
Abstract
Background
Clinical and preclinical studies have revealed that alterations in DNA damage response (DDR) pathways may play an important role in prostate cancer (PCa) etiology and progression. These alterations can influence PCa responses to radiotherapy and anti-androgen treatment. The identification of DNA repair gene aberrations in PCa has driven the interest for further evaluation whether these genetic changes may serve as biomarkers for patient stratification.
Methods
In this review, we summarize the current knowledge on DDR alterations in PCa, their potential impact on clinical interventions and prospects for improved management of PCa. We particularly focus on the influence of DDR gene mutations on PCa initiation and progression and describe the underlying mechanisms.
Results and Conclusions
A better understanding of these mechanisms, will contribute to better disease management as treatment strategies can be chosen based on the specific disease properties, since a growing number of treatments are targeting DDR pathway alterations (such as Poly(ADP-ribose) polymerase inhibitors). Furthermore, the recently discovered crosstalk between the DDR and androgen receptor signaling opens a new array of possible strategies to optimize treatment combinations. We discuss how these recent and ongoing studies will help to improve diagnostic, prognostic and therapeutic approaches for PCa management.
Similar content being viewed by others


Introduction
Prostate cancer (PCa) is the second most common cancer in men and the fourth most common tumor type worldwide [1]. Although organ-confined disease can be well managed, curative therapeutic options for disseminated disease are limited. First-line therapy for disseminated PCa is androgen deprivation therapy (ADT) that prevents androgen receptor (AR) pathway signaling as most PCas are dependent on activated AR signaling for cell survival [2, 3]. In time, patients under ADT may progress to castration-resistant PCa (CRPC), requiring first line chemotherapy (commonly docetaxel) [4]. New therapeutic strategies for CRPC are being offered to patients, such as new combinations and sequences of second-generation antiandrogen therapy (enzalutamide, abiraterone, apalutamide) or second line chemotherapy (cabazitaxel), which have shown notable benefit for patient survival [4]. In addition, promising new treatment modalities, such as Radium-223 and prostate-specific membrane antigen (PSMA)-directed radioligand therapy, are being exploited for patients with (bone) metastatic disease. Despite this progress in the development of new drugs, CRPC continues to be incurable, and drug resistance remains an issue.
Clinical and preclinical studies have revealed that alterations in DNA damage response (DDR) pathways play a role in PCa etiology and progression, especially in CRPC patients [5,6,7,8,9,10]. These DNA repair defects may be targeted by specific treatments, such as Poly(ADP-ribose) polymerase (PARP) inhibitors [11]. Moreover, several studies provided evidence that AR signaling links to the DDR in prostate cancer cells, which may have relevance for the first line disease management using ADT and AR-targeted agents [12, 13]. In this review, we summarize the current knowledge of DDR alterations in PCa, the AR-DDR crosstalk and the potential exploitation of DDR targeting drugs to improve clinical interventions.
DNA damage response pathways
DNA damage has emerged as a major culprit in cancer initiation and progression. The DNA is constantly damaged by exogenous sources such as genotoxic chemicals, ultraviolet (UV), and ionizing radiation (IR), as well as by endogenous DNA-damaging agents, such as reactive oxygen and nitrogen species [14, 15]. These sources will induce various damages to the DNA, including base oxidation, deamination, alkylation, interstrand crosslinks, adduct formation, single-strand breaks (SSBs), and double-strand breaks (DSBs). Additionally, spontaneous DNA damage is induced during replication. Collisions of the replication fork with DNA-binding proteins or the transcription machinery are the most common causes leading to replication fork stalling or collapse, which in turn induces DNA damage [16, 17]. Incorrect or failed repair of damaged DNA can lead to genetic alterations. Important consequences of genetic alterations are loss of tumor suppressor genes and activation of oncogenes, which may trigger the development of malignant cells or increase aggressiveness of tumor cells. Normal cells maintain genomic integrity using various DDR mechanisms to repair damaged DNA or induce cell death. The concept of DDR has been introduced to describe a series of biological reactions including DNA lesion site detection, repair protein recruitment, damage repair, cell cycle checkpoint control, and cell death pathways.
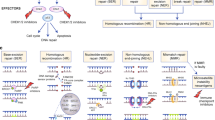
The highly diverse spectrum of DNA lesions can be repaired by a number of different DNA repair pathways, which have been reviewed extensively elsewhere [18,19,20]. In short, base excision repair (BER) involves multiple enzymes to excise and replace a single damaged nucleotide base, such as an oxidized base, but also an SSB [21]. Mismatch repair (MMR) is mainly involved in repair of base mismatches and insertions/deletions that can occur during replication and recombination [22]. The Fanconi anemia (FA) pathway repairs DNA interstrand crosslinks in the genome [23]. DSBs are resolved either by high-fidelity homologous recombination (HR) or error-prone non-homologous end joining (NHEJ). The HR pathway is only active when the cell is in the S/G2 cell cycle stage since it requires the presence of the sister chromatid as a repair template [24]. DSBs can be generated during replication when the replication fork encounters a DNA lesion and these breaks are exclusively repaired by HR. NHEJ is active during all cell cycle stages and functions by directly ligating broken DNA ends. Since no template is used during NHEJ, repair via this pathway is error prone (Fig. 1) [24]. After DNA damage induction, depending on the severity of the lesion and repair capacity, cells will continue to proliferate if damages are repaired, or cells stop proliferation, become senescent, or undergo programmed cell death (apoptosis) to remove damaged DNA from the cellular population [25]. Alterations in any of these pathways can result in genomic instability and consequently predispose to cancer, affect disease progression and/or influence therapy efficacy. Nonetheless, impaired DNA repair can also be a possible Achilles heel of the cancer that can be exploited for treatment [26].

DNA double strand (DSB) and single-strand break (SSB) repair pathways. The majority of the DSBs are repaired by the error-prone Non-Homologous End-Joining pathway (NHEJ, available during all cell cycle stages) and a smaller fraction of the DSBs are repaired via Homologous Recombination (HR, only during S/G2 cell cycle stages). SSBs are repaired by the Single Strand Break Repair pathway (available during all cell cycle stages). During DNA replication an unrepaired SSB can be converted into a DSB which can then only be repaired by HR
Molecular mechanisms underlying PCa risk
Multiple studies have indicated that germline mutations in DNA repair genes are associated with a higher risk of developing PCa. The individuals at risk have one inherited dysfunctional allele of the DNA repair gene and a second event (mutation or epigenetic silencing) can cause inactivation of the functional allele. The most common germline mutated DDR genes in primary PCa or CRPC are found in the Breast Cancer 1 and 2 (BRCA1 and BRCA2) genes. Similar to the role of mutations in BRCA1/2 in the development of breast cancer and ovarian cancer [14], various studies have shown that inactivating BRCA1/2 mutations, predominantly BRCA2, increase predisposition to PCa (Table 1) [7,8,9, 27,28,29]. BRCA1/2 are tumor suppressor genes and both encode large proteins which act in multiple cellular pathways. BRCA1 and BRCA2 are both involved in the HR pathway [30, 31], while BRCA1 has also been found to have other functions [32]. Loss-of-function mutations in BRCA1/2 lead to a deficiency in error-free HR repair. Therefore, DSBs will be repaired alternatively by other non-conservative and potentially mutagenic mechanisms, such as the NHEJ pathway. The resulting genomic instability (chromosomal translocations and deletions) and mutations may be the underlying mechanism of BRCA1/2 associated cancers [33, 34]. This could increase the risk of acquiring fusion genes, such as the TMPRSS2/ERG fusion that is found in 40-50% of PCa cases [35], although no solid evidence has been acquired to link BRCA1/2 mutation status to this fusion. Furthermore, the reason why BRCA1/2 mutations are particularly associated with specific cancer types, such as breast, ovarian and PCa remains unknown.
Francis et al. showed that BRCA2 can act as a tumor suppressor in the prostate [36]. Using a genetically engineered mouse model, it was found that deletion of Brca2 in prostate epithelia resulted in focal hyperplasia and low-grade prostate intraepithelial neoplasia (PIN) in animals over 12 months old. Epithelial cells in these lesions showed an increase in DNA damage. The evidence that other inherited gene mutations in DSB repair genes, such as BRCA1 Interacting Protein C-Terminal Helicase 1 (BRIP1) and Nibrin (NBS1), are also associated with PCa has been documented less extensively [6, 8].
Besides DSB gene alterations, mutations in the MMR genes MutS homolog 2 and 6 (MSH2 and MSH6) are also associated with increased PCa risk [7, 27]. MMR mutations would mainly cause point mutations or small insertions and deletions of short repetitive sequences of DNA which may result in microsatellite instability [37]. Therefore, underlying mechanisms of PCa can be linked to Lynch syndrome, a hereditary ‘non-polyposis’-colorectal carcinoma that is caused by MMR pathway mutations. The increased risk of PCa in MMR mutation carriers and in families with Lynch syndrome provide the rationale to include PCa in the Lynch syndrome tumor spectrum, which is relevant for risk estimates and surveillance recommendations in MMR mutation carriers [38].
DDR defects in PCa
DDR defects in primary PCa
The clinical behavior of localized PCa is highly variable: while some men have aggressive cancer leading to metastasis and death, many others have indolent cancers and these men can be cured by local therapy or may be safely observed without treatment [39]. Several studies have identified primary PCa tumors harboring a diversity of DDR gene alterations (summarized in Table 2) [40,41,42,43,44,45]. These studies identified a heterogeneous panel of repair defects caused by homozygous mutations or copy number alterations in primary prostate tumors compared to paired normal tissue in Ataxia–telangiectasia mutated (ATM), BRCA2, RAD51, mediator of DNA damage checkpoint 1 (MDC1), PARP1, and FA complementation group D2 (FANCD2), although the level of incidence varied between the studies. This considerable heterogeneity of repair defect prevalence among different studies could at least in part be attributed to the diversity of the study populations, as the genetic background can differ significantly between indolent, non-symptomatic and progressive PCa [46,47,48].
Loss-of-function DDR gene mutations can contribute to a more aggressive PCa phenotype with a higher probability of nodal involvement and distant metastasis [5, 49,50,51]. This aggressive phenotype was also reported in patients harboring BRCA1/2 and ATM combined mutations [52] and NBS1 mutations alone [6]. Recent clinical data have shown a strong prognostic value of a DDR mutation signature which may be used for risk stratification for high-risk PCa patients. Treatment outcome for BRCA1/2 mutation carriers showed worse outcomes for these patients than non-carriers when conventionally treated with surgery or radiation therapy [53].
The studies discussed above found DDR mutations in primary PCa, with a heterogeneous and overall low mutation rate. However, a direct (mechanistic) link between these mutations and PCa predisposition and treatment has not yet been established. As primary PCa is typically well managed and not lethal, it will therefore be of more interest to focus on the landscape of DDR defects in advanced PCa.
DDR defects in mCRPC
An enrichment of DDR gene alterations can be found during PCa progression, especially when the disease develops into metastatic CRPC (mCRPC) (summarized in Table 3) [54,55,56]. Heavily pre-treated mCRPC contained more genetic alterations in DDR genes (46%) than treatment-naive high grade localized tumors (27%) [54]. A multi-institutional clinical sequencing study revealed that the majority of affected individuals with CRPC harbor clinically actionable homozygous molecular alterations, with 23% of mCRPC harboring DDR aberrations and 8% harboring DDR germline mutations [55]. Aberrations in BRCA1, BRCA2, and ATM were observed at substantially higher frequencies (19.3% overall) in mCRPC compared to those in primary PCa. Among these DDR alterations, BRCA2 was the most frequently altered (12.7%), and ∼90% of these BRCA2 defective tumors exhibited biallelic loss. As aberrations in these genes are expected to confer sensitivity to PARP inhibitors [56], nearly 20% of mCRPC patients may potentially benefit from this therapy. Additionally, three out of four mCRPC tumors in this cohort which presented hypermutations are harboring defects in the MMR pathway genes MLH1 or MSH2 [55]. Whether this abundance of DDR alterations is specifically targeted to these genes or a general consequence of high mutational burden for advanced disease is still unclear.
DDR defects and response to PCa treatment
Various retrospective and prospective studies have been performed in which treatment outcome to conventional PCa treatment was compared in DDR mutation carriers and wild-type individuals. The prognostic and predictive impact related to standard therapies for DDR mutated mCRPC has yet to be determined, since these trials (summarized in Table 4) report inconsistent and conflicting outcomes: one study found no difference between the patient groups [57], while other studies reported DDR mutation carriers to have either inferior [58] or improved responses [59, 60] to the therapy. This inconsistency could be explained in several ways. First, the number of mCRPC patients harboring DDR mutations is very limited in each cohort. Second, the results can be biased due to different sampling, as metastatic biopsies are only feasible for patients with low-to-moderate tumor burden. This might exclude highly aggressive tumors and blood-based sequencing may underestimate the mutation rate as the somatic status is unknown for certain patients. Third, the disease showed extensive heterogeneity and patients had received various pre-treatments in the different cohorts. A recent prospective study showed that BRCA2 mutation carriers have a worse outcome in mCRPC disease and this may be affected by the first line treatment used [61]. However, future prospective studies are needed to shed further light on this issue and will hopefully resolve the above-mentioned controversy.
Radium-223, a bone-seeking α-particle emitter that induces DSBs, thereby killing cancer cells in the bone microenvironment, is commonly used for CRPC patients with symptomatic bone metastases [62]. Recently, a retrospective single-institution study showed that germline or somatic HR-deficient patients responded better to Radium-223 therapy compared to wild-type patients, with a better alkaline phosphatase responses (80% vs 39%, p = 0.04), and a trend toward longer overall survival (median 36.9 vs 19.0 months, p = 0.11) [63]. Synthetic lethality between HR mutations and Radium-223 activity maybe the underlying mechanism of a better efficacy, however these promising results need further (prospective) validation.
AR and DDR pathway crosstalk
Clinical trials have shown that the combination of ADT or anti-androgens with radiotherapy significantly increases patients survival and reduces distant metastases compared to radiotherapy alone [64,65,66,67,68,69]. It is widely perceived that suppression of the AR axis enhances the cytotoxic effects of radiotherapy and based on the beneficial effects, this combination is currently the standard of care for locally advanced PCa.
The molecular mechanism of radiosensitization induced by ADT was investigated in preclinical studies. Goodwin et al. reported that ADT potentiates the tumor-killing effect of ionizing radiation (IR) in AR proficient cells both in vitro and in vivo: ADT treated C4-2 (androgen independent) cells had a diminished capacity to repair IR induced DSBs. This study showed that the AR pathway directly regulates the NHEJ factor DNA-dependent protein kinase catalytic subunit (DNA-PKcs), resulting in a slight increase in NHEJ activity upon androgen addition in a plasmid-based functional assay [12]. The involvement of NHEJ was confirmed by Polkinghorn et al. who identified a set of 32 DDR genes as direct AR target genes [70]. Other studies using patients samples have demonstrated that castration primarily reduces Ku70 protein expression, which is essential for NHEJ [71, 72]. These studies suggest that ADT enhances IR effects by impairing NHEJ activity. Reciprocally, IR treatment caused marked induction of the androgen target genes TMPRSS2 and FKBP5 [12], suggesting that DNA damage induces AR activity (Fig. 2).

Interplay between androgen receptor (AR) and DNA damage repair in prostate cancer. Activation of AR by dihydrotestosterone (T) leads to transcriptional upregulation of DNA repair genes in various repair pathways. Reciprocally, irradiation results in upregulation of keys genes in the AR pathway via ROS. HR, homologous recombination; NHEJ, non-homologous end-joining; ROS, Reactive oxygen species; IR, Irradiation
In addition to direct regulation of the NHEJ pathway, other studies show that AR signaling plays a role in regulating genes involved in the HR, MMR, and FA pathways [12, 70, 73]. Enzalutamide treatment suppressed the expression of the HR genes BRCA1, RAD54L, and RecQ Mediated Genome Instability 2 (RMI2) [73]. A combination strategy in which enzalutamide pretreatment was followed by the PARP inhibitor olaparib resulted in significantly increased PCa cell apoptosis and inhibited colony formation in vitro. Further in vivo evaluation showed clear synergistic suppressive effects on PCa xenografts in hormone-sensitive models, but not in CRPC models [73]. However, from these studies, it is not yet clear whether enzalutamide directly induces HR deficiency, also called the BRCAness phenotype. A reduction of the S/G2 cell cycle fraction might also have caused reduction of HR gene expression, which resulted in reduced HR in the total cell population. Whatever the mechanistic explanation may be, this study warrants further clinical investigation into AR and PARP inhibitor combination therapies.
Based on these results, it is clear that both preclinical and clinical studies have found that AR signaling regulates the expression and/or function of DDR genes. Elucidation of the precise regulatory mechanisms and pathway interactions requires additional studies, which should focus on direct measurement of NHEJ and HR capacity in the presence and absence of AR signaling.
Exploiting DDR alterations for PCa treatment
As discussed above, 10–25% of PCa patients are harboring DDR mutations, especially among mCRPC patients. This section summarizes clinical and preclinical evidence how DDR alterations could be exploited therapeutically.
Immune checkpoint inhibitors
The successful development of immune checkpoint inhibitors such as programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) inhibitors revolutionized the field of cancer immunotherapy [74]. The interaction of PD-L1 on tumor cells with PD-1 on T-cells reduces T-cell functionality, preventing the immune system from attacking the tumor cells. Inhibitors that block this interaction can unleash a patient’s own T cells to kill tumors [75]. Immunotherapy responses appear to correlate with the mutational burden, presumably by the increase in neo-antigens [76]. PCa patients harboring MMR mutations, such as in MLH1 or MSH2, could be selected for PD-1 blockade immunotherapy, as a favorable response to PD-1 blockade therapy was observed previously in MMR-deficient tumors, as a result of the high level of neo-antigens in various solid tumors [77]. Interestingly, ductal adenocarcinoma, an aggressive histopathology of PCa, is associated with MMR defects, suggesting that these patients are possible candidates for this type of immunotherapy [78]. Interestingly, an increase in neo-antigens was also observed in patients who harbor a HR deficiency [79]. Altogether, these subgroups represent nearly 20% of mCRPC patients, making the use of PD-1/PD-L1 inhibitors a potentially attractive strategy for clinical trials in these patients.
PARP inhibitor treatments
Monotherapy
Tumors with compromised HR are highly sensitive to reduction of SSB repair by PARP1 inhibition, a phenomenon called synthetic lethality [80,81,82]. The mechanism of action of PARP inhibitors was originally described as inhibition of SSB repair via blocking the catalytic activity of PARP1. Unrepaired SSBs will be converted into the more genotoxic DSBs during DNA replication. These DSBs are repaired via HR in normal cells, but cannot be repaired in HR-deficient cancer cells, leading to tumor-specific cell death. Recently, this model has been updated as studies have shown that various PARP inhibitors are able to trap PARP1 at the DNA damage site [83,84,85]. Trapped PARP results in DSBs when the replication fork encounters this lesion, which require HR for resolution (Fig. 3). Considering the different PARP trapping abilities of the different PARP inhibitors, various therapeutic responses can be expected, with talazoparib having the most profound PARP trapping and cytotoxic effects [86].

Mechanism of action of Poly(ADP-ribose) polymerase (PARP) inhibitor. PARP enhances repair of single-strand breaks (SSBs) via base excision repair (BER). If SSBs remain unrepaired due to inhibition of PARP catalytic activity with PARP inhibitors (PARPi), double-strand breaks (DSBs) can be formed during replication. Alternatively, PARPi can trap the PARP protein on the DNA, which causes replication fork (RF) stalling and collapse. Homologous recombination (HR) is essential for repairing these DSBs
Following previous in vitro [80, 81] and in vivo studies in Brca2 knockout breast and ovarian tumor mouse models [87, 88], a number of trials evaluated PARP inhibitors as a single agent in CRPC patients with HR defects. The TOPARP study evaluated olaparib in a population of 50 mCRPC patients. Interestingly, 14 out of 16 DDR mutation carriers responded to olaparib treatment, compared to two of 33 patients in the non-DDR mutated group [56]. The promising results from this study led to the initiation of a large number of clinical trials targeting PARP by different inhibitors with or without HR gene mutation preselection in order to validate the effect, evaluate its safety profile and define the optimal timing of prescribing PARP inhibitors in mCRPC [89,90,91,92]. Interestingly, a recently published multicenter retrospective study including 23 mCRPC patients harboring DDR mutations (2 BRCA1, 15 BRCA2 an 6 ATM) showed that men with ATM mutations responded inferior to PARP inhibitor treatment compared to BRCA1/2 mutation carriers [93]. These data suggest that ATM mutated patients may not benefit from PARP inhibitor treatment as previously thought, and preselection of patients is importance to avoid unnecessary toxicity.
It is to be expected that due to increased use of next generation sequencing approaches, it is likely that more PCa patients with HR defects will be detected. However, the implementation and standardization of genomic testing still remains a major challenge. Besides blood-based germline mutation and biopsy based somatic mutation testing, new studies are looking into circulating tumor cells (CTC) or cell-free DNA based detection of a panel of clinically actionable genes to select eligible patients [90, 94, 95]. Moreover, the efforts made for identifying tumors with HR deficiency by using mutational signatures (HRDetect) or functional HRD tests will guide us to a more personalized cancer management approach [96,97,98,99,100].
Combination therapies
Besides PARP inhibition as monotherapy, trials have been initiated to evaluate combination of PARP inhibitors with other treatments in mCRPC patients. In view of the working mechanism of PARP inhibitors, an obvious strategy is to combine them with DNA-damaging agents, such as chemotherapy, radiotherapy and radioligand therapy (ongoing clinical trials are summarized in Table 5). Synergy with PARP inhibitors was identified in various clinical trials in other tumor types [101]. However overlapping hematological toxicities may represent a major hurdle when combining DNA-damaging agents and PARP inhibitors [102].
Previous preclinical work offered the rationale for the potential synergy of combining AR-targeting agents with PARP inhibitors. First, blockage of AR signaling and PARP inhibition cause downregulation of the DNA repair capacity of the cells via different complementary pathways (DSB repair and SSB repair) [73, 103]. According to preclinical studies, anti-androgen treatments may induce a BRCAness phenotype, which can be targeted by PARP inhibition. Second, PARP1 has been reported to promote AR-dependent transcription and PARP inhibitors will therefore reduce AR-functioning [104]. Unfortunately, a randomized multicenter trial failed to show a significant difference in prostate-specific antigen (PSA) response rate and median progression-free survival (PFS) between patients treated with abiraterone/prednisone plus the PARP inhibitor veliparib compared to abiraterone/prednisone alone [105]. Lack of effectivity can be explained by inefficient PARP trapping by veliparib. Interestingly, another recent randomized double-blind phase 2 trial showed significantly longer PFS for mCRPC patients receiving olaparib plus abiraterone treatment than single abiraterone therapy. Although the combination strategy showed more adverse events than monotherapy, the health-related quality of life did not decline [106]. These clinical data support the preclinical results in which synergy between olaparib and AR signaling inhibitor was found, regardless of the HR status [73, 103].
Other trials are combining PARP inhibitors with vascular endothelial growth factor (VEGF) inhibitors, which function by inhibiting tumor angiogenesis. Preclinical studies showed that restriction of angiogenesis induces hypoxia, which may create a BRCAness phenotype by reducing the expression of BRCA1 and RAD51 [107]. The VEGF inhibitors bevacizumab and cediranib were reported to induce severe hypoxia, causing a reduction of HR capacity and increased sensitivity to PARP inhibitors [108]. Based on these data, a clinical study targeting both processes in mCRPC patients is ongoing (Table 5).
Another approach that has been explored is the use of PARP inhibitors as radiosensitizer for patients with high-risk localized PCa (radiotherapy) or with metastatic lesions (radioligand therapy). Irradiation induces cell death by the production of reactive oxygen species (ROS) as well as by direct ionization of the DNA which leads to SSBs and DSBs. PARP inhibition is predicted to enhance this effect by preventing the repair of radiation-induced SSBs. In vitro models support the idea that PARP inhibitors can enhance radiation-induced cytotoxicity [109, 110]. Similar results were also found in targeted radioligand therapy for PCa [111], suggesting targeted radiotherapy can be further optimized in combination with PARP inhibitors.
As described above, the MMR pathway has been implicated in the immunotherapy response and alterations in other DDR genes may also increase efficacy of immunotherapy [79, 112]. Therefore, several studies were started in which PARP inhibitors were combined with immunotherapy. The PARP1 inhibitor talazoparib has been found to exhibit immunoregulatory effects in a Brca1 deficient ovarian cancer mouse model as the number of peritoneal CD8 (+) T cells and NK cells increased significantly after talazoparib treatment [113]. Furthermore, Higuchi et al. have shown that cytotoxic T-lymphocyte antigen-4 (CTLA-4) antibody synergized with PARP inhibitors therapeutically in the Brca1 deficient ovarian cancer mouse model and support the clinical testing of this combination regimen [114]. The first clinical trial with a small cohort of patients showed that the PD-L1 inhibitor durvalumab plus olaparib in mCRPC patients has acceptable toxicity and efficacy, and the therapeutic response is superior in men with DDR abnormalities [115]. This triggered other studies to investigate whether mCRPC patients with DDR defects would benefit from this particular combination therapy. Clinical trials are ongoing to evaluate its safety, optimal dosing and efficacy (Table 5).
Platinum-based chemotherapy
Platinum-based agents cause crosslinking of DNA, most notably interstrand crosslinks that covalently couple both DNA strands [116]. These crosslinks interfere with DNA replication and translation and induce apoptosis. Although platinum compounds have long been studied in advanced PCa patients in a large number of clinical trials, the various treatment regimens have not demonstrated a significant overall survival benefit in the overall patient population, and no treatment has received approval. Tumors with mutations in BRCA1/2 are specifically susceptible to platinum-based chemotherapy since the interstrand crosslinks can only be adequately repaired by HR-based DNA repair. Recent clinical trials provided evidence that breast and ovarian cancer patients with BRCA1/2 mutations are highly sensitive to platinum-based chemotherapy [99, 117, 118]. Pomerantz et al. retrospectively analyzed a single-institution cohort of mCRPC patients who received carboplatin-based chemotherapy and showed that BRCA2 mutation carriers had a higher response rate to carboplatin-based chemotherapy than non-BRCA2 associated patients [119]. Furthermore, a few case reports also highlighted exceptional responses to platinum-treatment in mCRPC patients with HR defects [120, 121]. With such promising results, more trials of carboplatin alone and in combination with docetaxel have been designed in advanced PCa harboring DDR aberrations (ongoing clinical trials are summarized in Table 6).
DNA-PKcs targeting treatment
Besides the discovery of the AR-DDR crosstalk via the key mediator DNA-PKcs, a following study has identified a new function of DNA-PKcs as a potent driver of PCa progression. Goodwin et al. found that DNA-PKcs functions as a selective modulator of transcriptional networks that induce cell migration, invasion and metastasis and suppression of DNA-PKcs inhibits tumor metastases. Moreover, DNA-PKcs levels are significantly increased in advanced disease and can be independently predictive for biochemical recurrence, poor overall survival [122]. Based on these findings, a phase I clinical trial is ongoing (NCT02833883) in which the combination of enzalutamide and DNA-PKcs inhibitor CC-115 is evaluated for treatment of mCRPC.
Conclusion
The identification of DDR defects in mCRPC has driven the interest for further evaluation of these gene deficiencies in patient stratification. PARP inhibitors may become part of the standard care of mCRPC patients who harbor HR deficiency; however the most optimal use of PARP inhibitors alone or in combination with other treatment modalities remains to be elucidated. Given the clearly aggressive course of DDR-deficient PCa, there is an urgent need to identify these patients at an early stage where the right treatment strategy could greatly improve prognosis. The discovery that the AR may regulate DDR factors opens a new array of possible strategies to optimize treatment combinations. Future studies are needed to broaden our understanding of DDR defects and interactions between DNA repair pathways and other processes in PCa, as well as to determine how this knowledge can be used to improve diagnostic, prognostic and therapeutic approaches.
Change history
17 July 2019
A Correction to this paper has been published: https://doi.org/10.1038/s41391-019-0162-1
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Cornford P, Bellmunt J, Bolla M, Briers E, De Santis M, Gross T, et al. EAU-ESTRO-SIOG guidelines on prostate cancer. Part II: treatment of relapsing, metastatic, and castration-resistant prostate cancer. Eur Urol. 2017;71:630–42.
Mottet N, Bellmunt J, Bolla M, Briers E, Cumberbatch MG, De Santis M, et al. EAU-ESTRO-SIOG guidelines on prostate cancer. Part 1: screening, diagnosis, and local treatment with curative intent. Eur Urol. 2017;71:618–29.
Nuhn P, De Bono JS, Fizazi K, Freedland SJ, Grilli M, Kantoff PW, et al. Update on systemic prostate cancer therapies: management of metastatic castration-resistant prostate cancer in the era of precision oncology. Eur Urol. 2019;75:88–99.
Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31:1748–57.
Cybulski C, Wokolorczyk D, Kluzniak W, Jakubowska A, Gorski B, Gronwald J, et al. An inherited NBN mutation is associated with poor prognosis prostate cancer. Br J Cancer. 2013;108:461–8.
Grindedal EM, Moller P, Eeles R, Stormorken AT, Bowitz-Lothe IM, Landro SM, et al. Germ-line mutations in mismatch repair genes associated with prostate cancer. Cancer Epidemiol Biomark Prev. 2009;18:2460–7.
Kote-Jarai Z, Jugurnauth S, Mulholland S, Leongamornlert DA, Guy M, Edwards S, et al. A recurrent truncating germline mutation in the BRIP1/FANCJ gene and susceptibility to prostate cancer. Br J Cancer. 2009;100:426–30.
Leongamornlert D, Mahmud N, Tymrakiewicz M, Saunders E, Dadaev T, Castro E, et al. Germline BRCA1 mutations increase prostate cancer risk. Br J Cancer. 2012;106:1697.
Mateo J, Boysen G, Barbieri CE, Bryant HE, Castro E, Nelson PS, et al. DNA repair in prostate cancer: biology and clinical implications. Eur Urol. 2017;71:417–25.
Tangutoori S, Baldwin P, Sridhar S. PARP inhibitors: a new era of targeted therapy. Maturitas. 2015;81:5–9.
Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3:1254–71.
Thompson TC, Li L, Broom BM. Combining enzalutamide with PARP inhibitors: pharmaceutically induced BRCAness. Oncotarget. 2017;8:93315.
Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110–20.
Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168:644–56.
Aguilera A, Gaillard H. Transcription and recombination: when RNA meets DNA. Cold Spring Harb Perspect Biol. 2014;6:a016543.
Syeda AH, Hawkins M, McGlynn P. Recombination and replication. Cold Spring Harb Perspect Biol. 2014;6:a016550.
Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204.
Hoeijmakers JHJ. DNA damage, aging, and cancer. New Engl J Med. 2009;361:1475–85.
Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8.
Wallace SS. Base excision repair: a critical player in many games. DNA Repair (Amst). 2014;19:14–26.
Li Z, Pearlman AH, Hsieh P. DNA mismatch repair and the DNA damage response. DNA Repair (Amst). 2016;38:94–101.
Moldovan GL, D’Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–49.
Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64.
d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22.
Dhawan M, Ryan CJ, Ashworth A. DNA repair deficiency is common in advanced prostate cancer: new therapeutic opportunities. Oncologist. 2016;21:940–5.
Dominguez-Valentin M, Joost P, Therkildsen C, Jonsson M, Rambech E, Nilbert M. Frequent mismatch-repair defects link prostate cancer to Lynch syndrome. BMC Urol. 2016;16:15.
Gallagher DJ, Gaudet MM, Pal P, Kirchhoff T, Balistreri L, Vora K, et al. Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin Cancer Res. 2010;16:2115–21.
Rosty C, Walsh MD, Lindor NM, Thibodeau SN, Mundt E, Gallinger S, et al. High prevalence of mismatch repair deficiency in prostate cancers diagnosed in mismatch repair gene mutation carriers from the colon cancer family registry. Fam Cancer. 2014;13:573–82.
Venkitaraman AR. Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J Cell Sci. 2001;114(Pt 20):3591–8.
Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–82.
Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene [Rev]. 2006;25:5864.
Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer. 2016;16:35–42.
Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12:68–78.
Wang M, Li Q, Gu C, Zhu Y, Yang Y, Wang J, et al. Polymorphisms in nucleotide excision repair genes and risk of primary prostate cancer in Chinese Han populations. Oncotarget. 2017;8:24362.
Francis JC, McCarthy A, Thomsen MK, Ashworth A, Swain A. Brca2 and Trp53 deficiency cooperate in the progression of mouse prostate tumourigenesis. PLoS Genet. 2010;6:e1000995.
Jiricny J. Postreplicative mismatch repair. Cold Spring Harb Perspect Biol. 2013;5:a012633.
Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, et al. Microsatellite instability is associated with the presence of lynch syndrome pan-cancer. J Clin Oncol. 2019;37:286–95.
Attard G, Parker C, Eeles RA, Schröder F, Tomlins SA, Tannock I, et al. Prostate cancer. Lancet. 2016;387:70–82. https://doi.org/10.1016/S0140-6736(14)61947-4
Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–77.
Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9.
Cancer Genome Atlas Research Network The molecular taxonomy of primary prostate. Cancer Cell. 2015;163:1011–25.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Ren S, Wei G-H, Liu D, Wang L, Hou Y, Zhu S, et al. Whole-genome and Transcriptome Sequencing of Prostate Cancer Identify New Genetic Alterations Driving Disease Progression. Eur Urol. 2018;73(3):322–39.
Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature. 2017;541:359–64.
Bangma CH, Roobol MJ. Defining and predicting indolent and low risk prostate cancer. Crit Rev Oncol Hematol. 2012;83:235–41.
Irshad S, Bansal M, Castillo-Martin M, Zheng T, Aytes A, Wenske S, et al. A molecular signature predictive of indolent prostate cancer. Sci Transl Med. 2013;5:202ra122.
Kamoun A, Cancel-Tassin G, Fromont G, Elarouci N, Armenoult L, Ayadi M, et al. Comprehensive molecular classification of localized prostate adenocarcinoma reveals a tumour subtype predictive of non-aggressive disease. Ann Oncol. 2018;29:1814–21. https://doi.org/10.1093/annonc/mdy224
Bancroft EK, Page EC, Castro E, Lilja H, Vickers A, Sjoberg D, et al. Targeted prostate cancer screening in BRCA1 and BRCA2 mutation carriers: results from the initial screening round of the IMPACT study. Eur Urol. 2014;66:489–99.
Carter BS, Beaty TH, Steinberg GD, Childs B, Walsh PC. Mendelian inheritance of familial prostate cancer. Proc Natl Acad Sci USA. 1992;89:3367–71.
Petrovics G, Price DK, Lou H, Chen Y, Garland L, Bass S, et al. Increased frequency of germline BRCA2 mutations associates with prostate cancer metastasis in a racially diverse patient population. Prostate Cancer Prostatic Dis. 2018 [Epub ahead of print].
Na R, Zheng SL, Han M, Yu H, Jiang D, Shah S, et al. Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur Urol. 2017;71:740–7.
Evans JR, Zhao SG, Chang SL, Tomlins SA, Erho N, Sboner A, et al. Patient-level DNA damage and repair pathway profiles and prognosis after prostatectomy for high-risk prostate cancer. JAMA Oncol. 2016;2:471–80.
Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43.
Robinson D, Van Allen Eliezer M, Wu Y-M, Schultz N, Lonigro Robert J, Mosquera J-M, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–708.
Mateo J, Cheng HH, Beltran H, Dolling D, Xu W, Pritchard CC, et al. Clinical outcome of prostate cancer patients with germline dna repair mutations: retrospective analysis from an International Study. Eur Urol. 2018;73:687–93.
Annala M, Struss WJ, Warner EW, Beja K, Vandekerkhove G, Wong A, et al. Treatment outcomes and tumor loss of heterozygosity in germline DNA repair-deficient prostate cancer. Eur Urol. 2017;72:34–42.
Antonarakis ES, Lu C, Luber B, Liang C, Wang H, Chen Y, et al. Germline DNA-repair gene mutations and outcomes in men with metastatic castration-resistant prostate cancer receiving first-line abiraterone and enzalutamide. Eur Urol. 2018;74:218–25.
Hussain M, Daignault-Newton S, Twardowski PW, Albany C, Stein MN, Kunju LP, et al. Targeting androgen receptor and dna repair in metastatic castration-resistant prostate cancer: results from NCI 9012. J Clin Oncol. 2018;36:991–9.
Castro E, Romero-Laorden N, Del Pozo A, Lozano R, Medina A, Puente J, et al. PROREPAIR-B: A prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration-resistant prostate Cancer. J Clin Oncol. 2019;37(6):490–503.
Poeppel TD, Handkiewicz-Junak D, Andreeff M, Becherer A, Bockisch A, Fricke E, et al. EANM guideline for radionuclide therapy with radium-223 of metastatic castration-resistant prostate cancer. Eur J Nucl Med Mol Imaging. 2018;45:824–45.
Isaacsson Velho P, Qazi F, Hassan S, Carducci MA, Denmeade SR, Markowski MC, et al. Efficacy of radium-223 in bone-metastatic castration-resistant prostate cancer with and without homologous repair gene defects. Eur Urol. 2018 [In press].
Bolla M, Collette L, Blank L, Warde P, Dubois JB, Mirimanoff RO, et al. Long-term results with immediate androgen suppression and external irradiation in patients with locally advanced prostate cancer (an EORTC study): a phase III randomised trial. Lancet. 2002;360:103–6.
Bolla M, de Reijke TM, Van Tienhoven G, Van den Bergh AC, Oddens J, Poortmans PM, et al. Duration of androgen suppression in the treatment of prostate cancer. N Engl J Med. 2009;360:2516–27.
Bolla M, Gonzalez D, Warde P, Dubois JB, Mirimanoff RO, Storme G, et al. Improved survival in patients with locally advanced prostate cancer treated with radiotherapy and goserelin. N Engl J Med. 1997;337:295–300.
Shipley WU, Seiferheld W, Lukka HR, Major PP, Heney NM, Grignon DJ, et al. Radiation with or without antiandrogen therapy in recurrent prostate cancer. N Engl J Med. 2017;376:417–28.
Warde P, Mason M, Ding K, Kirkbride P, Brundage M, Cowan R, et al. Combined androgen deprivation therapy and radiation therapy for locally advanced prostate cancer: a randomised, phase 3 trial. Lancet. 2011;378:2104–11.
Widmark A, Klepp O, Solberg A, Damber JE, Angelsen A, Fransson P, et al. Endocrine treatment, with or without radiotherapy, in locally advanced prostate cancer (SPCG-7/SFUO-3): an open randomised phase III trial. Lancet. 2009;373:301–8.
Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3:1245–53.
Al-Ubaidi FL, Schultz N, Loseva O, Egevad L, Granfors T, Helleday T. Castration therapy results in decreased Ku70 levels in prostate cancer. Clin Cancer Res. 2013;19:1547–56.
Tarish FL, Schultz N, Tanoglidi A, Hamberg H, Letocha H, Karaszi K, et al. Castration radiosensitizes prostate cancer tissue by impairing DNA double-strand break repair. Sci Transl Med. 2015;7:312re11.
Li L, Karanika S, Yang G, Wang J, Park S, Broom BM, et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal. 2017;10(480).
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer [Rev Artic]. 2012;12:252.
Weber J. Immune checkpoint proteins: a new therapeutic paradigm for cancer–preclinical background: CTLA-4 and PD-1 blockade. Semin Oncol. 2010;37:430–9.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015;348:124–8.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13.
Schweizer MT, Cheng HH, Tretiakova MS, Vakar-Lopez F, Klemfuss N, Konnick EQ, et al. Mismatch repair deficiency may be common in ductal adenocarcinoma of the prostate. Oncotarget. 2016;7:82504–10.
Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016;7:13587–98.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. Nature. 2005;434:913.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
O’Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet. 2017;18:613.
Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–8.
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012;72:5588–99.
Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13:433–43.
Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19:5003–15.
Hay T, Matthews JR, Pietzka L, Lau A, Cranston A, Nygren AO, et al. Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res. 2009;69:3850–5.
Kortmann U, McAlpine JN, Xue H, Guan J, Ha G, Tully S, et al. Tumor growth inhibition by olaparib in BRCA2 germline-mutated patient-derived ovarian cancer tissue xenografts. Clin Cancer Res. 2011;17:783–91.
Christenson ES, Antonarakis ES. PARP inhibitors for homologous recombination-deficient prostate cancer. Expert Opin Emerg Drugs. 2018;23:123–33. https://doi.org/10.1080/14728214.2018.1459563
Dhawan M, Ryan CJ. BRCAness and prostate cancer: diagnostic and therapeutic considerations. Prostate Cancer Prostatic Dis. 2018;21:488–98.
Palmbos PL, Hussain MH. Targeting PARP in prostate cancer: novelty, pitfalls, and promise. Oncol (Williston Park). 2016;30:377–85.
Ramakrishnan Geethakumari P, Schiewer MJ, Knudsen KE, Kelly WK. PARP inhibitors in prostate cancer. Curr Treat Options Oncol. 2017;18:37.
Marshall CH, Sokolova AO, McNatty AL, Cheng HH, Eisenberger MA, Bryce AH, et al. Differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur Urol. 2019 [In press].
Gordon V, Angel ED, Jerry L, Stephanie G, Laura L, Yipeng W, et al. A single cell genomic signature to detect homologous recombination deficiency (HRD) and PARP inhibitors sensitivity using patient’s circulating tumor cells (CTCs). J Clin Oncol. 2016;34(15_suppl):e23015–e.
Lanman RB, Mortimer SA, Zill OA, Sebisanovic D, Lopez R, Blau S, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS ONE. 2015;10:e0140712.
Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517–25.
Meijer TG, Verkaik NS, Sieuwerts AM, van Riet J, Naipal KAT, van Deurzen CHM, et al. Functional ex vivo assay reveals homologous recombination deficiency in breast cancer beyond BRCA gene defects. Clin Cancer Res. 2018;24:6277–87.
Naipal KA, Verkaik NS, Ameziane N, van Deurzen CH, Ter Brugge P, Meijers M, et al. Functional ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clin Cancer Res. 2014;20:4816–26.
Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22:3764–73.
Zhang W, van Weerden WM, de Ridder CMA, Erkens-Schulze S, Schonfeld E, Meijer TG, et al. Ex vivo treatment of prostate tumor tissue recapitulates in vivo therapy response. Prostate. 2018;79:390–402.
Dréan A, Lord CJ, Ashworth A. PARP inhibitor combination therapy. Crit Rev Oncol Hematol. 2016;108:73–85.
Hussain M, Carducci MA, Slovin S, Cetnar J, Qian J, McKeegan EM, et al. Targeting DNA repair with combination veliparib (ABT-888) and temozolomide in patients with metastatic castration-resistant prostate cancer. Investig New Drugs. 2014;32:904–12.
Asim M, Tarish F, Zecchini HI, Sanjiv K, Gelali E, Massie CE, et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun. 2017;8:374.
Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134–49.
Maha H, Stephanie D-N, Przemyslaw WT, Costantine A, Mark NS, Lakshmi PK, et al. Targeting androgen receptor and dna repair in metastatic castration-resistant prostate cancer: results from NCI 9012. J Clin Oncol. 2018;36:991–9.
Clarke N, Wiechno P, Alekseev B, Sala N, Jones R, Kocak I, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018;19:975–86.
Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, et al. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res. 2005;65:11597–604.
Chan N, Bristow RG. “Contextual” synthetic lethality and/or loss of heterozygosity: tumor hypoxia and modification of DNA repair. Clin Cancer Res. 2010;16:4553–60.
Barreto-Andrade JC, Efimova EV, Mauceri HJ, Beckett MA, Sutton HG, Darga TE, et al. Response of human prostate cancer cells and tumors to combining PARP inhibition with ionizing radiation. Mol Cancer Ther. 2011;10:1185–93.
Rae C, Mairs RJ. Evaluation of the radiosensitizing potency of chemotherapeutic agents in prostate cancer cells. Int J Radiat Biol. 2017;93:194–203.
Tesson M, Rae C, Nixon C, Babich JW, Mairs RJ. Preliminary evaluation of prostate-targeted radiotherapy using (131) I-MIP-1095 in combination with radiosensitising chemotherapeutic drugs. J Pharm Pharm. 2016;68:912–21.
Teo MY, Seier K, Ostrovnaya I, Regazzi AM, Kania BE, Moran MM, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J Clin Oncol. 2018;36:1685–94.
Huang J, Wang L, Cong Z, Amoozgar Z, Kiner E, Xing D, et al. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(−/−) murine model of ovarian cancer. Biochem Biophys Res Commun. 2015;463:551–6.
Higuchi T, Flies DB, Marjon NA, Mantia-Smaldone G, Ronner L, Gimotty PA, et al. CTLA-4 blockade synergizes therapeutically with PARP inhibition in BRCA1-deficient ovarian cancer. Cancer Immunol Res. 2015;3:1257–68.
Karzai F, VanderWeele D, Madan RA, Owens H, Cordes LM, Hankin A, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer. 2018;6:141.
Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharm. 2014;740:364–78.
Byrski T, Dent R, Blecharz P, Foszczynska-Kloda M, Gronwald J, Huzarski T, et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast cancer Res. 2012;14:R110.
Lesnock JL, Darcy KM, Tian C, Deloia JA, Thrall MM, Zahn C, et al. BRCA1 expression and improved survival in ovarian cancer patients treated with intraperitoneal cisplatin and paclitaxel: a Gynecologic Oncology Group Study. Br J Cancer. 2013;108:1231–7.
Pomerantz MM, Spisak S, Jia L, Cronin AM, Csabai I, Ledet E, et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer . 2017;123:3532–9.
Cheng HH, Pritchard CC, Boyd T, Nelson PS, Montgomery B. Biallelic inactivation of BRCA2 in platinum-sensitive metastatic castration-resistant prostate cancer. Eur Urol. 2016;69:992–5.
Zafeiriou Z, Bianchini D, Chandler R, Rescigno P, Yuan W, Carreira S, et al. Genomic analysis of three metastatic prostate cancer patients with exceptional responses to carboplatin indicating different types of DNA repair deficiency. Eur Urol. 2019;75:184–92.
Goodwin JF, Kothari V, Drake JM, Zhao S, Dylgjeri E, Dean JL, et al. DNA-PKcs-mediated transcriptional regulation drives prostate cancer progression and metastasis. Cancer Cell. 2015;28:97–113.
Funding
Chinese Scholarship Council (WZ, grant number 201506270172), the Dutch Cancer Foundation (DCvG, WMvW, JN, grant number 10317), the Daniel den Hoed Foundation (JN), the Erasmus University Rotterdam (JN).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, W., van Gent, D.C., Incrocci, L. et al. Role of the DNA damage response in prostate cancer formation, progression and treatment. Prostate Cancer Prostatic Dis 23, 24–37 (2020). https://doi.org/10.1038/s41391-019-0153-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41391-019-0153-2
This article is cited by
-
Structure based docking and biological evaluation towards exploring potential anti-cancerous and apoptotic activity of 6-Gingerol against human prostate carcinoma cells
BMC Complementary Medicine and Therapies (2024)
-
Novel frontiers in urogenital cancers: from molecular bases to preclinical models to tailor personalized treatments in ovarian and prostate cancer patients
Journal of Experimental & Clinical Cancer Research (2024)
-
Exploring potential therapeutic combinations for castration-sensitive prostate cancer using supercomputers: a proof of concept study
Scientific Reports (2024)
-
Homologous Recombination Repair Gene Mutations in Prostate Cancer: Prevalence and Clinical Value
Advances in Therapy (2024)
-
Antioxidative stress protein SRXN1 can be used as a radiotherapy prognostic marker for prostate cancer
BMC Urology (2023)
