
Abstract
In recent years, accumulating evidence has elucidated the role of lysosomes in dynamically regulating cellular and organismal homeostasis. Lysosomal changes and dysfunction have been correlated with the development of numerous diseases. In this review, we interpreted the key biological functions of lysosomes in four areas: cellular metabolism, cell proliferation and differentiation, immunity, and cell death. More importantly, we actively sought to determine the characteristic changes and dysfunction of lysosomes in cells affected by these diseases, the causes of these changes and dysfunction, and their significance to the development and treatment of human disease. Furthermore, we outlined currently available targeting strategies: (1) targeting lysosomal acidification; (2) targeting lysosomal cathepsins; (3) targeting lysosomal membrane permeability and integrity; (4) targeting lysosomal calcium signaling; (5) targeting mTOR signaling; and (6) emerging potential targeting strategies. Moreover, we systematically summarized the corresponding drugs and their application in clinical trials. By integrating basic research with clinical findings, we discussed the current opportunities and challenges of targeting lysosomes in human disease.
Similar content being viewed by others
Introduction
Since Christian de Duve discovered and named lysosomes in 1955, great progress has been made in understanding the structure and function of lysosomes and how they can be harnessed to improve clinical outcomes (Fig. 1).1,2,3,4,5 The richness in hydrolytic enzymes is an obvious feature that distinguishes lysosomes from other organelles.2,6,7 More than 60 acid hydrolases, which break down cell components and complex macromolecules into their constituent building blocks, have been identified in lysosomes.2,6,7 Therefore, lysosomes have long been regarded as “suicide bags” and the “garbage-disposal system” for cells.2,6 Nevertheless, in recent years, researchers have gained a better understanding of lysosomes by combining genomics, transcriptomics, proteomics, bioinformatics, and other methods and found that their functions are far more than digestion.7 Lysosomes are now regarded as regulators of cell and organismal homeostasis that mediate signal transduction, metabolic adaptation, cell proliferation, cell differentiation, cell secretion, and the quality control of proteins and organelles.2,6,8

Important events in the development of research into lysosomes as therapeutic targets. Since Christian de Duve discovered and named lysosomes in 1955, scientists have made significant contributions to reveal the structural characteristics and drugs of lysosomes and to connect lysosomes with important pathways such as autophagy, endocytosis, mTOR, and cell death, laying a foundation for the later use of lysosomes as therapeutic targets. Christian de Duve and Yoshinori Ohsumi won the Nobel Prize in 1974 and 2016, respectively, for their contributions to the discovery of lysosomes and the elucidation of autophagy mechanisms. CQ, chloroquine; LSDs, lysosomal storage disorders; CLEAR, coordinated lysosomal expression and regulation; NCCD, Nomenclature Committee on Cell Death
Lysosomal changes and dysfunction are have profound implications for the development of numerous human diseases.9,10 The prevalence of neurodegenerative and cardiovascular diseases in the elderly was thought to be closely related to the decline in lysosomal function with age.9,11 In contrast, cancer cells upregulate their metabolism by modulating lysosomal quantity, composition, and activity to meet their needs for cell growth and proliferation.2,8 Besides, the translocation and abnormal secretion of lysosomes are conducive to the invasion and metastasis of cancer cells, and upregulated autophagy is considered a vital means by which cancer cells develop resistance to chemotherapy and radiotherapy.5,10,12,13 Growing attention has been paid to the role of lysosomes in immunity.14 The abnormal degradation of major histocompatibility complex (MHC) molecules and immune checkpoints by lysosomes in cancer cells, as well as the defects in selective autophagy of tumor-infiltrating T lymphocytes, together contribute to tumor immune escape.15,16,17 In the cells of patients with autoimmune diseases, changes in lysosomal biogenesis, acidification and cathepsin activity have also been confirmed, and such changes are thought to be closely related to disease activity and progression.18,19,20,21,22,23
These lysosomal changes and dysfunction play a crucial role in the development of diseases, but they may also provide a therapeutic window for treatment.9,10,14 For example, increases in lysosomal size and capacity facilitate cell metabolism but also reduce the stability of lysosomal membranes, making cells more vulnerable to death.2,24 In addition to correcting these changes and abnormalities, we may also be able to use them to combat pathological cells.5,25,26 In this review, we summarized and discussed recent studies that clarified and supported the idea of targeting lysosomes in human disease and further explored the feasibility, opportunities, and challenges of such efforts by combining basic research with clinical research progress.
Lysosomal structure, biogenesis, and function
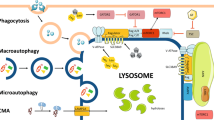
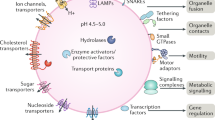
The lysosome is a membrane-enclosed vesicular organelle that contains two classes of proteins essential for the maintenance of structure and function: soluble lysosomal hydrolases that performs digestive functions and lysosomal membrane proteins with more complex functions, such as acting as proton pump and promoting intercellular interaction (Fig. 2).2,6,8 More than 60 acid hydrolases, including proteases, lipases, nucleases, have been found within the lysosome, and they require an acidic (pH ~4.5) environment maintained by the cooperation of an ATP-driven proton pump called the vacuolar H + -adenosine triphosphatase (v-ATPase) with other ion channels.7,27 A variety of lysosomal membrane proteins of mammalian cell have been identified, such as lysosome-associated membrane protein 1(LAMP1), LAMP2, lysosome integral membrane protein 2 (LIMP2; also known as SCARB2), v-ATPase, and acid sphingomyelinase (ASM).6,14,28 Of these proteins, LAMP1 and LAMP2 are the most abundant (accounting for 50% of all) lysosomal membrane proteins and are essential for metabolism, biogenesis, signal transduction, and cell homeostasis.2,6,29

Lysosomal structure and function. The lysosome is an acidic membrane-enclosed vesicular organelle containing a variety of hydrolases, and its activity and function are maintained by the channels or pump structures on its surface, such as v-ATPase, iron channels, and nutrient transporters. The lysosome acts not only as the endpoint of multiple trafficking routes, including autophagy, endocytosis, and phagocytosis, but also, as the platform for the recruitment and activation of mTOR, which regulates cell metabolism, growth, and differentiation. Ca2+ released from lysosomal calcium channels such as TRPML also regulates endocytic membrane trafficking, the nuclear transduction of TFEB, and the fusion of lysosomes with other cellular structures, such as autophagosomes and endosomes. However, the leakage of lysosomal contents such as cathepsins, ROS, Fe2+/3+, and Ca2+ contributes to multiple forms of cell death. Exogenous antigens are processed into peptides by lysosomal proteases, and the lysosome also acts as a bilateral switch that mediate both pro-inflammatory and anti-inflammatory processes. The central location of the lysosome in the communication and convergence of multiple pathways determines its pivotal and irreplaceable role in cell metabolism, proliferation, differentiation, immunity, and death. Black arrows indicate positive regulation or metabolite flux, while red arrows indicate negative regulation. mTORC1, mammalian target of rapamycin complex 1; mTORC2, mammalian target of rapamycin complex 2; Arg, arginine; TSC2, tuberous sclerosis complex; AMPK, AMP-activated protein kinase; TFEB, transcription factor EB; RTK, receptor tyrosine kinases; PP2A, protein phosphatase 2 A; TRPML1, transient receptor potential mucolipin 1; v-ATPase, vacuolar H + -adenosine triphosphatase; HSC70, heat shock cognate protein 70; LAMP2A, lysosome-associated membrane protein 2; LMP, lysosomal membrane permeability; MOMP, mitochondrial outer membrane permeabilization; CMA, chaperone-mediated autophagy; TGN, trans-Golgi network; CLEAR, coordinated lysosomal expression and regulation; ROS, reactive oxygen species; RAG, RAS-related GTP-binding protein; Unc-51-like kinase 1 (ULK1); PUMA, p53 upregulated modulator of apoptosis; BID, BH3 interacting domain death agonist; BAX, BCL2-associated X, apoptosis regulator; LDCD, lysosome-dependent cell death
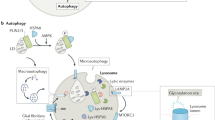
Lysosomal biogenesis is a combination of cellular biosynthesis and endocytosis pathways (Fig. 3).6,28 The expression of lysosomal genes is triggered by the binding of transcription factors (TFs) of microphthalmia/transcription factor E (MiT/TFE) family to the coordinated lysosomal expression and regulation (CLEAR) elements.2,30,31 Among these TFs, transcription factor EB (TFEB) is the first and most thoroughly studied TF known to directly bind to the CLEAR element.2,30,31 Transient receptor potential mucolipin channel (TRPML1), calcineurin, protein phosphatase 2A (PP2A), and mammalian target of rapamycin complex 1 (mTORC1) jointly regulate the activation and nuclear translocation of TFEB by modulating its phosphorylation status.2,8,32,33,34 After being synthesized in endoplasmic reticulum (ER), lysosomal proteins are transported to trans-Golgi network (TGN) and then be secreted to plasma membrane for subsequent endocytosis, or transmitted directly to the endo-lysosomal system.28 Sorting events in endo-lysosomal system eventually cause these compartments to be rich in lysosomal membrane proteins and lysosomal hydrolases, which constitute the major components of lysosomes.8,28

The biogenesis of lysosomes. Lysosomal biogenesis is a combination of cellular biosynthesis and endocytosis pathways. TRPML1 channel, calcineurin, PP2A, and mTORC1 jointly regulate the biosynthesis of lysosomal proteins by modulating the activation and nuclear translocation of TFEB. PP2A, protein phosphatase 2A; mTORC1, mammalian target of rapamycin complex 1; TGN, trans-Golgi network; TFEB, transcription factor EB; CLEAR, coordinated lysosomal expression and regulation
Lysosomes in cellular metabolism
Lysosomes are responsible for breaking down and recycling intracellular materials (through autophagy) and extracellular materials (through endocytosis and phagocytosis), which are then used to generate new cellular components and nutrients to meet the needs of cell metabolism and growth (Fig. 2).2,5,35,36
Lysosomes serve as the platforms for proper recruitment, assembly, and activation of mammalian target of rapamycin (mTOR) complex 1 (mTORC1), the mediator that coordinates the balance between anabolism and catabolism (Fig. 2).37,38 When nutrients are abundant, the stimulation of amino acids such as arginine and glutamine induce the activation of RAS-related GTP-binding proteins (RAGs), which interact with Ragulator and then trigger the recruitment of mTORC1 to lysosomal surface.4,37,39 Through the PI3K-AKT pathway, growth factors such as insulin activate Rheb, which binds to mTORC1 on the lysosomal surface and results in its activation.37,40,41 In return, mTORC1 inhibits lysosomal biogenesis though phosphorylating TFEB at Ser211 and inhibiting its nuclear transudtion42,43,44 and inhibits autophagy initiation by phosphorylating Unc-51-like kinase 1 (ULK1) at Ser757.45,46 The activated mTORC1 signaling also inhibits the lysosomal catabolism of extracellular proteins taken up through the macropinocytosis pathway, a nonselective form of endocytosis.46,47
When cells are deficient in nutrients, the inactivation of mTOR signaling and the formulation of the AMP-activated protein kinase (AMPK) complex contribute the upregulation of catabolism pathways.45 The deficiency of glucose sensed by aldolase promotes the interaction of axis inhibition protein 1 (AXIN)-live kinase B1 (LKB1) complex with v-ATPase and Ragulator, which then activates AMPK signaling (Fig. 2).48,49,50 Concurrently, AXIN cause the dissociation and inactivation of mTORC1 by interfering with the interaction between RAGs and the Ragulator, and the inactivation of mTORC1 restores lysosome biogenesis and autophagy.50 The activated AMPK not only promotes autophagy by phosphorylating the Ser317 and Ser777 of ULK1 but also inhibits mTOR pathway through phosphorylating the Raptor of mTORC1 and activating tuberous sclerosis complex (TSC2), which inhibits Rheb.45,51,52 As a scavenging pathway of extracellular proteins, the macropinocytosis pathway was also identified to be upregulated when inhibiting mTORC1.47,53 The amino acids obtained through catabolism pathways are then transported out of lysosomes by the lysosomal transmembrane protein called SLC38A9 in an arginine-regulated manner.54,55 By interacting with RAGs and Ragulator, SLC38A9 acts as an amino sensor essential for the activation the mTORC1 pathway.54,56 The increase of nutrients can lead to the reactivation of mTORC1, thereby inhibiting the catabolic pathway and promoting substance synthesis and cell growth and proliferation.47,54,56 When nutrients are scarce, the mTOR signaling is suppressed again.4,37,39 Therefore, mTOR signaling coordinates the activity of cell anabolism and catabolism and keeps them in a dynamic balance to meet the needs of cell growth and proliferation.46
Taken together, these findings indicate that lysosomes not only play a great role in cellular catabolism, which supplies nutrients for cell growth but also function as a platform for nutrient sensing and metabolic signal transduction (Fig. 2).
Lysosomes in cell proliferation and differentiation
In addition to mediating the adaptation of cell metabolism to meet the needs for cell growth and proliferation, lysosomes also mediate the turnover of cell surface receptors and other elements crucial to proliferation and differentiation signaling.2,27,57 Take epidermal growth factor receptor (EGFR), one of the most well-studied receptor tyrosine kinases (RTKs), as an example.27 After binding with ligands and activating downstream pathways, the clathrin adaptor protein complex AP2, growth factor receptor-bound protein 2 (Grb2), epsin, and eps15 together contribute to the endocytosis of EGFR, which then is and sorted for recycling or degradation.58 Suppressor of T-cell receptor (TCR) signaling (Sts)-1 and Sts-2 have been found to inhibit the endocytosis of activated EGFR through interacting with ubiquitin ligase Cbl.59 Besides, autophagy was reported to facilitate the recycling of EGFR by regulating early endosome homeostasis.60 Cells lacking ATG7 or ATG16L1, pivotal autophagy genes, were found to have an accumulation of early endosomal antigen-1 (EEA1)-positive endosomes resulting in the stalled trafficking of EGFR.60 Recently, Weber et al.61 used unbiased genetic screens and found that lysosomal acidity influenced cell proliferation by maintaining iron homeostasis. While mTORC1 facilitates cell growth and proliferation through promoting cell anabolism, mTORC2 mediates proliferation more directly by phosphorylating and activating the members the AGC (PKA/PKG/PKC) protein kinases, such as AKT, PKCα, PKCδ, PKCγ, and PKCε.38,62 It is worth noting that both mTORC2 and AKT signaling were found to be influenced by lysosome positioning.63
It has been proved that mTOR signaling (both mTORC1 and mTORC2) plays a pivotal role in the regulation of differentiation and function of numerous immune cells, such as T lymphocytes, macrophages, dendritic cells (DCs), and adipocytes.64,65,66,67,68,69,70 Mtor−/−T cells have been shown to be unable to differentiate into Th1, Th2 and Th17 cells, despite having normal activation markers and levels of IL-2 production under T-cell receptor (TCR) stimulation.71 Although mTOR signaling facilitates cell growth and proliferation, sustained activation of mTOR signaling has been found to result to the terminal differentiation and reduced proliferative capacity of T cells.72,73,74,75 Besides, auophagy plays different roles in cell differentiation, which may be correlated with the difference of stimulation and the stage of cell differentiation.69,76,77 While the differentiation of peripheral blood monocytes into macrophages induced by colony stimulating factor 1(CSF-1) was identified highly dependent on autophagy, Zhang et al.69 found that the inhibition of autophagy promoted the macrophagic differentiation of myeloid hematopoietic progenitor cells.76,77
Taken together, these findings suggest that the lysosome is a crucial regulatory hub for multiple pathways involved in cell proliferation and differentiation (Fig. 2).
Lysosomes in immunity
It is well-established that exogenous antigens are primarily processed into peptides by lysosomal proteases and loaded onto MHC-II molecules for recognition.14,78 In recent years, the high expression of MHC-II molecules has also been found to be correlated with a favorable prognosis of cancer patients.79 While autophagy facilitates the loading of MHC class II molecules by delivering cytoplasmic components to lysosomes, it also interferes with antigen presentation by MHC-I molecules through competing ubiquitinated proteins with proteasomes and degrading proteasomes.80,81,82,83 Expansion of the volume and protein levels of lysosomes and endosomes in phagocytes was found to promote antigen presentation in the immune response, and lysosomal recruitment and secretion was identified to facilitate the antigen extraction and the full activation of B lymphocytes.84,85,86 In addition to their role in antigen processing and presentation, lysosomes are responsible for the degradation and membrane presentation of immune checkpoints such as programmed cell death-ligand 1 (PD-L1),16,87 cytotoxic T-lymphocyte-associated protein 4 (CTLA-4),87 and CD70.10,88 Abnormalities in the degradation and presentation of immune checkpoints are closely associated with the progression and treatment failure of many diseases, especially cancer.16,87,89 After antigen stimulation, mTOR signaling is activated and then programs the differentiation of immune cells into functionally distinct lineages, such as Th1, Th2, Th17, Treg, cytotoxic CD8 + T cells, and memory CD8 + T cells.64,70,90
Lysosomes act as bilateral switches that regulate inflammatory process, an important part of the immune response that has both protective and disease-driving roles.14,30,91,92,93 On the one hand, lysosomes mediate the release of pro-inflammatory cytokines from immune cells, and the release of lysosomal hydrolase are of great benefit to the initiation and development of inflammation.30,92 Lysosomes have also been found to promote inflammation by degrading glucocorticoid receptors, which bind with glucocorticoid to modulate the expression of and inflammatory and pro-inflammatory factors.94,95 On the other hand, lysosomes mediate the release of anti-inflammatory cytokines and are responsible for breaking down inflammatory cytokines and elements such as PYCARD/ASC, a critical component of inflammasome.2,30,96
Taken together, these findings show that lysosomes are closely involved in the immune response and its strength modulation, and serve as the two-way switches that regulate inflammation (Fig. 2).
Lysosomes in cell death
Lysosomes mediate cell death at several levels. Under some unfavorable conditions, autophagy is activated to avoid cell death by degrading death-promoting factors such as (p53 upregulated modulator of apoptosis) PUMA and receptor-interacting protein kinases-1 (RIPK1) and promoting autophagy-dependent mitochondrial homeostasis.5,97,98 When exposed to extreme stress, lysosomal membrane permeability (LMP) occurs, and the leakage of cathepsins, reactive oxygen species (ROS), and Fe2+/3+ triggers multiple forms of cell death, such as apoptosis, necrosis, pyroptosis, ferroptosis, and lysosome-dependent cell death (LDCD) (Fig. 2).2,24,26,99 For instance, while cathepsins leaked from lysosomes can promote apoptosis through activating BID proteins or BAX channels, robust lysosomal cathepsin activities lead to cell necrosis by rapidly degrading essential cell components.100,101 Different from chaotic and drastic degradation of cell components that occurs in necrosis, LDCD was defined by the Nomenclature Committee on Cell Death (NCCD) as the regulated cell death demarcated by primary LMP and precipitated by cathepsins with or without the involvement of caspases and mitochondrial outer membrane permeabilization (MOMP).102
Although autophagy acts as a cytoprotective process most of the time, the pathway and its key components also participate lethal signaling.26,98,101,103 For example, it has been acknowledged that selective autophagy can promote ferroptosis through degrading ferritin and intracellular lipid droplets, causing iron accumulation and lipid peroxidation.103,104,105 Although much remains unclear, it has been confirmed that lysosomes play a crucial role in resisting and triggering cell death and the terminal clearance stage of cell death (Fig. 2).
Characteristic changes and dysfunction of lysosomes in human disease
Given the important roles of lysosomes in cell metabolism, cell proliferation and differentiation, immunity, and cell death, any lysosomal change or dysfunction may disrupt original cell and organismal homeostasis, causing or deteriorating human disease. As early as the 1960s, H.G. Hers discovered the relationship between the deficiency in lysosomal α‑glucosidase and Pompe disease and first proposed the concept of inborn lysosomal disease, the prototype of lysosomal storage disorders (LSDs) (Fig. 1).106,107 LSD are a group of rare metabolic disorders caused by inherited defects in genes that encode proteins involved in lysosomal homeostasis, such as lysosomal hydrolases or membrane proteins.108,109 In addition to LSDs, the initiation and development of numerous diseases, such as cancer, autoimmune disorders, neurodegenerative diseases, and cardiovascular diseases, have also been identified to have a close correlation with lysosomal changes and dysfunction.110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129 In this section, we started with several diseases and studied their lysosomal changes and dysfunction to lay the foundation for the selection of targeted strategies.
Cancer
Cancer cells always upregulate their metabolism by changing the quantity, localization, and activity of lysosomes to meet their needs for cell growth and proliferation.2,8 These changes have been correlated with the overexpression of some lysosomal proteins and lysosome-related proteins, such as lysosome catalase, lysosomal glycosidase, and kinesins.10,130,131,132 The abnormal activation of classical oncogenes, such as Kras and MYC, was found to increase the expression of lysosome catalase and glycosidase.10 Furthermore, several types of cancer, such as pancreatic adenocarcinoma,110,111 renal-cell carcinoma,110 melanoma,110,112 and breast cancer,113 have been found to have an overexpression of MIT/TFE genes, the TFs that facilitate the expression of lysosomal proteins.2
These lysosomal changes have profound effects on the proliferation, migration, and invasion of cancer cells, as well as their resistance to radiotherapy and chemotherapy.2,8,27 Upregulated nutrients-scavenging pathways such as autophagy and endocytosis allow cancer cells to compete for available nutrients and survive in unfavorable conditions, such as tumors with poor vascularization or undergoing radiotherapy or chemotherapy.8 Nutrients brought by upregulated nutrient-scavenging pathways activate mTOR signaling and promote cell synthesis of amino acids, glucose, nucleotides, fatty acids, and lipids, which are essential for cell proliferation.8,37,133 Aberrant hyperactivation of both catabolic and anabolic pathways facilitates the metabolism and proliferation of cancer cells.2 MTORC1 signaling and TFEB modulation constitute a feedback loop that coordinates the balance between lysosomal catabolism and anabolism to adapt to different metabolic conditions.2 In addition, lysosomes have also been found to contribute to the chemoresistance of cancer cells by sequestering drugs to prevent their action outside lysosomes.12 Besides, the upregulated autophagy pathway favors the invasion and metastasis of cancer cells through degrading epithelial-derived molecules such as E-cadherin.5,10 Furthermore, the redistribution of lysosomes to the periphery of cancer cells and their exocytosis of cathepsins, heparinase, and Neu1 also benefit cancer invasion, metastasis, and angiogenesis by affecting cell morphology and degrading their extracellular matrix and basement membrane.2,27
Lysosomal changes and dysfunction also play an important role in the escape of cancer cells from the host immune system. Lysosomal degradation is not only responsible for antigen processing but also controls the presentation of MHC-I at cell membrane.134,135,136 It has been reported that lysosomal degradation of MHC-I through autophagy-dependent pathways accounts for the decreased expression of MHC-I on the cell surface of pancreatic ductal adenocarcinoma (PDAC).135 The co-location of MHC-I with autophagosomes and lysosomes was observed in PDAC cells, and autophagy inhibition was identified to restore MHC-I levels and promote T-cell responses in mouse models.135 Besides, the deficiency or blockade of costimulatory molecules of tumor cells is one of the important mechanisms of tumor immune escape, and lysosomes are responsible for not only the degradation but also the membrane transportation and presentation of immune checkpoints such as CTLA-4, PD-L1, and CD47.10,27,89 The colocalization of CKLF-like MARVEL transmembrane domain-containing 6 (CMTM6) with PD-L1 in cell membrane and recycling endosomes was found to inhibit the lysosomal degradation of PD-L1, which interacts with PD-1 on T cells to evade T-cell-mediated immunosurveillance.16 Recently, the decreased activity of mitophagy, a type of selective autophagy, has been reported to lead to the accumulation of depolarized mitochondria in tumor-infiltrating T lymphocytes (TILs).17 The persistent metabolic insufficiency caused by defective mitophagy was thought to cause TLR exhaustion.17 These factors together contribute to the low immune response in tumors.
However, the changes mentioned above not only benefit cancer development but also lead to the reduced stability of lysosomal membranes and make lysosomes in cancer cells more susceptible to LMP, which may provide the therapeutic windows we seek.2,24,26,137 Considering the great role of these changes and dysfunctions in lysosomes in cancer cells, it is feasible to develop strategies targeting lysosomes to treat cancer (Fig. 4).

Available strategies for targeting lysosomes in human disease and their corresponding drugs. Different colors indicate different targeting strategies: red, targeting lysosomal acidification; yellow, targeting lysosomal cathepsins; blue, targeting lysosomal membrane permeability and integrity; green, targeting lysosomal calcium signaling; purple, targeting mTOR signaling; gray, emerging targeting strategies with great potential. The action mechanisms of these drugs are highlighted in bold. mTORC1, mammalian target of rapamycin complex 1; mTORC2, mammalian target of rapamycin complex 2; TRPML1, transient receptor potential mucolipin 1; LMP, lysosomal membrane permeability; ROS, reactive oxygen species; CMA, chaperone-mediated autophagy; CLEAR, coordinated lysosomal expression and regulation; TFEB, transcription factor EB; CQ, chloroquine; HCQ, hydroxychloroquine; QN, quinacrine; PLGA-aNP, poly(DL-lactide-co-glycolide) acidic nanoparticles; AAV, adeno-associated virus; rhCTSD, recombinant human pro-cathepsin D; rhPPCA, recombinant human protective protein/cathepsin A; ASM, acid sphingomyelinase; ZA, zoledronic acid; rhCTSD, recombinant human pro-Cathepsin D; DpdtC, Di-2-pyridylketone dithiocarbamate; Hsp70, heat shock protein 70; HspBP1, Hsp70 binding protein 1; 3,4-DC, 3,4-dimethoxychalcone; PI(3,5)P2, phosphatidyl-(3,5)-bisphosphate; MITF, melanogenesis-associated transcription factor; HPβCD, 2-Hydroxypropyl-β-cyclodextrin; PA, psoromic acid; 3-PEHPC, 3-(3-pyridyl)-2-hydroxy-2-phosphonopropanoic acid; RabGGTase, Rab geranylgeranyl transferase
Autoimmune disorders
Given that lysosomes play a crucial role in multiple stages of immunity, we took a close look at what happened to the lysosomes in the cells of patients with autoimmune disorders. Here, we take three kinds of rheumatic autoimmune disorders as examples to explain lysosomal changes and dysfunction in autoimmune disorders and the significance of these changes and dysfunction for the deterioration of these disorders.
One of the most obvious changes in the cells of autoimmune disorders is the upregulated expression and activity of cathepsins and their abnormal secretion (The details are provided in Table 1).14,114,115,116,117,118,119,138 The overexpression of cathepsin S, an enzyme responsible for degrading antigens, has been reported in all three diseases, and its inhibitors have been shown to be effective in mouse models.14,114,115,116,117,118,138 In addition, cathepsin S and L were found to have significant correlations with rheumatoid arthritis (RA)-associated autoantibodies, which may account for the chronic inflammatory response and destruction of human tissues.139 More directly, cathepsin B present in synovial fluid of the joint of patients with RA has been found to destroy joints by degrading bone collagen.140
Another change that greatly arouses our attention is the dysregulation of autophagy. Enhanced activation of autophagy within autoreactive cells and inflammatory cells is common in systemic lupus erythematosus (SLE), RA, and Sjögren’s syndrome (SS).19,141,142,143,144,145,146,147 It has been found that autophagy is not only the survival mechanism of autoreactive B cells in SLE patients, but also the key to plasmablast differentiation and the long-term autoantibody secretion of plasma cells.141,148 In patients with RA, upregulated autophagy was found to regulate the bone resorption of osteoclasts and promote the survival of fibroblast-like synovial cells, the main actor in RA pathogenesis.143,144,149,150 Upregulated autophagy was also observed in T and B cells that infiltrated minor salivary glands in SS, and was associated with histological severity.19,146,147 However, there are different views regarding the activation status of the autophagy pathway in the T cells of RA patients.151,152 While Yang et al. identified the autophagy defect associated with PFKFB3 deficiency in CD4 + T cells of RA patients, van Loosdregt et al.151 reported that autophagy in CD4 + T cells of RA patients was upregulated and promoted hyperactivation and apoptosis resistance of T cells.152 It is important to point out that the former study used naive CD4 + T cells while the latter used total CD4 + T cells. Therefore, the differences in the cell type might contribute to the difference in the results. In addition, Gros et al.153 supported the view that autophagy was upregulated in T cells, and believed that the reason for the difference in results might be the imperfect experimental design of the former study.
In theory, upregulated lysosomal biogenesis and reduced lysosomal pH are required for the maintenance of cathepsin activity and autophagy activation. Consistent with our expectation, the monocytes, B cells, and DCs from female SLE patients showed lower lysosomal pH than those of normal people.21,154 Nevertheless, while upregulated autophagy in macrophages has been reported to contribute to murine lupus, another study reported that macrophages from lupus-prone MRL/lpr mice exhibited impaired lysosomal maturation and acidification.142,155 Currently, there are few reports on the changes on lysosomal biogenesis and pH, and more research results are needed to clarify these issues.
Although much is still unknown about the lysosomal changes that occur in the cells of patients with autoimmune disorders, several lysosomal targeting agents have been shown to be effective in preclinical and clinical trials, such as P140, hydroxychloroquine, and RO5461111 (Fig. 4 and Supplementary Table S1).139,140,153,156 Therefore, targeting lysosomes in autoimmune disorders is still of great feasibility and potential.
Neurodegenerative diseases
The accumulation of modified or misfolded proteins is common in neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD).14,157 These proteins not only deposit in neurons, causing synapse destruction and neuronal death, but also impair lysosomal function, which aggravates their accumulation.157,158,159,160,161 For instance, α-synuclein, the main component of Lewy bodies in PD, has been reported to reduce lysosomal degradation capacity by disrupting hydrolase trafficking.14,162 In addition, increased oxidative and nitrative modifications of v-ATPase with age, as well as mutations in PS1,120 ATP6AP2,121 and ATP13A2/PARK9122 have been identified to promote neurodegenerative diseases by impairing lysosomal acidification and autophagy (reviewed elsewhere.160,163) Genetic or pharmacological activation of TFEB was shown to partially restore the degradation of misfolded proteins and ameliorate disease progression of AD,164,165,166 PD,167,168 and HD.169,170 In addition, chaperone-mediated autophagy (CMA) has been found to act as a compensatory pathway of macroautophagy that is activated in response to macroautophagy failure.171,172,173,174
The expression of cathepsins, the important executors of lysosomal degradation, was deregulated in cells of neurodegenerative diseases, and they showed reduced efficiency in degrading misfolded proteins and deregulated expression.14,175 For example, cathepsin D, the hydrolase responsible for degrading HTT, was identified to be less effective in degrading mHTT, the aggregation- prone HTT mutant in HD.14,176 It is worth noting that cathepsins do not always play a protective role. The accumulation of Aβ peptides and hyperphosphorylated Tau are hallmarks of AD, and cathepsin D is responsible for the generation of Aβ peptides and be correlated with the hyperphosphorylation of Tau.157,177,178 In addition, elevated cathepsin B in the serum has been shown to be identified significant correlated with cognitive dysfunction in patents with AD.175,179
Taken together, increasing evidence indicates the great role of lysosomal changes and dysfunction in the pathogenesis of neurodegenerative diseases. Therefore, they may serve as targets for the treatment neurodegenerative diseases.
Cardiovascular diseases
Accumulating studies have provided insights into lysosomal changes and dysfunction in cardiovascular diseases.123,124,125,126,127,128,129 During acute myocardial ischemia, upregulated autophagy protects cardiomyocytes from death in response to extreme hypoxia and nutritional stress.123 However, an increased abundance of autophagosome has been reported to contribute to cardiomyocyte death during ischemia reperfusion.124 Reactive oxygen species (ROS) released during the restoration of blood flow are thought to induce LAMP2 decline and BECLIN-1 upregulation, resulting in the impaired autophagosome clearance.124 Impaired autophagic flux and inadequate autophagosome clearance have also been confirmed to promote atherosclerosis,125,126 maladaptive post-infarction remodeling,127,128 and heart failure129 (reviewed elsewhere.160,180) In addition, the restoration of autophagosome clearance through forcing TFEB expression have been found to attenuate BNIP3-induced cardiomyocyte death.181 Upregulation of cathepsin D induced by myocardial infarction was reported to protect against cardiac remodeling and heart failure through promoting autophagic flux.182
Lysosome changes and dysfunction have also been characterized in hereditary cardiomyopathy and drug-induced cardiomyopathy. In the hearts of patients with Danon disease, an X-linked lysosomal storage disease characterized by life-threatening hypertrophic cardiomyopathy, cardiomyocytes showed a dramatically increase in autophagic vacuoles.183 It is generally believed that this increase in vacuoles is due to the loss-of-function mutations in the LAMP2 gene, which encodes the protein required for the maturation of autophagosomes.183 Drug-induced cardiotoxicity remains a major cause of concern in the application of numerous medicines.184 In recent years, a large number of studies have confirmed that the cardiotoxicity caused by doxorubicin, a classical chemotherapeutic drug, is related to its inhibition of cardiac autophagy via the impairment of lysosomal acidification and the suppression of TFEB expression.185,186,187 Li et al.185 posited that ROS accumulation resulting from compromised autophagy accounts for doxorubicin-induced cardiomyocyte injury.
In summary, many cardiovascular diseases have been recognized to be closely related to lysosomal alterations and dysfunction, which are mainly manifested as maladaptive autophagy. Exogenous supplementation with lysosomal enzymes or efforts to increase the expression of TFEB may be means to treat these diseases.
Overall, lysosomes in cancer cells, autoimmune disease cells, heart disease cells and neurodegenerative disease cells undergo some changes and disfunction, which are extremely important for the development of these disease. Lysosomal acidification, lysosomal cathepsins, lysosomal biogenesis and autophagy may serve as good targets for the treatment of these diseases.
Available strategies for targeting lysosomes in human disease
Target lysosomal acidification
The acidic environment in lysosomes is not only their structural characteristics but also the basis of their activities and functions.2,7,188 While low lysosomal pH is required for cancer cells to maintain their high metabolic state and is associated with the overactivation of autoimmune cells in autoimmune disorders, cells in neurodegenerative and cardiovascular disease always exhibit impaired lysosomal acidification and autophagy.163,185,189 Therefore, suitable targeting measures can be selected according to different lysosomal acidification states.
At present, agents inhibiting lysosomal acidification can be roughly divided into two categories: v-ATPase inhibitors and antimalarials and their derivatives (Fig. 4). The v-ATPase contains two essential domains, V0 and V1 domian, which function together to pump protons into the late endosome/lysosome.188,190 v-ATPase inhibitors including bafilomycin A1, concanamycin, archazolid A, and INDOL0 all exhibit great performance, but they compete with one another because they all work through interacting with V0 subunit c of v-ATPase.189,191,192 In addition, bafilomycin A1 was found to block autophagosome-lysosome fusion by targeting endoplasmic reticulum (ER) calcium pump Ca-P60A independent of v-ATPase-mediated lysosomal acidification.193,194 Salicylihalamide A acts through inhibiting the V0 domain and causing a dramatic redistribution of the V1 domain, allowing it to combine with the former class inhibitors to achieve better inhibition.189,195 The next generation of inhibitors including VoPQ, NiK12192, and FR177995 also exhibit effective v-ATPase inhibition, but their inhibition efficiency has only been confirmed in yeast cells.189,196
Another class of inhibitors is antimalarials and their derivatives, the only class of autophagy inhibitors that is currently available for clinical application.5,197 As representative cationic amphiphilic drugs (CADs), CQ and its derivatives share a common structure of a side-chain with a cationic amine group, which allows them to accumulate within lysosomes after protonation, resulting in lysosomal deacidification.5,26,198 In addition, CQ and HCQ have shown a strong ability of blocking the fusion of autophagosomes with lysosomes, which may be their main mechanism of autophagy inhibition.5,199 Nevertheless, their widespread application is restricted by their excessive dose-dependent effects, limited single-agent activity, and ocular toxicity at high doses or with long-term use.5,26,200,201 Quinacrine, another drug originally used for antimalarial therapy, show a 60-fold higher potency of lysosomal deacidification than CQ and may be a better candidate for autophagy inhibition.8 Their analogs and derivatives exhibit more potent abilities of lysosomal localization and autophagy inhibition than their prototype, and some derivatives, such as DQ661 and DC661, show additional mTORC1 inhibition.5,133,200,202,203,204 More importantly, Ravi K. Amaravadi et al. identified a target shared by monomeric and dimeric CQ derivatives called palmitoyl-protein thioesterase 1 (PPT1) and demonstrated that targeting PPT1 produced dramatic lysosomal deacidification and mTOR inhibition by modulating the lysosomal localization of v-ATPase subunits and disrupting the interaction between subunit V1A of v-ATPase and the Ragulator component called p18.203,204 Intriguingly, there are many similarities between retinopathy caused by long-term use of CQ and HCQ and retinopathy caused by PPT1 deficiency, such as maculopathy with pigmentary alterations and the presence of auto-fluorescent material throughout the retina.201,205 Therefore, there is a question of whether the use of these derivatives will cause more serious ocular side effects due to their stronger inhibition of PPT1.
The restoration of lysosomal acidification in cells with impaired autophagy can be achieved by targeting molecules that impede lysosomal acidification or exogenously supplementing acid.120,206 As we described above, mutated PS1 impaired lysosomal acidification through impeding V0a1 subunit of v-ATPase complex, which is one of the main causes of early-onset familial AD.120 Therefore, targeting mutated PS1 may partially restore lysosomal acidity and autophagy. Besides, Bourdenx et al.206 demonstrated that poly(DL-lactide-co-glycolide) acidic nanoparticles (PLGA-aNPs) were internalized into lysosomes within 24 h after the treatment and restored defective lysosomal acidification and autophagy-lysosomal pathways in three different pathological PD models, including fibroblasts from PD patients with ATP13A2 mutations, fibroblasts from PD patients with glucocerebrosidase (GBA) mutations, and BE-M17 cells with ATP13A2 knockdown.
In brief, lysosomal acidification plays different roles in different diseases, and suitable targeting measures can be chosen according to the lysosomal acidification state.
Target lysosomal cathepsins
Lysosomal cathepsins are among the most important components and functional executors of lysosomes.2,132 Accumulating findings have acknowledged that lysosomal cathepsins facilitate the proliferation, invasion, angiogenesis, and chemotherapy-resistance of cancer cells, and their expression and activities are frequently upregulated in leukemia and various solid tumors, such as melanoma, breast cancer, gastrointestinal cancer.160,207,208 In addition, as mentioned above, deregulated cathepsins also play a great role in the development and progression of autoimmune disorders and neurodegenerative diseases. Therefore, cathepsins have been proposed as good targets for the treatment of cancers, autoimmune disorders, and neurodegenerative diseases.22,207,209,210
Three families and 15 classes of cathepsins have been found in humans, and cathepsin B, cathepsin D, cathepsin K, cathepsin L, and cathepsin S are well-studied in the treatment disease.14,27,132,207,211,212 A variety of endogenous and reversible inhibitors show therapeutic potential in regulating cathepsins, such as stefin A and cystatin C.207,209 Besides, while the inhibitory effect of most synthesized cathepsin inhibitors is broad-spectrum and irreversible (shown in Fig. 4 and Supplementary Table S1), inhibitors such as CA074, odanacatib (MK-0822), KGP94, CLIK-148, and CLIK-195, are designed to have better specificity and efficiency.207,213 Cathepsin K is highly effective at degrading collagens of bone matrix, and its inhibitor odanacatib was once regarded as the most promising candidate for the treatment of bone destruction caused by inflammatory diseases and cancers.214,215 However, the development of odanacatib for the treatment of osteoporosis was finally terminated by the study’s sponsor because of its serious cardio-cerebrovascular adverse reactions observed in the phase III clinical trial of postmenopausal osteoporosis.215,216 The use of cathepsin antibodies or targeting cathepsin secretion also holds great promise as therapeutic agents to target abnormal activities of lysosomal cathepsins.2,27,207,217,218 For example, the nonreceptor tyrosine kinases Abl and Arg (Abl/Arg) were reported to promote the secretion of cathepsin B and cathepsin L, which facilitate melanoma invasion and metastasis by cleaving or degrading extracellular matrix proteins.218
As we described above, cathepsin D plays a protective role in HT and cardiac remodeling, so forced expression or exogenously supplementation of cathepsin D may be helpful for the alleviation of these diseases.176,182 Two studies in neuronal ceroid lipofuscinosis, a group of rare recessive lysosomal storage disorders with impaired lysosome-autophagy pathways, have provided some direction.219,220 The injection of adeno-associated virus encoding mouse cathepsin D into both cerebral ventricles and peritoneum have been proved to increase the lifespan of cathepsin D-knockout mice (Ctsd−/− mice).219 In addition, André R. A. Marques administered 25 mg/kg recombinant human pro-cathepsin D to Ctsd−/− mice through the tail vein and found a correction of lysosomal storage accumulation and impaired autophagic flux in their viscera and central nervous system.220 The lifespans of these mice were also longer than those of the control group.220 These data support the feasibility and efficiency of restoring lysosomal cathepsins in diseases characterized by reduced cathepsin efficiency.
In recent years, a variety of drugs have been synthesized, but few of them have been used in clinical studies (summarized in the clinical trial section). The complexity of the cathepsin web and our inadequate understanding of the integration and functionality of cathepsins within the web make it difficult to target cathepsins for clinical application.132,207,213 In addition, enzyme replacement therapy is not yet mature, and it is difficult to achieve accurate and efficient delivery of cathepsins to specific organs.108
Target lysosomal membrane permeability and integrity
Under stress conditions, lysosomal membrane permeabilization (LMP) or full rupture of lysosomes occurs, and the leakage of lysosomal contents into the cytoplasm triggers inflammatory responses and cell death.26,221,222 Therefore, defective membrane permeability and integrity may act not only causes of inflammatory diseases but also tools that we can use to treat cancer.26,223 Unlike other organelles, lysosomes lack antioxidant enzymes such as superoxide dismutase, which makes their membrane more vulnerable to the damage of ROS and the hydroxyl radicals they produce.24,224 Although ROS act as the byproduct of traditional chemotherapies in most cases, they can also be induced intentionally in lysosomes by photodynamic therapy or iron regulation.225,226 For example, sequestering iron in lysosomes with ironomycin (AM5) or enhancing the lysosomal degradation of ferritin and the release of iron by artemisinin compounds can evoke the ROS generation and LMP in cancer cells.227,228,229 Direct disrupting LAMP2, the constitutive protein of lysosomal membrane, with mycotoxin enniatin B1 may also be a good strategy to induce LMP.230
Furthermore, targeting acid sphingomyelinase (ASM) and its supporter, heat shock protein 70 (Hsp70), can induce LMP by causing sphingomyelin accumulation.26,231,232 Direct inhibitors of ASM such as zoledronic acid and Riccardin D-N, as well as functional inhibitors such as cationic amphiphilic drugs (CADs), all show highly efficient ASM inhibition and LMP induction.26,233,234 CADs are a wide group of chemicals that can permeate lysosomal membranes and accumulate within lysosomes after protonation, and antimalarials, antidepressants, antihistamines all fall into the CAD category.24,26,235,236 In addition to siramesine, CADs like terfenadine and amitriptyline have also been acknowledged to have a great inhibition of ASM and induce LMP in targeted cells (shown in Fig. 4 and Supplementary Table S1).235,237,238 The most significant advantages of this category of drugs are the safety and accessibility established by their long-term clinical use.239 HSP70 inhibitors such as 2-Phenylethynesulfonamide (PES),240,241 quercetin,242 triptolide,242 and etoposide243 show great performance in HSP70 inhibition and LMP induction, but none of them can specifically target lysosomal HSP70.26,231,240 Therefore, although these HSP70 inhibitors are of great significance for inducing LMP and subsequent cell death, they may also cause serious adverse reactions because of the simultaneous inhibition of cytoplasmic and membrane HSP70.231
Cells have developed numerous defensive mechanisms against lysosomal rupture and subsequent inflammatory responses and cell death.221,244,245 It is now generally believed that limited permeabilization of the lysosomal membrane can be repaired through the endosomal sorting complex required for transport (ESCRT) machinery, while badly damaged lysosomes can be engulfed and cleared through the lysophagy machinery, a selective autophagy process triggered by the ubiquitination of lysosomal proteins.221,246,247 Three subcomplexes with different functions (ESCRT-I, -II, and -III) have been identified to be involved in the ESCRT mechanism, and the recruitment and translocation of their components have been found heavily dependent on calcium (Ca2+) outflowing from lysosomes.244,245,248,249 The ubiquitination required for lysophagy is induced the exposure of lysosomal glycans, which are sensed by cytosolic lectins or ubiquitination enzymes.245 While cytosolic lectins bind the autophagy receptor NDP52 (nuclear dot protein 52 kDa) and recruit autophagic membranes, ubiquitination enzymes such as ubiquitin conjugating enzyme E2 Q family like 1 (UBE2QL1) and F-box protein 27 (FBXO27) directly mediate the ubiquitination of damaged lysosomal proteins.245,250,251,252,253 Recently, Gupta et al.254 used proteomic-based organelle profiling and identified the selective and high enrichment of myoferlin (MYOF) on the lysosomal membranes of pancreatic cancer cells.254 They suggested that MYOF provided early-acting protection against membrane damage by stabilizing the lipid bilayer or promoting the fusion of lysosomes with other vesicles acting as membrane donors rather than through the ESCRT machinery. Lysosomal dysfunction induced by knocking out MYOF was demonstrated to impair tumor growth both in vitro and in vivo in this study.254 It is possible to modulate the stability and integrity of lysosomal membranes by using these key molecules to intervene in the protective mechanism of lysosomal membranes.
In addition, targeting the microtubule cytoskeleton and inducing mitochondrial membrane permeabilization (MMP) were also found to induce LMP and cell death.26 However, the effectiveness of these two approaches seems to be ambiguous because the main cause-and-effect relationship is unclear.26,255,256 For example, although there are reports that microtubule regulators, including paclitaxel, vincristine, Deox b7, 4, and BpV (phen), can induce LMP and apoptosis, it is difficult to determine whether cell death is caused by LMP or the disruption of the mitotic spindle, a critical transition in the cell cycle.26,137,257 In addition, possible crosstalk between autophagy and LMP offers more options for targeting LMP. Trehalose, an effective autophagy inducer, was found to act by inducing lysosomal enlargement and LMP, while knocking down autophagy protein 5 (Atg5) ameliorated IMB-6G-induced LMP and apoptosis.258,259
Target lysosomal calcium signaling
While lysosomes are the main organelles that store intracellular calcium (Ca2+), Ca2+ mediates the mechanism of lysosomal biogenesis, acidification maintenance, reorganization, and almost all vesicle movements involving lysosomes such as autophagy and endocytosis.2,260,261,262 Growing attention has been paid to the role of lysosomes in the development of cancer and neurodegenerative diseases.262,263,264 Among the Ca2+ channels that have been verified in the lysosomal membranes of mammalian cells, there are two groups that are good targets because of their specific localization on the membranes of endo-lysosomal system: transient receptor potential mucolipin channels (TRPMLs) and two-pore channels (TPCs).2,265,266
TRPMLs (TRPML1-3) are six-transmembrane domain channels encoded respectively by Mucolipin (MCOLN) 1-3.261,265 TRPML1 is the best-studied channel and is correlated with lysosome biogenesis and various membrane fusion processes, such as lysosome-autophagosome fusion and plasma membrane repair.32,33,267,268,269,270 However, its role in cancer progression is much more ambiguous due to its heterogeneous expression.265,268 Cancers such as bladder urothelial carcinoma, melanoma, and triple-negative breast cancer, have an upregulated expression of TRPML1.268,271 However, there are several examples of cancers with low expression of TRPML1 whose viability can be inhibited by TRPML1 agonists, such as non-small-cell lung carcinoma and glioblastoma.268,271 The role of TRPML2 in chemokine trafficking and secretion in murine macrophages was identified, and a bioinformatics analysis correlated the gene encoding TRPML3 with the progression, aggressiveness, and prognosis of pancreatic ductal adenocarcinoma.272,273 Some inhibitors and activators with less selectivity for these channels have also been acknowledged or synthesized, such as phosphatidyl-(3,5)-bisphosphate (PI(3,5)P2), ML-SA1, MK6-83, and ML2-SA1 (Fig. 4 and Supplementary Table S1).265,268 Besides, agents that target PIKfyve, a phosphoinositide kinase phosphorylates PI(3)P to form PI(3,5)P2, showed good performance in inhibiting the malignant phenotype of autophagy-dependent cancer cells, such as apilimod, YM201636, WX8-family.274,275,276
TPCs are voltage-gated ion channels in the endo-lysosomal system that mediate Ca2+ signals through the Ca2+-mobilizing messenger nicotinic acid adenine dinucleotide phosphate (NAADP).266 Ned-19 and tetrandrine work by targeting NAADP and have shown a great performance in reducing the migration and adhesion of cancer cells such as T24, HUH7, and 4T1-Luc.265,266 Besides, Ned-19 and its analog Ned-K were also reported to correct morphological defects in lysosomes in PD caused by LRRK2 mutations.277
Although the important role of Ca2+ in neurodegenerative diseases has been established, the corresponding abnormalities in calcium channels have not been established in most neurodegenerative diseases.261,263,264 Therefore, it is difficult to select an appropriate calcium channel modulator. The unclear causes and effects of the heterogeneous expression in cancer cells and the lack of drugs with specific targeting also make it difficult to target calcium signaling for cancer therapy.
Target mTOR signaling
Lysosomes serve as platforms for the proper recruitment, assembly, and activation of mTOR signaling elements, and mTOR acts as a nutrient sensor that regulates the degradation activities of lysosomes.37,63 Functionally, lysosomes and mTOR form a tightly connected metabolic complex.37,63 While factors such as RAGs, Ragulator, and Rheb contribute to the recruitment of mTOR to lysosomes and its activation, the release of galectin-8 as a result of lysosomal injury leads to the dissociation and activity inhibition of mTOR.4,37,39,40,278 MTOR signaling, which modulates cell metabolism and proliferation, is frequently activated in cancer, so mTOR inhibitors can be applied to treat cancer.37,279 Since the inhibition of mTOR can induce lysosomal biogenesis and autophagy pathways, mTOR inhibitors can also be used in some diseases with impaired autophagy, such as neurodegenerative diseases (shown in Fig. 4 and Supplementary Table S1).42,43,44,45,46,280
While rapamycin (sirolimus) and its analogs (temsirolimus, everolimus, and ridaforolimus) mainly inhibit mTORC1, catalytic mTOR inhibitors (AZD2014, CC-223, TAK-228) inhibit both mTORC1 and mTORC2 through suppressing the catalytic activity of mTOR (shown in Fig. 4 and Supplementary Table S1).38,281 Since the PI3K-AKT pathway is involved in the activation and function of both mTOR1 and mTORC2, targeting PI3K or AKT can also achieve simultaneous inhibition of mTORC1 and mTORC2.38,282 Considerable progress has been made in the development of drugs targeting PI3K and AKT, and many of them show strong anti-tumor activity both in vivo and vitro, such as buparlisib (BKM120), pictilisib (GDC-0941), MK-2206, Ipatasertib (GDC-0068), and Capivasertib.281,282,283,284 However, it is worth noting that PI3K and AKT regulates multiple metabolic pathways, so the effect of targeting PI3K or AKT may not depend mainly on mTOR.
Selective inhibition of mTORC1 can be achieved by targeting the unique effector nodes responsible for its recruitment and activation, such as Ragulator, Rheb, and Raptor.38,285,286,287 As a guanine nucleotide exchange factor (GEF) for RAG GTPases, Ragulator signals amino acid levels and recruits mTORC1, and knocking out its essential component, p18, or disrupting its interaction with RAGs by overexpressing c17orf59 has been proven to attenuate aberrant mTORC1 activation.285,286 Furthermore, a small molecule called NR1 was reported to bind the switch II domain of Rheb and block mTORC1 signaling potently and selectively.288 In addition, a member of the MAP kinase (MAPK) subfamily called Nemo-like kinase (NLK) can phosphorylate Raptor, a distinctive component of mTORC1, resulting in the inhibition of the lysosomal localization of mTORC1 and its subsequent activation.289
Attention to the role of mTORC2 in cancer progression is emerging, but it has also been proposed that adverse reactions to the long-term application of mTOR inhibitors are the result of simultaneous mTORC2 inhibition.63,282,290 Autophagy induction was once thought to be responsible for weakening the tumor-inhibiting effect of mTORC1 inhibitors, but it now allows the use of rapamycin to treat diseases with impaired autophagy.38,291
Emerging potential targeting strategies
New insights into the mechanisms of the initiation and progression of human diseases associated with autophagic or lysosomal dysfunction have spawned several new targeting strategies. Here, we list several targeting strategies that we believe have great potential, but the lack of drugs with high specificity and efficiency curtails the application of most of these strategies.
Since lysosomal dysfunction is closely correlated with weakened immune signals in the cancer immune response, growing attention has been paid to improving the cancer immune response by targeting disturbed lysosomal degradation.15,16,292 While CMTM6 was reported to prevent the lysosomal degradation of PD-L1, which contributes to immune escape, Huntingtin-interacting protein 1 related protein (HIP1R) was found to interact with the conserved domain (771-867) of PD-L1 and transmit it to lysosomal degradation.16,87 Huanbin et al. designed and constructed a peptide called PD-LYSO that consists of the PD-L1-binding sequence and the lysosome sorting sequence of HIP1R and demonstrated that this peptide accelerated the lysosomal degradation of PD-L1.87 Besides, the proteolysis that targets chimeras called P22 has been identified to restored the immune response in an immunosuppressed coculture model of Hep3B/OS-8/hPD-L1 and CD3 T cells by inhibiting PD-L1 and promoting the lysosomal degradation of PD-L1.293 Furthermore, an aloperine derivative called SA-49 was found to decrease the expression of PD-L1 in non-small cell lung cancer cells though promoting the biogenesis of lysosomes and melanogenesis-associated transcription factor (MITF)-dependent lysosomal degradation of PD-L1.294 The application of SA-49 was proven to enhance the immune response of cocultured T and NK cells to cancer cells and to suppress the growth of Lewis tumor xenografts in C57BL/6 mice.294 Since autophagy was observed to promote the immune evasion of PDAC by degrading MHC-I, scientists tried to combine immune checkpoint blockade (ICB) therapy with autophagy inhibition.135 The addition of CQ to anti-PD1 and anti-CTLA-4 antibodies was proven to enhance achieved enhanced anti-tumor immune response in mice with orthotopic tumors.135
Targeting TFEB, the transcription factor that regulates the lysosomal–autophagic pathway, has been proved effective for slowing the progression of lysosome-related diseases such as cancer,295,296,297 neurodegenerative diseases,164,165,166,167,168,169,170 and cardiovascular diseases.181,298 Trehalose was found to induce autophagy through promoting the nuclear translocation of TFEB, and this induced autophagy showed protective effects against neurodegenerative diseases,299 atherosclerosis,300 and cisplatin-induced acute kidney injury.301 In addition, a recent review summarized a series of compounds that have been found to regulate the expression or nuclear translocation of TFEB in recent years, such as 3,4-dimethoxychalcone (3,4-DC),302 2-Hydroxypropyl-β-cyclodextrin (HPβCD),303 and Digoxin.298,304 However, most of these compounds are the modulators of Ca2+ signaling or pathways such as mTOR, AKT, and PKC, so it is difficult to determine whether the ultimate effect of these drugs occurs primarily through their effects on TFEB.298
Accumulating evidence has acknowledged the contributions of chaperone-mediated autophagy (CMA) to the development of cancers and autoimmune disorders, as well as the protective role of CMA in neurodegenerative diseases.171,172,173,174,305,306 While knocking down LAMP2A was demonstrated to reduce cell proliferation of numerous cancer cells, reduced transcription of heat shock cognate protein 70 (HSC70) and LAMP2A was reported to aggravate the accumulation of pathological proteins of neurodegenerative diseases, such as α-synuclein, mHTT, and Tau.172,305,307,308,309 In addition, a peptide called P140 was found to inhibit CMA in lupus-prone mice by reducing the expression of both LAMP2A and HSC70 and impairing the refolding properties of HSC70.310,311,312 It has been acknowledged that this inhibition of CMA affected the processing and presentation of autoantigens in B cells and accounts for a decrease in autoreactive T cells.310,312
Targeting Rab GTPases (Rabs) is attractive, because they are extensively involved in the biogenesis and function of endo-lysosomal systems.313,314 For example, the application of CID1067700, a receptor antagonist of Rab7 GTPase, was identified to inhibit reactive astrogliosis and attenuate brain atrophy of astrocytic injury models though inhibiting excessive transportation of cathepsin B from late endosomes to lysosomes.315 However, most Rabs lack specific inhibitors due to their low affinity for nucleotide-based competitive inhibitors and high similarity with each other in structure.314 Agents that target their post-translational modification or GTPase–GEF interactions may achieve indirect but efficient inhibition of rabs, such as psoromic acid (PA),316 3-(3-pyridyl)-2-hydroxy-2-phosphonopropanoic acid (3-PEHPC), StRIP3.314,317
Although research into the measures and their mechanisms for targeting lysosomes in cancer is still in the primary stage, continuous progress and increasing interest in lysosome research may result in profound developments in this field.
Representative clinical trials of the targeting strategies
The encouraging results of lysosome-related preclinical studies have aroused growing interest in the clinical transformation and application of targeting lysosomes in cancer. In fact, most of the targeting strategies and corresponding drugs that we summarized above have been tested or are being tested in clinical trials.
In malignancies
The clinical response to these targeting strategies varies greatly depending on the cancer type, cancer stage, drug type, combination efficacy, the expression of some cancer markers, and the lysosome profile that may have been previously overlooked (Table 2).
With incomparable easy-accessibility and safety as a result of long-term clinical use in antimalarial therapy, CQ and HCQ have been rapidly and widely repurposed as agents targeting lysosome acidification in anticancer clinical trials.5,200 However, their effectiveness as single drugs against cancer is very limited in practice, so researchers are now using them as sensitizers in combination with radiotherapy and chemotherapy drugs such as taxane, carboplatin, gemcitabine, temozolomide, and metformin.318,319,320,321,322 In a search of the ClinicalTrials.gov website, we found that numerous clinical trials (almost half) used glioblastomas or tumors with brain metastases as cancer targets for CQ, and most of the trials showed improved results, which might indicate the superiority and specificity of CQ in the treatment of cerebral tumors (Table 2).319,323 For example, adding CQ to whole-brain irradiation was confirmed to play a great role in the treatment of patients with brain metastases, with a 1-year progression-free survival rate of 83.9% compared with 55.1% in the control group.319
However, a trial that combined HCQ with temozolomide and radiotherapy to treat newly diagnosed glioblastoma multiforme ended up with no significant improvement.324 Some scholars believed that the inconsistent levels of autophagy inhibition were the main reasons for the unsatisfactory results.5 Besides, researchers are trying to use this effective sensitizer in the treatment of pancreatic cancer, a highly lethal cancer that requires high autophagy level to maintain metabolism and resist therapy.203,320,321,322 While adding HCQ to gemcitabine or gemcitabine plus nab-paclitaxel was determined to improve prognosis-related serum biomarker (CA19-9), opinions were divided regarding its ability to promote overall survival (OS) and progression-free survival (PFS) (Table 2).320,321,322 There are two clinical trials that respectively used SQSTM1/p62 and LC3-II as the marker of effective inhibition of autophagy, and one of the trials measured both markers, which showed inconsistent levels of autophagy inhibition.321,322 Consequently, the latter trial may not have achieved effective autophagy inhibition, which indicates the lack of a unified standard for autophagy inhibition. In addition, the imbalance in KRAS mutations between the experimental group and the control group may also affect the experimental results.320 Although the clinical response to HCQ is not always satisfactory, it remains the most widely used antimalarial drug in clinical anti-tumor research.5
Temsirolimus and everolimus are mTOR inhibitors that have been approved by the US FDA for cancer treatment.325,326,327 In the treatment of advanced renal-cell carcinoma, using temsirolimus as a single agent has been identified to achieve longer OS and PFS than interferon monotherapy, and showed no significant difference in OS compared with the combination-therapy of temsirolimus and interferon (Table 2).328 This clinical trial data was thought to contribute to the US FDA’s approval of temsirolimus for the treatment of advanced renal-cell carcinoma.325 Temsirolimus also showed great efficiency in metastatic endometrial cancer (Table 2).329 Although everolimus was approved by the US FDA for the treatment of numerous cancers, it was found to have very limited efficacy when used as a single agent (Table 2).326,330 Since the activities of mTORC2 may compensate for the inhibition of mTORC1, a series of inhibitors that simultaneously target mTORC1 and mTORC2 have been developed, such as vistusertib (AZD2014),331 CC-223,332 and TAK-228 (MLN0128).333 Among these drugs, vistusertib is the most clinically used, but it was shown to result in lower OS and PFS improvement than everolimus.331 The efficacy of TAK-228 in metastatic castration-resistant prostate cancer was also reported to be limited, and eight of nine patients discontinued the treatment early because of radiographic progression, drug toxicity, or investigator discretion.333 CC-223 was proven effective and safe for the treatment of non-pancreatic neuroendocrine tumors, and clinical trials of this drug in other cancers are underway.332
Although inhibitors of the PI3K-AKT pathway showed great efficiency in cancer therapy, mTOR signaling is not its only downstream signaling pathway, and the activation status of mTOR signaling was not reported in these clinical trials.334,335,336,337,338,339 Since the inhibition of mTOR always acts as a potent inducer of cytoprotective autophagy that greatly compromises therapeutic effects, researchers have tried to combine mTOR inhibitors with autophagy inhibitors.283,340,341 Temsirolimus was tolerable and efficient when combined with HCQ in the treatment of melanoma and multiple advanced solid tumors, and the combination of everolimus with HCQ in patients with renal-cell carcinoma also achieved the primary endpoint without dose-limiting toxicity observed.340,341
Inducing LMP directly leads to cell death in preclinical experiments, but the effectiveness of most drugs used for this strategy are not satisfactory. For example, little improvement has been achieved in the application of CADs for cancer therapy, except for antimalarials.342,343 Amitriptyline, a tricyclic antidepressant, failed to decrease chemotherapy-induced peripheral neuropathy (CIPN) symptoms in cancer survivors, and a trial of desipramine for the treatment of patients with small cell lung cancer was terminated early because of intolerable doses and a lack of clinical activity.342,343 Although mountains of HSP70 inhibitors have been synthesized and identified to be effective in vitro, few HSP70 inhibitors have been tested in clinical trials.231 MKT-077 is possibly the only Hsp70 inhibitor that has been tested in clinical trials against cancer currently, but the trial was halted because of irreversible renal toxicity.344 ASM inhibitors and agents that target microtubules, the other two classes of LMP inducers, have been much more widely used in clinical studies with good results.26,239,345,346 Among them, ZA, paclitaxel, and vincristine have been used as standard treatments for many cancers.26,239,346 Nevertheless, It is worth noting that their therapeutic effects in the treatment of cancers are not confined to their direct effects in lysosomes, and their induction of LMP in cancer cells is seldom examined in clinical trials.26,137,257
Few cathepsin inhibitors have been used in clinical trials, and only one trial of the cathepsin K inhibitor odanacatib in cancer therapy has been completed.347 Although odanacatib achieved bone resorption inhibition comparable to that of ZA in the treatment of patients with bone metastases of breast cancer, there were some limitations in this trial, such as the relatively small sample size and the lack of clinical outcomes as efficacy endpoints.348 In a multicenter phase 3 clinical trial of osteoporosis, the long-term use of odanacatib (median follow-up 47.6 months) was found to significantly increase the risk of cardio-cerebrovascular events, especially stroke.216 Therefore, the study’s sponsor decided not to develop it as a treatment for osteoporosis (in 2019).216 Since then, there have been no clinical trials or applications for clinical trials of odanacatib for cancer treatment.347 However, we still believe that this drug has potential for treating cancer bone metastases because patients diagnosed with cancer bone metastases generally have a short survival period and will not take the medication for such long periods.349
The understanding of lysosomal calcium channels is still in the primary stage, and there is an urgent need for targeted drugs with high specificity.14 The emerging targeting strategies that we summarized above faced run into similar dilemmas. More highly effective drugs and corresponding clinical trials are needed to judge the effectiveness, safety, and feasibility of these targeting strategies.
In non-malignant diseases
With milder gastrointestinal and skin complications than CQ, HCQ has been more widely used clinically and in clinical trials.156 According to the management recommendation of European League Against Rheumatism (EULAR) published in 2019, HCQ is recommended for all patients with SLE.350 In a clinical trial, patients with higher blood levels of HCQ (≥1000 ng/ml) were reported to be less likely to develop active SLE.351 Nevertheless, both EULAR and American Academy of Ophthalmology recommend that the daily dose of HCQ should not exceed 5 mg/kg actual body weight.350,352 Using HCQ as either a single or a combinatorial therapy has been proven to be effective in the treatment of RA, but the addition of HCQ has been found to decrease maximum concentration of methotrexate (MTX) and increase the risk of MTX-induced toxicities (Table 3).353,354 In a prospective, multicenter observational study of 4905 RA patients, Mary Wasko et al.355 found that using hydroxychloroquine was correlated with a low risk of suffering diabetes. However, in a clinical trial, RA patients who received HCQ 6.5 mg/kg daily for 8 weeks showed no difference in insulin resistance and a slight improvement in lipid levels (total cholesterol and low-density lipoprotein) compared with controls.356 A possible reason for the different conclusions of the trial may be the short observation time, and a longer trial time may make the improvement more obvious. Since HCQ may improve lipid metabolism in human body, some scholars believe that the use of HCQ in RA patients can reduce the frequency of cardiovascular events.357,358,359,360
P140, a phosphopetide that can inhibit CMA, was found to decrease the levels of IgG anti-dsDNA antibody in patients with SLE and effectively improve their SLE disease activity index (SLEDAI) score with no adverse effects.361 Intriguingly, in another phase 2 clinical trial of patients with SLE, the subcutaneous injection of 200 μg of P140 every 4 weeks was found to achieve a better SLE responder index (SRI) response at 12 weeks than the same dose given every 2 weeks or a placebo control.362 However, a phase III clinical trial reported that the SRI response rate of patients treated with P140 (200 μg every 4 weeks) showed no significant difference (52.5% vs. 44.6%, p = 0.2631) from that of the control group at week 52.362 Therefore, P140 is safe and well tolerated for the treatment of SLE, but its efficacy in achieving long-term remission and control of SLE may require further experimental evidence.361,362,363
Growing attention has been paid to the role of T cells in autoimmune diseases.364,365 As described above, mTOR signaling is involved in modulating the differentiation of T cells, so a number of clinical trials have been conducted to test the efficacy of its inhibitor in the treatment of autoimmune diseases.64,65,366,367 Low-dose sirolimus, an inhibitor that mainly acts on mTOC1, has been found to selectively upregulate Tregs and achieve better control of the disease activity of RA (Table 3).366 In patients with SLE, sirolimus was also effective for controlling disease activity and correcting pro-inflammatory T-cell lineage specification (Table 3).367 However, there are two issues that attract our attention. First, the use of mTOR inhibitors may induce autophagy, which is always activated in immune cells in autoimmune diseases and is closely related to disease development.283,368 Further studies are needed to determine autophagy activation at these doses and the necessity of combing sirolimus with autophagy inhibitors.283,368 Second, while the upregulation of nonselective IgG or autoantibodies is common in SLE, the promotion of Th2 cell differentiation as a result of mTORC1 inhibition may facilitate humoral immunity and aggravate the accumulation of autoimmune complexes.64,141,369
As we explained previously, mTORC1 inhibitors can also be used as autophagy inducers to treat neurodegenerative diseases. Several clinical trials of mTOR inhibitors for the treatment of PD and AD are actively recruiting, such as NCT04629495, NCT04200911, and NCT04127578. Trehalose, which induces autophagy via TFEB activation, are also testing in the clinical trial of AD (NCT04663854). These drugs may be valuable candidates for the treatment of neurodegenerative diseases.
Numerous cathepsin inhibitors have been developed, but few of them have been tested in clinical trials. A trial assessing the efficacy of RO5459072 in patients with primary SS was completed, but no significant improvement in the EULAR Sjogren’s Syndrome Disease Activity Index (ESSDAI) score was observed in the RO5459072 group.370 Other targeting strategies have only come to the forefront in recent years, and their corresponding drugs are still in development or undergoing preclinical testing.
Conclusion and perspectives
Although targeted lysosomes show great promise for treating human diseases such as malignancies, autoimmune diseases, neurodegenerative diseases and cardiovascular diseases, there are still many questions waiting to be answered. A key question that needs to be answered first is how to selectively target lysosomes, especially lysosomes in pathological cells. CADs, including CQ, have good lysosomal aggregation characteristics, but they may result to indiscriminate inhibition or deletion of all lysosomal function.24 Although lysosomal changes in diseases are the foundation of our targeting strategies, there is no clinical evidence that these changes allow currently available therapies to strike fatal blows to pathological cells far more often than to normal cells.2,14 Luckily, metabolic glycoengineering of unnatural sugars provides a powerful tool for selectively labeling cancer cells, and antibody-drug conjugates restrict the systemic delivery of antineoplastic agents to cells that express certain antigens.371,372 In addition, nanoscale drug delivery systems, such as cathepsin-sensitive drug delivery systems, possess the unique ability to penetrate cell barriers and locate in certain organelles such as lysosomes.14,373 These emerging approaches offer the possibility of specifically targeting lysosomes, especially lysosomes in pathological cells.
Another question arises from the heterogeneity and complexity of lysosomes and their associated pathways. Several commonly targeted molecules or pathways exert dual effects on cancer progression and adapt with time, and the broad-spectrum and complete suppression of these molecules and pathways may lead to unpredictable and irreparable side effects.2,14,235,374 Therefore, it is necessary to develop precise personalized treatment regimens and monitor the functional status of lysosomes in cancer patients in real-time. The expression of some specific molecules can be used to evaluate the activation of the pathways, and the introduction of microfluidic single-cell analysis technology is expected to achieve lysosomal level accuracy in the future.14,101,246 However, these methods have not currently achieved the real-time monitoring of lysosomal number and function in individuals. In addition, tremendous work is needed to develop uniform and implementable guidelines for standardizing research and diagnosis.14,321 For instance, a trial that evaluated both SQSTM1/p62 and LC3-II found inconsistent levels of autophagy inhibition, making it difficult to determine whether autophagy was effectively suppressed.321
The issue of drug specificity and efficacy also requires attention. The structural and functional similarities among molecules in the same family make it difficult to target specific molecules, and the complementary effects of these molecules may weaken the targeting effect.207,314 For example, when cathepsin L is inhibited, cathepsin B activity increases compensatively and partially offsets the effect of cathepsin L inhibition.207 However, if both are inhibited simultaneously, other related pathways of cathepsin B will be affected.207 Future research needs to not only develop targeting agents with high selectivity but also clarify the necessity and extent of the inhibition of compensatory molecules. In addition, many targeting agents have limited therapeutic effects as single agents and need to be used in combination.318,319,320,321,322,326,330 Although combination-therapy can improve the efficacy of these compounds, it may also increase the side effects of the drugs and aggravate the metabolic burden on patients’ liver and kidneys.97 Therefore, there is an urgent need to develop more potent drugs and identify the tumors that are sensitive to them.
In conclusion, there are still many difficulties and challenges to be overcome, but they are not completely unsolvable considering the improvement of lysosome-targeted drugs and the optimization of research instruments and methods. Lysosomes are important regulators of cell and organismal homeostasis that mediate energy metabolism, cell proliferation and differentiation, immunity, and cell death. The lysosomes of a variety of disease cells have been found to undergo some lysosomal changes and dysfunction that have a profound effect on disease progression. Lysosomal acidification, lysosomal cathepsins, lysosomal membrane permeability and integrity, lysosomal calcium signaling, mTOR signaling, lysosomal degradation of immune signals, TFEB, noncanonical autophagy, and vesicle movement are all promising targets for lysosomes, and some of these targeted drugs have been tested clinically effective and safe. Therefore, targeting lysosomes in human disease is a feasible, effective, and safe targeted strategy, and we can look forward to developing it as an excellent therapeutic intervention.
Data availability
All data generated or analyzed during this study are included in this published article.
References
De Duve, C., Pressman, B. C., Gianetto, R., Wattiaux, R. & Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 60, 604–617 (1955).
Ballabio, A. & Bonifacino, J. S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 21, 101–118 (2020).
Sardiello, M. et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009).
Sancak, Y. et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 (2010).
Levy, J. M. M., Towers, C. G. & Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542 (2017).
Piao, S. & Amaravadi, R. K. Targeting the lysosome in cancer. Ann. N. Y Acad. Sci. 1371, 45–54 (2016).
Lawrence, R. E. & Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142 (2019).
Perera, R. M. & Zoncu, R. The lysosome as a regulatory hub. Annu. Rev. Cell Dev. Biol. 32, 223–253 (2016).
Peng, W., Minakaki, G., Nguyen, M. & Krainc, D. Preserving lysosomal function in the aging brain: insights from neurodegeneration. Neurotherapeutics 16, 611–634 (2019).
Tang, T. et al. The role of lysosomes in cancer development and progression. Cell Biosci. 10, 131 (2020).
Sciarretta, S., Maejima, Y., Zablocki, D. & Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 80, 1–26 (2018).
Geisslinger, F., Müller, M., Vollmar, A. M. & Bartel, K. Targeting lysosomes in cancer as promising strategy to overcome chemoresistance-a mini review. Front. Oncol. 10, 1156 (2020).
Zhang, X., Wang, J., Li, X. & Wang, D. Lysosomes contribute to radioresistance in cancer. Cancer Lett. 439, 39–46 (2018).
Bonam, S. R., Wang, F. & Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 18, 923–948 (2019).
Xu, J., Brosseau, J. P. & Shi, H. Targeted degradation of immune checkpoint proteins: emerging strategies for cancer immunotherapy. Oncogene 39, 7106–7113 (2020).
Burr, M. L. et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105 (2017).
Yu, Y. R. et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat. Immunol. 21, 1540–1551 (2020).
Karami, J. et al. Role of autophagy in the pathogenesis of rheumatoid arthritis: Latest evidence and therapeutic approaches. Life Sci. 254, 117734 (2020).
Colafrancesco, S. et al. Autophagy occurs in lymphocytes infiltrating Sjögren’s syndrome minor salivary glands and correlates with histological severity of salivary gland lesions. Arthritis Res. Ther. 22, 238 (2020).
Qi, Y. Y., Zhou, X. J. & Zhang, H. Autophagy and immunological aberrations in systemic lupus erythematosus. Eur. J. Immunol. 49, 523–533 (2019).
Harris, V. M., Harley, I. T. W., Kurien, B. T., Koelsch, K. A. & Scofield, R. H. Lysosomal pH is regulated in a sex dependent manner in immune cells expressing CXorf21. Front. Immunol. 10, 578 (2019).
Tato, M. et al. Cathepsin S inhibition combines control of systemic and peripheral pathomechanisms of autoimmune tissue injury. Sci. Rep. 7, 2775 (2017).
Kim, S. J. et al. Increased cathepsin S in Prdm1(−/−) dendritic cells alters the T(FH) cell repertoire and contributes to lupus. Nat. Immunol. 18, 1016–1024 (2017).
Wang, F., Gomez-Sintes, R. & Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 19, 918–931 (2018).
Boya, P. Lysosomal function and dysfunction: mechanism and disease. Antioxid. Redox Signal. 17, 766–774 (2012).
Serrano-Puebla, A. & Boya, P. Lysosomal membrane permeabilization as a cell death mechanism in cancer cells. Biochem. Soc. Trans. 46, 207–215 (2018).
Davidson, S. M., Vander & Heiden, M. G. Critical functions of the lysosome in cancer biology. Annu Rev. Pharm. Toxicol. 57, 481–507 (2017).
Saftig, P. & Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10, 623–635 (2009).
Cui, L. et al. The lysosomal membrane protein lamp2 alleviates lysosomal cell death by promoting autophagic flux in ischemic cardiomyocytes. Front. Cell Dev. Biol. 8, 31 (2020).
Brady, O. A., Martina, J. A. & Puertollano, R. Emerging roles for TFEB in the immune response and inflammation. Autophagy 14, 181–189 (2018).
Raben, N. & Puertollano, R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell Dev. Biol. 32, 255–278 (2016).
Medina, D. L. et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299 (2015).
Medina, D. L. et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 21, 421–430 (2011).
Martina, J. A. & Puertollano, R. Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J. Biol. Chem. 293, 12525–12534 (2018).
Palm, W. & Thompson, C. B. Nutrient acquisition strategies of mammalian cells. Nature 546, 234–242 (2017).
Settembre, C., Fraldi, A., Medina, D. L. & Ballabio, A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296 (2013).
Mossmann, D., Park, S. & Hall, M. N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757 (2018).
Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976 (2017).
Lawrence, R. E. et al. A nutrient-induced affinity switch controls mTORC1 activation by its Rag GTPase-Ragulator lysosomal scaffold. Nat. Cell Biol. 20, 1052–1063 (2018).
Inoki, K., Li, Y., Xu, T. & Guan, K. L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 (2003).
Hoxhaj, G. & Manning, B. D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 20, 74–88 (2020).
Peña-Llopis, S. et al. Regulation of TFEB and V-ATPases by mTORC1. Embo J. 30, 3242–3258 (2011).
Martina, J. A., Chen, Y., Gucek, M. & Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 (2012).
Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo J. 31, 1095–1108 (2012).
Kim, J., Kundu, M., Viollet, B. & Guan, K. L. A. M. P. K. and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 (2011).
Florey, O. & Overholtzer, M. Macropinocytosis and autophagy crosstalk in nutrient scavenging. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374, 20180154 (2019).
Palm, W. et al. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162, 259–270 (2015).
González, A., Hall, M. N., Lin, S. C. & Hardie, D. G. AMPK and TOR: The Yin and Yang of cellular nutrient sensing and growth control. Cell Metab. 31, 472–492 (2020).
Zhang, C. S. et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116 (2017).
Zhang, C. S. et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 20, 526–540 (2014).
Van Nostrand, J. L. et al. AMPK regulation of Raptor and TSC2 mediate metformin effects on transcriptional control of anabolism and inflammation. Genes Dev. 34, 1330–1344 (2020).
Rogala, K. B. et al. Structural basis for the docking of mTORC1 on the lysosomal surface. Science 366, 468–475 (2019).
Nofal, M., Zhang, K., Han, S. & Rabinowitz, J. D. mTOR inhibition restores amino acid balance in cells dependent on catabolism of extracellular protein. Mol. Cell. 67, 936–46.e5 (2017).
Wang, S. et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347, 188–194 (2015).
Wyant, G. A. et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171, 642–54.e12 (2017).
Rebsamen, M. et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519, 477–481 (2015).
Fujimaki, K. et al. Graded regulation of cellular quiescence depth between proliferation and senescence by a lysosomal dimmer switch. Proc. Natl Acad. Sci. USA 116, 22624–22634 (2019).
Tomas, A., Futter, C. E. & Eden, E. R. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 24, 26–34 (2014).
Kowanetz, K. et al. Suppressors of T-cell receptor signaling Sts-1 and Sts-2 bind to Cbl and inhibit endocytosis of receptor tyrosine kinases. J. Biol. Chem. 279, 32786–32795 (2004).
Fraser, J. et al. Targeting of early endosomes by autophagy facilitates EGFR recycling and signalling. EMBO Rep. 20, e47734 (2019).
Weber, R. A. et al. Maintaining iron homeostasis is the key role of lysosomal acidity for cell proliferation. Mol. Cell. 77, 645–55.e7 (2020).
Sarbassov, D. D., Guertin, D. A., Ali, S. M. & Sabatini, D. M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 (2005).
Jia, R. & Bonifacino, J. S. Lysosome positioning influences mTORC2 and AKT signaling. Mol. Cell 75, 26–38.e3 (2019).
Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 12, 325–338 (2012).
Waickman, A. T. & Powell, J. D. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 249, 43–58 (2012).
Fernandez-Veledo, S., Vazquez-Carballo, A., Vila-Bedmar, R., Ceperuelo-Mallafre, V. & Vendrell, J. Role of energy- and nutrient-sensing kinases AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) in adipocyte differentiation. IUBMB Life 65, 572–583 (2013).
Sukhbaatar, N., Hengstschlager, M. & Weichhart, T. mTOR-mediated regulation of dendritic cell differentiation and function. Trends Immunol. 37, 778–789 (2016).
Zeng, H. & Chi, H. mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr. Opin. Immunol. 46, 103–111 (2017).
Zhang, M. et al. The MTOR signaling pathway regulates macrophage differentiation from mouse myeloid progenitors by inhibiting autophagy. Autophagy 15, 1150–1162 (2019).
Saravia, J., Raynor, J. L., Chapman, N. M., Lim, S. A. & Chi, H. Signaling networks in immunometabolism. Cell Res. 30, 328–342 (2020).
Delgoffe, G. M. et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844 (2009).
Rao, R. R., Li, Q., Odunsi, K. & Shrikant, P. A. The mTOR kinase determines effector versus memory CD8 + T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 32, 67–78 (2010).
Volkl, S. et al. Hyperactive mTOR pathway promotes lymphoproliferation and abnormal differentiation in autoimmune lymphoproliferative syndrome. Blood 128, 227–238 (2016).
Schietinger, A. & Greenberg, P. D. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 35, 51–60 (2014).
Akbar, A. N. & Henson, S. M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 11, 289–295 (2011).
Jacquel, A. et al. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 119, 4527–4531 (2012).
Obba, S. et al. The PRKAA1/AMPKα1 pathway triggers autophagy during CSF1-induced human monocyte differentiation and is a potential target in CMML. Autophagy 11, 1114–1129 (2015).
Germic, N., Frangez, Z., Yousefi, S. & Simon, H. U. Regulation of the innate immune system by autophagy: monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 26, 715–727 (2019).
Axelrod, M. L., Cook, R. S., Johnson, D. B. & Balko, J. M. Biological consequences of MHC-II expression by tumor cells in cancer. Clin. Cancer Res. 25, 2392–2402 (2019).
Schmid, D., Pypaert, M. & Münz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26, 79–92 (2007).
Wenger, T. et al. Autophagy inhibition promotes defective neosynthesized proteins storage in ALIS, and induces redirection toward proteasome processing and MHCI-restricted presentation. Autophagy 8, 350–363 (2012).
Marshall, R. S., Li, F., Gemperline, D. C., Book, A. J. & Vierstra, R. D. Autophagic degradation of the 26 S proteasome is mediated by the dual ATG8/ubiquitin receptor RPN10 in arabidopsis. Mol. Cell 58, 1053–1066 (2015).
Münz, C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol. Rev. 272, 17–27 (2016).
Hipolito, V. E. B. et al. Enhanced translation expands the endo-lysosome size and promotes antigen presentation during phagocyte activation. PLoS Biol. 17, e3000535 (2019).
Saez, J. J. et al. The exocyst controls lysosome secretion and antigen extraction at the immune synapse of B cells. J. Cell Biol. 218, 2247–2264 (2019).
Ibanez-Vega, J., Del Valle Batalla, F., Saez, J. J., Soza, A. & Yuseff, M. I. Proteasome dependent actin remodeling facilitates antigen extraction at the immune synapse of B Cells. Front. Immunol. 10, 225 (2019).
Wang, H. et al. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat. Chem. Biol. 15, 42–50 (2019).
Wang, H., Han, X. & Xu, J. Lysosome as the Black Hole for Checkpoint Molecules. Adv. Exp. Med. Biol. 1248, 325–346 (2020).
Feng, M. et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 19, 568–586 (2019).
Rostamzadeh, D. et al. mTOR signaling pathway as a master regulator of memory CD8(+) T-cells, Th17, and NK cells development and their functional properties. J. Cell Physiol. 234, 12353–12368 (2019).
Greten, F. R. & Grivennikov, S. I. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 51, 27–41 (2019).
Sendler, M. et al. Cathepsin B-Mediated Activation of Trypsinogen in Endocytosing Macrophages Increases Severity of Pancreatitis in Mice. Gastroenterology. 154, 704–718.e10 (2018).
Hou, J., Karin, M. & Sun, B. Targeting cancer-promoting inflammation-have anti-inflammatory therapies come of age? Nat. Rev. Clin. Oncol. 18, 261–279 (2021).
He, Y. et al. Identification of a lysosomal pathway that modulates glucocorticoid signaling and the inflammatory response. Sci. Signal. 4, ra44 (2011).
Ge, W., Li, D., Gao, Y. & Cao, X. The roles of lysosomes in inflammation and autoimmune diseases. Int. Rev. Immunol. 34, 415–431 (2015).
Shi, C. S. et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263 (2012).
Amaravadi, R., Kimmelman, A. C. & White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 30, 1913–1930 (2016).
Napoletano, F., Baron, O., Vandenabeele, P., Mollereau, B. & Fanto, M. Intersections between regulated cell death and autophagy. Trends Cell Biol. 29, 323–338 (2019).
Holland, L. K. K., Nielsen, I. O., Maeda, K. & Jaattela, M. SnapShot: lysosomal functions. Cell 181, 748–e1 (2020).
Kroemer, G. & Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 5, 886–897 (2005).
Carneiro, B. A. & El-Deiry, W. S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17, 395–417 (2020).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Zhou, B. et al. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 66, 89–100 (2020).
Hou, W. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016).
Bai, Y. et al. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 508, 997–1003 (2019).
Raivio, K. O. & Seegmiller, J. E. Genetic diseases of metabolism. Annu. Rev. Biochem. 41, 543–576 (1972).
Lieberman, A. P. et al. Autophagy in lysosomal storage disorders. Autophagy 8, 719–730 (2012).
Platt, F. M. Emptying the stores: lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 17, 133–150 (2018).
Myerowitz, R., Puertollano, R. & Raben, N. Impaired autophagy: The collateral damage of lysosomal storage disorders. Ebiomedicine 63, 103166 (2021).
Di Malta, C. et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 356, 1188–1192 (2017).
Perera, R. M. et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365 (2015).
Ballesteros-Álvarez, J. et al. MITF and TFEB cross-regulation in melanoma cells. PLoS ONE 15, e0238546 (2020).
Li, Y. et al. TFEB is a master regulator of tumor-associated macrophages in breast cancer. J. Immunother. Cancer. 8, e000543 (2020).
Hamm-Alvarez, S. F. et al. Tear cathepsin S as a candidate biomarker for Sjögren’s syndrome. Arthritis Rheumatol. 66, 1872–1881 (2014).
Rupanagudi, K. V. et al. Cathepsin S inhibition suppresses systemic lupus erythematosus and lupus nephritis because cathepsin S is essential for MHC class II-mediated CD4 T cell and B cell priming. Ann. Rheum. Dis. 74, 452–463 (2015).
Bernard, N. J. Connective tissue diseases. Inhibiting cathepsin S to treat SLE and lupus nephritis. Nat. Rev. Rheumatol. 10, 66 (2014).
Klinngam, W. et al. Inhibition of cathepsin S reduces lacrimal gland inflammation and increases tear flow in a mouse model of sjögren’s syndrome. Sci. Rep. 9, 9559 (2019).
Edman, M. C. et al. Increased Cathepsin S activity associated with decreased protease inhibitory capacity contributes to altered tear proteins in Sjögren’s Syndrome patients. Sci. Rep. 8, 11044 (2018).
Meng, Z. et al. Imbalanced Rab3D versus Rab27 increases cathepsin S secretion from lacrimal acini in a mouse model of Sjögren’s Syndrome. Am. J. Physiol. Cell Physiol. 310, C942–C954 (2016).
Lee, J. H. et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158 (2010).
Korvatska, O. et al. Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Hum. Mol. Genet. 22, 3259–3268 (2013).
Tsunemi, T. & Krainc, D. Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 23, 2791–2801 (2014).
Gustafsson, A. B. & Gottlieb, R. A. Autophagy in ischemic heart disease. Circ. Res. 104, 150–158 (2009).
Ma, X. et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation 125, 3170–3181 (2012).
Michiels, C. F., Fransen, P., De Munck, D. G., De Meyer, G. R. & Martinet, W. Defective autophagy in vascular smooth muscle cells alters contractility and Ca2+ homeostasis in mice. Am. J. Physiol. Heart Circulatory Physiol. 308, H557–H567 (2015).
Osonoi, Y. et al. Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy 14, 1991–2006 (2018).
Wu, X. et al. Impaired autophagy contributes to adverse cardiac remodeling in acute myocardial infarction. PLoS ONE 9, e112891 (2014).
Kubli, D. A. et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 288, 915–926 (2013).
Gottlieb, R. A. & Mentzer, R. M. Jr. Autophagy: an affair of the heart. Heart Fail Rev. 18, 575–584 (2013).
Cardoso, C. M. et al. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS ONE 4, e4424 (2009).
Li, T. F. et al. Overexpression of kinesin superfamily members as prognostic biomarkers of breast cancer. Cancer Cell Int. 20, 123 (2020).
Olson, O. C. & Joyce, J. A. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 15, 712–729 (2015).
Finicle, B. T., Jayashankar, V. & Edinger, A. L. Nutrient scavenging in cancer. Nat. Rev. Cancer 18, 619–633 (2018).
Chen, D. S. & Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017).
Yamamoto, K. et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105 (2020).
Yamamoto, K., Venida, A., Perera, R. M. & Kimmelman, A. C. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy 16, 1524–1525 (2020).
Bernard, D. et al. Select microtubule inhibitors increase lysosome acidity and promote lysosomal disruption in acute myeloid leukemia (AML) cells. Apoptosis 20, 948–959 (2015).
Cheng, L. et al. POS0458 IDENTIFICATION OF HUB GENES AND MOLECULAR PATHWAYS IN PATIENTS WITH RHEUMATOID ARTHRITIS BY BIOINFORMATICS ANALYSIS. Ann. Rheum. Dis. 80, 460 (2021).
Weitoft, T. et al. Cathepsin S and cathepsin L in serum and synovial fluid in rheumatoid arthritis with and without autoantibodies. Rheumatol. (Oxf., Engl.) 54, 1923–1928 (2015).
Hashimoto, Y. et al. Significance of cathepsin B accumulation in synovial fluid of rheumatoid arthritis. Biochem. Biophys. Res. Commun. 283, 334–339 (2001).
Clarke, A. J. et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann. Rheum. Dis. 74, 912–920 (2015).
Li, B., Yue, Y., Dong, C., Shi, Y. & Xiong, S. Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clin. Exp. Rheumatol. 32, 705–714 (2014).
Shin, Y. J. et al. Autophagy induction and CHOP under-expression promotes survival of fibroblasts from rheumatoid arthritis patients under endoplasmic reticulum stress. Arthritis Res. Ther. 12, R19 (2010).
Xu, K. et al. Autophagy induction contributes to the resistance to methotrexate treatment in rheumatoid arthritis fibroblast-like synovial cells through high mobility group box chromosomal protein 1. Arthritis Res. Ther. 17, 374 (2015).
An, Q., Yan, W., Zhao, Y. & Yu, K. Enhanced neutrophil autophagy and increased concentrations of IL-6, IL-8, IL-10 and MCP-1 in rheumatoid arthritis. Int. Immunopharmacol. 65, 119–128 (2018).
Alessandri, C. et al. CD4 T lymphocyte autophagy is upregulated in the salivary glands of primary Sjögren’s syndrome patients and correlates with focus score and disease activity. Arthritis Res. Ther. 19, 178 (2017).
Colafrancesco, S. et al. SAT0004 EVALUATION OF AUTOPHAGY IN INFILTRATING AND CIRCULATINGLYMPHOCYTES FROM PATIENTS WITH SJOGREN’S SYNDROME. Ann. Rheum. Dis. 78, 1066–1067 (2019).
Wilhelm, M. et al. Implication of a lysosomal antigen in the pathogenesis of lupus erythematosus. J. Autoimmun. 120, 102633 (2021).
Lin, N. Y. et al. Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann. Rheum. Dis. 72, 761–768 (2013).
Kim, E. K. et al. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis. 8, e2565 (2017).
van Loosdregt, J. et al. Increased autophagy in CD4(+) T cells of rheumatoid arthritis patients results in T-cell hyperactivation and apoptosis resistance. Eur. J. Immunol. 46, 2862–2870 (2016).
Yang, Z., Fujii, H., Mohan, S. V., Goronzy, J. J. & Weyand, C. M. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J. Exp. Med. 210, 2119–2134 (2013).
Gros, F. & Muller, S. Pharmacological regulators of autophagy and their link with modulators of lupus disease. Br. J. Pharm. 171, 4337–4359 (2014).
Harris, V. M. et al. Characterization of cxorf21 provides molecular insight into female-bias immune response in SLE pathogenesis. Front. Immunol. 10, 2160 (2019).
Monteith, A. J. et al. Defects in lysosomal maturation facilitate the activation of innate sensors in systemic lupus erythematosus. Proc. Natl Acad. Sci. USA 113, E2142–E2151 (2016).
Nirk, E. L., Reggiori, F. & Mauthe, M. Hydroxychloroquine in rheumatic autoimmune disorders and beyond. Embo Mol. Med. 12, e12476 (2020).
Hansson, O. Biomarkers for neurodegenerative diseases. Nat. Med. 27, 954–963 (2021).
Bloom, G. S. Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. Jama Neurol. 71, 505–508 (2014).
Rui, Y. N. et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 17, 262–275 (2015).
Zhang, Z. et al. Role of lysosomes in physiological activities, diseases, and therapy. J. Hematol. Oncol. 14, 79 (2021).
del Toro, D. et al. Mutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineurin/Rab8 complex from the Golgi apparatus. Mol. Biol. Cell 20, 1478–1492 (2009).
Mazzulli, J. R., Zunke, F., Isacson, O., Studer, L. & Krainc, D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl Acad. Sci. USA 113, 1931–1936 (2016).
Colacurcio, D. J. & Nixon, R. A. Disorders of lysosomal acidification-the emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 32, 75–88 (2016).
Polito, V. A. et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. Embo Mol. Med. 6, 1142–1160 (2014).
Xiao, Q. et al. Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing Aβ Generation and Amyloid Plaque Pathogenesis. J. Neurosci. 35, 12137–12151 (2015).
Xiao, Q. et al. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 34, 9607–9620 (2014).
Decressac, M. et al. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl Acad. Sci. USA 110, E1817–E1826 (2013).
Kilpatrick, K., Zeng, Y., Hancock, T. & Segatori, L. Genetic and chemical activation of TFEB mediates clearance of aggregated α-synuclein. PLoS ONE 10, e0120819 (2015).
Tsunemi, T. et al. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 4, 142ra97 (2012).
Napolitano, G. & Ballabio, A. TFEB at a glance. J. Cell Sci. 129, 2475–2481 (2016).
Koga, H., Martinez-Vicente, M., Macian, F., Verkhusha, V. V. & Cuervo, A. M. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2, 386 (2011).
Qi, L. et al. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS ONE 7, e46834 (2012).
Xilouri, M., Vogiatzi, T., Vekrellis, K., Park, D. & Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 4, e5515 (2009).
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T. & Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 (2004).
Hook, V. et al. Cathepsin B in neurodegeneration of Alzheimer’s disease, traumatic brain injury, and related brain disorders. Biochim. Biophys. Acta Proteins Proteom. 1868, 140428 (2020).
Qin, Z. H. et al. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 12, 3231–3244 (2003).
Siman, R. et al. Processing of the beta-amyloid precursor. Multiple proteases generate and degrade potentially amyloidogenic fragments. J. Biol. Chem. 268, 16602–16609 (1993).
Bi, X. et al. Novel cathepsin D inhibitors block the formation of hyperphosphorylated tau fragments in hippocampus. J. Neurochem. 74, 1469–1477 (2000).
Sun, Y. et al. Translational study of Alzheimer’s disease (AD) biomarkers from brain tissues in AβPP/PS1 mice and serum of AD patients. J. Alzheimers Dis. 45, 269–282 (2015).
Mialet-Perez, J. & Vindis, C. Autophagy in health and disease: focus on the cardiovascular system. Essays Biochem. 61, 721–732 (2017).
Ma, X., Godar, R. J., Liu, H. & Diwan, A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 8, 297–309 (2012).
Wu, P. et al. Myocardial upregulation of cathepsin D by ischemic heart disease promotes autophagic flux and protects against cardiac remodeling and heart failure. Circ. Heart Fail. 10, e004044 (2017).
Endo, Y., Furuta, A. & Nishino, I. Danon disease: a phenotypic expression of LAMP-2 deficiency. Acta Neuropathol. 129, 391–398 (2015).
Ferdinandy, P. et al. Definition of hidden drug cardiotoxicity: paradigm change in cardiac safety testing and its clinical implications. Eur Heart J. 40, 1771–1777 (2019).
Li, D. L. et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 133, 1668–1687 (2016).
Pizarro, M. et al. Basal autophagy protects cardiomyocytes from doxorubicin-induced toxicity. Toxicology 370, 41–48 (2016).
Bartlett, J. J., Trivedi, P. C., Yeung, P., Kienesberger, P. C. & Pulinilkunnil, T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem. J. 473, 3769–3789 (2016).
Liu, B. et al. STAT3 associates with vacuolar H(+)-ATPase and regulates cytosolic and lysosomal pH. Cell Res. 28, 996–1012 (2018).
Perez-Sayans, M., Somoza-Martin, J. M., Barros-Angueira, F., Rey, J. M. & Garcia-Garcia, A. V-ATPase inhibitors and implication in cancer treatment. Cancer Treat. Rev. 35, 707–713 (2009).
Santos-Pereira, C., Rodrigues, L. R. & Côrte-Real, M. Emerging insights on the role of V-ATPase in human diseases: Therapeutic challenges and opportunities. Med. Res. Rev. 41, 1927–1964 (2021).
Wiedmann, R. M. et al. The V-ATPase-inhibitor archazolid abrogates tumor metastasis via inhibition of endocytic activation of the Rho-GTPase Rac1. Cancer Res 72, 5976–5987 (2012).
Dixon, N. et al. Interaction of spin-labeled inhibitors of the vacuolar H + -ATPase with the transmembrane Vo-sector. Biophys. J. 94, 506–514 (2008).
Mauvezin, C., Nagy, P., Juhász, G. & Neufeld, T. P. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 6, 7007 (2015).
Mauvezin, C. & Neufeld, T. P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 11, 1437–1438 (2015).
Xie, X. S. et al. Salicylihalamide A inhibits the V0 sector of the V-ATPase through a mechanism distinct from bafilomycin A1. J. Biol. Chem. 279, 19755–19763 (2004).
Peng, W. et al. A distinct inhibitory mechanism of the V-ATPase by Vibrio VopQ revealed by cryo-EM. Nat. Struct. Mol. Biol. 27, 589–597 (2020).
Amaravadi, R. K. & Winkler, J. D. Lys05: a new lysosomal autophagy inhibitor. Autophagy 8, 1383–1384 (2012).
Salata, C., Calistri, A., Parolin, C., Baritussio, A. & Palù, G. Antiviral activity of cationic amphiphilic drugs. Expert Rev. Anti Infect. Ther. 15, 483–492 (2017).
Mauthe, M. et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14, 1435–1455 (2018).
Pascolo, S. Time to use a dose of Chloroquine as an adjuvant to anti-cancer chemotherapies. Eur. J. Pharmacol. 771, 139–144 (2016).
Marmor, M. F., Kellner, U., Lai, T. Y., Melles, R. B. & Mieler, W. F. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 Revision). Ophthalmology 123, 1386–1394 (2016).
Goodall, M. L. et al. Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib. Autophagy 10, 1120–1136 (2014).
Rebecca, V. W. et al. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 7, 1266–1283 (2017).
Rebecca, V. W. et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov. 9, 220–229 (2019).
Atiskova, Y. et al. Mice deficient in the lysosomal enzyme palmitoyl-protein thioesterase 1 (PPT1) display a complex retinal phenotype. Sci. Rep. 9, 14185 (2019).
Bourdenx, M. et al. Nanoparticles restore lysosomal acidification defects: Implications for Parkinson and other lysosomal-related diseases. Autophagy 12, 472–483 (2016).
Khaket, T. P., Kwon, T. K. & Kang, S. C. Cathepsins: potent regulators in carcinogenesis. Pharmacol. Therapeut. 198, 1–19 (2019).
Hadad, E. H. et al. Prognostic role and therapeutic susceptibility of cathepsin in various types of solid tumor and leukemia: a systematic review. J. Cell Physiol. 235, 7709–7730 (2020).
Breznik, B., Mitrović, A., T Lah, T. & Kos, J. Cystatins in cancer progression: More than just cathepsin inhibitors. Biochimie 166, 233–250 (2019).
Stoka, V., Turk, V. & Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 32, 22–37 (2016).
Patel, S., Homaei, A., El-Seedi, H. R. & Akhtar, N. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed. Pharmacother. 105, 526–532 (2018).
Vidak, E., Javoršek, U., Vizovišek, M. & Turk, B. Cysteine cathepsins and their extracellular roles: shaping the microenvironment. Cells. 8, 264 (2019).
Sudhan, D. R. & Siemann, D. W. Cathepsin L targeting in cancer treatment. Pharmacol. Therapeut. 155, 105–116 (2015).
Patel, S., Homaei, A., El-Seedi, H. R. & Akhtar, N. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed. Pharmacother.105, 526–532 (2018).
Dai, R. et al. Cathepsin K: the action in and beyond bone. Front. Cell Dev. Biol. 8, 433 (2020).
McClung, M. R. et al. Odanacatib for the treatment of postmenopausal osteoporosis: results of the LOFT multicentre, randomised, double-blind, placebo-controlled trial and LOFT Extension study. Lancet Diabetes Endocrinol. 7, 899–911 (2019).
Lopez, T. et al. Functional selection of protease inhibitory antibodies. Proc. Natl Acad. Sci. USA 116, 16314–16319 (2019).
Tripathi, R. et al. Abl and Arg mediate cysteine cathepsin secretion to facilitate melanoma invasion and metastasis. Sci. Signal. 11, eaao0422 (2018).
Pike, L. S. et al. Imaging gene delivery in a mouse model of congenital neuronal ceroid lipofuscinosis. Gene Ther. 18, 1173–1178 (2011).
Marques, A. R. A. et al. Enzyme replacement therapy with recombinant pro-CTSD (cathepsin D) corrects defective proteolysis and autophagy in neuronal ceroid lipofuscinosis. Autophagy 16, 811–825 (2020).
Kravic, B., Behrends, C. & Meyer, H. Regulation of lysosome integrity and lysophagy by the ubiquitin-conjugating enzyme UBE2QL1. Autophagy 16, 179–180 (2020).
Gabandé-Rodríguez, E. et al. Lipid-induced lysosomal damage after demyelination corrupts microglia protective function in lysosomal storage disorders. Embo J. 38, (2019).
Cash, T. P. et al. Induction of lysosome membrane permeabilization as a therapeutic strategy to target pancreatic cancer stem cells. Cancers (Basel). 12, 1790 (2020).
Krenn, M. A. et al. Ferritin-stimulated lipid peroxidation, lysosomal leak, and macroautophagy promote lysosomal “metastability” in primary hepatocytes determining in vitro cell survival. Free Radic. Biol. Med. 80, 48–58 (2015).
Huang, T. et al. Growth inhibition of a novel iron chelator, DpdtC, against hepatoma carcinoma cell lines partly attributed to ferritinophagy-mediated lysosomal ROS generation. Oxid. Med. Cell Longev. 2018, 4928703 (2018).
Martins, W. K. et al. Parallel damage in mitochondria and lysosomes is an efficient way to photoinduce cell death. Autophagy 15, 259–279 (2019).
Mai, T. T. et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 9, 1025–1033 (2017).
Sun, X. et al. Targeting autophagy enhances the anticancer effect of artemisinin and its derivatives. Med. Res. Rev. 39, 2172–2193 (2019).
Huang, L. et al. An artemisinin-mediated ROS evolving and dual protease light-up nanocapsule for real-time imaging of lysosomal tumor cell death. BIosens Bioelectron. 92, 724–732 (2017).
Oliveira, C. A. F., Ivanova, L., Solhaug, A. & Fæste, C. K. Enniatin B-induced lysosomal membrane permeabilization in mouse embryonic fibroblasts. Mycotoxin. Res. 36, 23–30 (2020).
Balogi, Z. et al. Hsp70 interactions with membrane lipids regulate cellular functions in health and disease. Prog Lipid Res. 74, 18–30 (2019).
Nylandsted, J. et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 200, 425–435 (2004).
Mitrofan, L. M., Castells, F. B., Pelkonen, J. & Mönkkönen, J. Lysosomal-mitochondrial axis in zoledronic acid-induced apoptosis in human follicular lymphoma cells. J. Biol. Chem. 285, 1967–1979 (2010).
Li, L. et al. Riccardin D-N induces lysosomal membrane permeabilization by inhibiting acid sphingomyelinase and interfering with sphingomyelin metabolism in vivo. Toxicol. Appl. Pharm. 310, 175–184 (2016).
Petersen, N. H. T. et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 24, 379–393 (2013).
Domagala, A. et al. Typical and atypical inducers of lysosomal cell death: a promising anticancer strategy. Int. J. Mol. Sci. 19, 2256 (2018).
Kornhuber, J. et al. Lipophilic cationic drugs increase the permeability of lysosomal membranes in a cell culture system. J. CELL PHYSIOL 224, 152–164 (2010).
Fucho, R. et al. ASMase regulates autophagy and lysosomal membrane permeabilization and its inhibition prevents early stage non-alcoholic steatohepatitis. J. Hepatol. 61, 1126–1134 (2014).
Canals, D., Perry, D. M., Jenkins, R. W. & Hannun, Y. A. Drug targeting of sphingolipid metabolism: sphingomyelinases and ceramidases. Brit. J. Pharmacol. 163, 694–712 (2011).
Elmallah, M. I. Y. et al. Membrane-anchored heat-shock protein 70 (Hsp70) in cancer. Cancer Lett. 469, 134–141 (2020).
Granato, M. et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. 4, e730 (2013).
Dudeja, V. et al. Heat shock protein 70 inhibits apoptosis in cancer cells through simultaneous and independent mechanisms. Gastroenterology 136, 1772–1782 (2009).
Tanimura, S. et al. Anticancer drugs up-regulate HspBP1 and thereby antagonize the prosurvival function of Hsp70 in tumor cells. J. Biol. Chem. 282, 35430–35439 (2007).
Radulovic, M. et al. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. Embo J. 37, e99753 (2018).
Papadopoulos, C., Kravic, B. & Meyer, H. Repair or lysophagy: dealing with damaged lysosomes. J. Mol. Biol. 432, 231–239 (2020).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 17, 1–382 (2021).
Repnik, U. et al. L-leucyl-L-leucine methyl ester does not release cysteine cathepsins to the cytosol but inactivates them in transiently permeabilized lysosomes. J. Cell Sci. 130, 3124–3140 (2017).
Vietri, M., Radulovic, M. & Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 21, 25–42 (2020).
Skowyra, M. L., Schlesinger, P. H., Naismith, T. V. & Hanson, P. I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360, eaar5078 (2018).
Thurston, T. L., Wandel, M. P., von Muhlinen, N., Foeglein, A. & Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418 (2012).
Fiskin, E., Bionda, T., Dikic, I. & Behrends, C. Global analysis of host and bacterial ubiquitinome in response to Salmonella typhimurium infection. Mol. Cell 62, 967–981 (2016).
Mizushima, N. The ubiquitin E2 enzyme UBE2QL1 mediates lysophagy. EMBO Rep. 20, e49104 (2019).
Koerver, L. et al. The ubiquitin-conjugating enzyme UBE2QL1 coordinates lysophagy in response to endolysosomal damage. EMBO Rep. 20, e48014 (2019).
Gupta, S. et al. Lysosomal retargeting of Myoferlin mitigates membrane stress to enable pancreatic cancer growth. Nat. Cell Biol. 23, 232–242 (2021).
Corona, A. K. & Jackson, W. T. Finding the middle ground for autophagic fusion requirements. Trends Cell Biol. 28, 869–881 (2018).
Liu, X. et al. The role of lysosomes in BDE 47-mediated activation of mitochondrial apoptotic pathway in HepG2 cells. Chemosphere 124, 10–21 (2015).
Chen, Q. et al. Potassium bisperoxo(1,10-phenanthroline)oxovanadate (bpV(phen)) induces apoptosis and pyroptosis and disrupts the P62-HDAC6 protein interaction to suppress the acetylated microtubule-dependent degradation of autophagosomes. J. Biol. Chem. 290, 26051–26058 (2015).
Rusmini, P. et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 15, 631–651 (2019).
Liu, L. et al. Lysosomal dysfunction and autophagy blockade contribute to IMB-6G-induced apoptosis in pancreatic cancer cells. Sci. Rep.-UK 7, 41862 (2017).
Rosato, A. S., Tang, R. & Grimm, C. Two-pore and TRPML cation channels: regulators of phagocytosis, autophagy and lysosomal exocytosis. Pharm. Ther. 220, 107713 (2021).
Lloyd-Evans, E. & Waller-Evans, H. Lysosomal Ca(2+) homeostasis and signaling in health and disease. Cold Spring Harbor. Perspect. Biol. 12, a035311 (2020).
Wu Y., Huang P. & Dong X. P. Lysosomal Calcium Channels in Autophagy and Cancer. Cancers (Basel). 13, (2021).
Schöndorf, D. C. et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5, 4028 (2014).
Gómez-Suaga, P. & Hilfiker, S. LRRK2 as a modulator of lysosomal calcium homeostasis with downstream effects on autophagy. Autophagy 8, 692–693 (2012).
Li, P., Gu, M. & Xu, H. Lysosomal ion channels as decoders of cellular signals. Trends Biochem. Sci. 44, 110–124 (2019).
Nguyen, O. N. et al. Two-pore channel function is crucial for the migration of invasive cancer cells. Cancer Res. 77, 1427–1438 (2017).
Boudewyn, L. C. & Walkley, S. U. Current concepts in the neuropathogenesis of mucolipidosis type IV. J. Neurochem. 148, 669–689 (2019).
Santoni, G., Santoni, M., Maggi, F., Marinelli, O. & Morelli, M. B. Emerging role of mucolipins TRPML channels in cancer. Front. Oncol. 10, 659 (2020).
Cheng, X., Shen, D., Samie, M. & Xu, H. Mucolipins: intracellular TRPML1-3 channels. Febs Lett. 584, 2013–2021 (2010).
Reddy, A., Caler, E. V. & Andrews, N. W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 106, 157–169 (2001).
Morelli, M. B. et al. Transient receptor potential mucolipin-1 channels in glioblastoma: role in patient’s survival. Cancers (Basel). 11, 525 (2019).
Plesch, E. et al. Selective agonist of TRPML2 reveals direct role in chemokine release from innate immune cells. Elife 7, (2018).
Wu, M., Li, X., Zhang, T., Liu, Z. & Zhao, Y. Identification of a nine-gene signature and establishment of a prognostic nomogram predicting overall survival of pancreatic cancer. Front. Oncol. 9, 996 (2019).
Gayle, S. et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood 129, 1768–1778 (2017).
Hou, J.-Z. et al. Inhibition of PIKfyve using YM201636 suppresses the growth of liver cancer via the induction of autophagy. Oncol. Rep. 41, 1971–1979 (2019).
Sharma, G. et al. A family of PIKFYVE inhibitors with therapeutic potential against autophagy-dependent cancer cells disrupt multiple events in lysosome homeostasis. Autophagy 15, 1694–1718 (2019).
Hockey, L. N. et al. Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J. Cell Sci. 128, 232–238 (2015).
Jia, J. et al. Galectins control MTOR and AMPK in response to lysosomal damage to induce autophagy. Autophagy 15, 169–171 (2019).
Guertin, D. A. & Sabatini, D. M. Defining the role of mTOR in cancer. Cancer Cell 12, 9–22 (2007).
Yu, L. et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946 (2010).
LoRusso, P. M. Inhibition of the PI3K/AKT/mTOR Pathway in Solid Tumors. J. Clin. Oncol. 34, 3803–3815 (2016).
Yang, J. et al. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol. Cancer 18, 26 (2019).
McKenna, M., McGarrigle, S. & Pidgeon, G. P. The next generation of PI3K-Akt-mTOR pathway inhibitors in breast cancer cohorts. Biochim. Biophys. Acta Rev. Cancer 1870, 185–197 (2018).
Revathidevi, S. & Munirajan, A. K. Akt in cancer: mediator and more. Semin. Cancer Biol. 59, 80–91 (2019).
Yao, Y. et al. Role of ragulator in the regulation of mechanistic target of rapamycin signaling in podocytes and glomerular function. J. Am. Soc. Nephrol. 27, 3653–3665 (2016).
Schweitzer, L. D., Comb, W. C., Bar-Peled, L. & Sabatini, D. M. Disruption of the rag-ragulator complex by c17orf59 Inhibits mTORC1. Cell Rep. 12, 1445–1455 (2015).
Bar-Peled, L., Schweitzer, L. D., Zoncu, R. & Sabatini, D. M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208 (2012).
Mahoney, S. J. et al. A small molecule inhibitor of Rheb selectively targets mTORC1 signaling. Nat. Commun. 9, 548 (2018).
Yuan, H.-X. et al. NLK phosphorylates Raptor to mediate stress-induced mTORC1 inhibition. Genes Dev. 29, 2362–2376 (2015).
Dumas, S. N. & Lamming, D. W. Next generation strategies for geroprotection via mTORC1 inhibition. J. Gerontol. Ser. A, Biol. Sci. Med. Sci. 75, 14–23 (2020).
Kaeberlein, M. & Galvan, V. Rapamycin and Alzheimer’s disease: time for a clinical trial? Sci. Transl. Med. 11, eaar4289 (2019).
Mao, Z. et al. CSN6 promotes the migration and invasion of cervical cancer cells by inhibiting autophagic degradation of cathepsin L. Int. J. Biol. Sci. 15, 1310–1324 (2019).
Cheng, B., Ren, Y., Cao, H. & Chen, J. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur. J. Med. Chem. 199, 112377 (2020).
Zhang, N. et al. SA-49, a novel aloperine derivative, induces MITF-dependent lysosomal degradation of PD-L1. Ebiomedicine 40, 151–162 (2019).
He, R. et al. TFEB-driven autophagy potentiates TGF-β induced migration in pancreatic cancer cells. J. Exp. Clin. Cancer Res.: CR 38, 340 (2019).
Liang, J., Jia, X., Wang, K. & Zhao, N. High expression of TFEB is associated with aggressive clinical features in colorectal cancer. Onco Targets Ther. 11, 8089–8098 (2018).
Giatromanolaki, A. et al. Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer 90, 98–105 (2015).
Lu, H., Sun, J., Hamblin, M. H., Chen, Y. E. & Fan, Y. Transcription factor EB regulates cardiovascular homeostasis. Ebiomedicine 63, 103207 (2021).
Rusmini, P. et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 15, 631–651 (2019).
Evans, T. D., Jeong, S. J., Zhang, X., Sergin, I. & Razani, B. TFEB and trehalose drive the macrophage autophagy-lysosome system to protect against atherosclerosis. Autophagy 14, 724–726 (2018).
Zhu, L. et al. Activation of TFEB-mediated autophagy by trehalose attenuates mitochondrial dysfunction in cisplatin-induced acute kidney injury. Theranostics 10, 5829–5844 (2020).
Chen, G. et al. 3,4-Dimethoxychalcone induces autophagy through activation of the transcription factors TFE3 and TFEB. Embo Mol. Med. 11, e10469 (2019).
Song, W., Wang, F., Lotfi, P., Sardiello, M. & Segatori, L. 2-Hydroxypropyl-β-cyclodextrin promotes transcription factor EB-mediated activation of autophagy: implications for therapy. J. Biol. Chem. 289, 10211–10222 (2014).
Wang, C. et al. Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat. Commun. 8, 2270 (2017).
Arias, E. & Cuervo, A. M. Pros and cons of chaperone-mediated autophagy in cancer biology. Trends Endocrinol. Metab. 31, 53–66 (2020).
Kaushik, S. & Cuervo, A. M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381 (2018).
Kon, M. et al. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 3, 109ra17 (2011).
Zhou, J. et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 12, 515–528 (2016).
Saha, T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 8, 1643–1656 (2012).
Page, N. et al. HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann. Rheum. Dis. 70, 837–843 (2011).
Wang, F., Tasset, I., Cuervo, A. M. & Muller, S. In vivo remodeling of altered autophagy-lysosomal pathway by a phosphopeptide in lupus. Cells 9, 2328 (2020).
Macri, C. et al. Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy 11, 472–486 (2015).
Langemeyer, L., Fröhlich, F. & Ungermann, C. Rab GTPase Function in endosome and lysosome biogenesis. Trends Cell Biol. 28, 957–970 (2018).
Qin, X. et al. Targeting Rabs as a novel therapeutic strategy for cancer therapy. Drug Discov. Today 22, 1139–1147 (2017).
Qin, Y. et al. CID1067700, a late endosome GTPase Rab7 receptor antagonist, attenuates brain atrophy, improves neurologic deficits and inhibits reactive astrogliosis in rat ischemic stroke. Acta Pharm. Sin. 40, 724–736 (2019).
Deraeve, C. et al. Psoromic acid is a selective and covalent Rab-prenylation inhibitor targeting autoinhibited RabGGTase. J. Am. Chem. Soc. 134, 7384–7391 (2012).
Spiegel, J. et al. Direct targeting of Rab-GTPase-effector interactions. Angew. Chem. Int. Ed. Engl. 53, 2498–2503 (2014).
Wolpin, B. M. et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 19, 637–638 (2014).
Rojas-Puentes, L. L. et al. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol. 8, 209 (2013).
Karasic, T. B. et al. Effect of gemcitabine and nab-paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: a phase 2 randomized clinical trial. Jama Oncol. 5, 993–998 (2019).
Zeh, H. J. et al. A randomized phase II preoperative study of autophagy inhibition with high-dose hydroxychloroquine and gemcitabine/nab-paclitaxel in pancreatic cancer patients. Clin. Cancer Res. 26, 3126–3134 (2020).
Boone, B. A. et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann. Surg. Oncol. 22, 4402–4410 (2015).
Sotelo, J., Briceño, E. & López-González, M. A. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 144, 337–343 (2006).
Rosenfeld, M. R. et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10, 1359–1368 (2014).
Huang, Z. et al. Clinical efficacy of mTOR inhibitors in solid tumors: a systematic review. Future Oncol. 11, 1687–1699 (2015).
U.S. food & drug administration. 2018 First Generic Drug Approvals. U.S. food & drug administration. https://www.fda.gov/drugs/first-generic-drug-approvals/2018-first-generic-drug-approvals (accessed August 5th 2021)
U.S. food & drug administration. 2019 First Generic Drug Approvals. U.S. food & drug administration. https://www.fda.gov/drugs/first-generic-drug-approvals/2018-first-generic-drug-approvals (accessed August 5th 2021)
Hudes, G. et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 356, 2271–2281 (2007).
Oza, A. M. et al. Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: a trial of the NCIC Clinical Trials Group. J. Clin. Oncol. 29, 3278–3285 (2011).
Ohtsu, A. et al. Everolimus for previously treated advanced gastric cancer: results of the randomized, double-blind, phase III GRANITE-1 study. J. Clin. Oncol. 31, 3935–3943 (2013).
Powles, T. et al. A randomised phase 2 study of AZD2014 versus everolimus in patients with VEGF-refractory metastatic clear cell renal cancer. Eur. Urol. 69, 450–456 (2016).
Wolin, E. et al. A phase 2 study of an oral mTORC1/mTORC2 kinase inhibitor (CC-223) for non-pancreatic neuroendocrine tumors with or without carcinoid symptoms. PLoS ONE 14, e0221994 (2019).
Graham, L. et al. A phase II study of the dual mTOR inhibitor MLN0128 in patients with metastatic castration resistant prostate cancer. Invest. N. Drugs 36, 458–467 (2018).
Baselga, J. et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 18, 904–916 (2017).
Yap, T. A. et al. Interrogating two schedules of the AKT inhibitor MK-2206 in patients with advanced solid tumors incorporating novel pharmacodynamic and functional imaging biomarkers. Clin. Cancer Res. 20, 5672–5685 (2014).
de Bono, J. S. et al. Randomized phase II study evaluating Akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN Loss. Clin. Cancer Res. 25, 928–936 (2019).
Jones, R. H. et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 21, 345–357 (2020).
André, F. et al. Alpelisib for -mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 380, 1929–1940 (2019).
Dent, S. et al. Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced breast cancer: the SANDPIPER trial. Ann. Oncol. 32, 197–207 (2021).
Rangwala, R. et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 10, 1391–1402 (2014).
Haas, N. B. et al. Autophagy inhibition to augment mtor inhibition: a phase I/II trial of everolimus and hydroxychloroquine in patients with previously treated renal cell carcinoma. Clin. Cancer Res. 25, 2080–2087 (2019).
Gewandter, J. S. et al. A phase III randomized, placebo-controlled study of topical amitriptyline and ketamine for chemotherapy-induced peripheral neuropathy (CIPN): a University of Rochester CCOP study of 462 cancer survivors. Support Care Cancer 22, 1807–1814 (2014).
Joel, N. & Stanford U. Phase 2a Desipramine in Small Cell Lung Cancer and Other High-Grade Neuroendocrine Tumors. ClinicalTrials.gov. https://ClinicalTrials.gov/show/NCT01719861 (2017).
Propper, D. J. et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann. Oncol. 10, 923–927 (1999).
Coleman, R. et al. Zoledronic acid (zoledronate) for postmenopausal women with early breast cancer receiving adjuvant letrozole (ZO-FAST study): final 60-month results. Ann. Oncol. 24, 398–405 (2013).
Terpos, E. et al. Treatment of multiple myeloma-related bone disease: recommendations from the Bone Working Group of the International Myeloma Working Group. Lancet Oncol. 22, e119–e130 (2021).
Studies found for cathepsin and cancer. ClinicalTrials.gov Search Results. https://clinicaltrials.gov/ct2/results?cond=Cancer&term=cathepsin&cntry=&state=&city=&dist= (accessed August 4th 2021).
Jensen, A. B. et al. The cathepsin K inhibitor odanacatib suppresses bone resorption in women with breast cancer and established bone metastases: results of a 4-week, double-blind, randomized, controlled trial. Clin. Breast Cancer 10, 452–458 (2010).
Wang, M., Xia, F., Wei, Y. & Wei, X. Molecular mechanisms and clinical management of cancer bone metastasis. Bone Res. 8, 30 (2020).
Fanouriakis, A. et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann. Rheum. Dis. 78, 736–745 (2019).
Costedoat-Chalumeau, N. et al. Hydroxychloroquine in systemic lupus erythematosus: results of a French multicentre controlled trial (PLUS Study). Ann. Rheum. Dis. 72, 1786–1792 (2013).
Marmor, M. F. et al. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 Revision). Ophthalmology 123, 1386–1394 (2016).
Clark, P. et al. Hydroxychloroquine compared with placebo in rheumatoid arthritis. A randomized controlled trial. Ann. Intern. Med. 119, 1067–1071 (1993).
Carmichael, S. J., Beal, J., Day, R. O. & Tett, S. E. Combination therapy with methotrexate and hydroxychloroquine for rheumatoid arthritis increases exposure to methotrexate. J. Rheumatol. 29, 2077–2083 (2002).
Wasko, M. C. et al. Hydroxychloroquine and risk of diabetes in patients with rheumatoid arthritis. JAMA 298, 187–193 (2007).
Solomon, D. H. et al. Effect of hydroxychloroquine on insulin sensitivity and lipid parameters in rheumatoid arthritis patients without diabetes mellitus: a randomized, blinded crossover trial. Arthritis Care Res. (Hoboken) 66, 1246–1251 (2014).
Rempenault, C. et al. Metabolic and cardiovascular benefits of hydroxychloroquine in patients with rheumatoid arthritis: a systematic review and meta-analysis. Ann. Rheum. Dis. 77, 98–103 (2018).
Liu, D. et al. Chloroquine and hydroxychloroquine are associated with reduced cardiovascular risk: a systematic review and meta-analysis. Drug Des. Dev. Ther. 12, 1685–1695 (2018).
Kerr, G. et al. Associations of hydroxychloroquine use with lipid profiles in rheumatoid arthritis: pharmacologic implications. Arthritis Care Res. (Hoboken) 66, 1619–1626 (2014).
Morris, S. J. et al. Hydroxychloroquine use associated with improvement in lipid profiles in rheumatoid arthritis patients. Arthritis Care Res. (Hoboken) 63, 530–534 (2011).
Muller, S. et al. Spliceosomal peptide P140 for immunotherapy of systemic lupus erythematosus: results of an early phase II clinical trial. Arthritis Rheumatism 58, 3873–3883 (2008).
ClinicalTrials.gov. A 52-Week, Randomized, Double-blind, Parallel-group, Placebo-controlled Study to Evaluate the Efficacy and Safety of a 200-mcg Dose of IPP-201101 Plus Standard of Care in Patients With Systemic Lupus Erythematosus. ClinicalTrials.gov. https://ClinicalTrials.gov/show/NCT02504645 (2019).
Zimmer, R., Scherbarth, H. R., Rillo, O. L., Gomez-Reino, J. J. & Muller, S. Lupuzor/P140 peptide in patients with systemic lupus erythematosus: a randomised, double-blind, placebo-controlled phase IIb clinical trial. Ann. Rheum. Dis. 72, 1830–1835 (2013).
Komatsu, N. & Takayanagi, H. Arthritogenic T cells in autoimmune arthritis. Int. J. Biochem. Cell Biol. 58, 92–96 (2015).
Noack, M. & Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev 13, 668–677 (2014).
Wen, H. Y. et al. Low-dose sirolimus immunoregulation therapy in patients with active rheumatoid arthritis: a 24-week follow-up of the randomized, open-label, parallel-controlled trial. J. Immunol. Res. 2019, 7684352 (2019).
Lai, Z. W. et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet 391, 1186–1196 (2018).
Rabanal-Ruiz, Y., Otten, E. G. & Korolchuk, V. I. mTORC1 as the main gateway to autophagy. Essays Biochem. 61, 565–584 (2017).
Lee, K. et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32, 743–753 (2010).
Hoffmann-La, R. A study to assess the efficacy of RO5459072 in participants with primary Sjogren’s syndrome. https://ClinicalTrials.gov/show/NCT02701985 (2018).
Wang, H. et al. Selective in vivo metabolic cell-labeling-mediated cancer targeting. Nat. Chem. Biol. 13, 415–424 (2017).
Drago, J. Z., Modi, S. & Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 18, 327–344 (2021).
Dheer, D., Nicolas, J. & Shankar, R. Cathepsin-sensitive nanoscale drug delivery systems for cancer therapy and other diseases. Adv. Drug Deliv. Rev. 151-152, 130–151 (2019).
Inpanathan, S. & Botelho, R. J. The lysosome signaling platform: adapting with the times. Front. Cell Dev. Biol. 7, 113 (2019).
Zhou, Y. et al. Cathepsin K deficiency ameliorates systemic lupus erythematosus-like manifestations in Fas(lpr) mice. J. Immunol. 198, 1846–1854 (2017).
Gros, F. et al. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy 8, 1113–1123 (2012).
Zhou, X. J., Klionsky, D. J. & Zhang, H. Podocytes and autophagy: a potential therapeutic target in lupus nephritis. Autophagy 15, 908–912 (2019).
Qi, Y. Y. et al. Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-α in lupus nephritis. Ann. Rheum. Dis. 77, 1799–1809 (2018).
Martinez, J. et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 533, 115–119 (2016).
Wei, W. et al. Inhibition of Ctsk modulates periodontitis with arthritis via downregulation of TLR9 and autophagy. Cell Prolif. 53, e12722 (2020).
Hummel, K. M. et al. Cysteine proteinase cathepsin K mRNA is expressed in synovium of patients with rheumatoid arthritis and is detected at sites of synovial bone destruction. J. Rheumatol. 25, 1887–1894 (1998).
Miyata, J. et al. Cathepsin G: the significance in rheumatoid arthritis as a monocyte chemoattractant. Rheumatol. Int. 27, 375–382 (2007).
Zhu, L. et al. The autophagy level is increased in the synovial tissues of patients with active rheumatoid arthritis and is correlated with disease severity. Mediators Inflamm. 2017, 7623145 (2017).
Steinfeld, S. et al. Prolactin up-regulates cathepsin B and D expression in minor salivary glands of patients with Sjögren’s syndrome. Lab. Invest. 80, 1711–1720 (2000).
Li, B., Wang, F., Schall, N. & Muller, S. Rescue of autophagy and lysosome defects in salivary glands of MRL/lpr mice by a therapeutic phosphopeptide. J. Autoimmun. 90, 132–145 (2018).
Gnant, M. et al. Zoledronic acid combined with adjuvant endocrine therapy of tamoxifen versus anastrozol plus ovarian function suppression in premenopausal early breast cancer: final analysis of the Austrian Breast and Colorectal Cancer Study Group Trial 12. Ann. Oncol. 26, 313–320 (2015).
Yokogawa, N. et al. Effects of hydroxychloroquine in patients with cutaneous lupus erythematosus: a multicenter, double-blind, randomized, parallel-group trial. Arthritis Rheumatol. 69, 791–799 (2017).
Wasko, M. C. et al. Antidiabetogenic effects of hydroxychloroquine on insulin sensitivity and beta cell function: a randomised trial. Diabetologia 58, 2336–2343 (2015).
Merk, H. et al. Inhibition of the V-ATPase by Archazolid A: a new strategy to inhibit EMT. Mol. Cancer Ther. 16, 2329–2339 (2017).
Peng, W. et al. A distinct inhibitory mechanism of the V-ATPase by Vibrio VopQ revealed by cryo-EM. Nat. Struct. Mol. Biol. 27, 589–597 (2020).
Sreelatha, A. et al. Vibrio effector protein, VopQ, forms a lysosomal gated channel that disrupts host ion homeostasis and autophagic flux. Proc. Natl Acad. Sci. USA 110, 11559–11564 (2013).
Sreelatha, A. et al. Vibrio effector protein VopQ inhibits fusion of V-ATPase-containing membranes. Proc. Natl Acad Sci. USA 112, 100–105 (2015).
McAfee, Q. et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl Acad. Sci. USA 109, 8253–8258 (2012).
Colella, R. et al. Induction of cell death in neuroblastoma by inhibition of cathepsins B and L. Cancer Lett. 294, 195–203 (2010).
Bell-McGuinn, K. M., Garfall, A. L., Bogyo, M., Hanahan, D. & Joyce, J. A. Inhibition of cysteine cathepsin protease activity enhances chemotherapy regimens by decreasing tumor growth and invasiveness in a mouse model of multistage cancer. Cancer Res. 67, 7378–7385 (2007).
Schurigt, U. et al. Trial of the cysteine cathepsin inhibitor JPM-OEt on early and advanced mammary cancer stages in the MMTV-PyMT-transgenic mouse model. Biol. Chem. 389, 1067–1074 (2008).
Mirković, B. et al. Nitroxoline impairs tumor progression in vitro and in vivo by regulating cathepsin B activity. Oncotarget 6, 19027–19042 (2015).
Maynadier, M. et al. Dipeptide mimic oligomer transporter mediates intracellular delivery of Cathepsin D inhibitors: a potential target for cancer therapy. J. Control. Release 171, 251–257 (2013).
Duong, L. T., Wesolowski, G. A., Leung, P., Oballa, R. & Pickarski, M. Efficacy of a cathepsin K inhibitor in a preclinical model for prevention and treatment of breast cancer bone metastasis. Mol. Cancer Ther. 13, 2898–2909 (2014).
Jerome, C., Missbach, M. & Gamse, R. Balicatib, a cathepsin K inhibitor, stimulates periosteal bone formation in monkeys. Osteoporos. Int. 23, 339–349 (2012).
Jakoš, T., Pišlar, A., Pečar Fonović, U., Švajger, U. & Kos, J. Cysteine cathepsins L and X differentially modulate interactions between myeloid-derived suppressor cells and tumor cells. Cancer Immunol. Immunother. 69, 1869–1880 (2020).
Chavarria, G. E. et al. Initial evaluation of the antitumour activity of KGP94, a functionalized benzophenone thiosemicarbazone inhibitor of cathepsin L. Eur. J. Med. Chem. 58, 568–572 (2012).
Zheng, X. et al. Cathepsin L inhibition suppresses drug resistance in vitro and in vivo: a putative mechanism. Am. J. Physiol. Cell Physiol. 296, C65–C74 (2009).
Vázquez, R. et al. Fsn0503h antibody-mediated blockade of cathepsin S as a potential therapeutic strategy for the treatment of solid tumors. Biochimie 108, 101–107 (2015).
Theron, M. et al. Pharmacodynamic Monitoring of RO5459072, a Small Molecule Inhibitor of Cathepsin S. Front. Immunol. 8, 806 (2017).
Mitrofan, L. M., Castells, F. B., Pelkonen, J. & Mönkkönen, J. Lysosomal-mitochondrial axis in zoledronic acid-induced apoptosis in human follicular lymphoma cells. J. Biol. Chem. 285, 1967–1979 (2010).
Beckmann, N., Sharma, D., Gulbins, E., Becker, K. A. & Edelmann, B. Inhibition of acid sphingomyelinase by tricyclic antidepressants and analogons. Front. Physiol. 5, 331 (2014).
Groth-Pedersen, L., Ostenfeld, M. S., Hoyer-Hansen, M., Nylandsted, J. & Jaattela, M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res. 67, 2217–2225 (2007).
Li, D. et al. Sulforaphane activates a lysosome-dependent transcriptional program to mitigate oxidative stress. Autophagy. 17, 872–887 (2021).
Lyu, L. et al. TBBPA regulates calcium-mediated lysosomal exocytosis and thereby promotes invasion and migration in hepatocellular carcinoma. Ecotox. Environ. SAFE 192, 110255 (2020).
Hua, H. et al. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 12, 71 (2019).
Mortensen, D. S. et al. CC-223, a potent and selective inhibitor of mTOR kinase: in vitro and in vivo characterization. Mol. Cancer Ther. 14, 1295–1305 (2015).
Sergin, I. et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat. Commun. 8, 15750 (2017).
Caza, T. N. et al. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann. Rheum. Dis. 73, 1888–1897 (2014).
Funding
This research was financially supported by the National Natural Science Foundation of China (No. 82073095).
Author information
Authors and Affiliations
Contributions
X.X.H. designed and finalized the study; M.D.C. wrote the review and generated the figures and tables; X.Y.L., K.M.W. and X.X.H. revised the review. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, M., Luo, X., Wu, K. et al. Targeting lysosomes in human disease: from basic research to clinical applications. Sig Transduct Target Ther 6, 379 (2021). https://doi.org/10.1038/s41392-021-00778-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00778-y
This article is cited by
-
Frontier knowledge and future directions of programmed cell death in clear cell renal cell carcinoma
Cell Death Discovery (2024)
-
Protein lipidation in health and disease: molecular basis, physiological function and pathological implication
Signal Transduction and Targeted Therapy (2024)
-
The role of lysosomes in metabolic and autoimmune diseases
Nature Reviews Nephrology (2023)
-
A nitric-oxide driven chemotactic nanomotor for enhanced immunotherapy of glioblastoma
Nature Communications (2023)
-
Phosphoinositides and intracellular calcium signaling: novel insights into phosphoinositides and calcium coupling as negative regulators of cellular signaling
Experimental & Molecular Medicine (2023)
