
Abstract
The response of immune cells in cardiac injury is divided into three continuous phases: inflammation, proliferation and maturation. The kinetics of the inflammatory and proliferation phases directly influence the tissue repair. In cardiac homeostasis, cardiac tissue resident macrophages (cTMs) phagocytose bacteria and apoptotic cells. Meanwhile, NK cells prevent the maturation and transport of inflammatory cells. After cardiac injury, cTMs phagocytose the dead cardiomyocytes (CMs), regulate the proliferation and angiogenesis of cardiac progenitor cells. NK cells prevent the cardiac fibrosis, and promote vascularization and angiogenesis. Type 1 macrophages trigger the cardioprotective responses and promote tissue fibrosis in the early stage. Reversely, type 2 macrophages promote cardiac remodeling and angiogenesis in the late stage. Circulating macrophages and neutrophils firstly lead to chronic inflammation by secreting proinflammatory cytokines, and then release anti-inflammatory cytokines and growth factors, which regulate cardiac remodeling. In this process, dendritic cells (DCs) mediate the regulation of monocyte and macrophage recruitment. Recruited eosinophils and Mast cells (MCs) release some mediators which contribute to coronary vasoconstriction, leukocyte recruitment, formation of new blood vessels, scar formation. In adaptive immunity, effector T cells, especially Th17 cells, lead to the pathogenesis of cardiac fibrosis, including the distal fibrosis and scar formation. CMs protectors, Treg cells, inhibit reduce the inflammatory response, then directly trigger the regeneration of local progenitor cell via IL-10. B cells reduce myocardial injury by preserving cardiac function during the resolution of inflammation.
Similar content being viewed by others

Introduction
In humans and animals, the heart is an indispensable organ for the survival. The cardiovascular system serves as the center of blood circulation, it pumps blood continuously by beating regularly to ensure energy supplies and material exchange in the body1. Cardiac injury is the current second major cause of death in modern trauma except brain trauma2. ischemic heart disease and stroke caused 12.9 million deaths in 2010, accounting for a quarter of the global deaths3, suggested by a statistics on the cause of death in 187 countries from 1980 to 2010. Therefore, it is important to clarify the detailed mechanism of the cardiac repair after injury for public health. The immune cells not only protect the host against invading pathogens, but also play vital roles in the repair and regeneration of damaged tissues4,5,6,7. Some researches on heart development reveal essential roles of immune cells in promoting cardiac homeostasis and injury repair, but these repair processes also increase “bystander damage” that overreacts to injury8.
Cardiac tissue injury and repair
Types of cardiac tissue injury
The types of researched cardiac tissue injury usually include ischemic injury, cryoinjury, resection, and gene ablation. Ischemic injury mainly includes ischemia-reperfusion injury (IRI) and permanent ligation injury (PLI)9,10. At present, the mouse heart IRI model is often used to study the pathological state of myocardial infarction (MI)11. MI is an ischemic cardiac disease. Ischemic cardiac disease is the main cause of human death worldwide. The first symptom of patients with ischemic cardiac disease is acute MI, and then myocarditis occurs because of infarction12,13,14. Myocarditis further leads to ventricular dysfunction and eventually causes heart failure (HF)15. IRI leads to the loss of myocardial cells, and during the healing process, the injured myocardial tissues are gradually replaced by fibrotic scar tissues16. IRI is commonly induced by ligation of left anterior descending coronary arteries to cause ischemic death of cardiomyocytes in downstream tissues. The ligation is then untied, and blood reperfusion is performed after ligation of the mouse artery for 30 min17, but PLI is induced by ligating coronary artery forever. Cryoinjury is usually performed by cauterization of ventricular tissue with a metal probe or a cryoprobe balanced with liquid nitrogen18, and it is administered for different times according to the experimental requirements to control the degree of injury. This is also a very common method in the treatment of cardiac injury19. Cryoinjury also leads to obvious cell necrosis at the injury site and the formation of fibrotic scars. This type of injury model is also similar to the normal cardiac pathological state in humans20,21,22. In the laboratory, a mouse resection model that is used to simulate surgical removal of part of the cardiac tissue or a small part of the ventricle is suitable for almost all animals, but although the resection method can effectively cause tissue loss, compared with MI, this method only causes a small amount of damage to the injury site and surrounding tissue by cell necrosis and fibrotic scar formation23,24. Gene ablation is a type of cardiac injury that causes cardiomyocyte death without surgical operation. The current method for gene ablation is to specifically express nitroreductase (NTR) from bacteria or diphtheria toxin receptor (DTR) on cardiomyocytes (CMs)25,26. NTR induces cytotoxic products such as metronidazole (Mtz), and NTR expression induces CMs to become sensitive to diphtheria, which leads to CM death27. However, the problem with gene ablation methods is that the fibrotic scars produced are difficult to quantify and compare (Fig. 1)6.
Repair of injured cardiac tissues
MI is the main ischemic cardiac disease after cardiac ischemic injury, which is defined as acute death of cardiac tissues28. There is a major unmet clinical need to treat cardiac ischemic injury, though preconditioning, postconditioning and remote conditioning of ischemic conditioning protect the heart from infarction with a lot of preclinical evidence, current phase II trials based on ischemic conditioning have different results for the treatment of cardiac ischemic injury29. Much effort aimed to temper the initial inflammatory response to preclinical models30 and patients has been done, because inflammatory response is vital to the repair process, but the clinical results are disappointing31. Most researches on cardiac ischemic injury and repair only focus on cytokines like interleukin-1β (IL1β) and tumor necrosis factors, antibodies to adhesion molecules of leukocyte invasion and the complement cascade30,32. Coronary ligation with 60 minutes can cause death of most cardiomyocytes in the subendocardial area33. Cardiomyocyte death cannot be confirmed for several hours after coronary ischemia, but irreversible changes have been induced in some cardiomyocytes of the subendocardial area after a 20–30 min interval of severe ischemia34. Current methods of repairing cardiac ischemic injury are divided into two categories: molecular therapy and cell therapy.
Molecular therapy
FGF2 reduces infarct size, and VEGFA increases angiogenesis
Growth factors are signaling molecules that contribute to various cellular processes. There are currently two growth factors that significantly improve cardiac function after MI: fibroblast growth factor 2 (FGF2) and the angiogenic factor vascular endothelial growth factor A (VEGFA). In contrast to wild-type mice, experimental mice with upregulated FGF2 after IRI exhibit reduced infarct sized and improved cardiac function. In addition, FGF2 deficiency exacerbates cardiac dysfunction after IRI35. In animal models of MI, the administration of VEGFA improves local coronary blood flow and restores cardiac function36. In addition, gene-based modification therapy, such as synthetically modified RNAs (modRNAs), modRNAs are nucleotides that one or more nucleotides replaced by modified nucleotides. In previous study, modRNAs have advantage of highly efficient expression of transient protein in vitro and in vivo, but they don’t elicit an innate immune response37,38,39,40,41,42. modRNAs increase the delivery and expression efficiency of VEGFA. First, in vitro experiments verified that VEGFA encoded by modRNAs has the new function of controlling the cell fate of pluripotent islet 1 + (Isl1 + ) human cardiac progenitor cells. VEGFA made these cells drive away from the fate of becoming cardiomyocytes but moved towards the fate of vascular endothelial cells43,44. Human VEGFA has been expressed by modRNAs in mouse hearts after MI. Compared with the application of DNA vectors45,46,47, modRNAs mediate the “pulse-like” expression of VEGFA in vivo, which has advantages in reducing infarct size, enhancing the function of myocardial perfusion and promoting survival48. To some extent, this effect is due to a new effect of VEGFA on epicardial progenitor cells: modRNA-mediated expression of VEGFA makes these progenitor cells proliferate, promotes their migration to cardiomyocytes, and redirects them to differentiate into vascular lineages49. These results indicate that modRNA-encoded VEGFA drives changes in the fate of cardiac progenitor cells in vivo50,51,52, thereby enhancing cardiac repair. In addition, the growth factor neuroregulatory protein 1 (NRG1) and its receptors, the tyrosine protein kinases ERBB2 and ERBB4, play key roles in formation of trabeculae and endocardial pads during cardiac development53. Activation of the NRG1-ERBB2/ERBB4 signaling pathway in the hearts of injured adult mice can induce cardiomyocyte proliferation and improve cardiac function54,55. Paired-like expression of homeodomain transcription factor 2 (Pitx2) in the hearts of newborn mice repairs cardiac injury after apical resection, while adult mouse cardiomyocytes expressing the Pitx2 gene effectively regenerate after myocardial infarction by regulating electron transport and scavenging reactive oxygen species (ROS)56. In addition, cardiac health and diseases are associated to the transcription factor NF-E2 related factor 2 (Nrf2). Multiple lines of evidence support the potential cardioprotective role of Nrf2. Nrf2 exerts a protective effect by reducing oxidative stress, apoptosis and inflammation57,58.
miRNAs mainly regulate cardiomyocyte proliferation
MiRNAs are a kind of noncoding small RNAs that are highly conserved, single-stranded RNAs. The coding gene is located in the noncoding region or the coding region containing the exons and introns. Mature miRNAs are approximately 22 bases in length. As new gene regulatory elements, miRNAs inhibit target gene transcription to regulate gene expression by binding to complementary sequences in the 3’-untranslated regions (UTRs) of mRNAs59. Some miRNAs activate DNA replication in CMs, and by using a library of 875 human miRNAs, a unbiased screening has detected a variety of miRNAs which induce the proliferation of neonatal rat cardiomyocytes60. In animal studies, it has been reported that other miRNAs, such as the miR-214, miR-302-367, miR-17-92 cluster, and the miR-222 cluster, also contribute to cardiac repair by inducing CM proliferation in vivo61,62,63,64. For example, overexpression of miR-199a or miR-590 induced the proliferation of adult mouse CMs in mouse hearts, and improved the cardiac function of adult mouse infarcted hearts, and reduced fibrosis. Therefore, the positive results of preclinical research prove the therapeutic potential of miRNAs.
Exosomes mediate communication between cardiomyocytes
Exosomes are a sort of small extracellular vesicles, their size is mainly 30–100 nm in diameter, which are produced by cells, they are characterized by some specific surface markers, such as CD9, CD63, and CD8165. Exosomes can act as mediators of communication between cells by carrying cell-specific mRNAs or miRNAs, and increasing research evidence supports the role of exosomes in cell communication between heart cells66. Today, small extracellular vesicles called exosomes are known as the key mediators of beneficial mesenchymal stem cell (MSC) paracrine effects66,67. Some miRNAs contained by exosomes have therapeutic significance by changing gene expression of recipient cells68. For example, miR-21a-5p is the main paracrine factor produced by MSCs, and plays a cardioprotective effect through the synergistic activity of multiple pathways69. The secretion of MSC-derived exosomes to the heart can reduce oxidative stress and promote the survival of cardiomyocytes after IRI in mice, further reducing infarct size and ameliorating cardiac function (Fig. 2)70.
Cell therapy
Currently, cell therapy usually involves the use of resident cardiomyocytes with stem-like characteristics for treatment. Preclinical studies used human embryonic stem cells (hESCs)71, and induced pluripotent stem cells (iPSCs)72, both are human pluripotent stem cells (hPSCs), they can differentiate into functional CMs in vitro73. In Clinical therapies, per patient dose may need 108 to 109 cells of hPSC-derived CMs, In addition, a large number of hPSC-derived CMs are needed to determine the safety or efficacy signals74. However, the current experimental data have always shown that hPSC-CM-mediated cardiac repair still has great challenges to clinical application, and the clinical goal seems difficult to achieve75. Heart-derived cells can expand, show pluripotency and differentiate into many heart-type cells in vitro76. Another method of repairing cardiac injury involves generating functional cardiomyocytes in vitro and then transplanting these cells into the injured heart. Before, to protect the failing heart, scientists initially adopted the first generation of cell-based therapies. Since sufficient cardiomyocytes were not available, first-generation cell therapy included the transfer of noncardiomyocytes. The initial candidate cells included skeletal muscle cells that were expected to promote cardiac contraction, as well as bone marrow-derived cells and MSCs that showed cardiogenic potential in vitro77,78. These cells have become the main source of cell-based therapy for HF. MSCs can differentiate into CMs by addition of DNA methyltransferase inhibitor 5-azacytidine (5-aza) or coculture with cardiac progenitor cells79,80. Though stem cell transplantation can form new blood vessels in animal models of MI, the clinical efficacy of stem cell transplantation in patients with MI is still unclear. Because of MSCs’ pluripotency, it may be dangerous if transplanting MSCs into human body73. “Paracrine hypothesis” is associated with stem cell-mediated cardiac repair, which is defined that stem cells release substances to improve injured and diseased myocardium and promote heart regeneration81. There are lots of evidence support that factors released by the autocrine and paracrine mechanisms from resident cardiac cells, could play an important role in repair process of the failing heart. Besides exosomes mentioned above, there are many other factors, which are released by the unconventional and the conventional secretory pathways82, also are in support of this hypothesis. Such as anti-apoptotic, pro-survival and angiogenic factors secreted by stem cells under particular condition83. It is found that nutritional factors by injecting a stem cells culture medium effectively promote heart repair in a mouse model of HF84. In the heart, the most studied paracrine factor is atrial natriuretic peptide because the ventricular myocytes in the diseased adult heart secrete atrial natriuretic peptides through constitutive conventional pathway in response to stretch and adrenaline stimulation, and healthy ventricular myocytes do not have the function of secreting granules85. In fact, if paracrine factors that improve heart function can be identified, pharmacological treatments based on these factors may be easier to translate into clinical applications than cell therapy86. Fibroblasts can be directly or partly reprogrammed to differentiate into cardiomyocyte-like cells or cardiac progenitor cells by overexpressing multiple transcription factors associated with cardiac development. One strategy for reprogramming fibroblasts into cardiomyocytes is to force the expression of five genes related to early heart factors, Mespl, Gata4, Tbx5, Nkx2-5, and Baf60c (also known as Smarcd3), to reprogram mouse fibroblasts into a scalable multipotent cardiac progenitor cell population, thus bypassing the multipotent state87. Induced expression of three genes related to cardiac transcription factors, Gata4, Mef2c and Tbx5, also called GMT, or a combination of GMT plus Hand2 (called GHMT) successfully reprogrammed mouse fibroblasts to differentiate into induced cardiomyocyte-like cells (iCMs) in vitro. iCMs express major cardiac genes and have cardiomyocyte characteristics, such as sarcomere structures, pulsatile contractions and spontaneous intracellular calcium oscillations, without undergoing cardiac precursor states88. However, GMT or GHMT reprogramming mixtures used for direct reprogramming of human fibroblasts require other factors, such as troponin, MESP1, estrogen-related receptor-γ (ESRRγ) and zinc finger protein ZFPM2, as well as some miRNAs such as miR-1 and miR-133, which are necessary to induce human fibroblasts to converse into cardiomyocyte-like cells89,90,91. To translate cellular reprogramming methods to the clinic, efforts are currently mainly focused on direct reprogramming of human cardiac cells. Previous studies found that the combined expression of the transcription factor c-ETS2 and mesodermal posterior protein 1 (MESP1) transformed human dermal fibroblasts into cardiac progenitor cells that express early cardiac factors, such as protein ISL1 and homeobox protein NKX2-5. This effect was not detected in directly reprogrammed mouse fibroblasts (Fig. 3)92.
Immune cells in the heart
Immune cells play important roles in heart of steady state, an increasing number of studies on cardiac injury repair also have shown that the various immune cells that reside or infiltrate the heart tissue play roles in the process of injury repair. Immune cells identified in heart, including macrophages, monocytes, neutrophils, dendritic cells (DCs), T cells and B cells, eosinophils, mast cells, which can reside or infiltrate cardiac tissue93 to maintain cardiac function.
Immune cells in cardiac homeostasis
cTMs phagocytose bacteria and apoptotic cells
Macrophages typically maintain tissue homeostasis and tissue repair, promote angiogenesis and phagocytosis of apoptotic cells and necrotic cells, clear and kill bacteria, and produce proinflammatory cytokines94. Macrophages are present in all tissues and participate in tissue growth and remodeling from the earliest stages of development95,96. Like in most tissues, macrophages are the main immune cells that reside in the heart. They are usually found near endothelial cells or in the interstitial space95,97. Cardiac tissue resident macrophages (cTMs) are spindle-shaped cells located in the interstitial space between muscle cells, fibroblasts and endothelial cells (ECs)98. cTMs have been widely characterized as an integral part of organ development. It has been shown that cTMs originate from the yolk sac early in development and are maintained until adulthood in mouse models99. cTMs have CCR2-MHC-low cell surface characteristics and are closely related to blood vessels in the myocardial wall during embryonic wall development, and their absence causes coronary vascular malformations. cTMs are located throughout cardiac tissues and are closely related to blood vessels to regulate the supply and discharge of blood. cTMs are also abundant in the cardiac conduction system97,98. In the steady state, cTMs phagocytose subcellular particles containing dysfunctional mitochondria ejected from CMs, which supports CM health100. In addition, cTMs help heart-specific processes and participate in maintaining proper electrical conduction98. A large number of cTMs present in the AV node are connected to cardiomyocytes through connexin 43 (CX43), and the destruction of Cx43 delays the electrical conduction of the AV node. To maintain homeostasis, cTMs interact closely with ECs and quickly internalize blood-borne fluorescein isothiocyanate dextran (FITC-dextran). cTMs in the adult heart phagocytose bacteria and apoptotic cells, indicating that cTMs have typical macrophage characteristics95,101. Many macrophages utilize macropinocytosis and phagocytosis to take up components of the local microenvironment. At the cellular physiological level, cTMs phagocytose fluorescently-labeled bacteria, indicating the ability of these cells to phagocytose bacteria101. The cTMs in the cardiac tissue are located between CMs and phagocytose some molecules produced by the surrounding CMs and absorb dead CMs95,97. cTMs also produce nutritional and immune-related factors and make contact with CMs to participate in the regulation of cardiac homeostasis102.
Monocytes and macrophages appear to play an important role in cardiac renewal. Macrophages have multiple roles in normal and injured hearts. cTMs are different from monocytes isolated from the adult mouse spleen and brain, and their gene expression profile is similar to that of anti-inflammatory M2 macrophages103. After gene ablation in adult mouse CMs, M2-like cTMs were replaced by macrophages derived from proinflammatory monocytes6. In a healthy state, macrophages express a large number of M2-specific markers97,104. This expression is also consistent with the characteristics of M2 macrophages, because M2 macrophages usually promote tissue reconstruction after injury, thereby helping to restore a steady state. cTMs are the most abundant cell populations in heart, they respond to injury signals by initiating neutrophil recruitment105 and secreting beneficial tissue and immunoregulatory factors97. Therefore, cTMs play an important role in maintaining cardiac homeostasis in steady state.
NK cells have an inhibitory effect on some cardiac diseases
NK cells are a large subset came from the innate lymphoid cell (ILC) family. The special markers expressed in mature mouse NK cells are DX5 and CD11b, there are also along with various organ-specific chemokine receptors on cell surface, and exhibit cytokine secretion profiles and inherent cytotoxic abilities that may be beneficial to migrate from the bone marrow to specific sites in the body106,107. NK cells are dysregulated in some cardiac diseases, including myocarditis, transplant rejection, and cardiac fibrosis, such as in a mouse model that limited myocarditis, NK cells are essential for suppressing cardiac viral infection and replication and reducing cardiac eosinophil infiltration. NK cells prevent the maturation and transport of inflammatory cells, which alter the environments of local cytokines108,109,110, such as IFNγ and other mediators, which are produced by NK cells, and induce apoptosis in nearby resident and hematopoietic cells.
DCs have a protective effect
DCs exist in the spleen and other lymphatic and nonlymphoid tissues and mainly serve as antigen-presenting cells. DCs are divided into two subtypes: cDCs, which exist in lymphoid tissue, blood and nonlymphoid tissue, and pDCs, Cardiac cDCs are divided into cDC1s and cDC2s according to their dependence and the expression of different transcription factors (TFs). Cardiac cDC1s express the markers CD103, CADM1, XCR-1, DNGR-1 (Clec9a) and IRF8, Similar to cDC1s in other organs. knockout of the transcription factor IRF8 in CD11c + cells in mice, cDC1s lack in all organs, including the heart111,112. DCs in the heart can produce different cytokines, such as tumor necrosis factor (TNF-α), IL-12 and IL-23, all of which have specific functions113,114. DCs upregulate cardiomyocyte hypertrophy and inflammation-related genes induced by advanced glycation end products (AGEs)115. Compared with cDCs in most other tissues, cardiac cDCs recruit to the heart by the chemokine receptor CCR2 without local proliferation116. Cardiac cDC1s in healthy heart migrate to draining mediastinal lymph nodes, to present cardiac autoantigens to specific αMyHC- CD4 + T cells, resulting in regulatory T cell (Treg cell) expansion. Compared to cDC2s phenotypes in other organs, cardiac cDC2s express the markers CD172α and CD11b and the transcription factor IRF4117. Although the heart is not in direct contact with the external environment, histological studies conducted on several nonlymphatic rat tissues in 1981 proved for the first time that the number of DCs in healthy hearts was comparable to that in barrier tissues such as skin118. It has been proposed to use cardiac DCs as gatekeepers against invasive heart disease and self-tolerance118.
Immune cells in cardiac injury repair
The response of immune cells to cardiac injury repair is divided into three overlapping phases: inflammation, proliferation, and maturation. Initially, resident and infiltrating immune cells activate inflammation and proliferation, and the relative balance between pathological inflammation and proliferation determines the development of HF119. The kinetics of the inflammatory and proliferation phases influence the repair results. Studies have shown that resident and recruited immune cells play roles in cardiac injury and appear in cardiac tissue in the early stage of disease120,121,122,123,124.
Immune cells embedded in injured heart tissue recognize and regulate inflammation by dynamically interacting with stromal cells in the interstitial heart, which may lead to the reappearance of heart morphology to support regeneration by rebuilding functional scaffolds in regenerative organisms or fail to resolve the inflammatory response and produce fibrotic scar tissue in adult mammals125. Immune cell stimulation is one of the earliest reactions that can be detected at the infarct site after MI, and the immune response plays an important role in coordinating multiple processes that control cardiac repair, including the survival of resident cells, the removal of fibrotic and dead cells, infarct area formation and vascular reconstruction. In fact, the early stage of inflammation is characterized by rapid sterile inflammation, immune cell infiltration and phagocytosis to remove damaged cells and extracellular matrix tissues, followed by the proliferation stage, processes that muscle layer fibroblasts proliferation, scar formation and neovascularization subside in the next few days126. Neutrophils, monocytes, endothelial cells and pericytes help to suppress and eliminate inflammatory responses. In addition, changes in the composition of the extracellular matrix are also involved in the suppression of inflammatory signals126. However, excessive infiltration of the myocardium by inflammatory cells exacerbates cardiac injury and worsens myocardial remodeling after MI through the release of proinflammatory cytokines, cytotoxic mediators, and reactive oxygen species (ROS)127,128,129. The recruitment of inflammatory cells must be strictly controlled to ensure tissue healing while avoiding excessive inflammatory responses after injury, which can otherwise lead to poorly adaptive remodeling and contractile dysfunction130. Cardiomyocytes secrete various anti-inflammatory signals, such as cytokine 1, MIC1, and growth differentiation factor 15 (GDF-15, which inhibits macrophages), to limit the infiltration of immune cells and abolish chronic inflammation.
cTMs promote CM proliferation and angiogenesis
After cardiac injury, cTMs are thought to play a key role in heart remodeling during the inflammatory phase. cTMs, especially the MHC-II-low subgroup, phagocytose dying CMs, contributing to local homeostasis. After myocardial injury, the activation of inflammatory bodies has weak tissue regeneration, and blockade of the CCR2 axis prevents ischemic injury131,132. After Ang II infusion, cTMs expanded without peripheral monocyte proliferation in situ, similar to the expansion of pleural macrophages after worm infection133.
cTMs regulate cardiac progenitor cell proliferation, especially in during the proliferation phase after heart injury. A genetic mouse model of myocardial loss was used to demonstrate that newborn mice have expanded populations of embryonic-derived cTMs, resulting in minimal inflammation and promoting cardiac recovery through the proliferation of myocardial cells and angiogenesis6. Using the macrophage depletion model, cardiac regeneration and neovascularization after MI were shown to depend on neonatal macrophages. Newborns lacking macrophages do not regenerate cardiac muscle and form fibrotic scars, resulting in reduced cardiac function and angiogenesis5. Macrophage depletion weakens the regenerative capacity of newborn animals and young mammals, highlighting the key role of macrophages in tissue repair after injury5,134. In addition, after systemic macrophage depletion, cardiac inflammation or senescence, CCR2 + Ly6C-high monocytes replace embryonic resident macrophages and coordinate heart inflammation95,135.
cTMs perform differently in the heart in different phases. In the neonatal heart, cTMs are essential for cardiac regeneration after MI injury, probably because cTMs have a phenotype of polarization and secrete necessary soluble factors that drive angiogenesis5. However, this protective mechanism disappeared within 2 weeks, approximately when monocyte-derived macrophages were recruited to the site of heart injury6. In the aging heart, the macrophage phenotype converts to the proinflammatory subtype, leading to inflammation136.
NK cells prevent cardiac fibrosis and promote vessel remodeling
NK cells play an important role in regulating the proliferation phase after cardiac injury. In the proliferation phase, NK cells prevent the development of cardiac fibrosis by directly restricting collagen formation of cardiac fibroblasts and the accumulation of specific inflammatory populations and profibrotic cell types such as eosinophils in the heart137. NK cells activated by IL-2 injection promote blood vessel remodeling through α4β7 integrin and killer lectin-like receptor subfamily G member 1 (KLRG1) without participating in the basic formation of blood vessels. Activated NK cells first bind to cardiac epithelial cells (CECs) through α4β7 integrin and vascular cell adhesion molecule 1 (VCAM-1) and disrupt the binding of N-cadherin through KLRG1. This process transfers β-catenin from the cytoplasm to the nucleus and eliminates the inhibitory effect of cell contact on proliferation138. In addition, NK cells interact with cardiac endothelial cells to increase vascularization and angiogenesis after MI138,139.
M1 stimulates myocardial fibrosis and then M2 promotes infarct scar formation and angiogenesis
Recruitment but not local proliferation is the main mechanism by which the number of monocytes and macrophages is regulated in the ischemic myocardium140. Macrophages in the hearts of adult mice are a heterogeneous population. In the heart, there are different subsets of macrophages with different functions and origins, including protective and pathogenic cells103. Injured cardiac tissues after MI or I/R have obvious myocardial macrophage and neutrophil infiltration141. After cardiac tissue injury, monocytes quickly mobilize from the bone marrow to the injured tissues, where they differentiate into macrophages or DCs and trigger an immune response. In MI, monocytes released by the spleen are recruited to the heart through the MCP-1/CCR2 interaction. When monocytes are recruited into cardiac tissue, these cells differentiate into inflammatory M1 macrophages and activated M2 macrophages142. Due to their abundance and phenotypic plasticity, macrophages are well suited to coordinate the repair response after MI. Macrophages have almost unlimited potential, and macrophage subpopulations are regulated to mediate protection of cells in the infarcted heart and are used to activate survival, repair, and regeneration responses143.
M1 macrophages are proinflammatory and usually induced by IFN-γ or TNF-α, while M2 macrophages are anti-inflammatory and usually induced by IL-4/IL-13/IL-1097,104. M1 macrophages are mainly in the early stage after MI. Infiltrating monocyte-derived M1 macrophages trigger the cardioprotective phenotype in the ischemic heart and play a positive role by activating antiapoptotic programs in cardiomyocytes143. A large number of M1 macrophages with proinflammatory characteristics are rapidly recruited to cardiac tissues, and M1 macrophages stimulate myocardial fibrosis by inducing the production of TGF-β1, while TGF-β1 stimulates Smad3 signaling, induces the production of collagen and MMP, and releases extracellular matrix from cardiac fibroblasts to promote tissue fibrosis and myocardial remodeling144. In this situation, Treg cells are important regulators of macrophage phenotype and function145,146,147. After ingesting dead cells and matrix fragments, macrophages release anti-inflammatory cytokines, which suppress inflammatory damage and reduce undesirable cardiac remodeling148. During the proliferation phase, M2 macrophages are the mainly subpopulations, which promote cardiac remodeling through infarct scar formation and angiogenesis. By inhibiting the expression of IL-6, MMP9, TNF-α and producing exogenous signals such as IL-10, macrophages are transformed into a repair-mediating M2 phenotype93. The result of this transformation is the emergence of M2 macrophages that produce IL-10, TGF-β and VEGF, thereby promoting fibrosis and angiogenesis, as well as the production of other factors, such as myeloid-derived growth factors126,149. Following the maturation phase, the wound begins to subside, and the infiltrating macrophages show features associated with inflammation inactivation/reduction, which promotes cardiac repair by mediating the fibrotic response150.
During the stage of infarct healing, monocytes/macrophages with strong phagocytosis abilities clear dead cells and matrix fragments; during this process, the scavenger receptor cluster protein 36 (CD36) can be detected on cardiac LY6C-high monocytes, and CD36 is essential for early phagocytosis and small infarct size of dying cardiomyocytes after permanent coronary ligation151. Hepcidin inhibits macrophage-induced cardiac repair and regeneration by regulating the IL-4/IL-13 pathway152. Overexpression of monocyte MCP-1 in the heart induces macrophage infiltration, new blood vessel formation, proliferation of myocardial fibroblasts and IL-6 secretion, which leads to the prevention of LV dysfunction and remodeling after myocardial infarction153. Early interference with macrophage recruitment is sufficient to disrupt the formation of new blood vessels, impeding neutrophil clearance and cardiac regeneration. In the mouse MI model, myocardial cells secrete REG3β to recruit macrophages after injury, and the loss of REG3β results in a large decrease in the number of ischemic cardiac macrophages, accompanied by insufficient neutrophil clearance and increased ventricular expansion154. Similarly, the loss of REG3β also leads delayed and reduced macrophage recruitment of the heart in a teleost medaka after injury and delayed neutrophil clearance. Observations obtained from mice, zebrafish, and medaka indicate that cardiac repair and regeneration require the timely recruitment of macrophages155.
The recruitment of M1 macrophages is triggered by a mechanism that partially involves CM-derived BMP4 in CM-conditioned medium. It has also been found that M1 macrophages affect the proliferation and differentiation potential of CMs, partly because of BMP molecules secreted by macrophages. BMP molecules involve the activation of classic inside-out signaling pathways, such as Smad1, 5, and 8, which are known to be activated during myocardial injury in vivo156. Angiotensin II (ANG II) is upregulated in the serum and cardiac tissue of mice with experimental autoimmune myocarditis (EAM), and ANG II significantly drives monocyte/macrophage recruitment through the CC chemokine receptor 2/5 (CCR2/5) axis. Two cytokines, IL-4 and IL-13, control the expression of chemokines157. Monocyte/macrophage infiltration is eliminated and development of EAM is improved by CCR2/5 antagonists and ANG II receptor inhibitors158. The macrophage gene expression pattern also changes when the myocardium is remodeled. Compared with that of the steady state, the expression of metalloproteinases in monocyte-derived macrophages and resident macrophages during HF is reduced, which may affect matrix renewal. More IL-1β is produced by monocyte-derived macrophages during HF than during normal conditions, while resident macrophages contribute more TNF. However, monocyte-derived macrophages also produce VEGF159, which may be related to cardiac hypertrophy-induced angiogenesis.
Neutrophils lead to cardiac damage early, but also promote cardiac injury later
CCR2 + monocytes and macrophages residing in the heart are essential for promoting neutrophil infiltration into injured myocardial tissue160. Necrotic cardiomyocytes lead to the activation of tissue-resident immune and nonimmune cells. Neutrophils are innate immune cells that migrate to damaged lesions within 24 h of cardiac injury in large numbers161,162. Neutrophils the first type of immune cell to infiltrate the infarcted myocardium and are also the first type of immune cell to be recruited to the myocardium in large numbers after ischemic cardiac injury or pressure overload8.
Sterile inflammation occurs when tissues are injured after infection. Neutrophils are recruited from the blood to inflammatory sites163, because they have pro-inflammatory and cytotoxic properties, they not only promote wound healing but also cause tissue damage164. Studies have found that the recruitment of neutrophils occurs after focal hepatic necrosis. The main mechanism is that adenosine triphosphate released by necrotic liver cells activates the Nlrp3 inflammasome, and the resulting inflammatory microenvironment promotes neutrophils adhering to the sinusoids165. Subsequently, the concentration of chemokines in the blood vessel changes, under the chemotaxis of these chemokines, neutrophils migrate from healthy tissues to foci of damage166,167.Inflammation induced by neutrophils initiates myocardial repair during MI127. In the early stage, macrophages and neutrophils secrete proinflammatory cytokines, which promote fibroblast differentiation and lead to sustained inflammation and myocardial injury168. The recruitment of neutrophils increases inflammation and leads to cardiac dysfunction.
This inflammatory response includes a series of events. First, neutrophils and monocytes/macrophages infiltrate to remove debris from necrotic cells by releasing high levels of ROS and secreting proteases and proinflammatory mediators, which not only induce tissue damage but also further recruit leukocytes; for example, neutrophils support the recruitment of proinflammatory LY6C-high monocytes and dominate between day 1 and day 4 after injury, maintaining inflammation and removing cellular debris169. Then, a proliferation phase follows the initial inflammatory phase, which is characterized by the expansion of neutrophils and macrophages, these cells remove dead cells and matrix fragments, as well as release cytokines and growth factors that can form highly vascularized structures consisting of connective tissues and new blood vessels and eventually heal tissues. The proliferation phase is characterized by fibroblast activation and endothelial cell proliferation, which ultimately leads to repairable myocardial fibrosis, angiogenesis, extracellular matrix deposition and growth factor release. Finally, inflammation and scar formation are resolved169. Defects in neutrophils lead to delayed collagen deposition, enhanced matrix degradation and increased sensitivity to heart rupture154. Therefore, neutrophil activation must be strictly controlled, although neutrophils are essential for initiating an acute inflammatory response170.
Neutrophils are recruited to the heart, and the response factors include DAMPs, cytokines including chemokines, endogenous lipid mediators (such as prostaglandin E2 and leukotriene B4), histamine and components171,172. The recruitment of neutrophils to the site of infection depends on CXC chemokines173. In mice, CXC chemokines control the recruitment of neutrophils through CXCR2, while in humans, neutrophil recruitment depends on both CXCR1 and CXCR2174. Neutrophils produce cytokines and chemokines, which attract spleen-derived macrophages to migrate into cardiac tissue175.
As regulators of immune responses, neutrophils affect chronic immune responses and the function of dendritic cells and lymphocytes. Neutrophils have been shown to improve the polarization of macrophages toward a repair phenotype by releasing lipoproteins associated with neutrophil gelatinase, thereby improving heart healing after MI126,176. The depletion of neutrophils does not affect the infarct size but worsens cardiac function and HF and increases cardiac fibrosis176.
DCs are immunoprotective in the heart
DCs are protective immunoregulators in post-MI healing177. During the healing process after MI, BM-derived activated CD11c + CD11b + DCs penetrate into the infarcted heart. It has been reported that after MI, DCs accumulate early in the infarct border area in rats and mice, and the number of cells reaches a peak at the 7th day177,178. DCs mediate the regulation of monocyte and macrophage homeostasis after MI, which indicates that DCs are beneficial to cardiac injury and repair. After activation, DCs recruit to lymphoid organs, where they accumulate and stimulate T cells179. The myocardium contains both pDCs and cDCs, which are necessary for maintaining the infiltration of congenital leukocytes180.
After MI, only cDCs participate and play a key pathological role181. By depleting cDCs exclusively, it has been demonstrated that the measured immune response and infiltration of macrophages, neutrophils and multiple T cell subpopulations are blunted, which are related to the indicators of improvements in cardiac structure and function. The depletion of cDCs is related to reduced mRNA expression of the proinflammatory cytokines IL-1β and IFN-γ in the heart182. In vivo analysis showed that in CD11c + DC-deficient mice, left ventricular function deteriorated after MI. After depleting bone marrow-derived DCs in CD11c-Bifidobacterium toxin receptor transgenic mice, left ventricular function and impaired remodeling were worsened after MI183. The DC depletion group showed long-acting inflammatory cytokines, such as IL-1β, IL-18 and TNFα. In addition, in the hearts of the DC depletion group, anti-inflammatory Ly-6C-low monocytes and other activated macrophages were also significantly infiltrated177. These results indicate that cardiac DCs have a strong immunoprotective function after MI. DCs prevent tissue-destructive autoimmunity after heart injury by activating conventional fork-head box protein P3 (FOXP3)– CD4 + T helper cells and FOXP3 + CD4 + Treg cells184. A reduction in the number of DCs is associated with heart rupture after MI, increasing the recruitment of proinflammatory monocytes that maintain the production of proinflammatory cytokines185. In short, DCs protect the heart by regulating the recruitment of different types of immune cells.
NKT cells protect the heart by producing IL-10
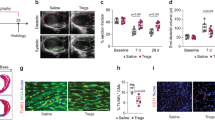
Natural killer T cells (NKT) are unique T lymphocyte subsets characterized by coexpression of NK receptors and unchanged T cell receptors (TCRs). TCRs recognize glycolipids presented by the major histocompatibility complex (MHC);186,187 these cells secrete T helper type 1 (Th1), Th2 or immunoregulatory cytokines188,189,190 such as IL-10 to modulate immune responses towards proinflammatory or regulatory profiles191,192. NKT cells involve in inflammation and tissue remodeling. These cells play a protective role in left ventricle (LV) remodeling and failure. After MI in mice, the recruitment of NKT cells in the non-infarcted area of the LV increased193. Pressure overload induced by transverse aortic constriction also increases the infiltration of invariant natural killer T (iNKT) cells in mouse hearts194. LV remodeling and the transition from hypertrophy to heart failure are exacerbated after the disruption of iNKT cells, and this process is associated with the activation of mitogen-activated protein kinase signaling195. The class I antigen-presenting molecule CD1d is mainly expressed on antigen-presenting cells, which combines to TCRs in NKT cells191. CD1d deficiency significantly accelerates Ang II-induced hypertrophy, causing cardiac remodeling and an inflammatory response196. DCs lacking CD1d reduce IL-10 production by NKT cells, and the administration of IL-10 to CD1d-KO mice may significantly reverse Ang II-induced hypertension and cardiac remodeling by activating STAT3 and inhibiting the TGF-β1 and NF-kB signaling pathways197,198. In addition, administration of the NKT cell activator α-galactosylceramide (α-GC) in the mouse myocardial infarction model resulted in enhanced infiltration of NKT cells in the non-infarcted area199, and it was found that α-GC administration significantly reduced left ventricle expansion and mortality caused by HF. It is suggested that these effects depend on NKT cells, and IL-10 is the most effective effector cytokine in this process (Fig. 4).
T cells promote heart remodeling through cytokines and growth factors
Antigen-activated T cells are recruited more efficiently than monocytes to infected cardiomyocytes, and early recruitment of effector T cells may also be involved in the maintenance of inflammatory cells at the site of chronic local infection200. The depletion of monocyte/macrophage lineage cells in the hypertensive heart leads to massive infiltration of inflammatory cells, mainly CD4 + T lymphocytes, in the area of cardiomyocyte loss201. These results suggest that monocytes/macrophages have a protective effect on adaptive immunity by inhibiting T cell recruitment of the heart. Antigen-activated CD4 + T cells are divided into four subtypes, including helper T cells (Th1 and Th2, Th17 cells) and Treg cells.
T lymphocytes have different roles in cardiac tissue injury and repair. The traditional view is that myocardial scar formation after MI only represents fibrosis with extracellular matrix proteins replacing necrotic tissues. Unlike the traditional view, it is now very clear that T cells are effective in coordinating this process. The resulting fibrosis indicates that T cell infiltration plays a central role in the pathogenesis of cardiac fibrosis202. Several experimental animal models have shown T cell regulatory mechanisms involving immune cell tolerance to cardiac antigens, including processes of T cell activation, recruitment of T cells to the heart, and cytokine production in response to ischemic or nonischemic sterile inflammation203. At present, the related mechanism of T cell recruitment of the heart involves c-Met signaling-mediated induction of the release of autocrine CCR5 ligand, which promotes the recruitment of T cells to the heart through the chemokine receptor CCR5204. The mechanism of T cell recruitment and myocardial infiltration in the MI experimental animal model seems to be time-dependent. For example, in myocarditis, the time after ischemia is different and may involve different responses. Primary myocarditis is only a small part of the cause of human HF, but in this case, the T cell infiltration mechanism is still observed in the more common form of HF, and T cells have a broad role in promoting cardiac fibrosis and failure.
In a mouse model of permanent coronary infarction, infiltrating CD4 + and CD8 + T cells gradually infiltrate the heart and reach a peak on the 7th day after MI. The infiltration of T cells into the myocardium may directly regulate the phenotype and function of fibroblasts at all stages of this process. In cardiac tissue, antigen-driven expansion of oligoclonal T cells may be an effector of rheumatic cardiac disease. Generally, Th1 cells secrete Th1 cytokines (such as IL-2, IFN-γ, and TNF-α) and promote the antifibrotic response in cardiac tissue, while Th2 cytokines (such as IL-4, IL-5, and IL-13) promote cardiac tissue fibrosis205. Th17 cells are CD4 + T cells that produce IL-17 and IL-22, play an important part in promoting inflammation during the cardiac tissue remodeling. Th17 cells assist in host resistance to infection by recruiting neutrophils and macrophages into infected tissues206. Th17 cells induce or enhance the expression of inflammatory cytokines, such as IL-6, IL-1β and TNFα, and promote extracellular matrix remodeling by producing repair-associated components such as matrix metalloproteinases (MMPs) or proteoglycans207. In addition to their effects on the early stages of infarct scar formation during chronic remodeling, Th17 cells in the heart seem to guide the distal fibrosis and scar formation that occur throughout the left ventricle of the heart202. In the rat model of HF induced by intraperitoneal injection of isoproterenol, the use of anti-IL-17 antibodies to block the production of IL-17 leads to a reduction in cardiac fibrosis. In this model, the expression of MMP-1 and receptor activator of NF-κB ligand (RANKL) and collagen synthesis in cardiac fibroblasts are inhibited, but MMPs and osteoprotegerin (OPG) are increased in tissue208. These results indicate that Th17 cells regulate cardiac fibrosis by the production of MMPs through the IL-17-RANKL/OPG system in cardiac fibroblasts, or by stabilizing the mRNA of proinflammatory cytokines in various cardiomyocytes and immune cells.
Treg cells are CD4 + CD25 + subpopulations of T lymphocytes that have strong inhibitory properties. Treg cells, as inhibitors of inflammation after MI, play an important role in immune homeostasis. Under physiological and pathological conditions, Treg cells both play a key role in control of innate and adaptive immune responses209. There are chronic inflammation appearing in adverse ventricular remodeling after MI210, almost all processes related to ventricular remodeling such as degradation of the interstitial matrix and collagen deposition, hypertrophy and apoptosis of the remaining cardiomyocytes, scar formation, and ventricular dilation are associated to inflammation211, Treg cells have beneficial effect on undergoing ventricular remodeling by CD28 superagonist-mediated expansion and adoptive cell migration after MI212. In the inflammatory phase, Treg cells protect the heart from adverse ventricular remodeling and maintain cardiac function by inhibiting proinflammatory cell infiltration and directly protecting cardiomyocytes212. In mice, Treg cells may reduce the inflammatory response during chronic cardiac remodeling after MI by inhibiting self-effector T cells, and effector T cells are controlled by the expansion of Treg cells. In the proliferation phase, Treg cells have been shown to directly activate and promote local progenitor cell regeneration213,214, and Treg cells promote myocardial recovery by the IL10-dependent pathway215,216. In zebrafish, Treg cells enhance precursor cell proliferation by activating neuromodulin-1 in the heart through IL-10 secretion. Treg cells indirectly regulate cardiac regeneration by controlling neutrophils217,218,219 and inducing macrophage polarization5,6,220.
Treg cells are also thought to be involved in M2 polarization after MI by producing IL-10221. In cardiac disease models, ANG II infusion and MI increase the number of CD4 + CD25 + Treg cells. Adoptively transferred Treg cells reduce cardiac hypertrophy, inflammation and fibrosis through IL-10 production and direct cell-cell interactions211,222. These results indicate that there is a close relationship between Treg cells, CMs and cardiac fibroblasts, which confirm the importance of Treg cells in cardiac remodeling.
The absence of effector T cells can increase the recruitment of proinflammatory monocytes and reduce neovascularization and collagen deposition184. The loss of IFN-γ expression in the injured myocardium due to insufficient Treg cell recruitment may affect other immune cell populations, including macrophages and neutrophils223,224,225.The depletion of Treg cells triggers autoimmunity and cardiac dysfunction and enhances the immune response to nonself antigens226. MHC class II-deficient mice and mice expressed a single transgenic T cell receptor show impaired wound healing and monocyte population expansion after ischemic injury. This finding indicates that self-antigens are presented to CD4 + T cells through MHC class II-expressing cells, such as macrophages and DCs, which then drives the immunosuppressive response in the myocardium125.
B cells regulate wound healing and tissue remodeling
Both T cells and B cells can regulate wound healing and tissue remodeling after myocardial injury. Effector T cells are activated in the proximal lymph nodes and quickly colonize the injured heart after MI227. First, while the number of B cells peaks after the onset of ischemia228, B cells produce proinflammatory cytokines to reduce cardiac contractility and promote cardiomyocyte apoptosis229; then, B cells accumulate in the infarcted heart, produce IL-10 affected by pericardial adipose tissues (PAT) and preferentially express cytokines, and B cells promote inflammation resolution and reduce myocardial injury to preserve cardiac function during the resolution of inflammation after MI. The specific loss of B cell-derived IL-10 after MI worsens cardiac function, exacerbates myocardial damage, and delays inflammation remission230.
Eosinophil recruitment by CCR3 and its ligands CCL11, CCL24, and CCL26
Eosinophils may play a pathogenic role in myocarditis. In mouse models of myocarditis and the hearts of patients with eosinophilic myocarditis, the recruitment of eosinophils to the injured heart depends on the expression of the chemokine receptor CCR3 and its ligands CCL11, CCL24, and CCL26. Cardiac fibroblasts are the source of CCL11 in the interstitium of the heart. CCL24 is produced by F4/80+ macrophages located at the focal foci of the heart. The expression of CCL11 and CCL24 is controlled by the cytokines IL-4 and IL-13 produced by Th2 cells. These findings are currently known as the precise pathway for eosinophil recruitment to the heart, and blocking this pathway prevents eosinophil-mediated heart injury157.
Mast cells induce myocardial remodeling and cardiac fibrosis
Mast cells (MCs) mainly regulate allergic reactions and host-pathogen interactions in the innate immune system, and myocardial MCs are known to infiltrate and proliferate in MI, arrhythmia, atherosclerosis, HF, and cardiomyopathy.
The number of MCs also increases when human cardiac muscle is hypertrophic231. These cells secrete some molecule such as platelet-derived growth factor A (PDGFA), TNF-α, TGF-β and histamine, which affect heart function. MCs are the main source of TNF-α and have different production methods. TNF-α induces MMP activation, leading to myocardial remodeling232. After pressure overload in the mouse heart, recruited mast cells induce PDGFA chain synthesis and promote cardiac fibroblast proliferation and collagen synthesis. In addition to PDGFA chain, ANG II-induced Rac 1 activation also leads to myocardial remodeling and atrial fibrillation through CTGF and lysyl oxidase-mediated miR-21 expression231. In myocardial ischemia and MI, local MCs release some mediators which contribute to coronary vasoconstriction, leukocyte recruitment, formation of new blood vessels, scar formation233. Among the mediators released by MCs, histamine and ET-1 promote severe arrhythmias, leading to sudden cardiac death234. In addition to coronary vasoconstriction and systolic failure, arrhythmias, including symptoms such as arrhythmia, premature beats, and atrial fibrillation, are also features of cardiac allergic reactions, which are caused by the release of several mast cell mediators235. In coronary atherosclerosis, mast cells release mast cell tryptase, chymotrypsin, and TNF-α to promote cholesterol accumulation and plaque instability236. Infiltrating MCs in the allergic dead heart are significantly increased, and the serum level of mast cell tryptase is high and is accompanied by severe pulmonary congestion and edema237. MCs are recruited and activated in the drug-related dead heart. The degree of MC degranulation, increase in tryptase levels and pathological changes in the victim’s heart related to the drug are similar to those of allergic death238. In HF, mast cell chymotrypsin causes progressive LV dysfunction by promoting cardiomyocyte apoptosis and fibroblast proliferation239. Chymotrypsin and tryptase also promote the characteristic fibrosis associated with cardiomyopathy. Myocardial remodeling and hypertrophy are induced by the release of cardiac MC mediators and proteases are typical characteristic of advanced HF associated with hypertension. In addition, recruited cardiac MCs contribute to fibrosis associated with autoimmunity and viral myocarditis240. MC-stabilizing drugs improve HF by reducing myocardial remodeling. Atrial fibrosis requires MCs to interact with fibroblasts in the heart and affects the sensitivity of atrial fibrillation, which is the most common type of arrhythmia in HF241. These results indicate that cardiac MCs play a key role in allergic cardiac diseases through certain mediators, such as TGF-β, TNF-α and histamine, and MCs also regulate atrial myocardial remodeling and communication between CMs and cardiac fibroblasts (Fig. 5).
Conclusion
There are four main types of cardiac tissue injury: ischemia-reperfusion injury (IRI), cryoinjury, resection, and gene ablation. Many studies have used IRI as a pathological model of MI. At present, there are two types of therapy for heart injury repair. One type of therapy functions in the molecular level. Heart function is restored through growth factors such as FGF2 and VEGFA, many types of miRNAs, exosomes that can regulate various physiological functions of myocardial cells by promoting vascular regeneration, myocardial cell proliferation, or inhibiting fibrosis, myocardial necrosis, and ROS levels. The other type of therapy functions in the cellular level and mainly uses cells with differentiation potential, such as MSCs, heart-derived cells and iPSCs, and induces these cells to differentiate into CMs or reprograms fibroblasts to differentiate into iCMs or cardiac progenitor cells.
Immune cells in the heart are mainly divided into resident and recruited cells. Resident immune cells, cTMs, NK cells and recruited immune cells play a role in the heart. Through cell-cell interactions, these cells phagocytose bacteria and necrotic cells and regulate proliferation, inflammation, fibrosis, and extracellular matrix and collagen formation to maintain normal heart functions.
The immune cell repair response to cardiac injury mainly manifests as three overlapping phases: inflammation, proliferation and maturation. In the inflammatory phase, inflammatory cells are recruited, proinflammatory factors are produced, and necrotic cardiomyocytes and cell debris are engulfed or cleared. In the proliferation phase, there is proliferation of cardiomyocytes and fibroblasts, angiogenesis, fibrosis and extracellular matrix formation. In the maturation stage, the recruitment of inflammatory cells is suppressed, anti-inflammatory cytokines are produced, and ultimately inflammation and scar formation are eliminated. Maintaining an appropriate balance between the inflammatory phase and the proliferation phase is the key to achieving the best repair outcome. Proper and timely limitation and elimination of inflammation are key elements in the quality of cardiac healing.
Though many therapies including molecular and cell methods have been applied, a lot of work and experimental data have accumulated in the role of the immune microenvironment in the cardiac injury repair, there still are some limitations on the current studies/data in cardiac injury repair. Most therapies of cardiac injury are only used in animals such as mice, zebrafish or animal and human cells, there is an unmet clinical need to treat cardiac injury. Studies mainly focus on cardiac ischemic injury such as IRI and MI, but there are other types of cardiac injury, for example, cardiac injury is caused trauma, heredity or virus infection. In the future, we should strengthen the transformation of current research into the clinical direction, build more different types of injury models, and expand the curable group of patients with cardiac injury t through the combination of basic research and clinical applications.

Types of cardiac tissue injury. Ischemia-reperfusion injury (IRI), permanent ligation injury (PLI), cryoinjury, resection, and gene ablation are the four main ways of studying cardiac injury and repair. IRI is used to simulate the pathological state of myocardial infarction (MI). The ligation is untied and blood reperfusion is performed after ligation of the mouse artery for 30 min Cryoinjury is caused by cauterization of ventricular tissue with a cryoprobe. Resection is used to surgically remove part of the cardiac tissues. The gene ablation method specifically expresses bacterial nitroreductase (NTR) or diphtheria toxin receptor (DTR) in cardiomyocytes (CMs) to result in CM death

Molecular therapy of injured cardiac tissues. Some molecules promote repair (blue line), while others inhibit repair (red line) after cardiac injury. Growth factors such as VEGFA, FGF2, and NRG1, the NRG1 receptors ERBB2 and ERBB4, exosomes, and miR-199a or miR-590 restore cardiac function. miR-199a or miR-590, the miR-17-92 cluster, miR-214, miR-302-367, and miR-222 induce CM proliferation. The miR-17-92 cluster, miR-214, miR-302-367, miR-222 and miR-199a or miR-590 reduce fibrosis. FGF2 and exosomes reduce infarct size. Exosomes, Nrf2 and Pitx2 scavenge ROS. Pitx2 also regulates electron transport. Exosomes can act as mediators of cell-to-cell communication. NRG1 and its receptors ERBB2 and ERBB4 and VEGFA improve vascular regeneration. VEGFA also improves local coronary blood flow

Cell therapy of injured cardiac tissues. iPSCs can differentiate into functional cardiomyocytes in vitro. Heart-derived cells can differentiate into many types of heart cells. GMT or GHMT mixtures reprogram mouse fibroblasts to differentiate into iCMs, and Smarcd3 reprograms mouse fibroblasts to differentiate into cardiac progenitor cells in vitro. MSCs differentiate into CMs in the presence of 5-aza or in coculture with cardiac progenitor cells. However, reprogramming human fibroblasts requires GMT or GHMT mixtures and troponin, MESP1, ESRRγ and ZFPM2, was well as miR-1 and miR-133. Human fibroblasts can be transformed into cardiac progenitor cells with c-ETS2 and MESP1

Immune cells in the heart. cTMs internalize blood-borne FITC-dextran; cTMs also have typical macrophage characteristics and phagocytose bacteria and apoptotic cells. cTMs regulate electrical conduction through CX43, and cTMs can also produce proinflammatory cytokines to induce inflammation in the aging heart and promote neutrophil infiltration. Resident NK cells reduce cardiac eosinophil infiltration. NK cells prevent the maturation and transport of inflammatory cells. Monocytes are recruited into cardiac tissue and differentiate into M1 and M2 macrophages. M1 macrophages affect the proliferation and differentiation of CMs by BMPs, and M2 macrophages are related to angiogenesis by producing VEGF. Recruited neutrophils secrete proinflammatory cytokines, which promote fibroblast differentiation and lead to sustained inflammation. DCs upregulate cardiomyocyte hypertrophy a. NKT cells secrete cytokines such as IL-10 to protect or regulate hypertrophy, cardiac remodeling and the inflammatory response induced by Ang II. Infiltrating CD4 + cells include helper T cells (Th1 and Th2), Th17 cells and Treg cells. Th1 cells reduce the fibrotic response, while Th2 and Th17 cells promote fibrosis. Th17 cells also promote inflammation and extracellular matrix remodeling, and Treg cells reduce inflammation. Mast cells promote fibrosis, angiogenesis and cardiac fibroblast proliferation and collagen synthesis through TNF-α

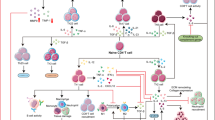
Three phases of immune response to cardiac injury repair. This diagram shows the inflammation, proliferation, and maturation phases after cardiac injury. In the Inflammatory phase, cTMs phagocytose dying cardiomyocytes. B cells secrete immunoregulatory factors to reduce cardiac contractility and promote cardiomyocyte apoptosis. DCs mediate the recruitment of inflammatory cells such as monocytes and M1 macrophages and homeostasis. In the proliferation phase, cTMs promote the proliferation of myocardial cells and angiogenesis. Neutrophils, monocytes and M2 macrophages also promote angiogenesis though VEGF, and M1 macrophages promote tissue fibrosis and myocardial remodeling by inducing extracellular matrix release from cardiac fibroblasts. NK cells protect against cardiac fibrosis by directly restricting collagen formation of cardiac fibroblasts and preventing the accumulation of specific inflammatory populations, and NK cells also promote blood vessel remodeling. Treg cells inhibit inflammation and fibrosis and promote precursor cell proliferation and macrophage polarization. In the maturation phase, the recruitment of inflammatory cells such as macrophages, neutrophils and eosinophils is inhibited, anti-inflammatory cytokines are secreted, infiltrating immune cells regulate inflammation inactivation/reduction by mediating the fibrotic response, and inflammation and scar formation are resolved. During this phase, hepcidin inhibits macrophage-induced cardiac repair and regeneration
References
Ma, X. et al. An exceptionally preserved arthropod cardiovascular system from the early Cambrian. Nat. Commun. 5, 3560 (2014).
Gosavi, S., Tyroch, A. H. & Mukherjee, D. Cardiac trauma. Angiology 67, 896–901 (2016).
Lozano, R. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128 (2012).
Godwin, J. W., Pinto, A. R. & Rosenthal, N. A. Macrophages are required for adult salamander limb regeneration. Proc. Natl. Acad. Sci. USA 110, 9415–9420 (2013).
Aurora, A. B. et al. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 124, 1382–1392 (2014).
Lavine, K. J. et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 111, 16029–16034 (2014).
Buchmann, K. Evolution of innate immunity: clues from invertebrates via fish to mammals. Front. Immunol. 5, 459 (2014).
Epelman, S., Liu, P. P. & Mann, D. L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 15, 117–129 (2015).
Xu, Z., McElhanon, K. E., Beck, E. X. & Weisleder, N. A murine model of myocardial ischemia-reperfusion injury. Methods Mol. Biol. 1717, 145–153 (2018).
Raucci, A. et al. The Janus face of HMGB1 in heart disease: a necessary update. Cell. Mol. life Sci.76, 211–229 (2019).
Haubner, B. J. et al. Complete cardiac regeneration in a mouse model of myocardial infarction. Aging 4, 966–977 (2012).
Nichols, M., Townsend, N., Scarborough, P. & Rayner, M. Cardiovascular disease in Europe 2014: epidemiological update. Eur. heart J. 35, 2950–2959 (2014).
Reed, G. W., Rossi, J. E. & Cannon, C. P. Acute myocardial infarction. Lancet 389, 197–210 (2017).
Yeh, R. W. et al. Population trends in the incidence and outcomes of acute myocardial infarction. N. Engl. J. Med. 362, 2155–2165 (2010).
Cooper, L. T. Myocarditis. N. Engl. J. Med. 360, 1526–1538 (2009).
Binder, A. et al. Myocardial protection from ischemia-reperfusion injury post coronary revascularization. Expert Rev. Cardiovascular Ther. 13, 1045–1057 (2015).
Hashmi, S. & Al-Salam, S. Acute myocardial infarction and myocardial ischemia-reperfusion injury: a comparison. Int. J. Clin. Exp. Pathol. 8, 8786–8796 (2015).
Darehzereshki, A. et al. Differential regenerative capacity of neonatal mouse hearts after cryoinjury. Developmental Biol. 399, 91–99 (2015).
Chablais, F., Veit, J., Rainer, G. & Jaźwińska, A. The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Developmental Biol. 11, 21 (2011).
González-Rosa, J. M. et al. Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Dev. (Camb., Engl.). 138, 1663–1674 (2011).
Schnabel, K., Wu, C.-C., Kurth, T. & Weidinger, G. Regeneration of cryoinjury induced necrotic heart lesions in zebrafish is associated with epicardial activation and cardiomyocyte proliferation. PloS ONE 6, e18503 (2011).
Strungs, E. G. et al. Cryoinjury models of the adult and neonatal mouse heart for studies of scarring and regeneration. Methods Mol. Biol. 1037, 343–353 (2013).
Porrello, E. R. et al. Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080 (2011).
Poss, K. D., Wilson, L. G. & Keating, M. T. Heart regeneration in zebrafish. Science 298, 2188–2190 (2002).
Curado, S. et al. Conditional targeted cell ablation in zebrafish: a new tool for regeneration studies. Developmental Dyn.: Off. Publ. Am. Assoc. Anatomists. 236, 1025–1035 (2007).
Curado, S., Stainier, D. Y. R. & Anderson, R. M. Nitroreductase-mediated cell/tissue ablation in zebrafish: a spatially and temporally controlled ablation method with applications in developmental and regeneration studies. Nat. Protoc. 3, 948–954 (2008).
Saito, M. et al. Diphtheria toxin receptor-mediated conditional and targeted cell ablation in transgenic mice. Nat. Biotechnol. 19, 746–750 (2001).
Frangogiannis, N. G. Pathophysiology of myocardial infarction. Comprehensive. Physiology 5, 1841–1875 (2015).
Wider, J. & Przyklenk, K. Ischemic conditioning: the challenge of protecting the diabetic heart. Cardiovasc. Diagnosis Ther. 4, 383–396 (2014).
Sager, H. B. et al. Targeting interleukin-1β reduces leukocyte production after acute myocardial infarction. Circulation 132, 1880–1890 (2015).
van Hout, G. P. et al. Translational failure of anti-inflammatory compounds for myocardial infarction: a meta-analysis of large animal models. Cardiovasc. Res. 109, 240–248 (2016).
Seropian, I. M., Toldo, S., Van Tassell, B. W. & Abbate, A. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J. Am. Coll. Cardiol. 63, 1593–1603 (2014).
Jennings, R. B. et al. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch. Pathol. 70, 68–78 (1960).
Jennings, R. B., Murry, C. E., Steenbergen, C. Jr. & Reimer, K. A. Development of cell injury in sustained acute ischemia. Circulation 82, Ii2–Ii12 (1990).
House, S. L. et al. Cardiac-specific overexpression of fibroblast growth factor-2 protects against myocardial dysfunction and infarction in a murine model of low-flow ischemia. Circulation 108, 3140–3148 (2003).
Harada, K. et al. Vascular endothelial growth factor administration in chronic myocardial ischemia. Am. J. Physiol. 270, H1791–H1802 (1996).
Karikó, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005).
Karikó, K., Muramatsu, H., Keller, J. M. & Weissman, D. Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol. Ther. 20, 948–953 (2012).
Karikó, K. et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 16, 1833–1840 (2008).
Kormann, M. S. et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 29, 154–157 (2011).
Mays, L. E. et al. Modified Foxp3 mRNA protects against asthma through an IL-10–dependent mechanism. J. Clin. Investig. 123, 1216–1228 (2013).
Warren, L. et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618–630 (2010).
Lui, K. O. et al. Driving vascular endothelial cell fate of human multipotent Isl1+ heart progenitors with VEGF modified mRNA. Cell Res. 23, 1172–1186 (2013).
Ptaszek, L. M., Mansour, M., Ruskin, J. N. & Chien, K. R. Towards regenerative therapy for cardiac disease. Lancet 379, 933–942 (2012).
Nagy, J. A. et al. Permeability properties of tumor surrogate blood vessels induced by VEGF-A. Lab. Investig. 86, 767–780 (2006).
Spilsbury, K. et al. Overexpression of vascular endothelial growth factor (VEGF) in the retinal pigment epithelium leads to the development of choroidal neovascularization. Am. J. Pathol. 157, 135–144 (2000).
Su, H. et al. Additive effect of AAV-mediated angiopoietin-1 and VEGF expression on the therapy of infarcted heart. Int. J. Cardiol. 133, 191–197 (2009).
Zangi, L. et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat. Biotechnol. 31, 898–907 (2013).
Malliaras, K. et al. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol. Med. 5, 191–209 (2013).
Cai, C.-L. et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 454, 104–108 (2008).
Loffredo, F. S., Steinhauser, M. L., Gannon, J. & Lee, R. T. Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell 8, 389–398 (2011).
Zhou, B. et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454, 109–113 (2008).
Gassmann, M. et al. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 378, 390–394 (1995).
Bersell, K., Arab, S., Haring, B. & Kühn, B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138, 257–270 (2009).
D’Uva, G. et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 17, 627–638 (2015).
Tao, G. et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 534, 119–123 (2016).
Ungvari, Z. et al. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. American journal of physiology. Heart Circ. Physiol. 299, H18–H24 (2010).
Zeng, C. et al. Curcumin protects hearts from FFA-induced injury by activating Nrf2 and inactivating NF-κB both in vitro and in vivo. J. Mol. Cell. Cardiol. 79, 1–12 (2015).
Bartel, D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009).
Eulalio, A. et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492, 376–381 (2012).
Chen, J. et al. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ. Res. 112, 1557–1566 (2013).
Tian, Y. et al. A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci. Transl. Med. 7, 279ra238 (2015).
Liu, X. et al. miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 21, 584–595 (2015).
Aurora, A. B. et al. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J. Clin. Investig. 122, 1222–1232 (2012).
Colombo, M., Raposo, G. & Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Developmental Biol. 30, 255–289 (2014).
Valadi, H. et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659 (2007).
Kinnaird, T. et al. Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ. Res. 94, 678–685 (2004).
Kishore, R. & Khan, M. More than tiny sacks: stem cell exosomes as cell-free modality for cardiac repair. Circ. Res. 118, 330–343 (2016).
Luther, K. M. et al. Exosomal miR-21a-5p mediates cardioprotection by mesenchymal stem cells. J. Mol. Cell. Cardiol. 119, 125–137 (2018).
Arslan, F. et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 10, 301–312 (2013).
Park, I.-H. et al. Generation of human-induced pluripotent stem cells. Nat. Protoc. 3, 1180–1186 (2008).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Mummery, C. L. et al. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: a methods overview. Circ. Res. 111, 344–358 (2012).
Braam, S. R. et al. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 4, 107–116 (2010).
Passier, R., van Laake, L. W. & Mummery, C. L. Stem-cell-based therapy and lessons from the heart. Nature 453, 322–329 (2008).
Beltrami, A. P. et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114, 763–776 (2003).
Menasché, P. et al. The myoblast autologous grafting in ischemic cardiomyopathy (MAGIC) trial: first randomized placebo-controlled study of myoblast transplantation. Circulation 117, 1189–1200 (2008).
Meyer, G. P. et al. Intracoronary bone marrow cell transfer after myocardial infarction: eighteen months’ follow-up data from the randomized, controlled BOOST (BOne marrOw transfer to enhance ST-elevation infarct regeneration) trial. Circulation 113, 1287–1294 (2006).
Planat-Bénard, V. et al. Spontaneous cardiomyocyte differentiation from adipose tissue stroma cells. Circ. Res. 94, 223–229 (2004).
Antonitsis, P., Ioannidou-Papagiannaki, E., Kaidoglou, A. & Papakonstantinou, C. In vitro cardiomyogenic differentiation of adult human bone marrow mesenchymal stem cells. The role of 5-azacytidine. Interact. Cardiovascular Thorac. Surg. 6, 593–597 (2007).
Glembotski, C. C. Expanding the paracrine hypothesis of stem cell-mediated repair in the heart: when the unconventional becomes conventional. Circ. Res. 120, 772–774 (2017).
Deretic, V., Jiang, S. & Dupont, N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 22, 397–406 (2012).
Gnecchi, M., Zhang, Z., Ni, A. & Dzau, V. J. Paracrine mechanisms in adult stem cell signaling and therapy. Circ. Res. 103, 1204–1219 (2008).
Shabbir, A., Zisa, D., Suzuki, G. & Lee, T. Heart failure therapy mediated by the trophic activities of bone marrow mesenchymal stem cells: a noninvasive therapeutic regimen. Am. J. Physiol. Heart Circ. Physiol. 296, H1888–H1897 (2009).
de Bold, A. J. Thirty years of research on atrial natriuretic factor: historical background and emerging concepts. Can. J. Physiol. Pharmacol. 89, 527–531 (2011).
Lionetti, V., Bianchi, G., Recchia, F. A. & Ventura, C. Control of autocrine and paracrine myocardial signals: an emerging therapeutic strategy in heart failure. Heart Fail. Rev. 15, 531–542 (2010).
Lalit, P. A. et al. Lineage reprogramming of fibroblasts into proliferative Induced cardiac progenitor cells by defined factors. Cell. Stem Cell. 18, 354–367 (2016).
Ieda, M. [Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors]. Nihon rinsho. Jpn. J. Clin. Med. 69, 524–527 (2011). Suppl 7.
Fu, J.-D. et al. Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Rep. 1, 235–247 (2013).
Nam, Y.-J. et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc. Natl Acad. Sci. USA 110, 5588–5593 (2013).
Wada, R. et al. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl Acad. Sci. USA 110, 12667–12672 (2013).
Islas, J. F. et al. Transcription factors ETS2 and MESP1 transdifferentiate human dermal fibroblasts into cardiac progenitors. Proc. Natl Acad. Sci. USA 109, 13016–13021 (2012).
Swirski, F. K. & Nahrendorf, M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 18, 733–744 (2018).
Varol, C., Mildner, A. & Jung, S. Macrophages: development and tissue specialization. Annu. Rev. Immunol. 33, 643–675 (2015).
Epelman, S. et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 40, 91–104 (2014).
Leid, J. et al. Primitive embryonic macrophages are required for coronary development and maturation. Circ. Res. 118, 1498–1511 (2016).
Pinto, A. R. et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PloS ONE 7, e36814 (2012).
Hulsmans, M. et al. Macrophages facilitate electrical conduction in the heart. Cell 169, 510–522 (2017).
Honold, L. & Nahrendorf, M. Resident and monocyte-derived macrophages in cardiovascular disease. Circ. Res. 122, 113–127 (2018).
Nicolás-Ávila, J. A. et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell. 183, 94–109 (2020).
Heidt, T. et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 115, 284–295 (2014).
Nicolás‐Ávila, J. A., Hidalgo, A. & Ballesteros, I. Specialized functions of resident macrophages in brain and heart. J. Leukoc. Biol. 104, 743–756 (2018).
Patel, B. et al. CCR2 monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. Jacc. Basic Transl. Sci. 3, 230–244 (2018).
Swirski, F. K., Robbins, C. S. & Nahrendorf, M. Development and function of arterial and cardiac macrophages. Trends Immunol. 37, 32–40 (2016).
Pinto, A. R., Godwin, J. W. & Rosenthal, N. A. Macrophages in cardiac homeostasis, injury responses and progenitor cell mobilisation. Stem Cell Res. 13, 705–714 (2014).
Shi, F.-D., Ljunggren, H.-G., La Cava, A. & Van Kaer, L. Organ-specific features of natural killer cells. Nat. Rev. Immunol. 11, 658–671 (2011).
Sharma, R. & Das, A. Organ-specific phenotypic and functional features of NK cells in humans. Immunologic Res. 58, 125–131 (2014).
Ayach, B. B. et al. Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc. Natl Acad. Sci. USA 103, 2304–2309 (2006).
Ong, S. et al. Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am. J. Pathol. 185, 847–861 (2015).
Knorr, M., Münzel, T. & Wenzel, P. Interplay of NK cells and monocytes in vascular inflammation and myocardial infarction. Front. Physiol. 5, 295 (2014).
Van der Borght, K. et al. Myocardial infarction primes autoreactive T cells through activation of dendritic cells. Cell Rep. 18, 3005–3017 (2017).
Sichien, D. et al. IRF8 transcription factor controls survival and function of terminally differentiated conventional and plasmacytoid dendritic cells, respectively. Immunity 45, 626–640 (2016).
Randolph, G. J., Ochando, J. & Partida-Sánchez, S. Migration of dendritic cell subsets and their precursors. Annu. Rev. Immunol. 26, 293–316 (2008).
Shortman, K. & Naik, S. H. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7, 19–30 (2007).
Cao, W. et al. Advanced glycation end products induced immune maturation of dendritic cells controls heart failure through NF-κB signaling pathway. Arch. Biochem. Biophys. 580, 112–120 (2015).
Clemente-Casares, X. et al. A CD103 conventional dendritic cell surveillance system prevents development of overt heart failure during subclinical viral myocarditis. Immunity 47, 974–989 (2017).
Guilliams, M. et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity 45, 669–684 (2016).
Hart, D. N. & Fabre, J. W. Demonstration and characterization of Ia-positive dendritic cells in the interstitial connective tissues of rat heart and other tissues, but not brain. J. Exp. Med. 154, 347–361 (1981).
Mouton, A. J., Rivera, O. J. & Lindsey, M. L. Myocardial infarction remodeling that progresses to heart failure: a signaling misunderstanding. American journal of physiology. Heart Circ. Physiol. 315, H71–H79 (2018).
Bond, A. R. et al. Changes in contractile protein expression are linked to ventricular stiffness in infants with pulmonary hypertension or right ventricular hypertrophy due to congenital heart disease. Open Heart 5, e000716 (2018).
Iacobazzi, D. et al. Cellular and molecular basis of RV hypertrophy in congenital heart disease. Heart 102, 12–17 (2016).
Liu, T. et al. Current understanding of the pathophysiology of myocardial fibrosis and its quantitative assessment in heart failure. Front. Physiol. 8, 238 (2017).
Vikhorev, P. G. & Vikhoreva, N. N. Cardiomyopathies and related changes in contractility of human heart muscle. Int. J. Mol. Sci. 19, 2234 (2018).
Humbert, M. et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur. Resp. J. 53, 1801887 (2019).
Forte, E., Furtado, M. B. & Rosenthal, N. The interstitium in cardiac repair: role of the immune-stromal cell interplay. Nat. Rev. Cardiol. 15, 601–616 (2018).
Prabhu, S. D. & Frangogiannis, N. G. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ. Res. 119, 91–112 (2016).
Frangogiannis, N. G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 11, 255–265 (2014).
Ben-Mordechai, T. et al. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J. Am. Coll. Cardiol. 62, 1890–1901 (2013).
Murray, P. J. & Wynn, T. A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 (2011).
Wilson, E. M., Diwan, A., Spinale, F. G. & Mann, D. L. Duality of innate stress responses in cardiac injury, repair, and remodeling. J. Mol. Cell. Cardiol. 37, 801–811 (2004).
Mezzaroma, E. et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl Acad. Sci. USA 108, 19725–19730 (2011).
Frangogiannis, N. G. et al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation 115, 584–592 (2007).
Jenkins, S. J. et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332, 1284–1288 (2011).
Godwin, J. W., Debuque, R., Salimova, E. & Rosenthal, N. A. Heart regeneration in the salamander relies on macrophage-mediated control of fibroblast activation and the extracellular landscape. NPJ Regen. Med. 2, 22 (2017).
Molawi, K. et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 211, 2151–2158 (2014).
Ma, Y., Mouton, A. J. & Lindsey, M. L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res.: J. Lab. Clin. Med. 191, 15–28 (2018).
Ong, S., Rose, N. R. & Čiháková, D. Natural killer cells in inflammatory heart disease. Clin. Immunol. 175, 26–33 (2017).
Bouchentouf, M. et al. Induction of cardiac angiogenesis requires killer cell lectin-like receptor 1 and α4β7 integrin expression by NK cells. J. Immunol. 185, 7014–7025 (2010).
Bouchentouf, M. et al. Interleukin-2 enhances angiogenesis and preserves cardiac function following myocardial infarction. Cytokine 56, 732–738 (2011).
Leuschner, F. et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 209, 123–137 (2012).
Morimoto, H. et al. MCP-1 induces cardioprotection against ischaemia/reperfusion injury: role of reactive oxygen species. Cardiovascular Res. 78, 554–562 (2008).
Moore, K. J. & Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355 (2011).
Frangogiannis, N. G. Emerging roles for macrophages in cardiac injury: cytoprotection, repair, and regeneration. J. Clin. Investig. 125, 2927–2930 (2015).
Zhang, F. et al. Cardiac contractility modulation attenuate myocardial fibrosis by inhibiting TGF-β1/Smad3 signaling pathway in a rabbit model of chronic heart failure. Cell. Physiol. Biochem. 39, 294–302 (2016).
Kubo, T. et al. Regulatory T cell suppression and anergy are differentially regulated by proinflammatory cytokines produced by TLR-activated dendritic cells. J. Immunol. 173, 7249–7258 (2004).
Yamazaki, S., Inaba, K., Tarbell, K. V. & Steinman, R. M. Dendritic cells expand antigen-specific Foxp3+ CD25+ CD4+ regulatory T cells including suppressors of alloreactivity. Immunol. Rev. 212, 314–329 (2006).
Ghiringhelli, F., Ménard, C., Martin, F. & Zitvogel, L. The role of regulatory T cells in the control of natural killer cells: relevance during tumor progression. Immunol. Rev. 214, 229–238 (2006).
Wan, E. et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 113, 1004–1012 (2013).
Korf-Klingebiel, M. et al. Myeloid-derived growth factor (C19orf10) mediates cardiac repair following myocardial infarction. Nat. Med. 21, 140–149 (2015).
Fernández-Velasco, M., González-Ramos, S. & Boscá, L. Involvement of monocytes/macrophages as key factors in the development and progression of cardiovascular diseases. Biochem. J. 458, 187–193 (2014).
Dehn, S. & Thorp, E. B. Myeloid receptor CD36 is required for early phagocytosis of myocardial infarcts and induction of Nr4a1-dependent mechanisms of cardiac repair. FASEB J. 32, 254–264 (2018).
Zlatanova, I. et al. Iron regulator hepcidin impairs macrophage-dependent cardiac repair after injury. Circulation 139, 1530–1547 (2019).
Morimoto, H. et al. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice prevents cardiac dysfunction and remodeling after myocardial infarction. Circ. Res. 99, 891–899 (2006).
Lörchner, H. et al. Myocardial healing requires Reg3β-dependent accumulation of macrophages in the ischemic heart. Nat. Med. 21, 353–362 (2015).
Lai, S.-L. et al. Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. eLife. 6, e25605 (2017).
Pallotta, I., Sun, B., Wrona, E. A. & Freytes, D. O. BMP protein-mediated crosstalk between inflammatory cells and human pluripotent stem cell-derived cardiomyocytes. J. Tissue Eng. Regen. Med. 11, 1466–1478 (2017).
Diny, N. L. et al. Macrophages and cardiac fibroblasts are the main producers of eotaxins and regulate eosinophil trafficking to the heart. Eur. J. Immunol. 46, 2749–2760 (2016).
Lu, H. et al. Angiotensin II-C-C chemokine receptor2/5 axis-dependent monocyte/macrophage recruitment contributes to progression of experimental autoimmune myocarditis. Microbiol. Immunol. 61, 539–546 (2017).
Sager, H. B. et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ. Res. 119, 853–864 (2016).
Li, W. et al. Heart-resident CCR2 macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight. 1, e87315 (2016).
Frangogiannis, N. G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 110, 159–173 (2012).
Yan, X. et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 62, 24–35 (2013).
Kono, H. & Rock, K. L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289 (2008).
Nathan, C. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 6, 173–182 (2006).
Middleton, J. et al. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell 91, 385–395 (1997).
Heit, B. et al. PTEN functions to’prioritize’chemotactic cues and prevent’distraction’in migrating neutrophils. Nat. Immunol. 9, 743 (2008).
McDonald, B. et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330, 362–366 (2010).
Hayashidani, S. et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 108, 2134–2140 (2003).
Nahrendorf, M. et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037–3047 (2007).
Ma, Y., Yabluchanskiy, A. & Lindsey, M. L. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenes. Tissue Repair. 6, 11 (2013).
Mantovani, A., Cassatella, M. A., Costantini, C. & Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 11, 519–531 (2011).
Serhan, C. N., Chiang, N. & Van Dyke, T. E. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361 (2008).
Chishti, A. D., Shenton, B. K., Kirby, J. A. & Baudouin, S. V. Neutrophil chemotaxis and receptor expression in clinical septic shock. Intensive Care Med. 30, 605–611 (2004).
Rios-Santos, F. et al. Down-regulation of CXCR2 on neutrophils in severe sepsis is mediated by inducible nitric oxide synthase-derived nitric oxide. Am. J. Respiratory Crit. Care Med. 175, 490–497 (2007).
Cheng, B. et al. Harnessing the early post-injury inflammatory responses for cardiac regeneration. J. Biomed. Sci. 24, 7 (2017).
Horckmans, M. et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 38, 187–197 (2017).
Anzai, A. et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 125, 1234–1245 (2012).
Gallego-Colon, E. et al. Cardiac-restricted IGF-1Ea overexpression reduces the early accumulation of inflammatory myeloid cells and mediates expression of extracellular matrix remodelling genes after myocardial infarction. Mediators Inflamm. 2015, 484357–484357 (2015).
Sage, A. P. et al. MHC Class II-restricted antigen presentation by plasmacytoid dendritic cells drives proatherogenic T cell immunity. Circulation 130, 1363–1373 (2014).
Liu, K. et al. In vivo analysis of dendritic cell development and homeostasis. Science 324, 392–397 (2009).
Schlitzer, A. et al. Identification of cDC1-and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat. Immunol. 16, 718–728 (2015).
Lee, J. S. et al. Conventional dendritic cells impair recovery after myocardial infarction. J. Immunol. 201, 1784–1798 (2018).
Frangogiannis, N. G. The immune system and cardiac repair. Pharmacol. Res. 58, 88–111 (2008).
Hofmann, U. et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125, 1652–1663 (2012).
Nagai, T. et al. Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans. J. Am. Heart Assoc. 3, e000839 (2014).
Kronenberg, M. & Gapin, L. The unconventional lifestyle of NKT cells. Nat. Rev. Immunol. 2, 557–568 (2002).
Godfrey, D. I. et al. NKT cells: facts, functions and fallacies. Immunol. today 21, 573–583 (2000).
Crowe, N. Y. et al. Glycolipid antigen drives rapid expansion and sustained cytokine production by NK T cells. J. Immunol. 171, 4020–4027 (2003).
Matsuda, J. L. et al. Mouse Vα14i natural killer T cells are resistant to cytokine polarization in vivo. Proc. Natl. Acad. Sci. USA 100, 8395–8400 (2003).
Matsuda, J. L. et al. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J. Exp. Med. 192, 741–754 (2000).
Godfrey, D. I. & Kronenberg, M. Going both ways: immune regulation via CD1d-dependent NKT cells. J. Clin. Investig. 114, 1379–1388 (2004).
Smyth, M. J. & Godfrey, D. I. NKT cells and tumor immunity—a double-edged sword. Nat. Immunol. 1, 459–460 (2000).
Fischer, P. & Hilfiker-Kleiner, D. Survival pathways in hypertrophy and heart failure: the gp130-STAT3 axis. Basic Res. Cardiol. 102, 393–411 (2007).
Sun, M. et al. Tumor necrosis factor-a mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation 115, 1398 (2007).
Takahashi, M. et al. The disruption of invariant natural killer T cells exacerbates cardiac hypertrophy and failure caused by pressure overload in mice. Exp. Physiol. 105, 489–501 (2020).
Cohn, J. N., Ferrari, R. & Sharpe, N. Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J. Am. Coll. Cardiol. 35, 569–582 (2000).
Wang, H.-X. et al. CD1d-dependent natural killer T cells attenuate angiotensin II-induced cardiac remodelling via IL-10 signalling in mice. Cardiovasc. Res. 115, 83–93 (2019).
van Puijvelde, G. H. et al. CD1d deficiency inhibits the development of abdominal aortic aneurysms in LDL receptor deficient mice. PloS ONE 13, e0190962 (2018).
Sköld, M. & Behar, S. M. Role of CD1d-restricted NKT cells in microbial immunity. Infect. Immun. 71, 5447–5455 (2003).
Ramjee, V. et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J. Clin. Investig. 127, 899–911 (2017).
Zandbergen, H. R. et al. Macrophage depletion in hypertensive rats accelerates development of cardiomyopathy. J. Cardiovasc. Pharmacol. Therap. 14, 68–75 (2009).
Blanton, R. M., Carrillo-Salinas, F. J. & Alcaide, P. T-cell recruitment to the heart: friendly guests or unwelcome visitors? American journal of physiology. Heart Circ. Physiol. 317, H124–H140 (2019).
Lichtman, A. H. The heart of the matter: protection of the myocardium from T cells. J. Autoimmun. 45, 90–96 (2013).
Komarowska, I. et al. Hepatocyte growth factor receptor c-Met instructs T cell cardiotropism and promotes T cell migration to the heart via autocrine chemokine release. Immunity 42, 1087–1099 (2015).
Marra, F., Aleffi, S., Galastri, S. & Provenzano, A. Mononuclear cells in liver fibrosis. Semin. Immunopathol. 31, 345–358 (2009).
Ivanov, I. I. et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006).
Afzali, B., Lombardi, G., Lechler, R. I. & Lord, G. M. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin. Exp. Immunol. 148, 32–46 (2007).
Feng, W. et al. IL-17 induces myocardial fibrosis and enhances RANKL/OPG and MMP/TIMP signaling in isoproterenol-induced heart failure. Exp. Mol. Pathol. 87, 212–218 (2009).
Sakaguchi, S. et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self‐tolerance and autoimmune disease. Immunol. Rev. 212, 8–27 (2006).
Gullestad, L. et al. Agents targeting inflammation in heart failure. Expert Opin. Investig. Drugs 14, 557–566 (2005).
Frangogiannis, N. G., Smith, C. W. & Entman, M. L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 53, 31–47 (2002).
Tang, T.-T. et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 107, 232 (2012).
Castiglioni, A. et al. FOXP3+ T cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and guide effective tissue regeneration. PloS ONE 10, e0128094 (2015).
Ali, N. et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell. 169, 1119–1129 (2017).
Clynes, R. Protective mechanisms of IVIG. Curr. Opin. Immunol. 19, 646–651 (2007).
Kaschina, E. et al. Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation 118, 2523–2532 (2008).
Carbone, F. et al. Pathophysiological role of neutrophils in acute myocardial infarction. Thrombosis Haemost. 110, 501–514 (2013).
D’Alessio, F. R. et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J. Clin. Investig. 119, 2898–2913 (2009).
Weirather, J. et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 115, 55–67 (2014).
Dombrowski, Y. et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 20, 674–680 (2017).
Liu, G. et al. Phenotypic and functional switch of macrophages induced by regulatory CD4+ CD25+ T cells in mice. Immunol. Cell Biol. 89, 130–142 (2011).
Kvakan, H. et al. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation 119, 2904–2912 (2009).
Sakaguchi, S., Yamaguchi, T., Nomura, T. & Ono, M. Regulatory T cells and immune tolerance. Cell 133, 775–787 (2008).
Tang, Q. & Bluestone, J. A. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat. Immunol. 9, 239–244 (2008).
Grossman, W. J. et al. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 21, 589–601 (2004).
Kim, J. M., Rasmussen, J. P. & Rudensky, A. Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 8, 191–197 (2007).
Hofmann, U. & Frantz, S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ. Res. 116, 354–367 (2015).
Zouggari, Y. et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 19, 1273–1280 (2013).
Nian, M., Lee, P., Khaper, N. & Liu, P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ. Res. 94, 1543–1553 (2004).
Wu, L. et al. IL-10-producing B cells are enriched in murine pericardial adipose tissues and ameliorate the outcome of acute myocardial infarction. Proc. Natl. Acad. Sci. USA 116, 21673–21684 (2019).
Brower, G. L. & Janicki, J. S. Pharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in rats. J. Card. Fail. 11, 548–556 (2005).
Hara, M. et al. Mast cells cause apoptosis of cardiomyocytes and proliferation of other intramyocardial cells in vitro. Circulation 100, 1443–1449 (1999).
Kubes, P. & Granger, D. N. Leukocyte-endothelial cell interactions evoked by mast cells. Cardiovasc. Res. 32, 699–708 (1996).
Ehrenreich, H. et al. Endothelins belong to the assortment of mast cell-derived and mast cell-bound cytokines. N. biologist. 4, 147–156 (1992).
Murray, D. B., Gardner, J. D., Brower, G. L. & Janicki, J. S. Endothelin-1 mediates cardiac mast cell degranulation, matrix metalloproteinase activation, and myocardial remodeling in rats. American journal of physiology. Heart Circ. Physiol. 287, H2295–H2299 (2004).
Wainwright, C. L., McCabe, C. & Kane, K. A. Endothelin and the ischaemic heart. Curr. Vasc. Pharmacol. 3, 333–341 (2005).
Perskvist, N. & Edston, E. Differential accumulation of pulmonary and cardiac mast cell-subsets and eosinophils between fatal anaphylaxis and asthma death: a postmortem comparative study. Forensic Sci. Int. 169, 43–49 (2007).
Perskvist, N., Söderberg, C., van Hage, M. & Edston, E. Pathogenic role of cardiac mast cell activation/degranulation,TNF-alpha, and cell death in acute drug-related fatalities. Vasc. Health Risk Manag. 3, 1053–1062 (2007).
Janicki, J. S. et al. Cardiac mast cell regulation of matrix metalloproteinase-related ventricular remodeling in chronic pressure or volume overload. Cardiovasc. Res. 69, 657–665 (2006).
Palaniyandi Selvaraj, S. et al. Involvement of mast cells in the development of fibrosis in rats with postmyocarditis dilated cardiomyopathy. Biol. Pharm. Bull. 28, 2128–2132 (2005).
Liao, C.-h. et al. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J. Clin. Investig. 120, 242–253 (2010).
Acknowledgements
This Review was supported by the National Key R&D Program of China (2018YFA0800503), excellent young scientist foundation of NSFC (31822017). This study was supported by Zhejiang Provincial Natural Science Foundation of China under Grant NO. LR19C080001. This study was supported by the National Natural Science Foundation of China (81572651/ 81771675). This study also supported by the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
S.K. wrote the manuscript. J.J. and L.Y. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, K., Li, Yy. & Jin, J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Sig Transduct Target Ther 6, 79 (2021). https://doi.org/10.1038/s41392-020-00455-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-00455-6
This article is cited by
-
Macrophages promote the transition from myocardial ischemia reperfusion injury to cardiac fibrosis in mice through GMCSF/CCL2/CCR2 and phenotype switching
Acta Pharmacologica Sinica (2024)
-
The gasdermin family: emerging therapeutic targets in diseases
Signal Transduction and Targeted Therapy (2024)
-
The roles of Th cells in myocardial infarction
Cell Death Discovery (2024)
-
Spatial analysis of tissue immunity and vascularity by light sheet fluorescence microscopy
Nature Protocols (2024)
-
Reward system activation improves recovery from acute myocardial infarction
Nature Cardiovascular Research (2024)
