
Abstract
Peptide drug development has made great progress in the last decade thanks to new production, modification, and analytic technologies. Peptides have been produced and modified using both chemical and biological methods, together with novel design and delivery strategies, which have helped to overcome the inherent drawbacks of peptides and have allowed the continued advancement of this field. A wide variety of natural and modified peptides have been obtained and studied, covering multiple therapeutic areas. This review summarizes the efforts and achievements in peptide drug discovery, production, and modification, and their current applications. We also discuss the value and challenges associated with future developments in therapeutic peptides.
Similar content being viewed by others
Introduction
Therapeutic peptides are a unique class of pharmaceutical agents composed of a series of well-ordered amino acids, usually with molecular weights of 500-5000 Da1. Research into therapeutic peptides started with fundamental studies of natural human hormones, including insulin, oxytocin, vasopressin, and gonadotropin-releasing hormone (GnRH), and their specific physiological activities in the human body2. Since the synthesis of the first therapeutic peptide, insulin, in 1921, remarkable achievements have been made resulting in the approval of more than 80 peptide drugs worldwide. The development of peptide drugs has thus become one of the hottest topics in pharmaceutical research.
The first half of the 20th century witnessed the discovery of several life-saving bioactive peptides, such as insulin and adrenocorticotrophic hormone, which were initially studied and isolated from natural sources. The discovery and development of insulin, a peptide with 51 amino acids, has been considered as one of the monumental scientific achievements in drug discovery. It was first isolated by Frederick Banting in 1921 and further developed by Frederick and Charles Best3,4, and was already available for patients with diabetes mellitus just a year after its first isolation. In 1923, insulin became the first commercial peptide drug and has since benefited thousands of diabetes patients to date. However, the production of human insulin during the 20th century could not keep up with the high market demand, and animal-derived insulins, such as bovine and porcine insulin, dominated the insulin market for almost 90 years until they were replaced by recombinant insulin5,6.
More peptide hormones and their receptors with therapeutic potential were identified and characterized from the 1950s to the 1990s7. Meanwhile, the technologies used for protein purification and synthesis, structure elucidation, and sequencing made substantial progress, thus accelerating the development of peptide drugs, leading to nearly 40 peptide drugs being approved worldwide. Notably, synthetic peptides such as synthetic oxytocin8, synthetic vasopressin9, and recombinant human insulin10,11 began to be developed in addition to natural peptides.
Peptide drug development entered a new era with the advent of the 21st century, since when advances in structural biology, recombinant biologics, and new synthetic and analytic technologies have significantly accelerated the process. A sophisticated system of peptide drug development has been established, including peptide drug discovery, drug design, peptide synthesis, structural modification, and activity evaluation. A total of 33 non-insulin peptide drugs have been approved worldwide since 2000 (Table 1). In addition, these peptide drugs are no longer simply hormone mimics or composed simply of natural amino acids. For example, enfuvirtide is a 36-amino acid biomimetic peptide mimicking human immunodeficiency virus (HIV) proteins used in combination therapy for the treatment of HIV-112,13; ziconotide14,15 is a neurotoxic peptide derived from the cone snail Conus magus, which was approved in 2004 and is used to manage severe chronic pain; teduglutide is a glucagon-like peptide 2 (GLP-2) analogue used to treat short bowel syndrome16,17, and is manufactured using a strain of Escherichia coli modified by recombinant DNA technology; and liraglutide is a chemically synthesized analogue of human glucagon-like peptide 1(GLP-1)18,19, made by attaching a C-16 fatty acid (palmitic acid) with a glutamic acid spacer on lysine residue (position 26 in the sequence), which acts as a GLP-1 receptor agonist to manage type 2 diabetes mellitus (T2DM). All these peptide drugs have been used in a wide range of therapeutic areas, such as urology, respiratory, pain, oncology, metabolic, cardiovascular, and antimicrobial applications20,21,22,23,24. To date, more than 170 peptides are in active clinical development (Table 2), with many more in preclinical studies1,7.
Peptide drugs account for a significant proportion of the pharmaceutical market, with worldwide sales of more than $70 billion in 201925, a more than two-fold increase compared with 201326. According to Njardarson et al., the top 200 drug sales in 201927, included 10 non-insulin peptide drugs. Interestingly, the top three sales of peptide drugs were all GLP-1 analogues for treating T2DM, including Trulicity (dulaglutide) ranked at 19 with $4.39 billion retail sales, Victoza (liraglutide), ranked at 32 with $3.29 billion sales, and Rybelsus (semaglutide), ranked at 83 with $1.68 billion sales (Fig. 1).

Top-selling non-insulin peptides worldwide in 2019. Data analysis according to Njardarson’s group27
In this article, we review the historical development of peptide drugs and current advances in peptide drug discovery. We focus on the pharmaceutical characteristics of therapeutic peptides and highlight new technologies that have improved the design, synthesis, modification, and evaluation of peptide drugs, and provide new perspectives in the applications of peptide drugs. We also refer readers to several recent reviews for further reading1,7,28.
Therapeutic peptides: advantages and drawbacks
Therapeutic peptides commonly act as hormones, growth factors, neurotransmitters, ion channel ligands, or anti-infective agents. They bind to cell surface receptors and trigger intracellular effects with high affinity and specificity, with a similar mode of action to biologics, including therapeutic proteins and antibodies. However, compared with biologics, therapeutic peptides show less immunogenicity and have lower production costs29,30,31,32.
Small molecule drugs are known to have an extended therapeutic history with inherent advantages, including low production costs and sale prices, oral administration, and good membrane penetration ability33. Both naturally extracted and chemically synthesized small molecules show competitive price advantages compared with peptides and biologics (proteins or antibodies)34,35. Oral administration of small molecules has the benefits of better safety and improved patient compliance, while their small size also enables them to penetrate the cell membrane to target intracellular molecules33,36. However, their small size also means that it is difficult for them to inhibit large surface interactions, such as protein-protein interactions (PPIs), effectively. PPIs usually occupy a contact area of 1500–3000 A2, while small molecules only cover 300–1000 A2 of the protein surface, due to their limited molecular size37. By contrast, the unique physiochemical properties of peptide drugs, including their larger size and more flexible backbone, enable them to act as potent inhibitors of PPIs38. The clinical use of small molecules is also limited by their low specificity compared with peptide drugs. For example, sorafenib and sunitinib are tyrosine kinase inhibitors that inhibit the tyrosine kinase domain activity of vascular endothelial growth factor (VEGF) receptors, resulting in anti-angiogenic effects that are used to treat cancer patients39,40,41; however, they also target other kinase receptors such as serine/threonine kinase receptors, leading to cytotoxicity42,43,44,45,46.
As natural amino acid-based therapeutics, therapeutic peptides have two intrinsic drawbacks (Fig. 2): membrane impermeability and poor in vivo stability, which represent major stumbling blocks for peptide drug development2,29.
-
1.
Peptides have weak membrane permeability. The membrane permeability of peptide drugs depends on multiple factors, including peptide length and amino acid composition. Peptides are generally unable to cross the cell membrane to target intracellular targets, thus limiting their applications in drug development. Lau et al. reported in 2018 that >90% of peptides in active clinical development targeted extracellular targets, including G-protein coupled receptors (GPCRs), gonadotropin-releasing hormone (GnRH) receptor, Glucagon-like peptide 1 (GLP-1) receptor7.
-
2.
Peptides have poor in vivo stability. Natural peptides consist of chains of amino acids joined by amide bonds, but lack the stability conferred by secondary or tertiary structures. The amide bonds can be easily hydrolyzed or destroyed by enzymes in vivo, upon exposure to the environment, without any protection. These inherent chemical properties make the peptides chemically and physically unstable, with a short half-life and fast elimination in vivo47.

Peptides versus small molecules and biologics. Comparison of advantages and drawbacks between peptides and small molecules or biologics
These intrinsic advantages and disadvantages of peptides present both challenges in peptide drug development and also opportunities and directions for peptide drug design and optimization.
Developmental path of therapeutic peptides: discovery, production, and optimization
Peptide drug discovery
Natural peptides/hormones in the human body
The history of peptide drug discovery started by exploiting natural hormones and peptides with well-studied physiological functions for treating diseases caused by hormone deficiencies, such as a lack of insulin required to regulate blood glucose levels in patients with T1DM or T2DM. Diabetes is treated either by insulin injection or by stimulating insulin secretion-related targets such as GLP-1 receptor, to produce insulin48. Searching for natural peptides and hormones or replace them by animal homologues, such as insulin, GLP-1, somatostatin, GnRH, 8-Arg-Vasopressin, and oxytocin, were the initial strategies used for peptide drug discovery and development (Fig. 3). However, the drawbacks associated with these natural peptides aroused interest in optimizing their natural sequences, leading to a series of natural hormone-mimetic peptide drugs.

Structure of human insulin and human insulin-derived drugs. Structure of human insulin (left, PDB: 1XDA). Modifications on its residues (B-Chain: B3: Asn, B28: Pro, B29: Lys; A-Chain: A21: Asn) resulted in several short- and long-acting insulin drugs (right, see table)
Peptides mimicking hormones
GLP-1 derived peptide drugs (Fig. 4a): GLP-1 is a 37-amino acid peptide that regulates insulin production and secretion49, with a very short half-life in vivo. Extensive efforts have been made to modify its sequence to enhance the stability of this hormone, while maintaining its potency and pharmacological effect50,51, leading to the development of the three top-selling anti-T2DM peptide drugs: Trulicity (dulaglutide), Victoza (liraglutide), and Ozempic (semaglutide).

Sequences and structures of natural hormones GLP-1 and GnRH and their peptidomimetic drugs. a Liraglutide is a GLP-1 derived peptide drug, modified on 26th residue (K) of its natural sequence. b Leuprolide and degarelix are modified from the natural sequence of GnRH
Gonadotropin-releasing hormone (GnRH) derived peptide drugs (Fig. 4b): GnRH is a peptide containing 10 amino acids that is produced by GnRH neurons in the hypothalamus52. Modification of the native sequence of GnRH has led to the development of several peptide drugs, such as leuprolide and degarelix. Leuprolide has the same biological activity as GnRH by activating GnRH receptors, and is used as a GnRH receptor agonist for treating hormone-responsive prostate cancer, endometriosis, uterine fibroids, and precocious puberty53,54. While the sequence of degarelix is optimized from GnRH, it acts as a GnRH antagonist by competitively binding to the GnRH receptor and is used to treat terminal prostate cancer55.
Many other approved peptide drugs are also derived from natural hormones1, including octreotide, a somatostatin mimic peptide drug, used for the treatment of growth hormone producing tumors and pituitary tumors56,57; desmopressin, an 8-Arg-vasopressin mimicking peptide drug, used for diabetes insipidus and nocturia58; carbetocin, an oxytocin homologue used to treat amenorrhea59 and atosiban, an oxytocin antagonist for suppressing premature labor60.
Peptides identified from natural products
Many bioactive peptides from bacteria, fungi, plants, and animals possess therapeutic properties, such as snake venom, which is considered as a vascular endothelial growth factor (VEGF) analogue, VEGF-F or svVEGF61,62,63. They are usually disulfide-rich cyclic peptides of no more than 80 residues, which can induce cytotoxicity by targeting ion channels and other membrane-bound receptors1,64. Venom peptides from snakes and scorpions have been modified for therapeutic applications. In addition, exenatide (Fig. 5a), optimized from Gila monster venom65 is a GLP-1 agonist and ziconotide, a venom peptide derived from Conus magus, has been used to treat chronic neuropathic pain66,67.

Sequences and structures. Exenatide (a) and lugdunin (b)
Non-ribosomal peptides (NRPs) comprise another class of peptides identified from natural products. The non-standard residues contained in the sequence mean that NRPs are not produced through the traditional biosynthesis pathways via ribosomes68, but are produced by non-ribosomal peptide synthetases via a pathway consisting of initiation, elongation, and termination modules69,70. Compared with peptides synthesized by ribosomes, NRPs are more resistant to hydrolases and show increased stability in vivo. The most-studied NRPs are mainly derived from bacteria and fungi, including vancomycin, cyclosporin, lugdunin71,72. (Fig. 5b), and teixobactin with antibacterial activities, and a-amanitin, nanocystin A, and actinomycin with anti-tumor activities73,74. In addition, cyclodepsipeptides are cyclic peptides that comprise a specific class of NRPs usually identified in plants75,76,77, such as enniatin B and emodepside78,79. These peptide drugs display enhanced plasma stability that enables their oral delivery. However, the synthesis and structure-activity relationships study of NRPs represent one of the most challenging and exciting areas of research for NRPs.
Rational design of peptides based on Protein–Protein Interactions
Developments in proteomics and structural biology have led to the discovery of many Protein-Protein interactions (PPIs) involved in most cellular processes and biological functions80,81. Over 14,000 PPIs, accounting for only about 1% of all PPIs in the human body, have been studied to date82. PPIs also regulate many essential cellular pathways in human diseases and are thus potential drug targets83. Peptides contain intrinsic advantages as inhibitors or activators of PPIs compared with small molecules and antibodies. Therefore, a new peptide drug discovery technology based on the known crystal structure of PPIs has thus been developed: the rational design of peptides. It is considered to be a promising strategy for the discovery of new peptide drug candidates84,85.
The rational design of peptides involves computer-assisted bioinformatics technology based on the resolved crystal structure of the target PPIs. Bioinformatic and computational analysis of the PPI binding interface enables the essential amino acids on the surface of the two interacting proteins to be identified. These essential amino acids contribute the major Gibbs energy of the PPIs and are commonly called “hotspots”86,87. Hotspots may be a continuous fragment of the protein or dispersed residues on different secondary structures of the protein. The design of peptide modulators for PPIs is based on these hotspots, either directly using the continuous fragment or using a strategy to link the dispersed residues as initial sequences88. However, further peptide development and structure optimization including peptide cyclization and backbone modification are required to improve their activity and physicochemical properties89,90. For example, identification of the essential peptide residues and the proposed substitution of non-essential residues via study of the structure-activity relationship, and chemical modification of the sequence to stabilize the peptide secondary structure, including turns, helices, hairpins, and extended conformations, can be applied to enhance the bioactivity and improve the physicochemical properties91,92.
Discovery of peptide drug candidates by phage display
Phage display is a highly effective and robust technology used to identify ligands of biological targets, first reported by Smith in 198593. Phage display uses recombinant technologies to engineer target ligands on the surface of the bacteriophage94. Only peptides containing proteinogenic amino acids, rather than NRPs, are produced in the phage. This high-throughput sequencing method can be used to identify drug leads, including antibodies and peptides95,96. Phage display has been widely used to discover new peptide ligands. Lerner et al. reported the discovery of potent peptide analogues of GLP-1 and other membrane receptor ligands by phage display, including proteins, peptides, and venoms, which mainly act as agonists97,98,99,100. In addition, peptides targeting transforming growth factor (TGF)-β1101 or epidermal growth factor receptor (EGFR)102, and peptide antagonists that disrupt the fibroblast growth factor (FGF)-1-FGFR1 interaction103 are good examples of peptide drugs discovered by phage display. Recent developments in phage display technology have focused on searching for more efficient screening protocols to simplify ligand selection among enormous amounts of data, such as by reducing phage panning cycles104. Heinis et al. used an “on-phage” modification technology to obtain chemically modified peptides from traditional phage display to obtain a bis-thioether cyclic peptide105. Another strategy involves developing novel display approaches. For example, Schumacher et al. developed a mirror-image phage display to explore D-chirality peptides106,107, and Szostak et al. performed mRNA display to discover and select macrocyclic peptides with unnatural amino acids108,109,110. Suga et al. used ribosomal display to exploit lead peptides, including bioactive macrocyclic peptides, containing D-amino acids and unnatural amino acids111,112,113. These developments have allowed the construction of numerous display libraries for the discovery of new peptide candidates.
Synthesis and modification of therapeutic peptides
The discovery of potential therapeutic peptides is the first step peptide drug development, followed by chemical or biological peptide synthesis and sequence modification to improve its pharmacological properties. Here we summarize the fundamental technologies utilized for peptide production and modification.
Chemical synthesis of peptides
The chemical synthesis of peptides is well-developed, particularly solid-phase peptide synthesis (SPPS) technology developed by Merrifield in 1963114. SPPS technology has since been remarkably improved in terms of its methodology and synthetic materials and plays a crucial role in modern peptide production. It facilitates peptide synthesis by combining amino acid coupling and deprotection in one simple reactor, which has further led to the invention of automatic peptide synthesizers. Compared with recombinant technology, the crude peptides obtained by SPPS are more monotonous, without other biological compounds such as enzymes, DNA and RNA fragments, non-related proteins, and peptides. Moreover, the impurities in the final SPPS product are easily identified because they are mainly derived from incomplete or side reactions during the synthesis procedure115, making subsequent purification relatively uncomplicated116.
SPPS consists of a cycle of coupling the carboxylic group of amino acids to a solid polymeric resin, and liberation of the amine group from the protection group (Fig. 6). Various resins, such as 4-methylbenzhydrylamine (HMBA) resin, Wang resin, 2-chlorotrityl chloride (CTC) resin, and Merrifield resin, are used to introduce either amide or free carboxylic groups into the C-terminal of peptide. The modern peptide industry has developed various functional resins by coupling the resins with different linkers, enabling the synthesis of long peptides and peptide cyclization in the solid phase117. During synthesis, the amine group of the amino acids and the side chains are usually protected by different chemical groups, which cause peptide aggregation and reduce the purity of the crude peptides. Two major SPPS strategies: Fmoc-SPPS and Boc-SPPS have been developed to remove the predominant amine protection groups, fluorenylmethyloxycarbonyl (Fmoc) and t-butyloxycarbonyl (Boc), respectively118,119.

A general process of solid-phase peptide synthesis (SPPS) with Fmoc protected amino acids (Fmoc-AA-OH). Fmoc-SPPS consists a cycle of coupling Fmoc-AA-OH to a solid polymeric resin and deprotection of Fmoc to liberate amino groups. The whole process can be carried out in a sieve reactor till the final peptide is cleaved from the resin
Boc-SPPS uses trifluoroacetic acid solution to remove the amine protection groups and hydrogen fluoride solution to cleave the final peptide, but these processes are associated with irritating odor and toxicity. Fmoc can be removed under milder conditions, and the Fmoc-SPPS strategy is thus often preferred120. However, Boc-SPPS has advantages for long peptide synthesis, because trifluoroacetic acid deprotection effectively destroys the aggregation during the peptide synthesis121. Fmoc-SPPS research is currently focused on resolving two major problems, including aggregation during long peptide synthesis and the formation of aspartimides for certain sequences118. Multiple methods have been utilized, including applying low-substitution resins to separate peptide chains122, microwaves to reduce the reaction time123, mixed solvents as a reaction solution124, and pseudoprolines to break the H-bond of the backbone to avoid or reduce aggregation during SPPS121. Aspartimide formation during Fmoc-SPPS significantly decreases the purity of the crude peptides. The solutions applied to reduce aspartimide formation were using microwaves to reduce the reaction time125, or using N-α-alkyl Asp–Gly dipeptide126, or adding 1-hydroxybenzotriazole (HOBt)127, Oxyma Pure128 during the deprotection process.
The synthesis of peptides of <50 residues by Fmoc-SPPS is relatively routine, but the chemical synthesis of longer peptides (>50 amino acids) is still challenging, especially in large-scale manufacture. Laboratory-scale peptide synthesis tends to be carried out automatically with the help of modern automated peptide synthesizers, such as CEM Liberty PRIME and CSBio II. These new automatic peptide synthesizers can provide sequential and multi-parallel peptide syntheses of up to 192 different sequences, using infrared or microwave heating to reduce the reaction time, and sometimes using ultraviolet monitoring to ensure the deprotection process129,130. Such synthesizers are extremely helpful for laboratory-scale peptide synthesis, producing the desired peptides rapidly for further structural and functional studies. However, there are limited applications of infrared and microwave heating to large-scale peptide manufacture due to a lack of large equipment and nonhomogeneous overheating, which may lead to the production of byproducts131. Most good manufacturing practice (GMP) thus prefers mild reaction conditions to minimize side reactions and relative impurities, and the large-scale production of long peptides (>50 amino acids) thus remains challenging.
The development of chemical peptide synthesis, especially by SPPS, has significantly accelerated the development of therapeutic peptides. Some recombinant peptide drugs, such as oxytocin and teriparatide, use chemical synthesis to produce active pharmaceutical ingredients. The chemical synthesis of peptides also permits their kaleidoscopic modification.
Chemical modification of peptide and peptidomimetics
As a particular class of therapeutic agents, the biological activity of peptides is intimately related to their chemical structure. Following the synthesis of peptides, they need to be modified using medicinal chemistry techniques to mimic, stabilize, or construct an ideal secondary structure to improve their biological activity and achieve selectivity, stability, and solubility of the peptide drugs132.
Before modification of the lead peptide drug candidate, it is necessary to identify the minimum active sequence with the desired biological properties. Classical sequence scanning, termed alanine-scan29,133,134, is then commonly used to replace each residue with alanine to produce a series of lead peptide analogues to determine which key residues confer the biological activity of the lead peptide: a decrease in activity suggests that the replaced residue was important, whereas a non-significant reduction of activity suggests that the replaced residue was redundant. Further modifications of the replaceable residues and C- and N-termini of the lead peptide are then carried out to produce the final peptide drug135.
Backbone modification of peptides
One of the main reasons for backbone modification is to improve the proteolytic stability of the peptide. Proteolytic sites in the peptide can be identified by stability studies and metabolite determination136. Backbone modification includes the substitution of L-amino acids by D-amino acids137,138, insertion of methyl-amino acids137,139, and the incorporation of β-amino acids140 and peptoids141,142,143. Introducing these non-natural amino acids into the peptide sequence, particularly at the proteolysis site, is an effective strategy for extending the plasma half-life of peptide drugs. A successful example is selepressin, which was derived from vasopressin and has similar target selectivity but a longer plasma half-life144,145.
Side chain modification of peptides
Side chain modification of peptides is achieved by replacing the natural amino acids with their analogues during peptide synthesis, to improve their binding affinity and target selectivity1,146. Variants of natural amino acid analogues such as homoarginine, benzyloxy-tyrosine, and β-phenylalanine are commonly commercially available147, and can be conveniently used to chemically modify the peptide side chain during peptide synthesis148. Several GLP-1 analogue drugs such as liraglutide and semaglutide have modified side chains48.
Mimicking and stabilization of secondary structures by backbone and side chain modification
The weak forces in peptides, such as hydrogen bonds, van der Waals forces, and intramolecular hydrophobic interactions are not adequate for a stable secondary structure conformation. Additional modifications of the backbone, N- or C-termini, or side-chains to mimic the structures of natural products or hot spots in PPI and stabilization of secondary structures are therefore needed to produce promising peptide drug candidates149,150.
-
Peptide cyclization. Cyclization is a common peptide modification technique that can include various strategies, such as head-to-tail, backbone-to-side chain, and side chain-to-side chain cyclization (Fig. 7)151,152,153. Peptide cyclization can increase proteolytic stability154,155, and cell-permeability156,157,158, and allows mimicking and stabilization of the peptide secondary structure. Without being connected to other peptides, a single peptide sequence cannot form loop or turn structures, but cyclization facilitates the formation of these secondary structures by pre-organizing intramolecular interactions159,160. Peptide cyclization is also commonly applied to stabilize other secondary structures, such as α-helixes and β-sheets161,162,163.
Fig. 7 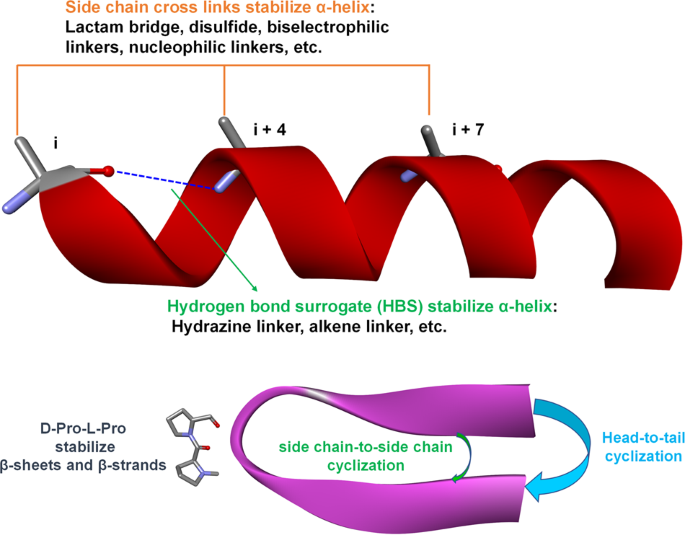
Strategies of peptide cyclization and stabilization of α-helices, β-sheets, and β-strands. The establishment of intramolecular cross-links can stabilize different secondary structures of peptides. Side chain cross-links between i and i + 4 or/and i + 7 and hydrogen bond surrogate cross-links can stabilize α-helices. Side chain-to-side chain, head-to-tail, and side chain-to-tail cyclization can stabilize turn, loop and β structures (β-sheets and β-strands). The D-Pro-L-Pro scaffold can specifically stabilize antiparallel β-hairpins
-
Peptide mimicking of α-helices and stabilization. Helices are one of the commonest types of protein secondary structures, representing about 30%-40% of all protein structures164. The α-helix is formed by intramolecular hydrogen bonds165 and accounts for 90% of helix structures165. Mimicking the α-helix in peptides enables the identification of modulators of PPIs. The α-helix can be stabilized either by building cross-links through side chains or replacing hydrogen bonds by covalent bonds (referred as hydrogen bond surrogates, HBS). In the α-helix structure, the side chains of amino acids at positions i, i + 4, and i +7 are on the same side, and building cross-links through i and i + 4 or i and i +7 effectively approach backbone atoms and help to form hydrogen bonds in helical structures166,167,168. Great efforts have been made to investigate different cross-links, such as lactam-based cross-links (Fig. 7), with the formation of a lactam bridge through the side chain of glutamic acid or aspartic acid with lysine169, the formation of disulfide bonds by replacing residues with cysteine or homocysteine170, and biselectrophilic linkers171,172. Stapled peptides represent a recent new cross-linking approach introduced to stabilize the α-helix structure, using non-natural electrophilic amino acids to replace residues at the i and i + 4 or i and i +7 position, and form ligations with nucleophilic cross-links154,173,174. The HBS modification strategy involves replacing one hydrogen bond of the α-helix peptide with a covalent bond to pre-organize the helical structure. Cabezas and Satterthwait first used hydrazine links to build an HBS peptide to mimic an α-helix175. The Arora group has also carried out extensive work on HBS peptides, using alkene linkers to stabilize the α-helix176,177,178,179. They recently started to use the HBS strategy to stabilize β-hairpins180,181,182, as well as the biological activities of these modified peptides181,183,184,185. We also used the HBS peptide modification strategy in our previous work, focusing on designing a full SPPS pathway to simplify the application of HBS in α-helix mimicking and stabilization186,187.
-
Peptide mimicking of β-strands and β-sheets. β-sheets and β-strands represent another class of protein secondary structures, based on turn mimics. The modification of peptides to stabilize β-sheets is usually achieved by the introduction of D-amino acids, such as D-Pro, to form a turn structure in the sequence. D-Pro-L-Pro templates are a well-known scaffold for stabilizing antiparallel β-hairpins in several successful PPI inhibitors188,189. Macrocyclization or amyloid beta-sheet mimics have also been applied to create β-sheets and β-strand structures190,191,192,193.

Chemical modification is an effective method of producing peptide analogues with the desired structures. The improved stability and activity have resulted in the introduction of several peptide drugs into the clinic, such as selepressin, liraglutide, and semaglutide. However, some modifications cannot improve the proteolytic stability and activity simultaneously. For example, the insertion of D-amino acid can usually help to extend the plasma half-life of the peptide, but peptides with D-amino acid modification rarely exhibit effective biological activity1,137,194.
Peptide production by recombinant technology
Chemical synthesis is the preferred method for the industrial preparation of peptides, because it can introduce versatile synthetic building blocks beyond the proteinogenic amino acids, such as unnatural amino acids, and biochemical or biophysical probes, allowing further modification or conjugation. Furthermore, the chemical synthesis process can be fully automated and easily scaled up. It provides a convenient and efficient approach for producing short- and medium-sized peptides, but the chemical synthesis of long peptides remains challenging, and alternative strategies are therefore required.
In addition to chemical synthesis, therapeutic peptides can be prepared by various biological methods, such as isolating bioactive peptides from natural sources by extraction195, enzymatic synthesis196, fermentation197,198, recombinant DNA technology199,200, and semisynthesis201,202. These approaches can be applied exclusively or in combination, depending on the complexity and difficulty of preparing the peptide203,204.
The practice of isolating peptide drugs from natural sources can be traced back to the 1920s, when insulin was first isolated from livestock pancreata and used to treat diabetes205,206, saving hundreds of thousands of lives. The pioneering success of insulin led to increasing public enthusiasm for peptide therapeutics, and several other animal-derived peptide drugs subsequently successfully entered clinical use, such as adrenocorticotropic hormone207 and calcitonin208. Non-ribosomally synthesized peptides represent another important family of natural sources for identifying and producing peptides with therapeutic potential, as exemplified by vancomycin and cyclosporin. Unlike ribosomally synthesized peptides or proteins, the synthesis of non-ribosomally synthesized peptides is controlled by clusters of genes encoding non-ribosomal peptide synthetases rather than the endogenous translational machinery, leading to the production of structurally and functionally diverse peptides, and allowing these molecules to overcome the inherent limitations of common peptide drugs. Venoms and toxins are recognized as valuable natural sources as starting points for identifying bioactive peptides208,209,210, and other natural sources, such as cyclotides and lantipeptides have also been studied and exploited211,212,213. Enzymatic synthesis is suitable for the synthesis of short peptides, such as dipeptides and tripeptides, and enzymatically synthesized peptides have been successfully applied for the production of food additives, pharmaceuticals, and agrochemicals. Fermentation has been well-documented as an eco-friendly approach for producing bioactive peptides, such as in the manufacture of cyclosporine214. Recombinant DNA technology enables the production of peptides and proteins with defined sequences and homogeneity. This approach is particularly useful for manufacturing long or complicated peptides with multiple disulfide bonds, which can otherwise be difficult to synthesize chemically. Human insulin and growth hormone are representative examples of the many available peptide drugs made using recombinant DNA technology. In addition, recombinant DNA technology can be combined with genetic code expansion and other novel technologies to introduce desired functional groups into the molecules via the incorporation of unnatural amino acids, as discussed below. Semi-synthesis provides a flexible approach for producing large bioactive polypeptides by linking synthetic peptides and recombinant DNA-expressed peptides215,216,217, and is a particularly useful approach when multiple artificial modifications are needed.
Peptides modification by genetic code expansion
Natural proteins are synthesized from 20 canonical amino acids, and this limited and conservative repertoire of amino acids significantly restricts the diversity and complexity of protein structures and functions. Genetic code expansion was developed two decades ago as a technology to overcome this limitation (Fig. 8)218,219. Genetic code expansion allows for the site-specific incorporation of non-canonical amino acids (ncAAs) with novel chemical and physical properties into a growing polypeptide during protein translation220,221. Four components are required to achieve this: 1) an ncAA with the desired chemical and physical properties; 2) a unique codon that specifies the ncAA, e.g., an amber stop codon (UAG) or quadruplet codon; 3) an orthogonal tRNA that suppresses the unique codon and does not crosstalk with its endogenous counterparts; and 4) an orthogonal amino-acyl tRNA synthetase that can specifically charge the ncAA onto the orthogonal tRNA and does not crosstalk with the endogenous amino-acyl tRNA synthetase/tRNA pairs218,222,223,224.

Scheme of genetic code expansion. Genetic code expansion enables the site-specific incorporation of an noncanonical amino acid (shown in green filled circle) into a growing peptide chain by suppressing an unique codon (e.g., amber stop codon)
More than 200 ncAAs with diverse functionalities have been genetically encoded into different organisms to date, such as Escherichia coli, yeast, mammalian cells, viruses, and even animals, providing an invaluable toolbox for protein studies and engineering225,226,227,228,229,230,231,232. This expanded set of building blocks, including bioorthogonal chemical conjugation partners, metal chelators, photo-crosslinkers, proximity-enabled crosslinkers, photocaged amino acids, amino acids with post-translational modifications (phosphorylation, sulfation, acylation, etc.), redox-active amino acids, and infra-red, nuclear magnetic resonance, fluorescent probes, has been widely used in the study, manipulation, and evolution of proteins233,234,235,236,237,238,239,240,241,242. The ability to genetically encode diverse ncAAs allows for the rational optimization and production of chemically modified recombinant proteins with defined structures, functions, and stoichiometries243,244. Here, we focus on the application of genetic code expansion in the evolution of therapeutic peptides and proteins.
PEGylation of peptides and proteins
Short protein and peptide therapeutics produced by genetic code expansion also have a short half-life because of their poor pharmacokinetics, including fast serum degradation and quick elimination. Attaching a polymer is one approach for extending the half-life of protein therapeutics245. PEG is formed by repetitive units of ethylene oxide and is a non-biodegradable, non-toxic, low-immunogenic polymer. PEGylation can increase the effective molecular weight of proteins to reduce their renal clearance by kidney filtration. The PEG moiety can also shield the proteins from digestion by proteolytic enzymes via increased steric hindrance, and help increase absorption by increasing the target protein’s water solubility246. These advantages make PEGylation a prevalent strategy for modifying therapeutic proteins, and PEGylation has been applied for optimizing protein therapeutics since the 1970s, with great success. There are currently >10 PEGylated protein therapeutics in the market, with more potential candidates in clinical trials247.
Conventional PEGylation often occurs at Lys or Cys residues248. However, if the target protein includes more than one reactive Lys or Cys residue, conjugation can occur randomly at any of these residues due to a lack of selectivity, leading to the generation of heterogeneous conjugation products that are hard to separate. Techniques allowing site-specific PEGylation in which the PEG moieties can be attached to proteins with selectivity and positional control are thus needed (Fig. 9).

PEGylation of therapeutic peptides and proteins via genetic code expansion. Azide or acetyl groups are introduced into therapeutic peptides and proteins by genetic code expansion to allow downstream PEGylation modifications
Genetic code expansion has provided a valuable tool for protein PEGylation. One approach involves genetically encoding an ncAA (Fig. 10) containing a bioorthogonal chemical handle at the desired location into the target protein, followed by conjugation to PEG via a bioorthogonal reaction. In 2004, Deiters et al. reported the first ncAA-mediated mono-PEGylation method based on the genetic incorporation of p-azido-phenylalanine (pAzF) in yeast249. An alkyne-derivatized PEG chain was site-specifically attached to superoxide dismutase (SOD) via a copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) click reaction with pAzF, and the resultant SOD showed similar enzyme activity to the wild-type protein. Zhang et al.250 showed that this mono-PEGylation method could also be applied to interferon (IFN)-α2b. An azide-bearing ncAA, NEAK, was site-specifically incorporated into IFN-α2b at distinct locations, allowing orthogonal and stoichiometric conjugation to PEG via a copper-free cycloaddition reaction. Three resultant IFN-α2b variants showed significantly higher biological activities and better pharmacokinetic profiles than other variants and the wild-type molecule in rodent models. The above examples demonstrate that mono-PEGylation can be generally applicable to various proteins.

Non-canonical amino acids described in this review
In 2011, Cho et al.251 reported the first clinical trial of recombinant proteins produced by genetic code expansion. Twenty human growth hormone (hGH) variants were made by incorporating p-acetylphenylalanine (pAcF) at distinct locations, followed by site-specific conjugation with PEG. One hGH variant with mono-PEGylation at residue 35, designated ARX201, presented favorable pharmacodynamic properties in GH-deficient rats, and comparable efficacy and safety to native hGH therapy, but with enhanced potency and reduced injection frequency, in adult GH-deficient patients. Wu et al.252 subsequently generated an hGH variant with combinatorial PEGylation at residues 35, 131, and 145 by genetically encoding multiple NEAK at these locations. The resultant multi-PEGylated hGH variant showed reduced immunogenicity and improved pharmacokinetic properties compared with mono-PEGylated hGH, without loss of bioactivity, and greater stability than mono-PEGylated hGH in rodent models. These examples illustrate the usefulness of genetic code expansion for optimizing therapeutic proteins and peptides.
An alternative site-specific PEGylation method involves directly introducing PEG-containing ncAAs into target proteins via genetic code expansion. Shozen et al. site-specifically incorporated ncAAs containing PEG4, PEG8, and PEG12 chains by suppressing a quadruplet codon using a cell-free translation system253. Tada et al. used a similar strategy to introduce longer PEG chains ranging from PEG4 to PEG24 into polypeptides by suppressing an amber stop codon254. Fu et al.255 recently introduced e-N-heptanoyl-l-lysine (HepoK) into GLP-1 (Fig. 10), and the resultant GLP-1 (HepoK) demonstrated stronger binding affinity towards human serum albumin (HSA) than wild-type GLP-1, as well as longer-lasting effects in terms of decreasing blood glucose levels, thus providing a powerful tool for studying protein lipidation.
Genetic code expansion and ncAAs have also been utilized to generate different types of vaccines, including peptide vaccines such as subunit, conjugated, and live-attenuated vaccines256,257. Grunewald et al.258 first demonstrated that incorporating an immunogenic ncAA into a protein of interest could break the immunological tolerance of self-proteins and evoke an immune response in animal models. Specifically, a single mutation of p-nitrophenylalanine (pNO2F) or phenylalanine was introduced into murine tumor necrosis factor-α (mTNF-α) at position 86 to generate mTNF-α (pNO2F) and mTNF-α (Phe), respectively. The resultant mTNF-α (pNO2F) induced a high-titer antibody response in mice, whereas mTNF-α (Phe) did not. In addition, the antibodies induced by mTNF-α (pNO2F) were found to be highly cross-reactive with native mTNF-α and protected mice against lipopolysaccharide (LPS)-induced death. In subsequent mechanistic studies, Grunewald et al.258 revealed that mTNF-α (pNO2F) mutants led to T cell-dependent polyclonal and anti-mTNF-α IgG antibody responses that were sustained for at least 40 weeks, and protected mice from severe endotoxemia induced by LPS challenge. This approach also elicited a high-titer IgG antibody response to murine retinol-binding protein, suggesting that this may be a generally applicable method for converting other weakly immunogenic self-proteins into vaccines. In a follow-up experiment259, besides ncAA (pNO2F), the incorporation of somatic mutations (Tyr in mTNF-α and Phe in EGF) and post-translational modifications (3NO2Tyr and SO3Tyr) at specific locations in self-proteins also elicited robust IgG antibody responses against the native proteins. The above results suggested that the site-specific incorporation of immunogenic ncAAs and certain natural post-translational modifications (PTMs) could break the immunological tolerance of self-proteins and produce therapeutic vaccines.
Wang et al.260 incorporated multiple ncAAs with a phenylalanine backbone into in Mycobacterium smegmatis, Bacillus Calmette-Guérin, and Mycobacterium tuberculosis to facilitate the study and development of tuberculosis vaccines. It is difficult to manipulate intact and live viruses using conventional chemical modification methods, due to the fragile nature and complicated assembly process of mammalian viruses. To overcome this challenge, Lin et al.261 reported the first example of site-specific incorporation of ncAAs into intact and live viruses followed by selective labelling, without loss of infectivity. Specifically, a panel of pyrrolysine analogues was genetically encoded into the envelope protein of hepatitis B virus (HBV) and assembled into live hepatitis D virus (HDV) in human hepatocytes, with stringent selectivity and high efficiency. By screening different incorporation sites, the viral infectivity was fully maintained. In addition, the ncAA-modified virus can be readily pulled down or conjugated via a copper(I)-catalyzed alkyne-azide cycloaddition click reaction. Wang et al.230 also applied an ncAA-mediated genetic switch to develop a live-attenuated HIV-1 vaccine. A panel of phenylalanine analogues was genetically encoded into the essential proteins of HIV-1 to control its replication, and HIV-1 replication could be precisely turned on and off via this approach. In a follow-up study, Yuan et al.262 merged the ncAA-mediated genetic switch into the viral genome and developed multi-cycle replicable HIV-1 based on amber suppression, representing a significant step towards the development of an HIV-1 vaccine. Chen et al.263 achieved precise control of HIV-1 replication via suppression of a quadruplet codon, which is not used by the native protein translation system, therefore minimizing the potential of proofreading and enhancing the safety of the vaccine. This method was also applied to influenza A virus231, and generated safe and effective live-attenuated vaccines that elicited robust protective immune responses in animal models, suggesting that ncAA-mediated live-attenuated vaccine is a generally applicable approach.
Covalent peptide/protein drugs
Small molecule covalent drugs have many advantages compared with noncovalent drugs, such as increased biochemical efficiency and potency, improved pharmacokinetics, prolonged duration of action, reduced dosage and dosing frequency, and potent inhibition of intractable targets264. Safety concerns about their low selectivity and the potential immunogenicity of covalent drug-protein adducts mean that the development of small molecule covalent drugs has been intentionally avoided265. However, the development of activity-based protein profiling and other recent technologies mean that small molecule covalent drugs have regained attention, and several small molecule drugs that act by a covalent binding mechanism have been approved for marketing266.
Theoretically, covalent protein drugs should offer similar advantages to small molecule drugs. However, due to their inherent inability to form covalent bonds of natural proteins, the therapeutic potential of covalent protein drugs has not been fully explored. Li et al.267 recently reported on a proximity-enabled reactive therapeutics (PERx) strategy to develop covalent protein drugs. They genetically incorporated the latent bioactive ncAA, fluorosulfate-L-tyrosine (FSY)268, into human programmed cell death protein 1 (PD-1) at position 129 and showed that the resulting PD-1(FSY) formed covalent bonds selectively with its natural ligand, PD-L1, in vitro and in vivo. Strikingly, PD-1(FSY) significantly enhanced the bioactivities of human naïve T cells and engineered chimeric antigen receptor T cells, compared with wild-type PD-1. PD-1(FSY) showed more potent inhibition of tumor growth and had equivalent or greater anti-tumor effects than a therapeutic anti-PD-L1 antibody in several immune-humanized mouse models. They then applied PERx to the covalent inhibition of the HER2 receptor by a FSY-modified affibody, illustrating that PERx could provide a general platform for developing covalent protein drugs. Compared with noncovalent protein drugs, PERx drugs can be used in their original form and do not require additional modifications to extend their half-life, because the covalent binding decouples the drug efficacy from its pharmacokinetics. Moreover, PERx allows small-protein biologics such as PD-1 (15.6 kDa) to be used as therapeutics, thus greatly expanding the scope of therapeutic proteins. In addition, PERx can minimize the off-target effect due to the inherent affinity between the protein drug and its target, as well as the proximity-driven crosslinking mechanism of the latent bioactive ncAA. These advantages mean that the PERx strategy has the potential to provide a general platform to develop novel covalent protein drugs. The chemistry behind the PERx strategy and more examples of covalent proteins have been reviewed in detail elsewhere269.
Lipid and larger proteins are frequently conjugated to improve the pharmacokinetics of covalent peptide drugs270,271,272. Approved peptide drugs, such as liraglutide, semaglutide, and insulin degludec, were conjugated with C14/16/18 fatty acids, which increased their plasma circulation times and reduced their degradation during kidney elimination270. Two plasma proteins, serum albumin and immunoglobulin, are also used to prolong the peptide-circulation times by increasing their molecular weight, thereby exceeding the molecular weight cut-off for glomerular filtration. For example, this strategy was used to extend the half-life of dulaglutide and albiglutide, administered by once-weekly injections273,274.
Developments in peptide drug delivery
Peptide modifications allow peptides to achieve better activity and plasma stability, and become more drug-like. However, the inherent properties of peptides mean that they are easily hydrolyzed by digestive enzymes in the stomach and intestine, and most peptide drugs are thus administrated by injection. Recent studies have investigated routes of peptide drug delivery to overcome these drawbacks275.
Co-formulation with permeation enhancers is a promising strategy to enable the oral administration of peptide drugs. Semaglutide conjugated with C18 fatty acid was approved for administration by once-weekly subcutaneous injection276,277, with greater plasma stability than other GLP-1 analogues. Even more encouragingly, the co-formulation of semaglutide with sodium N-[8-(2-hydroxybenzoyl amino]caprylate (SNAC) was approved for oral administration to treat T2DM. Co-formulation with SNAC prevents the destruction of semaglutide in the stomach by decreasing the efficacy of digestive enzymes. The hydrophobic SNAC molecules also increase the lipophilicity of semaglutide, thus improving its transcellular absorption through the gastric membrane and its transport into the systemic circulation278,279. Co-formulation with other permeation enhancers, enzyme inhibitors, and hydrogels have also been used to allow the oral administration of other peptide drugs, such as octreotide and insulin, which are now in clinical trials280,281. More strategies, including pulmonary administration, transdermal delivery, and the use of implantable pumps, are currently under investigation for the delivery of specific peptide drugs282,283, including the development of inhalable insulin and micro-implantable pumps for insulin delivery. We expect these technologies to be applied for more peptide drugs in the coming years.
Current development and application of therapeutic peptides in diseases
Therapeutic peptides in the treatment of diabetes mellitus
T2DM is caused by an acquired insulin deficiency and is common in middle-aged and older people. T2DM has been successfully treated with peptide drugs, including GLP-1 receptor agonists (GLP-1RAs) and the best-known peptide drug, insulin. GLP-1 is an endogenous growth hormone secreted by L-cells in the ileum. Its receptors are present in pancreatic β-cells, the peripheral and central nervous systems, heart and blood vessels, kidneys, lungs, and gastrointestinal mucosa (Fig. 11). GLP-1 interacts with its receptor to stimulate islet β-cells to secrete insulin, inhibit the release of glucagon by islet α-cells, increase satiety, and delay gastric emptying in a glucose-dependent manner284. Endogenous GLP-1 is degraded by dipeptidyl peptidase-4 (DPP-4) and is rapidly inactivated. In order to prolong the stimulation time of GLP-1 receptors, synthetic GLP-1RAs are required to prevent its degradation. Since the first GLP-1RA, exenatide, was approved by the US Food and Drug Administration (FDA) in 2005, several GLP-1RAs have entered the clinic, including liraglutide (2009), lixisenatide (2013), dulaglutide (2014), and semaglutide (2017)285. After injection, these GLP-1RAs effectively reduce glycosylated hemoglobin and average blood glucose levels and improve fasting blood glucose286.

Mechanisms of GLP-1 and GLP-1RA peptide drugs in regulation of T2DM. GLP-1 and GLP-1RA peptide drugs treat T2DM by regulating multiple organs functions, such as reducing gastric emptying and gastric acid secretion, reducing appetite, promoting cardiac glucose utilization, accelerating renal natriuresis and diuresis, minimizing glucose production in the liver and increasing insulin secretion in the pancreas
Some GLP-1RAs are also effective or the treatment of some complications of T2DM. Diabetic nephropathy is one of the most dangerous complications of T2DM, leading to severe effects on kidney function in diabetic patients, with clinical manifestations including proteinuria and decreased glomerular filtration rate (GFR). In a study of 35 patients with T2DM, lixisenatide reduced the absolute and partial excretion of magnesium, calcium, and phosphate by inhibiting the proximal tubule sodium-hydrogen antiporter 3 (NHE3) and thus increasing the absolute and partial excretion of sodium, chlorine, and potassium and increasing urine pH values compared with insulin glargine287. In addition, in a study of 30 T2DM patients, liraglutide significantly reduced GFR, urinary albumin excretion rate, and partial albumin excretion288. GLP-1RAs can reduce GFR by increasing sodium efflux to the macula densa, increasing tubulo-glomerular feedback and vasoconstriction of afferent arterioles, and may also reduce albuminuria by reducing plasma renin activity, reducing renal oxidative stress, and increasing natriuresis289. However, the extent to which these effects are mediated by GLP-1R remains to be determined. Recent studies have confirmed that the metabolites of GLP-1 retain important antioxidant and anti-apoptotic activities, which are independent of GLP-1R290. Cardiovascular disease remains the leading cause of death in patients with T2DM, and the prevention and treatment of cardiovascular complications should thus be considered when choosing treatments for T2DM. GLP-1RAs have been shown to play a beneficial role in cardiovascular disease. Recent clinical trials found that only liraglutide and semaglutide had advantages in terms of cardiovascular benefits, although the mechanism is still unclear and may be anti-atherosclerotic48. The protective effects of other GLP-1RAs on cardiovascular disease are not obvious, but they have no harmful effects on other safety parameters, and the risk-benefit distribution of GLP-1RAs is thus well-balanced291. GLP-1RAs also showed therapeutic effects on obesity symptoms in patients with T2DM. Thondam reported that a patient with severe hypothalamic obesity and various obesity-related complications, including T2DM, responded well to exenatide, with significantly improvements in weight and blood glucose control, possibly through a central regulatory mechanism increasing satiety and reducing energy intake292. A study of 25 obese patients with T2DM showed that patients treated with metformin and sulfonylurea/DPP-4 inhibitors for 6 months who took GLP-1RA (exenatide19, six cases) had significantly reduced average body weight, glycosylated hemoglobin, and intrahepatic lipids293. Body mass index and fat thickness also decreased significantly in 25 patients with T2DM treated with exenatide and liraglutide for 3 months294. T2DM can lead to bone brittleness and increase the risk of bone-related complications such as fractures and poor fracture healing. Experimental studies found that GLP-1RAs had a significant positive effect on bone quality and strength, possibly by improving the blood supply to the bone necessary for bone health295. In one study, liraglutide was applied to ovariectomized rats with T2DM, followed by high-throughput sequencing of bone marrow-derived exosome micro RNAs (miRNAs). Liraglutide was shown to cause significant changes in exosome miRNAs targeting the insulin signaling pathway, and changes in the Wnt/β-catenin signaling pathway mediated by bone marrow exosomes were implicated in the osteoprotective effect296.
The most common side effects of GLP-1RA treatment are gastrointestinal-related adverse reactions (i.e., nausea, vomiting, and diarrhea) and injection-site reactions, while long-acting GLP-1RAs have fewer side effects, a lower administration frequency. and better compliance. Metformin is still the first-line drug for the treatment of T2DM in the clinic. According to the European Diabetes Research Association and the American Diabetes Association, GLP-1RAs, sulfonylureas, thiazolidinediones, DPP-4 inhibitors, sodium-glucose cotransporter 2 inhibitors, and insulin are recommended as complementary drugs for patients whose blood sugar is not sufficiently controlled by metformin alone297. However, based on the many other benefits of GLP-1RAs in addition to blood glucose control, including renal protection, reduced risk of cardiovascular disease, weight control, no risk of hypoglycemia, benefits for skeletal symptoms, and low-frequency side effects, GLP-1RAs will play an essential role in the treatment of T2DM in the future.
Therapeutic peptides in the treatment of cardiovascular disease
Among non-communicable diseases, cardiovascular disease is now the leading cause of death and morbidity worldwide298. Hypertension is one of the main risk factors for the development of cardiovascular disease, and is considered to be caused by high activity of the renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system,as well as sodium retention299. The function of angiotensin-converting enzyme (ACE) in the RAAS is to cleave angiotensin I into angiotensin II, to contract blood vessels and indirectly increase blood pressure, while ACE2 hydrolyzes angiotensin II into vasodilator angiotensin (1-7) to indirectly reduce blood pressure300. Targeting the RAAS thus represents an ideal strategy for controlling cardiovascular diseases. Synthetic angiotensin II was approved by the FDA in 2017 for increasing blood pressure via intravenous infusion in adults with septicemia or other distributed shock301. Four peptides (WPRGYFL, GPDRPKFLGPF, WYGPDRPKFL, and SDWDRF) isolated and screened from Tetradesmus obliquus microalgae by Montone et al. showed inhibitory activity against ACE302. Liao et al. found that the tripeptide IRW, derived from egg white, reduced blood pressure in spontaneously hypertensive rats by up-regulating the expression of ACE2. These studies indicate the potential application of food-derived peptides targeting RAAS for the treatment of cardiovascular diseases303.
Natriuretic peptide (NPs)304,305, including atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP), are essential regulators of cardiac and vascular homeostasis (Fig. 12). Targeting NPs is thus another practical strategy for the prevention or treatment of cardiovascular diseases. Nesiritide is a recombinant human BNP that was approved by the FDA in 2001 for the treatment of acutely decompensated heart failure in patients with resting or mild dyspnea306; however, it has not been widely used due to its low specificity and limited safety307. NPs act mainly through NPR-A and/or NPR-B receptors, while NPR-C is mainly used for scavenging NPs308. Cenderitide is a dual NPR-A/NPR-B agonist composed of CNP and the C-terminal of dendroaspis natriuretic peptide isolated from the green mamba snake309. Cenderitide is currently in clinical research and has shown safety and potential for the treatment of heart failure and renal failure309,310. In addition, some peptides that are beneficial to cardiovascular disease are being tested in animals. For example, infusion of vasoactive intestinal peptide increased the concentration of myocardial vasoactive intestinal peptide and reversed existing myocardial fibrosis in rats311, and cyclopeptide RD808 neutralized the β1-adrenergic receptor, thus attenuating myocardial injury induced by the β1-adrenergic receptor in mice312. The central adrenocorticotropin-releasing factor (CRF)-related peptide system is currently attracting increasing attention as a target for the prevention of cardiovascular disease313. There is a complex relationship between the CRF-related peptide system and the cardiovascular system, but its exact regulatory role in cardiovascular function remains to be determined. In addition, the activity of circulating DPP-4 was increased and flow-mediated dilation was decreased in patients with T2DM. Flow-mediated dilation is a recognized alternative marker of endothelial dysfunction and a predictor of future cardiovascular events, suggesting that DPP-4 may be a potential target for preventing cardiovascular disease314.

Mechanism of natriuretic peptide (NPs) regulation. Atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and C-type natriuretic peptide (CNP) regulate cardiac and vascular homeostasis through binding to their receptors (NPR-A, -B and -C) to reduce sympathetic tone, fibrosis and renin secretion to treat cardiovascular diseases
Therapeutic peptides in the treatment of gastrointestinal diseases
Therapeutic peptides in the treatment of intestinal disease
In the human body, gastrointestinal flora constitutes a complex micro-ecosystem. Typically, the gastrointestinal flora in the human body constitutes a complex micro-ecosystem. Typically, the epithelium regulates the composition of the intestinal flora at the intestinal mucosal interface by providing a physical barrier and secreting various antimicrobial factors, including antimicrobial peptides (AMPs). The dominant flora (physiological flora) and weak flora (pathogenic bacteria) maintain a dynamic balance315, which is disrupted in various intestinal diseases caused by exotic bacteria, viruses, and parasites, food poisoning, adverse drug reactions, and genetic factors, such as enteritis, constipation, intestinal ulcers, and inflammatory bowel disease (IBD). The extensive use of antibiotics may further reduce the biodiversity of symbiotic bacteria, which is not conducive to treatment and may even aggravate the disease; for example, individuals affected by IBD are more likely to have used antibiotics within 2-5 years before diagnosis316. Peptide drugs have attracted much attention in this field because of their specificity, efficacy, and low toxicity.
Significant changes in the normal intestinal flora and the destruction of host-microbial symbiosis may be the key to the development of IBD317. IBD, including Crohn’s disease and ulcerative colitis, is caused by an intestinal immune response, and the associated inflammation is caused by the interaction between environmental and genetic factors318. However, the specific pathogenesis of IBD is still unclear and there is currently no effective cure. Intestinal microbial diversity is significantly reduced in patients with IBD319, and the two dominant phyla Firmicutes (Lachnospiraceae) and Bacteroidetes, were significantly decreased while the phylum Proteus was significantly increased320. Substantial evidence has indicated a key role for members of the phylum Proteus in IBD321. Proline-arginine-39, a small cationic AMP that is naturally secreted by porcine bone marrow and lymphoid tissue, has demonstrated antibacterial, immunomodulatory, and intestinal epithelial repair functions and may provide a safe alternative therapy for IBD322.
Patients with Crohn’s disease are often treated by bowel resection323, leading to short bowel syndrome (SBS). Damage to the small intestine and abnormal shortness of the small intestine at birth may also cause SBS, which is defined as symptoms associated with a persistent length of the residual small intestine of <200 cm324. GLP-2 is produced by intestinal endocrine L cells and various neurons in the central nervous system (Fig. 13)325 and has recently received extensive attention for the treatment of SBS. GLP-2 has demonstrated various beneficial effects, including stimulating crypt cell growth, reducing intestinal cell apoptosis, promoting intestinal mucosal dilatation, inhibiting gastric acid secretion and gastric emptying, stimulating intestinal blood flow, strengthening intestinal barrier function, reducing anti-inflammatory injury, and promoting nutrition and liquid absorption326,327,328. GLP-2 also regulated the expression of amino acid transporters and directly activated mTORC1 to increase the absorption of amino acids in the intestinal epithelium326. Some specific amino acids (including glutamine, glutamate, arginine, glycine, lysine, methionine, and sulfur-containing amino acids) have also been shown to play an important role in maintaining intestinal integrity, including preventing intestinal atrophy, improving intestinal barrier function, and reducing inflammation and apoptosis329. Endogenous GLP-2 is easily degraded by DPP-4; however, the GLP-2 analogue teduglutide prolongs the half-life from 7 minutes to about 2-3 hours by substitution of alanine by glycine in the second position of the N-terminal of GLP-2, effectively preventing its degradation by DPP-4330. Clinical studies have shown that teduglutide can effectively reduce or eliminate the need for parenteral nutrition and/or intravenous infusion support331, while the application of teduglutide in young pigs with distal ileectomy significantly increased the weight per unit weight and protein synthesis of the remnant intestine332. Teduglutide was approved by the FDA for clinical use in SBS patients in 2012. Wiśniewski et al. designed a series of GLP-2 analogues, including 2-glycine substitution, 10-norleucine substitution, 11- and/or 16-hydrophobic substitution, many of which were more effective against GLP-2R than natural hormones, showing good receptor selectivity and low systematic clearance. Among these, the peptide ([2Gly, 10Nle, 11DPhe, 16Leu] hGLP-2-(1−33)-NH2) was selected as a candidate for clinical development333. GLP-1 from the proglucagon family has similar functions to GLP-2 and has been suggested for the treatment of SBS. In one study, five patients with SBS showed improved stool frequency and morphology after treatment with the GLP-1 agonist exenatide334. Similarly, GLP-1 reduced diarrhea in nine SBS patients, but was less effective than GLP-2, while the combination of GLP-1 and GLP-2 was superior to administration of either alone335. Glicentin, another member of the proglucagon family, also appears to be involved in many processes such as enteral nutrition, exercise, and gastric acid secretion, indicating the prospect of developing glicentin-like peptides336. Other growth factors such as EGF, erythropoietin, and hepatocyte growth factor have also shown therapeutic potential in SBS. The combination of EGF and GLP-2 increased the length of the small intestine in three newborn piglet models of SBS, indicating that EGF has therapeutic potential in neonatal SBS337. Erythropoietin protected intestinal barrier function and protected the gastrointestinal tract from ischemia/reperfusion injury by stimulating the expression of tight junction proteins in animal models338, and enteral injection of hepatocyte growth factor reduced the incidence and severity of necrotizing enterocolitis in rats339.

The structure and sequence of GLP-2 (PDB: 2L63)325
Clostridium difficile toxin A produced by pathogenic strains of Clostridium difficile causes diarrhea and inflammation and even severe pseudomembranous colitis in infected people340. Periplanetasin-2 (YPCKLNLKLGKVPFH) is an AMP isolated from the American cockroach by Ji et al., which blocks the mucosal damage and inflammation induced by Clostridium difficile toxin A341, and was recently identified as a candidate drug for relieving/treating pseudomembranous colitis caused by Clostridium difficile toxin A341,342. The 9-mer disulfide dimer peptide CopA3 (LLCIALRKK) isolated from the Korean dung beetle significantly improved the small intestinal inflammatory response (acute enteritis) induced by Clostridium difficile toxin A and completely blocked the inflammatory response and subsequent fatal response of chronic colitis induced by sodium dextran sulfate in mice343. Food poisoning caused by Clostridium perfringens type A is related to several important human gastrointestinal diseases, and is thought to be mediated by the production of Clostridium perfringens enterotoxin (CPE) combined with human intestinal claudins. Archana et al. found that preincubation or co-incubation of CPE with the claudin-4 extracellular loop ECL-2 peptide significantly inhibited CPE-induced luminal fluid accumulation and histological lesions in rabbit intestinal loop344, suggesting that the synthetic peptide ECL-2 may represent a potential strategy for preventing intestinal histological damage caused by Clostridium perfringens type A. Cathelicidin secreted by human colonic epithelium is another AMP with a wide range of antimicrobial and immunomodulatory functions. Recent studies have shown that human cathelicidin helped early colonic epithelial cells defend against enterogenous Salmonella typhimurium by preventing bacterial invasion and maintaining the integrity of the epithelial barrier, possibly by producing Toll-like receptor-4 and pro-inflammatory cytokines345. In addition, enterovirus infection has also been shown to stimulate the expression of AMPs. Chen et al. found that small ribonucleic acid virus infection increased the expression and secretion of human β defensin-3 in intestinal epithelial cells, and human β defensin-3 had extracellular anti-enterovirus activity346.
Patients with cystic fibrosis (CF) usually also have intestinal obstruction and constipation, which may develop into distal intestinal obstruction syndrome347. The guanylate cyclase C (GCC) receptor agonist, linaclotide, was approved by the FDA in 2012 for the treatment of chronic constipation. Linaclotide has also been shown to improve intestinal transport in CF model mice, although further studies are required to evaluate its effects on the intestinal pathology in CF patients. The NHE3 inhibitor tenapanor improved gastrointestinal transport in CF mice by targeting inhibition of sodium absorption348, indicating that inhibition of GCC signal transduction and NHE3 may be a suitable target for the treatment of constipation in patients with CF.
In addition to drug-derived peptides, peptides may also be food-derived. Asn-Pro-Trp-Asp-Gln (NPWDQ), a peptide obtained by hydrolyzing casein (a major milk protein), significantly inhibited the penetration of the food allergen, ovalbumin, into human intestinal Caco-2 cells, suggesting that this peptide might improve the function of the intestinal epithelial barrier349. β-Casofensin is a peptide found in fermented milk, and in vivo experiments found that early administration of β-casofensin reduced indomethacin-induced intestinal injury and inflammation by protecting goblet cells and promoting wound healing350. Indomethacin-induced intestinal damage has the same clinical, histological, and pathophysiological characteristics as Crohn’s disease351, suggesting that β-casofensin may be a potential adjuvant therapy for Crohn’s disease.
Peptide drugs also have broad prospects in the treatment of intestinal diseases in livestock. Liu et al. developed a modified synthetic peptide KR-32 using natural snake AMP as the raw material. KR-32 improved the malabsorption of fatty acids, total digestibility of ether extract, and intestinal morphology in piglets treated with enterotoxigenic Escherichia coli K88, indicating the potential medicinal value of KR-32352. C-BF is a peptide derived from cathelicidins, as the most prominent AMP family, and is considered to be the most promising substitute for antibiotics. C-BF significantly improved the growth of weaned piglets and improved the structural and developmental damage to the small intestine caused by LPS, indicating that C-BF may be a potential treatment for intestinal damage caused by LPS/pathogens353.
Therapeutic peptides in the treatment of gastric disease
The gastric mucosa is one of the most vulnerable tissues in humans and animals, and gastric diseases are a common problem. Helicobacter pylori infection, non-steroidal anti-inflammatory drugs, alcohol, smoking, mood, and stress are the main factors responsible for stomach damage, which in turn leads to gastritis and ulcers. Stomach disease can develop into a chronic disease in the absence of timely treatment or with improper treatment, and sustained long-term damage greatly increases the risk of gastric cancer. Gastric cancer is currently the fourth most frequently diagnosed cancer worldwide, and the third and fifth leading causes of cancer-related deaths among men and women, respectively.
Although no peptides have yet been approved for the treatment of gastric diseases, the roles of peptides in gastric diseases, including endogenous and exogenous peptides, have been widely studied in the past decade. Calcitonin gene-related peptide (CGRP) is widely distributed in the gastrointestinal system and is the primary neurotransmitter of capsaicin-sensitive sensory nerves. These sensory nerves are involved in protecting the gastric mucosa from various stimuli, and CGRP acts as potential mediator in this process, increasing gastric mucosal blood flow, inhibiting gastric acid secretion, and preventing apoptosis and oxidative damage354. In addition to CGRP, the nitric oxide synthase-nitric oxide (NOS-NO) and cyclooxygenase-prostaglandin (COX-PG) systems have similar protective effects on the stomach355. CGRP, NO, and PG are considered to be the terminal mediators of gastric protection, and to mediate the gastroprotective effects of many endogenous peptides356. The primary pathogenesis of ethanol-induced gastric injury is gastric microvascular injury. As a peptide derived from the nerve growth factor inducible (VGF) gene, TLQP-21 mediated by constitutive NO, PGE2, and somatostatin, showed that central rather than peripheral injection could attenuate ethanol-induced gastric injury in a dose-dependent manner357,358. Novokinin (Arg-Pro-Leu-Lys-Pro-Trp) is an effective vasodilator and antihypertensive peptide modified by ovokinin, with high selective affinity for angiotensin II type 2 (AT2) receptors. Zhang et al. found that novokinin inhibited basal gastric acid secretion after intracerebroventricular in a dose-dependent manner and protected the gastric mucosa from alcohol-induced injury, by mediating the AT2 receptor-PG pathway359. These results indicated the value of TLQP-21 and novokinin for the treatment of gastric injury. A peptide extract obtained from the hydrolysis of waste beer yeast protein (especially < 3 kDa) reduced gastric mucosal injury in rats, indicating the potential value of yeast peptide extract for the treatment of gastric diseases360.
Animal stress-induced gastric injury is often used as a model to study the mechanism of stress-induced stomach diseases. The AMP hepcidin is thought to be produced by parietal cells regulating gastric acid production, and acid secretion was significantly decreased in hepcidin-knockout mice, suggesting that hepcidin may be related to the occurrence of gastric ulcers under stress conditions361. Nesfatin-1 belongs to the anorexia peptide family, which exists in neurons and endocrine cells of the intestinal tract. Studies by Alexandra et al. showed that nesfatin-1 had a significant protective effect on the stomach in rats exposed to water immersion restraint stress. The mechanism was related to decreased gastric juice secretion, hyperemia mediated by the COX-PG and NOS-NO systems, and activation of the vagus nerve, sensory nerve, and vanillin receptor355. Chronic mild stress can cause gastric ulcers in rats, and the somatostatin analogue octreotide can alleviate gastric ulcers by inhibiting apoptosis, inflammation, and oxidation362. Central rather than peripheral injection of oxytocin can eliminate the enhanced postprandial gastric contraction induced by restraint stress in rats, thus reducing delayed gastric emptying, suggesting that oxytocin may be a candidate drug for the treatment of stress-related gastrointestinal motility disorders363.
Gastric cancer is a severe stomach disease. Several peptides have shown therapeutic prospects in gastric cancer. GEBP11 is a new nine-amino acid homing peptide screened and identified by phage-display technology. GEBP11 selectively binds to human umbilical vein endothelial cells and tumor vessels, suggesting that it may be an important candidate for tumor imaging and targeted drug delivery364. Treatment with the iodine 131-labeled bifid PEGylated GEBP11 trimer (131Imur2PEG-(GEBP11)3) significantly inhibited the growth of human gastric cancer xenografts in nude mice and prolonged the survival time, indicating that 131Imur2PEG-(GEBP11)3 may be a suitable candidate for peptide-targeted therapy of gastric cancer and a drug carrier for antiangiogenic therapy of gastric cancer365. Helicobacter pylori infection is one of the most important causes of gastric cancer. H-P-6 (Pro-Gln-Pro-Lys-Val-Leu-Asp-Ser), an active peptide isolated from microbial hydrolysate of Chlamydomonas sp., has been shown to resist Helicobacter pylori-induced carcinogenicity. H-P-6 down-regulated phosphoinositide 3-kinase/Akt signal transduction and β-catenin nuclear translocation by inhibiting EGFR activation, and effectively inhibited Helicobacter pylori-induced human gastric adenocarcinoma cell (AGS) proliferation and migration without inhibiting bacterial viability or AGS cell invasion366. Zhang et al. synthesized the AMP pexiganan and its nanoparticles (PNPs), which demonstrated anti-Helicobacter pylori activity and stronger scavenging ability against Helicobacter pylori in mouse stomach than pexiganan, and showed potential for the treatment and prevention of Helicobacter pylori-related gastric diseases367. TFF1 is a mucin-related gastric mucosal cell secretory peptide. The expression of TFF1 was up-regulated in the gastric antrum in the acute phase rather than the chronic phase of Helicobacter pylori infection in mice, and was negatively correlated with the inflammatory response, indicating that TFF1 may help cells resist the development of bacteria and chronic inflammation368. TFF2, a member of the same family, has been shown to interact with gastrin MUC6 to stabilize the gastric mucus barrier and maintain gastric mucosal integrity369.
Peptides have also been shown to play a regulatory role in terms of gastric motility. Peripheral injection of GLP-2 increased gastrointestinal blood flow and gastric mucosal blood flow by increasing CGRP and endogenous PGs rather than NO370. Exogenous GLP-1 caused the release of NO into the gastric antrum through nerves in an isolated whole-stomach model, thus reducing gastric motility in mice371. However, whether the approved GLP-1/2 derived peptides have similar effects still needs to be investigated. BNP has the property of dilating blood vessels and can increase visceral perfusion and oxygenation, and recombinant BNP has been shown to increase hemoglobin oxygenation in the gastric mucosa microvasculature372. Motilin and ghrelin belong to the same peptide family, and these hormones play an important role in the regulation of gastrointestinal motility. Ghrelin and motilin can synergistically stimulate strong gastric contraction in vitro and in vivo373. Motilin and the combination of motilin and ghrelin stimulated gastric acid secretion in the shrew Suncus through the histamine-mediated pathway374.
Therapeutic peptides in the treatment of cancer
Traditional cancer treatments include surgery and radiotherapy, which have limited effects in patients with advanced cancer. The subsequent development of targeted therapy and immunotherapy have significantly improved the survival rate of cancer patients. Targeted therapy takes advantage of the reliance of tumor cells on specific molecules or signaling pathways to kill tumor cells using a “guided missile” approach375. Immunotherapy drugs do not attack tumor cells directly but modulate the patient’s own immune system and attack tumor cells by targeting immune checkpoints376. PD-1/PD-L1 is a well-known immune checkpoint, and five monoclonal antibodies against PD-1/PD-L1 interaction have been approved by the FDA for cancer treatment. However, antibodies have disadvantages including high cost, poor oral suitability, and high immunogenicity. Peptides have also attracted attention in the field of tumor diagnosis and treatment because of their small size, high affinity, easy modification, and low immunogenicity. Some modified peptides have also demonstrated good stability. For example, Carvajal et al. developed stable α-helical peptides as inhibitors of MDM2 and MDMX for the treatment of p53-dependent cancer377,378,379.
The short half-life of natural peptides in vivo means that peptides targeting various abnormally expressed receptors in tumor cells are usually modified peptide analogues. There are three main methods for the production of these peptides, each with its own strengths and weaknesses: 1) derivation from natural proteins; 2) chemical synthesis and reasonable engineering based on structure; and 3) screening of peptide libraries380. Among these, phage-display technology is a traditional and widely used method, with the advantages of simple operation and effective screening of a large number of different peptides381.
Peptides can be applied in tumor therapy in four main ways: 1) using radioisotopes, dyes, or other reported molecular-labeled peptides as probes for tumor diagnosis and imaging; 2) using peptide-coupled nanomaterials for tumor therapy; 3) using peptide vaccines to activate the immune system for prevention; and 4) using peptides alone as targeted drugs (Fig. 14)382,383,384.

Application of peptides in tumor therapy. a Screening and identification of peptide candidates from chemically synthesized peptide library and phage library. b Using radiolabeled, dye-labeled, or other designed peptides as probes for tumor diagnosis and imaging. c Application of peptide-conjugated nanomaterials in tumor therapy. d Using peptide vaccine and targeting peptides in tumor immunotherapy and targeted therapy
Peptide-based imaging probes bind to receptors specifically expressed in the tumor. These receptors can either be expressed on the cell surface, such as αvβ3 integrin (RGD peptide), EGF receptor, somatostatin receptor, neurotensin receptor, and transferrin receptor; intracellularly, such as Bcr/Abl, cyclin A, and cyclin kinase; or in extracellular matrices, such as fibronectin, matrix metalloproteinases, and prostate-specific antigen382. The locations of probes can be visualized by single-photon emission computed tomography/computed tomography scanning to indicate the tumor distribution. This technique has been applied for early tumor diagnosis and surgical resection. Several probes have been developed, such as octreoscan and depreotide, as radiolabeled conjugates of somatostatin-like peptides, have been approved by the FDA for imaging of tumors, such as neuroendocrine tumors and lung cancer385,386. Unfortunately, depreotide has been withdrawn. Radiolabeled peptides based on RGD peptides have recently received attention, and a series of probes have been synthesized, including (99m) Tc-3PRGD2, which can be used to detect differentiated thyroid cancer by negative whole-body scan of radioactive iodine387. Lutetium 177 dotatate is a radiolabeled somatostatin analog recently approved for the treatment of somatostatin receptor-positive gastroenteropancreatic neuroendocrine tumors388. It binds to somatostatin receptors and then releases radioactive Lutetium 177 into the tumor cells, which induces cellular damage via the formation of intracellular free radicals389,390.
Based on the same principle as peptide-based probes, internal radiotherapy can be achieved by labeling β-emitters on peptides; however, this is notably limited by radiation damage to normal target tissues with positive receptors, either near or far from the tumor384. A possible alternative to reduce the potential side effects involves delivering the peptides to tumors coupled to anti-cancer drugs, genes, and RNAs (small interfering RNA ([siRNA]/miRNA/mRNA)391. AN-152 and AN-207 are luteinizing hormone-releasing hormone analogues coupled to adriamycin, with anticancer activity against luteinizing hormone-releasing hormone receptor-positive cancers. The results of phase I and II clinical studies showed that the drug was effective for the treatment of breast cancer, endometrial cancer, and ovarian cancer, with moderate toxicity and side effects392. Chen et al. designed PEGylated liposome-polycation-DNA (LPD) nanoparticles and obtained LPD-PEG-NGR by modifying the NGR peptide targeting tumor-specific receptor aminopeptidase N. LPD-PEG-NGR delivered siRNA into solid tumors in mice with systemic, specific, and effective delivery, and by delivering c-myc siRNA, it effectively triggered the apoptosis of tumor cells by down-regulating the expression of c-myc, thus inhibiting the growth of some tumors393. In addition, tumor-penetrating peptides screened by phage screening in vivo can effectively deliver covalently coupled and co-administered drugs to the depths of tumor tissues394. These results indicate that peptide-based drug delivery systems have important potential for the treatment of tumors.
Antigenic peptides from specific target proteins can act as anticancer peptide vaccines by binding to the major histocompatibility complex (MHC) on antigen-presenting cells, to trigger the anti-tumor effects of helper or cytotoxic T cells. EGFRs, such as EGFR1 and HER2, are well-known targets for cancer treatment. The peptide vaccine TERT572Y, based on the HER2 structure, was used in 46 patients with advanced non-small-cell lung cancer. Subcutaneous injection of TERT572Y induced a TERT-specific immune response and significantly prolonged survival395. Manijeh et al. calculated and predicted potential epitopes by PEPOP and selected various peptide sequences from the extracellular domain of HER2 as candidate sequences. They then evaluated the binding affinity of these candidate peptides to MHC I and II molecules by molecular docking, to find the most stable binding structure between peptides and MHC I and II molecules, and selected MHC class I- and II-binding peptides as breast cancer peptide vaccines396. However, most clinical trials of peptide vaccines have failed to demonstrate excellent therapeutic effects, and peptide vaccines have thus received little attention. Nevertheless, Takumi et al. argued that the main reason for the lack of success in clinical studies of most cancer vaccines, including peptides, was due to their poor immunogenicity, and suggested that optimization of peptide formulations, adjuvants, and administration routes would achieve ideal results397. Peptides, known as cell-penetrating peptides (CPPs), can also be used as drug carriers to transport other peptides, proteins, DNAs, small RNAs, and drugs into cells398. The CPP-drug construct comprising nerinetide with the CPP Tat was used to deliver nerinetide across the blood–brain barrier and into neurons399. Peptides have also shown a promising delivery function by coupling to antigens to induce antigen-specific immune tolerance and reduce the risk of off-target responses400. Tsoras used peptide nanoclusters to improve peptide subunit vaccine immunogenicity for oncofetal antigen401.
In addition to being used as drug carriers and vaccines, peptides can also exert anti-tumor effects by binding to target receptors. Among these, the most popular peptides are those targeting the PD-1/PD-L1 signal pathway. Boohaker et al. designed a PD-L1 peptide mimic, PL120131, which can interfere with the interaction of PD-1/PD-L1 by binding to PD-1. PL120131 maintained the survival and activity of co-cultured T cells better than PD-1 antibody in a 3D co-culture model402. Based on peptides binding to PD-1 and PD-L1, Zhou et al. designed the self-inhibitory peptides DS-I and DS-II and their cyclic peptide forms, which showed strong affinity to PD-1403. Abbas et al. designed a new peptide targeting PD-1, FITC-YT-16, which significantly enhanced the anti-tumor activity of T cells in vitro404, while Sasikumar et al. designed the peptide NP-12 to bind PD-L1 competitively with PD-1. Moreover, NP-12 showed the same efficacy as commercial PD-1 targeted antibodies in inhibiting the growth and metastasis of primary tumors in preclinical models of melanoma, colon cancer, and renal cell carcinoma405. Although these peptides are not yet suitable for blocking PD-1/PD-L1 to treat tumors, they offer promising potential. Toxic peptides (VPs) from animals may also show an anti-tumor effect. Because VPs naturally target mammalian receptors, they show a high degree of specificity and selectivity for specific ion channels and receptors on the cell membrane. Hanatoxin-1, a peptide toxin isolated from Chilean spiders, specifically blocks the K+ channel on the membrane406. High expression of the K+ channel has been observed during the development of colon cancer, and Okada et al. found that the porogenic peptide LaFr26 purified from Lachesana sp. spider venom had a cytotoxic effect on the lung cancer cell lines LX22 and BEN, which expressed an endogenous K+ current407. Attention has also been paid to the role of AMPs in tumors408. Some AMPs have demonstrated anti-tumor activity, while others promoted tumor development409. The simplified θ-defensin analogue synthesized by Strzelecka et al. inhibited the growth of breast cancer cells in a 3D culture model, indicating that θ-defensin derivatives have anticancer potential410. Anticancer peptides are cationic amphiphilic molecules that preferentially kill cancer cells through folding-dependent membrane rupture. Referring to the membrane-specific interaction of anticancer peptides, Aronson et al. prepared a new class of peptide lipid particles that fuse rapidly with the tumor cell membrane and mediate cell killing, with little toxicity to normal cells, indicating a new tumor-lysis strategy411.
Although many peptides have shown the promising anti-tumor effects in preclinical and clinical studies. Only two peptides are currently approved for the treatment of tumors, mifamurtide for osteosarcoma and carfilzomib for multiple myeloma, and research into treatment strategies involving therapeutic peptides for more common tumors, such as lung cancer and gastric cancer, is still ongoing. The key is thus to identify more receptor targets that are specifically expressed in tumor cells and to strengthen their medical translation. In addition, the combination of peptides targeting various tumor receptors is also a potential strategy.
Antiviral peptides
Viruses parasitize all living creatures, including humans, animals, plants, bacteria, and archaea. Humans have always suffered from viral diseases, including Ebola hemorrhagic fever, influenza, and acquired immune deficiency syndrome (AIDS)412,413. Despite extensive efforts in antiviral drug development over the past two decades, leading to the approval and clinical use of multiple antiviral drugs414,415, there remains no effective treatment for some of these diseases such as AIDS.
Research into antiviral peptides has become a hot topic, because of the high specificity and activity of peptides416. Antiviral peptides act mainly by targeting the virus or its host to block infection417,418. Enfuvirtide, the first approved antiviral peptide, is a 36-amino acid peptide that blocks HIV infection by binding to the heptad-repeat domain of gp41 (HIV envelope protein) to prevent its fusion419. In 2011, the antiviral peptide drugs boceprevir and telaprevir were approved for clinical treatment of hepatitis C virus (HCV)420,421. They both bind to the HCV NS3/4A serine protease to inhibit protease activity, thus blocking HCV replication in the host422. More research on antiviral peptide drug candidates is being undertaken in pre-clinical and clinical studies, including myrcludex B against HBV and HDV423,424,425, flufirvitide against Influenza virus426,427, and sifuvirtide against HIV-1428,429,430.
Since 2020, the respiratory pandemic disease caused by the novel coronavirus SARS-CoV-2 has seriously disturbed people’s lives throughout the world431. Scientists have devoted extensive efforts to studying the mechanism of infection of COVID-19 since the start of the pandemic in early 2020, as well as searching for anti-COVID-19 treatments and drugs, including peptide drugs432,433,434,435,436. The COVID-19 genome was rapidly sequenced as an enveloped, positive single-stranded RNA coronavirus with a genome size of about 29.9 kb437,438, which is closely related to bat coronaviruses and the SARS-CoV virus439,440.
Vaccines are commonly considered as effective agents for preventing the spread of pandemic diseases. Vaccines have been approved and used in many countries, including vaccines based on mRNA441,442, recombinant adenoviral vectors443,444, and inactivated vaccines445,446. Peptide vaccines have certain advantages, such as high specificity, good safety, and easier production, and have thus become an active research area in development of vaccines against SARS-CoV-2447,448,449. Based on the infection mechanism, several research groups have designed and evaluated peptide vaccines against SARS-CoV-2. They used immunoinformatics technology to analyze and identify the key epitopes of B- and T-cell that specifically recognize the spike glycoprotein of SARS-CoV-2. Li et al. and Chakraborty et al. applied the natural epitope sequences as vaccine candidates against SARS-CoV-2450,451. While, Bhattacharya et al. and Waqas tried to construct new peptides as COVID-19 vaccine candidates based on the epitope fragments from B- and T-cell452,453. Similar work has also been carried out by other groups454,455,456. Herst et al. attempted to design COVID-19 peptide vaccines using the CTL peptide vaccine research platform for Ebola Zaire, and obtained a series of peptide vaccine candidates457. Many other studies have focused on blocking the infection process of SARS-CoV-2 using synthetic peptides or nucleotides458,459,460,461.
The development of antiviral peptides attracted wide attention during the COVID-19 pandemic, especially the development of peptide vaccines against SARS-CoV-2. Novel technologies, such as immunoinformatics characterization, epitope-based design, in silico identification, and molecular docking have been used expeditiously to design and identify peptide vaccine candidates. Although no peptide vaccines have yet been approved for the treatment of COVID-19, valuable experience has been gained in the development of peptide vaccines, not only against SARS-CoV-2, but also against new viruses in the future.
Conclusion and perspective
Peptides have become a unique class of therapeutic agents in recent years as a result of their distinct biochemical characteristics and therapeutic potential. Although peptides outperform small molecules and large biologics in some aspects, they often suffer from membrane impermeability and poor stability in vivo, due to the intrinsic limitations of amino acids. Extensive research has been carried out in terms of the discovery, production, and optimization of peptide drugs, in order to overcome these drawbacks. The integration of traditional lead peptide discovery methods with novel technologies, such as rational design and phage display, provides a reliable approach for the development of effective and selective lead peptides in a short period of time. The single or combined use of chemical and biological recombination synthetic approaches allows the efficient and reliable production of synthetic peptides on large scales. These peptides can be further modified in a site-specific manner through chemical synthesis or genetic code expansion to enhance their stability and physiological activity.
Although the field of therapeutic peptides started with natural hormones, the discovery and development trends have since shifted from simply mimicking natural hormones or peptides derived from nature to the rational design of peptides with desirable biochemical and physiological activities. Major breakthroughs in molecular biology, peptide chemistry and peptide delivery technologies have allowed significant progress in the fields of peptide drug discovery, peptide production, and their therapeutic applications. More than 80 therapeutic peptides have reached the global market to date, and hundreds of peptides are undergoing preclinical studies and clinical development. These peptide drugs have been applied to a wide range of diseases, such as diabetes mellitus, cardiovascular diseases, gastrointestinal diseases, cancer, infectious diseases, and vaccine development. Considering their huge therapeutic potentials, market prospects, and economic values, we expect therapeutic peptides to continue to attract investment and research efforts and to achieve long-term success.
References
Henninot, A., Collins, J. C. & Nuss, J. M. The current state of peptide drug discovery: back to the future? J. Med Chem. 61, 1382–1414 (2018).
Craik, D. J., Fairlie, D. P., Liras, S. & Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 81, 136–147 (2013).
Bliss, M. Banting’s, Best’s, and Collip’s accounts of the discovery of insulin. Bull. Hist. Med 56, 554–568 (1982).
Scott, D. A. & Best, C. H. The preparation of insulin. Ind. Eng. Chem. 17, 238–240 (1925).
Mathieu, C., Gillard, P. & Benhalima, K. Insulin analogues in type 1 diabetes mellitus: getting better all the time. Nat. Rev. Endocrinol. 13, 385–399 (2017).
Zaykov, A. N., Mayer, J. P. & DiMarchi, R. D. Pursuit of a perfect insulin. Nat. Rev. Drug Disco. 15, 425–439 (2016).
Lau, J. L. & Dunn, M. K. Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg. Med Chem. 26, 2700–2707 (2018).
Sawyer, W. H. & Manning, M. Synthetic analogs of oxytocin and the vasopressins. Annu Rev. Pharm. 13, 1–17 (1973).
Spiegelman, A. R. Synthetic vasopressin and diabetes insipidus. JAMA 187, 1035 (1964).
Keen, H. et al. Human insulin produced by recombinant DNA technology: safety and hypoglycaemic potency in healthy men. Lancet 2, 398–401 (1980).
Johnson, I. S. Human insulin from recombinant DNA technology. Science 219, 632–637 (1983).
Machado, E. S. et al. Successful desensitization of enfuvirtide after a first attempt failure. AIDS 20, 2130–2131 (2006).
de Castro, N. et al. Incidence and risk factors for liver enzymes elevations in highly treatment-experienced patients switching from enfuvirtide to raltegravir: a sub-study of the ANRS-138 EASIER trial. AIDS Res Ther. 13, 17 (2016).
Bourinet, E. & Zamponi, G. W. Block of voltage-gated calcium channels by peptide toxins. Neuropharmacology 127, 109–115 (2017).
Deer, T. R., Pope, J. E., Hanes, M. C. & McDowell, G. C. Intrathecal therapy for chronic pain: a review of morphine and ziconotide as firstline options. Pain. Med 20, 784–798 (2019).
Drucker, D. J. The discovery of GLP-2 and development of teduglutide for short bowel syndrome. ACS Pharm. Transl. Sci. 2, 134–142 (2019).
Burness, C. B. & McCormack, P. L. Teduglutide: a review of its use in the treatment of patients with short bowel syndrome. Drugs 73, 935–947 (2013).
Malm-Erjefalt, M. et al. Metabolism and excretion of the once-daily human glucagon-like peptide-1 analog liraglutide in healthy male subjects and its in vitro degradation by dipeptidyl peptidase IV and neutral endopeptidase. Drug Metab. disposition: Biol. fate Chem. 38, 1944–1953 (2010).
Hamad, F. et al. Systematic review of glucagon like peptide one receptor agonist liraglutide for subjects with heart failure with reduced left ventricular ejection fraction. Curr Diabetes Rev, 17, 280–292 (2021).
Fisher, E., Pavlenko, K., Vlasov, A. & Ramenskaya, G. Peptide-based therapeutics for oncology. Pharm. Med. 33, 9–20 (2019).
Iyengar, S., Ossipov, M. H. & Johnson, K. W. The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain 158, 543–559 (2017).
Sloan, L. A. Review of glucagon-like peptide-1 receptor agonists for the treatment of type 2 diabetes mellitus in patients with chronic kidney disease and their renal effects. J. Diabetes 11, 938–948 (2019).
Peterson, S. C. & Barry, A. R. Effect of glucagon-like peptide-1 receptor agonists on all-cause mortality and cardiovascular outcomes: a meta-analysis. Curr. Diabetes Rev. 14, 273–279 (2018).
Torres, M. D. T., Sothiselvam, S., Lu, T. K. & de la Fuente-Nunez, C. Peptide design principles for antimicrobial applications. J. Mol. Biol. 431, 3547–3567 (2019).
Research, T. M. Global IndustryAnalysis, Size, Share, Growth, Trends and Forecast. Pept. Mark. 2016–2024, (2016).
Peptide Therapeutics Market: Global Industry Analysis, Size, Share, Growth, Trends and Forecast 2012−2018. Transparency Market Research: Albany. NY, (2012).
group, N. Top 200 Pharmaceuticals by Retail Sales in 2019. University of Arizona, (2020).
Muttenthaler, M., King, G. F., Adams, D. J. & Alewood, P. F. Trends in peptide drug discovery. Nat. Rev. Drug Disco. 20, 309–325 (2021).
Fosgerau, K. & Hoffmann, T. Peptide therapeutics: current status and future directions. Drug Disco. Today 20, 122–128 (2015).
Giordano, C., Marchio, M., Timofeeva, E. & Biagini, G. Neuroactive peptides as putative mediators of antiepileptic ketogenic diets. Front Neurol. 5, 63 (2014).
Davda, J. et al. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. J. Immunother. Cancer. 7, 105 (2019).
Waldmann, H. Human monoclonal antibodies: the residual challenge of antibody immunogenicity. Methods Mol. Biol. 1060, 1–8 (2014).
Imai, K. & Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 6, 714–727 (2006).
Smith, A. J. New horizons in therapeutic antibody discovery: opportunities and challenges versus small-molecule therapeutics. J. Biomol. Screen 20, 437–453 (2015).
Lawson, A. D. Antibody-enabled small-molecule drug discovery. Nat. Rev. Drug Disco. 11, 519–525 (2012).
Li, X. F., Liu, C. F. & Rao, G. W. Monoclonal antibodies, small molecule inhibitors and antibody-drug conjugates as HER2 inhibitors. Curr. Med Chem. 28, 3339–3360 (2021).
Smith, M. C. & Gestwicki, J. E. Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev. Mol. Med 14, e16 (2012).
Petta, I. et al. Modulation of protein-protein interactions for the development of novel therapeutics. Mol. Ther. 24, 707–718 (2016).
Faivre, S., Demetri, G., Sargent, W. & Raymond, E. Molecular basis for sunitinib efficacy and future clinical development. Nat. Rev. Drug Disco. 6, 734–745 (2007).
White, P. T. & Cohen, M. S. The discovery and development of sorafenib for the treatment of thyroid cancer. Expert Opin. Drug Dis. 10, 427–439 (2015).
Wilhelm, S. et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 6, 126–126 (2007). (vol 5, pg 835, 2006).
Vuong, H. G. et al. Efficacy and toxicity of sorafenib in the treatment of advanced medullary thyroid carcinoma: A systematic review and meta-analysis. Head. Neck 41, 2823–2829 (2019).
Escudier, B., Worden, F. & Kudo, M. Sorafenib: key lessons from over 10 years of experience. Expert Rev. anticancer Ther. 19, 177–189 (2019).
Randrup Hansen, C. et al. Effects and side effects of using sorafenib and sunitinib in the treatment of metastatic renal cell carcinoma. Int J Mol Sci. 18, 461(2017).
Sehdev, S. Sunitinib toxicity management - a practical approach. Can. Urol. Assoc. J. 10, S248–S251 (2016).
Ferrara, N. & Adamis, A. P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Disco. 15, 385–403 (2016).
Diao, L. & Meibohm, B. Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin. pharmacokinetics 52, 855–868 (2013).
Del Olmo-Garcia, M. I. & Merino-Torres, J. F. GLP-1 receptor agonists and cardiovascular disease in patients with type 2 diabetes. J. Diabetes Res 2018, 4020492 (2018).
Reed, J., Bain, S. & Kanamarlapudi, V. Recent advances in understanding the role of glucagon-like peptide 1. F1000Res. 9, F1000 Faculty Rev-239 (2020).
Jones, L. H. & Price, D. A. Medicinal chemistry of glucagon-like peptide receptor agonists. Prog. Med Chem. 52, 45–96 (2013).
Alavi, S. E., Cabot, P. J. & Moyle, P. M. Glucagon-like peptide-1 receptor agonists and strategies to improve their efficiency. Mol. Pharm. 16, 2278–2295 (2019).
Marshall, J. C. & Kelch, R. P. Gonadotropin-releasing hormone: role of pulsatile secretion in the regulation of reproduction. N. Engl. J. Med 315, 1459–1468 (1986).
Wilson, A. C., Meethal, S. V., Bowen, R. L. & Atwood, C. S. Leuprolide acetate: a drug of diverse clinical applications. Expert Opin. Investig. Drugs 16, 1851–1863 (2007).
Hoda, M. R., Kramer, M. W., Merseburger, A. S. & Cronauer, M. V. Androgen deprivation therapy with Leuprolide acetate for treatment of advanced prostate cancer. Expert Opin. Pharmacother. 18, 105–113 (2017).
Uttley, L. et al. Degarelix for treating advanced hormone-dependent prostate cancer: an evidence review group perspective of a NICE single technology appraisal. Pharmacoeconomics 35, 717–726 (2017).
Lamberts, S. W. J. & Hofland, L. J. ANNIVERSARY REVIEW: octreotide, 40 years later. Eur. J. Endocrinol. 181, R173–R183 (2019).
Pokuri, V. K., Fong, M. K. & Iyer, R. Octreotide and lanreotide in gastroenteropancreatic neuroendocrine tumors. Curr. Oncol. Rep. 18, 7 (2016).
Svensson, P. J., Bergqvist, P. B., Juul, K. V. & Berntorp, E. Desmopressin in treatment of haematological disorders and in prevention of surgical bleeding. Blood Rev. 28, 95–102 (2014).
Theunissen, F. J., Chinery, L. & Pujar, Y. V. Current research on carbetocin and implications for prevention of postpartum haemorrhage. Reprod. Health 15, 94 (2018).
Kim, S. H. et al. The oxytocin receptor antagonist, Atosiban, activates pro-inflammatory pathways in human amnion via G(alphai) signalling. Mol. Cell Endocrinol. 420, 11–23 (2016).
Brown, M. C. et al. VEGF-related protein isolated from Vipera palestinae venom, promotes angiogenesis. Growth Factors 25, 108–117 (2007).
Yamazaki, Y. et al. Snake venom vascular endothelial growth factors (VEGF-Fs) exclusively vary their structures and functions among species. J. Biol. Chem. 284, 9885–9891 (2009).
Toivanen, P. I. et al. Snake venom VEGF Vammin induces a highly efficient angiogenic response in skeletal muscle via VEGFR-2/NRP specific signaling. Sci Rep-Uk. 7, 5525 (2017).
Wulff, H. et al. Antibodies and venom peptides: new modalities for ion channels. Nat. Rev. Drug Disco. 18, 339–357 (2019).
Miller, L. J., Sexton, P. M., Dong, M. & Harikumar, K. G. The class B G-protein-coupled GLP-1 receptor: an important target for the treatment of type-2 diabetes mellitus. Int J. Obes. Suppl. 4, S9–S13 (2014).
Schmidtko, A., Lotsch, J., Freynhagen, R. & Geisslinger, G. Ziconotide for treatment of severe chronic pain. Lancet 375, 1569–1577 (2010).
Pope, J. E. et al. The pharmacology of spinal opioids and ziconotide for the treatment of non-cancer pain. Curr. Neuropharmacol. 15, 206–216 (2017).
Walsh, C. T. Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Nat. Prod. Rep. 33, 127–135 (2016).
Marahiel, M. A. Working outside the protein-synthesis rules: insights into non-ribosomal peptide synthesis. J. Pept. Sci. 15, 799–807 (2009).
Awan, A. R., Shaw, W. M. & Ellis, T. Biosynthesis of therapeutic natural products using synthetic biology. Adv. Drug Deliv. Rev. 105, 96–106 (2016).
Schilling, N. A. et al. Synthetic lugdunin analogues reveal essential structural motifs for antimicrobial action and proton translocation capability. Angew. Chem. Int Ed. Engl. 58, 9234–9238 (2019).
Bitschar, K. et al. Lugdunin amplifies innate immune responses in the skin in synergy with host- and microbiota-derived factors. Nat. Commun. 10, 2730 (2019).
Niu, X., Thaochan, N. & Hu, Q. Diversity of linear non-ribosomal peptide in biocontrol fungi. J Fungi (Basel). 6, 61 (2020).
Brown, A. S., Calcott, M. J., Owen, J. G. & Ackerley, D. F. Structural, functional and evolutionary perspectives on effective re-engineering of non-ribosomal peptide synthetase assembly lines. Nat. Prod. Rep. 35, 1210–1228 (2018).
Gould, A., Ji, Y., Aboye, T. L. & Camarero, J. A. Cyclotides, a novel ultrastable polypeptide scaffold for drug discovery. Curr. Pharm. Des. 17, 4294–4307 (2011).
Sivanathan, S. & Scherkenbeck, J. Cyclodepsipeptides: a rich source of biologically active compounds for drug research. Molecules 19, 12368–12420 (2014).
Weidmann, J. & Craik, D. J. Discovery, structure, function, and applications of cyclotides: circular proteins from plants. J. Exp. Bot. 67, 4801–4812 (2016).
Prosperini, A. et al. A review of the mycotoxin enniatin B. Front Public Health 5, 304 (2017).
Martin, R. J. et al. Emodepside and SL0-1 potassium channels: a review. Exp. Parasitol. 132, 40–46 (2012).
Bonetta, L. Protein-protein interactions: Interactome under construction. Nature 468, 851–854 (2010).
Vidal, M., Cusick, M. E. & Barabasi, A. L. Interactome networks and human disease. Cell 144, 986–998 (2011).
Rolland, T. et al. A proteome-scale map of the human interactome network. Cell 159, 1212–1226 (2014).
Lee, A. C., Harris, J. L., Khanna, K. K. & Hong, J. H. A. Comprehensive review on current advances in peptide drug development and design. Int J Mol Sci. 20, 2383 (2019).
Hill, T. A., Shepherd, N. E., Diness, F. & Fairlie, D. P. Constraining cyclic peptides to mimic protein structure motifs. Angew. Chem. Int Ed. Engl. 53, 13020–13041 (2014).
Nevola, L. & Giralt, E. Modulating protein-protein interactions: the potential of peptides. Chem. Commun. (Camb.) 51, 3302–3315 (2015).
Geppert, T., Hoy, B., Wessler, S. & Schneider, G. Context-based identification of protein-protein interfaces and “hot-spot” residues. Chem. Biol. 18, 344–353 (2011).
Cukuroglu, E., Engin, H. B., Gursoy, A. & Keskin, O. Hot spots in protein-protein interfaces: towards drug discovery. Prog. Biophys. Mol. Biol. 116, 165–173 (2014).
Liu, D. G. et al. Small molecules engage hot spots through cooperative binding to inhibit a tight protein-protein interaction. Biochem.-Us 56, 1768–1784 (2017).
Villar, E. A. et al. How proteins bind macrocycles. Nat. Chem. Biol. 10, 723–731 (2014).
Wojcik, P. & Berlicki, L. Peptide-based inhibitors of protein-protein interactions. Bioorg. Med Chem. Lett. 26, 707–713 (2016).
Bhat, A., Roberts, L. R. & Dwyer, J. J. Lead discovery and optimization strategies for peptide macrocycles. Eur. J. Medicinal Chem. 94, 471–479 (2015).
Vinogradov, A. A., Yin, Y. & Suga, H. Macrocyclic peptides as drug candidates: recent progress and remaining challenges. J. Am. Chem. Soc. 141, 4167–4181 (2019).
Smith, G. P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315–1317 (1985).
Omidfar, K. & Daneshpour, M. Advances in phage display technology for drug discovery. Expert Opin. Drug Disco. 10, 651–669 (2015).
Frenzel, A., Schirrmann, T. & Hust, M. Phage display-derived human antibodies in clinical development and therapy. MAbs 8, 1177–1194 (2016).
Giustina, A. et al. EXPERT CONSENSUS DOCUMENT A consensus on the medical treatment of acromegaly. Nat. Rev. Endocrinol. 10, 243–248 (2014).
Zhang, H. K., Xie, J. & Lerner, R. A. A proximity based general method for identification of ligand and receptor interactions in living cells. Biochem Bioph Res Co. 454, 251–255 (2014).
Peng, Y. J. et al. A general method for insertion of functional proteins within proteins via combinatorial selection of permissive junctions. Chem. Biol. 22, 1134–1143 (2015).
Zhang, H. K. et al. Autocrine-based selection of drugs that target ion channels from combinatorial venom peptide libraries. Angew. Chem. Int Ed. 55, 9306–9310 (2016).
Zhang, H. K. et al. Autocrine selection of a GLP-1R G-protein biased agonist with potent antidiabetic effects. Nat Commun. 6, 8918 (2015).
Zong, X. L. et al. Phage Display, Peptide Production and Biological Assessment of Key Sequence of TGF-beta 1. Int J. Pept. Res Ther. 25, 1217–1223 (2019).
Yoshimoto, N. et al. High-throughput de novo screening of receptor agonists with an automated single-cell analysis and isolation system. Sci Rep-Uk. 4, 4242 (2014).
Lipok, M. et al. Identification of a peptide antagonist of the FGF1-FGFR1 signaling axis by phage display selection. Febs Open Bio 9, 914–924 (2019).
Matochko, W. L. & Derda, R. Next-generation sequencing of phage-displayed peptide libraries. Methods Mol. Biol. 1248, 249–266 (2015).
Heinis, C. & Winter, G. Encoded libraries of chemically modified peptides. Curr. Opin. Chem. Biol. 26, 89–98 (2015).
Schumacher, T. N. et al. Identification of D-peptide ligands through mirror-image phage display. Science 271, 1854–1857 (1996).
Diaz-Perlas, C. et al. Protein chemical synthesis combined with mirror-image phage display yields d-peptide EGF ligands that block the EGF-EGFR interaction. Chembiochem 20, 2079–2084 (2019).
Josephson, K., Ricardo, A. & Szostak, J. W. mRNA display: from basic principles to macrocycle drug discovery. Drug Disco. Today 19, 388–399 (2014).
Baggio, R. et al. Identification of epitope-like consensus motifs using mRNA display. J. Mol. Recognit. 15, 126–134 (2002).
Schlippe, Y. V. G., Hartman, M. C. T., Josephson, K. & Szostak, J. W. In vitro selection of highly modified cyclic peptides that act as tight binding inhibitors. J. Am. Chem. Soc. 134, 10469–10477 (2012).
Morioka, T. et al. Selection-based discovery of macrocyclic peptides for the next generation therapeutics. Curr. Opin. Chem. Biol. 26, 34–41 (2015).
Ohuchi, M., Murakami, H. & Suga, H. The flexizyme system: a highly flexible tRNA aminoacylation tool for the translation apparatus. Curr. Opin. Chem. Biol. 11, 537–542 (2007).
Passioura, T., Katoh, T., Goto, Y. & Suga, H. Selection-based discovery of druglike macrocyclic peptides. Annu Rev. Biochem. 83, 727–752 (2014).
Merrifield, R. B. Solid phase peptide synthesis. i. the synthesis of a tetrapeptide. J. Am. Chem. Soc. 85, 2149–2154 (1963).
Hackenberger, C. P. R., Dawson, P. E., Chen, Y. X. & Hojo, H. Modern peptide and protein chemistry: reaching new heights. J. Org. Chem. 85, 1328–1330 (2020).
Jaradat, D. M. M. Thirteen decades of peptide synthesis: key developments in solid phase peptide synthesis and amide bond formation utilized in peptide ligation. Amino Acids 50, 39–68 (2018).
Puentes, A. R., Morejon, M. C., Rivera, D. G. & Wessjohann, L. A. Peptide macrocyclization assisted by traceless turn inducers derived from Ugi peptide ligation with cleavable and resin-linked amines. Org. Lett. 19, 4022–4025 (2017).
Behrendt, R., White, P. & Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 22, 4–27 (2016).
Weidmann, J., Dimitrijevic, E., Hoheisel, J. D. & Dawson, P. E. Boc-SPPS: compatible linker for the synthesis of peptide o-aminoanilides. Org. Lett. 18, 164–167 (2016).
Wolczanski, G. & Lisowski, M. A general method for preparation of N-Boc-protected or N-Fmoc-protected alpha,beta-didehydropeptide building blocks and their use in the solid-phase peptide synthesis. J. Pept. Sci. 24, e3091 (2018).
Coin, I., Beyermann, M. & Bienert, M. Solid-phase peptide synthesis: from standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2, 3247–3256 (2007).
Garcia-Martin, F. et al. ChemMatrix, a poly(ethylene glycol)-based support for the solid-phase synthesis of complex peptides. J. Comb. Chem. 8, 213–220 (2006).
Pedersen, S. L., Tofteng, A. P., Malik, L. & Jensen, K. J. Microwave heating in solid-phase peptide synthesis. Chem. Soc. Rev. 41, 1826–1844 (2012).
Paradis-Bas, M., Tulla-Puche, J. & Albericio, F. The road to the synthesis of “difficult peptides”. Chem. Soc. Rev. 45, 631–654 (2016).
Palasek, S. A., Cox, Z. J. & Collins, J. M. Limiting racemization and aspartimide formation in microwave-enhanced Fmoc solid phase peptide synthesis. J. Pept. Sci. 13, 143–148 (2007).
Cardona, V. et al. Application of Dmb-dipeptides in the Fmoc SPPS of difficult and aspartimide-prone sequences. Int J. Pept. Res Ther. 14, 285–292 (2008).
Michels, T., Dolling, R., Haberkorn, U. & Mier, W. Acid-mediated prevention of aspartimide formation in solid phase peptide synthesis. Org. Lett. 14, 5218–5221 (2012).
Subiros-Funosas, R., El-Faham, A. & Albericio, F. Use of Oxyma as pH modulatory agent to be used in the prevention of base-driven side reactions and its effect on 2-chlorotrityl chloride resin. Biopolymers 98, 89–97 (2012).
Gentilucci, L., Tolomelli, A. & Squassabia, F. Peptides and peptidomimetics in medicine, surgery and biotechnology. Curr. Med Chem. 13, 2449–2466 (2006).
Varela, Y. F., Vanegas Murcia, M. & Patarroyo, M. E. Synthetic evaluation of standard and microwave-assisted solid phase peptide synthesis of a long chimeric peptide derived from four plasmodium falciparum proteins. Molecules. 23, 2877 (2018).
Hansen, A. M. et al. Microwave-assisted solid-phase synthesis of antisense acpP peptide nucleic acid-peptide conjugates active against colistin- and tigecycline-resistant E. coli and K. pneumoniae. Eur. J. Med Chem. 168, 134–145 (2019).
Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 17, 134–143 (2015).
Jamieson, A. G., Boutard, N., Sabatino, D. & Lubell, W. D. Peptide scanning for studying structure-activity relationships in drug discovery. Chem. Biol. Drug Des. 81, 148–165 (2013).
Eustache, S., Leprince, J. & Tuffery, P. Progress with peptide scanning to study structure-activity relationships: the implications for drug discovery. Expert Opin. Drug Disco. 11, 771–784 (2016).
Goodwin, D., Simerska, P. & Toth, I. Peptides as therapeutics with enhanced bioactivity. Curr. Medicinal Chem. 19, 4451–4461 (2012).
Miranda, L. P. et al. Identification of potent, selective, and metabolically stable peptide antagonists to the calcitonin gene-related peptide (CGRP) receptor. J. Med Chem. 51, 7889–7897 (2008).
Werner, H. M., Cabalteja, C. C. & Horne, W. S. Peptide backbone composition and protease susceptibility: impact of modification type, position, and tandem substitution. Chembiochem 17, 712–718 (2016).
Wei, X. et al. Retro-inverso isomer of Angiopep-2: a stable d-peptide ligand inspires brain-targeted drug delivery. Mol. Pharm. 11, 3261–3268 (2014).
Chatterjee, J., Rechenmacher, F. & Kessler, H. N-methylation of peptides and proteins: an important element for modulating biological functions. Angew. Chem. Int Ed. Engl. 52, 254–269 (2013).
Cheloha, R. W. et al. Backbone modification of a parathyroid hormone receptor-1 antagonist/inverse agonist. ACS Chem. Biol. 11, 2752–2762 (2016).
Liskamp, R. M. J., Rijkers, D. T. S., Kruijtzer, J. A. W. & Kemmink, J. Peptides and proteins as a continuing exciting source of inspiration for peptidomimetics. Chembiochem 12, 1626–1653 (2011).
Schneider, J. A. et al. Design of peptoid-peptide macrocycles to Inhibit the beta-catenin TCF interaction in prostate cancer. Nat Commun. 9, 4396 (2018).
Olsen, C. A., Montero, A., Leman, L. J. & Ghadiri, M. R. Macrocyclic peptoid-peptide hybrids as inhibitors of class I histone deacetylases. Acs Med Chem. Lett. 3, 749–753 (2012).
Wisniewski, K. et al. New, potent, selective, and short-acting peptidic V1a receptor agonists. J. Med Chem. 54, 4388–4398 (2011).
Maybauer, M. O. et al. The selective vasopressin type 1a receptor agonist selepressin (FE 202158) blocks vascular leak in ovine severe sepsis*. Crit. Care Med 42, e525–e533 (2014).
Masri, E., Ahsanullah, Accorsi, M. & Rademann, J. Side-chain modification of peptides using a phosphoranylidene amino acid. Org. Lett. 22, 2976–2980 (2020).
Frey, V. et al. Structure-activity relationships of Bak derived peptides: affinity and specificity modulations by amino acid replacement. Eur. J. Medicinal Chem. 43, 966–972 (2008).
Hone, A. J. et al. PeIA-5466: a novel peptide antagonist containing non-natural amino acids that selectively targets alpha 3 beta 2 nicotinic acetylcholine receptors. J. Medicinal Chem. 62, 6262–6275 (2019).
Milroy, L. G. et al. Modulators of protein-protein interactions. Chem. Rev. 114, 4695–4748 (2014).
Pelay-Gimeno, M., Glas, A., Koch, O. & Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: mimicking peptide binding epitopes. Angew. Chem. Int Ed. Engl. 54, 8896–8927 (2015).
White, C. J. & Yudin, A. K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 3, 509–524 (2011).
Pelay-Gimeno, M., Tulla-Puche, J. & Albericio, F. “Head-to-side-chain” cyclodepsipeptides of marine origin. Mar. Drugs 11, 1693–1717 (2013).
Mourao, C. B. F. et al. Head-to-tail cyclization after interaction with trypsin: a scorpion venom peptide that resembles plant cyclotides. J. Medicinal Chem. 63, 9500–9511 (2020).
Yang, P. Y. et al. Stapled, long-acting glucagon-like peptide 2 analog with efficacy in dextran sodium sulfate induced mouse colitis models. J. Med Chem. 61, 3218–3223 (2018).
Ricardo, M. G. et al. Multicomponent peptide stapling as a diversity-driven tool for the development of inhibitors of protein-protein interactions. Angew. Chem. Int Ed. 59, 5235–5241 (2020).
Dougherty, P. G. et al. Enhancing the cell permeability of stapled peptides with a cyclic cell-penetrating peptide. J. Medicinal Chem. 62, 10098–10107 (2019).
Perry, S. R. et al. Contiguous hydrophobic and charged surface patches in short helix-constrained peptides drive cell permeability. Org. Biomol. Chem. 16, 367–371 (2018).
Tian, Y. et al. Effect of stapling architecture on physiochemical properties and cell permeability of stapled alpha-helical peptides: a comparative study. Chembiochem 18, 2087–2093 (2017).
Li, C. Y. et al. Binding loop substitutions in the cyclic peptide SFTI-1 generate potent and selective chymase inhibitors. J. Medicinal Chem. 63, 816–826 (2020).
Miao, J. Y. et al. Designing cyclic-peptide inhibitors of protein-protein interactions by mimicking hot loops. Abstr Pap Am Chem S. 256, 434 (2018).
Neukirchen, S. et al. Cyclic peptides for targeting helical protein-protein interactions. J. Pept. Sci. 22, S50–S52 (2016).
Ricardo, M. G., Vasco, A. V., Rivera, D. G. & Wessjohann, L. A. Stabilization of Cyclic beta-Hairpins by Ugi-Reaction-Derived N-Alkylated Peptides: The Quest for Functionalized beta-Turns. Org. Lett. 21, 7307–7310 (2019).
van der Knaap, M. et al. Design, synthesis and structural analysis of mixed alpha/beta-peptides that adopt stable cyclic hairpin-like conformations. Tetrahedron 68, 2391–2400 (2012).
Koch, O., Cole, J., Block, P. & Klebe, G. Secbase: database module to retrieve secondary structure elements with ligand binding motifs. J. Chem. Inf. Model 49, 2388–2402 (2009).
Bullock, B. N., Jochim, A. L. & Arora, P. S. Assessing helical protein interfaces for inhibitor design. J. Am. Chem. Soc. 133, 14220–14223 (2011).
Jochim, A. L. & Arora, P. S. Systematic analysis of helical protein interfaces reveals targets for synthetic inhibitors. ACS Chem. Biol. 5, 919–923 (2010).
Bergey, C. M., Watkins, A. M. & Arora, P. S. HippDB: a database of readily targeted helical protein-protein interactions. Bioinformatics 29, 2806–2807 (2013).
Sawyer, N., Watkins, A. M. & Arora, P. S. Protein domain mimics as modulators of protein-protein interactions. Acc. Chem. Res 50, 1313–1322 (2017).
Khoo, K. K. et al. Lactam-stabilized helical analogues of the analgesic mu-conotoxin KIIIA. J. Med Chem. 54, 7558–7566 (2011).
Galande, A. K. et al. Thioether side chain cyclization for helical peptide formation: inhibitors of estrogen receptor-coactivator interactions. J. Pept. Res 63, 297–302 (2004).
Muppidi, A. et al. Rational design of proteolytically stable, cell-permeable peptide-based selective Mcl-1 inhibitors. J. Am. Chem. Soc. 134, 14734–14737 (2012).
Muppidi, A. et al. Design of antiviral stapled peptides containing a biphenyl cross-linker. Bioorg. Med Chem. Lett. 24, 1748–1751 (2014).
Sakagami, K., Masuda, T., Kawano, K. & Futaki, S. Importance of net hydrophobicity in the cellular uptake of all-hydrocarbon stapled peptides. Mol. Pharm. 15, 1332–1340 (2018).
Reguera, L. & Rivera, D. G. Multicomponent reaction toolbox for peptide macrocyclization and stapling. Chem. Rev. 119, 9836–9860 (2019).
Cabezas, E. & Satterthwait, A. C. The hydrogen bond mimic approach: Solid-phase synthesis of a peptide stabilized as an alpha-helix with a hydrazone link. J. Am. Chem. Soc. 121, 3862–3875 (1999).
Chapman, R. N., Dimartino, G. & Arora, P. S. A highly stable short alpha-helix constrained by a main-chain hydrogen-bond surrogate. J. Am. Chem. Soc. 126, 12252–12253 (2004).
Patgiri, A., Menzenski, M. Z., Mahon, A. B. & Arora, P. S. Solid-phase synthesis of short alpha-helices stabilized by the hydrogen bond surrogate approach. Nat. Protoc. 5, 1857–1865 (2010).
Patgiri, A., Witten, M. R. & Arora, P. S. Solid phase synthesis of hydrogen bond surrogate derived alpha-helices: resolving the case of a difficult amide coupling. Org. Biomol. Chem. 8, 1773–1776 (2010).
Chapman, R. N. & Arora, P. S. Optimized synthesis of hydrogen-bond surrogate helices: surprising effects of microwave heating on the activity of grubbs catalysts. Org. Lett. 8, 5825–5828 (2006).
Sawyer, N. & Arora, P. S. Hydrogen bond surrogate stabilization of beta-hairpins. Acs Chem. Biol. 13, 2027–2032 (2018).
Sawyer, N. & Arora, P. S. Hydrogen bond surrogate beta-hairpins to inhibit protein-protein interactions. Biophys. J. 114, 56a–57a (2018).
Sawyer, N. & Arora, P. S. Using hydrogen bond surrogate technology to stabilize beta-hairpins. Biophys. J. 112, 177a–177a (2017).
Kushal, S. et al. Hydrogen bond surrogate (HBS) helices as orthosteric regulator of hypoxia inducible transcription. Cancer Res. 72, 289 (2012).
Henchey, L. K., Porter, J. R., Ghosh, I. & Arora, P. S. High specificity in protein recognition by hydrogen-bond-surrogate alpha-helices: selective inhibition of the p53/MDM2 complex. Chembiochem 11, 2104–2107 (2010).
Bao, J., Dong, X. Y., Zhang, J. Z. H. & Arora, P. S. Dynamical binding of hydrogen-bond surrogate derived bak helices to antiapoptotic protein Bcl-x(L). J. Phys. Chem. B 113, 3565–3571 (2009).
Wang, L. et al. Synthesis and characterization of water-soluble macrocyclic peptides stabilizing protein alpha-turn. Org. Biomol. Chem. 16, 459–471 (2018).
Wang, L. et al. Solid phase synthesis of constrained 13-membered peptide macrocycles employing Fukuyama-Mitsunobu alkylations. Tetrahedron Lett. 56, 2456–2459 (2015).
Athanassiou, Z. et al. Structural mimicry of retroviral tat proteins by constrained beta-hairpin peptidomimetics: ligands with high affinity and selectivity for viral TAR RNA regulatory elements. J. Am. Chem. Soc. 126, 6906–6913 (2004).
Fasan, R. et al. Using a beta-hairpin to mimic an alpha-helix: Cyclic peptidomimetic inhibitors of the p53-HDM2 protein-protein interaction. Angew. Chem. Int Ed. 43, 2109–2112 (2004).
Cheng, P. N. et al. Amyloid beta-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nat. Chem. 4, 927–933 (2012).
Liu, C. et al. Characteristics of amyloid-related oligomers revealed by crystal structures of macrocyclic beta-sheet mimics. J. Am. Chem. Soc. 133, 6736–6744 (2011).
Konig, H. M. & Kilbinger, A. F. M. Learning from nature: beta-sheet-mimicking copolymers get organized. Angew. Chem. Int Ed. 46, 8334–8340 (2007).
Khakshoor, O., Demeler, B. & Nowick, J. S. Macrocyclic beta-sheet peptides that mimic protein quaternary structure through intermolecular beta-sheet interactions. J. Am. Chem. Soc. 129, 5558–5569 (2007).
Taylor, M. et al. Development of a proteolytically stable retro-inverso peptide inhibitor of beta-amyloid oligomerization as a potential novel treatment for Alzheimer’s disease. Biochem.-Us 49, 3261–3272 (2010).
Tian, Y. A. et al. Solid-phase extraction of N-linked glycopeptides. Nat. Protoc. 2, 334–339 (2007).
Kumar, D. & Bhalla, T. C. Microbial proteases in peptide synthesis: approaches and applications. Appl Microbiol Biot. 68, 726–736 (2005).
Sanjukta, S. & Rai, A. K. Production of bioactive peptides during soybean fermentation and their potential health benefits. Trends Food Sci. Tech. 50, 1–10 (2016).
Sultan, S. et al. Therapeutic potential of dairy bioactive peptides: a contemporary perspective. Crit. Rev. Food Sci. 58, 105–115 (2018).
Vlieghe, P., Lisowski, V., Martinez, J. & Khrestchatisky, M. Synthetic therapeutic peptides: science and market. Drug Discov. Today 15, 40–56 (2010).
Bristow, A. F. Recombinant-DNA-derived insulin analogs as potentially useful therapeutic agents. Trends Biotechnol. 11, 301–305 (1993).
Conibear, A. C., Watson, E. E., Payne, R. J. & Becker, C. F. W. Native chemical ligation in protein synthesis and semi-synthesis. Chem. Soc. Rev. 47, 9046–9068 (2018).
Hirasawa, S. et al. Facile and efficient chemoenzymatic semisynthesis of Fc-fusion compounds for half-life extension of pharmaceutical components. Bioconjugate Chem. 30, 2323–2331 (2019).
Sampaio de Oliveira, K. B. et al. Strategies for recombinant production of antimicrobial peptides with pharmacological potential. Expert Rev. Clin. Pharmacol. 13, 367–390 (2020).
Wibowo, D. & Zhao, C. X. Recent achievements and perspectives for large-scale recombinant production of antimicrobial peptides. Appl Microbiol Biotechnol. 103, 659–671 (2019).
Banting, F. G. et al. Pancreatic extracts in the treatment of diabetes mellitus. Can. Med Assoc. J. 12, 141–146 (1922).
Quianzon, C. C. & Cheikh, I. History of insulin. J Community Hosp Intern Med Perspect. 2, 2 (2012).
Elkinton, J. R. & Hunt, A. D. Jr Effects of pituitary adrenocorticotropic hormone therapy. J. Am. Med Assoc. 141, 1273–1279 (1949).
Copp, D. H. Calcitonin: discovery, development, and clinical application. Clin. Invest Med 17, 268–277 (1994).
Lewis, R. J. & Garcia, M. L. Therapeutic potential of venom peptides. Nat. Rev. Drug Disco. 2, 790–802 (2003).
King, G. F. Venoms as a platform for human drugs: translating toxins into therapeutics. Expert Opin. Biol. Th 11, 1469–1484 (2011).
de Veer, S. J., Kan, M. W. & Craik, D. J. Cyclotides: from structure to function. Chem. Rev. 119, 12375–12421 (2019).
Ojeda, P. G., Cardoso, M. H. & Franco, O. L. Pharmaceutical applications of cyclotides. Drug Disco. Today 24, 2152–2161 (2019).
Kim, D. R. et al. Function and distribution of a lantipeptide in strawberry fusarium wilt disease-suppressive soils. Mol. Plant Microbe Interact. 32, 306–312 (2019).
Murthy, M. V. R., Mohan, E. V. S. & Sadhukhan, A. K. Cyclosporin-A production by Tolypocladium inflatum using solid state fermentation. Process Biochem 34, 269–280 (1999).
Dik, D. A. et al. Semisynthesis of a bacterium with non-canonical cell-wall cross-links. J. Am. Chem. Soc. 142, 10910–10913 (2020).
Franck, C. et al. Semisynthesis of an evasin from tick saliva reveals a critical role of tyrosine sulfation for chemokine binding and inhibition. Proc. Natl Acad. Sci. USA 117, 12657–12664 (2020).
Deng, Y. et al. Semisynthesis of platensimycin derivatives with antibiotic activities in mice via Suzuki-Miyaura cross-coupling reactions. J. Med Chem. 61, 11341–11348 (2018).
Wang, L., Brock, A., Herberich, B. & Schultz, P. G. Expanding the genetic code of Escherichia coli. Science 292, 498–500 (2001).
Wang, L. & Schultz, P. G. Expanding the genetic code. Angew. Chem. Int Ed. Engl. 44, 34–66 (2004).
Oller-Salvia, B. & Chin, J. W. Efficient phage display with multiple distinct non-canonical amino acids using orthogonal ribosome-mediated genetic code expansion. Angew. Chem. Int Ed. Engl. 58, 10844–10848 (2019).
Wang, L. Engineering the genetic code in cells and animals: biological considerations and impacts. Acc. Chem. Res 50, 2767–2775 (2017).
Santoro, S. W. et al. An efficient system for the evolution of aminoacyl-tRNA synthetase specificity. Nat. Biotechnol. 20, 1044–1048 (2002).
Anderson, J. C. et al. An expanded genetic code with a functional quadruplet codon. Proc. Natl Acad. Sci. USA 101, 7566–7571 (2004).
Niu, W., Schultz, P. G. & Guo, J. An expanded genetic code in mammalian cells with a functional quadruplet codon. ACS Chem. Biol. 8, 1640–1645 (2013).
Valentini, T. D. et al. Bioorthogonal non-canonical amino acid tagging reveals translationally active subpopulations of the cystic fibrosis lung microbiota. Nat. Commun. 11, 2287 (2020).
Ernst, R. J. et al. Genetic code expansion in the mouse brain. Nat. Chem. Biol. 12, 776–778 (2016).
Liu, J. et al. Genetic code expansion in zebrafish embryos and its application to optical control of cell signaling. J. Am. Chem. Soc. 139, 9100–9103 (2017).
Brown, W., Liu, J. & Deiters, A. Genetic code expansion in animals. ACS Chem. Biol. 13, 2375–2386 (2018).
Chin, J. W. et al. An expanded eukaryotic genetic code. Science 301, 964–967 (2003).
Wang, N. X. et al. Construction of a live-attenuated HIV-1 vaccine through genetic code expansion. Angew. Chem. Int Ed. 53, 4867–4871 (2014).
Si, L. et al. Generation of influenza A viruses as live but replication-incompetent virus vaccines. Science 354, 1170–1173 (2016).
Zheng, Y. et al. Broadening the versatility of lentiviral vectors as a tool in nucleic acid research via genetic code expansion. Nucleic Acids Res 43, e73 (2015).
Lang, K. & Chin, J. W. Bioorthogonal reactions for labeling proteins. ACS Chem. Biol. 9, 16–20 (2014).
Hu, C. et al. Metalloprotein design using genetic code expansion. Chem. Soc. Rev. 43, 6498–6510 (2014).
Nguyen, T. A., Cigler, M. & Lang, K. Expanding the genetic code to study protein-protein interactions. Angew. Chem. Int Ed. Engl. 57, 14350–14361 (2018).
Courtney, T. & Deiters, A. Recent advances in the optical control of protein function through genetic code expansion. Curr. Opin. Chem. Biol. 46, 99–107 (2018).
Zhou, W. & Deiters, A. Chemogenetic and optogenetic control of post-translational modifications through genetic code expansion. Curr. Opin. Chem. Biol. 63, 123–131 (2021).
Debelouchina, G. T. & Muir, T. W. A molecular engineering toolbox for the structural biologist. Q Rev. Biophys. 50, e7 (2017).
Hoppmann, C. et al. Site-specific incorporation of phosphotyrosine using an expanded genetic code. Nat. Chem. Biol. 13, 842–844 (2017).
He, X. et al. Functional genetic encoding of sulfotyrosine in mammalian cells. Nat. Commun. 11, 4820 (2020).
Italia, J. S. et al. Genetically encoded protein sulfation in mammalian cells. Nat. Chem. Biol. 16, 379–382 (2020).
Neumann, H., Peak-Chew, S. Y. & Chin, J. W. Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat. Chem. Biol. 4, 232–234 (2008).
Krauskopf, K. & Lang, K. Increasing the chemical space of proteins in living cells via genetic code expansion. Curr. Opin. Chem. Biol. 58, 112–120 (2020).
Drienovska, I. & Roelfes, G. Expanding the enzyme universe with genetically encoded unnatural amino acids. Nat. Catal. 3, 193–202 (2020).
Jevsevar, S., Kunstelj, M. & Porekar, V. G. PEGylation of therapeutic proteins. Biotechnol. J. 5, 113–128 (2010).
Veronese, F. M. & Mero, A. The impact of PEGylation on biological therapies. BioDrugs 22, 315–329 (2008).
Gupta, V. et al. Protein PEGylation for cancer therapy: bench to bedside. J. Cell Commun. Signal 13, 319–330 (2019).
Zhang, C. et al. Site-specific PEGylation of therapeutic proteins via optimization of both accessible reactive amino acid residues and PEG derivatives. Biodrugs 26, 209–215 (2012).
Deiters, A. et al. Site-specific PEGylation of proteins containing unnatural amino acids. Bioorg. Med Chem. Lett. 14, 5743–5745 (2004).
Zhang, B. et al. Development of next generation of therapeutic IFN-alpha2b via genetic code expansion. Acta Biomater. 19, 100–111 (2015).
Cho, H. et al. Optimized clinical performance of growth hormone with an expanded genetic code. Proc. Natl Acad. Sci. USA 108, 9060–9065 (2011).
Wu, L. et al. Precise and combinatorial PEGylation generates a low-immunogenic and stable form of human growth hormone. J. Control Release 249, 84–93 (2017).
Shozen, N., Iijima, I. & Hohsaka, T. Site-specific incorporation of PEGylated amino acids into proteins using nonnatural amino acid mutagenesis. Bioorg. Med Chem. Lett. 19, 4909–4911 (2009).
Tada, S. et al. Genetic PEGylation. PLoS One 7, e49235 (2012).
Fu, C. et al. Genetically encoding a lipidated amino acid for extension of protein half-life. vivo. Angew. Chem. Int Ed. Engl. 58, 1392–1396 (2019).
Fok, J. A. & Mayer, C. Genetic-code-expansion strategies for vaccine development. Chembiochem 21, 3291–3300 (2020).
Huang, Y. & Liu, T. Therapeutic applications of genetic code expansion. Synth. Syst. Biotechnol. 3, 150–158 (2018).
Grunewald, J. et al. Immunochemical termination of self-tolerance. Proc. Natl Acad. Sci. USA 105, 11276–11280 (2008).
Gauba, V. et al. Loss of CD4 T-cell-dependent tolerance to proteins with modified amino acids. Proc. Natl Acad. Sci. USA 108, 12821–12826 (2011).
Wang, F. et al. Genetic incorporation of unnatural amino acids into proteins in mycobacterium tuberculosis. Plos One. 5, e9354 (2010).
Lin, S. X. et al. Site-specific engineering of chemical functionalities on the surface of live hepatitis D virus. Angew. Chem. Int Ed. 52, 13970–13974 (2013).
Yuan, Z. et al. Controlling multicycle replication of live-attenuated HIV-1 using an unnatural genetic switch. ACS Synth. Biol. 6, 721–731 (2017).
Chen, Y. et al. Controlling the replication of a genomically recoded HIV-1 with a functional quadruplet codon in mammalian cells. ACS Synth. Biol. 7, 1612–1617 (2018).
Lonsdale, R. & Ward, R. A. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 47, 3816–3830 (2018).
Singh, J., Petter, R. C., Baillie, T. A. & Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Disco. 10, 307–317 (2011).
Sutanto, F., Konstantinidou, M. & Domling, A. Covalent inhibitors: a rational approach to drug discovery. RSC Med Chem. 11, 876–884 (2020).
Li, Q. K. et al. Developing covalent protein drugs via proximity-enabled reactive therapeutics. Cell 182, 85 (2020). -+.
Wang, N. et al. Genetically encoding fluorosulfate-l-tyrosine to react with lysine, histidine, and tyrosine via SuFEx in proteins in Vivo. J. Am. Chem. Soc. 140, 4995–4999 (2018).
Berdan, V. Y., Klauser, P. C. & Wang, L. Covalent peptides and proteins for therapeutics. Bioorg. Med Chem. 29, 115896 (2021).
Pollaro, L. & Heinis, C. Strategies to prolong the plasma residence time of peptide drugs. Medchemcomm 1, 319–324 (2010).
Kaji, I., Karaki, S. & Kuwahara, A. Short-chain fatty acid receptor and its contribution to glucagon-like peptide-1 release. Digestion 89, 31–36 (2014).
Stewart, M. W. et al. Pharmacodynamics, pharmacokinetics, safety, and tolerability of albiglutide (Syncria (R)), a long-acting GLP-1 mimetic, in subjects with type 2 diabetes. Diabetes 57, A154–A154 (2008).
Fala, L. Trulicity (Dulaglutide): a new GLP-1 receptor agonist once-weekly subcutaneous injection approved for the treatment of patients with type 2 diabetes. Am. Health Drug Benefits 8, 131–134 (2015).
Fala, L. Tanzeum (Albiglutide): a once-weekly GLP-1 receptor agonist subcutaneous injection approved for the treatment of patients with type 2 diabetes. Am. Health Drug Benefits 8, 126–130 (2015).
Mitragotri, S., Burke, P. A. & Langer, R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat. Rev. Drug Disco. 13, 655–672 (2014).
Cosmi, F., Laini, R. & Nicolucci, A. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med 376, 890 (2017).
Knudsen, L. B. & Lau, J. The discovery and development of liraglutide and semaglutide. Front Endocrinol. (Lausanne) 10, 155 (2019).
Buckley, S. T. et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci Transl Med. 10, eaar7047 (2018).
Bucheit, J. D. et al. Oral semaglutide: a review of the first oral glucagon-like peptide 1 receptor agonist. Diabetes Technol. Ther. 22, 10–18 (2020).
Biermasz, N. R. New medical therapies on the horizon: oral octreotide. Pituitary 20, 149–153 (2017).
Eldor, R., Arbit, E., Corcos, A. & Kidron, M. Glucose-reducing effect of the ORMD-0801 oral insulin preparation in patients with uncontrolled type 1 diabetes: a pilot study. PLoS One 8, e59524 (2013).
Farra, R. et al. First-in-human testing of a wirelessly controlled drug delivery microchip. Sci. Transl. Med 4, 122ra121 (2012).
Kim, E. S. & Plosker, G. L. AFREZZA (R) (insulin human) inhalation powder: a review in diabetes mellitus. Drugs 75, 1679–1686 (2015).
Trujillo, J. M. & Nuffer, W. GLP-1 receptor agonists for type 2 diabetes mellitus: recent developments and emerging agents. Pharmacotherapy 34, 1174–1186 (2014).
George, C., Byun, A. & Howard-Thompson, A. New injectable agents for the treatment of type 2 diabetes part 2-glucagon-like peptide-1 (GLP-1) agonists. Am. J. Med 131, 1304–1306 (2018).
Rodbard, H. W. The clinical impact of GLP-1 receptor agonists in type 2 diabetes: focus on the long-acting analogs. Diabetes Technol. Ther. 20, S233–S241 (2018).
Tonneijck, L. et al. Renal tubular effects of prolonged therapy with the GLP-1 receptor agonist lixisenatide in patients with type 2 diabetes mellitus. Am. J. Physiol. Ren. Physiol. 316, F231–F240 (2019).
von Scholten, B. J. et al. Glucagon-like peptide 1 receptor agonist (GLP-1 RA): long-term effect on kidney function in patients with type 2 diabetes. J. Diabetes Complications 29, 670–674 (2015).
Greco, E. V. et al. GLP-1 Receptor Agonists and Kidney Protection. Medicina (Kaunas). 55, 233 (2019).
Thomas, M. C. The potential and pitfalls of GLP-1 receptor agonists for renal protection in type 2 diabetes. Diabetes Metab. 43, 2S20–22S27 (2017). Suppl 1.
Bethel, M. A. et al. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: a meta-analysis. Lancet Diabetes Endocrinol. 6, 105–113 (2018).
Thondam, S. K. et al. A glucagon-like peptide-1 (GLP-1) receptor agonist in the treatment for hypothalamic obesity complicated by type 2 diabetes mellitus. Clin. Endocrinol. (Oxf.) 77, 635–637 (2012).
Cuthbertson, D. J. et al. Improved glycaemia correlates with liver fat reduction in obese, type 2 diabetes, patients given glucagon-like peptide-1 (GLP-1) receptor agonists. PLoS One 7, e50117 (2012).
Morano, S. et al. Short-term effects of glucagon-like peptide 1 (GLP-1) receptor agonists on fat distribution in patients with type 2 diabetes mellitus: an ultrasonography study. Acta Diabetol. 52, 727–732 (2015).
Mabilleau, G., Pereira, M. & Chenu, C. Novel skeletal effects of glucagon-like peptide-1 (GLP-1) receptor agonists. J. Endocrinol. 236, R29–R42 (2018).
Li, J. et al. Glucagon-like peptide-1 (GLP-1) receptor agonist liraglutide alters bone marrow exosome-mediated miRNA signal pathways in ovariectomized rats with type 2 diabetes. Med Sci. Monit. 23, 5410–5419 (2017).
Ahren, B. Glucagon-like peptide-1 receptor agonists for type 2 diabetes: a rational drug development. J. Diabetes Investig. 10, 196–201 (2019).
Joseph, P. et al. Reducing the global burden of cardiovascular disease, part 1: the epidemiology and risk factors. Circ. Res 121, 677–694 (2017).
Goodfriend, T. L. & Calhoun, D. A. Resistant hypertension, obesity, sleep apnea, and aldosterone: theory and therapy. Hypertension 43, 518–524 (2004).
Simoes, E. S. A. C. & Teixeira, M. M. ACE inhibition, ACE2 and angiotensin-(1-7) axis in kidney and cardiac inflammation and fibrosis. Pharm. Res 107, 154–162 (2016).
Correa, T. D., Takala, J. & Jakob, S. M. Angiotensin II in septic shock. Crit. Care 19, 98 (2015).
Montone, C. M. et al. Peptidomic strategy for purification and identification of potential ACE-inhibitory and antioxidant peptides in Tetradesmus obliquus microalgae. Anal. Bioanal. Chem. 410, 3573–3586 (2018).
Liao, W., Fan, H., Davidge, S. T. & Wu, J. Egg white-derived antihypertensive peptide IRW (Ile-Arg-Trp) reduces blood pressure in spontaneously hypertensive rats via the ACE2/Ang (1-7)/Mas receptor axis. Mol. Nutr. Food Res 63, e1900063 (2019).
Kuwahara, K. The natriuretic peptide system in heart failure: diagnostic and therapeutic implications. Pharm. Ther. 227, 107863 (2021).
Burnett, J. C. Jr. Atrial natriuretic peptide, heart failure and the heart as an endocrine organ. Clin. Chem. 65, 1602–1603 (2019).
Shah, S. J. & Teerlink, J. R. Nesiritide: a reappraisal of efficacy and safety. Expert Opin. Pharmacother. 8, 361–369 (2007).
Sackner-Bernstein, J. D., Kowalski, M., Fox, M. & Aaronson, K. Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA 293, 1900–1905 (2005).
Forte, M. et al. Cardiovascular Pleiotropic Effects of Natriuretic Peptides. Int J Mol Sci. 20, 3874 (2019).
Ichiki, T., Dzhoyashvili, N. & Burnett, J. C. Jr. Natriuretic peptide based therapeutics for heart failure: Cenderitide: a novel first-in-class designer natriuretic peptide. Int J. Cardiol. 281, 166–171 (2019).
Lee, C. Y. et al. Pharmacodynamics of a novel designer natriuretic peptide, CD-NP, in a first-in-human clinical trial in healthy subjects. J. Clin. Pharm. 49, 668–673 (2009).
Duggan, K. A., Hodge, G., Chen, J. & Hunter, T. Vasoactive intestinal peptide infusion reverses existing myocardial fibrosis in the rat. Eur. J. Pharmacol. 862, 172629 (2019).
Dong, Y. et al. Cyclic peptide RD808 reduces myocardial injury induced by beta1-adrenoreceptor autoantibodies. Heart Vessels 34, 1040–1051 (2019).
Gao, H. R. & Gao, H. Y. Cardiovascular functions of central corticotropin-releasing factor related peptides system. Neuropeptides 75, 18–24 (2019).
Barchetta, I. et al. Greater circulating DPP4 activity is associated with impaired flow-mediated dilatation in adults with type 2 diabetes mellitus. Nutr. Metab. Cardiovasc Dis. 29, 1087–1094 (2019).
Bevins, C. L. & Salzman, N. H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol 9, 356–368 (2011).
Aroniadis, O. C. & Brandt, L. J. Fecal microbiota transplantation: past, present and future. Curr. Opin. Gastroenterol. 29, 79–84 (2013).
Hold, G. L. et al. Role of the gut microbiota in inflammatory bowel disease pathogenesis: what have we learnt in the past 10 years? World J. Gastroenterol. 20, 1192–1210 (2014).
Baumgart, D. C. & Carding, S. R. Inflammatory bowel disease: cause and immunobiology. Lancet 369, 1627–1640 (2007).
Nemoto, H. et al. Reduced diversity and imbalance of fecal microbiota in patients with ulcerative colitis. Dig. Dis. Sci. 57, 2955–2964 (2012).
Frank, D. N. et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl Acad. Sci. USA 104, 13780–13785 (2007).
Mukhopadhya, I. et al. Detection of Campylobacter concisus and other Campylobacter species in colonic biopsies from adults with ulcerative colitis. PLoS One 6, e21490 (2011).
Holani, R. et al. Proline-arginine rich (PR-39) cathelicidin: structure, expression and functional implication in intestinal health. Comp. Immunol. Microbiol Infect. Dis. 49, 95–101 (2016).
Limketkai, B. N., Parian, A. M., Shah, N. D. & Colombel, J. F. Short bowel syndrome and intestinal failure in Crohn’s disease. Inflamm. Bowel Dis. 22, 1209–1218 (2016).
Billiauws, L., Corcos, O. & Joly, F. What’s new in short bowel syndrome? Curr. Opin. Clin. Nutr. Metab. Care 21, 313–318 (2018).
Venneti, K. C. & Hewage, C. M. Conformational and molecular interaction studies of glucagon-like peptide-2 with its N-terminal extracellular receptor domain. FEBS Lett. 585, 346–352 (2011).
Lee, J. et al. Enteroendocrine-derived glucagon-like peptide-2 controls intestinal amino acid transport. Mol. Metab. 6, 245–255 (2017).
Nardini, P. et al. GLP-2 Prevents neuronal and glial changes in the distal colon of mice chronically treated with cisplatin. Int J Mol Sci. 21, 8875 (2020).
Pakarinen, M. P. GLP-2 cures the gut - What about the liver? EBioMedicine 46, 11–12 (2019).
Wang, W. W., Qiao, S. Y. & Li, D. F. Amino acids and gut function. Amino Acids 37, 105–110 (2009).
Marier, J. F. et al. Population pharmacokinetics of teduglutide following repeated subcutaneous administrations in healthy participants and in patients with short bowel syndrome and Crohn’s disease. J. Clin. Pharm. 50, 36–49 (2010).
Jeppesen, P. B. Pharmacologic options for intestinal rehabilitation in patients with short bowel syndrome. JPEN J. Parenter. Enter. Nutr. 38, 45S–52S (2014).
Thymann, T. et al. Acute effects of the glucagon-like peptide 2 analogue, teduglutide, on intestinal adaptation in short bowel syndrome. J. Pediatr. Gastroenterol. Nutr. 58, 694–702 (2014).
Wisniewski, K. et al. Synthesis and pharmacological characterization of novel glucagon-like peptide-2 (GLP-2) analogues with low systemic clearance. J. Med Chem. 59, 3129–3139 (2016).
Kunkel, D. et al. Efficacy of the glucagon-like peptide-1 agonist exenatide in the treatment of short bowel syndrome. Neurogastroenterol. Motil. 23, 739–e328 (2011).
Madsen, K. B. et al. Acute effects of continuous infusions of glucagon-like peptide (GLP)-1, GLP-2 and the combination (GLP-1+GLP-2) on intestinal absorption in short bowel syndrome (SBS) patients. A placebo-controlled study. Regul. Pept. 184, 30–39 (2013).
Raffort, J. et al. Insights on glicentin, a promising peptide of the proglucagon family. Biochem Med (Zagreb) 27, 308–324 (2017).
Lim, D. W. et al. Synergy of glucagon-like peptide-2 and epidermal growth factor coadministration on intestinal adaptation in neonatal piglets with short bowel syndrome. Am. J. Physiol. Gastrointest. Liver Physiol. 312, G390–G404 (2017).
Shiou, S. R. et al. Erythropoietin protects intestinal epithelial barrier function and lowers the incidence of experimental neonatal necrotizing enterocolitis. J. Biol. Chem. 286, 12123–12132 (2011).
Jain, S. K. et al. Amniotic fluid-borne hepatocyte growth factor protects rat pups against experimental necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 306, G361–369 (2014).
Di Bella, S. et al. Clostridium difficile Toxins A and B: insights into pathogenic properties and extraintestinal effects. Toxins (Basel). 8, 134 (2016).
Hong, J. et al. The American cockroach peptide periplanetasin-2 blocks clostridium difficile toxin a-induced cell damage and inflammation in the gut. J. Microbiol Biotechnol. 27, 694–700 (2017).
Yun, J., Hwang, J. S. & Lee, D. G. The antifungal activity of the peptide, periplanetasin-2, derived from American cockroach Periplaneta americana. Biochem J. 474, 3027–3043 (2017).
Kim, D. H. et al. The insect peptide CopA3 increases colonic epithelial cell proliferation and mucosal barrier function to prevent inflammatory responses in the gut. J. Biol. Chem. 291, 3209–3223 (2016).
Shrestha, A. et al. A synthetic peptide corresponding to the extracellular loop 2 region of claudin-4 protects against Clostridium perfringens enterotoxin in vitro and in vivo. Infect. Immun. 82, 4778–4788 (2014).
Marin, M. et al. Human cathelicidin improves colonic epithelial defenses against Salmonella typhimurium by modulating bacterial invasion, TLR4 and pro-inflammatory cytokines. Cell Tissue Res 376, 433–442 (2019).
Chen, W. et al. Induction and antiviral activity of human beta-defensin 3 in intestinal cells with picornavirus infection. Acta Virol. 62, 287–293 (2018).
Assis, D. N. & Freedman, S. D. Gastrointestinal disorders in cystic fibrosis. Clin. Chest Med 37, 109–118 (2016).
McHugh, D. R. et al. Linaclotide improves gastrointestinal transit in cystic fibrosis mice by inhibiting sodium/hydrogen exchanger 3. Am. J. Physiol. Gastrointest. Liver Physiol. 315, G868–G878 (2018).
Tanabe, S. et al. Identification of a peptide in enzymatic hydrolyzate of cheese that inhibits ovalbumin permeation in Caco-2 cells. J. Agric Food Chem. 54, 6904–6908 (2006).
Bessette, C. et al. Protective effects of beta-casofensin, a bioactive peptide from bovine beta-casein, against indomethacin-induced intestinal lesions in rats. Mol. Nutr. Food Res 60, 823–833 (2016).
Anthony, A., Pounder, R. E., Dhillon, A. P. & Wakefield, A. J. Similarities between ileal Crohn’s disease and indomethacin experimental jejunal ulcers in the rat. Aliment Pharm. Ther. 14, 241–245 (2000).
Liu, H. et al. Antimicrobial peptide KR-32 alleviates Escherichia coli K88-induced fatty acid malabsorption by improving expression of fatty acid transporter protein 4 (FATP4)1. J. Anim. Sci. 97, 2342–2356 (2019).
Zhang, H. et al. Effects of cathelicidin-derived peptide from reptiles on lipopolysaccharide-induced intestinal inflammation in weaned piglets. Vet. Immunol. Immunopathol. 192, 41–53 (2017).
Luo, X. J. et al. Stimulation of calcitonin gene-related peptide release through targeting capsaicin receptor: a potential strategy for gastric mucosal protection. Dig. Dis. Sci. 58, 320–325 (2013).
Szlachcic, A. et al. New satiety hormone nesfatin-1 protects gastric mucosa against stress-induced injury: mechanistic roles of prostaglandins, nitric oxide, sensory nerves and vanilloid receptors. Peptides 49, 9–20 (2013).
Tache, Y. Brainstem neuropeptides and vagal protection of the gastric mucosal against injury: role of prostaglandins, nitric oxide and calcitonin-gene related peptide in capsaicin afferents. Curr. Med Chem. 19, 35–42 (2012).
Sibilia, V. et al. Characterization of the mechanisms involved in the gastric antisecretory effect of TLQP-21, a vgf-derived peptide, in rats. Amino Acids 42, 1261–1268 (2012).
Sibilia, V. et al. TLQP-21, a VGF-derived peptide, prevents ethanol-induced gastric lesions: insights into its mode of action. Neuroendocrinology 92, 189–197 (2010).
Zhang, Y. et al. Novokinin inhibits gastric acid secretion and protects against alcohol-induced gastric injury in rats. Alcohol 56, 1–8 (2016).
Amorim, M. M. et al. Antiulcer and antiproliferative properties of spent brewer’s yeast peptide extracts for incorporation into foods. Food Funct. 7, 2331–2337 (2016).
Schwarz, P. et al. Hepcidin is localised in gastric parietal cells, regulates acid secretion and is induced by Helicobacter pylori infection. Gut 61, 193–201 (2012).
Nassar, N. N., Schaalan, M. F., Zaki, H. F. & Abdallah, D. M. Octreotide ameliorates gastric lesions in chronically mild stressed rats. World J. Gastroenterol. 17, 1135–1142 (2011).
Bulbul, M., Babygirija, R., Ludwig, K. & Takahashi, T. Central oxytocin attenuates augmented gastric postprandial motility induced by restraint stress in rats. Neurosci. Lett. 479, 302–306 (2010).
Liang, S. et al. Screening and identification of vascular-endothelial-cell-specific binding peptide in gastric cancer. J. Mol. Med (Berl.) 84, 764–773 (2006).
Zhang, J. et al. Targeted radiotherapy with tumor vascular homing trimeric GEBP11 peptide evaluated by multimodality imaging for gastric cancer. J. Control Release 172, 322–329 (2013).
Himaya, S. W., Dewapriya, P. & Kim, S. K. EGFR tyrosine kinase inhibitory peptide attenuates Helicobacter pylori-mediated hyper-proliferation in AGS enteric epithelial cells. Toxicol. Appl Pharm. 269, 205–214 (2013).
Zhang, X. L. et al. The synthetic antimicrobial peptide pexiganan and its nanoparticles (PNPs) exhibit the anti-helicobacter pylori activity in vitro and in vivo. Molecules 20, 3972–3985 (2015).
Esposito, R. et al. Gastric TFF1 expression from acute to chronic helicobacter infection. Front Cell Infect. Microbiol 7, 434 (2017).
Hoffmann, W. TFF2, a MUC6-binding lectin stabilizing the gastric mucus barrier and more (Review). Int J. Oncol. 47, 806–816 (2015).
Gulec Suyen, G., Isbil-Buyukcoskun, N., Cam, B. & Ozluk, K. Effects of centrally injected glucagon-like peptide-2 on gastric mucosal blood flow in rats: possible mechanisms. Peptides 64, 62–66 (2015).
Rotondo, A. et al. Glucagon-like peptide-1 relaxes gastric antrum through nitric oxide in mice. Peptides 32, 60–64 (2011).
Schwarte, L. A. et al. The differential effects of recombinant brain natriuretic peptide, nitroglycerine and dihydralazine on systemic oxygen delivery and gastric mucosal microvascular oxygenation in dogs. Anaesthesia 67, 501–507 (2012).
Takemi, S. et al. The important role of ghrelin on gastric contraction in Suncus murinus. Endocr. J. 64, S11–S14 (2017).
Goswami, C. et al. Motilin stimulates gastric acid secretion in coordination with ghrelin in suncus murinus. PLoS One 10, e0131554 (2015).
Wang, S. H. & Yu, J. Structure-based design for binding peptides in anti-cancer therapy. Biomaterials 156, 1–15 (2018).
Yang, Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J. Clin. Invest 125, 3335–3337 (2015).
Carvajal, L. A. et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Science Translational Medicine. 10, eaao3003 (2018).
Carvajal, L. A. et al. Dual inhibition of Mdmx and Mdm2 using an alpha-helical P53 stapled peptide (ALRN-6924) as a novel therapeutic strategy in acute myeloid leukemia. Blood. 130, 795 (2017).
Chang, Y. S. et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl Acad. Sci. USA 110, E3445–3454 (2013).
Sun, X. et al. Peptide-based imaging agents for cancer detection. Adv. Drug Deliv. Rev. 110-111, 38–51 (2017).
Deutscher, S. L. Phage display in molecular imaging and diagnosis of cancer. Chem. Rev. 110, 3196–3211 (2010).
Zhao, N., Qin, Y., Liu, H. & Cheng, Z. Tumor-targeting peptides: ligands for molecular imaging and therapy. Anticancer Agents Med Chem. 18, 74–86 (2018).
Xiao, Y. F. et al. Peptide-based treatment: a promising cancer therapy. J. Immunol. Res 2015, 761820 (2015).
Reubi, J. C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 24, 389–427 (2003).
Xu, C. & Zhang, H. Somatostatin receptor based imaging and radionuclide therapy. Biomed. Res Int 2015, 917968 (2015).
Maxwell, J. E. & Howe, J. R. Imaging in neuroendocrine tumors: an update for the clinician. Int J. Endocr. Oncol. 2, 159–168 (2015).
Gao, R. et al. Clinical Value of (99m)Tc-3PRGD2 SPECT/CT in Differentiated Thyroid Carcinoma with Negative (131)I Whole-Body Scan and Elevated Thyroglobulin Level. Sci. Rep. 8, 473 (2018).
Strosberg, J. et al. Phase 3 Trial of (177)Lu-dotatate for midgut neuroendocrine tumors. N. Engl. J. Med 376, 125–135 (2017).
Tagawa, S. T. et al. Phase 1/2 study of fractionated dose lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 ((177) Lu-J591) for metastatic castration-resistant prostate cancer. Cancer 125, 2561–2569 (2019).
Escala Cornejo, R. A. et al. Large cell neuroendocrine carcinoma of the lung with atypical evolution and a remarkable response to lutetium Lu 177 dotatate. Ann. Nucl. Med 32, 568–572 (2018).
Hagimori, M., Fuchigami, Y. & Kawakami, S. Peptide-based cancer-targeted DDS and molecular imaging. Chem. Pharm. Bull. (Tokyo) 65, 618–624 (2017).
Engel, J. B. et al. Targeting of peptide cytotoxins to LHRH receptors for treatment of cancer. Curr. Drug Targets 17, 488–494 (2016).
Chen, Y., Wu, J. J. & Huang, L. Nanoparticles targeted with NGR motif deliver c-myc siRNA and doxorubicin for anticancer therapy. Mol. Ther. 18, 828–834 (2010).
Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 110-111, 3–12 (2017).
Kotsakis, A. et al. A phase II trial evaluating the clinical and immunologic response of HLA-A2(+) non-small cell lung cancer patients vaccinated with an hTERT cryptic peptide. Lung Cancer 86, 59–66 (2014).
Mahdavi, M. & Moreau, V. In silico designing breast cancer peptide vaccine for binding to MHC class I and II: A molecular docking study. Comput Biol. Chem. 65, 110–116 (2016).
Kumai, T., Kobayashi, H., Harabuchi, Y. & Celis, E. Peptide vaccines in cancer-old concept revisited. Curr. Opin. Immunol. 45, 1–7 (2017).
Guidotti, G., Brambilla, L. & Rossi, D. Cell-penetrating peptides: from basic research to clinics. Trends Pharm. Sci. 38, 406–424 (2017).
Hill, M. D. et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): a multicentre, double-blind, randomised controlled trial. Lancet 395, 878–887 (2020).
Li, J. et al. Cytomembrane infused polymer accelerating delivery of myelin antigen peptide to treat experimental autoimmune encephalomyelitis. Acs Nano 12, 11579–11590 (2018).
Tsoras, A. N. & Champion, J. A. Cross-linked peptide nanoclusters for delivery of oncofetal antigen as a cancer vaccine. Bioconjug Chem. 29, 776–785 (2018).
Boohaker, R. J. et al. Rational design and development of a peptide inhibitor for the PD-1/PD-L1 interaction. Cancer Lett. 434, 11–21 (2018).
Zhou, K. et al. Structure-based derivation and intramolecular cyclization of peptide inhibitors from PD-1/PD-L1 complex interface as immune checkpoint blockade for breast cancer immunotherapy. Biophys. Chem. 253, 106213 (2019).
Abbas, A. B. et al. Design and synthesis of A PD-1 binding peptide and evaluation of its anti-tumor activity. Int J Mol Sci. 20, 572 (2019).
Sasikumar, P. G. et al. A rationally designed peptide antagonist of the PD-1 signaling pathway as an immunomodulatory agent for cancer therapy. Mol. Cancer Ther. 18, 1081–1091 (2019).
Mahadevappa, R., Ma, R. & Kwok, H. F. Venom peptides: improving specificity in cancer therapy. Trends Cancer 3, 611–614 (2017).
Okada, M., Ortiz, E., Corzo, G. & Possani, L. D. Pore-forming spider venom peptides show cytotoxicity to hyperpolarized cancer cells expressing K+ channels: A lentiviral vector approach. PLoS One 14, e0215391 (2019).
Zhang, Q. Y. et al. Antimicrobial peptides: mechanism of action, activity and clinical potential. Mil. Med Res 8, 48 (2021).
Jin, G. & Weinberg, A. Human antimicrobial peptides and cancer. Semin Cell Dev. Biol. 88, 156–162 (2019).
Strzelecka, P. et al. Simplified, serine-rich theta-defensin analogues as antitumour peptides. Chem. Biol. Drug Des. 90, 52–63 (2017).
Aronson, M. R. et al. Lipopeptisomes: anticancer peptide-assembled particles for fusolytic oncotherapy. Acta Biomater. 80, 269–277 (2018).
Howard, C. R. & Fletcher, N. F. Emerging virus diseases: can we ever expect the unexpected? Emerg Microbes Infec. 1, e46 (2012).
Rider, T. H. et al. Broad-Spectrum Antiviral Therapeutics. Plos One. 6, e22572 (2011).
Agarwal, G. & Gabrani, R. Antiviral Peptides: Identification and Validation. Int J Pept Res Ther, 18, 1–20 (2020).
Boas, L. C. P. V. et al. Antiviral peptides as promising therapeutic drugs. Cell Mol. Life Sci. 76, 3525–3542 (2019).
Kaconis, Y. et al. A new and promising therapeutic approach against gram-negative sepsis based on the antimicrobial peptide Aspidasept (R) and its derivatives. Infection 41, S15–S15 (2013).
Chew, M. F., Poh, K. S. & Poh, C. L. Peptides as therapeutic agents for dengue virus. Int J. Med Sci. 14, 1342–1359 (2017).
Su, X. et al. Protein- and peptide-based virus inactivators: inactivating viruses before their entry into cells. Front Microbiol 11, 1063 (2020).
Teissier, E., Penin, F. & Pecheur, E. I. Targeting cell entry of enveloped viruses as an antiviral strategy. Molecules 16, 221–250 (2011).
Perry, C. M. Telaprevir: a review of its use in the management of genotype 1 chronic hepatitis C. Drugs 72, 619–641 (2012).
Garnock-Jones, K. P. Boceprevir: a review of its use in the management of chronic hepatitis C genotype 1 infection. Drugs 72, 2431–2456 (2012).
Divyashree, M. et al. Clinical applications of antimicrobial peptides (AMPs): where do we stand now? Protein Pept. Lett. 27, 120–134 (2020).
Bogomolov, P. et al. A proof-of-concept Phase IIa clinical trial to treat chronic HBV/HDV with the entry inhibitor myrcludex B. Hepatology 64, 121a–121a (2016).
Loglio, A. et al. Myrcludex monotherapy in compensated cirrhotics with delta hepatitis: safety and effectiveness beyond two years of treatment in a real-life setting. Hepatology 72, 624a–624a (2020).
Wedemeyer, H. et al. Final results of a multicenter, open-label phase 2 clinical trial (MYR203) to assess safety and efficacy of myrcludex B in cwith PEG-interferon Alpha 2a in patients with chronic HBV/HDV co-infection. J. Hepatol. 70, E81–E81 (2019).
Badani, H. et al. Mechanism of action of flufirvitide, a peptide inhibitor of influenza virus infection. Biophys. J. 106, 707a–707a (2014).
Skalickova, S. et al. Perspective of use of antiviral peptides against influenza. Virus Viruses-Basel. 7, 5428–5442 (2015).
Yu, D. et al. Molecular mechanism of HIV-1 resistance to sifuvirtide, a clinical trial-approved membrane fusion inhibitor. J. Biol. Chem. 293, 12703–12718 (2018).
Li, L. et al. Efficacy, stability, and biosafety of sifuvirtide gel as a microbicide candidate against HIV-1. PLoS One 7, e37381 (2012).
Yao, X. et al. Broad antiviral activity and crystal structure of HIV-1 fusion inhibitor sifuvirtide. J. Biol. Chem. 287, 6788–6796 (2012).
Phelan, A. L., Katz, R. & Gostin, L. O. The novel coronavirus originating in wuhan, China: challenges for global health governance. JAMA 323, 709–710 (2020).
Xia, S. et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res 30, 343–355 (2020).
Jiang, S. Don’t rush to deploy COVID-19 vaccines and drugs without sufficient safety guarantees. Nature 579, 321 (2020).
Ledford, H. Coronavirus breakthrough: dexamethasone is first drug shown to save lives. Nature 582, 469 (2020).
Pulla, P. India expands use of controversial drug for coronavirus despite safety concerns. Nature, Epub ahead of print (2020).
Nelde, A. et al. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat Immunol, 22, 74–85 (2020).
Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574 (2020).
Wu, F. et al. Author Correction: A new coronavirus associated with human respiratory disease in China. Nature 580, E7 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Andersen, K. G. et al. The proximal origin of SARS-CoV-2. Nat. Med 26, 450–452 (2020).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med 383, 2603–2615 (2020).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med 384, 403–416 (2021).
Voysey, M. et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 397, 99–111 (2021).
Sadoff, J. et al. Safety and efficacy of single-dose Ad26.COV2.S vaccine against Covid-19. N. Engl. J. Med. 384, 2187–2201 (2021).
Xia, S. et al. Safety and immunogenicity of an inactivated COVID-19 vaccine, BBIBP-CorV, in people younger than 18 years: a randomised, double-blind, controlled, phase 1/2 trial. Lancet Infect Dis, 22, 196–208 (2021).
Sapkal, G. N. et al. Inactivated COVID-19 vaccine BBV152/COVAXIN effectively neutralizes recently emerged B.1.1.7 variant of SARS-CoV-2. J Travel Med. 28, taab051 (2021).
Topuzogullari, M. et al. An insight into the epitope-based peptide vaccine design strategy and studies against COVID-19. Turk. J. Biol. 44, 215–227 (2020).
Mahdavi, M., Moreau, V. & Kheirollahi, M. Identification of B and T cell epitope based peptide vaccine from IGF-1 receptor in breast cancer. J. Mol. Graph Model 75, 316–321 (2017).
Yasmin, T. & Nabi, A. H. M. N. B and T cell epitope-based peptides predicted from evolutionarily conserved and whole protein sequences of Ebola virus as vaccine targets. Scand. J. Immunol. 83, 321–337 (2016).
Lin, L. et al. Epitope-based peptide vaccines predicted against novel coronavirus disease caused by SARS-CoV-2. Virus Res 288, 198082 (2020).
Chakraborty, C. et al. Immunoinformatics approach for the identification and characterization of T Cell and B cell epitopes towards the peptide-based vaccine against SARS-CoV-2. Arch. Med. Res. 52, 362–370 (2021).
Bhattacharya, M. et al. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. J. Med Virol. 92, 618–631 (2020).
Waqas, M. et al. Determine the potential epitope based peptide vaccine against novel SARS-CoV-2 targeting structural proteins using immunoinformatics approaches. Front. in Mol. Biosci. 7, 227 (2020).
Kalita, P., Padhi, A. K., Zhang, K. Y. J. & Tripathi, T. Design of a peptide-based subunit vaccine against novel coronavirus SARS-CoV-2. Micro. Pathog. 145, 104236 (2020).
Lim, H. X. et al. Development of multi-epitope peptide-based vaccines against SARS-CoV-2. Biomed. J. 44, 18–30 (2021).
Alam, A. et al. Design of an epitope-based peptide vaccine against the SARS-CoV-2: a vaccine-informatics approach. Brief. Bioinforma. 22, 1309–1323 (2021).
Herst, C. V. et al. An effective CTL peptide vaccine for Ebola Zaire Based on Survivors’ CD8+ targeting of a particular nucleocapsid protein epitope with potential implications for COVID-19 vaccine design. Vaccine 38, 4464–4475 (2020).
Bhattacharjee, S. Toward COVID-19 Therapeutics: a viewpoint from the nonprotein amino acid based synthetic peptide design approach. ACS Chem Neurosci, 11, 3701–3703 (2020).
Kabra, R. & Singh, S. Evolutionary artificial intelligence based peptide discoveries for effective Covid-19 therapeutics. Biochim Biophys. Acta Mol. Basis Dis. 1867, 165978 (2020).
Sahu, B. et al. Design and in-silico screening of Peptide Nucleic Acid (PNA) inspired novel pronucleotide scaffolds targeting COVID-19. Curr Comput Aided Drug Des, 18, 26–40 (2020).
Zhao, Q. Y. et al. Synthesis and immunological evaluation of synthetic peptide based anti-SARS-CoV-2 vaccine candidates. Chem. Commun. 57, 1474–1477 (2021).
Knop, F. K., Bronden, A. & Vilsboll, T. Exenatide: pharmacokinetics, clinical use, and future directions. Expert Opin. Pharmacother. 18, 555–571 (2017).
Ladenheim, E. E. Liraglutide and obesity: a review of the data so far. Drug Des., Dev. Ther. 9, 1867–1875 (2015).
Petersen, A. B., Knop, F. K. & Christensen, M. Lixisenatide for the treatment of type 2 diabetes. Drugs today 49, 537–553 (2013).
Bronden, A., Naver, S. V., Knop, F. K. & Christensen, M. Albiglutide for treating type 2 diabetes: an evaluation of pharmacokinetics/pharmacodynamics and clinical efficacy. Expert Opin. drug Metab. Toxicol. 11, 1493–1503 (2015).
Jendle, J. et al. Efficacy and safety of dulaglutide in the treatment of type 2 diabetes: a comprehensive review of the dulaglutide clinical data focusing on the AWARD phase 3 clinical trial program. Diabetes/Metab. Res. Rev. 32, 776–790 (2016).
Pratley, R. et al. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet 394, 39–50 (2019).
Kim, E. S. & Keam, S. J. Teduglutide: a review in short bowel syndrome. Drugs 77, 345–352 (2017).
Love, B. L., Johnson, A. & Smith, L. S. Linaclotide: a novel agent for chronic constipation and irritable bowel syndrome. Am. J. Health-Syst. Pharm.: AJHP: Off. J. Am. Soc. Health-Syst. Pharmacists 71, 1081–1091 (2014).
Younk, L. M., Mikeladze, M. & Davis, S. N. Pramlintide and the treatment of diabetes: a review of the data since its introduction. Expert Opin. Pharmacother. 12, 1439–1451 (2011).
Kirby, R. S., Fitzpatrick, J. M. & Clarke, N. Abarelix and other gonadotrophin-releasing hormone antagonists in prostate cancer. BJU Int 104, 1580–1584 (2009).
Sonesson, A. & Rasmussen, B. B. In vitro and in vivo human metabolism of degarelix, a gonadotropin-releasing hormone receptor blocker. Drug Metab. disposition: Biol. fate Chem. 41, 1339–1346 (2013).
Mushtaq, A. et al. Efficacy and toxicity profile of carfilzomib based regimens for treatment of multiple myeloma: A systematic review. Crit. Rev. Oncol./Hematol. 125, 1–11 (2018).
Meyers, P. A. Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma. Expert Rev. anticancer Ther. 9, 1035–1049 (2009).
Keijzers, G. B. Aviptadil (Senatek). Curr. Opin. investigational drugs 2, 545–549 (2001).
Wisniewski, K. Design of oxytocin analogs. Methods Mol. Biol. 2001, 235–271 (2019).
Thirunarayanan, N., Raaka, B. M. & Gershengorn, M. C. Taltirelin is a superagonist at the human thyrotropin-releasing hormone receptor. Front Endocrinol. (Lausanne) 3, 120 (2012).
Dhillon, S. & Keam, S. J. Bremelanotide: first approval. Drugs 79, 1599–1606 (2019).
Minisola, S. et al. Update on the safety and efficacy of teriparatide in the treatment of osteoporosis. Ther. Adv. Musculoskelet. Dis. 11, 1759720X19877994 (2019).
Bhattacharyya, S., Pal, S. & Chattopadhyay, N. Abaloparatide, the second generation osteoanabolic drug: Molecular mechanisms underlying its advantages over the first-in-class teriparatide. Biochem Pharmacol. 166, 185–191 (2019).
DeMicco, M. et al. Randomized clinical trial: efficacy and safety of plecanatide in the treatment of chronic idiopathic constipation. Therapeutic Adv. Gastroenterol. 10, 837–851 (2017).
O’Connor, C. M. et al. Effect of nesiritide in patients with acute decompensated heart failure. N. Engl. J. Med 365, 32–43 (2011).
Jadhav, A. P. & Sadaka, F. G. Angiotensin II in septic shock. Am. J. Emerg. Med 37, 1169–1174 (2019).
Jeon, J., Lee, Y. J. & Lee, S. Y. Effect of icatibant on angiotensin-converting enzyme inhibitor-induced angioedema: a meta-analysis of randomized controlled trials. J. Clin. Pharm. therapeutics. 44, 685–692 (2019).
Pu, J. et al. Development of protein- and peptide-based HIV entry inhibitors targeting gp120 or gp41. Viruses. 11, 705 (2019).
Grunfeld, C., Dritselis, A. & Kirkpatrick, P. Tesamorelin. Nat. Rev. Drug Disco. 10, 95–96 (2011).
Williams, J. A., Day, M. & Heavner, J. E. Ziconotide: an update and review. Expert Opin. Pharmacother. 9, 1575–1583 (2008).
Neunert, C. E. & Rose, M. J. Romiplostim for the management of pediatric immune thrombocytopenia: drug development and current practice. Blood Adv. 3, 1907–1915 (2019).
Schmid, H. Peginesatide for the treatment of renal disease-induced anemia. Expert Opin. Pharmacother. 14, 937–948 (2013).
Piehl, E. & Fernandez-Bustamante, A. Lucinactant for the treatment of respiratory distress syndrome in neonates. Drugs today 48, 587–593 (2012).
Hamano, N., Komaba, H. & Fukagawa, M. Etelcalcetide for the treatment of secondary hyperparathyroidism. Expert Opin. Pharmacother. 18, 529–534 (2017).
Minder, E. I., Barman-Aksoezen, J. & Schneider-Yin, X. Pharmacokinetics and Pharmacodynamics of Afamelanotide and its Clinical Use in Treating Dermatologic Disorders. Clin. pharmacokinetics 56, 815–823 (2017).
Wildemberg, L. E. & Gadelha, M. R. Pasireotide for the treatment of acromegaly. Expert Opin. Pharmacother. 17, 579–588 (2016).
Lutetium lu. 177 dotatate (Lutathera) for gastroenteropancreatic neuroendocrine tumors. Med Lett Drugs Ther. 60, e152–e153, (2018).
Lutetium Lu. 177 Dotatate Approved by FDA. Cancer Discov. 8, OF2, (2018).
Evangelista, L. et al. Ga-68 DOTA-peptides and F-18 FDG PET/CT in patients with neuroendocrine tumor: a review. Clin. Imaging 67, 113–116 (2020).
Moyade, P. & Vinjamuri, S. The heart matters: a review of incidental cardiac uptake on Ga-68 DOTA peptide PET-CT scans. Nucl. Med Commun. 40, 1081–1085 (2019).
Haws, R. et al. Effect of setmelanotide, a melanocortin-4 receptor agonist, on obesity in Bardet-Biedl syndrome. Diabetes Obes Metab, 22, 2133–2140 (2020).
Kamermans, A. et al. Setmelanotide, a novel, selective melanocortin receptor-4 agonist exerts anti-inflammatory actions in astrocytes and promotes an anti-inflammatory macrophage phenotype. Front Immunol. 10, 2312 (2019).
Craig, C. M. et al. Efficacy and pharmacokinetics of subcutaneous exendin (9-39) in patients with post-bariatric hypoglycaemia. Diabetes Obes. Metab. 20, 352–361 (2018).
Schuster, N. M. & Rapoport, A. M. Calcitonin gene-related peptide-targeted therapies for migraine and cluster headache: a review. Clin. Neuropharmacol. 40, 169–174 (2017).
Panickar, K. S. Corticorelin, a synthetic human corticotropin-releasing factor analog, for the treatment of peritumoral brain edema. Curr. Opin. Mol. Ther. 12, 780–789 (2010).
Izquierdo, A. G., Crujeiras, A. B., Casanueva, F. F. & Carreira, M. C. Leptin, Obesity, and leptin resistance: where are we 25 years later? Nutrients. 11, 2704 (2019).
Sjogren, M. H. Thymalfasin: an immune system enhancer for the treatment of liver disease. J. Gastroenterol. Hepatol. 19, S69–72 (2004).
Nguyen, T. T. et al. Expression of active matrix metalloproteinase-9 as a likely contributor to the clinical failure of aclerastide in treatment of diabetic foot ulcers. Eur. J. Pharmacol. 834, 77–83 (2018).
Rodgers, K. E., Bolton, L. L., Verco, S. & diZerega, G. S. NorLeu(3)-Angiotensin (1-7) [DSC127] as a Therapy for the Healing of Diabetic Foot Ulcers. Adv. wound care 4, 339–345 (2015).
Cohen-Barak, O. et al. Assessment of the pharmacokinetics, pharmacodynamics, and safety of single doses of TV-1106, a long-acting growth hormone, in healthy Japanese and Caucasian subjects. Clin. Pharm. Drug Dev. 6, 331–342 (2017).
Temel, J. S. et al. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials. Lancet Oncol. 17, 519–531 (2016).
Rocha-Lima, C. M. et al. A multicenter phase II study of G17DT immunogen plus irinotecan in pretreated metastatic colorectal cancer progressing on irinotecan. Cancer Chemother. Pharm. 74, 479–486 (2014).
Hirose, T. et al. Open-label, randomized study comparing basal insulin peglispro and insulin glargine, in combination with oral antihyperglycemic medications, in insulin-naive Asian patients with type 2 diabetes. J. Diabetes Investig. 9, 100–107 (2018).
Lin, T. C. & Hsiao, M. Ghrelin and cancer progression. Biochim Biophys. Acta Rev. Cancer 1868, 51–57 (2017).
Laterre, P. F. et al. Effect of selepressin vs placebo on ventilator- and vasopressor-free days in patients with septic shock: the SEPSIS-ACT randomized clinical trial. JAMA, 322, 1476-1485 (2019).
Johannsson, G. et al. Safety and convenience of once-weekly somapacitan in adult GH deficiency: a 26-week randomized, controlled trial. Eur. J. Endocrinol. 178, 491–499 (2018).
Dong, J. Z. et al. Discovery and characterization of taspoglutide, a novel analogue of human glucagon-like peptide-1, engineered for sustained therapeutic activity in type 2 diabetes. Diabetes Obes. Metab. 13, 19–25 (2011).
Sosne, G. & Ousler, G. W. Thymosin beta 4 ophthalmic solution for dry eye: a randomized, placebo-controlled, Phase II clinical trial conducted using the controlled adverse environment (CAE) model. Clin. Ophthalmol. 9, 877–884 (2015).
Frias, J. P. et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 392, 2180–2193 (2018).
Anker, S. D. et al. Ularitide for the treatment of acute decompensated heart failure: from preclinical to clinical studies. Eur. Heart J. 36, 715–723 (2015).
Cales, P. et al. Early administration of vapreotide for variceal bleeding in patients with cirrhosis. N. Engl. J. Med 344, 23–28 (2001).
Lorget, F. et al. Evaluation of the therapeutic potential of a CNP analog in a Fgfr3 mouse model recapitulating achondroplasia. Am. J. Hum. Genet 91, 1108–1114 (2012).
Emons, G. et al. Efficacy and safety of AEZS-108 (INN: zoptarelin doxorubicin acetate) an LHRH agonist linked to doxorubicin in women with platinum refractory or resistant ovarian cancer expressing LHRH receptors: a multicenter phase II trial of the ago-study group (AGO GYN 5). Gynecol. Oncol. 133, 427–432 (2014).
Savage, P. D. et al. Phase II trial of angiotensin-(1-7) for the treatment of patients with metastatic sarcoma. Sarcoma 2016, 4592768 (2016).
Escudero-Castellanos, A. et al. Synthesis and preclinical evaluation of the 177Lu-DOTA-PSMA(inhibitor)-Lys3-bombesin heterodimer designed as a radiotheranostic probe for prostate cancer. Nucl. Med Commun. 40, 278–286 (2019).
Lucas, X. Clinical use of deslorelin (GnRH agonist) in companion animals: a review. Reprod. Domest. Anim. 49, 64–71 (2014).
Sekar, R., Singh, K., Arokiaraj, A. W. & Chow, B. K. Pharmacological actions of glucagon-like peptide-1, gastric inhibitory polypeptide, and glucagon. Int Rev. Cell Mol. Biol. 326, 279–341 (2016).
Hewitt, D. J. et al. Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. Cephalalgia 31, 712–722 (2011).
Recober, A. & Russo, A. F. Olcegepant, a non-peptide CGRP1 antagonist for migraine treatment. IDrugs 10, 566–574 (2007).
Rabiee, A. et al. Pancreatic polypeptide administration enhances insulin sensitivity and reduces the insulin requirement of patients on insulin pump therapy. J. Diabetes Sci. Technol. 5, 1521–1528 (2011).
Ukkola, O. H. et al. High serum fasting peptide YY (3-36) is associated with obesity-associated insulin resistance and type 2 diabetes. Regul. Pept. 170, 38–42 (2011).
Shailubhai, K. et al. Phase II clinical evaluation of SP-304, a guanylate cyclase-c agonist, for treatment of chronic constipation: 1322. 105, S487–S488, (2010).
Koch, L. Pharmacotherapy: somatoprim versus octreotide in acromegaly. Nat. Rev. Endocrinol. 8, 66 (2011).
de Schepper, J. et al. Long-acting pegylated human GH in children with GH deficiency: a single-dose, dose-escalation trial investigating safety, tolerability, pharmacokinetics and pharmacodynamics. Eur. J. Endocrinol. 165, 401–409 (2011).
Nielsen, V. E. et al. Stimulation with 0.3-mg recombinant human thyrotropin prior to iodine 131 therapy to improve the size reduction of benign nontoxic nodular goiter: a prospective randomized double-blind trial. Arch. Intern Med 166, 1476–1482 (2006).
Tejeda, M. et al. Evaluation of the antitumor efficacy of the somatostatin structural derivative TT-232 on different tumor models. Anticancer Res 26, 3477–3483 (2006).
Ding, L. et al. BPI-3016, a novel long-acting hGLP-1 analogue for the treatment of Type 2 diabetes mellitus. Pharm. Res 122, 130–139 (2017).
Walter, M. et al. No effect of the altered peptide ligand NBI-6024 on beta-cell residual function and insulin needs in new-onset type 1 diabetes. Diabetes Care 32, 2036–2040 (2009).
Funding
This work was supported by Zhejiang Provincial Natural Science Foundation of China under Grant No. LD22H310004, the National Natural Science Foundation of China (No. 81770176), the “Pioneer” and “Leading Goose” R&D Program of Zhejiang (No. 2022C03005), the special support plan for Zhejiang Province high-level talents (No. 2019R52011), the Zhejiang Provincial Natural Science Foundation of China under Grant No. LQ20H300005 and Program of Xinmiao Talents in Zhejiang Province (2021R406062).
Author information
Authors and Affiliations
Contributions
C.F. and R.W.: conception and co-design of this work; L.W., N.W., W.Z: preparation and writing of manuscript; X.C., Z.Y., G.S., X.W.: Data collection, interpretation and preparation of figures and tables. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
C.F. is the editorial board member of Signal Transduction and Targeted Therapy, but she has not been involved in the process of the manuscript handling. The remaining authors declare no competing interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., Wang, N., Zhang, W. et al. Therapeutic peptides: current applications and future directions. Sig Transduct Target Ther 7, 48 (2022). https://doi.org/10.1038/s41392-022-00904-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-00904-4
This article is cited by
-
Computational design of anti-cancer peptides tailored to target specific tumor markers
BMC Chemistry (2024)
-
Death-associated protein kinase 1 as a therapeutic target for Alzheimer's disease
Translational Neurodegeneration (2024)
-
Applications of peptides in nanosystems for diagnosing and managing bacterial sepsis
Journal of Biomedical Science (2024)
-
Intracellular delivery of Parkin-RING0-based fragments corrects Parkin-induced mitochondrial dysfunction through interaction with SLP-2
Journal of Translational Medicine (2024)
-
Non-symmetric stapling of native peptides
Nature Reviews Chemistry (2024)
