
Abstract
As an essential micronutrient, copper is required for a wide range of physiological processes in virtually all cell types. Because the accumulation of intracellular copper can induce oxidative stress and perturbing cellular function, copper homeostasis is tightly regulated. Recent studies identified a novel copper-dependent form of cell death called cuproptosis, which is distinct from all other known pathways underlying cell death. Cuproptosis occurs via copper binding to lipoylated enzymes in the tricarboxylic acid (TCA) cycle, which leads to subsequent protein aggregation, proteotoxic stress, and ultimately cell death. Here, we summarize our current knowledge regarding copper metabolism, copper-related disease, the characteristics of cuproptosis, and the mechanisms that regulate cuproptosis. In addition, we discuss the implications of cuproptosis in the pathogenesis of various disease conditions, including Wilson’s disease, neurodegenerative diseases, and cancer, and we discuss the therapeutic potential of targeting cuproptosis.
Similar content being viewed by others
Introduction
The essential micronutrient copper (Cu) functions as a key catalytic cofactor in a wide array of biological processes, including mitochondrial respiration, antioxidant defense, and bio-compound synthesis. Importantly, the intracellular Cu concentration is kept at a relatively low range, and moderate increases can cause cytotoxicity and even lead to cell death; thus, the uptake, distribution, and elimination of Cu are tightly regulated. Moreover, in humans, genetic mutations that cause Cu accumulation have been linked to severe, potentially life-threatening pathological conditions.1 Thus, understanding the process by which Cu accumulation causes cellular toxicity is an important first step toward developing new, effective therapies.
Cuproptosis is a recently discovered form of regulated cell death triggered by excess Cu2+.2 This Cu-induced form of programmed cell death is distinct from other cell death pathways, including apoptosis, ferroptosis, and necroptosis. Intracellular Cu targets and binds to lipoylated components in the tricarboxylic acid (TCA) cycle, and aggregation of these Cu-bound lipoylated mitochondrial proteins—and the subsequent reduction in Fe–S (iron–sulfur) clusters—induces proteotoxic stress and ultimately cell death.
Here, we review our current knowledge regarding the features of cuproptosis and the underlying mechanisms. In addition, we discuss new perspectives with respect to the putative pathophysiological role of cuproptosis in various diseases, as well as the therapeutic potential of targeting this novel form of cell death.
Systemic copper metabolism
Cu is an essential element in virtually all living organisms, and numerous studies have shown that Cu serves as a cofactor for a variety of key metabolic enzymes that drive a broad range of physiological processes.3,4 Thus, the systemic Cu levels must be maintained within a narrow range to ensure normal biochemical processes.
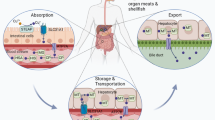
The absorption of dietary Cu occurs primarily at the duodenum and small intestine.5 The uptake of Cu into intestinal epithelial cells is mediated mainly by Cu transport protein 1 (CTR1), located on the apical side of the enterocytes. This process is facilitated by the activity of the metalloreductases six-transmembrane epithelial antigen of the prostate (STEAP) and duodenal cytochrome b (DCYTB),6,7 which reduce divalent Cu2+ to monovalent Cu+; the ionic state in which CTR1 transports Cu. Following absorption by the gastrointestinal tract, Cu is secreted into the bloodstream and bound to soluble chaperones, such as albumin, transcuprein, histidines, and macroglobulins.8,9,10,11 Upon reaching the liver, the hepatocytes mediate the uptake of Cu via CTR1. Inside the cytoplasm, Cu is then either delivered by Cu chaperones to specific proteins or chelated by metallothionein (MT) for storage.12,13,14,15 The principal Cu chaperones include COX17 (which delivers Cu to cytochrome c oxygenase), CCS (Cu chaperone for superoxide dismutase, which delivers Cu to superoxide dismutase 1), and ATOX1 (which delivers Cu to ATP7A and ATP7B). The Cu-ATPases (ATP7B in the case of hepatocytes) pump Cu ions from the liver back into the blood, where it again binds to soluble chaperones and is transported to specific tissues and organs.16 Upon reaching its target tissues, Cu catalyzes reactions in a wide range of physiological processes, including mitochondrial energy production, tyrosine and neurotransmitter metabolism, redox homeostasis, and extracellular matrix remodeling.17,18,19,20
In the body, Cu is stored primarily in the liver,21,22 and excess of Cu is eliminated in the feces, either through biliary excretion—the major form of endogenous Cu elimination—or as unabsorbed metal ions.23 Other pathways for Cu elimination such as urine, sweat, and menses play a relatively minor role in Cu loss. Taken together, the systemic Cu status is regulated by duodenal absorption and/or biliary excretion. In the context of high Cu intake, the absorption of Cu decreases and elimination of Cu increases; conversely, during periods of low Cu intake, endogenous Cu excretion via the bile decreases, and the retention of absorbed Cu increases.24
Copper homeostasis is tightly regulated in cells
To maintain Cu homeostasis at the cellular level, intracellular Cu content is regulated by a complex network of Cu-dependent proteins, including cuproenzymes, Cu chaperones, and membrane transporters. These proteins work together to coordinate the import, export, and intracellular utilization of Cu, thus maintaining cellular Cu levels within a specific range, which helps prevent the consequences of Cu overload and Cu deficiency.
Cu uptake into cells
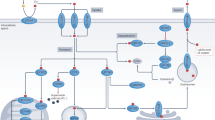
The high-affinity Cu transporter CTR1 (also known as SLC31A1) is structurally and functionally conserved from yeast to humans and is responsible for the majority of Cu uptake into cells.25 A growing body of evidence suggests that CTR1 transports Cu+, and Ctr1 may function together with metalloreductases such as STEAP and/or DCYTB to transport Cu across the membrane26,27 (Fig. 1). Moreover, in vitro studies have shown that the expression of CTR1 is regulated in a Cu-dependent manner, in which it is down-regulated under Cu-overload conditions and up-regulated under Cu-depleted conditions.28 Similarly, increased expression of Ctr1 has been observed in the intestines of mice fed a Cu-deficient diet, suggesting a negative-feedback loop of Ctr1 regulation.29

Summary of the pathways that mediate cellular Cu metabolism. Extracellular Cu2+ is reduced by the reductase STEAP to Cu+, which is transported into the cell by the Cu transporter CTR1, where it is delivered to cytosolic Cu chaperones such as CCS and SOD1 and then delivered to specific subcellular compartments such as the mitochondria, TGN, and nucleus. In the mitochondria, Cu is involved in the respiratory chain and redox pathways via binding to CCO. In the mitochondrial intermembrane space, COX17 binds to and delivers Cu to either SCO1 or COX11, which transfers Cu to the cytochrome oxidase subunit. In the nucleus, Cu can bind to transcription factors and drive gene expression. Finally, in the TGN the Cu+-ATPase transporters ATP7A and ATP7B transfer Cu from the cytosol to the TGN lumen, where it activates Cu-dependent enzymes in the secretory pathway. When cytosolic Cu levels are high, ATP7A and ATP7B exit the TGN and facilitate Cu export. Created with BioRender. ATOX1, antioxidant 1 copper chaperone; ATP7A and ATP7B, ATPase copper transporter 7A and 7B, respectively; CCO, cytochrome c oxidase; CCS, copper chaperone for superoxide dismutase; COX17 cytochrome c oxidase copper chaperone 17, COX11 cytochrome c oxidase copper chaperone 11, SCO1 synthesis of cytochrome c oxidase 1, SOD1 superoxide dismutase 1, STEAP the six-transmembrane epithelial antigen of the prostate, SLC31A1 solute carrier family 31 member 1, TGN trans-Golgi network
Several lines of evidence suggest that CTR1 is required for the transport of Cu into specific organs/tissues. For example, intestine-specific Ctr1 knockout mice develop Cu deficiency in the majority of peripheral tissues, suggesting that Ctr1 functions as the major factor driving intestinal Cu absorption.30 Interestingly, global Ctr1 global knockout (Ctr1–/–) mice die in utero a mid-way through gestation, and embryonic fibroblasts prepared from Ctr1–/– embryos have drastically reduced levels of Cu and Cu-dependent enzyme activity, suggesting that Ctr1 plays an essential role in embryonic development in mammals.31
Intracellular Cu distribution
Within the cytoplasm, Cu trafficking is tightly coordinated by a fine-tuned network of high-affinity Cu chaperones. The chaperone antioxidant-1 (ATOX1) is responsible for transferring Cu to ATP7A and ATP7B in the trans-Golgi network (TGN) and facilitates the synthesis of cuproenzymes such as lysyl oxidase, tyrosinase, and ceruloplasmin32 (Fig. 1). In embryonic mouse fibroblasts, Atox1 has also been shown to serve as a Cu-dependent transcriptional regulator, and contribute to cell proliferation.33 Mice lacking the gene encoding Atox1 are observed to be perinatal lethal due to impaired Cu balance, which likely reflects the central role that this chaperone plays in Cu transport and homeostasis.34
The chaperone CCS delivers Cu to superoxide dismutase 1 (SOD1) to detoxify ROS and maintain Cu homeostasis (Fig. 1). Studies have shown that organisms lacking SOD1 have increased oxidative stress. For example, yeast with mutated sod1 accumulate DNA mutations,35 and SOD1 knockout mice develop hepatocellular carcinoma, presumably as a result of oxidative damage of hepatocytes.36 Moreover, CCS expression is regulated by cellular Cu status; when Cu content decreases, CCS levels increase, and when Cu content is high, the degradation of CCS increases.37,38 Both CCS and SOD1 are localized in both the cytoplasm and mitochondrial intermembrane space (IMS), where they detoxify mitochondria-derived superoxides.39,40
In addition to delivering Cu to the secretory compartment and cytosolic proteins, Cu is targeted to the mitochondria, where cytochrome oxidase (COX) uses Cu for oxidative phosphorylation and mitochondrial function. In humans, COX consists of two core subunits, COX1 and COX2, which bind Cu at the conserved CuB and CuA sites, respectively.41 The Cu chaperone COX17, located in the IMS, is responsible for transporting Cu from the cytosol to the mitochondrial IMS and contributes to the assembly of COX.15,42 In the IMS, Cu+ is bound to COX17 and delivered either to the chaperone synthesis of cytochrome c oxidase 1 (SCO1) for transfer to the COX2 subunit, or to COX11 for delivery to the COX1 subunit43,44,45 (Fig. 1). Finally, mutations in COX17, SCO1, and SCO2 have been associated with decreased COX activity and can even be fatal.46,47
Cu efflux mediated by Cu-ATPases
The Cu-ATPases ATP7A and ATP7B act as the major transporter for exporting cellular Cu. The regulation of ATP7A/7B localization and function is important for mediating this process and is multifaceted48 (Fig. 1). Under physiological cellular Cu levels, these transporters are found to be located in the TGN where they pump Cu from the cytosol into the lumen of the TGN. When intracellular Cu increases, these transporters translocate from the TGN to vesicular compartments and fuse with the plasma membrane to export Cu; when Cu content returns to physiological levels, they are recycled back to the TGN.49
ATP7A and ATP7B have distinct expression patterns. ATP7A is expressed in most tissues/organs, with the exception of the liver, where ATP7B is found to be predominantly expressed.32 At the basolateral membrane of enterocytes, Cu is pumped by ATP7A into the portal circulation for delivery to the liver, the main organ for Cu storage.50 In other tissues such as the placenta and the blood–brain barrier (BBB), ATP7A mediates the transport of Cu across polarized cells to ensure adequate Cu is delivered for the development of the fetus and brain.32 In the liver, ATP7B mobilizes Cu in secretory vesicles to the bile in order to prevent the accumulation of Cu. Consistent with their important roles in maintaining health, mutations in ATP7A and ATP7B cause inherited disorders of Cu metabolism, which have been recognized as Menkes disease (MD) and Wilson’s disease (WD), respectively.51
Diseases related to copper dysregulation
Given that either Cu deficiency or accumulation exhibit adverse health effect, it is important to note that the inability to maintain Cu balance has been associated with a wide range of pathologic conditions, including MD, WD, neurodegenerative diseases, cancer, and cardiovascular disease.
Menkes disease
MD is an X-linked recessive disease that affects Cu metabolism and is caused by mutations in the ATP7A gene.52 The resulting loss of functional ATP7A in the intestine leads to reduced Cu efflux into the blood, an accumulation of Cu in enterocytes, and generalized severe systemic Cu deficiency.53 MD is a fatal disorder, and affected patients usually die in early childhood. Children with MD exhibit severe symptoms such as mental retardation, hypothermia, neuronal degeneration, bone fractures, hair and skin abnormalities, and widespread vascular abnormalities.51
In the brain of patients with MD, the deficiency of Cu adversely affects the physiological function of Cu-dependent enzymes such as dopamine-β-hydroxylase (DβH), which is essential for synthesizing the neurotransmitter.54 Infants with MD present with high levels of dihydroxyphenylalanine, the precursor of catecholamines, suggesting reduced DβH activity.54,55 The impaired function of DβH may lead to impaired synaptogenesis and axonal growth, manifesting as hypotonia and seizures. In addition, impaired mitochondrial function and intracerebral lactic acidosis caused by reduced COX activity in the brain may also contribute to the degeneration of neurons in these patients.56
Low levels of Cu have also been reported in the heart, liver, muscle, skin, and connective tissues of patients with MD. For example, Cu deficiency in the heart has correlated with congenital cardiovascular abnormalities such as tetralogy of Fallot, which can present in newborn infants with MD.57 In the skin, muscle, and connective tissues, reduced ATP7A function results in decreased activity of lipoxygenase (LOX), which oxidizes the lysine and hydroxylysine in elastin and collagen.58 Decreased LOX activity reduces the formation of the covalent cross-links that provide tensile strength and elasticity to skeletal, muscular, and cardiovascular connective tissues.59 Thus, patients with MD often present with abnormalities in their connective tissue, including osteopenia, skin laxity, arterial aneurysms, and spontaneous fractures.60
Currently, patients with MD are diagnosed primarily through a genetic screen and treated using Cu salts such as Cu-histidine complexes.61,62 Subcutaneously injected Cu-histidinate delivers Cu directly through the bloodstream to various tissue/organs, bypassing the malfunctioning mechanism by which Cu is normally absorbed through the intestine, thereby helping restore systemic Cu levels in patients with MD.61,63 Notably, treatment with Cu-histidine has been shown to be effective in patients in which the mutated ATP7A protein retains the ability to transport Cu across the BBB.64 In addition, patients who are treated with Cu-histidine in an early stage of the disease can exhibit gross improvements, with fine neuronal development and motor movement, whereas patients who are diagnosed and treated in a later stage generally have a poor prognosis.64 Moreover, treatment with Cu-histidine appears to result in a better general neurological outcome in asymptomatic patients compared to symptomatic patients.65
Wilson disease
A classic example of a Cu-overload condition is WD, an autosomal recessive disorder characterized by a variety of mutations in the ATP7B gene. The resulting impaired ATP7B function impairs Cu excretion and leads to persistent Cu accumulation in the brain, liver, and other tissues.66 In healthy individuals, hepatic Cu content is generally <55 μg/g dry weight; in patients with WD, hepatic Cu can exceed 250 μg/g dry weight.67 Most frequently, WD patients present with hepatic and/or neuropsychiatric symptoms.
Cu toxicity is considered as the primary cause of organ damage in patients with WD.68,69,70 Moreover, DNA damage, lipid peroxidation, and dysfunction of mitochondrial are typical findings in the liver of patients with WD.71,72 Morphological changes in hepatic mitochondria—including widening of the interstitial space, separation and enlargement of the inner and outer membranes, and/or the occurrence of large vacuoles—usually occur in the early stages of WD, and these changes are considered a hallmark feature of hepatic injury in WD.67 Patients with WD can present with liver symptoms that include fulminant hepatic failure (also known as acute liver failure), persistently elevated levels of serum aminotransferases, jaundice, and chronic hepatitis.73 Interestingly, some patients do not manifest these hepatic changes, but instead present with neurological symptoms; however, hepatic Cu accumulation has also been observed in these particular patients and may lead to the development of hepatic cirrhosis.74 In most cases of WD, Cu deposition can be observed in the cornea known as Kayser–Fleischer rings, representing as an ophthalmic manifestation of the disease.66
Approximately 40–50% of patients with WD present with neurological and neuropsychiatric signs.75 Moreover, high levels of Cu have been observed in nearly all brain regions in patients with WD.76 The main neurological symptoms include tremors, akinetic-rigid syndrome (Parkinsonism), ataxia, and dystonia; other common neurological findings can include dysarthria, spasticity, and a lack of motor coordination.77 Structural brain MRI scans in patients with MD have shown widespread lesions in the midbrain, putamen, pons, globus pallidus, thalamus, and cerebellum, as well as cortical atrophy.67,78 Loss of neurons and the occurrence of abnormal astrocytes are typical neuropathological features in patients with WD.79 Non-hepatic and non-neuronal complications associated with WD include osteomalacia, osteoarthritis, hemolysis, cardiac arrhythmia, and renal abnormalities.80,81,82
Evidence is limited regarding the strategy of restricting dietary Cu for treating WD.83 Current strategies for treating WD primarily focus on oral zinc to decrease Cu absorption and the use of chelating agents such as d-penicillamine and trientine.84,85,86 The choice of treatment generally depends on the disease stage, with Cu chelators recommended for patients with advanced symptomatology.84,87 Weiss et al. previously evaluated the efficacy of the chelators d-penicillamine and trientine in patients with WD; they found that although treatment led to hepatic improvement in >90% of patients, the response rate among neurological patients was considerably less favorable.88 However, after 4 years of therapy, around 60% of patients showed improvement in neurological symptoms.88 Another chelating agent, tetrathiomolybdate (TTM), has also been shown to reduce circulating levels of free Cu ions.89 A randomized trial comparing the efficacy of TTM versus trientine in patients with neurological WD has found that neurological deterioration was less common in the TTM-treated group.86 In addition, another clinical trial has shown that the use of WTX101, the bis-choline salt of TTM, exhibits beneficial effects and significant improvements in controlling Cu-induced neurological symptoms.90
Neurodegenerative diseases
Alzheimer’s disease (AD)
AD is one of the most common neurodegenerative disorders. The pathological hallmark of AD is the deposition of amyloid plaques and neurofibrillary tangles in the gray matter due to aberrant processing of amyloid precursor protein (APP) leading to the aggregation of amyloid-β (Aβ) peptides and tau protein, respectively.91
Mounting evidence has implicated that altered Cu homeostasis may be associated with the pathogenesis of AD, as Cu may interact with several key pathogenic factors such as Aβ and tau. Moreover, serum-free Cu levels are known to be elevated with aging,92 and both total and free Cu levels are increased in the serum of patients with AD compared to healthy controls.93,94,95 Importantly, high levels of Cu have been measured in senile plaques in patients with AD.96 Excess Cu could bind directly to Aβ peptides with high affinity, further increasing aggregation of Aβ and driving increased neurotoxicity.97,98 Binding of Cu to Aβ leads to the formation of neurotoxic dityrosine-linked β-amyloid dimers, which prevents the dimers from degrading to monomers and may therefore be relevant to the formation of amyloid deposits.97,99,100 In vitro, sequestration of Cu from Aβ peptides prevents its accumulation and leads to degradation of Aβ, suppression of hydroxyl radical (•OH) production, and oxidative damage, eventually reducing cell death.101,102,103
Cu also plays an essential role in the activation of microglia, a process linked to neurodegeneration. In the BV2 cells (a mouse microglial cell line), Cu could trigger activation of the NF-κB signaling pathway and increase the release of inflammatory factors such as nitric oxide (NO) and the cytokine tumor necrosis factor-α (TNF-α).104 Cu in excess may also reduce the brain’s physiological ability to remove Aβ peptides.105 Kitazawa et al. have reported that the existence of Cu-Aβ complexes reduce the expression of LRP1 (lipoprotein receptor-related protein 1), further reducing the clearance of neurotoxic Aβ and increasing brain Aβ deposits.104 Moreover, Cu may play a pathogenic role in tau proteins in the context of AD. For example, Cu could trigger the phosphorylation and aggregation of tau protein, enhancing the neurotoxicity of tau aggregates.106,107
Aiming to evaluate the effect of lowering-Cu therapy in treating AD, several Cu chelators have been developed and tested in mouse models of AD, showing promising beneficial effects such as antioxidant properties, reduced Aβ aggregation, and amelioration of neurological symptoms. For example, using an APP transgenic mouse model of AD, Cherny et al. have shown that the chelator clioquinol could decrease Aβ deposits and improve the capacities in both learning and memory.101 Moreover, several phase II trials have shown that clioquinol is capable of reducing Aβ aggregation and improving cognitive function, but fail to provide sufficient evidence of a positive benefit in a larger trial.108,109 The clioquinol derivative PBT2 has been shown to inhibit Cu-induced Aβ accumulation and possess greater BBB permeability and solubility.110,111 In a mouse model of AD, the treatment of PBT2 decreases the level of interstitial brain Aβ and the phosphorylation level of tau protein, and lead to the restoration of cognition.108 Finally, positive results have also been obtained from several phase Ib/IIa trials, which demonstrate that PBT2 could reduce Aβ levels and improve cognitive performance in patients with AD.112,113
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a progressive disease characterized by the selective degeneration of motor neurons leading to muscular weakness, muscle atrophy, and eventually death. Approximately 10–20% of cases present as an autosomal dominant familial form of ALS (FALS), with the remaining cases presenting as a non-inherited, sporadic form of ALS (SALS). One of the principal causes of FALS is the mutation in the SOD1 gene.114 In the context of ALS, the interaction of CCS with mutant SOD1 is faulty, resulting in decreased delivery of Cu to the mitochondria and the accumulation of the less-stabilized SOD1, which is prone to acquire pro-oxidants, consequently causing a toxic gain-of-function effect in motor neurons.115 Interestingly, a previous study has shown that overexpressing CCS accelerates neurological deficits and shortens the life span of mice carrying the G93A mutation in SOD1 (SOD1G93A mice).116
The role of Cu in the pathogenesis of ALS is unclear and currently under study. For example, Cu deficiency has been shown to promote the aberrant hydrophobicity of mutant SOD1, contributing to the impaired interaction of SOD1 and its neuronal toxicity, and this effect could be reversed by the supplementation of Cu.117,118 In the spinal cord of SOD1G37R mutant mice, more than 50% of the mutant SOD1 protein is not fully Cu-bound, and treatment with a Cu-containing compound significantly decreased the levels of metal-deficient SOD1 and increased the pool of fully Cu-bound SOD1.119 Additional studies have shown that aggregated mutant SOD1 has low Cu content both in cultured cells and in the spinal cord of transgenic SOD1 mice, regardless of the mutant protein’s ability to bind Cu120,121; moreover, the degree of Cu deficiency in mutant SOD1 aggregates is proportional to the clinical severity of ALS.122
In addition, the dysregulated cellular Cu homeostasis and impaired function of Cu-dependent enzymes may also contribute to the toxic effects of mutant SOD1 proteins and disease progression. For example, in SOD1G93A-mutant mice, elevated concentrations of Cu have been observed at the presymptomatic stage in skeletal muscle and the spinal cord, and this condition aggravates further during disease progression.123,124 Elevated Cu levels have also been observed in the cerebrospinal fluid of patients with ALS.121,125 Further, as discussed above overexpressing CCS in SOD1G93A mice accelerate disease progression, and this effect could be attenuated by treating the mice with a complex that delivers Cu.116,126 Specifically, CCS-overexpressing SODG93A mice exhibit significantly decreased COX activity when these mice reached the end stage of disease, whereas SODG93A mice not overexpressing CCS had no apparent decrease in COX activity at the same stage,126 supporting the hypothesis that overexpressing CCS increases the delivery of Cu to SOD1 while reducing the delivery of Cu to other enzymes (e.g., mitochondrial cytochrome c oxidase), thus leading to additional toxic effects.
It is interesting to note both Cu chelators such as d-penicillamine and TTM and Cu-delivery agents such as CuII(atsm) have shown beneficial effects in several mouse models.119,127,128 As an example, treatment with CuII(atsm) has been shown to improve motor function and increase survival in both the SOD1G93A and SOD1G37R mouse models of ALS.119,129,130 In SOD1G37R mice, CuII(atsm) increased the delivery of Cu to the metal-deficient mutant form of apo-SOD1, allowing its conversion to the more stable, less toxic holo-SOD1 form.119 Treatment with TTM could prolong the survival of both presymptomatic and symptomatic SOD1G93A mice, decrease the loss of motor neurons and attenuate the severity of skeletal muscle atrophy. Notably, TTM is capable of both suppressing the activity and reducing the aggregation of mutant SOD1 proteins.119,131 Chelating Cu using d-penicillamine has also been reported to have beneficial effects in transgenic SOD1 mouse models, delaying disease progression and prolonging survival.127
Huntington’s disease (HD)
HD is a rare autosomal-dominant neurological disorder characterized by a progressive loss of psychiatric, cognitive, and motor function. HD has been hypothesized to be caused by an abnormal expansion of the polyglutamine repeat at the N-terminus of the mutant Huntingtin (HTT) protein, leading to brain atrophy primarily within the striatum and cerebral cortex.132 The formation of aggregated mutant HTT proteins in neurons leads to reduced energy production, oxidative stress, and neurodegeneration.133,134
Cu has been suggested to play a role in HD. Abnormally high concentrations of Cu have been found in the striatum in patients with HD and in mouse models of HD.135,136 Moreover, several reports have suggested that an accumulation of Cu promotes aggregation of the HTT protein and interacts with histidine residues at the N-terminus of the protein.136 In vitro studies have shown that Cu binds to HTT proteins with 17–68 glutamine residues, whereas neither Fe nor Zn exhibits binding affinity. In addition, Cu chelators can inhibit the formation of mutant HTT aggregates, whereas Cu supplementation promotes aggregate formation.136 Moreover, Cu may contribute to the progression of HD by inhibiting mitochondrial dehydrogenases, including succinate dehydrogenase (SDH) and lactate dehydrogenase (LDH), both of which are susceptible to Cu-mediated inactivation.137,138 In the brain, LDH is critical for lactate metabolism,139 and neurons use lactate released by astrocytes as an energy substrate. In the striatum of patients with HD, reduced lactate clearance has been measured using magnetic resonance spectroscopy,140 reminiscent of the neuronal degeneration observed in mice treated with LDH inhibitors.136 These findings suggest that Cu may contribute to the pathogenesis of HD by inhibiting key enzymes involved in lactate metabolism.
With respect to treatment options, the use of Cu chelators, Clioquinol, or tetrathiomolybdate, mitigates the pathology and behavioral abnormalities of the R6/2 HD mouse model.141,142 In addition, the dietary intervention of Cu uptake with the treatment of bathocuproine disulfonate (BCS), the water-soluble Cu chelator, could significantly improve the survival rate in a Drosophila model of HD.143
Cancer
Cu status in cancer
The role of Cu in the progression of cancer has long been a topic of study, as Cu ions may be involved in the activation of cell proliferation-related signaling pathways. Evidence has suggested that cancer cells generally have a higher demand for Cu compared to healthy, resting cells.144 Moreover, increased Cu concentrations have been reported in tumor tissues and/or serum obtained from patients with various types of cancers, including breast,145,146 lungs, gastrointestinal,147 oral,148 thyroids,149 gall bladder,150 gynecologic,151,152 pancreatic,153 and prostate cancer.154 In addition, elevated levels of serum Cu have been correlated with both tumor stage and disease progression in patients with colorectal, lung, and breast cancer.155,156,157
The role of Cu in carcinogenesis
Given that copper acts as a key factor in cellular signaling, it is not surprising that Cu is involved in the development and progression of cancer by promoting cell proliferation, angiogenesis, and metastasis.
A plethora of studies investigated the putative effects of Cu on the growth of cancer cells. For instance, daily administration of Cu sulfate (CuSO4) has been shown to increase tumor growth in a rat model of chemically induced mammary tumorigenesis.158 In a mouse model of pancreatic islet cell carcinoma, chronic exposure to elevated levels of Cu by adding 20 μM CuSO4 to the drinking water also accelerated tumor growth.159 Mice bearing BRAFV600E-driven lung cancer also have increased tumor proliferation when supplemented with high levels of Cu.160 Mechanistically, Cu can increase the production of ROS, which are intrinsically linked to malignant cell transformation. Moreover, studies have shown that CTR1-dependent Cu import activates the mitogen-activated protein kinase (MAPK) signaling cascade.161 In addition, Cu directly binds to MEK1 with high affinity, which in turn promotes tumor growth by activating downstream ERK1/2 phosphorylation.160,162 Notably, Cu can drive carcinogenesis via its functional role as an essential regulator of the autophagic kinases ULK1/2. The loss of Ctr1 results in impaired ULK1 activation and its downstream signaling, thus reducing the growth and survival of xenografted KRASG12D lung tumors.163
Angiogenesis, a process that involves the migration and proliferation of endothelial cells, as well as the formation of the vascular tube and new blood vessels, acts as an important factor in tumor progression. The notion that Cu possesses pro-angiogenic properties was first raised by McAuslan, who found that Cu salts could induce endothelial cell migration, an early step in angiogenesis.164 In support of this hypothesis, delivering Cu to the cornea in rabbits could induce the formation of new blood vessels, and Cu has been shown to enhance the proliferation and mobility of endothelial cells.165 In in vitro studies, silencing CTR1 expression in endothelial cells using siRNA blocks Cu entry, decreases cell migration and reduces vascular tube formation.166 The pro-angiogenic properties of Cu may be attributed to its ability to regulate various factors involved in angiogenesis. For instance, Cu could regulate the secretion of angiogenic molecules such as fibroblast growth factor (FGF) and the inflammatory cytokine IL-1α.167,168 Cu can also modulate the affinity of angiogenin with endothelial cells by binding directly to this angiogenic factor.169 Deficiency of Cu has been shown to suppress transcriptional activity of NF-κB, thus inhibiting the expression of pro-angiogenic factors such as bFGF, vascular endothelial growth factor (VEGF), IL-8, IL-6, and IL-1α.170 Cu is also required for the activation of hypoxia-inducible factor 1 (HIF-1) activity, and the chelation of Cu blocks the HIF-1-mediated expression of VEGF, a potent angiogenic factor.171,172 Overexpressing the Cu-binding protein SOD1 markedly increases the production of VEGF and enhances angiogenesis induced by FGF and tumor development. Similarly, the Cu chaperone ATOX1 has also been found to serve as a modulator of angiogenesis, the depletion of Atox1 inhibit the migration of vascular smooth muscle cell that is stimulated by platelet-derived growth factor (PDGF), suggesting that ATOX1 likely plays a role in vascular remodeling and tumor angiogenesis.173
Evidence has shown that Cu could activate metastasis-related enzymes and signaling cascades, thus promoting the spread of cancer. For instance, the cuproenzyme LOX is involved in the invasion and metastasis of tumor cells.174 In estrogen receptor (ER) negative breast cancer, high expression of LOX is associated with bone metastasis and leads to the formation of tumor-driven osteolytic lesions.175 In tumor endothelial cells, decreased LOX expression inhibits cell migration and tube formation, and both angiogenesis and metastasis were suppressed by LOX inhibitors in an in vivo model.176 In an orthotopic breast cancer mouse model, knocking down ATP7A reduced LOX activity and reduced the recruitment of myeloid cells to the lungs, suppressing tumor growth and metastasis.177 The Cu chaperone ATOX1 may also be involved in the metastasis-related ATP7A-LOX pathway. A recent study using single-cell tracking analysis suggests that ATOX1 may be required for the migration of breast cancer cells, as silencing ATOX1 expression result in reduced activity of LOX and reduce the velocity and directionality of cell migration.178 In addition, the secreted Cu-binding glycoprotein SPARC (secreted protein acidic and cysteine-rich, also known as osteonectin) has been shown to modulate cell–matrix interactions and promote the invasion and metastasis of tumor cells.179 Cu also plays a role in modulating the expression of programmed death-ligand 1 (PD-L1), an immune checkpoint inhibitor associated with cancer immune evasion; specifically, depleting Cu promotes the degradation of PD-L1, thereby reducing tumor growth and improving survival in a neuroblastoma xenograft mouse model.180
Cu complexes in cancer therapy
The altered regulation of Cu homeostasis in tumors, coupled with the important role that Cu may play in promoting the progression of cancer, has led to the development of Cu-coordinating compounds for use in anticancer therapies. Two main strategies for targeting Cu homeostasis have been proposed, including the use of Cu chelators to decrease Cu bioavailability, and the use of Cu ionophores to deliver Cu into cells in order to increase intracellular Cu levels.
Cu chelators
A number of Cu-chelating agents such as TTM, trientine, and d-penicillamine have been developed and tested for their anti-tumor activity in both animal models as well as in clinical trials. Depletion of Cu using chelators has been shown to delay cancer metastasis by inhibiting the vascularization of lesions across several animal models, including a rabbit VX2 carcinoma model,181 a mouse model of hepatocellular carcinoma,182 and head and neck squamous cell carcinoma.183 The antimetastatic activity of Cu chelators may be attributed to their ability to prevent the recruitment of endothelial progenitor cells, which play an important role in angiogenesis and in the development of macroscopic metastases.184 In addition, depleting Cu in HER2/neu transgenic mice causes tumor shrinkage, suppresses angiogenesis, and inhibits the progression from microscopic to macroscopic tumors.170,185 In a mouse model of mesothelioma tumor, the use of trientine, d-penicillamine, and TTM could also exhibit inhibitory effects on tumor growth and impede tumor angiogenesis.186
With respect to the specificity of Cu chelators, the use of trientine has been shown to inhibit the expression of IL-8 and exhibits anti-tumor effects in hepatocellular carcinoma.187 Trientine also decreases CD31 expression and inhibits endothelial cell proliferation.182 In addition, d-penicillamine has been shown to inhibit the activity of LOX, thereby resulting in impaired collagen cross-link formation, reduced VEGF expression, and delay the progression of glioblastoma in vivo.188 Brem et al. have reported that combining d-penicillamine treatment with a low Cu diet reduced tumor weight and vascular density in a gliosarcoma xenograft model.189
Among the various Cu chelators developed to date, TTM has been well-studied in several models. For example, in RIP1-Tag2 mice, a model of pancreatic neuroendocrine tumor, administration of TTM delays the angiogenesis in premalignant lesions as well as reduces late-stage tumor growth.159 TTM may exert its anti-tumor effects by suppressing the transcriptional activity of NF-κB, which in turn decreases the production and secretion of downstream angiogenic factors, such as VEGF and cytokines such as IL-1α and IL-8, thereby reducing angiogenesis.190 In addition, TTM has been shown to induce the degradation of HIF-1α and reduce the expression of pro-angiogenic factors.191 TTM can also suppress Cu chaperone proteins, inhibiting the delivery of Cu to cuproenzymes such as LOX.192 Moreover, lowering Cu levels using TTM affects the activity of MEK1/2 kinase and BRAF-driven tumorigenesis, thus decreasing the growth of xenografted BRAFV600E tumors. With respect to its clinical relevance, a phase II trial to investigate the effects of TTM in patients with malignant mesothelioma found that TTM has anti-angiogenic effects and delays disease progression in patients with stage I or stage II mesothelioma.193 Notably, another phase II trial found that although TTM may not improve survival in patients with kidney cancer when used as a monotherapy, it may be more effective when combined with other anti-angiogenic agents.194
Cu ionophores
Several Cu ionophores such as elesclomol, bis(thiosemicarbazone) analogs [CuII(atsm) and CuII(gtsm)], and disulfiram (DSF) have been shown to raise intracellular Cu levels and exert anticancer activity. The cytotoxic properties of ionophores toward cancer cells may be attributed in part to their ability to increase ROS production and/or inhibit the proteasome.195,196 Specifically, treatment with DSF and Cu could decrease expression of the tumor suppressor gene PTEN and activate AKT signaling in human breast cancers, providing a rationale for testing combination therapies using Cu ionophores and PI3K-AKT inhibitors in future studies.197 With respect to glioblastoma, the cytotoxic effects of the standard-of-care drug temozolomide could be augmented by the addition of a DSF–Cu complex, and the combination of DSF–Cu with temozolomide is capable of inhibiting tumor growth and improving survival in a xenograft mouse model.198 The DSF–Cu complex has also been shown to display cytotoxicity towards aldehyde dehydrogenase (ALDH)+ cancer stem cells due to its inhibitory effects on ALDH activity.199,200 In addition, for Cu bis(thiosemicarbazone) analogs, the anticancer activity of CuII(atsm) has been demonstrated in hamsters bearing colon cancer tumors,201 and CuII(gtsm) has been shown to exert tumor-killing effects on prostate cancer cells in vitro and significantly reduce the prostate cancer burden in an orthotopic mouse model.202
Elesclomol is a novel Cu ionophore that has been shown to bring Cu to the mitochondria, and subsequently, increase oxidative stress and cause cell death.203 As a drug candidate, elesclomol has been shown to enhance the therapeutic efficacy of paclitaxel (Taxol) in patients with refractory solid tumors and in patients with stage IV metastatic melanoma.204 It is noted that in the phase III clinical trial consisting of patients with advanced melanoma, the treatment of elesclomol lead to a more favorable outcome in patients with normal LDH levels compared with those patients with high LDH levels, suggesting that elesclomol may be less effective in treating cancer types with a high rate of glycolysis.205
Cardiovascular disease
Cu in relation to atherosclerosis
Cu has also been suggested to play an important role in the pathogenesis of atherosclerosis. Epidemiological evidence has linked high serum Cu levels with an increased risk of atherosclerotic disease.206,207 In addition, in human atherosclerotic plaques, elevated levels of Cu have been observed.208 The local release of Cu ions around rat carotid arteries has been shown to promote the thickening of the neointima and the generation of arteriosclerotic lesions in response to vascular injury,209 whereas the use of Cu chelators exhibit inhibitory effects on vascular inflammation, the development of atherosclerotic lesions, and neointimal formation in response to vascular injury in ApoE knockout mice.167,210,211,212 Notably, in neointimal vascular smooth muscle cells (VSMCs) or in the intimal lesions of atherosclerotic vessels, the Cu chaperone ATOX1 and exporter ATP7A are found to be highly expressed and colocalized.209,213,214,215 Moreover, in Atox1−/− mice, the expansion of extracellular matrix and neointimal formation were inhibited in response to vascular injury, accompanied by decreased accumulation of VSMCs in the neointima and reduced LOX activity.173
The potential mechanism by which Cu promotes atherosclerosis is currently unclear. One possibility is that Cu may play role in inflammatory responses involved in atherosclerosis. Cu deficiency reduces the expression of adhesion molecules such as ICAM-1 and VCAM-1, which mediate the adhesion of leukocytes to activated endothelial cells.216 In addition, Cu is capable of interacting with risk factors for the atherogenic process, such as triggering the oxidation of LDL and interacting with homocysteine to enhance hydrogen peroxidation.217,218,219 Deficiency of Cu could increase the level of total cholesterol, a key factor to increase atherosclerosis risk. Furthermore, Cu deficiency may lead to reduced NO levels by decreasing SOD1 levels, in turn promoting atherosclerosis via impaired endothelial function, reduced vasodilation, and increased oxidative stress.220 Given that both Cu accumulation and Cu deficiency are potentially detrimental to vascular integrity and function, maintaining Cu homeostasis is essential for preventing atherosclerosis and related cardiovascular disease.
Cu in relation to cardiac hypertrophy
Cardiac tissue requires high levels of Cu to maintain mitochondrial function and produce large amounts of energy needed for normal cardiac function. Indeed, it has been demonstrated in experimental animal models, the deficiency of Cu could lead to hypertrophic cardiomyopathy.221,222 In humans, cardiomyopathy has been observed in individuals with mutations in the synthesis of cytochrome c oxidase 2 (SCO2), a chaperone involved in trafficking Cu to cytochrome c oxidase (COX).223 Changes in cardiac morphology due to cardiac Cu deficiency generally include mitochondrial swelling and fragmentation, myofibril derangement, and enlarged myocytes.224 Functionally, impaired mitochondrial respiratory function and electrocardiographic abnormalities have also been observed in Cu-deficient hearts.224,225 In Cu-deficient animals and humans, cardiovascular lesions could be presented as aortic fissures and aortic rupture, possibly due to an altered architecture of elastic fibers.226 Cu deficiency can also alter the expression of myocardial gene profile, for example by upregulating the inflammatory cytokine TNF-α and genes involved in regulating cardiac contractility, fibrosis, and calcium cycling, all of which may underlie the changes in cardiac function under Cu-deficient conditions.227,228
Repletion of Cu via supplementation can reverse many of the adverse effects of Cu deficiency in the heart.227 In Cu-deficient mice, lipids are deposited in the myocardial tissue, with cardiac hypertrophy, mitochondrial swelling, and a blunted response to the isoproterenol, an agonist of beta-adrenergic; The repletion of Cu could reverse these changes in the heart and improve electrical conduction.229,230 Interestingly, in a patient with SCO2 mutations who presented with severe hypertrophic cardiomyopathy, Cu-histidine supplementation has been reported to improve heart function, showing attenuated cardiomyopathy as well as the normalization of blood pressure.231
Given that altered regulation of Cu, homeostasis has been associated with a variety of disease conditions, future studies are warranted in order to determine the precise mechanism by which Cu imbalance causes cellular damage, thus providing important new information that can be used to design treatments for preventing disease and/or slowing disease progression.
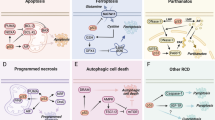
Cuproptosis is distinct from other known cell death pathways
Previous studies have shown that excessive levels of trace elements can cause cell death via specific pathways. Ferroptosis, an iron-dependent form of cell death, is one such example in which excess iron triggers the accumulation of lipid peroxides in membranes, leading to programmed cell death.232,233 In addition to pathways involved in iron metabolism, several other molecular pathways have also been implicated in ferroptosis, including the guanosine triphosphate cyclohydrolase 1/tetrahydrobiopterin (GCH1/BH4) axis,234 the cysteine/glutathione/glutathione peroxidase 4 (GSH/GPX4) axis,235,236 and the ferroptosis suppressor protein 1/coenzyme Q (FSP1/CoQ) axis.234,237,238 Moreover, recent studies suggest that ferroptosis is involved in a wide range of diseases such as hemochromatosis, neurodegenerative diseases, liver fibrosis, and cardiovascular disease.239,240,241,242,243,244,245,246,247,248
In contrast, the mechanisms that underlie Cu-induced cell death are poorly understood. Indeed, contradictory findings suggest that excess Cu2+ may induce cell death via apoptosis,249 caspase-independent cell death, or an accumulation of ROS.250,251,252 Notably, Tsvetkov et al. recently showed that intracellular Cu induces a novel form of regulated cell death characterized by the aggregation of lipoylated mitochondrial enzymes and a loss of Fe–S proteins; they called this Cu-dependent form of cell death cuproptosis.2 Specifically, the authors found that treating cells with the Cu ionophore elesclomol at concentrations as low as 40 nM increased intracellular Cu levels and triggered cuproptosis.2 Importantly, they also showed that pharmacologically inhibiting all other known cell death pathways—including necroptosis (with necrostatin-1), ferroptosis (with ferrostatin-1), oxidative stress (with N-acetyl cysteine), and apoptosis (with Z-VAD-FMK)—failed to suppress elesclomol-induced cell death; in contrast, only Cu chelator treatment show potent rescue effect on such excess Cu2+-induced cell death, suggesting that cuproptosis is distinct from other forms of cell death.
Cuproptosis is linked to altered mitochondrial enzymes
The mitochondrion is a major target of Cu-induced cell death, with oxidative damage to the mitochondrial membrane and impaired function of enzymes in the TCA cycle.138,253 In patients with Cu overload, aconitase inactivation has been attributed to reduced CCO activity and inhibition of the TCA cycle.71 Interestingly, metabolite profiling of cells treated with Cu ionophore presents a time-dependent increase in the dysregulation of many TCA cycle-associated metabolites, and inhibiting the electron transport chain complexes I and II significantly reduced Cu-induced cell death.2 Furthermore, Cu treatment selectively alters a series of metabolic enzymes via lipoylation, a highly conserved post-translational modification. Although relatively few proteins are lipoylated in mammalian cells, all lipoylated proteins are involved in the TCA cycle.254,255 One such lipoylated protein is dihydrolipoamide S-acetyltransferase (DLAT), a subunit of the pyruvate dehydrogenase complex.256 Cu can bind directly to DLAT, promoting the disulfide bond-dependent aggregation of lipoylated DLAT. Using a genome-wide CRISPR screen, mitochondrial ferredoxin (FDX1), and lipoyl synthase (LIAS) were identified as key regulators of Cu toxicity,2,257 and genetic knockout of either FDX1 or LIAS leads to an accumulation of pyruvate and α-ketoglutarate, reducing protein lipoylation and inhibiting Cu-induced cell death (Fig. 2).

Schematic model of cuproptosis. Cu ionophores such as elesclomol bind extracellular Cu and transport it to intracellular compartments. Cu then binds to lipoylated mitochondrial enzymes in the TCA cycle such as DLAT, inducing the aggregation of these proteins. FDX1/LIAS is an upstream regulator of protein lipoylation, facilitating the aggregation of mitochondrial proteins and loss of Fe–S clusters. Together, these aberrant processes lead to proteotoxic stress and ultimately cell death. Cu chelators such as TTM inhibit cuproptosis, while inhibitors of ferroptosis (Fer-1), necroptosis (Nec-1), and oxidative stress (NAC) have no effect on cuproptosis. The solid orange circles in the TCA cycle indicate metabolites that are relevant to the lipoic acid pathway. Created with BioRender. α-KG α-ketoglutarate, DLAT dihydrolipoamide S-acetyltransferase, FDX1 ferredoxin-1, Fe–S iron–sulfur, Fer-1 ferrostatin-1, LIAS lipoic acid synthetase, NAC N-acetyl cysteine, Nec-1 necrostatin-1, TCA tricarboxylic acid, TTM tetrathiomolybdate
Importantly, Cu toxicity has also been linked to a disruption of iron–sulfur (Fe–S)-containing enzymes. For example, in Saccharomyces cerevisiae, Cu-mediated damage to the labile Fe–S pool in mitochondrial ferredoxin leads to downstream growth inhibition.258 In vitro, Cu has been shown to block Fe–S cluster formation by inhibiting the activity of relevant mitochondrial assembly proteins.259 Moreover, the loss of the mitochondrial ABC transporter ATM1, which transports intermediates required for Fe–S cluster formation, exacerbates Cu-induced toxicity.260 Notably, a recent study has found that treating cells with a Cu ionophore resulted in an FDX1-dependent loss of Fe–S cluster proteins1 (Fig. 2). The majority of Fe–S proteins are essential cofactors for enzymes involved in the electron transport chain and other biochemical processes; thus, the aggregation of mitochondrial enzymes may disrupt the function of these Fe–S clusters and ultimately lead to cell death. Given that Cu has also been shown to destabilize Fe–S proteins in bacteria and yeast, it is reasonable to speculate that a Cu-containing complex might be developed to trigger cuproptosis in bacteria as an antimicrobial agent.261,262,263
Cuproptosis as a putative therapeutic target for Wilson’s disease
As mentioned above, mutations in the ATP7B gene, which encodes the Cu-transporting P-type ATPase ATP7B, are associated with Wilson’s disease, a life-threatening disorder in which patients present with progressive Cu accumulation in several tissues, particularly the liver, brain, and cornea. Importantly, Cu deposition in the liver and brain can cause cellular damage and severe hepatic and neurological symptoms.67 Disrupted mitochondrial membranes and increased oxidative damage have been observed in the Long–Evans Cinnamon rat, an animal model of Wilson disease.264 In addition, Tsvetkov et al. have shown that Atp7b knockout mice exhibit reduced levels of lipoylated enzymes and Fe–S cluster proteins compared to wild-type mice,2 reminiscent of the cellular phenotype induced by Cu ionophores (Fig. 3).

The putative role of cuproptosis in Wilson’s disease. a Patients with Wilson’s disease present with Cu overload in their hepatocytes. Mutations in ATP7B impair Cu loading into secretory vesicles and into cuproproteins such as ceruloplasmin. Cu excretion via the biliary tract is also impaired, resulting in an accumulation of Cu in the liver. The resulting Cu-induced toxicity leads to chronic liver disease and cirrhosis. Cu also accumulates in other tissues, including the brain, cornea, and kidneys, resulting in neurological impairment, Kayser–Fleischer rings, and impaired kidney function, respectively. b Atp7b knockout mice (a model of Wilson’s disease) develop Cu overload, with a loss of lipoylated and Fe–S cluster proteins in the liver, suggesting that this Cu overload has similar cellular effects as those induced by Cu ionophores, suggesting that cuproptosis may play a pathogenic role in Wilson’s disease. Created with BioRender. ATOX1 antioxidant 1 copper chaperone-1, ATP7B ATPase Cu transporter 7B, Fe–S iron–sulfur, Slc3a1 solute carrier family 3 member 1, STEAP six-transmembrane epithelial antigen of prostate
Cu chelation, such as trientine/d-Penicillamine, serves as an effective treatment for Wilson’s disease.265,266 Moreover, the Cu chelator TTM has also been evaluated in several clinical trials. For example, Brewer et al. studied 48 patients with neurological presentation and found that disease progression was reduced in the TTM-treated group compared to the trientine-treated group.86 In another phase II clinical trial, Weiss et al. evaluated the effect of the oral Cu–protein-binding molecule WTX101 (the bis-choline salt of TTM) and found that WTX101 rapidly improved Cu homeostasis, leading to the improved neurological outcome and stabilizing liver function90 (Table 1). Given that TTM inhibits the cytotoxic effects of Cu ionophores, it is reasonable to speculate that cuproptosis may play a role in the progression of Wilson’s disease. It is also worth noting that α-lipoic acid, a metabolite in the cuproptosis pathway, has shown promising results in both cell-based and animal models of Wilson’s disease.267 Therefore, it would be interesting to test whether novel compounds that target cuproptosis may be effective in treating this intractable disease.
Cuproptosis in relation to neurodegenerative diseases
Many studies have suggested that altered Cu homeostasis is directly related to the progression of several neurodegenerative diseases, including Alzheimer’s disease (AD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS).268 For example, high levels of Cu have been found in senile plaques and serum in patients with AD,269,270 and high concentrations of Cu have also been measured in Aβ plaques in the dentate gyrus subregion of the hippocampus in a mouse model of AD.271 On the other hand, Cu chelation treatment and knocking down the Cu transporter Ctr1 were shown to rescue neurotoxicity in a Drosophila model of AD.272 With respect to HD, an accumulation of Cu in the brain has been suggested to promote the aggregation of mutant HTT proteins, thus accelerating disease progression.136,273 In addition, a number of studies have suggested that restricting dietary Cu, treatment with Cu chelators, and genetic manipulation of Cu transporters may delay disease progression in animal models of HD.141,142,143 With respect to ALS, mutations in the SOD1 gene (which encodes Cu/Zn superoxide dismutase, a physiologically important Cu-binding protein) are the principal cause of the familial form of ALS.274,275 Interestingly, relatively low levels of Cu have been measured in mutant SOD1 proteins, whereas increased levels of Cu have been reported in the cerebrospinal fluid of patients with ALS and mouse models of ALS.120,125 Notably, Roberts et al. previously reported that treating SOD1G37R mice (a model of ALS) with the therapeutic Cu-delivery agent CuII(atsm) restored Cu content in mutant SOD1 proteins and increased survival.119 The therapeutic efficacy of CuII(atsm) in treating ALS has also been tested in patients276 (Table 1). It is also interesting to note that the Cu chelator TTM has also been shown to decrease Cu levels in the spinal cord of transgenic mice expressing the human SOD1G93A mutant protein, extending their lifespan.128 Taken together, these findings indicate that the precise role of Cu in the progression of ALS and other neurodegenerative diseases warrants further study.
With respect to the underlying mechanism, evidence suggests that mitochondrial dysfunction may play a role in Cu-induced neurotoxicity. For example, exposing neuroblastoma cells to Cu markedly increases the production of mitochondrial ROS, followed by decreased production of pyruvate dehydrogenase in the TCA cycle and respiratory complex I.253 In addition, adding Cu to neuronal/glial cultures leads to a collapse in mitochondrial membrane potential (ΔΨm) and inhibits the production of α-ketoglutarate dehydrogenases and mitochondrial pyruvate, whereas the antioxidant dihydrolipoic acid attenuates these Cu-induced effects and subsequent cell death.138 Nevertheless, additional studies are clearly needed in order to determine whether cuproptosis plays a pathogenic role in neurodegenerative diseases discussed here and whether blocking cuproptosis may serve as a novel therapeutic strategy for treating patients with these devastating, progressive conditions.
Potential therapeutic strategies for targeting cuproptosis in cancer
In recent decades, a growing body of evidence suggests that the Cu-complex might serve as a potential therapeutic target for treating cancer, as Cu has been shown to promote the death of cancer cells via apoptosis and/or an accumulation of free radicals.277,278 These findings regarding cuproptosis provide new insights into potential applications for treating cancer. Notably, Xu et al. reported the development of a glucose oxidase (GOx)-engineered nonporous Cu-coordination nanomaterial called GOx@[Cu(tz)] as a cuproptosis-based therapy in cancer.279 Using an in vivo system, the authors showed that GOx@[Cu(tz)] inhibited tumor growth by 92% in athymic mice with bladder tumors, with negligible systemic toxicity.279 In addition, Cu ionophores such as elesclomol have been shown to have anticancer activity by inducing ROS production in cancer cells.280 This novel finding suggests that elesclomol may have additional killing potency in cancer cells that express high levels of lipoylated mitochondrial proteins and a hyperactive respiratory state (Fig. 4). Interestingly, a randomized double-blind phase III clinical trial to evaluate elesclomol in combination with chemotherapy in patients with melanoma showed that elesclomol had stronger anti-tumor activity in patients with low plasma levels of lactate dehydrogenase205 (Table 1). Given that low lactate dehydrogenase levels represent a high mitochondrial metabolic state, this finding is consistent with the observation that cells with high levels of lipoylated TCA enzymes and a hyperactive mitochondrial respiratory rate are more sensitive to elesclomol. Further clinical trials are needed in order to explore the feasibility of using Cu ionophores to treat tumors with these metabolic profiles.

Two potential therapeutic strategies to target cuproptosis in cancer. a The Cu ionophore elesclomol is believed to induce cuproptosis in cancer cells that either express high levels of lipoylated mitochondrial enzymes or are in a hyperactive respiratory state. b Disulfiram combined with Cu selectively targets cancer cells with high ALDH expression. Created with BioRender. ALDH aldehyde dehydrogenase, DSF disulfiram
Disulfiram (DSF), another Cu-binding compound, has also been shown to be able to induce cuproptosis.2 Moreover, in vitro studies, have shown that when combined with cupric ion [Cu(II)], DSF has anti-tumor activity in a variety of cancers.281 Building on the previous finding that DSF inhibits aldehyde dehydrogenase (ALDH), preclinical studies have shown that the combination of DSF and Cu(II) selectively targets and kills ALDH+ cancer stem cells, reducing the risk of tumor recurrence.199,282,283 In clinical trials, the anticancer and/or chemosensitizing effects of DSF, which can cross the BBB, have been demonstrated in patients with glioblastoma.284,285,286 Moreover phase I clinical trial has shown that the combination of DSF–Cu and temozolomide has an acceptable safety profile and increases progression-free survival in patients with glioblastoma287 (Table 1). These findings suggest that ionophore-mediated Cu delivery to the intracellular compartment might be a promising therapeutic strategy for a subset of tumors, and additional Cu–ionophore complexes that target cuproptosis warrant further development.
Conclusions and future perspectives
In cells, Cu acts as a double-edged sword: on one hand, Cu is an essential cofactor for many enzymes; on the other hand, excess Cu can induce oxidative stress and drive cell death. Recent studies have revealed that cuproptosis, a Cu-dependent form of cell death, is mediated by the lipoylation of mitochondrial enzymes. This novel finding provides new perspectives regarding the link between Cu-induced cell death and mitochondrial metabolism, advancing our understanding of Cu biology, and shedding new light on cell death pathways.2
Pioneering studies have revealed that a variety of metal ions can trigger cell death via distinct signaling pathways. For example, ferroptosis, an iron-dependent form of cell death, is characterized by excessive lipid peroxidation on cell membranes. Comparing ferroptosis and cuproptosis, it is interesting to note that mitochondria play a critical role in these two different types of cell death. Recent work has shown that mitochondrial glutathione (GSH) can slow Cu-induced cell death by suppressing enzyme lipoylation and promoting the oligomerization of DLAT. With respect to ferroptosis, the use of mitochondria-targeted ROS scavenger mitoquinone (MitoQ), which also increases GSH levels, could preserve mitochondria integrity and protect cells from lipid peroxide accumulation and subsequent cell death. On the other hand, a series of morphological changes in mitochondria, including mitochondrial shrinkage, increased membrane density, and mitochondrial fragmentation, have been observed during ferroptosis, but not in cuproptosis. In light of the finding that Cu can affect iron homeostasis and even induce ferroptosis, further study is needed in order to determine the precise morphological features of cuproptosis and to determine whether potentially relevant crosstalk exists between these two pathways.
As a newly discovered form of cell death, the precise mechanisms that underlie cuproptosis are poorly understood, although the lipoic acid pathway has been shown to play a key role in mediating cuproptosis. An interesting question is whether other metabolic pathways are also involved in cuproptosis. In addition, based on currently available data, another open question is how the aggregation of lipoylated mitochondrial enzymes triggers the Cu-dependent signaling cascades that lead to cell death. Additional studies are therefore warranted in order to identify the key players and explore the mechanisms that underlie cuproptosis, thereby providing a clear picture of cuproptosis at the cellular, tissue, and systemic levels. Identifying and characterizing the signaling pathways that regulate this newly discovered form of Cu-dependent cell death will likely offer new opportunities for clinical applications.
Given that Cu accumulation occurs in a variety of diseases and conditions, including Wilson’s disease, certain neurodegenerative diseases, and cancer, it is reasonable to speculate that cuproptosis may play a pathogenic role in these diseases and may therefore serve as a potential therapeutic target. Based on the finding that Cu chelator exhibit a potent suppression effect on Cu-induced cell death, it would be important to carry out appropriate Cu-deprivation strategies to reduce the level of intracellular Cu2+ ion in Cu-overload conditions, for example, using a Cu chelator, reducing dietary Cu intake, and/or genetically modifying Cu transporters. More evidence is clearly needed in order to better understand the dynamic processes by which cuproptosis initiates and facilitates disease progression.
As cuproptosis was identified only recently, no reliable biomarkers are currently available, limiting our ability to determine whether cuproptosis is involved in human pathological conditions. In this fast-growing field, the lack of cuproptosis-specific biomarkers would be a long-standing bottleneck limiting the development of cuproptosis-targeted clinical applications. Thus, reliable and sensitive biomarkers of cuproptosis are needed to be identified in different disease settings.
With respect to treatment options, both high-throughput functional screening and AI-based approaches will likely accelerate the development of new compounds that target cuproptosis. To maximize the safety and efficacy of these therapeutic compounds, pharmaceutical studies should confirm their targeted delivery to affected organs. Tackling these key hurdles will improve our understanding of the role that cuproptosis plays in various pathophysiological conditions, thus providing a clear scientific rationale for the clinical development of strategies designed to target cuproptosis in order to treat and/or prevent Cu-related diseases.
References
Walshe, J. M. Wilson’s disease. Lancet 369, 902 (2007).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375, 1254–1261 (2022).
Maung, M. T. et al. The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB J. 35, e21810 (2021).
Festa, R. A. & Thiele, D. J. Copper: an essential metal in biology. Curr. Biol. 21, R877–R883 (2011).
Mason, K. E. A conspectus of research on copper metabolism and requirements of man. J. Nutr. 109, 1979–2066 (1979).
Georgatsou, E., Mavrogiannis, L. A., Fragiadakis, G. S. & Alexandraki, D. The yeast Fre1p/Fre2p cupric reductases facilitate copper uptake and are regulated by the copper-modulated Mac1p activator. J. Biol. Chem. 272, 13786–13792 (1997).
Dancis, A., Roman, D. G., Anderson, G. J., Hinnebusch, A. G. & Klausner, R. D. Ferric reductase of Saccharomyces cerevisiae: molecular characterization, role in iron uptake, and transcriptional control by iron. Proc. Natl Acad. Sci. USA 89, 3869–3873 (1992).
Weiss, K. C. & Linder, M. C. Copper transport in rats involving a new plasma protein. Am. J. Physiol. 249, E77–E88 (1985).
Ramos, D. et al. Mechanism of copper uptake from blood plasma ceruloplasmin by Mammalian cells. PLoS ONE 11, e0149516 (2016).
Moriya, M. et al. Copper is taken up efficiently from albumin and alpha2-macroglobulin by cultured human cells by more than one mechanism. Am. J. Physiol. Cell Physiol. 295, C708–C721 (2008).
Liu, N. et al. Transcuprein is a macroglobulin regulated by copper and iron availability. J. Nutr. Biochem. 18, 597–608 (2007).
Luza, S. C. & Speisky, H. C. Liver copper storage and transport during development: implications for cytotoxicity. Am. J. Clin. Nutr. 63, 812S–820S (1996).
Boyd, S. D., Ullrich, M. S., Skopp, A. & Winkler, D. D. Copper sources for Sod1 activation. Antioxidants 9, 500 (2020).
Pufahl, R. A. et al. Metal ion chaperone function of the soluble Cu(I) receptor Atx1. Science 278, 853–856 (1997).
Heaton, D. N., George, G. N., Garrison, G. & Winge, D. R. The mitochondrial copper metallochaperone Cox17 exists as an oligomeric, polycopper complex. Biochemistry 40, 743–751 (2001).
La Fontaine, S., Ackland, M. L. & Mercer, J. F. Mammalian copper-transporting P-type ATPases, ATP7A and ATP7B: emerging roles. Int. J. Biochem. Cell Biol. 42, 206–209 (2010).
Lutsenko, S. Copper trafficking to the secretory pathway. Metallomics 8, 840–852 (2016).
Lutsenko, S., Bhattacharjee, A. & Hubbard, A. L. Copper handling machinery of the brain. Metallomics 2, 596–608 (2010).
Csiszar, K. Lysyl oxidases: a novel multifunctional amine oxidase family. Prog. Nucleic Acid Res. Mol. Biol. 70, 1–32 (2001).
Lutsenko, S., LeShane, E. S. & Shinde, U. Biochemical basis of regulation of human copper-transporting ATPases. Arch. Biochem. Biophys. 463, 134–148 (2007).
Thiele, D. J. Integrating trace element metabolism from the cell to the whole organism. J. Nutr. 133, 1579S–1580S (2003).
Pena, M. M., Lee, J. & Thiele, D. J. A delicate balance: homeostatic control of copper uptake and distribution. J. Nutr. 129, 1251–1260 (1999).
Aggett, P. J. An overview of the metabolism of copper. Eur. J. Med. Res. 4, 214–216 (1999).
Turnlund, J. R., Keyes, W. R., Anderson, H. L. & Acord, L. L. Copper absorption and retention in young men at three levels of dietary copper by use of the stable isotope 65Cu. Am. J. Clin. Nutr. 49, 870–878 (1989).
Puig, S. & Thiele, D. J. Molecular mechanisms of copper uptake and distribution. Curr. Opin. Chem. Biol. 6, 171–180 (2002).
Ohgami, R. S., Campagna, D. R., McDonald, A. & Fleming, M. D. The Steap proteins are metalloreductases. Blood 108, 1388–1394 (2006).
McKie, A. T. et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 291, 1755–1759 (2001).
Liang, Z. D., Tsai, W. B., Lee, M. Y., Savaraj, N. & Kuo, M. T. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol. Pharmacol. 81, 455–464 (2012).
Kuo, Y. M., Gybina, A. A., Pyatskowit, J. W., Gitschier, J. & Prohaska, J. R. Copper transport protein (Ctr1) levels in mice are tissue specific and dependent on copper status. J. Nutr. 136, 21–26 (2006).
Nose, Y., Kim, B. E. & Thiele, D. J. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 4, 235–244 (2006).
Lee, J., Petris, M. J. & Thiele, D. J. Characterization of mouse embryonic cells deficient in the ctr1 high affinity copper transporter. Identification of a Ctr1-independent copper transport system. J. Biol. Chem. 277, 40253–40259 (2002).
Lutsenko, S., Barnes, N. L., Bartee, M. Y. & Dmitriev, O. Y. Function and regulation of human copper-transporting ATPases. Physiol. Rev. 87, 1011–1046 (2007).
Itoh, S. et al. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J. Biol. Chem. 283, 9157–9167 (2008).
Hamza, I., Prohaska, J. & Gitlin, J. D. Essential role for Atox1 in the copper-mediated intracellular trafficking of the Menkes ATPase. Proc. Natl Acad. Sci. USA 100, 1215–1220 (2003).
Gralla, E. B. & Valentine, J. S. Null mutants of Saccharomyces cerevisiae Cu,Zn superoxide dismutase: characterization and spontaneous mutation rates. J. Bacteriol. 173, 5918–5920 (1991).
Elchuri, S. et al. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 24, 367–380 (2005).
Bertinato, J. & L’Abbe, M. R. Copper modulates the degradation of copper chaperone for Cu,Zn superoxide dismutase by the 26 S proteosome. J. Biol. Chem. 278, 35071–35078 (2003).
Prohaska, J. R., Geissler, J., Brokate, B. & Broderius, M. Copper, zinc-superoxide dismutase protein but not mRNA is lower in copper-deficient mice and mice lacking the copper chaperone for superoxide dismutase. Exp. Biol. Med. (Maywood) 228, 959–966 (2003).
Okado-Matsumoto, A. & Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 276, 38388–38393 (2001).
Sturtz, L. A., Diekert, K., Jensen, L. T., Lill, R. & Culotta, V. C. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 276, 38084–38089 (2001).
Nyvltova, E., Dietz, J. V., Seravalli, J., Khalimonchuk, O. & Barrientos, A. Coordination of metal center biogenesis in human cytochrome c oxidase. Nat. Commun. 13, 3615 (2022).
Prohaska, J. R. Role of copper transporters in copper homeostasis. Am. J. Clin. Nutr. 88, 826S–829S (2008).
Horng, Y. C., Cobine, P. A., Maxfield, A. B., Carr, H. S. & Winge, D. R. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J. Biol. Chem. 279, 35334–35340 (2004).
Cobine, P. A., Pierrel, F. & Winge, D. R. Copper trafficking to the mitochondrion and assembly of copper metalloenzymes. Biochim. Biophys. Acta 1763, 759–772 (2006).
Banci, L. et al. Mitochondrial copper(I) transfer from Cox17 to Sco1 is coupled to electron transfer. Proc. Natl Acad. Sci. USA 105, 6803–6808 (2008).
Leary, S. C. et al. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum. Mol. Genet. 13, 1839–1848 (2004).
Takahashi, Y. et al. Mammalian copper chaperone Cox17p has an essential role in activation of cytochrome C oxidase and embryonic development. Mol. Cell. Biol. 22, 7614–7621 (2002).
Palmgren, M. G. & Nissen, P. P-type ATPases. Annu. Rev. Biophys. 40, 243–266 (2011).
La Fontaine, S. & Mercer, J. F. Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch. Biochem. Biophys. 463, 149–167 (2007).
Linz, R. & Lutsenko, S. Copper-transporting ATPases ATP7A and ATP7B: cousins, not twins. J. Bioenerg. Biomembr. 39, 403–407 (2007).
Shim, H. & Harris, Z. L. Genetic defects in copper metabolism. J. Nutr. 133, 1527S–1531S (2003).
Moller, L. B., Mogensen, M. & Horn, N. Molecular diagnosis of Menkes disease: genotype–phenotype correlation. Biochimie 91, 1273–1277 (2009).
Tumer, Z. & Moller, L. B. Menkes disease. Eur. J. Hum. Genet. 18, 511–518 (2010).
Kaler, S. G., Goldstein, D. S., Holmes, C., Salerno, J. A. & Gahl, W. A. Plasma and cerebrospinal fluid neurochemical pattern in Menkes disease. Ann. Neurol. 33, 171–175 (1993).
Kaler, S. G., Gahl, W. A., Berry, S. A., Holmes, C. S. & Goldstein, D. S. Predictive value of plasma catecholamine levels in neonatal detection of Menkes disease. J. Inherit. Metab. Dis. 16, 907–908 (1993).
Ojha, R. & Prasad, A. N. Menkes disease: what a multidisciplinary approach can do. J. Multidiscip. Healthc. 9, 371–385 (2016).
Hicks, J. D. et al. Increased frequency of congenital heart defects in Menkes disease. Clin. Dysmorphol. 21, 59–63 (2012).
Byers, P. H. et al. X-linked cutis laxa: defective cross-link formation in collagen due to decreased lysyl oxidase activity. N. Engl. J. Med. 303, 61–65 (1980).
Szauter, K. M., Cao, T., Boyd, C. D. & Csiszar, K. Lysyl oxidase in development, aging and pathologies of the skin. Pathol. Biol. (Paris) 53, 448–456 (2005).
Royce, P. M., Camakaris, J. & Danks, D. M. Reduced lysyl oxidase activity in skin fibroblasts from patients with Menkes’ syndrome. Biochem. J. 192, 579–586 (1980).
Sarkar, B., Lingertat-Walsh, K. & Clarke, J. T. Copper-histidine therapy for Menkes disease. J. Pediatr. 123, 828–830 (1993).
Kim, J. H. et al. Novel mutations and clinical outcomes of copper-histidine therapy in Menkes disease patients. Metab. Brain Dis. 30, 75–81 (2015).
George, D. H. & Casey, R. E. Menkes disease after copper histidine replacement therapy: case report. Pediatr. Dev. Pathol. 4, 281–288 (2001).
Kaler, S. G. et al. Neonatal diagnosis and treatment of Menkes disease. N. Engl. J. Med. 358, 605–614 (2008).
Kaler, S. G. Neurodevelopment and brain growth in classic Menkes disease is influenced by age and symptomatology at initiation of copper treatment. J. Trace Elem. Med. Biol. 28, 427–430 (2014).
Czlonkowska, A. et al. Wilson disease. Nat. Rev. Dis. Prim. 4, 21 (2018).
Ala, A., Walker, A. P., Ashkan, K., Dooley, J. S. & Schilsky, M. L. Wilson’s disease. Lancet 369, 397–408 (2007).
Huster, D. et al. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J. Biol. Chem. 282, 8343–8355 (2007).
Halliwell, B. Oxidative stress and neurodegeneration: where are we now? J. Neurochem. 97, 1634–1658 (2006).
Scheinberg, I. H. & Sternlieb, I. Wilson disease and idiopathic copper toxicosis. Am. J. Clin. Nutr. 63, 842s–845s (1996).
Gu, M. et al. Oxidative-phosphorylation defects in liver of patients with Wilson’s disease. Lancet 356, 469–474 (2000).
Huster, D. Structural and metabolic changes in Atp7b-/- mouse liver and potential for new interventions in Wilson’s disease. Ann. N. Y. Acad. Sci. 1315, 37–44 (2014).
Ferenci, P. et al. Diagnosis and phenotypic classification of Wilson disease. Liver. Int. 23, 139–142 (2003).
Przybylkowski, A. et al. Liver cirrhosis in patients newly diagnosed with neurological phenotype of Wilson’s disease. Funct. Neurol. 29, 23–29 (2014).
Das, S. K. & Ray, K. Wilson’s disease: an update. Nat. Clin. Pract. Neurol. 2, 482–493 (2006).
Dusek, P., Litwin, T. & Czlonkowska, A. Wilson disease and other neurodegenerations with metal accumulations. Neurol. Clin. 33, 175–204 (2015).
Poujois, A. & Woimant, F. Wilson’s disease: a 2017 update. Clin. Res. Hepatol. Gastroenterol. 42, 512–520 (2018).
Sinha, S. et al. Wilson’s disease: cranial MRI observations and clinical correlation. Neuroradiology 48, 613–621 (2006).
Bertrand, E. et al. Neuropathological analysis of pathological forms of astroglia in Wilson’s disease. Folia Neuropathol. 39, 73–79 (2001).
Factor, S. M., Cho, S., Sternlieb, I., Scheinberg, I. H. & Goldfischer, S. The cardiomyopathy of Wilson’s disease. Myocardial alterations in nine cases. Virchows Arch. A Pathol. Anat. Histol. 397, 301–311 (1982).
Sozeri, E., Feist, D., Ruder, H. & Scharer, K. Proteinuria and other renal functions in Wilson’s disease. Pediatr. Nephrol. 11, 307–311 (1997).
Misra, A. K. et al. Arthropathic presentation of Wilson’s disease. J. Assoc. Physicians India 52, 246–248 (2004).
Russell, K., Gillanders, L. K., Orr, D. W. & Plank, L. D. Dietary copper restriction in Wilson’s disease. Eur. J. Clin. Nutr. 72, 326–331 (2018).
Brewer, G. J. et al. Treatment of Wilson’s disease with zinc: XV long-term follow-up studies. J. Lab. Clin. Med. 132, 264–278 (1998).
Van Caillie-Bertrand, M., Degenhart, H. J., Luijendijk, I., Bouquet, J. & Sinaasappel, M. Wilson’s disease: assessment of d-penicillamine treatment. Arch. Dis. Child. 60, 652–655 (1985).
Brewer, G. J. et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch. Neurol. 63, 521–527 (2006).
Bandmann, O., Weiss, K. H. & Kaler, S. G. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 14, 103–113 (2015).
Weiss, K. H. et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin. Gastroenterol. Hepatol. 11, 1028–1035 (2013).
Brewer, G. J. et al. Treatment of Wilson’s disease with tetrathiomolybdate: V. Control of free copper by tetrathiomolybdate and a comparison with trientine. Transl. Res. 154, 70–77 (2009).
Weiss, K. H. et al. Bis-choline tetrathiomolybdate in patients with Wilson’s disease: an open-label, multicentre, phase 2 study. Lancet Gastroenterol. Hepatol. 2, 869–876 (2017).
Guzior, N., Wieckowska, A., Panek, D. & Malawska, B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr. Med. Chem. 22, 373–404 (2015).
Eskici, G. & Axelsen, P. H. Copper and oxidative stress in the pathogenesis of Alzheimer’s disease. Biochemistry 51, 6289–6311 (2012).
Squitti, R. et al. Meta-analysis of serum non-ceruloplasmin copper in Alzheimer’s disease. J. Alzheimers Dis. 38, 809–822 (2014).
Squitti, R. et al. Elevation of serum copper levels in Alzheimer’s disease. Neurology 59, 1153–1161 (2002).
Bucossi, S. et al. Copper in Alzheimer’s disease: a meta-analysis of serum,plasma, and cerebrospinal fluid studies. J. Alzheimers Dis. 24, 175–185 (2011).
Noda, Y. et al. Copper enhances APP dimerization and promotes Abeta production. Neurosci. Lett. 547, 10–15 (2013).
Atwood, C. S. et al. Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-beta. Biochemistry 43, 560–568 (2004).
Cheignon, C. et al. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464 (2018).
Al-Hilaly, Y. K. et al. A central role for dityrosine crosslinking of Amyloid-beta in Alzheimer’s disease. Acta Neuropathol. Commun. 1, 83 (2013).
Streltsov, V. A. et al. The structure of the amyloid-beta peptide high-affinity copper II binding site in Alzheimer disease. Biophys. J. 95, 3447–3456 (2008).
Cherny, R. A. et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30, 665–676 (2001).
Behbehani, G. R., Barzegar, L., Mohebbian, M. & Saboury, A. A. A comparative interaction between copper ions with Alzheimer’s beta amyloid peptide and human serum albumin. Bioinorg. Chem. Appl. 2012, 208641 (2012).
Wu, W. H. et al. Sequestration of copper from beta-amyloid promotes selective lysis by cyclen-hybrid cleavage agents. J. Biol. Chem. 283, 31657–31664 (2008).
Kitazawa, M., Hsu, H. W. & Medeiros, R. Copper exposure perturbs brain inflammatory responses and impairs clearance of amyloid-beta. Toxicol. Sci. 152, 194–204 (2016).
Singh, I. et al. Low levels of copper disrupt brain amyloid-beta homeostasis by altering its production and clearance. Proc. Natl Acad. Sci. USA 110, 14771–14776 (2013).
Voss, K. et al. Modulation of tau phosphorylation by environmental copper. Transl. Neurodegener. 3, 24 (2014).
Du, X. et al. Inhibitory act of selenoprotein P on Cu(+)/Cu(2+)-induced tau aggregation and neurotoxicity. Inorg. Chem. 53, 11221–11230 (2014).
Bush, A. I. Drug development based on the metals hypothesis of Alzheimer’s disease. J. Alzheimers Dis. 15, 223–240 (2008).
Jenagaratnam, L. & McShane, R. Clioquinol for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 1, CD005380 (2006).
Adlard, P. A. et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 59, 43–55 (2008).
Crouch, P. J. et al. The Alzheimer’s therapeutic PBT2 promotes amyloid-beta degradation and GSK3 phosphorylation via a metal chaperone activity. J. Neurochem. 119, 220–230 (2011).
Faux, N. G. et al. PBT2 rapidly improves cognition in Alzheimer’s disease: additional phase II analyses. J. Alzheimers Dis. 20, 509–516 (2010).
Lannfelt, L. et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 7, 779–786 (2008).
Swinnen, B. & Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 10, 661–670 (2014).
Gil-Bea, F. J., Aldanondo, G., Lasa-Fernandez, H., Lopez de Munain, A. & Vallejo-Illarramendi, A. Insights into the mechanisms of copper dyshomeostasis in amyotrophic lateral sclerosis. Expert Rev. Mol. Med. 19, e7 (2017).
Son, M. et al. Overexpression of CCS in G93A-SOD1 mice leads to accelerated neurological deficits with severe mitochondrial pathology. Proc. Natl Acad. Sci. USA 104, 6072–6077 (2007).
Lynch, S. M. & Colon, W. Dominant role of copper in the kinetic stability of Cu/Zn superoxide dismutase. Biochem. Biophys. Res. Commun. 340, 457–461 (2006).
Tiwari, A. et al. Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J. Biol. Chem. 284, 27746–27758 (2009).
Roberts, B. R. et al. Oral treatment with Cu(II)(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 34, 8021–8031 (2014).
Lelie, H. L. et al. Copper and zinc metallation status of copper-zinc superoxide dismutase from amyotrophic lateral sclerosis transgenic mice. J. Biol. Chem. 286, 2795–2806 (2011).
Bourassa, M. W., Brown, H. H., Borchelt, D. R., Vogt, S. & Miller, L. M. Metal-deficient aggregates and diminished copper found in cells expressing SOD1 mutations that cause ALS. Front. Aging Neurosci. 6, 110 (2014).
Pratt, A. J. et al. Aggregation propensities of superoxide dismutase G93 hotspot mutants mirror ALS clinical phenotypes. Proc. Natl Acad. Sci. USA 111, E4568–E4576 (2014).
Tokuda, E., Okawa, E. & Ono, S. Dysregulation of intracellular copper trafficking pathway in a mouse model of mutant copper/zinc superoxide dismutase-linked familial amyotrophic lateral sclerosis. J. Neurochem. 111, 181–191 (2009).
Enge, T. G., Ecroyd, H., Jolley, D. F., Yerbury, J. J. & Dosseto, A. Longitudinal assessment of metal concentrations and copper isotope ratios in the G93A SOD1 mouse model of amyotrophic lateral sclerosis. Metallomics 9, 161–174 (2017).
Roos, P. M., Vesterberg, O., Syversen, T., Flaten, T. P. & Nordberg, M. Metal concentrations in cerebrospinal fluid and blood plasma from patients with amyotrophic lateral sclerosis. Biol. Trace Elem. Res. 151, 159–170 (2013).
Williams, J. R. et al. Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD(G93A) mice co-expressing the Copper-Chaperone-for-SOD. Neurobiol. Dis. 89, 1–9 (2016).
Hottinger, A. F., Fine, E. G., Gurney, M. E., Zurn, A. D. & Aebischer, P. The copper chelator d-penicillamine delays onset of disease and extends survival in a transgenic mouse model of familial amyotrophic lateral sclerosis. Eur. J. Neurosci. 9, 1548–1551 (1997).
Tokuda, E. et al. Ammonium tetrathiomolybdate delays onset, prolongs survival, and slows progression of disease in a mouse model for amyotrophic lateral sclerosis. Exp. Neurol. 213, 122–128 (2008).
Hilton, J. B. et al. Cu(II)(atsm) improves the neurological phenotype and survival of SOD1(G93A) mice and selectively increases enzymatically active SOD1 in the spinal cord. Sci. Rep. 7, 42292 (2017).
Soon, C. P. W. et al. Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J. Biol. Chem. 286, 44035–44044 (2011).
Tokuda, E., Okawa, E., Watanabe, S., Ono, S. & Marklund, S. L. Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities. Neurobiol. Dis. 54, 308–319 (2013).
Sturrock, A. & Leavitt, B. R. The clinical and genetic features of Huntington disease. J. Geriatr. Psychiatry Neurol. 23, 243–259 (2010).
DiFiglia, M. et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 (1997).
Arrasate, M. & Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11 (2012).
Dexter, D. T. et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114(Part 4), 1953–1975 (1991).
Fox, J. H. et al. Mechanisms of copper ion mediated Huntington’s disease progression. PLoS ONE 2, e334 (2007).
Pamp, K., Bramey, T., Kirsch, M., De Groot, H. & Petrat, F. NAD(H) enhances the Cu(II)-mediated inactivation of lactate dehydrogenase by increasing the accessibility of sulfhydryl groups. Free Radic. Res. 39, 31–40 (2005).
Sheline, C. T. & Choi, D. W. Cu2+ toxicity inhibition of mitochondrial dehydrogenases in vitro and in vivo. Ann. Neurol. 55, 645–653 (2004).
Kasischke, K. A., Vishwasrao, H. D., Fisher, P. J., Zipfel, W. R. & Webb, W. W. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 305, 99–103 (2004).
Harms, L., Meierkord, H., Timm, G., Pfeiffer, L. & Ludolph, A. C. Decreased N-acetyl-aspartate/choline ratio and increased lactate in the frontal lobe of patients with Huntington’s disease: a proton magnetic resonance spectroscopy study. J. Neurol. Neurosurg. Psychiatry 62, 27–30 (1997).
Cherny, R. A. et al. PBT2 reduces toxicity in a C. elegans model of polyQ aggregation and extends lifespan, reduces striatal atrophy and improves motor performance in the R6/2 mouse model of Huntington’s disease. J. Huntingt.’s. Dis. 1, 211–219 (2012).
Tallaksen-Greene, S. J. et al. Evaluation of tetrathiomolybdate in the R6/2 model of Huntington disease. Neurosci. Lett. 452, 60–62 (2009).
Xiao, G., Fan, Q., Wang, X. & Zhou, B. Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc. Natl Acad. Sci. USA 110, 14995–15000 (2013).
Denoyer, D., Masaldan, S., La Fontaine, S. & Cater, M. A. Targeting copper in cancer therapy: ’Copper That Cancer’. Metallomics 7, 1459–1476 (2015).
Pavithra, V. et al. Serum levels of metal ions in female patients with breast cancer. J. Clin. Diagn. Res. 9, BC25–c27 (2015).
Feng, J. F. et al. Serum total oxidant/antioxidant status and trace element levels in breast cancer patients. Int. J. Clin. Oncol. 17, 575–583 (2012).
Nayak, S. B., Bhat, V. R., Upadhyay, D. & Udupa, S. L. Copper and ceruloplasmin status in serum of prostate and colon cancer patients. Indian J. Physiol. Pharmacol. 47, 108–110 (2003).
Baharvand, M., Manifar, S., Akkafan, R., Mortazavi, H. & Sabour, S. Serum levels of ferritin, copper, and zinc in patients with oral cancer. Biomed. J. 37, 331–336 (2014).
Shen, F. et al. The association between serum levels of selenium, copper, and magnesium with thyroid cancer: a meta-analysis. Biol. Trace Elem. Res. 167, 225–235 (2015).
Basu, S. et al. Heavy and trace metals in carcinoma of the gallbladder. World J. Surg. 37, 2641–2646 (2013).
Margalioth, E. J., Schenker, J. G. & Chevion, M. Copper and zinc levels in normal and malignant tissues. Cancer 52, 868–872 (1983).
Yaman, M., Kaya, G. & Simsek, M. Comparison of trace element concentrations in cancerous and noncancerous human endometrial and ovary tissues. Int. J. Gynecol. Cancer 17, 220–228 (2007).
Lener, M. R. et al. Serum concentrations of selenium and copper in patients diagnosed with pancreatic cancer. Cancer Res. Treat. 48, 1056–1064 (2016).
Saleh, S. A. K., Adly, H. M., Abdelkhaliq, A. A. & Nassir, A. M. Serum levels of selenium, zinc, copper, manganese, and iron in prostate cancer patients. Curr. Urol. 14, 44–49 (2020).
Gupta, S. K., Shukla, V. K., Vaidya, M. P., Roy, S. K. & Gupta, S. Serum and tissue trace elements in colorectal cancer. J. Surg. Oncol. 52, 172–175 (1993).
Diez, M. et al. Serum and tissue trace metal levels in lung cancer. Oncology 46, 230–234 (1989).
Sharma, K., Mittal, D. K., Kesarwani, R. C., Kamboj, V. P. & Chowdhery, K. Diagnostic prognostic significance of serum and tissue trace elements in breast malignancy. Indian J. Med. Sci. 48, 227–232 (1994).
Skrajnowska, D. et al. Copper and resveratrol attenuates serum catalase, glutathione peroxidase, and element values in rats with DMBA-induced mammary carcinogenesis. Biol. Trace Elem. Res. 156, 271–278 (2013).
Ishida, S., Andreux, P., Poitry-Yamate, C., Auwerx, J. & Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl Acad. Sci. USA 110, 19507–19512 (2013).
Brady, D. C. et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 509, 492–496 (2014).
Turski, M. L. et al. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol. Cell. Biol. 32, 1284–1295 (2012).
Brady, D. C., Crowe, M. S. & Greenberg, D. N. & Counter, C. M. Copper chelation inhibits BRAF(V600E)-driven melanomagenesis and counters resistance to BRAF(V600E) and MEK1/2 inhibitors. Cancer Res. 77, 6240–6252 (2017).
Tsang, T. et al. Copper is an essential regulator of the autophagic kinases ULK1/2 to drive lung adenocarcinoma. Nat. Cell Biol. 22, 412–424 (2020).
McAuslan, B. R. & Reilly, W. Endothelial cell phagokinesis in response to specific metal ions. Exp. Cell Res. 130, 147–157 (1980).
Raju, K. S., Alessandri, G., Ziche, M. & Gullino, P. M. Ceruloplasmin, copper ions, and angiogenesis. J. Natl Cancer Inst. 69, 1183–1188 (1982).
Narayanan, G., R, B. S., Vuyyuru, H. & Muthuvel, B. & Konerirajapuram Natrajan, S. CTR1 silencing inhibits angiogenesis by limiting copper entry into endothelial cells. PLoS ONE 8, e71982 (2013).
Mandinov, L. et al. Copper chelation represses the vascular response to injury. Proc. Natl Acad. Sci. USA 100, 6700–6705 (2003).
Prudovsky, I. et al. The intracellular translocation of the components of the fibroblast growth factor 1 release complex precedes their assembly prior to export. J. Cell Biol. 158, 201–208 (2002).
Badet, J. et al. Specific binding of angiogenin to calf pulmonary artery endothelial cells. Proc. Natl Acad. Sci. USA 86, 8427–8431 (1989).
Pan, Q. et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 62, 4854–4859 (2002).
Feng, W., Ye, F., Xue, W., Zhou, Z. & Kang, Y. J. Copper regulation of hypoxia-inducible factor-1 activity. Mol. Pharmacol. 75, 174–182 (2009).
Qiu, L., Ding, X., Zhang, Z. & Kang, Y. J. Copper is required for cobalt-induced transcriptional activity of hypoxia-inducible factor-1. J. Pharmacol. Exp. Ther. 342, 561–567 (2012).
Kohno, T. et al. Novel role of copper transport protein antioxidant-1 in neointimal formation after vascular injury. Arterioscler. Thromb. Vasc. Biol. 33, 805–813 (2013).
Erler, J. T. et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44 (2009).
Cox, T. R. et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 522, 106–110 (2015).
Osawa, T. et al. Lysyl oxidase secreted by tumour endothelial cells promotes angiogenesis and metastasis. Br. J. Cancer 109, 2237–2247 (2013).
Shanbhag, V. et al. ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis. Proc. Natl Acad. Sci. USA 116, 6836–6841 (2019).
Blockhuys, S., Zhang, X. & Wittung-Stafshede, P. Single-cell tracking demonstrates copper chaperone Atox1 to be required for breast cancer cell migration. Proc. Natl Acad. Sci. USA 117, 2014–2019 (2020).
Nagaraju, G. P., Dontula, R., El-Rayes, B. F. & Lakka, S. S. Molecular mechanisms underlying the divergent roles of SPARC in human carcinogenesis. Carcinogenesis 35, 967–973 (2014).
Voli, F. et al. Intratumoral copper modulates PD-L1 expression and influences tumor immune evasion. Cancer Res. 80, 4129–4144 (2020).
Brem, S., Tsanaclis, A. M. & Zagzag, D. Anticopper treatment inhibits pseudopodial protrusion and the invasive spread of 9L gliosarcoma cells in the rat brain. Neurosurgery 26, 391–396 (1990).
Yoshii, J. et al. The copper-chelating agent, trientine, suppresses tumor development and angiogenesis in the murine hepatocellular carcinoma cells. Int. J. Cancer 94, 768–773 (2001).
Cox, C. et al. The role of copper suppression as an antiangiogenic strategy in head and neck squamous cell carcinoma. Laryngoscope 111, 696–701 (2001).
Donate, F. et al. Identification of biomarkers for the antiangiogenic and antitumour activity of the superoxide dismutase 1 (SOD1) inhibitor tetrathiomolybdate (ATN-224). Br. J. Cancer 98, 776–783 (2008).
Pan, Q., Rosenthal, D. T., Bao, L., Kleer, C. G. & Merajver, S. D. Antiangiogenic tetrathiomolybdate protects against Her2/neu-induced breast carcinoma by hypoplastic remodeling of the mammary gland. Clin. Cancer Res. 15, 7441–7446 (2009).
Crowe, A., Jackaman, C., Beddoes, K. M., Ricciardo, B. & Nelson, D. J. Rapid copper acquisition by developing murine mesothelioma: decreasing bioavailable copper slows tumor growth, normalizes vessels and promotes T cell infiltration. PLoS ONE 8, e73684 (2013).
Moriguchi, M. et al. The copper chelator trientine has an antiangiogenic effect against hepatocellular carcinoma, possibly through inhibition of interleukin-8 production. Int. J. Cancer 102, 445–452 (2002).
Mammoto, T. et al. Role of collagen matrix in tumor angiogenesis and glioblastoma multiforme progression. Am. J. Pathol. 183, 1293–1305 (2013).
Brem, S. S. et al. Inhibition of angiogenesis and tumor growth in the brain. Suppression of endothelial cell turnover by penicillamine and the depletion of copper, an angiogenic cofactor. Am. J. Pathol. 137, 1121–1142 (1990).
Pan, Q., Bao, L. W. & Merajver, S. D. Tetrathiomolybdate inhibits angiogenesis and metastasis through suppression of the NFkappaB signaling cascade. Mol. Cancer Res. 1, 701–706 (2003).
Kim, K. K. et al. Tetrathiomolybdate inhibits mitochondrial complex IV and mediates degradation of hypoxia-inducible factor-1alpha in cancer cells. Sci. Rep. 5, 14296 (2015).
Alvarez, H. M. et al. Tetrathiomolybdate inhibits copper trafficking proteins through metal cluster formation. Science 327, 331–334 (2010).
Pass, H. I., Brewer, G. J., Dick, R., Carbone, M. & Merajver, S. A phase II trial of tetrathiomolybdate after surgery for malignant mesothelioma: final results. Ann. Thorac. Surg. 86, 383–389 (2008).
Redman, B. G. et al. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin. Cancer Res. 9, 1666–1672 (2003).
Xiao, Y. et al. Molecular study on copper-mediated tumor proteasome inhibition and cell death. Int. J. Oncol. 37, 81–87 (2010).
Denoyer, D. et al. Copper as a target for prostate cancer therapeutics: copper-ionophore pharmacology and altering systemic copper distribution. Oncotarget 7, 37064–37080 (2016).
Zhang, H. et al. Disulfiram treatment facilitates phosphoinositide 3-kinase inhibition in human breast cancer cells in vitro and in vivo. Cancer Res. 70, 3996–4004 (2010).
Lun, X. et al. Disulfiram when combined with copper enhances the therapeutic effects of temozolomide for the treatment of glioblastoma. Clin. Cancer Res. 22, 3860–3875 (2016).
Liu, P. et al. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. Br. J. Cancer 109, 1876–1885 (2013).
Liu, P. et al. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br. J. Cancer 107, 1488–1497 (2012).
Lewis, J. et al. Copper-64-diacetyl-bis(N4-methylthiosemicarbazone): an agent for radiotherapy. Proc. Natl Acad. Sci. USA 98, 1206–1211 (2001).
Cater, M. A. et al. Increasing intracellular bioavailable copper selectively targets prostate cancer cells. ACS Chem. Biol. 8, 1621–1631 (2013).
Blackman, R. K. et al. Mitochondrial electron transport is the cellular target of the oncology drug elesclomol. PLoS ONE 7, e29798 (2012).
Berkenblit, A. et al. Phase I clinical trial of STA-4783 in combination with paclitaxel in patients with refractory solid tumors. Clin. Cancer Res. 13, 584–590 (2007).
O’Day, S. J. et al. Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J. Clin. Oncol. 31, 1211–1218 (2013).
Ford, E. S. Serum copper concentration and coronary heart disease among US adults. Am. J. Epidemiol. 151, 1182–1188 (2000).
Kok, F. J. et al. Serum copper and zinc and the risk of death from cancer and cardiovascular disease. Am. J. Epidemiol. 128, 352–359 (1988).
Stadler, N., Lindner, R. A. & Davies, M. J. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: evidence for the presence of elevated levels of iron and copper. Arterioscler. Thromb. Vasc. Biol. 24, 949–954 (2004).
Volker, W. et al. Copper-induced inflammatory reactions of rat carotid arteries mimic restenosis/arteriosclerosis-like neointima formation. Atherosclerosis 130, 29–36 (1997).
Wei, H., Frei, B., Beckman, J. S. & Zhang, W. J. Copper chelation by tetrathiomolybdate inhibits lipopolysaccharide-induced inflammatory responses in vivo. Am. J. Physiol. Heart Circ. Physiol. 301, H712–H720 (2011).
Wei, H., Zhang, W. J., Leboeuf, R. & Frei, B. Copper induces-and copper chelation by tetrathiomolybdate inhibits-endothelial activation in vitro. Redox Rep. 19, 40–48 (2014).
Wei, H., Zhang, W. J., McMillen, T. S., Leboeuf, R. C. & Frei, B. Copper chelation by tetrathiomolybdate inhibits vascular inflammation and atherosclerotic lesion development in apolipoprotein E-deficient mice. Atherosclerosis 223, 306–313 (2012).
Qin, Z., Itoh, S., Jeney, V., Ushio-Fukai, M. & Fukai, T. Essential role for the Menkes ATPase in activation of extracellular superoxide dismutase: implication for vascular oxidative stress. FASEB J. 20, 334–336 (2006).
Jeney, V. et al. Role of antioxidant-1 in extracellular superoxide dismutase function and expression. Circ. Res. 96, 723–729 (2005).
Ashino, T. et al. Unexpected role of the copper transporter ATP7A in PDGF-induced vascular smooth muscle cell migration. Circ. Res. 107, 787–799 (2010).
Schuschke, D. A., Saari, J. T. & Miller, F. N. Leukocyte-endothelial adhesion is impaired in the cremaster muscle microcirculation of the copper-deficient rat. Immunol. Lett. 76, 139–144 (2001).
Starkebaum, G. & Harlan, J. M. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J. Clin. Investig. 77, 1370–1376 (1986).
Steinberg, D. Low density lipoprotein oxidation and its pathobiological significance. J. Biol. Chem. 272, 20963–20966 (1997).
Ziouzenkova, O., Sevanian, A., Abuja, P. M., Ramos, P. & Esterbauer, H. Copper can promote oxidation of LDL by markedly different mechanisms. Free Radic. Biol. Med. 24, 607–623 (1998).
Al-Bayati, M. A., Jamil, D. A. & Al-Aubaidy, H. A. Cardiovascular effects of copper deficiency on activity of superoxide dismutase in diabetic nephropathy. N. Am. J. Med. Sci. 7, 41–46 (2015).
Medeiros, D. M. & Wildman, R. E. Newer findings on a unified perspective of copper restriction and cardiomyopathy. Proc. Soc. Exp. Biol. Med. 215, 299–313 (1997).
DiNicolantonio, J. J., Mangan, D. & O’Keefe, J. H. Copper deficiency may be a leading cause of ischaemic heart disease. Open Heart 5, e000784 (2018).
Jaksch, M. et al. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet. 9, 795–801 (2000).
Kim, B. E. et al. Cardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organs. Cell Metab. 11, 353–363 (2010).
Kang, Y. J., Wu, H. & Saari, J. T. Alterations in hypertrophic gene expression by dietary copper restriction in mouse heart. Proc. Soc. Exp. Biol. Med. 223, 282–287 (2000).
Harris, E. D. Basic and clinical aspects of copper. Crit. Rev. Clin. Lab. Sci. 40, 547–586 (2003).
Elsherif, L., Jiang, Y., Saari, J. T. & Kang, Y. J. Dietary copper restriction-induced changes in myocardial gene expression and the effect of copper repletion. Exp. Biol. Med. (Maywood) 229, 616–622 (2004).
Chao, J. C., Medeiros, D. M., Davidson, J. & Shiry, L. Low levels of ATP synthase and cytochrome c oxidase subunit peptide from hearts of copper-deficient rats are not altered by the administration of dimethyl sulfoxide. J. Nutr. 124, 789–803 (1994).
Jiang, Y. et al. Dietary copper supplementation reverses hypertrophic cardiomyopathy induced by chronic pressure overload in mice. J. Exp. Med. 204, 657–666 (2007).
Elsherif, L., Wang, L., Saari, J. T. & Kang, Y. J. Regression of dietary copper restriction-induced cardiomyopathy by copper repletion in mice. J. Nutr. 134, 855–860 (2004).
Freisinger, P., Horvath, R., Macmillan, C., Peters, J. & Jaksch, M. Reversion of hypertrophic cardiomyopathy in a patient with deficiency of the mitochondrial copper binding protein Sco2: is there a potential effect of copper? J. Inherit. Metab. Dis. 27, 67–79 (2004).
Xie, Y. et al. Ferroptosis: process and function. Cell Death Differ. 23, 369–379 (2016).
Stockwell, B. R. et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Zheng, J. & Conrad, M. The metabolic underpinnings of ferroptosis. Cell Metab. 32, 920–937 (2020).
Yan, R. et al. The structure of erastin-bound xCT-4F2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res. 32, 687–690 (2022).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019).
Yu, Y. et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 136, 726–739 (2020).
Fang, X. et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ. Res. 127, 486–501 (2020).
Wang, H. et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 66, 449–465 (2017).
Lei, P., Ayton, S. & Bush, A. I. The essential elements of Alzheimer’s disease. J. Biol. Chem. 296, 100105 (2021).
Tuo, Q. Z. et al. Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct. Target Ther. 7, 59 (2022).
Luoqian, J. et al. Ferroptosis promotes T-cell activation-induced neurodegeneration in multiple sclerosis. Cell. Mol. Immunol. 19, 913–924 (2022).
Yan, H. F. et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct. Target Ther. 6, 49 (2021).
Fang, X., Ardehali, H., Min, J. & Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 1-17, https://doi.org/10.1038/s41569-022-00735-4 (2022).
Chen, J., Li, X., Ge, C., Min, J. & Wang, F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 29, 467–480 (2022).
Fang, X. et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl Acad. Sci. USA 116, 2672–2680 (2019).
Cen, D., Brayton, D., Shahandeh, B., Meyskens, F. L. Jr. & Farmer, P. J. Disulfiram facilitates intracellular Cu uptake and induces apoptosis in human melanoma cells. J. Med. Chem. 47, 6914–6920 (2004).
Nagai, M. et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic. Biol. Med. 52, 2142–2150 (2012).
Tardito, S. et al. Copper binding agents acting as copper ionophores lead to caspase inhibition and paraptotic cell death in human cancer cells. J. Am. Chem. Soc. 133, 6235–6242 (2011).
Shimada, K. et al. Copper-binding small molecule induces oxidative stress and cell-cycle arrest in glioblastoma-patient-derived cells. Cell Chem. Biol. 25, 585–594e587 (2018).
Arciello, M., Rotilio, G. & Rossi, L. Copper-dependent toxicity in SH-SY5Y neuroblastoma cells involves mitochondrial damage. Biochem. Biophys. Res. Commun. 327, 454–459 (2005).
Solmonson, A. & DeBerardinis, R. J. Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 293, 7522–7530 (2018).
Mayr, J. A., Feichtinger, R. G., Tort, F., Ribes, A. & Sperl, W. Lipoic acid biosynthesis defects. J. Inherit. Metab. Dis. 37, 553–563 (2014).
Rowland, E. A., Snowden, C. K. & Cristea, I. M. Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease. Curr. Opin. Chem. Biol. 42, 76–85 (2018).
Tsvetkov, P. et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 15, 681–689 (2019).
Vallieres, C., Holland, S. L. & Avery, S. V. Mitochondrial ferredoxin determines vulnerability of cells to copper excess. Cell Chem. Biol. 24, 1228–1237e1223 (2017).
Brancaccio, D. et al. [4Fe-4S] Cluster assembly in mitochondria and Its impairment by copper. J. Am. Chem. Soc. 139, 719–730 (2017).
Garcia-Santamarina, S., Uzarska, M. A., Festa, R. A., Lill, R. & Thiele, D. J. Cryptococcus neoformans iron–sulfur proteinbiogenesis machinery is a novel layer of protection against Cu stress. mBio 8, e01742–17 (2017).
Patteson, J. B. et al. Biosynthesis of fluopsin C, a copper-containing antibiotic from Pseudomonas aeruginosa. Science 374, 1005–1009 (2021).
Raffa, N. et al. Dual-purpose isocyanides produced by Aspergillus fumigatus contribute to cellular copper sufficiency and exhibit antimicrobial activity. Proc. Natl. Acad. Sci. USA 118, e2015224118 (2021).
Mao, X. & Schimmer, A. D. The toxicology of Clioquinol. Toxicol. Lett. 182, 1–6 (2008).
Sternlieb, I., Quintana, N., Volenberg, I. & Schilsky, M. L. An array of mitochondrial alterations in the hepatocytes of Long–Evans Cinnamon rats. Hepatology 22, 1782–1787 (1995).
Walshe, J. M. Treatment of Wilson’s disease with trientine (triethylene tetramine) dihydrochloride. Lancet 1, 643–647 (1982).
Walshe, J. M. Penicillamine, a new oral therapy for Wilson&v;s disease. Am. J. Med. 21, 487–495 (1956).
Smirnova, J. et al. Copper(I)-binding properties of de-coppering drugs for the treatment of Wilson disease. alpha-Lipoic acid as a potential anti-copper agent. Sci. Rep. 8, 1463 (2018).
Maffia, M. et al. Copper dyshomeostasis in neurodegenerative diseases. Acta Physiol. 227, 58–58 (2019).
Ji, M. B. et al. Label-free imaging of amyloid plaques in Alzheimer’s disease with stimulated Raman scattering microscopy. Sci. Adv. 4, eaat7715 (2018).
Li, D. D., Zhang, W., Wang, Z. Y. & Zhao, P. Serum copper, zinc, and iron levels in patients with Alzheimeras disease: a meta-analysis of case-control studies. Front. Aging Neurosci. 9, 300 (2017).
James, S. A. et al. Iron, copper, and zinc concentration in A beta plaques in the APP/PS1 mouse model of Alzheimer’s disease correlates with metal levels in the surrounding neuropil. ACS Chem. Neurosci. 8, 629–637 (2017).
Lang, M. et al. Inhibition of human high-affinity copper importer Ctr1 orthologous in the nervous system of Drosophila ameliorates Abeta42-induced Alzheimer’s disease-like symptoms. Neurobiol. Aging 34, 2604–2612 (2013).
Hands, S. L., Mason, R., Sajjad, M. U., Giorgini, F. & Wyttenbach, A. Metallothioneins and copper metabolism are candidate therapeutic targets in Huntington&v;s disease. Biochem. Soc. Trans. 38, 552–558 (2010).
Rosen, D. R. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 (1993).
Tafuri, F., Ronchi, D., Magri, F., Comi, G. P. & Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 9, 336 (2015).
Rowe, D. et al. Treatment Continuation Study for Patients With ALS/MND Who Completed Study CMD-2019-001 https://ClinicalTrials.gov/show/NCT04313166 (2020).
Rezaei, A. et al. Effect of a copper (II) complex on the induction of apoptosis in human hepatocellular carcinoma cells. Asian Pac. J. Cancer Prev. 19, 2877–2884 (2018).
Gupte, A. & Mumper, R. J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 35, 32–46 (2009).
Xu, Y. et al. Enzyme-engineered nonporous copper(I) coordination polymer nanoplatform for cuproptosis-based synergistic cancer therapy. Adv. Mater. e2204733, https://doi.org/10.1002/adma.202204733 (2022).
Kirshner, J. R. et al. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol. Cancer Ther. 7, 2319–2327 (2008).
Jiao, Y., Hannafon, B. N. & Ding, W. Q. Disulfiram’s anticancer activity: evidence and mechanisms. Anti-Cancer Agents Med. Chem. 16, 1378–1384 (2016).
Wang, Y. et al. Blocking the formation of radiation-induced breast cancer stem cells. Oncotarget 5, 3743–3755 (2014).
Wu, L. et al. Disulfiram and BKM120 in combination with chemotherapy impede tumor progression and delay tumor recurrence in tumor initiating cell-rich TNBC. Sci. Rep. 9, 236 (2019).
Viola-Rhenals, M. et al. Recent advances in Antabuse (Disulfiram): the importance of its metal-binding ability to its anticancer activity. Curr. Med. Chem. 25, 506–524 (2018).
Yang, H. et al. Repurposing old drugs as new inhibitors of the ubiquitin–proteasome pathway for cancer treatment. Semin. Cancer Biol. 68, 105–122 (2021).
Jakola, A. S. et al. Disulfiram in Recurrent Glioblastoma https://ClinicalTrials.gov/show/NCT02678975 (2017).
Huang, J. et al. A phase I study to repurpose disulfiram in combination with temozolomide to treat newly diagnosed glioblastoma after chemoradiotherapy. J. Neuro-Oncol. 128, 259–266 (2016).
Acknowledgements
This work was supported by research grants from the National Natural Science Foundation of China (31930057 to F.W. and 31970689 to J.M.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, L., Min, J. & Wang, F. Copper homeostasis and cuproptosis in health and disease. Sig Transduct Target Ther 7, 378 (2022). https://doi.org/10.1038/s41392-022-01229-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01229-y
This article is cited by
-
A cuproptosis score model and prognostic score model can evaluate clinical characteristics and immune microenvironment in NSCLC
Cancer Cell International (2024)
-
Prognostic analysis of hepatocellular carcinoma based on cuproptosis -associated lncRNAs
BMC Gastroenterology (2024)
-
Immunomodulation of cuproptosis and ferroptosis in liver cancer
Cancer Cell International (2024)
-
Augmenting MEK inhibitor efficacy in BRAF wild-type melanoma: synergistic effects of disulfiram combination therapy
Journal of Experimental & Clinical Cancer Research (2024)
-
Copper homeostasis and cuproptosis in atherosclerosis: metabolism, mechanisms and potential therapeutic strategies
Cell Death Discovery (2024)
