
Abstract
Nicotinamide adenine dinucleotide (NAD+) and its metabolites function as critical regulators to maintain physiologic processes, enabling the plastic cells to adapt to environmental changes including nutrient perturbation, genotoxic factors, circadian disorder, infection, inflammation and xenobiotics. These effects are mainly achieved by the driving effect of NAD+ on metabolic pathways as enzyme cofactors transferring hydrogen in oxidation-reduction reactions. Besides, multiple NAD+-dependent enzymes are involved in physiology either by post-synthesis chemical modification of DNA, RNA and proteins, or releasing second messenger cyclic ADP-ribose (cADPR) and NAADP+. Prolonged disequilibrium of NAD+ metabolism disturbs the physiological functions, resulting in diseases including metabolic diseases, cancer, aging and neurodegeneration disorder. In this review, we summarize recent advances in our understanding of the molecular mechanisms of NAD+-regulated physiological responses to stresses, the contribution of NAD+ deficiency to various diseases via manipulating cellular communication networks and the potential new avenues for therapeutic intervention.
Similar content being viewed by others

Introduction
NAD+ was first described in 1906 as a component that could increase the fermentation rate in yeast.1 Years later, NAD+ was determined to play a vital role for hydrogen transfer in redox reaction.2 As an essential redox carrier, NAD+ receives hydride from metabolic processes including glycolysis, the TCA cycle, and fatty acid oxidation (FAO) to form NADH. NADH, therefore, serves as a central hydride donor to ATP synthesis through mitochondrial OXPHOS, along with the generation of ROS. Beyond its vital role as a coenzyme in energy metabolism, the important role of NAD+ has expanded to be a co-substrate for various enzymes including sirtuins, PARPs, CD157, CD73, CD38 and SARM1.3,4,5,6 Recently, it has been found that NAD+ serves as a nucleotide analog in DNA ligation and RNA capping.7,8 Therefore, the dynamic NAD+ and its metabolites levels, in response to diverse cellular stress and physiological stimuli, rewire biological processes via post-synthesis modification of fundamental biomolecules, including DNA, RNA and proteins.9,10,11,12,13 Through these activities, NAD+ impact energy metabolism, DNA repair, epigenetic modification, inflammation, circadian rhythm and stress resistance. NAD+ deficiency, however, contributes to a spectrum of diseases including metabolic diseases, cancer, aging and neurodegeneration disorders.
Here, we summarize recent advances in our understanding of the NAD+ homeostasis in response to growth conditions or environmental stimuli, highlighting the actions of NAD+ in coordinating metabolic reprogramming and maintaining cellular physiologic biology, which enables the plastic cells to adapt to environmental changes. Furthermore, we will discuss the NAD+ and its metabolites serving as an essential hub in both physiological and pathophysiological processes and explore the potential of NAD+ modulation in the clinical treatment of diseases.
NAD+ homeostasis
NAD+, one of the most common metabolites in the human body, is in a homeostatic status of biosynthesis, consumption, recycling and degradation at both cellular and systemic levels.14
NAD+ biosynthesis
De novo pathway
Mammalian cells can generate NAD+ de novo from dietary tryptophan (Trp) by the kynurenine pathway (KP), which is initialized by either TDO or IDO. The intermediate ACMS can cyclize spontaneously to QA. However, ACMSD converts ACMS to picolinic acid, limiting the flux from tryptophan to NAD+.15 Another critical step catalyzes the conversion of QA to NAMN by QPRT, which commits the pathway to NAD+ biosynthesis.16,17 The Preiss-Handler pathway can convert dietary NA to NAMN by NAPRT.18 NAMN derived from both the tryptophan and NA is catalyzed by NMNATs to yield NAAD, which is then amidated to NAD+ by NAD synthase (NADSYN) using glutamine as nitrogen donor (Fig. 1).19,20

Overview of the NAD+ metabolism and its physiological function. Mammalian cells can synthesize NAD+ de novo from tryptophan by the kynurenine pathway or from NA by the Preiss‐Handler pathway, while most NAD+ is recycled via salvage pathways from nicotinamide (NAM), a by-production of NAD+-consuming reactions. NAD+ can be reduced into NADH in the metabolic processes, including glycolysis, fatty acid oxidation and the TCA cycle. NADH, in turn, drives the generation of ATP via OXPHOS, the production of lactic acid from pyruvate, and the desaturation of PUFAs. NADPH plays an essential role in antioxidant defense and regulates cellular signaling via NADPH oxidases (NOXs). Moreover, NAD+ is found to decorate various RNAs in different organisms as nucleotide analog and serves as an alternative adenylation donor for DNA ligation in NHEJ repair. NAD+ also acts as a co-substrate for a wide variety of enzymes, including PARPs, sirtuins, CD38/CD157 and SARM1, impacting metabolism, genomic stability, gene expression, inflammation, circadian rhythm and stress resistance. Using NAD+ as a co-substrate, both PARPs and sirtuins regulate their target molecules, generating NAM as a by-product. The CD38/CD157 and SARM1 also catalyze NAD+ to NAM, producing ADPR and cADPR. Abbreviations: IDOs, indoleamine 2,3-dioxygenase; QA, quinolinic acid; NAMN, nicotinate mononucleotide; QPRT, quinolinate phosphoribosyl-transferase; NAPRT, nicotinic acid phosphoribosyltransferase; NMNATs, nicotinamide mononucleotide adenylyl transferases; NADSYN, NAD synthase; NR, nicotinamide riboside; Trp, tryptophan; NADKs, NAD+ kinases; PARPs, poly (ADP-ribose) polymerases; NNT, nicotinamide nucleotide transhydrogenase; TDO, tryptophan 2,3-dioxygenase; SARM1, sterile alpha and TIR motif containing 1; NNMT, Nicotinamide N-methyltransferase; NMN, nicotinamide mononucleotide; PUFAs, polyunsaturated fatty acids; NAM, nicotinamide
Salvage pathway
Rather than generated de novo, most NAD+ is recycled from NAM, NA, NR and NMN in the salvage pathway to maintain the cellular NAD+ levels.21 Among these precursors, NAM could be recycled from NAD+ consumption reactions, including both NAD+-dependent deacylation and ADP-ribosylation, into NMN by NAMPT, which catalyzes the rate-limiting reaction in the salvage pathway.22 The precursor NR is imported by ENTs and transformed to NMN by NRK1/2.23 Ultimately, NMN is adenylylated by NMNAT to yield NAD+24,25 (Fig. 1).
NAD+ degradation
NAD+ consumption
As a co-substrate important to various postsynthesis modifications of fundamental macromolecules, NAD+ can be cleaved by NAD+-consuming enzymes including PARPs, sirtuins, CD38 and SARM1 to generate NAM and ADP-ribose (ADPR) (Fig. 1). The sirtuins are NAD+-dependent deacetylases that are distributed in the nucleus (e.g., SIRT1, SIRT6, and SIRT7), the cytoplasm (e.g., SIRT2) and mitochondria (e.g., SIRT3-5), respectively.26 Through mediating the post-translational modification dependent on NAD+, sirtuins modulate the adaptation to the altered cellular energetic status, especially the activation of oxidative metabolism and stress resistance in mitochondria in various physiological or pathological conditions.26 PARPs catalyze reversible ADP-ribosylation of macromolecular targets including proteins, DNA and RNA, utilizing NAD+ as a cofactor to provide monomer or polymers of ADP-ribose nucleotide.27,28 PARP members can be categorized into several groups, the poly-ADP-ribosyl transferases (e.g., PARP1, 2, and 5), the mono-ADP-ribosyl transferases (e.g., including PARP 3, 4, 6–8, and 10–16) and RBPs (e.g., PARP7, 10, and 12–14).27,29 PARPs-mediated ADP-ribosylation (ADPr) plays an essential role in cellular physiological processes in response to stimuli, particularly oxidative stress-induced DNA damage. Sustained PARP activation triggered by intense insults can cause NAD+ depletion and subsequent cell death.30 CD38 consumes NAD+ to make the calcium-releasing second messengers including ADPR (major product), 20-deoxy-ADPR (2dADPR), NAADP and cADPR, contributing to age-related NAD+ decline.31,32 SARM1 is an important NAD+ consumer in neurons. The dimerization of TIR domain cleaves NAD+ into ADP-ribose, cADPR, and nicotinamide.33,34,35
NAD+-consuming enzymes seem to have a different Michaelis constant (Km) value for NAD+. The Km of SIRT1 and SIRT3 ranges from 94 to 888 μM, which renders their activity tightly fluctuating with the dynamic physiological cellular NAD+ levels. Other sirtuins, including SIRT2, SIRT4, SIRT5 and SIRT6, have a Km for NAD+ below the physiological range, implying that NAD+ might not necessarily be the rate-limiting of their activity.5,36,37,38,39,40,41,42,43,44,45,46 PARP-1, accounting for approximately 90% of the NAD+ used by the PARP family, has a lower Km for NAD+ in the range of 20–97 μM.47,48,49 Of note, the CD38 and SARM1 display Km for NAD+ in a markedly low micromolar range (15–25 μM).50 Based on their different Km values, NAD+-consuming enzymes display various potential of reducing NAD+. Under normal homeostatic conditions, CD38 is expressed at low levels, whereas rising expression of CD38 with aging plays a vital role in age-associated NAD+ reduction.51,52 78c, a highly potent and specific CD38 inhibitor, increases NAD+ levels, leading to activation of sirtuins and PARPs.53 Generally, the reported Km of PARP1 and CD38 for NAD+ are lower than those of the sirtuins, suggesting that elevated activation of PARP1 or CD38 may limit endogenous SIRT activation by reducing NAD+ content. This notion is confirmed by the observation that PARP1 and CD38 inhibition effectively increases total NAD+ availability, leading to SIRT1 activation.54
NAD+ methylation
Excess NAM that is not recycled is metabolized through two enzymatic systems and eventually excreted from the body.55 The first system methylates the NAM into MNAM by NNMT, which utilizes the SAM as methyl donor.56 The MNAM together with their oxidized compounds, 4py and 2py, are eventually eliminated in the urine.57 This methylation system is quantitatively by far the predominant NAM scavenging pathway under most conditions. While an acute pharmacological dose of NAM can be converted by CYP2E1 to nicotinamide N-oxide, which is then excreted to the urine.55,58,59 Therefore, NNMT and CYP2E1 divert NAM from recycling to NAD+, restraining NAM accumulation and inhibition of NAD+-dependent signaling.60 The Km of the human NNMT enzyme for NAM (approximately 430 μM) is much higher than the affinity of NAMPT for NAM (<1 μM), suggesting an unsaturated NNMT under normal conditions. Increasing dietary NAM intake can lead to a proportional increase in NAM methylation.61,62 Further, elevated NNMT expression or increased MNAM levels in the liver can stabilize SIRT 1, which in turn promotes glucose and cholesterol metabolism (Fig. 1).63
Subcellular distribution of NAD+
NAD+/NADH
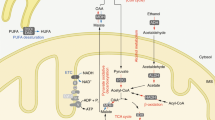
Both the oxidized NAD(P)+ and the reduced NAD(P)H have redox and signaling functions with an uneven subcellular distribution. As listed in Box 1, a portfolio of approaches is developed to map the total quantification or cellular concentrations of the four NAD+ coenzymes. Semisynthetic fluorescent biosensor-based analysis of U2OS cells exhibits around 70 μM for cytoplasmic NAD+, ~110 μM for nuclear NAD+ and ~90 μM for mitochondrial NAD+, respectively. Meanwhile, the concentration of free cytosolic NAD+ detected in cell lines including U2OS, HEK293T, NIH/3T3 and HeLa is ranging from 40 to 70 µM.64,65,66,67 The similar depletion rate of free NAD+ in the cytoplasm and nucleus supports the notion regarding a probable exchange of NAD+ between these compartments, while a slower rate of free NAD+ depletion in mitochondrial suggests that the mitochondrial NAD+ pool is segregated from the cytosolic and nuclear pools.65,68 In agreement with these reports, mounting evidence implies that the distinct fluctuation of NAD+ in mitochondria may be attributed to the membrane impermeability of NAD(H).69,70 Controversially, isotope-tracer method analysis shows that mammalian mitochondria are capable of taking up intact NAD+ as well as its precursors, such as NMN and NR.37,71,72,73 Although NAD+ transporter has been identified in bacteria, yeast and plants, no mammalian homolog has been discovered so far to validate the import of NAD+ into mitochondria.74,75,76,77,78
The NAD+ pool in each cellular compartment can also be maintained independently via recycling the NAD+ from NAM, dependent on various forms of NMNATs, e.g., the nucleic NMNAT1, cytosolic NMNAT2 and mitochondrial NMNAT3.79 Nevertheless, the independent mechanism for maintaining NAD+ through salvaging NMN in mitochondria is challenged by the controversy around the presence or absence of NAMPT and NMNAT3 in mitochondria.80,81 The electrons of NADH, rather than NADH itself, generated from glycolysis in the cytoplasm could be transferred across the mitochondrial membrane through the NADH shuttle systems.66,82 The glycerol-3-phosphate (G-3-P) shuttle and malate-aspartate shuttle transfer the electrons from cytosolic NADH to mitochondrial FADH2 or NADH, respectively. Then, the electrons are finally transferred to the ETC83,84,85,86,87,88,89,90 (Fig. 2).

Subcellular equilibrium of NAD+. The NAD+ homeostasis is maintained by the biosynthesis, consumption and recycling in differentiate subcellular compartments including the cytosol, the nucleus and the mitochondria. NAD+ precursors including Trp, NA, NR, NMN and NAM are metabolized into NAD+ via Preiss-Handler pathway, de novo pathway and salvage pathway, respectively. NAD+ can receive hydride to yield the reduced form NADH in the metabolic processes including glycolysis, FAO, and the TCA cycle. NADH provides an electron pair to drive the mitochondrial OXPHOS for the generation of ATP and the conversion of lactic acid to pyruvate. The cytosolic and mitochondrial NADH is exchanged through the malate-aspartate shuttle and glycerol-3-phosphate shuttle, while the cytosolic and mitochondrial NADPH is exchanged by the isocitrate-a-KG shuttle. NAD+ can also be phosphorylated into NADP+ by NAD+ kinases including nicotinamide nucleotide transhydrogenase (NNT) and NAD kinases (NADKs). Cytosolic NADP+ is reduced into NADPH by G6PD and 6PGD in the pentose phosphate pathway, and by ME1 in the conversion of malate to pyruvate. Mitochondrial NADPH is produced by IDH2, GLUD, NNT and ME3. The NADPH is required for the activation of NOXs and the synthesis of palmitate. Abbreviation: α-KGDH, alpha-ketoglutarate dehydrogenase; GLUD, glutamate dehydrogenase; NNT, nicotinamide nucleotide transhydrogenase; G3PDH, glyceraldehyde 3-phosphate dehydrogenase; 6PGD, 6-phosphogluconate dehydrogenase; G6PD, glucose-6-phosphate dehydrogenase; GPx, glutathione peroxidases; IDH1/2, isocitrate dehydrogenase 1 and 2; MDH, malate dehydrogenase; ME1/3, malic enzyme; NADK, NAD+ kinase; NOXs, NADPH oxidases; OXPHOS, oxidative phosphorylation; PPP, pentose phosphate pathway; PRx, peroxiredoxin; SDH, succinate dehydrogenase; SOD1-3, superoxide dismutase type 1-3; TCA cycle, tricarboxylic acid cycle; GSH, Glutathione; LDH, Lactate dehydrogenase
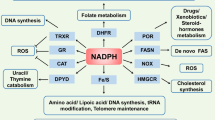
NADP+/NADPH
Approximately 10% of cellular NAD+ may be phosphorylated by NAD+ kinases into NADP+, which can be dephosphorylated to NAD+ by NADP+ phosphatases.91,92 Cytosolic NADPH, the reduced form of NADP+, is mainly generated in the pentose phosphate pathway (PPP) involving G6PD and 6PGD. The mitochondrial NADPH can be produced by ME3 that converts pyruvate to malate and by IDH2 that catalyzes isocitrate to α-ketoglutarate.93,94 Additionally, NADP+ can also receive the electrons from NADH to form NADPH by NNT that locates in the mitochondrial inner membrane.95 These distinct synthesis pathways might contribute to the differential subcellular NADPH/NADP+ ratio, such as a significantly higher ratio in mitochondria (~170) than that of the cytosol and the nucleus (40~50) in U2OS cells.64 NADPH is required for both the reductive biosynthetic reactions of cholesterol and palmitate and the oxidative reactions catalyzed by NADPH oxidases (NOXs), nitric oxide synthases (NOS), cytochrome P-450, and so on.95,96 Most importantly, NADPH provides the primary reducing power for the thioredoxin (Trx) and glutathione (GSH) systems to eliminate ROS (Fig. 2). In line with that, the free NADPH/NADP+ ratio that indicates the reduction potential is normally sustained at a high level inside cells and is significantly reduced following pro-oxidant agents or H2O2 exposure.64,96,97 Given its essential role in metabolism and antioxidant defense, NADPH/NADP+ ratio in cancer cells exhibiting high metabolic rate and ROS contents (50-70) is much higher than that in the embryonic kidney immortalized cell line HEK293 (~20).64 Albeit further research is needed, quantification of NADPH/NADP+ ratios provides an effort to map metabolic and redox state of different cell types and organelles.
NAD+ homeostasis at the systemic level
The NAD+ and its metabolites systemically flux and exchange across tissues, with a tissue-specific distribution of NAD+ biosynthetic enzymes and a tissue-specific preference for specific NAD+ precursors. It is reported that the de novo biosynthesis of NAD+ from tryptophan mainly occur in the liver and, to a lesser extent, kidney, which is attributed to the exclusively expressed enzymes involved in de novo NAD+ synthesis in these tissues. Therefore, the concentration of tryptophan in the diet affects the liver NAD+ levels. Tryptophan also compensates the NAD+ biosynthesis when the salvage pathway is blocked.35,69,91 NAM is the main NAD+ source in both cell lines and most murine tissues. The circulating NAM, 95% of which is released by the liver, is the main NAD+ source for the rest of the body. The NAM uptake preference differs dramatically, ranging from the highest 70 μM in spleen and small intestine to the lowest 2–9 μM in the white adipose and skeletal muscle. Besides tryptophan and NAM, NA is the third NAD+ precursor with concentrations >0.1 mM in mammalian plasma, which can only be used by spleen, small intestine, pancreas, kidney and liver.91 Accordingly, these tissues have been observed with a remarkable expression of NAPRT that guides NA into NAD+ biosynthesis.
Additionally, NMN and NR are capable of efficiently elevating tissue NAD+ concentrations. Given that NMN itself is a non-cell penetrating biosynthetic intermediates, NMN or its metabolites may be actively transported across the membrane. The solute carrier family 12 member 8 (Slc12a8) has been reported as a specific NMN transporter that is responsible for the uptake of NMN and maintenance of NAD+ level in the murine small intestine.98,99 However, it has been reported that the dephosphorylation of NMN into NR by extracellular 5’-nucleotidases is required for the uptake and utilization of NMN in cellular NAD+ synthesis.98 Similarly, the modification of the phosphate group in NMN allows its transportation to activate SARM1 in cells.100 Therefore, whether NMN is directly transported by Slc12a8 remains unclear, which needs further investigation. NR, which can cross the cell membrane through a dypiridamole-inhibitable nucleoside transporter, is preferentially used by muscle to synthesize NAD+.98 Accordingly, the NR-using enzyme, NRK2, is usually specifically expressed in the skeletal muscle.
Beyond the systemic regulation of NAD+ homeostasis across various tissues, it has recently been described that bacteria contribute to mammalian host NAD+ biosynthesis, especially following oral intake of the amidated precursors. The oral NAM and NR can be deamidated by gut microbiota into NA, NAR, NAAD and NAMN in the small intestine and the colon. These deamidated NAD+ metabolites are circulated to the liver and kidney, significantly contributing to the bulk of NAD+ biosynthesis.101
Despite major advances in the acknowledgment of tissue-specific NAD+ homeostasis, further work will be needed to fully characterize the subcellular and systemic modulation of NAD+ metabolism, which can improve the preventive and therapeutic strategies based on maintaining healthy NAD+ homeostasis.
NAD+ metabolism in physiological function
Serving as crucial co-enzymes for redox reactions and co-substrates for NAD+-dependent enzymes, NAD+ and its metabolites function as a regulatory hub controlling a broad range of physiological processes, including redox homeostasis, genomic stability, gene expression, RNA processing, energy metabolism, immunity and inflammation, and circadian clock.
NAD+ metabolism maintains the redox homeostasis
Cells continuously generate oxidants and produce antioxidants. An imbalance between the oxidant formation and the antioxidant capacity in favor of the former causes oxidative stress.102 Maintenance of a physiological (low-level) oxidative stress, also denoted as oxidative eustress, is pivotal for governing biological processes and physiological functions including cell cycle and proliferation, circadian clock, innate immunity, self-renewal of stem cells and neurogenesis.103,104,105,106 However, a variety of stimuli, including nutrient perturbation, genotoxic stress, infection, pollutants and xenobiotics, trigger ROS overproduction, resulting in excessive oxidant challenge (oxidative distress). Oxidative distress causes damage to fundamental macromolecules including proteins, lipids, RNA and DNA at a cellular level, which promotes abnormal cell death and inflammation, often culminating in additional oxidative stress at a systemic level.95,107 Through giving rise to fast, barrier-less and non-selective oxidation reactions that are responsible for a severe insult of both cell and systematic tissues, oxidative stress is involved in a myriad of pathologies. Of note, NAD+ deficiency exerts effects on the emergence of oxidative stress in multiple diseases, while boosting NAD+ has protective effects due to enhancement of antioxidant capacity via increasing the GSH levels and the activity of antioxidant enzymes.108 To counteract the detrimental effects of oxidants, cells can heighten the production of reducing equivalents such as NADPH.109 Moreover, NAD+-consuming enzymes, such as SIRT3, can also manipulate the cellular redox status via regulating the activity of enzymes for ROS generation and antioxidant factors for ROS eradication.110,111,112 Therefore, NAD(P)+/NAD(P)H represents the switching hub that controls prooxidant-antioxidant balance and determines the redox biology (Fig. 3).

NAD+ metabolism controls the redox homeostasis. ROS could be produced from either metabolic reaction in mitochondria, such as OXPHOS, or from a range of cytosolic enzymes, including NOXs, XO, LOX, CYPs, all of which need the NADH/NADPH serving as the electron donor. To maintain the redox homeostasis, both enzymatic and non-enzymatic antioxidant system components exhibit their effects in coordination with each other to contract with the ROS. GSH, the most abundant of non-enzymatic antioxidants, is synthesized from glutamate, cysteine and glycine catalyzed by two consecutive cytosolic enzymes, GCL and GS. Importantly, NADPH serves as the reductive power for ROS-detoxifying enzymes including glutathione reductases (GR) and thioredoxin reductases (TrxR) to maintain the reduced forms of GSH and Trx (SH)2 in response to ROS produced from mitochondria or NOXs. Abbreviations: 6PGD, 6-phosphogluconate dehydrogenase; CYPs, Cytochromes P450; G6PD, glucose-6-phosphate dehydrogenase; GCL; GR, glutathione reductases; GS; LOX; NAD, nicotinamide adenine dinucleotide; NOXs, NADPH oxidases; NADPH, nicotinamide adenine dinucleotide phosphate; OXPHOS, oxidative phosphorylation; PRx, peroxiredoxin; GPx, glutathione peroxidases; SOD1/2, superoxide dismutase 1 and 2; Trx, thioredoxin; TrxR, thioredoxin reductases; XO, xanthine oxidase
NADH/NADPH as the electron donor in ROS generation
Major endogenous ROS via superoxide radicals is constantly produced by both non-enzymatic reactions such as the mitochondrial respiration that needs NADH and enzyme-catalyzed reactions including NOXs that require NADPH.113
In a physiological context, the vast majority of cellular ROS is produced in mitochondria using NADH as electron donor.114,115 Mitochondrial NADH supplies NADH dehydrogenase (complex I) with electrons, which along with the electrons obtained from FADH2 via complex II drive the mitochondrial ETC to generate a proton (H+) gradient across the IMM for the production of ATP. The complex I and complex III of ETC are able to produce the superoxide anion radical (O2–) and release it to both the matrix and the intermembrane space.114,115,116 Additionally, NADH or FADH2 is the electron carrier for the mitochondrial membrane proteins, such as GPDM and FQR, and the metabolic enzymes in matrix, such as OGDH and PDH, all of which contribute to ROS production.115
Another significant intracellular source of ROS is the NOXs, especially in response to physiological stimuli, including growth factors, hormones and cytokines, and pathological impulse, such as bacterial and viral infections.114 Rather than generating ROS as a by-product, NOXs produce superoxide as a primary product using NADPH as the electron donor.117 The NOX proteins, including NOX1-5 and DUOX1/2, have the conserved NADPH-binding site at the C-terminus, which extracts electrons from NADPH. The presence of FAD-binding region and transmembrane hemes enable NOXs to act as an electron-transportation chain that transfers two electrons from cytosolic NADPH to extracellular O2, resulting in the generation of O2–.95,116
Beyond mitochondria and the NOX family, a variety of enzymes including xanthine oxidase (XO), NOS, lipoxygenase and cytochromes P450 (CYP) can all produce ROS using NAD(P)H as electron donor.115 Mammalian xanthine oxidoreductase (XOR), one enzyme in purine catabolism, can exist in both dehydrogenase (XDH) form, which prefers NAD+ as the electron acceptor, and XO form, which transfers the electrons directly to O2, leading to the formation of O2– and H2O2.118,119 Receiving electrons from NADPH, NOS catalyzes the production of NO from L-arginine participating in a number of biological processes, including neurotransmission, vasodilation and immune response.120,121 ROS are also produced via the metabolism of arachidonic acid by lipoxygenases in the presence of NADH or NADPH.122,123,124 Mammalian CYPs are a family of heme-containing NAD(P)H-dependent monooxygenases that metabolize numerous endogenous metabolites, including fatty acids and steroids, and exogenous substrates, including carcinogens, pesticides and drugs, resulting in continuous production of ROS.115,125,126
NADPH as the final reducing power for antioxidant defense
Besides functioning as the electron donor for ROS production, NADPH also supplies the reducing power for antioxidant defense. To fine-tune the redox homeostasis that can either prohibit the damage by oxidative distress or maintain the physiologic ROS to sustain normal cellular processes, organisms have evolved a complex antioxidant defense system consisting of both enzymatic and non-enzymatic scavengers.106,127,128 Intriguingly, both enzymatic and non-enzymatic antioxidant system components exhibit their effects in coordination with each other to contribute to redox homeostasis and cell fate using NADPH as the ultimate donor of reductive power.116,129,130 Two classes of enzymatic components, glutathione reductases (GRs) and thioredoxin reductases (TrxRs), are homologous flavoenzymes that use electrons from NADPH to reduce a disulfide to a dithiol. Then, the active site dithiol in GRs reduces the oxidized GSH (the disulfide GSSG) into reduced GSH, the most important non-enzymatic scavenger. GSH is able to reduce the disulfide bonds and hydroperoxides by glutathione peroxidases (GPxs) or promote the glutathionylation at cysteine residues by Glutathione S‐Transferase (GST) to protect protein from oxidation.107,131,132,133 Similarly, mammalian TrxRs maintain the reduced thioredoxin (Trx) concentration that supports peroxiredoxin (Prx) to remove H2O2.94,134 Therefore, through supplying electrons for bioreductive synthesis and the regeneration of GSH and reduced thioredoxin, NADPH plays critical roles in the maintenance of redox homeostasis and modulating redox signaling.133
NAD+-dependent enzymes control redox homeostasis
SIRT3 acts as an essential modulator of oxidative stress via deacetylation of substrates associated with both ROS generation and detoxification. SIRT3 deacetylates and activates the multiple components of ETC including NDUFA9 of Complex I, SDHA of Complex II and core I subunit of complex III. The alteration of ETC, therefore, might contribute to an increased ROS generation.135,136,137 SIRT3 also enhances cellular antioxidant capacity through augmenting the reducing power, NADPH, and increasing the activity of antioxidant enzymes, such as SOD2 and catalase. SIRT3-mediated deacetylation of IDH2 increases the generation of mitochondrial NADPH, which elevates the reduced GSH levels.138 Simultaneously, SIRT3 can not only induce the expression of antioxidant enzymes by activating the FOXO3a, but also enhance the activation of SOD2 and catalase via NAD+-dependent deacetylation.139,140 Besides SIRT3-mediated deacetylation, SIRT5-dependent desuccinylation improves ROS detoxification via increasing SOD1 activity.141 These findings reveal a new redox regulation of NAD+ by SIRT3‐dependent deacetylation in response to oxidative stress, improving the resistance to the detrimental effects of oxidative damage.
NAD+ sustains genomic stability
The constant challenges from endogenous ROS/RNS or exogenous insults, such as radiation, chemical mutagens and carcinogens, render the DNA damage a relatively common cellular event. Notably, DNA damage and subsequent genome instability are major driving forces for tumorigenesis and aging via driving mutation. To sustain the genome stability, cells have evolved a complicated fine-tuning mechanism, termed as DNA-damage response (DDR), to detect and repair DNA lesions.142,143,144,145 As key regulators of multiple DNA repair pathways, PARPs and sirtuins modulate the post-modification of repair components using NAD+ as co-substrate (Fig. 4). Consistently, NAD+ deficiency leads to an impaired DDR and an increased genomic instability, suggesting an interplay between genomic stability and NAD+ metabolism.145,146,147

NAD+ serves as a pivotal regulator of gene expression. NAD+ and its metabolites are used as substrates and cofactors for reactions that coordinate genomic stability, epigenetic status and RNA processing through NAD+-dependent enzymes. NAD+-dependent histone-deacetylases, especially SIRT1, possess deacetylase activities on multiple transcription coactivators as well as histones, resulting in epigenome remodeling. The lower activity of sirtuins upon lower level of NAD+ may contribute to histone hyperacetylation and aberrant gene transcription. Using NAD+ as a (ADP)-ribose donor, PARPs mediate ADP-ribosylation on itself or on a variety of nuclear target proteins such as topoisomerases, DNA polymerases, histones and DNA ligases, playing roles in genome stability and gene regulation, from chromatin to RNA biology. Recently, it has been found that NAD+ can also serve as a nucleotide analog in DNA ligation and RNA capping in response to stresses. Abbreviations: CTCF, CCCTC-binding factor
DNA ligation
The pathological DSBs are primarily repaired by the NHEJ, a process involves synapsis, end-processing and ligation.148,149 DNA ligases-mediated DNA end ligation is initialized by adenylating the ligase with an AMP moiety. In prokaryotes, both ATP and NAD+ are adenylation donor for DNA ligases, while in eukaryotes, only ATP is known to be used by DNA ligases for the adenylation.150 Recently, it has been reported that human DNA ligase IV, a crucial enzyme in NHEJ, can acquire AMP moiety from NAD+ for DNA end ligation. The BRCA1 C-terminal (BRCT) domain of ligase IV is required to recognize of NAD+ for subsequently ligation in NHEJ.7 Although future studies will be required to fully characterize the structure of NAD+-associated human DNA Ligase IV, these findings reveal that like ATP, NAD+ can serve as a provider of adenylation for DNA ligation in the NHEJ DNA repair pathway.
DNA repair
Beyond regulating the NHEJ pathway acting as an adenylation donor, NAD+ also modulates other DNA repair pathways via activating NAD+-consuming enzymes, PARPs and sirtuins.151,152 As sensitive DNA damage sensors, PAPRs are recruited and immediately activated by DNA breaks. The DNA-bound PARPs, such as PARP1-3, can attach the mono-ADP-ribose (MAR) or poly-ADP-ribose (PAR) moieties directly to the DNA breaks.153,154,155,156 Meanwhile, PARPs also catalyze the ADP-ribosylation of various proteins that facilitate the chromatin relaxation and the recruitment of repair factors.157,158,159,160,161,162 The effect of PARPs to stimulate chromatin decompaction might be exerted via the steric hindrance of PAR chain, the negative charge of DNA and PAR, or the displacement of core histones.162,163 Simultaneously, the accumulated PARs at DNA break sites are required for the recruitment of DNA repairs, including XRCC1, DDB2, ALC1, RECQ1, CHD2, BRCA1, Ligase V, MRE11 and NBS1, to initiate DNA repair.164,165,166,167 Similarly, DNA damage induces the relocalization of NAD+-dependent deacetylase SIRT1 to DNA breaks, which promotes DNA repair via opening the chromatin and recruiting the main DNA repair factors including KU70, NBS1, WRN, KAP1, XPA and APEX1.168,169,170,171,172,173,174,175,176,177 Additionally, PARPs and sirtuins also simulate genomic damage-signaling kinases, including ATM, p53, DNA-PK, CIRBP and FOXOs, to accelerate DNA repair.178,179,180,181,182
Given that DNA damage-activated PARPs account for up to 90% of cellular NAD+ consumption, the DNA repair activity is highly dependent on the cellular NAD+ concentration.133,183,184 As expected, decreased NAD+ levels induce the accumulation of DNA damage, whereas replenishing intracellular NAD+ stimulates the DNA repair.185,186,187 In contrast to the positive effect of NAD+ on DNA repair, NADP+ suppresses the ADP-ribosylation-mediated DNA damage repair via functioning as an endogenous inhibitor of PARPs. The structure of NADP+ similar to that of NAD+ renders its binding to PARPs. However, NADP+ has an additional phosphate group on the 2’ position of the ribose ring, which abolishes the formation of linear PAR chain.184
NAD+ manipulates the gene expression
Cellular metabolism, such as NAD+ metabolism, is directly connected to gene expression through regulating the post-translational modifications (PTMs) of histones, the covalent chemical modifications of DNA, the activity of transcription factor and RNA processing.188,189
Histone modification
Histone modification is one of the most crucial epigenetic modification that affects DNA structure and gene expression. The post-translational modifications of histones include acetylation, ADP-ribosylation, phosphorylation and methylation. Among these modifications, the acetylation and ADP-ribosylation are regulated by NAD+-dependent enzymes, sirtuins and PARPs, respectively. Sirtuins, also known as NAD+-dependent class III HDACs, remove the acetyl groups from histone, which restores the electrostatic affinity between DNA and histones to stabilize the chromatin architecture.190,191 SIRT1-3 maintain the chromatin structure via deacetylation of a crucial histone residue, H4K16. The reduced intracellular NAD+ concentration limits the deacetylase activity of SIRT1, resulting in elevated H4K16Ac and gene expression.192,193 SIRT6 can coordinate NF-κB to deacetylate the H3K9Ac, sequestering the expression of glucocorticoid receptors (GRs).194 SIRT7 is able to selectively deacetylate the H3K18Ac, which represses the expression of a specific set of gene targets that is linked to oncogenic transformation.195 Histones also serve as acceptors of ADP-ribose upon DNA damage to initiate DNA repair.183 The ADP-ribosylation of histones by PARP-1 induces the dissociation of nucleosomes, leading to the decompaction of chromatin. Furthermore, PARP-1-mediated PARylation of KDM5B prevents the demethylation of H3K4me3, rendering the exclusion of H1 and the opening of chromatin. The decompensation of chromatin structure, therefore, allows the loading of the transcriptional machinery and facilitates gene transcription.
DNA modification
DNA methylation is another widely studied epigenetic modification that is often involved in the regulation of gene expression. NAD+ deficiency can promote the DNA methylation, resulting in gene silencing. NAD+ depletion elevates the methylation of BDNF promoter, triggering the dissociation of the DNA methylation-sensitive nuclear factor CCCTC-binding factor (CTCF) and cohesin from the BDNF locus. The detachment of these factors causes the altered methylation and acetylation of histone at this locus, leading to chromatin compaction and gene silencing.196 The NAD+-consuming enzymes, PARPs, are associated with the regulation of DNA modification. Inhibition of the PARPs-mediated ADP-ribosylation causes a chromatin compaction DNA hypermethylation.197 PARP-1 can be activated by the chromatin insulator CTCF even without niched DNA and efficiently automodified, dependent on NAD+. The PARs of PARP‐1 compete with DNA for the noncovalently binding DNMT1, causing suppression of its methyltransferase activity.198,199 Therefore, the NAD+-dependent enzymatic activity of PARP-1 is a crucial regulator of gene expression via protecting the genome from aberrant hypermethylation.
Another evidence linking the NAD+ metabolism with DNA methylation is the NNMT, which transfers the methyl group from SAM to NAM, producing S-adenosylhomocysteine (SAH) and a stable metabolites 1-methylnicotinamide (MNA). Given that the SAM is the universal methyl donor for methylation of substrates including proteins, nucleotide acids and lipids, the NNMT induced methyl sink in the form of MNA impairs the genome methylation.200,201 Moreover, the methionine metabolism catalyzed by NNMT diverts the NAM from the NAD+ salvage pathway. As a consequence, the reduced cellular NAD+ content restricts the PARP1-catalyzed ADP-ribosylation and the following DNA methylation. Collectively, increased NNMT expression inhibits the DNA methylation through not only decreasing the cellular NAD+, the donor for ADP-ribosylation required for methylation, but also reducing the level of SAM, the methyl donor for methylation.
NAD+ modulates RNA processing
Decorating of RNA as a nucleotide analog
Chemical modifications of the RNA 5’-end play a pivotal role in various biological functions including the protection of RNA from exonuclease cleavage, the recruitment of proteins for pre-mRNA processing and nuclear export and the initiation of protein synthesis. Recently, NAD+ has been found to be incorporated into RNAs as an initiating nucleotide during transcription to form NAD+ cap in different organisms including bacterial, yeast, human and virus.8,9,10,11,12,202 Unlike the well-characterized m7G-capped mRNA, which maintains highly stability of mRNA for translation, the NAD+-capped RNAs are vulnerable to decay and are inefficiently translated in cells.9,10,203
NAD+ capping can be catalyzed by eukaryotic nuclear RNAP II using NAD+ and NADH as non-canonical initiating nucleotides (NCINs) in de novo transcription initiation.8 Besides the RNAs produced by nuclear RNAP II, RNAs synthesized by mitochondrial RNAP (mtRNAP) can also be NAD+ capped. The human mtRNAP conducts a higher efficiency of NAD+ capping than the nuclear RNAP II, leading to ~15% NAD+ capping of mitochondrial transcripts.204 The 5′ end NAD+ cap of RNAs in the cytoplasm will be removed by two mammalian hydrolases, DXO and Nudt12. Reported as a deNADding enzymes, the Nudt12 contributes to the decapping of RNA following exposure to nutrient stress, such as glucose deprivation, while DXO is responsible for the environmental stress, such as heat shock.12,205,206,207 In line with that NAD+-capped RNA levels respond to the cellular total NAD+ concentrations, the capping efficiencies of NAD+ capping and NADH capping are also regulated by intracellular NAD+/NADH ratio.204,207 These results raise the possibility that RNAP II and mtRNAPs might function as both sensors and executors, which sense NAD+/NADH ratios and induce the NAD+ capping to regulate the gene expression, leading to the crosstalk between cellular NAD+ metabolism and transcriptional activity.
ADP-ribosylation of RNA
Beyond decorating RNA as a nucleotide analog, NAD+ also provides the ADPR groups for the reversible mono-ADP-ribosylation of RNA phosphorylated ends. This RNA modification is catalyzed by PARP10 with a preference for 5′ ends, depending on NAD+ concentration. In addition to PARP10, TRPT1, a PARP homolog, also catalyzes the ADP-ribosylation of RNA. The ADP-ribosylation renders RNA resistant to phosphatase, which might protect the RNA from the nuclease attack. Similar to the reversible ADP-ribosylation of proteins and DNA, the ADP-ribosylation of RNA can also be efficiently reversed by several cellular hydrolases including TARG1, MACROD1-2, PARG, NUDT16 and ARH3 viruses.29 Besides human hydrolases, macrodomain-containing hydrolases from VEEV and SARS can remove the ADP-ribosylation of RNA catalyzed by PARPs, suggesting a potential mechanism of pathogenesis via inhibiting the antiviral activity of IFN-stimulated genes, PARPs. Altogether, the ADP-ribose moieties attached to RNA end might protect RNA against degradation or serve as a platform for recruiting proteins, controlling the functional state of RNA.
NAD+ facilitates cellular energy metabolism
NAD+/NADH as hydride-donating coenzyme for metabolism
Acting as a coenzyme, NAD+ plays pivotal roles in energy metabolism pathways including glycolysis, the TCA cycle, OXPHOS, FAO and alcohol (ethanol) metabolism.66 The glycolysis process begins with one glucose molecule and ends with two molecules of pyruvate, which are subsequently transported into the mitochondria to begin the TCA cycle. NAD+ promotes glycolysis by facilitating the enzymatic reactions catalyzed by GAPDH and lactate dehydrogenase (LDH), which use NAD+ as a coenzyme.208,209 NAD+ is reduced to NADH coupled with the oxidation of G3P to 1,3-BP by GAPDH.210 Cytosolic pyruvate can also be converted to lactate by LDH, coupled with the oxidation of NADH to NAD+.211 This process helps maintain the cytosolic level of NAD+, thus contributing to the continuity of glycolysis. When transported into the mitochondria, the glycolytic end-product pyruvate is decarboxylated to produce acetyl-CoA by PDH complex, which reduces NAD+ to NADH simultaneously.210 Acetyl-CoA then starts the TCA cycle, where NAD+ serves as a coenzyme for three rate-limiting enzymes, α-ketoglutarate dehydrogenase (KGDH), isocitrate dehydrogenase 3 (IDH3) and malate dehydrogenase (MDH2), to generate NADH. Thus, the TCA cycle can convert four molecules of NAD+ to NADH using one molecule of pyruvate in the mitochondria under aerobic conditions.212 As an electron donor, NADH produced in the TCA cycle plays a crucial role in ATP synthesis by OXPHOS, which generates most of the energy through the H+ gradient in animal cells.213
FAO breaks down a long-chain acyl-CoA, which is generated from fatty acid and coenzyme A by acyl-CoA synthetase in the cytosol, to generate acetyl-coA, NADH and FADH2 in the mitochondria.214 This process is performed in repeated cycles, each of which removes a two-carbon acetyl-coA from the acyl-CoA via four enzymes, the enoyl-CoA hydratase, ketoacyl-CoA thiolase, acyl-CoA dehydrogenase (ACADs) and hydroxyacyl-CoA dehydrogenase (HADH). The last cycle generates two molecules of acetyl-coA. FADH2 is generated by ACADs, while NADH is produced from NAD+ in the reaction catalyzed by HADH. Both NADH and FADH2 generated in the FAO are utilized to synthesize ATP by the ETC. NAD+ is also a cofactor in alcohol oxidation metabolism taking place mainly in liver cells. Alcohol oxidation is completed in a two-step reaction by two enzymes, alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH), which catalyzes the reduction of NAD+ to NADH.215 Both the sufficient glycolysis and the effective oxidation of alcohol require fast reoxidation of NADH to NAD+ through the coordinated reduction of pyruvate to lactate by LDH or production of ATP by mitochondrial ETC.213,216
NAD+-dependent modification of metabolic enzymes
Beyond serving as a hydride-donating coenzyme for metabolism, NADH/NAD+ also acts as co-substrate for the sirtuins-mediated post-translational modification of metabolic enzymes including acetylation, ADP-ribosylation, succinylation and malonylation. A large number of enzymes that participated in cytosolic glycolysis, gluconeogenesis, the urea cycle, nitrogen metabolism, glycogen metabolism, mitochondrial fatty acid oxidation, the TCA cycle and amino acid catabolism can be regulated by sirtuins.217,218
The mitochondrial sirtuin-related acetylome covers almost all the mitochondrial metabolism, including the enriched SIRT3-related TCA cycle, ETC, FAO, the SIRT4-associated anion transporters, the translation and energy metabolism, the SIRT5-regulated TCA cycle and branched chain amino acid catabolism (BCAA) metabolism.219,220 The mitochondria SIRT3 function as a metabolic sensor that links the cellular energy status with the mitochondrial protein acetylation patterns. In healthy mitochondria, SIRT3 interacts with ATP5O, while the low pH owing to the loss of membrane potential weakens the binding affinity between SIRT3 and ATP5O, leading to the redistribution of SIRT3 to other mitochondria substrates. The pH-insensitive association between SIRT3 and ATP5O provides a fundamental role for SIRT3 in resetting mitochondrial acetylation in response to stress.219 The transition to fasting enhances both the cellular NAD+ level and the SIRT3 expression, which, in turn, catalyzes the deacetylation of LCAD to promote fatty-acid oxidation.221 SIRT3 orchestrates the metabolism reprogramming via controlling the balance between cytosolic glycolytic metabolism and mitochondrial oxidative metabolism.222 SIRT3 also plays a regulatory role in proline metabolism via deacetylation of PYCR1.223
SIRT4 modulates mitochondria energy homeostasis and longevity based on its lysine deacetylase, lysine deacylase, lipoamidase and ribosylase activity. Under nutrient-replete conditions, the deacetylation of malonyl CoA decarboxylase (MCD) by SIRT4 plays a pivotal role in lipid homeostasis via suppressing fatty acid oxidation and inducing lipid anabolism.224 The lysine deacylase activity of SIRT4 is involved in the control of leucine metabolism and insulin secretion through regulating the acylation status of enzymes in these pathways.225 SIRT4 also acts as a cellular lipoamidase with a preferred catalytic efficiency for lipoyl- and biotinyl-lysine modifications to its deacetylation activity. SIRT4 hydrolyzes the lipoamide cofactors from the E2 component dihydrolipoyllysine acetyltransferase (DLAT), leading to diminished PDH activity.226 Furthermore, SIRT4 uses NAD+ to ADP-ribosylate and reduce GDH activity, thereby inhibits insulin secretion in response to amino acids in β cells.227
SIRT5 is an NAD+-dependent lysine desuccinylase, demalonylase, and deglutarylase.228 BAT specific deletion of Sirt5 exhibits hypersuccinylation of proteins involved in the amino acid metabolism, ETC and FAO. A bunch of mitochondrial proteins have succinylation modification, such as UCP1 in thermogenic function, GLUD1 in glutamate metabolism, SDH in ETC, the TCA cycle, GLS2 and CPS1 in glutaminolysis, ECHA and VLCAD in FAO, HMGCS2 in ketogenesis and SHMT2 in serine catabolism.229,230,231,232,233 SIRT5-mediated desuccinylation also participates in protection against peroxisome-induced oxidative stress via targeting ACOX1.230 Moreover, SIRT5 functions as a leading regulator of protein malonylation in both cytosolic metabolisms including glycolysis, gluconeogenesis and mitochondria FAO. For instance, SIRT5 increases the activity of GAPDH by demalonylation, thereby controlling the energetic flux through glycolysis.227,234,235 Collectively, sirtuins orchestrate an integrated modulation of metabolic pathways via NAD+-dependent post-translational regulation in response to diverse nutrient signals.
Rhythmic NAD+ oscillates circadian clock
Organisms have developed internal clocks as a timekeeping mechanism to collaborate biological processes with the exogenous environmental and endogenous factors. NAD+ functions as a metabolic driver of circadian transcription via epigenetic mechanisms, transducing signals originated by environmental stimuli to the circadian clock. The linkage of NAD+ metabolism to the internal clocks is firstly evidenced by that the NAD(P)+/NAD(P)H ratio modulates the DNA-binding activity of the core oscillators, such as CLOCK: BMAL1 and NPAS2: BMAL1 heterodimers. The redox state of FAD and NADPH also displays an oscillation pattern in organotypic slices of suprachiasmatic nucleus (SCN).236 The circadian control of intracellular NAD+ levels by the clock is attributed to the oscillation expression of NAMPT, a rate-limiting enzyme in the salvage of NAD+ with a 24-hour rhythm.36,38,237,238,239 The E-boxes in the promoter of Nampt gene allow the direct transcriptional control by the CLOCK: BMAL1 chromatin complex.240 Furthermore, the expression of enzymes in the NAD+ salvage pathway, including Nmrk1, Nampt, and Nadk, has circadian oscillation patterns in WT and Liver-RE mice that exclusively express BMAL1 in the liver, suggesting the circadian clock might reprogram NAD+ salvage synthesis to maintain the fluctuation of NAD+.241
The oscillation of NAD+, in turn, coordinates the transcription and behavior through the circadian clock. The reduction of NAD+ in old mice dampens the circadian transcription, which can be rescued by NAD+ repletion to youthful levels with NR.242 The regulatory effect of NAD+ on circadian reprogramming is mediated by changing the activity of sirtuins and PARPs, which determines the transcriptional activity of core oscillators. SIRT1/6 can be recruited into the core clock CLOCK: BMAL1 complex, which renders the rhythmic acetylation of BMAL1 and the cyclic H3K9/14Ac at circadian promoters on their target genes.38,238,243 Besides, the oscillation activation of SIRT1 also regulates the circadian dynamics via deacetylation of the core clock repressor PER2K680 and mixed-lineage leukemia 1 (MLL1), thereby controlling rhythmic chromatin property and the activity of BMAL1: CLOCK complex.36,38,238,242,244 Similar to sirtuins, the activity of PARPs is also regulated by the circadian clock. The oscillation activation of PARP-1 interacts with and poly(ADP-ribosyl)ates CLOCK, leading to suppressed binding of CLOCK: BMAL1 to DNA and altered circadian gene expression.245 Moreover, PARP1 interacts with CTCF in a circadian manner, regulating lamina-associated chromatin and circadian oscillations in transcription.246,247 These reports indicate a connection between NAD+-dependent epigenetic modification and the core circadian clockwork circuitry.
The interplay of NAD+/NADP+ metabolism with circadian clock is further evidenced by the oscillating redox, in which ROS levels display a different liver pattern compared to other tissues due to the unique NAD+ oscillation in response to the autonomous hepatic clock. Circadian disruption in beta-Bmal1(-/-) mice and arrhythmic ClockΔ19 mice decrease the Nrf2 expression and subsequently impair the antioxidant defense system, contributing to increased ROS accumulation, oxidative damage and mitochondrial uncoupling.248,249 Prxs, the most critical H2O2-removing enzymes, exhibit rhythmic cycles of oxidation.250 The circadian clock system can also regulate the production and consumption of GSH through circadian regulation of the rate-limiting enzymes in GSH biosynthesis and cellular detoxification.236 The oxidation cycle of both Prxs and GSH is directly influenced by the availability of redox cofactor NADPH, suggesting that NADPH metabolism might play a vital role in controlling redox rhythmic and transcriptional oscillations. In line with this notion, it has been demonstrated that inhibition of NADPH production from PPP alters circadian rhythms through changing the activity of CLOCK: BMAL1.251,252,253 Thus, NAD(P)+/NAD(P)H acts as an important modulator of cellular energetic status, enabling the reset of redox rhythmic and transcriptional oscillations based on metabolic signals.254
NAD+ metabolism programs immunity and inflammation
NAD+, along with citrate and succinate, is a novel class of metabolites with inflammatory signaling capacity, linking the NAD+ metabolism to the programming of immune responses.255 Restoring the NAD+ levels via de novo biosynthesis in the liver prevents hepatic lipid accumulation and attenuates inflammation in mice on a high-fat diet (HFD).15 Similarly, increased generation of NAD+ via the KP in resting, aged or immune-challenged macrophages restores OXPHOS and homeostatic immune responses, whereas inhibition of de novo NAD+ synthesis induces an increased inflammation-associated TCA-cycle metabolite succinate and elevated mitochondria-generated ROS, resulting in rising innate immune dysfunction in aging and age-associated diseases.256 Mitochondrial complex III produces ROS immediately after stimulation, which has an essential role in inflammatory macrophage activation. However, the mitochondrial ROS are also responsible for DNA damage, which causes the abundant consumption of NAD+ by PARPs. The NAD+ abundance as well as the NAD+/NADH ratio, therefore, decline significantly even with the induction of the de novo synthesis from the KP in response to the lipopolysaccharide (LPS) challenge.256,257 To maintain the cellular NAD+ level, NAD+ salvage enzyme NAMPT has been activated by LPS to boost the salvage pathway.258 Elevated expression of NAMPT maintains the NAD+ content to drive the glycolysis, which supports the activation of inflammatory macrophages.258 While in the mitochondrial respiration-impaired cells, NAD+ could reduce the exacerbated inflammatory response via improving lysosomal function. The addition of nicotinamide precursor NAM in mitochondrial respiration-impaired cells restores the lysosomal function and limits the increased proinflammatory profile.259 Furthermore, endotoxin dose-dependent switch of NAD+ biosynthesis pathways from NAMPT-dependent salvage to IDO1-dependent de novo biosynthesis maintains the nuclear NAD+ pool, which promotes SIRT1-directed epigenetic regulation of immune tolerance.260,261
Owing to its rate-limiting enzymatic activity in NAD+ salvage pathway, the elevated expression of NAMPT in the innate immune cells, including macrophages and dendritic cells, further proposed a link between intracellular NAD+ levels and inflammation.262,263,264 Specific competitive inhibitor of NAMPT could ameliorate the immunity or inflammatory disorders, including DSS-induced colitis, arthritis via reducing intracellular NAD+ levels in inflammatory cells and circulating inflammatory cytokines, including IL-1beta, TNF-alpha, and IL-6.265,266,267 Cellular levels of NAD+ regulated by NAMPT also impacts NAD+-dependent enzymes, such as sirtuins. For example, sirtuins modulated the optimal TNF translation.268 The elevated NAD+ levels concomitant with SIRT1 switches the NF-κB-dependent transcription into the RelB-dependent transcription of the TNF-α in endotoxin tolerant sepsis blood leukocytes.269 Additionally, SIRT6 can modulate TNF production by regulating the TNF mRNA translational efficiency.269 In a pancreatic cell line, SIRT6 induces the production of cytokines including IL‐8 and TNF, which promote cell migration.26 Sirtuins control immune responses via directly regulating inflammatory transcription factors, including deacetylation of FOXP3 to repress Treg cell responses, deacetylation of RORγt to promoteTH17 cell responses, and suppression of NF-κB to reduce inflammatory responses.188
Besides NAD+, NADPH also plays essential roles in immunity and inflammation, mainly dependent on the NADPH oxidases and redox signaling.270 In an inflammatory response, activation of epithelial and immune cells triggers NOXs to generate ROS, which can directly kill microorganisms.271,272,273 NOXs-derived ROS can also act as a second messenger in signaling transduction. It has been reported that NOX4 directly interacts with TLR4, which is pivotal for LPS-mediated NF-κB activation.274 In the nasal airway epithelium, the interaction of TLR5 and another NOX isozyme, Duox2, induces the ROS generation and IL-8 expression in response to flagellin exposure.275,276 The phagocytic NADPH oxidase complex can also be activated by Rubicon to induce a ROS burst, inflammatory cytokine production and potent antimicrobial activities.277
Abnormal NAD+ metabolism in the pathophysiological condition
Given the essential regulatory role of NAD+ in fundamental physiologies, NAD+ metabolic abnormalities contribute to the pathophysiology of various diseases, such as infection, cancers, metabolic diseases, aging and age-associated neurodegeneration disorders.
Perturbed NAD+ metabolism in response to infection
Microbial infection, including viruses and bacteria, causes an imbalance in the cellular redox environment, thus inducing different responses, e.g., antioxidant defenses, cell signaling, immune response and other processes. NAD+ or NADPH level determines the role of ROS in infections, either protecting against invading microorganisms or causing tissue damage during the resulting excessive inflammation (Fig. 5).

Physiological actions of NAD+ in the host response to infection. Microbial infection, including viruses and bacteria, causes oxidative stress that has a critical effect on both the microbe and host cells. The production of ROS from NOXs depending on NADPH termed respiratory burst is a powerful antimicrobial weapon and a major component of the innate immune defense against bacterial and fungal infections. Meanwhile, oxidative stress causes the host DNA damage that enhances the consumption of NAD+ by elevated PARPs. The intracellular NAD+ can also be reduced by activation of CD38 that is required for the inflammation against infection. The NAD+ deficiency therefore might not be able to support the clearance of microbial by autophagy or phagolysosome, the innate immune and inflammation response. Abbreviations: EBV, Epstein-Barr virus; HCV, hepatitis C virus; HRV, human rhinovirus; HRSV, human orthopneumovirus; iNOS, inducible nitric oxide synthase; ISGs, interferon-stimulated genes; IV, Influenza virus; KSHV, Kaposi’s sarcoma-associated herpesvirus; MPO, myeloperoxidase; Mtb, Mycobacterium tuberculosis; NOXs, NADPH oxidases
NAD+ mitigates viral infection-induced oxidative damage
Oxidative stress is implicated as a pathogenic factor in viral infection.278 It can be caused by diverse virus families ranging from DNA (i.e., HBV, EBV, HSV-1) to RNA viruses (i.e., HCV, RSV, DENV, Influenza, ZIKA, HIV).279,280,281,282,283 The increased cellular ROS by viral infection cause DNA damage, gene mutation, cell death, viral DNA integration and tumorigenesis.284,285,286,287,288,289,290 For instance, acute phase of HCV infection induces oxidative stress via enhancing NOXs expression and activity to generate ROS generation and decreasing GSH, which supports the high rates of viral replication and apoptotic cell death. On the other hand, the chronic infection maintains a reduced environment to establish viral persistence.291 Moreover, NOX-induced ROS play various roles in the mechanisms of oncogenesis by HCV, including genome instability, epigenetic regulation, inflammation and modulation of cell growth and death.292 In RSV-infected airway epithelial cells, NOX-generated ROS trigger the activation of the transcription factors IRF and STAT, thereby inducing the expression of chemokines and cytokines involved in the immune/inflammatory responses of the lung.293 NQO1, an enzyme involved in the elimination of ROS, is inhibited by HBx, leading to decreased GSH levels and increased susceptibility of hepatoma cells to oxidative damage, cumulating in HBV-associated pathogenesis of chronic liver diseases.294
To repair the oxidative stress-induced DNA damage, a large amount of NAD+ were consumed by elevated PARPs in response to virus infection, i.e., HSV-1, ZIKV and New Sindbis virus (SV).281,282,295 Beyond the important role in DNA repair, PARP-1 also acts as a modulator of NF-κB, inducing the downstream CCL2-CCR2 signaling, which is required for the recruitment of NK cell to the infection site and viral control.296 Therefore, PARPs have antiviral activity against multiple classes of viruses, including retroviruses, alphaviruses, filoviruses, herpesviruses, adenoviruses and coronavirus through enhancing the innate immunity.297,298,299,300 Sirtuins are another class of NAD+-consuming enzyme, which have broad-range antiviral properties on diverse viruses including HCV, HIV-1, HCMV and influenza A (H1N1) virus.301,302,303,304,305 Besides controlling the virus replication, PARPs or sirtuins also contributes to the oncogenic virus infection, such as oncogenic gamma herpesviruses KSHV and Epstein Barr virus (EBV) infection, and the tumorigenesis through the epigenetic remodeling.306,307,308 CD38 is the third NAD+-consuming enzyme that is upregulated in response to a number of viral infections.309,310,311,312 CD38 is under the transcriptional control of RSV‐induced IFNs. The CD38‐generated cADPR, in turn, augments the IFNs-induced ISGs and NF-κB-mediated inflammation, leading to the antiviral hyperinflammation response.311 In addition to the excessively increased consumption of NAD+, multiple viruses cause a decline in NAD+ concentration through reducing the protein levels of crucial enzymes in the NAD+ biosynthesis pathway, including QPRT and NAMPT.304,313 Regarding the redox role of NADH/NADPH in eliminating ROS, the depletion of NAD+/NADP+ pool exacerbates the oxidative damage during virus infection.306,308,314,315,316
NAD+ contributes to the bactericidal activity
Bacterial infection induces rapid production of intracellular ROS either by NOXs or mitochondria that are, in turn, crucially required by macrophages to clear bacteria.105,317 Elimination of the ROS results in defective bactericidal activity, allowing bacteria to survive and repeatedly colonize various tissue sites.318,319 NOXs in immune cells, such as macrophages and neutrophils, are primarily responsible for ROS production and termed respiratory bursts during phagocytic bacterial killing.320,321 Additionally, NOX2-generated ROS are necessary for LC3 recruitment to phagosomes, revealing an autophagy-dependent antibacterial activity of NOX2 in phagocytes.322 Mycobacterium tuberculosis (Mtb) can trigger the production of ROS via depletion of NAD+. TNT, a major cytotoxicity factor of Mtb, hydrolyzes the cellular NAD+ to NAM and ADPR, thereby activating the necroptosis effectors MLKL and RIPK3. Moreover, the NAD+ depletion or the NAD+ hydrolysis products induced signaling contributes to the TNT-triggered ROS production.323
Moreover, the NOX-dependent oxidative burst caused by phagocytosis of bacterial cells activates CD38, producing NAADP in the maturing phagosome. NAADP induces the lysosomal Ca2+ efflux and calcineurin-mediated TFEB activation, which enhances the expression of pro-inflammatory cytokines including IL-1β, IL-1α and IL-6.324 CD38 also exerts bactericidal activity in an NAD+-dependent manner.325,326 CD38 controls neutrophil chemotaxis to bacterial chemoattractants via producing cyclic ADP-ribose.326 In macrophage, high levels of CD38 induced by LXR agonists reduce NAD+ levels and interfere with cytoskeletal rearrangements triggered by invasive bacteria, protecting host macrophages from substantial infection.327 However, in T-cell, the NAD+ depletion by elevated CD38 expression increases the acetylation of EZH2 by in a SIRT1-dependent manner, leading to reduced cytotoxicity of CD8 T cell and enhanced inclination to infections in patients with systemic lupus erythematosus (SLE).328 Additionally, the NAD+ concentration and NAD+/NADH ratio are significantly elevated in response to Group A streptococcus (GAS)-infection. The addition of NAM remarkably enhances the intracellular NAD+ content that promotes the autophagosomal acidification and clearance of GAS in endothelial cells.323 Therefore, NAD(P)+/NAD(P)H exerts the bactericidal activity by promoting the ROS generation, the pro-inflammatory response and the anti-infection autophagy.
NAD+ deficiency accelerates aging
Multiply evidence elucidates that NAD+ and NAD+-related metabolites govern biological functions in aging, including metabolism, redox homeostasis, mitochondria function and the circadian clock. The NAD+ decline during normal aging results in oxidative damage, metabolic disorder, circadian rhythm abnormalities, and mitochondrial dysfunction through regulating signaling pathways, such as p53, NF-κB, PGC-1α and HIF-1α, by sirtuins and PARPs.329,330,331,332 Accordingly, boosting NAD+ provides a therapeutic option for improving the health lifespan and treating aging-related diseases.
NAD+ deficiency accelerates aging
NAD+ levels display steadily reduction in old worms, which causes a further shorter lifespan.333 Similarly, mice and rats exhibit an NAD+ decline during aging in a variety of tissues, such as muscle, adipose tissue, brain, skin, liver and pancreas.334 The reduced NAD+ is also observed in the aged human brain and liver.335 In line with that, the plasma levels of NAD+ and its metabolites, NADP+ and NAAD also remarkably decline during aging.336
The age-dependent decline of NAD+ might be due to either enhanced consumption or reduced biosynthesis. The NAD+ levels and NAMPT expression are severely inhibited in various tissues including liver, skeletal muscle, WAT and pancreas in age induced T2D models. The decreased NAMPT might be due to the chronic inflammation and impaired circadian clock during aging.337,338 However, another study describes no alteration of the NAMPT mRNA or protein levels in aged mice and human tissues.52 Thus, these controversial findings of NAMPT-catalyzed NAD+ biosynthesis overage might come from the differential cell type and tissue context, which will be elucidated in future studies. Another explanation for NAD+ decline with age is the increased NAD+ consumption by PARP or CD38. In contrast to the unchanged levels of PARP1, both the protein levels and enzymatic activity of CD38 are enhanced during aging, contributing to the age-related NAD+ decline in mammals. CD38 is also responsible for the mitochondrial dysfunction by regulation of SIRT3 activity.52 Nevertheless, CD38-deficient old mice preserve the NAD+ levels, mitochondrial respiration and metabolic functions.339 CD38 expression might be induced by chronic inflammation, one characteristic during aging.52,340
The NAD+ decline is a primary driver for the progressive of biological dysfunction and age-related pathologies. Thus, the genetic or pharmacological modulation of NAD+ provides a therapeutic option for multiple age-related diseases. Indeed, genetic and pharmacological replenishment of NAD+ improves the age-related biologic function and increases lifespan at least in worms and mice.341,342 The increased expression of NAMPT in aging human SMCs prolonged lifespan via delaying senescence and enhancing resistance to oxidative stress.343 Supplementation with NAD+ precursors, NR and NMN, elevated NAD+ levels that can maintain the mitochondrial and metabolic functions by activating SIRT1 in mice, leading to an extensive lifespan of mice.337,344,345 Augmentation of NAD+ by β-lap, a potent substrate of NQO1, effectively prevents ARHL and its accompanying harmful effects by preventing oxidative stress and inflammation and improving mitochondrial function in rodents.346 Moreover, mounting evidence has shown that NAD+-dependent sirtuins can extend the lifespan of yeast, worms, flies and mice and alleviate many diseases of aging-related pathologies. For instance, both brain-specific or whole-body SIRT1-overexpressing transgenic mice exhibit a slowed aging and a prolonged lifespan.347,348
Aging-related NAD+ decline causes mitochondrial dysfunction
Combining the evidence that mitochondrial dysfunction is a hallmark of aging and NAD+ plays a crucial role in the maintenance of mitochondrial function.341,345, we can hypothesize that aging-related NAD+ decline might be the cause of mitochondria dysfunction. NAD+ boosters play a preventive role in aging via the early-phase activated UPRmt, and the late-phase induced antioxidant defense. Regulation of NAD+ availability by PARP inhibitors and NAD+ precursor modulates mitochondrial function through sir-2.1 in worm to extend lifespan.341 The PARylation is also markedly increased in muscle and the liver of aged mice in parallel with robustly decline of NAD+ levels. Since CSB can limit the activity of PARP1 via displacing it from damaged DNA, in CSB-deficient cells and mice, the PAPRPs-mediated PARylation is increased and accounts for the majority of cellular NAD+ consumption. This aberrant activation of PARPs represses the SIRT1 activity and mitochondrial dysfunction, which can be rescued by both PARP inhibitor and NAD+ precursors.349 Aging-related nuclear NAD+ decline inhibits the mitochondrially encoded genes via the SIRT1-HIF-1α-c-Myc pathway, while boosting NAD+ levels rescues the mitochondrial function in old mice in a SIRT1-dependent manner.345 NAD+ also affects the acetylation and activity of oxidative enzymes in mitochondria via altering SIRT3 activity. The circadian activity of SIRT3 induced by NAD+ oscillation regulates the rhythmic acetylation and activation of oxidative enzymes and respiration in isolated mitochondria.350
NAD+ ameliorates the oxidative damage during aging
There is a growing awareness that oxidative damage is an essential driver of age-related deterioration in cell function.351,352 The DNA oxidative damage and protein oxidation in the aged human brain are associated with declined antioxidant enzyme activities.353,354 Age-related increase in oxidative stress and cell senescence leads cells/tissues to be more prone to undergo necroptosis, thereby releasing DAMPs that trigger the chronic inflammation observed with aging.355 The pro-inflammatory cytokines, in turn, augment both mitochondrial and NOX-generated ROS, contributing to further accumulation of oxidative damage (Fig. 6).356,357,358,359

NAD+ deficits in aging-associated dysfunction and cancer. Environmental stimuli, including nutrient perturbation, infection, radiation and inflammation, induce oxidative stress, which causes the damage of cellular biomolecules and organelles. NAD+ and its metabolites function as crucial regulators to maintain cellular redox homeostasis through replenishing the reducing power or modulating the activity of NAD+-consuming enzymes including sirtuins and PARPs. However, disequilibrium of NAD+ metabolism could disturb physiological processes, including mitochondria function, circadian rhythm, inflammation, DNA repair and metabolism, leading to aging-associated dysfunction and cancer. NAD+ levels could be augmented by dietary NAD+ precursor, inhibitors of NAD+-consuming enzymes, caloric restriction and exercise. NAD+ boosters restore the bioenergetics, redox balance and signaling pathways, ameliorating the adverse effects of pathophysiological conditions, including infection, aging and cancer. Abbreviations: 2-HG, 2-hydroxyglutarate; α-KG, α-ketoglutarate; CCGs, clock-controlled genes; FOXO1, Forkhead Box O1; GSH, Glutathione; IDH1Mt, mutant isocitrate dehydrogenase 1; NOXs, NADPH oxidase; PER2, period circadian clock 2; PPP, pentose phosphate pathway; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator alpha; ROS, reactive oxygen species; OXPHOS, Oxidative phosphorylation; TCA cycle, tricarboxylic acid cycle
NADH/NADPH is a powerful reduce source for buffering oxidative stress, thereby protecting cells/tissues from oxidative stress during aging. The remarkable reduction of NAD+ concentration and NAD+/NADH ratio in aged rats occurs in parallel with enhanced oxidative stress and diminished antioxidant capacity.334 NMN addition in isolated aortas elevates the NAD+ and MnSOD levels, thus enhancing the antioxidant capacity.344 The overexpression of Nmnat3 efficiently boosts NAD+ in a variety of murine tissues, which significantly suppresses the ROS generation, and protects from aging-related insulin resistance.360 Overexpression of G6PD promotes NADPH production, preventing tissue from oxidative damage to improve mice health span.361 NAD+ also regulates oxidative stress in cellular senescence by regulating sirtuins and PARPs. NAD+-dependent SIRT1 is significantly upregulated in response to oxidative stress, protecting heart from oxidative damage, contributing to retard of aging.362
NAD+ deficiency correlates with disturbed circadian clocks during aging
Besides mitigating the oxidative damage, NAD+ can extend lifespan by driving the circadian rhythms. The misalignment of the circadian clocks, the internal timekeeper mechanism that links metabolism with the exogenous and endogenous factors, has been associated with the acceleration of aging.240 The circadian sirtuins link the NAD+ metabolism to the circadian clock machinery during aging. SIRT1 induces the circadian transcription of core clock genes, such as Cry1, Per2, Rorγ, and Bmal1 via either rhythmically deacetylating BMAL1 or PER2.36,38 SIRT1 also modulates CLOCK-mediated chromatin remodeling at H3 Lys9/Lys14 at circadian promoters to control circadian.38 In the SCN of aged mice, SIRT1 level is significantly decreased, resulting in a reduction of BMAL1 and other circadian proteins.363 The autonomous hepatic clock induces the NAD+ salvage pathway to partially restore NAD+ oscillation, driving the SIRT1 circadian function in the liver even without inputs from other clocks.241 Therefore, NAD+-dependent SIRT1 regulates the aging-dependent decline in central circadian function.
The critical role of NAD+ pool in tumorigenesis
NAD+ not only acts as a co-enzyme for metabolic redox reactions, but also functions as a co-substrate to modulate the activity of NAD+-consuming enzymes that govern the critical steps in tumorigenesis, including genome stability, metabolism, cell growth, cell death, redox homeostasis and immune response. The sirtuins and PARPs in tumorigenesis exhibit both oncogenic and tumor suppressor activity, which might be determined by their sublocalization and cell type (Fig. 6).
NAD+-related metabolic reprogramming and redox homeostasis in tumorigenesis
Cancer cells undergo metabolic reprogramming that provides the substrates and energy for biomass generation, to sustain the stress response and continuous proliferation.364 The metabolic reprogramming is characterized by shifting glucose metabolism to aerobic glycolysis, including enhanced cytosolic lactate fermentation and PPP and decreased OXPHOS. This shift not only allows for rapid production of energy but also for maintenance of NADH/NAD+ redox ratio, which is required for metabolic processes, such as aerobic glycolysis, the TCA cycle, OXPHOS, FAO, serine biosynthesis and antioxidant defense.365,366,367 Cytosolic NAD+ is required for glycolysis, in which GAPDH converts NAD+ to NADH. In health cells, cytosolic NADH is shuttled into mitochondria, where it is turned into NAD+ by OXPHOS whereas in cancer cells, the conversion of NAD+ to NADH in mitochondria is not sufficient for the high rate of glycolysis due to reduced OXPHOS. Therefore, cancer cells enhance the cytosolic lactate fermentation to generate NADH by LDHA. Activation of LDHA by oncogenic receptor tyrosine kinase FGFR1 promotes glycolysis and tumor growth by increasing the NAD+/NADH ratio,368 while the aberration of NAD+/NADH due to reduced activity of mitochondrial complex I promotes the aggressiveness of human breast cancer cells.369
The ‘hyper-metabolism’ of cancer cells causes the excessive generation of ROS.370 ROS contribute to tumorigenesis through multiple processes, including causing oxidative DNA damage, genomic instability and inflammatory stress to drive malignant transformation, and acting as a messenger to regulate signaling pathways to support tumor initiation, development, and angiogenesis.142,261,272,371,372,373,374 Cancer cells build a complicate and powerful antioxidant system, such as the GSH and Trx systems, to adapt to the high ROS levels. Notably, both GSH and Trx systems rely on the reducing power of NADPH, which is generated by G6PD in PPP, ME1 in glutamine metabolism and NNT. In cancer cells, increased ROS will oxidize the specific isoform of pyruvate kinase (PKM2), diverting glucose flux towards the PPP and generation of NADPH for GSH recycling.96,375,376 Similarly, nutrition stress or oxidative stress induces the expression of enzymes, including NAMPT, ME1 and NNT, which augments the NADPH to support the cell survival under glucose deprival and anoikis conditions, thereby promoting tumor growth and metastasis.377,378,379 IDH1 mutations in human cancers favor consuming the NADPH for 2-HG synthesis at the expense of other NADPH-requiring pathways essential for cell viability even when NADPH is limiting.96 Additionally, AMPK activation by reduced ATP levels maintains NADPH through inhibiting NADPH consumption in fatty-acid synthesis and enhancing NADPH production from fatty-acid oxidation instead of PPP to inhibit cell death.380 However, NAD+ depletion exacerbates oxidative damage via reducing the antioxidant defense capacity, resulting in impaired cell proliferation and increased cell death.365,366
The NAD+/NADH ratio emerges as a fine-tuned signal to regulate redox status through sirtuins.26 Sirtuins manipulate the metabolism reprogramming via directedly altering the activity of metabolic enzymes by NAD+-dependent modification or changing their expression by regulating transcription factors.217 All the enzymes but PGI in the glycolysis and the TCA cycle can be acetylated under the control of sirtuins. GAPDH and PKM2 are two major enzymes in glycolysis that regulated by sirtuins. SIRT1 can bind and retain GAPDH in the cytosol, but SIRT5 actives GAPDH via demalonylation, thereby elevating glycolytic flux.381 Both SIRT2-catalyzed deacetylation and SIRT5-mediated desuccinylation of PKM2 reduce the activity of PKM2, preventing the carbon entry into the TCA cycle. In contrast, SIRT3 deacetylates and activates PDHA1 acetylation, linking glycolysis and OXPHOS.382 SIRT3 and SIRT5 stabilize and activate the enzymatic activity of SHMT2 by its deacetylase and desuccinylase activity, respectively, thereby promoting the serine catabolism to drive carcinogenesis.383 SIRT5-catalyzed demalonylation and inactivation of SDHA block the TCA cycle and induce succinate accumulation, promoting tumorigenesis and drug resistance.383,384 Additionally, sirtuins also regulate the expression of metabolic enzymes via transcription factors, such as HIF-1α. SIRT3 enhances the enzymatic activities of SOD2 and IDH2 to limit ROS levels, which repress the metabolism reprogramming in cancer cell via destabilizing HIF-1α, thereby repressing tumor growth.142,222,225,385,386,387 It has also been demonstrated that the enzymes in glycolysis (e.g., GLUT4, HK1, GSK3B, and GAPDH), the enzymes (e.g., PDPR PDHA1, and PDHX) in carbohydrate metabolism, and most enzymes in ETC and ATP synthesis are assigned as ADP-ribose amino acid acceptor.388 However, whether the ADP-ribosylation of metabolic enzymes contributes to the metabolism reprogramming in cancer cells requires further investigation.
NAD+-regulated genome stability and gene transcription in carcinogenesis
NAD+ metabolism is not only essential for metabolic and redox homeostasis, but also required for epigenetic reprogramming in tumorigenesis. Genomic instability and altered transcriptional pattern are well-known hallmarks of cancer.364 Sirtuins and PARPs control the genome stability and gene transcription by regulating the histone modification, DNA repair, as well as the recruitment and activation of transcription factors.169,389 SIRT1 is responsible for the histone acetylation patterns, including the H4K4ac, H3K9ac, H4K16ac and H1K26ac, associated with tight chromatin compaction. The activity of SIRT1 also modulates the formation of H3K9me3, H4K20me3 and H3K79me2 to regulate chromatin. For instance, SIRT1 alters the acetylation patterns of histones H3 and H4, including H3k4ac, H3k9ac and H4k16ac, to regulate the expression of cancer-related genes in breast cancer.390 Besides the impact on chromatin state, SIRT1 modulates the non-histone proteins to initiate DNA repair and gene transcription. SIRT1 induces the recruitment of DNA repair factors, including NBS1 and Rad51, in keratinocytes to maintain genome stability and gene transcription.174 Oxidative stress-induced SIRT1 can deacetylase hMOF to reduce the expression of DNA repair proteins, including RAD50, BRCA2 and FANCA in human colorectal cancer cells.169 hMOF also plays crucial roles in transcription activation by H4K16 acetylation in HeLa and glioma cancer cells.391,392 The contradictory role of SIRT1 in cancer is manifested as their overexpression in neoplasms, such as prostate, AML, non-melanoma or melanoma skin cancer, and colon carcinomas, and the reduced expression in other cancers, including breast cancer and hepatic cell carcinomas.393,394 Thus, the mechanisms underlying the regulatory role of SIRT1 in DNA repair and gene transcription in cancer development need further exploration.
As aforementioned, PARPs govern the genome stability and gene transcription via NAD+-dependent ADP-ribosylation. PARPs-induced ADP-ribose marks elevate 10- to 27-fold in response to the oxidative genome damage by H2O2 in human osteosarcoma cells.133,183,184 A variety of cancers have somatic mutations resulting in genomic disability and defective DNA repair, including BRCA1/2, ATM, CHK2 and TP53.395 The loss of double-strand repair pathway due to BRCA1 or BRCA2 mutations renders cancer cells more dependent on the PARPs-mediated repair, and more sensitive to PARP inhibition, raising a possibility of a wider application of PARPi in cancer therapy.396,397,398
NAD+ metabolism is also linked to epigenetic modification by NNMT that transfers the methyl group of SAM to NAM. NNMT is increased in a broad range of cancers, such as papillary thyroid cancer, renal clear cell carcinoma, glioblastoma tumors, bladder cancer, colorectal cancers, gastric cancers, and oral squamous cell carcinoma.200,399,400,401,402 Elevated NNMT inhibits the methylation potential of cancer cells through inducing methyl sink in the form of MNA. Reducing the NNMT expression impairs the cell proliferation and tumor growth of mesenchymal glioblastoma stem cells (GSCs), accompanied by reduced methylation ability.403 Besides, NNMT promotes HCC cell invasion and metastasis by changing the H3K27me patterns and transcriptionally activating CD44. NNMT-mediated CD44 mRNA m6A methylation produces a CD44v3 splice variant, while MNA stabilizes CD44 protein by inhibiting the ubiquitin-mediated degradation.404 Furthermore, NNMT depletion elevates the NAD(H)+ levels that result in an enhanced expression of sirtuin target genes and a reduced H3K9Ac.405 Therefore, NNMT acts as a crucial metabolic modulator of epigenetic modification, promoting the migration, invasion, proliferation and survival of cancer cells.
The oncometabolite, 2-hydroxyglutarate (2-HG), also couples the NADP+/NADPH to epigenetic modification, including histone and DNA demethylases, in tumorigenesis. Mutant IDHs, accounting for 80% of lower-grade gliomas and secondary GBM, continuously produce 2-HG. To support 2-HG synthesis, cancer cells with IDH1R132H mutation enhance NADPH production via the PPP.406,407 Interestingly, IDH1 mutants compete for NADPH to synthesize 2-HG with other pathways that are critical for cell viability, resulting in further disruption of cellular metabolism and redox homeostasis in tumorigenesis.36
NAD+-dependent cancer cell proliferation and metastasis
Given the massive demand for NAD+ to support the metabolism reprogramming, the genome integrity and gene transcription in tumorigenesis, cancer cells enhance the capacity of NAD+ production through various pathways. It has been demonstrated that tumors that arise from cells with highly NAPRT expression will rely on NAD+ de novo synthesis for survival. While cancers derived from tissues with normal NAPRT levels are entirely dependent on the NAD+ salvage pathway for survival.408 Both the upregulated NAPRT in ovarian cancer and the high expression of NAMPT in glioblastoma, colorectal cancer tumors, and breast cancer, increase intracellular NAD+ levels, contributing to cancer cell metabolism and DNA repair process in tumors.409 Moreover, resistant CCRF-CEM cells with high QPRT activity exploit amino acid catabolism as a substitute pathway for NAD+ generation.410
High expression of NAMPT or NAPRT is associated with tumor progression, invasion, and drug resistance.411,412,413,414 This effect is mediated by NAD+-dependent PARPs and SIRT1.415,416,417 SIRT6 induces the NAMPT activity to increase NAD+ content, thereby preventing oxidative damage. Activation of the c-MYC-NAMPT-SIRT1 feedback loop may crucially contribute to the initiation and development of both routes to colorectal cancer.413,417 Declining NAD+ levels reduced the SIRT1-mediated inhibition of STAT3, which induces the secretion of IL-6 and TFG-β to sustain the signaling required for EMT.418 CD38 expression inversely correlates with prostate cancer progression due to its ability to lower intracellular NAD+, resulting in cell-cycle arrest, reduced glycolytic and mitochondrial metabolism, and impaired fatty acid and lipid synthesis.419 These findings demonstrated a pro-tumor activity of NAMPT, suggesting a promising therapeutic target for cancer treatment. NAMPT inhibitors, FK866, STF-118804 and KPT-9274, can reduce the viability and growth of different cancer cells and have an additive effect in combination with main current chemotherapeutic drugs.420,421,422,423
NAD+ metabolism and metabolic diseases
Diabetes
Diabetes is a chronic metabolic disease characterized by hyperglycemia. The human pancreas cannot produce enough insulin, or the body cannot effectively use the produced insulin, which causing a pathological increase in blood sugar. Pancreatic β-cells maintain systemic glucose homeostasis by controlling the release of insulin, thereby responding to changes in metabolic demand. The high capacity, low-affinity GLUT2 and high KM glucokinase (GK) in β-cell ensure the proximal glucose-sensing. The glucose fluxes through the glycolytic pathway and the TCA cycle, enable the production of NADH and ATP. Thus, the elevated blood glucose levels lead to more production of NADH and ATP, resulting in closure of ATP-sensitive potassium channels, cell depolarization, Ca2+ influx and culminating in insulin secretion.424,425,426,427 In addition to NADH produced in the mitochondria by the TCA cycle, cytoplasmic NAD+ generation is essential for insulin secretion.82,88 Given that β-cells only have extremely low LDHA activity to regenerate NAD+ for glycolysis, NADH generated by glycolysis must be transferred into the mitochondria to be oxidized by complex I.424 Cytoplasmic NADH from the glycolytic pathway is delivered to mitochondria through two NADH shuttles, G3P shuttle and MA shuttle, allowing NAD+ recycling to sustain glycolytic flux. As evidenced, at maximal glucose stimulation, the rising of NAD(P)H levels is estimated to be approximately 30 mM in whole pancreatic islet beta cells, with a 7 mM in the cytoplasmic domain and an approximately 20 second delayed 60 mM in mitochondrial domain.428 The NADH shuttle thus promotes the increase of Ca2+ after the formation of mitochondrial membrane potential and sufficient ATP generation from ETC, concomitantly triggering glucose-stimulated insulin secretion (GSIS) (Fig. 7).429,430,431

Pathophysiological role of NAD+ disarrangement in metabolic diseases. a The liver is a master organ of NAD+ metabolism and may facilitate the NAD+ biosynthesis in other tissues. NAD+ metabolism plays a critical role in the lipid metabolism through modulating the activity of sirtuins. The reduced NAMPT expression and NAD+ levels contribute to the development of NAFLD through manipulating dysmetabolic imbalance, hepatic energy homeostasis, glucose homeostasis, hepatic inflammation and insulin resistance. b Decreased NAD+/NADH ratio by the mismatch between NADH production and oxidation inhibits the activity of sirtuins in the failing heart. Elevated protein acetylation weakens the energy metabolism through negative feedback to OXPHOS and substrate metabolism, impairing antioxidant defense and sensitizing the mPTP to ROS or calcium. c The deduced NAD+ levels in kidney are attributed to the decreased expression of enzymes in NAD+ de novo synthesis and increased consumption by DNA damage activated PARPs. NAD+ depletion inhibits the SIRT1/PGC1α mediated mitochondrial quality control, ATP production and NAD+ de novo biosynthesis. The phosphorylation of NAD+ to NADP+ enhances the antioxidant defense against oxidant stress. NAD+-dependent defect in FAO results in intracellular lipid accumulation. In addition, the defected FAO and increased desaturation of PUFAs to HUFAs due to NAD+ deficiency and impaired mitochondrial function result in the accumulation of HUFA-containing triglycerides and cellular lipid in renal tubular cells. d The insulin secretion is adjusted by the dynamic glucose concentration in blood. As a master regulator of insulin secretion, glucose is metabolized via the glycolysis and TCA cycle to produce NADH and ATP. The increased NADH and ATP induces the closure of ATP-dependent K+ channels, the opening of voltage-gated L-type Ca2+ channels, the raising of cytosolic Ca2+ and culminating in insulin secretion in pancreatic β-cells. The activity of mitochondrial shuttles including the glycerophosphate and malate/aspartate shuttles allows the reoxidation of cytosolic NADH into NAD+, which is required for maintenance of the glycolysis. Purple representants the downregulated proteins or activated biological functions, while brown labels the upregulated proteins and repressed physiological activities. Abbreviations: ACMSD, alpha-amino-beta-carboxy-muconate-semialdehyde decarboxylase; AR, Aldose reductase; ETC, electron transport chain; Grxs, glutaredoxins; HUFAs, highly unsaturated fatty acids; KMO, kynurenine 3-monooxygenase; FAO, fatty acid oxidation; PUFAs, polyunsaturated fatty acids; SDH, Sorbitol dehydrogenase; Trxs, thioredoxins. 3-HK, 3-hydroxykynurenine
Sustained high levels of insulin demand will eventually give rise to functionally compromised or physical loss of β-cells, which culminate in hyperglycemia and diabetes.432,433,434 However, β-cells exposed to diabetes and hyperglycemia exhibit striking changes in metabolism.433,435 Importantly, the increase of Krebs’ cycle in the mitochondrial that generally responds to glucose is terminated, which will cause glucose to fail to escalate the NADH and ATP content in the pancreatic islets of diabetic patients.433 Rather than producing from mitochondrial TCA cycle under controlled conditions, the NADH in diabetic islets is generated by cytoplasmic sorbitol oxidation and mitochondrial pyruvate oxidation in response to diabetes and hyperglycemia stimulate.433,436 In diabetes, when GPDH is inhibited due to the reduced utilization of NAD+, about 30% of glucose is involved in the polyol pathway. When β cells are over-nutrient and hyperglycemic, the excessive NADH produced by the polyol pathway will promote the production of superoxide through the overload complex I of ETC, resulting in cell dysfunction and impaired insulin secretion.424 Moreover, the two components of the G3P shuttle (including GPD1 in the cytoplasm and GPD2 in the mitochondria) are up-regulated in mRNA and protein levels in diabetic islets, thus ensuring the transfer of electrons from NADH produced in glycolysis to mitochondria.429,433 In line with that, the overexpression of cytoplasmic malic enzyme (ME1) enhanced the GSIS and anaplerosis in insulinoma cells.437 Selective reduction of cytosolic ME1 expression and enzyme activity significantly reduces GSIS and amino acid-stimulated insulin secretion (AASIS).438 There is growing evidence indicating that cytosolic NADPH is one of the effectory metabolic coupling factors, a variety of critical intermediates and cofactors involved in the GSIS. Although glucose causes a dose-dependent inhibition of pentose phosphate pathway activity in beta cells, the major pathway for NADPH production, the cytosolic ratio of NADPH/NADP+ increases during glucose-stimulated insulin release.439,440,441 NADPH stimulates the exocytotic machinery by the redox proteins glutaredoxin and thioredoxin and has a local redox reaction, thereby accelerating exocytosis and promoting the secretion of insulin in pancreatic β-cells.442,443
The NAD+ level is associated with insulin resistance. HFD significantly impairs the role of NAMPT-regulated NAD+ biosynthesis in metabolic organs.337 Mice that specifically knock out the Nampt gene of adipocytes have serious insulin resistance, which is manifested by an increase in plasma free fatty acid content and a decrease in plasma content of the main insulin-sensitive adipokine, adiponectin. These deleterious alterations can be normalized by administering NMN.444 Furthermore, NMN alienates glucose intolerance and lipid profiles by recovering NAD+ levels in age induced T2D mouse model.337 Conversely, overexpression of Nmnat3 in mouse can effectively increase NAD+ levels in a variety of tissues and prevent aging-related insulin resistance caused by diet.360 Owing to its expression and activity increase with age, CD38 is essential for age-associated NAD+ decrease through degradation of NMN in vivo. CD38 deficiency has improved glucose intolerance with HFD, which could be further ameliorated by supplement of NR.52 78c, as a highly specific and effective CD38 inhibitor, can reverse age-associated NAD+ reduction and improve some metabolic and physiological parameters of aging, such as glucose tolerance, cardiac function, muscle function and exercise capacity in both natural aging and accelerated aging mice models.53
Obesity
The pathological expansion of adipose tissue is specifically manifested in the dysregulated production of adipokines and lipid, low-grade inflammation and enrichment of extracellular matrix. Insulin resistance is a critical whole-body abnormal metabolism closely related to obesity.445,446 A reduction of NAD+ levels in cells is observed in many tissues with obesity, like the skeletal muscles, hypothalamus, liver and adipose tissue.337,447 Supplementation of NR or NMN can protect against the decrease of NAD+ levels, and partially inhibit the weight gain of the mice fed with HFD by enhancing energy expenditure.448 The NAD+ biosynthesis regulated by NAMPT in adipocytes plays an important role in the pathogenesis of obesity-related metabolic complications.444 Both the expression of NAMPT in visceral fat and the level of NAMPT in serum are positively correlated with the degree of obesity.449,450,451,452,453 In contrast, obese subjects have lower levels of NAMPT in subcutaneous fat tissue.454,455,456 The upregulation of NAMPT by the activation of the HIF1-α pathway under hypoxic conditions plays a vital role in processing dietary lipids to regulate the plasticity of adipose tissue, whole-body glucose homeostasis and food intake. The deficiency of adipose Nampt can partially reduce food intake, thereby preventing obesity caused by diet. In addition, NAMPT-mediated NAD+ biosynthesis plays a vital role in adipose by promoting weight gain caused by HFD, which can be proved by the inability of HFD-fed FANKO mice that can expand adipose tissue normally.453,457
Several studies have shown that in different adipocyte models, such as primary adipocytes, 3T3-L1 preadipocyte cell and SGBS cell, NAMPT can be secreted directly into the supernatant through non-classical pathways. These results indicate that the adipose tissue is one of the main sources of secreted extracellular NAMPT (eNAMPT).458 Treatment with eNAMPT can increase the expression of lipoprotein lipase and PPARγ in preadipocytes and promote the expression of fatty acid synthase in differentiated adipocytes, which indicates that eNAMPT may be a positive regulator in adipocytes lipid metabolism.459 Adipose tissue-specific Nampt knockin and knockout mice (ANKI and ANKO) showed opposite alterations of circulating eNAMPT, which accordingly affected hypothalamic NAD+/SIRT1 signaling and physical activity. Treatment with NMN can improve physical activity deficits in ANKO mice.460 The biosynthesis of NAD+ in adipocytes is crucial for the extension of HDF-induced white fat depots and may have more specific effects in lipid accumulation and processing.457 Based on these observations, the effect of NAMPT on obesity depends on its enzymatic activity. Increasing NAD+ levels by supplementing NR in mouse tissues and mammalian cells activates SIRT1 and SIRT3, which ultimately leads to increased oxidative metabolism and prevents metabolic abnormalities induced by HFD.37 Moreover, adding leucine to HFD can increase the expression of NAMPT and SIRT1 and elevate the level of NAD+ in cells, which will reduce the acetylation of FoxO1 and PPAR-γ co-activator 1α (PGC1α). Deacetylation of PGC1α may promote the upregulation of genes related to fatty acid oxidation and biogenesis in mitochondria, thereby restoring mitochondrial function and protecting against HFD-induced obesity.461
Non-alcoholic fatty liver disease (NAFLD)
Alterations of hepatic metabolism are critical to the development of liver diseases, in which the NAFLD is the most common chronic liver disease and is strongly related to metabolic syndrome. NAFLD might eventually cause more severe liver diseases, such as liver fibrosis, liver cirrhosis, liver failure and hepatocellular carcinoma (HCC).462,463,464 It is reported that reduced NAD+ concentration caused a dysmetabolic imbalance, leading to the development of NAFLD.465 Oral administration of NR halts the progression of NAFLD through rescuing the NAD+ reduction, reducing the total cholesterol and triglyceride levels, decreasing the AST level, and reversing Kupffer cells accumulated and inflammatory in aged group.466,467 Troxerutin, as a derivative from natural bioflavonoid rutin, could promote NAMPT expression to restore the NAD+ level depleted by oxidative stress in the HFD-induced NAFLD mouse model (Fig. 7).468 In a transgenic mouse model of DN-NAMPT, the researchers found that middle-aged mice had a systemic reduction of NAD+ and showed a moderate NAFLD phenotype, like triggered inflammation, lipid accumulation, increased oxidative stress and impaired insulin sensitivity of liver. Some of these phenotypes are further exacerbated after feeding a high-fat diet. However, oral NR can completely reverse these phenotypes caused by NAD+ deficiency or high-fat diet.335 Significantly, knockdown of Nampt gene increases, while over-expression reduces hepatic triglyceride both in vitro and in vivo models. The expression of NAMPT in patients with NAFLD has decreased systemically both in serum and within the hepatic tissue, which is regulated by PPARα activation and glucose.469 Meanwhile, the NAMPT is also a target of FoxO transcription factors that control the NAD+ signaling in the regulation of hepatic triglyceride homeostasis.470 Additionally, the hepatic microRNA-34a, which is increased in obesity, reduces NAD+ levels and SIRT1 activity by targeting NAMPT. The antagonism of microRNA-34a could moderate inflammation, steatosis and glucose intolerance, and recover NAMPT/NAD+ levels in diet-induced obese mouse model.447 It was found that higher level of NAMPT in serum in women is correlated with a much lower hepatic de novo lipogenesis (DNL) index, although they do not correlate with the DNL index, but had a correlation with a higher hepatic fat in men, implying a sex-dependent interpretation role of serum NAMPT level for NAFLD prognosis.471 Mechanistically, reduced NAMPT/NAD+ inhibits SIRT1’s function, thereby attenuating the deacetylation activity of SREBP1, leading to the expression of ACC and FASN.463 Conversely, stabilization of SIRT1 by increasing NNMT expression or MNAM levels improves lipid parameters.63,472,473
Beyond the metabolic activity, NAMPT may also participate in NAFLD’s pathogenesis by controlling hepatic inflammation, insulin resistance and glucose homeostasis.474,475 eNAMPT regulates glucose production via the PKA/CREB signaling in HepG2 cells.476 The expression of NAMPT is closely related to the expression of pro-inflammatory cytokines in the inflammation induced by free fatty acids and is remarkably reduced by the inhibition of NF-κB in HepG2 cells.477 To date, the clinical analysis reveals controversy regarding the relationship of circulating NAMPT with NAFLD. Several studies report no statistically significant difference in NAMPT levels between NAFLD and healthy controls, as well as among different histological NAFLD groups.478,479 Another study shows that NAFLD patients have systemically decreased the expression of NAMPT in both serum and hepatic tissue.469 In contrast, the liver and serum NAMPT of morbidly obese women with NAFLD are significantly higher than that of morbidly obese women with healthy livers.480 The serum NAMPT level and its expression in hepatic tissues are positively correlated with pro-inflammatory factors.480 Moreover, the expression of NAMPT is notably higher in fibrosis patients and is correlated positively with the stage of fibrosis in NAFLD patients.481 Elevated serum NAMPT, together with inflammatory factors, such as IL-6, IL-8 and TNF-a, is associated with an increased likelihood of exhibiting NAFLD and NASH.482 Given that hepatocytes are only one of the sources of eNAMPT, circulating NAMPT levels may not represent its actually concentration in local liver or adipose tissues, thus requiring further research to determine its exact role in NAFLD.
NAD+ metabolism in neurodegenerative disorders
Neurodegenerative diseases are a heterogeneous group of diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS), which are characterized by progressive degeneration of the structure and function of the peripheral and central nervous system, with the characteristics like the accumulation of misfolded and aggregated proteins that are associated with severe proteotoxic stress (Fig. 8).

Linkages between NAD+ depletion and neurodegenerative disorders. Most neurodegenerative disorders, including axonal degeneration, Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (PD) and Amyotrophic lateral sclerosis (ALS), are associated with mitochondrial dysfunction, lowered antioxidant capacity and heightened mitophagy, all of which are converged into the age-related NAD+ depletion induced by either enhanced consumption or impaired biosynthesis. These neural pathologies can be rescued by NAD+ boosting. Purple representants the downregulated proteins or activated biological functions, while brown labels the upregulated proteins and repressed physiological activities in neurodegenerative disorders. Abbreviations: mHtt, mutant Huntingtin; Aβ, amyloid beta; NFTs, neurofibrillary tangles; 3-HAA, 3-hydroxyanthranilic acid; QA, quinolinic acid; WldS, slow Wallerian degeneration; 3-HK, 3-hydroxykynurenine
Axonal degeneration
Axonal degeneration is an early and prominent feature of many neurological disorders, including AD, PD, ALS, ischemic brain and spinal cord injuries, diabetic neuropathy and traumatic brain injury.483,484
SARM1, as the evolutionary conservative executor of degradation cascade, is required for the progression of rapid Wallerian degradation. The TIR domain of SARM1 has inherent NADase activity, which can cleave NAD+ into nicotinamide, cADPR and ADPR. The nicotinamide acts as a feedback inhibitor of SARM1. Axons require the NADase activity of the full length SARM1 to facilitate axonal degeneration and NAD+ consumption after injury.33,34,485 Similarly, the loss of endogenous NMNAT2 is an important cause of axon degeneration after injury.486 The axon damages caused by SARM1 or NMNAT2 can be restored by increasing NAD + synthesis, like over-expressing the NMNAT2 enzyme.34,487 The naturally occurring mutant mice, Wallerian degeneration slow (WldS) with chimeric gene containing the N-terminal 70AA of UBE4B and full length NMNAT1, show a delayed Wallerian degeneration phenotype.488,489,490,491,492
Several mechanisms are underlying the protective role of NMNAT on severed axons.493 Firstly, NMNAT acts as a stress-response protein that aids the clearance or refolding of misfolded proteins like a chaperone.494,495 Secondly, NMNAT and WldS proteins facilitate axon preservation by suppressing the accumulation of NMN. The activity of NMNAT1 is essential for axon survival because activity reduced mutants have no axon protection effect. The protection effect can also be abolished by the expression of exogenous NMN and ectopic expression of NMN deamidase.489,496 Thirdly, NMNAT1 does not change the NAD+ biosynthesis, but prevents the SARM1-dependent NAD+ depletion caused by injury, which is important for axon degeneration.490 Furthermore, Sir2, as a mammalian homolog of SIRT1, is the downstream effector of increased Nmnat activity, which can lead to axon protection in Wallerian degeneration slow mice.491 Additionally, SARM1 protein is required for NMN to promote axon degeneration and Ca2+ influx. SARM1 and NMN play a role in common signaling, which ultimately leads to the increase and breakage of Ca2+ in axons and the dissociation of mitochondrial dysfunction.497 Although the inhibitor of NMN synthase NAMPT reduces NAD+ level, it can still provide strong morphological and functional protection for damaged synapses and axons.489
Alzheimer’s disease (AD)
AD is a long-term chronic disease in the prodromal and preclinical stages with an average course of 8 to 10 years, which is the most common neurodegenerative disorder.
Currently, amyloid β peptide (Aβ), APOE and microtubule-associated protein tau are three important factors that have sufficient evidence as the etiology of AD.498 The key pathological features of AD are the accumulation of Aβ-enriched neuritic plaques and neurofibrillary tangles (NFTs) (consisting of tau protein).499 Aβ and phosphorylated Tau (p-Tau) neurofibrillary lesions lead to the pathology of neurons that display oxidative damage, impaired Ca2+ processing, reduced DNA repair, dysfunction of lysosomal and mitophagy, all of which have a positive correlation with the age-related NAD+ decline.500,501,502,503,504 Mounting evidence supports the promotion of NAD+ consumption on the process of AD, whereas the accumulation of NAD+ suppresses the AD-related pathological progress and the decline of cognitive function in different AD model ranging from C. elegans to mice.187,500,505,506 The increased activity and expression of CD38 following aging is responsible for the age-associated decrease in NAD+ level and defective mitochondrial function, which is at least partially regulated by the NAD+/SIRT3 signaling pathway.52 CD38 deficiency in APPswePS1DE9 mouse reduces soluble Aβ concentration and Aβ plaques, correlated with improved spatial cognition.507
The brains of individuals with preclinical AD (PCAD) and AD exhibit oxidative damage to a variety of molecules, e.g., accumulation of protein carbonyls (PCs) in regions that are rich in Aβ-peptide-containing SPs, increased lipid peroxidation in AD and PCAD hippocampi and elevated DNA damage.508 Recent studies have found that damaged cellular energy expenditure and DNA repair are related to the AD’s pathogenesis.
In the novel AD mice with DNA repair defects (3xTgAD /Polβ+/-), the content of NAD+ is reduced. Increasing NAD+ by supplementing NR can remarkably normalize DNA damage, p-Tau, synaptic transmission and neuroinflammation, improve the ability of motor function, memory and learning and increase the activity of SIRT3 in the brain.187 Notably, NAD+ augmentation improves DNA repair through improving the neuronal NHEJ activity in AD mice.186,187,509,510 The accumulated DNA oxidative damage in AD hyper-activates the DNA damage sensor PARP-1, thereby reducing the concentration of cellular NAD+ and suppressing the function of NAD+-SIRT1-PGC-1α axis, which in turn causes abnormal mitochondria.511 Replenishing cellular NAD+ can promote DNA repair in neurons and restore mitochondrial function through mitophagy.511 Mitophagy diminishes insoluble Aβ1-42, Aβ1-40, and hyper-phosphorylated tau, preventing cognitive or memory impairment in mouse model.500,512 Additionally, NAD+ protects neurons against p-Tau pathologies. NAD+ accumulation may inhibit the phosphorylation of different Tau protein sites by inhibiting the activity of Cdk5-p25 complex.501 Nicotinamide, as a competitive inhibitor of sirtuins, specifically reduces the phosphorylation of tau (Thr231), which is related to microtubule depolymerization in an analogous manner to that of SIRT1.513 NMNAT, as a binding partner of HSP90, can specifically recognize p-Tau to inhibit its amyloid aggregation in vitro and reduce its symptoms in the fly tauopathy model, and this effect could be competitively destroyed by its enzymatic substrate.514
Parkinson’s disease (PD)
PD is a common neurodegenerative disease, mainly including motor and non-motor symptoms.515 The neurons of PD patients exhibit symptoms, such as mitochondrial dysfunction, oxidative stress and NAD+ metabolic changes. Maintenance of NAD+ levels is vital for proficient neuronal function.333 It is reported that LRRK2 G2019S dopaminergic neurons exhibit a decreased NAD+ pool and a reduced sirtuin deacetylase activity, correlating with elevated acetylation of sirtuin substrates p53, α-tubulin and SOD2.516 In the primary cell (sPD cell) derived from patients with sporadic PD, the cytosolic conversion of pyruvate to lactic acid resulted in a significant increase in the nuclear NAD+ level and cellular NAD+/NADH ratio. The alteration of NAD+ metabolism in sPD cells contributes to the activation of SIRT2 and subsequently reduces the acetylation level of α-tubulin. Inhibition of the deacetylase function of sirtuin-2 can enhance the acetylation of α-tubulin and facilitate the clearance and transport of misfolded protein.517 The redox status of NAD+/NADH is remarkably decreased in iPSC neurons from GBA mutation associated PD, which may be due to the decrease of NMNAT2, as evidenced by the significant increase after NMN treatment.518 Furthermore, increasing the level of available NAD+ by supplementing diet containing NAD+ precursors or inhibiting the activity of NAD+-dependent enzyme (e.g., PARP that snatches NAD+ competing with mitochondria) is a feasible strategy to avoid the neurotoxicity related to mitochondrial dysfunction.518,519,520 Besides, through maintaining the rate of NAD+ production and normalizing the NADH/NAD+ balance, both pentylenetetrazole (PTZ) and niacin (NA) exhibit neuroprotective properties.521,522
SARM1 participates in the occurrence of PD mainly through its enzyme activity of NAD+ degradation. Compared with healthy people, the phosphorylation level of SARM1 is significantly increased in the neuronal cells from PD patients, with a high sensitivity to oxidative stress. In the case of oxidative stress, JNK increases the phosphorylation of SARM1, resulting in enhanced NAD+ degradation activity, which in turn promotes the suppression of mitochondrial respiration.523 The binding of SARM1 and PINK1 can facilitate TRAK6-induced ubiquitination of lysine 433 on PINK1, which is essential for keeping the stabilized status of PINK1 and bringing it into the outer membrane of mitochondria. Down-regulation of SARM1 and TRAF6 reduce the level of PINK1 and then recruits Parkin to the impaired mitochondria, indicating that SARM1 plays a crucial role in mitophagy via modulating PINK1.524
There is evidence showing that supplementation of high-dose NAD+ precursors in cells or Drosophila can alleviate the pathological phenotype by reducing oxidative stress and mitochondrial damage and improving motor function, which provides a feasible solution for the prevention and improvement of PD.525,526
Huntington’s disease (HD)
HD, also known as Huntington’s chorea (Huntington’s chorea), is an autosomal dominant hereditary neurodegenerative disorder that has a tremendous devastating effect on patients and their families, characterized by chorea-like movements, cognitive decline and psychotic-like symptoms. It is caused by repeated amplification of the CAG trinucleotide in the huntingtin (Htt) gene on the short arm of chromosome 4, which leads to a prolonged polyglutamine stretch at the N-terminus of the protein.527,528,529,530 Mitochondrial defection and increased oxidative stresses are the most prominent features in the cells of HD patients.
SIRT3, as a deacetylase in mitochondria, modulates the transcription of mitochondrial in response to oxidative stress.531 Notably, the expression of the Htt mutant reduces the deacetylase effect of SIRT3. In the HD model, reduced expression and deacetylase activity of SIRT3, which in turn prevents the deacetylation of LKB1 and SOD2, leading to a decrease in the level of NAD+, defects in mitochondrial biogenesis and accumulation of ROS.532 However, the overexpression of SIRT1 displays protection against abnormal motor function, cortical and striatal atrophy and loss of striatal neurons in the transgenic HD mice via regulating mitochondrial function.531 Both SIRT1 and SIRT3 exert their function through PGC-1α, which acts as a modifier in HD and ALS patients and other models. SIRT1/3-PGC-1α pathway in HD transgenic mice attenuates motor deficits and neurodegeneration by alleviating oxidative stress, eliminating huntingtin aggregates and restoring mitochondrial function.531,533,534,535,536,537
It has been reported that the kynurenine pathway (KP) is closely related to the pathogenesis of HD. The degradation process of tryptophan in KP produces a variety of neuroactive metabolites with amino acid-like structures, such as an N-methyl-D-aspartate (NMDA) receptor agonist quinolinic acid (QA), and a neuroprotective NMDA receptor agonist kynurenic acid (KYNA).538,539 Compared with the control group, the ratio of kynurenine (KYN) to KYNA in HD increases significantly, which is related to the decrease in the production of KYNA in HD patients.539 Genetic and pharmacological inhibition of TDO and KMO can increase KYNA, but not the level of neurotoxic product 3-HK, thereby improving the symptoms of neurodegeneration.538
Amyotrophic lateral sclerosis (ALS)
ALS is a neurological disorder that causes progressive degeneration of the motor neurons in the brainstem, spinal cord and cerebral cortex.540,541 More than 25 gene mutations have been reported to be closely related to ALS, of which C9orf72 repetitive amplification mutations and SOD1 mutations are the most common causes. Among them, ALS caused by mutations in the antioxidant enzyme SOD1 accounts for about 1–2% of sporadic ALS (SALS) and 20% of familial ALS (FALS).542,543,544,545,546,547 Astrocytes are specific contributors to spinal motor neuron degeneration in SOD1-related ALS.548 The spinal cord astrocytes isolated from SOD1G93A transgenic rats were co-cultured with motor neurons, resulting in the induction of motor neuron death.549 The reactive astrocytes can promote excitotoxic injury of motor neurons by producing nitric oxide and peroxynitrite, which cause mitochondrial damage and apoptosis in cultured neurons, decreasing glutamate transport, releasing pro-apoptotic mediators selectively toxic to motor neurons.550 Enhancing NAD+ availability can abrogate the neurotoxicity of astrocytes from diverse ALS models. Either overexpression of NAMPT or supplementation of NMN can increase the mitochondria and total cellular NAD+ levels of ALS astrocytes, thereby enhancing the ability to resist oxidative stress and restoring the toxicity of astrocytes to motor neurons.551 The NR repletion increases the levels of UPRmt-related proteins and promotes the clearance of mutant hSOD1 neurotoxic protein.552 Moreover, supplementation of NR can reduce the expression of neuroinflammation biomarkers in the spinal cord, alleviate the degeneration of motor neurons and appropriately extend the survival time of hSOD1G93A transgenic mice.540,541
Studies have found that ALS is associated with NAD+ metabolism through KP pathway damage. The impairment of KP in ALS is confirmed by the following evidence: higher serum tryptophan, cerebrospinal fluid (CSF), KYN and QA and a decrease in serum picolinic acid levels in ALS patients. Moreover, both the inflammation of microglia in the motor cortex and spinal cord of ALS patients and the expression of IDO and QA in neuronal cells and microglia increased significantly.501,553 In parallel with the impaired de novo pathway, the NAD+ decline in the ALS might also be due to the inadequate NAMPT-mediated NAD+ salvage synthesis pathway. NAM, a metabolite in the salvage pathway, is reduced in both CSF and serum from ALS patients compared with healthy people. NR supplement can increase the NAD+ concentration, dependent on NRKs (NR kinases), thereby avoiding the need for NAMPT in the salvage synthesis pathway.552,554
The protective effect of NAD+ on ALS might also be linked to the altered activities of SIRTs, however, the conclusions of many studies are quite different. It has been found that the expression level and function of Sirt3 is selectively reduced in the spinal cord of SOD1G93A mice at the end of ALS course, while the level of Sirt3 is increased in the human spinal cord after autopsy.555
The overexpression of NAD+ dependent deacetylase SIRT6 and SIRT3 can eliminate the neurotoxicity in the astrocyte cultured in vitro by activating the Nrf2-mediated antioxidant defenses.556 Interestingly, the expression of SIRT1 in the spinal cord of WT mouse is reduced during normal aging, while the expression of SIRT1 in different regions of the brain (including the spinal cord, hippocampus, thalamus and cerebral cortex) in SOD1G93A mouse is increased.557 Besides, overexpression of SIRT1 in motor neurons slows down the progression of ALS in severely phenotypic SOD1G93A (with high copy numbers) mice, partly by activating the HSF1/HSP70i molecular chaperone system.558,559 However, SIRT1 and SIRT2 are generally reduced in ALS primary motor cortex while they are upregulated in the spinal cord in human post-mortem tissues. In contrast to the neuroprotection role of SIRT1, SIRT2 upregulation is toxic to neuronal cells.540,557,560,561,562,563,564 A preliminary clinical trial has confirmed the importance of SIRT1 activation and NAD+ metabolism in ALS. The drug used in this trail is EH301 (a mixture of pterostilbene and NR), which contains the SIRT1 activator and the precursor of NAD+. Compared with the placebo control group, EH301 can significantly alleviate the development of ALS.501,565
NAD+ in cardiac and renal diseases
NAD+ and heart failure (HF)
HF is a complex clinical syndrome caused by various initial heart damage and subsequent disturbance in compensatory effects and pathogenesis mechanisms. It is manifested through a number of complex molecular and systemic dysfunctions from the subcellular level to the multi-organ system of the whole body.566,567 There are many kinds of evidence indicating that the imbalance of myocardial NAD+ pool is causally linked to metabolic remodeling and mitochondrial dysfunction in HF.568 A wide range of tissues display a reduction of NAD+ in the aging mice model. The down-regulation of NAD+ levels in the heart is most significant. NAD+ is reduced by 70% within 3 to 24 months, and as a compensation, the concentration of NADH has increased by 50%.334,569 The increased NADH/NAD+ ratio and protein hyperacetylation are found in the HF patients’ hearts and pathologically hypertrophied mice.570 It is important to note that hyperacetylation of mitochondrial proteins is considered to be an inducer for cardiac dysfunction.570,571,572 Increasing NAD+ levels by activating the NAD+ salvage pathway can inhibit mitochondrial protein hyperacetylation and cardiac hypertrophy and improve cardiac function under stress. Proteomics analysis identified a subgroup of mitochondrial proteins, particularly sensitive to the changes of NADH/NAD+ ratio. It is reported that the hyperacetylation modification of mitochondrial proteins caused by the imbalance of NAD+ redox mainly promotes the pathological remodeling of the heart through two different mechanisms. First, the hyperacetylation of the mitochondrial malate-aspartate shuttle protein inhibits the oxidation and transport of NADH in the mitochondria, resulting in an imbalance of redox in the cytoplasm. Second, the acetylation modification of the oligomycin-sensitive conferring protein increases its binding to cyclophilin D and enhances the sensitivity of the mitochondrial permeability transition pore. Both of these conditions can be restored by regulating the normalization of NAD+ redox balance at the genetic or pharmacological level.570
It has been reported that hyperacetylation of cardiac protein in mice with HFD is closely related to the decreased expression of SIRT3.573 Exogenous supplementation of NAD+ can maintain the intracellular NAD+ level and block the symptoms of agonist-induced cardiac hypertrophy in vitro and in vivo by regulating the activation of SIRT3.574 As a negative regulator in cardiac hypertrophy, SIRT3 protects the heart by inhibiting intracellular ROS.140 Mice lacking SIRT3 become more sensitive to the stimulation of several pharmacological or non-pharmacological stressors, showing symptoms, such as fibrosis, cardiac hypertrophy and high mortality.575 In the SIRT3 knockout mouse model, the progression of fibrosis and cardiac hypertrophy also accelerated with age. In addition, mitochondrial swelling also appears to increase with age due to the increased opening of mitochondrial permeability transition pore (mPTP).575 Similarly, the loss of mitochondrial complex I leads to a decrease in the ratio of NAD+/NADH and can inhibit the activity of Sirt3, thereby enhancing the protein acetylation and mPTP sensitivity. Supplementing cKO mice with NAD+ precursors can partially normalize the NAD+/NADH ratio, acetylation of protein and sensitivity of mPTP.576 It is noteworthy that compared with normal mice, SIRT3-deficient mice can cause increased oxidation of fatty acid in heart, which is due to the high acetylation modification and high activity of β-HAD and LCAD.573 Moreover, in the absence of sirt5, the succinylation of protein lysine mainly occurs in the heart. When fasting, Sirt5 knockout mouse has reduced ECHA (an enzyme regulates the oxidation of fatty acid) activity, increased long-chain acyl-CoAs content and decreased ATP content.229 Compared with control mice in the same litter, after 20 min of ischemia and 90 min of reperfusion, the area of cardiac infarction in Sirt5 knockout mice was larger. The ischemia-reperfusion injury (I/R injury) of Sirt5 knockout mouse heart is restored to normal levels by dimethyl malonate (a succinate dehydrogenase (SDH) inhibitor) pretreatment, which implies that the change of SDH activity is the leading cause of the damage.577 Clinically, I/R injury occurs during myocardial infarction or blood supply stop during cardiovascular surgery. I/R injury is related to the decrease of endogenous NAMPT and the down-regulated expression of SIRT1, SIRT3 and SIRT4. NAMPT strictly controls the NAD+ and ATP content, thus playing an important part in regulating cell survival by suppressing apoptosis and increasing autophagy flux in cardiomyocyte.578,579,580 SIRT3 reduction can increase the sensitivity of heart-derived cells and the adult heart to I/R injury and may cause more severe I/R injury in the elderly heart.581,582 Exogenous expression of NMN can activate Sirt1-mediated FoxO1 deacetylation, which can protect the heart from I/R injury during ischemia and reperfusion. Similarly, calorie restriction promotes NAD+ to stimulate the Nampt-Sirt1 signaling pathway, which can protect the heart from I/R injury by up-regulating antioxidants and down-regulating pro-apoptotic molecules by activating FoxO.579,580,583,584,585
It is reported that the human cardiac fibroblasts with high expression of NOX5 and NOX4 are the main source of cardiac fibrosis related to heart failure and cardiac hypertrophy. NADPH produced by G6PD increases the level of NOXs, thereby producing most of the superoxide during the course of heart failure in patients with ischemic cardiomyopathy. Under the acceleration of TGF-β1, Nox4 mRNA is significantly upregulated and mediates the transformation of fibroblasts into myofibroblasts by activating TGF-β1-Smad2/3 signaling, which leads to cardiac fibrosis.586,587
NAD+ and kidney failure
Acute kidney injury (AKI) is a common clinical syndrome, and its prevalence and mortality increase with age. In the AKI mouse model, compared with the kidneys from 3-month-old mice, NAD+ levels in the kidneys from 20-month-old mice were significantly reduced.588 Renal ischemia impairs de novo NAD+ biosynthesis via reducing the renal expression of QPRT. Knockout of one allele of QPRT recapitulates these effects and increases susceptibility to AKI compared with control mice, which could be prevented by augmenting NAD+ metabolism with oral NAM supplementation.589,590 The robust finding that the early rise of urinary quinolinate levels and the urinary quinolinate/tryptophan ratio are related to the probability of AKI and adverse outcomes in a cohort of >300 patients indicates that impaired NAD+ metabolism leads to kidney injury in patients.591 Additionally, boosting NAD+ levels via inhibiting ACMSD (an enzyme restricts the de novo synthesis of NAD+ from tryptophan) also protects against AKI after renal I/R injury.15 The decrease in enzymes related to NAD+ de novo synthesis is due to the inhibition of the activity of PPARγ coactivator 1α (PGC-1α), which is a crucial determinant of renal recovery from AKI.592,593 Moreover, due to the decrease of 3-hydroxykynurenine (a cytotoxic metabolite of KMO), the mice that lack active kynurenine 3-monooxygenase (KMO) will not develop into AKI after I/R injury.590
The expression of PARPs in the injured kidney’s proximal tubules is upregulated from 6–12 h after I/R injury.594 Oxidative stress and DNA damage caused by I/R injury lead to excessive activation of PARPs.595 A study suggests that the activation of PARP may lead to cell death through ATP consumption and enhancement of the inflammatory cascade in mice.596 Inhibition of excessive activation of PARP can protect renal function from abnormal hemodynamics, renal metabolic disorders and renal cell apoptosis during AKI.594,597 Meanwhile, knocking out the Parp1 gene can protect mice from ischemic kidney damage.596
The simultaneous effects of impaired NAD+ biosynthesis and over depletion of NAD+ by PARPs can lead to a decrease in the level of NAD+ in the kidney during AKI. The decline in NAD+ levels therefore results in impaired metabolism and mitochondrial function via SIRTs and PGC-1α.593,598,599 SIRT3 protects against mitochondrial damage in the kidneys by attenuating oxidative stress, inhibiting inflammation and inducing autophagy through regulation of the AMPK/mTOR pathway.600,601 SIRT1 regulates the gluconeogenesis/glycolysis pathway by executing fasting signals via PGC-1α. Augmenting NAD+ induces SIRT1-mediated deacetylation of PGC-1α, thereby increasing the production of lipolysis product β-hydroxybutyrate and the production of PGE2, a prostaglandin to maintain kidney function.593,599 Additionally, supplementation with NMN restores the activity of kidney SIRT1, thereby reducing the stress response by regulating the JNK pathway and protecting mice from cisplatin-induced AKI.588
Boosting NAD+ as a therapeutic strategy
In general, intracellular NAD+ levels are maintained between 0.2 and 0.5 mM, depending on the cell type or tissue. However, the concentration and distribution of NAD+ can fluctuate in response to diverse physiological stimuli and cellular stresses. Altered NAD+ homeostasis has been linked to multiple diseases affecting different organs, including the brain and nervous system, liver, heart and kidney. NAD+ depletion is a hallmark of ageing and numerous age-related disorders.237,238,599,602,603,604,605 Therefore, boosting NAD+ offers a promising option for enhancing resilient to aging or diseases, thereby extending a healthy lifespan.23 The NAD+ level can be elevated by dietary supplementation of NAD+ precursors, such as Trp, NA, NMN and NR, inhibition of NAD+-consuming enzymes, including PARP1 and CD38, management of the NAD+ biosynthesis via controlling NAD+-biosynthesis enzymes, or improving NAD+ bioavailability through exercise and caloric restriction.
Supplementation of NAD+ precursors
NAD+ precursors can be used as a nutritional supplement to improve a broad spectrum of physiological functions and pathological processes.606,607,608,609,610,611 As highlighted in Table 1, the therapeutic and preventive efficacy of NAD+ boosters, especially the soluble and orally bioavailable endogenous molecules NR, NAM and Niacin, have been assessed in a series of clinical trials in humans.
NAD+ precursor: NMN
NMN administration can effectively and rapidly enhance NAD+ biosynthesis in various tissues, even in the brain, with a promising safety.612 Aged animals are more responsive to NAD+ replenishment by NMN treatment than the young one due to the age-related decline in NAD+ availability. NMN treatment exerts beneficial effects on insulin secretion and insulin sensitivity in age- and diet-induced diabetes by restoring NAD+ biosynthesis. Long-term NMN administration rescues the age-associated decline in physiological function, including mitochondrial function, energy metabolism, gene expression changes, insulin sensitivity and plasma lipid profile, thereby improving physical activities, such as bone density and eye function.612 NMN treatment improves the neuronal functions, including rescuing the memory and cognition in rodent models for Alzheimer’s disease, protecting neurons from cell death after intracerebral hemorrhage or ischemia, recovering severe retinal degeneration and restoring the age-associated loss of the neural stem/progenitor pool. Besides, the NMN administration also exerts a pleiotropic effect on acute heart failure and renal injury.23,613,614 It is worth noting that clinical trials of NAM have been initiated in patients with cancers including bladder cancer, non-small-cell lung carcinoma, non-melanoma skin cancer, non-Hodgkin’s lymphoma and multiple myeloma (Table 1).
Despite that the NMN bioavailability is evidenced by the rapid absorption and convention of administered NMN to NAD+ in various organs, including skeletal muscle, kidney and liver, the transportation of NMN into cells remains unclear. NMN may be directly taken up by specific transporters, as the NAD+ content in peripheral organs, such as the gut, is immediately increased by NMN administration.23,614 However, in vitro studies demonstrate that Nrk1/2 depletion abandons the incorporation of NMN into NAD+ synthesis. Moreover, NMN administration can significantly elevate NAD+ biosynthesis in white adipose tissue, heart and the liver, but not the NAD+ content in brown adipose tissue and kidney, suggesting a tissue- and cell-type-specific transportation of NMN to cells or tissues for NAD+ biosynthesis presumably due to the deferent expression pattern of NRK1.612,615 Therefore, the identification of the putative NMN transporter and its tissue specific expression pattern will help assess the cell type or tissue-specific preference of NMN, paving the way for precious clinical application of NMN in different conditions.
NAD+ precursor: NR
NR is another natural compound that displays a surprisingly robust effect on systemic NAD+ metabolism. A large phase clinical trial of NR has been registered on a broad range of pathologies, including infection, neoplasms, aging-related diseases and disorders that occur in the circulatory system, genitourinary system, nervous system and skin (Table 1). Oral NR supplementation in aged participants elevates the muscle NAD+ metabolome, ameliorates metabolic dysfunction, depresses levels of circulating inflammatory cytokines and increases the anti-inflammatory molecule adiponectin in aged human.606,616,617 Dietary administration with NR improves cold tolerance, endurance and energy expenditure. NR protects mice from HFD-induced body weight gain, enhances the liver weight regain by promoting hepatocyte replication and increasing hepatic ATP content in the regenerating liver.618 NR exhibits beneficial effects in several muscle disorders through improving mitochondrial function and decreasing the UPRmt in heart failure mice. NR boosts the NAD+ biosynthesis to prevent DNA damage and tumorigenesis. NAD+ repletion with NR may reverse NAFLD by improving mitochondrial function in both HFHS-fed mice and HFC‐fed Apoe−/− mice.619 It has been reported that NR has a variety of compelling benefits in the nervous system, including improving the cognitive function and synaptic plasticity in Alzheimer’s disease and preventing noise-induced hearing loss. NR restores the age-associated decline in the metabolic cycle and circadian behavioral, including the BMAL1 activity, oscillation mitochondrial respiration, rhythmic transcription and late evening activity to youthful levels.242
NR can be directly transported by ENTs into cells and enhance NAD+ biosynthesis bypassing the NAMPT-mediated salvage pathway. However, the short stability of NR in circulation and rate-limited utilization by the expression of NRKs restrict its clinical application. Dihydronicotinamide riboside (NRH), a reduced form of NR with oral bioavailability, is developed to overcome these limitations. NRH provides better efficacy to boost NAD+ synthesis using an NRK1/2-independent pathway compared with NR and NMN, preventing cisplatin-induced acute kidney injury. The potent and efficient NRH that serve as an NAD+ booster, offers a promising option to increase NAD+ levels.620,621
NAD+ precursors: NAM and NA
NAM is an uncharged molecule that can diffuse rapidly across the plasma, supporting the NAD+ biosynthesis for most tissues in vivo.91 Oral administrated NAM is converted into NA in the small intestine and colon by nicotinamidase PncA of gut microbiota. Gut microbiota-mediated deamidation of NAM is necessary and responsible for the NAD+ biosynthesis in various organs, including the kidney, the liver and the colon.101 NAM protects against streptozotocin (STZ)-induced diabetes by recovering the NAD+ decline in pancreatic islet cells. NAM treatment exhibits profound metabolic improvements in obesity and type 2 diabetes mouse model. However, there are several side effects caused by NAM, limiting the application of NAM. Firstly, NAM exerts feedback inhibition on SIRT1 activity. Secondly, it has been indicated that high doses or long-term NAM reduces the methyl group availability and cellular methylation potential via promoting the methyl sink in the form of 1‐MNA. Consistent with this hypothesis, dietary methionine supplementation attenuates the development of steatohepatitis induced by high doses of NAM.66,622
NA is effective in treating dyslipidemia due to its cholesterol lowering actions. NA treatment decreases the serum low-density lipoprotein and triglyceride content and elevates the high-density lipoprotein levels. Nevertheless, the clinical application of NA is limited because the pharmacological dosing of it induces cutaneous flushing via activation of a G protein-coupled receptor, GPR109A. Given this undesirable effect, several niacin derivatives with prolonged release time, including enduracin, niaspan and acipimox, have been developed. Therefore, niacin has been replaced by its derivatives in the clinical treatment of hyperlipidemia. The Acipimox can directly affect mitochondrial function in skeletal muscle of patients with type 2 diabetes.623
The side effects of NAD+ boosting
The aforementioned findings suggest that elevating NAD+ levels by administration of NAD+ precursors, including NMN, NR, NAM, and NA, is a rational therapeutic strategy to improve a healthy lifespan. Given that NAD+-depleting drugs exhibit anti-tumor potential due to their impact on DNA repair and inflammation, long-term boosting NAD+ might increase the risk of driving tumor growth. Moreover, the detrimental side effects of NAD+ and its intermediates may be caused by the NAD+-dependent sirtuins that have both oncogenic and tumor suppressive activity in different contexts. Consistent with this hypothesis, NMN treatment accelerates pancreatic cancer progression via creating an inflammatory environment.66,260 Thus, future clinical studies are necessary to assess the long-term safety of NAD+ precursors in human therapeutics.
Inhibition of NAD+ consumption
The excessive activation of PARPs or CD38 causes a NAD+ consumption up to the extent that leads to ATP decline, energy loss and cell death.52,184,624,625 Thus, reducing the NAD+ consumption via suppressing PARPs or CD38 is also a strategy to boost NAD+.53,626
Accumulating evidence demonstrates that aberrant PARPs activation by DNA damage causes NAD+ depletion, contributing to the progression of tumorigenesis and neurodegenerative disorders that involve DNA repair defects. To date, PARP inhibitors, including niraparib, rucaparib and olaparib have been approved by US-FDA to treat cancers, including prostate cancer, breast cancer and ovarian cancer, through disrupting DNA repair and replication pathways.627,628,629 PARPs-mediated ADP-ribosylation accounts for up to 90% of the cellular intracellular NAD+ consumption, leading to reduced NAD+ availability for sirtuins. Therefore, genetic ablation or pharmacological inhibition of Parp-1 enhances the Sirt1 activity through restoring the NAD+ content, providing a protection benefit for various tissues, including the liver, muscle and brown adipose tissue.630,631 NADP+ has been demonstrated as an endogenous inhibitor of PARPs, which extend the therapeutic effect of PARP inhibitors on cancers with higher levels of NADP+.184
A variety of flavonoids, including apigenin, quercetin, luteolin, kuromanin, and luteolinidin, exhibit inhibitory effect on CD38 activity.23 78c is a highly specific CD38 inhibitor, which has greater potency than the flavonoids in reversing NAD+ decline during aging, thereby improving several age-associated physiological functions, including cardiac function, muscle function and glucose tolerance.53 Interestingly, 78c elevates NAD+ to a higher level in mouse liver than that in muscle, arguing a tissue specific CD38 activity.632 Thus, further studies uncovering the tissue specific CD38 activities will facilitate the development of clinical application of CD38 inhibitors.
Controlling NAD+-biosynthesis pathway
Enhancing NAD+-biosynthesis is one alternative approach to elevate NAD+ concentration through either increasing the activity of enzymes in NAD+-biosynthetic pathway or inhibiting the activity of enzymes in the side branch pathway.633
NAD+ is synthesized from both de novo pathway and the salvage pathway.634 The distinct expression level of NAPRT in various healthy tissues determines the choice of NAD+ biosynthetic pathway for survival. Cancers arising from tissue with a highly NAPRT are expression completely and irreversibly dependent on the NAPRT-regulated de novo pathway, while cancers deriving from tissues with the low level of NAPRT mainly rely on the NAMPT-mediated NAD+ salvage pathway. This deferent dependence renders cancer cells resistant to inhibition of NAMPT by other NAD+ synthesis.408 In line with this hypothesis, the loss of NAPRT in both RCC cell lines and EMT-subtype gastric cell lines renders the cells hypersensitive to NAMPT inhibitors, such as FK866, and KPT-9274, suggesting that NAMPT inhibitors may be a promising strategy for NAPRT deficient tumors.421,423 Moreover, bacteria-mediated deamidated NAD+ biosynthesis also rescues NAMPTi-induced toxicity in cancer cells and xenograft tumors.101
The enzymatic ability of NAMPT can be enhanced by pharmacological agents, P7C3 or SBI-797812. P7C3 is a NAMPT enhancer with good bioavailability and nontoxicity. It has been demonstrated that P7C3 and its analogs have neuroprotective efficacy in a broad range of preclinical rodent and nonhuman primate models relying on the activation of NAMPT.635,636,637,638 Therefore, the neuroprotective activity of P7C3 offers a new pharmacotherapy for age-associated ALS, Alzheimer’s disease and Parkinson’s disease.635,637,638 SBI-797812 activates NAMPT via stabilizing the NAMPT phosphorylation at His247, enhancing the efficiency of NMN generation, providing another option to raise NAD+.639 Together, the NAMPT enhancers, P7C3 and SBI-797812, warrant further study for the clinical treatment of neuron related diseases.
The NAD+-biosynthetic pathway can also be increased by blocking the side branch to prevent the escape of intermediates. Overexpressing ACMSD reduces the NAD+ level by dissipating ACMS from the de novo NAD+ synthesis into the side branch pathway for acetyl-CoA production, while inhibiting ACMSD elevates NAD+ concentration. The high expression of ACMSD in the kidney and liver offers the therapeutic potential of ACMSD inhibitors, such as TES-991 and TES-1025, for renal and hepatic dysfunction.15,640 ACMSD may also be a novel target for Parkinson’s disease, as it inhibits the generation of neurotoxin quinolinic acid in the kynurenine pathway.641 Similarly, NNMT shifts the NAM into producing 1-methylnicotinamide, leading to impaired salvage pathway. WAT- and liver-specific knockdown of NNMT prevent diet-induced obesity through enhancing the energy expenditure. The effect of NNMT is achieved by its effect on histone methylation. Pharmacological inhibition of NNMT significantly shows benefits diet-induced obese mice, including reducing the body weight gain and adipocyte size, and decreasing serum cholesterol levels.642,643 These results suggest that NNMT is an appealing target for obesity and type 2 diabetes treatment.642 Together, ACMSD and NNMT provide novel targets for modulating NAD+ homeostasis, which will be of great importance to determine whether ACMSD and NNMT inhibitors can increase NAD+ levels and achieve therapeutic effects.
Increasing NAD+ bioavailability
Intracellular NAD+ levels can also be increased by energy stress, including fasting, glucose restriction, caloric restriction (CR) and exercise.599 CR-mediated NAD+ boosting depends on the NAD+ salvage biosynthesis rather than de novo pathway via elevating the NAMPT expression.237,238,644,645,646 CR restores the age-associated circadian decline via sharpening circadian control of NAD+ metabolism and NAD+/SIRT1-modulated epigenetic modification.647,648,649,650 It has been shown that both long-term and short-term CR rescue the large elastic artery stiffening and endothelial dysfunction.651,652 Similarly, CR-boosted NAD+ level protects the brain against aging and diseases through attenuating plasma membrane lipid peroxidation, protein carbonyls, nitrotyrosine and oxidative stress.653
Beyond CR, the exercise has attracted growing attention due to its benefits on health. In this context, exercise could also increase the NAD+ level and SIRT1 activity due to an increase in NAMPT.605 It has been reported that the NAMPT protein levels in athletes’ skeletal muscle is much higher than in type 2 diabetic, sedentary obese and nonobese subjects. Furthermore, exercise training induces a robust increase in the skeletal muscle of NAMPT protein in sedentary nonobese subjects.605 The exercise-mediated NAD+ boosting by inducing Nampt is a response strategy for energy stress, which is abolished by depletion of the energy sensor AMPK.603 Intriguingly, the gut microenvironment, especially the host-bacteria interactions also contributes to NAD+ metabolism. The gut microbiota-derived deamidated pathway facilitates the utilization of NAM or NR for hepatic NAD+ synthesis, suggesting that manipulation of microbiota might offer a new option to manipulate NAD+ metabolism.101 Taken together, both caloric restriction and exercise provide the potential therapeutic strategies in therapy against pathologies related to NAD+ decline.
Concluding remarks
The levels and compartmentalization of NAD+ dictate energy state that impinges on normal physiological and biological responses, as indicated by the regulatory role of NAD+ in proper redox homeostasis, genomic stability, gene expression, circadian clock, inflammation, metabolism, cellular bioenergetics, mitochondrial homeostasis and adaptive stress responses. A healthy lifestyle and exercise are non-pharmacologic strategies to improve the body’s resilience and extend healthy lifespan through enhancing NAD+ levels. NAD+ boosters can be applied for a broad spectrum of NAD+ deficiency related pathologies, such as infection, cancer, metabolic diseases, acute injury, aging and aging-related neurodegenerative disorders. Conceivably, this could be achieved by boosting NAD+ via enhancing the NAD+ generation and diminishing NAD+ consumption.
Despite exciting and emerging strides in NAD+ biology, there are a variety of outstanding questions that warrant future systematic exploitation to accelerate the translation of remarkable bench work to effective clinical application in humans. The first interesting question is that the precise mechanisms executing the beneficial effects of NAD+ and its metabolites on pathologies and lifespan remain elusive. Further investigation understanding the landscape of NAD+ in response to diseases and identifying the specific effector molecules for each NAD+ precursors at different time points provide critical insights into development of effective interventions for various physiologies. Secondly, the systemic NAD+ metabolome is largely unexplored. Are there any tissue specificities for NAD+ boosting, such tissue preferences of distinct NAD+ precursors? What is the crosstalk with the NAD+ systems of each organ? What is the distinct NAD+ metabolome in each tissue? In spite of growing interest in the use of NAD+ precursors as a strategy for healthy aging, the in vivo pharmacokinetics remain poorly understood. The efficacy of NAD+ boosters, the therapeutic dosages and favorable administration routes should be optimized for different diseases in humans. It is also essential to fully assess the unforeseen side effects of long-term NAD+ boosting. This task requires the development of new technologies to enable the simplifying, accurate and reproducible monitoring of dynamic NAD+ and its metabolites in patients and healthy individuals.
References
Harden, A. & Y., W. J. The alcoholic ferment of yeast-juice part II.–the coferment of yeast-juice. Proc. R. Soc. Lond. B Biol. Sci 78, 7 (1906).
Warburg, O. & Christian, W. J. B. Z. Pyridin, the hydrogen-transferring component of the fermentation enzymes (pyridine nucleotide). Biochem. Z. 287, 291–328 (1936).
Chambon, P., Weill, J. D. & Mandel, P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 11, 39–43 (1963).
Frye, R. A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 260, 273–279 (1999).
Imai, S., Armstrong, C. M., Kaeberlein, M. & Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 (2000).
Landry, J. et al. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl Acad. Sci. USA 97, 5807–5811 (2000).
Chen, S. H. & Yu, X. Human DNA ligase IV is able to use NAD+ as an alternative adenylation donor for DNA ends ligation. Nucleic Acids Res. 47, 1321–1334 (2019).
Bird, J. G. et al. The mechanism of RNA 5’ capping with NAD+, NADH and desphospho-CoA. Nature 535, 444–447 (2016).
Chen, Y. G., Kowtoniuk, W. E., Agarwal, I., Shen, Y. & Liu, D. R. LC/MS analysis of cellular RNA reveals NAD-linked RNA. Nat. Chem. Biol. 5, 879–881 (2009).
Cahova, H., Winz, M. L., Hofer, K., Nubel, G. & Jaschke, A. NAD captureSeq indicates NAD as a bacterial cap for a subset of regulatory RNAs. Nature 519, 374–377 (2015).
Walters, R. W. et al. Identification of NAD+ capped mRNAs in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA 114, 480–485 (2017).
Jiao, X. et al. 5’ End nicotinamide adenine dinucleotide cap in human cells promotes RNA decay through DXO-mediated deNADding. Cell 168, 1015–1027 e1010 (2017).
Wang, J. et al. Quantifying the RNA cap epitranscriptome reveals novel caps in cellular and viral RNA. Nucleic Acids Res. 47, e130 (2019).
Anderson, R. M., Bitterman, K. J., Wood, J. G., Medvedik, O. & Sinclair, D. A. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181–185 (2003).
Katsyuba, E. et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 563, 354–359 (2018).
Badawy, A. A. Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int. J. Tryptophan Res. 10, 1178646917691938 (2017).
Youn, H. S. et al. Structural insights into the quaternary catalytic mechanism of hexameric human quinolinate phosphoribosyltransferase, a key enzyme in de novo NAD biosynthesis. Sci. Rep. 6, 19681 (2016).
Marletta, A. S. et al. Crystal structure of human nicotinic acid phosphoribosyltransferase. FEBS Open Bio 5, 419–428 (2015).
Brazill, J. M., Li, C., Zhu, Y. & Zhai, R. G. NMNAT: It’s an NAD(+) synthase… It’s a chaperone… It’s a neuroprotector. Curr. Opin. Genet. Dev. 44, 156–162 (2017).
Rizzi et al. A novel deamido-NAD+-binding site revealed by the trapped NAD-adenylate intermediate in the NAD+ synthetase structure. Structure 6, 1129–1140 (1998).
Braidy, N. et al. Role of nicotinamide adenine dinucleotide and related precursors as therapeutic targets for age-related degenerative diseases: rationale, biochemistry, pharmacokinetics, and outcomes. Antioxid. Redox Signal. 30, 251–294 (2019).
Wang, T. et al. Structure of Nampt/PBEF/visfatin, a mammalian NAD+ biosynthetic enzyme. Nat. Struct. Mol. Biol. 13, 661–662 (2006).
Rajman, L., Chwalek, K. & Sinclair, D. A. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 27, 529–547 (2018).
Zhou, T. et al. Structure of human nicotinamide/nicotinic acid mononucleotide adenylyltransferase. Basis for the dual substrate specificity and activation of the oncolytic agent tiazofurin. J. Biol. Chem. 277, 13148–13154 (2002).
Werner, E., Ziegler, M., Lerner, F., Schweiger, M. & Heinemann, U. Crystal structure of human nicotinamide mononucleotide adenylyltransferase in complex with NMN. FEBS Lett. 516, 239–244 (2002).
Chalkiadaki, A. & Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 15, 608–624 (2015).
Ke, Y., Zhang, J., Lv, X., Zeng, X. & Ba, X. Novel insights into PARPs in gene expression: regulation of RNA metabolism. Cell. Mol. Life Sci.76, 3283–3299 (2019).
Gupte, R., Liu, Z. & Kraus, W. L. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 31, 101–126 (2017).
Munnur, D. et al. Reversible ADP-ribosylation of RNA. Nucleic Acids Res. 47, 5658–5669 (2019).
Larsen, S. C., Hendriks, I. A., Lyon, D., Jensen, L. J. & Nielsen, M. L. Systems-wide analysis of serine ADP-ribosylation reveals widespread occurrence and site-specific overlap with phosphorylation. Cell Rep. 24, 2493–2505 e2494 (2018).
Zhao, Y. J., Lam, C. M. & Lee, H. C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal. 5, ra67 (2012).
Fliegert, R. et al. Adenine nucleotides as paracrine mediators and intracellular second messengers in immunity and inflammation. Biochem. Soc. Trans. 47, 329–337 (2019).
Summers, D. W., Gibson, D. A., DiAntonio, A. & Milbrandt, J. SARM1-specific motifs in the TIR domain enable NAD+ loss and regulate injury-induced SARM1 activation. Proc. Natl Acad. Sci. USA 113, E6271–e6280 (2016).
Essuman, K. et al. The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron 93, 1334–1343.e1335 (2017).
Katsyuba, E., Romani, M., Hofer, D. & Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2, 9–31 (2020).
Asher, G. et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134, 317–328 (2008).
Canto, C. et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 15, 838–847 (2012).
Nakahata, Y. et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 134, 329–340 (2008).
Smith, B. C., Hallows, W. C. & Denu, J. M. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal. Biochem. 394, 101–109 (2009).
Pacholec, M. et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 285, 8340–8351 (2010).
Gerhart-Hines, Z. et al. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol. Cell 44, 851–863 (2011).
Jin, L. et al. Biochemical characterization, localization, and tissue distribution of the longer form of mouse SIRT3. Protein Sci. 18, 514–525 (2009).
Hirschey, M. D. et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 44, 177–190 (2011).
Madsen, A. S. et al. Investigating the sensitivity of NAD+-dependent sirtuin deacylation activities to NADH. J. Biol. Chem. 291, 7128–7141 (2016).
Roessler, C., Tüting, C., Meleshin, M., Steegborn, C. & Schutkowski, M. A novel continuous assay for the deacylase sirtuin 5 and other deacetylases. J. Med. Chem. 58, 7217–7223 (2015).
Kugel, S. et al. Identification of and molecular basis for SIRT6 loss-of-function point mutations in cancer. Cell Rep. 13, 479–488 (2015).
Mendoza-Alvarez, H. & Alvarez-Gonzalez, R. Poly(ADP-ribose) polymerase is a catalytic dimer and the automodification reaction is intermolecular. J. Biol. Chem. 268, 22575–22580 (1993).
Amé, J. C. et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J. Biol. Chem. 274, 17860–17868 (1999).
Bai, P. & Cantó, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 16, 290–295 (2012).
Cakir-Kiefer, C., Muller-Steffner, H., Oppenheimer, N. & Schuber, F. Kinetic competence of the cADP-ribose-CD38 complex as an intermediate in the CD38/NAD+ glycohydrolase-catalysed reactions: implication for CD38 signalling. Biochem. J. 358, 399–406 (2001).
Horenstein, A. L. et al. NAD+-metabolizing ectoenzymes in remodeling tumor-host interactions: the human myeloma model. Cells 4, 520–537 (2015).
Camacho-Pereira, J. et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 23, 1127–1139 (2016).
Tarrago, M. G. et al. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD(+) decline. Cell Metab. 27, 1081–1095 e1010 (2018).
Pehar, M., Harlan, B. A., Killoy, K. M. & Vargas, M. R. Nicotinamide adenine dinucleotide metabolism and neurodegeneration. Antioxid. Redox Signal. 28, 1652–1668 (2018).
Real, A. M., Hong, S. & Pissios, P. Nicotinamide N-oxidation by CYP2E1 in human liver microsomes. Drug Metab. Dispos. 41, 550–553 (2013).
Aksoy, S., Szumlanski, C. L. & Weinshilboum, R. M. Human liver nicotinamide N-methyltransferase. cDNA cloning, expression, and biochemical characterization. J. Biol. Chem. 269, 14835–14840 (1994).
Felsted, R. L. & Chaykin, S. N1-methylnicotinamide oxidation in a number of mammals. J. Biol. Chem. 242, 1274–1279 (1967).
Chaykin, S., Dagani, M., Johnson, L. & Samli, M. T. H. E. Fate of nicotinamide in the mouse. Urinary metabolites. J. Biol. Chem. 240, 932–938 (1965).
Shibata, K., Kakehi, H. & Matsuo, H. Niacin catabolism in rodents. J. Nutr. Sci. Vitaminol. 36, 87–98 (1990).
Bockwoldt, M. et al. Identification of evolutionary and kinetic drivers of NAD-dependent signaling. Proc. Natl Acad. Sci. USA 116, 15957–15966 (2019).
Burgos, E. S. & Schramm, V. L. Weak coupling of ATP hydrolysis to the chemical equilibrium of human nicotinamide phosphoribosyltransferase. Biochemistry 47, 11086–11096 (2008).
Kang-Lee, Y. A. et al. Metabolic effects of nicotinamide administration in rats. J. Nutr. 113, 215–221 (1983).
Hong, S. et al. Nicotinamide N-methyltransferase regulates hepatic nutrient metabolism through Sirt1 protein stabilization. Nat. Med. 21, 887–894 (2015).
Sallin, O. et al. Semisynthetic biosensors for mapping cellular concentrations of nicotinamide adenine dinucleotides. eLife 7, e32638 (2018).
Cambronne, X. A. et al. Biosensor reveals multiple sources for mitochondrial NAD+. Science 352, 1474–1477 (2016).
Canto, C., Menzies, K. J. & Auwerx, J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 22, 31–53 (2015).
Nakagawa, T., Lomb, D. J., Haigis, M. C. & Guarente, L. SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137, 560–570 (2009).
Houtkooper, R. H., Cantó, C., Wanders, R. J. & Auwerx, J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 31, 194–223 (2010).
Dall, M. et al. Mitochondrial function in liver cells is resistant to perturbations in NAD(+) salvage capacity. J. Biol. Chem. 294, 13304–13326 (2019).
van Roermund, C. W., Elgersma, Y., Singh, N., Wanders, R. J. & Tabak, H. F. The membrane of peroxisomes in Saccharomyces cerevisiae is impermeable to NAD(H) and acetyl-CoA under in vivo conditions. EMBO J. 14, 3480–3486 (1995).
Nikiforov, A., Dölle, C., Niere, M. & Ziegler, M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 286, 21767–21778 (2011).
Brown, K. et al. SIRT3 reverses aging-associated degeneration. Cell Rep. 3, 319–327 (2013).
Davila, A. et al. Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. eLife 7, e33246 (2018).
de Souza Chaves, I. et al. The mitochondrial NAD(+) transporter (NDT1) plays important roles in cellular NAD(+) homeostasis in Arabidopsis thaliana. Plant J. 100, 487–504 (2019).
Todisco, S., Agrimi, G., Castegna, A. & Palmieri, F. Identification of the mitochondrial NAD+ transporter in Saccharomyces cerevisiae. J. Biol. Chem. 281, 1524–1531 (2006).
van Roermund, C. W. et al. The peroxisomal NAD carrier from arabidopsis imports NAD in exchange with AMP. Plant Physiol. 171, 2127–2139 (2016).
Palmieri, F. et al. Molecular identification and functional characterization of Arabidopsis thaliana mitochondrial and chloroplastic NAD+ carrier proteins. J. Biol. Chem. 284, 31249–31259 (2009).
Haferkamp, I. et al. A candidate NAD+ transporter in an intracellular bacterial symbiont related to Chlamydiae. Nature 432, 622–625 (2004).
Berger, F., Lau, C., Dahlmann, M. & Ziegler, M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 280, 36334–36341 (2005).
Yang, H. et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130, 1095–1107 (2007).
Pittelli, M. et al. Inhibition of nicotinamide phosphoribosyltransferase: cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J. Biol. Chem. 285, 34106–34114 (2010).
Eto, K. et al. Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science 283, 981–985 (1999).
Mráček, T., Drahota, Z. & Houštěk, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 401-410, 2013 (1827).
Pardo, B. & Contreras, L. Redox shuttles in the brain. Neural Metab. In Vivo 4, 841–883 (2012).
Contreras, L. & Satrústegui, J. Calcium signaling in brain mitochondria: interplay of malate aspartate NADH shuttle and calcium uniporter/mitochondrial dehydrogenase pathways. J. Biol. Chem. 284, 7091–7099 (2009).
Kauppinen, R. A., Sihra, T. S. & Nicholls, D. G. Aminooxyacetic acid inhibits the malate-aspartate shuttle in isolated nerve terminals and prevents the mitochondria from utilizing glycolytic substrates. Biochim. Biophys. Acta 930, 173–178 (1987).
Ramos, M. et al. Developmental changes in the Ca2+-regulated mitochondrial aspartate-glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Brain Res. Dev. Brain Res. 143, 33–46 (2003).
Rubi, B., del Arco, A., Bartley, C., Satrustegui, J. & Maechler, P. The malate-aspartate NADH shuttle member Aralar1 determines glucose metabolic fate, mitochondrial activity, and insulin secretion in beta cells. J. Biol. Chem. 279, 55659–55666 (2004).
Kane, D. A. Lactate oxidation at the mitochondria: a lactate-malate-aspartate shuttle at work. Front. Neurosci. 8, 366 (2014).
Gellerich et al. Cytosolic Ca2+ regulates the energization of isolated brain mitochondria by formation of pyruvate through the malate-aspartate shuttle. Biochem. J. 443, 747–755 (2012).
Liu et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab. 27, 1067–1080.e1065 (2018).
Kawai, S. & Murata, K. Structure and function of NAD kinase and NADP phosphatase: key enzymes that regulate the intracellular balance of NAD(H) and NADP(H). Biosci. Biotechnol. Biochem. 72, 919–930 (2008).
Cracan, V., Titov, D. V., Shen, H., Grabarek, Z. & Mootha, V. K. A genetically encoded tool for manipulation of NADP(+)/NADPH in living cells. Nat. Chem. Biol. 13, 1088–1095 (2017).
Miller, C. G. & Schmidt, E. E. Disulfide reductase systems in liver. Br. J. Pharmacol. 176, 532–543 (2019).
Sies, H., Berndt, C. & Jones, D. P. Oxidative stress. Annu. Rev. Biochem. 86, 715–748 (2017).
Gelman, S. J. et al. Consumption of NADPH for 2-HG synthesis increases pentose phosphate pathway flux and sensitizes cells to oxidative stress. Cell Rep. 22, 512–522 (2018).
Zhao, Y. et al. SoNar, a highly responsive NAD+/NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. Cell Metab.21, 777–789 (2015).
Ratajczak, J. et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 7, 13103 (2016).
Grozio, A. et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 1, 47–57 (2019).
Zhao, Z. Y. et al. A cell-permeant mimetic of NMN activates SARM1 to produce cyclic ADP-ribose and induce non-apoptotic cell death. iScience 15, 452–466 (2019).
Shats, I. et al. Bacteria boost mammalian host NAD metabolism by engaging the deamidated biosynthesis pathway. Cell Metab. 31, 564–579.e567 (2020).
Sies, H. Oxidative stress: introductory remarks. In Oxidative Stress. 1–8 (Academic Press, 1985).
Luo, H., Chiang, H. H., Louw, M., Susanto, A. & Chen, D. Nutrient sensing and the oxidative stress response. Trends Endocrinol. Metab. 28, 449–460 (2017).
Schieber, M. & Chandel, N. S. ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462 (2014).
Scaturro, P. & Pichlmair, A. Oxeiptosis: a discreet way to respond to radicals. Curr. Opin. Immunol.56, 37–43 (2019).
Le Belle, J. E. et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 8, 59–71 (2011).
Tan, B. L., Norhaizan, M. E., Liew, W. P. & Sulaiman Rahman, H. Antioxidant and oxidative stress: a mutual interplay in age-related diseases. Front. Pharmacol. 9, 1162 (2018).
Wang et al. NAD(+) administration decreases doxorubicin-induced liver damage of mice by enhancing antioxidation capacity and decreasing DNA damage. Chem. Biol. Interact. 212, 65–71 (2014).
Blacker et al. Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat. Commun. 5, 3936 (2014).
Sultana et al. Garlic activates SIRT-3 to prevent cardiac oxidative stress and mitochondrial dysfunction in diabetes. Life Sci. 164, 42–51 (2016).
Lu et al. A small molecule activator of SIRT3 promotes deacetylation and activation of manganese superoxide dismutase. Free Radic. Biol. Med. 112, 287–297 (2017).
Bause, A. S. & Haigis, M. C. SIRT3 regulation of mitochondrial oxidative stress. Exp. Gerontol. 48, 634–639 (2013).
Gorrini, C., Harris, I. S. & Mak, T. W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947 (2013).
Tiganis, T. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol. Sci. 32, 82–89 (2011).
Holmstrom, K. M. & Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421 (2014).
Moldogazieva, N. T., Mokhosoev, I. M., Feldman, N. B. & Lutsenko, S. V. ROS and RNS signalling: adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 52, 507–543 (2018).
Bedard, K. & Krause, K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313 (2007).
Nishino et al. The C-terminal peptide plays a role in the formation of an intermediate form during the transition between xanthine dehydrogenase and xanthine oxidase. FEBS J. 282, 3075–3090 (2015).
Battelli, M. G., Polito, L. & Bolognesi, A. Xanthine oxidoreductase in atherosclerosis pathogenesis: not only oxidative stress. Atherosclerosis 237, 562–567 (2014).
Tejero, J. & Stuehr, D. Tetrahydrobiopterin in nitric oxide synthase. IUBMB Life 65, 358–365 (2013).
Smith, B. C., Underbakke, E. S., Kulp, D. W., Schief, W. R. & Marletta, M. A. Nitric oxide synthase domain interfaces regulate electron transfer and calmodulin activation. Proc. Natl Acad. Sci. USA 110, E3577–E3586 (2013).
Roy, P., Roy, S. K., Mitra, A. & Kulkarni, A. P. Superoxide generation by lipoxygenase in the presence of NADH and NADPH. Biochim. Biophys. Acta 1214, 171–179 (1994).
Cho, K. J., Seo, J. M. & Kim, J. H. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol. Cells 32, 1–5 (2011).
Shintoku et al. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 108, 2187–2194 (2017).
Bradfield, J. Y., Lee, Y. H. & Keeley, L. L. Cytochrome P450 family 4 in a cockroach: molecular cloning and regulation by regulation by hypertrehalosemic hormone. Proc. Natl Acad. Sci. USA 88, 4558–4562 (1991).
Zangar, R. C., Davydov, D. R. & Verma, S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol. 199, 316–331 (2004).
Dickinson, B. C., Peltier, J., Stone, D., Schaffer, D. V. & Chang, C. J. Nox2 redox signaling maintains essential cell populations in the brain. Nat. Chem. Biol. 7, 106–112 (2011).
Levine, R. L., Mosoni, L., Berlett, B. S. & Stadtman, E. R. Methionine residues as endogenous antioxidants in proteins. Proc. Natl Acad. Sci. USA 93, 15036–15040 (1996).
Fernandez-Marcos, P. J. & Nóbrega-Pereira, S. NADPH: new oxygen for the ROS theory of aging. Oncotarget 7, 50814–50815 (2016).
Adrian, M. D. & Alan, G. H. Why antioxidant therapies have failed in clinical trials. J. Theor. Biol. 457, 1–5 (2018).
Weydert, C. J. & Cullen, J. J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 5, 51–66 (2010).
Ren, X. et al. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. redox Signal. 27, 989–1010 (2017).
Filomeni, G., De Zio, D. & Cecconi, F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 22, 377–388 (2015).
Zhong, L. & Holmgren, A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J. Biol. Chem. 275, 18121–18128 (2000).
Ahn, B. H. et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl Acad. Sci. USA 105, 14447–14452 (2008).
Ding, Y. et al. Sirtuin 3 is required for osteogenic differentiation through maintenance of PGC-1ɑ-SOD2-mediated regulation of mitochondrial function. Int. J. Biol. Sci. 13, 254–264 (2017).
Finley, L. W. et al. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PloS ONE 6, e23295 (2011).
Someya, S. et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812 (2010).
Chen, Y. et al. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 12, 534–541 (2011).
Sundaresan, N. R. et al. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 119, 2758–2771 (2009).
Bringman-Rodenbarger, L. R., Guo, A. H., Lyssiotis, C. A. & Lombard, D. B. Emerging roles for SIRT5 in metabolism and cancer. Antioxid. Redox Signal. 28, 677–690 (2018).
Kim, H. S. et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17, 41–52 (2010).
Niedernhofer, L. J. et al. Nuclear genomic instability and aging. Annu. Rev. Biochem. 87, 295–322 (2018).
Sancar, A., Lindsey-Boltz, L. A., Unsal-Kaçmaz, K. & Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 (2004).
Lagunas-Rangel, F. A. Current role of mammalian sirtuins in DNA repair. DNA Repair 80, 85–92 (2019).
Fania, L. et al. Role of nicotinamide in genomic stability and skin cancer chemoprevention. Int. J. Mol. Sci. 20, 5946 (2019).
Hurtado-Bagès, S., Knobloch, G., Ladurner, A. G. & Buschbeck, M. The taming of PARP1 and its impact on NAD(+) metabolism. Mol. Metab. 38, 100950 (2020).
Chang, H. H. Y., Pannunzio, N. R., Adachi, N. & Lieber, M. R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 18, 495–506 (2017).
Hnízda, A. & Blundell, T. L. Multicomponent assemblies in DNA-double-strand break repair by NHEJ. Curr. Opin. Struct. Biol. 55, 154–160 (2019).
Feng, H., Parker, J. M., Lu, J. & Cao, W. Effects of deletion and site-directed mutations on ligation steps of NAD+-dependent DNA ligase: a biochemical analysis of BRCA1 C-terminal domain. Biochemistry 43, 12648–12659 (2004).
Satoh, M. S., Poirier, G. G. & Lindahl, T. NAD(+)-dependent repair of damaged DNA by human cell extracts. J. Biol. Chem. 268, 5480–5487 (1993).
Satoh, M. S., Poirier, G. G. & Lindahl, T. Dual function for poly(ADP-ribose) synthesis in response to DNA strand breakage. Biochemistry 33, 7099–7106 (1994).
Munnur, D. & Ahel, I. Reversible mono-ADP-ribosylation of DNA breaks. FEBS J. 284, 4002–4016 (2017).
Talhaoui, I. et al. Poly(ADP-ribose) polymerases covalently modify strand break termini in DNA fragments in vitro. Nucleic Acids Res. 44, 9279–9295 (2016).
Matta, E., Kiribayeva, A., Khassenov, B., Matkarimov, B. T. & Ishchenko, A. A. Insight into DNA substrate specificity of PARP1-catalysed DNA poly(ADP-ribosyl)ation. Sci. Rep. 10, 3699 (2020).
Eustermann, S. et al. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol. Cell 60, 742–754 (2015).
Suskiewicz, M. J. et al. HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 579, 598–602 (2020).
Durkacz, B. W., Omidiji, O., Gray, D. A. & Shall, S. (ADP-ribose)n participates in DNA excision repair. Nature 283, 593–596 (1980).
Sims, J. L., Berger, S. J. & Berger, N. A. Effects of nicotinamide on NAD and poly(ADP-ribose) metabolism in DNA-damaged human lymphocytes. J. Supramol. Struct. Cell. Biochem. 16, 281–288 (1981).
Adamietz, P. Poly(ADP-ribose) synthase is the major endogenous nonhistone acceptor for poly(ADP-ribose) in alkylated rat hepatoma cells. Eur. J. Biochem. 169, 365–372 (1987).
Ray Chaudhuri, A. & Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621 (2017).
Caron, M. C. et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 10, 2954 (2019).
Strickfaden, H. et al. Poly(ADP-ribosyl)ation-dependent transient chromatin decondensation and histone displacement following laser microirradiation. J. Biol. Chem. 291, 1789–1802 (2016).
Haince et al. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 283, 1197–1208 (2008).
Hanzlikova, H., Gittens, W., Krejcikova, K., Zeng, Z. & Caldecott, K. W. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 45, 2546–2557 (2017).
Breslin, C. et al. The XRCC1 phosphate-binding pocket binds poly (ADP-ribose) and is required for XRCC1 function. Nucleic Acids Res. 43, 6934–6944 (2015).
Pascal, J. M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 71, 177–182 (2018).
Oberdoerffer, P. et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918 (2008).
Alves-Fernandes, D. K. & Jasiulionis, M. G. The role of SIRT1 on DNA damage response and epigenetic alterations in cancer. Int. J. Mol. Sci. 20, 3153 (2019).
Vidal, A. E., Boiteux, S., Hickson, I. D. & Radicella, J. P. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 20, 6530–6539 (2001).
Brunet, A. et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015 (2004).
Yuan, Z., Zhang, X., Sengupta, N., Lane, W. S. & Seto, E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell 27, 149–162 (2007).
Kahyo, T., Mostoslavsky, R., Goto, M. & Setou, M. Sirtuin-mediated deacetylation pathway stabilizes Werner syndrome protein. FEBS Lett. 582, 2479–2483 (2008).
Langsfeld, E. S., Bodily, J. M. & Laimins, L. A. The deacetylase sirtuin 1 regulates human papillomavirus replication by modulating histone acetylation and recruitment of DNA damage factors NBS1 and Rad51 to viral genomes. PLoS Pathog. 11, e1005181 (2015).
Roth, M., Wang, Z. & Chen, W. Y. SIRT1 and LSD1 competitively regulate KU70 functions in DNA repair and mutation acquisition in cancer cells. Oncotarget 7, 50195–50214 (2016).
Yu, W. et al. KU70 inhibition impairs both non-homologous end joining and homologous recombination DNA damage repair through SHP-1 induced dephosphorylation of SIRT1 in T-cell acute lymphoblastic leukemia (T-ALL) [corrected]. Cell. Physiol. Biochem. 49, 2111–2123 (2018).
Jarrett, S. G. et al. Sirtuin 1-mediated deacetylation of XPA DNA repair protein enhances its interaction with ATR protein and promotes cAMP-induced DNA repair of UV damage. J. Biol. Chem. 293, 19025–19037 (2018).
Haince, J. F. et al. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 282, 16441–16453 (2007).
Ruscetti, T. et al. Stimulation of the DNA-dependent protein kinase by poly(ADP-ribose) polymerase. J. Biol. Chem. 273, 14461–14467 (1998).
Chen, J. K., Lin, W. L., Chen, Z. & Liu, H. W. PARP-1-dependent recruitment of cold-inducible RNA-binding protein promotes double-strand break repair and genome stability. Proc. Natl Acad. Sci. USA 115, E1759–e1768 (2018).
Ariumi, Y. et al. Suppression of the poly(ADP-ribose) polymerase activity by DNA-dependent protein kinase in vitro. Oncogene 18, 4616–4625 (1999).
Singh, C. K. et al. The role of sirtuins in antioxidant and redox signaling. Antioxid. Redox Signal. 28, 643–661 (2018).
Leidecker, O. et al. Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat. Chem. Biol. 12, 998–1000 (2016).
Bian, C. et al. NADP(+) is an endogenous PARP inhibitor in DNA damage response and tumor suppression. Nat. Commun. 10, 693 (2019).
Wilk, A. et al. Extracellular NAD(+) enhances PARP-dependent DNA repair capacity independently of CD73 activity. Sci. Rep. 10, 651 (2020).
Fang, E. F. et al. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 24, 566–581 (2016).
Hou, Y. et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl Acad. Sci. USA 115, E1876–e1885 (2018).
Gaber, T., Strehl, C. & Buttgereit, F. Metabolic regulation of inflammation. Nat. Rev. Rheumatol. 13, 267–279 (2017).
Caiafa, P., Guastafierro, T. & Zampieri, M. Epigenetics: poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. FASEB J. 23, 672–678 (2009).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Gräff, J. & Tsai, L. H. Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 14, 97–111 (2013).
Vaquero, A., Sternglanz, R. & Reinberg, D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 26, 5505–5520 (2007).
Ryall, J. G. et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 16, 171–183 (2015).
Tran, L., Schulkin, J., Ligon, C. O. & Greenwood-Van Meerveld, B. Epigenetic modulation of chronic anxiety and pain by histone deacetylation. Mol. Psychiatry 20, 1219–1231 (2015).
Barber, M. F. et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 487, 114–118 (2012).
Chang, J. et al. Nicotinamide adenine dinucleotide (NAD)-regulated DNA methylation alters CCCTC-binding factor (CTCF)/cohesin binding and transcription at the BDNF locus. Proc. Natl Acad. Sci. USA 107, 21836–21841 (2010).
Cramer, T., Rosenberg, T., Kisliouk, T. & Meiri, N. PARP inhibitor affects long-term heat-stress response via changes in DNA methylation. Neuroscience 399, 65–76 (2019).
Guastafierro, T. et al. CCCTC-binding factor activates PARP-1 affecting DNA methylation machinery. J. Biol. Chem. 283, 21873–21880 (2008).
Reale, A., Matteis, G. D., Galleazzi, G., Zampieri, M. & Caiafa, P. Modulation of DNMT1 activity by ADP-ribose polymers. Oncogene 24, 13–19 (2005).
Ulanovskaya, O. A., Zuhl, A. M. & Cravatt, B. F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 9, 300 (2013).
Eckert, M. A. et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 569, 723–728 (2019).
Wang, Y. et al. NAD( + )-capped RNAs are widespread in the Arabidopsis transcriptome and can probably be translated. Proc. Natl Acad. Sci. USA 116, 12094–12102 (2019).
Ramanathan, A., Robb, G. B. & Chan, S. H. mRNA capping: biological functions and applications. Nucleic Acids Res. 44, 7511–7526 (2016).
Bird, J. G. et al. Highly efficient 5' capping of mitochondrial RNA with NAD(+) and NADH by yeast and human mitochondrial RNA polymerase. eLife 7, e42179 (2018).
Grudzien-Nogalska, E. & Kiledjian, M. New insights into decapping enzymes and selective mRNA decay. Wiley Interdiscip. Rev. RNA 8, 1379 (2017).
Frindert, J. et al. Identification, biosynthesis, and decapping of NAD-capped RNAs in B. subtilis. Cell Rep. 24, 1890–1901 e1898 (2018).
Grudzien-Nogalska et al. Structural and mechanistic basis of mammalian Nudt12 RNA deNADding. Nat. Chem. Biol. 15, 575–582 (2019).
Yamamoto, M., Inohara, H. & Nakagawa, T. Targeting metabolic pathways for head and neck cancers therapeutics. Cancer Metastasis Rev. 36, 503–514 (2017).
Tan, B. et al. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, leads to altered carbohydrate metabolism in cancer cells. J. Biol. Chem. 290, 15812–15824 (2015).
Lunt, S. Y. & Vander Heiden, M. G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464 (2011).
Xiao, W., Wang, R. S., Handy, D. E. & Loscalzo, J. NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid. Redox Signal. 28, 251–272 (2018).
Yang, Y. & Sauve, A. A. NAD(+) metabolism: bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 1864, 1787–1800 (2016).
Saraste, M. Oxidative phosphorylation at the fin de siècle. Science 283, 1488–1493 (1999).
Adeva-Andany, M. M., Carneiro-Freire, N., Seco-Filgueira, M., Fernández-Fernández, C. & Mouriño-Bayolo, D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 46, 73–90 (2019).
Zakhari, S. Overview: how is alcohol metabolized by the body? Alcohol Res. Health 29, 245–254 (2006).
Yellen, G. Fueling thought: management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell Biol. 217, 2235–2246 (2018).
Guan, K. L. & Xiong, Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem. Sci. 36, 108–116 (2011).
Shakespear, M. R. et al. Lysine deacetylases and regulated glycolysis in macrophages. Trends Immunol. 39, 473–488 (2018).
Yang, W. et al. Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell 167, 985–1000.e1021 (2016).
Carrico, C., Meyer, J. G., He, W., Gibson, B. W. & Verdin, E. The mitochondrial acylome emerges: proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab. 27, 497–512 (2018).
Hirschey, M. D. et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125 (2010).
Finley, L. W. et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 19, 416–428 (2011).
Chen, S. et al. SIRT3 regulates cancer cell proliferation through deacetylation of PYCR1 in proline metabolism. Neoplasia 21, 665–675 (2019).
Laurent, G. et al. SIRT4 represses peroxisome proliferator-activated receptor α activity to suppress hepatic fat oxidation. Mol. Cell. Biol. 33, 4552–4561 (2013).
Anderson, K. A. et al. SIRT4 is a lysine deacylase that controls leucine metabolism and insulin secretion. Cell Metab. 25, 838–855 e815 (2017).
Mathias, R. A. et al. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159, 1615–1625 (2014).
Haigis, M. C. et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126, 941–954 (2006).
Du, J. et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 (2011).
Sadhukhan, S. et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl Acad. Sci. USA 113, 4320–4325 (2016).
Yang, X. et al. SHMT2 desuccinylation by SIRT5 drives cancer cell proliferation. Cancer Res. 78, 372–386 (2018).
Wang, G. et al. Regulation of UCP1 and mitochondrial metabolism in brown adipose tissue by reversible succinylation. Mol. Cell 74, 844–857.e847 (2019).
Rardin, M. J. et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 18, 920–933 (2013).
Greene, K. S. et al. SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc. Natl Acad. Sci. USA 116, 26625–26632 (2019).
Nishida, Y. et al. SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol. Cell 59, 321–332 (2015).
Park, J. et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 50, 919–930 (2013).
Stangherlin, A. & Reddy, A. B. Regulation of circadian clocks by redox homeostasis. J. Biol. Chem. 288, 26505–26511 (2013).
Ramsey, K. M. et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324, 651–654 (2009).
Nakahata, Y., Sahar, S., Astarita, G., Kaluzova, M. & Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324, 654–657 (2009).
Luna, A., McFadden, G. B., Aladjem, M. I. & Kohn, K. W. Predicted role of NAD utilization in the control of circadian rhythms during DNA damage response. PLoS Computat. Biol. 11, e1004144 (2015).
Orozco-Solis, R. & Sassone-Corsi, P. Circadian clock: linking epigenetics to aging. Curr. Opin. Genet. Dev. 26, 66–72 (2014).
Koronowski, K. B. et al. Defining the independence of the liver circadian clock. Cell 177, 1448–1462 e1414 (2019).
Levine, D. C. et al. NAD(+) controls circadian reprogramming through PER2 nuclear translocation to counter aging. Mol. Cell 78, 835–849.e837 (2020).
Masri, S. et al. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell 158, 659–672 (2014).
Aguilar-Arnal, L., Katada, S., Orozco-Solis, R. & Sassone-Corsi, P. NAD(+)-SIRT1 control of H3K4 trimethylation through circadian deacetylation of MLL1. Nat. Struct. Mol. Biol. 22, 312–318 (2015).
Asher, G. et al. Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell 142, 943–953 (2010).
Reinke, H. & Asher, G. Crosstalk between metabolism and circadian clocks. Nat. Rev. Mol. Cell Biol. 20, 227–241 (2019).
Zhao, H. et al. PARP1- and CTCF-mediated interactions between active and repressed chromatin at the lamina promote oscillating transcription. Mol. Cell 59, 984–997 (2015).
Lee, J. et al. Bmal1 and beta-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced beta-cell failure in mice. Mol. Cell. Biol. 33, 2327–2338 (2013).
Pekovic-Vaughan et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. 28, 548–560 (2014).
Rhee, S. G. & Kil, I. S. Mitochondrial H2O2 signaling is controlled by the concerted action of peroxiredoxin III and sulfiredoxin: linking mitochondrial function to circadian rhythm. Free Radical Biol. Med. 100, 73–80 (2016).
Rey, G. et al. The pentose phosphate pathway regulates the circadian clock. Cell Metab. 24, 462–473 (2016).
O’Neill, J. S. & Reddy, A. B. Circadian clocks in human red blood cells. Nature 469, 498–503 (2011).
Aguilar-Arnal, L. & Sassone-Corsi, P. Chromatin landscape and circadian dynamics: Spatial and temporal organization of clock transcription. Proc. Natl Acad. Sci. USA 112, 6863–6870 (2015).
Kondratov, R. V., Kondratova, A. A., Gorbacheva, V. Y., Vykhovanets, O. V. & Antoch, M. P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 20, 1868–1873 (2006).
Mills, E. & O’Neill, L. A. Succinate: a metabolic signal in inflammation. Trends Cell Biol. 24, 313–320 (2014).
Minhas, P. S. et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat. Immunol. 20, 50–63 (2019).
Mills, E. L. et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e413 (2016).
Cameron, A. M. et al. Inflammatory macrophage dependence on NAD(+) salvage is a consequence of reactive oxygen species-mediated DNA damage. Nat. Immunol. 20, 420–432 (2019).
Baixauli, F. et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 22, 485–498 (2015).
Nacarelli, T. et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 21, 397–407 (2019).
Zhang, J. et al. Switch of NAD salvage to de novo biosynthesis sustains SIRT1-RelB-dependent inflammatory tolerance. Front. Immunol. 10, 2358 (2019).
Gallí, M., Gool, Van, Rongvaux, F., Andris, A. & Leo, F. O. The nicotinamide phosphoribosyltransferase: a molecular link between metabolism, inflammation, and cancer. Cancer Res. 70, 8–11 (2010).
Halvorsen, B. et al. Increased expression of NAMPT in PBMC from patients with acute coronary syndrome and in inflammatory M1 macrophages. Atherosclerosis 243, 204–210 (2015).
Jia, S. H. et al. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J. Clin. Investig. 113, 1318–1327 (2004).
Gerner, R. R. et al. NAD metabolism fuels human and mouse intestinal inflammation. Gut 67, 1813–1823 (2018).
Busso, N. et al. Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PloS ONE 3, e2267 (2008).
Luk, T., Malam, Z. & Marshall, J. C. Pre-B cell colony-enhancing factor (PBEF)/visfatin: a novel mediator of innate immunity. J. Leukocyte Biol. 83, 804–816 (2008).
Van Gool, F. et al. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat. Med. 15, 206–210 (2009).
Liu, T. F., Yoza, B. K., El Gazzar, M., Vachharajani, V. T. & McCall, C. E. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 286, 9856–9864 (2011).
Sorgdrager, F. J. H., Naude, P. J. W., Kema, I. P., Nollen, E. A. & Deyn, P. P. Tryptophan metabolism in inflammaging: from biomarker to therapeutic target. Front. Immunol. 10, 2565 (2019).
Landskron, G., la Fuente, De, Thuwajit, M., Thuwajit, P. & Hermoso, C. M. A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 149185 (2014).
Klaunig, J. E. Oxidative stress and cancer. Curr. Pharm. Design 24, 4771–4778 (2018).
Panday, A., Sahoo, M. K., Osorio, D. & Batra, S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 12, 5–23 (2015).
Park, H. S. et al. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J. Immunol. 173, 3589–3593 (2004).
Joo, J. H. et al. Dual oxidase 2 is essential for the toll-like receptor 5-mediated inflammatory response in airway mucosa. Antioxid. Redox Signal. 16, 57–70 (2012).
Kawahara, T. et al. Role of nicotinamide adenine dinucleotide phosphate oxidase 1 in oxidative burst response to Toll-like receptor 5 signaling in large intestinal epithelial cells. J. Immunol. 172, 3051–3058 (2004).
Yang, C. S. et al. Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host Microbe 11, 264–276 (2012).
Beck, M. A., Handy, J. & Levander, O. A. The role of oxidative stress in viral infections. Ann. NY Acad. Sci. 917, 906–912 (2000).
Dan, Y. et al. Hepatitis B virus X protein (HBx)-induced abnormalities of nucleic acid metabolism revealed by (1)H-NMR-based metabonomics. Sci. Rep. 6, 24430 (2016).
Roe, B., Kensicki, E., Mohney, R. & Hall, W. W. Metabolomic profile of hepatitis C virus-infected hepatocytes. PloS ONE 6, e23641 (2011).
Vastag, L., Koyuncu, E., Grady, S. L., Shenk, T. E. & Rabinowitz, J. D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 7, e1002124 (2011).
Xu, G. et al. PARP-1 mediated cell death is directly activated by ZIKV infection. Virology 537, 254–262 (2019).
Murray, M. F., Nghiem, M. & Srinivasan, A. HIV infection decreases intracellular nicotinamide adenine dinucleotide [NAD]. Biochem. Biophys. Res. Commun. 212, 126–131 (1995).
Yen, Y. T., Chen, H. C., Lin, Y. D., Shieh, C. C. & Wu-Hsieh, B. A. Enhancement by tumor necrosis factor alpha of dengue virus-induced endothelial cell production of reactive nitrogen and oxygen species is key to hemorrhage development. J. Virol. 82, 12312–12324 (2008).
Jan, J. T. et al. Potential dengue virus-triggered apoptotic pathway in human neuroblastoma cells: arachidonic acid, superoxide anion, and NF-kappaB are sequentially involved. J. Virol.74, 8680–8691 (2000).
Marianneau, P., Cardona, A., Edelman, L., Deubel, V. & Despres, P. Dengue virus replication in human hepatoma cells activates NF-kappaB which in turn induces apoptotic cell death. J. Virol. 71, 3244–3249 (1997).
Machida, K. et al. Hepatitis C virus infection activates the immunologic (type II) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J. Virol. 78, 8835–8843 (2004).
Machida, K. et al. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J. Virol. 80, 866–874 (2006).
Lin, R. J., Liao, C. L. & Lin, Y. L. Replication-incompetent virions of Japanese encephalitis virus trigger neuronal cell death by oxidative stress in a culture system. J. General Virol. 85, 521–533 (2004).
Dandri, M. et al. Increase in de novo HBV DNA integrations in response to oxidative DNA damage or inhibition of poly (ADP-ribosyl) ation. Hepatology 35, 217–223 (2002).
Anticoli, S. et al. Counteraction of HCV-induced oxidative stress concurs to establish chronic infection in liver cell cultures. Oxid. Med. Cell Longev. 2019, 6452390 (2019).
Choi, J., Corder, N. L., Koduru, B. & Wang, Y. Oxidative stress and hepatic Nox proteins in chronic hepatitis C and hepatocellular carcinoma. Free Radical Biol. Med. 72, 267–284 (2014).
Liu, T. et al. Reactive oxygen species mediate virus-induced STAT activation: role of tyrosine phosphatases. J. Biol. Chem. 279, 2461–2469 (2004).
Wu, Y. L. et al. Epigenetic silencing of NAD(P)H:quinone oxidoreductase 1 by hepatitis B virus X protein increases mitochondrial injury and cellular susceptibility to oxidative stress in hepatoma cells. Free Radical Biol.Med. 65, 632–644 (2013).
Ubol, S. et al. Temporal changes in chromatin, intracellular calcium, and poly(ADP-ribose) polymerase during Sindbis virus-induced apoptosis of neuroblastoma cells. J. Virol. 70, 2215–2220 (1996).
Shou, Q., Fu, H., Huang, X. & Yang, Y. PARP-1 controls NK cell recruitment to the site of viral infection. JCI Insight 4, e121291 (2019).
Liu, S. Y., Sanchez, D. J., Aliyari, R., Lu, S. & Cheng, G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl Acad. Sci. USA 109, 4239–4244 (2012).
Atasheva, S., Frolova, E. I. & Frolov, I. Interferon-stimulated poly(ADP-Ribose) polymerases are potent inhibitors of cellular translation and virus replication. J. Virol.88, 2116–2130 (2014).
Li, L. et al. PARP12 suppresses Zika virus infection through PARP-dependent degradation of NS1 and NS3 viral proteins. Sci. Signal 11, eaas9332 (2018).
Grunewald, M. E. et al. The coronavirus macrodomain is required to prevent PARP-mediated inhibition of virus replication and enhancement of IFN expression. PLoS Pathog. 15, e1007756 (2019).
Koyuncu, E. et al. Sirtuins are evolutionarily conserved viral restriction factors. mBio 5, e02249–14 (2014).
Yu, J. W., Sun, L. J., Zhao, Y. H., Kang, P. & Yan, B. Z. Inhibition of silent information regulator 1 induces glucose metabolism disorders of hepatocytes and enhances hepatitis C virus replication. Hepatol. Int. 7, 524–532 (2013).
Zhang, H. S., Zhou, Y., Wu, M. R., Zhou, H. S. & Xu, F. Resveratrol inhibited Tat-induced HIV-1 LTR transactivation via NAD(+)-dependent SIRT1 activity. Life Sci. 85, 484–489 (2009).
Wang, Z. et al. Quinolinate phosphoribosyltransferase is an antiviral host factor against hepatitis C virus infection. Sci. Rep. 7, 5876 (2017).
Muscolini, M. et al. SIRT1 Modulates the sensitivity of prostate cancer cells to vesicular stomatitis virus oncolysis. J. Virol. 93, e00626–19 (2019).
Lupey-Green, L. N. et al. PARP1 Stabilizes CTCF binding and chromatin structure to maintain Epstein-Barr virus latency type. J. Virol. 92, e00755–18 (2018).
Li, Q., He, M., Zhou, F., Ye, F. & Gao, S. J. Activation of Kaposi’s sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: Identification of sirtuin 1 as a regulator of the KSHV life cycle. J. Virol. 88, 6355–6367 (2014).
He, M. & Gao, S. J. A novel role of SIRT1 in gammaherpesvirus latency and replication. Cell Cycle 13, 3328–3330 (2014).
Savarino, A. et al. Human CD38 interferes with HIV-1 fusion through a sequence homologous to the V3 loop of the viral envelope glycoprotein gp120. FASEB J. 17, 461–463 (2003).
Wang, Z. et al. Clonally diverse CD38(+)HLA-DR(+)CD8(+) T cells persist during fatal H7N9 disease. Nat. Commun. 9, 824 (2018).
Schiavoni, I. et al. CD38 modulates respiratory syncytial virus-driven proinflammatory processes in human monocyte-derived dendritic cells. Immunology 154, 122–131 (2018).
Maponga, T. G. et al. HBV and HIV viral load but not microbial translocation or immune activation are associated with liver fibrosis among patients in South Africa. BMC Infect. Dis. 18, 214 (2018).
Zhang, H. S., Sang, W. W., Wang, Y. O. & Liu, W. Nicotinamide phosphoribosyltransferase/sirtuin 1 pathway is involved in human immunodeficiency virus type 1 Tat-mediated long terminal repeat transactivation. J. Cell. Biochem. 110, 1464–1470 (2010).
Moore, P. S. & Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 10, 878–889 (2010).
Cheng, S. T., Ren, J. H., Cai, X. F., Jiang, H. & Chen, J. HBx-elevated SIRT2 promotes HBV replication and hepatocarcinogenesis. Biochem. Biophys. Res. Commun. 496, 904–910 (2018).
Piracha, Z. Z. et al. Sirtuin 2 isoform 1 enhances Hepatitis B virus RNA transcription and DNA synthesis through the AKT/GSK-3beta/beta-catenin signaling pathway. J. Virol. 92, e00955–18 (2018).
West, A. P. et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480 (2011).
Nguyen, G. T., Green, E. R. & Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial resistance. Front. Cell. Infect. Microbiol. 7, 373 (2017).
Laroux, F. S., Romero, X., Wetzler, L., Engel, P. & Terhorst, C. Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of gram-negative bacteria. J. Immunol. 175, 5596–5600 (2005).
Sun, K. & Metzger, D. W. Influenza infection suppresses NADPH oxidase-dependent phagocytic bacterial clearance and enhances susceptibility to secondary methicillin-resistant Staphylococcus aureus infection. J. Immunol. 192, 3301–3307 (2014).
Chakraborty, S. et al. Glycolytic reprograming in Salmonella counters NOX2-mediated dissipation of ΔpH. Nat. Commun. 11, 1783 (2020).
Huang, J. et al. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl Acad. Sci. USA 106, 6226–6231 (2009).
Pajuelo, D., Gonzalez-Juarbe, N. & Niederweis, M. NAD hydrolysis by the tuberculosis necrotizing toxin induces lethal oxidative stress in macrophages. Cell. Microbiol. 22, e13115 (2020).
Najibi, M., Moreau, J. A., Honwad, H. H. & Irazoqui, J. E. A novel PHOX/CD38/MCOLN1/TFEB axis important for macrophage activation during bacterial phagocytosis. bioRxiv 669325 (2019).
Lischke, T. et al. CD38 controls the innate immune response against Listeria monocytogenes. Infect. Immunity 81, 4091–4099 (2013).
Partida-Sánchez et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med. 7, 1209–1216 (2001).
Matalonga, J. et al. The nuclear receptor LXR limits bacterial infection of host macrophages through a mechanism that impacts cellular NAD metabolism. Cell Rep. 18, 1241–1255 (2017).
Katsuyama, E. et al. The CD38/NAD/SIRTUIN1/EZH2 axis mitigates cytotoxic CD8 T cell function and identifies patients with SLE prone to infections. Cell Rep. 30, 112–123 e114 (2020).
Imai, S. & Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471 (2014).
Powell, R. D. et al. Resveratrol attenuates hypoxic injury in a primary hepatocyte model of hemorrhagic shock and resuscitation. J. Trauma Acute Care Surg. 76, 409–417 (2014).
Aguirre-Rueda, D. et al. Astrocytes protect neurons from Aβ1-42 peptide-induced neurotoxicity increasing TFAM and PGC-1 and decreasing PPAR-γ and SIRT-1. Int. J. Med. Sci. 12, 48–56 (2015).
Busch, F., Mobasheri, A., Shayan, P., Stahlmann, R. & Shakibaei, M. Sirt-1 is required for the inhibition of apoptosis and inflammatory responses in human tenocytes. J. Biol. Chem. 287, 25770–25781 (2012).
Fang, E. F. et al. NAD(+) in aging: Molecular mechanisms and translational implications. Trends Mol. Med. 23, 899–916 (2017).
Braidy, N. et al. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PloS ONE 6, e19194 (2011).
Zhou, C. C. et al. Hepatic NAD(+) deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br. J. Pharmacol. 173, 2352–2368 (2016).
Clement, J., Wong, M., Poljak, A., Sachdev, P. & Braidy, N. The plasma NAD(+) metabolome is dysregulated in “normal” aging. Rejuvenation Res. 22, 121–130 (2019).
Yoshino, J., Mills, K. F., Yoon, M. J. & Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 14, 528–536 (2011).
Stein, L. R. & Imai, S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 33, 1321–1340 (2014).
Johnson, S. & Imai, S. I. NAD (+) biosynthesis, aging, and disease. F1000Research 7, 132 (2018).
Tirumurugaan, K. G. et al. TNF-alpha induced CD38 expression in human airway smooth muscle cells: role of MAP kinases and transcription factors NF-kappaB and AP-1. Am. J. Physiol. Lung Cell. Mol. Physiol.292, L1385–L1395 (2007).
Mouchiroud, L. et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441 (2013).
Zhang, H. et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443 (2016).
van der Veer, E. et al. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J. Biol. Chem. 282, 10841–10845 (2007).
de Picciotto, N. E. et al. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15, 522–530 (2016).
Gomes, A. P. et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638 (2013).
Kim, H. J. et al. Augmentation of cellular NAD(+) by NQO1 enzymatic action improves age-related hearing impairment. Aging Cell 18, e13016 (2019).
Satoh, A. et al. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 18, 416–430 (2013).
Kanfi, Y. et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature 483, 218–221 (2012).
Scheibye-Knudsen, M. et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 20, 840–855 (2014).
Peek, C. B. et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342, 1243417 (2013).
Venkataraman, K., Khurana, S. & Tai, T. C. Oxidative stress in aging-matters of the heart and mind. Int. J. Mol. Sci.14, 17897–17925 (2013).
Barnham, K. J., Masters, C. L. & Bush, A. I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 3, 205–214 (2004).
Jeong, Y. J. et al. Impact of long-term RF-EMF on oxidative stress and neuroinflammation in aging brains of C57BL/6 mice. Int. J. Mol. Sci. 19 (2018).
Sun, M. et al. The need to incorporate aged animals into the preclinical modeling of neurological conditions. Neurosci. Biobehav. Rev. 109, 114–128 (2020).
Royce, G. H., Brown-Borg, H. M. & Deepa, S. S. The potential role of necroptosis in inflammaging and aging. GeroScience 41, 795–811 (2019).
Yang, D. et al. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res. 85, 462–472 (2007).
Fan, L. M. et al. Nox2 contributes to age-related oxidative damage to neurons and the cerebral vasculature. J. Clin. Investig. 129, 3374–3386 (2019).
Stefanatos, R. & Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 592, 743–758 (2018).
Li, S. Y. et al. Aging induces cardiac diastolic dysfunction, oxidative stress, accumulation of advanced glycation endproducts and protein modification. Aging Cell 4, 57–64 (2005).
Gulshan, M. et al. Overexpression of Nmnat3 efficiently increases NAD and NGD levels and ameliorates age-associated insulin resistance. Aging Cell 17, e12798 (2018).
Nóbrega-Pereira, S. et al. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 7, 1–9 (2016).
Alcendor, R. R. et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 100, 1512–1521 (2007).
Chang, H. C. & Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 153, 1448–1460 (2013).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Kennedy, B. E. et al. NAD(+) salvage pathway in cancer metabolism and therapy. Pharmacol. Res. 114, 274–283 (2016).
Sharif, T. et al. The NAD(+) salvage pathway modulates cancer cell viability via p73. Cell Death Differ. 23, 669–680 (2016).
Olmos, Y., Brosens, J. J. & Lam, E. W. Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist. Updat. 14, 35–44 (2011).
Fan, J. et al. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol. Cell. Biol. 31, 4938–4950 (2011).
Santidrian, A. F. et al. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin. Investig. 123, 1068–1081 (2013).
Cairns, R. A., Harris, I. S. & Mak, T. W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 (2011).
Peiris-Pages, M., Martinez-Outschoorn, U. E., Sotgia, F. & Lisanti, M. P. Metastasis and oxidative stress: are antioxidants a metabolic driver of progression? Cell Metab. 22, 956–958 (2015).
Sanchez, M. et al. Cross talk between eIF2alpha and eEF2 phosphorylation pathways optimizes translational arrest in response to oxidative. Stress. iScience 20, 466–480 (2019).
Grivennikov, S. I., Greten, F. R. & Karin, M. Immunity, inflammation, and cancer. Cell 140, 883–899 (2010).
Liu, X. et al. NADPH oxidase 1-dependent ROS is crucial for TLR4 signaling to promote tumor metastasis of non-small cell lung cancer. Tumour Biol. 36, 1493–1502 (2015).
Anastasiou, D. et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283 (2011).
Perl, A., Hanczko, R., Telarico, T., Oaks, Z. & Landas, S. Oxidative stress, inflammation and carcinogenesis are controlled through the pentose phosphate pathway by transaldolase. Trends Mol. Med. 17, 395–403 (2011).
Li, S. et al. Nicotinamide nucleotide transhydrogenase-mediated redox homeostasis promotes tumor growth and metastasis in gastric cancer. Redox Biol. 18, 246–255 (2018).
Hong, S. M. et al. NAMPT suppresses glucose deprivation-induced oxidative stress by increasing NADPH levels in breast cancer. Oncogene 35, 3544–3554 (2016).
Lu, Y. X. et al. ME1 regulates NADPH homeostasis to promote gastric cancer growth and metastasis. Cancer Res. 78, 1972–1985 (2018).
Jeon, S. M., Chandel, N. S. & Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665 (2012).
Joo, H. Y. et al. SIRT1 interacts with and protects glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from nuclear translocation: implications for cell survival after irradiation. Biochem. Biophys. Res. Commun. 424, 681–686 (2012).
Ozden, O. et al. SIRT3 deacetylates and increases pyruvate dehydrogenase activity in cancer cells. Free Radical Biol. Med. 76, 163–172 (2014).
Zhu, Y., Liu, J., Park, J., Rai, P. & Zhai, R. G. Subcellular compartmentalization of NAD(+) and its role in cancer: a sereNADe of metabolic melodies. Pharmacol. Therap. 200, 27–41 (2019).
Ma, Y. et al. SIRT5-mediated SDHA desuccinylation promotes clear cell renal cell carcinoma tumorigenesis. Free Radical Biol. Med. 134, 458–467 (2019).
Ren, T. et al. MCU-dependent mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 36, 5897–5909 (2017).
Bell, E. L., Emerling, B. M., Ricoult, S. J. & Guarente, L. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 30, 2986–2996 (2011).
Zou, X. et al. SIRT3-mediated dimerization of IDH2 directs cancer cell metabolism and tumor growth. Cancer Res. 77, 3990–3999 (2017).
Hopp, A. K., Gruter, P. & Hottiger, M. O. Regulation of glucose metabolism by NAD(+) and ADP-ribosylation. Cells 8, 890 (2019).
Zhang, T. & Kraus, W. L. SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim. Biophys. Acta 1804, 1666–1675 (2010).
Rifaï, K. et al. SIRT1-dependent epigenetic regulation of H3 and H4 histone acetylation in human breast cancer. Oncotarget 9, 30661–30678 (2018).
Lu, L. et al. Modulations of hMOF autoacetylation by SIRT1 regulate hMOF recruitment and activities on the chromatin. Cell Res. 21, 1182–1195 (2011).
Saidi, D. et al. Glioma-induced SIRT1-dependent activation of hMOF histone H4 lysine 16 acetyltransferase in microglia promotes a tumor supporting phenotype. Oncoimmunology 7, e1382790 (2018).
Bradbury, C. A. et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 19, 1751–1759 (2005).
Wang, R. H. et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14, 312–323 (2008).
Narod, S. A. & Foulkes, W. D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer 4, 665–676 (2004).
Sachdev, E., Tabatabai, R., Roy, V., Rimel, B. J. & Mita, M. M. PARP inhibition in cancer: an update on clinical development. Target Oncol. 14, 657–679 (2019).
George, A., Kaye, S. & Banerjee, S. Delivering widespread BRCA testing and PARP inhibition to patients with ovarian cancer. Nat. Rev. Clin. Oncol. 14, 284–296 (2017).
Ashworth, A. & Lord, C. J. Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 15, 564–576 (2018).
Pissios, P. Nicotinamide N-methyltransferase: more than a vitamin B3 clearance enzyme. Trends Endocrinol. Metab. 28, 340–353 (2017).
Sartini, D. et al. Upregulation of tissue and urinary nicotinamide N-methyltransferase in bladder cancer: potential for the development of a urine-based diagnostic test. Cell Biochem. Biophys. 65, 473–483 (2013).
Xu, Y. et al. Expression profile and prognostic value of NNMT in patients with pancreatic cancer. Oncotarget 7, 19975–19981 (2016).
Tomida, M., Mikami, I., Takeuchi, S., Nishimura, H. & Akiyama, H. Serum levels of nicotinamide N-methyltransferase in patients with lung cancer. J. Cancer Res. Clin. Oncol. 135, 1223–1229 (2009).
Jung, J. et al. Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight 2, e90019 (2017).
Li, J. et al. Elevated N-methyltransferase expression induced by hepatic stellate cells contributes to the metastasis of hepatocellular carcinoma via regulation of the CD44v3 isoform. Mol. Oncol.13, 1993–2009 (2019).
Eckert, M. A. et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 569, 723–728 (2019).
Jiang, L. et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258 (2016).
Michalak, E. M., Burr, M. L., Bannister, A. J. & Dawson, M. A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 20, 573–589 (2019).
Chowdhry, S. et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 569, 570–575 (2019).
Piacente, F. et al. Nicotinic acid phosphoribosyltransferase regulates cancer cell metabolism, susceptibility to NAMPT Inhibitors, and DNA Repair. Cancer Res. 77, 3857–3869 (2017).
Thongon, N. et al. Cancer cell metabolic plasticity allows resistance to NAMPT inhibition but invariably induces dependence on LDHA. Cancer Metab. 6, 1 (2018).
Li, X. Q. et al. NAMPT and NAPRT, key enzymes in NAD salvage synthesis pathway, are of negative prognostic value in colorectal cancer. Front. Oncol. 9, 736 (2019).
Zhang, H. et al. Epigenetic regulation of NAMPT by NAMPT-AS drives metastatic progression in triple-negative breast cancer. Cancer Res. 79, 3347–3359 (2019).
Brandl, L. et al. The c-MYC/NAMPT/SIRT1 feedback loop is activated in early classical and serrated route colorectal cancer and represents a therapeutic target. Med. Oncol. 36, 5 (2018).
Ge, X. et al. NAMPT regulates PKM2 nuclear location through 14-3-3zeta: Conferring resistance to tamoxifen in breast cancer. J. Cell. Physiol.234, 23409–23420 (2019).
Lucena-Cacace, A., Otero-Albiol, D., Jimenez-Garcia, M. P., Peinado-Serrano, J. & Carnero, A. NAMPT overexpression induces cancer stemness and defines a novel tumor signature for glioma prognosis. Oncotarget 8, 99514–99530 (2017).
Lucena-Cacace, A., Otero-Albiol, D., Jimenez-Garcia, M. P., Munoz-Galvan, S. & Carnero, A. NAMPT is a potent oncogene in colon cancer progression that modulates cancer stem cell properties and resistance to therapy through Sirt1 and PARP. Clin. Cancer Res. 24, 1202–1215 (2018).
Sociali, G. et al. SIRT6 deacetylase activity regulates NAMPT activity and NAD(P)(H) pools in cancer cells. FASEB J. 33, 3704–3717 (2019).
Wang, W. et al. Decreased NAD activates STAT3 and integrin pathways to drive epithelial-mesenchymal transition. Mol. Cell. Proteomics 17, 2005–2017 (2018).
Chmielewski, J. P. et al. CD38 inhibits prostate cancer metabolism and proliferation by reducing cellular NAD(+) pools. Mol. Cancer Res. 16, 1687–1700 (2018).
Espindola-Netto, J. M. et al. Preclinical efficacy of the novel competitive NAMPT inhibitor STF-118804 in pancreatic cancer. Oncotarget 8, 85054–85067 (2017).
Abu Aboud, O. et al. Dual and specific inhibition of NAMPT and PAK4 By KPT-9274 decreases kidney cancer growth. Mol. Cancer Ther. 15, 2119–2129 (2016).
Nagaya, M., Hara, H., Kamiya, T. & Adachi, T. Inhibition of NAMPT markedly enhances plasma-activated medium-induced cell death in human breast cancer MDA-MB-231 cells. Arch. Biochem. Biophys. 676, 108155 (2019).
Lee, J. et al. Selective cytotoxicity of the NAMPT inhibitor FK866 toward gastric cancer cells with markers of the epithelial-mesenchymal transition, due to loss of NAPRT. Gastroenterology 155, 799–814.e713 (2018).
Luo, X., Li, R. & Yan, L. J. Roles of pyruvate, NADH, and mitochondrial complex I in redox balance and imbalance in β cell function and dysfunction. J. Diabetes Res. 2015, 512618 (2015).
Prentki, M., Matschinsky, F. M. & Madiraju, S. R. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 18, 162–185 (2013).
Maechler, P. & Wollheim, C. B. Mitochondrial signals in glucose-stimulated insulin secretion in the beta cell. J. Physiol. 529(Pt 1), 49–56 (2000).
MacDonald, P. E., Joseph, J. W. & Rorsman, P. Glucose-sensing mechanisms in pancreatic beta-cells. Philos. Trans. R. Soc. London 360, 2211–2225 (2005).
Patterson, G. H., Knobel, S. M., Arkhammar, P., Thastrup, O. & Piston, D. W. Separation of the glucose-stimulated cytoplasmic and mitochondrial NAD(P)H responses in pancreatic islet beta cells. Proc. Natl Acad. Sci. USA 97, 5203–5207 (2000).
Lawlor, N. et al. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res. 27, 208–222 (2017).
Pralong, W. F., Bartley, C. & Wollheim, C. B. Single islet beta-cell stimulation by nutrients: relationship between pyridine nucleotides, cytosolic Ca2+ and secretion. EMBO J. 9, 53–60 (1990).
Heart, E. et al. Ca2+, NAD(P)H and membrane potential changes in pancreatic beta-cells by methyl succinate: comparison with glucose. Biochem. J. 403, 197–205 (2007).
Boland, B. B., Rhodes, C. J. & Grimsby, J. S. The dynamic plasticity of insulin production in β-cells. Mol. Metab. 6, 958–973 (2017).
Haythorne, E. et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 10, 2474 (2019).
Basile, G., Kulkarni, R. N. & Morgan, N. G. How, when, and where do human β-cells regenerate? Curr. Diabetes Rep. 19, 48 (2019).
Lamontagne, J. et al. Identification of the signals for glucose-induced insulin secretion in INS1 (832/13) β-cells using metformin-induced metabolic deceleration as a model. J. Biol. Chem. 292, 19458–19468 (2017).
Ido et al. Early neural and vascular dysfunctions in diabetic rats are largely sequelae of increased sorbitol oxidation. Antioxid. Redox Signal. 12, 39–51 (2010).
Heart, E. et al. Role for malic enzyme, pyruvate carboxylation, and mitochondrial malate import in glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 296, E1354–E1362 (2009).
Guay, C., Madiraju, S. R., Aumais, A., Joly, E. & Prentki, M. A role for ATP-citrate lyase, malic enzyme, and pyruvate/citrate cycling in glucose-induced insulin secretion. J. Biol. Chem. 282, 35657–35665 (2007).
Schuit, F. et al. Metabolic fate of glucose in purified islet cells glucose-regulated anaplerosis in beta cells. J. Biol. Chem. 272, 18572–18579 (1997).
MacDonald, M. J., Chaplen, F. W., Triplett, C. K., Gong, Q. & Drought, H. Stimulation of insulin release by glyceraldehyde may not be similar to glucose. Arch. Biochem. Biophys. 447, 118–126 (2006).
Ashcroft, S. J. & Christie, M. R. Effects of glucose on the cytosolic ration of reduced/oxidized nicotinamide-adenine dinucleotide phosphate in rat islets of Langerhans. Biochem. J. 184, 697–700 (1979).
Ivarsson, R. et al. Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 54, 2132–2142 (2005).
Reinbothe et al. Glutaredoxin-1 mediates NADPH-dependent stimulation of calcium-dependent insulin secretion. Mol. Endocrinol. 23, 893–900 (2009).
Stromsdorfer, K. L. et al. NAMPT-mediated NAD(+) biosynthesis in adipocytes regulates adipose tissue function and multi-organ insulin sensitivity in mice. Cell Rep. 16, 1851–1860 (2016).
Yach, D., Stuckler, D. & Brownell, K. D. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nat. Med. 12, 62–66 (2006).
Yamaguchi, S. & Yoshino, J. Adipose tissue NAD(+) biology in obesity and insulin resistance: From mechanism to therapy. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology 39, https://doi.org/10.1002/bies.201600227 (2017).
Choi, S. E. et al. Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell 12, 1062–1072 (2013).
Garten, A. et al. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol. 11, 535–546 (2015).
Berndt, J. et al. Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes 54, 2911–2916 (2005).
Terra, X. et al. Increased levels and adipose tissue expression of visfatin in morbidly obese women: the relationship with pro-inflammatory cytokines. Clin. Endocrinol. 77, 691–698 (2012).
Varma, V. et al. Human visfatin expression: relationship to insulin sensitivity, intramyocellular lipids, and inflammation. J. Clin. Endocrinol. Metab. 92, 666–672 (2007).
Chang, Y. H., Chang, D. M., Lin, K. C., Shin, S. J. & Lee, Y. J. Visfatin in overweight/obesity, type 2 diabetes mellitus, insulin resistance, metabolic syndrome and cardiovascular diseases: a meta-analysis and systemic review. Diabetes Metab. Res. Rev. 27, 515–527 (2011).
Segawa, K. et al. Visfatin in adipocytes is upregulated by hypoxia through HIF1alpha-dependent mechanism. Biochem. Biophys. Res. Commun. 349, 875–882 (2006).
Barth, S. et al. Expression of neuropeptide Y, omentin and visfatin in visceral and subcutaneous adipose tissues in humans: relation to endocrine and clinical parameters. Obes. Facts 3, 245–251 (2010).
Jukarainen, S. et al. Obesity is associated with low NAD(+)/SIRT pathway expression in adipose tissue of BMI-discordant monozygotic twins. J. Clin. Endocrinol. Metab. 101, 275–283 (2016).
Rappou, E. et al. Weight Loss Is Associated With Increased NAD(+)/SIRT1 Expression But Reduced PARP Activity in White Adipose Tissue. J. Clin. Endocrinol. Metab. 101, 1263–1273 (2016).
Nielsen, K. N. et al. NAMPT-mediated NAD(+) biosynthesis is indispensable for adipose tissue plasticity and development of obesity. Mol. Metab. 11, 178–188 (2018).
Tanaka, M. et al. Visfatin is released from 3T3-L1 adipocytes via a non-classical pathway. Biochem. Biophys. Res. Commun. 359, 194–201 (2007).
Yang, C. C. et al. Visfatin regulates genes related to lipid metabolism in porcine adipocytes. J. Anim. Sci. 88, 3233–3241 (2010).
Yoon, M. J. et al. SIRT1-mediated eNAMPT secretion from adipose tissue regulates hypothalamic NAD+ and function in mice. Cell Metab. 21, 706–717 (2015).
Li, H., Xu, M., Lee, J., He, C. & Xie, Z. Leucine supplementation increases SIRT1 expression and prevents mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice.Am. J. Physiol. Endocrinol. Metab. 303, E1234–E1244 (2012).
Engin, A. Non-Alcoholic Fatty liver disease. Adv. Exp. Med. Biol. 960, 443–467 (2017).
Wang, L. F. et al. Inhibition of NAMPT aggravates high fat diet-induced hepatic steatosis in mice through regulating Sirt1/AMPKalpha/SREBP1 signaling pathway. Lipids Health Dis. 16, 82 (2017).
Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome.Lancet Diabetes Endocrinol. 2, 901–910 (2014).
Guarino, M. & Dufour, J. F. Nicotinamide and NAFLD: is there nothing new under the Sun? Metabolites 9, 180 (2019).
Han, X. et al. Nicotinamide riboside exerts protective effect against aging-induced NAFLD-like hepatic dysfunction in mice. PeerJ 7, e7568 (2019).
von Schönfels, W. et al. Metabolomic tissue signature in human non-alcoholic fatty liver disease identifies protective candidate metabolites. Liver Int. 35, 207–214 (2015).
Zhang, Z. F. et al. Troxerutin improves hepatic lipid homeostasis by restoring NAD(+)-depletion-mediated dysfunction of lipin 1 signaling in high-fat diet-treated mice. Biochem. Pharmacol. 91, 74–86 (2014).
Dahl, T. B. et al. Intracellular nicotinamide phosphoribosyltransferase protects against hepatocyte apoptosis and is down-regulated in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 95, 3039–3047 (2010).
Tao, R. et al. Hepatic FoxOs regulate lipid metabolism via modulation of expression of the nicotinamide phosphoribosyltransferase gene. J. Biol. Chem. 286, 14681–14690 (2011).
Amirkalali, B. et al. Association between nicotinamide phosphoribosyltransferase and de novo lipogenesis in nonalcoholic fatty liver disease.Med. Princ. Pract. 26, 251–257 (2017).
Trammell, S. A. & Brenner, C. NNMT: a bad actor in fat makes good in liver. Cell Metab. 22, 200–201 (2015).
Song, Q. et al. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. J. Hepatol. 73, 783–793 (2020).
Boutari, C., Perakakis, N. & Mantzoros, C. S. Association of adipokines with development and progression of nonalcoholic fatty liver disease. Endocrinol. Metab. 33, 33–43 (2018).
Saxena, N. K. & Anania, F. A. Adipocytokines and hepatic fibrosis. Trends Endocrinol. Metab. 26, 153–161 (2015).
Choi, Y. J. et al. Extracellular visfatin activates gluconeogenesis in HepG2 cells through the classical PKA/CREB-dependent pathway. Hormone Metab. Res. 46, 233–239 (2014).
Choi, Y. J. et al. Involvement of visfatin in palmitate-induced upregulation of inflammatory cytokines in hepatocytes. Metabolism 60, 1781–1789 (2011).
Genc, H. et al. Association of plasma visfatin with hepatic and systemic inflammation in nonalcoholic fatty liver disease. Ann. Hepatol. 12, 548–555 (2013).
Polyzos, S. A. et al. Adipocytokines and cytokeratin-18 in patients with nonalcoholic fatty liver disease: Introduction of CHA index. Ann. Hepatol. 12, 749–757 (2013).
Auguet, T. et al. Plasma visfatin levels and gene expression in morbidly obese women with associated fatty liver disease. Clin. Biochem. 46, 202–208 (2013).
Kukla, M. et al. Liver visfatin expression in morbidly obese patients with nonalcoholic fatty liver disease undergoing bariatric surgery. Polish J. Pathol. 61, 147–153 (2010).
Jamali, R., Arj, A., Razavizade, M. & Aarabi, M. H. Prediction of nonalcoholic fatty liver disease via a novel panel of serum adipokines. Medicine 95, e2630 (2016).
Lingor, P., Koch, J. C., Tönges, L. & Bähr, M. Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res. 349, 289–311 (2012).
Johnson, V. E., Stewart, W. & Smith, D. H. Axonal pathology in traumatic brain injury. Exp. Neurol. 246, 35–43 (2013).
Osterloh, J. M. et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484 (2012).
Conforti, L., Gilley, J. & Coleman, M. P. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 15, 394–409 (2014).
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A. & Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348, 453–457 (2015).
Coleman, M. P. & Freeman, M. R. Wallerian degeneration, wld(s), and nmnat. Annu. Rev. Neurosci. 33, 245–267 (2010).
Di Stefano, M. et al. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 22, 731–742 (2015).
Sasaki, Y., Nakagawa, T., Mao, X., DiAntonio, A. & Milbrandt, J. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD(+) depletion. eLife 5, e19749 (2016).
Araki, T., Sasaki, Y. & Milbrandt, J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013 (2004).
Mack, T. G. et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 4, 1199–1206 (2001).
Ali, Y. O., Li-Kroeger, D., Bellen, H. J., Zhai, R. G. & Lu, H. C. NMNATs, evolutionarily conserved neuronal maintenance factors. Trends Neurosci. 36, 632–640 (2013).
Zhai, R. G. et al. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 452, 887–891 (2008).
Ali, Y. O. et al. NMNAT2:HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 14, e1002472 (2016).
Sasaki, Y., Vohra, B. P., Lund, F. E. & Milbrandt, J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J. Neurosci. 29, 5525–5535 (2009).
Loreto, A., Di Stefano, M., Gering, M. & Conforti, L. Wallerian degeneration is executed by an NMN-SARM1-dependent late Ca(2+) influx but only modestly influenced by mitochondria. Cell Rep. 13, 2539–2552 (2015).
Masters, C. L. et al. Alzheimer’s disease. Nat. Rev. Dis. Primers 1, 15056 (2015).
Corbett, A. et al. Drug repositioning for Alzheimer’s disease. Nat. Rev. Drug Discov. 11, 833–846 (2012).
Fang, E. F. et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412 (2019).
Lautrup, S., Sinclair, D. A., Mattson, M. P. & Fang, E. F. NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655 (2019).
Canter, R. G., Penney, J. & Tsai, L. H. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 539, 187–196 (2016).
Jo, D. G. et al. Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer’s disease. Neurobiol. Aging 31, 917–925 (2010).
Wang, J., Xiong, S., Xie, C., Markesbery, W. R. & Lovell, M. A. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 93, 953–962 (2005).
Dong, Y. & Brewer, G. J. Global metabolic shifts in age and Alzheimer’s disease mouse brains pivot at NAD+/NADH redox sites. J. Alzheimer’s Dis. 71, 119–140 (2019).
Liu, D. et al. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 34, 1564–1580 (2013).
Blacher, E. et al. Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Ann. Neurol. 78, 88–103 (2015).
Butterfield, D. A. & Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160 (2019).
Hou, Y., Song, H., Croteau, D. L., Akbari, M. & Bohr, V. A. Genome instability in Alzheimer disease. Mech. Ageing Dev. 161, 83–94 (2017).
Fang, E. F. et al. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 17, 308–321 (2016).
Fang, E. F. et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157, 882–896 (2014).
Fang, E. F. Mitophagy and NAD(+) inhibit Alzheimer disease. Autophagy 15, 1112–1114 (2019).
Green, K. N. et al. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 28, 11500–11510 (2008).
Ma, X. et al. Nicotinamide mononucleotide adenylyltransferase uses its NAD(+) substrate-binding site to chaperone phosphorylated Tau. eLife 9, e51859 (2020).
Schneider, R. B., Iourinets, J. & Richard, I. H. Parkinson’s disease psychosis: presentation, diagnosis and management. Neurodegen. Dis. Manag. 7, 365–376 (2017).
Schwab, A. J. et al. Decreased sirtuin deacetylase activity in LRRK2 G2019S iPSC-derived dopaminergic neurons. Stem Cell Rep. 9, 1839–1852 (2017).
Esteves, A. R. et al. Mitochondrial metabolism regulates microtubule acetylome and autophagy trough sirtuin-2: impact for Parkinson’s disease. Mol. Neurobiol. 55, 1440–1462 (2018).
Schondorf, D. C. et al. The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease. Cell Rep. 23, 2976–2988 (2018).
Lehmann, S., Loh, S. H. & Martins, L. M. Enhancing NAD(+) salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 6, 141–147 (2017).
Lehmann, S., Costa, A. C., Celardo, I., Loh, S. H. & Martins, L. M. Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson’s disease. Cell Death Dis. 7, e2166 (2016).
Tapias, V., McCoy, J. L. & Greenamyre, J. T. Phenothiazine normalizes the NADH/NAD(+) ratio, maintains mitochondrial integrity and protects the nigrostriatal dopamine system in a chronic rotenone model of Parkinson’s disease. Redox Biol. 24, 101164 (2019).
Wakade, C. & Chong, R. A novel treatment target for Parkinson’s disease. J. Neurol. Sci. 347, 34–38 (2014).
Murata, H. et al. c-Jun N-terminal kinase (JNK)-mediated phosphorylation of SARM1 regulates NAD(+) cleavage activity to inhibit mitochondrial respiration. J. Biol. Chem. 293, 18933–18943 (2018).
Murata, H., Sakaguchi, M., Kataoka, K. & Huh, N. H. SARM1 and TRAF6 bind to and stabilize PINK1 on depolarized mitochondria. Mol. Biol. Cell 24, 2772–2784 (2013).
Jia, H. et al. High doses of nicotinamide prevent oxidative mitochondrial dysfunction in a cellular model and improve motor deficit in a Drosophila model of Parkinson’s disease. J. Neurosci. Res. 86, 2083–2090 (2008).
Hikosaka, K., Yaku, K., Okabe, K. & Nakagawa, T. Implications of NAD metabolism in pathophysiology and therapeutics for neurodegenerative diseases. Nutr. Neurosci. 1–13 (2019).
Ghosh, R. & Tabrizi, S. J. Clinical features of Huntington’s disease. Adv. Exp. Med. Biol. 1049, 1–28 (2018).
Wyant, K. J., Ridder, A. J. & Dayalu, P. Huntington’s disease-update on treatments. Curr. Neurol. Neurosci. Rep. 17, 33 (2017).
McColgan, P. & Tabrizi, S. J. Huntington’s disease: a clinical review. Eur. J. Neurol. 25, 24–34 (2018).
Snowden, J. S. The neuropsychology of Huntington’s disease. Arch. Clin. Neuropsychol. 32, 876–887 (2017).
Lloret, A. & Beal, M. F. PGC-1α, sirtuins and PARPs in Huntington’s disease and other neurodegenerative conditions: NAD+ to rule them all. Neurochem. Res. 44, 2423–2434 (2019).
Fu, J. et al. trans-(-)-ε-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington disease. J. Biol. Chem. 287, 24460–24472 (2012).
Weydt, P. et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 4, 349–362 (2006).
Chaturvedi, R. K. et al. Impaired PGC-1alpha function in muscle in Huntington’s disease. Human Mol. Genetics 18, 3048–3065 (2009).
Tsunemi, T. et al. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 4, 142ra197 (2012).
Hathorn, T., Snyder-Keller, A. & Messer, A. Nicotinamide improves motor deficits and upregulates PGC-1α and BDNF gene expression in a mouse model of Huntington’s disease. Neurobiol. Dis. 41, 43–50 (2011).
Ravikumar, B. et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595 (2004).
Campesan, S. et al. The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s disease. Curr. Biol. 21, 961–966 (2011).
Beal, M. F., Matson, W. R., Swartz, K. J., Gamache, P. H. & Bird, E. D. Kynurenine pathway measurements in Huntington’s disease striatum: evidence for reduced formation of kynurenic acid. J. Neurochem. 55, 1327–1339 (1990).
Carrera-Julia, S., Moreno, M. L., Barrios, C., de la Rubia Orti, J. E. & Drehmer, E. Antioxidant alternatives in the treatment of amyotrophic lateral sclerosis: a comprehensive review. Front. Physiol. 11, 63 (2020).
Harlan, B. A. et al. Evaluation of the NAD(+) biosynthetic pathway in ALS patients and effect of modulating NAD(+) levels in hSOD1-linked ALS mouse models. Exp. Neurol. 327, 113219 (2020).
Nguyen, H. P., Van Broeckhoven, C. & van der Zee, J. ALS genes in the genomic era and their implications for FTD. Trends Genet. 34, 404–423 (2018).
Saccon, R. A., Bunton-Stasyshyn, R. K., Fisher, E. M. & Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136, 2342–2358 (2013).
Niedzielska, E. et al. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 53, 4094–4125 (2016).
Rosen, D. R. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 (1993).
Wei, R., Bhattacharya, A., Hamilton, R. T., Jernigan, A. L. & Chaudhuri, A. R. Differential effects of mutant SOD1 on protein structure of skeletal muscle and spinal cord of familial amyotrophic lateral sclerosis: role of chaperone network. Biochem. Biophys. Res. Commun. 438, 218–223 (2013).
Al-Chalabi, A. et al. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 124, 339–352 (2012).
Nagai, M. et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622 (2007).
Vargas, M. R., Pehar, M., Cassina, P., Beckman, J. S. & Barbeito, L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J. Neurochem. 97, 687–696 (2006).
Barbeito, L. H. et al. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res. Brain Res. Rev. 47, 263–274 (2004).
Harlan, B. A. et al. Enhancing NAD+ salvage pathway reverts the toxicity of primary astrocytes expressing amyotrophic lateral sclerosis-linked mutant superoxide dismutase 1 (SOD1). J. Biol. Chem. 291, 10836–10846 (2016).
Zhou, Q. et al. Nicotinamide riboside enhances mitochondrial proteostasis and adult neurogenesis through activation of mitochondrial unfolded protein response signaling in the brain of ALS SOD1(G93A) mice. Int. J. Biol. Sci. 16, 284–297 (2020).
Chen, Y. et al. The kynurenine pathway and inflammation in amyotrophic lateral sclerosis. Neurotox. Res. 18, 132–142 (2010).
Blacher, E. et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 572, 474–480 (2019).
Buck, E. et al. Comparison of sirtuin 3 levels in ALS and Huntington’s disease-differential effects in human tissue samples vs. transgenic mouse models. Front. Mol. Neurosci. 10, 156 (2017).
Harlan, B. A., Pehar, M., Killoy, K. M. & Vargas, M. R. Enhanced SIRT6 activity abrogates the neurotoxic phenotype of astrocytes expressing ALS-linked mutant SOD1. FASEB J. 33, 7084–7091 (2019).
Lee, J. C. et al. Region-specific changes in the immunoreactivity of SIRT1 expression in the central nervous system of SOD1(G93A) transgenic mice as an in vivo model of amyotrophic lateral sclerosis. Brain Res. 1433, 20–28 (2012).
Herskovits, A. Z. et al. SIRT1 deacetylase in aging-induced neuromuscular degeneration and amyotrophic lateral sclerosis. Aging Cell 17, e12839 (2018).
Watanabe, S. et al. SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol. Brain 7, 62 (2014).
Körner, S. et al. Differential sirtuin expression patterns in amyotrophic lateral sclerosis (ALS) postmortem tissue: neuroprotective or neurotoxic properties of sirtuins in ALS? Neurodegener. Dis. 11, 141–152 (2013).
Kim, D. et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179 (2007).
Tang, B. L. Could sirtuin activities modify ALS onset and progression? Cell. Mol. Neurobiol. 37, 1147–1160 (2017).
Carrì, M. T., D’Ambrosi, N. & Cozzolino, M. Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem. Biophys. Res. Commun. 483, 1187–1193 (2017).
D’Amico, E., Factor-Litvak, P., Santella, R. M. & Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Rad. Biol. Med. 65, 509–527 (2013).
de la Rubia, J. E. et al. Efficacy and tolerability of EH301 for amyotrophic lateral sclerosis: a randomized, double-blind, placebo-controlled human pilot study. Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 115–122 (2019).
Tang, W. H. W., Li, D. Y. & Hazen, S. L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 16, 137–154 (2019).
Ziaeian, B. & Fonarow, G. C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 13, 368–378 (2016).
Walker, M. A. & Tian, R. Raising NAD in heart failure: time to translate? Circulation 137, 2274–2277 (2018).
Mericskay, M. Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: Pathophysiological implications and therapeutic potential. Arch. Cardiovasc. Dis. 109, 207–215 (2016).
Lee, C. F. et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation 134, 883–894 (2016).
Wettersten, H. I., Aboud, O. A., Lara, P. N. Jr. & Weiss, R. H. Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 13, 410–419 (2017).
Horton, J. L. et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2, e84897 (2016).
Alrob, O. A. et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc. Res. 103, 485–497 (2014).
Pillai, V. B. et al. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 285, 3133–3144 (2010).
Hafner, A. V. et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2, 914–923 (2010).
Karamanlidis, G. et al. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell. Metab. 18, 239–250 (2013).
Boylston, J. A. et al. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 88, 73–81 (2015).
Hsu, C. P., Oka, S., Shao, D., Hariharan, N. & Sadoshima, J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ. Res. 105, 481–491 (2009).
Yamamoto, T. et al. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS ONE 9, e98972 (2014).
Hsu, C. P. et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 122, 2170–2182 (2010).
Porter, G. A., Urciuoli, W. R., Brookes, P. S. & Nadtochiy, S. M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts. Am. J. Physiol. Heart Circ. Physiol. 306, H1602–H1609 (2014).
Kane, A. E. & Sinclair, D. A. Sirtuins and NAD(+) in the development and treatment of metabolic and cardiovascular diseases. Circ. Res. 123, 868–885 (2018).
Yang, Y. et al. SIRT1 activation by curcumin pretreatment attenuates mitochondrial oxidative damage induced by myocardial ischemia reperfusion injury. Free Rad. Biol. Med. 65, 667–679 (2013).
Shalwala, M. et al. Sirtuin 1 (SIRT1) activation mediates sildenafil induced delayed cardioprotection against ischemia-reperfusion injury in mice. PLoS ONE 9, e86977 (2014).
Chen, C. J. et al. Resveratrol protects cardiomyocytes from hypoxia-induced apoptosis through the SIRT1-FoxO1 pathway. Biochem. Biophys. Res. Commun. 378, 389–393 (2009).
Cucoranu, I. et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 97, 900–907 (2005).
Gupte, R. S. et al. Upregulation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase activity increases oxidative stress in failing human heart. J. Card. Fail. 13, 497–506 (2007).
Guan, Y. et al. Nicotinamide mononucleotide, an NAD(+) precursor, rescues age-associated susceptibility to AKI in a sirtuin 1-dependent manner. J. Am. Soc. Nephrol. 28, 2337–2352 (2017).
Poyan Mehr, A. et al. De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat. Med. 24, 1351–1359 (2018).
Zheng, X. et al. Kynurenine 3-monooxygenase is a critical regulator of renal ischemia-reperfusion injury. Exp. Mol. Med. 51, 1–14 (2019).
Carney, E. F. Augmenting NAD(+) may combat kidney stress. Nat. Rev. Nephrol. 14, 657 (2018).
Tran, M. et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 121, 4003–4014 (2011).
Tran, M. T. et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531, 528–532 (2016).
Martin, D. R., Lewington, A. J., Hammerman, M. R. & Padanilam, B. J. Inhibition of poly(ADP-ribose) polymerase attenuates ischemic renal injury in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1834–1840 (2000).
Pressly, J. D. & Park, F. DNA repair in ischemic acute kidney injury. Am. J. Physiol. Renal Physiol. 312, F551–f555 (2017).
Zheng, J., Devalaraja-Narashimha, K., Singaravelu, K. & Padanilam, B. J. Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. Am. J. Renal Physiol. 288, F387–F398 (2005).
Liu, S. B., Liu, J., Liu, D. W., Wang, X. T. & Yang, R. L. Inhibition of poly-(ADP-ribose) polymerase protects the kidney in a canine model of endotoxic shock. Nephron 130, 281–292 (2015).
Morigi, M. et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 125, 715–726 (2015).
Rodgers, J. T. et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434, 113–118 (2005).
Zhao, W. et al. SIRT3 protects against acute kidney injury via AMPK/mTOR-regulated autophagy. Front. Physiol. 9, 1526 (2018).
Zhao, W. Y., Zhang, L., Sui, M. X., Zhu, Y. H. & Zeng, L. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Scientific Rep. 6, 33201 (2016).
Fulco, M. et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 14, 661–673 (2008).
Canto, C. et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219 (2010).
Chen, D. et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 22, 1753–1757 (2008).
Costford, S. R. et al. Skeletal muscle NAMPT is induced by exercise in humans. Am. J Physiol. Endocrinol. Metab. 298, E117–E126 (2010).
Elhassan, Y. S. et al. Nicotinamide riboside augments the aged human skeletal muscle NAD(+) metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. 28, 1717–1728 e1716 (2019).
Kohsaka, A. et al. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab. 6, 414–421 (2007).
Ando, H. et al. Impairment of peripheral circadian clocks precedes metabolic abnormalities in ob/ob mice. Endocrinology 152, 1347–1354 (2011).
Verlande, A. & Masri, S. Circadian clocks and cancer: timekeeping governs cellular metabolism. Trends Endocrinol. Metab. 30, 445–458 (2019).
Roh, E. et al. Effects of chronic NAD supplementation on energy metabolism and diurnal rhythm in obese mice. Obesity 26, 1448–1456 (2018).
Zheng, M. et al. Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J. Cell. Mol. Med. 23, 3995–4004 (2019).
Mills, K. F. et al. Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 24, 795–806 (2016).
Yao, Z., Yang, W., Gao, Z. & Jia, P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci. Lett. 647, 133–140 (2017).
Hosseini, L., Vafaee, M. S., Mahmoudi, J. & Badalzadeh, R. Nicotinamide adenine dinucleotide emerges as a therapeutic target in aging and ischemic conditions. Biogerontology 20, 381–395 (2019).
Yoshino, J., Baur, J. A. & Imai, S. I. NAD(+) Intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 27, 513–528 (2018).
Lee, H. J. & Yang, S. J. Nicotinamide riboside regulates inflammation and mitochondrial markers in AML12 hepatocytes. Nutr. Res. Pract. 13, 3–10 (2019).
Madeo, F., Carmona-Gutierrez, D., Hofer, S. J. & Kroemer, G. Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential. Cell. Metab. 29, 592–610 (2019).
Trammell, S. A. et al. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci. Rep. 6, 26933 (2016).
Gariani, K. et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 63, 1190–1204 (2016).
Giroud-Gerbetant, J. et al. A reduced form of nicotinamide riboside defines a new path for NAD(+) biosynthesis and acts as an orally bioavailable NAD(+) precursor. Mol. Metab. 30, 192–202 (2019).
Yang, Y., Mohammed, F. S., Zhang, N. & Sauve, A. A. Dihydronicotinamide riboside is a potent NAD(+) concentration enhancer in vitro and in vivo. J. Biol. Chem. 294, 9295–9307 (2019).
Kuchmerovska, T., Shymanskyy, I., Bondarenko, L. & Klimenko, A. Effects of nicotinamide supplementation on liver and serum contents of amino acids in diabetic rats. Eur. J. Med. Res. 13, 275–280 (2008).
van de Weijer, T. et al. Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans. Diabetes 64, 1193–1201 (2015).
Pillai, J. B., Isbatan, A., Imai, S. & Gupta, M. P. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J. Biol. Chem. 280, 43121–43130 (2005).
Alano, C. C. et al. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 30, 2967–2978 (2010).
Zhang, D. et al. DNA damage-induced PARP1 activation confers cardiomyocyte dysfunction through NAD(+) depletion in experimental atrial fibrillation. Nat. Commun. 10, 1307 (2019).
Yap, T. A., Plummer, R., Azad, N. S. & Helleday, T. The DNA damaging revolution: PARP inhibitors and beyond. Am. Soc. Clin. Oncol. Educ. Book 39, 185–195 (2019).
Adashek, J. J., Jain, R. K. & Zhang, J. Clinical development of PARP inhibitors in treating metastatic castration-resistant prostate cancer. Cells 8, 860 (2019).
Lin, K. Y. & Kraus, W. L. PARP inhibitors for cancer therapy. Cell 169, 183 (2017).
Ellisen, L. W. PARP inhibitors in cancer therapy: promise, progress, and puzzles. Cancer Cell 19, 165–167 (2011).
Pirinen, E. et al. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 19, 1034–1041 (2014).
Haffner, C. D. et al. Discovery, synthesis, and biological evaluation of thiazoloquin(az)olin(on)es as potent CD38 inhibitors. J. Med. Chem. 58, 3548–3571 (2015).
Migliavacca, E. et al. Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 10, 5808 (2019).
Schultz, M. D. et al. Inhibition of the NAD salvage pathway in schistosomes impairs metabolism, reproduction, and parasite survival. PLoS Pathog.16, e1008539 (2020).
Bauman, M. D. et al. Neuroprotective efficacy of P7C3 compounds in primate hippocampus. Trans. Psychiatry 8, 202 (2018).
Blaya, M. O. et al. Neurotherapeutic capacity of P7C3 agents for the treatment of traumatic brain injury. Neuropharmacology 145, 268–282 (2019).
Voorhees, J. R. et al. (-)-P7C3-S243 protects a rat model of Alzheimer’s disease from neuropsychiatric deficits and neurodegeneration without altering amyloid deposition or reactive glia. Biol. Psychiatry 84, 488–498 (2018).
Wang, G. et al. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell 158, 1324–1334 (2014).
Gardell, S. J. et al. Boosting NAD(+) with a small molecule that activates NAMPT. Nat. Commun. 10, 3241 (2019).
Yoshino, J. ACMSD: a novel target for modulating NAD(+) homeostasis. Trends Endocrinol. Metab. 30, 229–232 (2019).
Thirtamara-Rajamani, K. et al. Is the enzyme ACMSD a novel therapeutic target in Parkinson’s disease? J. Parkinson’s Dis. 7, 577–587 (2017).
Neelakantan, H. et al. Selective and membrane-permeable small molecule inhibitors of nicotinamide N-methyltransferase reverse high fat diet-induced obesity in mice. Biochem. Pharmacol. 147, 141–152 (2018).
Kraus, D. et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 508, 258–262 (2014).
Lin, S. J., Defossez, P. A. & Guarente, L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128 (2000).
Lin, S. J., Ford, E., Haigis, M., Liszt, G. & Guarente, L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 18, 12–16 (2004).
Lin, S. J. et al. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418, 344–348 (2002).
Masri, S. et al. Lung adenocarcinoma distally rewires hepatic circadian homeostasis. Cell 165, 896–909 (2016).
Damiola, F. et al. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 14, 2950–2961 (2000).
Stokkan, K. A., Yamazaki, S., Tei, H., Sakaki, Y. & Menaker, M. Entrainment of the circadian clock in the liver by feeding. Science 291, 490–493 (2001).
Vollmers, C. et al. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc. Natl Acad. Sci. USA 106, 21453–21458 (2009).
Rippe, C. et al. Short-term calorie restriction reverses vascular endothelial dysfunction in old mice by increasing nitric oxide and reducing oxidative stress. Aging Cell 9, 304–312 (2010).
Donato, A. J. et al. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell 12, 772–783 (2013).
Hyun, D. H., Emerson, S. S., Jo, D. G., Mattson, M. P. & de Cabo, R. Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc. Natl Acad. Sci. USA 103, 19908–19912 (2006).
Lu, M., Zhu, X. H., Zhang, Y. & Chen, W. Intracellular redox state revealed by in vivo (31) P MRS measurement of NAD(+) and NADH contents in brains. Magn. Reson. Med. 71, 1959–1972 (2014).
Zerez, C. R., Lee, S. J. & Tanaka, K. R. Spectrophotometric determination of oxidized and reduced pyridine nucleotides in erythrocytes using a single extraction procedure. Anal. Biochem. 164, 367–373 (1987).
Rongvaux, A. et al. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J. Immunol. 181, 4685–4695 (2008).
Zhang, Q. et al. Redox sensor CtBP mediates hypoxia-induced tumor cell migration. Proc. Natl Acad. Sci. USA 103, 9029–9033 (2006).
Xie, W., Xu, A. & Yeung, E. S. Determination of NAD(+) and NADH in a single cell under hydrogen peroxide stress by capillary electrophoresis. Anal. Chem. 81, 1280–1284 (2009).
Casey, T. M., Dufall, K. G. & Arthur, P. G. An improved capillary electrophoresis method for measuring tissue metabolites associated with cellular energy state. Eur. J. Biochem. 261, 740–745 (1999).
Stephanson, C. J. & Flanagan, G. P. Differential metabolic effects on mitochondria by silica hydride using capillary electrophoresis. J. Med. Food 7, 79–83 (2004).
Demarest, T. G. et al. Assessment of NAD(+)metabolism in human cell cultures, erythrocytes, cerebrospinal fluid and primate skeletal muscle. Anal. Biochem. 572, 1–8 (2019).
Pálfi, M., Halász, A. S., Tábi, T., Magyar, K. & Szöko, E. Application of the measurement of oxidized pyridine dinucleotides with high-performance liquid chromatography-fluorescence detection to assay the uncoupled oxidation of NADPH by neuronal nitric oxide synthase. Anal. Biochem. 326, 69–77 (2004).
Yoshino, J. & Imai, S. Accurate measurement of nicotinamide adenine dinucleotide (NAD+) with high-performance liquid chromatography. Methods Mol. Biol. 1077, 203–215 (2013).
Petucci, C. et al. Measurement of pyridine nucleotides in biological samples using LC-MS/MS. Methods Mol. Biol. 1996, 61–73 (2019).
Balducci, E. et al. Assay methods for nicotinamide mononucleotide adenylyltransferase of wide applicability. Anal. Biochem. 228, 64–68 (1995).
Chance, B., Cohen, P., Jobsis, F. & Schoener, B. Intracellular oxidation-reduction states in vivo. Science 137, 499–508 (1962).
Zhao, Y. et al. In vivo monitoring of cellular energy metabolism using SoNar, a highly responsive sensor for NAD(+)/NADH redox state. Nat. Protoc. 11, 1345–1359 (2016).
Zou, Y. et al. Analysis of redox landscapes and dynamics in living cells and in vivo using genetically encoded fluorescent sensors. Nat. Protoc. 13, 2362–2386 (2018).
Zhao, Y. et al. Genetically encoded fluorescent sensors for intracellular NADH detection. Cell. Metab. 14, 555–566 (2011).
Blacker, T. S. et al. Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat. Commun. 5, 3936 (2014).
Wilkening, S. et al. Characterization of Frex as an NADH sensor for in vivo applications in the presence of NAD(+) and at various pH values. Photosynth. Res. 133, 305–315 (2017).
Bilan, D. S. et al. Genetically encoded fluorescent indicator for imaging NAD(+)/NADH ratio changes in different cellular compartments. Biochim. Biophys. Acta 1840, 951–957 (2014).
Cameron, W. D. et al. Apollo-NADP(+): a spectrally tunable family of genetically encoded sensors for NADP(+). Nat. Methods 13, 352–358 (2016).
Tao, R. et al. Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. Nat. Methods 14, 720–728 (2017).
Chouinard, V. A. et al. Brain bioenergetics and redox state measured by (31)P magnetic resonance spectroscopy in unaffected siblings of patients with psychotic disorders. Schizophr. Res. 187, 11–16 (2017).
Valkovič, L., Chmelík, M. & Krššák, M. In-vivo(31)P-MRS of skeletal muscle and liver: a way for non-invasive assessment of their metabolism. Anal. Biochem. 529, 193–215 (2017).
Lu, M., Zhu, X. H. & Chen, W. In vivo (31) P MRS assessment of intracellular NAD metabolites and NAD(+) /NADH redox state in human brain at 4 T. NMR Biomed. 29, 1010–1017 (2016).
Peeters, T. H., van Uden, M. J., Rijpma, A., Scheenen, T. W. J. & Heerschap, A. 3D (31) P MR spectroscopic imaging of the human brain at 3 T with a (31) P receive array: an assessment of (1) H decoupling, T(1) relaxation times, (1) H-(31) P nuclear Overhauser effects and NAD. NMR Biomed. e4169 (2019).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81821002, 81790251, 81430071, 81672381, 81972665 and 81772487), Guangdong Basic and Applied Basic Research Foundation (2019B030302012), the National 973 Basic Research Program of China (2013CB911300), the Science and Technology Department of Sichuan Province (No. 2017SZ0057, 2019YJ0050).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xie, N., Zhang, L., Gao, W. et al. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Sig Transduct Target Ther 5, 227 (2020). https://doi.org/10.1038/s41392-020-00311-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-00311-7
This article is cited by
-
Changes in the innate immune response to SARS-CoV-2 with advancing age in humans
Immunity & Ageing (2024)
-
Diminished NAD+ levels and activation of retrotransposons promote postovulatory aged oocyte (POAO) death
Cell Death Discovery (2024)
-
Insights into the modulation of bacterial NADase activity by phage proteins
Nature Communications (2024)
-
Adipose tissue macrophages secrete small extracellular vesicles that mediate rosiglitazone-induced insulin sensitization
Nature Metabolism (2024)
-
Nicotinamide mononucleotide, a potential future treatment in ocular diseases
Graefe's Archive for Clinical and Experimental Ophthalmology (2024)
