
Abstract
Ferroptosis is an iron-dependent cell death, which is different from apoptosis, necrosis, autophagy, and other forms of cell death. The process of ferroptotic cell death is defined by the accumulation of lethal lipid species derived from the peroxidation of lipids, which can be prevented by iron chelators (e.g., deferiprone, deferoxamine) and small lipophilic antioxidants (e.g., ferrostatin, liproxstatin). This review summarizes current knowledge about the regulatory mechanism of ferroptosis and its association with several pathways, including iron, lipid, and cysteine metabolism. We have further discussed the contribution of ferroptosis to the pathogenesis of several diseases such as cancer, ischemia/reperfusion, and various neurodegenerative diseases (e.g., Alzheimer’s disease and Parkinson’s disease), and evaluated the therapeutic applications of ferroptosis inhibitors in clinics.
Similar content being viewed by others

Introduction
Ferroptosis is a newly identified iron-dependent cell death that is different from other cell death forms, including apoptosis and necrosis. The program involves three primary metabolisms involving thiol, lipid, and iron, leading to an iron-dependent generation of lipid peroxidation and, ultimately, cell death. Ferroptosis can be prevented by the enzymatic reaction of two major antioxidant systems involving glutathione peroxidase 4 (GPx4) that catalyzes the reduction of lipid peroxides in a glutathione-dependent reaction and the recently identified ferroptosis suppressor protein (FSP1) that catalyzes the regeneration of ubiquinone (Coenzyme Q10, CoQ10), which act as a lipid peroxyl radical trap.1,2 Specific inhibitors can prevent ferroptosis, e.g., ferrostatin-1 acts as a radical-trapping antioxidant (RTA).1
Ferroptotic cell death is accompanied by a series of variations in cell morphology, metabolism, and protein expression that allows discrimination from other forms of cell death. At the cellular and subcellular levels, cells undergoing ferroptosis adopt a characteristic rounded shape before cell death similar to necrotic cells, but there is no cytoplasmic and organelle swelling, or plasma membrane rupture.1,3 The nuclei in ferroptotic cells conserve its structural integrity, without condensation, chromatin margination, plasma membrane blebbing, or formation of apoptotic bodies,1 which are characteristic features of apoptosis.4 Also, morphological features such as double-membrane enclosed vesicles from autophagic cells and intensive blebbing and loss of plasma membrane integrity shown in pyroptosis, are not observed in ferroptotic cells.5 The lone distinctive morphological feature is mitochondria that appeared smaller than normal with increased membrane density.1
Ferroptosis is regulated by a set of genes and shows a variety of metabolic changes. The detection of these changes, as evidence of ferroptosis, is essential for further research. Iron is an essential part of driving intracellular lipid peroxidation and ferroptosis.6 Ferroptosis can be prevented by using iron chelators (e.g., deferoxamine), whereas supplying exogenous iron (e.g., ferric ammonium citrate) enhances ferroptosis.1,7 Several studies have shown that the regulation of genes related to iron metabolism can also regulate ferroptotic cell death, such as transferrin, nitrogen fixation 1 (NFS1), iron response element-binding protein 2 (IREB2), Nuclear receptor coactivator 4 (NCOA4), etc.1,8,9 So, iron abundance is an essential indicator for monitoring ferroptosis. FRET Iron Probe 1 (FIP-1), a fluorescence probe, is widely used to detect the change of labile iron status during ferroptosis.10 Also, iron concentration can be measured with inductively coupled plasma-MS (ICP-MS) or Perls’ Prussian Blue staining.5 Lipid peroxidation level is also one of the most critical indicators of ferroptosis.1 A significant increase in peroxidized phospholipids was observed in many ferroptosis models.11,12 BODIPY-C11 (or C11-BODIPY) and LiperFluo are currently two major assays used to measure lipid peroxidation in ferroptosis, while the latter is considered to be a more reliable probe owing to its higher specificity.11 Also, changes in GPx4 activity can be used as an indicator for ferroptosis, which can be monitored by either the nicotinamide adenine dinucleotide phosphate (NADPH) activity assay13 or quantification of phosphatidylcholine hydroperoxide with LC-MS.5
The discovery of ferroptosis
Ferroptosis inducers were discovered in another high-throughput small molecule-screening study, as selectively lethal compounds to RAS mutant tumor cells, before the notion of ferroptosis was developed (Fig. 1). Back in 2003, erastin was found to be lethal with the expression of the engineered mutant Ras oncogene in human foreskin fibroblasts (BJeLR cells).14 However, subsequent studies have not identified sufficient targets for erastin-induced cell death.3 Ras-selective lethal small molecule (RSL)-3 and RSL5 were later identified in 2008 as synthetic compounds that selectively killed BJeLR cells in a non-apoptotic manner.7 It was not until 2012 that the form of cell death was named ferroptosis and erastin was found to inhibit cystine uptake by the cystine/glutamate antiporter (system \({\mathrm{X}}_c^ -\)) leading to cell death (Fig. 1).1

The discovery of ferroptosis starts with identification of system xCT, which was published in 1980. However, the term ‘ferroptosis' was only named in 2012.
System\({\mathrm{X}}_c^ -\) was found to function by transporting cystine into the cell in exchange for glutamate in 1980.15 An early report has indicated that glutamate toxicity in a neuronal cell line is triggered by inhibition of cystine transport, leading to oxidative stress.16 Subsequently, it was discovered that antioxidant supplementation (e.g., alpha-tocopherol, α-toc) prevented glutamate-induced cell death in neuronal cell lines lacking N-methyl-D-aspartate receptor.17 Soon thereafter, it was shown that inhibition of arachidonate 12-lipoxygenase (Alox12), an iron-containing lipid dioxygenase, effectively inhibited cell death induced by glutamate in the hippocampal cell line HT22 and primary cortical neurons.18 Treatment of cells with exogenous arachidonic acid (AA), an Alox12 substrate, further accelerated cell death.18
In 2014, Yang et al. suggested that GPx4 plays a crucial role in protecting against ferroptosis by reducing phospholipid hydroperoxide and hence repressing lipoxygenase-mediated lipid peroxidation.13 In the extracellular milieu, the iron-carrier protein transferrin and glutamine were identified as essential factors required to induce ferroptosis. Conversely, inhibition of glutaminolysis and cell surface transferrin receptor can reduce heart injury triggered by ischemia/reperfusion (I/R) against ferroptosis.19 On the oxidation pathway, lipoxygenases (Lox) catalyze PUFA oxidation via a phosphorylase kinase G2 (PHKG2)-dependent iron pool,19 whereas compounds with RTA activity ameliorate ferroptosis via blocking lipid autoxidation.20
In 2017, it was shown that acyl-CoA synthetase long-chain family member 4 (ACSL4) is a biomarker and critical contributor to ferroptosis, that is required for the production of polyunsaturated fatty acids (PUFA) required for the execution of ferroptosis.21 A further study by Ingold et al. (2018), depicted the requirement for selenium utilization by GPx4 to inhibit ferroptosis.22 Recently, a new ferroptosis suppression pathway has been identified with the discovery that FSP1, CoQ10 oxidoreductase, can inhibit ferroptosis in a glutathione-independent pattern.2,23 In a further study of the ferroptosis sensitivity gene via genome-wide CRISPR–Cas9 suppressor screens, oxidative organelles peroxisomes were found to help the cancer cells escape and increase the susceptibility to ferroptosis through synthesizing polyunsaturated ether phospholipids (PUFA-ePL).24
Regulation of ferroptosis
Oxidation mechanisms
Summary of polyunsaturated fatty acids
Fatty acids are essential components of cellular lipid metabolism and fulfill several cellular functions, including energy supply, cell membrane formation, and serve as a precursor for several signaling molecules.25 However, tight regulation of fatty acid metabolism is required to prevent toxicity as observed in cell death pathways such as pyroptosis26 and ferroptosis. AMP-activated protein kinase (AMPK), a sensor of cellular energy status, can regulate ferroptosis via AMPK-mediated phosphorylation of acetyl-CoA carboxylase (ACC) and polyunsaturated fatty acid biosynthesis.27 Liver kinase B1 (LKB1) is a main upstream kinase responsible for the activation of AMPK in response to energy stress. The depletion of LKB1 also can sensitize mouse embryonic fibroblasts to lipid hydroperoxidation and ferroptosis.28 Long-chain fatty acids are mainly obtained from the diet and are named PUFA when they include more than two double bonds.29
PUFAs are components of the cell membrane and regulate several biological functions, including inflammation, immunity, synaptic plasticity, and cellular growth.30 The structure of PUFA is prone to oxidation because of the weak C–H bond at the bis-allylic positions.31 Furthermore, membrane PUFA is the primary target of reactive oxygen species (ROS) attack.32 In general, a higher number of double bonds in PUFA increases its susceptibility to oxidation.33 After the initial oxidation step, the free radicals can shift within the same molecule or oxidize further molecules.34 Therefore, PUFAs are the main substrate of lipid peroxidation during ferroptosis. Exogenous administration of the monounsaturated fatty acid (MUFA) oleic acid (OA, C18:1) can effectively inhibit erastin induced ferroptosis by competing with PUFAs for incorporation into phospholipids (PLs).35 This fact suggests that MUFAs are not the substrate of lipid peroxidation during ferroptosis. In addition, sterol lipids, including cholesterols, can be oxidized in membranes or low-density lipoprotein particles,36 and oxidized cholesterol is also the active substrate of GPx4.37 However, exogenous cholesterol treatment is not enough to regulate the lethality of RSL3 in human cancer cells. All evidence highlights the critical role of PUFA in ferroptosis.38
Research on PUFA mainly focuses on ω-6 and ω-3 eicosanoid.30 In vivo, the most common PUFA is AA, which is present in all tissues.39 While the composition of PUFA changes with the environment in many tissues, docosahexaenoic acid (DHA) and AA are the most abundant isoform of PUFA in the brain and retina,40 and DHA supplementation in childhood may improve cognitive and motor function in children with attention deficit/hyperactivity disorder.41 Due to its high lipid composition, the brain is particularly vulnerable to oxidative damage through lipid ROS. Therefore long-chain PUFA (LCPUFA) plays also an essential role in neurocognitive disorder diseases.42,43
The process of lipid peroxidation
PUFA is a double-edged sword, and its peroxidation may cause damage to cells. It can be integrated into the membrane by ACLS421 and lysophosphatidylcholine acyltransferase 3 (LPCAT3).44 PUFA oxidation can occur either by non-enzymatic free radical chain reaction or enzyme catalysis (Fig. 2).

The indicated pathways control ferroptosis sensitivity via lipid ROS generation. Phosphatidylethanolamines (PE); phospholipid (PL-H); phospholipid alkoxyl radical (PL-O·); phospholipid peroxyl radical (PL-OO·); phospholipid hydroperoxide (PL-OOH); transferrin (TF). The symbols used in the figure have been marked with names of the biomolecules.
AA and adrenic acid (AdA) are the main PUFAs to induce ferroptosis.11 Taking AA as an example, ACSL4 catalyzes the ligation of CoA into AA to form a CoA-AA intermediate, which is esterified into phosphatidylethanolamine by LPCAT3 to form arachidonic acid-phosphatidylethanolamines (PE-AA). The oxidation of the formed PE-AA may follow either enzymatically through the action of Lox, or non-enzymatically through autoxidation to form PE-AA-OOH, both of which ultimately causes cell death.1,38,45,46 It has been reported that this reaction may occur on the mitochondrial membrane1 or the mitochondrial and endoplasmic reticulum membrane.11 BID (a pro-apoptotic protein) links mitochondria with ferroptosis,47 and the mitochondrial TCA cycle promotes ferroptosis.48
There are several hypotheses about the mechanism of ferroptotic cell death caused by lipid peroxides. At the structural level, PUFAs act as critical components of the cell membranes, and the extensive lipid peroxidation might transform the chemical and geometric structures of the lipid bilayer. Also, the accumulation of peroxidative lipid leads to membrane pores formation and destroys the barrier function, resulting in membrane thickness decrease and the change of membrane permeabilization.49 In a molecular dynamics study, lipid peroxidation increased the curvature of bio-membranes and acyl tails of peroxidative lipids, which are more hydrophilic, would bend to the water phase, causing membrane instability and micelle formation.49 These changes will eventually affect cell survival through inducing permeabilization. Lipid peroxides may be decomposed into toxic derivatives such as 4-hydroxynonenal (4-HNEs) and malondialdehyde (MDA). These by-products are produced by the decomposition of AA and other PUFAs through enzymatic and nonenzymatic pathways.50 4-HNE and MDA were reported as the major toxic products which could react with DNA bases, proteins, and other nucleophilic molecules leading to serious cytotoxicity.51 Besides, once lipid peroxides are formed, they may further amplify ROS signaling and drive the mitochondrial caspase signaling pathway observed in pyroptosis,52 suggesting a potential link between ferroptosis and pyroptosis.
In this pathway, it is ACSL4 but not other ACSLs that changes the sensitivity of cells to ferroptosis by affecting the lipid composition,21 and ACSL4 reduction enhances resistance to ferroptotic cell death.53 I/R injury, ionizing radiation, and the inhibition of the NF2-YAP pathway can promote ferroptosis by rising ACSL4 expression.54,55,56 On the contrary, integrin α6β4 can mediate the activation of Src and STAT3, resulting in decreased expression of ACSL4 and suppression of ferroptosis.57
LPCAT3 is the most abundant subtype of acyltransferase and participates in transferring PUFA to the sn-2 position of cell membranes.45,58 The primary target of LPCAT3 is acetylated AA, which is inserted into membrane PLs in RSL3-induced ferroptosis.44 Liver X receptor can promote the expression of LPCAT3, facilitate the binding between AA and PLs, and increase the abundance of polyunsaturated phospholipids.59
The role of iron
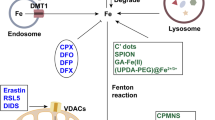
Transferrin is the primary protein responsible for iron transport.60 The iron import starts by binding of iron-bound transferrin (Fe3+) to transferrin receptor 1 (TFR1, recently introduced as a specific ferroptosis marker61) and subsequent endocytosis in endosomes. In acidic endosomes, Fe3+ is reduced to Fe2+ by six-transmembrane epithelial antigens of the prostate 3 (STEAP3), and transported to the cytoplasm through divalent metal transporter 1 (DMT1).62 Cytosolic and mitochondrial labile iron pool (LIP), the intracellular nonprotein-bound redox-active iron, that can be used in cellular processes or stored into ferritin in a process mediated by the chaperones: Poly-(rC)-binding protein 1 (PCBP1) and PCBP2 to ferritin.63 Ferroportin (FPN) is the only known protein that exports intracellular iron in mammals,64 and the iron homeostasis is severely disturbed in FPN-deficient mice.65
The imbalance between iron import, storage, and export, may affect the cell susceptibility to ferroptosis. It has been shown that increased expression of transferrin receptor, induced by pseudolaric acid B, reinforces iron import and then triggers ferroptosis in glioma cells.66 Enhanced ferritin degradation in a process termed ferritinophagy, could increase the level of LIP and enhance ferroptosis.67 Recently, the Prominin2 protein has been shown to enhance ferroptosis resistance by promoting ferritin export.68 Iron and iron derivatives, such as heme or [Fe-S] clusters, are the essential active centers of many enzymes that are involved in ROS generation (Lipoxygenases, cytochrome P450, NADPH oxidases et al.).6
Electrons may escape from oxidation-reduction reaction and be captured by O2 to form superoxide (O2•), peroxides (H2O2 and ROOH), and free radicals (HO• and RO•).69 The oxidation by Fe2+ and H2O2, which is called Fenton reaction, would provide hydroxyl radicals that subtract hydrogen (H) from lipid to form a lipid radical (L•) as the start of the non-enzymatic reaction of lipid peroxidation.70 Lipid radicals combine with O2 to form lipid peroxyl radical (LOO•), which then snatches hydrogen from adjacent PUFA to form LOOH and a new lipid radical, and develops another oxidation reaction71(Fig. 2).
The role of lipoxygenases
Lox, a dioxygenase containing non-heme iron, catalyzes the oxidation of PUFA (with a 1-cis,4-cis-pentadiene structure) via stereospecific peroxidation,72 and the nomenclature of different Lox accounts for the specific site of their oxygenation product.73 There are six Lox isoforms in humans: 15-Lox-1, 15-Lox-2, 12-Lox-1, 12-Lox-2, E3-Lox, and 5-Lox, of which 12/15-Lox are widely distributed in different tissues.74 For tumor protein 53 (p53)-dependent cancer suppression,12-Lox-induced ferroptosis is crucial.75 15-Lox selectively catalyzes PE-AA oxidation and executes ferroptotic cell death.76 The classical substrate of Lox is PUFA, and the sn2-15-hydroperoxy-eicasotetraenoyl-phosphatidylethanolamines (sn2-15-HpETE-PE) catalyzed by 15-Lox can be used as a signal of ferroptosis.77 Whereas, phosphatidylethanolamine-binding protein 1 (PEBP1), a scaffold protein inhibitor of protein kinase cascade, combines with 15-Lox after it is dissociated from RAF1 kinase, shows high selectivity and specificity for ETE-PE and promotes ferroptosis by generation of lipid death signals.12 NO•, a reactive free radical, was found to interact with other free radicals, disturb the lipid peroxidation caused by 15-Lox, and lead to the oxidative truncation of 15-HpETE.78
However, the key role of Lox in ferroptosis is still in debate. The preferred substrate of Lox is free PUFAs, so the first step for Lox is to cleave PUFA acyl chains from PLs through the activity of phospholipase.79 However, this model is inconsistent with ferroptosis, where the lipid peroxidation in ferroptosis occurs on esterified PUFA-PLs rather than free type, implicated by the role of LPCAT3 in ferroptosis44,58 and the facts as mentioned earlier that the MUFA OA strongly suppresses erastin-induced ferroptosis by competing with PUFAs for incorporation into PLs.35 In some studies, 12/15-Lox deletion cannot rescue the embryonic lethality of GPx4 knockout mice, nor can it eliminate the cell death following whole-body GPx4 deletion in adult mice.80,81 In addition, some cell lines sensitive to ferroptosis did not express any major Lox enzyme.82 Therefore, Lox may not be necessary in ferroptosis, or it may play a role in some more complex environments or situations by complementing the autoxidation pathway, which should be further investigated.
Antioxidant mechanisms
The GPx4 pathway
GPx4, a selenocysteine-containing, and glutathione-dependent enzyme, catalyzes the reduction of specific lipid hydroperoxides into lipid alcohols.83 GPx4 belongs to the family of Glutathione peroxidases (GPxs),84 but in contrast to other GPxs, GPx4 lacks a dimerization interface and exists as a monomeric species.85 GPx4 is a multifunctional protein capable of reducing peroxidized lipids either in free form or in complex with lipids such as PLs, with proteins such as lipoproteins or within membranes.86 This characteristic of reducing lipid peroxidation within membrane lipids determines its predominant role in preventing ferroptosis (Fig. 3). This catalytic reaction of GPx4 follows a ping-pong mechanism, whereby the enzyme active site shuttles between an oxidized and reduced state. First, the active site selenolate (Se–H) in GPx4 is oxidized to selenic acid (Se–OH) by a peroxide substrate. Then, the first glutathione (GSH) is used to reduce the selenic acid-generating an intermolecular selenylsulfide bond, which is reduced by a second GSH to form oxidized glutathione (GSSG) and regenerate the enzyme87 (Fig. 3).

Schematic description of the two defense mechanisms identified in ferroptosis, the GSH-dependent GPx4 pathway, and the NADPH-dependent FSP1 pathway. Glutamine (Gln); glutamate (Glu); cysteine (Cys); glycine (Gly); glutathione-disulfide reductase (GSR); stimulating proteins 1 and 3 (SP1/3); nuclear factor Y (NF-Y); cAMP-response element modulator-tau (CREM-tau); early growth response protein 1 (EGR1); nuclear factor κB (NF-κB); sterol regulatory-binding element 1 (SREBP1).
GPx4 is also involved in the development and maintenance of a variety of physiological functions,88 and its genetic ablation or expression of an inactive form causes early embryonic lethality.89 However, disruption of mitochondrial GPx4 in mice is not lethal and causes infertility in males through abnormal sperm development.90 Neurons-specific deletion of GPx4 is neonatally lethal,91 while conditional GPx4 deletion in adult mice results in mitochondrial damage, hippocampal neurodegeneration, and increases astrogliosis.92 The ablation of GPx4 can trigger lipid-oxidation-induced acute renal failure and associated death.80
GPx4 regulation—GSH axis: GSH is essential for the GPx4-catalyzed reaction as it serves as an electron donor for reducing toxic phospholipid hydroperoxides (such as PE-AA-OOH/PE-AdA-OOH) to nontoxic phospholipid alcohols (such as PE-AA-OH, PE-AdA-OH), and the GSSG is generated as a by-product.87 GSH can be regenerated by reducing GSSG using glutathione reductase (GR). During this progress, reduced NADPH acts as the electron donor.5 NADPH abundance may be used as a predictor for the outcome of a ferroptotic event.93 Meanwhile, the regulation of cytosolic NADPH levels via Metazoan SpoT Homologue 1 (MESH1) can control ferroptosis.94 Therefore, GSH is considered as a critical factor for maintaining GPx4 activity.
GSH is synthesized from glutamate, cysteine, and glycine in two steps under the catalysis of the cytosolic enzymes glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS) to participate in the regulation of ferroptosis5 (Fig. 3). Cysteine is the most limiting amino acid for GSH synthesis and inhibition of its import through the system \({\mathrm{X}}_c^-\) is sufficient to trigger ferroptosis in vitro.6 System \({\mathrm{X}}_c^-\) is a cystine/glutamate antiporter that facilitates the exchange of cystine and glutamate across the plasma membrane.1,15 Upon transport inside the cell, cystine is reduced to cysteine by GSH or thioredoxin reductase 1 (TXNRD1).95 Cysteine is a semi essential amino acid as it can be produced from methionine through the transsulfuration pathway converting methionine to homocysteine, cystathionine, and in a final step to cysteine.96 It has been shown that hepatocellular carcinoma cells are resistant to sorafenib, a novel multi-targeted oral drug for the treatment of tumors,97 which is acquired through transcriptional regulation of genes involved in lipid ROS and iron metabolism.98 Recently, inhibition of DJ-1 (also known as PARK7-parkinsonism-associated deglycase) has been shown to enhance the sensitivity of tumor cells to ferroptosis induced by sorafenib and evaluated the antitumor activity of erastin in a xenograft nude mouse model via inhibition of the transsulfuration pathway and limiting the supply of cysteine.99 P53, a tumor suppressor gene, is mutated in at least 80% of the most difficult-to-treat cancers, such as high-grade serous ovarian cancers, or triple-negative breast cancers.100 Cell-cycle arrest, apoptosis, and senescence are widely accepted as the major mechanisms by which p53 inhibits tumor formation. P53(3KR), an acetylation-defective mutant that fails to induce the above process, retains the ability to suppression of early-onset spontaneous tumorigenesis.101 The function of p53(3KR) tumorigenesis suppression is realized by inhibiting SLC7A11 expression and inducing ferroptosis in xenograft models.102 Similarly, BRCA1 associated protein 1 (BAP1) and OTU deubiquitinase, ubiquitin aldehyde-binding 1 (OTUB1), both involved in tumor regulation, can control ferroptosis via regulating the expression of system \({\mathrm{X}}_c^ -\).103,104 These facts indicate that the modulation of ferroptosis may be involved in the resistance of tumor cells to cell death and that system \({\mathrm{X}}_c^ -\) plays a central role in this process.
GPx4 Regulation—Selenium axis: Selenium is indispensable for mammalian life, and the deficiency of the selenocysteine- (Sec-) -specific tRNA gene Trsp (nuclear-encoded tRNA selenocysteine 2) is embryonic lethal.105 Selenium has been involved in many biological processes such as cancer prevention and promotion, protecting against cardiovascular diseases, and treating certain muscle disorders.106 The GPx4 mRNA contains a selenocysteine insertion sequence element in the 3’ untranslated region that encodes an active site of selenocysteine via a UGA codon.107 It has been suggested that selenium can regulate GPx4 expression through increasing ribosome density in downstream of UGA-Sec codons and sec incorporation efficiency in part by the degree of Sec-tRNA[Ser]SecUm34 methylation.108 The Sec-tRNA must first be activated by the addition of an isopentenyl lipid group, a product of the mevalonate (MVA) pathway.109,110 This may explain how disruption of the MVA pathway by statins leads to reduced GPx4 expression and increased ferroptosis in certain cells.111
The catalytic function of selenocysteine is due to its rapid deprotonation,87 while thiol groups remain protonated at neutral pH.88 It has been shown that under selenium deficiency conditions, cysteine can replace selenocysteine in various selenoproteins.112,113 However, replacing selenocysteine by cysteine in recombinant GPx4 leads to a 1000-fold reduction in catalytic activity.114 These facts suggest that selenocysteine as the catalytic moiety is necessary to guarantee a rapid reduction of hydroperoxide by GPx4 and prevent ferroptosis.86
The FSP1 pathway
FSP1 was primal disclosed as a p53‐responsive gene (PRG) in p53-mediated apoptosis, designated as PRG3. Because of the similarity in amino acid sequences between it and the human apoptosis-inducing factor (AIF), it was also named apoptosis-inducing factor (AIF)-like mitochondrion-associated inducer of death (AMID)115,116 or apoptosis-inducing factor mitochondria-associated 2 (AIFM2).117 Unlike AIF, FSP1 is predominantly cytosolic and may have an affinity for the cytosolic surface of the mitochondrial outer membrane. This is consistent with the apparent lack of an extended N-terminal mitochondrial targeting sequence analogous to that found in AIF.116 FSP1 was then found to be a flavoprotein oxidoreductase, and its proapoptotic function may be through inhibiting the redox activity. FSP1 can combine with DNA, nicotinamide coenzyme, and the modified flavin 6-hydroxy-FAD. When it binds to dsDNA (e.g., bacterial and (retro)viral), its function as an oxidoreductase will be antagonistic, leading to the accumulation of ROS.117 Doxorubicin treatments significantly enhance cardiac levels of 4-HNE and FSP1, and the 4-HNE adduction of FSP1 facilitates its translocation from mitochondria, which can change the activity of FSP1 to a proapoptotic protein.118 However, it has been shown that the expression of FSP1 induces cellular apoptosis at much lower levels than AIF in several different cell lines.119 A study has previously described AIF and FSP1 as type 2 NADH ubiquinone oxidoreductase (NDH-2) enzymes in mammalian mitochondria, reporting the possible interaction between FSP1 and ubiquinone.120 However, FSP1 lacks a mitochondrial localization sequence in AIF, so the role of FSP1 in cell death remains unclear.
Several studies have suggested that the sensitivity to GPx4 inhibitors varies greatly across different cell lines in ferroptosis.121 To explore the possible additional regulatory pathways, CRISPR/Cas9-based screens have been performed, and revealed that FSP1 is a previously unrecognized anti-ferroptotic gene.2,23 FSP1 catalyzes the regeneration of non-mitochondrial CoQ10 using NAD(P)H to block ferroptosis by inhibiting the propagation of lipid peroxides.2,23 Interestingly, membrane targeting of FSP1 via the myristoylation motif of N terminus is essential for its anti-ferroptotic activity. And selectively targetting FSP1(G2A), a mutant that lacks the myristoylation site, to the endoplasmic reticulum did not impact ferroptosis sensitivity.2 Indeed, it has been reported that the supplementation of cells with CoQ10 effectively suppresses ferroptosis as early as 2016,122 without a detailed mechanism.
CoQ10, as the main effector of the FSP1 pathway, is widely distributed among membranes of mammalian cells.123 It is composed of a benzoquinone ring and a polyisoprenoid tail containing between 6 and 10 subunits that are species-specific.124 Non-mitochondrial CoQ10 plays an essential role as a reversible redox carrier in the plasma membrane and Golgi apparatus membrane electron transport,125 and represents an essential endogenous lipid-soluble antioxidant by directly scavenging lipid peroxyl radicals.126 CoQ10 can reduce lipid hydroperoxides more effectively than α-toc.127 Moreover, oral administration of CoQ10 is reported in the treatment of various human diseases such as cardiomyopathy,128 Parkinson’s disease,129 and diabetes.130 In contrast, low CoQ10 levels due to mutations in CoQ10 biosynthetic enzymes or associated enzymes are associated with several diseases.131,132,133
CoQ10 biosynthesis pathway is tightly regulated both at the transcriptional and translational levels. CoQ10 can be synthesized using acetyl-CoA via MVA pathway134 (Fig. 3). MVA pathway enzymes condense three acetyl-CoA molecules in a two-step reaction to produce 3-hydroxy-3-methylglutaryl CoA (HMG-CoA). Then HMG-CoA reductase (HMGCR) reduces HMG-CoA to MVA via an irreversible reaction.135 MVA is then converted into isopentenyl-diphosphate (IPP) through a series of enzymatic steps, which serves as a monomeric unit for the consequent synthesis of all downstream metabolites.136 IPP is catalyzed into CoQ10 through various enzymatic steps,137 including two major intermediates, farnesyl diphosphate (FPP) and geranylgeranyl-diphosphate (GGPP).138
Other antioxidant pathways
In addition to NAD(P)H-FSP1-CoQ10 and NAD(P)H-GSH-GPx4 as two parallel pathways that suppress ferroptosis, other natural antioxidants can also play a part in preventing ferroptosis, such as vitamin E,139 thioredoxin,140 and mitoquinone.141 Recent research discovered that inducible nitric oxide synthase (iNOS)/NO• abundance modulates susceptibility to ferroptosis in macrophages/microglia.78 BH4 is a potent radical-trapping antioxidant that protects cells from ferroptosis upon GPX4 inhibition by reducing lipid peroxidation and is regenerated by dihydrofolate reductase (DHFR).142 Besides, nuclear factor erythroid 2-related factor 2 (Nrf2) may play a role in modulating the cellular ferroptosis response. Nrf2 is responsible for regulating several antioxidant genes.143 Importantly, almost all genes implicated in ferroptosis are transcriptionally regulated by Nrf2, including genes of glutathione regulation, NADPH regeneration, and iron regulation.144,145,146,147 Moreover, Nrf2 indirectly modulates the lipids, whose abundance contributes to ferroptosis sensitivity.6,148 Consequently, Nrf2 activation results in resistance to ferroptosis in cancer cells,98,149 and other cell types.150
The link between ferroptosis and other cell death pathways
Oxytosis
Oxytosis was introduced as a form of non-apoptotic regulated cell death in 2001, which was characterized by oxidative stress and GSH depletion.151 In the early studies, oxidative glutamate toxicity served as a specific example of the more general oxytosis pathway. The link between glutamate toxicity and GSH depletion was established by the glutamate-mediated inhibition of cystine uptake by the system \({\mathrm{X}}_c^ -\).16 GSH depletion during oxytosis can be expected to impair GPx4 activity as GSH is required for GPx4 activity.152 Most of the oxytosis studies have been carried out in HT22 cells that were explicitly sensitive to glutamate toxicity.91 The defective GPx4 expression can enhance cytotoxicity by glutamate-induced oxytosis in the retina.153 Interestingly, the lipophilic antioxidant α-toc can efficiently inhibit oxytosis.154 Exogenous AA potentiates oxytotic cell death, while multiple Lox inhibitors can protect from GSH depletion.91
The characteristics of oxytosis are consistent with ferroptosis. Oxytosis in HT22 cells can be inhibited by iron chelators and exacerbated by different sources of iron.155 Calcium entry into cells is a necessary step in oxytosis.151 Glutamate induces a significant increase in intracellular Ca2+ about 30–50 fold,156 and inhibitors of calcium entry could effectively inhibit the occurrence of oxytosis.151 However, the role of calcium in ferroptosis has not been established. Both oxytosis and ferroptosis induce the expression of eIF2α.151,157 Bid knockout using CRISPR/Cas9 approaches can protect neurons against both ferroptosis and oxytosis.47 The pharmacological inhibition of double-stranded RNA-dependent protein kinase-mediated neuroprotective effects against both ferroptosis and oxytosis.158
Autophagic cell death
The process that removes intracellular components such as unused proteins and damaged organelles through lysosomes, was named “autophagy” in 1963.159 Autophagy plays a multifaceted role in regulating both the quality and quantity of proteins and organelles,160 therefore it determines cell fate via various pathways.161,162,163 Recent studies have placed the autophagy process in ferroptosis since it regulates the abundance of ferritin, the major iron storage protein. NCOA4 is a selective cargo receptor for the autophagic turnover of ferritin by lysosomes.67 Genetic and pharmacological inhibition of NCOA4 can protect cells from ferroptosis via reducing cellular labile iron.9 Inhibition of lysosomal function, the endpoint of autophagy flux, can significantly block erastin-induced ferroptosis in both MEFs and HT1080 cells.9 Dihydroartemisinin also promotes ferroptosis by inducing ferritinophagy and increasing the labile iron pool in acute myeloid leukemia.164
Similarly, lipophagy, which is a form of selective autophagy that leads to the autophagic degradation of intracellular lipid droplets (LDs), also can regulate ferroptosis.165 The knockdown of the LD cargo receptor RAB7A can inhibit ferroptosis.166 In contrast, the overexpression of TPD52 (tumor protein D52) limits RSL3-induced ferroptosis by increasing lipid storage.167 These studies have strengthened the link between autophagy and ferroptosis.
In Tables 1 and 2, we summarized some of the currently used small molecules and drugs that interfere with ferroptosis, the postulated mechanism and the corresponding cellular/animal experimental model.
Links between ferroptosis and disease
Cancer
Cancer cells accumulate high levels of iron as compared to normal cells.168 Research has advocated the abnormality of iron homeostasis in several cancer types, including breast cancer, ovarian cancer, renal cancer, and lung cancer.62 Non-thermal plasma (NTP) breaks ferritin and induces reduction from Fe3+ to Fe2+, accompanied by lipid peroxidation and mitochondrial superoxide generation, which selectively eliminates oral squamous cell carcinoma cells.169 FINO2, an endoperoxide-containing 1,2-dioxolane, can oxidize Fe2+ leading to lipid peroxidation and kill BJeLR cancer cells via ferroptosis.170 Silencing the expression of prominin2 decreases the cellular iron export of RSL3-treated mammary epithelial (MCF10A cells) and breast carcinoma cells (Hs578t cells) via reducing the formation of ferritin-containing multivesicular bodies (MVBs).68 Inhibition of NFS1, a [Fe–S] cluster biosynthetic enzyme, stimulates the expression of transferrin receptor but restrains ferritin, causing iron-starvation response and leading to ferroptosis in lung cancer cells.8 Salinomycin has been reported to kill cancer stem cells in a mechanism involving iron sequestration within lysosomes, leading to ferroptosis and lysosomal membrane permeabilization.171 In high-risk neuroblastoma, withaferin A (WA) blocking the function of Kelch-like ECH-associated protein 1 (KEAP1) can reduce the inhibition of Nrf2 that indirectly increases LIP through heme oxygenase-1 (HO-1) and kills tumors via the KEAP1-Nrf2 pathway, a noncanonical pathway of ferroptosis.172
Cancer cells need a high metabolic rate to maintain their rapid proliferation, accompanied by an increase in ROS production.173 Therefore, high ROS levels are an inherent feature of tumors, and cancer cells have to boost their antioxidant defense capacity to overcome this enhanced oxidative stress.174 Targeting the antioxidant defense mechanism of cancer cells may be an effective potential treatment strategy by predisposing them to oxidative stress-induced cell death, such as apoptosis and ferroptosis. The cytotoxicity induced by chemotherapeutic drugs such as 5-FU, oxaliplatin, and paclitaxel is linked with elevated ROS,175,176,177 and depletion of intracellular GSH using RNAi against the anti-oxidant transcription factor Nrf2 leads to increased ROS and increased sensitivity to chemotherapy in preclinical studies.178 ROS initiate the oxidation of PUFAs and play an important role in non-enzymatic lipid peroxidation or auto-oxidation of lipids.32 In several arsenic trioxide-resistant human leukemic cell lines, the DHA can enhance the cytotoxic effect of As2O3 through an increase of intracellular lipid peroxidation products.179 WA inactivates GPx4 and induces ferroptosis via accumulating lipid peroxides to toxic levels, which might also explain its ability to kill high-risk neuroblastoma cells and inhibit tumor growth of neuroblastoma xenografts.172 Interestingly, peroxisomes may contribute to ferroptosis through synthesizing PUFA-ePLs, and 786-O tumor xenografts can evade GPx4 knockout induced ferroptosis in mice by downregulating PUFA-ePLs.24 These facts highlight the potential value of lipid peroxidation and ferroptosis in tumor suppression strategies.
Ferroptosis is suggested to be a good approach to circumvent the therapy resistance of cancer cells. Once ‘epithelial-to-mesenchymal’ transition (EMT) occurs in tumor cells, which will acquire drug resistance and be intractable for treatment. By gene-signature, proteomic, and lineage-based correlation analyses, therapy-resistant high mesenchymal state cancer cells are found to be dependent on GPx4 for their survival and thus vulnerable to ferroptosis.111 ZEB1, which promotes tumor invasion and therapy-resistant by inducing EMT in carcinoma cells, was found to regulate the sensitivity of mesenchymal state cancer cells to GPx4 inhibition as a lipogenic factor.111 In a recent study, the loss of tumor suppressor Merlin, a frequent tumourigenic event in mesothelioma, dictates GPx4 dependency in murine models of mesothelioma through the upregulation of multiple ferroptosis modulators, including ACSL4 and transferrin receptor.54 Coincidentally, the residual persister cancer cells, which contribute to tumor relapse, acquire a dependency on GPx4 to survive.180 In these tumor cells, the antioxidant genes, such as NADPH, GSH, and Nrf2, were significantly downregulated, so the inhibition of GPx4 results in selective persister cell ferroptotic death in vitro and prevents tumor relapse in vivo.180 Melanoma usually metastases to lymph nodes before forming distant metastases due to the low cell survival rate in the blood, which is one of the most important predictors of distant metastasis and death in patients with cutaneous melanomas.181,182 It is recently shown that the lymphatic environment protects metastasizing melanoma cells from ferroptosis to increase their survival rate during subsequent metastasis through blood.183 By pretreating with ferroptosis inhibitors, the melanoma cells can form more metastases than untreated cells after intravenous injection in immunocompetent mice.183 In addition, pharmacological and genetic regulation of GSH can also effectively kill tumor cells. Imidazole ketone erastin (IKE) can alleviate tumor growth via interfering with cystine uptake in a diffuse large B cell lymphoma (DLBCL) xenograft model.184 Combinational treatment with ferroptosis inducing drugs with vemurafenib, a BRAF kinase inhibitor, results in a substantial decrease in long-term persisting melanoma cell.185
Furthermore, ferroptosis may participate in tumor immunity. Dendritic cells (DCs) appear essential for antitumor immunity via conditioning the tumor microenvironment and regulating priming of antitumor T cells.186 It has been reported that ALox15-derived lipid peroxide regulates DCs maturation and modulates adaptive immune responses.150 Oxidized phosphatidylcholine inhibited DC maturation via the activation of the transcription factor Nrf2 and dampened the differentiation of T helper 17 (TH17) cells.150 Similarly, oxidized phosphatidylethanolamines are also involved in the immune response, which can clear apoptotic cells by mouse inflammatory macrophages in vitro and in vivo.187 IFNγ plays an essential role in antitumor immune response, and Mauguso et al. confirmed that resistance to immunotherapy is attributed to defects in IFNγ signaling.188 Interestingly, IFN-γ can suppress the expression of SLC7A11A and SLC3A2, two essential proteins for the synthesis of GSH, leading to the transport deficit of cystine, which lipid peroxidation and ferroptosis in cancer.189
Adaptive immunity is an indispensable and powerful ‘weapon’ in tumor immunity, and ferroptosis may also be involved in adaptive immunity.190 In T cell-specific GPx4-deficient mice, healthy thymic T cell development and T cell responses upon secondary infection were observed. However, both antigen-specific CD8+ and CD4+ T cells failed to expand and to protect from acute infections, and CD8+ T cells had an intrinsic defect in maintaining homeostatic balance in the periphery,190 which can be rescued with a high dosage of vitamin E.190 Ferroptosis also plays a significant role in CD8+ T cell-induced cancer cell death in cancer immunotherapy.189 Mechanistically, interferon-gamma (IFNγ) from CD8+ T cells triggers lipid peroxidation and ferroptosis by the inhibition of system \({\mathrm{X}}_c^ -\).189 Cyst(e)inase, an engineered enzyme that degrades cystine, efficiently induces oxidative stress and ferroptosis. A combination of cyst(e)inase and PD-L1 can strongly inhibit the growth of ID8 cell-derived tumors,191 accompanied by increased lipid peroxidation in tumor cells and increased percentages of IFNγ+ and TNF+ CD8+ and CD4+ T cells in the tumor microenvironment.189 Notably, In cancer patients, the expression of system \({\mathrm{X}}_c^ -\) was negatively associated with CD8+ T cell signature, IFNγ expression, and patient outcome.189 In humoral immunity, GPx4 is essential to prevent ferroptosis during development, maintenance, and responses of innate-like B cells.192 However, in germinal center reactions, and antibody responses of follicular B2 cells, it is not GPx4-dependent.192 Therefore, the synergy of ferroptosis and tumor immunotherapy may be a potential cancer treatment strategy.
Ferroptosis is involved in many anti-tumor therapies, including radiotherapy, one of the standard methods used in clinical cancer treatment. The antitumor effects of irradiation are attributed to the microparticles released by irradiated cells, which were shown to induce immunogenic death mainly through ferroptosis.193 The expression of the Ataxia-Telangiectasia mutated gene in irradiated tumor cells would inhibit the expression of SLC7A11, blocking cystine uptake and resulting in lipid peroxidation accumulation.194 Ionizing radiation also upregulates the expression of ACSL4 in human cancer cells.55 Erastin treatment in HeLa and NCI-H1975 adenocarcinoma cell lines aggravates radiation-induced cell death.195 Clinical drugs, such as sorafenib,196 sulfasalazine,197 artemisinin,198 and ibuprofen,199 may induce ferroptosis in cancer cells, and ferroptosis inducers combined with temozolomide, cisplatin, haloperidol, and doxorubicin can enhance the chemotherapy effect of these drugs in the treatment of tumors.200,201,202 There are to date numerous studies on inducing ferroptosis in cancer cells by increasing lipid peroxidation; however, they remain in preclinical stages.
Over the past decade, nanotechnology has made significant contributions to oncology, and it has great potential in cancer treatment. Several studies have applied nanotechnology to induce ferroptosis for the development of cancer therapies. The first ferroptosis-inducing nanoparticles were described in 2016, where the authors have shown that intravenous injection of ultrasmall poly(ethylene glycol) (PEG)-coated silica nanoparticles can reduce the growth of the tumor by inducing ferroptosis in mice.203 In 2018, FeGd-HN@Pt@LF/RGD2 nanoparticles that deliver Fe2+ and Fe3+ were used for the treatment of orthotopic brain tumors.204 In 2019, up-conversion nanoparticles (UCNP) that can release Fe2+ and induce ferroptosis had been invented and tested in 4T1 xenograft mice.205 Subsequent studies have used several different strategies to induce ferroptosis in cancer cells, including particles called salinomycin-loaded gold nanoparticles(AuNPs), which leads to iron accumulation and intracellular GSH exhaustion,206 a lipid peroxidation generator consisting of a novel GSH and iron redox couple207 and oxygen-boosted phototherapy.208
Neurodegeneration
The growing incidence of neurodegenerative diseases has brought a considerable burden to society and significant distress to both patients and caregivers. However, there are still limited treatment strategies for these diseases.209 Therefore, it is urgent to impel further exploration of the relationship between pathological characteristics, disease mechanism, and neuronal death. Here, we mainly focus on the relationship of ferroptosis with Alzheimer’s disease (AD) and Parkinson’s disease (PD).
Iron dyshomeostasis and lipid peroxidation, hallmarks of ferroptosis, have long been noted in AD and PD pathology. Aging is the major risk factor for neurodegenerative diseases and is accompanied by brain iron accumulation.210 Similarly, iron accumulation in affected brain regions of diseases has been reported in various neurodegenerative diseases.211,212,213 Thus, iron has been suggested as an essential factor contributing to the neurodegenerative processes. Iron can lead to the dissociation of IRPs from the IRE by binding to IRPs, altered translation of target transcripts.214 Interesting, IREs are found in the 5’-UTR of amyloid precursor protein (APP) and α-synuclein (α-Syn) transcripts, and iron accumulation can upregulate the levels of α-Syn, APP and amyloid β-peptide (Aβ).215 And deferoxamine (DFO), as a widely used iron chelator, can inhibit amyloidogenic APP processing and Aβ aggregation in animal studies.216,217 In separate randomized controlled trials, deferiprone (DFP), an orally bioavailable brain permeable iron chelator, was shown to alleviate nerve function scores and ameliorate iron-related neurological symptoms.218,219,220 Similarly, DFP has reported beneficial effects for PD patients in phase II studies.221,222 The loss of ceruloplasmin (Cp), a protein responsible for iron export, is also associated with iron-dependent parkinsonism in mice,223 and in the cerebrospinal fluid (CSF) of PD patients.224 As discussed earlier, iron is also involved in ROS production and lipid peroxidation, and the iron accumulation is always accompanied by oxidative stress.
Due to its high metabolic activity, brain tissues are particularly vulnerable to oxidative stress.225 Increased oxidative stress is a feature of several neurodegenerative diseases, including Alzheimer’s disease226 and Parkinson’s disease.227 Piceid, as a natural antioxidant, can protect the vulnerable SNc neurodegeneration via correcting several major anti-oxidant pathways/parameters, including GSH, MDA and the SOD, selectively in three rodent models of PD.228 Similarly, the use of antioxidants, such as MitoQ and SOD2, can alleviate the pathological characteristics in AD animal models.229,230 Furthermore, neuronal membranes are rich in PUFAs, that are prone to oxidation,231 and consequently, lipid peroxidation is likely to contribute to oxidative stress associated with neurodegeneration. Isotope-reinforced (deuterated) PUFA (D-PUFA) is effective in reducing lipid peroxidation and Aβ level in the APP/PS1 transgenic mouse model of Alzheimer’s disease.232 Alpha-Lipoic acid (ALA), a fat-soluble and water-soluble antioxidant and also a naturally occurring enzyme cofactor with reducing lipid peroxidation properties, can significantly alleviate AD pathology in P301S tau transgenic mice with alleviated properties of ferroptosis.233 Similarly, 2000 IU/d of α-toc compared with placebo resulted in a slower functional decline among patients with mild to moderate AD.234
Copper(II)-diacetyl-bis(4-methylthiosemicarbazonato) (Cu-ATSM) is a PET tracer initially developed for hypoxia imaging but has recently shown neuronal protection in multiple PD models and prevent lipid peroxidation without altering the oxidation state of iron.235 Cu-ATSM obtains the anti-ferroptotic activity like liproxstatin-1 by preventing the propagation of lipid radicals rather than preventing iron oxidation. Combined with its ability to enter the brain, Cu-ATSM may be an attractive investigational product for clinical trials of ferroptosis and neurodegeneration.236 The maintenance of glutathione GSH is a key antioxidant element in brain redox homeostasis. In the AD model, N-acetyl cysteine (NAC) can protect neurons function and improving learning and memory deficits via increasing GSH levels along with the reduced MDA,237 which may also be related to the anti-amyloid efficacy of NAC.238 Nigral GSH loss and oxidative stress are predispositions to PD,239 and in a randomized, double-blind placebo-controlled clinical trial, the oral adjunction of omega-3 fatty acids and vitamin E for three months improves GSH level and Unified Parkinson’s Disease Rating Stage (UPDRS).240 In a recent epigenetic study on blood-based methylome-wide association study of PD, it has been identified that hypermethylation in the promoter region of the SLC7A11 gene can downregulate system \({\mathrm{X}}_c^-\) along with the reduced GSH synthesis and increased sensitivity to ferroptosis.241 Similarly, there is a linear correlation between GSH loss and neurodegeneration in neurons cultured from aged 3xTg-AD mice, and 3xTg-AD neurons are more dependent on GSH availability than the non-Tg neurons.242
These data implicate a potential link between ferroptosis and neurodegenerative diseases, while several subsequent studies have provided more direct evidence. In 2018, it was shown that tau overexpression and hyperphosphorylation can induce neuronal loss via ferroptosis, and ALA supplementation effectively inhibited cognitive decline through reducing tau-induced iron overload, lipid peroxidation, and upregulating GPx4 expression in P301S tau transgenic mice.243 Moreover, a targeted mutation of GPx4 (selenocysteine to cysteine substitution) or GPx4 conditional deletion in neurons causes neuronal toxicity and rapid neuronal death in mice, which are accompanied by multiple ferroptotic characteristics.22,92 GPx4 is also critical for maturation and survival of photoreceptor cells; photoreceptor-specific rapidly underwent severe degeneration and completely disappeared by conditional knock-out GPx4.244 Targeted conditional knockout of GPx4 in forebrain neurons of adult mice causes AD-like cognitive impairments and neurodegeneration that can be attenuated by ferroptosis inhibitors.245 Also, the expression of GPx4 might play a neuroprotective role in PD pathology.246 It is shown that GPx4 colocalizes with AS-positive nigral Lewy bodies and dystrophic TH-positive fibers in the putamen, and it is increased relative to cell density, probably because of an increase in survival of cells expressing GPx4.246 There is a correlation between selenium level, a key factor for GPx4 activity, and susceptibility to ferroptosis.247 In AD patients, the selenium level can be related to the pathological progress of the disease.248,249 The treatment of selenium attenuates a beta production by reduced 4-HNE-induced transcription of beta-secretase (BACE1) and protects against Aβ-mediated toxicity in primary cultured neurons.250 In a paraquat-induced rat PD model, selenium feeding also can reduce bradykinesia and DNA damage.251 Depletion of DJ-1, a known cause of early-onset autosomal recessive Parkinson’s disease,252 can also increase the sensitivity of neurons to ferroptosis by markedly reduce GSH levels through inhibition of the transsulfuration pathway.99
Besides, ferroptosis inhibitors are protective in cellular models of Huntington’s disease,253 and an MPTP mouse model of Parkinson’s disease.254 Recently promising results were reported for the use of an anti-ferroptotic compound in phase I clinical trials in amyotrophic lateral sclerosis (ALS) patients and PD patients.236 Therefore, ferroptosis may play an essential role in the pathogenesis of various neurodegenerative diseases, and anti-ferroptotic strategy should be further investigated.
Ischemia/reperfusion
Ischemia/reperfusion (I/R) is a pathological condition contributing to morbidity and mortality in a wide range of conditions. When the tissue experiences obtunded blood flow by blockage or rupture of an artery, the ischemia occurs. The interruption of blood supply means the exhaustion of energy and cell death. So it is necessary to restore blood supply as soon as possible. However, more grave functional and structural changes become evident in the process of blood flow recovery.255 This pathological progress is ischemia/reperfusion injury(IRI), and it can trigger myocardial infarction, acute kidney injury, circulatory arrest, and even sleep apnea. However, IRI is also a significant challenge in organ transplantation,256 and the adverse effects of IRI in clinical situations are difficult to limit.
Iron is a potential therapeutic target for IRI (Fig. 4). Clinical studies have shown that children with following severe ischemic-anoxic insult have significantly increased iron levels in multiple areas of the brain.257 Also, increased iron levels during I/R were proposed to mediate tissue damage in IRI.258,259,260,261,262 Supporting evidence was provided by reduced IRI damage following iron chelation in several animal models of IRI.263,264,265 It has also been proved that adjunctive DFO treatment can ameliorate oxidative stress injury in ST-elevation-myocardial infarction.266

The relationships between iron, ROS, and ferroptosis in ischemia/reperfusion has been illustrated, where ferroptosis may be a result of accumulated ROS, induced by impaired iron export. Iron chelators, anti-oxidants, and inhibitors of ferroptosis may prevent the toxic reaction.
In addition, it is well known that reperfusion of ischemic tissue can lead to a “burst” of ROS, which leads to further deterioration and tissue damage267 (Fig. 4). Accordingly, antioxidants were shown to protect from IRI in various conditions.268,269,270,271 It is also known that the increase in oxidation is also accompanied by lipid peroxidation272 (Fig. 4). ALA, as described before, can raise glutathione intracellularly, and it has a protective effect on ischemia-reperfusion injury in a variety of clinical conditions, such as simultaneous kidney-pancreas transplantation,273 human liver transplantation,274 and liver resection.275 The levels of lipid peroxide is markedly increased, while the levels of GSH and GPx4 are significantly reduced in rodent models of ischemic stroke, and carvacrol protected hippocampal neurons by increasing GPx4 expression.276 Knockout of GPx4 induces kidney failure in mice, which can be inhibited by lipid peroxidation inhibitors.80 Similarly, cardiac IRI can be alleviated by mitochondria-specific overexpression of GPx4.277
Intriguingly, the regulation of iron and lipid peroxidation can impact ferroptosis sensitivity, and accumulated evidence shows that the regulation of these two vital factors can affect IRI by controlling the ferroptosis (Fig. 4). I/R in the brain can acutely suppress tau expression, an Alzheimer’s disease protein that can facilitate iron export,278,279 causing iron accumulation accompanied by ferroptosis in the infarct zone, which aggravated neuronal damage.262 Iron chelation and ferrostatin-1 have similar protective effects on heart failure induced by both acute and chronic I/R.280 Pharmacological selenium significantly reduces infarct volume by driving an adaptive transcriptional program to block ferroptosis.247 The inhibition of glutaminolysis, involved in the NADPH-GSH-GPx4 pathway, can attenuate cardiac IRI by blocking ferroptosis.19
In addition, ACSL4 was upregulated in human ischemic intestinal tissues compared with that in healthy tissues, and liproxstatin-1 and siRNA to inhibit ischemia/hypoxia-induced ACSL4 ameliorated I/R-induced intestinal injury.56 12/15-LOX knockout mice can protect neurons against cerebral ischemic injury.281 The heart after acute and chronic I/R is accompanied by severe cardiomyopathy. In DOX-treated murine hearts, HO-1 was significantly upregulated, promoting systemic accumulation of nonheme iron via heme degradation and accompanied by lipid peroxidation and ferroptosis.280 Ferrostatin-1 can significantly reduce DOX cardiomyopathy.280 And ferrostatin-1 also has been shown to ameliorate heart failure induced by I/R.280
Similarly, the inhibition of ferroptosis was effective in attenuating I/R-associated renal injury.80,282 Augmenter of liver regeneration (ALR) can also affect kidney injury by regulating ferroptosis in renal I/R.283 The inhibition of ALR using short hairpin RNA lentiviral (shRNA) aggravates pathology progression and leads to increased ROS, mitochondrial damage, and ferroptosis.283 Moreover, in intestinal IRI and testicular IRI models, the inhibition of ferroptosis produces a significant protective effect.56,284 These data reinforce the relevance of ferroptosis in I/R and present new avenues for using ferroptosis inhibition as a therapeutic strategy for I/R related damage.
Other pathological conditions
Doxorubicin (DOX) is a commonly used chemotherapeutic drug for the treatment of breast cancer, leukemia, and other malignancies, but its use is limited by the severe toxic side effects, which may cause cardiomyopathy and heart failure.285 Ferroptosis inhibitors can protect against DOX-induced cardiomyopathy.280 Besides, ferroptosis is involved in other pathological conditions, such as hemochromatosis, cystic fibrosis, chronic obstructive pulmonary disease, and Pelizaeus–Merzbacher disease.76,286,287,288
Concluding remarks
Ferroptosis research still faces its challenges as several mechanistic aspects of ferroptotic cell death are not well understood. Notably, the role of iron and lipoxygenases in triggering or propagating lipid peroxidation and the contribution of organelles such as mitochondria are under extensive investigation.
Ferroptosis lead to an imbalance of redox state and to a sequence of events different from other types of cell death, which includes iron liberation from ferritin and lipid peroxidation. However, why does this imbalance not simply trigger apoptosis? It was previously found that ferroptosis and necroptosis can be alternatives, in that necroptosis drives basal resistance to ferroptosis through depleting PUFAs and ferroptosis also drives basal resistance to necroptosis by reducing membrane permeabilization.289 It is yet to define whether ferroptosis is a specific mechanism to inhibit other death pathways.
Also, whether ferroptosis occurs as an automatic response to diverse stimuli that destabilize the metabolic balance, or it is the stimuli that directly disrupt the balance and cause ferroptosis. Is ferroptosis achieved “actively” or “passively”? Increasing evidence has shown the crosstalk between ferroptosis and other cell death. The further illumination of this interrelation also is a requisite for exploring the associated mechanisms and developing treatments.
Therefore, it is vital to selectively label cells undergoing ferroptosis, which will facilitate the exploration of the role of ferroptosis in pathological and physiological contexts. Most probes are limited on biochemical assays; thus, specific indicators to identify cells that are explicitly undergoing ferroptosis in tissue sections would greatly facilitate our understanding of ferroptosis and its potential therapeutic use in diseases.
References
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
Yagoda, N. et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868 (2007).
Ke, B. et al. Targeting programmed cell death using small-molecule compounds to improve potential cancer therapy. Med. Res. Rev. 36, 983–1035 (2016).
Liang, C., Zhang, X., Yang, M. & Dong, X. Recent progress in ferroptosis inducers for cancer therapy. Adv. Mater. 31, e1904197 (2019).
Doll, S. & Conrad, M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life 69, 423–434 (2017).
Yang, W. S. & Stockwell, B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245 (2008).
Alvarez, S. W. et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643 (2017).
Gao, M. et al. Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032 (2016).
Aron, A. T., Loehr, M. O., Bogena, J. & Chang, C. J. An endoperoxide reactivity-based FRET probe for ratiometric fluorescence imaging of labile iron pools in living cells. J. Am. Chem. Soc. 138, 14338–14346 (2016).
Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017).
Wenzel, S. E. et al. PEBP1 Wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628–641. e626 (2017).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Dolma, S., Lessnick, S. L., Hahn, W. C. & Stockwell, B. R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296 (2003).
Bannai, S. & Kitamura, E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J. Biol. Chem. 255, 2372–2376 (1980).
Murphy, T. H. et al. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558 (1989).
Schubert, D., Kimura, H. & Maher, P. Growth factors and vitamin E modify neuronal glutamate toxicity. Proc. Natl Acad. Sci. USA 89, 8264–8267 (1992).
Li, Y., Maher, P. & Schubert, D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 19, 453–463 (1997).
Gao, M. et al. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308 (2015).
Zilka, O. et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci. 3, 232–243 (2017).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Ingold, I. et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422. e421 (2018).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019).
Zou, Y. et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585, 603–608 (2020).
DeBose-Boyd, R. A. Significance and regulation of lipid metabolism. Semin. Cell Dev. Biol. 81, 97–97 (2018).
Kang, R. et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal Polymicrobial Sepsis. Cell Host Microbe 24, 97–108. e104 (2018).
Lee, H. et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 22, 225–234 (2020).
Li, C. et al. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct. Target Ther. 5, 187 (2020).
Gill, I. & Valivety, R. Polyunsaturated fatty acids, Part 2: biotransformations and biotechnological applications. Trends Biotechnol. 15, 470–478 (1997).
Gill, I. & Valivety, R. Polyunsaturated fatty acids, Part 1: occurrence, biological activities and applications. Trends Biotechnol. 15, 401–409 (1997).
Porter, N. A., Wolf, R. A., Yarbro, E. M. & Weenen, H. The autoxidation of arachidonic acid: formation of the proposed SRS-A intermediate. Biochem. Biophys. Res. Commun. 89, 1058–1064 (1979).
Yin, H., Xu, L. & Porter, N. A. Free radical lipid peroxidation: mechanisms and analysis. Chem. Rev. 111, 5944–5972 (2011).
Rouzer, C. A. & Marnett, L. J. Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem. Rev. 103, 2239–2304 (2003).
Frank, C. E. Hydrocarbon autoxidation. Chem. Rev. 46, 155–169 (1950).
Magtanong, L. et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Biol. 26, 420–432.e429 (2019).
Girotti, A. W. & Korytowski, W. Cholesterol hydroperoxide generation, translocation, and reductive turnover in biological systems. Cell Biochem. Biophys. 75, 413–419 (2017).
Thomas, J. P. et al. Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins. Biochim. Biophys. Acta 1045, 252–260 (1990).
Yang, W. S. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl Acad. Sci. USA 113, E4966–E4975 (2016).
Janssen, C. I. & Kiliaan, A. J. Long-chain polyunsaturated fatty acids (LCPUFA) from genesis to senescence: the influence of LCPUFA on neural development, aging, and neurodegeneration. Prog. Lipid Res. 53, 1–17 (2014).
Bazinet, R. P. & Laye, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 15, 771–785 (2014).
Bos, D. J. et al. Reduced symptoms of inattention after dietary omega-3 fatty acid supplementation in boys with and without attention deficit/hyperactivity disorder. Neuropsychopharmacology 40, 2298–2306 (2015).
Coluccia, A. et al. Developmental omega-3 supplementation improves motor skills in juvenile-adult rats. Int. J. Dev. Neurosci. 27, 599–605 (2009).
Pu, H. et al. Repetitive and prolonged omega-3 fatty acid treatment after traumatic brain injury enhances long-term tissue restoration and cognitive recovery. Cell Transplant. 26, 555–569 (2017).
Dixon, S. J. et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609 (2015).
Hishikawa, D. et al. Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc. Natl Acad. Sci. USA 105, 2830–2835 (2008).
Küch, E.-M. et al. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. et. Biophys. Acta 1841, 227–239 (2014).
Neitemeier, S. et al. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 12, 558–570 (2017).
Gao, M. et al. Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363 e353 (2019).
Wong-Ekkabut, J. et al. Effect of lipid peroxidation on the properties of lipid bilayers: a molecular dynamics study. Biophys. J. 93, 4225–4236 (2007).
Feng, H. & Stockwell, B. R. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. 16, e2006203 (2018).
Ayala, A., Munoz, M. F. & Arguelles, S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 360438 (2014).
Zhou, B. et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 28, 1171–1185 (2018).
Yuan, H. et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 478, 1338–1343 (2016).
Wu, J. et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406 (2019).
Lei, G. et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 30, 146–162 (2020).
Li, Y. et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 26, 2284–2299 (2019).
Brown, C. W., Amante, J. J., Goel, H. L. & Mercurio, A. M. The α6β4 integrin promotes resistance to ferroptosis. J. Cell Biol. 216, 4287–4297 (2017).
Li, Z. et al. Deficiency in lysophosphatidylcholine acyltransferase 3 reduces plasma levels of lipids by reducing lipid absorption in mice. Gastroenterology 149, 1519–1529 (2015).
Rong, X. et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 18, 685–697 (2013).
Andrews, N. C. & Schmidt, P. J. Iron homeostasis. Annu Rev. Physiol. 69, 69–85 (2007).
Feng, H. et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 30, 3411–3423.e3417 (2020).
El Hout, M., Dos Santos, L., Hamai, A. & Mehrpour, M. A promising new approach to cancer therapy: targeting iron metabolism in cancer stem cells. Semin. Cancer Biol. 53, 125–138 (2018).
Ryu, M.-S. et al. PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J. Clin. Investig. 127, 1786–1797 (2017).
Ganz, T. Cellular iron: ferroportin is the only way out. Cell Metab. 1, 155–157 (2005).
Drakesmith, H., Nemeth, E. & Ganz, T. Ironing out ferroportin. Cell Metab. 22, 777–787 (2015).
Wang, Z. et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 428, 21–33 (2018).
Mancias, J. D. et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109 (2014).
Brown, C. W. et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev. Cell 51, 575–586. e574 (2019).
Latunde-Dada, G. O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 1861, 1893–1900 (2017).
Diggle, C. P. In vitro studies on the relationship between polyunsaturated fatty acids and cancer: tumour or tissue specific effects? Prog. Lipid Res. 41, 240–253 (2002).
Rice-Evans, C. & Burdon, R. Free radical-lipid interactions and their pathological consequences. Prog. lipid Res. 32, 71–110 (1993).
Kuhn, H. et al. The evolutionary hypothesis of reaction specificity of mammalian ALOX15 orthologs. Prog. Lipid Res. 72, 55–74 (2018).
Joshi, Y. B., Giannopoulos, P. F. & Praticò, D. The 12/15-lipoxygenase as an emerging therapeutic target for Alzheimer’s disease. Trends Pharmacol. Sci. 36, 181–186 (2015).
Singh, N. K. & Rao, G. N. Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog. Lipid Res. 73, 28–45 (2019).
Chu, B. et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 21, 579–591 (2019).
Dar, H. H. et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J. Clin. Invest. 128, 4639–4653 (2018).
Anthonymuthu, T. S. et al. Empowerment of 15-lipoxygenase catalytic competence in selective oxidation of membrane ETE-PE to ferroptotic death signals, HpETE-PE. J. Am. Chem. Soc. 140, 17835–17839 (2018).
Kapralov, A. A. et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 16, 278–290 (2020).
Kuhn, H., Banthiya, S. & van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 1851, 308–330 (2015).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014).
Brütsch, S. H. et al. Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the Alox15 gene does not rescue such knock-in mice. Antioxid. Redox Signal. 22, 281–293 (2015).
Shah, R., Shchepinov, M. S. & Pratt, D. A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 4, 387–396 (2018).
Chen, J. J. & Galluzzi, L. Fighting resilient cancers with iron. Trends Cell Biol. 28, 77–78 (2018).
Ursini, F. et al. Diversity of glutathione peroxidases. Methods Enzymol. 252, 38–53 (1995).
Kelner, M. J. & Montoya, M. A. Structural organization of the human selenium-dependent phospholipid hydroperoxide glutathione peroxidase gene (GPX4): chromosomal localization to 19p13.3. Biochem. Biophys. Res. Commun. 249, 53–55 (1998).
Brigelius-Flohe, R. & Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 1830, 3289–3303 (2013).
Forcina, G. C. & Dixon, S. J. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics 19, e1800311 (2019).
Cardoso, B. R., Hare, D. J., Bush, A. I. & Roberts, B. R. Glutathione peroxidase 4: a new player in neurodegeneration? Mol. Psychiatry 22, 328–335 (2017).
Ingold, I. et al. Expression of a catalytically inactive mutant form of glutathione peroxidase 4 (Gpx4) confers a dominant-negative effect in male fertility. J. Biol. Chem. 290, 14668–14678 (2015).
Schneider, M. et al. Mitochondrial glutathione peroxidase 4 disruption causes male infertility. FASEB J. 23, 3233–3242 (2009).
Seiler, A. et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248 (2008).
Yoo, S. E. et al. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic. Biol. Med. 52, 1820–1827 (2012).
Shimada, K., Hayano, M., Pagano, N. C. & Stockwell, B. R. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem. Biol. 23, 225–235 (2016).
Ding, C.-K. C. et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Na. Metabolism 2, 270–277 (2020).
Mandal, P. K. et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 285, 22244–22253 (2010).
Hayano, M. et al. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 23, 270–278 (2016).
Chen, Z. et al. Recent progress in treatment of hepatocellular carcinoma. Am. J. Cancer Res. 10, 2993–3036 (2020).
Sun, X. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184 (2016).
Cao, J. et al. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat. Commun. 11, 1251–1251 (2020).
Duffy, M. J., Synnott, N. C. & Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 83, 258–265 (2017).
Li, T. et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149, 1269–1283 (2012).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Zhang, Y. et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 20, 1181–1192 (2018).
Liu, T., Jiang, L., Tavana, O. & Gu, W. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res 79, 1913–1924 (2019).
Bosl, M. R. et al. Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (Trsp). Proc. Natl Acad. Sci. USA 94, 5531–5534 (1997).
Hatfield, D. L., Tsuji, P. A., Carlson, B. A. & Gladyshev, V. N. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem. Sci. 39, 112–120 (2014).
Christensen, M. J. & Burgener, K. W. Dietary selenium stabilizes glutathione peroxidase mRNA in rat liver. J. Nutr. 122, 1620–1626 (1992).
Howard, M. T., Carlson, B. A., Anderson, C. B. & Hatfield, D. L. Translational redefinition of UGA codons is regulated by selenium availability. J. Biol. Chem. 288, 19401–19413 (2013).
Warner, G. J. et al. Inhibition of selenoprotein synthesis by selenocysteine tRNA[Ser]Sec lacking isopentenyladenosine. J. Biol. Chem. 275, 28110–28119 (2000).
Kryukov, G. V. et al. Characterization of mammalian selenoproteomes. Science 300, 1439–1443 (2003).
Viswanathan, V. S. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017).
Mannes, A. M. et al. Cysteine mutant of mammalian GPx4 rescues cell death induced by disruption of the wild-type selenoenzyme. FASEB J. 25, 2135–2144 (2011).
Xu, X. M. et al. Targeted insertion of cysteine by decoding UGA codons with mammalian selenocysteine machinery. Proc. Natl Acad. Sci. USA 107, 21430–21434 (2010).
Yu, Y. et al. Characterization and structural analysis of human selenium-dependent glutathione peroxidase 4 mutant expressed in Escherichia coli. Free Radic. Biol. Med. 71, 332–338 (2014).
Horikoshi, N., Cong, J., Kley, N. & Shenk, T. Isolation of differentially expressed cDNAs from p53-dependent apoptotic cells: activation of the human homologue of the Drosophila peroxidasin gene. Biochem. Biophys. Res. Commun. 261, 864–869 (1999).
Wu, M. et al. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J. Biol. Chem. 277, 25617–25623 (2002).
Gong, M. et al. DNA binding suppresses human AIF-M2 activity and provides a connection between redox chemistry, reactive oxygen species, and apoptosis. J. Biol. Chem. 282, 30331–30340 (2007).
Miriyala, S. et al. Novel role of 4-hydroxy-2-nonenal in AIFm2-mediated mitochondrial stress signaling. Free Radic. Biol. Med. 91, 68–80 (2016).
Kumar, S. & Miller, L. H. Cellular mechanisms in immunity to blood stage infection. Immunol. Lett. 25, 109–114 (1990).
Elguindy, M. M. & Nakamaru-Ogiso, E. Apoptosis-inducing factor (AIF) and its family member protein, amid, are rotenone-sensitive NADH:ubiquinone oxidoreductases (NDH-2). J. Biol. Chem. 290, 20815–20826 (2015).
Zou, Y. et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 10, 1617 (2019).
Shimada, K. et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503 (2016).
Morre, D. J. & Morre, D. M. Non-mitochondrial coenzyme Q. Biofactors 37, 355–360 (2011).
Shukla, S. & Dubey, K. K. CoQ10 a super-vitamin: review on application and biosynthesis. 3 Biotech 8, 249 (2018).
Farquhar, M. G. & Palade, G. E. The Golgi apparatus: 100 years of progress and controversy. Trends Cell Biol. 8, 2–10 (1998).
Ernster, L. & Forsmark-Andree, P. Ubiquinol: an endogenous antioxidant in aerobic organisms. Clin. Investig. 71, S60–S65 (1993).
Stocker, R., Bowry, V. W. & Frei, B. Ubiquinol-10 protects human low density lipoprotein more efficiently against lipid peroxidation than does alpha-tocopherol. Proc. Natl Acad. Sci. USA 88, 1646–1650 (1991).
Mortensen, S. A. et al. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO: a randomized double-blind trial. JACC Heart Fail 2, 641–649 (2014).
Liu, J., Wang, L., Zhan, S. Y. & Xia, Y. Coenzyme Q10 for Parkinson’s disease. The Cochrane database of systematic reviews CD008150, (2011).
Alehagen, U. et al. Increase in insulin-like growth factor 1 (IGF-1) and insulin-like growth factor binding protein 1 after supplementation with selenium and coenzyme Q10. A prospective randomized double-blind placebo-controlled trial among elderly Swedish citizens. PLoS ONE 12, e0178614 (2017).
Lopez, L. C. et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 79, 1125–1129 (2006).
Mollet, J. et al. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 117, 765–772 (2007).
Quinzii, C. et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 78, 345–349 (2006).
Friedmann Angeli, J. P., Krysko, D. V. & Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 19, 405–414 (2019).
Bloch, K. The biological synthesis of cholesterol. Science 150, 19–28 (1965).
Miziorko, H. M. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch. Biochem. Biophys. 505, 131–143 (2011).
Ernster, L. & Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1271, 195–204 (1995).
Mullen, P. J. et al. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 16, 718–731 (2016).
Carlson, B. A. et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 9, 22–31 (2016).
Llabani, E. et al. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat. Chem. 11, 521–532 (2019).
Jelinek, A. et al. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 117, 45–57 (2018).
Soula, M. et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 16, 1351–1360 (2020).
Gao, B., Doan, A. & Hybertson, B. M. The clinical potential of influencing Nrf2 signaling in degenerative and immunological disorders. Clin. Pharm. 6, 19–34 (2014).
Lee, J. M. et al. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 278, 12029–12038 (2003).
Kerins, M. J. & Ooi, A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid. Redox Signal. 29, 1756–1773 (2018).
Sasaki, H. et al. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 277, 44765–44771 (2002).
Wu, K. C., Cui, J. Y. & Klaassen, C. D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 123, 590–600 (2011).
Lee, C. Collaborative power of Nrf2 and PPARgamma activators against metabolic and drug-induced oxidative injury. Oxid. Med. Cell Longev. 2017, 1378175 (2017).
Roh, J.-L., Kim, E. H., Jang, H. & Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 11, 254–262 (2017).
Rothe, T. et al. 12/15-Lipoxygenase-mediated enzymatic lipid oxidation regulates DC maturation and function. J. Clin. Investig. 125, 1944–1954 (2015).
Tan, S., Schubert, D. & Maher, P. Oxytosis: a novel form of programmed cell death. Curr. Top. Med Chem. 1, 497–506 (2001).
Stockwell, B. R. et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Sakai, O. et al. Role of glutathione peroxidase 4 in glutamate-induced oxytosis in the retina. PLoS ONE 10, e0130467 (2015).
Ishige, K., Schubert, D. & Sagara, Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic. Biol. Med. 30, 433–446 (2001).
Kang, Y. et al. Cellular protection using Flt3 and PI3Kalpha inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat. Commun. 5, 3672 (2014).
Tan, S. et al. The regulation of reactive oxygen species production during programmed cell death. J. Cell Biol. 141, 1423–1432 (1998).
Lee, Y. S. et al. Ferroptosis-induced endoplasmic reticulum stress: cross-talk between ferroptosis and apoptosis. Mol. Cancer Res. 16, 1073–1076 (2018).
Hirata, Y. et al. Inhibition of double-stranded RNA-dependent protein kinase prevents oxytosis and ferroptosis in mouse hippocampal HT22 cells. Toxicology 418, 1–10 (2019).
Klionsky, D. J. & Emr, S. D. Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721 (2000).
Marino, G., Niso-Santano, M., Baehrecke, E. H. & Kroemer, G. Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 15, 81–94 (2014).
Berry, D. L. & Baehrecke, E. H. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 131, 1137–1148 (2007).
Seillier, M. et al. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ. 19, 1525–1535 (2012).
Yu, S. W. et al. Autophagic death of adult hippocampal neural stem cells following insulin withdrawal. Stem Cells 26, 2602–2610 (2008).
Du, J. et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 131, 356–369 (2019).
Zhou, B. et al. Ferroptosis is a type of autophagy-dependent cell death. In Seminars in Cancer Biology. (Academic Press, 2019).
Bai, Y. et al. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 508, 997–1003 (2019).
Kamili, A. et al. TPD52 expression increases neutral lipid storage within cultured cells. J. Cell Sci. 128, 3223–3238 (2015).
Spangler, B. et al. A reactivity-based probe of the intracellular labile ferrous iron pool. Nat. Chem. Biol. 12, 680–685 (2016).
Sato, K. et al. Non-thermal plasma specifically kills oral squamous cell carcinoma cells in a catalytic Fe(II)-dependent manner. J. Clin. Biochem. Nutr. 65, 8–15 (2019).
Gaschler, M. M. et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 14, 507–515 (2018).
Mai, T. T. et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 9, 1025–1033 (2017).
Hassannia, B. et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 128, 3341–3355 (2018).
Sosa, V. et al. Oxidative stress and cancer: an overview. Ageing Res. Rev. 12, 376–390 (2013).
Gorrini, C., Harris, I. S. & Mak, T. W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947 (2013).
Afzal, S. et al. Oxidative damage to guanine nucleosides following combination chemotherapy with 5-fluorouracil and oxaliplatin. Cancer Chemother. Pharm. 69, 301–307 (2012).
Alexandre, J. et al. Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Cancer Res. 67, 3512–3517 (2007).
Ramanathan, B. et al. Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res. 65, 8455–8460 (2005).
Jiang, T. et al. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 70, 5486–5496 (2010).
Baumgartner, M. et al. Enhancement of arsenic trioxide-mediated apoptosis using docosahexaenoic acid in arsenic trioxide-resistant solid tumor cells. Int. J. Cancer 112, 707–712 (2004).
Hangauer, M. J. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017).
Alitalo, A. & Detmar, M. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene 31, 4499–4508 (2012).
Morton, D. L. et al. Final trial report of sentinel-node biopsy versus nodal observation in melanoma. N. Engl. J. Med. 370, 599–609 (2014).
Ubellacker, J. M. et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118 (2020).
Zhang, Y. et al. Imidazole Ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem. Biol. 26, 623–633. e629 (2019).
Tsoi, J. et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell 33, 890–904. e895 (2018).
Wculek, S. K. et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20, 7–24 (2020).
Uderhardt, S. et al. 12/15-lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunity 36, 834–846 (2012).
Manguso, R. T. et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418 (2017).
Wang, W. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019).
Matsushita, M. et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 212, 555–568 (2015).
Cramer, S. L. et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 23, 120–127 (2017).
Muri, J., Thut, H., Bornkamm, G. W. & Kopf, M. B1 and marginal zone B cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep. 29, 2731–2744. e2734 (2019).
Wan, C. et al. Irradiated tumor cell-derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci. Adv. 6, eaay9789 (2020).
Lang, X. et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 9, 1673–1685 (2019).
Shibata, Y. et al. Erastin, a ferroptosis-inducing agent, sensitized cancer cells to X-ray irradiation via glutathione starvation in vitro and in vivo. PLoS ONE 14, e0225931 (2019).
Li, Y. et al. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol. Lett. 19, 323–333 (2020).
Kim, E. H. et al. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 432, 180–190 (2018).
Efferth, T. From ancient herb to modern drug: artemisia annua and artemisinin for cancer therapy. Semin Cancer Biol. 46, 65–83 (2017).
Gao, X. et al. Ibuprofen induces ferroptosis of glioblastoma cells via downregulation of nuclear factor erythroid 2-related factor 2 signaling pathway. Anticancer Drugs 31, 27–34 (2020).
Bai, T. et al. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 491, 919–925 (2017).
Chen, L. et al. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lyase function. Oncol. Rep. 33, 1465–1474 (2015).
Yamaguchi, H. et al. Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clin. Cancer Res. 19, 845–854 (2013).
Kim, S. E. et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 11, 977–985 (2016).
Shen, Z. et al. Fenton-reaction-acceleratable magnetic nanoparticles for ferroptosis therapy of orthotopic brain tumors. ACS Nano 12, 11355–11365 (2018).
Bao, W. et al. Nanolongan with multiple on-demand conversions for ferroptosis-apoptosis combined anticancer therapy. ACS Nano 13, 260–273 (2019).
Zhao, Y., Zhao, W., Lim, Y. C. & Liu, T. Salinomycin-loaded gold nanoparticles for treating cancer stem cells by ferroptosis-induced cell death. Mol. Pharmaceut. 16, 2532–2539 (2019).
He, Y.-J. et al. Fenton reaction-independent ferroptosis therapy via glutathione and iron redox couple sequentially triggered lipid peroxide generator. Biomaterials 241, 119911–119911 (2020).
Xu, T. et al. Enhanced ferroptosis by oxygen-boosted phototherapy based on a 2-in-1 nanoplatform of ferrous hemoglobin for tumor synergistic therapy. ACS nano. https://doi.org/10.1021/acsnano.1029b09426 (2020).
Heemels, M. T. Neurodegenerative diseases. Nature 539, 179 (2016).
Buijs, M. et al. In vivo assessment of iron content of the cerebral cortex in healthy aging using 7-Tesla T2*-weighted phase imaging. Neurobiol. Aging 53, 20–26 (2017).
Belaidi, A. A. & Bush, A. I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J. Neurochem. 139(Suppl 1), 179–197 (2016).
Belaidi, A. A. et al. Marked age-related changes in brain iron homeostasis in amyloid protein precursor knockout mice. Neurotherapeutics 15, 1055–1062 (2018).
Masaldan, S., Belaidi, A. A., Ayton, S. & Bush, A. I. Cellular senescence and iron dyshomeostasis in Alzheimer’s disease. Pharmaceuticals. 12, 93 (2019).
Bogdan, A. R., Miyazawa, M., Hashimoto, K. & Tsuji, Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem. Sci. 41, 274–286 (2016).
Zhou, Z. D. & Tan, E. K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 12, 75 (2017).
Guo, C. et al. Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 562–575 (2013).
Guo, C. et al. Intranasal deferoxamine attenuates synapse loss via up-regulating the P38/HIF-1alpha pathway on the brain of APP/PS1 transgenic mice. Front. Aging Neurosci. 7, 104 (2015).
Abbruzzese, G. et al. A pilot trial of deferiprone for neurodegeneration with brain iron accumulation. Haematologica 96, 1708–1711 (2011).
Klopstock, T. et al. Safety and efficacy of deferiprone for pantothenate kinase-associated neurodegeneration: a randomised, double-blind, controlled trial and an open-label extension study. Lancet Neurol. 18, 631–642 (2019).
Moreau, C. et al. Could conservative iron chelation lead to neuroprotection in amyotrophic lateral sclerosis? Antioxid. Redox Signal. 29, 742–748 (2018).
Devos, D. et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 21, 195–210 (2014).
Martin-Bastida, A. et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 7, 1398 (2017).
Ayton, S. et al. Iron accumulation confers neurotoxicity to a vulnerable population of nigral neurons: implications for Parkinson’s disease. Mol. Neurodegener. 9, 27 (2014).
Barbariga, M. et al. Ceruloplasmin functional changes in Parkinson’s disease-cerebrospinal fluid. Mol. Neurodegener. 10, 59 (2015).
Belanger, M., Allaman, I. & Magistretti, P. J. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738 (2011).
Lopez, N. et al. Oxidative stress in Alzheimer’s disease and mild cognitive impairment with high sensitivity and specificity. J. Alzheimers Dis. 33, 823–829 (2013).
Yoritaka, A. et al. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl Acad. Sci. USA 93, 2696–2701 (1996).
Chen, Y. et al. Anti-oxidant polydatin (piceid) protects against substantia nigral motor degeneration in multiple rodent models of Parkinson’s disease. Mol. Neurodegener. 10, 4 (2015).
Dumont, M. et al. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB J. 23, 2459–2466 (2009).
Young, M. L. & Franklin, J. L. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol. Cell Neurosci. 101, 103409 (2019).
Conrad, M. & Pratt, D. A. The chemical basis of ferroptosis. Nat. Chem. Biol. 15, 1137–1147 (2019).
Raefsky, S. M. et al. Deuterated polyunsaturated fatty acids reduce brain lipid peroxidation and hippocampal amyloid β-peptide levels, without discernable behavioral effects in an APP/PS1 mutant transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 66, 165–176 (2018).
Zhang, Y. H. et al. alpha-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 14, 535–548 (2018).
Dysken, M. W. et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA 311, 33–44 (2014).
Hung, L. W. et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med. 209, 837–854 (2012).
Southon, A. et al. Cu(II) (atsm) inhibits ferroptosis: implications for treatment of neurodegenerative disease. Br. J. Pharm. 177, 656–667 (2020).
Fu, A. L., Dong, Z. H. & Sun, M. J. Protective effect of N-acetyl-L-cysteine on amyloid beta-peptide-induced learning and memory deficits in mice. Brain Res. 1109, 201–206 (2006).
Tucker, S. et al. RNA therapeutics directed to the non coding regions of APP mRNA, in vivo anti-amyloid efficacy of paroxetine, erythromycin, and N-acetyl cysteine. Curr. Alzheimer Res. 3, 221–227 (2006).
Coles, L. D. et al. Repeated-dose oral n-acetylcysteine in parkinson’s disease: pharmacokinetics and effect on brain glutathione and oxidative stress. J. Clin. Pharm. 58, 158–167 (2018).
Taghizadeh, M. et al. The effects of omega-3 fatty acids and vitamin E co-supplementation on clinical and metabolic status in patients with Parkinson’s disease: a randomized, double-blind, placebo-controlled trial. Neurochem. Int. 108, 183–189 (2017).
Vallerga, C. L. et al. Analysis of DNA methylation associates the cystine-glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat. Commun. 11, 1238 (2020).
Ghosh, D., Levault, K. R. & Brewer, G. J. Relative importance of redox buffers GSH and NAD(P)H in age-related neurodegeneration and Alzheimer disease-like mouse neurons. Aging Cell 13, 631–640 (2014).
Zhang, Y. H. et al. α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 14, 535–548 (2018).
Ueta, T. et al. Glutathione peroxidase 4 is required for maturation of photoreceptor cells. J. Biol. Chem. 287, 7675–7682 (2012).
Hambright, W. S. et al. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17 (2017).
Bellinger, F. P. et al. Glutathione peroxidase 4 is associated with neuromelanin in substantia nigra and dystrophic axons in putamen of Parkinson’s brain. Mol. Neurodegener. 6, 8 (2011).
Alim, I. et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177, 1262–1279. e1225 (2019).
B, R. C. et al. The APOE epsilon4 allele is associated with lower selenium levels in the brain: implications for Alzheimer’s disease. ACS Chem. Neurosci. 8, 1459–1464 (2017).
Cardoso, B. R. et al. Supranutritional sodium selenate supplementation delivers selenium to the central nervous system: results from a randomized controlled pilot trial in Alzheimer’s disease. Neurotherapeutics 16, 192–202 (2019).
Gwon, A. R. et al. Selenium attenuates A beta production and A beta-induced neuronal death. Neurosci. Lett. 469, 391–395 (2010).
Ellwanger, J. H. et al. Selenium reduces bradykinesia and DNA damage in a rat model of Parkinson’s disease. Nutrition 31, 359–365 (2015).
Bonifati, V. et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259 (2003).
Skouta, R. et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 136, 4551–4556 (2014).
Do Van, B. et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 94, 169–178 (2016).
Soares, R. O. S. et al. Ischemia/reperfusion injury revisited: an overview of the latest pharmacological strategies. Int. J. Mol. Sci. 20, 5034 (2019).
Eltzschig, H. K. & Eckle, T. Ischemia and reperfusion-from mechanism to translation. Nat. Med. 17, 1391–1401 (2011).
Dietrich, R. B. & Bradley, W. G. Jr. Iron accumulation in the basal ganglia following severe ischemic-anoxic insults in children. Radiology 168, 203–206 (1988).
Bulluck, H. et al. Residual myocardial iron following intramyocardial hemorrhage during the convalescent phase of reperfused st-segment-elevation myocardial infarction and adverse left ventricular remodeling. Circ. Cardiovasc. Imaging. 9, e004940 (2016).
Fang, K. M. et al. Trace element, antioxidant activity, and lipid peroxidation levels in brain cortex of gerbils after cerebral ischemic injury. Biol. Trace Elem. Res. 152, 66–74 (2013).
Linkermann, A. Nonapoptotic cell death in acute kidney injury and transplantation. Kidney Int. 89, 46–57 (2016).
Park, U. J. et al. Blood-derived iron mediates free radical production and neuronal death in the hippocampal CA1 area following transient forebrain ischemia in rat. Acta Neuropathol. 121, 459–473 (2011).
Tuo, Q. Z. et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 22, 1520–1530 (2017).
Galaris, D., Barbouti, A. & Korantzopoulos, P. Oxidative stress in hepatic ischemia-reperfusion injury: the role of antioxidants and iron chelating compounds. Curr. Pharm. Des. 12, 2875–2890 (2006).
Hanson, L. R. et al. Intranasal deferoxamine provides increased brain exposure and significant protection in rat ischemic stroke. J. Pharm. Exp. Ther. 330, 679–686 (2009).
Scindia, Y. et al. Hepcidin mitigates renal ischemia-reperfusion injury by modulating systemic iron homeostasis. J. Am. Soc. Nephrol. 26, 2800–2814 (2015).
Chan, W. et al. Effect of iron chelation on myocardial infarct size and oxidative stress in ST-elevation-myocardial infarction. Circ. Cardiovasc. Inter. 5, 270–278 (2012).
Hess, M. L. & Manson, N. H. Molecular oxygen: friend and foe. The role of the oxygen free radical system in the calcium paradox, the oxygen paradox and ischemia/reperfusion injury. J. Mol. Cell Cardiol. 16, 969–985 (1984).
Dare, A. J. et al. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 5, 163–168 (2015).
Jiang, Y. et al. miR-210 mediates vagus nerve stimulation-induced antioxidant stress and anti-apoptosis reactions following cerebral ischemia/reperfusion injury in rats. J. Neurochem. 134, 173–181 (2015).
Zhou, H. et al. Protective role of melatonin in cardiac ischemia-reperfusion injury: from pathogenesis to targeted therapy. J Pineal Res. 64, e12471 (2018).
Ni, D. et al. Ceria nanoparticles meet hepatic ischemia-reperfusion injury: the perfect imperfection. Adv. Mater. 31, e1902956 (2019).
Farmer, E. E. & Mueller, M. J. ROS-mediated lipid peroxidation and RES-activated signaling. Annu. Rev. Plant Biol. 64, 429–450 (2013).
Ambrosi, N. et al. Alpha-lipoic acid protects against ischemia-reperfusion injury in simultaneous kidney-pancreas. Transplantation 100, 908–915 (2016).
Casciato, P. et al. alpha-lipoic acid reduces postreperfusion syndrome in human liver transplantation-a pilot study. Transpl. Int. 31, 1357–1368 (2018).
Dunschede, F. et al. Reduction of ischemia reperfusion injury after liver resection and hepatic inflow occlusion by alpha-lipoic acid in humans. World J. Gastroenterol. 12, 6812–6817 (2006).
Guan, X. et al. The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci. 235, 116795 (2019).
Dabkowski, E. R., Williamson, C. L. & Hollander, J. M. Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Radic. Biol. Med. 45, 855–865 (2008).
Lei, P. et al. Lithium suppression of tau induces brain iron accumulation and neurodegeneration. Mol. Psychiatry 22, 396–406 (2017).
Lei, P. et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 18, 291–295 (2012).
Fang, X. et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl Acad. Sci. USA 116, 2672–2680 (2019).
van Leyen, K. et al. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke 37, 3014–3018 (2006).
Linkermann, A. et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl Acad. Sci. USA 111, 16836–16841 (2014).
Huang, L. L. et al. Augmenter of liver regeneration protects the kidney from ischaemia-reperfusion injury in ferroptosis. J. Cell Mol. Med. 23, 4153–4164 (2019).
Li, L. et al. Ferroptosis is associated with oxygen-glucose deprivation/reoxygenation-induced Sertoli cell death. Int. J. Mol. Med. 41, 3051–3062 (2018).
Singal, P. K. & Iliskovic, N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 339, 900–905 (1998).
Nobuta, H. et al. Oligodendrocyte death in Pelizaeus-Merzbacher disease is rescued by iron chelation. Cell Stem Cell 25, 531–541. e536 (2019).
Wang, H. et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 66, 449–465 (2017).
Yoshida, M. et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 10, 3145 (2019).
Muller, T. et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol. Life Sci. 74, 3631–3645 (2017).
Wang, Y. Q. et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 8, 308 (2016).
Gout, P. W., Buckley, A. R., Simms, C. R. & Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 15, 1633–1640 (2001).
Louandre, C. et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 133, 1732–1742 (2013).
Dixon, S. J. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 3, e02523–e02523 (2014).
Mãrtensson, J. & Meister, A. Glutathione deficiency decreases tissue ascorbate levels in newborn rats: ascorbate spares glutathione and protects. Proc. Natl Acad. Sci. USA 88, 4656–4660 (1991).
Basit, F. et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 8, e2716–e2716 (2017).
Ellinghaus, P. et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2, 611–624 (2013).
Kong, Z., Liu, R. & Cheng, Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomedicine Pharmacother. 109, 2043–2053 (2019).
Gao, J. et al. Selenium-encoded isotopic signature targeted profiling. ACS Cent. Sci. 4, 960–970 (2018).
Eaton, J. K. et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 16, 497–506 (2020).
Woo, J. H. et al. Elucidating compound mechanism of action by network perturbation analysis. Cell 162, 441–451 (2015).
Wen, Q. et al. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 510, 278–283 (2019).
Li, Q. et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2, e90777–e90777 (2017).
NaveenKumar, S. K., Hemshekhar, M., Kemparaju, K. & Girish, K. S. Hemin-induced platelet activation and ferroptosis is mediated through ROS-driven proteasomal activity and inflammasome activation: protection by melatonin. Biochim. et Biophys. Acta 1865, 2303–2316 (2019).
Turcu, A. L. et al. DMT1 inhibitors kill cancer stem cells by blocking lysosomal iron translocation. Chemistry. https://doi.org/10.1002/chem.202000159 (2020).
Ma, S., Henson, E. S., Chen, Y. & Gibson, S. B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 7, e2307–e2307 (2016).
Villalpando-Rodriguez, G. E., Blankstein, A. R., Konzelman, C. & Gibson, S. B. Lysosomal destabilizing drug siramesine and the dual tyrosine kinase inhibitor lapatinib induce a synergistic ferroptosis through reduced heme oxygenase-1 (HO-1) levels. Oxid. Med. Cell. Longev. 2019, 9561281–9561281 (2019).
Abrams, R. P., Carroll, W. L. & Woerpel, K. A. Five-membered ring peroxide selectively initiates ferroptosis in cancer cells. ACS Chem. Biol. 11, 1305–1312 (2016).
Chang, L.-C. et al. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 416, 124–137 (2018).
Lee, J. H. et al. Brusatol, a Nrf2 inhibitor targets STAT3 signaling cascade in head and neck squamous cell carcinoma. Biomolecules. 9, 550 (2019).
Ooko, E. et al. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 22, 1045–1054 (2015).
Beaudoin-Chabot, C. et al. Deuterated polyunsaturated fatty acids reduce oxidative stress and extend the lifespan of C. elegans. Front. Physiol. 10, 641–641 (2019).
Nieva-Echevarria, B., Goicoechea, E. & Guillen, M. D. Polyunsaturated lipids and vitamin A oxidation during cod liver oil in vitro gastrointestinal digestion. Antioxidant effect of added BHT. Food Chem. 232, 733–743 (2017).
Sun, Z. et al. Perturbation of 3-tert-butyl-4-hydroxyanisole in adipogenesis of male mice with normal and high fat diets. Sci. total Environ. 703, 135608–135608 (2020).
Krainz, T. et al. A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Cent. Sci. 2, 653–659 (2016).
Probst, L., Dachert, J., Schenk, B. & Fulda, S. Lipoxygenase inhibitors protect acute lymphoblastic leukemia cells from ferroptotic cell death. Biochem. Pharm. 140, 41–52 (2017).
Kenny, E. M. et al. Ferroptosis contributes to neuronal death and functional outcome after traumatic brain injury. Crit. Care Med. 47, 410–418 (2019).
Gregus, A. M. et al. Systematic analysis of rat 12/15-lipoxygenase enzymes reveals critical role for spinal eLOX3 hepoxilin synthase activity in inflammatory hyperalgesia. FASEB J. 27, 1939–1949 (2013).
Li, L. et al. Inhibition of 5-lipoxygenase pathway attenuates acute liver failure by inhibiting macrophage activation. J. Immunol. Res. 2014, 697560–697560 (2014).
Liu, Y. et al. The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol. Pharm. Bull. 38, 1234–1239 (2015).
Rush, T. et al. Glutathione-mediated neuroprotection against methylmercury neurotoxicity in cortical culture is dependent on MRP1. Neurotoxicology 33, 476–481 (2012).
Sha, L. K. et al. Loss of Nrf2 in bone marrow-derived macrophages impairs antigen-driven CD8(+) T cell function by limiting GSH and Cys availability. Free Radic. Biol. Med. 83, 77–88 (2015).
Wang, D. et al. Antiferroptotic activity of non-oxidative dopamine. Biochem. Biophys. Res. Commun. 480, 602–607 (2016).
Xie, Y. et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 20, 1692–1704 (2017).
Acknowledgements
Supported by funds from the Ministry of Science and Technology of China (2018YFC1312300), The National Natural Science Foundation of China (81722016, 82071191), The Alzheimer’s Association (AARFD-16-442821).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yan, Hf., Zou, T., Tuo, Qz. et al. Ferroptosis: mechanisms and links with diseases. Sig Transduct Target Ther 6, 49 (2021). https://doi.org/10.1038/s41392-020-00428-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-00428-9
