
Abstract
Introduction
Using pre-procedure analgesia with the risk of apnoea may complicate the Less Invasive Surfactant Administration (LISA) procedure or reduce the effect of LISA.
Methods
The NONA-LISA trial (ClinicalTrials.gov, NCT05609877) is a multicentre, blinded, randomised controlled trial aiming at including 324 infants born before 30 gestational weeks, meeting the criteria for surfactant treatment by LISA. Infants will be randomised to LISA after administration of fentanyl 0.5–1 mcg/kg intravenously (fentanyl group) or isotonic saline solution intravenously (saline group). All infants will receive standardised non-pharmacological comfort care before and during the LISA procedure. Additional analgesics will be provided at the clinician’s discretion. The primary outcome is the need for invasive ventilation, meaning mechanical or manual ventilation via an endotracheal tube, for at least 30 min (cumulated) within 24 h of the procedure. Secondary outcomes include the modified COMFORTneo score during the procedure, bronchopulmonary dysplasia at 36 weeks, and mortality at 36 weeks.
Discussion
The NONA-LISA trial has the potential to provide evidence for a standardised approach to relief from discomfort in preterm infants during LISA and to reduce invasive ventilation. The results may affect future clinical practice.
Impact
-
Pre-procedure analgesia is associated with apnoea and may complicate procedures that rely on regular spontaneous breathing, such as Less Invasive Surfactant Administration (LISA).
-
This randomised controlled trial addresses the effect of analgesic premedication in LISA by comparing fentanyl with a placebo (isotonic saline) in infants undergoing the LISA procedure. All infants will receive standardised non-pharmacological comfort.
-
The NONA-LISA trial has the potential to provide evidence for a standardised approach to relief from discomfort or pain in preterm infants during LISA and to reduce invasive ventilation. The results may affect future clinical practice regarding analgesic treatment associated with the LISA procedure.
Similar content being viewed by others
Introduction
Respiratory distress syndrome (RDS) caused by surfactant deficiency remains a significant reason for neonatal mortality and short- and long-term morbidity in preterm infants.1 RDS usually develops during the first 24 h of the delivery, and most infants with RDS present with breathing difficulties and an increased need for oxygen supplementation within the first few hours of birth. Surfactant decreases alveolar surface tension and helps to keep the lungs aerated, allowing pulmonary gas exchange. Exogenous surfactant treatment of preterm infants with RDS reduces mortality and long-term morbidity.2,3,4
Less Invasive Surfactant Administration (LISA) is a strategy to administer surfactant to infants with worsening RDS despite optimised non-invasive ventilation. LISA aims to prevent alveolar collapse and avoid endotracheal intubation and invasive ventilation before, during, and after surfactant administration.4,5,6 LISA involves surfactant administration via a thin catheter placed in the trachea using laryngoscopy on a spontaneously breathing infant. Before LISA was introduced, surfactant was administered via an endotracheal tube followed by invasive ventilation and later via the INtubation-SURfactant-Extubation (INSURE) method with immediate extubation following surfactant administration. Studies have shown that using LISA reduces the incidence of intraventricular haemorrhage (IVH), mortality, the need for invasive ventilation, and the risk of bronchopulmonary dysplasia (BPD) in preterm infants.7,8 LISA is currently considered the preferred method of surfactant administration in spontaneously breathing infants according to the European Consensus Guidelines on Management of RDS,4 and LISA is increasingly used in NICUs worldwide.9
However, the need for analgesic premedication in LISA is debated. In a recent study of 153 LISA experts,10 41% indicated no use of pre-procedure sedatives or analgesics, and 49% reported using fentanyl as a pre-procedure treatment. On the contrary, 4% indicated no use of non-pharmacological treatment. There is a delicate balance between the desired effect of analgesia reducing discomfort and pain associated with laryngoscopy as opposed to acute cardiovascular side effects and the risk of over-sedation and apnoea. This may potentially impede the procedure and result in the need for positive pressure ventilation, which may harm the surfactant-deficient lung.11,12 Observational data suggests the ease of performing LISA to be unaffected by whether opiates are used or not.13
The NONA-LISA trial (ClinicalTrials.gov, NCT05609877) compares LISA with saline to LISA with 0.5–1 mcg/kg fentanyl in infants born before 30 gestational weeks and evaluate the need for invasive ventilation via an endotracheal tube for at least 30 min (cumulated) within 24 h of the procedure. Secondary outcomes include the number of laryngoscopy attempts, duration of the procedure, modified COMFORTneo score during the procedure, bronchopulmonary dysplasia at 36 weeks, and mortality at 36 weeks.
Methods
Design and setting
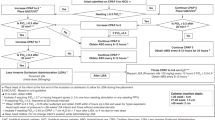
The NONA-LISA trial is a multicentre, blinded, randomised controlled trial of fentanyl 0.5–1 mcg/kg or placebo (isotonic saline), administered to infants born before 30 gestational weeks who meet the criteria for surfactant treatment by LISA. A total of 324 infants will be randomised in a 1:1 ratio to one of the two arms. They will be followed up at 36 weeks PMA and at hospital discharge (shown in Fig. 1). The trial protocol conforms with the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT),14 and the trial results will be reported in compliance with the Consolidated Standards of Reporting Trials (CONSORT) Statement.15 The trial will be initiated at (but not limited to) the Danish level III Neonatal Intensive Care Units (NICUs).

Abbreviations: NONA-LISA NON-pharmacologic Approach Less Invasive Surfactant Administration, LISA Less Invasive Surfactant Administration, GA gestational age.
Population
Infants will be eligible for inclusion if they are born before 30 gestational weeks at one of the trial sites and meet the criteria for first-choice surfactant treatment by LISA as described by Sweet et al.4: worsening babies with RDS and FiO2 > 0.30 on CPAP pressure ≥6 cm H2O. The primary respiratory support is CPAP in all four units.
Infants will be excluded if they meet any of the exclusion criteria; 1) suspicion of lung hypoplasia, 2) endotracheal intubation at any time before randomisation, 3) suspicion of pneumothorax, pulmonary haemorrhage or pleural effusion before LISA, 4) major congenital anatomical anomalies as described by the European Surveillance of Congenital Anomalies (EUROCAT).16
Randomisation
Participants will be randomly assigned to the saline or fentanyl group in a 1:1 ratio using computer-generated random allocation sequences with permuted blocks of varying sizes (two and four). The randomisation sequence will be stratified by trial site and gestational age at birth (more or less than 28 completed gestational weeks). Once randomised in the Research Electronic Data Capture (REDCap) system, all entry data will automatically be transferred to the electronic case record forms (eCRF) linked to the infant’s unique identification number. The allocation sequence is pre-coded and generated from the randomisation programme. A person not involved in the trial will generate the allocation sequence. The clinical staff will enrol participants.
Blinding
Blinding will be secured by the following means: The patient will be randomised after entering stratification variables in REDCAP. The randomisation sequence will be generated by one person unrelated to the study. The trial medication (fentanyl or isotonic saline) will be prepared once per day in sealed syringes with similar appearance and weight by the pharmacy or staff not involved in patient care. Thus, neither staff involved in screening or treatment of patients nor the parents will know the treatment allocation. Primary analyses will be performed blinded to the group allocation (Group A compared with Group B) and presented to all authors, who will agree on two alternative written interpretations before the randomisation code is unblinded to reduce the risk of interpretation bias.
Interventions
Participants are randomised to receive LISA with fentanyl 0.5-1 mcg/kg (fentanyl group) or LISA with isotonic saline (saline group). All infants will receive standardised non-pharmacological comfort care before and during the LISA procedure (Online Supplement, Appendix A). All LISA procedures will be performed using video laryngoscopes according to the Hobart method.2,3 Both groups will receive the neonatal unit’s standard pre-procedure care (e.g., atropine and caffeine), which will not be standardised across trial sites. Naloxone may be administered at the clinician’s discretion and will not be used routinely. In both groups, the infant’s level of discomfort or pain will be monitored continuously during the procedure by vital signs and a modified COMFORTneo score (Online Supplement, Appendix A).17 Open-label analgesics can be administered to all participants at the clinician’s discretion by indications of non-tolerance (e.g., modified COMFORTneo score >14).
All administered medications will be registered. A single dose of 200 mg/kg porcine surfactant (Curosurf®, Chiesi Pharma AB, Italy) will be used in this trial.
Outcomes
Primary outcomes
The primary outcome of this trial is the need for invasive ventilation, meaning mechanical or manual ventilation via an endotracheal tube for at least 30 min (cumulated) within 24 h of the procedure. Non-invasive ventilation (NIV) is not included in the primary outcome.
Secondary outcomes
The secondary outcomes include:
-
Unique and composite outcome of death or moderate/severe BPD18 at 36 weeks PMA.
-
Adverse events during the procedure (from the introduction of the laryngoscope blade into the oral cavity to the removal of the catheter) in terms of apnoea that require bag and mask ventilation, desaturation with an absolute decrease in peripheral oxygen saturation >20% from pre-procedure baseline, and bradycardia <100 BPM individual outcomes.19
-
Pain or discomfort during the procedure (according to modified COMFORTneo score >1417,20,21).
-
Highest modified COMFORTneo score during the procedure.
-
Need for a second dose of surfactant.
-
Incidence of LISA procedures resulting in the INSURE procedure.
-
Incidence of observed surfactant reflux.
-
Incidence of observed injury to the upper airway.
-
Incidence of observed injury to the lower airway.
-
Incidence of invasive ventilation within 48 h after LISA.
-
Cumulated duration of invasive ventilation during hospitalisation.
-
Cumulated duration of any non-invasive respiratory support during hospitalisation.
-
Cumulated duration of any respiratory support during hospitalisation.
-
Procedural duration from the introduction of the laryngoscope blade over the lips to the removal of the catheter.
-
Number of attempts to visualise vocal cords (laryngoscopy) where the laryngoscope is completely removed from the oral cavity between attempts.
-
Incidence of pneumothorax within 48 h after LISA.
-
Incidence of massive pulmonary haemorrhage within 48 h after LISA (aspiration of haemorrhagic secretions from the trachea concurrent with the need for escalated respiratory support).22
-
Duration of hospitalisation.
-
Morbidities in terms of necrotising enterocolitis (according to the radiographic signs of Bell’s Staging Criteria),23 treatment-demanding retinopathy of prematurity, intraventricular haemorrhage grade 3–4 and periventricular leukomalacia (according to the Papile classification)24 as individual outcomes.
Data collection
Baseline characteristics will include infant gestational age at birth, delivery mode, singleton or multiple births, birth weight, sex, APGAR score (1, 5, and 10 min after delivery), resuscitation measures during the first 30 min after birth, first blood gas, umbilical arterial and venous pH, standard base excess, and lactate. Information on vital signs (oxygen saturation, FiO2, and heart rate) will be collected 15 min before and after the procedure. Information on treatment with antibiotics, fluids, vasopressors, and early caffeine before the LISA procedure will be recorded. We will calculate the time from meeting the criteria for surfactant treatment until the procedure starts. We will also include maternal information regarding smoking status, diseases (diabetes mellitus, amnionitis as per the obstetrician, preeclampsia, and eclampsia), and antenatal administration of steroids, magnesium sulphate, indomethacin, and antibiotics, including timing.
A generic procedure description will be created for the clinician to use in the medical records to ensure that all information, including modified COMFORTneo score before the procedure, administration of atropine, early caffeine and naloxone, and indications for additional fentanyl administration and cumulated dosage, is recorded uniformly.
If the infant is discharged to a step-down unit, information will be retrieved from that unit.
Data management
Study data will be collected and managed using REDCap electronic data capture tools hosted at the Capital Region of Denmark.25 A password will protect access to the online forms and dataset, and infants will be identified by number only to protect confidentiality before, during, and after the trial. A clinical trial coordinator will be responsible for monitoring the study’s progress and the data’s completeness.
Patient and public involvement
A parent representative provided feedback resulting in minor revisions of the parent information material and consent forms before trial initiation (Online Supplement, Appendices B and C). A parent representative will also be involved in the dissemination plans of the trial. However, parents were not involved in the study design and will not be involved in recruiting or data collection during the study.
Sample size estimation and inclusion timeline
The sample size was calculated according to the primary outcome. Previous studies on preterm infants born before 30 gestational weeks report a frequency of invasive ventilation after the LISA procedure from 33%26 and 41%27 up to 75%.28 Based on the incidence of invasive ventilation following LISA, we anticipate that the primary outcome will have an incidence of around 45% in the fentanyl group in our trial. Considering an alpha of 0.05 and a power of 0.8 to detect an anticipated incidence of about 30% for the saline group, this trial will enrol 324 infants in a 1:1 ratio. Based on an article by Wiingreen et al., ~123 infants are born before 30 gestational weeks each year in Denmark with the need for surfactant.29 The inclusion will last three to four years, considering an expected inclusion rate of 80%.
Statistical methods
Baseline characteristics will be presented for each group in the trial population. Categorical variables will be presented as frequencies (counts and percentages). Continuous variables will be presented as medians with interquartile ranges [IQR] or as means with standard deviations [SD].
The primary analysis will use the intention-to-treat principle. A secondary analysis will use the per-protocol principle and exclude infants if they receive additional analgesics associated with the procedure.
The primary outcome will be reported as frequencies (counts and percentages) per treatment group with unadjusted absolute risk difference and relative risks with 95% confidence intervals (CI). The number needed to treat for benefit or harm will be reported if the results of the primary analyses show a statistically significant difference between the two exposure groups.
Secondly, a logistic regression model will be performed adjusting for inclusion site and gestational age (<28 weeks vs more than 28 weeks) and the following independent covariates, which are predictors of assisted ventilation and length of ventilation:30 repeat doses of prenatal corticosteroids, 5-min APGAR scores, sex, admission illness severity (according to the Score for Neonatal Acute Physiology/SNAP score31), oxygenation index, and small-for-gestational-age status.32
All secondary dichotomous outcomes will be described and analysed per the same strategy as the primary outcome. At the same time, mean and percentage differences will be calculated for continuous outcomes.
All analyses will be conducted by use of the statistical software R Studio. Two-sided P values less than 0.05 will be considered statistically significant.
Discussion
This randomised controlled trial compares infants exposed to the LISA procedure with pre-procedure fentanyl or placebo (isotonic saline) by the risk of invasive ventilation, meaning mechanical or manual ventilation via an endotracheal tube, for at least 30 min (cumulated) within 24 h of the procedure. All infants will receive standardised non-pharmacological comfort care before and during the LISA procedure (Online Supplement, Appendix A). In this trial, we plan to test the hypothesis that there is no difference between the need for invasive ventilation within 24 h of the LISA procedure performed with or without pre-procedure analgesia (i.e., fentanyl).
The LISA procedure is performed worldwide without consensus regarding non-pharmacological and pharmacological approaches. There is clinical equipoise regarding the positive and negative effects of pharmacological analgesic treatment when performing the LISA procedure. There is a delicate balance between the desired effect of reduction in pain and discomfort and the risk of over-sedation and apnoea requiring positive pressure ventilation.11,12 Comfort and pain relief using a non-pharmacological approach has yet to be thoroughly investigated in the context of LISA and how it compares with pharmacological analgesic treatment. Some studies indicate that a non-pharmacological approach, including facilitated tucking,33 swaddling,34 and skin-to-skin care35 compared to pharmacological analgesic treatment, may reduce the incidence of apnoea and, ultimately, the use of invasive ventilation,20,21 while achieving the same level of comfort. Hence, some centres make the first LISA attempt without any premedication, as studies report that the LISA procedure is generally well tolerated without analgesia,1,36,37 especially in preterm infants.38 Therefore, some studies advise using pharmacological agents only if non-pharmacological methods are insufficient to ensure patient comfort.39 It is essential to investigate pharmacological and non-pharmacological approaches to reduce pain, discomfort, and adverse effects due to medication during neonatal procedures, including the LISA procedure, as the insertion of a laryngoscope and intratracheal administration of surfactant obviously contradicts the term “less invasive”.40
As a safety precaution in the NONA-LISA trial, the staff will continuously monitor the infant’s level of discomfort or pain during the procedure by an objective score (modified COMFORTneo). The COMFORTneo score is the national standard for assessing pain and discomfort in newborns. Most NICU nurses are COMFORTneo certified. Previous studies have demonstrated the ability of the COMFORTneo score to assess pain and discomfort during the LISA procedure.20,21 However, the COMFORTneo score formally requires a two-minute assessment of the infant and was not designed for intra-procedure assessments as planned in the NONA-LISA trial. To avoid interrupting the procedure flow, performing the score over 2 min is not feasible. Thus, intra-procedure assessments of COMFORTneo in this trial will be based on the standard COMFORTneo items assessed faster than 2 min. The use of COMFORTneo in this trial is described in detail in Online Supplement, Appendix A. Importantly, if the modified COMFORTneo score is ≥14 during the procedure, the procedure may be paused, and non-pharmacological measures must be improved. If improved non-pharmacological measures are ineffective, the team may opt for open-label fentanyl administration. The infant will still be given the highest level of care and attention by the staff.
This trial has certain limitations. The pragmatic design of this trial allowing fentanyl dosages between 0.5–1.0 mcg/kg will imply difficulty interpreting the specific results, as this trial will not have the necessary power to determine causality between an exact dosage of fentanyl and adverse events (e.g., apnoea). Despite this, a pragmatic design was necessary to ensure participation from all feasible Danish trial sites. As the dosing interval reflects clinical practice, it ensures generalisability and relevance for neonatal intensive care outside trial settings.
Non-pharmacological measures in neonatal care are complex and multifactorial, where all factors are relevant for the potential success of the intervention. Several studies have shown synergy when combining several non-pharmacological measures. However, there is no evidence favouring one specific approach. This study will use a protocolised non-pharmacological approach to standardise the treatment across inclusion sites.
The incidence of invasive ventilation following LISA varies significantly in other studies, and the anticipated incidences in the fentanyl and saline groups may cause an inadequate sample size to achieve the necessary power. However, compared to ongoing clinical trials like PRELISA (NCT05065424), we expect to include more than five times as many infants in the NONA-LISA trial. Other trials like the PROLISA (NCT04016246) include more infants but compare propofol to placebo.
The NONA-LISA trial received approval for deferred consent, which may significantly improve the inclusion rates. Nevertheless, with all the Danish NICUS as inclusion sites, the inclusion period will last up to four years, making the NONA-LISA trial vulnerable to changes in best practices regarding surfactant treatment. We expect to include other trial sites.
Conclusion
In conclusion, the NONA-LISA trial has the potential to provide evidence for a standardised approach to relief from discomfort or pain in preterm infants during LISA (NONA-LISA) and to reduce the need for invasive ventilation during the first day after the procedure.
Data availability
The datasets generated during and analysed during this trial are available from the corresponding author upon reasonable request.
References
Kribs, A. Minimally invasive surfactant therapy and noninvasive respiratory support. Clin. Perinatol. 43, 755–771 (2016).
Polin, R. A. & Carlo, W. A., COMMITTEE ON FETUS AND NEWBORN. Surfactant replacement therapy for preterm and term neonates with respiratory distress. Pediatrics 133, 156–163 (2014).
Herting, E., Härtel, C. & Göpel, W. Less invasive surfactant administration: best practices and unanswered questions. Curr. Opin. Pediatr. 32, 228–234 (2020).
Sweet, D. G. et al. European consensus guidelines on the management of respiratory distress syndrome: 2022 update. Neonatology 120, 3–23 (2023).
Herting, E. Less Invasive Surfactant Administration (LISA) — ways to deliver surfactant in spontaneously breathing infants. Early Hum. Dev. 89, 875–880 (2013).
Kribs, A., Pillekamp, F., Hünseler, C., Vierzig, A. & Roth, B. Early administration of surfactant in spontaneous breathing with nCPAP: feasibility and outcome in extremely premature infants (postmenstrual age ≤27 weeks). Pediatr. Anesth. 17, 364–369 (2007).
Isayama, T., Iwami, H., McDonald, S. & Beyene, J. Association of noninvasive ventilation strategies with mortality and bronchopulmonary dysplasia among preterm infants: a systematic review and meta-analysis. JAMA 316, 611 (2016).
Gortner, L., Schüller, S. S. & Herting, E. Review demonstrates that less invasive surfactant administration in preterm neonates leads to fewer complications. Acta Paediatr. 107, 736–743 (2018).
Beltempo, M. et al. Respiratory management of extremely preterm infants: an international survey. Neonatology 114, 28–36 (2018).
Breindahl, N. et al. Curriculum and assessment tool for less invasive surfactant administration: an international Delphi consensus study. Pediatr. Res. https://doi.org/10.1038/s41390-023-02621-2 (2023).
Moschino, L. et al. Sedation for less invasive surfactant administration in preterm infants: a systematic review and meta-analysis. Pediatr. Res. 93, 471–491 (2023).
Björklund, L. J. et al. Manual ventilation with a few large breaths at birth compromises the therapeutic effect of subsequent surfactant replacement in immature lambs. Pediatr. Res. 42, 348–355 (1997).
Krajewski, P., Szpecht, D. & Hożejowski, R. Premedication practices for less invasive surfactant administration—results from a nationwide cohort study. J. Matern. Fetal Neonatal Med. 35, 4750–4754 (2022).
Chan, A.-W. et al. SPIRIT 2013 explanation and elaboration: guidance for protocols of clinical trials. BMJ 346, e7586–e7586 (2013).
Altman, D. M. CONSORT 2010 guideline. (2010).
Dolk, H., Loane, M. & Garne, E. The prevalence of congenital anomalies in Europe. In: Rare Diseases Epidemiology (eds. Posada de la Paz, M. & Groft, S. C.) vol. 686, 349–364 (Springer Netherlands, 2010).
van Dijk, M. et al. Taking up the challenge of measuring prolonged pain in (Premature) neonates: the COMFORTneo scale seems promising. Clin. J. Pain. 25, 607–616 (2009).
Jobe, A. H. & Bancalari, E. Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 163, 1723–1729 (2001).
Hodgson, K. A. et al. A multicentre, randomised trial of stabilisation with nasal high flow during neonatal endotracheal intubation (the SHINE trial): a study protocol. BMJ Open 10, e039230 (2020).
Dekker, J. et al. Sedation during minimal invasive surfactant therapy in preterm infants. Neonatology 109, 308–313 (2016).
Dekker, J. et al. Sedation during minimal invasive surfactant therapy: a randomised controlled trial. Arch. Dis. Child. Fetal Neonatal Ed. 104, F378–F383. https://doi.org/10.1136/archdischild-2018-315015 (2019).
Welde, M. A., Sanford, C. B., Mangum, M., Paschal, C. & Jnah, A. J. Pulmonary hemorrhage in the neonate. Neonatal Netw. 40, 295–304 (2021).
Bell, M. J. et al. Neonatal necrotizing enterocolitis: therapeutic decisions based upon clinical staging. Ann. Surg. 187, 1–7 (1978).
Papile, L.-A., Burstein, J., Burstein, R. & Koffler, H. Incidence and evolution of subependymal and intraventricular hemorrhage: a study of infants with birth weights less than 1500 gm. J. Pediatr. 92, 529–534 (1978).
Harris, P. A. et al. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 42, 377–381 (2009).
Göpel, W. et al. Avoidance of mechanical ventilation by surfactant treatment of spontaneously breathing preterm infants (AMV): an open-label, randomised, controlled trial. Lancet 378, 1627–1634 (2011).
Göpel, W. et al. Less invasive surfactant administration is associated with improved pulmonary outcomes in spontaneously breathing preterm infants. Acta Paediatr. 104, 241–246 (2015).
Kribs, A. et al. Nonintubated surfactant application vs conventional therapy in extremely preterm infants: a randomized clinical trial. JAMA Pediatr. 169, 723 (2015).
Wiingreen, R. et al. Surfactant need by gestation for very preterm babies initiated on early nasal CPAP: a Danish observational multicentre study of 6,628 infants born 2000-2013. Neonatology 111, 331–336 (2017).
Wilson, A., Gardner, M. N., Armstrong, M. A., Folck, B. F. & Escobar, G. J. Neonatal assisted ventilation: predictors, frequency, and duration in a mature managed care organization. Pediatrics 105, 822–830 (2000).
Richardson, D. K., Gray, J. E., McCormick, M. C., Workman, K. & Goldmann, D. A. Score for Neonatal Acute Physiology: a physiologic severity index for neonatal intensive care. Pediatrics 91, 617–623 (1993).
Arbuckle, T. E. & Sherman, G. J. An analysis of birth weight by gestational age in Canada. CMAJ 140, 157–160, 165 (1989).
Liaw, J.-J. et al. Effects of combined use of non-nutritive sucking, oral sucrose, and facilitated tucking on infant behavioural states across heel-stick procedures: a prospective, randomised controlled trial. Int. J. Nurs. Stud. 50, 883–894 (2013).
Shu, S.-H., Lee, Y.-L., Hayter, M. & Wang, R.-H. Efficacy of swaddling and heel warming on pain response to heel stick in neonates: a randomised control trial. J. Clin. Nurs. 23, 3107–3114 (2014).
Chermont, A. G., Falcao, L. F. M., de Souza Silva, E. H. L., de Cassia Xavier Balda, R. & Guinsburg, R. Skin-to-skin contact and/or oral 25% dextrose for procedural pain relief for term newborn infants. Pediatrics 124, e1101–e1107 (2009).
Dargaville, P. A., Aiyappan, A., Cornelius, A., Williams, C. & De Paoli, A. G. Preliminary evaluation of a new technique of minimally invasive surfactant therapy. Arch. Dis. Child Fetal Neonatal Ed. 96, F243–F248 (2011).
Dargaville, P. A. et al. Minimally-invasive surfactant therapy in preterm infants on continuous positive airway pressure. Arch. Dis. Child Fetal Neonatal Ed. 98, F122–F126 (2013).
Mehler, K. et al. Survival among infants born at 22 or 23 weeks’ gestation following active prenatal and postnatal care. JAMA Pediatr. 170, 671 (2016).
Peterson, J., Den Boer, M. C. & Roehr, C. C. To sedate or not to sedate for less invasive surfactant administration: an ethical approach. Neonatology 118, 639–646 (2021).
De Luca, D. et al. Less invasive surfactant administration: a word of caution. Lancet Child Adolesc. Health 4, 331–340 (2020).
Acknowledgements
The authors would like to acknowledge the help of neonatal nurses Linda Lundstrøm and Rikke Louise Stenkjær (Department of Neonatal and Paediatric Intensive Care, Copenhagen University Hospital, Rigshospitalet, Copenhagen, Denmark), Eva Jørgensen and Lene Bøjgaard Bak (Division of Neonatology, Department of Paediatrics, Aarhus University Hospital, Aarhus, Denmark) for providing indispensable input on the protocolised non-pharmacological approach for both treatments arms in the NONA-LISA trial (Online Supplement, Appendix A).
Funding
Chiesi Denmark, a subsidiary of Chiesi Farmaceutici, Italy, supported this trial. The funding body will not be involved in the trial design, data collection, analysis, interpretation, writing, or decision to submit the manuscript for publication. Open access funding provided by Copenhagen University.
Author information
Authors and Affiliations
Contributions
N.B., M.G.T., T.B.H., and L.A. participated in the trial conception and design and constructed the first protocol draft. All authors revised the protocol critically for important content. All co-authors will be involved in the data acquisition, analysis, and interpretation. NB drafted the protocol and the protocol article. After revising them critically for important intellectual content, all co-authors approved the protocol and this manuscript. All co-authors read and approved this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent to participate
Written informed consent will be sought from all participants’ parents or legal guardians. All participants’ parents or legal guardians will be presented with oral and written information by a member of the trial group at an appropriate time after enrolment. Any questions will be answered. Participation will be voluntary and unpaid. We respect the parents’ decision to join or leave the study at any time. Important protocol modifications will be communicated to the relevant parties.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Breindahl, N., Henriksen, T.B., Heiring, C. et al. NON-pharmacological Approach Less Invasive Surfactant Administration (NONA-LISA) trial: protocol for a randomised controlled trial. Pediatr Res (2024). https://doi.org/10.1038/s41390-023-02998-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41390-023-02998-0
