
Abstract
Background
Kawasaki disease (KD) is the most prevailing cause of acquired heart disease in children, due to permanent coronary artery damage. Recently, the role of long noncoding RNAs (lncRNAs) in human diseases has been highlighted. However, the role of lncRNA SOCS2 antisense RNA 1 (SOCS2-AS1) on the function of human umbilical vein endothelial cells (HUVECs) in KD remains elusive.
Methods
SOCS2-AS1 expression was examined via RT-qPCR. CCK-8, EdU, caspase-3 activity, flow cytometry and TUNEL assays were conducted for exploring the function of SOCS2-AS1 in HUVECs of KD. The interaction among RNAs (SOCS2-AS1, miR-324-5p and CUEDC2) was validated via luciferase reporter, RIP and RNA pull-down assays.
Results
SOCS2-AS1 was highly expressed in serum and tissues of KD patients. SOCS2-AS1 depletion repressed the proliferation of HUVECs, whereas it facilitated apoptosis. Further, SOCS2-AS1 could bind with miR-324-5p and negatively regulated miR-324-5p expression in HUVECs. Besides, CUE domain containing 2 (CUEDC2) was the downstream target of miR-324-5p, and SOCS2-AS1 could release CUEDC2 expression via sponging miR-324-5p in HUVECs. Furthermore, downregulating miR-324-5p or upregulating CUEDC2 could rescue the progression of HUVECs restrained by SOCS2-AS1 knockdown.
Conclusions
SOCS2-AS1 upregulates CUEDC2 via inhibiting miR-324-5p to promote the progression of HUVECs in KD, providing new insights for KD treatment.
Impact
-
SOCS2-AS1 is highly expressed in the serum of KD patients.
-
SOCS2-AS1 contributes to cell proliferation in HUVECs of KD through elevating CUEDC2 expression by sequestering miR-324-5p.
-
SOCS2-AS1/miR-324-5p/CUEDC2 axis exerts a progression-facilitating function in KD.
-
These findings suggest SOCS2-AS1 as a novel potential target for KD treatment.
Similar content being viewed by others


Introduction
Kawasaki disease (KD) is also called Kawasaki syndrome. Mucocutaneous lymph node syndrome was first discovered by a Japanese man named Tomisaku Kawasaki in 1967.1,2 Kawasaki disease is described as an acquired cardiac disease with unknown etiology, mainly occurring in infants and young children. It has been reported that vascular injury is the predominant cause of KD incidence and mortality.3,4 As a self-limited childhood systemic vasculitis, KD is featured with a particular preference for the coronary arteries. If not treated, a quarter of children may develop KD-induced abnormality of coronary arteries.5 In spite of recent optimal therapies, approximately 5−15% of children with KD may develop perpetual vascular injury. However, the knowledge of the molecular mechanisms underlying KD is limited.
Defined as a subclass of noncoding RNAs (ncRNAs) with more than 200 bases in length but without protein-coding capacity,6 long noncoding RNAs (lncRNAs) have been investigated in diverse human diseases. Researchers have revealed the regulatory role of lncRNAs in a variety of physiological processes, including cell proliferation, apoptosis as well as inflammation.7 Increasing evidence has proved that abnormally expressed lncRNAs elicit pivotal effects on the initiation and progression of diverse human diseases. For instance, LINC00341/miR-214/FOXO4 feedback loop regulates the proliferation and migration of vascular smooth muscle cells in atherosclerosis.8 LncRNA AK088388 facilitates autophagy by sponging miR-30a to modulate cardiomyocyte damage.9 Although lncRNA SOCS2 antisense RNA 1 (SOCS2-AS1) has been uncovered as a key regulator in prostate cancer,10 little is known about its underlying effect on the progression of human umbilical vein endothelial cells (HUVECs) in KD.
MicroRNAs (miRNAs) have been demonstrated to play a part in multiple diseases. For instance, serum miR-186 induces endothelial cell apoptosis by targeting SMAD6 in Kawasaki disease.11 MiR-451a might act as a serum diagnosis marker for endometriosis.12 Upregulation of miR-34a impairs cognition in Alzheimer’s disease.13 Inhibiting miR-29b is related to airway inflammation in chronic obstructive pulmonary disease.14 Additionally, miR-324-5p protects against endothelial progenitor cell injury induced by oxidative stress through modulating Mtfr1.15 Overexpression of miR-324-5p suppresses cell proliferation and invasion in colorectal cancer via targeting ELAVL1,16 and miR-324-5p obstructs replication of H5N1 virus via regulating viral PB1 and host CUEDC2.17 More importantly, miR-324-5p was the potential downstream target of SOCS2-AS1 in our research. Nonetheless, whether the interaction between SOCS2-AS1 and miR-324-5p affected the activities of HUVECs remained to be investigated.
In this study, we applied a series of in vitro assays, including functional assays and molecular mechanism assays, to explore the potential function and regulatory mechanism of SOCS2-AS1 in HUVECs of KD.
Materials and methods
Clinical specimens
The serum samples of 48 KD patients (24 males and 24 females; mean age: 2.05 years old) and 30 healthy people (18 males and 12 females; mean age: 1.61 years old) as normal control were obtained from Yancheng Maternal and Child Health Care Hospital and stored at −80 °C. All 48 KD patients fulfilled with the diagnostic criteria of KD. Patients with KD who received the specific treatments before study were excluded. The control group consisted of 30 healthy children who underwent physical examination in the Child Health Care of our hospital at the same time. They were matched for age and sex with the study patients. The healthy control group was excluded from KD, cardiovascular disease and other related medical history after asking clinical history and conducting physical and laboratory examinations. This study was conducted with the ethical approval from Ethics Committee of the Yancheng Maternal and Child Health Care Hospital (approval number: KY2019032) and the written informed consents of all participants.
Cell lines
Human umbilical vein endothelial cells (Cambrex, Walkersville, MD) were cultivated in M199 medium at 37 °C in 95% air and 5% CO2. Human umbilical vein endothelial cells were incubated with patients’ serum as the vascular inflammation model of KD.18,19,20 Ten percent fetal bovine serum (Invitrogen, Carlsbad, CA), 1% of penicillin/streptomycin (Invitrogen) and 20 ng/mL of bovine endothelial cell growth supplement were added to the medium for cell culture.
RNA isolation and real-time quantitative PCR (RT-qPCR)
Human umbilical vein endothelial cells were plated in six-well plates for extracting total RNA samples and synthesizing cDNA using TRIzol (Invitrogen) and Oligo dT (Invitrogen) as per the standard procedures. The first-strand cDNA was reversely transcribed by Super-Script First-Strand cDNA System (Invitrogen), and then amplified by SYBR Green qPCR SuperMix-UDG (Invitrogen). Thermal cycling and fluorescence detection were conducted on StepOnePlus™ real-time PCR System (Applied Biosystems, Foster City, CA), followed by analysis via 2−ΔΔCT method.
Transfection
Human umbilical vein endothelial cells at about 80% cell density were seeded into six-well plates for 48 h of transfection using Lipofectamine 2000 (Invitrogen). The short hairpin RNAs (shRNAs) against SOCS2-AS1 (sh-SOCS2-AS1#1/2) and negative control (sh-NC), miR-324-5p mimics and NC mimics, pcDNA3.1/CUEDC2 and negative control (pcDNA3.1), miR-324-5p inhibitor and NC inhibitor, these transfection plasmids were all procured from Genepharma (Shanghai, China).
Cell counting kit-8 (CCK-8)
5 × 103 HUVECs were treated with 10 μL of CCK-8 (Dojindo Laboratories, Kumamoto, Japan) in the 96-well culture plate for 3 h. Microplate reader was employed to measure the absorbance at 450 nm for cell viability.
EdU staining assay
Human umbilical vein endothelial cells were first incubated with the EdU kit (Ribobio, Guangzhou China), and then mixed with the 4% paraformaldehyde and 0.5% Troxin X-100 and 1× Apollo® 488 fluorescent solution, in succession. Following 4′,6-diamidino-2-phenylindole (DAPI) staining, the stained cells were subjected to fluorescent microscope (Leica, Wetzlar, Germany).
Caspase-3 activity detection
Caspase-3 activity kit (Solarbio, Beijing, China) was utilized for this assay. The protein samples of HUVECs were mixed with the reaction buffer and caspase-3 substrate for 4 h in the 96-well culture plates, followed by detection by microplate reader at 405 nm.
Flow cytometry analysis of cell apoptosis
The transfected HUVECs were reaped after 48 h and rinsed in cold phosphate buffer saline, followed by culturing in 1× Binding Buffer containing Annexin V-labeled with fluorescein isothiocyanate and propidium iodide solution (BD Biosciences, San Jose, CA) for 15 min. The apoptosis rate was assayed by flow cytometry.
TUNEL staining assay
Transfected HUVECs were subjected to dUTP-end labeling solution (Clontech, Mountain View, CA) and DAPI staining after fixation and permeabilization with 1% formaldehyde and 0.2% Triton X-100. The stained HUVECs were observed by fluorescence microscope (NIKON, Tokyo, Japan).
Subcellular fraction assay
Subcellular fraction assay in HUVECs was achieved by PARIS™ Kit (Invitrogen) to isolate nuclear and cytoplasmic RNAs based on the user manual. After lysing in cell fraction buffer, cells were centrifuged for obtaining the supernatant (cytoplasmic fraction). Cell disruption buffer was used to incubate the remaining pellets for acquiring the nuclear fraction. RNA in nucleus or cytoplasm was analyzed through RT-qPCR.
RNA pull-down
Pierce Magnetic RNA-Protein Pull-Down Kit (Thermo Fisher Scientific, Waltham, MA) was procured for this study. Protein extracts from HUVECs were incubated with the biotin-labeled SOCS2-AS1 biotin probe or control SOCS2-AS1 no-biotin probe, along with the magnetic beads for 1 h. RT-qPCR was conducted for determining the RNA enrichment in the pull-down complexes.
Dual-luciferase reporter assay
The reporter vectors pmirGLO-SOCS2-AS1/CUEDC2-WT (wild-type) were constructed by inserting the full-length SOCS2-AS1 or CUEDC2 3′UTR containing the predictive miR-324-5p binding sites to the pmirGLO Dual-Luciferase Vectors (Promega, Madison, WI). The reporter vector pmirGLO-SOCS2-AS1/CUEDC2-Mut (mutant) was cloned by using the mutant sequences of SOCS2-AS1/CUEDC2 3′UTR containing the point mutations of miR-324-5p binding sites. Human umbilical vein endothelial cells were cotransfected with the SOCS2-AS1-WT/Mut or CUEDC2-WT/Mut and indicated transfection plasmids for 48 h. After that, the luciferase activities were analyzed by Dual-Luciferase Reporter Assay System (Promega).
RNA immunoprecipitation (RIP)
Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore, Bedford, MA) was procured for RIP assay. 1 × 107 HUVECs were lysed in RIP lysis buffer and then cultivated with the anti-Ago2 or anti-IgG (Millipore) as the negative control. The RNAs recovered by beads were analyzed by RT-qPCR.
Statistical analysis
SPSS Vision 19.0 (SPSS, Chicago, IL) was applied for statistical analysis in this study with Student’s t test and one-way analysis of variance (ANOVA). All results of at least three independent experiments were exhibited as the mean ± SD. The group difference was thought as significant with the p value < 0.05.
Results
SOCS2-AS1 is highly expressed in the serum of KD patients and SOCS2-AS1 knockdown represses the progression of HUVECs
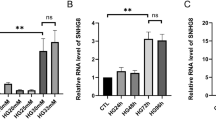
In comparison with the serum from healthy volunteers, SOCS2-AS1 was highly expressed in the serum of KD patients (Fig. 1a, n = 3). Besides, SOCS2-AS1 expression was much higher in tissues from KD patients with coronary aneurysms (CA) than in those from KD patients without coronary aneurysms (N-CA) (Fig. S1a, n = 3). To analyze the biological function of SOCS2-AS1 on the progression of HUVECs in KD, we first silenced SOCS2-AS1 in HUVECs by transfection with sh-SOCS2-AS1#1/2, and SOCS2-AS1 expression was obviously decreased under such conditions (Fig. 1b, n = 3). Given that sh-SOCS2-AS1#1 presented with better knockdown efficiency in HUVECs, it was utilized for further analysis. Subsequent CCK-8 and EdU assays revealed that SOCS2-AS1 knockdown significantly suppressed the proliferation of HUVECs, as downregulating SOCS2-AS1 remarkably reduced cell viability and the proportion of EdU positive cells (Fig. 1c, d, n = 4). Moreover, decreased expression of SOCS2-AS1 in HUVECs led to a notable elevation in the caspase-3 activity (Fig. 1e, n = 4), suggesting that SOCS2-AS1 depletion could facilitate cell apoptosis. Similarly, flow cytometry and TUNEL assays testified that silenced SOCS2-AS1 gave rise to the enhanced capability of cell apoptosis (Fig. 1f, g, n = 4). In sum, SOCS2-AS1 expression is dramatically upregulated in the serum of KD patients and SOCS2-AS1 deficiency impairs the progression of HUVECs.

a SOCS2-AS1 expression in the serum from patients with KD (n = 48) and healthy volunteers (n = 30) was detected via RT-qPCR (in triplicate, n = 3). b SOCS2-AS1 expression in HUVECs transfected with sh-SOCS2-AS1#1/2 or sh-NC was examined by RT-qPCR (in triplicate, n = 3). c, d Cell proliferation ability in HUVECs transfected with sh-SOCS2-AS1#1 or sh-NC was analyzed via CCK-8 and EdU assays (n = 4). e–g Cell apoptosis ability in transfected cells was analyzed after applying caspase-3 activity, flow cytometry and TUNEL analyses (n = 4). Error bars represent the mean ± SD of at least three independent experiments. **P < 0.01.
SOCS2-AS1 directly targets miR-324-5p in HUVECs
To explore the molecular mechanism of SOCS2-AS1 underlying KD, we first employed subcellular fractionation assay to determine the subcellular distribution of SOCS2-AS1 in HUVECs. The result demonstrated that SOCS2-AS1 was majorly scattered in cytoplasm of HUVECs (Fig. 2a, n = 3), indicating the posttranscriptional regulation potential of SOCS2-AS1 in HUVECs. Thus, we hypothesized that SOCS2-AS1 might act as a competing endogenous RNA (ceRNA) in HUVECs. After searching lncRNASNP2 (http://bioinfo.life.hust.edu.cn/lncRNASNP/#!/) and DIANA tools (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2/index-predicted), we obtained 14 miRNAs predicted to have the capacity to bind with SOCS2-AS1 (Fig. 2b). Besides, 14 miRNAs (miR-942-5p, miR-324-5p, miR-3158-5p, miR-4763-5p, miR-4722-3p, miR-6867-3p, miR-6892-3p, miR-6784-3p, miR-4423-5p, miR-4469, miR-3154, miR-6840-3p, miR-6804-5p and miR-6867-5p) were exhibited in Fig. 2c. Later on, only miR-324-5p was uncovered to be markedly enriched in SOCS2-AS1 biotin probe group through conducting RNA pull-down assay (Fig. 2d, n = 3). Afterwards, the binding sites between SOCS2-AS1 and miR-324-5p were speculated via lncRNASNP2 website (Fig. 2e). The efficiency of miR-324-5p overexpression was validated and miR-324-5p expression was remarkably upregulated in miR-324-5p mimics transfected HUVECs (Fig. 2f, n = 3). Afterwards, luciferase reporter assay detected that the luciferase activity of SOCS2-AS1-WT was signally reduced by miR-324-5p upregulation, whereas that of SOCS2-AS1-Mut was not affected (Fig. 2g, n = 4). In addition, the expression of miR-324-5p was remarkably increased after knocking down SOCS2-AS1 in HUVECs (Fig. 2h, n = 3). To sum up, SOCS2-AS1 binds with miR-324-5p and negatively regulates miR-324-5p expression in HUVECs.

a The subcellular distribution of SOCS2-AS1 in HUVECs was determined by subcellular fractionation (in triplicate, n = 3). b, c Total 14 miRNAs predicted to have the capacity to bind with SOCS2-AS1 were illustrated after utilizing lncRNASNP2 and DIANA tools. d The binding capacity between SOCS2-AS1 and these miRNAs was tested through RNA pull-down assay (in triplicate, n = 3). e The binding sites between SOCS2-AS1 and miR-324-5p predicted by lncRNASNP2 were displayed. f The efficiency of miR-324-5p overexpression in HUVECs was evaluated by RT-qPCR (in triplicate, n = 3). g The interaction between SOCS2-AS1 and miR-324-5p was verified via luciferase reporter assay (n = 4). h RT-qPCR was utilized to measure the expression of miR-324-5p in transfected cells (in triplicate, n = 3). Error bars represent the mean ± SD of at least three independent experiments. **P < 0.01.
SOCS2-AS1 elevates CUEDC2 expression by sponging miR-324-5p in HUVECs
To make further exploration of ceRNA mechanism of SOCS2-AS1 in HUVECs, we utilized miRWalk (http://mirwalk.umm.uni-heidelberg.de/) and found that CUEDC2 and RAN had the binding potential with miR-324-5p (Fig. 3a). After miR-324-5p was overexpressed in HUVECs, a conspicuous decline was observed in the expression of CUEDC2 whereas no clear changes could be noted in that of RAN (Fig. 3b, n = 3). Besides, SOCS2-AS1 depletion resulted in the notably decreased expression of CUEDC2 in HUVECs (Fig. 3c, n = 3). Moreover, the binding sites between CUEDC2 and miR-324-5p were predicted via TargetScan (http://www.targetscan.org/vert_72/) (Fig. 3d). Then, RT-qPCR detected a significant increase in the expression of CUEDC2 after transfecting HUVECs with pcDNA3.1/CUEDC2 (Fig. 3e, n = 3). Subsequently, luciferase reporter assay demonstrated that overexpression of CUEDC2 could reverse the inhibitive effect of miR-324-5p overexpression on the luciferase activity of SOCS2-AS1-WT, but not that of SOCS2-AS1-Mut (Fig. 3f, n = 4). It was then revealed through RIP assay that three RNA molecules, including SOCS2-AS1, miR-324-5p and CUEDC2, were all differentially enriched in anti-Ago2 group (Fig. 3g, n = 3), suggesting that they coexisted in RNA-induced silencing complexes. Subsequently, miR-324-5p expression was downregulated by miR-324-5p inhibitor in HUVECs (Fig. 3h, n = 3). Furthermore, the suppressive effect induced by SOCS2-AS1 deficiency on CUEDC2 expression was countervailed by miR-324-5p inhibitor (Fig. 3i, n = 3). Taken together, SOCS2-AS1 increases CUEDC2 expression by competitively binding with miR-324-5p in HUVECs.

a CUEDC2 and RAN were predicted to be able to bind with miR-324-5p after searching the miRWalk database. b The expression of CUEDC2 and RAN in HUVECs transfected with miR-324-5p mimics or NC mimics was detected via RT-qPCR (in triplicate, n = 3). c The expression of CUEDC2 in HUVECs transfected with sh-SOCS2-AS1#1 or sh-NC was examined via RT-qPCR (in triplicate, n = 3). d The binding sites between CUEDC2 and miR-324-5p were predicted via TargetScan. e The efficiency of CUEDC2 overexpression in HUVECs was assessed by RT-qPCR (in triplicate, n = 3). f, g The interactions among the three RNA molecules (SOCS2-AS1, miR-324-5p and CUEDC2) were testified by luciferase reporter (n = 4) and RIP assays (in triplicate, n = 3). h The efficiency of miR-324-5p inhibition was analyzed by RT-qPCR (in triplicate, n = 3). i RT-qPCR was applied to detect CUEDC2 expression in different groups (in triplicate, n = 3). Error bars represent the mean ± SD of at least three independent experiments. **P < 0.01.
SOCS2-AS1 facilitates proliferation but inhibits apoptosis of HUVECs by targeting miR-324-5p/CUEDC2 axis
After understanding the ceRNA mechanism of SOCS2-AS1 in HUVECs, we proceeded to test the effect of this regulatory mechanism on the proliferation and apoptosis of HUVECs in KD. As illustrated in Fig. 4a, b (n = 4), inhibited expression of miR-324-5p or increased expression of CUEDC2 led to a reverse in the restraining effect of SOCS2-AS1 silence on the proliferation ability of HUVECs. Furthermore, the enhanced caspase-3 activity induced by silenced SOCS2-AS1 could be offset by downregulating miR-324-5p or upregulating CUEDC2 in HUVECs (Fig. 4c, n = 4). Similarly, flow cytometry and TUNEL assays testified that the promoting effect of SOCS2-AS1 knockdown on the apoptosis of HUVECs could be countervailed by miR-324-5p inhibition or CUEDC2 upregulation (Fig. 4d, e, n = 4). In summary, SOCS2-AS1 regulates the progression of HUVECs in KD via miR-324-5p/CUEDC2 axis.

a, b Cell proliferation ability of HUVECs transfected with different plasmids was analyzed via CCK-8 and EdU assays (n = 4). c–e The apoptosis ability of HUVECs in different groups was analyzed after utilizing caspase-3 activity, flow cytometry and TUNEL analyses (n = 4). Error bars represent the mean ± SD of at least three independent experiments. **P < 0.01.
Discussion
In recent years, researches have paid much attention to the pathogenesis of KD and revealed that ncRNAs are implicated in the occurrence and progression of KD.21,22,23,24 Moreover, the proliferation and apoptosis of HUVECs have been manifested to be related to KD formation.25,26 Also, the significance of lncRNAs in KD has been revealed previously. As an example, THRIL expression is associated with the severity of symptoms in KD patients.27 In this study, SOCS2-AS1 was highly expressed in the serum of KD patients compared with in that of healthy volunteers. In previous studies, similar in vitro models were constructed, in which HUVECs were incubated with serum samples of KD patients.18,19,20 Currently, we proved that silenced SOCS2-AS1 impaired cell proliferation whereas facilitated cell apoptosis in HUVECs, suggesting the promoting role of SOCS2-AS1 in KD progression. In the reported studies, SOCS2-AS1 inhibited cell apoptosis and enhanced castration-resistance in prostate cancer.10 Our study was the first to uncover the regulatory function of SOCS2-AS1 in HUVECs of KD.
Based on previous investigations, transcriptional and posttranscriptional regulation have been accepted as the primary molecular mechanisms of lncRNAs in human diseases.28,29 In this research, owing to the finding that SOCS2-AS1 was majorly distributed in cytoplasm of HUVECs, we speculated that SOCS2-AS1 might act as a ceRNA in HUVECs by sponging certain miRNA. More importantly, the implication of miRNAs in KD has been reported. For instance, miR-186 overexpression induces the apoptosis of HUVECs in KD through activating MAPK via inhibiting SMAD6.11 Existing evidence has validated that miR-324-5p elicits suppressive effects on the progression of some cancers, such as gastric cancer,30 cervical cancer31 and colorectal cancer.16 However, the role of miR-324-5p in KD has not been illustrated before. In the current study, we validated that miR-324-5p could bind with SOCS2-AS1 and its expression was negatively regulated by SOCS2-AS1 in HUVECs.
CUE domain containing 2 (CUEDC2) has been researched and verified to be a promising biomarker in serous ovarian carcinoma32,33 and colorectal cancer.34 This study confirmed that CUEDC2 was a direct target of miR-324-5p in HUVECs. More interestingly, the interaction between miR-324-5p and CUEDC2 has been validated in H5N1 virus infection.17 Additionally, SOCS2-AS1 could regulate CUEDC2 expression by competitively binding with miR-324-5p in HUVECs. Final rescue assays depicted that miR-324-5p inhibition or CUEDC2 overexpression could reverse the effects of SOCS2-AS1 knockdown on the proliferation and apoptosis of HUVECs.
All in all, SOCS2-AS1 promotes the progression of HUVECs in KD through sponging miR-324-5p to upregulate CUEDC2. This finding provides new clues for further exploration of the relation between lncRNA and KD development, suggesting a novel molecular marker for the treatment of KD. Additionally, present findings need to be supported by animal experiments, and the upstream regulatory mechanism of SOCS2-AS1 in KD and need to be further researched in the future.
References
Makino, N. et al. Descriptive epidemiology of Kawasaki disease in Japan, 2011-2012: from the results of the 22nd nationwide survey. J. Epidemiol. 25, 239–245 (2015).
Singh, S., Vignesh, P. & Burgner, D. The epidemiology of Kawasaki disease: a global update. Arch. Dis. Child. 100, 1084–1088 (2015).
Newburger, J. W., Takahashi, M. & Burns, J. C. Kawasaki disease. J. Am. Coll. Cardiol. 67, 1738–1749 (2016).
Chen, K. Y. et al. Kawasaki disease and cardiovascular risk: a comprehensive review of subclinical vascular changes in the longer term. Acta Paediatr. 105, 752–761 (2016).
McCrindle, B. W. et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation 135, e927–e999 (2017).
Dey, B. K., Mueller, A. C. & Dutta, A. Long non-coding RNAs as emerging regulators of differentiation, development, and disease. Transcription 5, e944014 (2014).
Yang, B. et al. Down-regulation of the long noncoding RNA-HOX transcript antisense intergenic RNA inhibits the occurrence and progression of glioma. J. Cell Biochem. 119, 2278–2287 (2018).
Liu, X. et al. Long noncoding RNA LINC00341 promotes the vascular smooth muscle cells proliferation and migration via miR-214/FOXO4 feedback loop. Am. J. Transl. Res. 11, 1835–1842. (2019).
Wang, J. J., Bie, Z. D. & Sun, C. F. Long noncoding RNA AK088388 regulates autophagy through miR-30a to affect cardiomyocyte injury. J. Cell Biochem. 120, 10155–10163 (2019).
Misawa, A., Takayama, K., Urano, T. & Inoue, S. Androgen-induced long noncoding RNA (lncRNA) SOCS2-AS1 promotes cell growth and inhibits apoptosis in prostate cancer cells. J. Biol. Chem. 291, 17861–17880 (2016).
Wu, R. et al. miR186, a serum microRNA, induces endothelial cell apoptosis by targeting SMAD6 in Kawasaki disease. Int. J. Mol. Med. 41, 1899–1908 (2018).
Nothnick, W. B. et al. Serum miR-451a levels are significantly elevated in women with endometriosis and recapitulated in baboons (Papio anubis) with experimentally-induced disease. Reprod. Sci. 24, 1195–1202 (2017).
Sarkar, S. et al. Over-expression of miR-34a induces rapid cognitive impairment and Alzheimer’s disease-like pathology. Brain Res. 1721, 146327 (2019).
Tang, K., Zhao, J., Xie, J. & Wang, J. Decreased miR-29b expression is associated with airway inflammation in chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell Mol. Physiol. 316, L621–L629 (2019).
Chen, P. et al. miR-324-5p protects against oxidative stress-induced endothelial progenitor cell injury by targeting Mtfr1. J. Cell Physiol. 234, 22082–22092 (2019).
Gu, C., Zhang, M., Sun, W. & Dong, C. Upregulation of miR-324-5p inhibits proliferation and invasion of colorectal cancer cells by targeting ELAVL1. Oncol. Res. 27, 515–524 (2019).
Kumar, A. et al. MicroRNA hsa-miR-324-5p suppresses H5N1 virus replication by targeting the viral PB1 and host CUEDC2. J. Virol. 92, e01057-18 (2018).
Jia, C. et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 10, 778 (2019).
Rong, X. et al. miR-27b suppresses endothelial cell proliferation and migration by targeting Smad7 in Kawasaki disease. Cell Physiol. Biochem. 48, 1804–1814 (2018).
Ueno, K. et al. Disruption of endothelial cell homeostasis plays a key role in the early pathogenesis of coronary artery abnormalities in Kawasaki disease. Sci. Rep. 7, 43719 (2017).
Ko, T. M. et al. Genome-wide transcriptome analysis to further understand neutrophil activation and lncRNA transcript profiles in Kawasaki disease. Sci. Rep. 9, 328 (2019).
Lin, Z. et al. Long noncoding RNA gastric cancer-related lncRNA1 mediates gastric malignancy through miRNA-885-3p and cyclin-dependent kinase 4. Cell Death Dis. 9, 607 (2018).
Chen, R. et al. Quantitative proteomics reveals that long non-coding RNA MALAT1 interacts with DBC1 to regulate p53 acetylation. Nucleic Acids Res. 45, 9947–9959 (2017).
Chu, M. et al. Bone marrow-derived microRNA-223 works as an endocrine genetic signal in vascular endothelial cells and participates in vascular injury from Kawasaki disease. J. Am. Heart Assoc. 6, e004878 (2017).
Li, Z. et al. A plasma mir-125a-5p as a novel biomarker for Kawasaki disease and induces apoptosis in HUVECs. PLoS ONE 12, e0175407 (2017).
Jiang, C. et al. TNF-alpha induces vascular endothelial cells apoptosis through overexpressing pregnancy induced noncoding RNA in Kawasaki disease model. Int. J. Biochem. Cell Biol. 72, 118–124 (2016).
Li, Z. et al. The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL. Proc. Natl. Acad. Sci. USA 111, 1002–1007 (2014).
Yang, S. & Sun, J. LncRNA SRA deregulation contributes to the development of atherosclerosis by causing dysfunction of endothelial cells through repressing the expression of adipose triglyceride lipase. Mol. Med. Rep. 18, 5207–5214 (2018).
Yari, M. et al. Association between long noncoding RNA ANRIL expression variants and susceptibility to coronary artery disease. Int. J. Mol. Cell Med. 7, 1–7 (2018).
Lin, H. et al. MiR-324-5p reduces viability and induces apoptosis in gastric cancer cells through modulating TSPAN8. J. Pharm. Pharm. 70, 1513–1520 (2018).
Jiang, H. et al. Long non-coding RNA TPT1-AS1 promotes cell growth and metastasis in cervical cancer via acting AS a sponge for miR-324-5p. J. Exp. Clin. Cancer Res. 37, 169 (2018).
Wang, A. et al. Overexpression of CUEDC2 predicts poor prognosis in ovarian serous carcinomas. J. Cancer 6, 542–547 (2015).
Wang, A. et al. CUEDC2 contributes to cisplatin-based chemotherapy resistance in ovarian serious carcinoma by regulating p38 MAPK signaling. J. Cancer 10, 1800–1807 (2019).
Hu, S. et al. Expression of CUEDC2 in colorectal cancer with different invasion and migration abilities. J. Int. Med. Res. 47, 905–914 (2019).
Acknowledgements
We thank the contributions from all participators in this research.
Author information
Authors and Affiliations
Contributions
Q.Z. designed the subject and contributed to the manuscript. D.C. performed and analyzed all experiments. Throughout the project, all authors contributed intellectually. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was conducted with the ethical approval from the Ethics Committee of the Yancheng Maternal and Child Health Care Hospital (approval number: KY2019032) and the written informed consents of all participants.
Informed consents
This study was conducted with the written informed consents of all participants.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhao, J., Chen, D. Kawasaki disease: SOCS2-AS1/miR-324-5p/CUEDC2 axis regulates the progression of human umbilical vein endothelial cells. Pediatr Res 92, 388–395 (2022). https://doi.org/10.1038/s41390-020-1029-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-1029-9
This article is cited by
-
LncRNAs in Kawasaki disease and Henoch-Schönlein purpura: mechanisms and clinical applications
Molecular and Cellular Biochemistry (2023)
