
Abstract
Bronchopulmonary dysplasia (BPD) is the most prevalent chronic lung disease in infants and presents as a consequence of preterm birth. Due to the lack of effective preventive and treatment strategies, BPD currently represents a major therapeutic challenge that requires continued research efforts at the basic, translational, and clinical levels. However, not all very low birth weight premature babies develop BPD, which suggests that in addition to known gestational age and intrauterine and extrauterine risk factors, other unknown factors must be involved in this disease’s development. One of the main goals in BPD research is the early prediction of very low birth weight infants who are at risk of developing BPD in order to initiate the adequate preventive strategies. Other benefits of determining the risk of BPD include providing prognostic information and stratifying infants for clinical trial enrollment. In this article, we describe new opportunities to address BPD’s complex pathophysiology by identifying prognostic biomarkers and develop novel, complex in vitro human lung models in order to develop effective therapies. These therapies for protecting the immature lung from injury can be developed by taking advantage of recent scientific progress in -omics, 3D organoids, and regenerative medicine.
Similar content being viewed by others


Background
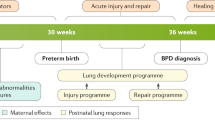
Bronchopulmonary dysplasia (BPD) is the most prevalent chronic lung disease in infants and presents as a consequence of preterm birth. First described in 1967,1 its definition and pathophysiology have changed over the past two decades due to the development of new ventilation strategies, advances in neonatal care, surfactant therapy, and use of prenatal steroids. Currently, the most accepted diagnostic criteria dictates a severity assessment at 36 weeks of postmenstrual age, which is too late to implement an efficient treatment strategy2 (Fig. 1).

Timeline of bronchopulmonary dysplasia (BPD) development, since birth until 36 weeks gestational age. At preterm birth (23–28 weeks) the lung is still immature, at the sacular stage of development. During the first month the premature lung is damaged by postnatal neonatal intensive care unit (NICU)-related factors that increase the risk of developing BPD. At 36 weeks postmenstrual age the damage is already established. Therefore, the optimal time for preventive treatment with mesenchymal stromal cells (MSC) is in the first 2 weeks of life
BPD incidence is directly correlated with the degree of infant immaturity, and this may explain why improvements in the survival of extremely low birth weight infants seem to correlate with an increased BPD incidence despite the development of new therapeutic approaches.3,4,5 However, BPD rates show wide variability with incidence values oscillating between 15 and 50% in infants <1500 g.6
Current prevention strategies such as antenatal steroid therapy, surfactant administration, less invasive respiratory strategies, lower oxygen saturation targets, vitamin A, methylxanthines, macrolides, inhaled nitric oxide, erythropoietin, and/or diet supplements have not been able to diminish the increasing BPD incidence (Table 1).7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22 In addition, many therapies (diuretics, bronchodilators, postnatal corticosteroids, patent ductus arteriosus (PDA) treatment, improved nutrition, and fluid management) currently used for BPD treatment remain controversial due to their side effects and a lack of evidence (Table 2).13,15,16,18,23,24
BPD is no longer a disease limited to the perinatal period. Recent data have shown that premature babies will have not only respiratory morbidities in their childhood and adult life (decreased lung function, increased incidence of emphysema, higher risk of wheezing) but also present decreased right ventricular function, increased cardiovascular risk, and higher incidence of arterial hypertension in adolescence and adulthood.24,25,26,27 In addition, BPD is associated with retinopathy of prematurity and neurological morbidities like developmental delay and cerebral palsy.28,29
BPD currently represents a major therapeutic challenge that requires continued research efforts at the basic, translational, and clinical levels. However, not all very low birth weight premature babies develop BPD, which suggests that in addition to known gestational age and intrauterine and extrauterine risk factors, other unknown factors must be implicated in the development of this disease. In this review, we will discuss the actual and future challenges in the field.
BPD: a lung developmental problem
The first description of BPD was based on the clinical and histological evaluation of premature babies with a mean birth weight of 1660 g and 31 weeks of gestational age who were dying from this complication.1 The pathology of the so-called old BPD was characterized by alternating areas of alveolar emphysema and atelectasis, marked inflammation, fibrosis, prominent airway injury, and smooth muscle hyperplasia. These chronic lesions were thought to be secondary to mechanical ventilation and oxygen toxicity. Advances in neonatal critical care and increased survival of extremely premature infants have modified the physiopathology of BPD to a new disease of predominantly disrupted lung development with less lung injury.4
The lungs of very low birth weight infants (VLBWIs) at birth are in the saccular or alveolar phase of their airway branching morphogenesis after assuming a survival limit of 24 postmenstrual weeks gestation. During the first weeks of life, these infants are exposed to harmful stimuli that can alter lung development16 (Fig. 1). The pathology of this new BPD is characterized by a constant reduction in alveolarization with enlarged air spaces and a highly variable degree of inflammation and fibrosis.28 Evidence suggests that vascular development and alveolarization are closely related, which explains the simplified lung vasculature found in clinical and experimental BPD. Lung capillaries are reduced in number, dysmorphic, abnormally distributed, present a decrease in branching, and are separated from the air surface.29
This new BPD is considered a multi-factorial disease in which the combination of prenatal and postnatal risk factors and lung developmental arrest, in addition to an unknown role of genetics, appear to be responsible for BPD development (Fig. 2). Prenatal factors contributing to BPD include placental abnormalities (preeclampsia, maternal smoking), intrauterine growth restriction, intrauterine infections (chorioamnionitis), and multiple births in addition to others.16,30 Postnatal factors directly implicated in BPD development include mechanical ventilation and oxygen supplementation, prematurity-related comorbidities, and infections (sepsis, necrotizing enterocolitis, lung infections, and others).16,30 Cardiac and vascular factors such as ventricular dysfunction, intracardiac shunts (both of which generate a lung volume overload), or pulmonary vein stenosis also play an important role in BPD development.31,32,33

Risk factors for bronchopulmonary dysplasia (BPD). ASD atrial septal defect, PDA patent ductus arteriosus
The challenge of predicting BPD in preterm newborns
Considering the lack of effective treatments for BPD and a diagnosis that is established at 36 weeks postmenstrual age,2 the main goal in this area is to prevent BDP development by identifying those VLBWI at higher risk of developing BPD and to implement early postnatal strategies in these infants (Fig. 1). Other benefits of determining the risk of BPD include providing prognostic information and stratifying infants for clinical trial enrollment.30
Gestational age, birth weight, gender, clinical parameters (PDA, respiratory distress syndrome, pulmonary interstitial emphysema, and others), chest X-rays, ventilator settings, oxygenation indexes, and blood gases, as well as other risk factors for BPD have been used to create these clinical predictive models. Despite these efforts, a reliable and reproducible model that could be easily applicable across neonatal intensive care units has not yet been found.
In 2013, Onland et al.34 published the first systematic review and external validation of the 26 most relevant clinical predictive models of BPD in preterm infants >30 years. Eighteen were derivation models (such as studies developing a novel prediction model), and eight were validation studies (such as studies evaluating a single predictor or a known model). The external validation analysis showed that most prediction models do not perform sufficiently well to be considered part of routine care.
Finding an appropriate prediction model or risk score that can establish the risk of presenting BPD at an early stage of the disease in the first week of life prior to the start of the disease or even at birth has been and still is a major challenge in BPD. Many clinical prediction models have been developed and published over the past few decades.35,36,37,38 However, despite these efforts, a reliable and reproducible model that could be easily applicable across neonatal intensive care units has not yet been found. Therefore, the search for predictive factors for early identification of infants at high risk of BPD is an important area of research. Additional variables such as echocardiographic parameters and/or biomarkers in addition to innovative molecular diagnosis methods will be required to increase the prediction accuracy of these models.
New tools in translational research for BPD
Animal models of BPD
Animal models play an important role in understanding disease pathogenesis and in preclinical drug development.
Both large (baboons, sheep, and pigs) and small (rabbits, rats, and mice) animals have been exposed to known risk factors for BPD (hyperoxia, perinatal inflammation, perinatal hypoxia, or mechanical ventilation).39,40,41,42 However, the majority of the BPD model studies have been performed using mice and rats delivered at term and exposed to high levels of oxygen, ranging from 40% to 100% O2.43,44 In terms of mimicking human disease, exposure to different oxygen concentrations allows induction of different severity degrees.45 However, this advantage also represents one of the main weaknesses of this model since the beneficial effects of therapeutic interventions might go unnoticed in severe hyperoxia-based BPD models (such as 85% O2), whereas small effects might be overestimated in less severe hyperoxia models (such as <60% O2).45 Researchers also need to consider that in addition to the oxygen concentration, the timing and duration of this exposure may influence the histopathological findings and long-term consequences of experimental BPD.46,47 In addition, VLBWI also suffer from periods of hypoxia, which are often not considered when developing a BPD model. Therefore, new BPD models incorporating periods of either intermittent or transient hypoxia are being developed to increase the translational potential of the hyperoxia BPD model.48,49 Finally, there is an urgent need to develop clinically relevant BPD models based on combined exposure to other injurious stimuli such as mechanical ventilation and inflammation that are involved in the development of BPDn.44,50
One of the main problems faced during BPD animal model development is the definition of BPD, which is an operational definition (patient on oxygen therapy at 28 days or 36 weeks postmenstrual age), and it does not consider the pathophysiology, which is very complex. BPD is not a single entity; it is a more extensive disease that results in alveolar simplification, severe pulmonary vascular remodeling and pulmonary hypertension, and airway problems such as tracheobronchomalacia or stenosis in airway areas secondary to fibrosis and inflammation.51 This heterogeneity in the disease is very difficult to reproduce in an animal model and, indeed, most animal models present only mild or moderate BPD rather than severe BPD.
Another major disadvantage of current BPD animal models is the lack of genetic effect reproduction, which is partially responsible for the broad spectrum of BPD and of variance for moderate-to-severe BPD.52,53,54,55
Despite these and other limitations of current BPD animal models, they are still the most appropriate tool available for investigating possible BPD-related mechanisms in addition to potential therapeutic targets and strategies. However, our ability to translate these results to the clinical setting is limited; therefore, there is a critical need for new in vitro models of human lungs to accelerate the translation of basic research findings into applicable clinical situations.
Novel human tissue-based and cell-based in vitro models for the discovery and development of new drugs in BPD
Lung development involves complex cross-talk between the different cell types present in the lung; therefore, in vitro studies can be very complex. However, in vitro assays based on whole or partial organ culture (such as the use of precision-cut lung slices) allow for the preservation of highly differentiated functions, including vascular and airway reactivity56 or ex vivo alveolarization when harvested from newborn mice.57 These parameters can be studied following in vitro exposure to relevant injurious stimuli, including mechanical stretch.58 Similarly, numerous studies have successfully applied the use of murine and human embryonic lung explants to study the role of physiological and pathophysiological mediators on airway morphogenesis;59,60,61 however, studies analyzing vascular development are scarce.62 Furthermore, with the development of three-dimensional (3D) cell culture assays, we can now generate lung organoids which are being used to analyze the impact of harmful stimuli on processes such as branching morphogenesis or fibrosis.63,64,65,66 Finally, the new micro-engineered models facilitate the development of the lung-on-a-chip model, a biomimetic microdevice that can replicate the alveolar–capillary barrier in the human lung that represents a promising tool for understanding the interactions between the different types of cells present in the lung with relevant physiological and pathophysiological stimuli67 (Fig. 3). However, these techniques are in the early stages of development, and they still need to demonstrate whether they reflect better the clinical entity than the animal models already available. In this regard, increasing evidence supports their value for studying airway morphogenesis and recapitulating pulmonary edema and lung fibrosis formation following exposure to toxic agents; however, given that disrupted vascular development is also a hallmark of BPD, efforts directed at better reassembly of the vascular portion of the lung must be a priority in this research area.

New tools in translational research for bronchopulmonary dysplasia (BPD). Integrating in vitro assays based on organ culture (such as precision-cut lung slices) with advances in pluripotent stem cell technology (notably induced pluripotent stem cells (iPSCs)), three-dimensional (3D) or multicellular devices (such as lung-on-a-chip) is critical to develop more relevant models for BPD and to accelerate the translation of basic research findings into the clinics. Traditional functional assays and candidate marker analyses should be combined with “omics” and system biology approaches in order to identify novel therapeutic targets and new diagnostic and prognostic biomarkers which allow us to identify those patients who could benefit from an early intervention. BALF bronchoalveolar lavage
All of these technologies can be applied to lung tissue obtained at BPD patient autopsies and can provide deeper insight into the pathophysiology of late-stage disease and be used to test the potential benefit of new therapeutic strategies. However, the lack of availability of lung tissue from early-stage disease and BPD survivors is a major limitation to understanding the pathophysiology of this disease and the development of new therapies. Nevertheless, the unexpected discovery that somatic cells can be reprogrammed into a pluripotent state68 has triggered the possibility for generating pluripotent stem cell-based models of human disease with unlimited applications in the development of new therapeutics and personalized medicine.
These and other stem cell-based models of cardiovascular disease are already under development. An in vitro bioassay has recently been developed in which co-cultured endothelial cells grown from stem cells in the peripheral blood (blood outgrowth endothelial cells) and peripheral blood mononuclear cells from the same donor can be used to predict the patient response to biological drugs.69
OMICS IN BPD
BDP susceptibility and outcomes are influenced by the interactions between genetic and environmental factors. This complexity may underlie the limited success in identifying prognostic biomarkers as discussed above. Thus, using an unbiased approach can identify new networks and targets that can be used to stratify BPD patients and potentially develop specific therapies.
Genomics
It is now evident that BPD has a strong hereditary compound; several strategies used to define the genetic background of BPD have failed. Thus, genome-wide association studies (GWAS) that have been successfully applied to other human diseases have been unsuccessful in BPD patients. Indeed, a study by Wang et al.70 with 1726 infants could not identify any BPD-associated single-nucleotide polymorphisms (SNPs). It could also not validate the SNP identified in previous studies.71 However, combining GWAS with other approaches seems to increase our chances of success.72 Although no single SNP achieved genomic-wide significance, in cases in which the GWAS-derived data were combined with pathway-based analysis, the authors identified two novel pathways (miR-219 and CD44) that were potentially involved in the genetic predisposition to BPD or death.72 Similarly, a recent functional genomics study of BPD patient blood samples identified rare exome mutations in the genes involved in the control of pulmonary structure and function.55
Microbiomics
Some pilot studies have attempted to characterize the lung microbiome in preterm babies, and a clinical trial to evaluate its potential association with the development of BPD is currently ongoing (NCT03229967).37 To study the active surveillance and screening of lower respiratory tract bacterial colonization in newborns admitted to the neonatal intensive care unit is challenging; thus, stratification of BPD risk is currently being examined.38 In relation to the microbiome, proinflammatory and chemotactic factors have been detected in high concentrations in infants who develop BPD and have been implied in early lung injury and systemic inflammation.39 Therefore, it is crucial to study how bacterial colonization and inflammation sustain the detrimental effects toward BPD even if the effects are independent of each other.
Transcriptomics
The characterization of genome-wide transcriptional profiling via high-throughput technologies such as microarrays or next-generation RNA sequencing technologies offer novel insight into the molecular mechanisms of normal murine and human lung development in addition to experimental models of BPD.73,74,75,76,77,78,79 However, studies using samples from patients with BPD are rare. Bhattacharya et al.79 studied altered pathways from lung tissues obtained at autopsies from BPD patients. They included both pathways previously identified in animal models (such as insulin-like growth factor (IGF) or the Sonic hedgehog (Shh) pathway) in addition to novel mechanisms of disease, including those indicating mast cell accumulation in pulmonary tissue from BPD patients.
More recently, microRNAs (miRNAs) have emerged as key players of gene regulation during lung development and in pulmonary disease pathogenesis.80,81 miRNAs are a class of small non-coding RNA with relatively few nucleotides (≈22), but that can repress multiple gene targets concurrently. A number of miRNAs are differentially expressed during lung development.82,83,84 Some studies have also analyzed the pattern of miRNA expression in clinical samples from BPD patients and identified dysregulation of the miR-17/92 cluster, miR-21, miR-206, miR-219, or Let-7f.72,85,86 A recent study has suggested that a decrease in the expression of miR-876-3p in airway exosomes might be associated with a higher risk of developing severe BPD in preterm infants.87 However, further mechanistic and validation studies are needed to confirm the potential of these miRNAs as BPD biomarkers or pathogenic mediators/therapeutic targets.
Proteomics and metabolomics
Proteomics can characterize the global expression of proteins in a biological sample; metabolomics can be used to quantify metabolites. Many studies have tried to identify single or pre-defined groups of protein biomarkers in clinical samples from patients with BPD with minimal success.88 To the best of our knowledge, there is only one study using a proteomic approach with bronchoaspiration lung fluid samples from BPD patients.89 Using this approach, an increase in several serum proteins (albumin, serotransferrin, clusterin) and a decrease in calcium signaling-related proteins (calcyphosine, calcium, and integrin-binding protein 1) was found in babies with higher risk for BPD development. Studies applying metabolomics to BPD also suggest that metabolic profiling of amniotic fluid or urine collected within 24 h of life may identify those patients at a higher risk of developing BPD.90,91 However, further studies applying bioinformatics and integrating the massive amount of data generated by -omic approaches with clinical data are needed to efficiently identify those infants at high risk of BDP development. In this regard, initiatives such as those promoted by the LungMAP consortium to create an integrated and publicly accessible molecular atlas of the developing lung will undoubtedly facilitate advancements in this area.92 In the near future, microRNA studies could complement new clinical trials and help elucidate the pathogenic mechanisms of the disease and effects of new therapies.
Innovative therapies in the prevention of BPD
Cell-based therapies For BPD
Advances in stem cell biology over the past two decades have now positioned stem cells as a possible paradigm shift in medicine. Unlike conventional drugs, cells can sense local environmental signals and then proliferate, migrate, or differentiate in response to those specific signals.93 These properties are harnessed by numerous investigators in order to test the repair potential of various stem cells in experimental models and in human diseases, including BPD.
Mesenchymal stromal cells (MSCs) are the most extensively studied cells because of their ease of isolation and culture and their pleiotropic effects (anti-inflammatory, pro-angiogenic, anti-apoptotic, anti-oxidant, and anti-fibrotic activities).94 Evidence suggests that the depletion or dysfunction of MSC in the developing lung is associated with BPD.95,96,97,98 These findings, in combination with the pleiotropic effects of MSC, offer a strong rationale for testing the therapeutic benefits of these cells in BPD.99,100
MSCs have already been used in other pediatric diseases such as graft-versus-host, lysosomal storage, and orthopedic diseases in addition to spinal muscular atrophy with promising results and safe and feasible administration.101 Allogeneic MSC therapy is feasible due to their low allergenic profile in which they express human leukocyte antigen class I but not class II.100,102
MSC and lung repair in experimental BPD
In rodent models of hyperoxia-induced lung damage, proof of concept studies with bone marrow (BM)-derived MSC administered intratracheally, intravenously, or intraperitoneally attenuate lung inflammation and minimizes oxidative stress, alveolar growth, and lung vascular damage.96,103,104,105 Furthermore, BM-derived MSC improve survival, prevent vascular growth arrest, diminish lung inflammation, inhibit lung fibrosis, and reduce pulmonary hypertension in these rodent models.99 Likewise, human umbilical cord/cord blood-derived MSC preserved and rescued lung histology and lung function.106 These cells were found to be safe (no tumor formation) in murine models and had therapeutic benefits in lung structures persisting up to 6 months;106 however, additional precautions should be taken in clinical trials in order to avoid the risk of anomalous proliferation in infants with an immature immune system, including premature newborns.106 A recent systematic review and meta-analysis has evaluated all preclinical studies with MSC in experimental BPD models.107
Interestingly, low engraftment rates (<5%) of MSC in the lung suggest that the therapeutic benefits of MSC are mediated by a paracrine mechanism.99,108 These observations are supported by experiments demonstrating that the conditioned media from MSC cultures attenuate lung injury.96,103,104,105,108,109 These findings may open many new therapeutic options including cell-free therapies based on individual active compounds or extracellular vesicles shed by MSC.109 Purified exosomes from MSC derived from human umbilical cord Wharton's jelly and basement membrane (BM) can restore lung architecture and improve lung development and function in hyperoxia-induced BPD animal models.110 Extracellular vesicles can transfer a range of biologically active compounds, including proteins, lipids, oligonucleotides, and drugs and represent a potentially safer alternative to cell-based therapies. However, the development of exosome-based therapies still present major challenges, including a lack of standardization in isolation and purification methods.107,111,112
Early phase clinical trials with MSC for BPD
In a phase I, dose-escalation trial with human umbilical cord blood-derived MSC conducted in nine infants born between 23- and 29-week gestational age, a single intra-tracheal administration within the first 2 weeks of life was shown to be feasible with no adverse effects113 (NCT01632475). A phase II trial to test the safety and efficacy of human umbilical cord blood-derived MSC for the prevention of BPD in a larger number of patients is currently underway (NCT02381366).
While more laboratory research is definitely required, the next 5 years will provide an early indication as to whether the benefits seen in animal research can be translated to clinical settings. A variety of other stem/progenitor cells are currently being investigated in lung injury animal models in addition to clinical trials of neonatal lung disease, including cord blood-derived mononuclear cells,114 endothelial progenitor cells,115 and amniotic epithelial cells.100,116
A major challenge will be the standardization of quality control for the practical manufacturing of cell products that have uniform quality and efficacy.117 Also, questions regarding the dosing, timing, and administration route will need to be answered in appropriately designed clinical trials. The introduction of new therapies in this vulnerable patient population must be accompanied by rigorous assessment of the short- and long-term outcomes. The establishment of registries of all treated patients will be imperative to ensure long-term follow-up.
Recombinant human IGF-1/recombinant human IGFBP-3
IGF-1 levels normally increase in the third trimester of pregnancy but decrease in preterm infants.118 IGF-1 is upregulated by insulin and stimulates cell proliferation, maturation, and differentiation.118 Hyperglycemia is common in preterm babies as a result of insulin resistance secondary to acute stress and relative insulin deficiency. Lower IGF-1 levels in preterm infants are associated with reduced weight gain and increased risk of chronic lung disease, retinopathy of prematurity, and necrotizing enterocolitis. Early insulin treatment during the first week of life induces a late increase in IGF-1 levels between days 7 and 28 of life, improved weight gain, and may improve the outcomes of these infants.118 Intravenous infusion of recombinant human IGF-1 complexed with its binding protein recombinant human IGFBP-3 (rhIGF-1/rhIGFBP-3) has been investigated as a therapy since IGF-1 replacement in extremely preterm infants has been demonstrated to be safe and well tolerated.119,120 In a recent phase II clinical trial, IGFBP was shown to reduce the incidence of retinopathy of prematurity with no significant reduction in the incidence of BPD in the treatment group and no safety issues.121 An extension of this trial is now ongoing (NCT02386839). A phase 2b/3 clinical trial will start at the end of 2018 and will recruit extremely preterm infants to prove the efficacy and safety of rhIGF-1/rhIGFBP-3 (NCT03253263).
IGF-1 is a potential therapy for BPD, a topic that still requires further investigation. The potential therapeutic power of IGF-1 for retinopathy of prematurity and necrotizing enterocolitis makes this treatment quite attractive for VLBWI. In addition, the absence of toxicity encourages the performance of clinical trials.
Recombinant human Clara cell 10 kDa protein
Clara cell-specific 10-kDa (0CC10) is a major secretory protein 0CC10 binds to lipid components of the pulmonary surfactant such as phosphatidylcholine and phosphatidylinositol, suggesting that it may transport or protect these phospholipids from degradation. It also negatively regulates airway inflammatory responses and also regulates surfactant phospholipids catabolism.122 The Clara cell protein (CC)10 reduces lung inflammation in premature infants with respiratory distress syndrome.123 CC10 is oxidized in premature infants who have developed BPD, suggesting that the CC10 structure and function are critical for normal bronchoalveolar fluid homeostasis.124 It is diminished in neonates with respiratory distress syndrome.122 Treatment with recombinant human CC10 (rhCC10) in a premature lamb model of respiratory distress showed a significant rhCC10 dose-dependent increase in respiratory compliance and ventilation efficiency index.125 Early intervention with rhCC10 up-regulates surfactant proteins and vascular endothelial growth factor expression. This finding suggests that CC10 can protect against hyperoxia and mechanical ventilation in the immature lung.125 Despite these promising findings in animal models, further research on the therapeutic effects of rhCC10 I in neonatal chronic lung diseases is needed. An active clinical trial is ongoing to prove the efficacy of treatment with rhCC10 in premature neonates with respiratory distress (NCT01941745).
Summary
Given the complexity and heterogeneity of BPD, cooperative research aimed to integrate animal and clinical studies is crucial to improve our ability for early detection and to develop new treatments for BPD. New diagnostic technology, “omics”, and systems biology should be increasingly applied to neonatal critical care medicine and should be combined with data from multi-centric clinical trials to develop a prognostic index to identify those babies at a higher risk of developing the disease. The combination of biological samples’ analyses (amniotic fluid, urine, blood, and bronchoaspiration fluid) in addition to these new technologies could identify those patients who will benefit from early intervention. Therefore, advances in BPD will require multidisciplinary teams, not only for clinical management and follow- up, but also to conduct research projects that can integrate the data obtained from animal models, metabolomics, proteomics, genetics, and stem cell-based therapies. These new therapies and/or predictive models will have to be tested with the proper clinical trials. Several clinical trials exploring some of these innovative therapies are underway; therefore, the old dream of preventing BPD and its sequels in extremely preterm babies could shortly become a reality
References
Northway, W. H. Jr., Rosan, R. C. & Porter, D. Y. Pulmonary disease following respiratory therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N. Engl. J. Med. 276, 357–368 (1967).
Jobe, A. H. & Bancalari, E. Bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 163, 1723–1729 (2001).
Fanaroff, A. A. et al. Trends in neonatal morbidity and mortality for very low birthweight infants. Am. J. Obstet. Gynecol. 196, 147.e141–148 (2007).
Van Marter, L. J. Epidemiology of bronchopulmonary dysplasia. Semin. Fetal Neonatal Med. 14, 358–366 (2009).
Jobe, A. H. The new bronchopulmonary dysplasia. Curr. Opin. Pediatr. 23, 167–172 (2011).
Bancalari, E., Claure, N. & Sosenko, I. R. Bronchopulmonary dysplasia: changes in pathogenesis, epidemiology and definition. Semin. Neonatol. 8, 63–71 (2003).
Schulze, A. et al. Proportional assist ventilation in low birth weight infants with acute respiratory disease: a comparison to assist/control and conventional mechanical ventilation. J. Pediatr. 135, 339–344 (1999).
Schmidt, B. et al. Caffeine therapy for apnea of prematurity. N. Engl. J. Med. 354, 2112–2121 (2006).
Wheeler, K., Klingenberg, C., McCallion, N., Morley, C. J. & Davis, P. G. Volume-targeted versus pressure-limited ventilation in the neonate. Cochrane Database Syst. Rev. 11, Cd003666 (2010).
Darlow, B. A. & Graham, P. J. Vitamin A supplementation to prevent mortality and short- and long-term morbidity in very low birthweight infants. Cochrane Database Syst. Rev. 10, Cd000501 (2011).
Donohue, P. K. et al. Inhaled nitric oxide in preterm infants: a systematic review. Pediatrics 127, e414–e422 (2011).
Bahadue, F. L. & Soll, R. Early versus delayed selective surfactant treatment for neonatal respiratory distress syndrome. Cochrane Database Syst. Rev. 11, Cd001456 (2012).
Ghanta, S., Leeman, K. T. & Christou, H. An update on pharmacologic approaches to bronchopulmonary dysplasia. Semin. Perinatol. 37, 115–123 (2013).
Schmidt, B. et al. Effects of targeting higher vs lower arterial oxygen saturations on death or disability in extremely preterm infants: a randomized clinical trial. JAMA 309, 2111–2120 (2013).
Barrington, K. J. Management during the first 72 h of age of the periviable infant: an evidence-based review. Semin. Perinatol. 38, 17–24 (2014).
Jain, D. & Bancalari, E. Bronchopulmonary dysplasia: clinical perspective. Birth Defects Res. A 100, 134–144 (2014).
Nair, V., Loganathan, P. & Soraisham, A. S. Azithromycin and other macrolides for prevention of bronchopulmonary dysplasia: a systematic review and meta-analysis. Neonatology 106, 337–347 (2014).
Iyengar, A. & Davis, J. M. Drug therapy for the prevention and treatment of bronchopulmonary dysplasia. Front. Pharmacol. 6, 12 (2015).
Manja, V., Lakshminrusimha, S. & Cook, D. J. Oxygen saturation target range for extremely preterm infants: a systematic review and meta-analysis. JAMA Pediatr. 169, 332–340 (2015).
Collins, C. T. et al. Docosahexaenoic acid and bronchopulmonary dysplasia in preterm infants. N. Engl. J. Med. 376, 1245–1255 (2017).
Jain, D. & Bancalari, E. Prevention of bronchopulmonary dysplasia: current strategies. Zhongguo dang dai er ke za zhi=Chin. J. Contemp. Pediatr. 19, 841–851 (2017).
Tarnow-Mordi, W. et al. Outcomes of two trials of oxygen-saturation targets in preterm infants. N. Engl. J. Med. 374, 749–760 (2016).
Baveja, R. & Christou, H. Pharmacological strategies in the prevention and management of bronchopulmonary dysplasia. Semin. Perinatol. 30, 209–218 (2006).
Wilson-Costello, D. et al. Impact of postnatal corticosteroid use on neurodevelopment at 18 to 22 months’ adjusted age: effects of dose, timing, and risk of bronchopulmonary dysplasia in extremely low birth weight infants. Pediatrics 123, e430–e437 (2009).
Lewandowski, A. J. et al. Right ventricular systolic dysfunction in young adults born preterm. Circulation 128, 713–720 (2013).
Sipola-Leppanen, M. et al. Cardiovascular risk factors in adolescents born preterm. Pediatrics 134, e1072–e1081 (2014).
Gough, A. et al. Impaired lung function and health status in adult survivors of bronchopulmonary dysplasia. Eur. Respir. J. 43, 808–816 (2014).
Baraldi, E. & Filippone, M. Chronic lung disease after premature birth. N. Engl. J. Med. 357, 1946–1955 (2007).
Nakanishi, H., Uchiyama, A. & Kusuda, S. Impact of pulmonary hypertension on neurodevelopmental outcome in preterm infants with bronchopulmonary dysplasia: a cohort study. J. Perinatol. 36, 890–896 (2016).
Guimaraes, H. et al. Risk factors for bronchopulmonary dysplasia in five Portuguese neonatal intensive care units. Rev. Port. Pneumol. 16, 419–430 (2010).
del Cerro, M. J. et al. Pulmonary hypertension in bronchopulmonary dysplasia: clinical findings, cardiovascular anomalies and outcomes. Pediatr. Pulmonol. 49, 49–59 (2014).
Coalson, J. J. Pathology of bronchopulmonary dysplasia. Semin. Perinatol. 30, 179–184 (2006).
Mourani, P. M. et al. Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 191, 87–95 (2015).
Onland, W. et al. Clinical prediction models for bronchopulmonary dysplasia: a systematic review and external validation study. BMC Pediatr. 13, 207 (2013).
Sinkin, R. A., Cox, C. & Phelps, D. L. Predicting risk for bronchopulmonary dysplasia: selection criteria for clinical trials. Pediatrics 86, 728–736 (1990).
Bhering, C. A., Mochdece, C. C., Moreira, M. E., Rocco, J. R. & Sant’Anna, G. M. Bronchopulmonary dysplasia prediction model for 7-day-old infants. J. Pediatr. (Rio J.) 83, 163–170 (2007).
Ambalavanan, N. et al. Predictors of death or bronchopulmonary dysplasia in preterm infants with respiratory failure. J. Perinatol. 28, 420–426 (2008).
Laughon, M. M. et al. Prediction of bronchopulmonary dysplasia by postnatal age in extremely premature infants. Am. J. Respir. Crit. Care Med. 183, 1715–1722 (2011).
Albertine, K. H. Utility of Large-animal models of BPD: chronically ventilated preterm lambs. Am. J. Physiol. Lung Cell Mol. Physiol. https://doi.org/10.1152/ajplung.00178 (2015).
Berger, J. & Bhandari, V. Animal models of bronchopulmonary dysplasia. The term mouse models. Am. J. Physiol. Lung Cell Mol. Physiol. 307, L936–L947 (2014).
D’Angio, C. T. & Ryan, R. M. Animal models of bronchopulmonary dysplasia. The preterm and term rabbit models. Am. J. Physiol. Lung Cell Mol. Physiol. 307, L959–L969 (2014).
Yoder, B. A. & Coalson, J. J. Animal models of bronchopulmonary dysplasia. The preterm baboon models. Am. J. Physiol. Lung Cell Mol. Physiol. 307, L970–L977 (2014).
Hartnett, M. E. Pathophysiology and mechanisms of severe retinopathy of prematurity. Ophthalmology 122, 200–210 (2015).
Nardiello, C., Mizikova, I. & Morty, R. E. Looking ahead: where to next for animal models of bronchopulmonary dysplasia? Cell Tissue Res. 367, 457–468 (2017).
Nardiello, C. et al. Standardisation of oxygen exposure in the development of mouse models for bronchopulmonary dysplasia. Dis. Model Mech. 10, 185–196 (2017).
O’Reilly, M. & Thebaud, B. Animal models of bronchopulmonary dysplasia. The term rat models. Am. J. Physiol. Lung Cell Mol. Physiol. 307, L948–L958 (2014).
Grisafi, D. et al. Human amniotic fluid stem cells protect rat lungs exposed to moderate hyperoxia. Pediatr. Pulmonol. 48, 1070–1080 (2013).
Li, H. et al. The 50/10 oxygen-induced retinopathy model serves as a hyperoxia and hypoxia model of bronchopulmonary dysplasia. Am. J. Med. Sci. 355, 581–587 (2018).
Mankouski, A. et al. Intermittent hypoxia during recovery from neonatal hyperoxic lung injury causes long-term impairment of alveolar development: a new rat model of BPD. Am. J. Physiol. Lung Cell Mol. Physiol. 312, L208–L216 (2017).
Ambalavanan, N. & Morty, R. E. Searching for better animal models of BPD: a perspective. Am. J. Physiol. Lung Cell Mol. Physiol. 311, L924–l927 (2016).
Lal, C. V. & Ambalavanan, N. Biomarkers, early diagnosis, and clinical predictors of bronchopulmonary dysplasia . Clin. Perinatol. 42, 739–754 (2015).
Bhandari, V. B. M. et al. Familial and genetic susceptibility to major neonatal morbidities in preterm twins. Pediatrics 117, 1901–1906 (2006).
Lavoie, P. M. P. C. & Jang, K. L. Heritability of bronchopulmonary dysplasia, defined according to the consensus statement of the national institutes of health. Pediatrics 122, 479–485 (2008).
Carrera, P. et al. Exome sequencing and pathway analysis for identification of genetic variability relevant for bronchopulmonary dysplasia (BPD) in preterm newborns: apilot study. Clin. Chim. Acta 451, 39–45 (2015).
Li, J. et al. Exome sequencing of neonatal blood spots and the identification of genes implicated in bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 192, 589–596 (2015).
Moreno, L. et al. Pharmacology of airways and vessels in lung slices in situ: role of endogenous dilator hormones. Respir. Res. 7, 111 (2006).
Pieretti, A. C., Ahmed, A. M., Roberts, J. D. Jr. & Kelleher, C. M. A novel in vitro model to study alveologenesis. Am. J. Respir. Cell Mol. Biol. 50, 459–469 (2014).
Davidovich, N., Chhour, P. & Margulies, S. S. Uses of remnant human lung tissue for mechanical stretch studies. Cell Mol. Bioeng. 6, 175–182 (2013).
Blackwell, T. S. et al. NF-kappaB signaling in fetal lung macrophages disrupts airway morphogenesis. J. Immunol. 187, 2740–2747 (2011).
Brown, K. R., England, K. M., Goss, K. L., Snyder, J. M. & Acarregui, M. J. VEGF induces airway epithelial cell proliferation in human fetal lung in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L1001–L1010 (2001).
Young, S. L., Evans, K. & Eu, J. P. Nitric oxide modulates branching morphogenesis in fetal rat lung explants. Am. J. Physiol. Lung Cell Mol. Physiol. 282, L379–L385 (2002).
Seedorf, G. et al. Hepatocyte growth factor as a downstream mediator of vascular endothelial growth factor-dependent preservation of growth in the developing lung. Am. J. Physiol. Lung Cell Mol. Physiol. 310, L1098–L1110 (2016).
Wilkinson, D. C. et al. Development of a three-dimensional bioengineering technology to generate lung tissue for personalized disease modeling. Stem Cells Transl. Med. 6, 622–633 (2017).
Sucre, J. M. et al. A three-dimensional human model of the fibroblast activation that accompanies bronchopulmonary dysplasia identifies Notch-mediated pathophysiology. Am. J. Physiol. Lung Cell Mol. Physiol. 310, L889–L898 (2016).
Branchfield, K. et al. A three-dimensional study of alveologenesis in mouse lung. Dev. Biol. 409, 429–441 (2016).
Mowes, A., de Jongh, B. E., Cox, T., Zhu, Y. & Shaffer, T. H. A translational cellular model to study the impact of high-frequency oscillatory ventilation on human epithelial cell function. J. Appl. Physiol. (1985) 122, 198–205 (2017).
Huh, D. D. A human breathing lung-on-a-chip. Ann. Am. Thorac. Soc. 12(Suppl. 1), S42–S44 (2015).
Yu, J. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 (2007).
Reed, D. M. et al. An autologous endothelial cell:peripheral blood mononuclear cell assay that detects cytokine storm responses to biologics. FASEB J. 29, 2595–2602 (2015).
Wang, H. et al. A genome-wide association study (GWAS) for bronchopulmonary dysplasia. Pediatrics 132, 290–297 (2013).
Piersigilli, F. & Bhandari, V. Biomarkers in neonatology: the new “omics” of bronchopulmonary dysplasia. J. Matern.-Fetal Neonatal Med. 29, 1758–1764 (2016).
Cuna, A. et al. Alterations in gene expression and DNA methylation during murine and human lung alveolar septation. Am. J. Respir. Cell Mol. Biol. 53, 60–73 (2015).
Mariani, T. J., Reed, J. J. & Shapiro, S. D. Expression profiling of the developing mouse lung: insights into the establishment of the extracellular matrix. Am. J. Respir. Cell Mol. Biol. 26, 541–548 (2002).
Bonner, A. E., Lemon, W. J. & You, M. Gene expression signatures identify novel regulatory pathways during murine lung development: implications for lung tumorigenesis. J. Med. Genet. 40, 408–417 (2003).
Clerch, L. B., Baras, A. S., Massaro, G. D., Hoffman, E. P. & Massaro, D. DNA microarray analysis of neonatal mouse lung connects regulation of KDR with dexamethasone-induced inhibition of alveolar formation. Am. J. Physiol. Lung Cell. Mol. Physiol. 286, L411–L419 (2004).
Wagenaar, G. T. et al. Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radic. Biol. Med. 36, 782–801 (2004).
Cox, B. et al. Integrated proteomic and transcriptomic profiling of mouse lung development and Nmyc target genes. Mol. Syst. Biol. 3, 109 (2007).
Kho, A. T. et al. Transcriptomic analysis of human lung development. Am. J. Respir. Crit. Care Med. 181, 54–63 (2010).
Bhattacharya, S. et al. The genome-wide transcriptional response to neonatal hyperoxia identifies Ahr as a key regulator. Am. J. Physiol. Lung Cell. Mol. Physiol. 307, L516–L523 (2014).
Khoshgoo, N., Kholdebarin, R., Iwasiow, B. M. & Keijzer, R. MicroRNAs and lung development. Pediatr. Pulmonol. 48, 317–323 (2013).
Sessa, R. & Hata, A. Role of microRNAs in lung development and pulmonary diseases. Pulm. Circ. 3, 315–328 (2013).
Williams, A. E., Moschos, S. A., Perry, M. M., Barnes, P. J. & Lindsay, M. A. Maternally imprinted microRNAs are differentially expressed during mouse and human lung development. Dev. Dyn. 236, 572–580 (2007).
Lu, Y., Okubo, T., Rawlins, E. & Hogan, B. L. Epithelial progenitor cells of the embryonic lung and the role of microRNAs in their proliferation. Proc. Am. Thorac. Soc. 5, 300–304 (2008).
Bhaskaran, M. et al. MicroRNA-127 modulates fetal lung development. Physiol. Genom. 37, 268–278 (2009).
Yang, Y., Qiu, J., Kan, Q., Zhou, X. G. & Zhou, X. Y. MicroRNA expression profiling studies on bronchopulmonary dysplasia: a systematic review and meta-analysis. Genet. Mol. Res. 12, 5195–5206 (2013).
Rogers, L. K. et al. Attenuation of miR-17 approximately 92 Cluster in bronchopulmonary dysplasia. Ann. Am. Thorac. Soc. 12, 1506–1513 (2015).
Lal, C. V. et al. Exosomal microRNA predicts and protects against severe bronchopulmonary dysplasia in extremely premature infants. JCI Insight 3, e93994 (2018).
Bhandari, A. & Bhandari, V. Biomarkers in bronchopulmonary dysplasia. Paediatr. Respir. Rev. 14, 173–179 (2013).
Magagnotti, C. et al. Calcium signaling-related proteins are associated with broncho-pulmonary dysplasia progression. J. Proteom. 94, 401–412 (2013).
Fanos, V. et al. Urinary metabolomics of bronchopulmonary dysplasia (BPD): preliminary data at birth suggest it is a congenital disease. J. Matern.-Fetal Neonatal Med. 27(Suppl. 2), 39–45 (2014).
Baraldi, E. et al. Untargeted metabolomic analysis of amniotic fluid in the prediction of preterm delivery and bronchopulmonary dysplasia. PLoS ONE 11, e0164211 (2016).
Ardini-Poleske, M. E. et al. LungMAP: The Molecular Atlas of Lung Development Program. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L733–l740 (2017).
Fischbach, M. A., Bluestone, J. A. & Lim, W. A. Cell-based therapeutics: the next pillar of medicine. Sci. Transl. Med. 5, 179ps177 (2013).
Caplan, A. I. & Correa, D. The MSC: an injury drugstore. Cell Stem Cell 9, 11–15 (2011).
Irwin, D. et al. Neonatal lung side population cells demonstrate endothelial potential and are altered in response to hyperoxia-induced lung simplification. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L941–L951 (2007).
van Haaften, T. et al. Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am. J. Respir. Crit. Care Med. 180, 1131–1142 (2009).
Popova, A. P. et al. Isolation of tracheal aspirate mesenchymal stromal cells predicts bronchopulmonary dysplasia. Pediatrics 126, e1127–e1133 (2010).
Collins, J. J. & Thebaud, B. Lung mesenchymal stromal cells in development and disease: to serve and protect? Antioxid. Redox Signal. 21, 1849–1862 (2014).
O’Reilly, M. & Thebaud, B. Cell-based therapies for neonatal lung disease. Cell Tissue Res. 367, 737–745 (2017).
Kang, M. & Thebaud, B. Stem cell biology and regenerative medicine for neonatal lung diseases. Pediatr. Res. 83, 291–297 (2018).
Nitkin, C. R. & Bonfield, T. L. Concise review: mesenchymal stem cell therapy for pediatric disease: perspectives on success and potential improvements. Stem Cells Transl. Med. 6, 539–565 (2017).
Lesage, F., Jimenez, J., Toelen, J. & Deprest, J. Preclinical evaluation of cell-based strategies to prevent or treat bronchopulmonary dysplasia in animal models: a systematic review. J. Matern.-Fetal Neonatal Med. 31, 958–966 (2018).
Aslam, M. et al. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am. J. Respir. Crit. Care Med. 180, 1122–1130 (2009).
Waszak, P. et al. Preconditioning enhances the paracrine effect of mesenchymal stem cells in preventing oxygen-induced neonatal lung injury in rats. Stem Cells Dev. 21, 2789–2797 (2012).
Sutsko, R. P. et al. Long-term reparative effects of mesenchymal stem cell therapy following neonatal hyperoxia-induced lung injury. Pediatr. Res. 73, 46–53 (2013).
Pierro, M. et al. Short-term, long-term and paracrine effect of human umbilical cord-derived stem cells in lung injury prevention and repair in experimental bronchopulmonary dysplasia. Thorax 68, 475–484 (2013).
Augustine, S. et al. Mesenchymal stromal cell therapy in bronchopulmonary dysplasia: systematic review and meta-analysis of preclinical studies. Stem Cells Transl. Med. 6, 2079–2093 (2017).
Lee, C. et al. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 126, 2601–2611 (2012).
Fung, M. E. & Thebaud, B. Stem cell-based therapy for neonatal lung disease: it is in the juice. Pediatr. Res. 75, 2–7 (2014).
Willis, G. R. et al. Mesenchymal stromal cell exosomes ameliorate experimental bronchopulmonary dysplasia and restore lung function through macrophage immunomodulation. Am. J. Respir. Crit. Care Med. 197, 104–116 (2018).
Lener, T. et al. Applying extracellular vesicles based therapeutics in clinical trials—an ISEV position paper. J. Extracell. Vesicles 4, 30087 (2015).
Willis, G. R., Kourembanas, S. & Mitsialis, S. A. Toward exosome-based therapeutics: isolation, heterogeneity, and fit-for-purpose potency. Front. Cardiovasc. Med. 4, 63 (2017).
Chang, Y. S. et al. Mesenchymal stem cells for bronchopulmonary dysplasia: phase 1 dose-escalation clinical trial. J. Pediatr. 164, 966–972.e966 (2014).
Monz, D. et al. Human umbilical cord blood mononuclear cells in a double-hit model of bronchopulmonary dysplasia in neonatal mice. PLoS ONE 8, e74740 (2013).
Alphonse, R. S. et al. Existence, functional impairment, and lung repair potential of endothelial colony-forming cells in oxygen-induced arrested alveolar growth. Circulation 129, 2144–2157 (2014).
Vosdoganes, P., Lim, R., Moss, T. J. & Wallace, E. M. Cell therapy: a novel treatment approach for bronchopulmonary dysplasia. Pediatrics 130, 727–737 (2012).
Sharma, R. R., Pollock, K., Hubel, A. & McKenna, D. Mesenchymal stem or stromal cells: a review of clinical applications and manufacturing practices. Transfusion 54, 1418–1437 (2014).
Beardsall, K. et al. Relationship between insulin-like growth factor I levels, early insulin treatment, and clinical outcomes of very low birth weight infants. J. Pediatr. 164, 1038–1044.e1031 (2014).
Lofqvist, C. et al. A pharmacokinetic and dosing study of intravenous insulin-like growth factor-I and IGF-binding protein-3 complex to preterm infants. Pediatr. Res. 65, 574–579 (2009).
Ley, D. et al. Longitudinal infusion of a complex of insulin-like growth factor-I and IGF-binding protein-3 in five preterm infants: pharmacokinetics and short-term safety. Pediatr. Res. 73, 68–74 (2013).
Hansen-Pupp, I. et al. Continuous longitudinal infusion of rhIGF-1/rhIGFBP-3 in extremely preterm infants: Evaluation of feasibility in a phase II study. Growth Horm. IGF Res. 36, 44–51 (2017).
Arias-Martinez, J. et al. Clara cell protein expression in human neonates during respiratory distress syndrome. Cell. Physiol. Biochem. 29, 753–760 (2012).
Levine, C. R. et al. The safety, pharmacokinetics, and anti-inflammatory effects of intratracheal recombinant human Clara cell protein in premature infants with respiratory distress syndrome. Pediatr. Res. 58, 15–21 (2005).
Ramsay, P. L. et al. Clara cell secretory protein oxidation and expression in premature infants who develop bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 164, 155–161 (2001).
Wolfson, M. R. et al. Recombinant human Clara cell secretory protein treatment increases lung mRNA expression of surfactant proteins and vascular endothelial growth factor in a premature lamb model of respiratory distress syndrome. Am. J. Perinatol. 25, 637–645 (2008).
Acknowledgements
This study (PI14/00219, PI15/01100) was supported by the Instituto de Salud Carlos III (Plan Estatal de I+D+i) and cofinanced by the European Development Regional Fund “A way to achieve Europe” (ERDF). This article has been funded by a national public Spanish grant from the Instituto de Salud Carlos III: PI14/00219.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Álvarez-Fuente, M., Moreno, L., Mitchell, J.A. et al. Preventing bronchopulmonary dysplasia: new tools for an old challenge. Pediatr Res 85, 432–441 (2019). https://doi.org/10.1038/s41390-018-0228-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-018-0228-0
This article is cited by
-
Lung ultrasound score as a tool to predict severity of bronchopulmonary dysplasia in neonates born ≤25 weeks of gestational age
Journal of Perinatology (2024)
-
A glucocorticoid-receptor agonist ameliorates bleomycin-induced alveolar simplification in newborn rats
Pediatric Research (2023)
-
Prematurity and BPD: what general pediatricians should know
European Journal of Pediatrics (2023)
-
Präventionskonzepte in der Frühgeborenenmedizin
Monatsschrift Kinderheilkunde (2022)
-
Prematurity alters the progenitor cell program of the upper respiratory tract of neonates
Scientific Reports (2021)
