wang, Y. et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4(+) T cells. Immunity 44, 1337–1349 (2016).
Lee, Y. G. et al. Sirtuin 2 enhances allergic asthmatic inflammation. JCI Insight 4, e124710 (2019).
Kim, Y. Y. et al. AGK2 ameliorates mast cell-mediated allergic airway inflammation and fibrosis by inhibiting FcεRI/TGF-β signaling pathway. Pharm. Res 159, 105027 (2020).
Song, J. & Wang, J. SIRT3 regulates bronchial epithelium apoptosis and aggravates airway inflammation in asthma. Mol. Med. Rep. 25, 144 (2022).
Elias, J. A. Airway remodeling in asthma unanswered questions. Am. J. Respir. Crit. Care Med 161, S168–S171 (2000).
Wawrzyniak, P. et al. Regulation of bronchial epithelial barrier integrity by type 2 cytokines and histone deacetylases in asthmatic patients. J. Allergy Clin. Immunol. 139, 93–103 (2017).
Liu, F. & Shang, Y. X. Sirtuin 6 attenuates epithelial-mesenchymal transition by suppressing the TGF-β1/Smad3 pathway and c-Jun in asthma models. Int. Immunopharmacol. 82, 106333 (2020).
Fang, P. et al. SIRT7 regulates the TGF-β1-induced proliferation and migration of mouse airway smooth muscle cells by modulating the expression of TGF-β receptor I. Biomed. Pharmacother. 104, 781–787 (2018).
Qu, L. et al. Caveolin-1 identified as a key mediator of acute lung injury using bioinformatics and functional research. Cell Death Dis. 13, 686 (2022).
Li, T. et al. Resveratrol reduces acute lung injury in a LPS-induced sepsis mouse model via activation of Sirt1. Mol. Med. Rep. 7, 1889–1895 (2013).
Matthay, M. A. et al. Acute respiratory distress syndrome. Nat. Rev. Dis. Prim. 5, 18 (2019).
Weng, C. et al. Nanoscale porphyrin metal-organic frameworks deliver siRNA for alleviating early pulmonary fibrosis in acute lung injury. Front Bioeng. Biotechnol. 10, 939312 (2022).
Yang, F. et al. MicroRNA-499-5p targets SIRT1 to aggravate lipopolysaccharide-induced acute lung injury. Free Radic. Res 55, 71–82 (2021).
Gao, R. et al. Sirt1 restrains lung inflammasome activation in a murine model of sepsis. Am. J. Physiol. Lung Cell Mol. Physiol. 308, L847–L853 (2015).
Huang, J. et al. The SIRT1 inhibitor EX-527 suppresses mTOR activation and alleviates acute lung injury in mice with endotoxiemia. Innate Immun. 23, 678–686 (2017).
Tian, Y. G. & Zhang, J. Protective effect of SIRT3 on acute lung injury by increasing manganese superoxide dismutase-mediated antioxidation. Mol. Med. Rep. 17, 5557–5565 (2018).
Wang, Q. L. et al. Sirtuin 6 regulates macrophage polarization to alleviate sepsis-induced acute respiratory distress syndrome via dual mechanisms dependent on and independent of autophagy. Cytotherapy 24, 149–160 (2022).
Elangovan, V. R. et al. Endotoxin- and mechanical stress-induced epigenetic changes in the regulation of the nicotinamide phosphoribosyltransferase promoter. Pulm. Circ. 6, 539–544 (2016).
Li, S. et al. Resveratrol protects mice from paraquat-induced lung injury: The important role of SIRT1 and NRF2 antioxidant pathways. Mol. Med. Rep. 13, 1833–1838 (2016).
Zhang, R. et al. Ginkgolide C alleviates acute lung injury caused by paraquat poisoning via regulating the Nrf2 and NF-κB signaling pathways. Oxid. Med. Cell Longev. 2022, 7832983 (2022).
Moore, J. B. & June, C. H. Cytokine release syndrome in severe COVID-19. Science 368, 473–474 (2020).
Bordoni, V. et al. The unbalanced p53/SIRT1 axis may impact lymphocyte homeostasis in COVID-19 patients. Int. J. Infect. Dis. 105, 49–53 (2021).
Zhang, P., Zhao, T. & Zhou, W. The clinical significance of SIRT3 in COVID-19 patients: a single center retrospective analysis. Ann. Clin. Lab. Sci. 51, 686–693 (2021).
Georgieva, A. M. et al. Inactivation of Sirt6 ameliorates muscular dystrophy in mdx mice by releasing suppression of utrophin expression. Nat. Commun. 13, 4184 (2022).
Wu, S. & Liu, H. Sirtuins-novel regulators of epigenetic alterations in airway inflammation. Front Genet 13, 862577 (2022).
Chen, H. Y. et al. Artificial intelligence: Emerging player in the diagnosis and treatment of digestive disease. World J. Gastroenterol. 28, 2152–2162 (2022).
Mathews, S. C. et al. Prevalence and financial burden of digestive diseases in a commercially insured population. Clin. Gastroenterol. Hepatol. 20, 1480–1487.e1487 (2022).
Zhang, J. et al. Sirt6 alleviated liver fibrosis by deacetylating conserved lysine 54 on Smad2 in hepatic stellate cells. Hepatology 73, 1140–1157 (2021).
Yu, Y., Cai, J., She, Z. & Li, H. Insights into the epidemiology, pathogenesis, and therapeutics of nonalcoholic fatty liver diseases. Adv. Sci. (Weinh.) 6, 1801585 (2019).
Younossi, Z. & Henry, L. Contribution of alcoholic and nonalcoholic fatty liver disease to the burden of liver-related morbidity and mortality. Gastroenterology 150, 1778–1785 (2016).
Cotter, T. G. & Rinella, M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology 158, 1851–1864 (2020).
Asrani, S. K., Devarbhavi, H., Eaton, J. & Kamath, P. S. Burden of liver diseases in the world. J. Hepatol. 70, 151–171 (2019).
Guo, L. et al. Acetylation of mitochondrial trifunctional protein α-subunit enhances its stability to promote fatty acid oxidation and is decreased in nonalcoholic fatty liver disease. Mol. Cell Biol. 36, 2553–2567 (2016).
Wu, T., Liu, Y. H., Fu, Y. C., Liu, X. M. & Zhou, X. H. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 44, 410–418 (2014).
Kundu, A. et al. EX-527 Prevents the progression of high-fat diet-induced hepatic steatosis and fibrosis by upregulating SIRT4 in Zucker rats. Cells 9, 1101 (2020).
Linden, A. G. et al. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 59, 475–487 (2018).
Ponugoti, B. et al. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 285, 33959–33970 (2010).
Li, M. et al. SIRT1 antagonizes liver fibrosis by blocking hepatic stellate cell activation in mice. Faseb J. 32, 500–511 (2018).
Derdak, Z. et al. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J. Hepatol. 58, 785–791 (2013).
Yin, H. et al. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 146, 801–811 (2014).
Centers for Disease Control (CDC). Changing patterns of acquired immunodeficiency syndrome in hemophilia patients-United States. MMWR Morb. Mortal. Wkly. Rep. 34, 241–243 (1985).
Kim, H. G. et al. The epigenetic regulator SIRT6 protects the liver from alcohol-induced tissue injury by reducing oxidative stress in mice. J. Hepatol. 71, 960–969 (2019).
Ren, H. et al. Sirtuin 2 prevents liver steatosis and metabolic disorders by deacetylation of hepatocyte nuclear factor 4α. Hepatology 74, 723–740 (2021).
Liu, Y. et al. Retinol-binding protein 4 induces hepatic mitochondrial dysfunction and promotes hepatic steatosis. J. Clin. Endocrinol. Metab. 101, 4338–4348 (2016).
Nassir, F., Arndt, J. J., Johnson, S. A. & Ibdah, J. A. Regulation of mitochondrial trifunctional protein modulates nonalcoholic fatty liver disease in mice. J. Lipid Res. 59, 967–973 (2018).
Goetzman, E. S. et al. Impaired mitochondrial medium-chain fatty acid oxidation drives periportal macrovesicular steatosis in sirtuin-5 knockout mice. Sci. Rep. 10, 18367 (2020).
Wang, H. et al. miRNA-130b-5p promotes hepatic stellate cell activation and the development of liver fibrosis by suppressing SIRT4 expression. J. Cell Mol. Med. 25, 7381–7394 (2021).
Arteaga, M. et al. Inhibition of SIRT2 suppresses hepatic fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G1155–G1168 (2016).
Ma, Y. et al. Hepatic SIRT3 upregulation in response to chronic alcohol consumption contributes to alcoholic liver disease in mice. Front Physiol. 10, 1042 (2019).
Roychowdhury, S. et al. An early complement-dependent and TLR-4-independent phase in the pathogenesis of ethanol-induced liver injury in mice. Hepatology 49, 1326–1334 (2009).
Zhou, Z. et al. Intestinal SIRT1 deficiency protects mice from ethanol-induced liver injury by mitigating ferroptosis. Am. J. Pathol. 190, 82–92 (2020).
Li, S. et al. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology 66, 936–952 (2017).
Zhai, Y., Petrowsky, H., Hong, J. C., Busuttil, R. W. & Kupiec-Weglinski, J. W. Ischaemia-reperfusion injury in liver transplantation-from bench to bedside. Nat. Rev. Gastroenterol. Hepatol. 10, 79–89 (2013).
Nakamura, K. et al. Sirtuin 1 attenuates inflammation and hepatocellular damage in liver transplant ischemia/Reperfusion: From mouse to human. Liver Transpl. 23, 1282–1293 (2017).
Nakamura, K. et al. Heme oxygenase-1 regulates sirtuin-1-autophagy pathway in liver transplantation: From mouse to human. Am. J. Transpl. 18, 1110–1121 (2018).
Kadono, K. et al. Myeloid Ikaros-SIRT1 signaling axis regulates hepatic inflammation and pyroptosis in ischemia-stressed mouse and human liver. J. Hepatol. 76, 896–909 (2022).
Wang, Q. et al. TGR5 deficiency aggravates hepatic ischemic/reperfusion injury via inhibiting SIRT3/FOXO3/HIF-1ɑ pathway. Cell Death Disco. 6, 116 (2020).
Wang, J. et al. Sirtuin 2 aggravates postischemic liver injury by deacetylating mitogen-activated protein kinase phosphatase-1. Hepatology 65, 225–236 (2017).
Prifti, G. M. et al. Recent advances in hepatitis B treatment. Pharm. (Basel) 14, 417 (2021).
Ren, J. H. et al. SIRT3 restricts hepatitis B virus transcription and replication through epigenetic regulation of covalently closed circular DNA involving suppressor of variegation 3-9 homolog 1 and SET domain containing 1A histone methyltransferases. Hepatology 68, 1260–1276 (2018).
Yuan, S. et al. Multiomics interrogation into HBV (Hepatitis B virus)-host interaction reveals novel coding potential in human genome, and identifies canonical and non-canonical proteins as host restriction factors against HBV. Cell Disco. 7, 105 (2021).
Jenke, A. C. et al. Restitution of gene expression and histone acetylation signatures altered by hepatitis B virus through antiviral microRNA-like molecules in nontransformed murine hepatocytes. Clin. Epigenetics 6, 26 (2014).
Deng, J. J. et al. Interplay between SIRT1 and hepatitis B virus X protein in the activation of viral transcription. Biochim. Biophys. Acta Gene Regul. Mech. 1860, 491–501 (2017).
Yu, H. B. et al. SIRT7 restricts HBV transcription and replication through catalyzing desuccinylation of histone H3 associated with cccDNA minichromosome. Clin. Sci. (Lond.) 135, 1505–1522 (2021).
Cao, P., Chen, Q., Shi, C. X., Wang, L. W. & Gong, Z. J. Sirtuin1 attenuates acute liver failure by reducing reactive oxygen species via hypoxia inducible factor 1α. World J. Gastroenterol. 28, 1798–1813 (2022).
Zhao, X. et al. MicroRNA-128-3p aggravates doxorubicin-induced liver injury by promoting oxidative stress via targeting Sirtuin-1. Pharm. Res 146, 104276 (2019).
LoBianco, F. V. et al. The role of sirtuin 3 in radiation-induced long-term persistent liver injury. Antioxid. (Basel) 9, 409 (2020).
Kemelo, M. K., Wojnarová, L., Kutinová Canová, N. & Farghali, H. D-galactosamine/lipopolysaccharide-induced hepatotoxicity downregulates sirtuin 1 in rat liver: role of sirtuin 1 modulation in hepatoprotection. Physiol. Res 63, 615–623 (2014).
Kemelo, M. K., Kutinová Canová, N., Horinek, A. & Farghali, H. Sirtuin-activating compounds (STACs) alleviate D-galactosamine/lipopolysaccharide-induced hepatotoxicity in rats: involvement of sirtuin 1 and heme oxygenase 1. Physiol. Res 66, 497–505 (2017).
Zhou, Y. et al. Protection of CD4+ T cells from hepatitis C virus infection-associated senescence via DeltaNp63-miR-181a-Sirt1 pathway. J. Leukoc. Biol. 100, 1201–1211 (2016).
Sun, L. J. et al. Hepatitis C virus core protein induces dysfunction of liver sinusoidal endothelial cell by down-regulation of silent information regulator 1. J. Med. Virol. 90, 926–935 (2018).
Zhang, C. et al. Hepatitis C virus core protein induces hepatic steatosis via Sirt1-dependent pathway. Liver Int 38, 803–812 (2018).
Mohamed, G. A. et al. Cucurbitacin E glucoside alleviates concanavalin A-induced hepatitis through enhancing SIRT1/Nrf2/HO-1 and inhibiting NF-kB/NLRP3 signaling pathways. J. Ethnopharmacol. 292, 115223 (2022).
Huang, T. H., Chen, C. C., Liu, H. M., Lee, T. Y. & Shieh, S. H. Resveratrol pretreatment attenuates concanavalin a-induced hepatitis through reverse of aberration in the immune response and regenerative capacity in aged mice. Sci. Rep. 7, 2705 (2017).
Steinmetz, C. et al. Activation of silent mating type information regulation 2 homolog 1 by human chorionic gonadotropin exerts a therapeutic effect on hepatic injury and inflammation. Hepatology 65, 2074–2089 (2017).
Chenari, S., Safari, F. & Moradi, A. Curcumin enhances liver SIRT3 expression in the rat model of cirrhosis. Iran. J. Basic Med. Sci. 20, 1306–1311 (2017).
Zhang, Y. et al. Quercetin attenuates NLRP3 inflammasome activation and apoptosis to protect INH-induced liver injury via regulating SIRT1 pathway. Int Immunopharmacol. 85, 106634 (2020).
Liu, C. et al. Hepatic SIRT6 modulates transcriptional activities of FXR to alleviate acetaminophen-induced hepatotoxicity. Cell Mol. Gastroenterol. Hepatol. 14, 271–293 (2022).
Cui, X. et al. Inactivation of Sirt1 in mouse livers protects against endotoxemic liver injury by acetylating and activating NF-κB. Cell Death Dis. 7, e2403 (2016).
Kazgan, N. et al. Intestine-specific deletion of SIRT1 in mice impairs DCoH2-HNF-1α-FXR signaling and alters systemic bile acid homeostasis. Gastroenterology 146, 1006–1016 (2014).
Yu, L. et al. Protective effects of SRT1720 via the HNF1α/FXR signalling pathway and anti-inflammatory mechanisms in mice with estrogen-induced cholestatic liver injury. Toxicol. Lett. 264, 1–11 (2016).
Isaacs-Ten, A. et al. MetaBolic Regulation Of Macrophages by SIRT1 determines activation during cholestatic liver disease in mice. Cell Mol. Gastroenterol. Hepatol. 13, 1019–1039 (2022).
Petrov, M. S. & Yadav, D. Global epidemiology and holistic prevention of pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 16, 175–184 (2019).
Liu, Y., Sun, Y., Xue, B. H., Wang, X. D. & Yu, W. L. Negative regulation of SIRT1 by IRF9 involved in hyperlipidemia acute pancreatitis associated with kidney injury. Dig. Dis. Sci. 66, 1063–1071 (2021).
Wang, N. et al. Resveratrol protects against L-arginine-induced acute necrotizing pancreatitis in mice by enhancing SIRT1-mediated deacetylation of p53 and heat shock factor 1. Int. J. Mol. Med. 40, 427–437 (2017).
Deng, S. et al. Chronic pancreatitis and pancreatic cancer demonstrate active epithelial-mesenchymal transition profile, regulated by miR-217-SIRT1 pathway. Cancer Lett. 355, 184–191 (2014).
Collaborators, G.B.D.I.B.D. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 5, 17–30 (2020).
Liu, F. et al. Sirtuin-6 preserves R-spondin-1 expression and increases resistance of intestinal epithelium to injury in mice. Mol. Med 23, 272–284 (2017).
Lo Sasso, G. et al. SIRT2 deficiency modulates macrophage polarization and susceptibility to experimental colitis. PLoS One 9, e103573 (2014).
Xu, K. et al. Protective effects of SIRT6 overexpression against DSS-induced Colitis in mice. Cells 9, 1513 (2020).
Zhang, Y. et al. Crosstalk between gut microbiota and Sirtuin-3 in colonic inflammation and tumorigenesis. Exp. Mol. Med. 50, 1–11 (2018).
Wang, F. et al. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1β production and to prevent DSS-induced Colitis in mice. Cell Rep. 19, 2331–2344 (2017).
Liu, X., Li, C., Li, Q., Chang, H. C. & Tang, Y. C. SIRT7 facilitates CENP-A nucleosome assembly and suppresses intestinal tumorigenesis. iScience 23, 101461 (2020).
Caruso, R. et al. Defective expression of SIRT1 contributes to sustain inflammatory pathways in the gut. Mucosal Immunol. 7, 1467–1479 (2014).
Wellman, A. S. et al. Intestinal epithelial sirtuin 1 regulates intestinal inflammation during aging in mice by altering the intestinal microbiota. Gastroenterology 153, 772–786 (2017).
Ren, M. T. et al. Sirtuin 1 alleviates endoplasmic reticulum stress-mediated apoptosis of intestinal epithelial cells in ulcerative colitis. World J. Gastroenterol. 25, 5800–5813 (2019).
Akimova, T. et al. Targeting sirtuin-1 alleviates experimental autoimmune colitis by induction of Foxp3+ T-regulatory cells. Mucosal Immunol. 7, 1209–1220 (2014).
Bennett, C. L. et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet 27, 20–21 (2001).
Hu, Y. et al. MicroRNA-351-5p aggravates intestinal ischaemia/reperfusion injury through the targeting of MAPK13 and Sirtuin-6. Br. J. Pharm. 175, 3594–3609 (2018).
Wang, Z. et al. SIRT3-mediated deacetylation of PRDX3 alleviates mitochondrial oxidative damage and apoptosis induced by intestinal ischemia/reperfusion injury. Redox Biol. 28, 101343 (2020).
Dong, W. et al. Resveratrol ameliorates subacute intestinal ischemia-reperfusion injury. J. Surg. Res. 185, 182–189 (2013).
Li, H. et al. Lipopolysaccharide upregulates miR-132/212 in Hirschsprung-associated enterocolitis, facilitating pyroptosis by activating NLRP3 inflammasome via targeting Sirtuin 1 (SIRT1). Aging (Albany NY) 12, 18588–18602 (2020).
Bai, M. et al. SIRT1 relieves Necrotizing Enterocolitis through inactivation of Hypoxia-inducible factor (HIF)-1a. Cell Cycle 19, 2018–2027 (2020).
Chen, Y. et al. Circulating exosomal microRNA-18a-5p accentuates intestinal inflammation in Hirschsprung-associated enterocolitis by targeting RORA. Am. J. Transl. Res. 13, 4182–4196 (2021).
Zhu, H., Lin, Y. & Liu, Y. miR34a increases inflammation and oxidative stress levels in patients with necrotizing enterocolitis by downregulating SIRT1 expression. Mol. Med. Rep. 24, 664 (2021).
Scheijen, E. E. M. & Wilson, D. M. 3rd genome integrity and neurological disease. Int. J. Mol. Sci. 23, 4142 (2022).
Bergen, D. C. & Silberberg, D. Nervous system disorders: a global epidemic. Arch. Neurol. 59, 1194–1196 (2002).
Soria Lopez, J. A., Gonzalez, H. M. & Leger, G. C. Alzheimer’s disease. Handb. Clin. Neurol. 167, 231–255 (2019).
Scheltens, P. et al. Alzheimer’s disease. Lancet 397, 1577–1590 (2021).
Abozaid, O. A. R. et al. Resveratrol-selenium nanoparticles alleviate neuroinflammation and neurotoxicity in a rat model of Alzheimer’s disease by regulating Sirt1/miRNA-134/GSK3beta expression. Biol. Trace Elem. Res 200, 5104–5114 (2022).
Fagiani, F., Lanni, C., Racchi, M. & Govoni, S. (Dys)regulation of synaptic activity and neurotransmitter release by beta-amyloid: a look beyond Alzheimer’s disease pathogenesis. Front. Mol. Neurosci. 14, 635880 (2021).
Guo, J., Cheng, J., North, B. J. & Wei, W. Functional analyses of major cancer-related signaling pathways in Alzheimer’s disease etiology. Biochim. Biophys. Acta Rev. Cancer 1868, 341–358 (2017).
An, Y., Li, Y., Hou, Y., Huang, S. & Pei, G. Alzheimer’s amyloid-beta accelerates cell senescence and suppresses the SIRT1/NRF2 pathway in human microglial cells. Oxid. Med. Cell Longev. 2022, 3086010 (2022).
Cao, L., Liu, C., Wang, F. & Wang, H. SIRT1 negatively regulates amyloid-beta-induced inflammation via the NF-kappaB pathway. Braz. J. Med. Biol. Res. 46, 659–669 (2013).
Feng, X. et al. Resveratrol inhibits beta-amyloid-induced neuronal apoptosis through regulation of SIRT1-ROCK1 signaling pathway. PLoS One 8, e59888 (2013).
Perone, I. et al. Mitochondrial SIRT3 deficiency results in neuronal network hyperexcitability, accelerates age-related abeta pathology, and renders neurons vulnerable to abeta toxicity. Neuromolecular Med (2022).
Wang, Y. et al. RTN4B-mediated suppression of Sirtuin 2 activity ameliorates beta-amyloid pathology and cognitive impairment in Alzheimer’s disease mouse model. Aging Cell 19, e13194 (2020).
Gu, J. L. & Liu, F. Tau in Alzheimer’s disease: pathological alterations and an attractive therapeutic target. Curr. Med Sci. 40, 1009–1021 (2020).
Alquezar, C., Arya, S. & Kao, A. W. tau post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front Neurol. 11, 595532 (2020).
Wesseling, H. et al. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell 183, 1699–1713.e1613 (2020).
Carroll, T., Guha, S., Nehrke, K. & Johnson, G. V. W. Tau post-translational modifications: potentiators of selective vulnerability in sporadic Alzheimer’s disease. Biol. (Basel) 10, 1047 (2021).
Lu, S. et al. SIRT1 regulates O-GlcNAcylation of tau through OGT. Aging (Albany NY) 12, 7042–7055 (2020).
Esteves, A. R. et al. Acetylation as a major determinant to microtubule-dependent autophagy: Relevance to Alzheimer’s and Parkinson disease pathology. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 2008–2023 (2019).
Li, S. et al. Sirtuin 3 mediates tau deacetylation. J. Alzheimers Dis. 69, 355–362 (2019).
Palomer, E. et al. Epigenetic repression of Wnt receptors in AD: a role for Sirtuin2-induced H4K16ac deacetylation of Frizzled1 and Frizzled7 promoters. Mol. Psychiatry 27, 3024–3033 (2022).
Li, Q., Liu, Y. & Sun, M. Autophagy and Alzheimer’s disease. Cell Mol. Neurobiol. 37, 377–388 (2017).
Ruankham, W. et al. Sesamin and sesamol attenuate H2O2-induced oxidative stress on human neuronal cells via the SIRT1-SIRT3-FOXO3a signaling pathway. Nutr. Neurosci. 24, 90–101 (2021).
Lee, J. et al. SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell 17, e12679 (2018).
Liu, Y. et al. SIRT3 mediates hippocampal synaptic adaptations to intermittent fasting and ameliorates deficits in APP mutant mice. Nat. Commun. 10, 1886 (2019).
Han, P. et al. Pituitary adenylate cyclase-activating polypeptide protects against beta-amyloid toxicity. Neurobiol. Aging 35, 2064–2071 (2014).
Weir, H. J. et al. CNS SIRT3 expression is altered by reactive oxygen species and in Alzheimer’s disease. PLoS One 7, e48225 (2012).
Pradhan, R. et al. Blood circulatory level of seven sirtuins in Alzheimer’s disease: potent biomarker based on translational research. Mol. Neurobiol. 59, 1440–1451 (2022).
Pukhalskaia, A. E. et al. Sirtuins as possible predictors of aging and Alzheimer’s disease development: verification in the hippocampus and saliva. Bull. Exp. Biol. Med. 169, 821–824 (2020).
Chen, X. et al. Rs2015 Polymorphism in miRNA Target Site of Sirtuin2 gene is associated with the risk of Parkinson’s disease in Chinese Han population. Biomed. Res. Int. 2019, 1498034 (2019).
Jesus, S. et al. Genetic association of sirtuin genes and Parkinson’s disease. J. Neurol. 260, 2237–2241 (2013).
Guo, Y. J. et al. Resveratrol alleviates MPTP-induced motor impairments and pathological changes by autophagic degradation of alpha-synuclein via SIRT1-deacetylated LC3. Mol. Nutr. Food Res. 60, 2161–2175 (2016).
Park, J. H. et al. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol. Neurodegener. 15, 5 (2020).
Zhang, X. et al. PGC-1alpha/ERRalpha-Sirt3 pathway regulates DAergic neuronal death by directly deacetylating SOD2 and ATP synthase beta. Antioxid. Redox Signal 24, 312–328 (2016).
Shi, H. et al. Sirt3 protects dopaminergic neurons from mitochondrial oxidative stress. Hum. Mol. Genet. 26, 1915–1926 (2017).
Jeong, H. et al. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 18, 159–165 (2011).
Baldo, B. et al. SIRT1 is increased in affected brain regions and hypothalamic metabolic pathways are altered in Huntington disease. Neuropathol. Appl. Neurobiol. 45, 361–379 (2019).
Luthi-Carter, R. et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl Acad. Sci. USA 107, 7927–7932 (2010).
Enzmann, G., Kargaran, S. & Engelhardt, B. Ischemia-reperfusion injury in stroke: impact of the brain barriers and brain immune privilege on neutrophil function. Ther. Adv. Neurol. Disord. 11, 1756286418794184 (2018).
Pantazi, E. et al. Role of sirtuins in ischemia-reperfusion injury. World J. Gastroenterol. 19, 7594–7602 (2013).
Zhang, X. S. et al. Sirtuin 1 activation protects against early brain injury after experimental subarachnoid hemorrhage in rats. Cell Death Dis. 7, e2416 (2016).
Liu, R. et al. QKI 6 ameliorates CIRI through promoting synthesis of triglyceride in neuron and inhibiting neuronal apoptosis associated with SIRT1-PPARgamma-PGC-1alpha axis. Brain Behav. 11, e2271 (2021).
Wang, P. et al. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 8, 77–87 (2012).
Kim, S. H., Lu, H. F. & Alano, C. C. Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture. PLoS One 6, e14731 (2011).
Yang, X. et al. Sirt3 protects against ischemic stroke injury by regulating HIF-1alpha/VEGF signaling and blood-brain barrier integrity. Cell Mol. Neurobiol. 41, 1203–1215 (2021).
Zhao, H. et al. Sirt3 inhibits cerebral ischemia-reperfusion injury through normalizing Wnt/beta-catenin pathway and blocking mitochondrial fission. Cell Stress Chaperones 23, 1079–1092 (2018).
Diaz-Canestro, C. et al. Sirtuin 5 as a novel target to blunt blood-brain barrier damage induced by cerebral ischemia/reperfusion injury. Int. J. Cardiol. 260, 148–155 (2018).
Zhang, Y., Yan, Q. & Zhang, Y. Overexpression of sirtuin 2 and its association with prognosis in acute ischemic stroke patients. J. Clin. Lab. Anal. 35, e23707 (2021).
Pennisi, G. et al. Redox regulation of cellular stress response in multiple sclerosis. Biochem. Pharmacol. 82, 1490–1499 (2011).
De-Bernardi-Ojuel, L., Torres-Collado, L. & Garcia-de-la-Hera, M. Occupational therapy interventions in adults with multiple sclerosis or amyotrophic lateral sclerosis: a scoping review. Int. J. Environ. Res. Public Health 18, 1432 (2021).
Deeb, O. & Nabulsi, M. Exploring multiple sclerosis (MS) and amyotrophic lateral scler osis (ALS) as neurodegenerative diseases and their treatments: a review study. Curr. Top. Med. Chem. 20, 2391–2403 (2020).
Molnar-Kasza, A. et al. Evaluation of neuropathological features in the SOD1-G93A low copy number transgenic mouse model of amyotrophic lateral sclerosis. Front Mol. Neurosci. 14, 681868 (2021).
Lee, J. C. et al. Region-specific changes in the immunoreactivity of SIRT1 expression in the central nervous system of SOD1(G93A) transgenic mice as an in vivo model of amyotrophic lateral sclerosis. Brain Res 1433, 20–28 (2012).
Buck, E. et al. Comparison of Sirtuin 3 levels in ALS and Huntington’s disease-differential effects in human tissue samples vs. transgenic mouse models. Front. Mol. Neurosci. 10, 156 (2017).
Harlan, B. A., Pehar, M., Killoy, K. M. & Vargas, M. R. Enhanced SIRT6 activity abrogates the neurotoxic phenotype of astrocytes expressing ALS-linked mutant SOD1. FASEB J. 33, 7084–7091 (2019).
Yuen, A. W. C., Keezer, M. R. & Sander, J. W. Epilepsy is a neurological and a systemic disorder. Epilepsy Behav. 78, 57–61 (2018).
Wallace, E. et al. Altered circadian rhythms and oscillation of clock genes and sirtuin 1 in a model of sudden unexpected death in epilepsy. Epilepsia 59, 1527–1539 (2018).
Li, F. & Liu, L. SIRT5 deficiency enhances susceptibility to kainate-induced seizures and exacerbates hippocampal neurodegeneration not through mitochondrial antioxidant enzyme SOD2. Front Cell Neurosci. 10, 171 (2016).
Lin, W. et al. Inhibition of miR-134-5p protects against kainic acid-induced excitotoxicity through Sirt3-mediated preservation of mitochondrial function. Epilepsy Res 176, 106722 (2021).
Jester, D. J., Lee, S., Molinari, V. & Volicer, L. Cognitive deficits in Parkinson’s disease with excessive daytime sleepiness: a systematic review. Aging Ment. Health 24, 1769–1780 (2020).
Vinter, K., Callesen, M. B., Kristensen, T. E. & Jakobsen, A. V. [Cognitive deficits in epilepsy]. Ugeskr. Laege. 184, V01220071 (2022).
Vermeulen, T. et al. Cognitive deficits in older adults with psychotic depression: a meta-analysis. Am. J. Geriatr. Psychiatry 27, 1334–1344 (2019).
Sun, Z., Zhao, S., Suo, X. & Dou, Y. Sirt1 protects against hippocampal atrophy and its induced cognitive impairment in middle-aged mice. BMC Neurosci. 23, 33 (2022).
Fang, Q. J. et al. Surgery-induced downregulation of hippocampal sirtuin-1 contributes to cognitive dysfunction by inhibiting autophagy and activating apoptosis in aged mice. Am. J. Transl. Res. 12, 8111–8122 (2020).
Wang, D. et al. Sirt3-mediated mitochondrial dysfunction is involved in fluoride-induced cognitive deficits. Food Chem. Toxicol. 158, 112665 (2021).
Wang, D. et al. Mitigation of honokiol on fluoride-induced mitochondrial oxidative stress, mitochondrial dysfunction, and cognitive deficits through activating AMPK/PGC-1alpha/Sirt3. J. Hazard Mater. 437, 129381 (2022).
Zhu, K., Xu, W., Han, D. & Mei, X. Effect of Sirtuin-1 and Wnt/beta-catenin signaling pathway in rat model of spinal cord injury. Comput. Math. Methods Med 2022, 1799607 (2022).
Lu, P. et al. Effects of Sirtuin 1 on microglia in spinal cord injury: involvement of Wnt/beta-catenin signaling pathway. Neuroreport 30, 867–874 (2019).
Zhaohui, C. & Shuihua, W. Protective effects of SIRT6 against inflammation, oxidative stress, and cell apoptosis in spinal cord injury. Inflammation 43, 1751–1758 (2020).
Chen, J. & Qin, R. MicroRNA1385p regulates the development of spinal cord injury by targeting SIRT1. Mol. Med. Rep. 22, 328–336 (2020).
Wang, X. et al. MicroRNA-30c abrogation protects against spinal cord ischemia reperfusion injury through modulating SIRT1. Eur. J. Pharm. 851, 80–87 (2019).
Ellis, A. & Bennett, D. L. Neuroinflammation and the generation of neuropathic pain. Br. J. Anaesth. 111, 26–37 (2013).
Cong, L. et al. Resveratrol attenuates manganese-induced oxidative stress and neuroinflammation through SIRT1 signaling in mice. Food Chem. Toxicol. 153, 112283 (2021).
Zhang, Y. & Chi, D. Overexpression of SIRT2 alleviates neuropathic pain and neuroinflammation through deacetylation of transcription factor nuclear factor-kappa B. Inflammation 41, 569–578 (2018).
Sa de Almeida, J. et al. Microglial sirtuin 2 shapes long-term potentiation in hippocampal slices. Front Neurosci. 14, 614 (2020).
Pais, T. F. et al. The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J. 32, 2603–2616 (2013).
Zhou, D. & Jiang, Y. Sirtuin 3 attenuates neuroinflammation-induced apoptosis in BV-2 microglia. Aging (Albany NY) 11, 9075–9089 (2019).
Li, H. et al. TREM2 ameliorates lipopolysaccharide-induced oxidative stress response and neuroinflammation by promoting sirtuin3 in BV2 cells. Neurotox. Res 40, 56–65 (2022).
Kikusui, T., Nagasawa, M., Nomoto, K., Kuse-Arata, S. & Mogi, K. Endocrine regulations in human-dog coexistence through domestication. Trends Endocrinol. Metab. 30, 793–806 (2019).
Estes, C. et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J. Hepatol. 69, 896–904 (2018).
Lantier, L. et al. SIRT3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high-fat-fed mice. Diabetes 64, 3081–3092 (2015).
Li, P. et al. SIRT1 attenuates renal fibrosis by repressing HIF-2alpha. Cell Death Disco. 7, 59 (2021).
Hong, Q. et al. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int 93, 1330–1343 (2018).
Lo, C. S. et al. Heterogeneous nuclear ribonucleoprotein f stimulates sirtuin-1 gene expression and attenuates nephropathy progression in diabetic mice. Diabetes 66, 1964–1978 (2017).
Moschen, A. R. et al. Adipose tissue and liver expression of SIRT1, 3, and 6 increase after extensive weight loss in morbid obesity. J. Hepatol. 59, 1315–1322 (2013).
Ceolotto, G. et al. Sirtuin 1 stabilization by HuR represses TNF-alpha- and glucose-induced E-selectin release and endothelial cell adhesiveness in vitro: relevance to human metabolic syndrome. Clin. Sci. (Lond.) 127, 449–461 (2014).
International Diabetes Federation. IDF Diabetes Atlas. 8th ed. IDF, 2017 World Health Organization. Global Report on Diabetes. (2016).
Bourne, R. R. et al. Causes of vision loss worldwide, 1990-2010: a systematic analysis. Lancet Glob. Health 1, e339–e349 (2013).
Moxey, P. W. et al. Lower extremity amputations-a review of global variability in incidence. Diabet. Med 28, 1144–1153 (2011).
Dyck, P. J. et al. The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population-based cohort: the Rochester Diabetic Neuropathy Study. Neurology 43, 817–824 (1993).
United States Renal Data System. International Comparisons. In:United States Renal Data System. 2014 USRDS annual data report: epidemiology of kidney disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 188-210 (2014).
Guarente, L. & Franklin, H. Epstein Lecture: Sirtuins, aging, and medicine. N. Engl. J. Med. 364, 2235–2244 (2011).
Gomes, P., Fleming Outeiro, T. & Cavadas, C. Emerging role of Sirtuin 2 in the regulation of Mammalian metabolism. Trends Pharm. Sci. 36, 756–768 (2015).
Cui, Y. et al. Isoliquiritigenin inhibits non-small cell lung cancer progression via m(6)A/IGF2BP3-dependent TWIST1 mRNA stabilization. Phytomedicine 104, 154299 (2022).
Calabrese, V. et al. Oxidative stress, glutathione status, sirtuin and cellular stress response in type 2 diabetes. Biochim. Biophys. Acta 1822, 729–736 (2012).
Gui, J. et al. Gestational diabetes induces alterations of sirtuins in fetal endothelial cells. Pediatr. Res 79, 788–798 (2016).
Bian, C. et al. Association of SIRT6 circulating levels with urinary and glycometabolic markers in pre-diabetes and diabetes. Acta Diabetol. 58, 1551–1562 (2021).
Cote, C. D. et al. Resveratrol activates duodenal Sirt1 to reverse insulin resistance in rats through a neuronal network. Nat. Med. 21, 498–505 (2015).
Jimenez-Gomez, Y. et al. Resveratrol improves adipose insulin signaling and reduces the inflammatory response in adipose tissue of rhesus monkeys on high-fat, high-sugar diet. Cell Metab. 18, 533–545 (2013).
Cornejo, P. J. et al. The Stress-Responsive microRNA-34a alters insulin signaling and actions in adipocytes through induction of the tyrosine phosphatase PTP1B. Cells 11 (2022).
Erion, D. M. et al. SirT1 knockdown in liver decreases basal hepatic glucose production and increases hepatic insulin responsiveness in diabetic rats. Proc. Natl Acad. Sci. USA 106, 11288–11293 (2009).
Watanabe, H. et al. Sirt2 facilitates hepatic glucose uptake by deacetylating glucokinase regulatory protein. Nat. Commun. 9, 30 (2018).
Arora, A. & Dey, C. S. SIRT2 regulates insulin sensitivity in insulin resistant neuronal cells. Biochem. Biophys. Res. Commun. 474, 747–752 (2016).
Choubey, S. K., Prabhu, D., Nachiappan, M., Biswal, J. & Jeyakanthan, J. Molecular modeling, dynamics studies and density functional theory approaches to identify potential inhibitors of SIRT4 protein from Homo sapiens: a novel target for the treatment of type 2 diabetes. J. Biomol. Struct. Dyn. 35, 3316–3329 (2017).
Dominy, J. E. Jr. et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol. Cell 48, 900–913 (2012).
Zhang, P. et al. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc. Natl Acad. Sci. USA 111, 10684–10689 (2014).
Spaeth, J. M. et al. Defining a novel role for the Pdx1 transcription factor in islet beta-cell maturation and proliferation during weaning. Diabetes 66, 2830–2839 (2017).
Fujitani, Y. Transcriptional regulation of pancreas development and beta-cell function [Review]. Endocr. J. 64, 477–486 (2017).
Feldman, E. L. et al. Diabetic neuropathy. Nat. Rev. Dis. Prim. 5, 42 (2019).
Cole, J. B. & Florez, J. C. Genetics of diabetes mellitus and diabetes complications. Nat. Rev. Nephrol. 16, 377–390 (2020).
Li, L., Zeng, H., He, X. & Chen, J. X. Sirtuin 3 alleviates diabetic cardiomyopathy by regulating TIGAR and cardiomyocyte metabolism. J. Am. Heart Assoc. 10, e018913 (2021).
Yang, H. et al. Hippocampal insulin resistance and the Sirtuin 1 signaling pathway in diabetes-induced cognitive dysfunction. Neural Regen. Res 16, 2465–2474 (2021).
Tang, G. et al. Clinical efficacies, underlying mechanisms and molecular targets of Chinese medicines for diabetic nephropathy treatment and management. Acta Pharm. Sin. B 11, 2749–2767 (2021).
Heerspink, H. J. L. et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): a double-blind, randomised, placebo-controlled trial. Lancet 393, 1937–1947 (2019).
Thomas, B. The global burden of diabetic kidney disease: time trends and gender gaps. Curr. Diab. Rep. 19, 18 (2019).
Huang, K. et al. Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-beta1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic. Biol. Med 65, 528–540 (2013).
Pop-Busui, R. et al. Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes Care 40, 136–154 (2017).
Cheng, X. R., Zhou, W. X. & Zhang, Y. X. The behavioral, pathological and therapeutic features of the senescence-accelerated mouse prone 8 strain as an Alzheimer’s disease animal model. Ageing Res. Rev. 13, 13–37 (2014).
Nagao, K., Jinnouchi, T., Kai, S. & Yanagita, T. Pterostilbene, a dimethylated analog of resveratrol, promotes energy metabolism in obese rats. J. Nutr. Biochem. 43, 151–155 (2017).
Zhang, Z. et al. Sirtuin 1 alleviates diabetic neuropathic pain by regulating synaptic plasticity of spinal dorsal horn neurons. Pain 160, 1082–1092 (2019).
Chandrasekaran, K. et al. Overexpression of Sirtuin 1 protein in neurons prevents and reverses experimental diabetic neuropathy. Brain 142, 3737–3752 (2019).
Cheung, N., Mitchell, P. & Wong, T. Y. Diabetic retinopathy. Lancet 376, 124–136 (2010).
Mishra, M., Duraisamy, A. J. & Kowluru, R. A. Sirt1: a guardian of the development of diabetic retinopathy. Diabetes 67, 745–754 (2018).
Prakoso, D. et al. Gene therapy targeting cardiac phosphoinositide 3-kinase (p110alpha) attenuates cardiac remodeling in type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 318, H840–H852 (2020).
Li, K. et al. Tetrahydrocurcumin ameliorates diabetic cardiomyopathy by attenuating high glucose-induced oxidative stress and fibrosis via activating the SIRT1 Pathway. Oxid. Med. Cell Longev. 2019, 6746907 (2019).
Song, S. et al. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharm. Sin. 42, 230–241 (2021).
Collaborators, G. B. D. O. et al. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 377, 13–27 (2017).
Deng, T., Lyon, C. J., Bergin, S., Caligiuri, M. A. & Hsueh, W. A. Obesity, inflammation, and cancer. Annu. Rev. Pathol. 11, 421–449 (2016).
Hotamisligil, G. S. Inflammation and metabolic disorders. Nature 444, 860–867 (2006).
Gregor, M. F. & Hotamisligil, G. S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 29, 415–445 (2011).
Ahn, J., Lee, H., Jung, C. H., Jeon, T. I. & Ha, T. Y. MicroRNA-146b promotes adipogenesis by suppressing the SIRT1-FOXO1 cascade. EMBO Mol. Med. 5, 1602–1612 (2013).
Cyr, N. E. et al. Central Sirt1 regulates body weight and energy expenditure along with the POMC-derived peptide alpha-MSH and the processing enzyme CPE production in diet-induced obese male rats. Endocrinology 156, 961–974 (2015).
Zhang, T., Liu, J., Tong, Q. & Lin, L. SIRT3 acts as a positive autophagy regulator to promote lipid mobilization in adipocytes via activating AMPK. Int. J. Mol. Sci. 21 (2020).
Timmers, S. et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 14, 612–622 (2011).
Li, H., Xu, M., Lee, J., He, C. & Xie, Z. Leucine supplementation increases SIRT1 expression and prevents mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 303, E1234–E1244 (2012).
Krishnan, J. et al. Dietary obesity-associated Hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 26, 259–270 (2012).
Tang, Q. et al. Sirtuin 6 supra-physiological overexpression in hypothalamic pro-opiomelanocortin neurons promotes obesity via the hypothalamus-adipose axis. FASEB J. 35, e21408 (2021).
Pfluger, P. T., Herranz, D., Velasco-Miguel, S., Serrano, M. & Tschop, M. H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl Acad. Sci. USA 105, 9793–9798 (2008).
Tao, R., Xiong, X., DePinho, R. A., Deng, C. X. & Dong, X. C. Hepatic SREBP-2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6. J. Lipid Res. 54, 2745–2753 (2013).
Bhargava, P. & Schnellmann, R. G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13, 629–646 (2017).
Wang, R. et al. Potential adverse effects of nanoparticles on the reproductive system. Int. J. Nanomed. 13, 8487–8506 (2018).
Li, H. et al. Depleting microRNA-183-3p improves renal tubulointerstitial fibrosis after acute kidney injury via SIRT1/PUMA/FOXO3a deacetylation. Life Sci. 269, 119017 (2021).
Li, L., Fu, H. & Liu, Y. The fibrogenic niche in kidney fibrosis: components and mechanisms. Nat. Rev. Nephrol. 18, 545–557 (2022).
He, W. et al. Deletion of soluble epoxide hydrolase suppressed chronic kidney disease-related vascular calcification by restoring Sirtuin 3 expression. Cell Death Dis. 12, 992 (2021).
Castillo, R. L. et al. Protective effects of polyunsatutared fatty acids supplementation against testicular damage induced by intermittent hypobaric hypoxia in rats. J. Biomed. Sci. 22, 8 (2015).
Levey, A. S., Titan, S. M., Powe, N. R., Coresh, J. & Inker, L. A. Kidney disease, race, and GFR estimation. Clin. J. Am. Soc. Nephrol. 15, 1203–1212 (2020).
Kellum, J. A. et al. Acute kidney injury. Nat. Rev. Dis. Prim. 7, 52 (2021).
Meersch, M., Schmidt, C. & Zarbock, A. Perioperative acute kidney injury: an under-recognized problem. Anesth. Analg. 125, 1223–1232 (2017).
Susantitaphong, P. et al. World incidence of AKI: a meta-analysis. Clin. J. Am. Soc. Nephrol. 8, 1482–1493 (2013).
Hoste, E. A. J. et al. Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 14, 607–625 (2018).
Mehta, R. L. et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet 385, 2616–2643 (2015).
Shen, H. et al. Sirtuin-3 mediates sex differences in kidney ischemia-reperfusion injury. Transl. Res 235, 15–31 (2021).
Morigi, M. et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Invest 125, 715–726 (2015).
Wang, Q. et al. Sirt3 modulate renal ischemia-reperfusion injury through enhancing mitochondrial fusion and activating the ERK-OPA1 signaling pathway. J. Cell Physiol. 234, 23495–23506 (2019).
Emma, F., Montini, G., Parikh, S. M. & Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 12, 267–280 (2016).
Guan, Y. et al. Nicotinamide mononucleotide, an NAD(+) precursor, rescues age-associated susceptibility to AKI in a sirtuin 1-dependent manner. J. Am. Soc. Nephrol. 28, 2337–2352 (2017).
Fan, H. et al. The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion-induced acute kidney injury. Kidney Int 83, 404–413 (2013).
Lempiainen, J., Finckenberg, P., Levijoki, J. & Mervaala, E. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br. J. Pharm. 166, 1905–1915 (2012).
He, S. et al. NAD(+) ameliorates endotoxin-induced acute kidney injury in a sirtuin1-dependent manner via GSK-3beta/Nrf2 signalling pathway. J. Cell Mol. Med. 26, 1979–1993 (2022).
Jung, Y. J., Park, W., Kang, K. P. & Kim, W. SIRT2 is involved in cisplatin-induced acute kidney injury through regulation of mitogen-activated protein kinase phosphatase-1. Nephrol. Dial. Transpl. 35, 1145–1156 (2020).
Chiba, T. et al. Sirtuin 5 regulates proximal tubule fatty acid oxidation to protect against AKI. J. Am. Soc. Nephrol. 30, 2384–2398 (2019).
Sanchez-Navarro, A. et al. Sirtuin 7 deficiency reduces inflammation and tubular damage induced by an episode of acute kidney injury. Int. J. Mol. Sci. 23, 2573 (2022).
Ping, H., Zhang, X. & Xing, N. Prevalence of chronic kidney disease in China. Lancet 380, 216 (2012).
Levin, A. et al. Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet 390, 1888–1917 (2017).
Webster, A. C., Nagler, E. V., Morton, R. L. & Masson, P. Chronic kidney disease. Lancet 389, 1238–1252 (2017).
Duffield, J. S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Invest 124, 2299–2306 (2014).
Djudjaj, S. & Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Asp. Med. 65, 16–36 (2019).
Schnaper, H. W. Renal fibrosis. Methods Mol. Med 117, 45–68 (2005).
United States Renal Data System 2003 Annual Data Report. Division of Kidney, Urologic and Hematologic Diseases, NIDDK/NIH, Department of Health and Human Services, Bethesda, MD (2003).
Isaka, Y. Targeting TGF-beta signaling in kidney fibrosis. Int. J. Mol. Sci. 19, 2532 (2018).
Leask, A. & Abraham, D. J. TGF-beta signaling and the fibrotic response. FASEB J. 18, 816–827 (2004).
Ren, Y. et al. CTGF siRNA ameliorates tubular cell apoptosis and tubulointerstitial fibrosis in obstructed mouse kidneys in a Sirt1-independent manner. Drug Des. Devel Ther. 9, 4155–4171 (2015).
Ren, Y. et al. The Sirt1 activator, SRT1720, attenuates renal fibrosis by inhibiting CTGF and oxidative stress. Int. J. Mol. Med. 39, 1317–1324 (2017).
He, F. F. et al. Inhibition of SIRT2 alleviates fibroblast activation and renal tubulointerstitial fibrosis via MDM2. Cell Physiol. Biochem 46, 451–460 (2018).
Zhang, Y. et al. Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis. 12, 847 (2021).
Jin, J. et al. Loss of proximal tubular sirtuin 6 aggravates unilateral ureteral obstruction-induced tubulointerstitial inflammation and fibrosis by regulation of beta-catenin acetylation. Cells 11, 1477 (2022).
Ruiz-Ortega, M., Rayego-Mateos, S., Lamas, S., Ortiz, A. & Rodrigues-Diez, R. R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 16, 269–288 (2020).
Vasko, R. et al. Endothelial sirtuin 1 deficiency perpetrates nephrosclerosis through downregulation of matrix metalloproteinase-14: relevance to fibrosis of vascular senescence. J. Am. Soc. Nephrol. 25, 276–291 (2014).
Yang, S. Y. et al. Downregulation of angiotensin type 1 receptor and nuclear factor-kappaB by sirtuin 1 contributes to renoprotection in unilateral ureteral obstruction. Sci. Rep. 6, 33705 (2016).
Ponnusamy, M. et al. Blocking sirtuin 1 and 2 inhibits renal interstitial fibroblast activation and attenuates renal interstitial fibrosis in obstructive nephropathy. J. Pharm. Exp. Ther. 350, 243–256 (2014).
Xi, J. et al. SIRT3 inhibited the formation of calcium oxalate-induced kidney stones through regulating NRF2/HO-1 signaling pathway. J Cell Biochem (2018).
Ye, Q. L. et al. Sirt1 inhibits kidney stones formation by attenuating calcium oxalate-induced cell injury. Chem. Biol. Interact. 347, 109605 (2021).
Ryu, D. R. et al. Sirt1-hypoxia-inducible factor-1alpha interaction is a key mediator of tubulointerstitial damage in the aged kidney. Aging Cell 18, e12904 (2019).
Chuang, P. Y. et al. Reduction in podocyte SIRT1 accelerates kidney injury in aging mice. Am. J. Physiol. Ren. Physiol. 313, F621–F628 (2017).
Huang, W. et al. Sirt6 deficiency results in progression of glomerular injury in the kidney. Aging (Albany NY) 9, 1069–1083 (2017).
Li, W. et al. SIRT6 protects vascular smooth muscle cells from osteogenic transdifferentiation via Runx2 in chronic kidney disease. J. Clin. Invest 132, e150051 (2022).
Wei, W. et al. Bone marrow mesenchymal stem cell exosomes suppress phosphate-induced aortic calcification via SIRT6-HMGB1 deacetylation. Stem Cell Res. Ther. 12, 235 (2021).
Sener, T. E. et al. Resveratrol treatment may preserve the erectile function after radiotherapy by restoring antioxidant defence mechanisms, SIRT1 and NOS protein expressions. Int. J. Impot Res. 30, 179–188 (2018).
Francisco, C. M. et al. Resveratrol reverses male reproductive damage in rats exposed to nicotine during the intrauterine phase and breastfeeding. Andrology 10, 951–972 (2022).
Iniesta-Cuerda, M., Havrankova, J., Rimnacova, H., Garcia-Alvarez, O. & Nevoral, J. Male SIRT1 insufficiency leads to sperm with decreased ability to hyperactivate and fertilize. Reprod. Domest. Anim. 57, 72–77 (2022).
Bello, J. H. et al. Dysregulation of mitochondrial sirtuin genes is associated with human male infertility. Andrologia 54, e14274 (2022).
Kiyak Caglayan, E., Engin-Ustun, Y., Gocmen, A. Y., Polat, M. F. & Aktulay, A. Serum sirtuin 1 levels in patients with polycystic ovary syndrome. J. Obstet. Gynaecol. 35, 608–611 (2015).
Emidio, G. D. et al. Methylglyoxal-dependent glycative stress and deregulation of SIRT1 functional network in the ovary of PCOS mice. Cells 9, 209 (2020).
Zhang, Q. et al. Mitochondrial and glucose metabolic dysfunctions in granulosa cells induce impaired oocytes of polycystic ovary syndrome through Sirtuin 3. Free Radic. Biol. Med 187, 1–16 (2022).
Yoo, J. Y. et al. KRAS activation and over-expression of SIRT1/BCL6 contributes to the pathogenesis of endometriosis and progesterone resistance. Sci. Rep. 7, 6765 (2017).
Rezk, N. A., Lashin, M. B. & Sabbah, N. A. MiRNA 34-a regulate SIRT-1 and Foxo-1 expression in endometriosis. Noncoding RNA Res 6, 35–41 (2021).
Giorgio, I. et al. On mechanically driven biological stimulus for bone remodeling as a diffusive phenomenon. Biomech. Model Mechanobiol. 18, 1639–1663 (2019).
Chen, Y. et al. SIRT1, a promising regulator of bone homeostasis. Life Sci. 269, 119041 (2021).
McCulloch, K., Litherland, G. J. & Rai, T. S. Cellular senescence in osteoarthritis pathology. Aging Cell 16, 210–218 (2017).
Sulzbacher, I. Osteoarthritis: histology and pathogenesis. Wien. Med. Wochenschr. 163, 212–219 (2013).
Liu, S., Yang, H., Hu, B. & Zhang, M. Sirt1 regulates apoptosis and extracellular matrix degradation in resveratrol-treated osteoarthritis chondrocytes via the Wnt/beta-catenin signaling pathways. Exp. Ther. Med. 14, 5057–5062 (2017).
Zhang, M., Mou, L., Liu, S., Sun, F. & Gong, M. Circ_0001103 alleviates IL-1beta-induced chondrocyte cell injuries by upregulating SIRT1 via targeting miR-375. Clin. Immunol. 227, 108718 (2021).
Ma, C. H. et al. Homocysteine causes dysfunction of chondrocytes and oxidative stress through repression of SIRT1/AMPK pathway: A possible link between hyperhomocysteinemia and osteoarthritis. Redox Biol. 15, 504–512 (2018).
Wang, Y., Zhao, X., Lotz, M., Terkeltaub, R. & Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor gamma coactivator 1alpha. Arthritis Rheumatol. 67, 2141–2153 (2015).
Lu, Q. et al. SIRT1 restoration enhances chondrocyte autophagy in osteoarthritis through PTEN-mediated EGFR ubiquitination. Cell Death Disco. 8, 203 (2022).
Li, W., Xiong, Y., Chen, W. & Wu, L. Wnt/beta-catenin signaling may induce senescence of chondrocytes in osteoarthritis. Exp. Ther. Med. 20, 2631–2638 (2020).
Wang, J. et al. SIRT3 activation by dihydromyricetin suppresses chondrocytes degeneration via maintaining mitochondrial homeostasis. Int. J. Biol. Sci. 14, 1873–1882 (2018).
Fu, Y. et al. Aging promotes sirtuin 3-dependent cartilage superoxide dismutase 2 acetylation and osteoarthritis. Arthritis Rheumatol. 68, 1887–1898 (2016).
Zhang, C., Lu, Y., Yuan, F. & Jiang, S. Circular RNA CCDC66 regulates osteoarthritis progression by targeting miR-3622b-5p. Gerontology 68, 431–441 (2022).
Consensus development conference: diagnosis, prophylaxis, and treatment of osteoporosis. Am. J. Med. 94, 646-650 (1993).
Martin, T., Gooi, J. H. & Sims, N. A. Molecular mechanisms in coupling of bone formation to resorption. Crit. Rev. Eukaryot. Gene Expr. 19, 73–88 (2009).
Chang, H. C. & Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 153, 1448–1460 (2013).
Zainabadi, K., Liu, C. J., Caldwell, A. L. M. & Guarente, L. SIRT1 is a positive regulator of in vivo bone mass and a therapeutic target for osteoporosis. PLoS One 12, e0185236 (2017).
Barbour, K. E. et al. Inflammatory markers and risk of hip fracture in older white women: the study of osteoporotic fractures. J. Bone Miner. Res. 29, 2057–2064 (2014).
Sun, W. et al. Overexpression of Sirt1 in mesenchymal stem cells protects against bone loss in mice by FOXO3a deacetylation and oxidative stress inhibition. Metabolism 88, 61–71 (2018).
Stegen, S. et al. Osteocytic oxygen sensing controls bone mass through epigenetic regulation of sclerostin. Nat. Commun. 9, 2557 (2018).
Moon, Y. J. et al. Sirtuin 6 in preosteoclasts suppresses age- and estrogen deficiency-related bone loss by stabilizing estrogen receptor alpha. Cell Death Differ. 26, 2358–2370 (2019).
Feng, J. et al. Protective effects of resveratrol on postmenopausal osteoporosis: regulation of SIRT1-NF-kappaB signaling pathway. Acta Biochim Biophys. Sin. (Shanghai) 46, 1024–1033 (2014).
Wang, X., Chen, L. & Peng, W. Protective effects of resveratrol on osteoporosis via activation of the SIRT1-NF-kappaB signaling pathway in rats. Exp. Ther. Med. 14, 5032–5038 (2017).
Dowdell, J. et al. Intervertebral disk degeneration and repair. Neurosurgery 80, S46–S54 (2017).
Shi, Q., Xu, L. & Zeng, X. Sirtuin 1 participates in intervertebral disc degeneration via the nicotinamide phosphoribosyl transferase/nicotinamide adenine dinucleotide/sirtuin 1 pathway responsible for regulating autophagy of nucleus pulposus cells. Exp. Ther. Med. 23, 267 (2022).
Cai, W. T., Guan, P., Lin, M. X., Fu, B. & Wu, B. Sirt1 suppresses MCP-1 production during the intervertebral disc degeneration by inactivating AP-1 subunits c-Fos/c-Jun. Eur. Rev. Med. Pharmacol. Sci. 24, 5895–5904 (2020).
Yang, M. et al. Sirtuin 2 expression suppresses oxidative stress and senescence of nucleus pulposus cells through inhibition of the p53/p21 pathway. Biochem. Biophys. Res. Commun. 513, 616–622 (2019).
Song, Y. et al. Sirtuin 3-dependent mitochondrial redox homeostasis protects against AGEs-induced intervertebral disc degeneration. Redox Biol. 19, 339–353 (2018).
Wang, J. et al. Small molecule natural compound agonist of SIRT3 as a therapeutic target for the treatment of intervertebral disc degeneration. Exp. Mol. Med. 50, 1–14 (2018).
Kang, L., Hu, J., Weng, Y., Jia, J. & Zhang, Y. Sirtuin 6 prevents matrix degradation through inhibition of the NF-kappaB pathway in intervertebral disc degeneration. Exp. Cell Res. 352, 322–332 (2017).
Zhang, Y. et al. The autophagic-lysosomal and ubiquitin proteasome systems are simultaneously activated in the skeletal muscle of gastric cancer patients with cachexia. Am. J. Clin. Nutr. 111, 570–579 (2020).
Ou, H. C. et al. Low-level laser prevents doxorubicin-induced skeletal muscle atrophy by modulating AMPK/SIRT1/PCG-1alpha-mediated mitochondrial function, apoptosis and up-regulation of pro-inflammatory responses. Cell Biosci. 11, 200 (2021).
Han, Z. et al. Role of SIRT2 in regulating the dexamethasone-activated autophagy pathway in skeletal muscle atrophy. Biochem. Cell Biol. 99, 562–569 (2021).
Zheng, J. et al. Sirtuin 3 deficiency accelerates Angiotensin II-induced skeletal muscle atrophy. Connect Tissue Res 61, 586–593 (2020).
Cruz-Jentoft, A. J. et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 39, 412–423 (2010).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Wu, X., Cao, N., Fenech, M. & Wang, X. Role of sirtuins in maintenance of genomic stability: relevance to cancer and healthy aging. DNA Cell Biol. 35, 542–575 (2016).
Wang, T. et al. Research progress on sirtuins family members and cell senescence. Eur. J. Med. Chem. 193, 112207 (2020).
Yan, P. et al. LARP7 ameliorates cellular senescence and aging by allosterically enhancing SIRT1 deacetylase activity. Cell Rep. 37, 110038 (2021).
Oberdoerffer, P. et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918 (2008).
Diao, Z. et al. SIRT3 consolidates heterochromatin and counteracts senescence. Nucleic Acids Res 49, 4203–4219 (2021).
Green, C. L., Lamming, D. W. & Fontana, L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell Biol. 23, 56–73 (2022).
Rodgers, J. T. et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434, 113–118 (2005).
Peterson, C. Semantic and pragmatic uses of ‘but’. J. Child Lang. 13, 583–590 (1986).
Jung-Hynes, B., Reiter, R. J. & Ahmad, N. Sirtuins, melatonin and circadian rhythms: building a bridge between aging and cancer. J. Pineal Res. 48, 9–19 (2010).
Wang, R. H. et al. Negative reciprocal regulation between Sirt1 and Per2 modulates the circadian clock and aging. Sci. Rep. 6, 28633 (2016).
Sun, S. et al. Sirt6 deacetylase activity regulates circadian rhythms via Per2. Biochem. Biophys. Res. Commun. 511, 234–238 (2019).
Lee, S. H., Lee, J. H., Lee, H. Y. & Min, K. J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 52, 24–34 (2019).
Wang, Y. et al. An overview of Sirtuins as potential therapeutic target: Structure, function and modulators. Eur. J. Med. Chem. 161, 48–77 (2019).
Howitz, K. T. et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196 (2003).
Hoseini, A. et al. The effects of resveratrol on metabolic status in patients with type 2 diabetes mellitus and coronary heart disease. Food Funct. 10, 6042–6051 (2019).
Saul, N., Pietsch, K., Menzel, R. & Steinberg, C. E. Quercetin-mediated longevity in Caenorhabditis elegans: is DAF-16 involved? Mech. Ageing Dev. 129, 611–613 (2008).
Zhu, Y. et al. SIRT1 activation by butein attenuates sepsis-induced brain injury in mice subjected to cecal ligation and puncture via alleviating inflammatory and oxidative stress. Toxicol. Appl. Pharm. 363, 34–46 (2019).
Wolter, F., Clausnitzer, A., Akoglu, B. & Stein, J. Piceatannol, a natural analog of resveratrol, inhibits progression through the S phase of the cell cycle in colorectal cancer cell lines. J. Nutr. 132, 298–302 (2002).
Yerra, V. G., Kalvala, A. K. & Kumar, A. Isoliquiritigenin reduces oxidative damage and alleviates mitochondrial impairment by SIRT1 activation in experimental diabetic neuropathy. J. Nutr. Biochem. 47, 41–52 (2017).
Milne, J. C. et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716 (2007).
Yee, Ng,P. et al. The identification of the SIRT1 activator SRT2104 as a clinical candidate. Lett. Drug Des. Discov. 10, 793–797 (2013).
US8343997B2 Sirtris Pharmaceuticals Inc. (2013).
Chini, C. C. et al. SIRT1-activating compounds (STAC) negatively regulate pancreatic cancer cell growth and viability through a SIRT1 lysosomal-dependent pathway. Clin. Cancer Res. 22, 2496–2507 (2016).
Minor, R. K. et al. SRT1720 improves survival and healthspan of obese mice. Sci. Rep. 1, 70 (2011).
Hubbard, B. P. & Sinclair, D. A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharm. Sci. 35, 146–154 (2014).
Dai, H., Sinclair, D. A., Ellis, J. L. & Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharm. Ther. 188, 140–154 (2018).
Noh, R. M. et al. Cardiometabolic effects of a novel SIRT1 activator, SRT2104, in people with type 2 diabetes mellitus. Open Heart 4, e000647 (2017).
Venkatasubramanian, S. et al. Effects of the small molecule SIRT1 activator, SRT2104 on arterial stiffness in otherwise healthy cigarette smokers and subjects with type 2 diabetes mellitus. Open Heart 3, e000402 (2016).
Feldman, J. L., Baeza, J. & Denu, J. M. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 288, 31350–31356 (2013).
You, W. et al. Structural basis of sirtuin 6 activation by synthetic small. Mol. Angew. Chem. Int. Ed. Engl. 56, 1007–1011 (2017).
Iachettini, S. et al. Pharmacological activation of SIRT6 triggers lethal autophagy in human cancer cells. Cell Death Dis. 9, 996 (2018).
Huang, Z. et al. Identification of a cellularly active SIRT6 allosteric activator. Nat. Chem. Biol. 14, 1118–1126 (2018).
Yoshino, M. et al. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science 372, 1224–1229 (2021).
Yuan, H. et al. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 119, 1904–1914 (2012).
Tervo, A. J. et al. An in silico approach to discovering novel inhibitors of human sirtuin type 2. J. Med. Chem. 47, 6292–6298 (2004).
Jung-Hynes, B., Nihal, M., Zhong, W. & Ahmad, N. Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition? J. Biol. Chem. 284, 3823–3832 (2009).
Audrito, V. et al. Nicotinamide blocks proliferation and induces apoptosis of chronic lymphocytic leukemia cells through activation of the p53/miR-34a/SIRT1 tumor suppressor network. Cancer Res 71, 4473–4483 (2011).
Taylor, D. M. et al. A brain-permeable small molecule reduces neuronal cholesterol by inhibiting activity of sirtuin 2 deacetylase. ACS Chem. Biol. 6, 540–546 (2011).
Chopra, V. et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2, 1492–1497 (2012).
Hisada, R. et al. The deacetylase SIRT2 contributes to autoimmune disease pathogenesis by modulating IL-17A and IL-2 transcription. Cell Mol. Immunol. 19, 738–750 (2022).
Bedalov, A., Gatbonton, T., Irvine, W. P., Gottschling, D. E. & Simon, J. A. Identification of a small molecule inhibitor of Sir2p. Proc. Natl Acad. Sci. USA 98, 15113–15118 (2001).
Grozinger, C. M., Chao, E. D., Blackwell, H. E., Moazed, D. & Schreiber, S. L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 276, 38837–38843 (2001).
Breitenstein, A. et al. Sirt1 inhibition promotes in vivo arterial thrombosis and tissue factor expression in stimulated cells. Cardiovasc Res 89, 464–472 (2011).
Nadtochiy, S. M., Redman, E., Rahman, I. & Brookes, P. S. Lysine deacetylation in ischaemic preconditioning: the role of SIRT1. Cardiovasc Res 89, 643–649 (2011).
Mai, A. et al. Design, synthesis, and biological evaluation of sirtinol analogues as class III histone/protein deacetylase (Sirtuin) inhibitors. J. Med. Chem. 48, 7789–7795 (2005).
Wang, J. et al. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int. J. Oncol. 41, 1101–1109 (2012).
Ota, H. et al. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 25, 176–185 (2006).
Lara, E. et al. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene 28, 781–791 (2009).
Rotili, D. et al. Discovery of salermide-related sirtuin inhibitors: binding mode studies and antiproliferative effects in cancer cells including cancer stem cells. J. Med. Chem. 55, 10937–10947 (2012).
Pagans, S. et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 3, e41 (2005).
Garg, G., Singh, A. K., Singh, S. & Rizvi, S. I. Promising drug discovery strategies for sirtuin modulators: what lessons have we learnt? Expert Opin. Drug Disco. 16, 915–927 (2021).
Portmann, S. et al. Antitumor effect of SIRT1 inhibition in human HCC tumor models in vitro and in vivo. Mol. Cancer Ther. 12, 499–508 (2013).
Marshall, G. M. et al. SIRT1 promotes N-Myc oncogenesis through a positive feedback loop involving the effects of MKP3 and ERK on N-Myc protein stability. PLoS Genet 7, e1002135 (2011).
Napper, A. D. et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 48, 8045–8054 (2005).
Solomon, J. M. et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell Biol. 26, 28–38 (2006).
Broussy, S., Laaroussi, H. & Vidal, M. Biochemical mechanism and biological effects of the inhibition of silent information regulator 1 (SIRT1) by EX-527 (SEN0014196 or selisistat). J. Enzym. Inhib. Med. Chem. 35, 1124–1136 (2020).
Chen, G. et al. Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis. Oncogene 36, 6863–6872 (2017).
Zhang, Y. et al. Identification of a small molecule SIRT2 inhibitor with selective tumor cytotoxicity. Biochem. Biophys. Res. Commun. 386, 729–733 (2009).
Karwaciak, I. et al. AC-93253 triggers the downregulation of melanoma progression markers and the inhibition of melanoma cell proliferation. Chem. Biol. Interact. 236, 9–18 (2015).
Zhang, Q. et al. A small molecule Inauhzin inhibits SIRT1 activity and suppresses tumour growth through activation of p53. EMBO Mol. Med. 4, 298–p312 (2012).
Zhang, Q. et al. The role of IMP dehydrogenase 2 in Inauhzin-induced ribosomal stress. Elife 3, e03077 (2014).
Trapp, J. et al. Adenosine mimetics as inhibitors of NAD+-dependent histone deacetylases, from kinase to sirtuin inhibition. J. Med. Chem. 49, 7307–7316 (2006).
Shim, K. H. et al. Small-molecule drug screening identifies drug Ro 31-8220 that reduces toxic phosphorylated tau in Drosophila melanogaster. Neurobiol. Dis. 130, 104519 (2019).
Rumpf, T. et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 6, 6263 (2015).
Qu, Z. A. et al. SIRT2 inhibits oxidative stress and inflammatory response in diabetic osteoarthritis. Eur. Rev. Med. Pharmacol. Sci. 24, 2855–2864 (2020).
Xu, D., He, H., Liu, D., Geng, G. & Li, Q. A novel role of SIRT2 in regulating gap junction communications via connexin-43 in bovine cumulus-oocyte complexes. J. Cell Physiol. 235, 7332–7343 (2020).
Lain, S. et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 13, 454–463 (2008).
Voogd, T. E., Vansterkenburg, E. L., Wilting, J. & Janssen, L. H. Recent research on the biological activity of suramin. Pharm. Rev. 45, 177–203 (1993).
Trapp, J. et al. Structure-activity studies on suramin analogues as inhibitors of NAD+-dependent histone deacetylases (sirtuins). Chem. Med. Chem. 2, 1419–1431 (2007).
Schuetz, A. et al. Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure 15, 377–389 (2007).
Zoltner, M. et al. Suramin exposure alters cellular metabolism and mitochondrial energy production in African trypanosomes. J. Biol. Chem. 295, 8331–8347 (2020).
Gey, C. et al. Phloroglucinol derivatives guttiferone G, aristoforin, and hyperforin: inhibitors of human sirtuins SIRT1 and SIRT2. Angew. Chem. Int. Ed. Engl. 46, 5219–5222 (2007).
Kim, Y. Y. et al. AGK2 ameliorates mast cell-mediated allergic airway inflammation and fibrosis by inhibiting FcepsilonRI/TGF-beta signaling pathway. Pharm. Res 159, 105027 (2020).
Carafa, V. et al. The Pan-Sirtuin inhibitor MC2494 regulates mitochondrial function in a leukemia cell line. Front Oncol. 10, 820 (2020).
Rotili, D. et al. Benzodeazaoxaflavins as sirtuin inhibitors with antiproliferative properties in cancer stem cells. J. Med. Chem. 55, 8193–8197 (2012).
Tervo, A. J. et al. Discovering inhibitors of human sirtuin type 2: novel structural scaffolds. J. Med. Chem. 49, 7239–7241 (2006).
Abedimanesh, N. et al. Effects of crocin and saffron aqueous extract on gene expression of SIRT1, AMPK, LOX1, NF-kappaB, and MCP-1 in patients with coronary artery disease: A randomized placebo-controlled clinical trial. Phytother. Res 34, 1114–1122 (2020).
Abedimanesh, S., Bathaie, S. Z., Ostadrahimi, A., Asghari Jafarabadi, M. & Taban Sadeghi, M. The effect of crocetin supplementation on markers of atherogenic risk in patients with coronary artery disease: a pilot, randomized, double-blind, placebo-controlled clinical trial. Food Funct. 10, 7461–7475 (2019).
Heshmati, J. et al. The effects of curcumin supplementation on oxidative stress, Sirtuin-1 and peroxisome proliferator activated receptor gamma coactivator 1alpha gene expression in polycystic ovarian syndrome (PCOS) patients: A randomized placebo-controlled clinical trial. Diabetes Metab. Syndr. 14, 77–82 (2020).
Gautam, S., Kumar, M., Kumar, U. & Dada, R. Effect of an 8-week yoga-based lifestyle intervention on psycho-neuro-immune axis, disease activity, and perceived quality of life in rheumatoid arthritis patients: a randomized controlled trial. Front Psychol. 11, 2259 (2020).
Bisht, S., Chawla, B., Tolahunase, M., Mishra, R. & Dada, R. Impact of yoga based lifestyle intervention on psychological stress and quality of life in the parents of children with retinoblastoma. Ann. Neurosci. 26, 66–74 (2019).
Tolahunase, M. R., Sagar, R., Faiq, M. & Dada, R. Yoga- and meditation-based lifestyle intervention increases neuroplasticity and reduces severity of major depressive disorder: A randomized controlled trial. Restor. Neurol. Neurosci. 36, 423–442 (2018).
Gautam, S., Kumar, U., Kumar, M., Rana, D. & Dada, R. Yoga improves mitochondrial health and reduces severity of autoimmune inflammatory arthritis: A randomized controlled trial. Mitochondrion 58, 147–159 (2021).
Fiore, D. et al. PDE5 inhibition ameliorates visceral adiposity targeting the miR-22/SIRT1 pathway: Evidence from the CECSID trial. J. Clin. Endocrinol. Metab. 101, 1525–1534 (2016).
de Kreutzenberg, S. V. et al. Metformin improves putative longevity effectors in peripheral mononuclear cells from subjects with prediabetes. A randomized controlled trial. Nutr. Metab. Cardiovasc. Dis. 25, 686–693 (2015).
Noureldein, M. H., Abd El-Razek, R. S., El-Hefnawy, M. H. & El-Mesallamy, H. O. Fenofibrate reduces inflammation in obese patients with or without type 2 diabetes mellitus via sirtuin 1/fetuin A axis. Diabetes Res. Clin. Pract. 109, 513–520 (2015).
Ferraz-Bannitz, R. et al. Bariatric surgery can acutely modulate ER-stress and inflammation on subcutaneous adipose tissue in non-diabetic patients with obesity. Diabetol. Metab. Syndr. 13, 19 (2021).
Fiorentino, F. et al. Sirtuin modulators: past, present, and future perspectives. Fut. Med. Chem. 14, 915–939 (2022).
Roggerio, A. et al. Gene expression of sirtuin-1 and endogenous secretory receptor for advanced glycation end products in healthy and slightly overweight subjects after caloric restriction and resveratrol administration. Nutrients 10, 937 (2018).
Mansur, A. P. et al. Serum concentrations and gene expression of sirtuin 1 in healthy and slightly overweight subjects after caloric restriction or resveratrol supplementation: A randomized trial. Int. J. Cardiol. 227, 788–794 (2017).
Bo, S. et al. Impact of sirtuin-1 expression on H3K56 acetylation and oxidative stress: a double-blind randomized controlled trial with resveratrol supplementation. Acta Diabetol. 55, 331–340 (2018).
van der Meer, A. J. et al. The selective sirtuin 1 activator SRT2104 reduces endotoxin-induced cytokine release and coagulation activation in humans. Crit. Care Med 43, e199–e202 (2015).
Beijers, R. J. et al. Resveratrol and metabolic health in COPD: A proof-of-concept randomized controlled trial. Clin. Nutr. 39, 2989–2997 (2020).
Yoshino, J. et al. Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab. 16, 658–664 (2012).
Asghari, S., Asghari-Jafarabadi, M., Somi, M. H., Ghavami, S. M. & Rafraf, M. Comparison of calorie-restricted diet and resveratrol supplementation on anthropometric indices, metabolic parameters, and serum sirtuin-1 levels in patients with nonalcoholic fatty liver disease: a randomized controlled clinical trial. J. Am. Coll. Nutr. 37, 223–233 (2018).
Clasen, B. F. et al. Growth hormone signaling in muscle and adipose tissue of obese human subjects: associations with measures of body composition and interaction with resveratrol treatment. J. Clin. Endocrinol. Metab. 99, E2565–E2573 (2014).
Poulsen, M. M. et al. High-dose resveratrol supplementation in obese men: an investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 62, 1186–1195 (2013).
Sands, B. E. et al. Assessing colonic exposure, safety, and clinical activity of SRT2104, a novel oral SIRT1 activator, in patients with mild to moderate ulcerative colitis. Inflamm. Bowel Dis. 22, 607–614 (2016).
Wu, J. et al. Boosting NAD+ blunts TLR4-induced type I IFN in control and systemic lupus erythematosus monocytes. J. Clin. Invest. 132, e139828 (2022).
El Ters, M. et al. Biological efficacy and safety of niacinamide in patients with ADPKD. Kidney Int. Rep. 5, 1271–1279 (2020).
Dollerup, O. L. et al. Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J. Physiol. 598, 731–754 (2020).
De Jong, N. P. et al. Short-term adaptations in skeletal muscle mitochondrial oxidative capacity and metabolic pathways to breaking up sedentary behaviors in overweight or obese adults. Nutrients 14, 454 (2022).
Kalhori, A., Rafraf, M., Navekar, R., Ghaffari, A. & Jafarabadi, M. A. Effect of turmeric supplementation on blood pressure and serum levels of sirtuin 1 and adiponectin in patients with nonalcoholic fatty liver disease: a double-blind, randomized, placebo-controlled trial. Prev. Nutr. Food Sci. 27, 37–44 (2022).
Goncalinho, G. H. F. et al. Effects of coffee on sirtuin-1, homocysteine, and cholesterol of healthy adults: does the coffee powder matter? J. Clin. Med. 11, 2985 (2022).
Deus, L. A. et al. Metabolic and hormonal responses to chronic blood-flow restricted resistance training in chronic kidney disease: a randomized trial. Appl. Physiol. Nutr. Metab. 47, 183–194 (2022).
Opstad, T. B., Sundfor, T., Tonstad, S. & Seljeflot, I. Effect of intermittent and continuous caloric restriction on Sirtuin1 concentration depends on sex and body mass index. Nutr. Metab. Cardiovasc. Dis. 31, 1871–1878 (2021).
Wasserfurth, P. et al. Impact of dietary modifications on plasma sirtuins 1, 3 and 5 in older overweight individuals undergoing 12-weeks of circuit training. Nutrients 13, 3824 (2021).
Abiri, B., Sarbakhsh, P. & Vafa, M. Randomized study of the effects of vitamin D and/or magnesium supplementation on mood, serum levels of BDNF, inflammation, and SIRT1 in obese women with mild to moderate depressive symptoms. Nutr. Neurosci. 25, 2123–2135 (2021).
Ghadimi, M. et al. Decreased insulin resistance in diabetic patients by influencing Sirtuin1 and Fetuin-A following supplementation with ellagic acid: a randomized controlled trial. Diabetol. Metab. Syndr. 13, 16 (2021).
Safarpour, P. et al. Vitamin D supplementation improves SIRT1, Irisin, and glucose indices in overweight or obese type 2 diabetic patients: a double-blind randomized placebo-controlled clinical trial. BMC Fam. Pr. 21, 26 (2020).
Davari, M. et al. Effects of cinnamon supplementation on expression of systemic inflammation factors, NF-kB and Sirtuin-1 (SIRT1) in type 2 diabetes: a randomized, double blind, and controlled clinical trial. Nutr. J. 19, 1 (2020).
Aslani, M. R. et al. Effects of conjugated linoleic acid supplementation on serum levels of interleukin-6 and sirtuin 1 in COPD patients. Avicenna J. Phytomed. 10, 305–315 (2020).
Mohammadshahi, M., Zakizadeh, E., Ahmadi-Angali, K., Ravanbakhsh, M. & Helli, B. The synergic effects of alpha-lipoic acid supplementation and electrical isotonic contraction on anthropometric measurements and the serum levels of VEGF, NO, sirtuin-1, and PGC1-alpha in obese people undergoing a weight loss diet. Arch. Physiol. Biochem. 128, 1195–1201 (2020).
Khazdouz, M. et al. Effect of selenium supplementation on expression of SIRT1 and PGC-1alpha genes in ulcerative colitis patients: a double blind randomized clinical trial. Clin. Nutr. Res. 9, 284–295 (2020).
Aghasi, M. et al. Beneficial effects of green cardamom on serum SIRT1, glycemic indices and triglyceride levels in patients with type 2 diabetes mellitus: a randomized double-blind placebo controlled clinical trial. J. Sci. Food Agric. 99, 3933–3940 (2019).
Aliashrafi, S., Arefhosseini, S. R., Lotfi-Dizaji, L. & Ebrahimi-Mameghani, M. Effect of vitamin D supplementation in combination with weight loss diet on lipid profile and sirtuin 1 in obese subjects with vitamin D deficiency: a double blind randomized clinical trial. Health Promot Perspect. 9, 263–269 (2019).
Alayon, A. N., Ortega Avila, J. G. & Echeverri Jimenez, I. Carbohydrate metabolism and gene expression of sirtuin 1 in healthy subjects after Sacha inchi oil supplementation: a randomized trial. Food Funct. 9, 1570–1577 (2018).
Lopez-Domenech, S. et al. Pinitol alleviates systemic inflammatory cytokines in human obesity by a mechanism involving unfolded protein response and sirtuin 1. Clin. Nutr. 37, 2036–2044 (2018).
Sardu, C. et al. Inflammatory cytokines and SIRT1 levels in subcutaneous abdominal fat: relationship with cardiac performance in overweight pre-diabetics patients. Front Physiol. 9, 1030 (2018).
Daneshi-Maskooni, M. et al. Green cardamom increases Sirtuin-1 and reduces inflammation in overweight or obese patients with non-alcoholic fatty liver disease: a double-blind randomized placebo-controlled clinical trial. Nutr. Metab. (Lond.) 15, 63 (2018).
Sohrab, G., Nasrollahzadeh, J., Tohidi, M., Zand, H. & Nikpayam, O. Pomegranate juice increases sirtuin1 protein in peripheral blood mononuclear cell from patients with type 2 diabetes: a randomized placebo controlled clinical trial. Metab. Syndr. Relat. Disord. 16, 446–451 (2018).
Margolis, L. M., Murphy, N. E., Carrigan, C. T., McClung, H. L. & Pasiakos, S. M. Ingesting a combined carbohydrate and essential amino acid supplement compared to a non-nutritive placebo blunts mitochondrial biogenesis-related gene expression after aerobic exercise. Curr. Dev. Nutr. 1, e000893 (2017).
Jakubowicz, D. et al. Influences of breakfast on clock gene expression and postprandial glycemia in healthy individuals and individuals with diabetes: a randomized clinical trial. Diabetes Care 40, 1573–1579 (2017).
Krumpolec, P. et al. Aerobic-strength exercise improves metabolism and clinical state in parkinson’s disease patients. Front Neurol. 8, 698 (2017).
Mazaherioun, M. et al. Beneficial effects of n-3 polyunsaturated fatty acids on adiponectin levels and AdipoR gene expression in patients with type 2 diabetes mellitus: a randomized, placebo-controlled, double-blind clinical trial. Arch. Med. Sci. 13, 716–724 (2017).
Lopez-Moreno, J. et al. Dietary fat quantity and quality modifies advanced glycation end products metabolism in patients with metabolic syndrome. Mol. Nutr. Food Res. 61 (2017).
Vlassara, H. et al. Oral AGE restriction ameliorates insulin resistance in obese individuals with the metabolic syndrome: a randomised controlled trial. Diabetologia 59, 2181–2192 (2016).
Wegman, M. P. et al. Practicality of intermittent fasting in humans and its effect on oxidative stress and genes related to aging and metabolism. Rejuvenation Res 18, 162–172 (2015).
Saboori, S. et al. Beneficial effects of omega-3 and vitamin E coadministration on gene expression of SIRT1 and PGC1alpha and serum antioxidant enzymes in patients with coronary artery disease. Nutr. Metab. Cardiovasc Dis. 26, 489–494 (2016).
Traba, J. et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J. Clin. Invest. 125, 4592–4600 (2015).
Mendham, A. E. et al. Rugby-specific small-sided games training is an effective alternative to stationary cycling at reducing clinical risk factors associated with the development of type 2 diabetes: a randomized, controlled trial. PLoS One 10, e0127548 (2015).
Di Renzo, L. et al. Intake of red wine in different meals modulates oxidized LDL level, oxidative and inflammatory gene expression in healthy people: a randomized crossover trial. Oxid. Med. Cell Longev. 2014, 681318 (2014).
Amengual, J. E. et al. Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood 122, 2104–2113 (2013).
Krueger, J. G. et al. A randomized, placebo-controlled study of SRT2104, a SIRT1 activator, in patients with moderate to severe psoriasis. PLoS One 10, e0142081 (2015).
Baksi, A. et al. A phase II, randomized, placebo-controlled, double-blind, multi-dose study of SRT2104, a SIRT1 activator, in subjects with type 2 diabetes. Br. J. Clin. Pharm. 78, 69–77 (2014).
Funding
This work was supported by the National Key R&D Program of China (No. 2017YFC0907401 to Y. -H. Z. and No. 2022YFC2704200 to Q. -J. W.), Natural Science Foundation of China (No. 82073647 to Q. -J.-W. No. 82102254 to T. -N. Z., and No. 81902607 to Y. -X. Z.), LiaoNing Revitalization Talents Program (No. XLYC1907102 to Q. -J. W. and No. XLYC1802095 to Y. -H. Z.), the JieBangGuaShuai Project of Liaoning Province (No. 2021JH1/1040050 to Y. -H. Z.), Outstanding Scientific Fund of Shengjing Hospital (Q. -J. W. and Y. -H. Z.), and 345 Talent Project of Shengjing Hospital of China Medical University (Q. -J. W., T. N. Z., and Y. -X. Z.).
Author information
Authors and Affiliations
Contributions
Q.- J. W., T. -N. Z., and Y. -H. Z. provided the conceptual idea and design of this study and wrote the manuscript. H. -H. C., X. -F. Y., J. -L. L., Y. -Y. L., Y. -S. L., G. Z., J. -Q. Z., Y. -F. W,. J. -Y. G., F.-H. L., Q. C., Y. -X. Z., and C. -G. L. performed literature searching and wrote the manuscript. H. -H. C., X. -F. Y., J. -L. L., Y. -Y. L., Y. -S. L., G. Z., J. -Q. Z., Y. -F. W., and J. -Y. G. made the figures and tables and wrote the manuscript. All authors listed have made a substantial contribution to this work. All authors have read and approved the article. Q. -J. W. and T. -N. Z. contributed equally to this work. We thank the research team (Ya-Lin Zhang, Lu Zhao, Zhu-Xi Liu, and Si-Tian Zang from Shengjing Hospital of China Medical University) for their efforts in data collection and preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, QJ., Zhang, TN., Chen, HH. et al. The sirtuin family in health and disease. Sig Transduct Target Ther 7, 402 (2022). https://doi.org/10.1038/s41392-022-01257-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01257-8
This article is cited by
-
SIRT7 promotes the proliferation and migration of anaplastic thyroid cancer cells by regulating the desuccinylation of KIF23
BMC Cancer (2024)
-
The effect of weight loss therapies on sirtuin 1 regulation: a systematic review and meta-analysis of randomized controlled trials
BMC Nutrition (2024)
-
Sirtuins in intervertebral disc degeneration: current understanding
Molecular Medicine (2024)
-
Targeting histone deacetylases in head and neck squamous cell carcinoma: molecular mechanisms and therapeutic targets
Journal of Translational Medicine (2024)
-
Sirtuins in kidney diseases: potential mechanism and therapeutic targets
Cell Communication and Signaling (2024)
