
Abstract
Interaction of the T cell receptor (TCR) with an MHC-antigenic peptide complex results in changes at the molecular and cellular levels in T cells. The outside environmental cues are translated into various signal transduction pathways within the cell, which mediate the activation of various genes with the help of specific transcription factors. These signaling networks propagate with the help of various effector enzymes, such as kinases, phosphatases, and phospholipases. Integration of these disparate signal transduction pathways is done with the help of adaptor proteins that are non-enzymatic in function and that serve as a scaffold for various protein–protein interactions. This process aids in connecting the proximal to distal signaling pathways, thereby contributing to the full activation of T cells. This review provides a comprehensive snapshot of the various molecules involved in regulating T cell receptor signaling, covering both enzymes and adaptors, and will discuss their role in human disease.
Similar content being viewed by others


Introduction
T cells are key mediators in mounting an effective adaptive cell-mediated immune response.1,2 T cells continuously screen lymphoid and peripheral tissues for antigens such as peptides or lipids displayed by major histocompatibility complex (pMHC) molecules of other cells. Normal T cell development in the thymus undergoes a major developmental checkpoint in which T cell receptor (TCR) signaling is involved. Thymocytes bearing TCR with a high affinity for self-peptide MHC complexes undergo apoptosis (negative selection), whereas those bearing low-affinity TCR survive and differentiate into mature T cells (positive selection).3 This ensures that only those T cells that are self-tolerant survive while eliminating the self-reactive T cells.4 These naïve single positive mature T cells then leave the thymus and enter the peripheral lymphoid organs, such as the spleen and lymph nodes, where they get exposed to foreign peptides presented by the MHC molecules of antigen-presenting cells (APCs), such as the macrophages, dendritic cells, and B cells, during pathogenic infection.5 Upon engagement of TCR with the antigenic peptide, T cells get activated, undergoing clonal expansion and differentiation to perform their effector functions, due to a complex series of molecular changes at the plasma membrane, cytoplasm, and nucleus.6,7 T cell signaling is thus important for efficient T cell development, activation, and immune tolerance. TCR signaling dysregulation can thus lead to anergy or autoimmunity.8
In general, the transmission of external cues to the interior of the cell occurs through binding of a ligand to the extracellular domain of the receptor, leading to receptor aggregation or conformational changes. Once this is accomplished, protein tyrosine kinases (PTKs) phosphorylate various tyrosine residues present in the cytoplasmic tail of the receptor, which serve as a docking site for various signaling molecules containing specific phosphotyrosine recognition domains such as the SRC homology 2 (SH2) and phosphotyrosine-binding (PTB) domains.6 This initiates proximal biochemical signals mediated by the key effector enzymes, such as kinases, phosphatases, and phospholipases, that culminate in distal signaling by activating numerous transcription factors required for translating these events into gene activation.9 However, both the proximal and distal signaling need to be integrated, and this is done by various adaptor proteins whose main function is to form multiprotein complexes.10
Adaptors are proteins that usually lack intrinsic enzymatic activity and instead possess multiple binding domains for phosphotyrosine, proline-rich region and lipid interactions, and sequence motifs that in turn are involved in binding to such domains.11,12 The phosphorylated tyrosine residues on various adaptor proteins serve as binding sites for many critical effector enzymes and other adaptor proteins.6 Thus, they behave as scaffolds, facilitating protein–protein interactions that aid in forming multiprotein complexes, thereby integrating signaling cascades necessary for efficient T cell biology. Apart from this, adaptors also interact with other adaptors present at the plasma membrane microdomains and even play important roles in the regulation of the cytoskeleton.6,10,13 Hematopoietic-specific adaptor proteins have led to a better understanding of T cell signaling.13 Those adaptor proteins can regulate signal transduction both positively and negatively.10,13 In this review, we discuss the role of TCR signaling in human health and disease.
Components and structure of TCR complex
The core TCR complex consists of two TCR chains and six cluster of differentiation 3 (CD3) chains. Several other components include coreceptors, kinases, and ligands.14,15
TCR-CD3 chains
The human genome expresses four TCR genes known as TCRα, TCRβ, TCRγ, and TCRδ, which forms two distinct heterodimers: TCRα/TCRβ or TCRγ/TCRδ.16,17,18 The majority of mature T cells expresses TCRα and TCRβ isoforms, generally referred to as T cells (or αβ T cells), while a small portion (0.5–5%) of T lymphocytes (γδ T cells) expresses TCRγ and TCRδ isoforms.19 In this review, we will focus on αβ T cells, and henceforth the nomenclature T cells will refer to αβ T cells.
Both heterodimers form multiprotein complexes with CD3 δ, γ, ε, and ζ chains. TCR chains consist of an extracellular region, transmembrane region, and a shorter cytoplasmic tail. The extracellular region contains a variable immunoglobulin-like (V) domain, a constant immunoglobulin-like (C) domain, and connecting peptide.20 The RAG1 and RAG2 recombinases facilitate the assembly of the V domain from gene segments that serve as the antigen recognition site. The C domain is used for the interactions with CD3 chains.
There are considerable structural differences between αβ and γδ chains in terms of C domain and connecting peptide, which are also reflected in the assembly of the TCR complexes, surface shape, and charge distribution.21,22,23,24 However, in both complexes, three dimers of CD3 proteins, δε and γε heterodimers and ζζ homodimers, are present.23,25 These CD3 proteins associate with TCR via non-covalent hydrophobic interactions and are required for a complete TCR localization on the cell surface (Fig. 1a).

a TCRα/TCRβ and TCRγ/TCRδ heterodimers form complexes with the CD3 molecules. Heterodimers of CD3ε/CD3δ and CD3γ/CD3ε, and a homodimer of CD3ζ/CD3ζ form complexes with TCR dimers. TCR heterodimers contain intramolecular and intermolecular disulfide bonds. CD3 chains contain 10 ITAMs distributed in different CD3 molecules. The variable region (V) of TCR heterodimers recognize the antigen peptide-loaded on MHC (pMHC). In the absence of pMHC, the intracellular part of the CD3 molecules forms a close conformation in which ITAMs are inaccessible to the kinases for phosphorylation. b Coreceptor CD4 acts as a single molecule while CD8α and CD8β can form homodimers or heterodimers. c MCH-I consists of an α-chain containing three immunoglobulin domains (α1, α2, α3) and β2-microglobulin (β2m). MCH-2 is the heterodimer of an α chain and a β-chain containing two immunoglobulin domains (α1, α2, and β1, β2) in each chain. d LCK-loaded CD4 molecules bind to the MHC-II bound TCR (TCRα/TCRβ) complex. This allows LCK to phosphorylate two distinct sites on ITAMs. Then ZAP-70 interacts with the phosphotyrosine sites and mediates more tyrosine phosphorylation. CD4 and MHC-II interaction is mediated through the membrane-proximal α2 and β2 domains of MHC-II and the membrane-distal D1 domain of CD4.
TCR co-receptors
Initial studies demonstrated that T cells expressing common TCRα/TCRβ heterodimer with distinct functions—for example, cytotoxic T cells that directly destroy infected cells and a subset of helper T cells that help B cells—may easily be distinguishable by the expression of mutually exclusive cell surface molecules CD8 and CD4.26,27,28,29,30 Later studies indicated that these two receptors may play important roles in the association of MHC molecules and thus are referred to as co-receptors.31,32,33 Both CD4 and CD8 molecules play important roles during the development of T cells by helping the TCR complex select a different class of MHC molecules.34 Like TCR molecules, both CD4 and CD8 molecules contain an extracellular domain, transmembrane domain, and a short intracellular tail.35 Although both CD8 and CD4 act as coreceptors with similar functionality, they share a minimal structural similarity. The extracellular domain of CD4 contains two V domains (D1 and D3) and two C domains (Fig. 1b). CD4 acts as a monomer on the T cell surface where it uses the D1 domain for MHC recognition and the cytoplasmic tail for interaction with non-receptor tyrosine kinase LCK.36 On the other hand, the CD8 extracellular domain contains only a single V domain. However, two CD8 isoforms, CD8α and CD8β, are expressed and can form homo- or heterodimers (Fig. 1b). Most CD8-positive T cells express as a heterodimer; some CD8-positive T cells—for example, intraepithelial lymphocytes and memory precursors—express as an αα homodimer.37,38,39
TCR ligands
Ligands for T cells are divided into two classes: MHC class I (MHCI) and MHC class II (MHCII) (Fig. 1c). Human MHCIs are complexes of human leukocyte antigens (HLAs: HLA-A, HLA-B, and HLA-C) and β2-microglobulin while MHCIIs are heterodimers of several HLAs (HLA-DP, HLA-DQ, and HLA-DR).40 Antigen peptide-bound MHCI (pMHC-I) molecules can be presented on any nucleated cells recognized by CD8+ T cells. On the other hand, CD4+ T cells recognize antigen peptide-bound MHCII (pMHC-II) molecules that are presented on the APCs, such as B cells, macrophages, and dendritic cells.40 Besides peptide presentation by MHC molecules, lipid antigens present similarly structurally to MHCII molecules, such as CD1 family proteins.41,42
Lymphocyte-specific PTK (LCK)
LCK is a member of the SRC family kinase (SFK). The SFKs are a family of ten structurally similar non-receptor PTKs which have been implicated in various cellular functions.43,44,45 All SFKs contain highly conserved regulatory domains (SH3 and SH2), a protein tyrosine kinase domain (SH1), a C-terminal tail, and a poorly conserved N-terminal region (SH4 domain). The SH4 domain contains a myristoylation site by which SFKs anchor to the membrane.46 The SH3 domain, in general, recognizes proline-rich motifs (PxxP), and the SH2 domain interacts with phosphotyrosine residues.47,48 However, the function of the SH4 domain cannot be generalized for all SFKs except that it holds the myristoylation site. The SH4 domain of some SFKs also contains a palmitoylation in addition to the myristoylation site. Likewise, other SFKs’ LCK activity is tightly controlled by phosphorylation/dephosphorylation cycles (Fig. 1d). C-terminal SRC kinase (CSK) phosphorylates LCK on Y505 residue that interacts with the LCK-SH2 domain, keeping it inactive.49 The leukocyte common antigen (CD45), also known as protein tyrosine phosphatase receptor type C (PTPRC), removes the regulatory phosphotyrosine residue that releases the kinase domain from autoinhibitory states (priming) and also makes the SH2 domain available for interaction with other proteins.50,51,52 This priming step results in autophosphorylation of Y,394 leading to full activation of LCK.52 Besides a role in LCK activation, several studies have pointed out that CD45 can negatively regulate LCK function by removing tyrosine phosphorylation from LCK-Y394.53,54 T cells maintaining a certain level of CD45 expression uphold appropriate LCK activation whereas low levels of CD45 expression correlate with the lower TCR activation, and higher CD45 expression reduces LCK activity by removing tyrosine phosphorylation from LCK-Y394.53 Thus, T cells can regulate TCR activity by modulating CD45 expression.55 Several cytosolic phosphatases, including PTPN6(SHP-1), and PTPN22, also control LCK activity by removing phosphate group from Y394.56,57 Therefore, LCK activity is probably dynamically regulated by cellular abundance and activity of CSK, CD45, PTPN6, and PTPN22.
TCR activation and proximal signaling
Early T cell signaling takes place within a few seconds, and the first step is TCR activation.58 An early event in the proximal signaling of TCR is the involvement and activation of a set of PTKs.6 Several PTKs, such as LCK, FYN, and ZAP-70, are important signaling components for T cell development and activation of TCR signaling through tyrosine phosphorylation on CD3.8,59,60,61 The cytosolic tail of the CD3 proteins contains a unique motif, the immunoreceptor tyrosine-based activation motifs (ITAMs), that consists of two tyrosine residues flanked by leucine/isoleucine and spaced by bulky aromatic amino acid, thus having a consensus sequence of D/Ex(0-2)YxxI/Lx(6-8)YxxI/L.62,63,64 Each of the CD3δ, γ, and ε chains contain one ITAM each, whereas each CD3ζ chains contain three ITAMs, thus each TCR-CD3 complex contains ten ITAMs.6,65,66 For TCR activation, tyrosine residues in ITAMs need to be phosphorylated, which is initiated by LCK and, to some degree, by FYN.67,68,69 Although FYN can induce phosphorylation of ITAMs, its role is dispensable for T cell development.70,71 Thus, tyrosine phosphorylation on CD3 ITAMs by LCK during T cell development probably cannot be replaced by other tyrosine kinases.72
LCK is known to be associated with several growth factor receptors, including KIT, FLT3, and AXL, in a phosphorylation-dependent manner.73,74,75 The interaction between LCK and growth factor receptors is mediated via the SH2 domain of LCK that interacts with the phosphotyrosine residue of the activated receptor. Since the TCR-CD3 complex lacks intrinsic kinase activity, the pMHC-loaded TCR-CD3 complex remains unphosphorylated, and therefore LCK cannot directly interact with the inactive complex through an SH2 domain. However, to facilitate the phosphorylation of ITAMs, LCK needs to be localized to the cell membrane. LCK can anchor to the cell membrane via its myristoylation (serine 2) and palmitoylation (cysteine 3/5) sites present in the SH4 domain or through the interaction with the cytoplasmic tail of coreceptors CD4 and CD8.69,76,77,78,79,80,81,82,83,84,85,86,87,88 The C-terminal tail of CD4 and CD8α contains a conserved CxCP motif, which is absent in CD8β, required for this interaction.81,89,90 This motif interacts with the CxxC motif present in the LCK SH4 domain, mediating the interaction in a zinc-ion-dependent manner.89,90,91,92,93,94 Therefore, only the homodimer CD8α/CD8α and heterodimeric CD8α/CD8β can load LCK to the TCR complex, and although a CD8β/CD8β homodimer can be formed, it cannot recruit LCK to the TCR complex and thereby does not play a role in TCR signaling.
T cells usually express CD4 or CD8 coreceptors, and therefore pMHC-bound TCR browses for LCK-loaded coreceptors where the non-polymorphic part of pMHCs interacts with the distal membrane part of the coreceptors.76,77,78,79 The membrane-distal D1 domain of CD4 associates with the membrane-proximal α2 and β2 domains of MHC-II (Fig. 2), but it does not directly interact with the TCR complex.95,96,97,98 Similar to the CD4–MHC-II interaction, binding with the CD8α/CD8α homodimer or CD8α/CD8β heterodimer to the MHC-I complex is mediated through the membrane-distal D1 domain of CD8 and membrane-proximal β2M and α3 of the MHCI complex.99 Additionally, the membrane distal α2 domain of the MHC-I complex also participates in interaction with CD8.99 Such interaction keeps TCR and coreceptors orthogonal, which is likely important for the stability of the complex, LCK loading, and TCR activation.100,101,102,103 Although both the CD8α/CD8α homodimer or CD8α/CD8β heterodimer binds with MHC-1 complex with a similar affinity, the CD8β/CD8β homodimer does not bind with MHC-1.104,105,106

In resting T cells, CD3ζ and CD3ε remain membrane-embedded. Perhaps membrane-bound CD3ζ might be released to the cytosol, where free LCK induces tyrosine phosphorylation on at least two sites in ITAMs. This basal tyrosine phosphorylation creates docking sites for ZAP-70 interaction. After antigen engagement, the TCR complex recruits coreceptor-bound LCK that phosphorylates ZAP-70 and interacts with it through the SH2 domain facilitating tyrosine phosphorylation on other residues on ITAMs.
Interaction between coreceptors and LCK has two important functions: it brings LCK in close proximity to the TCR complex and it stabilizes coreceptors by preventing clathrin-mediated endocytosis.94 However, once LCK phosphorylates CD3 proteins, it leaves coreceptors that result in the internalization of coreceptors.107,108,109,110,111 Nevertheless, the interaction between LCK-loaded coreceptor and pMHC acts as a rate-limiting step to initiate TCR signaling.112
Initial studies suggest that coreceptor-bound LCK mediates tyrosine phosphorylation on all four CD3 chains.113,114,115,116 However, those studies have not suggested any site-specificity or whether LCK loaded to the CD4 or CD8 makes any difference in CD3 tyrosine phosphorylation dynamics. To address the sequence of CD3 tyrosine phosphorylation, several attempts have been taken with a specific focus on CD3ζ, which has six tyrosine phosphorylation sites.117,118 In resting T cells, at least two tyrosine residues in second and third ITAMs of CD3ζ remain to be phosphorylated.117 When activated, N-terminal tyrosine residue in the third ITAM displays dependency on the N-terminal tyrosine residue of the first ITAM, and C-terminal tyrosine residue in the second ITAM needs the C-terminal tyrosine residue to be phosphorylated on the first ITAM.117 Furthermore, LCK phosphorylates all six tyrosine residues of recombinant CD3ζ ITAMs in a specific order, starting from the N-terminal tyrosine residue of the first ITAM.118 These studies suggest that tyrosine phosphorylation in ITAMs is a controlled molecular event that has further been shown to be regulated by TCR ligand.117
LCK-induced phosphorylation of both tyrosine residues in ITAM has been shown to be required for interaction with ZAP-70 where ZAP-70 SH2 domains mediate the interactions.67,119 This interaction stabilizes tyrosine phosphorylation of CD3ζ and induces ZAP-70 tyrosine phosphorylation, which was independent of ZAP-70 kinase activity, suggesting that ZAP-70 does not play a role in CD3ζ tyrosine phosphorylation but that interaction with tyrosine residues probably limits phosphatase access, protecting tyrosine phosphorylation. Nevertheless, the interaction is important for ZAP-70 activation and downstream signaling.
The mechanism through which CD3 ITAMs remain unphosphorylated in an inactive TCR complex remains to be debated. Using CD3ε as a model, it has been depicted that the positively charged cytoplasmic domain remains embedded in the negatively charged inner membrane, which sequestrates tyrosine residues.120,121 Similarly, the cytoplasmic part of CD3ζ remains lipid-bound, preventing tyrosine phosphorylation.122 However, the role of the inner membrane in the prevention of tyrosine phosphorylation was questioned by another study.123 This study demonstrated that removal of positively charged residues did not enhance tyrosine phosphorylation but that pervanadate enhanced CD3 tyrosine phosphorylation, concluding that phosphatase might be involved in the prevention of tyrosine phosphorylation.123 Pervanadate prevents tyrosine phosphatases by oxidizing the catalytic cysteine of phosphatase.124 Those claims were later contested by the fact that removal of positively charged residues decreases LCK-mediated CD3 tyrosine phosphorylation, and pervanadate can prevent membrane association of CD3 with lipid membranes.125
Combining early studies, a model has been proposed suggesting that interaction between the cytoplasmic chain and lipid membrane prevents ITAM phosphorylation, and pMHC association to the TCR induces structural changes that release CD3 from the inner membrane.126,127 This model has further been supported by the fact that positively charged ions, such as Ca2+, can release sequestered CD3 chains, facilitating tyrosine phosphorylation,128 and that defects in Mg2+ transport are linked to the defective T cell activation due to impaired Ca2+ influx in T cells.129 Antigen engagement increases Ca2+ intake86 as well as TCR proximal Ca2+ concentration.130
Although this model provides a simplified overview of TCR activation, the model fails to explain how ITAMs in CD3δ and CD3γ remain protected from tyrosine phosphorylation, as they lack membrane-binding residues,126,131 and how a basal level of CD3ζ tyrosine phosphorylation is maintained if they remain to be sequestered in the plasma membrane.60,117,120,132,133,134 Thus, complete sequestering to the plasma membrane might be an unlikely event. Rather, dynamic switching between membrane binding and cytosolic release might happen, and other forces may also be involved.135
Constitutive association of ZAP-70 to the CD3ζ might also go against the model of complete sequestering, as ZAP-70 association is mediated through the SH2 domain and phosphotyrosine residues.132 Furthermore, constitutively active LCK localized at the cell membrane was detected in up to 40% of resting T cells.53 Constitutively LCK activation is probably required for maintaining basal tyrosine phosphorylation of CD3ζ, as T cells with reduced constitutively LCK activation displayed undetectable levels of CD3ζ tyrosine phosphorylation.53 But why is TCR signaling not triggered in resting cells if free LCKs are available and apparently are more active than coreceptor-bound LCKs136? Perhaps membrane association CD3ζ limits the accessibility to the tyrosine sites in ITAMs, and, if they are accessible in resting cells, CD3ζ orientation allows phosphatases to remove tyrosine phosphorylation.123 Therefore, the association of pMHC to the TCR complex that mediates structural changes of the cytosolic part of CD3 is important for TCR activation.
A two-stage kinetics of TCR–pMHC–CD8 interaction has been suggested, where the first TCR binds with the pMHC (within <0.1 s), and then the tri-molecular complex is formed.137 The tri-molecular complex was affected by proximal signaling, as pharmacological inhibition of SFKs or CD45 abolished initiation of high-affinity binding.137 LCK association to the coreceptor stabilizes coreceptors by preventing endocytosis.94 However, how the kinase activity of LCK stabilizes the tri-molecular complex remains to be determined. As an early event, CD8 interacts with CD3ζ, which is independent of pMHC but LCK-dependent, and in the later events, tri-molecular complex is required to maintain the CD8–CD3ζ interaction.138 As LCK can be either free or CD8 bound,138 CD3ζ might get a chance to meet either free LCK or coreceptor bound LCK before ligand association, probably further explaining CD3ζ tyrosine phosphorylation in resting cells.
A single-molecule analysis suggests that the movement of LCK during TCR activation is not directed but is rather a Brownian movement.139 Therefore, it would be challenging for a TCR complex to find LCK instantly unless LCK is already recruited to the complex in resting cells. Free LCKs which are also membrane-bound display higher mobility than coreceptor-associated LCKs, probably due to the difference in molecular size.136 This might also explain why free LCKs are recruited in the early TCR complexes.138 Besides the ability to move faster, free LCKs display higher catalytic activity as measured by tyrosine phosphorylation (LCK-Y394).136 However, in any case, coreceptor-bound LCK is required for TCR activation, and the number of coreceptors-bound LCK increases during the maturation process.140 Finally, it has been demonstrated that LCKs directly interact with CD3ε in which the interaction is ionic and is mediated through the juxtamembrane basic residue-rich sequence (BRS) of CD3ε and the unique domain (UD) of LCK.141
The early steps of the TCR activation process seem to be highly debated. Current models for TCR activation either considered CD3 membrane sequestering and ignored the basal level of CD3ζ or the other way around.126,127,135 Perhaps both of the conditions simultaneously occur, and therefore probably CD3ζ holds its states as membrane-embedded and outside the membrane, allowing constitutively active LCK to phosphorylate tyrosine residues on CD3ζ ITAMs (Fig. 2). Then ZAP-70 binds to the phosphotyrosine residues in CD3 ITAMs. Once bound, ZAP-70 is phosphorylated by LCK, which leads to its activation.142 This results in the formation of a multi-nucleated signaling complex as further phosphorylation of ZAP-70 allows binding of additional proteins and adaptors, thereby itself behaving as a scaffold.142,143 Thus, the activated state of TCR is characterized by phosphorylation of ITAMs, followed by phosphorylation and activation of ZAP-70.
Distal TCR signaling
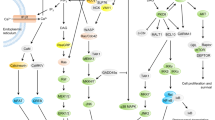
Engagement of TCR with the MHC-antigenic peptide complex of APCs triggers the formation of multi-molecular signalosomes at TCR. This leads to the generation of proximal signaling, followed by the activation of multiple distal signaling cascades, such as Ca2+–calcineurin–NFAT, PKCθ–IKK–NFκβ, RASGRP1–RAS–ERK1/2, and TSC1/2–mTOR, with the help of secondary messengers, enzymes, and various adaptor proteins (Fig. 3). These signaling cascades finally bring out the diverse phenotypic effects, as they control many aspects of T cell biology.1
Ca2+–calcineurin–NFAT pathway
Phospholipase Cγ1 (PLCγ1) is the main molecule connecting the TCR proximal to distal signaling cascades.144 The membrane-bound phosphatidylinositol 4,5-bisphosphate (PIP2) gets hydrolyzed by activated PLCγ1 into diacylglycerol (DAG) and inositol-3-phosphate (IP3).145 Both of these essential secondary messengers initiate a variety of distal signaling cascades important for T cell activation. Membrane-bound DAG can activate PKCθ, RASGRP1, and PDK1-mediated pathways. On the other hand, IP3 triggers the activation of a Ca2+-dependent calcineurin NFAT pathway.145,146,147
IP3 generated from PIP2 binds to the Ca2+-permeable ion channel receptors (IP3R) on the endoplasmic reticulum (ER), thereby releasing ER Ca2+ stores in the cytoplasm.148 It has been found that ERs can sense the intracellular Ca2+ levels through the constitutive expression of a transmembrane protein called stromal interaction molecule (STIM). Depletion of intracellular Ca2+ levels thus triggers an influx of extracellular Ca2+ into T cells from Orai1 type plasma membrane calcium-release activated calcium (CRAC) channel.149,150,151,152 Increased intracellular Ca2+ activates a protein phosphatase, calcineurin, that dephosphorylates the nuclear factor of activated T cells (NFAT), thereby causing its nuclear translocation. Nuclear NFAT forms a complex with AP-1 transcriptional factors (JUN/FOS) derived from the DAG–RAS–MAPK–ERK1/2 pathway. This transcriptional complex is responsible for inducing the expression of various genes, like IL-2 and other effector molecules, that are responsible for T cell activation. In contrast, in the absence of AP-1, NFAT alone activates various genes, like several ubiquitin ligases and diacylglycerol kinase α (DGKα), that are responsible for T cell anergy, a state of T cell unresponsiveness, one of the processes to induce immune tolerance.153,154,155,156 Thus, two opposite T cell functions—activation and anergy—are controlled by NFAT proteins.153
In addition to calcineurin, Ca2+ also activates a Ca2+/calmodulin-dependent kinase (CaMK) that mediates T cell activation through activation of transcription factors, such as cyclic-AMP-responsive-element-binding protein and myocyte enhancer factor 2.157 A missense mutation in Orai1 can lead to impaired Ca2+ signaling, which affects nuclear translocation of NFAT, and thereby NFAT-induced cytokines production causes severe combined immunodeficiency (SCID) in humans.158,159 Thus, the universal secondary messenger Ca2+ regulates several important functions, including proliferation, differentiation, and cytokine production in T cells.160
PKCθ–IKK–NF-κβ pathway
Protein kinase C (PKC) is a family of ten protein serine/threonine kinases that plays numerous roles in physiological and pathological conditions.161,162,163,164 PKC family proteins are divided into three subfamilies: classical (PKCα, PKCβ1, PKCβ2, and PKCγ), novel (PKCδ, PKCε, PKCη, and PKCθ), and atypical (PKCζ and PKCι). DAG and Ca2+ regulate activation of classical PKC isoforms, and novel PKC isoforms are regulated by DAG while atypical PKC isoforms are independent of DAG and Ca2+ for activation.161 Several PKC isoforms have been implicated in T cell functions.165 For example, the novel isoform PKCθ binds to DAG through the PKC conserved region 1 (C1) domain, which is required for its recruitment to the lipid raft after TCR engagement. PKCθ plays major non-redundant roles in T cell activation, even though T cells express several other PKCs.166,167
Nuclear factor κβ (NF-κβ) is an evolutionarily conserved transcription factor that plays important roles in regulating genes involved in inflammatory and immune responses, cell growth, survival, and differentiation.168 TCR-mediated T cell activation involves both the canonical (classical) and the non-canonical (alternative) NF-κβ pathway. An essential factor required for complete T cell activation via the non-canonical NF-κβ pathway is MAP3K14 (also known as NF-κβ-inducing kinase; NIK).169 However, more studies are required regarding this. On the other hand, PKCθ–IKKβ–NF-κβ forms the canonical branch of the NF-κβ pathway and is widely studied.170
Once PKCθ is activated following TCR stimulation, it triggers the formation of a tri-molecular complex of adaptor proteins in the cytoplasm called the CBM complex, which is composed of the caspase recruitment domain-containing membrane-associated guanylate kinase protein-1 (CARMA1), B cell lymphoma/leukemia 10 (BCL10), and mucosa-associated lymphoid tissue translocation protein-1 (MALT1).171 This is initiated by phosphorylation of CARMA1 by activated PKCθ,172 which is required for its oligomerization and association with BCL10.173 MALT1 then binds to BCL10, and this association recruits an E3 ubiquitin ligase, called the tumor necrosis factor receptor-associated factor 6 (TRAF6), that polyubiquitinates and degrades IKKγ, or the NF-κβ essential modifier (NEMO), a regulatory protein of the IKK complex.174,175 Consequently, the catalytic subunits of Iκβ kinases (IKK), α and β, are no longer inhibited, and they phosphorylate Iκβ, thereby inducing its ubiquitination and degradation. NF-κβ is thus released from its inhibitory Iκβ complex in the cytoplasm, and it translocates into the nucleus to regulate gene expression.176,177,178 This canonical PKCθ–IKKβ–NF-κβ pathway is extremely important for T cell survival, homeostasis, activation, and effector function.179 Deregulation of this pathway can cause defective T cell survival and activation, autoimmunity, SCID, and lymphoma.180,181,182
RASGRP1–RAS–ERK1/2–AP1 pathway
DAG from PIP2 induces the activation of another key molecule, a RAS guanyl nucleotide-releasing protein (RASGRP1), and recruits it to the plasma membrane.183,184 RASGRP1 and Son of Sevenless (Sos) are two known guanine nucleotide exchange factors (GEFs) responsible for RAS activation in T cells.1 RAS, a small G protein, binds to GTP in the activated state and initiates the RAS-MAPK cascade by activating the serine/threonine kinase Raf1.183,185 Raf1, a mitogen-activated protein kinase (MAPK) kinase kinase (MAPKKK), then phosphorylates and activates MAPK kinases (MAPKKs), such as MEK1/2, that further phosphorylate and activate MAPK extracellular signal-regulated kinase-1 & 2 (ERK1/2).6,185,186 T cell development, differentiation, and TCR-induced signal strength are all controlled by ERK1/2 signaling.187,188,189 Furthermore, ERKs trigger the phosphorylation and activation of their downstream target Elk, a transcription factor responsible for inducing the expression of c-Fos transcription factor. The VAV1–Rac pathway induces the expression of Jun.190 Thus, the formation and activation of a dimeric complex, activator protein-1 (AP-1) composed of Jun/Fos, that plays a critical role in immune response, followed by IL-2 transcription, is sustained by the DAG–RAS pathway.1,6 Further, the signal transducer and activator of transcription (STAT3) and LCK get phosphorylated by ERKs.191,192
RASGRP1 is extremely important for the development of conventional αβ T cells but not the meager population of γδ T cells.193,194,195 However, it is important for the activation of both types of T cell population as well as for the expression of IL-17.195 RASGRP1 deficiency can cause defects in the activation of various signaling pathways, such as RAS–MAPK–ERK1/2, mTOR, and PI3K/AKT.193,196 Moreover, abnormal expression of both RASGRP1 and RAS was described in T cells of systemic lupus erythematosus (SLE) patients, thereby implicating the involvement of this pathway in the generation of SLE.197
p38 and JNK pathways
The other MAPKs such as p38 and JNK family proteins play important roles in the proliferation, differentiation, and function of different subsets of T cells.198,199,200,201,202 p38 is a family of four highly structurally homolog proteins including p38α (MAPK14), p38β (MAPK11), p38γ (MAPK12), and p38δ (MAPK13).203 p38α is widely known as p38 and is one of the most studied isoforms. On the other hand, the JNK family is composed of three members which include JNK1 (MAPK8), JNK2 (MAPK9), and JNK3 (MAPK10).204 Three dual-specificity MAP2Ks including MKK3 (MAP2K3, MEK3), MKK4 (MAP2K4, MEK4), and MKK6 (MAP2K6, MEK6) are involved in p38 activation through phosphorylation on the conserved T180-X-Y182 motif in the loop of the substrate recognition site.203 Among those three MAP2Ks, MKK3 and MKK6 display higher specificity to p38 while MKK4 also activates JNKs.203,204,205,206 The conserved T180-X-Y182 motif is required to be phosphorylated on both threonine and tyrosine residues for p38 activation.207 In mammalian cells, the classical p38 pathway is regulated by ten different MAP3Ks that allow for the integration of various signaling nodes. However, in T cells, p38 activity is mediated by a non-classical pathway which is downstream of proximal TCR signaling and probably independent of MAPK cascades.203 In such a case, activation of TCR proximal signaling results in the phosphorylation of p38 at Y323 residue by ZAP-70, which triggers autophosphorylation on regulatory residues (T180-X-Y182) followed by p38 activation.208 Additionally, activated p38 mediated phosphorylation of ZAP-70 on T293 residue may act as a negative feedback loop possibly by limiting excessive TCR signaling.209 JNK activation is likely mediated through the activation of PKCθ and CBM complex upon the activation of TCR proximal signaling (reviewed in ref. 210). BCL10 oligomers in the CBM complex can recruit TAK1 (MAP3K7), MKK7 (MAP2K7), and JNK2, leading to the activation of JNK.210,211
TSC1/2–mTOR pathway
Upon TCR engagement, both DAG–RASGRP1–RAS–ERK1/2 and PI3K–AKT pathways induce the activation of mTORC1 and mTORC2 (refs. 196,212) that differentially regulate the generation of CD4+ helper effector T cell types (Th).213 mTORC1 down-regulates SOCS5 to promote STAT3 activity, thereby promoting Th17 differentiation.214 Resultingly, mice having Rheb or raptor-deficient T cells show defects in Th17 differentiation.214,215 On the other hand, mTORC2 phosphorylates AKT at S473 and PKCθ at S660/676 to induce Th1 and Th2 differentiation respectively.215 Thus, mice having rictor-deficient T cells show defects in differentiation of IFNγ-producing Th1 and IL-4-producing Th2 effector cells.214,215 The activity of mTOR subsequently needs to be tightly controlled, as it regulates T cell activation, differentiation, and function.216
As discussed above, the CBM complex plays a crucial role in TCR signaling by recruiting key signaling mediators. Hamilton et al.217 demonstrated that CARMA1 and MALT1 in CBM complex, but not BCL10, are required for optimal activation of mTOR in T cells. Furthermore, the CBM complex is involved in TCR-induced glutamine uptake and the activation of mTOR pathways.218 Nevertheless, mTOR regulates intracellular metabolic signaling which links to biosynthetic and bioenergetic metabolisms (reviewed in refs. 219,220,221).
Signaling by the co-stimulatory molecules
TCR engagement leads to activation of proximal and distal signaling pathways. However, productive T cell activation also involves the engagement of additional cell surface receptors, i.e., co-stimulatory molecules like CD28. This is required to avoid anergy, a state of T cell unresponsiveness where T cells become refractory to restimulation by IL-2. If the TCR signals are weak, it results in cell death or anergy. These weak TCR signals are amplified strongly by CD28 engagement, thereby resulting in cell proliferation and differentiation. However, only CD28 engagement results in the expression of a few genes transiently with no biological consequences.222
The key event coupling CD28 to several downstream signaling pathways is the recruitment of phosphatidylinositol-3-kinase (PI3K) to the phosphorylated cytoplasmic tail of CD28, which converts PIP2 to PIP3. Once AKT is recruited to PIP3, it acts on several substrates. AKT facilitates prolonged nuclear localization of NFAT, and thus IL-2 transcription, by inactivating GSK-3. IL-2-inducible T cell kinase (ITK) is also associated with PIP3, and this kinase is important for phosphorylation and activation of PLCγ1.170 Apart from this, NF-κβ is one of the major signaling pathways regulated by co-stimulation signaling in T cells. AKT associates with CARMA1 and hence facilitates the formation of the CBM complex, which enhances the nuclear translocation and activation of NF-κβ. However, AKT is non-essential for NF-κβ signaling in T cells.223 The most important mediator of the NF-κβ signaling pathway is phosphoinositide-dependent kinase-1 (PDK1), whose recruitment and phosphorylation enable its efficient binding to both CARMA1 and PKCθ, thus inducing NF-κβ activation.224,225 Indeed, activation of NF-κβ and PKCθ was found abrogated upon deletion of PDK1 in T cells.224 VAV1 is a GEF for small GTPases, such as Rac1, Rac2, and Rhog, where it plays a crucial role in strongly amplifying CD28-mediated activation of NFAT and NF-κβ signaling pathways.226,227 Thus, signaling by co-stimulatory molecules quantifies the signals that are already activated by TCR ligation, thereby strongly sustaining T cell activation. These include the PI3K–AKT–mTOR, NFAT, NF-κβ, and MAPK pathways (Fig. 3). While PI3K signaling is primarily mediated by CD28, initial activation of PI3K results in upregulation of phosphoinositide-3-kinase adaptor protein-1 (PIK3AP1, also known as BCAP).228 PIK3AP1 potentiates PI3K signaling in response to CD3 engagement in CD8+ T cells.228
Positive regulators of T cell signaling
Two of the most important adaptors that are phosphorylated by activated ZAP-70 and play a critical role in positively regulating TCR signaling are a transmembrane adaptor protein, linker for activation of T cells (LAT), and a cytosolically localized SH2 domain-containing leukocyte phosphoprotein of 76 kDa (SLP-76)229,230 (Fig. 3). These adaptor proteins form the backbone of the proximal signaling complex (proximal signalosome) that recruits various other effector proteins,231,232 along with phospholipase Cγ1 (PLCγ1), which links the proximal with several distal signaling pathways upon TCR engagement.144 This results in a stable and dynamic zone of contact between APCs and T cells, designated as the immunological synapse (IS).
Lipid rafts are microdomains located within the plasma membrane that are enriched with cholesterol, glycosphingolipids, and sphingomyelin, and these rafts accumulate at the IS. They are also known as glycolipid-enriched microdomains (GEMs), detergent-resistant membranes (DRMs), or detergent-insoluble glycolipid-enriched membranes (DIGs). Key components of the TCR signaling pathway, such as LCK, LAT, RAS, CD4, and FYN, along with some others, are located within the lipid rafts.6 Post-translational modification of lipids in these molecules is very important for their localization in the lipid rafts. RAS is both palmitoylated and farnesylated, whereas most of the SRC PTKs, including the T cell-specific LCK, undergoes myristoylation and palmitoylation, necessary for its localization in the lipid rafts and subsequent targeting and phosphorylation of the CD3 ζ chain.233,234,235 TCR engagement and activation result in its rapid association with the lipid rafts, and this localization is important for the early tyrosine phosphorylation events of the TCR subunits by the SRC family PTKs.236,237 This can be achieved by the PTK LCK that is present in the rafts, where its SH2 domain binds to the phosphorylated tyrosine residues in activated ZAP-70, thereby bringing TCRs bearing activated ZAP-70 to the lipid rafts.238,239
The GM-CSF/IL3/IL5 common β-chain-associated protein (CBAP) is involved in the regulation of TCR downstream signaling, as CBAP-deficient cells display reduced phosphorylation of PLCγ1, LAT, JNK1/2, and ZAP-70,240 suggesting that it may play a role in both proximal and distal signaling. Besides its role in normal physiology, CBAP plays an important role in T cell acute lymphoblastic leukemia (T-ALL) pathology. CBAP was found to be highly expressed in T-ALL, and its expression enhanced T-ALL cell growth.241 Similar to TCR signaling, loss of CBAP decreased ERK1/2, S6K, RSK, and TSC2 phosphorylation and thereby decreased aerobic glycolysis and energy metabolism.241
LAT
The first adaptor essential for the successful transmission of TCR signals is the LAT.13 It is a transmembrane protein of 36–38 kDa242 consisting of a tyrosine-rich cytoplasmic tail and a short extracellular region.229 It requires palmitoylation on its two cysteine residues (C26 and C29) for localization to the lipid rafts. Mutation of C26 fully inhibited the localization of LAT to the lipid rafts, whereas C29 mutation had a partial effect. Moreover, no tyrosine phosphorylation of LAT was detected when C26 was mutated, indicating that raft localization of LAT is vital for its phosphorylation.243 Several molecules have been proposed that link TCR and LAT by binding to both of them. PLCγ1 binds to phosphorylated tyrosine residue 132 of LAT244 via its N-terminal SH2 domain and to phosphorylated tyrosine residues on activated ZAP-70 via its C-terminal SH2 domain.245 An Abl-SH3 interacting protein, 3BP2 has also been found to interact with both LAT and ZAP-70 via its SH2 domain, probably in a multimeric form.246 Another important molecule found to link TCR and LAT is a small adaptor protein, Shb, that binds to the phosphorylated tyrosine residues on the CD3 ζ chain via its SH2 domain and also binds phosphorylated LAT via its non-SH2 phosphotyrosine binding domain.247 TCR engagement results in rapid phosphorylation of LAT on its tyrosine residues by ZAP-70.248 Thus, LAT phosphorylation and distal signaling events were inhibited when mutant Shb was expressed with a defective SH2 domain.247 Once phosphorylated, LAT then binds to several proteins, such as enzymes and adaptor molecules, via diverse binding sites as discussed below, therein bringing them to the plasma membrane.
Overexpression of LAT did not augment TCR-mediated downstream signaling pathways.249 Moreover, LAT-deficient Jurkat cells also displayed TCR-induced receptor phosphorylation and ZAP-70 activation but were found to be defective in all steps distal from this. There was no activation of PLCγ1 with reduction of Ca2+ mobilization, ERK activation, NFAT activation, and reduced IL-2 gene transcription.249,250 Reconstitution with LAT restored these defects. Moreover, LAT-deficient mice blocked thymic differentiation at the pre-TCR stage, thereby showing no T cells in the lymph nodes and spleen.250 LAT, then, is important for TCR-mediated signaling and intra-thymic development of T cells.
Growth factor receptor-bound protein 2 (GRB2) and GRB2-related adaptor downstream of Shc (GADS)
GRB2 and GADS are cytosolic adaptor proteins, with the former expressed ubiquitously and the latter expressed only in the hematopoietic cells, playing an important role in hematopoietic growth factor receptors signaling.251,252 Activated/phosphorylated LAT binds to both the GRB2 family proteins via their SH2 domains, thereby translocating them to the plasma membrane, along with their SH3 domain-associated proteins.6 GRB2 is constitutively associated with Sos, a dual-specific GEF for small GTPases such as RAS and Rho. Upon TCR activation, Grb2-Sos complex associates with LAT, leading to activation of RAS. However, Grb2 seems insufficient for RAS activation in T cells, as LAT mutants failed to induce complete RAS activation.183,253 An additional small linker molecule, Shc, was found to mediate the association of Sos with GRB2 in T cells.254 An E3 ubiquitin ligase, CBL (discussed later), is another GRB2 associated protein that binds to phosphorylated LAT in T cells.255 Upon TCR stimulation, another member of the GRB2 family, GADS not only binds to phosphorylated LAT but also specifically binds to a critical adaptor molecule of T cells, SLP-76,256 thereby associating LAT with SLP-76. GADS has also been found to associate with a serine/threonine kinase, hematopoietic progenitor kinase-1 (HPK1), involved in JNK pathway activation.256,257 T cell development was found impaired, with specific defects in both positive and negative selection of thymocytes, in GADS-deficient mice.258
The three distal tyrosine residues of LAT (171, 191, and 226) are involved in binding to Grb2, whereas Tyr 171 and 191 are involved in binding to GADS.244 Mutation in any one of the tyrosine residues did not affect either Grb2 or GADS binding, whereas loss of both 171 and 191 decreased GRB2 binding, and mutation of both these residues completely abolished GADS binding. The binding of GRB2 was only abolished when all three tyrosine residues were mutated. Since these tyrosine sites might directly interact with PLCγ1 through its C-terminal SH2 domain or indirectly via GADS–SLP-76–PLCγ1 interaction, mutations of these tyrosine residues also impacted PLCγ1 binding due to loss of SLP-76 binding, with PLCγ1 activation completely inhibited, calcium flux partially inhibited, and PLCγ1-LAT association being undetected.244
SH2 domain-containing leukocyte phosphoprotein of 76 kDa (SLP-76)
SLP-76 is another crucial multidomain adaptor protein of 76 kDa, localized in the cytoplasm13,230 and expressed only in cells of the hematopoietic system, such as thymocytes, mature T cells, natural killer cells, megakaryocytes, and macrophages but not B cells.259 It plays a very important role by linking LAT, activating PLCγ1, and other downstream signaling pathways.13 The proline-rich region of SLP-76 binds to the SH3 domain of PLCγ1,260 leading to the formation of the LAT–GADS–SLP76–PLCγ1 complex. Thus, the two complexes described above, LAT-GADS-SLP-76 and LAT-PLCγ1, interact with each other via binding of SLP-76 to PLCγ1.261 SLP-76-deficient Jurkat cells subsequently displayed severe impairment of PLCγ1 phosphorylation, resulting in decreased calcium flux and IL-2 production, following TCR engagement.262 Overexpression of SLP-76 in Jurkat cells increased TCR-mediated NFAT activity and IL-2 transcription as well as ERK activation, but calcium flux remained unchanged.263,264 Moreover, SLP-76-deficient mice showed an intra-thymic block at an early developmental stage of T cells (double negative stage), thereby failing to generate normal, peripheral T cells, displaying the same phenotype as the LAT-deficient mice.265,266 Such findings demonstrate that SLP-76, like LAT, is important for TCR-mediated signaling and intra-thymic development of T cells.
Upon TCR stimulation, SLP-76 gets phosphorylated by ZAP-70 at its multiple tyrosine residues, which serve as binding sites for various SH2 domain-containing proteins. These include proteins involved in cytoskeletal rearrangements, such as VAV1, non-catalytic tyrosine kinase (NCK), and the PTK ITK (described below).267,268,269 On the other hand, the SH2 domain of SLP-76 associates with the phosphorylated tyrosine residues of a 130 kDa multidomain adaptor protein, named SLP-76-associated phosphoprotein (SLAP)/FYN-binding protein (FYB)/Adhesion and degranulation promoting adaptor protein (ADAP).270,271 SLP-76 thus regulates cytoskeletal changes in activated T cells by coordinated and precise loading of the effector molecules VAV, NCK, and ADAP into the complex, vital for the stability of the complex and its optimal activation.1
Connecting link PLCγ1
Since both LAT and SLP-76 form the backbone of the proximal signaling complex, deficiency of both the adaptors in Jurkat cells and mouse models showed diminished activation of RAS signaling due to impairment in formation of the proximal signalosome.265,272 As the connecting link between proximal and distal signaling pathways, PLCγ1 is the central signaling molecule in T cells, and phosphorylation on its multiple tyrosine residues is required for its full activation.273 This is mediated by the TEC PTK family members, such as IL-2 inducible T cell kinase (ITK) and resting lymphocyte kinase (RLK)/TXK. Deletion of both ITK and RLK exhibited complete loss of PLCγ1 activity along with defects in calcium flux following TCR engagement.274,275,276,277 In contrast, overexpression of RLK enhanced PLCγ1 phosphorylation and calcium flux.278 Plasma membrane localization of ITK and RLK/TXK is possible via its N-terminal pleckstrin homology (PH) domain and palmitoylation respectively. ITK interacts with many different molecules via its various binding domains. The Tec homology (TH) region of ITK is a proline-rich region that interacts with the SH3 domain of Grb2. The SH3 domain of ITK, in turn, interacts with the proline-rich regions of PLCγ1.260 LAT interactions with ITK have also been reported; nevertheless, the exact mechanisms are still unknown.279 Moreover, the SH2 domain of ITK interacts with tyrosine-phosphorylated SLP-76.268,269 Thus, ITKs can interact with the LAT-associated molecules via multiple mechanisms. ITKs in turn are activated by both SLP-76 and LCK.280,281 When ITK associates with SLP-76, it is present in close proximity to its substrate PLCγ1, and direct phosphorylation of ITK at Y511 by LCK promotes its activation.269,282 Moreover, apart from ITK, the association of PLCγ1 with LAT, GADS, and SLP-76 is also required for its optimal activation.253 In response to TCR engagement, PLCγ1 activation is thus regulated by the signaling complex (signalosome) composed of LAT, GADS, SLP-76, PLCγ1, and ITK.6
Signaling lymphocyte activation molecule (SLAM)-associated protein (SAP)
Signals from both the TCR-CD3 complex and co-stimulatory receptors, such as CD28, CD2, and the CD150/SLAM (signaling lymphocyte activation molecule) family, are required for full activation of T cells.283 SAP is a small cytoplasmic protein of 128 amino acids284 that associates via its SH2 domain with the immunoreceptor tyrosine-based switch motifs (ITSMs) present in the cytoplasmic tail of the SLAM family of receptors. Once bound to a specific ITSM, it may prevent binding of the SH2 domain-containing protein tyrosine phosphatase 2 (SHP-2) and thereby compete with it. On the other hand, it may favor the recruitment of SH2 domain-containing inositol phosphatase (SHIP), causing the switch between these two signaling pathways.285,286 The SLAM family consists of a number of transmembrane costimulatory receptors, such as CD150/SLAM, CD244/2B4, CD84, CD229/Ly-9, CD319/CRACC, and NTB-A.287 Thus, SAP can bind via its SH2 domain to the ITSMs of various SLAM families of receptors, and this interaction plays a crucial role in mediating the costimulatory signals necessary for T cell activation.283 Moreover, SAP also exerts its adaptor role by binding to various SH3 domain-containing proteins, such as FYN, PKCθ, βPix, and NCK1,288,289,290,291 thus recruiting them to the SLAM family of transmembrane receptors.292
Along with SLAM, SAP has also been found to directly associate with the first ITAM (Y72-Y83) of the CD3ζ chain in various T cell lines and peripheral blood lymphocytes. Knockdown of SAP resulted in a decrease of several canonical T cell signaling pathways, such as AKT and ERK; reduced the recruitment of PLCγ1, SLP76, and Grb2 to the phosphotyrosine containing complex; and also reduced IL-2 and IL-4 mRNA induction. Through its direct association with the CD3ζ chain, SAP was found to play a central role in T cell activation.292 Indeed, mutations or deletions of the SH2D1A gene encoding SAP resulted in X-linked lymphoproliferative syndrome-1 (XLP1), which is characterized by immunodeficiency due to a specific defect in T cells (apoptosis resistance and impaired interaction with B cell), reduced cytotoxicity of natural killer cells, a decrease in B cell functions, and defective NKT cell development.293,294
SAP and NTB-A (SLAMF6) are essential proteins that potentiate the strength of proximal TCR signals required for restimulation-induced cell death (RICD).295 RICD is an important consequence of repeated TCR signaling essential for TCR-induced apoptosis in thymocytes, mature T cells, T cell malignancies, and T cell therapies (reviewed in refs. 296,297,298). Therefore, T cells with impaired SAP function display resistance to RICD, which likely explains severe CD8+ T cell lymphoproliferation in XLP1 patients.295
Negative regulators of T cell signaling
Inappropriate activation of T cells is prevented by the termination of TCR signals, and this is mediated by certain proteins that negatively regulate TCR signaling (Fig. 4).
Adaptors serving as negative regulators
Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG)/CSK-binding protein (CBP)
An important transmembrane adaptor protein negatively regulating TCR signaling is PAG/CBP which is found in the lipid rafts.299,300 In the absence of TCR engagement or in resting T cells, PAG is constitutively tyrosine phosphorylated in its cytoplasmic tail. This serves as a docking site for the SH2 domain of the major negative regulator of SRC kinases, the tyrosine kinase c-terminal SRC kinase (CSK), thereby localizing to the rafts and activating.301,302,303 Once activated, CSK phosphorylates LCK at the C-terminal Y505 residue, which leads to its kinase domain inactivation as it causes LCK to bind to its internal SH2 domain.49,69,301,302,303 Thus, CSK gets activated upon binding to PAG in the lipid rafts, and it inhibits the activity of SRC family kinases. However, upon TCR activation, tyrosine phosphatase CD45 transiently dephosphorylates PAG. This results in the dissociation of CSK from the glycosphingolipid-enriched microdomains (GEMs), relieving the inhibition of SRC kinases for signal transmission.304 Moreover, the inhibitory Y505 residue of LCK also gets dephosphorylated by CD45 tyrosine phosphatase, which, furthermore, slightly dephosphorylates positive regulatory autophosphorylation at Y394.53,305 Thus, the PAG-CSK complex maintains T cell quiescence by transmitting negative regulatory signals.13
SH2 or SHP-2-interacting transmembrane adaptor protein (SIT)
Another transmembrane adaptor protein negatively regulating TCR signaling is SIT expressed in lymphocytes.306,307,308 It associates with the TCR complex as a disulfide-linked homodimer.306 The cytoplasmic tail of SIT contains immunoreceptor tyrosine-based inhibition motifs (ITIMs) that, upon tyrosine phosphorylation, associate with SHP-2. SIT mediates its negative regulation of TCR signaling through the inhibition of NFAT activity. That, however, remains unaffected after mutation of the tyrosine residue within the ITIM motif, which completely abrogates binding to SHP-2. Thus, SIT-SHP-2 interaction seems unimportant for SI-mediated negative regulation of T cell signaling.306 GRB2 was also found to be associated with SIT via two consensus YxN motifs whose mutations abrogated the binding. This also had no effect on the inhibitory function of SIT.308 Moreover, the effector molecule that might mediate the negative regulatory function of SIT was found to be CSK via co-precipitation experiments.308 However, the precise mechanism of SIT-mediated negative regulation of TCR signaling needs to be elucidated,13 as its role in lymphocyte function seems to be more complex.10
Enzymes serving as negative regulators
Enzymes such as phosphatases, kinases, and ligases also play important roles in negatively regulating TCR signaling.
Phosphatases
Apart from CD45 and SHP-2 already mentioned above, there are other tyrosine phosphatases that mediate negative regulation. Adhesion molecules called carcinoembryonic antigen-related cell adhesion molecule-1 (CEACAM1) are expressed at later time points of TCR stimulation.309 The SH2 domain-containing protein tyrosine phosphatase 1 (SHP-1) is recruited to the phosphorylated ITIMs of CEACAM1, and it dephosphorylates LCK at Y394, inactivating it57,309,310 and thus terminating TCR signaling. The binding of TCR to antagonists or weak antigens induces LCK-mediated phosphorylation of SHP-1 at Y564, thereby activating it, which, in turn, dephosphorylates and inactivates LCK.309,310 Moreover, SHP-1 binding to LCK is prevented by ERK1/2-mediated phosphorylation of LCK at S59, sustaining TCR signaling. SHP-1 activity is thus indirectly regulated by ERK1/2.311 Additional phosphatases that negatively regulate TCR signaling include PTEN, which dephosphorylates PIP3, and dual-specificity phosphatases, which dephosphorylate MAPKs.312,313
Diacylglycerol kinases
Subcellular levels of DAG are regulated by lipid kinases, DGKs that phosphorylate DAG to produce phosphatidic acid (PA).314,315 Consequently, the increase in DGK activity attenuates RAS–MEK–ERK–AP1 signaling induced by TCR-mediated DAG activation.316,317 Ten DGK isoforms are expressed in mammals,315 with DGK α and ζ being expressed at high levels in T cells.318 Both isoforms negatively regulate the DAG–RASGRP1–RAS–ERK1/2 pathway and thus inhibit activation of mTORC1 and mTORC2 complexes.196 Indeed, the genetic ablation of these isoforms resulted in increased activation of the RAS–MEK–ERK–AP1 pathway, mTOR signaling, and PKCθ–NF-κβ pathway. This led to the loss of T cell anergy and increased T cell hyperactivation.145,155,156,319,320 Both DGK α and ζ perform redundant roles in T cells, as their deficiency resulted in severe T cell developmental blockade at the DP stage, which was partially restored with the phosphatidic acid treatment.321 DGK activity can be regulated by SAP. Overexpression of SAP reduces DGKα activity which was shown to be dependent on the SH3-binding ability of SAP in T cells, suggesting that SAP acts as a negative regulator of DGKα.322 As SAP-deficient XLP1 displays resistance to RICD, pharmacological inhibition of DGKα in SAP-deficient cells can restore RICD, indicating that XLP1 patients will likely benefit from DGKα-targeted therapy.295,323,324
E3 ubiquitin ligases
E3 ubiquitin ligases are enzymes that ubiquitinate different proteins and target them for proteasomal- or lysosomal-mediated degradation.325 Some of these ligases regulate T cell tolerance, and T cells can become autoreactive upon their deletion or mutation, leading to autoimmunity.326 TRAF6, as mentioned before, is one such E3 ubiquitin ligase that plays an important role in the activation of the NF-κβ signaling pathway.174,175 Itch is another ubiquitin ligase that not only targets PLCγ1 and PKCθ327,328 but also Jun, thereby causing diminished activation of AP-1.329 Itch thus regulates T cell anergy by degrading certain components of TCR signaling.330 Itch-deficient mouse models are therefore prone to autoimmune and pro-inflammatory phenotypes.1
A well-studied E3 ligase that marks various proteins for ubiquitin-mediated degradation is Casitas B cell lymphoma (CBLB). This enzyme, along with c-CBL, another member of the CBL family, negatively regulates TCR signaling.331 In activated T cells, the CD3ζ chain gets ubiquitinated by CBLB at its multiple lysine residues and induces degradation of surface TCRs.332,333,334,335 Some other targets for CBL-mediated ubiquitination and protein degradation include members of the proximal signaling complex, SRC- and Syk-family PTKs,336,337 the regulatory p85 subunit of PI3K,338 and the adaptor molecule VAV1.337 All these events result in the attenuation of TCR signaling. In another mode of action, T cell activation leads to dissociation of CBLB from Grb2, and it then binds to CRKL, an adaptor molecule required for T cell adhesion and migration. CRKL is in turn constitutively associated with C3G, a GEF for the small GTPases such as RAP1, RAP2, and R-RAS.339,340,341,342,343,344,345 The CRKL–C3G–RAP1 signaling pathway increases the affinity of β1-integrins to the extracellular matrix (ECM), regulating and mediating the adherence of the hematopoietic cells to ECM and stromal cells.346,347 Cell proliferation, cytoskeletal reorganization, and cell-to-cell contact are some of the most critical biological effects of the CRKL–C3G–Rap1 signaling pathway.341,346,348 The binding of CBL to CRKL results in the ubiquitination of CRKL, thus disrupting the CRKL–C3G–Rap1 signaling. On the other hand, an increase in CRKL–C3G–RAP1 signaling, along with clustering of the integrin, lymphocyte function-associated antigen 1 (LFA-1), was observed in response to TCR engagement upon knockdown of CBLB.349,350 CBLB thus serves as the negative regulator of CRKL–C3G–RAP1-mediated signaling events that promote T lymphocyte adhesion, migration, and homing.351 Both LCK and FYN seem to be involved in TCR downregulation, as pharmacological inhibition of LCK and FYN led to stabilization of the TCR complex.352
Adaptor proteins in cytoskeletal reorganization
Early events leading to T cell activation also involve cytoskeletal changes required for lymphocyte migration and mediating cell-to-cell adhesion.353 Interaction of T cells and APCs results in T cell activation, which involves supramolecular rearrangement of a number of receptors at the contact zone, thus forming a synapse. Initially, the integrin receptors of T cells and integrin receptor ligands of APCs are present in the center surrounded by a ring of MHC-peptide complexes. However, this pattern completely reverses within a few minutes, and MHC-peptide complexes form the central region known as the central supramolecular activation cluster (cSMAC), surrounded by integrin receptors in the periphery, forming the peripheral supramolecular activation cluster (pSMAC).354,355,356 These structures are stable for a few hours where specific molecules can be detected. For example, PKCθ has been detected in cSMAC,354 whereas CD45 is initially excluded from cSMAC only to migrate back to it later.357 These molecular rearrangements are partly regulated by the cytoskeleton,354,355 where a ring of polymerized actin accumulates in T cell-APC conjugates or at the interface between T cells stimulated with anti-TCR antibodies.358 LAT and all its binding partners, such as PLCγ1, GADS, and GRB2, are essential for efficient actin polymerization, as the absence of LAT or mutation in binding sites of either of the components inhibits polymerization of actin.358 SLP-76 is one of the molecular adaptors present in a complex with LAT in activated T cells. It binds to several different adaptors involved in regulating the cytoskeleton. Two such adaptors, VAV and NCK, bind to the amino-terminal phosphotyrosine residues of SLP-76 (refs. 359,360) to promote cytoskeletal reorganization, whereas ADAP binds to the carboxyl-terminal phosphotyrosine residues of SLP-76 to promote integrin signaling361 (Fig. 1).
VAV
VAV structurally is a multidomain adaptor protein and functionally a GEF for the activation of Rac and Cdc42, members of the Rho/Rac family of small GTPases.362 Defects in IL-2 production and partial blocks in calcium mobilization were seen upon targeted disruption of VAV. VAV-deficient T cells also showed defects in cytoskeletal function,363,364 along with impaired SMAC formation.365 LAT-deficient T cells would thus fail in the recruitment of VAV via SLP-76, thereby decreasing the amount of activated Rac and Cdc42. This could result in inadequate activation of phosphatidylinositol 4-phosphate 5-kinase, an enzyme responsible for generating PIP2, a substrate of PLCγ1.366 Moreover, there could be inadequate activation of Wiskott–Aldrich syndrome protein (WASP) due to insufficient Rac activation.6
Wiskott–Aldrich syndrome protein
WASP, as the name suggests, was initially identified as a defective protein from Wiskott-Aldrich syndrome patients.367 Decreased IL-2 production, calcium flux, and defective actin polymerization were seen in VAV−/− animals, and a similar phenotype was observed in T cells from WASP patients and murine cells from WAS−/− animals.368,369 This could be explained by the fact that WASP normally exists in an autoinhibited state in resting T cells, where the GTPase-binding domain interacts with the C-terminus that contains the Arp2/3 complex responsible for actin polymerization. The effectors of VAV, RAC, and CDC42, along with PIP2, upon activation, associate with WASP, thereby synergistically activating it and stimulating actin polymerization through the Arp2/3 complex.370,371 WASP is also associated with the NCK, which in turn binds SLP-76.372 Thus, SLP-76 might act as a scaffold by binding to both the NCK and VAV, thereby bringing WASP, RAC, and CDC42-GTP into close proximity so that they can interact with each other.373
NCK and T cell-specific adaptor protein (TSAd)
NCK is another adaptor protein known to regulate the actin cytoskeleton that constitutively interacts with VAV1.374 Both NCK and VAV1 further interact with SLP-76 upon TCR engagement, thereby forming a complex that associates with components of the TCR-CD3 complex, leading to reorganization of actin at the T cell-APC interface.360,373,375 A T cell-specific adaptor protein, TSAd, mediates the association of NCK with LCK and SLP-76 in T cells, thus controlling actin polymerization events in activated T cells. Both NCK and TSAd were found to co-localize in Jurkat cells, where NCK, via its SH2 and SH3 domains, interacts with pTyr280 and pTyr305 and the proline-rich region (PRR) of TSAd respectively. Further, increased polymerization of actin was observed in Jurkat cells expressing TSAd, and this was due to the presence of TSAd exon 7, which encodes interaction sites for both NCK and LCK.376
Moreover, many proteins tend to associate with NCK, as more than 60 binding partners have been identified.377,378 Thus, TSAd may influence the actin cytoskeleton by bringing LCK in the vicinity of different NCK binding partners.376 Moreover, CXCL-12-induced migration and cytoskeletal rearrangements in T cells are regulated via TSAd by promoting LCK-mediated tyrosine phosphorylation of ITK.379 ITK not only binds to TSAd281,379 but also SLP-76,375 thereby forming a multiprotein complex of NCK, TSAd, LCK, SLP-76, and ITK that may interact with each other and several other molecules in a cooperative manner.376 NCK thus plays a vital role in the regulation of the actin cytoskeleton, IS formation after TCR engagement, and cell proliferation and migration.377,380,381
Adhesion and degranulation promoting adaptor protein (ADAP)
Another component of the actin polymerization machinery in T cells is the SLP-76-associated phosphoprotein of 130 kDa (SLAP-130)/FYN-binding protein (FYB)/adhesion and degranulation promoting adaptor protein (ADAP).6 It is a multidomain adaptor protein that, upon TCR engagement, gets phosphorylated by SRC family kinases, such as FYN, enabling its binding to the SH2 domains of SLP-76 and FYN.382,383 Peripheral T cells deficient in ADAP demonstrated defects in cell proliferation, cytokine production, and clustering of the integrin LFA-1 upon TCR stimulation,270,271 whereas TCR-driven IL-2 transcription was increased upon co-transfection of ADAP with SLP-76 and FYN.383,384 Integrin clustering with the help of ADAP facilitates T cell migration in response to stromal cell-derived factor 1 alpha (SDF1α)385 and enhances T cell-APC conjugate formation.386 ADAP thus couples TCR-mediated actin cytoskeletal changes to integrin activation.13 ADAP associates with proteins of the Ena (enabled)/VASP (vasodilator-stimulated phosphoprotein) family, important for the regulation of actin dynamics and T cell polarization, thereby regulating cell adhesion mediated by integrins.387 ADAP also interacts with a multiprotein complex composed of WASP, Arp2/3, VAV, NCK, and SLP-76. TCR-mediated actin rearrangement was inhibited when the binding between the WASP and Arp2/3 complex or ADAP and Ena/VASP proteins was hindered, suggesting that TCR signaling is linked to cytoskeletal remodeling by these interactions.388
SRC kinase-associated phosphoprotein of 55 kDa (SKAP-55)
SKAP-55 or Scap1 is a T cell-specific adaptor protein that is constitutively associated with ADAP.389,390 It enhances cellular adhesion by not only promoting the clustering of LFA-1 but also enhancing its binding to intercellular adhesion molecule-1 (ICAM-1) and fibronectin. SKAP-55 also increases T cell/APC conjugate formation, thereby inducing its translocation to the lipid rafts.386 This brings it into close proximity with the SRC kinase FYN that phosphorylates SKAP-55.391 Both ADAP and SKAP-55 might control the formation of SMACs since they enhance LFA-1-mediated adhesion during T cell/APC interactions, which are important for SMAC formation.13
Mammalian CT10 (chicken tumor virus number 10) regulator of kinase (CRK)
The CRK adaptor proteins are ubiquitously expressed and regulate proliferation, differentiation, adhesion, migration, and apoptosis of immune cells by integrating signals from various effector molecules, such as ECM, growth factors, pathogens, and apoptotic cells.392,393,394,395,396 There are three members belonging to the family of CRK adaptor proteins, CRKI, CRKII, and CRK-like (CRKL), that mediate various protein interactions through their SH2 and SH3 domains. Transient interactions with STAT5, ZAP-70, CBL, and CASL (CRK-associated substrate lymphocyte type) are mediated by the CRK SH2 domains, thus activating the lymphocytes. Cytokines secreted upon TCR activation can induce STAT5 tyrosine phosphorylation, possibly through Janus kinase 3 (JAK3), which was shown to be required for T cell proliferation.397 Furthermore, the STAT5 function is required for amino acid biosynthesis.398 Cell adhesion of lymphocytes, their extravasation, and recruitment to the sites of inflammation are mediated by the constitutive association of CRK with C3G via their SH3 domains. A detailed function of CRKL-C3G is mentioned in the ubiquitin ligase section of this paper, while the reader is also referred to the comprehensive review on the role of CRK adaptor proteins in T cell adhesion and migration.399
TCR signaling dysregulation in diseases
Dysregulation of TCR signaling can lead to the generation of various diseases, given its importance in executing different functions of T cell biology. Thus, defects in TCR signaling can lead to immune deficiency. On the other hand, its hyperactivation can lead to autoimmune diseases. TCR signal transduction is thus tightly regulated via multiple mechanisms by various enzymes and non-enzymatic proteins that serve as scaffolds for efficient signal transmission.183 Mutations in any of these mediators can contribute to the dysregulation of TCR signaling, leading to various disorders.
Both immune deficiency and autoimmunity have been observed when tyrosine phosphatase CD45 is misexpressed. SCID which is characterized by the absence or defective function of T cells, displays deficiency of CD45 expression,400,401 whereas multiple sclerosis (MS) is an end result of certain CD45 polymorphisms.402 SCID was also generated due to mutations in genes coding for CD3 δ, ε, and ζ chains.403 Both mice and humans showed immunodeficiency due to defective expression of LCK.404,405 Furthermore, a rare form of SCID was observed in humans with functionally impaired CD4+ T cells and the absence of CD8+ T cells due to deficiency or mutation of ZAP-70.406,407 In contrast, the development of T cells at the CD4+ CD8+ double-positive stage in mice was blocked due to deficiency of ZAP-70, thereby having a complete absence of single positive CD4+ and CD8+ T cells.1 On the other hand, autoimmune disorders can be caused by an abnormal thymic selection of T cells or their uncontrolled proliferation due to dysregulated TCR signaling. Rheumatoid arthritis (RA) and SLE have been found to be associated with reduced expression of CD3ζ.408,409 Similar to human RA, autoimmune arthritis has been observed in mice having spontaneous mutations in the SH3 domain of ZAP-70.410 Non-T cell activation linker (NTAL) is a transmembrane adaptor molecule that enhances methylprednisolone and TCR-induced apoptosis in T-ALL through increased ERK phosphorylation.411 NTAL thus serves as a tumor suppressor in T-ALL, where its high mRNA expression correlates with a good response to prednisone and vice versa.412 Accordingly, NTAL−/− mice displayed activated T cells characteristic of an autoimmune syndrome.411
Peripheral T cell lymphomas (PTCL) and T cell acute lymphoblastic leukemias or lymphomas (T-ALL) both constitute different groups of T cell malignancies; PTCL arises from post-thymic mature T cells whereas T-ALL arises from thymic immature T cells blocked at various stages of development.413 Multiple molecular aberrations have been described in genes involved in TCR signaling in PTCL,413 with 84% of Sezary syndrome samples414 and 90% of adult T cell leukemia/lymphoma samples415 showing mutations in TCR signaling components. Upregulation of the LAT adaptor along with frequent activating mutations (gain-of-function alterations) in the adaptor CARD11 and PLCγ1 have been observed in most cases of Sezary syndrome cutaneous T cell lymphoma,414 contributing to cell survival and proliferation and disease progression. Thus, the TCR signaling in this context is oncogenic. In contrast, translocations involving TCR genes have been identified in T-ALL416 with no recurrent mutations in any of the TCR signaling components.413 In a landmark study, Trinquand et al. in 2016417 identified that TCR engagement with an MHC-restricted TCR-specific antigen or via CD3 stimulation with anti-CD3 antibody OKT3 made TCR-positive T-ALL cells undergo apoptosis in a similar transcriptional program as the thymic negative selection. However, leukemia recurrence was observed in TCR-positive T-ALL xenografts due to the presence and selection of TCR-negative subclones as a mechanism of tumor relapse from OKT3-mediated therapy. Nevertheless, it is quite encouraging to see that mature T-ALL cells can be induced to undergo apoptosis by TCR activation, using the gene signature for negative selection that is reminiscent in these cells. Subsequently, when assessing the potential of novel anticancer therapies, it is necessary to also assess the importance of cell and disease, as TCR signaling supports oncogenesis in PTCL whereas it appears to have an anti-oncogenic effect in T-ALL.413
Applications of TCR-based immunotherapy
The emergence of T cell-based immunotherapy has revolutionized the understanding of the role of T cells in mitigating a wide variety of diseases, including viral, autoimmune, and malignant diseases. TCR engineering has provided a compelling approach to fight cancer, disrupting the immuno-oncology research field and introducing a new class of impressive cancer immuno-therapeutic strategies, including adoptive cellular therapy (ACT), checkpoint blockade, tumor microenvironment (TME) regulation, and cancer therapeutic vaccines.

The figure depicts the activation of various enzymes and adaptor molecules upon engagement of TCR with the MHC antigenic peptide complex. The phosphorylation events carried out are depicted as small, blue-colored circles. Black lines with arrows indicate activation.
Adoptive T cell transfer therapy
Experimental research in T cells adoptive transfer has revealed the superior capabilities of T cells to identify tumor antigens and to harness the immune system, contributing to anti-tumor activity.418 This type of therapy was first demonstrated clinically by Southam et al.419 in 1966 when patients with unresectable cancer displayed tumor regression upon co-transplantation with patient-derived leukocytes and autologous tumor cells. Although this strategy has been successfully applied, adoptive T cell transfer has not been generalized widely due to the fact that the number of infiltrated T cells was insufficient to exert a full potential of the anti-tumor activity or to boost the body’s immune response against cancer. In addition, the validated immune response in patients receiving this type of therapy was found to be cancer-type and patient-dependent.420,421 Therefore, the engineering of T cells has provided an effective alternative to activate and expand T cells ex vivo with defined specificity against tumor antigens. In this context, TCR-engineered lymphocytes have garnered considerable attention over the past decade, offering significant curative outcomes in patients with cancer. Because tumor cells downregulate MHC molecules, also known as HLA, this posed a challenge for proper T cell response directed against tumor antigen presentation resulting in immune tolerance.422 However, the development of synthetic chimeric antigen receptors (CARs) has overcome this challenge by redirecting T cell specificity to recognize and lyse tumor antigens on the surface of the malignant cell independently of MHC molecules.423

The figure depicts various adaptors and enzymes, like kinases and phosphatases, involved in negatively regulating TCR signaling. The phosphorylation events carried out are depicted as small, blue-colored circles. Black lines with arrows indicate activation. Dotted black lines with arrows indicate dephosphorylation events.
To date, different types of ACTs have been developed, including TCR engineered T cell therapy (TCR-T), tumor-infiltrating lymphocytes (TILs), and CAR T therapy.424,425,426,427 These strategies allow the fast entry of T cell-receptor-based immunotherapies to clinical trials with encouraging clinical outcomes.
TILs therapy
TILs were the first classical attempt for ACT in which infiltrating T cells are isolated from the tumor mass and then expanded ex vivo, activated, and subsequently reinfused into the patient. Several reports have shown that TILs therapy induced a significant durable response in melanoma, including in patients resistant to immune-checkpoint blockade (ICB), as well as objective response in different types of cancers, such as gastrointestinal, colon, and breast cancers.428,429,430,431,432 However, this strategy has been hindered by limited access to solid tumors localized at restricted areas or that have non-resectable metastases as well as long ex vivo processing time and insufficient anti-tumor immunity.433 Completed clinical trials which used TILs-based therapy are summarized briefly in Table 1.
TCR-T therapy
TCR-T therapy was developed to overcome some drawbacks of TILs therapy. This strategy utilizes the same principle as TILs but with genetic modification through retroviral transduction of TCRs to recognize tumor-specific antigens via MHC (Fig. 5). Despite the success of this therapeutic approach, the specificity remains challenging because tumors usually escape such attacks by downregulating MHC. The first clinical outcomes of TCR-T therapy were reported in 2006 when Morgan et al.434 demonstrated durable response among patients with melanoma after transducing autologous T cells with a TCR recognizing the melanocyte differentiation antigen (MART-1). Subsequent clinical trials in 2009 and 2014 confirmed TCR-mediated tumor regression in 30% and 69% of metastatic melanoma patients using MART-1 and gp100 TCR-engineered T cells, respectively.435,436 Parallel clinical responses were also documented using cancer-testis antigens, such as MAGE-A3 and NY-ESO-1. Robbins et al.437 observed that 6 out of 11 patients with synovial cell sarcoma and 5 out of 11 patients with melanoma treated with TCR targeting NY-ESO-1 antigen displayed objective responses. Moreover, targeting NY-ESO-1 antigen in multiple myeloma (MM) using TCR-T therapy has achieved similar robust clinical responses.438 These clinical data suggest that TCR-T therapy can potentially harness the immune system to target and eliminate cancer cells. Although the most commonly reported clinical outcomes were from clinical trials targeting melanoma, other clinical trials have started to introduce this type of therapy more frequently in other solid tumors. This is based on the fact that melanoma incidence has been increasing over the past few years more than other cancers.439,440 Furthermore, melanoma lesions are relatively accessible compared to other solid tumors; therefore, the means for ex vivo expansion of T cells can be readily available, making melanoma one of the best models for immuno-oncology not only in therapy but also in research purposes. A list of current active clinical trials using TCR-T is summarized in Table 2.

T cells are isolated from the patient’s cancer tissue or peripheral blood and genetically modified by retroviral transduction to express antigen-specific TCR or CAR on T cells. Cells are then expanded ex vivo until sufficient cell numbers are achieved and reinfused into the patient’s body, where they can fight cancer cells.
CAR T cell therapy
CAR T cell therapy is lauded as a major step in the development of personalized cancer treatment. The patient’s own T cells are collected by leukapheresis (or peripheral blood) and genetically modified to express a synthetic receptor that binds a specific tumor antigen. These cells are then activated and expanded ex vivo and reinfused into the patient to target and attack cancer cells (Fig. 5). Unlike traditional T cells, CAR T cells recognize antigens independently of MHC presentation due to CAR’s unique structure, containing a transmembrane region with antigen-binding domain and intracellular signaling and co-signaling domains, allowing MHC-independent CAR T cells to bind to their target. Thus, CAR-T cell therapy can overcome cancer-mediated immune tolerance response. Unprecedented clinical response with a high remission rate has been observed using anti-CD19 CAR-T cell therapy in treating patients with B cell malignancies, including B cell acute lymphoblastic leukemia (B-ALL), chronic lymphocytic leukemia (CLL), and B cell non-Hodgkin lymphoma (B-NHL).441,442,443,444,445 In a phase II multicenter clinical trial, anti-CD19 CAR-T therapy was conducted on patients with refractory B cell lymphomas where 82% of patients displayed significant tumor regression and 54% showed complete response rate.446 Moreover, a systematic review and meta-analysis of all published clinical trials conducted by Irbaz Bin Riaz et al. to study the efficacy and safety of anti-CD19 and anti-CD20 CAR-T therapy for B cell hematologic malignancies showed that, among 16 eligible studies, the overall response rate was 61% with complete and partial responses of 42% and 19% respectively. Another clinical trial aimed at determining long-term follow-up of anti-CD19 CAR- T cell therapy has reported the longest durable remission in patients with B cell lymphoma—up to 113 months after treatment—suggesting that anti-CD19 CAR T cells may be curative for B cell lymphoma.447 The success of anti-CD19 CAR-T therapy could be related to the high expression of CD19 in some B cell malignancies and its specificity to the B cell lineage. However, clinical studies showed that the loss of CD19 antigen following treatment is a common cause of disease relapse.448 Thus, anti-CD22 CAR-T has emerged as a potential alternative to anti-CD19 CAR-T therapy. Clinical trials have shown that anti-CD22 CAR-T cells could overcome resistance mediated by anti-CD19 CAR-T cell immunotherapy in patients with B-ALL.449
B cell maturation antigen (BCMA/CD269) targeted therapy has emerged as a promising target for CAR-T cell immunotherapy in multiple myeloma (MM). Although this target is still under investigation, clinical trials phase I exhibited parallel safety and toxicity profiles and suggest its clinical activity against MM.450,451,452 In addition, CAR-T has been shown to target prostate cancer through directing CAR-T cells against prostate-specific membrane antigen (PSMA) and displayed an acceptable safety and efficacy profile.453 In a phase I clinical study, Yao Wang et al.454 targeted patients with CD133-positive and late-stage metastasis malignancies by CAR-T cells in which three patients exhibited partial remission and 14 achieved stable disease.
Despite the success of CAR-T therapy in hematological malignancies, solid tumors have introduced a greater challenge owing to their immunosuppressive microenvironment. While CAR-T therapy has rendered the TME more immunogenic, CAR-T has generated a significant toxic profile; for example, cytokine release syndrome, neurotoxicity, therapy-related mortality, and manufacturing issues have complicated CAR-T cell therapy for solid tumors.455,456,457,458 Efforts to overcome these challenges to generate a more favorable toxicity with CAR-T cell therapy are ongoing.
Immune-checkpoint blockade
A growing body of evidence indicates that peripheral T cell tolerance is an essential factor of the specific immune response to tumor cells. The low cytotoxic capabilities of T cells may be related to the high expression levels of a number of inhibitory molecules including Cytotoxic T lymphocyte antigen 4 (CTLA4) and programmed cell death 1 (PD1). These evolutionarily conserved negative T cell activation regulators act as checkpoint molecules. CTLA4 and PD1 are highly expressed by various types of cancers, and their binding to their respective ligands contribute to T cell functional impairment, which fails to elicit the required immunity against minimal residual disease, and thereby play an important role in cancer recurrence.459 The discovery of immune-checkpoint’s role in cancer has changed the paradigm of cancer therapeutics and added immunotherapy to the list of common three cancer pillars including surgery, targeted therapy, radiotherapy, and chemotherapy.
PD1 and CTLA4 are the most extensively studied immune-checkpoint negative regulators due to their prominent role in fine-tuning tumor-infiltrating T cells. Targeting PD1 and its ligand programmed death-ligand 1 (PD-L1), as well as CTLA4, have gained immense attention after they had shown an unprecedented objective and durable responses across many clinical trials in a subset of patients of metastatic and unresectable cancers leading to different lines of FDA-approved immune-checkpoint inhibitors, and thereby translating checkpoint blockade therapy into an integral part of clinical standard therapy.460,461,462,463,464,465,466 For example, PD-1/PDL-1 inhibitors are now considered as a first-line treatment for patients with melanoma after they have demonstrated a significant increase in overall survival compared to dacarbazine chemotherapy.467 PD-1 inhibitors such as nivolumab and pembrolizumab have shown clinical efficacy in several lines of solid and hematological neoplasms including non-small-cell lung (NSCLC), bladder, pancreatic, follicular B cell, and non-Hodgkin lymphoma.468,469 In addition, it is worth noting that most of the observed effects were correlated with the extent of tumor-infiltrating T cells. Furthermore, in NSCLC, pembrolizumab displayed an improved objective response in patients harboring a high nonsynonymous mutational burden due to a defect in the DNA repair pathway, molecular smoking signature, and higher neoantigen burden.470 In a clinical study conducted to evaluate the correlation between immune cell infiltration and the clinical outcomes in pancreatic ductal adenocarcinoma, with respect to immune-checkpoint molecules, Rong Liu et al.471 found that increase infiltration of PD-1-positive T cells is associated with favorable patient’s prognosis and overall survival. Moreover, patients with melanoma who respond to anti-PD-1 therapy displayed increased intratumoral CD8+ T cells which were associated with tumor regression.472 Caroline Robert et al.473 reported significantly longer overall survival in patients with previously untreated metastatic melanoma using combination therapy of ipilimumab and dacarbazine. Previous reports also indicated that ipilimumab and nivolumab combination therapy exhibited a significant survival benefit in patients with advanced renal cell carcinoma and metastatic melanoma.474,475 Although this line of evidence supports the beneficial role of checkpoint blockade combination therapy, it increases the risk of drug-induced toxicity and therefore should be evaluated with caution.
Targeting the TME
One of the most common challenges in TCR-based immunotherapy is TME. The TME promotes an immunosuppressive nest through: (1) tumor tissue remodeling by regulation of ECM and inhibition of T cells migration476; (2) recruitment of tumor-associated stromal cells, such as T regulatory cells (T reg), myeloid-derived suppressor cells, and tumor-associated fibroblasts477; (3) production of immunosuppressive cytokines and chemokines, such as TGFβ, IL-10, indoleamine 2,3-deoxygenase (IDO), CCL2, and CCL22 (ref. 478); (4) the metabolic state of the tumor tissue that is tightly regulated by oxygen, amino acids, and glucose levels479; (5) expression of ligands that activate inhibitory receptors, such as CTLA4 or PD-1 (ref. 476); (6) epigenetic regulation of the tumor stromal cells—for example, CXCL9 and CXCL10 silencing caused by DNA methylation inhibit T cells homing, thereby cause resistance to immune-checkpoint drugs480; (7) promoting T cells anergy, a process of self-inhibition that results from TCR activation in low levels or absence of appropriate co-stimulation481 (Fig. 6). Taken altogether, these mechanisms reflect the high degree of the tumor heterogeneity that is implicated in tumor evasion, prevent tumor destruction by T cells, and contribute to the development of dysfunctional antitumor immune responses.

The immunosuppressive microenvironment induced by cancer-associated stromal cells modulates cancer progression and therapy resistance. Infiltration of immune cells, such as T reg cells, N2 neutrophils, tumor-associated macrophages, MDSC cells, the transformation of malignant fibroblasts, release of pro-inflammatory cytokines and chemokines, dysregulated vasculature and extracellular matrix remodeling, overexpression of negative immune-checkpoint regulators, metabolic status of the tumor including O2 and nutrients deprivation, the genetic composition of the tumor cells, all this heterogeneous ecosystem of the TME contributes to the tumor therapy resistance.
Several immuno-modulatory strategies have been proposed to target the TME by which the T cell-immune response can be reactivated. For example, it has been found that targeting CTLA4 by ipilimumab reduced the number of infiltrating T reg cells.482,483 However, it is still unclear whether the predominant effect observed by ipilimumab therapy is due to T reg cells depletion or due to effector T cells (eff T cells) infiltration. Studies that demonstrated modest clinical outcomes of ipilimumab in cancer patients support the notion of depleting intratumoral T reg cells484,485 whereas other studies failed to observe such findings because tremelimumab, an IgG2 isotype CTLA4 antibody with minimal potential antibody-dependent cytotoxicity, displayed similar clinical activity as ipilimumab, suggesting that Treg depletion is not crucial for CTLA4 antibodies-mediated cancer regression.486 It is believed that CTLA4-antibodies-mediated depletion of Treg cells in tumors is regulated to a large extent by the host Fc receptor polymorphisms and the availability of effectors of antibody-dependent cellular cytotoxicity in the TME.487 Thus, engineered CTLA4 antibodies with optimized Fc receptors to selectively deplete T reg cells represent an interesting approach towards developing specific anti-tumor immunity.
Lymphocyte-activation gene 3 (LAG-3) is a cell surface molecule expressed on activated eff T and Treg cells in the TME. Binding LAG-3 to its ligand MHC class II mediates suppression of eff T cell activity and upregulation of Treg activity, contributing to the immune tolerance of the TME.488 Antibodies directed against LAG-3 have shown a modest impact on the clinical outcomes in patients with renal, pancreatic, and metastatic breast cancers.489 The evaluation of LAG-3 alone or in combination with other immune checkpoints in clinical trials is ongoing.
Another avenue to manipulate the TME is by promoting T cells response while binding other immune effectors simultaneously. In this context, bispecific antibodies have emerged as a successful strategy to target co-stimulatory and co-inhibitory molecules, including PD-L1 and LAG-3 to dampen the suppressive TME. Bispecific blinatumomab, which binds CD19 on the tumor cells and CD3 on the T cells, showed promising success in patients with acute lymphoblastic leukemia.490
Several signaling pathways have been implicated in tumor-associated molecular alterations that contribute to the immune suppression of the TME, such as Kirsten rat sarcoma viral oncogene (KRAS), focal adhesion kinase (FAK), and Janus kinase-1/2 (JAK1/2). KRAS and FAK signaling promote the recruitment of myeloid suppressor and T reg cells through the granulocyte–monocyte colony-stimulating factor (GM-CSF) in KRAS-associated cancers and CCL5 in squamous cell carcinoma, leading to T cell exhaustion.491,492 FAK has also been reported to mediate SRC kinase negative TCR regulation following T cell activation.493 Furthermore, JAK1 and JAK2 loss-of-function mutations were found to be correlated to acquired PD-1 blockade resistance in melanoma.494 Thus, these data suggest that aberrant signaling pathways are critical regulators of the TME, and targeting these pathways using TCR-based immunotherapy with selective pathway inhibitors might provide a rationale for a combinatorial approach and could overcome the immune activation resistance posed by the TME.
Cancer vaccines
Cancer therapeutic vaccines have become an attractive tool to specifically direct T cell response towards tumor cells. Patients with cancer are exposed to tumor antigens in which T cells get activated, amplified, and elicit a tumor-directed immune response that can induce long-lasting memory T cells, mediating durable clinical responses. While most of the previous cancer vaccines utilized tumor-associated antigens (TAA), the currently developed approach is based on tumor-specific antigens (TSA), known as neoantigens, which result from somatic cancer cell mutations. Unlike TAAs, neoantigens are exclusively expressed by cancer cells with high immunogenicity without being negatively subjected to central or peripheral tolerance, and therefore they elicit specific tumor T cell immune response and prevent “off-target” that can damage the healthy tissues.495 Moreover, the advancement of newly emerging technologies such as next-generation sequencing and mass spectrometry-based algorithms have accelerated the translational and manufacturing aspects of vaccinomics and identified different cancer neoantigens for personalized immunotherapy, meaning it can be tailored individually to each cancer patient.
Several reports showed that TCR-based therapy combined with neoantigen vaccines provoke an efficient antitumor response. For example, neoantigen-reactive T cells have been found to express a high amount of PD-1 following neoantigen vaccines.496,497,498 In a study conducted by Patrick A. Ott et al., vaccination with neoantigens was associated with neoantigen-specific T cell expansion and induction of polyfunctional CD4+ and CD8+ T cells, which targeted 58 out of 97 unique neoantigens in melanoma patients. Moreover, four out of six patients were recurrence-free at 25 months after vaccination while the two with cancer recurrence were subjected to subsequent anti-PD-1 therapy and exhibited a complete cancer regression.499 In a phase Ib clinical trial, Keskin et al.500 demonstrated that neoantigen-specific infiltrating T cells express co-inhibitory receptors following cancer vaccination in glioblastoma patients, providing a rationale for neoantigen vaccines and immune-checkpoint blockade combination therapy. One study which used autologous tumor lysate-dendritic cell vaccine generated neoantigen-specific T cell responses following the vaccination in patients with ovarian carcinoma.501 The use of peptide vaccines and dendritic cells along with TCR-T therapy has also been reported. Anti-melanoma antigens recognized by T cells (MART-1) and anti-glycoprotein (GP-100) peptide vaccines have been shown to stimulate the immune cells and induce MART-1 and GP-100 specific TCR-T cells in patients with metastatic melanoma.434,502 Other clinical trials combining neoantigen vaccines with different cancer immunotherapies are summarized elsewhere.503 These clinical data indicate that cancer vaccines in combination with TCR-based immunotherapies could potentially boost the immune system to eradicate cancer cells while leaving behind long-lasting immune protection against cancer recurrence.
Concluding remarks
The breakthrough made by TCR-based therapeutic applications is rapidly transforming the paradigm of immunotherapies. PD-1 and CTLA4 are the most extensively studied immune-checkpoint negative regulators due to their prominent role in fine-tuning tumor-infiltrating T cells. However, cancer cells exploit this negative regulation and escape from the immune system surveillance. Moreover, due to a wide variety of PD-1 or PD-L1 expression among different types of cancers, not all patients are eligible to undergo this type of treatment. On the other hand, although CAR-T therapy has emerged as a potential strategy to target hematological malignancies, this type of therapy is still hindered by the challenges posed by the TME heterogeneity in solid tumors and by the accompanied therapy-related toxicities. While TCR-T cell therapy has provided impressive clinical results, given its ability to target solid tumors, incorporating cancer vaccines, and recognition of intracellular antigens, the development and proliferation of such therapy encounters numerous obstacles, such as manufacturing issues, tailoring treatment for each patient based on identified genetic mutations, and the tumor immunogenicity. However, we believe that the development of the next generation of TCR-based therapy will overcome these dilemmas, and more groundbreaking applications for cancer immunotherapy are expected to be revealed in the near future.
References
Gorentla, B. K. & Zhong, X. P. T cell receptor signal transduction in T lymphocytes. J. Clin. Cell Immunol. 2012, 5 (2012).
Marshall, J. S., Warrington, R., Watson, W. & Kim, H. L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 14, 49 (2018).
Quang, C. T., Zaniboni, B. & Ghysdael, J. A TCR-switchable cell death pathway in T-ALL. Oncoscience 4, 17–18 (2017).
Zuniga-Pflucker, J. C. T-cell development made simple. Nat. Rev. Immunol. 4, 67–72 (2004).
Sallusto, F., Geginat, J. & Lanzavecchia, A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 22, 745–763 (2004).
Samelson, L. E. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu. Rev. Immunol. 20, 371–394 (2002).
Cantrell, D. A. T-cell antigen receptor signal transduction. Immunology 105, 369–374 (2002).
Hwang, J. R., Byeon, Y., Kim, D. & Park, S. G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 52, 750–761 (2020).
Courtney, A. H., Lo, W. L. & Weiss, A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem. Sci. 43, 108–123 (2018).
Koretzky, G. A. & Boerth, N. J. The role of adapter proteins in T cell activation. Cell Mol. Life Sci. 56, 1048–1060 (1999).
Gerth, E. & Mattner, J. The role of adaptor proteins in the biology of natural killer T (NKT) cells. Front. Immunol. 10, 1449 (2019).
Flynn, D. C. Adaptor proteins. Oncogene 20, 6270–6272 (2001).
Wilkinson, B., Wang, H. & Rudd, C. E. Positive and negative adaptors in T-cell signalling. Immunology 111, 368–374 (2004).
Wucherpfennig, K. W. et al. Structural biology of the T-cell receptor: insights into receptor assembly, ligand recognition, and initiation of signaling. Cold Spring Harb. Perspect. Biol. 2, a005140 (2010).
Kuhns, M. S. & Badgandi, H. B. Piecing together the family portrait of TCR-CD3 complexes. Immunol. Rev. 250, 120–143 (2012).
Rast, J. P. et al. alpha, beta, gamma, and delta T cell antigen receptor genes arose early in vertebrate phylogeny. Immunity 6, 1–11 (1997).
Gaulard, P. et al. Expression of the alpha/beta and gamma/delta T-cell receptors in 57 cases of peripheral T-cell lymphomas. Identification of a subset of gamma/delta T-cell lymphomas. Am. J. Pathol. 137, 617–628 (1990).
Bruno, L., Fehling, H. J. & von Boehmer, H. The alpha beta T cell receptor can replace the gamma delta receptor in the development of gamma delta lineage cells. Immunity 5, 343–352 (1996).
Zhao, Y., Niu, C. & Cui, J. Gamma-delta (gammadelta) T cells: friend or foe in cancer development? J. Transl. Med. 16, 3 (2018).
van Boxel, G. I., Holmes, S., Fugger, L. & Jones, E. Y. An alternative conformation of the T-cell receptor alpha constant region. J. Mol. Biol. 400, 828–837 (2010).
Allison, T. J. et al. Structure of a human gammadelta T-cell antigen receptor. Nature 411, 820–824 (2001).
Touma, M. et al. The TCR C beta FG loop regulates alpha beta T cell development. J. Immunol. 176, 6812–6823 (2006).
Morath, A. & Schamel, W.W. Alphabeta and gammadelta T cell receptors: similar but different. J. Leukoc. Biol. 107, 1045–1055 (2020).
Dong et al. Structural basis of assembly of the human T cell receptor-CD3 complex. Nature 573, 546–552 (2019).
Alcover, A., Alarcon, B. & Di Bartolo, V. Cell biology of T cell receptor expression and regulation. Annu. Rev. Immunol. 36, 103–125 (2018).
Crotty, S. A brief history of T cell help to B cells. Nat. Rev. Immunol. 15, 185–189 (2015).
Bach, F. H., Bach, M. L. & Sondel, P. M. Differential function of major histocompatibility complex antigens in T-lymphocyte activation. Nature 259, 273–281 (1976).
Cantor, H. & Boyse, E. A. Regulation of cellular and humoral immune responses by T-cell subclasses. Cold Spring Harb. Symp. Quant. Biol. 41, 23–32 (1977).
Shiku, H. et al. Expression of T-cell differentiation antigens on effector cells in cell-mediated cytotoxicity in vitro. Evidence for functional heterogeneity related to the surface phenotype of T cells. J. Exp. Med. 141, 227–241 (1975).
Janeway, C. A. Jr. T-cell development. Accessories or coreceptors? Nature 335, 208–210 (1988).
Ellmeier, W., Sawada, S. & Littman, D. R. The regulation of CD4 and CD8 coreceptor gene expression during T cell development. Annu. Rev. Immunol. 17, 523–554 (1999).
Ellmeier, W., Haust, L. & Tschismarov, R. Transcriptional control of CD4 and CD8 coreceptor expression during T cell development. Cell Mol. Life Sci. 70, 4537–4553 (2013).
Lustgarten, J., Waks, T. & Eshhar, Z. CD4 and CD8 accessory molecules function through interactions with major histocompatibility complex molecules which are not directly associated with the T cell receptor-antigen complex. Eur. J. Immunol. 21, 2507–2515 (1991).
Tikhonova, A. N. et al. Alphabeta T cell receptors that do not undergo major histocompatibility complex-specific thymic selection possess antibody-like recognition specificities. Immunity 36, 79–91 (2012).
Leahy, D. J. A structural view of CD4 and CD8. FASEB J. 9, 17–25 (1995).
Hernandez-Hoyos, G., Sohn, S. J., Rothenberg, E. V. & Alberola-Ila, J. Lck activity controls CD4/CD8 T cell lineage commitment. Immunity 12, 313–322 (2000).
Zamoyska, R. The CD8 coreceptor revisited: one chain good, two chains better. Immunity 1, 243–246 (1994).
Kim, S. V. & Flavell, R. A. Immunology. CD8alphaalpha and T cell memory. Science 304, 529–530 (2004).
Madakamutil, L. T. et al. CD8alphaalpha-mediated survival and differentiation of CD8 memory T cell precursors. Science 304, 590–593 (2004).
Wieczorek, M. et al. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front. Immunol. 8, 292 (2017).
Cohen, N. R., Garg, S. & Brenner, M. B. Antigen presentation by CD1 lipids, T cells, and NKT cells in microbial immunity. Adv. Immunol. 102, 1–94 (2009).
Jackman, R. M., Moody, D. B. & Porcelli, S. A. Mechanisms of lipid antigen presentation by CD1. Crit. Rev. Immunol. 19, 49–63 (1999).
Zhai, Y. et al. Src-family protein tyrosine kinases: a promising target for treating cardiovascular diseases. Int. J. Med. Sci. 18, 1216–1224 (2021).
Thomas, S. M. & Brugge, J. S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 13, 513–609 (1997).
Kazi, J. U. & Ronnstrand, L. The role of SRC family kinases in FLT3 signaling. Int. J. Biochem. Cell Biol. 107, 32–37 (2019).
Patwardhan, P. & Resh, M. D. Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol. Cell Biol. 30, 4094–4107 (2010).
Saksela, K. & Permi, P. SH3 domain ligand binding: what’s the consensus and where’s the specificity? FEBS Lett. 586, 2609–2614 (2012).
Kaneko, T., Joshi, R., Feller, S. M. & Li, S. S. Phosphotyrosine recognition domains: the typical, the atypical and the versatile. Cell Commun. Signal. 10, 32 (2012).
Bergman, M. et al. The human p50csk tyrosine kinase phosphorylates p56lck at Tyr-505 and down regulates its catalytic activity. EMBO J. 11, 2919–2924 (1992).
Mustelin, T. & Altman, A. Dephosphorylation and activation of the T cell tyrosine kinase pp56lck by the leukocyte common antigen (CD45). Oncogene 5, 809–813 (1990).
Yamaguchi, H. & Hendrickson, W. A. Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature 384, 484–489 (1996).
Bommhardt, U., Schraven, B. & Simeoni, L. Beyond TCR signaling: emerging functions of Lck in cancer and immunotherapy. Int. J. Mol. Sci. 20, 3500 (2019).
McNeill, L. et al. The differential regulation of Lck kinase phosphorylation sites by CD45 is critical for T cell receptor signaling responses. Immunity 27, 425–437 (2007).
Zikherman, J. et al. CD45-Csk phosphatase-kinase titration uncouples basal and inducible T cell receptor signaling during thymic development. Immunity 32, 342–354 (2010).
Zamoyska, R. Why is there so much CD45 on T cells? Immunity 27, 421–423 (2007).
Wu, J. et al. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J. Biol. Chem. 281, 11002–11010 (2006).
Chiang, G. G. & Sefton, B. M. Specific dephosphorylation of the Lck tyrosine protein kinase at Tyr-394 by the SHP-1 protein-tyrosine phosphatase. J. Biol. Chem. 276, 23173–23178 (2001).
Huse, M. et al. Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity 27, 76–88 (2007).
Molina, T. J. et al. Profound block in thymocyte development in mice lacking p56lck. Nature 357, 161–164 (1992).
van Oers, N. S., Killeen, N. & Weiss, A. Lck regulates the tyrosine phosphorylation of the T cell receptor subunits and ZAP-70 in murine thymocytes. J. Exp. Med. 183, 1053–1062 (1996).
Guirado, M. et al. Phosphorylation of the N-terminal and C-terminal CD3-epsilon-ITAM tyrosines is differentially regulated in T cells. Biochem. Biophys. Res. Commun. 291, 574–581 (2002).
Weiss, A. & Littman, D. R. Signal transduction by lymphocyte antigen receptors. Cell 76, 263–274 (1994).
Wange, R. L. & Samelson, L. E. Complex complexes: signaling at the TCR. Immunity 5, 197–205 (1996).
Letourneur, F. & Klausner, R. D. A novel di-leucine motif and a tyrosine-based motif independently mediate lysosomal targeting and endocytosis of CD3 chains. Cell 69, 1143–1157 (1992).
Yao, X. R., Flaswinkel, H., Reth, M. & Scott, D. W. Immunoreceptor tyrosine-based activation motif is required to signal pathways of receptor-mediated growth arrest and apoptosis in murine B lymphoma cells. J. Immunol. 155, 652–661 (1995).
Bu, J. Y., Shaw, A. S. & Chan, A. C. Analysis of the interaction of ZAP-70 and syk protein-tyrosine kinases with the T-cell antigen receptor by plasmon resonance. Proc. Natl Acad. Sci. USA 92, 5106–5110 (1995).
Iwashima, M. et al. Sequential interactions of the TCR with two distinct cytoplasmic tyrosine kinases. Science 263, 1136–1139 (1994).
Gascoigne, N. R., Casas, J., Brzostek, J. & Rybakin, V. Initiation of TCR phosphorylation and signal transduction. Front. Immunol. 2, 72 (2011).
Palacios, E. H. & Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 23, 7990–8000 (2004).
Stein, P. L., Lee, H. M., Rich, S. & Soriano, P. pp59fyn mutant mice display differential signaling in thymocytes and peripheral T cells. Cell 70, 741–750 (1992).
Appleby, M. W. et al. Defective T cell receptor signaling in mice lacking the thymic isoform of p59fyn. Cell 70, 751–763 (1992).
Straus, D. B. & Weiss, A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell 70, 585–593 (1992).
Krystal, G. W., DeBerry, C. S., Linnekin, D. & Litz, J. Lck associates with and is activated by Kit in a small cell lung cancer cell line: inhibition of SCF-mediated growth by the Src family kinase inhibitor PP1. Cancer Res. 58, 4660–4666 (1998).
Marhall, A., Kazi, J. U. & Ronnstrand, L. The Src family kinase LCK cooperates with oncogenic FLT3/ITD in cellular transformation. Sci. Rep. 7, 13734 (2017).
Braunger, J. et al. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene 14, 2619–2631 (1997).
Laugel, B. et al. Different T cell receptor affinity thresholds and CD8 coreceptor dependence govern cytotoxic T lymphocyte activation and tetramer binding properties. J. Biol. Chem. 282, 23799–23810 (2007).
Singer, A. & Bosselut, R. CD4/CD8 coreceptors in thymocyte development, selection, and lineage commitment: analysis of the CD4/CD8 lineage decision. Adv. Immunol. 83, 91–131 (2004).
Zamoyska, R. CD4 and CD8: modulators of T-cell receptor recognition of antigen and of immune responses? Curr. Opin. Immunol. 10, 82–87 (1998).
Janeway, C. A. Jr. The T cell receptor as a multicomponent signalling machine: CD4/CD8 coreceptors and CD45 in T cell activation. Annu. Rev. Immunol. 10, 645–674 (1992).
Veillette, A., Bookman, M. A., Horak, E. M. & Bolen, J. B. The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck. Cell 55, 301–308 (1988).
Shaw, A. S. et al. The lck tyrosine protein kinase interacts with the cytoplasmic tail of the CD4 glycoprotein through its unique amino-terminal domain. Cell 59, 627–636 (1989).
Zlatkine, P., Mehul, B. & Magee, A. I. Retargeting of cytosolic proteins to the plasma membrane by the Lck protein tyrosine kinase dual acylation motif. J. Cell Sci. 110, 673–679 (1997).
Bijlmakers, M. J., Isobe-Nakamura, M., Ruddock, L. J. & Marsh, M. Intrinsic signals in the unique domain target p56(lck) to the plasma membrane independently of CD4. J. Cell Biol. 137, 1029–1040 (1997).
Yasuda, K. et al. Serine 6 of Lck tyrosine kinase: a critical site for Lck myristoylation, membrane localization, and function in T lymphocytes. J. Immunol. 165, 3226–3231 (2000).
Stephen, T. L., Wilson, B. S. & Laufer, T. M. Subcellular distribution of Lck during CD4 T-cell maturation in the thymic medulla regulates the T-cell activation threshold. Proc. Natl Acad. Sci. USA 109, 7415–7420 (2012).
Irvine, D. J., Purbhoo, M. A., Krogsgaard, M. & Davis, M. M. Direct observation of ligand recognition by T cells. Nature 419, 845–849 (2002).
Zimmermann, L. et al. Direct observation and quantitative analysis of Lck exchange between plasma membrane and cytosol in living T cells. J. Biol. Chem. 285, 6063–6070 (2010).
Akimzhanov, A. M. & Boehning, D. Rapid and transient palmitoylation of the tyrosine kinase Lck mediates Fas signaling. Proc. Natl Acad. Sci. USA 112, 11876–11880 (2015).
Shaw, A. S. et al. Short related sequences in the cytoplasmic domains of CD4 and CD8 mediate binding to the amino-terminal domain of the p56lck tyrosine protein kinase. Mol. Cell Biol. 10, 1853–1862 (1990).
Rudd, C. E. et al. The CD4 receptor is complexed in detergent lysates to a protein-tyrosine kinase (pp58) from human T lymphocytes. Proc. Natl Acad. Sci. USA 85, 5190–5194 (1988).
Turner, J. M. et al. Interaction of the unique N-terminal region of tyrosine kinase p56lck with cytoplasmic domains of CD4 and CD8 is mediated by cysteine motifs. Cell 60, 755–765 (1990).
Lin, R. S., Rodriguez, C., Veillette, A. & Lodish, H. F. Zinc is essential for binding of p56(lck) to CD4 and CD8alpha. J. Biol. Chem. 273, 32878–32882 (1998).
Huse, M., Eck, M. J. & Harrison, S. C. A Zn2+ ion links the cytoplasmic tail of CD4 and the N-terminal region of Lck. J. Biol. Chem. 273, 18729–18733 (1998).
Kim, P. W. et al. A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8. Science 301, 1725–1728 (2003).
Wang, J. H. et al. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proc. Natl Acad. Sci. USA 98, 10799–10804 (2001).
Yin, Y., Wang, X. X. & Mariuzza, R. A. Crystal structure of a complete ternary complex of T-cell receptor, peptide-MHC, and CD4. Proc. Natl Acad. Sci. USA 109, 5405–5410 (2012).
Wang, X. X. et al. Affinity maturation of human CD4 by yeast surface display and crystal structure of a CD4-HLA-DR1 complex. Proc. Natl Acad. Sci. USA 108, 15960–15965 (2011).
Li, Y., Yin, Y. & Mariuzza, R. A. Structural and biophysical insights into the role of CD4 and CD8 in T cell activation. Front. Immunol. 4, 206 (2013).
Gao, G. F. et al. Crystal structure of the complex between human CD8alpha(alpha) and HLA-A2. Nature 387, 630–634 (1997).
van der Merwe, P. A. & Davis, S. J. Molecular interactions mediating T cell antigen recognition. Annu. Rev. Immunol. 21, 659–684 (2003).
Kim, S. T. et al. The alphabeta T cell receptor is an anisotropic mechanosensor. J. Biol. Chem. 284, 31028–31037 (2009).
Kim, S. T. et al. Distinctive CD3 heterodimeric ectodomain topologies maximize antigen-triggered activation of alpha beta T cell receptors. J. Immunol. 185, 2951–2959 (2010).
Yoon, S. T., Dianzani, U., Bottomly, K. & Janeway, C. A. Jr. Both high and low avidity antibodies to the T cell receptor can have agonist or antagonist activity. Immunity 1, 563–569 (1994).
Devine, L., Kieffer, L. J., Aitken, V. & Kavathas, P. B. Human CD8 beta, but not mouse CD8 beta, can be expressed in the absence of CD8 alpha as a beta beta homodimer. J. Immunol. 164, 833–838 (2000).
Rybakin, V. et al. CD8alphaalpha and -alphabeta isotypes are equally recruited to the immunological synapse through their ability to bind to MHC class I. EMBO Rep. 12, 1251–1256 (2011).
Yachi, P. P., Ampudia, J., Gascoigne, N. R. & Zal, T. Nonstimulatory peptides contribute to antigen-induced CD8-T cell receptor interaction at the immunological synapse. Nat. Immunol. 6, 785–792 (2005).
Weyand, C. M., Goronzy, J. & Fathman, C. G. Modulation of CD4 by antigenic activation. J. Immunol. 138, 1351–1354 (1987).
Shin, J. et al. Structural features of the cytoplasmic region of CD4 required for internalization. EMBO J. 9, 425–434 (1990).
Sleckman, B. P. et al. Disruption of the CD4-p56lck complex is required for rapid internalization of CD4. Proc. Natl Acad. Sci. USA 89, 7566–7570 (1992).
Acres, R. B., Conlon, P. J., Mochizuki, D. Y. & Gallis, B. Rapid phosphorylation and modulation of the T4 antigen on cloned helper T cells induced by phorbol myristate acetate or antigen. J. Biol. Chem. 261, 16210–16214 (1986).
Pelchen-Matthews, A. et al. The protein tyrosine kinase p56lck inhibits CD4 endocytosis by preventing entry of CD4 into coated pits. J. Cell Biol. 117, 279–290 (1992).
Stepanek, O. et al. Coreceptor scanning by the T cell receptor provides a mechanism for T cell tolerance. Cell 159, 333–345 (2014).
Barber, E. K. et al. The CD4 and CD8 antigens are coupled to a protein-tyrosine kinase (p56lck) that phosphorylates the CD3 complex. Proc. Natl Acad. Sci. USA 86, 3277–3281 (1989).
Burgess, K. E. et al. Biochemical identification of a direct physical interaction between the CD4:p56lck and Ti(TcR)/CD3 complexes. Eur. J. Immunol. 21, 1663–1668 (1991).
Weissman, A. M. et al. Tyrosine phosphorylation of the human T cell antigen receptor zeta-chain: activation via CD3 but not CD2. J. Immunol. 141, 3532–3536 (1988).
Monostori, E. et al. Activation of human T lymphocytes via the CD2 antigen results in tyrosine phosphorylation of T cell antigen receptor zeta-chains. J. Immunol. 144, 1010–1014 (1990).
Kersh, E. N., Shaw, A. S. & Allen, P. M. Fidelity of T cell activation through multistep T cell receptor zeta phosphorylation. Science 281, 572–575 (1998).
Housden, H. R. et al. Investigation of the kinetics and order of tyrosine phosphorylation in the T-cell receptor zeta chain by the protein tyrosine kinase Lck. Eur. J. Biochem. 270, 2369–2376 (2003).
Isakov, N. et al. ZAP-70 binding specificity to T cell receptor tyrosine-based activation motifs: the tandem SH2 domains of ZAP-70 bind distinct tyrosine-based activation motifs with varying affinity. J. Exp. Med. 181, 375–380 (1995).
Xu, C. et al. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell 135, 702–713 (2008).
Deford-Watts, L. M. et al. The cytoplasmic tail of the T cell receptor CD3 epsilon subunit contains a phospholipid-binding motif that regulates T cell functions. J. Immunol. 183, 1055–1064 (2009).
Aivazian, D. & Stern, L. J. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat. Struct. Biol. 7, 1023–1026 (2000).
Fernandes, R. A. et al. What controls T cell receptor phosphorylation? Cell 142, 668–669 (2010).
Huyer, G. et al. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 272, 843–851 (1997).
Gagnon, E. et al. Response multilayered control of T cell receptor phosphorylation. Cell 142, 669–671 (2010).
Kuhns, M. S. & Davis, M. M. The safety on the TCR trigger. Cell 135, 594–596 (2008).
Xu, X., Li, H. & Xu, C. Structural understanding of T cell receptor triggering. Cell Mol. Immunol. 17, 193–202 (2020).
Shi, X. et al. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature 493, 111–115 (2013).
Li, F. Y. et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 475, 471–476 (2011).
Lioudyno, M. I. et al. Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc. Natl Acad. Sci. USA 105, 2011–2016 (2008).
Sigalov, A. B., Aivazian, D. A., Uversky, V. N. & Stern, L. J. Lipid-binding activity of intrinsically unstructured cytoplasmic domains of multichain immune recognition receptor signaling subunits. Biochemistry 45, 15731–15739 (2006).
van Oers, N. S., Killeen, N. & Weiss, A. ZAP-70 is constitutively associated with tyrosine-phosphorylated TCR zeta in murine thymocytes and lymph node T cells. Immunity 1, 675–685 (1994).
van Oers, N. S. et al. Constitutive tyrosine phosphorylation of the T-cell receptor (TCR) zeta subunit: regulation of TCR-associated protein tyrosine kinase activity by TCR zeta. Mol. Cell Biol. 13, 5771–5780 (1993).
Dorfman, J. R., Stefanova, I., Yasutomo, K. & Germain, R. N. CD4+ T cell survival is not directly linked to self-MHC-induced TCR signaling. Nat. Immunol. 1, 329–335 (2000).
Chakraborty, A. K. & Weiss, A. Insights into the initiation of TCR signaling. Nat. Immunol. 15, 798–807 (2014).
Wei, Q. et al. Lck bound to coreceptor is less active than free Lck. Proc. Natl Acad. Sci. USA 117, 15809–15817 (2020).
Jiang, N. et al. Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity 34, 13–23 (2011).
Casas, J. et al. Ligand-engaged TCR is triggered by Lck not associated with CD8 coreceptor. Nat. Commun. 5, 5624 (2014).
Ike, H. et al. Mechanism of Lck recruitment to the T-cell receptor cluster as studied by single-molecule-fluorescence video imaging. Chemphyschem 4, 620–626 (2003).
Horkova, V. et al. Dynamics of the coreceptor-LCK interactions during T cell development shape the self-reactivity of peripheral CD4 and CD8 T cells. Cell Rep. 30, 1504–1514 e1507 (2020).
Li, L. et al. Ionic CD3-Lck interaction regulates the initiation of T-cell receptor signaling. Proc. Natl Acad. Sci. USA 114, E5891–E5899 (2017).
Wang, H. et al. ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb. Perspect. Biol. 2, a002279 (2010).
Neumeister, E. N. et al. Binding of ZAP-70 to phosphorylated T-cell receptor zeta and eta enhances its autophosphorylation and generates specific binding sites for SH2 domain-containing proteins. Mol. Cell Biol. 15, 3171–3178 (1995).
Beach, D. et al. Dual role of SLP-76 in mediating T cell receptor-induced activation of phospholipase C-gamma1. J. Biol. Chem. 282, 2937–2946 (2007).
Zhong, X. P. et al. Diacylglycerol kinases in immune cell function and self-tolerance. Immunol. Rev. 224, 249–264 (2008).
Oh-hora, M. & Rao, A. Calcium signaling in lymphocytes. Curr. Opin. Immunol. 20, 250–258 (2008).
Berridge, M. J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 1793, 933–940 (2009).
Kania, E. et al. IP3 receptor-mediated calcium signaling and its role in autophagy in cancer. Front. Oncol. 7, 140 (2017).
Vig, M. et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223 (2006).
Penna, A. et al. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature 456, 116–120 (2008).
Weidinger, C., Shaw, P. J. & Feske, S. STIM1 and STIM2-mediated Ca(2+) influx regulates antitumour immunity by CD8(+) T cells. EMBO Mol. Med. 5, 1311–1321 (2013).
Zhang, S. L. et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905 (2005).
Macian, F. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 5, 472–484 (2005).
Macian, F. et al. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 109, 719–731 (2002).
Zha, Y. et al. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-alpha. Nat. Immunol. 7, 1166–1173 (2006).
Olenchock, B. A. et al. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat. Immunol. 7, 1174–1181 (2006).
Savignac, M., Mellstrom, B. & Naranjo, J. R. Calcium-dependent transcription of cytokine genes in T lymphocytes. Pflugers Arch. 454, 523–533 (2007).
Le Deist, F. et al. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood 85, 1053–1062 (1995).
Feske, S. et al. Severe combined immunodeficiency due to defective binding of the nuclear factor of activated T cells in T lymphocytes of two male siblings. Eur. J. Immunol. 26, 2119–2126 (1996).
Joseph, N., Reicher, B. & Barda-Saad, M. The calcium feedback loop and T cell activation: how cytoskeleton networks control intracellular calcium flux. Biochim Biophys. Acta 1838, 557–568 (2014).
Kazi, J. U. The mechanism of protein kinase C regulation. Front. Biol. 6, 328–336 (2011).
Parker, P. J. et al. Equivocal, explicit and emergent actions of PKC isoforms in cancer. Nat. Rev. Cancer. 21, 51–63 (2020).
Kabir, N. N., Rönnstrand, L. & Kazi, J. U. Protein kinase C expression is deregulated in chronic lymphocytic leukemia. Leuk. Lymphoma 54, 2288–2290 (2013).
Kazi, J. U., Kabir, N. N. & Rönnstrand, L. Protein kinase C (PKC) as a drug target in chronic lymphocytic leukemia. Med. Oncol. 30, 757 (2013).
Pfeifhofer-Obermair, C., Thuille, N. & Baier, G. Involvement of distinct PKC gene products in T cell functions. Front. Immunol. 3, 220 (2012).
Szamel, M. & Resch, K. T-cell antigen receptor-induced signal-transduction pathways-activation and function of protein kinases C in T lymphocytes. Eur. J. Biochem. 228, 1–15 (1995).
Sun, Z. Intervention of PKC-theta as an immunosuppressive regimen. Front. Immunol. 3, 225 (2012).
Blanchett, S., Boal-Carvalho, I., Layzell, S. & Seddon, B. NF-kappaB and extrinsic cell death pathways—entwined do-or-die decisions for T cells. Trends Immunol. 42, 76–88 (2020).
Matsumoto, M. et al. Essential role of NF-kappa B-inducing kinase in T cell activation through the TCR/CD3 pathway. J. Immunol. 169, 1151–1158 (2002).
Yamagishi, M. W. & New, T. Paradigm of T cell signaling: learning from malignancies. J. Clin. Cell Immunol. S12, 007 (2012).
Lu, H. Y. et al. The CBM-opathies—a rapidly expanding spectrum of human inborn errors of immunity caused by mutations in the CARD11-BCL10-MALT1 complex. Front. Immunol. 9, 2078 (2018).
Matsumoto, R. et al. Phosphorylation of CARMA1 plays a critical role in T cell receptor-mediated NF-kappaB activation. Immunity 23, 575–585 (2005).
Weil, R. & Israel, A. Deciphering the pathway from the TCR to NF-kappaB. Cell Death Differ. 13, 826–833 (2006).
Sun, L. et al. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14, 289–301 (2004).
Zhou, H. et al. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature 427, 167–171 (2004).
Hayden, M. S., West, A. P. & Ghosh, S. NF-kappaB and the immune response. Oncogene 25, 6758–6780 (2006).
Hoffmann, A., Natoli, G. & Ghosh, G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 25, 6706–6716 (2006).
So, T. & Croft, M. Regulation of the PKCtheta-NF-kappaB axis in T lymphocytes by the tumor necrosis factor receptor family member OX40. Front. Immunol. 3, 133 (2012).
Schulze-Luehrmann, J. & Ghosh, S. Antigen-receptor signaling to nuclear factor kappa B. Immunity 25, 701–715 (2006).
Krappmann, D. et al. Molecular mechanisms of constitutive NF-kappaB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene 18, 943–953 (1999).
Staudt, L. M. Oncogenic activation of NF-kappaB. Cold Spring Harb. Perspect. Biol. 2, a000109 (2010).
Krishna, S. et al. Chronic activation of the kinase IKKbeta impairs T cell function and survival. J. Immunol. 189, 1209–1219 (2012).
Smith-Garvin, J. E., Koretzky, G. A. & Jordan, M. S. T cell activation. Annu. Rev. Immunol. 27, 591–619 (2009).
Ebinu, J. O. et al. RasGRP, a Ras guanyl nucleotide- releasing protein with calcium- and diacylglycerol-binding motifs. Science 280, 1082–1086 (1998).
Janknecht, R., Ernst, W. H., Pingoud, V. & Nordheim, A. Activation of ternary complex factor Elk-1 by MAP kinases. EMBO J. 12, 5097–5104 (1993).
Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6, 827–837 (2005).
Jia, H. et al. Ras-ERK1/2 signaling accelerates the progression of colorectal cancer via mediation of H2BK5ac. Life Sci. 230, 89–96 (2019).
Bertin, S. et al. Dual-specificity phosphatase 6 regulates CD4+ T-cell functions and restrains spontaneous colitis in IL-10-deficient mice. Mucosal Immunol. 8, 505–515 (2015).
Damasio, M. P. et al. Extracellular signal-regulated kinase (ERK) pathway control of CD8+ T cell differentiation. Biochem. J. 478, 79–98 (2020).
Kaminuma, O. et al. Vav-Rac1-mediated activation of the c-Jun N-terminal kinase/c-Jun/AP-1 pathway plays a major role in stimulation of the distal NFAT site in the interleukin-2 gene promoter. Mol. Cell Biol. 21, 3126–3136 (2001).
Chung, J., Uchida, E., Grammer, T. C. & Blenis, J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell Biol. 17, 6508–6516 (1997).
Rohrs, J. A., Siegler, E. L., Wang, P. & Finley, S. D. ERK activation in CAR T cells is amplified by CD28-mediated increase in CD3zeta phosphorylation. iScience 23, 101023 (2020).
Dower, N. A. et al. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat. Immunol. 1, 317–321 (2000).
Shen, S. et al. Critical roles of RasGRP1 for invariant NKT cell development. J. Immunol. 187, 4467–4473 (2011).
Chen, Y. et al. Differential requirement of RasGRP1 for gammadelta T cell development and activation. J. Immunol. 189, 61–71 (2012).
Gorentla, B. K., Wan, C. K. & Zhong, X. P. Negative regulation of mTOR activation by diacylglycerol kinases. Blood 117, 4022–4031 (2011).
Mor, A., Philips, M. R. & Pillinger, M. H. The role of Ras signaling in lupus T lymphocytes: biology and pathogenesis. Clin. Immunol. 125, 215–223 (2007).
Rincon, M., Flavell, R. A. & Davis, R. J. Signal transduction by MAP kinases in T lymphocytes. Oncogene 20, 2490–2497 (2001).
Rincon, M. & Davis, R. J. Regulation of the immune response by stress-activated protein kinases. Immunol. Rev. 228, 212–224 (2009).
Dodeller, F. et al. The p38 mitogen-activated protein kinase regulates effector functions of primary human CD4 T cells. Eur. J. Immunol. 35, 3631–3642 (2005).
Conze, D. et al. c-Jun NH(2)-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8(+) T cell activation. J. Exp. Med. 195, 811–823 (2002).
Dong, C. et al. Defective T cell differentiation in the absence of Jnk1. Science 282, 2092–2095 (1998).
Canovas, B. & Nebreda, A. R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 22, 346–366 (2021).
Bogoyevitch, M. A. et al. c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim. Biophys. Acta 1804, 463–475 (2010).
Chang, L. & Karin, M. Mammalian MAP kinase signalling cascades. Nature 410, 37–40 (2001).
Davis, R. J. Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 (2000).
Bellon, S. et al. The structure of phosphorylated p38gamma is monomeric and reveals a conserved activation-loop conformation. Structure 7, 1057–1065 (1999).
Salvador, J. M. et al. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat. Immunol. 6, 390–395 (2005).
Giardino Torchia, M. L. et al. Intensity and duration of TCR signaling is limited by p38 phosphorylation of ZAP-70(T293) and destabilization of the signalosome. Proc. Natl Acad. Sci. USA 115, 2174–2179 (2018).
Blonska, M. & Lin, X. CARMA1-mediated NF-kappaB and JNK activation in lymphocytes. Immunol. Rev. 228, 199–211 (2009).
Blonska, M. et al. The CARMA1-Bcl10 signaling complex selectively regulates JNK2 kinase in the T cell receptor-signaling pathway. Immunity 26, 55–66 (2007).
Sinclair, L. V. et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat. Immunol. 9, 513–521 (2008).
Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 12, 325–338 (2012).
Delgoffe, G. M. et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 12, 295–303 (2011).
Lee, K. et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32, 743–753 (2010).
Pollizzi, K. N. & Powell, J. D. Regulation of T cells by mTOR: the known knowns and the known unknowns. Trends Immunol. 36, 13–20 (2015).
Hamilton, K. S. et al. T cell receptor-dependent activation of mTOR signaling in T cells is mediated by Carma1 and MALT1, but not Bcl10. Sci. Signal 7, ra55 (2014).
Nakaya, M. et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40, 692–705 (2014).
Liu, C. et al. mTOR and metabolic regulation of conventional and regulatory T cells. J. Leukoc. Biol. 97, 837–847 (2015).
Procaccini, C. & Matarese, G. Regulatory T cells, mTOR kinase, and metabolic activity. Cell Mol. Life Sci. 69, 3975–3987 (2012).
Pierdominici, M., Vacirca, D., Delunardo, F. & Ortona, E. mTOR signaling and metabolic regulation of T cells: new potential therapeutic targets in autoimmune diseases. Curr. Pharm. Des. 17, 3888–3897 (2011).
Xia, F. et al. TCR and CD28 concomitant stimulation elicits a distinctive calcium response in naive T cells. Front. Immunol. 9, 2864 (2018).
Narayan, P., Holt, B., Tosti, R. & Kane, L. P. CARMA1 is required for Akt-mediated NF-kappaB activation in T cells. Mol. Cell Biol. 26, 2327–2336 (2006).
Park, S. G. et al. The kinase PDK1 integrates T cell antigen receptor and CD28 coreceptor signaling to induce NF-kappaB and activate T cells. Nat. Immunol. 10, 158–166 (2009).
Ishimaru, N., Kishimoto, H., Hayashi, Y. & Sprent, J. Regulation of naive T cell function by the NF-kappaB2 pathway. Nat. Immunol. 7, 763–772 (2006).
Turner, M. & Billadeau, D. D. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat. Rev. Immunol. 2, 476–486 (2002).
Hehner, S. P. et al. Tyrosine-phosphorylated Vav1 as a point of integration for T-cell receptor- and CD28-mediated activation of JNK, p38, and interleukin-2 transcription. J. Biol. Chem. 275, 18160–18171 (2000).
Singh, M. D. et al. B cell adaptor for PI3-kinase (BCAP) modulates CD8(+) effector and memory T cell differentiation. J. Exp. Med. 215, 2429–2443 (2018).
Zhang, W. et al. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92, 83–92 (1998).
Bubeck Wardenburg, J. et al. Phosphorylation of SLP-76 by the ZAP-70 protein-tyrosine kinase is required for T-cell receptor function. J. Biol. Chem. 271, 19641–19644 (1996).
Sommers, C. L., Samelson, L. E. & Love, P. E. LAT: a T lymphocyte adapter protein that couples the antigen receptor to downstream signaling pathways. Bioessays 26, 61–67 (2004).
Koretzky, G. A., Abtahian, F. & Silverman, M. A. SLP76 and SLP65: complex regulation of signalling in lymphocytes and beyond. Nat. Rev. Immunol. 6, 67–78 (2006).
Resh, M. D. Myristylation and palmitylation of Src family members: the fats of the matter. Cell 76, 411–413 (1994).
van’t Hof, W. & Resh, M. D. Rapid plasma membrane anchoring of newly synthesized p59fyn: selective requirement for NH2-terminal myristoylation and palmitoylation at cysteine-3. J. Cell Biol. 136, 1023–1035 (1997).
Kabouridis, P. S., Magee, A. I. & Ley, S. C. S-acylation of LCK protein tyrosine kinase is essential for its signalling function in T lymphocytes. EMBO J. 16, 4983–4998 (1997).
Xavier, R. et al. Membrane compartmentation is required for efficient T cell activation. Immunity 8, 723–732 (1998).
Montixi, C. et al. Engagement of T cell receptor triggers its recruitment to low-density detergent-insoluble membrane domains. EMBO J. 17, 5334–5348 (1998).
Duplay, P., Thome, M., Herve, F. & Acuto, O. p56lck interacts via its src homology 2 domain with the ZAP-70 kinase. J. Exp. Med. 179, 1163–1172 (1994).
Pelosi, M. et al. Tyrosine 319 in the interdomain B of ZAP-70 is a binding site for the Src homology 2 domain of Lck. J. Biol. Chem. 274, 14229–14237 (1999).
Ho, K. C. et al. CBAP promotes thymocyte negative selection by facilitating T-cell receptor proximal signaling. Cell Death Dis. 5, e1518 (2014).
Chiang, Y. J. et al. CBAP modulates Akt-dependent TSC2 phosphorylation to promote Rheb-mTORC1 signaling and growth of T-cell acute lymphoblastic leukemia. Oncogene 38, 1432–1447 (2019).
June, C. H., Fletcher, M. C., Ledbetter, J. A. & Samelson, L. E. Increases in tyrosine phosphorylation are detectable before phospholipase C activation after T cell receptor stimulation. J. Immunol. 144, 1591–1599 (1990).
Zhang, W., Trible, R. P. & Samelson, L. E. LAT palmitoylation: its essential role in membrane microdomain targeting and tyrosine phosphorylation during T cell activation. Immunity 9, 239–246 (1998).
Zhang, W. et al. Association of Grb2, Gads, and phospholipase C-gamma 1 with phosphorylated LAT tyrosine residues. Effect of LAT tyrosine mutations on T cell angigen receptor-mediated signaling. J. Biol. Chem. 275, 23355–23361 (2000).
Williams, B. L. et al. Phosphorylation of Tyr319 in ZAP-70 is required for T-cell antigen receptor-dependent phospholipase C-gamma1 and Ras activation. EMBO J. 18, 1832–1844 (1999).
Deckert, M. et al. Adaptor function for the Syk kinases-interacting protein 3BP2 in IL-2 gene activation. Immunity 9, 595–605 (1998).
Lindholm, C. K. et al. Requirement of the Src homology 2 domain protein Shb for T cell receptor-dependent activation of the interleukin-2 gene nuclear factor for activation of T cells element in Jurkat T cells. J. Biol. Chem. 274, 28050–28057 (1999).
Williams, B. L. et al. Genetic evidence for differential coupling of Syk family kinases to the T-cell receptor: reconstitution studies in a ZAP-70-deficient Jurkat T-cell line. Mol. Cell Biol. 18, 1388–1399 (1998).
Finco, T. S. et al. LAT is required for TCR-mediated activation of PLCgamma1 and the Ras pathway. Immunity 9, 617–626 (1998).
Zhang, W. et al. Functional analysis of LAT in TCR-mediated signaling pathways using a LAT-deficient Jurkat cell line. Int. Immunol. 11, 943–950 (1999).
Kazi, J. U. & Rönnstrand, L. FMS-like tyrosine kinase 3/FLT3: from basic science to clinical implications. Physiol. Rev. 99, 1433–1466 (2019).
Chougule, R. A. et al. Expression of GADS enhances FLT3-induced mitogenic signaling. Oncotarget 7, 14112–14124 (2016).
Sommers, C. L. et al. Mutation of the phospholipase C-gamma1-binding site of LAT affects both positive and negative thymocyte selection. J. Exp. Med. 201, 1125–1134 (2005).
Ravichandran, K. S., Lorenz, U., Shoelson, S. E. & Burakoff, S. J. Interaction of Shc with Grb2 regulates association of Grb2 with mSOS. Mol. Cell Biol. 15, 593–600 (1995).
Joazeiro, C. A. et al. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science 286, 309–312 (1999).
Liu, S. K., Fang, N., Koretzky, G. A. & McGlade, C. J. The hematopoietic-specific adaptor protein gads functions in T-cell signaling via interactions with the SLP-76 and LAT adaptors. Curr. Biol. 9, 67–75 (1999).
Liu, Z. G., Hsu, H., Goeddel, D. V. & Karin, M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 87, 565–576 (1996).
Yoder, J. et al. Requirement for the SLP-76 adaptor GADS in T cell development. Science 291, 1987–1991 (2001).
Clements, J. L. et al. SLP-76 expression is restricted to hemopoietic cells of monocyte, granulocyte, and T lymphocyte lineage and is regulated during T cell maturation and activation. J. Immunol. 161, 3880–3889 (1998).
Yablonski, D., Kadlecek, T. & Weiss, A. Identification of a phospholipase C-gamma1 (PLC-gamma1) SH3 domain-binding site in SLP-76 required for T-cell receptor-mediated activation of PLC-gamma1 and NFAT. Mol. Cell Biol. 21, 4208–4218 (2001).
Lin, J. & Weiss, A. Identification of the minimal tyrosine residues required for linker for activation of T cell function. J. Biol. Chem. 276, 29588–29595 (2001).
Kumar, L. et al. Differential role of SLP-76 domains in T cell development and function. Proc. Natl Acad. Sci. USA 99, 884–889 (2002).
Motto, D. G. et al. Implication of the GRB2-associated phosphoprotein SLP-76 in T cell receptor-mediated interleukin 2 production. J. Exp. Med. 183, 1937–1943 (1996).
Musci, M. A. et al. Three domains of SLP-76 are required for its optimal function in a T cell line. J. Immunol. 159, 1639–1647 (1997).
Pivniouk, V. et al. Impaired viability and profound block in thymocyte development in mice lacking the adaptor protein SLP-76. Cell 94, 229–238 (1998).
Clements, J. L. et al. Requirement for the leukocyte-specific adapter protein SLP-76 for normal T cell development. Science 281, 416–419 (1998).
Bubeck Wardenburg, J. et al. Regulation of PAK activation and the T cell cytoskeleton by the linker protein SLP-76. Immunity 9, 607–616 (1998).
Su, Y. W. et al. Interaction of SLP adaptors with the SH2 domain of Tec family kinases. Eur. J. Immunol. 29, 3702–3711 (1999).
Bunnell, S. C. et al. Biochemical interactions integrating Itk with the T cell receptor-initiated signaling cascade. J. Biol. Chem. 275, 2219–2230 (2000).
Griffiths, E. K. et al. Positive regulation of T cell activation and integrin adhesion by the adapter Fyb/Slap. Science 293, 2260–2263 (2001).
Peterson, E. J. et al. Coupling of the TCR to integrin activation by Slap-130/Fyb. Science 293, 2263–2265 (2001).
Zhang, W. et al. Essential role of LAT in T cell development. Immunity 10, 323–332 (1999).
Nishibe, S. et al. Increase of the catalytic activity of phospholipase C-gamma 1 by tyrosine phosphorylation. Science 250, 1253–1256 (1990).
Atherly, L. O. et al. The Tec family tyrosine kinases Itk and Rlk regulate the development of conventional CD8+ T cells. Immunity 25, 79–91 (2006).
Readinger, J. A. et al. Tec kinases regulate T-lymphocyte development and function: new insights into the roles of Itk and Rlk/Txk. Immunol. Rev. 228, 93–114 (2009).
Liu, K. Q., Bunnell, S. C., Gurniak, C. B. & Berg, L. J. T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. J. Exp. Med. 187, 1721–1727 (1998).
Schaeffer, E. M. et al. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science 284, 638–641 (1999).
Sommers, C. L. et al. A role for the Tec family tyrosine kinase Txk in T cell activation and thymocyte selection. J. Exp. Med. 190, 1427–1438 (1999).
Shan, X. & Wange, R. L. Itk/Emt/Tsk activation in response to CD3 cross-linking in Jurkat T cells requires ZAP-70 and Lat and is independent of membrane recruitment. J. Biol. Chem. 274, 29323–29330 (1999).
Sela, M. et al. Sequential phosphorylation of SLP-76 at tyrosine 173 is required for activation of T and mast cells. EMBO J. 30, 3160–3172 (2011).
Granum, S. et al. The kinase Itk and the adaptor TSAd change the specificity of the kinase Lck in T cells by promoting the phosphorylation of Tyr192. Sci. Signal. 7, ra118 (2014).
Bogin, Y., Ainey, C., Beach, D. & Yablonski, D. SLP-76 mediates and maintains activation of the Tec family kinase ITK via the T cell antigen receptor-induced association between SLP-76 and ITK. Proc. Natl Acad. Sci. USA 104, 6638–6643 (2007).
Sanzone, S. et al. SLAM-associated protein deficiency causes imbalanced early signal transduction and blocks downstream activation in T cells from X-linked lymphoproliferative disease patients. J. Biol. Chem. 278, 29593–29599 (2003).
Veillette, A. et al. Importance and mechanism of ‘switch’ function of SAP family adapters. Immunol. Rev. 232, 229–239 (2009).
Sayos, J. et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature 395, 462–469 (1998).
Shlapatska, L. M. et al. CD150 association with either the SH2-containing inositol phosphatase or the SH2-containing protein tyrosine phosphatase is regulated by the adaptor protein SH2D1A. J. Immunol. 166, 5480–5487 (2001).
Chen, S. & Dong, Z. NK cell recognition of hematopoietic cells by SLAM-SAP families. Cell Mol. Immunol. 16, 452–459 (2019).
Cannons, J. L. et al. Biochemical and genetic evidence for a SAP-PKC-theta interaction contributing to IL-4 regulation. J. Immunol. 185, 2819–2827 (2010).
Gu, C. et al. The X-linked lymphoproliferative disease gene product SAP associates with PAK-interacting exchange factor and participates in T cell activation. Proc. Natl Acad. Sci. USA 103, 14447–14452 (2006).
Latour, S. et al. Binding of SAP SH2 domain to FynT SH3 domain reveals a novel mechanism of receptor signalling in immune regulation. Nat. Cell Biol. 5, 149–154 (2003).
Li, C., Schibli, D. & Li, S. S. The XLP syndrome protein SAP interacts with SH3 proteins to regulate T cell signaling and proliferation. Cell Signal. 21, 111–119 (2009).
Proust, R., Bertoglio, J. & Gesbert, F. The adaptor protein SAP directly associates with CD3zeta chain and regulates T cell receptor signaling. PLoS ONE 7, e43200 (2012).
Canna, S. W. & Marsh, R. A. Pediatric hemophagocytic lymphohistiocytosis. Blood 135, 1332–1343 (2020).
Panchal, N., Booth, C., Cannons, J. L. & Schwartzberg, P. L. X-Linked lymphoproliferative disease type 1: a clinical and molecular perspective. Front. Immunol. 9, 666 (2018).
Snow, A. L. et al. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J. Clin. Invest. 119, 2976–2989 (2009).
Zheng, L., Li, J. & Lenardo, M. Restimulation-induced cell death: new medical and research perspectives. Immunol. Rev. 277, 44–60 (2017).
Snow, A. L. et al. The power and the promise of restimulation-induced cell death in human immune diseases. Immunol. Rev. 236, 68–82 (2010).
ElTanbouly, M. A. & Noelle, R. J. Rethinking peripheral T cell tolerance: checkpoints across a T cell’s journey. Nat. Rev. Immunol. 21, 257–267 (2021).
Brdicka, T. et al. Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J. Exp. Med. 191, 1591–1604 (2000).
Kawabuchi, M. et al. Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature 404, 999–1003 (2000).
Leo, A. et al. Adapters in lymphocyte signaling. J. Clin. Invest. 109, 301–309 (2002).
Takeuchi, S. et al. Transmembrane phosphoprotein Cbp positively regulates the activity of the carboxyl-terminal Src kinase, Csk. J. Biol. Chem. 275, 29183–29186 (2000).
Torgersen, K. M. et al. Release from tonic inhibition of T cell activation through transient displacement of C-terminal Src kinase (Csk) from lipid rafts. J. Biol. Chem. 276, 29313–29318 (2001).
Davidson, D. et al. Phosphorylation-dependent regulation of T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor. Mol. Cell Biol. 23, 2017–2028 (2003).
Salmond, R. J. et al. T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol. Rev. 228, 9–22 (2009).
Marie-Cardine, A. et al. SHP2-interacting transmembrane adaptor protein (SIT), a novel disulfide-linked dimer regulating human T cell activation. J. Exp. Med. 189, 1181–1194 (1999).
Hubener, C. et al. Complete sequence, genomic organization, and chromosomal localization of the human gene encoding the SHP2-interacting transmembrane adaptor protein (SIT). Immunogenetics 53, 337–341 (2001).
Pfrepper, K. I. et al. Structural and functional dissection of the cytoplasmic domain of the transmembrane adaptor protein SIT (SHP2-interacting transmembrane adaptor protein). Eur. J. Immunol. 31, 1825–1836 (2001).
Nagaishi, T. et al. SHP1 phosphatase-dependent T cell inhibition by CEACAM1 adhesion molecule isoforms. Immunity 25, 769–781 (2006).
Lorenz, U. S. H. P.-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol. Rev. 228, 342–359 (2009).
Stefanova, I. et al. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat. Immunol. 4, 248–254 (2003).
Caunt, C. J. et al. Spatiotemporal regulation of ERK2 by dual specificity phosphatases. J. Biol. Chem. 283, 26612–26623 (2008).
Stambolic, V. et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39 (1998).
Shulga, Y. V., Topham, M. K. & Epand, R. M. Regulation and functions of diacylglycerol kinases. Chem. Rev. 111, 6186–6208 (2011).
Sim, J. A., Kim, J. & Yang, D. Beyond lipid signaling: pleiotropic effects of diacylglycerol kinases in cellular signaling. Int. J. Mol. Sci. 21, 6861 (2020).
Zhong, X. P. et al. Regulation of T cell receptor-induced activation of the Ras-ERK pathway by diacylglycerol kinase zeta. J. Biol. Chem. 277, 31089–31098 (2002).
Carrasco, S. & Merida, I. Diacylglycerol-dependent binding recruits PKCtheta and RasGRP1 C1 domains to specific subcellular localizations in living T lymphocytes. Mol. Biol. Cell 15, 2932–2942 (2004).
Riese, M. J., Moon, E. K., Johnson, B. D. & Albelda, S. M. Diacylglycerol kinases (DGKs): novel targets for improving T cell activity in cancer. Front. Cell Dev. Biol. 4, 108 (2016).
Zhong, X. P. et al. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat. Immunol. 4, 882–890 (2003).
Zhong, X. P., Olenchock, B. A. & Koretzky, G. A. The role of diacylglycerol kinases in T cell anergy. Ernst Schering Found. Symp. Proc. 139–149 (2007).
Guo, R. et al. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase alpha and zeta. Proc. Natl Acad. Sci. USA 105, 11909–11914 (2008).
Baldanzi, G. et al. SAP-mediated inhibition of diacylglycerol kinase alpha regulates TCR-induced diacylglycerol signaling. J. Immunol. 187, 5941–5951 (2011).
Booth, C. et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood 117, 53–62 (2011).
Ruffo, E. et al. Inhibition of diacylglycerol kinase alpha restores restimulation-induced cell death and reduces immunopathology in XLP-1. Sci. Transl. Med 8, 321ra327 (2016).
Buetow, L. & Huang, D. T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 17, 626–642 (2016).
Hu, H. & Sun, S. C. Ubiquitin signaling in immune responses. Cell Res 26, 457–483 (2016).
Heissmeyer, V. et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat. Immunol. 5, 255–265 (2004).
Scharschmidt, E. et al. Degradation of Bcl10 induced by T-cell activation negatively regulates NF-kappa B signaling. Mol. Cell Biol. 24, 3860–3873 (2004).
Gao, M. et al. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 306, 271–275 (2004).
Liu, Y. C. The E3 ubiquitin ligase Itch in T cell activation, differentiation, and tolerance. Semin. Immunol. 19, 197–205 (2007).
Naramura, M. et al. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat. Immunol. 3, 1192–1199 (2002).
Wang, H. Y. et al. Cbl promotes ubiquitination of the T cell receptor zeta through an adaptor function of Zap-70. J. Biol. Chem. 276, 26004–26011 (2001).
Murphy, M. A. et al. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol. Cell Biol. 18, 4872–4882 (1998).
Fournel, M., Davidson, D., Weil, R. & Veillette, A. Association of tyrosine protein kinase Zap-70 with the protooncogene product p120c-cbl in T lymphocytes. J. Exp. Med. 183, 301–306 (1996).
Lupher, M. L. Jr. et al. A novel phosphotyrosine-binding domain in the N-terminal transforming region of Cbl interacts directly and selectively with ZAP-70 in T cells. J. Biol. Chem. 271, 24063–24068 (1996).
Ota, Y. et al. Characterization of Cbl tyrosine phosphorylation and a Cbl-Syk complex in RBL-2H3 cells. J. Exp. Med. 184, 1713–1723 (1996).
Bachmaier, K. et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature 403, 211–216 (2000).
Hartley, D. & Corvera, S. Formation of c-Cbl.phosphatidylinositol 3-kinase complexes on lymphocyte membranes by a p56lck-independent mechanism. J. Biol. Chem. 271, 21939–21943 (1996).
Knudsen, B. S., Feller, S. M. & Hanafusa, H. Four proline-rich sequences of the guanine-nucleotide exchange factor C3G bind with unique specificity to the first Src homology 3 domain of Crk. J. Biol. Chem. 269, 32781–32787 (1994).
Tanaka, S. et al. C3G, a guanine nucleotide-releasing protein expressed ubiquitously, binds to the Src homology 3 domains of CRK and GRB2/ASH proteins. Proc. Natl Acad. Sci. USA 91, 3443–3447 (1994).
Gotoh, T. et al. Identification of Rap1 as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol. Cell Biol. 15, 6746–6753 (1995).
Buday, L. et al. Interactions of Cbl with two adapter proteins, Grb2 and Crk, upon T cell activation. J. Biol. Chem. 271, 6159–6163 (1996).
Reedquist, K. A. et al. Stimulation through the T cell receptor induces Cbl association with Crk proteins and the guanine nucleotide exchange protein C3G. J. Biol. Chem. 271, 8435–8442 (1996).
Ichiba, T. et al. Enhancement of guanine-nucleotide exchange activity of C3G for Rap1 by the expression of Crk, CrkL, and Grb2. J. Biol. Chem. 272, 22215–22220 (1997).
Ohashi, Y. et al. T cell receptor-mediated tyrosine phosphorylation of Cas-L, a 105-kDa Crk-associated substrate-related protein, and its association of Crk and C3G. J. Biol. Chem. 273, 6446–6451 (1998).
Bos, J. L. Linking Rap to cell adhesion. Curr. Opin. Cell Biol. 17, 123–128 (2005).
Katagiri, K. et al. Rap1 is a potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol. Cell Biol. 20, 1956–1969 (2000).
Tanaka, S., Ouchi, T. & Hanafusa, H. Downstream of Crk adaptor signaling pathway: activation of Jun kinase by v-Crk through the guanine nucleotide exchange protein C3G. Proc. Natl Acad. Sci. USA 94, 2356–2361 (1997).
Shao, Y., Elly, C. & Liu, Y. C. Negative regulation of Rap1 activation by the Cbl E3 ubiquitin ligase. EMBO Rep. 4, 425–431 (2003).
Zhang, W. et al. Negative regulation of T cell antigen receptor-mediated Crk-L-C3G signaling and cell adhesion by Cbl-b. J. Biol. Chem. 278, 23978–23983 (2003).
Uemura, N. et al. Involvement of the adapter protein CRKL in integrin-mediated adhesion. Oncogene 18, 3343–3353 (1999).
van der Donk, L. E. H. et al. Separate signaling events control TCR downregulation and T cell activation in primary human T cells. Immun. Inflamm. Dis. 9, 223–238 (2020).
Dustin, M. L. & Cooper, J. A. The immunological synapse and the actin cytoskeleton: molecular hardware for T cell signaling. Nat. Immunol. 1, 23–29 (2000).
Monks, C. R. et al. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 395, 82–86 (1998).
Grakoui, A. et al. The immunological synapse: a molecular machine controlling T cell activation. Science 285, 221–227 (1999).
Dustin, M. L. & Chan, A. C. Signaling takes shape in the immune system. Cell 103, 283–294 (2000).
Johnson, K. G., Bromley, S. K., Dustin, M. L. & Thomas, M. L. A supramolecular basis for CD45 tyrosine phosphatase regulation in sustained T cell activation. Proc. Natl Acad. Sci. USA 97, 10138–10143 (2000).
Bunnell, S. C. et al. Dynamic actin polymerization drives T cell receptor-induced spreading: a role for the signal transduction adaptor LAT. Immunity 14, 315–329 (2001).
Tuosto, L., Michel, F. & Acuto, O. p95vav associates with tyrosine-phosphorylated SLP-76 in antigen-stimulated T cells. J. Exp. Med. 184, 1161–1166 (1996).
Wunderlich, L., Farago, A., Downward, J. & Buday, L. Association of Nck with tyrosine-phosphorylated SLP-76 in activated T lymphocytes. Eur. J. Immunol. 29, 1068–1075 (1999).
Wang, H. et al. ADAP-SLP-76 binding differentially regulates supramolecular activation cluster (SMAC) formation relative to T cell-APC conjugation. J. Exp. Med. 200, 1063–1074 (2004).
Rohatgi, R. et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97, 221–231 (1999).
Fischer, K. D. et al. Vav is a regulator of cytoskeletal reorganization mediated by the T-cell receptor. Curr. Biol. 8, 554–562 (1998).
Holsinger, L. J. et al. Defects in actin-cap formation in Vav-deficient mice implicate an actin requirement for lymphocyte signal transduction. Curr. Biol. 8, 563–572 (1998).
Wulfing, C., Bauch, A., Crabtree, G. R. & Davis, M. M. The vav exchange factor is an essential regulator in actin-dependent receptor translocation to the lymphocyte-antigen-presenting cell interface. Proc. Natl Acad. Sci. USA 97, 10150–10155 (2000).
O’Rourke, L. M. et al. CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity 8, 635–645 (1998).
Snapper, S. B. & Rosen, F. S. The Wiskott-Aldrich syndrome protein (WASP): roles in signaling and cytoskeletal organization. Annu. Rev. Immunol. 17, 905–929 (1999).
Snapper, S. B. et al. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity 9, 81–91 (1998).
Zhang, J. et al. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein-deficient lymphocytes. J. Exp. Med. 190, 1329–1342 (1999).
Higgs, H. N. & Pollard, T. D. Regulation of actin polymerization by Arp2/3 complex and WASp/Scar proteins. J. Biol. Chem. 274, 32531–32534 (1999).
Mullins, R. D. How WASP-family proteins and the Arp2/3 complex convert intracellular signals into cytoskeletal structures. Curr. Opin. Cell Biol. 12, 91–96 (2000).
Rivero-Lezcano, O. M., Marcilla, A., Sameshima, J. H. & Robbins, K. C. Wiskott-Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol. Cell Biol. 15, 5725–5731 (1995).
Zeng, R. et al. SLP-76 coordinates Nck-dependent Wiskott-Aldrich syndrome protein recruitment with Vav-1/Cdc42-dependent Wiskott-Aldrich syndrome protein activation at the T cell-APC contact site. J. Immunol. 171, 1360–1368 (2003).
Pauker, M. H. et al. Studying the dynamics of SLP-76, Nck, and Vav1 multimolecular complex formation in live human cells with triple-color FRET. Sci. Signal. 5, rs3 (2012).
Barda-Saad, M. et al. Cooperative interactions at the SLP-76 complex are critical for actin polymerization. EMBO J. 29, 2315–2328 (2010).
Hem, C. D. et al. T cell specific adaptor protein (TSAd) promotes interaction of Nck with Lck and SLP-76 in T cells. Cell Commun. Signal. 13, 31 (2015).
Buday, L., Wunderlich, L. & Tamas, P. The Nck family of adapter proteins: regulators of actin cytoskeleton. Cell Signal. 14, 723–731 (2002).
Lettau, M., Pieper, J. & Janssen, O. Nck adapter proteins: functional versatility in T cells. Cell Commun. Signal. 7, 1 (2009).
Berge, T. et al. T cell specific adapter protein (TSAd) interacts with Tec kinase ITK to promote CXCL12 induced migration of human and murine T cells. PLoS ONE 5, e9761 (2010).
Lettau, M. et al. The adapter protein Nck: role of individual SH3 and SH2 binding modules for protein interactions in T lymphocytes. Protein Sci. 19, 658–669 (2010).
Labelle-Cote, M. et al. Nck2 promotes human melanoma cell proliferation, migration and invasion in vitro and primary melanoma-derived tumor growth in vivo. BMC Cancer 11, 443 (2011).
da Silva, A. J. et al. Cloning of a novel T-cell protein FYB that binds FYN and SH2-domain-containing leukocyte protein 76 and modulates interleukin 2 production. Proc. Natl Acad. Sci. USA 94, 7493–7498 (1997).
Raab, M. et al. FYN-T-FYB-SLP-76 interactions define a T-cell receptor zeta/CD3-mediated tyrosine phosphorylation pathway that up-regulates interleukin 2 transcription in T-cells. J. Biol. Chem. 274, 21170–21179 (1999).
Geng, L., Raab, M. & Rudd, C. E. Cutting edge: SLP-76 cooperativity with FYB/FYN-T in the Up-regulation of TCR-driven IL-2 transcription requires SLP-76 binding to FYB at Tyr595 and Tyr651. J. Immunol. 163, 5753–5757 (1999).
Hunter, A. J. et al. Cutting edge: a novel function for the SLAP-130/FYB adapter protein in beta 1 integrin signaling and T lymphocyte migration. J. Immunol. 164, 1143–1147 (2000).
Wang, H. et al. SKAP-55 regulates integrin adhesion and formation of T cell-APC conjugates. Nat. Immunol. 4, 366–374 (2003).
Romero, S., Le Clainche, C. & Gautreau, A. M. Actin polymerization downstream of integrins: signaling pathways and mechanotransduction. Biochem. J. 477, 1–21 (2020).
Krause, M. et al. Fyn-binding protein (Fyb)/SLP-76-associated protein (SLAP), Ena/vasodilator-stimulated phosphoprotein (VASP) proteins and the Arp2/3 complex link T cell receptor (TCR) signaling to the actin cytoskeleton. J. Cell Biol. 149, 181–194 (2000).
Liu, J. et al. FYB (FYN binding protein) serves as a binding partner for lymphoid protein and FYN kinase substrate SKAP55 and a SKAP55-related protein in T cells. Proc. Natl Acad. Sci. USA 95, 8779–8784 (1998).
Marie-Cardine, A. et al. Molecular cloning of SKAP55, a novel protein that associates with the protein tyrosine kinase p59fyn in human T-lymphocytes. J. Biol. Chem. 272, 16077–16080 (1997).
Wu, L., Yu, Z. & Shen, S. H. SKAP55 recruits to lipid rafts and positively mediates the MAPK pathway upon T cell receptor activation. J. Biol. Chem. 277, 40420–40427 (2002).
Birge, R. B., Kalodimos, C., Inagaki, F. & Tanaka, S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun. Signal 7, 13 (2009).
Gelkop, S. et al. Involvement of crk adapter proteins in regulation of lymphoid cell functions. Immunol. Res. 28, 79–91 (2003).
Feller, S. M. Crk family adaptors-signalling complex formation and biological roles. Oncogene 20, 6348–6371 (2001).
Kumar, S., Fajardo, J. E., Birge, R. B. & Sriram, G. Crk at the quarter century mark: perspectives in signaling and cancer. J. Cell Biochem. 115, 819–825 (2014).
Liu, D. The adaptor protein Crk in immune response. Immunol. Cell Biol. 92, 80–89 (2014).
Bitar, M. et al. Evaluating STAT5 phosphorylation as a mean to assess T cell proliferation. Front. Immunol. 10, 722 (2019).
Jones, N. et al. Akt and STAT5 mediate naive human CD4+ T-cell early metabolic response to TCR stimulation. Nat. Commun. 10, 2042 (2019).
Braiman, A. & Isakov, N. The role of Crk adaptor proteins in T-cell adhesion and migration. Front. Immunol. 6, 509 (2015).
Kung, C. et al. Mutations in the tyrosine phosphatase CD45 gene in a child with severe combined immunodeficiency disease. Nat. Med. 6, 343–345 (2000).
Tchilian, E. Z. et al. A deletion in the gene encoding the CD45 antigen in a patient with SCID. J. Immunol. 166, 1308–1313 (2001).
Wilkinson, B., Downey, J. S. & Rudd, C. E. T-cell signalling and immune system disorders. Expert Rev. Mol. Med. 7, 1–29 (2005).
Fischer, A. Severe combined immunodeficiencies (SCID). Clin. Exp. Immunol. 122, 143–149 (2000).
Sawabe, T. et al. Defect of lck in a patient with common variable immunodeficiency. Int. J. Mol. Med. 7, 609–614 (2001).
Goldman, F. D. et al. Defective expression of p56lck in an infant with severe combined immunodeficiency. J. Clin. Invest. 102, 421–429 (1998).
Elder, M. E. et al. Human severe combined immunodeficiency due to a defect in ZAP-70, a T cell tyrosine kinase. Science 264, 1596–1599 (1994).
Arpaia, E. et al. Defective T cell receptor signaling and CD8+ thymic selection in humans lacking zap-70 kinase. Cell 76, 947–958 (1994).
Berg, L., Ronnelid, J., Klareskog, L. & Bucht, A. Down-regulation of the T cell receptor CD3 zeta chain in rheumatoid arthritis (RA) and its influence on T cell responsiveness. Clin. Exp. Immunol. 120, 174–182 (2000).
Takeuchi, T. et al. CD3 zeta defects in systemic lupus erythematosus. Ann. Rheum. Dis. 71(Suppl. 2), i78–i81 (2012).
Sakaguchi, N. et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature 426, 454–460 (2003).
Svojgr, K. et al. The adaptor protein NTAL enhances proximal signaling and potentiates corticosteroid-induced apoptosis in T-ALL. Exp. Hematol. 40, 379–385 (2012).
Svojgr, K. et al. Adaptor molecules expression in normal lymphopoiesis and in childhood leukemia. Immunol. Lett. 122, 185–192 (2009).
Lemonnier, F. & Mak, T. W. Activating TCR Signaling to Thwart T-ALL. Cancer Discov. 6, 946–948 (2016).
Wang, L. et al. Genomic profiling of Sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat. Genet. 47, 1426–1434 (2015).
Kataoka, K. et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 47, 1304–1315 (2015).
Van Vlierberghe, P. & Ferrando, A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Invest. 122, 3398–3406 (2012).
Trinquand, A. et al. Triggering the TCR developmental checkpoint activates a therapeutically targetable tumor suppressive pathway in T-cell leukemia. Cancer Discov. 6, 972–985 (2016).
Restifo, N. P., Dudley, M. E. & Rosenberg, S. A. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat. Rev. Immunol. 12, 269–281 (2012).
Southam, C. M., Brunschwig, A., Levin, A. G. & Dizon, Q. S. Effect of leukocytes on transplantability of human cancer. Cancer 19, 1743–1753 (1966).
Geukes Foppen, M. H., Donia, M., Svane, I. M. & Haanen, J. B. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol. Oncol. 9, 1918–1935 (2015).
Perica, K., Varela, J. C., Oelke, M. & Schneck, J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med. J. 6, e0004 (2015).
Axelrod, M. L., Cook, R. S., Johnson, D. B. & Balko, J. M. Biological consequences of MHC-II expression by tumor cells in cancer. Clin. Cancer Res. 25, 2392–2402 (2019).
Jena, B., Dotti, G. & Cooper, L. J. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood 116, 1035–1044 (2010).
Dembic, Z. et al. Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature 320, 232–238 (1986).
Gross, G., Waks, T. & Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl Acad. Sci. USA 86, 10024–10028 (1989).
Mazumder, A. & Rosenberg, S. A. Successful immunotherapy of natural killer-resistant established pulmonary melanoma metastases by the intravenous adoptive transfer of syngeneic lymphocytes activated in vitro by interleukin 2. J. Exp. Med. 159, 495–507 (1984).
Rosenberg, S. A., Spiess, P. & Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 233, 1318–1321 (1986).
Andersen, R. et al. T cells isolated from patients with checkpoint inhibitor-resistant melanoma are functional and can mediate tumor regression. Ann. Oncol. 29, 1575–1581 (2018).
Rosenberg, S. A. et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 17, 4550–4557 (2011).
Tran, E. et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390 (2015).
Tran, E. et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 375, 2255–2262 (2016).
Zacharakis, N. et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 24, 724–730 (2018).
Benmebarek, M. R. et al. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int. J. Mol. Sci. 20, 1283 (2019).
Morgan, R. A. et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314, 126–129 (2006).
Chodon, T. et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin. Cancer Res. 20, 2457–2465 (2014).
Johnson, L. A. et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114, 535–546 (2009).
Robbins, P. F. et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 29, 917–924 (2011).
Rapoport, A. P. et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 21, 914–921 (2015).
Maio, M. Melanoma as a model tumour for immuno-oncology. Ann. Oncol. 23, viii10–viii14 (2012). Suppl 8.
Smylie, M. G. Use of immuno-oncology in melanoma. Curr. Oncol. 27, S51–S58 (2020).
Brentjens, R. J. et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 9, 279–286 (2003).
Grupp, S. A. et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 368, 1509–1518 (2013).
Maude, S. L., Teachey, D. T., Porter, D. L. & Grupp, S. A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 125, 4017–4023 (2015).
Porter, D. L. et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 7, 303ra139 (2015).
Porter, D. L. et al. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 365, 725–733 (2011).
Neelapu, S. S. et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017).
Cappell, K. M. et al. Long-term follow-up of anti-cd19 chimeric antigen receptor T-cell therapy. J. Clin. Oncol. 38, 3805–3815 (2020).
Boyiadzis, M. M. et al. Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective and significance. J. Immunother. Cancer 6, 137 (2018).
Fry, T. J. et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 24, 20–28 (2018).
Cohen, A. D. et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Invest. 129, 2210–2221 (2019).
Sellner, L. et al. B-cell maturation antigen-specific chimeric antigen receptor T cells for multiple myeloma: clinical experience and future perspectives. Int. J. Cancer 147, 2029–2041 (2020).
Shah, N. et al. B-cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia 34, 985–1005 (2020).
Yu, Y. D. & Kim, T. J. Chimeric antigen receptor-engineered T cell therapy for the management of patients with metastatic prostate cancer: a comprehensive review. Int. J. Mol. Sci. 22, 640 (2021).
Wang, Y. et al. CD133-directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology 7, e1440169 (2018).
Faramand, R. et al. Tumor microenvironment composition and severe cytokine release syndrome (crs) influence toxicity in patients with large B-cell lymphoma treated with Axicabtagene Ciloleucel. Clin. Cancer Res. 26, 4823–4831 (2020).
Seimetz, D., Heller, K. & Richter, J. Approval of first CAR-Ts: have we solved all hurdles for ATMPs? Cell Med. 11, 2155179018822781 (2019).
Siegler, E. L. & Kenderian, S. S. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies. Front. Immunol. 11, 1973 (2020).
Tully, S. et al. Impact of increasing wait times on overall mortality of chimeric antigen receptor T-cell therapy in large B-cell lymphoma: a discrete event simulation model. JCO Clin. Cancer Inf. 3, 1–9 (2019).
Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res 38, 255 (2019).
Cohen, E. E. W. et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet 393, 156–167 (2019).
Hargadon, K. M., Johnson, C. E. & Williams, C. J. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 62, 29–39 (2018).
Herbst, R. S. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550 (2016).
Powles, T. et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet 391, 748–757 (2018).
Reck, M. et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 375, 1823–1833 (2016).
Robert, C. et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 372, 320–330 (2015).
Schmid, P. et al. Atezolizumab and Nab-Paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 379, 2108–2121 (2018).
Mahoney, K. M., Freeman, G. J. & McDermott, D. F. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin. Ther. 37, 764–782 (2015).
Makuku, R. et al. Current and future perspectives of PD-1/PDL-1 blockade in cancer immunotherapy. J. Immunol. Res. 2021, 6661406 (2021).
Zhao, B., Zhao, H. & Zhao, J. Efficacy of PD-1/PD-L1 blockade monotherapy in clinical trials. Ther. Adv. Med Oncol. 12, 1758835920937612 (2020).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Liu, R., Liao, Y. Z., Zhang, W. & Zhou, H. H. Relevance of immune infiltration and clinical outcomes in pancreatic ductal adenocarcinoma subtypes. Front. Oncol. 10, 575264 (2020).
Tumeh, P. C. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014).
Robert, C. et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 364, 2517–2526 (2011).
Motzer, R. J. et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 20, 1370–1385 (2019).
Wolchok, J. D. et al. Overall survival with combined Nivolumab and Ipilimumab in advanced melanoma. N. Engl. J. Med 377, 1345–1356 (2017).
Kunert, A. & Debets, R. Engineering T cells for adoptive therapy: outsmarting the tumor. Curr. Opin. Immunol. 51, 133–139 (2018).
Yang, Y. et al. Myeloid-derived suppressor cells in tumors: from mechanisms to antigen specificity and microenvironmental regulation. Front. Immunol. 11, 1371 (2020).
Paluskievicz, C. M. et al. T regulatory cells and priming the suppressive tumor microenvironment. Front. Immunol. 10, 2453 (2019).
Anderson, K. G., Stromnes, I. M. & Greenberg, P. D. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell 31, 311–325 (2017).
Zhuang, Y., Liu, C., Liu, J. & Li, G. Resistance mechanism of PD-1/PD-L1 blockade in the cancer-immunity cycle. Onco Targets Ther. 13, 83–94 (2020).
Staveley-O’Carroll, K. et al. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc. Natl Acad. Sci. USA 95, 1178–1183 (1998).
Hodi, F. S. et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc. Natl Acad. Sci. USA 105, 3005–3010 (2008).
Liakou, C. I. et al. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl Acad. Sci. USA 105, 14987–14992 (2008).
Romano, E. et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl Acad. Sci. USA 112, 6140–6145 (2015).
Tarhini, A. A. et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS ONE 9, e87705 (2014).
Sharma, A. et al. Anti-CTLA-4 immunotherapy does not deplete FOXP3(+) regulatory T cells (Tregs) in human cancers. Clin. Cancer Res. 25, 1233–1238 (2019).
Arce Vargas, F. et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. 33, 649–663 e644 (2018).
Huard, B. et al. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. Eur. J. Immunol. 24, 3216–3221 (1994).
Puhr, H. C. & Ilhan-Mutlu, A. New emerging targets in cancer immunotherapy: the role of LAG3. ESMO Open 4, e000482 (2019).
Kantarjian, H. et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 376, 836–847 (2017).
de Oliveira, C. E. et al. CCR5-dependent homing of T regulatory cells to the tumor microenvironment contributes to skin squamous cell carcinoma development. Mol. Cancer Ther. 16, 2871–2880 (2017).
Pylayeva-Gupta, Y. et al. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21, 836–847 (2012).
Chapman, N. M., Connolly, S. F., Reinl, E. L. & Houtman, J. C. Focal adhesion kinase negatively regulates Lck function downstream of the T cell antigen receptor. J. Immunol. 191, 6208–6221 (2013).
Zaretsky, J. M. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016).
Hollingsworth, R. E. & Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 4, 7 (2019).
Kinkead, H. L. et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 3, (2018).
Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017).
Zhu, G. et al. Albumin/vaccine nanocomplexes that assemble in vivo for combination cancer immunotherapy. Nat. Commun. 8, 1954 (2017).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Keskin, D. B. et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019).
Kandalaft, L. E. et al. Autologous lysate-pulsed dendritic cell vaccination followed by adoptive transfer of vaccine-primed ex vivo co-stimulated T cells in recurrent ovarian cancer. Oncoimmunology 2, e22664 (2013).
Ribas, A. et al. Dendritic cell vaccination combined with CTLA4 blockade in patients with metastatic melanoma. Clin. Cancer Res 15, 6267–6276 (2009).
Lee, K. L., Schlom, J. & Hamilton, D. H. Combination therapies utilizing neoepitope-targeted vaccines. Cancer Immunol. Immunother. 70, 875–885 (2021).
Acknowledgements
This research was supported by the Crafoord Foundation, the Swedish Cancer Society, and the Swedish Childhood Cancer Foundation.
Funding
Open access funding provided by Lund University.
Author information
Authors and Affiliations
Contributions
K.S., A.A.-H., J.S., and J.U.K. reviewed the literature and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shah, K., Al-Haidari, A., Sun, J. et al. T cell receptor (TCR) signaling in health and disease. Sig Transduct Target Ther 6, 412 (2021). https://doi.org/10.1038/s41392-021-00823-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00823-w
This article is cited by
-
Association of TRAIL receptor with phosphatase SHP-1 enables repressing T cell receptor signaling and T cell activation through inactivating Lck
Journal of Biomedical Science (2024)
-
Integrative temporal multi-omics reveals uncoupling of transcriptome and proteome during human T cell activation
npj Systems Biology and Applications (2024)
-
The phosphatase DUSP22 inhibits UBR2-mediated K63-ubiquitination and activation of Lck downstream of TCR signalling
Nature Communications (2024)
-
Dietary factors and their influence on immunotherapy strategies in oncology: a comprehensive review
Cell Death & Disease (2024)
-
Role of Sam68 as an adaptor protein in inflammatory signaling
Cellular and Molecular Life Sciences (2024)
