
Abstract
Background
Identification and functional annotations of regulatory sequences play a pivotal role in heart development and function.
Methods
To generate a map of human heart-specific enhancers, we performed an integrative analysis of 148 chromatin immunoprecipitation coupled to massively parallel sequencing (ChIP-seq) samples with enhancer-associated epigenetic marks from the heart, liver, brain, and kidney. Functional validation of heart-specific enhancer activity was then performed using cultured cells.
Results
A 144.6-Mb candidate heart-specific enhancer compendium was generated by integrating the analysis of 148 epigenomic data sets from human and mouse hearts and control tissues. To validate in vivo enhancer activity, we tested 12 of these sequences around 45 CHD-related genes in cultured cells and found that 8 (67%) have reproducible heart-specific enhancer activity. A functional analysis demonstrated that the identified human heart-specific enhancer wf1 regulates the FBN1 gene which is involved in heart disease.
Conclusions
Our study provides an integrative analysis pipeline for ChIP-seq data and identified a comprehensive catalog of human heart-specific enhancers for clinical CHD-related studies.
Impact
-
Establishing an efficient way to analyze regulatory regions in CHD is very important.
-
A highly qualified heart-specific enhancer compendium was generated by integrating 148 online ChIP-seq samples.
-
Sixty-seven percent of predicted regulatory sequences have reproducible heart-specific enhancer activity in vivo.
-
Human heart-specific enhancer wf1 regulates the CHD-related FBN1 gene.
Similar content being viewed by others


Introduction
Congenital heart disease (CHD) is one of the most common birth defects, with a worldwide occurrence of 7 per 1000 live births; 1.35 million infants are born with CHD each year.1 Genetic factors play an important role in the cause of CHD. Since the application of linkage analysis and chromosomal microarray technology, many of the CHD-associated genes, including NKX2-5, GATA4, TBX5, TBX20, and NOTCH, have been identified, and some family-specific mutations have been revealed.2,3,4,5,6 Decades of extensive genetics research have led to a deeper understanding of CHD causation than ever before with the rapid development of genome-wide association studies (GWASs) and massively parallel sequencing.7,8 However, there is an approximately 31–46% chance of identifying causal genetic variants in patients with accurately phenotypic familial CHD, and these variants often reside in known CHD genes.9 In patients with sporadic CHD, de novo coding variants and copy number variations (CNVs) in CHD genes accounted for only 20% of all cases. Nearly 80% of mutations found by whole-genome sequencing (WGS) in the isolated CHDs beyond regions of known CHD genes cannot be appropriately explained. In practice, mutations found contributing to CHD are mainly limited to exons and the surrounding regions, which account for only approximately 5% of the whole genome. However, except for new CHD genes, the majority of putative causative mutations identified with WGS reside in the noncoding region of the genome, which accounts for nearly 95% of the whole genome.10 Approximately 5% of the regulatory elements reside in large noncoding regions that regulate gene expression independently or together. Without changing gene expression proteins, these regions participate in the occurrence of diseases by affecting the time, space, and yield of protein expression. The genomic location and function of regulatory elements that orchestrate gene expression in heart development remain obscure, hindering research progress on their contributions to CHD. Thus, establishing an efficient way to analyze these regions is the most urgent problem to be solved.
The rapidly growing availability of sequenced genomes and advanced bioinformatic tools have enabled us to carry out more sophisticated analyses at the whole-genome level. ChIP-seq is a promising approach for the genome-wide mapping of protein binding and epigenetic marks.11 This approach can be used for the identification of putative regulatory elements by using a regulation-specific antibody. More than 300 ChIP-seq assays using different histone protein markers or tissue-specific antibodies in the Gene Expression Omnibus (GEO) database have been performed in human or mouse hearts in recent years according to the public database. However, integrative analyses of these online ChIP-seq data are rare.
The Encyclopedia of DNA Elements (ENCODE) Project suggests that nearly 37% of the human genome might have functional and regulatory effects on tissue-specific expression patterns.12 Enhancers are a major category of noncoding regulatory elements that activate gene expression from an unrestricted distance in a cell type-specific manner.13 Researchers have indicated that the in vivo mapping of p300 binding is a highly accurate means for identifying enhancers and their associated activities in embryonic forebrain, midbrain, and limb tissue.11 Subsequently, a ChIP-seq study concentrating on three different mouse tissues via H3K27ac to examine the genome-wide utilization of enhancers across different developmental stages ultimately identified nearly 90,000 putative distal enhancers.14 Then, ~6200 putative enhancers from fetal and adult human heart tissue were identified using an epigenomic approach.15 These studies performed ChIP-seq via different histone markers and created different groups of putative enhancers on different tissues. Little attention was given to the difference between different groups or the integration of large sets of ChIP-seq data. Research published in 2016 indeed integrated 35 ChIP-seq data sets from human and mouse hearts to generate >8000 putative human heart enhancers. However, they focused on only human enhancers, and not in a heart-specific manner. Thus, integrating large amounts of ChIP-seq data from different tissues and different histone markers remains a challenge.
In the present study, we creatively integrated more than one hundred ChIP-seq data sets mapping enhancer-associated chromatin marks in heart tissue and control tissue from mice and humans to identify heart-specific enhancers. This “virtual heart-specific enhancer panel” includes approximately predicted putative human enhancer elements with importance scores. Subsequently, a comparison with the published heart-enhancer catalogs was performed to analyze our method’s efficiency. Finally, in vitro validation showed that the percent of enhancer activity was significantly higher in the heart cell line than in the control cell line. Our study provides a foundation for heart-specific enhancers, as well as the potential value for creating human enhancer panels for CHD-related mutation screening.
Materials and methods
ChIP-seq data enrollment and preparation
The ChIP-seq data on 3 enhancer histone modifications (H3K4me1, H3K4me3, and H3K27ac) and from 4 tissues (heart, liver, kidney, brain) in humans or mice were obtained from the GEO database (http://www.ncbi.nlm.nih.gov/geo/) or the ENCODE website for the following analysis and calculation. We used antibodies against H3K4me1, a modification preferentially associated with enhancers;16 H3K4me3, a modification associated with promoters and enhancers;17 and H3K27ac, associated with active regulatory regions.13 Each profile was manually assessed, and those with unhealthy or diseased statuses were excluded. Public ChIP-seq processing differs between each laboratory and uses different annotation tools. The Cistrome DB website is a resource of human and mouse cis-regulatory information derived from ChIP-seq data with a standard analysis pipeline. It is a useful tool to obtain standardized ChIP-seq data. After the collection of ChIP-seq IDs in GEO and ENCODE, we downloaded all the annotated profiles from the Cistrome DB. Before downloading, we assessed the quality of each public data set on the website. Every data set met at least three of the six quality control indicators (raw sequence median quality score, % reads uniquely mapped, PCR bottleneck coefficient, number of merged total/fold 10/fold 20 peaks, fraction of reads in peaks, % peaks in promoter/exon/intron/intergenic, and % top 5k peaks overlapping with union DHS). Then, the files that met the requirements were downloaded locally for further assembly conversion. On the Cistrome DB website, original raw data were mapped to the human (hg38) or mouse (mm10) genome. To integrate and compare these data with human genomic sequencing data, we converted all the peak segments from the human (hg38) and mouse (mm10) genomes to the human (hg19) genome using the LiftOver tool. The mouse samples (mm10) were mapped to the human (hg19) genome with a 0.5 minimum ratio of bases that must remap. Each peak with a length change rate greater than 1 after LiftOver analysis was excluded.
Data integration and score
Downloaded data were divided into six files according to species and histone marks: group 1, hg19FromHg38_H3K4me1; group 2, hg19FromHg38_H3K4me3; group 3, hg19FromHg38_H3K27ac; group 4, hg19FromMm10_H3K4me1; group 5, hg19FromMm10_H3K4me3; and group 6, hg19FromMm10_H3K27ac. All the peaks in each group were virtually linearized along the genome according to the genomic location, and the frequency difference in each base site between the heart and the control subgroup was calculated. Each base site with the same frequency difference was combined with a new updated peak to obtain the score. For data visualization, we multiplied each score with the same modulus and ultimately obtained 6 tables with scores ranging from 0 to 1000. Then, the total lengths and total peaks from each table were measured to identify the score cutoff values. Finally, the same score cutoff values in the six tables were combined to obtain the final table and final scores.
Intersecting heart-specific enhancer catalog with the VISTA Enhancer Browser
The VISTA Enhancer Browser is a publicly available genomic database that provides experimentally validated human and mouse noncoding sequences with enhancers in transgenic mice. Transgenic mice with heart expression were defined as having heart-enhancer activity. Heart-specific enhancer activity indicates that all the transgenic mice had enhancer activity only in the heart. After searching all the online published experimental data, regions meeting our criteria were downloaded from the VISTA Enhancer Browser (http://enhancer.lbl.gov/) for further analyses.
Cell culture, transfections, and reporter assays
HEK293 cells, SH-SY5Y cells, and AC16 cells derived from human embryonic kidney cells, brain cells, and cardiomyocytes, respectively, were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, 1% glutamine, and 1% penicillin/streptomycin in an atmosphere with 5% CO2 at 37 °C. For the transfection experiment, the insert regions were generated by PCR using genomic DNA from HEK293 cells except for wf5 and wf10 (primers in Table 1). Wf5 and wf10 were synthesized by the self-combination of two oligos. Considering the active coverage of mild enhancer activity, we replaced the SV40 promoter of the pGL3-promoter vector (Promega, Madison, WI) with a minimal E1b promoter. A peak fragment was then cloned into the newly reconstructed pGL3-promoter vector (named pGL3-E1b vector) using the KpnI and XhoI sites, and direct sequencing was subsequently performed to confirm proper insertion. Cells were seeded in 48-well plates, and transient transfections were performed following the manufacturer’s recommendations. The transfection mixtures contained 400 ng of the expression vectors, 8 ng of pRL-TK, 0.3 μL of Lipofectamine 3000 (Invitrogen, Carlsbad, CA), and 0.8 μL of P3000. pRL-TK, which expresses Renilla luciferase, was used as an internal control. The cells were lysed at the indicated hours after transfection, and the luciferase activities were measured using a dual-luciferase reporter assay system (Promega) with a BioTek Synergy2 instrument. The results were calculated as the ratio of luciferase activity to Renilla luciferase activity. For the target gene identification experiments, the related plasmids were constructed by Jieli Science and Technology Co. Ltd. The 2-kb regions upstream of possible target genes were cloned into the pGL3-basic vector before ATG of the luciferase gene. The experiment was performed three times independently.
Statistical analysis
Statistical analyses were performed and graphs were constructed using GraphPad Prism 6. Luciferase data are expressed as the means ± standard deviation of three independent experiments. Student’s t test was used to compare luciferase activities between the two groups. Statistical significance criteria were defined as *P < 0.05, **P < 0.01, †P < 0.001, ‡P < 0.0001.
Results
ChIP-seq data collection and characterization
To comprehensively identify heart-specific enhancers in the human genome that can be incorporated into future CHD studies, we thoroughly included ChIP-seq data from different species and developmental stages. Enhancers are enriched in several histone modifications, including the monomethylation of H3K4me1, H3K4me3, and H3K27ac.16,18 The workflow of our study is shown in Fig. 1. A total of 148 eligible epigenomic data sets for these three histone proteins (see Supplementary Table 1 for detailed information) were downloaded from the Cistrome Data Browser (DB) website. A total of 148 data sets were divided into different groups according to their species, sources, and histone marks (Fig. 2a). The number of H3K4me1-related data sets was 40, which included 20 from human tissues (5 from heart tissues and 15 from control tissues) and 20 from mouse tissues (7 from heart tissues and 13 from control tissues). The number of H3K4me3-related data sets was 51, which included 31 from human tissues (8 from heart tissues and 23 from control tissues) and 20 from mouse tissues (10 from heart tissues and 10 from control tissues). The number of H3K27ac-related data sets was 57, which included 13 from human tissues (7 from heart tissues and 6 from control tissues) and 44 from mouse tissues (12 from heart tissues and 32 from control tissues).

More than 100 ChIP-seq data sets mapping three enhancer-associated chromatin marks in heart tissue and control tissue from mice and humans were integrated to identify heart-specific enhancers. We then demonstrate the importance of each putative enhancer by scoring. We also show the functional importance of enhancers by comparing two different published enhancer catalogs and validated the enhancer activities and targeted heart genes in vitro.

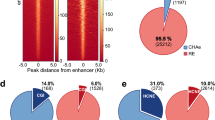
a The number of included ChIP-seq data sets in each group. b, c LiftOver analyses of different species and assembly into the human GRCh37/hg19 assembly. b Average number of total peaks and average cover range (kb) before and after LiftOver in human and mouse data. c Average largest and smallest peak lengths before and after LiftOver in human and mouse data, respectively. d, e Summary of cover range in genome and total peak number using cutoff scores of 501 (cutoff_0%), 600 (cutoff_20%), 700 (cutoff_40%), 800 (cutoff_60%), 900 (cutoff_80%), and 1000 (cutoff_100%). Regardless of cover range (d) or total peak number (e), they significantly decreased as the cutoff score increased.
Converting different species and assemblies to the human GRCh37/hg19 assembly
All the profiles downloaded from the Cistrome DB website were analyzed through a uniform processing pipeline based on the human GRCh38/hg38 assembly and the mouse GRCm38/mm10 assembly. However, most genomic sequencing of CHD samples was aligned to the human GRCh37/hg19 assembly for data annotation. To unify all the genome coordinates and genome annotations between different assemblies, we transferred all the downloaded profiles from the original version to the human GRCh37/hg19 assembly using the LiftOver tool. The average number of total peaks in 64 downloaded ChIP-seq data sets in humans was 42,013, and after LiftOver analysis, this number became 41,811. The average number of total peaks in 84 downloaded ChIP-seq data sets in mice was 44,232, and after LiftOver analysis, this number became 30,003 (32% of the total peaks were lost during the process). The average cover ranges in each profile were 18,119 kb in humans and 18,373 kb in mice (Fig. 2b). After LiftOver analysis, these numbers became 18,037 kb and 14,381 kb, respectively. The average cover size in the mouse data was decreased by nearly 22%. The average largest and smallest peaks in the human data were 2468 bp and 173 bp, respectively, and 2328 bp and 159 bp were the respective transformed peak lengths. Before LiftOver analysis, 2816 and 164 bp were the average largest and smallest peaks, respectively, in the mouse data, and 3343 bp and 68 bp were the respective LiftOver results (Fig. 2c). In summary, from four aspects, the data before and after LiftOver changed little between different assemblies in humans. However, great changes occurred in the LiftOver process from mouse assembly to human assembly.
Putative human heart-specific enhancer virtual catalog designed at the whole-genome level
Peaks from all the processed data sets were merged according to the different groups. The peak number distributions of scores greater than 500 in the six groups are summarized in Supplementary Table 2. The total peak numbers of the six groups ranged from 400,000 to 703,000 (average 563,000). In each group, the peak number in each score interval increased with decreasing score. The total length in the six groups ranged from 19 to 53 Mb, with an average of 35 Mb, which accounted for nearly 1% of the whole human genome (Supplementary Table 3). Peak lengths with scores ranging from 501 to 700 bp accounted for >80% in each group (except for the group hg19FromMm10_H3K4me3 group, which accounted for 52.5%), which roughly coincided with a normal distribution. In addition, we merged the six tables using cutoff scores of 501, 600, 700, 800, 900, and 1000. Finally, a 144.6-Mb candidate heart-specific enhancer compendium was generated using a 501 cutoff score. Regardless of cover range or peak number, they significantly decreased as the cutoff score increased (Fig. 2d, e).
Comparison with reported heart-enhancer catalogs
To validate the efficiency of our catalog with those of other enhancer catalogs, we reviewed and compared two different catalogs of heart-related enhancers. One is from a published research article in the Journal of Nature Communication, and the other is heart-expressed validated enhancers reported in the VISTA Enhancer Browser. Diane E’s catalog has integrated 35 ChIP-seq data sets from only the heart and generated a 264-Mb catalog of human putative enhancers. The size of our present compendium across the entire genome is 144.6 Mb, which is approximately only 25% of the reported size, but we included many more ChIP-seq original files, which indicated that our set is much more heart-specific and could be an efficient way to identify core enhancer regions. In addition, 54 heart-specific enhancers, which accounted for 155,468 bp of the total length, were identified in the VISTA Enhancer Browser until 12/30/2019 (Supplementary Table 4). Interestingly, all 54 heart-specific enhancers overlapped with our putative heart-specific enhancer virtual catalog. An example of candidate heart enhancers identified through this integrative analysis was validated with VISTA (Fig. 3). Six validated enhancers named hs1862, hs2161, hs1760, mm138, mm75, and mm172 were randomly chosen in the VISTA and enhancer activity in the heart was downloaded and shown. In each VISTA locus, the relative locations of the integrative candidate enhancer elements and the importance score in our study are also shown. All six validated enhancers in VISTA overlap with our candidate enhancers with high scores.

For each locus, we show the relative locations of the integrative candidate enhancer elements and the tested elements in VISTA. The importance score and in vivo activity of each locus are also shown. The hs/mm numbers indicate the VISTA identifier. The heart is identified by a red arrow. Scale bars, 1 cm = 1 kb.
In vitro validation of human heart-specific enhancers near disease genes
To generate a more powerful human heart-specific enhancer screening catalog, we chose a cutoff score of 900 for further analyses. The total lengths of those with scores no less than 900 ranged from 1422 bp to 1.9 Mb (average 0.55 Mb). Ultimately, we obtained a 3-Mb virtual putative heart-specific enhancer set with 48,923 peaks in the whole genome (Table 2). The chromosome distribution of peaks with scores no less than 900 is displayed in Fig. 4. It visualizes the peak locations over the whole genome and calculates the coverage of peak regions over chromosomes. Each peak had its own score greater than 900. Such a large-sized data set is suitable for panel design and virtual analyses of CHD-related mutations, but it is still an obstacle for us to choose for further validation experiments. We then narrowed our study scale to 50 kb upstream and downstream of the 45 CHD-related core genes (Table 3). Crossing with the 3-Mb cutoff 900 panel, we obtained a 46.6-kb heart-specific core enhancer panel. A total of 239 peaks were enrolled, with an average length of 195 bp (Supplementary Table 5).

The chromosome distribution of peaks with scores no less than 900 is displayed.
In the captured regions, we randomly chose 12 regions, considering their distance to CHD-related genes, scores, and average lengths, which were named wf1 to wf12 (Table 4). The regions ranged from 23 to 795 bp (average 350 bp), and the distance to near genes ranged from inside to 33 kb. The 12 regions were then examined for enhancer activity discrepancies in HEK293, SH-SY5Y, and AC16 cells. The results in the heart cell line AC16 showed that 8/12 of the tested plasmids had enhancer activity relative to that of the vector pGL3-E1b (Fig. 5a). However, none of these 12 tested regions showed increased transcriptional activity compared with that of pGL3-E1b in the nonheart (control) cell lines HEK293 and SH-SY5Y (Fig. 5b, c). These results suggest that the 8 regions we identified have relatively heart-specific enhancer activity.

Wf1 to wf12 expression vectors, along with a Renilla reporter construct (internal control), were cotransfected into AC16 cells (a), HEK293 cells (b), and SH-SY5Y cells (c). a The transcriptional activity levels of wf1, wf2, wf3, wf5, wf7, wf9, wf10, and wf11 were higher than that of pGL3-E1b. *P < 0.05, **P < 0.01, †P < 0.001, ‡P < 0.0001. b, c The transcriptional activity levels of wf1, wf2, wf3, wf5, wf7, wf9, wf10, and wf11 were lower than that of pGL3-E1b.
The human heart-specific enhancer wf1 can directly regulate FBN1
We applied an in situ replacement strategy to identify whether the possible CHD-related gene FBN1 is the real target gene of the enhancer wf1. All putative enhancers located within the 100-kb range around the FBN1 gene are shown in Fig. 6a. The region between the multiple cloning site and the ATG before the luciferase gene in the pGL3-basic vector was replaced with a 2-kb fragment immediately behind the ATG of the human FBN1 gene (named Basic-wf1). The Basic-wf1-deletion plasmid was derived from the Basic-wf1 plasmid with the deletion of wf1. These expression vectors were tested for transcriptional activity in HEK293 cells and AC16 cells. The results showed that the transcriptional activity of Basic-wf1-deletion was significantly decreased compared with that of Basic-wf1 in both cell lines (Fig. 6b, c), suggesting that the 237-bp enhancer fragment indeed functionally targets the FBN1 gene.

a Distribution of putative heart-specific enhancers near the FBN1 gene. The FBN1 gene is shown as gray rectangles, and the orientation of arrows indicates the translational initiation sites of the gene. Peaks are indicated by lines. The horizontal line at the bottom indicates the studied range of the DNA sequence. b The transcriptional activity of pGL3-Basic-wf1 was significantly higher than that of pGL3-Basic-wf1-deleted in AC16 cells (‡P < 0.0001). c The transcriptional activity of pGL3-basic-wf1 was significantly higher than that of pGL3-Basic-wf1-deleted in HEK293 cells (†P < 0.001).
Discussion
In our study, we creatively generated a human heart-specific enhancer catalog in a human genome spectrum. Approximately 400 genes have been discovered to be involved in the causation of CHD by the advent of whole-exon sequencing (WES) and WGS.19 However, initial WGS analyses have focused primarily on the <2% of the genome that encodes proteins.20 Tissue-specific enhancers, especially distant-acting enhancers, vastly outnumber protein-coding genes in mammalian genomes. The strong contribution of enhancers in many human diseases is now widely recognized.21 Researchers were the first to identify that even point mutations in a long-range SHH enhancer are associated with preaxial polydactyly.22 In CHD, researchers found that a single base-pair mutation in a TBX5 distant enhancer abrogated the ability of the enhancer to drive TBX5 expression within the heart in an animal model of CHD.23 Taken together, these results suggest that disease-predisposing variants should be expanded to the regulatory regions of the whole genome. However, the possible contribution of enhancers in heart disease has been difficult to evaluate because their genomic locations relative to genes remain largely obscure.15 To date, several large-scale discoveries of heart enhancers have been performed through ChIP-seq.11,15,24 All of them concentrated on the construction of a putative heart-enhancer catalog, with little concentration on the integration of heart-specific enhancers in the genomic spectrum. The accurate control of tissue-specific gene expression plays an important role in heart development, but few cardiac-specific enhancers have thus far been identified. Our virtual catalog of heart-specific enhancers at the genomic scale can be used as an annotative and analytical tool in the analysis of WGS data. This catalog will enable more sophisticated analyses to assess the burden of variations predisposing toward CHD from specific aspects.
To date, several CHD-related sequencing methods have been used in the clinic, including targeted sequencing (exome sequencing and disease-specific gene panels) or nontargeted sequencing, such as WGS. None of the regulatory panels were designed or used in clinical mutation screening. Our project provides a new prospective for CHD-related mutation screening in clinical CHD patients with no mutations found with WES or in samples with no disease-related single nucleotide polymorphisms (SNPs) found with WGS. The virtual heart-specific enhancer catalogs range from 19 to 53 Mb, with an average 35 Mb, which accounts for approximately 1% of the whole human genome. With the superiority of our integration approach, the accurate size of the virtual heart-specific enhancer compendium is flexible and depends on the needs of researchers. The catalog could be much smaller with an increasing cutoff score. In particular, every single base pair has its own exact score with our integration method, so it is indeed an efficient way for us to analyze the important relationship between even a single base pair and disease. Therefore, the pipeline for regulatory region analysis is a new approach for developing disease-susceptible mutations screening systems, which could be a complement to the current CHD-related sequencing methods.
In this study, we creatively developed a heart-specific enhancer compendium that counted 25% of the published heart-enhancer catalog. Our study included ChIP-seq data from three histone markers and analyzed data from heart and control organs to identify heart-specific enhancers, while the published article included only heart-related ChIP-seq data, mostly from H3K27ac, to screen for putative heart enhancers. The average length in our putative heart-specific compendium is 61 bp, while the average length in this published article is 3212 bp, which means that except for heart specificity, our strategy might be a good way to identify the core enhancer regions. In addition, the 100% overlap with heart-specific enhancers reported in VISTA highlights the sensitivity of our integrative approach in the present study and strengthens our belief toward further animal research. Except for efficiency and specificity, this integrating approach can be used for a wide range of extension applications in regulatory-related studies. Promoters, enhancers, and silencers are all regulatory elements. We could choose appropriate histone markers to establish different candidate regulatory compendiums using this pipeline. Particularly, for enhancers, we could generate candidate active or poised enhancer groups by defining histone markers, which greatly enhances our research scope toward regulatory-related studies.
Enhancer or heart-specific enhancer prediction and the annotation of noncoding sequences by high-throughput approaches is the first step for interpreting WGS data in CHD. Functional testing of enhancer activity and disease-related mutation validation is generally the second step to explain the cause of disease. In our study, we initially tested heart-specific enhancer activity in two different cell lines and then chose one enhancer near the candidate heart gene to confirm its target gene. We successfully identified a cluster of heart-specific enhancers, and enhancer wf1 indeed targets the predicted CHD-related gene FBN1. The in vitro method is a much higher-throughput and efficient method than the in vivo method. However, developmental information on enhancers and target genes may be lost to some degree. In the future, genomic deletion of enhancers or mutations inside enhancers could be constructed to assess phenotypic robustness in animal models.
In addition, we included three enhancer-related histone protein markers.18,25 It is possible that if we included more enhancer histone markers, such as EP300-related ChIP-seq data, the overlapping length could be improved. Furthermore, during data processing, we discarded some data that met our criteria but could not be found on the Cistrome website. In the future, if we could perform a bioinformatics analysis workflow from raw reads to peak calling, these profiles could remain and improve our credibility. Our present study was based on online bioinformatics sources. In the near future, we will perform more complicated bioinformatics analyses.
Conclusion
We creatively integrated more than 100 ChIP-seq data sets mapping enhancer-associated chromatin marks in heart tissue and control tissue from mice and humans to identify heart-specific enhancers. Overall, our study highlights the important role of heart-specific enhancers in heart development and provides a valuable catalog of human heart-specific enhancers that can be easily and widely integrated into CHD-related studies.
References
van der Bom, T., Bouma, B. J., Meijboom, F. J., Zwinderman, A. H. & Mulder, B. J. The prevalence of adult congenital heart disease, results from a systematic review and evidence based calculation. Am. Heart J. 164, 568–575 (2012).
Schott, J. J. et al. Congenital heart disease caused by mutations in the transcription factor Nkx2-5. Science 281, 108–111 (1998).
Kirk, E. P. et al. Mutations in cardiac T-box factor gene Tbx20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am. J. Hum. Genet. 81, 280–291 (2007).
Garg, V. et al. Mutations in Notch1 cause aortic valve disease. Nature 437, 270–274 (2005).
Garg, V. et al. Gata4 mutations cause human congenital heart defects and reveal an interaction with Tbx5. Nature 424, 443–447 (2003).
Basson, C. T. et al. Mutations in human Tbx5 [Corrected] cause limb and cardiac malformation in Holt-Oram Syndrome. Nat. Genet. 15, 30–35 (1997).
Sifrim, A. et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 48, 1060–1065 (2016).
Wu, Y. et al. Multi-trait analysis for genome-wide association study of five psychiatric disorders. Transl. Psychiatry 10, 209 (2020).
Blue, G. M. et al. Targeted next-generation sequencing identifies pathogenic variants in familial congenital heart disease. J. Am. Coll. Cardiol. 64, 2498–2506 (2014).
Postma, A. V., Bezzina, C. R. & Christoffels, V. M. Genetics of congenital heart disease: the contribution of the noncoding regulatory genome. J. Hum. Genet. 61, 13–19 (2016).
Visel, A. et al. Chip-Seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858 (2009).
The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Heintzman, N. D. et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 (2009).
Nord, A. S. et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531 (2013).
May, D. et al. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 44, 89–93 (2011).
Heintzman, N. D. et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318 (2007).
Guenther, M. G., Levine, S. S., Boyer, L. A., Jaenisch, R. & Young, R. A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77–88 (2007).
Creyghton, M. P. et al. Histone H3k27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA 107, 21931–21936 (2010).
Zaidi, S. et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223 (2013).
Gilissen, C. et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347 (2014).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Lettice, L. A. et al. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 12, 1725–1735 (2003).
Smemo, S. et al. Regulatory variation in a Tbx5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 21, 3255–3263 (2012).
Dickel, D. E. et al. Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat. Commun. 7, 12923 (2016).
Zhao, X. D. et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 1, 286–298 (2007).
D’Amato, G., Luxán, G. & de la Pompa, J. L. Notch signalling in ventricular chamber development and cardiomyopathy. FEBS J. 283, 4223–4237 (2016).
Funding
This work was supported by the Natural Science Foundations of Shanghai [grant numbers 21ZR1410100 to Q.L.], the National Natural Science Foundations of China (NSFC) [grant numbers 81771632 to Q.L. and 81873481 and 81741081 to Y.H.G.], and the National Key Research and Development Program [grant number 2016YFC1000500, to Q.L.].
Author information
Authors and Affiliations
Contributions
(I) Conception and design: F.W., Q.L., Y.H.G.; (II) administrative support: Y.W.Z., Y.T.G.; (III) provision of study materials: X.D.C., F.W., F.W.; (IV) collection and assembly of data: Y.H.W., X.W.; (V) data analysis and interpretation: all authors; (VI) manuscript writing: all authors; (VII) final approval of manuscript: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, F., Zhang, Y., Wu, F. et al. Functional assessment of heart-specific enhancers by integrating ChIP-seq data. Pediatr Res 92, 1332–1340 (2022). https://doi.org/10.1038/s41390-022-01981-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-022-01981-5
