
Abstract
Background
Previous genetic research in pediatric cardiomyopathy (CM) has focused on pathogenic variants for diagnostic purposes, with limited data evaluating genotype-outcome correlations. We explored whether greater genetic variant burden (pathogenic or variants of unknown significance, VUS) correlates with worse outcomes.
Methods
Children with dilated CM (DCM) and hypertrophic CM (HCM) who underwent multigene testing between 2010 and 2018 were included. Composite endpoint was freedom from major adverse cardiac event (MACE).
Results
Three hundred and thirty-eight subjects were included [49% DCM, median age 5.7 (interquartile range (IQR) 0.2–13.4) years, 51% HCM, median age 3.0 (IQR 0.1–12.5) years]. Pathogenic variants alone were not associated with MACE in either cohort (DCM p = 0.44; HCM p = 0.46). In DCM, VUS alone [odds ratio (OR) 4.0, 95% confidence interval (CI) 1.9–8.3] and in addition to pathogenic variants (OR 5.2, 95% CI 1.7–15.9) was associated with MACE. The presence of VUS alone or in addition to pathogenic variants were not associated with MACE in HCM (p = 0.22 and p = 0.33, respectively).
Conclusion
Increased genetic variant burden (pathogenic variants and VUS) is associated with worse clinical outcomes in DCM but not HCM. Genomic variants that influence DCM onset may be distinct from those driving disease progression, highlighting the potential value of universal genetic testing to improve risk stratification.
Impact
-
In pediatric CM, inconsistent findings historically have been shown between genotype and phenotype severity when only pathogenic variants have been considered.
-
Increased genetic variant burden (including both pathogenic variants and VUS) is associated with worse clinical outcomes in DCM but not HCM.
-
Genomic variants that influence CM onset may be distinct from those variants that drive disease progression and influence outcomes in phenotype-positive individuals.
-
Incorporation of both pathogenic variants and VUS may improve risk stratification models in pediatric CM.
Similar content being viewed by others

Introduction
Pediatric cardiomyopathy (CM) has an incidence of 1 to 1.5 cases per 100,000 person-years and has a wide phenotypic spectrum ranging from minimal cardiac involvement to severe ventricular dysfunction with the need for advanced heart failure therapy.1,2 With advances in next-generation sequencing technology, an increasing number of genetic variants are emerging as potential causes of CM, including both pathogenic variants and variants of unknown significance (VUS).3,4,5,6,7 In familial dilated CM (DCM), pathogenic variants have been identified in 73% of cases,8 and in pediatric patients with hypertrophic CM (HCM), genetic variants are present in over half of sporadic HCM cases and nearly two-thirds of familial cases.9
Despite significant research focused on identifying genetic variants in CM for diagnostic purposes, there have been limited data evaluating genotype for prognostic purposes. Previous data are inconsistent from studies evaluating the correlation between genotype and clinical outcomes, but this may be explained by the fact that previous research has primarily focused only on pathogenic mutations as causative variants. For example, a recent study using the Pediatric Cardiomyopathy Registry demonstrated that the presence of a pathogenic variant was not associated with worse outcome at 1 year follow-up in pediatric DCM.10 However, VUS have historically not been considered when evaluating genotype-outcome association, as the pathogenicity of these variants is uncertain. However, VUS may represent important genetic modifiers that influence cardiac outcomes in pediatric CM. Notably, a recent study of pediatric HCM found that greater variant burden, including both pathogenic variants and VUS, was associated with worse outcomes, a novel finding in the CM literature.11,12 Another recent study of a small cohort of pediatric CM also suggested that increased variant burden, including VUS, was associated with adverse outcomes.13 The concept of VUS as a risk modifier is not confined to CM; for example, in congenital heart disease, there is increasing evidence that greater genetic variant burden is associated with worse cardiovascular and neurodevelopmental outcomes.14,15,16 Thus, consideration of variant burden incorporating both pathogenic variants and VUS as potential risk modifiers may help generate a better genetic risk stratification model for pediatric CM. Such a model could have significant clinical implications, including risk stratification for adverse events, prophylactic interventions including implantable cardioverter-defibrillator (ICD) placement, and advanced heart failure therapies.
The purpose of this study is to determine whether genetic variant burden (defined as either pathogenic variants or VUS) has prognostic value by correlating with clinical outcomes in pediatric patients with DCM or HCM who underwent clinical evaluation and genetic testing at our institution. This study tested the hypothesis that the presence of a greater number of genetic variants (including both pathogenic variants and VUS) is associated with worse clinical outcome in a longitudinally followed large pediatric CM cohort.
Methods
Study cohort
This is a retrospective, single-center cohort study that evaluated outcomes in pediatric patients (age < 21 years at diagnosis) with a history of DCM or HCM who were evaluated within the cardiomyopathy program at the Children’s Hospital of Philadelphia between 2010 and 2018. Patients were excluded from endpoint analysis if they had not undergone clinical multigene testing. Those that only underwent single-gene targeted genetic testing were excluded due to risk for under-identifying variant burden if only single-gene testing was performed. Patients were also excluded if they had negative phenotype on screening evaluation. Baseline demographic, clinical, echocardiographic, and cardiac magnetic resonance imaging (MRI) were reviewed and the data were abstracted by the lead investigator (D.S.B.) through review of the medical record. The Children’s Hospital of Philadelphia Institutional Review Board (IRB 18-015616) approved this study with waiver of consent.
CM was classified based on phenotype defined by standard imaging criteria (either echocardiographic or cardiac MRI).17 DCM was defined by left ventricular end-diastolic z-score > +2.0 with diminished left ventricular systolic function.1,18 HCM was defined by end-diastolic interventricular septal thickness or posterior wall z-score > +2.0 or absolute value ≥ 1.5 cm.19,20 Subjects with neuromuscular disease were included if they had multigene testing performed. Secondary CM including congenital heart disease, myocarditis, pulmonary disease, endocrine disease, cardiotoxic exposure, and systemic hypertension were excluded.
Classification of variants
All clinical genetic testing was reviewed retrospectively. Molecular results were obtained from a variety of clinical diagnostic labs. The composition of cardiomyopathy panels evolved over the time period of the study. Representative panel compositions are shown in Supplementary Table 1. Exome sequencing was completed in a subset of patients including many trio samples that included unaffected parental controls. Each diagnostic lab utilized a different proprietary analysis pipeline; therefore, after data abstraction from the clinical record, each variant was analyzed by a clinical geneticist (R.C.A.-N.) to reassess pathogenicity. Specifically, for each of the single-nucleotide variants (SNVs), ClinVar was queried. ClinVar is a National Institute of Health-supported public archive of 430,000 unique human variants and their associated reported phenotype. If multiple pathogenicity classifications were reported, the most common variant annotation was adopted, if the clinical and variant features supported this classification. Features used to validate classification included population frequency in a large genomic dataset (gnomAD version 3.0),21 inheritance pattern, and accompanying supportive clinical data (such as biochemical testing for mutations in metabolic genes). These features incorporated into the final variant classification as recommended by the American College of Medical Genetics (ACMG) variant classification guidelines.22 If multiple genetic tests were performed, unique genetic variants were recorded from each testing modality and incorporated into the final analysis.
For the 1098 variants identified in the total population (Supplementary Table 2), classification was unchanged for 896 variants after reanalysis. One hundred and thirty-five variants underwent minor reclassification defined as a changing between the categories of likely pathogenic and pathogenic or between likely benign and benign. One hundred twenty-seven variants had more significant reclassifications between the categories of pathogenic/likely pathogenic variants and VUS, benign/likely benign and VUS, or pathogenic/likely pathogenic and benign/likely benign. For the purposes of this study, pathogenic variants included both pathogenic/likely pathogenic, and no variants included no/benign/likely benign variants.
Statistical analysis
The composite endpoint was freedom from major adverse cardiac event (MACE), defined as mechanical circulatory support [ventricular assist device (VAD) or extracorporeal membrane oxygenation (ECMO)], transplant, and aborted cardiac arrest or death. Need for transplant evaluation with decision to not list due to patient or family request was included in the composite endpoint. Secondary endpoints included freedom from individual MACE endpoints. Analysis was performed by individual CM categories.
Demographic, clinical, imaging, and genetic variables were summarized using standard descriptive statistics. Non-normally distributed continuous data were reported as median [with interquartile range (IQR)] and categorical variables were reported as counts (with percentages). To explore differences in characteristics between patients with and without genetic testing, as well as genetic variant and composite endpoints, we used Chi-square, Fisher’s exact, and Wilcoxon rank-sum/Kruskal–Wallis tests. Due to potential interaction between genotype and severity of phenotype (including age at presentation or presence of heart failure at presentation), bivariate analysis was selected for an exploratory analytic approach to understand whether a relationship exists between genotype and outcome in this pediatric cardiomyopathy population. Bivariate odds ratios (ORs) and 95% confidence intervals (95% CIs) are reported for overall MACE and each component of MACE. Kaplan–Meier curves are presented as time to MACE stratified by genetic variant type. Statistical significance was determined a priori at a two-tailed p value ≤ 0.05. All analyses were conducted in STATA Version 15.1.
Results
Patient characteristics
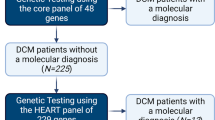
During the study period, 338 subjects were included in the final analysis, of which 165 (49%) had DCM and 173 (51%) had HCM (Fig. 1). Median age at positive phenotype was 5.7 (0.2–13.4) years in DCM and 3.0 (0.1–12.5) years in HCM (Table 1). Median follow-up was 4.7 (1.5–8.4) years in DCM and 3.6 (0.9–7.3) years in HCM. The most common reason for presentation in the DCM cohort were heart failure (46%) or clinical screening (35%), while in HCM the most common reason for presentation were clinical screening (33%) or murmur (26%). About one-quarter of the cohort had a family history of CM, which was more common in HCM than DCM.

Subject flow chart.
Table 2 compares patient characteristics between those with and without genetic testing. The median follow-up was similar across DCM and HCM cohorts. In DCM, those who underwent genetic testing were similar age at presentation to those who did not undergo genetic testing, while in HCM those who underwent genetic testing were younger. In DCM, those that underwent genetic testing presented with similar rates of heart failure and cardiac arrest, but more often diagnosed based on clinical screening. In HCM, those that underwent genetic testing presented more often with heart failure or clinical screening. Notably, there was not a significant difference in family history between genetic testing cohorts in either DCM or HCM.
Variant spectrum
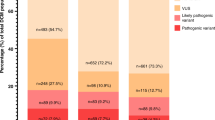
Multigene testing was performed in 72% of DCM and 65% of HCM cases. The most common test was a CM panel performed in 52% in both DCM and HCM patients (Supplementary Table 3). Exome sequencing was performed in 18% in DCM and 11% in HCM, and microarray analysis in 25% of DCM and 22% in HCM. Six percent underwent targeted familial genetic testing exclusively and were excluded from analysis. The distribution of variant burden across the DCM and HCM cohorts is shown in Fig. 2 and specific variants are shown in Supplementary Table 2. Over half of DCM patients had at least one variant present, while nearly two-thirds of HCM patients had at least one variant (variants shown in Supplementary Table 2). The HCM cohort had a higher proportion of pathogenic variants, while VUS were more common in DCM. Inheritance patterns were not significantly different across phenotypes. Supplementary Table 4 shows differences in demographics and phenotype based on variant burden. Those with either pathogenic variants or VUS were younger at positive phenotype (p = 0.001) and more often presented with heart failure (p = 0.002), while those without genetic variants presented more frequently through clinical screening (p < 0.001).

Distribution of genetic variants across overall cohort.
Genetic variant burden and clinical outcomes
The composite MACE endpoint occurred in 52% in DCM and 20% in HCM. In the DCM cohort, the most common MACE were transplant (19%), death (11%), VAD implant (10%), and aborted cardiac arrest (10%). In the HCM cohort, the most common MACE were aborted cardiac death (5%) and death (8%).
In DCM, the presence of a pathogenic variant alone was not associated with worse outcomes (p = 0.658) (Table 3 and Fig. 3). The presence of VUS alone was associated with MACE (OR 4.0, 95% CI 1.9–8.3; p < 0.001), primarily driven by cardiac transplantation (OR 2.9, 95% CI 1.2–6.8; p = 0.019. The presence of VUS in addition to pathogenic variant was associated with further increased association with MACE (OR 5.2, 95% CI 1.7–15.9; p = 0.004). This was primarily driven by increased VAD support (OR 4.6, 95% CI 1.3–15.0; p = 0.016), ECMO (OR 7.0, 95% CI 1.1–45.6; p = 0.041), and transplantation (OR 6.0, 95% CI 2.0–18.5; p = 0.002). In HCM the presence of a pathogenic variant alone was not associated with worse outcomes (p = 0.458) (Table 3 and Fig. 4). In addition, VUS alone (p = 0.215) and pathogenic variant with a VUS were also not associated with worse outcomes (p = 0.332).

Kaplan–Meier survival estimates for freedom from MACE-based variant burden in dilated cardiomyopathy.

Kaplan–Meier survival estimates for freedom from MACE-based variant burden in hypertrophic cardiomyopathy.
Discussion
Unlike prior genotype–phenotype studies in pediatric CM that primarily focused on using genotype for diagnostic purposes, this study illustrates the novel utility of genotype for predicting clinical outcome in children. Consistent with prior studies, our study demonstrates that the presence of pathogenic variants alone is insufficient to predict clinical outcomes and adverse events in pediatric CM.5 However, in our cohort of children with DCM, increased variant burden including both pathogenic variants and VUS was associated with worse outcomes, although this finding was not seen in HCM. This exploratory analysis highlights the potential clinical utility of comprehensive genetic testing (from large multigene panels or exome sequencing) that incorporates both known pathogenic variants and VUS in future risk stratification models in pediatric DCM.
Genetic testing is now recommended for all types of CM for diagnostic purposes in both patient and at-risk family members.6,23,24 With rapidly developing genetic testing techniques, genetic testing is being employed more widely and increasing number of genetic variants in CM are being identified.5,6 Historically, genotyping has been used for diagnostic purposes in CM, but not for prognostication due to the limited data regarding the variants that drive disease progression and therefore clinical outcomes in CM. However, recent literature in pediatric CM and in congenital heart disease has demonstrated that greater genetic variant burden, including both pathogenic and VUS variants, correlates with worse clinical outcomes.13,14,15,16 Although genetic modifiers may play a different role in congenital heart disease in regard to disease expression and progression due to its polygenic development process, similar preliminary findings have been identified in small pediatric CM populations.25 In our study, the correlation between variant burden, including VUS, and outcomes highlights the need for further mechanistic studies of genomic factors that contribute to disease progression. Our data support the hypothesis that the variants that predispose an individual to a poor outcome may in fact be distinct from those factors associated with risk of disease onset. VUS may represent genetic modifiers that affect outcomes in pediatric CM, particularly DCM, and genetic modifiers are important because they may lead to better risk stratification models and, potentially, targets for novel therapies.
Several studies have evaluated whether pathogenic variants are associated with outcomes in pediatric CM. A small, single-center study of non-HCM pediatric CM demonstrated that presence of a pathogenic variant was associated with higher rates of transplantation and mortality.25 However, in a large cohort study of pediatric DCM patients in the Pediatric Cardiomyopathy Registry, the presence of pathogenic mutations was not associated with disease severity or adverse outcomes.10 Our study validates this finding, demonstrating that the presence of pathogenic variants alone is not associated with worse outcomes.
A recent study by Mathew et al.12 evaluated genetic testing for pediatric HCM and found that greater variant burden was associated with more severe clinical phenotype and worse clinical outcomes. In their study, they identified either a pathogenic variant or VUS as variant positive. However, in our study, greater variant burden was not associated with decreased freedom from MACE. The differences in our study’s findings may be related to differences in study definitions of MACE, with the Mathew et al.12 study including ICD implantation as a MACE, while ours did not. This endpoint was not selected in our study due to variability in ICD implantation practice patterns, particularly for primary prevention.
When considering future care for pediatric CM, incorporation of a variant burden quantification that considers both VUS and known pathogenic variants may help move beyond diagnostic purposes and potentially improve our risk stratification models. Specific consideration for VUS as genetic modifiers may lead to targets for novel therapeutic strategies. A future model using genome-wide polygenic risk scores for pediatric CM would be highly valuable to a clinician when counseling patients and determining need for advanced heart failure therapies.26,27 Multicenter registries that have incorporated genome-wide genetic testing across large pediatric CM cohorts, such as the Pediatric Cardiomyopathy Registry, would provide a robust platform to further study whether a genome-wide polygenic risk score that incorporates both VUS and pathogenic variants would accurately risk stratify pediatric CM patients.
Study limitations
This study has several limitations. First, although the cohorts seemed generally similar, there may be selection bias among those that who underwent genetic testing. Also, there may be selection bias within the study cohort because our study population was derived from a large referral center. Thus, generalizability of our findings to the broader pediatric CM population should be the aim for future studies. In addition, we do not know which VUS are potential causative variants or genetic modifiers, and we do not know the underlying biological mechanism by which they modify cardiovascular risk. Since this was an exploratory analysis to look at simple associations between characteristics, genetic characteristics and outcomes, future analyses may look into the directionality of associations via alternative statistical modeling techniques. To further investigate this, one potential next step may be to take identified VUS and perform a pathway analysis to determine if there are common genetic pathways.
In addition, genetic testing was completed at a number of diagnostic labs and panels evolved during the study period. This may have influenced genetic yield, although earlier testing panels incorporated genes with the highest known number of pathogenic variants. The type of genetic testing was not standardized across patients as it was based on a convenience sample of clinically indicated genetic testing, potentially underestimating the true genetic variant burden. Finally, the median age at last encounter in our genetically tested cohort was 12.9 years; therefore, this study did not capture adverse events that occurred in adulthood. Future larger studies using comprehensive testing are needed to further validate the preliminary findings of this study.
Conclusion
Consistent with prior data, our study demonstrates presence of pathogenic variants alone is not associated with worse outcomes in pediatric CM. However, our study found that increased genetic variant burden incorporating both VUS and pathogenic variants is associated with worse clinical outcomes in pediatric DCM but not HCM. Although studies validating the genotype-outcome association findings of this study in a larger pediatric population are needed, this exploratory analysis highlights the potential utility of universal genetic testing in pediatric DCM care beyond diagnostic purposes. Evaluating genetic variant burden in pediatric DCM may also provide prognostic value that could ultimately help improve risk stratification models in this complex population.
References
Lipshultz, S. E. et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N. Engl. J. Med. 348, 1647–1655 (2003).
Lee, T. M. et al. Pediatric cardiomyopathies. Circ. Res. 121, 855–873 (2017).
Pugh, T. J. et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 16, 601–608 (2014).
Fatkin, D. et al. Precision medicine in the management of dilated cardiomyopathy: JACC state-of-the-art review. J. Am. Coll. Cardiol. 74, 2921–2938 (2019).
Herkert, J. C. et al. Toward an effective exome-based genetic testing strategy in pediatric dilated cardiomyopathy. Genet. Med. 20, 1374–1386 (2018).
Vasilescu, C. et al. Genetic basis of severe childhood-onset cardiomyopathies. J. Am. Coll. Cardiol. 72, 2324–2338 (2018).
Kuhnisch, J. et al. Targeted panel sequencing in pediatric primary cardiomyopathy supports a critical role of TNNI3. Clin. Genet. 96, 549–559 (2019).
Cuenca, S. et al. Genetic basis of familial dilated cardiomyopathy patients undergoing heart transplantation. J. Heart Lung Transplant. 35, 625–635 (2016).
Morita, H. et al. Shared genetic causes of cardiac hypertrophy in children and adults. N. Engl. J. Med. 358, 1899–1908 (2008).
Kantor Paul, F. et al. Abstract 14210: phenotype, but not genotype, determines survival in pediatric dilated cardiomyopathy: a study from the NHLBI-Funded Pediatric Cardiomyopathy Registry. Circulation 138(Suppl_1), A14210–A14210 (2018).
Limongelli, G. et al. Clinical and genetic characterization of patients with hypertrophic cardiomyopathy and right atrial enlargement. J. Cardiovasc. Med. (Hagerstown) 18, 249–254 (2017).
Mathew, J. et al. Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin. Genet. 93, 310–319 (2018).
Al-Wakeel-Marquard, N. et al. RIKADA study reveals risk factors in pediatric primary cardiomyopathy. J. Am. Heart Assoc. 8, e012531 (2019).
Kim, D. S. et al. Patient genotypes impact survival after surgery for isolated congenital heart disease. Ann. Thorac. Surg. 98, 104–110 (2014). discussion 110–1.
Kim, D. S. et al. Burden of potentially pathologic copy number variants is higher in children with isolated congenital heart disease and significantly impairs covariate-adjusted transplant-free survival. J. Thorac. Cardiovasc. Surg. 151, 1147–51 e4 (2016).
Blue, G. M. et al. Genetic burden and associations with adverse neurodevelopment in neonates with congenital heart disease. Am. Heart J. 201, 33–39 (2018).
Lipshultz, S. E. et al. Cardiomyopathy in children: classification and diagnosis: a scientific statement from the American Heart Association. Circulation 140, e9–e68 (2019).
Kampmann, C. et al. Normal values of M mode echocardiographic measurements of more than 2000 healthy infants and children in central Europe. Heart 83, 667–672 (2000).
Grenier, M. A. et al. Design and implementation of the North American Pediatric Cardiomyopathy Registry. Am. Heart J. 139(Part 3), S86–S95 (2000).
Lopez, L. et al. Recommendations for quantification methods during the performance of a pediatric echocardiogram: a report from the Pediatric Measurements Writing Group of the American Society of Echocardiography Pediatric and Congenital Heart Disease Council. J. Am. Soc. Echocardiogr. 23, 465–495 (2010). quiz 576-7.
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Hershberger, R. E. et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 20, 899–909 (2018).
Kindel, S. J. et al. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J. Card. Fail. 18, 396–403 (2012).
Ellepola, C. D. et al. Genetic testing in pediatric cardiomyopathy. Pediatr. Cardiol. 39, 491–500 (2018).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Torkamani, A., Wineinger, N. E. & Topol, E. J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 19, 581–590 (2018).
Acknowledgements
This work was supported, in part, by the Cardiac Center Clinical Research Core at the Children’s Hospital of Philadelphia. No specific support was provided for this project.
Author information
Authors and Affiliations
Contributions
D.S.B. conceptualized and designed the study, drafted the initial manuscript, and reviewed and revised the manuscript. R.C.A.-N. reviewed and classified all genetic variants. M.J.O’C., H.G., A.R., K.Y.L., and J.W.R. designed the data collection instruments, collected data, carried out the initial analyses, and reviewed and revised the manuscript. J.W.G. and R.C.A.-N. conceptualized and designed the study, coordinated and supervised data collection, and critically reviewed the manuscript for important intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Burstein, D.S., Gaynor, J.W., Griffis, H. et al. Genetic variant burden and adverse outcomes in pediatric cardiomyopathy. Pediatr Res 89, 1470–1476 (2021). https://doi.org/10.1038/s41390-020-1101-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-1101-5
This article is cited by
-
Electrocardiographic Findings in Genotype-Positive and Non-sarcomeric Children with Definite Hypertrophic Cardiomyopathy and Subclinical Variant Carriers
Pediatric Cardiology (2023)
-
Exercise and Sports Participation in Children with Cardiomyopathy: A Review
Current Treatment Options in Cardiovascular Medicine (2023)
