
Abstract
Lung cancer is the leading cause of cancer-related death across the world. Unlike lung adenocarcinoma, patients with lung squamous cell carcinoma (LSCC) have not benefitted from targeted therapies. Although immunotherapy has significantly improved cancer patients’ outcomes, the relatively low response rate and severe adverse events hinder the clinical application of this promising treatment in LSCC. Therefore, it is of vital importance to have a better understanding of the mechanisms underlying the pathogenesis of LSCC as well as the inner connection among different signaling pathways, which will surely provide opportunities for more effective therapeutic interventions for LSCC. In this review, new insights were given about classical signaling pathways which have been proved in other cancer types but not in LSCC, including PI3K signaling pathway, VEGF/VEGFR signaling, and CDK4/6 pathway. Other signaling pathways which may have therapeutic potentials in LSCC were also discussed, including the FGFR1 pathway, EGFR pathway, and KEAP1/NRF2 pathway. Next, chromosome 3q, which harbors two key squamous differentiation markers SOX2 and TP63 is discussed as well as its related potential therapeutic targets. We also provided some progress of LSCC in epigenetic therapies and immune checkpoints blockade (ICB) therapies. Subsequently, we outlined some combination strategies of ICB therapies and other targeted therapies. Finally, prospects and challenges were given related to the exploration and application of novel therapeutic strategies for LSCC.
Similar content being viewed by others

Introduction
Lung cancer is the leading cause of cancer-related death across the world.1 In 2020, lung cancer remained the leading cause of cancer deaths, with an estimated 1.8 million deaths.2 Lung cancer was traditionally classified into two primary groups, small versus non-small-cell type. Approximately 85% of lung cancers are non-small-cell lung cancer (NSCLC). Lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LSCC) are the most common subtypes, with the latter accounting for approximately 30% of all NSCLC.3,4 The 5-year survival rate for NSCLC is 26%, and only 24% of lung cancers are diagnosed at a localized stage, for which the 5-year survival rate is 60%.5 The mortality rate of lung cancer has declined largely in the past few decades, with the pace accelerating in recent years due to major advances in treatment for NSCLC.5,6 Over the past 20 years, the treatment of NSCLC has evolved from the empirical use of cytotoxic therapy to effective and better-tolerated regimens by targeting specific molecular subtypes in both LUAD7 and LSCC.4 This improvement is largely attributed to the accumulation of molecular knowledge and the discovery of targeted molecular abnormalities. Previously, the treatment options for these two subtypes of NSCLC were historically similar.8 In 2004, two studies found out that a subgroup of NSCLC patients with specific mutations in the epidermal growth factor receptor (EGFR) gene were correlated with clinical responses to the tyrosine-kinase inhibitor (TKI) gefitinib.9,10 These findings ushered in a widening divide in the management of these two diseases.
While the steady identification of new oncogenic alterations and development of new targeted drugs continue to prolong the progression-free survival (PFS) and the overall survival (OS) in LUAD patients, it was of note that the use of targeted therapy in LSCC patients has been associated with negative outcomes in early-phase studies.11,12,13,14,15,16,17,18,19 In fact, both the mutated genes and the recurrent somatic copy numbers vary widely between these two diseases.4,20 The common driver mutations in LUAD, like EGFR and KRAS, are rarely mutated in LSCC.21,22 The biomarker-driven therapies for LSCC evaluated in the Lung Cancer Master Protocol (Lung-MAP; S1400) failed to show an improvement of therapeutic effect of currently available targeted therapy with an overall response rate of only 7%.15,17,19,23 This has changed with the advent of immune-checkpoint blockade (ICB) therapy, which has the potential to transcend the histological boundaries and, most importantly, achieve sustained remission in patients.24 Immunotherapies, such as anti-programmed cell death 1 (PD-1)/programmed cell death ligand 1 (PD-L1) therapy, lead to durable responses and manageable adverse effects.25 Currently, pembrolizumab and atezolizumab were both approved by the US Food and Drug Administration (FDA) as the first-line monotherapy in patients with advanced LSCC with PD-L1 levels of 50% or more.26,27,28 In addition, pembrolizumab plus chemotherapy is also recommended as a first-line therapy in patients with metastatic LSCC.29 Other ICB therapies, including nivolumab,30,31 ipilimumab,30,31 cemiplimab,32 sintilimab,33 tislelizumab,34 camrelizumab,35 and sugemalimab,36 have also significantly improved the outcomes of patients with advanced LSCC. However, a large proportion of LSCC patients still do not respond to current immunotherapy. Identification of biomarkers for immunotherapy and exploration of more effective therapeutics remains to be an unmet need for LSCC patients.
In this review, we will discuss the current knowledge of potentially targetable molecular alterations in LSCC as well as providing some updated information about ongoing or finished clinical trials which may change or have changed the therapeutic landscapes of LSCC.
Genomic landscapes of LSCC and therapeutic targets
LSCC is strongly associated with smoking and is characterized by a high overall mutation rate of 8.1 mutations per megabase and marked genomic complexity.4 A number of significantly mutated genes have been identified in LSCC: TP53, CDKN2A, PTEN, PIK3CA, KEAP1, MLL2, HLA-A, NFE2L2, NOTCH1, and RB1, all of which demonstrated robust evidence of gene expression.4,37 Almost all LSCC displays somatic mutation of TP53. The amplification between chromosome 3q26 and 3q28 represents a hallmark for LSCC, which harbors key squamous differentiation markers SOX2 and TP63.4,37,38,39,40 In addition, other significantly amplified regions include 8p11 (FGFR1, WHSC1L1), 7p11 (EGFR), 11q13 (CCDN1) and 4q12 (KDR, KIT, PDGFRA).37,41 Although EGFR mutations were found in 7% of the LSCC cases, there were no activating mutations of exon 19 deletions or Leu858Arg substitution.4 These mutated genes cause frequent alterations in the following signaling pathways: CDKN2A/RB1, NFE2L2/KEAP1/CUL3, PI3K/AKT, and SOX2/TP63/NOTCH1 pathways, some of which play an important role in cell-cycle control, response to oxidative stress, apoptotic signaling, and squamous cell differentiation.4,37 These signaling pathways are interconnected with each other. It has been shown that both mutated genes and recurrent somatic copy-number alterations are largely distinct in LSCC and LUAD.20 In fact, an analysis of 12 cancer types revealed the convergence of squamous-like subtype, suggesting similarities in genomic- and pathway-based determinants in four different tumor types: LSCC, head and neck squamous cell carcinoma (HNSCC), some bladder urothelial carcinoma and a very few LUAD.42,43 Besides frequent alterations in different signaling pathways caused by mutated genes and amplification in the chromosome 3q region, aberrant protein lysine methylation modification also influences other cellular pathways in LSCC. Recently, NSD3—a histone dimethyltransferase encoded by an FGFR1-neighboring gene—was identified as a major mutational driver in LSCC.44 Other histone modifiers, including SETD8,45 LSD138,46,47 and EZH2,38,48,49 have also been identified to be involved in LSCC tumorigenesis and the inhibition of these targets could produce potent antitumoral effects. In the last 10 years, the great benefits that ICB therapy have demonstrated in several clinical trials heralded a new era in the management of LSCC. For resectable NSCLC, compared to traditional cytotoxic chemotherapy alone, the addition of ICB therapy both in the neoadjuvant and adjuvant settings has significantly improved the outcomes of patients.50,51 We briefly summarized some of the pivotal clinical studies and discoveries that might change or have changed the management of LSCC in the last thirty years (Fig. 1).

Timeline illustrating the evolving treatment landscapes and research history of LSCC. Timeline highlights some of the pivotal clinical studies and discoveries that might change or have changed the management of LSCC. 1 L first line, 2 L second line, ICB therapy immune-checkpoint blockade therapy, OS overall survival, TCGA The Cancer Genome Atlas
Targeting signaling pathways in LSCC
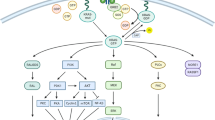
In the past few decades, multiple mutated genes in LSCC have been discovered. As we mentioned in the last section, these mutated genes cause alterations in several signaling pathways. In this section, we would discuss pathways proved in other cancer types but not in LSCC, including PI3K signaling pathway, VEGF/VEGFR signaling, and CDK4/6 pathway. Other signaling pathways in LSCC which may be potentially targeted were also discussed, including FGFR1 pathway, EGFR pathway, and KEAP1/NRF2 pathway. It is of note that these signaling pathways are also closely connected with each other (Fig. 2).

Mutated driver genes and downstream signal pathways in LSCC. FGFR1 amplification is observed in 20% of LSCC patients. FGFR1 amplifications can lead to overexpression of wild-type FGFR1 proteins on the cell membrane, resulting in increased sensitivity to FGF and the promotion of tumor growth via increased activation of four key downstream signaling pathways: PLCγ, PI3K-AKT, RAS-MAPK, and STAT (green). Although mutations in EGFR gene are relatively rare in LSCC, in certain cases of LSCC these mutations are responsible for constitutive ligand-independent receptor activation and downstream signaling, promoting cell survival and proliferation. Mitogenic signaling, including activation of upstream PI3K and MAPK signaling, could drive cyclin D upregulation, which leads to CDK4/6 activation. The phosphorylation of RB by cyclin D-CDK4/6 complexes and cyclin E-CDK2 complexes releases E2F transcriptional factors to activate genes required for G1-S transition. The CDKN2A gene encodes p16INK4A and p14ARF, which regulate cell cycle by inhibiting CDK4/6 and CDK2, respectively. In LSCC, the inactivated CDKN2A caused by genomic alterations may upregulate this pathway. DAG diacylglycerol, E2F E2 family, FRS2 FGFR substrate 2, GRB2 growth factor receptor-bound 2, IP3 inositol triphosphate, MDM2 murine double minute 2, P phosphorylation, PIP2 phosphatidylinositol-4,5-biphosphate, PKC protein kinase C, PLCγ phospholipase Cγ, PTEN phosphatase and tensin homolog, STAT signal transducer and activator of transcription
The classical PI3K pathway
The PI3K signaling pathway is one of the most frequently altered signaling pathways in human cancer,52,53,54,55 which can be activated by various growth factors/ligands specific to different RTKs, including members of the EGFR family, and the insulin and insulin-like growth factor 1 (IGF-1) receptor, FGF, etc.56 LSCC has high rates of alterations in the PI3K pathway, and alterations were found in 68% of LSCC samples.57 Activation of PI3K signaling pathway mediated through molecular aberrations is instrumental in promoting tumor development as well as resistance to antitumor therapies.55,58 PI3K belongs to a family of lipid kinases, which are classified into three different classes based on structural features and lipid substrate preferences.59 Class I PI3K is frequently implicated in cancer. Class IA consists of the PIK3CA, PIK3CB, and PIK3CD genes, and encode for the catalytic subunit of p110α, p110β, and p110δ, respectively. Class IB includes PIK3CG coding for p110γ.54 Whereas p110α and p110β show broad tissue distribution, p110δ and p110γ are highly enriched in all leukocyte subtypes.60
A common mechanism of PI3K activation in cancers is through the presence of mutations in the PIK3CA gene.61 The reported incidence of PIK3CA alterations in LSCC varies between 8 and 20% and the two main mutation types are canonical PIK3CA mutations and PIK3CA amplification.62,63,64 The canonical PIK3CA mutations affect two different domains of p110α, the kinase domain and the helical domain.65 These two types of mutations can activate the downstream signaling through two distinctive mechanisms. The kinase domain mutations can change the dynamics of the membrane-binding surface and affect the PIP2 substrate.66 The helical domain mutations (e.g., E542K, E545K) abrogate the inhibitory interactions between p110α and the N-terminal SH2 domain of the p85 regulatory subunit, leading to constitutive activity that mimics pTyr stimulation.66,67
Mutations in other genes may also lead to abnormal activation of the PI3K pathway. Phosphatase and tensin homolog (PTEN) is a 9-exon tumor-suppressor gene located on chromosome 10q23. This gene encodes for a 403-amino acid protein with dual lipid and protein phosphatase utility which contains four functional domains: an N-terminal PI(4,5)P2-binding/phosphatase, domain, a C2 domain, a carboxyl-terminal tail domain (C-tail), and a PDZ-binding domain (PDZ-BD).68 This protein classically dampens the PI3K/AKT/mTOR growth-promoting signaling cascade by directly dephosphorylating phosphatidylinositol-3,4,5-trisphosphate (PIP3) and converting it back to the phosphatidylinositol-4,5-bisphosphate (PIP2) inactive state.69,70 Accordingly, PTEN dysfunction causes dysregulation of this and other pathways, resulting in tumorigenesis and cancer progression.71,72,73 PTEN is mutated in 7–10% of LSCC62,63,74 and these somatic mutations tend to be distributed across its 9 exons. Some tumor-associated missense mutations may lead to complete loss or severe impairment of the phosphatase activity of the encoded enzyme.75,76,77 Many tumor-derived PTEN mutants retain partial or complete catalytic function, suggesting that alternative mechanisms can lead to the inactivation of PTEN.78 In addition to its own genetic alterations, PTEN gene expression is also regulated at epigenetic,79 transcriptional,80,81,82 post-transcriptional83,84,85,86, and post-translational87,88,89,90,91,92 levels as well as by protein–protein interactions.93,94
Partial loss of PTEN function can have dramatic effects on tumorigenesis and cancer progression,71,72 reflecting the fact that PTEN is a haploinsufficient tumor suppressor. PTEN function is not often completely lost in cancer, providing an opportunity to reactivate its function as a mode of cancer treatment. It has been reported that a tumor-suppressive metabolic state is induced in transgenic mouse lines with the systemic elevation of PTEN.95 In 2019, Lee et al.96 reported a way to reactivate PTEN by inhibiting the MYC-WWP1 inhibitory pathway. In the study, they identified the HECT-type E3 ubiquitin ligase WWP1 as a physical PTEN interactor, the amplification, and overexpression of which may lead to pleiotropic inactivation of PTEN. A natural and potent WWP1 inhibitor, indole-3-carbinol (I3C), was also found to effectively suppress tumorigenesis driven by the PI3K-AKT pathway. Therefore, both genetic and pharmacological targeting of the MYC-WWP1 axis may be a viable approach for cancer patients driven by impaired PTEN function.
Although extensive research has been conducted on the PI3K pathway component genes as potential molecular therapeutic targets in human cancers in the past two decades, clinical success to date has been limited to the approval of the PI3K inhibitors for hematological malignancies and breast cancer.34,97,98,99,100,101 Even among the four current FDA-approved PI3K inhibitors for the treatment of hematological malignancies, some of the indications have been withdrawn from the marketing authorization application.102,103,104,105 Although some of the decisions were made, according to the manufacturing companies, based on business needs, this will certainly have implications for the future of PI3K inhibitors. In NSCLC, the early-phase clinical trials of PI3K inhibitors and dual PI3K/mTOR inhibitors have yielded negative results.16,17,18 The modest therapeutic efficacy of PI3K inhibitors may be attributed to various reasons, including insufficient target inhibition, intrinsic and acquired drug resistance, and tolerability.106 Unlike other oncogenes, such as EGFR in LUAD, the correlation between specific PI3K pathway mutations and drug sensitivity is not absolute.107 This makes patient selection more complicated based on PI3K pathway mutation status. Another issue that cannot be overlooked about PI3K inhibitors is their on-target, off-tumor toxicity, particularly hyperglycemia and hyperinsulinemia which are observed as major dose-limiting toxicities.98,108,109 Despite the benefits of PFS shown in several randomized clinical trials, the increased toxicities of PI3K inhibitors have raised concerns about the potential detriments of OS in the PI3K inhibitor arm.34,105,110,111,112,113 In blood cancers, future approvals of PI3K inhibitors by FDA should be supported by randomized data.114
It has been suggested that alternative dosing regimens which offer intermittent pathway inhibition can increase the therapeutic window without compromising therapeutic efficacy.115,116,117 Another solution is to develop selective compounds that are more selective for mutant PI3K than wild-type PI3K. In the future, more durable therapeutic responses could be achieved by a more tailored PI3K-based therapies with a better understanding of the role of PI3K in cancer and surrounding environments.
Cell cycle in LSCC: the CDK4/6 pathway
The CDKN2A locus, located on human chromosome 9p21, is one of the most common genetic losses in human cancer.118,119 TCGA profiling of 178 LSCC samples revealed that CDKN2A is inactivated in 72% cases of LSCC.4 The CDKN2A locus encodes two alternatively spliced proteins, p16INK4a (p16) and p14ARF (p14), which function as cell-cycle inhibitors. These two tumor-suppressor proteins function in distinct anticancer pathways: p16 regulates retinoblastoma (RB), and p14 regulates p53. RB is a tumor-suppressor protein which controls cell cycle by preventing entry into the DNA synthesis (S) phase of the cell-division cycle.120 The p16 protein directly inhibits the activities of the cyclin D-dependent kinases, cyclin-dependent kinase (CDK) 4 and CDK6, thus maintaining RB in its dephosphorylated, anti-proliferative state, and leading to cell growth arrest.121 The tumor-suppressor protein p53 plays a pivotal role in regulating cell growth following exposure of cells to various stress stimuli.122 The p14 protein associates directly with murine double minute 2 (MDM2), a negative regulator of p53, preventing the export and degradation of p53.123,124,125,126
At present, the therapeutic focus has been on leveraging CDK4/6 inhibition to activate RB and limit tumor cell proliferation to delay disease progression.127,128 Interesting to note, the pan-caner analysis of the CDK4/6 pathway showed that CDKN2A loss and RB1 loss were mutually exclusive in most cancers that lose these genes at a significant level (>5%).129 The proteogenomic portrait of LSCC revealed that loss of one of these two key CDK4/6 pathway inhibitors is a universal feature of LSCC.38 However, CDK4/6 inhibitors have shown minimal efficacy in LSCC clinical trials.19,130,131,132 Phospho-RB levels have been shown to be correlated with response to CDK4/6 inhibitors in various LSCC cell lines.38 The heterogeneity of RB expression and phosphorylation may provide a reasonable explanation for the diverse responses toward CDK4/6 inhibitors. The screening of tumors based on the downstream functional assessment (i.e., RB expression and phosphorylation) may identify tumors that are sensitive to CDK4/6 inhibitors.
VEGF-VEGFR signaling in LSCC
Vascular endothelial growth factor (VEGF, here referred to as VEGF-A) is a member of a protein family that also includes VEGF-B, VEGF-C, VEGF-D, VEGF-E (a virally encoded protein), and placental growth factor (PIGF, also known as PGF).133 VEGF-B has multifaceted and context-dependent functions that safeguard the balance between blood vessel growth and degeneration to ensure normal blood vessel density and integrity.134 VEGF-C and VEGF-D are mainly implicated in lymphangiogenesis.135 As VEGF-A plays a dominant role in regulating angiogenesis and disease, it is referred to as VEGF in this review. Alternative exon splicing causes multiple isoforms of VEGF which are characterized by their differential ability to bind heparin.136 VEGF binds to both VEGF receptor 1 (R1) and VEGFR2 while VEGFR2 is the main receptor for VEGF.137,138 VEGF isoforms can also interact with the neuropilin co-receptors (NRP1 and NRP2).139,140 During tumorigenesis, angiogenesis plays a key role in maintaining the expansion in tumor. Most human tumors overexpressed VEGF mRNA, and its expression correlates with invasiveness, increased vascular density, metastasis, tumor recurrence and poor prognosis.141 Accordingly, several strategies that target this VEGF-VEGFR signaling has been devised.142,143
Neutralizing monoclonal antibodies (mAbs) against VEGF have shown great effect in preclinical studies144 and were the first type of antiangiogenic drugs that entered the market. In 2004, bevacizumab was approved by the FDA for the treatment of metastatic colorectal cancer based on the results of AVF2107 clinical trial.145 However, as the benefits of bevacizumab extended to other malignancies, including non-squamous NSCLC, renal cell carcinoma, ovarian cancer, and cervical cancer,142 LSCC is not one of them, as clinical trials have shown that bevacizumab increases the risk of life-threatening pulmonary hemorrhages in squamous cell carcinomas.146,147 Another antiangiogenic agent, ramucirumab, a human IgG1 monoclonal antibody targeting the extracellular domain of VEGFR2, is currently the only antiangiogenic agent that is approved by FDA for the treatment of LSCC. Based on the results of phase III REVEL clinical trial,147 ramucirumab plus docetaxel is recommended as a subsequent therapy option for metastatic NSCLC following disease progression on or after platinum-based chemotherapy.148
FGFR1 pathway
Fibroblast growth factor receptor 1 (FGFR1) belongs to the FGFR family of receptor tyrosine kinases (RTKs), which consists of four members: FGFR1 to FGFR4. All these four members share a canonical RTK architecture, consisting of a large ligand-binding extracellular domain that comprises three immunoglobulin-like domains (D1-3) followed by a single transmembrane helix and an intracellular domain containing the catalytically active “split” tyrosine-kinase domain.149,150 There is also a fifth related receptor, FGFR5 (also known as FGFRL1), which lacks the cytoplasmic tyrosine-kinase domain.151 The native ligand of FGFRs is fibroblast growth factors (FGFs), which can be divided into two categories: hormone-like FGFs (i.e., FGF19, 21, and 23) and canonical FGFs (i.e., FGF1-10, 16–18, and 20).150,152 The intracellular signaling of the FGFR pathway is primarily mediated mainly through four key downstream pathways: RAS-RAF-MAPK pathway, PI3K-AKT, signal transducer and activator of transcription (STAT), and phospholipase Cγ (PLCγ)153,154,155 (Fig. 1). Dysregulation of FGFR signaling promotes the proliferation,156 survival157 and development of drug resistance158 in tumor cells, as well as the development of angiogenesis159 and immune evasion in the tumor microenvironment (TME).160 These findings make FGFR pursued as a potential therapeutic target and support the development of FGFR-targeting anticancer agents.
FGFR1 amplifications are the predominant type of FGFR mutation, occurring in nearly 20% of LSCC patients.160,161 Although the studies in preclinical models have suggested that FGFR inhibitors may be a viable therapeutic option in this cohort of patients,161,162 a number of FGFR-specific small molecular inhibitors tested in phase I and phase II trials have shown modest effects with overall response rates of 8–15%.11,12,13,14,15 The results from these trials suggest that FGFR1 amplification is not a reliable predictor of response to FGFR1 inhibitors and that FGFR1 mutations have a more complex impact in NSCLC than EGFR-mutated or ALK-rearranged NSCLC.163 A previous study has found that elevated FGFR1 mRNA and/or protein expression was often independent of FGFR1 amplification.164 Future studies are needed to clarify the role of FGFR1 signaling in the pathogenesis of LSCC.
EGFR pathway
EGFR belongs to the HER/erbB family of RTKs, which includes HER1 (EGFR/erbB1), HER2 (neu, erbB2), HER3 (erbB3), and HER4 (erbB4). All members display similar structures: an extracellular, cysteine-rich ligand-binding region, a single alpha-helix membrane-spanning region and a cytoplasmic tyrosine-kinase-containing domain.165 The intracellular signaling of EGFR pathway is mediated mainly through the RAS/MAPK pathway, the PI3K pathway, and the STAT pathway.166,167 Downstream EGFR signaling ultimately leads to increased proliferation,168 angiogenesis,169 metastasis,170 and decreased apoptosis.171 Alterations in EGFR signaling pathways result in constitutive activation of its kinase activity and the inhibition of tumor apoptosis, leading to a poor clinical outcome.172,173 All these findings make EGFR pursued as therapeutic targets and support the development of EGFR-targeting anticancer agents.174
The reported rate of EGFR mutation in LSCC patients is 4.2–7%,4,175,176 which is much lower compared with LUAD patients. In previous prospective phase III clinical trials assessing the efficacy of first-line EGFR-TKIs in the treatment of NSCLC, only 27 cases of LSCC patients with EGFR mutation were identified in six clinical trials, which were further randomized into two groups.177,178,179,180,181,182 This limited number of LSCC cases makes it hard to assess the benefits of EGFR-TKIs for EGFR-mutated LSCC in prospective studies. Subgroup analysis in the BR.21 and SATURN clinical trials showed that erlotinib was effective in unselected LSCC patients.183,184 A meta-analysis also confirmed that EGFR-TKIs demonstrated an improved OS and PFS compared to placebo in unselected patients with advanced LSCC.185 Based on previous retrospective matched-pair studies,186,187 EGFR-TKIs were less effective in EGFR-mutant LSCC than in LUAD but still had clinical benefits for LSCC patients. Another retrospective study found that in Chinese female EGFR-mutant LSCC, EGFR-TKIs conferred longer PFS and OS than chemotherapy, but the survival was similar with patients without EGFR mutations.188 In conclusion, for EGFR-mutant LSCC, EGFR-TKIs can improve the outcomes of these patients compared with chemotherapy, but its efficacy is not as robust as that of EGFR-TKIs for EGFR-mutant LUAD.
Notably, EGFR protein was significantly upregulated in the squamous cancers but not in LUAD,38 although many activating EGFR mutations occurred in LUAD. This EGFR amplified LSCC cohort did not show elevated EGFR pathway activity,189 but displayed a high correlation with mRNA abundance of the five EGFR ligands. This is consistent with the results in HNSCC,190 which indicates a squamous cell carcinoma feature that EGFR ligand abundance drives the activity of EGFR pathways. It suggests that EGFR ligand abundance, rather than EGFR amplification, might be a better predictor for EGFR inhibitor response in this population of LSCC patients.
KEAP1/NRF2 pathway
The Kelch-like ECH-associated protein 1 (KEAP1, encoded by KEAP1)/nuclear factor erythroid 2-related factor 2 (NRF2, encoded by NFE2L2) pathway plays a physiologic protective role against environmental insults.191 This KEAP1-NRF2 system operates as a typical two-component system: KEAP1 as a sensor for insults, NRF2 as an effector for the coordinated activation of cytoprotective genes (Fig. 3a). The NRF2 signaling is primarily regulated by KEAP1 in response to reactive oxygen species (ROS) and electrophiles,192,193 but also by the PI3K signaling pathway,194 with glycogen synthase kinase 3 (GSK3) acting as a key mediator195 (Fig. 3a). In addition to crosstalk with the PI3K-AKT signaling pathway, the KEAP1-NRF2 system also interacts with the autophagy pathway through the adaptor p62.196 Hyperactivation of NRF2 plays a critical role in promoting both tumorigenesis and resistance to multiple therapies,197,198,199,200,201 resulting from the mutually exclusive loss-of-function (LOF) mutations in KEAP1 or gain-of-function (GOF) mutations in NRF2. Furthermore, there is also evidence that NRF2 has its tumor-preventive role during initiation,202 suggesting that the dual stage-specific pro- and anti-tumorigenic effects of NRF2 are context-dependent.

Physiologic activation and regulation of NRF2 and metabolic reprogramming by NRF2 in LSCC cells. a In unstressed conditions, KEAP1 forms a ubiquitin E3 ligase complex with CULLIN3 (CUL3) and binds with NFR2 via the DLG and ETGE motifs in the Neh2 domain of NRF2 in the cytoplasm. NRF2 is then polyubiquitinated and degraded through the proteasome system after its synthesis. When cells are exposed to electrophiles or ROS, KEAP1 is modified and the KEAP1-CUL3 ubiquitin E3 ligase activity declines, which stabilizes NRF2. Stabilized and accumulated NRF2 translocates to the nucleus and functions as a transcriptional factor. NRF2 is also regulated through a KEAP1-independent mechanism in which GSK3 plays an important role. NRF2 is phosphorylated by GSK3 and then recognized by β-TrCP. By contrast, the Neh6 domain of NRF2 serves as the degron exploited in this β-TrCP-CUL1-dependent degradation of NRF2. Following its ubiquitination by the β-TrCP-CUL1 E3 ubiquitin ligase complex, NRF2 is degraded by the proteasome. b LSCC cells displayed a dual reliance on glucose and glutamine metabolism. Activation of NRF2 increases the synthesis of GSH from intracellular glutamate, cysteine, and glycine. GLS1 catalyzes the transformation of glutamine to glutamate. Cystine is imported by the xc– antiporter system (xCT). Serine and glycine are synthesized via NRF2-dependent processes. Under chronic mTOR inhibition which suppresses glycolysis, LSCC cells could upregulate glutaminolysis through the GSK3 signaling pathway which developed acquired resistance to mTOR inhibition. β-TrCP β-transducin repeat-containing protein genes, GLS1 glutaminase 1, GSH glutathione, PDK phosphoinositide-dependent kinase, ROS reactive oxygen species, TCA cycle tricarboxylic acid cycle
Alterations in the KEAP1/NRF2 pathway are significantly enriched in lung and upper airway cancers. In LSCC, mutations in NRF2 are more prevalent while KEAP1 mutations are more common in LUAD.4,203,204,205 KEAP1/NRF2 pathway mutations were found in approximately one third of patients with LSCC. As we mentioned earlier, NRF2 has a dual function during carcinogenesis: preventing cancer initiation and promoting tumor progression. This is supported by the fact that NRF2-null mice exhibit an increased level of lung metastasis compared with wild-type mice after cancer cell incubation,206 while activated NRF2 in tumor cells is associated with poor prognosis and more aggressive disease.199,207 This contribution of NRF2 to the malignant phenotype of tumor cells is independent of its antioxidant activities and is associated with its key role in metabolic reprogramming of cancer cells. LSCC cells displayed a dual reliance on glucose and glutamine metabolism.208 The metabolic enzymes that are involved in this pentose phosphate pathway and glutamine metabolism were found to be directly activated through the antioxidant response element (ARE).209 NRF2 also regulates the intracellular abundance of these amino acids. SLC7A11 is a NRF2 target gene encoding SLC7A11, which dimerizes with SLC3A2 to form the xc− antiporter system (xCT).210 xCT functions as a concentration-dependent antiporter, which exports glutamate while importing cystine, the dimerized form of cysteine. Three amino acids were required in the synthesis of glutathione: glutamate, cysteine, and glycine, and xCT can keep intracellular stores of cysteine for glutathione synthesis.209,210 This NRF2-mediated depletion of intracellular glutamate stores either through export (xCT) or consumption (glutathione synthesis) makes highly metabolic LSCC cells dependent on extracellular glutamine import, which is transformed to glutamate by GLS1211 (Fig. 3b). This vulnerability of tumor cells can be targeted through the inhibition of the activity of GLS1. Another possible therapeutic target is targeting the PI3K-AKT pathway. PI3K-AKT signaling is a major proliferative signal that inactivates GSK3 by phosphorylation, which increases NRF2 in a KEAP1-independent way (Fig. 3a). The GSK3 pathway is also a key regulator of adaptive glutamine metabolism.208
Based on this preclinical evidence, therapeutic interventions targeting the KEAP/NRF2 signaling have been tested in clinical trials. Three treatment approaches were used to target this signaling, including glutaminase inhibition, glutamine antagonism, and PI3K-AKT signaling inhibition. However, the phase II KEAPSAKE study which evaluated telaglenastat, a glutaminase inhibitor, in advanced non-squamous NSCLC with KEAP1/NRF2 mutations was terminated due to lack of clinical benefits among patients treated with telaglenasta in the interim analysis.212 For PI3K-AKT signaling inhibition, an mTOR kinase inhibitor sapanisertib was evaluated in a phase II clinical trial for advanced or recurrent LSCC with KEAP1/NRF2 mutations (NCT02417701). The preliminary results showed an objective response rate (ORR) of 25% (3/12) in the NFEL2 mutant squamous cohort and 16.7% (1/6) in the KEAP1 mutant squamous cohort.213 The relatively low response rate in these two cohorts may be attributed to the circumvention of mTOR inhibition by upregulation of glutamine metabolism through GSK3 signaling axis in LSCC cells (Fig. 3b).208 Glutamine blockade seems to be a promising strategy, which has been shown to induce divergent metabolic programs between cancer cells and effector T cells, acting as a “metabolic checkpoint” for tumor immunotherapy.214 Currently, sirpiglenastat, a broad glutamine antagonist, is being evaluated in combination with atezolizumab for patients with advanced solid tumors in phase I/IIa clinical trial (NCT04471415).215
Therapeutic targets on chromosome 3Q in LSCC
SOX2 and TP63 are both regarded as key squamous differentiation markers located on chromosome 3q.38,39 The suppression of SOX2 gene in LSCC cell lines had the highest anti-proliferative effects among the immediate genes on chromosome 3q26.33, including PIK3CA, TP63, DCUND1.40 It was also found that LSCC cell lines amplified for SOX2 and TP63 were highly dependent on them, supporting oncogene addiction.38
SOX2 belongs to the sex-determining region Y (SRY)-related high-mobility-group (HMG) box family characterized by a DNA-binding HMG domain, which shares at least 46% of sequence homology across all sox proteins.216 As a transcription factor, SOX2 recognizes and binds to the promoter of various target genes with complex, important, and pleiotropic impacts in multiple tissues in development and maintaining homeostasis.217,218 Dysregulation of SOX2 expression is an important factor contributing to cancer pathogenesis,219,220 associated with several tumor features, including cancer stemness,221,222,223,224 cell proliferation and migration,225,226,227 apoptosis,228,229,230 and drug resistance.231,232,233,234
TP63 encodes p63, a transcription factor that belongs to the tumor-suppressor p53 superfamily of transcription factors, including two additional paralogs, p53 and p73.235,236 The three members of the p53 family share very significant homology both at the genomic and at the protein level. Each contains a transactivation domain (TAD), a DNA-binding domain (DBD) and an oligomerization domain (OD).237 A common feature of all p53 family members is that they can be expressed in many different isoforms.236,238 For p63, three different splice variants are characterized by their differences in their C-termini: a full-length α form; a β form that is truncated after exon XII; and a γ form that lacks exons XII-XIV and uses an additional exon XV.238 Each of these isoforms can be further divided into TA forms and ∆N forms, based on the presence of the TAD or not, which is decided by whether transcription of the precursor mRNA starts from exon I (TA forms) or from exon III′ (∆N forms). Physiologically, p63 plays a critical role in sustaining epithelial development and morphogenesis in the regulation of epithelial proliferation.239,240 However, in the control of tumorigenesis, TA-isoforms and ∆N-isoforms shared antagonistic properties. TAp63 functions as tumor suppressors to halt tumorigenesis241 while ∆Np63 is more associated with promoting tumorigenesis.242,243
SOX2
Given that SOX2 is amplified in various types of cancer and involved in tumorigenesis via complicated signaling pathways and protein–protein interactions, targeting SOX2 is a promising strategy for anticancer therapy.40 Previously, as a transcription factor, SOX2 was deemed undruggable because of its absence of active sites or allosteric regulatory pockets to be targeted by small molecule inhibitors (SMIs).244 Therefore, studies targeting SOX2 in anticancer therapy has been focusing on the upstream and downstream signaling of SOX2. Recently, Liu et al.245 reported the development of a platform using the technique of proteolysis-targeting chimeras (PROTACs), which is able to selectively degrade the transcription factors of interest. This generalizable platform may help target SOX2 as an effective anticancer therapy.
∆Np63
The main isoform of p63 expressed in adult squamous tissues is ∆Np63.246 For squamous cell carcinomas (SCCs), ∆Np63 acts as a proto-oncogenic transcription factor and the master regulator of SCC formation.247,248,249,250 The oncogenic potential of ∆Np63 is related to its direct competition with p53, TAp63, and TAp73 on the same p53 responsive elements and the consequent inhibition.251,252 High levels of endogenous ΔNp63 protein abundance are essential to induce and maintain SCC tumors.247,253 Acute gene ablation of ΔNp63 in an autochthonous SCC model could induce rapid tumor regression.253 Besides, ΔNp63 is also found to regulate chemoresistance in SCCs by controlling the expression of DNA repair genes.254,255 Collectively, these findings implicate that ΔNp63 is a promising therapeutic target in LSCC. As a transcription factor, ΔNp63 was considered undruggable, as with most transcription factors which lack suitable domains for the binding of SMIs.244 The development of a generalizable platform by Liu et al.245 based on the technique of PROTACs, which is able to selectively degrade the transcription factors of interest, may provide new strategies to target ∆Np63. However, it is of note that ∆Np63 is associated with the regulation of a massive subset of different genes and cellular processes, which makes complete blocking of ∆Np63 almost impossible.
USP28
Ubiquitin-specific peptidase 28 (USP28) belongs to the largest deubiquitinating enzyme family, which removes ubiquitin from the ubiquitin conjugates.256 ΔNp63 is tightly regulated at the protein level by the ubiquitin-proteasome system, which can be targeted by multiple E3 ligases.257 USP28 is highly abundant in SCCs and correlates with poor prognosis.258 In SCCs, USP28 could stabilize ΔNp63 and maintain elevated ΔNp63 levels by counteracting its proteasome-mediated degradation.258 The researchers further confirmed that the pharmacologic inhibition of USP28 showed a selective anti-proliferative response of SCC cells.258 In addition to its tumor-suppressive function, inhibition of USP28 in ΔNp63 expressing SCC could sensitize SCC cells to cisplatin treatment by toning down the DNA damage response pathways.259 Taken together, these data show that USP28-ΔNp63 axis is required in the maintenance of SCC identity and control of SCC marker gene.
USP28 stabilizes ΔNp63 independently of FBXW7,258 which is a component of SCF (complex of SKP1, CUL1, and F-box protein)-type ubiquitin ligases.260 FBXW7 is a tumor suppressor that binds to key regulators of cell division and growth, including cyclin E, MYC, JUN, and Notch, most of which are proto-oncogenes that are closely related to the pathogenesis of human cancers.261 Recurrent mutations in the FBXW7 tumor-suppressor gene have been reported in LSCC.20,262 FBXW7 and USP28 are closely related in that USP28 could lead to FBXW7 substrate accumulation (either via destabilization of FBXW7 or via stabilization of both FBXW7 and its substrates).263,264 Therefore, targeting USP28 to destabilize the substrates of FBXW7 represents a promising strategy to inhibit the function of MYC and other oncogenic regulators.
Inhibition of USP28 is particularly effective in mouse LSCC models, resulting in dramatic tumor regression.258,265 The USP28 inhibitor used by Prieto-Garcia et al.258 was AZ1, a dual USP25/USP28 inhibitor, while the USP28 inhibitor FT206 used by Ruiz et al.,265 preferentially inhibits USP28 compared to USP25. Despite evidence that USP25 is an oncoprotein,266 its oncogenic function in LSCC is still enigmatic. There is currently no specific inhibitor of USP28 mainly due to the highly similar catalytic structure of USP25 and USP28. In the future, with the help of novel drug development technologies, USP28 inhibitors may become a promising therapeutic option for LSCC, but further clinical trials are still needed.
Survivin
Survivin (also known as BIRC5) has been a well-known cancer therapeutic target since its discovery over 20 years ago.267 Because of its essential role in cell mitosis and inhibition of apoptosis,268,269,270 as well as its variable expression levels in cancer and normal cells,271 survivin appears be a ideal candidate for anticancer therapy. However, no survivin-specifc drugs have yet reached the clinic. SMIs and inhibitory peptides targeting survivin for NSCLC have been explored in clinical trials but have shown modest or no improvement.272,273,274
Recently, Satpathy et al.38 identified ΔNp63-low LSCC which showed no elevation at RNA or protein levels. Accordingly, they also discovered a substantial number of LSCC cell lines with low ΔNp63 expression which were significantly more vulnerable to the survivin inhibitor YM-155. These findings may provide new strategies for selecting LSCC patients based on the TP63 status, which may have better response to survivin inhibition.
TNIK
TRAF2-/NCK-interacting kinase (TNIK) is a member of germinal center kinase (GCK) family, which was found previously involved in the promotion of colorectal cancer, triple-negative breast cancer (TNBC), prostate cancer, and chronic myelogenous leukemia.275,276,277,278,279 Furthermore, this protein kinase was identified as a potential genetic dependency in tumors with distal amplification of the 3q chromosome.280 TNIK gene is amplified in ~50% of LSCC cases.63 In LSCC cell lines with high TNIK expression, depleting TNIK can significantly reduce their growth.281 In addition, TNIK inhibition also showed antitumor activity and increased apoptosis in established LSCC patient-derived xenografts. These findings suggest TNIK as a potential therapeutic target for LSCC patients with TNIK gene copy-number gains.
Epigenetic therapeutic targets in LSCC
Epigenetics is defined as the DNA sequence-independent inheritance of phenotype or gene expression.282 There are four major mechanisms of epigenetic regulation: DNA methylation, histone post-translational modifications, chromatin structure regulation, and noncoding RNA regulation.282 In cancer cells, the epigenetic features are commonly dysregulated. High rate of alterations in many epigenetic regulator genes was observed in cancer genome-sequencing studies.283 This plethora of genetic lesions in epigenetic regulators has attracted much attention as possible targets for the development of epidrugs. Most epidrugs that has been approved by FDA are for the treatment of hematopoietic malignancies.284 Tazemetostat, a EZH2 inhibitor, was approved by FDA for advanced epithelioid sarcoma, making it the first epidrug to treat solid tumors.285 In this chapter, we will discuss some potential epigenetic therapeutic targets in LSCC.
NSD3: the neighboring gene of FGFR1
A recent proteogenomic portrait of LSCC suggested that WHSC1L1 (NSD3), but not FGFR1, may be the critical driver oncogene within the recurrent focal amplicon (8p11.23).38 NSD1, NSD2, NSD3, and ASH1L are four related enzymes in mammals which can synthesize the euchromatin-associated H3K36me2 modification.286 NSD3 dimethylates (adds two methyl groups to) the 36th amino acid residue in histone H3 (a lysine residue dubbed H3K36).287 This process, in which various chemical groups are covalently added to, or removed from, the DNA bases and the tails of the histones is referred to as epigenetic modifications.288 Amplification of NSD3 and its immediate neighbors (e.g., FGFR1), located on the chromosomal region 8p11-12, is one of the frequent molecular alterations in LSCC289 and has been implicated in the etiology of LSCC.290,291 In contrast to FGFR1, gene amplification of NSD3 correlates strongly with increased mRNA expression.291 Accordingly, a recent study has also shown that depletion of NSD3 in the 8p11-12 amplified LSCC cell lines and mouse model significantly attenuated tumor growth.44 This study also confirmed the ability of NSD3 to cooperate with SOX2 to transform human tracheobronchial epithelial (AALE) cells which further verified that NSD3 could promote human LSCC tumorgenesis40 (Fig. 4a).

Schematic diagram of the different roles for epigenetic therapeutic targets in LSCC. a A recent study suggested that NSD3, the neighboring gene of FGFR1, rather than FGFR1, was the critical driver oncogene within this recurrent focal amplicon of 8p11-12 genomic region. The amplification of NSD3 leads to increased NSD3 expression, thus increasing the synthesis of H3K36me2. Less common than the amplification of 8p11-12 and NSD3 expression, the GOF variant NSD3 was also present in LSCC. These two works together to increase H3K36me2, stimulating transcription of oncogenic targets, including mTOR pathways and MYC-associated pathways. This process rendered the tumor NSD3-addicted, which could be inhibited by BETi. b SOX2 and BCL11A are both identified as LSCC oncogenes. The BCL11A-SOX2 transcriptional program is crucial for the maintenance of a squamous phenotype. SETD8 is a monomethyltransferase, whose gene is regulated by SOX2 and BCL11A. The inhibition of SETD8 selectively limits LSCC tumor growth. c LSD1 could promote tumorigenesis in two different ways. The first way is demethylase-dependent. In SOX2-expressing tumor cells, LSD1 inhibition will induce increased H3K9me1/me2. The repressive H3K9 methylations act on the SOX2 gene, leading to SOX2 downregulation, reduced oncogenic potential, and increased cellular differentiation. The second way is demethylase-independent. In cells with a low level of LSD1, FBXW7 forms a dimer, which promotes ubiquitylation for proteasomal degradation of oncoprotein substrates, thus suppressing cell outgrowth. In cancer cells with overexpressed LSD1, the FBXW7 dimerization is blocked by LSD1 binding to FBXW7 in a demethylase-independent manner. FBXW7 self-ubiquitylation will then be triggered, followed by degradation by proteasome as well as lysosome in a p62-dependent pathway. d EZH2 is an enzymatic subunit of PRC2, which also includes EED, SUZ12, and RBBP4/7. The SET domain of EZH2 is responsible for the catalyzes the mono-, di-, and trimethylation of H3K27 from the universal methyl donor SAM, after which SAM becomes SAH. EZH2 also has noncanonical functions with its hidden TAD. The EZH2 TAD directly interacts with cMyc and other activators, including p300 and SWI/SNF. GOF gain-of-function, PRC2 polycomb repressive complex 2, SAH S-adenosyl-l-homocysteine, SAM S-adenosyl-l-methionine, TAD transactivation domain
Given that currently there is no catalytic inhibitors of NSD3 available in physiological settings, the researchers in that study also found that the four bromodomain inhibitors (BETi) exhibited the highest differential lethality over cells with mutated NSD3.44 This clinical actionable vulnerability, accompanied with findings of Li et al.292 who used cryo-electron microscopy to solve the structures of normal and oncogenic mutant forms of NSD3 bound to a nucleosome, will certainly provide valuable information for the design and development of drugs for treating LSCC as well as other NSD-driven diseases.
SETD8
SETD8 (also known as PR-Set7, SET8, and KMT5A) is currently the only known H4K20me1 monomethyltransferase, which is implicated in the regulation of multiple biological activities, including DNA replication, DNA damage repair, cell-cycle progression, and transcription regulation.293,294 During mitosis, SETD8 is concentrated in the nucleus during G1 and G2 phases and is degraded through ubiquitination at G1/S transition.295 Besides H4K20, SETD8 can also regulate the tumor-suppressor protein p53 and proliferating cell nuclear antigen (PCNA), which are closely related to carcinogenesis.296,297,298 SETD8 is implicated in cancer proliferation, migration, invasiveness, and oncogenesis, associated with a poor outcome.299,300
In the study by Lazarus et al.,45 BCL11A, which encodes a transcriptional regulator, was identified and characterized as a LSCC oncogene. Along with SOX2, which was also regarded as an oncogene in LSCC,40 this BCL11A-SOX2 transcriptional program provides a potential therapeutic window for LSCC. To disrupt this BCL11A-SOX2 transcriptional program, a Gene Ontology (GO) was performed and the SETD8 gene is selected, which is regulated by both BCL11A and SOX2.45 Knockdown of SETD gene could selectively inhibit LSCC tumor growth, but not LUAD cell. Besides, SETD8 inhibition also sensitizes LSCC cell lines to chemotherapy. Collectively, this study highlights the BCL11A-SOX2 transcriptional program as a novel target for LSCC and suggests the monomethyltransferase SETD8 as a potential downstream target45 (Fig. 4b).
LSD1
Lysine-specific demethylase 1 (LSD1, also known as KDM1A, KIAA0601, BHC110, and AOF2) is one of the SOX2-related targets that has been extensively studied. LSD1 is the first identified histone demethylase, which has the dual substrate specificity to catalyze the demethylation of histone 3 lysine 4 (H3K4me1/2) and H3K9me1/2 for transcriptional repression.301,302,303 The expression of LSD1 histone demethylase was reported to be significantly elevated in SOX2-expressing LSCC.46 LSCC cell lines with amplified SOX2 gene are particularly sensitive to LSD1 inactivation, whereas SOX2-negative cells are not. The regulation of SOX2 gene by LSD1 is directly through the bivalent H3K4 and H3K9 methylations. As a key regulator of SOX2, which is a lineage-survival oncogene of LSCC,40 LSD1 can serve as a specific and selective target for the treatment of LSCC.
In addition to its demethylase activity, the demethylase-independent activity of LSD1 has also been implicated in carcinogenesis.47,304,305 LSD1 can act as a pseudosubstrate of FBXW7. FBXW7 is a typical tumor suppressor that targets many oncoproteins for ubiquitylation and degradation.261 FBXW7 dimerization is disrupted by the binding of FBXW7 and LSD1 which promotes FBXW7 self-ubiquitylation and degradation through proteasome and lysosomal pathways, independent of the demethylase activity of LSD1, thus leading to accelerated growth47 (Fig. 4c). The discovery of this demethylase-independent activity of LSD1 implicates that the efforts to develop LSD1 inhibitors should be extended to directly target LSD1 rather than just inhibit its demethylase activity, which should harbor broader utility in anticancer therapy.
Currently, many LSD1 inhibitors are tested in phase I/II clinical trials,306 although most inhibitors were based on blocking its demethylase activity. However, the ineffectiveness of catalytic inhibition of LSD1 has been noticed in certain cancers.307,308 Therefore, targeting LSD1-involved protein interactions with the emerging technologies of PROTACs,309 not confined to the inhibition of its demethylate activity, may be a novel anticancer therapy in cancers with LSD1 overexpression like LSCC.
EZH2
Enhancer of zeste homolog 2 (EZH2) is a histone methyltransferase associated with transcriptional repression.310 As an enzymatic subunit of the polycomb repressive complex 2 (PRC2), EZH2 catalyzes the addition of methyl group to histone 3 lysine 27 (H3K27), which serves as an anchor point for the recruitment of additional polycomb group proteins, contributing to formation of a repressive chromatin state.311,312 Overexpression of EZH2 is observed in numerous tumor entities313,314,315,316,317,318 and associated with cancer progression and poor outcomes in patients.319,320 In LSCC, EZH2 dependency was observed in SOX2 amplified LSCC cell lines.38 Accordingly, the inhibition of EZH2 in TNBC could lead to the downregulation of SOX2 expression,48 suggesting that the inhibition of EZH2 may be an alternative strategy to depress the expression of SOX2 in LSCC.
EZH2 is usually found in actively proliferating tissues, whereas its homolog EZH1 is present in both dividing and differentiated cells.321 Compared to PRC2-EZH2, PRC2-EZH1 demonstrates a lower histone methyltransferase activity, and its knockdown alone does not lead to global reduction of H3K27 methylation.322 This suggests that EZH2 plays a predominant role in H3K27 methyltransferase in malignant cells and provides a rationale for the development of EZH2-specific inhibitors. On the other hand, the residual H3K27 after EZH2 inhibition also suggests the rationale for dual EZH1 and EZH2 inhibition. Apart from its catalytic function, EZH2 could also modulate gene expression during carcinogenesis in a PRC2-independent way.323 This PRC2-independent functions is associated with other non-PRC2 partners, the interactions with which are often methyltransferase-independent.324,325,326,327 For example, EZH2 could exert its oncogenic function as a transcriptional coactivator of androgen receptor in cells of castration-resistant prostate cancer.324 In acute leukemia, the noncanonical functions of EZH2 were fulfilled by binding cMyc at non-PRC2 targets and using a hidden transactivation domain (TAD) for activator recruitment and gene activation49 (Fig. 4d). The discovery of the noncanonical function of EZH2 provides rationale for the development of EZH2 SMIs, without necessarily inhibiting its catalytic function.
Currently, there is only one EZH2 inhibitor tazemetostat approved by FDA for advanced epithelioid sarcoma and follicular lymphoma328,329 while other EZH2 inhibitors are still being tested in phase I/II clinical trials. Most EZH2 inhibitors developed are catalytic inhibitors of EZH2 which targets its methyltransferase function. To suppress the multifaceted activities of EZH2, including both the canonical and noncanonical activities, a PROTACs technique-based degrader, MS177, has been shown to be fast-acting and more potent in suppressing tumor growth.49 This surely represents a promising therapeutic strategy for the development of EZH2 inhibitors.
Targeting the immune checkpoint in LSCC
Cancer immunotherapy has emerged as a powerful tool in the armamentarium against cancer, especially for LSCC which is refractory to currently available chemotherapy and targeted therapies. The field of oncology has been revolutionized by the emergence of cancer immunotherapy with significantly prolonged survival of patients in several fatal cancer types. Immunotherapy is increasingly being used as first-line treatment for many cancer indications. The idea of cancer immunotherapy against cancer is to deploy the immune system as a tool to treat neoplastic diseases. The first well-documented attempt to tackle cancer via the immune system dates back to 1890s, when Dr. William Coley, known as the “Father of Cancer Immunotherapy”, injected streptococcal organisms into a patient with inoperable cancer.330 We now understand that this effect is achieved by nonspecific immune stimulation, an approach that, while working well, received a lot of criticism at that time. Compared with traditional chemotherapy and targeted therapy, immunotherapy has potential efficacy across the boundaries of histology and driver mutational status and can lead to sustained remissions for those patients who achieve a response with fewer side effects.24,25,331 In this part, we will mainly discuss ICB therapies in LSCC. The current application of cancer immunotherapy in LSCC was summarized in Table 1. We will also present new insights into current immunotherapeutic targets as well as new targets for ICB therapies.
ICB therapy
Immune-checkpoint blockade is one of the most promising approaches to activating antitumor immunity. The immune-checkpoint pathways are involved in the major mechanisms underlying tumor immune evasion. Physiologically, these immunosuppressive signaling pathways play important roles in maintaining self-tolerance to prevent autoimmunity, limit immune-mediated tissue damage, and control the resolution of inflammation.332,333 Cancer cells may take advantage of these immune checkpoints to disguise themselves from body immune system.334,335 Among these immune checkpoints, cytotoxic T-lymphocyte antigen 4 (CTLA-4) and PD-1/PD-L1 axis are the most potent examples of T-cell immune-checkpoint molecules. The ICB therapies which were approved by FDA or National Medical Products Administration (NMPA) for LSCC are summarized in Table 1.
CTLA-4: the first clinically targeted immune-checkpoint receptor
CTLA-4 is a homolog of CD28 and binds both B7-1 (also known as CD80) and B7-2 (also known as CD86) with much higher affinity than CD28.336,337,338,339 The CTLA-4 and CD28 genes are located in the same region of chromosome 2 (2q33.2) and are expressed by both CD4+ and CD8+ T cells with opposing functions in T-cell activation.337,339 Through interacting with a pair of ligands (B7-1 and B7-2) expressed on antigen-presenting cells (APCs), including macrophages, dendritic cells (DCs) and B cells, CD28 mediates T-cell activation by co-stimulating T-cell receptor (TCR) signaling while the interaction of the ligands with CTLA-4 serves to inhibit T-cell response.340 These regulatory effects of CTLA-4 mainly restrict the expansion of CD4+ helper T cells while boosting regulatory T cells (Tregs),334,341 thus leading to a pro-tumor immunosuppressive phenotype.342
The recognition of CTLA-4 as a negative regulator of T-cell activation makes antagonizing CTLA-4 a reasonable method to enhance the antitumor immunity of T cells.343 Initial preclinical studies found that CTLA-4 blockade enhanced antitumor immunity and caused regression of immunogenic tumors without inducing substantial autoimmunity.344,345 Based on these preclinical findings, several clinical trials have been conducted to evaluate the therapeutic efficacy of CTLA-4 antibodies in tumors,346,347,348,349 which finally led to the FDA approval of ipilimumab by FDA for the treatment of advanced melanoma. However, the impressive effects of ipilimumab in melanoma patients did not proceed in renal cell carcinoma,350 NSCLC,351 small-cell lung cancer352 and prostate cancer.353 Another CTLA-4-blocking antibody, tremelimumab, has not received FDA approval since it did not improve survival compared to chemotherapy in metastatic melanoma.354 As the first immune-checkpoint inhibitor, ipilimumab is also currently the only CTLA-4-blocking antibody that has gained approval for anticancer treatment. No CTLA-4 inhibitors have been approved as monotherapy or in combination therapy with chemotherapy for the treatment of NSCLC (Table 1).
The fact that anti-CTLA-4 antibodies are capable to induce long-term immunity in cancer patients demonstrates that CTLA-4 remains an important immunotherapy target.355,356 Nevertheless, CTLA-4-targeting inhibitors have not reached its full potential, as evidenced by high rates of immunotherapy-related adverse effects (irAEs) and relatively low response rates. The strong irAEs of ipilimumab limit the doses tolerated by cancer patients. Both anti-PD-1/PD-L1 antibodies and anti-CTLA-4 antibodies have irAEs, while the effects of anti-CTLA-4 therapy are generally more severe.357,358,359,360 The dose-limiting toxicity of ipilimumab presented an opportunity of developing the next-generation molecules with wider therapeutic window.361,362,363 Recently, additional mechanisms were raised to explain the immunotherapeutic effects of anti-CTLA-4 mAbs, including depletion of regulatory T cells (Tregs) in TME.341,364,365,366,367 According to Du et al.,368 ipilimumab remains full activity without blocking B7-CTLA-4 interaction. In their studies, the humanized antibodies they developed without blockade of the B7-CTLA-4 interaction were as effective as ipilimumab at causing rejection of cancer. To further confirm that this tumor rejection was induced by Tregs depletion through antibody-dependent cellular cytotoxicity (ADCC), concurrent administration of anti-FcR antibodies treatment completely abolished the anticancer effect of ipilimumab. Collectively, these findings suggest that the selective Treg depletion in the tumors may be the primary mechanism of antitumor effect of anti-CTLA-4 antibody rather than the blockade of B7-CTLA-4 interactions369 (Fig. 5).

Roles of Fcγ receptors in anti-CTLA-4 function. Selective deletion of Tregs in the tumor microenvironment results in tumor immunity (left). Expressing higher levels of CTLA-4 than effector T cells, intratumoral Tregs are selectively depleted through ADCP by macrophages and/or ADCC by NK cells. In T-effector cells, T-cell activity is enhanced by the recognition of MHC-Ag by the TCR in the presence of an anti-CTLA-4 antibody that had co-engaged with FcγR on APCs (right). ADCC antibody-dependent cellular cytotoxicity, ADCP antibody-dependent cellular phagocytosis, APC antigen-presenting cells, MHC-Ag major histocompatibility complex-antigen peptide complexes major histocompatibility complex-antigen peptide complexes, NK cells natural killer cells, TCR T-cell receptor
Many new types of anti-CTLA-4 antibodies have been developed to increase antitumor effect, reduce side effects, or both. Increasing the ability of Fc to bind to FcR is one of the strategies to enhance the antitumor effect which can be achieved through a non-fucosylated derivative of ipilimumab (BMS986218) or an engineered Fc variant of an anti-CTLA-4 antibody (AGEN-1181or its mouse surrogate).370 The next-gen anti-CTLA-4 mAb, ONC-392, which effectively and selectively eliminates Tregs, has been granted Fast Track designation granted by FDA for monotherapy in PD-(L)1-resistant NSCLC.371 Different from other anti-CTLA-4 mAbs being tested, the pH-sensitivity nature of ONC-392 avoids antibody-triggered lysosomal degradation of CTLA-4, thereby reducing toxicity and exerting its anticancer potential.363 ONC-392 is currently being evaluated in Phase I clinical trial (PRESERVE-001; NCT04140526) for advanced solid tumors and NSCLC. An additional approach to moderate the adverse event profile of anti-CTLA-4 is to limit the CTLA-4 blockade within the tumor. For example, a “proform” of ipilimumab (BMS-986249) was synthesized, which was designed to remain inert in the periphery, but have activity restored when unmasked by tumor-associated proteases.370 Another approach is to generate a pH-selective form of ipilimumab, which could preferentially and reversibly target the acidic TME over the neutral periphery.362
PD-1 axis
PD-1 axis was the second immune-checkpoint pathway targeted for ICB therapy. In 2014, fully humanized anti-PD-1 mAbs pembrolizumab and nivolumab became the first PD-1 targeted therapeutics approved by FDA for refractory and advanced melanoma.357,372,373,374 Although anti-PD-(L)1 therapy entered the market later than anti-CTLA-4 therapy, PD (L)-1 blockade have shown broader clinical utility than anti-CTLA-4 treatment. For LSCC, a number of anti-PD-(L)1 therapeutics have been approved by FDA and NMPA as monotherapy or in combination therapy with chemotherapy (Table 1).
PD-1 was first identified as a putative mediator of apoptosis in 1992,375 and its role in maintaining peripheral tolerance by serving as a negative regulator of immune responses was elucidated in 1999 when Nishimura et al.376 found that PD-1-deficient mice developed a late onset of lupus-like autoimmune disease. Nearly at the same time, Dong et al.377 revealed a new member of the B7 family which might be involved in the negative regulation of cell-mediated immune responses. In the next year, this new member of the B7 family was confirmed to be the ligand of PD-1 (PD-L1) and an inhibitor of T-cell activation.335 PD-L2, a second ligand with higher affinity for PD-1, was also identified.378,379 Subsequent work found out that PD-L2 could have both co-stimulatory and co-inhibitory functions depending on the receptor and context.380 After being implicated in the negative regulation of T cells, the PD-1 axis was regarded as an active target of developing anticancer therapies. Multiple preclinical studies have showed that the PD-1 axis in the tumor causes the resistance to immune-mediated cytolysis, while blocking PD-L1 or PD-1 with specific mAbs in tumors could reverse tumors’ inherent resistance to cytotoxicity by T cells.381,382,383,384 However, solely blocking PD-L2 did not demonstrate any antitumor effect.385 Following the success of preclinical studies, mAbs targeting the PD-1 axis were designed and showed remarkable efficacy in clinical trials. In a head-to-head comparison for PD-L1 expressing advanced NSCLC, monotherapy with the PD-1 inhibitor pembrolizumab showed significantly better OS and lower incidence of adverse events than chemotherapy.386,387 The PD-L1 inhibitor atezolizumab also resulted in significantly longer OS than platinum-based chemotherapy in NSCLC patients with high PD-L1 expression.27,388
As studies in immunotherapy increase, difference between the clinical effect of anti-PD-1 and anti-PD-L1 has been reported. Such disparities have drawn the attention of clinicians and a better understanding of this discrepancy may guide us for a better administration of these drugs. Currently there are no head-to-head comparisons of anti-PD-1 mAbs and anti-PD-L1 mAbs in clinical trials. In a systematic review and meta-analysis, Duan et al.389 adjusted indirect comparisons based on a well-designed mirror principle to minimize the potential bias and found out that anti-PD-1 mAbs appeared to exhibit significantly greater OS compared with anti-PD-L1 with a comparable safety profile in patients with solid tumors. The possible reason for the improved efficacy of anti-PD-1 mAbs compared with anti-PD-L1 mAbs may come from the mechanisms of PD-1 and PD-L1 blockade in anticancer therapy. Anti-PD-1 mAbs can bind to PD-1 and further block the interaction between PD-1 and its ligands (PD-L1 and PD-L2), while the PD-1/PD-L2 axis remains intact and exerts its immune suppressive functions when PD-L1 is blocked by anti-PD-L1 mAbs. Nevertheless, the blockade of PD-1 may shift the balance of the binding of PD-L2 with its other partner, repulsive guidance molecule b (RGMb), which can lead to pneumonitis.380 This is also confirmed by the fact that patients treated with PD-1 inhibitors have a higher incidence of pneumonitis than patients who received PD-L1 inhibitors.390,391
Although great success has been achieved in the treatment of LSCC with the advent of PD-1 axis inhibitors, the ORR of PD-1 axis inhibitor in the treatment of advanced NSCLC is ~30%.27,387 Therefore, it is of utmost importance for the establishment of effective biomarkers for predicting the efficacy of anti-PD-1 axis agents. The assessment of PD-L1 expression on tumor cells is a logical biomarker for the prediction of treatment response to anti-PD-1 or anti-PD-L1 therapies. A real-world study in China has found out that LSCC patients were associated with higher incidence rate of positive PD-L1 expression, suggesting a benefit of using ICIs in LSCC patients.392 Although PD-L1 immunohistochemistry (IHC) plays an important role in patient stratification in clinical trials of anti-PD-1 or anti-PD-L1 therapies, it has poor reliability as a biomarker for anti-PD-1 or anti-PD-L1 therapies, as patients with negative PD-L1 expression can still benefit from anti-PD-1 or anti-PD-L1 therapies.393,394,395 Beyond PD-L1 expression, several other biomarkers have also successively predicted the efficacy of ICB therapy to certain extent. Among them, tumor mutational burden (TMB), gene expression profiling (GEP), and multiplex immunohistochemistry/immunofluorescence (mIHC/IF) are mostly used.396 Due to the lack of accurate assessment of response, future improvements in diagnostic accuracy may be achieved through a multiple incorporation of existing markers and newly discovered markers.396,397,398,399,400
LAG3
Lymphocyte activating gene 3 (LAG3, also known as CD223), first discovered in 1990,401 is a transmembrane molecule that is expressed on CD4+ and CD8+ T cells, natural killer T (NKT) cells, natural killer (NK) cells, plasmacytoid dendritic cells (pDCs) and Tregs.402,403 The LAG3 gene is located on human chromosome 12 (12p13.31), adjacent to the coding region of CD4.404 The LAG3 protein and CD4 protein share approximately 20% similarities in their amino acid sequences, which is mostly pronounced on their extracellular regions.404,405 Due to this similarity in extracellular structures, like CD4, LAG3 can also bind to major histocompatibility complex class II (MHC-II) proteins, but with higher affinity, which is also the canonical ligand for LAG3.406 Once LAG3 binds to MHC-II proteins, the inhibitory signals are transmitted through its cytoplasmic domain, thereby downregulating T-cell function.407 Several other ligands were also found to interact with LAG3, including Galectin-3408 (Gal-3), liver sinusoidal endothelial cell lectin409,410 (LSECtin), and fibrinogen-like protein 1411 (FGL1).
The fact that LAG3-deficient T cells show enhanced homeostatic expansion suggests the inhibitory role of LAG3 in immune responses.412,413 LAG3 is co-expressed with other inhibitory receptors, such as PD-1, on CD8+ tumor antigen-specific T cells under chronic tumor antigen stimulation, which leads to T-cell exhaustion.403,414 LAG3 expression was also confirmed to play an important role in supporting Tregs activity.402 In intratumoral Tregs, LAG3 is expressed at a higher level than in Tregs found in peripheral or normal tissue.415,416 Multiple LAG3-modulating candidates have been developed, including LAG3-inhibiting antibody and LAG3 fusion protein.417 However, LAG3 monotherapy in several mouse models has shown limited antitumor effect with slightly reduced tumor growth, whereas LAG3/PD-1 co-blockade has shown much stronger synergistic antitumor effects.418,419,420,421,422 Several anti-LAG3 antibodies are currently being evaluated in clinical trials, in which relatlimab is the furthest along in clinical development among all the anti-LAG3 mAb. On March 18, 2022, the combination therapy of relatlimab and nivolumab was approved by FDA for the treatment of unresectable or metastatic melanoma, making LAG3 the third FDA-approved immune checkpoint that was approved by FDA after CTLA-4 and PD-1 axis.423,424 This approval of LAG3 mAb marks an exciting beginning for this inhibitory receptor but many aspects of its biological functions still remain enigmatic. New ligands of LAG3 are still emerging and the effects of LAG3 on immune cells remain to be fully characterized.417 For LSCC, several anti-LAG3 antibodies and bispecific antibodies are being evaluated in phase I and phase II clinical trials (Table 2).
Other targets for ICB
More negative regulators of T-cell activation have been discovered which are potential targets for ICB, including T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain (TIGIT), T-cell immunoglobulin 3 (TIM3), V-domain immunoglobulin suppressor of T-cell activation (VISTA).370
TIGIT is a member of the immunoglobulin superfamily and was first identified in 2009.425 Highly expressed on human and murine tumor-infiltrating T cells,426 dual PD-1/PD-L1 and TIGIT blockade is a promising combination immunotherapy for cancer. Co-targeting of TIGIT with PD-1 axis is supported by preclinical studies, which demonstrated a synergistic effect in augmenting proliferation and function of antitumor CD8+ T cells than that shown in each single blockade.426,427,428 Tiragolumab is the first anti-TIGIT mAb tested in a phase II study. In the phase II CITYSCAPE study,429 tiragolumab plus atezolizumab as a first-line treatment for PD-L1-positive NSCLC have shown significantly improved efficacy compared with atezolizumab alone. Despite the success in this phase II study, the phase III SKYSCRAPER-01 study, which evaluated tiragolumab plus atezolizumab for PD-L1-high metastatic NSCLC, did not meet its co-primary endpoint of PFS while the other co-primary endpoint of OS was immature.430 Despite this discouraging news, it is also possible that this combination of immunotherapy may have benefits in long-term efficacy indicators like OS, which has been confirmed in previous immunotherapy clinical trials.431 Before the results of OS came out, it might be too early to judge this combination therapy. Currently, three other combination therapies of anti-TIGIT agents and anti-PD-1 axis agents are being evaluated in phase III clinical trials (Table 2).
TIM3 was originally found to be expressed on differentiated Th1 cells, which has also been defined as a marker for terminally differentiated effector Th1 cells.432,433 There is a firm connection between elevated TIM3 expression and exhausted CD8+ T cell.434,435,436 This elevated expression level of TIM3 in exhausted T cells is also associated with PD-1 expression, suggesting a correlation between TIM3 and PD-1 in T-cells exhaustion.437,438,439 Most clinical trials of TIM3 inhibitors are assessing the efficacy of the combination of TIM3 inhibitors and anti-PD-1 axis mAbs (e.g., NCT03680508, NCT03099109).
Like TIM3, VISTA (also known as PD-1H, B7-H5) is also a promising target for combination immunotherapy.440 VISTA shares significant sequence homology with the B7 family ligands PD-L1 and PD-L2 and imposes quiescence on mammalian myeloid and naïve T cells.441,442 The interaction of VISTA and its ligand P-selectin glycoprotein ligand 1 (PSGL-1) is governed by pH, selectively at acidic pH such as that found in TME.443 Most antibodies that target VISTA are being evaluated in preclinical studies. Only a few anti-VISTA drugs are currently being assessed in phase I studies (e.g., NCT05082610, NCT04564417).
Activating receptors on T cells have also been extensively studied as targets for immunotherapy, including inducible co-stimulator (ICOS, also known as CD278), tumor necrosis factor receptor superfamily member 4 (TNFRSF4, also known as CD134, OX40), tumor necrosis factor receptor superfamily member 9 (TNFRSF9, also known as 4-1BB).444 However, the distinct nature of agonist antibodies targeting immune co-stimulatory receptors rendered them unique among other antibody therapies in cancer.445 Some next-generation approaches, such as recombinant ligands and bispecific antibodies, may help unlock the full therapeutic potential of such targets.
Multi-target combination therapeutic strategies
CTLA-4 and PD-1 axis
The distinct functions of CTLA-4 and PD-1 axis are reflected in the different toxicity seen in their respective knockout mouse models. Mice lacking the CTLA-4 gene developed lymphoproliferative diseases and died by 3–4 weeks of age,446,447 whereas mice lacking PD-1 had more limited and variable, model-dependent autoimmunity, including glomerulonephritis, arthritis and cardiomyopathy.376,448,449,450 Spatially, CTLA-4 regulation occurs primarily within lymphoid organs, whereas PD-1 limits T-cell activation locally within peripheral tissues.451,452 Temporally, PD-1 acts later during T-cell activation for long-term tolerance. The distinct functions of CTLA-4 and PD-1 axis provide a rationale for the combination therapy of CTLA-4 and PD-1 axis blockade. Combinations of anti-CTLA-4 and anti-PD-1, or anti-CTLA-4 and anti-PD-L1, have shown improved efficacy than either agent alone in clinical trials or preclinical models.358,453,454,455 In NSCLC, based on the results of checkmate 227 clinical trial,30 nivolumab plus ipilimumab has been approved by FDA for the first-line treatment of patients with tumors expressing PD-L1(≥1%), which was also the first chemotherapy-free regimen for NSCLC. Besides, nivolumab plus ipilimumab and two cycles of platinum-doublet chemotherapy is also FDA-approved for the first-line treatment of advanced NSCLC, regardless of tumor PD-L1 expression.31
Despite the success of nivolumab plus ipilimumab in the treatment of NSCLC, there were also negative results from clinical trials evaluating the combination ICB therapy. In the phase III MYSTIC study, durvalumab plus tremelimumab did not significantly improve OS or PFS compared with chemotherapy in metastatic NSCLC.456 For advanced, pretreated, immune-checkpoint inhibitor-naive LSCC, the addition of ipilimumab to nivolumab did not improve outcomes.457 These results demonstrate the need for a better mechanistic understanding of the crosstalk among anti-PD-1, anti-PD-L1, and anti-CTLA-4. The cis-PD-L1/CD80 interactions were found to have implications in the synergy of anti-PD-L1 and anti-CTLA-4 combination therapy.458,459 Recognized as the ligands of PD-1 and CD28/CTLA-4 respectively, PD-L1 and CD80 were also found to interact with each other.460,461 Recent studies reported that PD-L1 and CD80 could heterodimerize in cis when these molecules are overexpressed on the same cell.458,462 This PD-L1:CD80 cis-heterodimerization could inhibit both PD-L1:PD-1 and CD80:CTLA-4 interactions through distinct mechanisms while preserving the ability of CD80 to activate the T-cell co-stimulatory receptor CD28. Therefore, by disrupting PD-L1:CD80 heterodimers, anti-PD-L1 mAbs licenses high-avidity CD80:CTLA-4 interactions which triggers Treg-mediated depletion of CD80 from APCs and inhibits CD28 co-stimulation.458 Since this CD80 depletion by anti-PD-L1 is CTLA-4 dependent and can be reversed by CTLA-4 blockade,341,463 it provides a rationale for co-blocking PD-L1 and CTLA-4 in cancer immunotherapy. In another study, Tekguc et al.464 also found that the Treg-mediated depletion of CD80 from APCs via CTLA-4-dependent trogocytosis can also increase free PD-L1 available for the inhibition of PD-1 expressing effector T cells. Therefore, the combination of blocking CTLA-4 and PD-1 axis may synergistically hinder this Treg-mediated immunosuppression and enhance antitumor efficacy.
Some bispecific antibodies, capable of simultaneously binding PD-1/PD-L1 and CTLA-4 with high affinity, are also being evaluated in ongoing clinical trials (Table 2).465,466 KN046, a novel bispecific antibody that blocks PD-L1 interaction with PD-1 and CTLA-4 interaction with CD80/CD86, was well tolerated and effective in treating advanced NSCLC, with promising PFS and OS benefits in LSCC.465 Pivotal Phase III clinical trials in advanced unresectable or metastatic LSCC is currently ongoing for this bispecific antibody (NCT04474119).
ICB and PI3K pathway inhibition
The hyperactive PI3K signaling, whether it is the consequence of PI3KCA mutations or PTEN deletions, can promote the establishment of tumor suppression by developing tumors,467 suggesting the potential use of PI3K inhibitor to enhance the efficacy of immunotherapy in the clinic (Fig. 6a). In preclinical models of melanoma, loss of PTEN in tumor cells inhibits T-cell-mediated tumor killing and restricts T-cell trafficking into tumors.468 A number of immunosuppressive cytokines, including CCL12 and VEGF are elevated in melanoma patients harboring PTEN loss. In this study, the lipidation of autophagosome protein LC3 and autophagy in tumor cells, which can decrease T-cell priming and modulate resistance to T-cell-mediated apoptosis, is also inhibited due to the loss of PTEN protein and activation of PI3K.468 Consistent with these findings, previous studies have also found that inactivation of PI3Kδ could break Tregs-mediated tumor immune tolerance, resulting in the activation of CD8+ T-cell responses and subsequent tumor regression.469,470,471,472 The Tregs T-cell receptor (TCR) downstream signaling, proliferation, and survival are dominantly dependent on PI3Kδ, but not PI3Kα or PI3Kβ.470 This potential adjuvant role for PI3Kδ inhibition in cancer immunotherapy was confirmed in a neoadjuvant, phase II clinical trial treatment-naive patients with resectable HNSCC.117 The inhibition of PI3Kδ by AMG319 decreased the number of tumor-infiltrating Tregs and activated intratumoral CD4+ and CD8+ T cells. However, the unfavorable safety profile should also be noticed, with frequent and severe grade 3/4 irAEs, probably driven by the systemic effect on Tregs in non-malignant tissues.117 Besides, as PI3K signaling is also essential in maintaining effector T-cell function,473,474,475 a systemic inhibition of PI3Kδ impairs the function of CD8+ cytotoxic T lymphocytes, which antagonizes ICB therapy intending to boost the CD8+ T-cell response, counteracting any advantages brought by impairing intratumoral Tregs.476 The protocols of administrating PI3Kδ inhibitors were considered as an essential part. A modified treatment regimen with intermittent dosing of PI3Kδ inhibitors has shown a comparable antitumor efficacy while limiting toxicity.115,116,117 In addition, given that PI3Kδ signaling might be required for signaling reactivation in exhausted T cells by ICB therapy,477,478 sequential combination treatment might be more effective. The study of Isoyama et al.472 confirmed that the combination protocol with anti-PD-1 mAb administrated first, followed by anti-PD-1 mAb plus PI3Kδ inhibitor induced the most effective and durable antitumor activity.

Impact of oncogenic signaling on tumor immune response. a Loss of PTEN protein function and improper PI3K activation inhibit efficient LC3 lipidation, which further promote resistance to T-cell-mediated killing by inhibiting autophagy. PTEN loss could also induce expression of immunosuppressive cytokines, including CCL12 and VEGF. b CDK4/6 inhibition enhances T-cell activation through the derepression of NFAT family proteins and their target genes, which encodes critical regulators of T-cell function. CDK4/6 inhibition could also induce Rb-mediated G1-arrest and promote the phenotypic and functional acquisition of immunologic T-cell memory. Besides, the PD-L1 protein stability is regulated by the CDK4-SPOP-FZR1 signaling pathway. Physiologically, PD-L1 protein stability is negatively regulated through phosphorylating its upstream physiological E3 ligase SPOP. This phosphorylation promotes SPOP binding to 14-3-3γ, which subsequently disrupts FZR1-mediated destruction of SPOP. The inhibition of CDK4/6 inhibits the phosphorylation of SPOP, thus promoting its degradation by FZR1, thus increasing PD-L1 protein levels. c Tumor-derived VEGF limits NF-κB activation in immature DCs, which in turn leads to defective functional maturation of DCs and insufficient induction of tumor immunity. VEGF could also impact the endothelial cells expression of immunological molecules. It decreases the expression of VCAM-1, which is important for the antitumor T cells adhesion and infiltration into tumors. Besides, VEGF also increases the expression of FAS ligand on endothelial cells, triggering apoptosis of T cells. VEGF also promotes the expansion of immune suppressive MDSCs, which further promotes the recruitment of Tregs. d EZH2 inhibition increases the production of CXCL9 and CXCL10, which are attractant cytokines promoting trafficking of T cells to tumor. Besides, EZH2 inhibition could selectively target intratumeral Tregs and reduce its immunosuppressive capacity. e In tumor cells, the ablation of LSD1 in cancer cells increases repetitive element expression, including ERVs, and decreases expression of RISC components. This leads to dsRNA stress and activation of type 1 interferon, which stimulates antitumor T-cell infiltration. In addition, inhibiting LSD1 in CD8+ T cells unleashes the transcription program mediated by TCF1, which is critical for the maintenance of the progenitor subset of intratumoral CD8+ T cells for persistent tumor control. dsRNA double-stranded RNA, ERVs endogenous retroviral elements, MDSCs myeloid-derived suppressor cells, RISC RNA-induced silencing complex, TCF1 T-cell factor 1, VCAM-1 vascular cell adhesion molecule-1
ICB and CDK4/6 inhibition
CDK4/6 is regarded as a promising target in treating LSCC when appropriate candidate patients are identified by the downstream functional assessment.38 Recent studies have found that inhibition of CDK4/6 not only induces tumor cell-cycle arrest, but also increases T-cell inflammatory signature in tumors, which may act synergistically with ICB therapies479,480 (Fig. 6b). In mouse models, combining CDK4/6 inhibitors with PD-1 axis inhibitors resulted in significantly improved antitumor efficacy compared with either treatment alone.480,481,482,483 The mechanisms of this synergistic effects have been under extensive studies recently. Goel et al.111 found that the inhibition of CDK4/6 could increase the functional capacity of tumor cells to present antigens. Besides, CDK4/6 inhibitors could markedly and selectively reduce the immunosuppressive Tregs population.111 This preference of CDK4/6 inhibitor may be attributed to the higher expression level of RB by Tregs (3.1-fold higher in Tregs than in CD8+ T cells,484) a key modulator of CDK4/6 pathway.485 The inhibition of CDK4/6 in tumor cells also increases PD-L1 protein levels,481 which could be one of the mechanisms leading to the resistance of CDK4/6 inhibitor via evasion of immune checkpoints surveillance. This provides the rationale for the combination of PD-L1 blockade treatment and CDK4/6 inhibitors as a more potent antitumor treatment option. Properly timed and sequenced doses of CDK4/6 inhibitors could enhance T-cell activation483 and induce T-cell memory for maintaining long-term antitumor immunity.486,487 Despite the great efficacy shown in preclinical models, some clinical trials co-administering CDK4/6 inhibitors and ICB therapy were halted because of severe toxicity (e.g., NCT02779751,488,489 NCT04118036, NCT04075604). A different administrating method of CDK4/6 inhibitors has been proposed which may mitigate the toxicity risk.486,487 The capacity of CDK4/6 inhibitors to promote T-cell memory gives rationales for using CDK4/6 inhibitors as a preconditioning tool, priming the T-cell pool before the application of ICB. In mouse models, preconditioning tumor-bearing mice with a CDK4/6 inhibitor significantly improved the efficacy of anti-PD-1 ICB therapy.486 Taken together, these results suggest CDK4/6 inhibition in combination with immunotherapy is a promising therapeutic strategy but still needs further investigation.
ICB and VEGF-VEGFR signaling blockade
With over 30 years of extensive research on VEGF, the biological role of VEGF has extended beyond its impact on neovascularization and angiogenesis, which also functions as an immunomodulator (Fig. 6c). It has been shown that multiple immune cells could be influenced by VEGF, including DCs, T cells, Tregs, and MDSCs.490 Both in animal models and humans, the inhibition of VEGF could increase the number of tumor-infiltrating lymphocytes.491,492 VEGF is also important for the expansion of the immunosuppressive MDSCs via its binding to VEGFR1, which further promotes an immunosuppressive microenvironment by de novo development of Tregs.493 In renal cell carcinoma, the inhibition of VEGF-VEGFR signaling by either mAbs or VEGF-TKIs could reverse MDSC-mediated immunosuppression.494,495 This immunosuppressive profile of the VEGF/VEGFR axis is also confirmed by the fact that VEGF-A-VEGFR pathway blockade inhibits tumor-induced proliferation of Tregs.496,497 Aberrant angiogenesis within the TME can mediate immune escape and reduce the efficacy of immunotherapy by hampering the delivery of drugs, oxygen, and effector T cells.498,499 This tumor hypoxia promotes the recruitment of Tregs, which facilitates angiogenesis through excess production of VEGF.500 In hand with other tolerogenic leukocyte populations such as MDSCs501 and pDCs502 which also produce VEGF and support angiogenesis, this leads to further tumor tolerance and growth. During adaption to the hypoxic TME, tumor-infiltrating cytotoxic T cells are characterized by high glycolytic rates.503,504 A recent study has found that hypoxia-inducible transcription factor 1α (HIF1α) is essential for the effector state in CD8+ T cells.505 VEGF-deficient CD8+ T cells showed lower efficiency on infiltrating tumors. An HIF1α/VEGF axis has been proposed in cytotoxic T cells to regulate tumor progression.505 The above-mentioned findings provide further evidence for the ongoing clinical evaluation of combined immunotherapies and antiangiogenic approaches. Currently, the only approved combination therapy of immunotherapy and antiangiogenic approach for NSCLC is the PD-L1 inhibitor atezolizumab in combination with bevacizumab, paclitaxel, and carboplatin for the first-line treatment of patients with metastatic non-squamous NSCLC with no EGFR or ALK genomic tumor aberrations.506 AK112, a tetrameric bispecific antibody targeting PD-1 and VEGF, has shown in a phase I dose-escalation study with a manageable safety profile.507 It is currently being evaluated in a phase II clinical trial for stage IIIB/C or IV NSCLC (NCT04736823).
ICB and epigenetic therapies
Recent studies revealed the prominent role for modulating immune cells and regulating anticancer immune response by epigenetic therapy, which nominates these epidrugs a new category of immune modulators.508,509,510,511,512,513 These preclinical findings have led to the evaluation of various epidrugs and immunotherapy combinations in clinical trials for treatment of LSCC (Table 2). Some combinations of epidrugs and ICB therapy in early-stage clinical trials have demonstrated significant clinical activity and acceptable safety.514 In this part, we will discuss the combination of ICB therapy with inhibition of potentially effective epigenetic targets in LSCC.
An important epigenetic modulator in LSCC is the H3K27 methyltransferase EZH2, whose dependency was observed in SOX2 amplified LSCC cell lines.38 Like other epigenetic modulators, EZH2 also plays an important role in mediating the immune response to the tumor both in tumor cells and immune cells (Fig. 6d). Previous studies have found that EZH2 can repress the expression and subsequent production of Th1-type chemokines CXCL9 and CXCL10, thus preventing efficient T-cell trafficking to tumors, a process that can be reversed by EZH2 inhibition in combination with anti-PD-L1 checkpoint blockade.513,515 Through its epigenetic reprogramming of T-cell antigen-presenting gene, EZH2 has been shown to be a driver of resistance to cancer immunotherapy.516,517,518,519 Besides, EZH2 plays an important role in maintaining the identity of immunosuppressive Tregs during cellular activation, where it is recruited by Foxp3 to repress key genes.520,521 Disruption of EZH2 activity in Tregs can reprogram the tumor-infiltrating Tregs for pro-inflammatory activities, thereby enhancing the recruitment and function of CD8+ and CD4+ effector T cells to eliminate tumors.522 The dual functions of EZH2 in repressing antigen presentation and altering Tregs functions makes the EZH2 inhibition a rational strategy in combination with CTLA-4 inhibitors or PD-1 axis inhibitors, respectively. Indeed, the additional efficacy of both combination strategies was confirmed in preclinical mouse models.523,524,525 This synergistic effect of EZH2 blockade and ICB therapy is currently being validated in multiple clinical trials (e.g., NCT02220842,526 NCT03854474,527 NCT04407741).
As a direct regulator of SOX2, which is a lineage-survival oncogene of LSCC,40 inhibition of LSD1 is regarded as a promising treatment strategy for LSCC.38 Recently, studies have also found that this epigenetic regulator is important in regulating tumor immunity (Fig. 6e). In 2018, Sheng et al.510 found that the inhibition of this histone demethylase in cancer cells resulted in double-stranded RNA (dsRNA) stress and activation of type 1 interferon, thereby stimulating potent antitumor T-cell immunity. Furthermore, the inhibition of LSD1 could elicit significant response to anti-PD-1 therapy in ICB-refractory mouse melanoma.510 Consistently, TCGA data analysis reveals an inverse correlation between LSD1 expression and CD8+ T-cell infiltration in various human cancers.510 In the same year, Qin et al.528 also found that combining LSD1 inhibitors with anti-PD-1 mAbs significantly suppressed tumor growth and pulmonary metastasis in mice bearing TNBC xenograft tumors, whereas anti-PD-1 mAbs alone failed to elicit an obvious anticancer effect. Over the next few years, the enhancement of immunotherapy efficacy through inhibiting LSD1 was also demonstrated in several other tumors.529,530,531 The mechanisms of LSD1 ablation in tumor immunity have not been clearly elucidated. According to Liu et al.,532 the inhibition of LSD1 in T cells increases the persistence of the progenitor-exhausted CD8+ T cells through augmenting the transcriptional network controlled by T-cell factor 1 (TCF1), which is essential for maintaining the progenitor phenotype. These progenitor-exhausted CD8+ T cells are characterized by high proliferation capacity, which gives rise to more differentiated cells with strong cytotoxicity. These properties make progenitor-exhausted CD8+ T cells a major determinant of responses to PD-1 axis blockade.533,534,535,536 Based on this synergistic effect of LSD1 inhibitors and PD-1 axis inhibitors, there is already an ongoing clinical trial evaluating this combination therapy in SCLC and LSCC patients (NCT04350463). Another study has proposed the addition of blocking TGF-β in this combination therapy.537 The efficacy of this triple combination therapy has been validated in certain poorly immunogenic or “cold” tumors.537 However, the safety and efficacy of this combination strategy still need to be verified in future studies.
Conclusions and future perspectives
Accounting for ~30% of all NSCLC, LSCC remains a leading cause of death with few therapeutic options.3,4 While targeted therapies demonstrated significant benefits in LUAD patients, patients with LSCC have not benefited from targeted therapy due to the distinct nature of LSCC.20,538 The advent of immunotherapies has significantly improved the prognosis of patients with LSCC, and this burgeoning field of cancer immunotherapy continues to grow as new druggable targets are discovered. With comprehensive proteogenomic data, the established LSCC biology can be more deeply elucidated, potentially uncovering new potential implicated therapeutics targets.38
In this review, we discussed some new insights in some signaling pathways which have been proved in other cancer types, like PI3K pathway, CDK4/6 pathway and VEGF/VEGFR signaling. The two key squamous differentiation markers SOX2 and TP63 offer the chance of therapeutic targets in LSCC. SOX2 was considered undruggable before, thus intensifying therapeutic interests in upstream or downstream targets, including LSD1 and EZH2. Newly identified epigenetic targets, like NSD3, were also emerging as potential targets in treating LSCC. Having shown great benefits in LSCC, ICB therapies still faced the problems of a relatively low response rate and high rate of irAEs in some cases. We discussed some newly discovered mechanisms of these immune checkpoints which may be useful in tackling these problems. Combinations of different ICB therapies or ICB therapy and other targeted therapies have emerged as an appealing treatment paradigm. Whether it is the combination of ICB therapies plus epigenetic therapies, or ICB therapies plus VEGF-VEGFR inhibitor, it represents an innate inner connection among these different signaling pathways. With the persistent exploration of these complex biological interactions among different signaling pathways, it will surely provide exciting opportunities for new, improved and personalized therapeutic interventions in LSCC patients. Multi-omics clustering has identified five LSCC molecular subtypes38 and this heterogeneity of LSCC reveals the fact that combination therapies targeting more than one target or signaling pathway may yield more therapeutic choices. In the last decade, ICB therapies have made a major breakthrough in improving the prognosis of LSCC patients both in the first- and second-line settings. We believe immunotherapy will remain the pillar of LSCC treatment. Meanwhile, the clinical translation of other novel therapeutic targets is still in a great demand which may improve the efficacy of current ICB therapy-based regimens.
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Herbst, R. S., Morgensztern, D. & Boshoff, C. The biology and management of non-small cell lung cancer. Nature 553, 446–454 (2018).
Heist, R. S., Sequist, L. V. & Engelman, J. A. Genetic changes in squamous cell lung cancer: a review. J. Thorac. Oncol. 7, 924–933 (2012).
Society, A. C. Cancer facts & figures 2022. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2022/2022-cancer-facts-and-figures.pdf (2022).
Cheng, Y., Zhang, T. & Xu, Q. Therapeutic advances in non-small cell lung cancer: focus on clinical development of targeted therapy and immunotherapy. MedComm 2, 692–729 (2021).
Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Non-small Cell Lung Cancer Collaborative Group. Chemotherapy in non-small cell lung cancer: a meta-analysis using updated data on individual patients from 52 randomised clinical trials. BMJ 311, 899–909 (1995).
Paez, J. G. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 (2004).
Lynch, T. J. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139 (2004).
Paik, P. K. et al. A phase Ib open-label multicenter study of AZD4547 in patients with advanced squamous cell lung cancers. Clin. Cancer Res. 23, 5366–5373 (2017).
Lim, S. H. et al. Efficacy and safety of dovitinib in pretreated patients with advanced squamous non-small cell lung cancer with FGFR1 amplification: a single-arm, phase 2 study. Cancer 122, 3024–3031 (2016).
Nogova, L. et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose-escalation and dose-expansion study. J. Clin. Oncol. 35, 157–165 (2017).
Tabernero, J. et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 33, 3401–3408 (2015).
Aggarwal, C. et al. SWOG S1400D (NCT02965378), a phase II study of the fibroblast growth factor receptor inhibitor AZD4547 in previously treated patients with fibroblast growth factor pathway-activated stage IV squamous cell lung cancer (Lung-MAP Substudy). J. Thorac. Oncol. 14, 1847–1852 (2019).
Vansteenkiste, J. F. et al. Safety and efficacy of buparlisib (BKM120) in patients with PI3K pathway-activated non-small cell lung cancer: results from the phase II BASALT-1 study. J. Thorac. Oncol. 10, 1319–1327 (2015).
Langer, C. J. et al. SWOG S1400B (NCT02785913), a phase II study of GDC-0032 (taselisib) for previously treated PI3K-positive patients with stage IV squamous cell lung cancer (Lung-MAP Sub-Study). J. Thorac. Oncol. 14, 1839–1846 (2019).
Bendell, J. C. et al. A first-in-human phase 1 study of LY3023414, an oral PI3K/mTOR dual inhibitor, in patients with advanced cancer. Clin. Cancer Res. 24, 3253–3262 (2018).
Edelman, M. J. et al. SWOG S1400C (NCT02154490)—a phase II study of palbociclib for previously treated cell cycle gene alteration-positive patients with stage IV squamous cell lung cancer (Lung-MAP Substudy). J. Thorac. Oncol. 14, 1853–1859 (2019).
Campbell, J. D. et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 48, 607–616 (2016).
Sun, Y. et al. EGFR mutations subset in Chinese lung squamous cell carcinoma patients. Mol. Med. Rep. 17, 7575–7584 (2018).
Acker, F. et al. KRAS mutations in squamous cell carcinomas of the lung. Front. Oncol. 11, 788084 (2021).
Redman, M. W. et al. Biomarker-driven therapies for previously treated squamous non-small-cell lung cancer (Lung-MAP SWOG S1400): a biomarker-driven master protocol. Lancet Oncol. 21, 1589–1601 (2020).
Forde, P. M., Kelly, R. J. & Brahmer, J. R. New strategies in lung cancer: translating immunotherapy into clinical practice. Clin. Cancer Res. 20, 1067–1073 (2014).
Sharma, P. & Allison, J. P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214 (2015).
Reck, M. et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 375, 1823–1833 (2016).
Herbst, R. S. et al. Atezolizumab for first-line treatment of PD-L1-selected patients with NSCLC. N. Engl. J. Med. 383, 1328–1339 (2020).
Herbst, R. S. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550 (2016).
Paz-Ares, L. et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med. 379, 2040–2051 (2018).
Hellmann, M. D. et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N. Engl. J. Med. 381, 2020–2031 (2019).
Paz-Ares, L. et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 22, 198–211 (2021).
Sezer, A. et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet 397, 592–604 (2021).
Zhou, C. et al. Sintilimab plus platinum and gemcitabine as first-line treatment for advanced or metastatic squamous NSCLC: results from a randomized, double-blind, phase 3 trial (ORIENT-12). J. Thorac. Oncol. 16, 1501–1511 (2021).
Matasar, M. J. et al. Copanlisib plus rituximab versus placebo plus rituximab in patients with relapsed indolent non-Hodgkin lymphoma (CHRONOS-3): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 22, 678–689 (2021).
Ren, S. et al. Camrelizumab plus carboplatin and paclitaxel as first-line treatment for advanced squamous NSCLC (CameL-Sq): a phase 3 trial. J. Thorac. Oncol. 17, 544–557 (2022).
Zhou, C. et al. Sugemalimab versus placebo, in combination with platinum-based chemotherapy, as first-line treatment of metastatic non-small-cell lung cancer (GEMSTONE-302): interim and final analyses of a double-blind, randomised, phase 3 clinical trial. Lancet Oncol. 23, 220–233 (2022).
Kim, Y. et al. Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients. J. Clin. Oncol. 32, 121–128 (2014).
Satpathy, S. et al. A proteogenomic portrait of lung squamous cell carcinoma. Cell 184, 4348–4371.e4340 (2021).
Qian, J. & Massion, P. P. Role of chromosome 3q amplification in lung cancer. J. Thorac. Oncol. 3, 212–215 (2008).
Bass, A. J. et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 41, 1238–1242 (2009).
Ramos, A. H. et al. Amplification of chromosomal segment 4q12 in non-small cell lung cancer. Cancer Biol. Ther. 8, 2042–2050 (2009).
Hoadley, K. A. et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158, 929–944 (2014).
Dotto, G. P. & Rustgi, A. K. Squamous cell cancers: a unified perspective on biology and genetics. Cancer Cell 29, 622–637 (2016).
Yuan, G. et al. Elevated NSD3 histone methylation activity drives squamous cell lung cancer. Nature 590, 504–508 (2021).
Lazarus, K. A. et al. BCL11A interacts with SOX2 to control the expression of epigenetic regulators in lung squamous carcinoma. Nat. Commun. 9, 3327 (2018).
Zhang, X. et al. Pluripotent stem cell protein Sox2 confers sensitivity to LSD1 inhibition in cancer cells. Cell Rep. 5, 445–457 (2013).
Lan, H. et al. LSD1 destabilizes FBXW7 and abrogates FBXW7 functions independent of its demethylase activity. Proc. Natl Acad. Sci. USA 116, 12311–12320 (2019).
Yomtoubian, S. et al. Inhibition of EZH2 catalytic activity selectively targets a metastatic subpopulation in triple-negative breast cancer. Cell Rep. 30, 755–770.e756 (2020).
Wang, J. et al. EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat. Cell Biol. 24, 384–399 (2022).
Felip, E. et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non-small-cell lung cancer (IMpower010): a randomised, multicentre, open-label, phase 3 trial. Lancet 398, 1344–1357 (2021).
Forde, P. M. et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N. Engl. J. Med. 386, 1973–1985 (2022).
Arafeh, R. & Samuels, Y. PIK3CA in cancer: the past 30 years. Semin. Cancer Biol. 59, 36–49 (2019).
Murugan, A. K., Munirajan, A. K. & Tsuchida, N. Genetic deregulation of the PIK3CA oncogene in oral cancer. Cancer Lett. 338, 193–203 (2013).
Zhao, L. & Vogt, P. K. Class I PI3K in oncogenic cellular transformation. Oncogene 27, 5486–5496 (2008).
Fruman, D. A. & Rommel, C. PI3K and cancer: lessons, challenges and opportunities. Nat. Rev. Drug Discov. 13, 140–156 (2014).
Lemmon, M. A. & Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 (2010).
Sanchez-Vega, F. et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 173, 321–337.e310 (2018).
Burris, H. A. 3rd Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother. Pharmacol. 71, 829–842 (2013).
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M. & Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11, 329–341 (2010).
Okkenhaug, K. & Vanhaesebroeck, B. PI3K in lymphocyte development, differentiation and activation. Nat. Rev. Immunol. 3, 317–330 (2003).
Castel, P., Toska, E., Engelman, J. A. & Scaltriti, M. The present and future of PI3K inhibitors for cancer therapy. Nat. Cancer 2, 587–597 (2021).
AACR Project Genie Consortium, et al. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 7, 818–831 (2017).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disco. 2, 401–404 (2012).
COSMIC (Catalogue of Somatic Mutations in Cancer). https://cancer.sanger.ac.uk/cosmic (2022).
Samuels, Y. et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554 (2004).
Burke, J. E. et al. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA). Proc. Natl Acad. Sci. USA 109, 15259–15264 (2012).
Miled, N. et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 317, 239–242 (2007).
Li, J. et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 (1997).
Álvarez-Garcia, V., Tawil, Y., Wise, H. M. & Leslie, N. R. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin. Cancer Biol. 59, 66–79 (2019).
Worby, C. A. & Dixon, J. E. PTEN. Annu. Rev. Biochem. 83, 641–669 (2014).
Alimonti, A. et al. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 42, 454–458 (2010).
Carracedo, A., Alimonti, A. & Pandolfi, P. P. PTEN level in tumor suppression: how much is too little? Cancer Res. 71, 629–633 (2011).
Trotman, L. C. et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 1, E59 (2003).
COSMIC (Catalogue of Somatic Mutations in Cancer). https://cancer.sanger.ac.uk/cosmic (2022).
Mighell, T. L., Evans-Dutson, S. & O’Roak, B. J. A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. Am. J. Hum. Genet. 102, 943–955 (2018).
Han, S. Y. et al. Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res. 60, 3147–3151 (2000).
Rodríguez-Escudero, I. et al. A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum. Mol. Genet. 20, 4132–4142 (2011).
Song, M. S., Salmena, L. & Pandolfi, P. P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296 (2012).
Hollander, M. C., Blumenthal, G. M. & Dennis, P. A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 11, 289–301 (2011).
Lu, J. et al. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS ONE 4, e5577 (2009).
Escrivà, M. et al. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol. Cell. Biol. 28, 1528–1540 (2008).
Lee, J. Y. et al. Id-1 activates Akt-mediated Wnt signaling and p27(Kip1) phosphorylation through PTEN inhibition. Oncogene 28, 824–831 (2009).
Cai, J. et al. miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non-small cell lung cancer. Cancer Res. 73, 5402–5415 (2013).
Guo, H., Ingolia, N. T., Weissman, J. S. & Bartel, D. P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840 (2010).
Johnsson, P. et al. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat. Struct. Mol. Biol. 20, 440–446 (2013).
Poliseno, L. et al. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465, 1033–1038 (2010).
Al-Khouri, A. M. et al. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J. Biol. Chem. 280, 35195–35202 (2005).
Kwon, J. et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl Acad. Sci. USA 101, 16419–16424 (2004).
Okumura, K. et al. PCAF modulates PTEN activity. J. Biol. Chem. 281, 26562–26568 (2006).
Ding, L. et al. CBP loss cooperates with PTEN haploinsufficiency to drive prostate cancer: implications for epigenetic therapy. Cancer Res. 74, 2050–2061 (2014).
Wang, X. et al. Crucial role of the C-terminus of PTEN in antagonizing NEDD4-1-mediated PTEN ubiquitination and degradation. Biochem. J. 414, 221–229 (2008).
González-Santamaría, J. et al. Regulation of the tumor suppressor PTEN by SUMO. Cell Death Dis. 3, e393 (2012).
Chagpar, R. B. et al. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc. Natl Acad. Sci. USA 107, 5471–5476 (2010).
Taniguchi, C. M. et al. The phosphoinositide 3-kinase regulatory subunit p85alpha can exert tumor suppressor properties through negative regulation of growth factor signaling. Cancer Res. 70, 5305–5315 (2010).
Garcia-Cao, I. et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 149, 49–62 (2012).
Lee, Y. R. et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 364, eaau0159 (2019).
André, F. et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: final overall survival results from SOLAR-1. Ann. Oncol. 32, 208–217 (2021).
André, F. et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 380, 1929–1940 (2019).
Furman, R. R. et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 370, 997–1007 (2014).
Fowler, N. H. et al. Umbralisib, a dual PI3Kδ/CK1ε inhibitor in patients with relapsed or refractory indolent lymphoma. J. Clin. Oncol. 39, 1609–1618 (2021).
Flinn, I. W. et al. The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood 132, 2446–2455 (2018).
Aliqopa: Withdrawal of the marketing authorisation application. https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/aliqopa (2021).
Secura Bio Announces Copiktra® (duvelisib) Strategic Focus On T-cell Lymphoma And Voluntary U.S. Withdrawal Of The Relapsed Or Refractory Follicular Lymphoma Indication. https://www.prnewswire.com/news-releases/secura-bio-announces-copiktra-duvelisib-strategic-focus-on-t-cell-lymphoma-and-voluntary-us-withdrawal-of-the-relapsed-or-refractory-follicular-lymphoma-indication-301436834.html (2021).
Gilead Statement on Zydelig® U.S. Indication for Follicular Lymphoma and Small Lymphocytic Leukemia. https://www.gilead.com/news-and-press/company-statements/gilead-statement-on-zydelig-us-indication-for-follicular-lymphoma-and-small-lymphocytic-leukemia (2022).
FDA Drug Safety Communication: FDA investigating possible increased risk of death with lymphoma medicine Ukoniq (umbralisib). https://www.fda.gov/drugs/development-approval-process-drugs/fda-investigating-possible-increased-risk-death-lymphoma-medicine-ukoniq-umbralisib (2022).
Okkenhaug, K., Graupera, M. & Vanhaesebroeck, B. Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discov. 6, 1090–1105 (2016).
Weigelt, B. & Downward, J. Genomic determinants of PI3K pathway inhibitor response in cancer. Front. Oncol. 2, 109 (2012).
Baselga, J. et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 18, 904–916 (2017).
Krop, I. E. et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 17, 811–821 (2016).
Gribben, J. G. et al. Umbralisib plus ublituximab (U2) is superior to obinutuzumab plus chlorambucil (O plus Chl) in patients with treatment naive (TN) and relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL): results from the Phase 3 Unity-CLL Study. Blood 136, 37–39 (2020).
Goel, S. et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475 (2017).
Important drug warning: decreased overall survival and increased risk of serious infections in patients receiving ZYDELIG (idelalisib). http://cllsociety.org/docs/Zydelig%20Safety%20Update.pdf (2016).
FDA Oncologic Drugs Advisory Committee Meeting Briefing Document: PI3K Inhibitors in Hematologic Malignancies. https://www.fda.gov/media/157762/download (2022).
FDA’s ODAC votes unanimously that future PI3K inhibitors should include randomized data for blood cancers. https://endpts.com/fdas-odac-votes-unanimously-that-future-pi3k-inhibitors-should-include-randomized-data-for-blood-cancers/ (2022).
Yang, W. et al. Strategically timing inhibition of phosphatidylinositol 3-kinase to maximize therapeutic index in estrogen receptor alpha-positive, PIK3CA-mutant breast cancer. Clin. Cancer Res. 22, 2250–2260 (2016).
Will, M. et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov. 4, 334–347 (2014).
Eschweiler, S. et al. Intermittent PI3Kδ inhibition sustains anti-tumour immunity and curbs irAEs. Nature 605, 741–746 (2022).
Sharpless, N. E. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat. Res. 576, 22–38 (2005).
Zhao, R. et al. Implications of genetic and epigenetic alterations of CDKN2A (p16(INK4a)) in cancer. EBioMedicine 8, 30–39 (2016).
Weinberg, R. A. The retinoblastoma protein and cell cycle control. Cell 81, 323–330 (1995).
Serrano, M., Hannon, G. J. & Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366, 704–707 (1993).
Sionov, R. V. & Haupt, Y. The cellular response to p53: the decision between life and death. Oncogene 18, 6145–6157 (1999).
Juven-Gershon, T. & Oren, M. Mdm2: the ups and downs. Mol. Med. 5, 71–83 (1999).
Zhang, Y. & Xiong, Y. Control of p53 ubiquitination and nuclear export by MDM2 and ARF. Cell Growth Differ. 12, 175–186 (2001).
Weber, J. D. et al. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1, 20–26 (1999).
Honda, R. & Yasuda, H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 18, 22–27 (1999).
Sherr, C. J., Beach, D. & Shapiro, G. I. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 6, 353–367 (2016).
Asghar, U., Witkiewicz, A. K., Turner, N. C. & Knudsen, E. S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 14, 130–146 (2015).
Knudsen, E. S. et al. Pan-cancer molecular analysis of the RB tumor suppressor pathway. Commun. Biol. 3, 158 (2020).
Patnaik, A. et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 6, 740–753 (2016).
Ahn, E. R. et al. Palbociclib in patients with non-small-cell lung cancer with CDKN2A alterations: results from the targeted agent and profiling utilization registry study. JCO Precis. Oncol. 4, 757–766 (2020).
Middleton, G. et al. The national lung matrix trial of personalized therapy in lung cancer. Nature 583, 807–812 (2020).
Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2, 795–803 (2002).
Li, X. et al. Complicated life, complicated VEGF-B. Trends Mol. Med. 18, 119–127 (2012).
Alitalo, K., Tammela, T. & Petrova, T. V. Lymphangiogenesis in development and human disease. Nature 438, 946–953 (2005).
Houck, K. A. et al. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 267, 26031–26037 (1992).
de Vries, C. et al. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 255, 989–991 (1992).
Terman, B. I. et al. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem. Biophys. Res. Commun. 187, 1579–1586 (1992).
Soker, S. et al. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 92, 735–745 (1998).
Olsson, A. K., Dimberg, A., Kreuger, J. & Claesson-Welsh, L. VEGF receptor signalling—in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371 (2006).
Kerbel, R. S. Tumor angiogenesis. N. Engl. J. Med. 358, 2039–2049 (2008).
Ferrara, N. & Adamis, A. P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 15, 385–403 (2016).
Jayson, G. C., Kerbel, R., Ellis, L. M. & Harris, A. L. Antiangiogenic therapy in oncology: current status and future directions. Lancet 388, 518–529 (2016).
Kim, K. J. et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 362, 841–844 (1993).
Hurwitz, H. et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 350, 2335–2342 (2004).
Sandler, A. B. et al. Retrospective evaluation of the clinical and radiographic risk factors associated with severe pulmonary hemorrhage in first-line advanced, unresectable non-small-cell lung cancer treated with Carboplatin and Paclitaxel plus bevacizumab. J. Clin. Oncol. 27, 1405–1412 (2009).
Johnson, D. H. et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J. Clin. Oncol. 22, 2184–2191 (2004).
Larkins, E. et al. U.S. Food and Drug Administration approval summary: ramucirumab for the treatment of metastatic non-small cell lung cancer following disease progression on or after platinum-based chemotherapy. Oncologist 20, 1320–1325 (2015).
Farrell, B. & Breeze, A. L. Structure, activation and dysregulation of fibroblast growth factor receptor kinases: perspectives for clinical targeting. Biochem. Soc. Trans. 46, 1753–1770 (2018).
Dai, S. et al. Fibroblast growth factor receptors (FGFRs): structures and small molecule inhibitors. Cells 8, 614 (2019).
Wiedemann, M. & Trueb, B. Characterization of a novel protein (FGFRL1) from human cartilage related to FGF receptors. Genomics 69, 275–279 (2000).
Dieci, M. V., Arnedos, M., Andre, F. & Soria, J. C. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 3, 264–279 (2013).
Turner, N. & Grose, R. Fibroblast growth factor signalling: from development to cancer. Nat. Rev. Cancer 10, 116–129 (2010).
Ornitz, D. M. & Itoh, N. The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266 (2015).
Chen, H. et al. Elucidation of a four-site allosteric network in fibroblast growth factor receptor tyrosine kinases. eLife 6, e21137 (2017).
Abate-Shen, C. & Shen, M. M. FGF signaling in prostate tumorigenesis-new insights into epithelial-stromal interactions. Cancer Cell 12, 495–497 (2007).
Ruotsalainen, T., Joensuu, H., Mattson, K. & Salven, P. High pretreatment serum concentration of basic fibroblast growth factor is a predictor of poor prognosis in small cell lung cancer. Cancer Epidemiol. Biomark. Prev. 11, 1492–1495 (2002).
Casanovas, O., Hicklin, D. J., Bergers, G. & Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8, 299–309 (2005).
Czubayko, F. et al. A secreted FGF-binding protein can serve as the angiogenic switch in human cancer. Nat. Med. 3, 1137–1140 (1997).
Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 16, 105–122 (2019).
Weiss, J. et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2, 62ra93 (2010).
Zhang, J. et al. Translating the therapeutic potential of AZD4547 in FGFR1-amplified non-small cell lung cancer through the use of patient-derived tumor xenograft models. Clin. Cancer Res. 18, 6658–6667 (2012).
Weeden, C. E., Solomon, B. & Asselin-Labat, M. L. FGFR1 inhibition in lung squamous cell carcinoma: questions and controversies. Cell Death Discov. 1, 15049 (2015).
Wynes, M. W. et al. FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI sensitivity across all lung cancer histologies. Clin. Cancer Res. 20, 3299–3309 (2014).
Wells, A. EGF receptor. Int. J. Biochem. Cell Biol. 31, 637–643 (1999).
Jimeno, A. & Hidalgo, M. Pharmacogenomics of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. Biochim. Biophys. Acta 1766, 217–229 (2006).
Citri, A. & Yarden, Y. EGF-ERBB signalling: towards the systems level. Nat. Rev. Mol. Cell Biol. 7, 505–516 (2006).
Sibilia, M. et al. A strain-independent postnatal neurodegeneration in mice lacking the EGF receptor. EMBO J. 17, 719–731 (1998).
de Jong, J. S., van Diest, P. J., van der Valk, P. & Baak, J. P. Expression of growth factors, growth-inhibiting factors, and their receptors in invasive breast cancer. II: correlations with proliferation and angiogenesis. J. Pathol. 184, 53–57 (1998).
Wells, A. Tumor invasion: role of growth factor-induced cell motility. Adv. Cancer Res. 78, 31–101 (2000).
Gibson, E. M. et al. Epidermal growth factor protects epithelial-derived cells from tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by inhibiting cytochrome c release. Cancer Res. 62, 488–496 (2002).
Woodburn, J. R. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol. Ther. 82, 241–250 (1999).
Kumar, A., Petri, E. T., Halmos, B. & Boggon, T. J. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J. Clin. Oncol. 26, 1742–1751 (2008).
Holbro, T. & Hynes, N. E. ErbB receptors: directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 44, 195–217 (2004).
Imyanitov, E. N. et al. Distribution of EGFR mutations in 10,607 Russian patients with lung cancer. Mol. Diagn. Ther. 20, 401–406 (2016).
Fiala, O. et al. Gene mutations in squamous cell NSCLC: insignificance of EGFR, KRAS and PIK3CA mutations in prediction of EGFR-TKI treatment efficacy. Anticancer Res. 33, 1705–1711 (2013).
Rosell, R. et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 13, 239–246 (2012).
Zhou, C. et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 12, 735–742 (2011).
Zhong, W. Z. et al. Gefitinib versus vinorelbine plus cisplatin as adjuvant treatment for stage II-IIIA (N1-N2) EGFR-mutant NSCLC (ADJUVANT/CTONG1104): a randomised, open-label, phase 3 study. Lancet Oncol. 19, 139–148 (2018).
Mitsudomi, T. et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 11, 121–128 (2010).
He, J. et al. Icotinib versus chemotherapy as adjuvant treatment for stage II-IIIA EGFR-mutant non-small-cell lung cancer (EVIDENCE): a randomised, open-label, phase 3 trial. Lancet Respir. Med. 9, 1021–1029 (2021).
Kelly, K. et al. Adjuvant erlotinib versus placebo in patients with stage IB-IIIA non-small-cell lung cancer (RADIANT): a randomized, double-blind, phase III trial. J. Clin. Oncol. 33, 4007–4014 (2015).
Clark, G. M. et al. Smoking history and epidermal growth factor receptor expression as predictors of survival benefit from erlotinib for patients with non-small-cell lung cancer in the National Cancer Institute of Canada Clinical Trials Group study BR.21. Clin. Lung Cancer 7, 389–394 (2006).
Cappuzzo, F. et al. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: a multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol. 11, 521–529 (2010).
Ameratunga, M. et al. Epidermal growth factor receptor-tyrosine kinase inhibitors in advanced squamous cell carcinoma of the lung: a meta-analysis. Asia Pac. J. Clin. Oncol. 10, 273–278 (2014).
Liu, Y. et al. Efficacy of epidermal growth factor receptor-tyrosine kinase inhibitors for lung squamous carcinomas harboring EGFR mutation: a multicenter study and pooled analysis of published reports. Oncotarget 8, 49680–49688 (2017).
Liang, S. et al. Efficacy of icotinib in advanced lung squamous cell carcinoma. Cancer Med. 7, 4456–4466 (2018).
Chang, Q. et al. Epidermal growth factor receptor mutation status and response to tyrosine kinase inhibitors in advanced Chinese female lung squamous cell carcinoma: a retrospective study. Front. Oncol. 11, 652560 (2021).
Schubert, M. et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 9, 20 (2018).
Huang, C. et al. Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 39, 361–379.e316 (2021).
Yamamoto, M., Kensler, T. W. & Motohashi, H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 98, 1169–1203 (2018).
Itoh, K. et al. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8, 379–391 (2003).
Suzuki, T. et al. Molecular mechanism of cellular oxidative stress sensing by Keap1. Cell Rep. 28, 746–758.e744 (2019).
Martin, D. et al. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 279, 8919–8929 (2004).
Salazar, M. et al. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 281, 14841–14851 (2006).
Ichimura, Y. et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 51, 618–631 (2013).
DeNicola, G. M. et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 (2011).
Le Gal, K. et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 7, 308re308 (2015).
Binkley, M. S. et al. KEAP1/NFE2L2 mutations predict lung cancer radiation resistance that can be targeted by glutaminase inhibition. Cancer Discov. 10, 1826–1841 (2020).
Homma, S. et al. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. 15, 3423–3432 (2009).
Shibata, T. et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 135, 1358–1368 (2008). 1368.e1351-1354.
Satoh, H. et al. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 73, 4158–4168 (2013).
Cloer, E. W. et al. NRF2 activation in cancer: from DNA to protein. Cancer Res. 79, 889–898 (2019).
Pillai, R., Hayashi, M., Zavitsanou, A. M. & Papagiannakopoulos, T. NRF2: KEAPing tumors protected. Cancer Discov. 12, 625–643 (2022).
Imielinski, M. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120 (2012).
Satoh, H. et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 31, 1833–1843 (2010).
Solis, L. M. et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 16, 3743–3753 (2010).
Momcilovic, M. et al. The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell 33, 905–921.e905 (2018).
Mitsuishi, Y. et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79 (2012).
Lewerenz, J. et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 18, 522–555 (2013).
Sayin, V. I. et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 6, e28083 (2017).
Calithera Biosciences Announces Decision to Discontinue KEAPSAKE Clinical Trial. https://www.globenewswire.com/news-release/2021/11/05/2328406/32478/en/Calithera-Biosciences-Announces-Decision-to-Discontinue-KEAPSAKE-Clinical-Trial.html (2021).
Sapanisertib in Treating Patients With Stage IV or Recurrent Lung Cancer. https://clinicaltrials.gov/ct2/show/results/NCT02417701?term=Sapanisertib&cond=NSCLC&draw=2&rank=6 (2022).
Leone, R. D. et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021 (2019).
Johnson, M. L. et al. Phase 1 and phase 2a, first-in-human (FIH) study, of DRP-104, a broad glutamine antagonist, in adult patients with advanced solid tumors. J. Clin. Oncol. 39, (2021).
Bowles, J., Schepers, G. & Koopman, P. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol. 227, 239–255 (2000).
Nowling, T. K., Johnson, L. R., Wiebe, M. S. & Rizzino, A. Identification of the transactivation domain of the transcription factor Sox-2 and an associated co-activator. J. Biol. Chem. 275, 3810–3818 (2000).
Schaefer, T. & Lengerke, C. SOX2 protein biochemistry in stemness, reprogramming, and cancer: the PI3K/AKT/SOX2 axis and beyond. Oncogene 39, 278–292 (2020).
Weina, K. & Utikal, J. SOX2 and cancer: current research and its implications in the clinic. Clin. Transl. Med. 3, 19 (2014).
Hüser, L. et al. Targeting SOX2 in anticancer therapy. Expert Opin. Ther. Targets 22, 983–991 (2018).
Nakatsugawa, M. et al. SOX2 is overexpressed in stem-like cells of human lung adenocarcinoma and augments the tumorigenicity. Lab. Investig. 91, 1796–1804 (2011).
Santini, R. et al. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene 33, 4697–4708 (2014).
Rybak, A. P. & Tang, D. SOX2 plays a critical role in EGFR-mediated self-renewal of human prostate cancer stem-like cells. Cell. Signal. 25, 2734–2742 (2013).
Lundberg, I. V. et al. SOX2 expression is associated with a cancer stem cell state and down-regulation of CDX2 in colorectal cancer. BMC Cancer 16, 471 (2016).
Herreros-Villanueva, M. et al. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis 2, e61 (2013).
Liu, P. et al. SOX2 promotes cell proliferation and metastasis in triple negative breast cancer. Front. Pharmacol. 9, 942 (2018).
Xiang, R. et al. Downregulation of transcription factor SOX2 in cancer stem cells suppresses growth and metastasis of lung cancer. Br. J. Cancer 104, 1410–1417 (2011).
Feng, R. et al. Sox2 protects neural stem cells from apoptosis via up-regulating survivin expression. Biochem. J. 450, 459–468 (2013).
Chen, S. et al. SOX2 regulates apoptosis through MAP4K4-survivin signaling pathway in human lung cancer cells. Carcinogenesis 35, 613–623 (2014).
Ren, C. et al. Inhibition of SOX2 induces cell apoptosis and G1/S arrest in Ewing’s sarcoma through the PI3K/Akt pathway. J. Exp. Clin. Cancer Res. 35, 44 (2016).
Bora-Singhal, N. et al. Novel HDAC11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing Sox2. Sci. Rep. 10, 4722 (2020).
Piva, M. et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 6, 66–79 (2014).
He, J. et al. Sox2 inhibits Wnt-β-catenin signaling and metastatic potency of cisplatin-resistant lung adenocarcinoma cells. Mol. Med. Rep. 15, 1693–1701 (2017).
Tripathi, S. C. et al. MCAM mediates chemoresistance in small-cell lung cancer via the PI3K/AKT/SOX2 signaling pathway. Cancer Res. 77, 4414–4425 (2017).
Yang, A. et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 2, 305–316 (1998).
Murray-Zmijewski, F. & Lane, D. P. & Bourdon, J. C. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 13, 962–972 (2006).
Costanzo, A. et al. TP63 and TP73 in cancer, an unresolved “family” puzzle of complexity, redundancy and hierarchy. FEBS Lett. 588, 2590–2599 (2014).
Levrero, M. et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J. Cell Sci. 113(Pt 10), 1661–1670 (2000).
Mills, A. A. et al. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398, 708–713 (1999).
Yang, A. et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398, 714–718 (1999).
Guo, X. et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat. Cell Biol. 11, 1451–1457 (2009).
Wu, G. et al. DeltaNp63alpha up-regulates the Hsp70 gene in human cancer. Cancer Res. 65, 758–766 (2005).
Westfall, M. D. & Pietenpol, J. A. p63: molecular complexity in development and cancer. Carcinogenesis 25, 857–864 (2004).
Lambert, S. A. et al. The human transcription factors. Cell 172, 650–665 (2018).
Liu, J. et al. TF-PROTACs enable targeted degradation of transcription factors. J. Am. Chem. Soc. 143, 8902–8910 (2021).
Vanbokhoven, H., Melino, G., Candi, E. & Declercq, W. p63, a story of mice and men. J. Invest. Dermatol. 131, 1196–1207 (2011).
Rocco, J. W. et al. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 9, 45–56 (2006).
Somerville, T. D. D. et al. TP63-mediated enhancer reprogramming drives the squamous subtype of pancreatic ductal adenocarcinoma. Cell Rep. 25, 1741–1755.e1747 (2018).
Soares, E. & Zhou, H. Master regulatory role of p63 in epidermal development and disease. Cell. Mol. Life Sci. 75, 1179–1190 (2018).
Napoli, M. et al. ΔNp63 regulates a common landscape of enhancer associated genes in non-small cell lung cancer. Nat. Commun. 13, 614 (2022).
Su, X., Chakravarti, D. & Flores, E. R. p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat. Rev. Cancer 13, 136–143 (2013).
Papoutsaki, M. et al. A p38-dependent pathway regulates DeltaNp63 DNA binding to p53-dependent promoters in UV-induced apoptosis of keratinocytes. Oncogene 24, 6970–6975 (2005).
Ramsey, M. R. et al. FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J. Clin. Invest. 123, 3525–3538 (2013).
Bretz, A. C. et al. ΔNp63 activates the Fanconi anemia DNA repair pathway and limits the efficacy of cisplatin treatment in squamous cell carcinoma. Nucleic Acids Res. 44, 3204–3218 (2016).
Sen, T. et al. DeltaNp63alpha confers tumor cell resistance to cisplatin through the AKT1 transcriptional regulation. Cancer Res. 71, 1167–1176 (2011).
Amerik, A. Y. & Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 1695, 189–207 (2004).
Armstrong, S. R. et al. The regulation of tumor suppressor p63 by the ubiquitin-proteasome system. Int. J. Mol. Sci. 17, 2041 (2016).
Prieto-Garcia, C. et al. Maintaining protein stability of ∆Np63 via USP28 is required by squamous cancer cells. EMBO Mol. Med. 12, e11101 (2020).
Prieto-Garcia, C. et al. Inhibition of USP28 overcomes Cisplatin-resistance of squamous tumors by suppression of the Fanconi anemia pathway. Cell Death Differ. 29, 568–584 (2022).
Welcker, M. & Clurman, B. E. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8, 83–93 (2008).
Davis, R. J., Welcker, M. & Clurman, B. E. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 26, 455–464 (2014).
Kan, Z. et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–873 (2010).
Schülein-Völk, C. et al. Dual regulation of Fbw7 function and oncogenic transformation by Usp28. Cell Rep. 9, 1099–1109 (2014).
Popov, N. et al. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 9, 765–774 (2007).
Ruiz, E. J. et al. USP28 deletion and small-molecule inhibition destabilizes c-MYC and elicits regression of squamous cell lung carcinoma. eLife 10, e71596 (2021).
Wang, X. M. et al. The deubiquitinase USP25 supports colonic inflammation and bacterial infection and promotes colorectal cancer. Nat. Cancer 1, 811–825 (2020).
Ambrosini, G., Adida, C. & Altieri, D. C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 3, 917–921 (1997).
Dohi, T., Xia, F. & Altieri, D. C. Compartmentalized phosphorylation of IAP by protein kinase A regulates cytoprotection. Mol. Cell 27, 17–28 (2007).
Rosa, J. et al. Survivin modulates microtubule dynamics and nucleation throughout the cell cycle. Mol. Biol. Cell. 17, 1483–1493 (2006).
Verhagen, A. M., Coulson, E. J. & Vaux, D. L. Inhibitor of apoptosis proteins and their relatives: IAPs and other BIRPs. Genome Biol. 2, Reviews3009 (2001).
Velculescu, V. E. et al. Analysis of human transcriptomes. Nat. Genet. 23, 387–388 (1999).
Giaccone, G. et al. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J. Clin. Oncol. 27, 4481–4486 (2009).
Kelly, R. J. et al. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol. 24, 2601–2606 (2013).
Li, F., Aljahdali, I. & Ling, X. Cancer therapeutics using survivin BIRC5 as a target: what can we do after over two decades of study? J. Exp. Clin. Cancer Res. 38, 368 (2019).
Masuda, M. et al. TNIK inhibition abrogates colorectal cancer stemness. Nat. Commun. 7, 12586 (2016).
Fu, C. A. et al. TNIK, a novel member of the germinal center kinase family that activates the c-Jun N-terminal kinase pathway and regulates the cytoskeleton. J. Biol. Chem. 274, 30729–30737 (1999).
Schürch, C. et al. CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J. Clin. Invest. 122, 624–638 (2012).
Li, Z., Lim, S. K., Liang, X. & Lim, Y. P. The transcriptional coactivator WBP2 primes triple-negative breast cancer cells for responses to Wnt signaling via the JNK/Jun kinase pathway. J. Biol. Chem. 293, 20014–20028 (2018).
Lee, R. S. et al. Characterization of the ERG-regulated kinome in prostate cancer identifies TNIK as a potential therapeutic target. Neoplasia 21, 389–400 (2019).
Hagerstrand, D. et al. Systematic interrogation of 3q26 identifies TLOC1 and SKIL as cancer drivers. Cancer Discov. 3, 1044–1057 (2013).
Torres-Ayuso, P. et al. TNIK is a therapeutic target in lung squamous cell carcinoma and regulates FAK activation through Merlin. Cancer Discov. 11, 1411–1423 (2021).
Cavalli, G. & Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 571, 489–499 (2019).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014).
Bates, S. E. Epigenetic therapies for cancer. N. Engl. J. Med. 383, 650–663 (2020).
Hoy, S. M. Tazemetostat: first approval. Drugs 80, 513–521 (2020).
Husmann, D. & Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 26, 880–889 (2019).
Li, Y. et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 284, 34283–34295 (2009).
Sroka, M. W. & Vakoc, C. R. An epigenetic tipping point in cancer comes under the microscope. Nature 590, 399–400 (2021).
Rooney, C. et al. Characterization of FGFR1 locus in sqNSCLC reveals a broad and heterogeneous amplicon. PLoS ONE 11, e0149628 (2016).
Balsara, B. R. et al. Comparative genomic hybridization analysis detects frequent, often high-level, overrepresentation of DNA sequences at 3q, 5p, 7p, and 8q in human non-small cell lung carcinomas. Cancer Res. 57, 2116–2120 (1997).
Tonon, G. et al. High-resolution genomic profiles of human lung cancer. Proc. Natl Acad. Sci. USA 102, 9625–9630 (2005).
Li, W. et al. Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature 590, 498–503 (2021).
Beck, D. B., Oda, H., Shen, S. S. & Reinberg, D. PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 26, 325–337 (2012).
Milite, C. et al. The emerging role of lysine methyltransferase SETD8 in human diseases. Clin. Epigenetics 8, 102 (2016).
Yin, Y., Yu, V. C., Zhu, G. & Chang, D. C. SET8 plays a role in controlling G1/S transition by blocking lysine acetylation in histone through binding to H4 N-terminal tail. Cell Cycle 7, 1423–1432 (2008).
Shi, X. et al. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 27, 636–646 (2007).
Dhami, G. K. et al. Dynamic methylation of Numb by Set8 regulates its binding to p53 and apoptosis. Mol. Cell 50, 565–576 (2013).
Takawa, M. et al. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 72, 3217–3227 (2012).
Herviou, L. et al. Targeting the methyltransferase SETD8 impairs tumor cell survival and overcomes drug resistance independently of p53 status in multiple myeloma. Clin. Epigenetics 13, 174 (2021).
Liu, B. et al. MiR-502/SET8 regulatory circuit in pathobiology of breast cancer. Cancer Lett. 376, 259–267 (2016).
Shi, Y. et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004).
Metzger, E. et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437, 436–439 (2005).
Laurent, B. et al. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol. Cell 57, 957–970 (2015).
Vinyard, M. E. et al. CRISPR-suppressor scanning reveals a nonenzymatic role of LSD1 in AML. Nat. Chem. Biol. 15, 529–539 (2019).
Sehrawat, A. et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc. Natl Acad. Sci. USA 115, E4179–e4188 (2018).
Fang, Y., Liao, G. & Yu, B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J. Hematol. Oncol. 12, 129 (2019).
Romo-Morales, A. et al. Catalytic inhibition of KDM1A in Ewing sarcoma is insufficient as a therapeutic strategy. Pediatr. Blood Cancer 66, e27888 (2019).
Sonnemann, J. et al. LSD1 (KDM1A)-independent effects of the LSD1 inhibitor SP2509 in cancer cells. Br. J. Haematol. 183, 494–497 (2018).
Lai, A. C. & Crews, C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16, 101–114 (2017).
Viré, E. et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874 (2006).
Cao, R. et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043 (2002).
Czermin, B. et al. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111, 185–196 (2002).
Li, H., Cai, Q., Godwin, A. K. & Zhang, R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol. Cancer Res. 8, 1610–1618 (2010).
Kleer, C. G. et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl Acad. Sci. USA 100, 11606–11611 (2003).
Zhang, H. et al. Oncogenic deregulation of EZH2 as an opportunity for targeted therapy in lung cancer. Cancer Discov. 6, 1006–1021 (2016).
Kondo, Y. et al. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol. Res. 37, 974–983 (2007).
Wagener, N. et al. Enhancer of zeste homolog 2 (EZH2) expression is an independent prognostic factor in renal cell carcinoma. BMC Cancer 10, 524 (2010).
Bachmann, I. M. et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 24, 268–273 (2006).
Kim, K. H. & Roberts, C. W. Targeting EZH2 in cancer. Nat. Med. 22, 128–134 (2016).
Xu, B., Konze, K. D., Jin, J. & Wang, G. G. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp. Hematol. 43, 698–712 (2015).
Margueron, R. & Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011).
Margueron, R. et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 32, 503–518 (2008).
Wang, J. & Wang, G. G. No easy way out for EZH2: its pleiotropic, noncanonical effects on gene regulation and cellular function. Int. J. Mol. Sci. 21, 9501 (2020).
Xu, K. et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338, 1465–1469 (2012).
Lawrence, C. L. & Baldwin, A. S. Non-canonical EZH2 transcriptionally activates RelB in triple negative breast cancer. PLoS ONE 11, e0165005 (2016).
Yan, J. et al. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood 121, 4512–4520 (2013).
Kim, J. et al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 25, 2808–2820.e2804 (2018).
FDA approves tazemetostat for advanced epithelioid sarcoma. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tazemetostat-advanced-epithelioid-sarcoma. (2020).
FDA granted accelerated approval to tazemetostat for follicular lymphoma. https://www.fda.gov/drugs/fda-granted-accelerated-approval-tazemetostat-follicular-lymphoma. (2020).
McCarthy, E. F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 26, 154–158 (2006).
Tan, S., Li, D. & Zhu, X. Cancer immunotherapy: pros, cons and beyond. Biomed. Pharmacother. 124, 109821 (2020).
Baumeister, S. H., Freeman, G. J., Dranoff, G. & Sharpe, A. H. Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol. 34, 539–573 (2016).
Wykes, M. N. & Lewin, S. R. Immune checkpoint blockade in infectious diseases. Nat. Rev. Immunol. 18, 91–104 (2018).
Doyle, A. M. et al. Induction of cytotoxic T lymphocyte antigen 4 (CTLA-4) restricts clonal expansion of helper T cells. J. Exp. Med. 194, 893–902 (2001).
Freeman, G. J. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 (2000).
Brunet, J. F. et al. A new member of the immunoglobulin superfamily-CTLA-4. Nature 328, 267–270 (1987).
Harper, K. et al. CTLA-4 and CD28 activated lymphocyte molecules are closely related in both mouse and human as to sequence, message expression, gene structure, and chromosomal location. J. Immunol. 147, 1037–1044 (1991).
Linsley, P. S. et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 174, 561–569 (1991).
Linsley, P. S. et al. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1, 793–801 (1994).
Qureshi, O. S. et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603 (2011).
Wing, K. et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008).
Morad, G., Helmink, B. A., Sharma, P. & Wargo, J. A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 185, 576 (2022).
Grosso, J. F. & Jure-Kunkel, M. N. CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun. 13, 5 (2013).
van Elsas, A., Hurwitz, A. A. & Allison, J. P. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 190, 355–366 (1999).
Leach, D. R., Krummel, M. F. & Allison, J. P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 (1996).
Camacho, L. H. et al. Phase I/II trial of tremelimumab in patients with metastatic melanoma. J. Clin. Oncol. 27, 1075–1081 (2009).
Kirkwood, J. M. et al. Phase II trial of tremelimumab (CP-675,206) in patients with advanced refractory or relapsed melanoma. Clin. Cancer Res. 16, 1042–1048 (2010).
O’Day, S. J. et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann. Oncol. 21, 1712–1717 (2010).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Yang, J. C. et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J. Immunother. 30, 825–830 (2007).
Lynch, T. J. et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J. Clin. Oncol. 30, 2046–2054 (2012).
Reck, M. et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann. Oncol. 24, 75–83 (2013).
Kwon, E. D. et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 15, 700–712 (2014).
Ribas, A. et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 31, 616–622 (2013).
Maio, M. et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J. Clin. Oncol. 33, 1191–1196 (2015).
Schadendorf, D. et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 33, 1889–1894 (2015).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015).
Larkin, J. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015).
Khoja, L. et al. Tumour- and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann. Oncol. 28, 2377–2385 (2017).
Michot, J. M. et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur. J. Cancer 54, 139–148 (2016).
Du, X. et al. Uncoupling therapeutic from immunotherapy-related adverse effects for safer and effective anti-CTLA-4 antibodies in CTLA4 humanized mice. Cell Res. 28, 433–447 (2018).
Lee, P. S. et al. Improved therapeutic index of an acidic pH-selective antibody. MAbs 14, 2024642 (2022).
Zhang, Y. et al. Hijacking antibody-induced CTLA-4 lysosomal degradation for safer and more effective cancer immunotherapy. Cell Res 29, 609–627 (2019).
Selby, M. J. et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 1, 32–42 (2013).
Simpson, T. R. et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 210, 1695–1710 (2013).
Bulliard, Y. et al. Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 210, 1685–1693 (2013).
Lo, B. et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349, 436–440 (2015).
Du, X. et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 28, 416–432 (2018).
Tang, F. et al. Anti-CTLA-4 antibodies in cancer immunotherapy: selective depletion of intratumoral regulatory T cells or checkpoint blockade? Cell Biosci. 8, 30 (2018).
Korman, A. J., Garrett-Thomson, S. C. & Lonberg, N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat. Rev. Drug Discov. 21, 509–528 (2021).
ONCOC4 ANNOUNCES FAST TRACK DESIGNATION GRANTED BY THE U.S. FDA FOR ONC-392 MONOTHERAPY IN PD(L)1-RESISTANT NSCLC. https://oncoc4.com/index.php/blog/news-releases/item/11-oncoc4-announces-fast-track-designation-granted-by-the-u-s-fda-for-onc-392-monotherapy-in-pd-l-1-resistant-nsclc. (2022).
Weber, J. S. et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 16, 375–384 (2015).
Weber, J. et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N. Engl. J. Med. 377, 1824–1835 (2017).
Schachter, J. et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 390, 1853–1862 (2017).
Ishida, Y., Agata, Y., Shibahara, K. & Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895 (1992).
Nishimura, H. et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11, 141–151 (1999).
Dong, H., Zhu, G., Tamada, K. & Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 5, 1365–1369 (1999).
Latchman, Y. et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261–268 (2001).
Tseng, S. Y. et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med 193, 839–846 (2001).
Xiao, Y. et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J. Exp. Med. 211, 943–959 (2014).
Hirano, F. et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 65, 1089–1096 (2005).
Iwai, Y. et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl Acad. Sci. USA 99, 12293–12297 (2002).
Strome, S. E. et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 63, 6501–6505 (2003).
He, Y. F. et al. Blocking programmed death-1 ligand-PD-1 interactions by local gene therapy results in enhancement of antitumor effect of secondary lymphoid tissue chemokine. J. Immunol. 173, 4919–4928 (2004).
Tanegashima, T. et al. Immune suppression by PD-L2 against spontaneous and treatment-related antitumor immunity. Clin. Cancer Res. 25, 4808–4819 (2019).
Reck, M. et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. J. Clin. Oncol. 37, 537–546 (2019).
Mok, T. S. K. et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet 393, 1819–1830 (2019).
Jassem, J. et al. Updated overall survival analysis from IMpower110: atezolizumab versus platinum-based chemotherapy in treatment-naive programmed death-ligand 1-selected NSCLC. J. Thorac. Oncol. 16, 1872–1882 (2021).
Duan, J. et al. Use of immunotherapy with programmed cell death 1 vs programmed cell death ligand 1 inhibitors in patients with cancer: a systematic review and meta-analysis. JAMA Oncol. 6, 375–384 (2020).
Pillai, R. N. et al. Comparison of the toxicity profile of PD-1 versus PD-L1 inhibitors in non-small cell lung cancer: a systematic analysis of the literature. Cancer 124, 271–277 (2018).
Khunger, M. et al. Incidence of pneumonitis with use of programmed death 1 and programmed death-ligand 1 inhibitors in non-small cell lung cancer: a systematic review and meta-analysis of trials. Chest 152, 271–281 (2017).
Zheng, Q. et al. Clinicopathological and molecular characteristics associated with PD-L1 expression in non-small cell lung cancer: a large-scale, multi-center, real-world study in China. J. Cancer Res. Clin. Oncol. 147, 1547–1556 (2021).
Mahoney, K. M. & Atkins, M. B. Prognostic and predictive markers for the new immunotherapies. Oncology 28(Suppl 3), 39–48 (2014).
Gibney, G. T., Weiner, L. M. & Atkins, M. B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 17, e542–e551 (2016).
Doroshow, D. B. et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 18, 345–362 (2021).
Lu, S. et al. Comparison of biomarker modalities for predicting response to PD-1/PD-L1 checkpoint blockade: a systematic review and meta-analysis. JAMA Oncol. 5, 1195–1204 (2019).
Chen, G. et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 560, 382–386 (2018).
Walk, E. E. et al. The cancer immunotherapy biomarker testing landscape. Arch. Pathol. Lab. Med. 144, 706–724 (2020).
Jardim, D. L., Goodman, A., de Melo Gagliato, D. & Kurzrock, R. The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell 39, 154–173 (2021).
Gohil, S. H. et al. Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat. Rev. Clin. Oncol. 18, 244–256 (2021).
Triebel, F. et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 171, 1393–1405 (1990).
Huang, C. T. et al. Role of LAG-3 in regulatory T cells. Immunity 21, 503–513 (2004).
Blackburn, S. D. et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 10, 29–37 (2009).
Huard, B. et al. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 25, 2718–2721 (1995).
Dijkstra, J. M. et al. Identification and characterization of a second CD4-like gene in teleost fish. Mol. Immunol. 43, 410–419 (2006).
Huard, B. et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl Acad. Sci. USA 94, 5744–5749 (1997).
Workman, C. J., Dugger, K. J. & Vignali, D. A. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol. 169, 5392–5395 (2002).
Kouo, T. et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol. Res. 3, 412–423 (2015).
Liu, W. et al. Characterization of a novel C-type lectin-like gene, LSECtin: demonstration of carbohydrate binding and expression in sinusoidal endothelial cells of liver and lymph node. J. Biol. Chem. 279, 18748–18758 (2004).
Xu, F. et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 74, 3418–3428 (2014).
Wang, J. et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 176, 334–347.e312 (2019).
Workman, C. J. & Vignali, D. A. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur. J. Immunol. 33, 970–979 (2003).
Workman, C. J. & Vignali, D. A. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J. Immunol. 174, 688–695 (2005).
Andrews, L. P., Marciscano, A. E., Drake, C. G. & Vignali, D. A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 276, 80–96 (2017).
Camisaschi, C. et al. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J. Immunol. 184, 6545–6551 (2010).
Wei, T. et al. Increased expression of immunosuppressive molecules on intratumoral and circulating regulatory T cells in non-small-cell lung cancer patients. Am. J. Cancer Res. 5, 2190–2201 (2015).
Mullard, A. LAG3 pushes immuno-oncology’s leading edge. Nat. Rev. Drug Discov. 21, 167–169 (2022).
Woo, S. R. et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 72, 917–927 (2012).
Huang, R. Y. et al. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 6, 27359–27377 (2015).
Goding, S. R. et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J. Immunol. 190, 4899–4909 (2013).
Matsuzaki, J. et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl Acad. Sci. USA 107, 7875–7880 (2010).
Jing, W. et al. Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. J. Immunother. Cancer 3, 2 (2015).
Tawbi, H. A. et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 386, 24–34 (2022).
FDA approves Opdualag for unresectable or metastatic melanoma. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-opdualag-unresectable-or-metastatic-melanoma (2022).
Yu, X. et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 10, 48–57 (2009).
Johnston, R. J. et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 26, 923–937 (2014).
He, W. et al. CD155T/TIGIT Signaling Regulates CD8(+) T-cell Metabolism and Promotes Tumor Progression in Human Gastric Cancer. Cancer Res. 77, 6375–6388 (2017).
Zhang, Q. et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 19, 723–732 (2018).
Cho, B. C. et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 23, 781–792 (2022).
Genentech Reports Interim Results for Phase III SKYSCRAPER-01 Study in PD-L1-High Metastatic Non-Small Cell Lung Cancer. https://www.gene.com/media/press-releases/14951/2022-05-10/genentech-reports-interim-results-for-ph (2022).
Kaen, D. L. et al. Immunotherapy in lung cancer: are the promises of long-term benefit finally met? Adv. Exp. Med. Biol. 1342, 113–142 (2021).
Monney, L. et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541 (2002).
Sánchez-Fueyo, A. et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 4, 1093–1101 (2003).
Golden-Mason, L. et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 83, 9122–9130 (2009).
Jin, H. T. et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl Acad. Sci. USA 107, 14733–14738 (2010).
Wu, W. et al. Blockade of Tim-3 signaling restores the virus-specific CD8+ T-cell response in patients with chronic hepatitis B. Eur. J. Immunol. 42, 1180–1191 (2012).
Sakuishi, K. et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194 (2010).
Fourcade, J. et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 207, 2175–2186 (2010).
Zhou, Q. et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 117, 4501–4510 (2011).
Yuan, L., Tatineni, J., Mahoney, K. M. & Freeman, G. J. VISTA: a mediator of quiescence and a promising target in cancer immunotherapy. Trends Immunol. 42, 209–227 (2021).
Wang, L. et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 208, 577–592 (2011).
ElTanbouly, M. A. et al. VISTA is a checkpoint regulator for naïve T cell quiescence and peripheral tolerance. Science 367, eaay0524 (2020).
Johnston, R. J. et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 574, 565–570 (2019).
Waldman, A. D., Fritz, J. M. & Lenardo, M. J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 651–668 (2020).
Mayes, P. A., Hance, K. W. & Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 17, 509–527 (2018).
Tivol, E. A. et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547 (1995).
Waterhouse, P. et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988 (1995).
Nishimura, H. et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291, 319–322 (2001).
Wang, J. et al. Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc. Natl Acad. Sci. USA 102, 11823–11828 (2005).
Wang, J. et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 22, 443–452 (2010).
Fife, B. T. & Bluestone, J. A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 224, 166–182 (2008).
Topalian, S. L., Taube, J. M., Anders, R. A. & Pardoll, D. M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 16, 275–287 (2016).
Callahan, M. K., Postow, M. A. & Wolchok, J. D. CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front. Oncol. 4, 385 (2014).
Duraiswamy, J., Kaluza, K. M., Freeman, G. J. & Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 73, 3591–3603 (2013).
Lim, S. O. et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 30, 925–939 (2016).
Rizvi, N. A. et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non-small cell lung cancer: the MYSTIC phase 3 randomized clinical trial. JAMA Oncol. 6, 661–674 (2020).
Gettinger, S. N. et al. Nivolumab plus ipilimumab vs nivolumab for previously treated patients with stage IV squamous cell lung cancer: the lung-MAP S1400I phase 3 randomized clinical trial. JAMA Oncol. 7, 1368–1377 (2021).
Zhao, Y. et al. PD-L1:CD80 cis-heterodimer triggers the co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity 51, 1059–1073.e1059 (2019).
Sugiura, D. et al. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 364, 558–566 (2019).
Butte, M. J. et al. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 27, 111–122 (2007).
Butte, M. J. et al. Interaction of human PD-L1 and B7-1. Mol. Immunol. 45, 3567–3572 (2008).
Chaudhri, A. et al. PD-L1 binds to B7-1 only in cis on the same cell surface. Cancer Immunol. Res. 6, 921–929 (2018).
Onishi, Y., Fehervari, Z., Yamaguchi, T. & Sakaguchi, S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl Acad. Sci. USA 105, 10113–10118 (2008).
Tekguc, M. et al. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc. Natl Acad. Sci. USA 118, e2023739118 (2021).
Zhou, C. et al. A Phase II study of KN046 (bispecific anti-PD-L1/CTLA-4) in patients (pts) with metastatic non-small cell lung cancer (NSCLC). J. Thorac. Oncol. 16, S636–S636 (2021).
Wu, L. et al. A phase Ib/II trial of AK104 (PD-1/CTLA-4 bispecific antibody) in combination with anlotinib in advanced NSCLC. Ann. Oncol. 32, S1006–S1006 (2021).
Spranger, S. & Gajewski, T. F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 18, 139–147 (2018).
Peng, W. et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 6, 202–216 (2016).
Ali, K. et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 510, 407–411 (2014).
Ahmad, S. et al. Differential PI3Kδ signaling in CD4(+) T-cell subsets enables selective targeting of T regulatory cells to enhance cancer immunotherapy. Cancer Res. 77, 1892–1904 (2017).
Abu-Eid, R. et al. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol. Res. 2, 1080–1089 (2014).
Isoyama, S. et al. Cancer immunotherapy with PI3K and PD-1 dual-blockade via optimal modulation of T cell activation signal. J. Immunother. Cancer 9, e002279 (2021).
Kaech, S. M. & Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 12, 749–761 (2012).
Kim, E. H. & Suresh, M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front. Immunol. 4, 20 (2013).
Carnevalli, L. S. et al. PI3Kα/δ inhibition promotes anti-tumor immunity through direct enhancement of effector CD8(+) T-cell activity. J. Immunother. Cancer 6, 158 (2018).
Lim, E. L. et al. Phosphoinositide 3-kinase δ inhibition promotes antitumor responses but antagonizes checkpoint inhibitors. JCI Insight 3, e120626 (2018).
Parry, R. V. et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 25, 9543–9553 (2005).
Hui, E. et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433 (2017).
Schaer, D. A. et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. 22, 2978–2994 (2018).
Zhang, Q. F. et al. CDK4/6 inhibition promotes immune infiltration in ovarian cancer and synergizes with PD-1 blockade in a B cell-dependent manner. Theranostics 10, 10619–10633 (2020).
Zhang, J. et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95 (2018).
Jerby-Arnon, L. et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997.e924 (2018).
Deng, J. et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 8, 216–233 (2018).
Heng, T. S. & Painter, M. W. The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094 (2008).
Finn, R. S. et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 11, R77 (2009).
Lelliott, E. J. et al. CDK4/6 Inhibition Promotes Antitumor Immunity through the Induction of T-cell Memory. Cancer Discov. 11, 2582–2601 (2021).
Heckler, M. et al. Inhibition of CDK4/6 promotes CD8 T-cell memory formation. Cancer Discov. 11, 2564–2581 (2021).
Rugo, H. S. et al. A phase lb study of abemaciclib in combination with pembrolizumab for patients with hormone receptor positive (HR plus), human epidermal growth factor receptor 2 negative (HER2-) locally advanced or metastatic breast cancer (MBC) (NCT02779751): interim results. J. Clin. Oncol. 38, 1051 (2020).
Pujol, J. L. et al. A phase Ib study of abemaciclib in combination with pembrolizumab for patients (pts) with stage IV Kirsten rat sarcoma mutant (KRAS-mut) or squamous non-small cell lung cancer (NSCLC) (NCT02779751): interim results. J. Clin. Oncol. 38, 9562 (2020).
Khan, K. A. & Kerbel, R. S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 15, 310–324 (2018).
Shrimali, R. K. et al. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 70, 6171–6180 (2010).
Wallin, J. J. et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 7, 12624 (2016).
Huang, B. et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 66, 1123–1131 (2006).
Kusmartsev, S. et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J. Immunol. 181, 346–353 (2008).
Ko, J. S. et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 15, 2148–2157 (2009).
Terme, M. et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 73, 539–549 (2013).
Hansen, W. et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J. Exp. Med. 209, 2001–2016 (2012).
Jain, R. K. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26, 605–622 (2014).
Fukumura, D. et al. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat. Rev. Clin. Oncol. 15, 325–340 (2018).
Facciabene, A. et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 475, 226–230 (2011).
Yang, L. et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 6, 409–421 (2004).
Curiel, T. J. et al. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res. 64, 5535–5538 (2004).
Cham, C. M. & Gajewski, T. F. Metabolic mechanisms of tumor resistance to T cell effector function. Immunol. Res. 31, 107–118 (2005).
Chang, C. H. et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013).
Palazon, A. et al. An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell 32, 669–683.e665 (2017).
Socinski, M. A. et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med. 378, 2288–2301 (2018).
Coward, J. et al. Safety and efficacy of AK112, an anti-PD-1/VEGF-A bispecific antibody, in patients with advanced solid tumors in a phase I dose escalation study. J. Clin. Oncol. 39, 2515–2515 (2021).
Roulois, D. et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973 (2015).
Chiappinelli, K. B. et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986 (2015).
Sheng, W. et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174, 549–563.e519 (2018).
Wu, L. et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. 16, e2006134 (2018).
Dawson, M. A. The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science 355, 1147–1152 (2017).
Peng, D. et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253 (2015).
Sullivan, R. J. et al. Efficacy and safety of entinostat (ENT) and pembrolizumab (PEMBRO) in patients with melanoma previously treated with anti-PD1 therapy. Cancer Res. 79, 9530 (2019).
Nagarsheth, N. et al. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. 76, 275–282 (2016).
Guo, B., Tan, X. & Cen, H. EZH2 is a negative prognostic biomarker associated with immunosuppression in hepatocellular carcinoma. PLoS ONE 15, e0242191 (2020).
Kim, H. J., Cantor, H. & Cosmopoulos, K. Overcoming immune checkpoint blockade resistance via EZH2 inhibition. Trends Immunol. 41, 948–963 (2020).
Guo, W. et al. LincRNA-immunity landscape analysis identifies EPIC1 as a regulator of tumor immune evasion and immunotherapy resistance. Sci. Adv. 7, eabb3555 (2021).
Dersh, D. et al. Genome-wide screens identify lineage- and tumor-specific genes modulating MHC-I- and MHC-II-restricted immunosurveillance of human lymphomas. Immunity 54, 116–131.e110 (2021).
DuPage, M. et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity 42, 227–238 (2015).
Arvey, A. et al. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat. Immunol. 15, 580–587 (2014).
Wang, D. et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep. 23, 3262–3274 (2018).
Goswami, S. et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J. Clin. Invest. 128, 3813–3818 (2018).
Hong, Y. K. et al. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell. Immunol. 336, 66–74 (2019).
Zhou, L. et al. Targeting EZH2 enhances antigen presentation, antitumor immunity, and circumvents Anti-PD-1 resistance in head and neck cancer. Clin. Cancer Res. 26, 290–300 (2020).
Palomba, M. L. et al. Combination of atezolizumab and tazemetostat in patients with relapsed/refractory diffuse large B-cell lymphoma: results from a phase Ib study. Clin. Lymphoma Myeloma Leuk. 22, 504–512 (2021).
Meeks, J. J. et al. A pilot study of tazemetostat and MK-3475 (pembrolizumab) in advanced urothelial carcinoma (ETCTN 10183). J. Clin. Oncol. 38, TPS607 (2020).
Qin, Y. et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 38, 390–405 (2019).
Xu, S. et al. LSD1 silencing contributes to enhanced efficacy of anti-CD47/PD-L1 immunotherapy in cervical cancer. Cell Death Dis. 12, 282 (2021).
Shen, D. D. et al. LSD1 deletion decreases exosomal PD-L1 and restores T-cell response in gastric cancer. Mol. Cancer 21, 75 (2022).
Cheng, W. et al. Growth differentiation factor 1-induced tumour plasticity provides a therapeutic window for immunotherapy in hepatocellular carcinoma. Nat. Commun. 12, 7142 (2021).
Liu, Y. et al. LSD1 inhibition sustains T cell invigoration with a durable response to PD-1 blockade. Nat. Commun. 12, 6831 (2021).
Blackburn, S. D., Shin, H., Freeman, G. J. & Wherry, E. J. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc. Natl Acad. Sci. USA 105, 15016–15021 (2008).
Im, S. J. et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016).
Miller, B. C. et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336 (2019).
Siddiqui, I. et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e110 (2019).
Sheng, W. et al. Simultaneous inhibition of LSD1 and TGFβ enables eradication of poorly immunogenic tumors with anti-PD-1 treatment. Cancer Discov. 11, 1970–1981 (2021).
Paik, P. K. et al. New treatment options in advanced squamous cell lung cancer. Am. Soc. Clin. Oncol. Educ. Book 39, e198–e206 (2019).
Acknowledgements
This study was supported by National Key Research and Development Program of China (2021YFC2500903, 2021YFC2500905), Natural Science Foundation of Shanghai (22ZR1439200), National Natural Science Foundation of China (82072557, 81871882), Shanghai Municipal Commission of Health and Family Planning Outstanding Academic Leaders Training Program (2017BR055), and Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant (20172005). Some icons or graphic elements in Figs. 2, 3, 4, 5, and 6 are adapted from BioRender.com (2022), retrieved from https://app.biorender.com/. Final schematic illustrations were created and integrated by our original design.
Author information
Authors and Affiliations
Contributions
H.C.L. and Y.Z. conceived and revised the paper. Z.Y.N. and R.S.J. drafted the manuscript and prepared the figures. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Niu, Z., Jin, R., Zhang, Y. et al. Signaling pathways and targeted therapies in lung squamous cell carcinoma: mechanisms and clinical trials. Sig Transduct Target Ther 7, 353 (2022). https://doi.org/10.1038/s41392-022-01200-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01200-x
This article is cited by
-
Cost-effectiveness of immunotherapies for advanced squamous non-small cell lung cancer: a systematic review
BMC Cancer (2024)
-
PHF12 regulates HDAC1 to promote tumorigenesis via EGFR/AKT signaling pathway in non-small cell lung cancer
Journal of Translational Medicine (2024)
-
Implications of ZNF334 gene in lymph node metastasis of lung SCC: potential bypassing of cellular senescence
Journal of Translational Medicine (2024)
-
Wee1 inhibitor PD0166285 sensitized TP53 mutant lung squamous cell carcinoma to cisplatin via STAT1
Cancer Cell International (2024)
-
CTDSPL2 promotes the progression of non-small lung cancer through PI3K/AKT signaling via JAK1
Cell Death Discovery (2024)
