
Abstract
Cachexia is a severe complication of cancer that adversely affects the course of the disease, with currently no effective treatments. It is characterized by a progressive atrophy of skeletal muscle and adipose tissue, resulting in weight loss, a reduced quality of life, and a shortened life expectancy. Although the cachectic condition primarily affects the skeletal muscle, a tissue that accounts for ~40% of total body weight, cachexia is considered a multi-organ disease that involves different tissues and organs, among which the cardiac muscle stands out for its relevance. Patients with cancer often experience severe cardiac abnormalities and manifest symptoms that are indicative of chronic heart failure, including fatigue, shortness of breath, and impaired exercise tolerance. Furthermore, cardiovascular complications are among the major causes of death in cancer patients who experienced cachexia. The lack of effective treatments for cancer cachexia underscores the need to improve our understanding of the underlying mechanisms. Increasing evidence links the wasting of the cardiac and skeletal muscles to metabolic alterations, primarily increased energy expenditure, and to increased proteolysis, ensuing from activation of the major proteolytic machineries of the cell, including ubiquitin-dependent proteolysis and autophagy. This review aims at providing an overview of the key mechanisms of cancer cachexia, with a major focus on those that are shared by the skeletal and cardiac muscles.
Similar content being viewed by others

Introduction
Cachexia is a devastating syndrome, often announcing the onset of the terminal phase of several diseases, including respiratory and cardiac failure, AIDS, sepsis as well as cancer. It is defined as an unstoppable weight loss of at least 5% of body mass in 6 months, mostly affecting lean mass, while fat tissue wasting occurs at a variable penetrance. In some cases, cachexia might be masked by concomitant obesity, in which loss of lean mass is counteracted by fat deposition1. Cachexia occurs in at least 80% of metastatic cancer patients, thus representing a highly penetrant complication and the primary cause of death in at least one-third of cancer patients. To date, cachexia still represents an unmet medical need, because a substantial portion of patients suffering from chronic diseases succumb to this complication, due to the lack of therapeutic options.
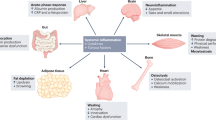
Despite its major burden on life quality and healthcare systems, our knowledge of the disease is still limited. Cachexia is indeed a complex syndrome affecting several organs, promoting systemic metabolic rewiring, and a diffuse inflammatory condition. Cachectic patients present increased resting energy expenditure, mostly due to systemic lipolysis and mitochondrial dysfunction, while systemic inflammation contributes to local tissue dysfunctions such as anorexia and fat tissue browning. Furthermore, cachexia is often associated with gut dysbiosis and intestinal membrane permeabilization, resulting in elevated levels of circulating proinflammatory molecules that further worsen systemic inflammation2 (Fig. 1).

Cancer cells, together with the activation of the inflammatory response and the toxic effects of chemotherapy, contribute to concomitant and interconnected alterations in multiple distant organs, including the cardiac and skeletal muscles and the gut, in the course of cancer-induced cachexia. ILs interleukins, TNF tumor necrosis factor, TGF transforming growth factor, TLRs toll-like receptors, DAMPs damage-associated molecular patterns, FAO fatty acid oxidation, PAMPs pathogen-associated molecular patterns.
Nevertheless, the major manifestation of cachexia is skeletal muscle wasting, which results in lean mass loss and frailty. Importantly, muscle wasting results in a severe drop in quality of life, causing respiratory distress and fatigue3. Moreover, muscle mass loss is an important sign of suffering, hence many pharmacological regimens are normally interrupted once systemic weight loss occurs.
During cachexia, cardiac wasting can also occur, primarily as a consequence of cardiac proteins loss2. This is for example the case of cancer patients where cardiac wasting is often secondary to therapy with cardiotoxic anti-cancer drugs or the presence of the tumor that, by producing circulating mediators, promotes atrophy of cardiomyocytes and negatively impacts on cardiac contractility4. Vice versa, cardiac dysfunction itself promotes skeletal muscle wasting, a complication known as cardiac cachexia. Furthermore, cardiac cachexia is an independent predictor of survival in chronic heart failure patients5 in experimental models6,7,8,9 and in up to 19.5% of heart failure patients, particularly those with reduced ejection fraction10.
Understanding the complex crosstalk between the heart, the skeletal muscle, and the host in chronic disease conditions, particularly in cancer, is of utmost importance for the identification of novel potential targets for therapeutic approaches. Here, we will discuss the current knowledge of the common biological basis of muscle and heart wasting, with particular reference to cancer-induced cachexia.
Inter- and intracellular mediators of skeletal and heart muscle cachexia
Skeletal muscle and cardiac wasting has been demonstrated in some cancer types, including lung, pancreatic, and gastrointestinal tumors11. Although cancer cells rarely metastasize to skeletal and cardiac muscle, factors secreted by either the primary tumor, metastases, or activated immune cells can induce extensive muscle wasting. Different from other types of muscle atrophy, like those induced by fasting, denervation, or disuse, cancer cachexia is characterized by massive systemic inflammation12,13. Cytokines and other pro-cachectic mediators can be directly released by some types of cancer cells into the bloodstream, however the majority of catabolic cytokines is generated by immune cells in response to cancer14. Furthermore, organ damage, induced by metastatic erosion or chemo- and radiotherapy, may lead to the secretion of danger-associated molecular patterns (DAMPs), endogenous signals of cell damage15 that contribute to inflammation and the development of the cachectic syndrome. Finally, both cardiac and skeletal muscle can act as endocrine organs, by releasing signaling molecules called myokines and cardiokines, respectively, which include members of the transforming growth factor (TGF) superfamily (like Myostatin and Activin A)16. All these circulating mediators are involved in the promotion of skeletal and/or cardiac muscle catabolism and convey the pro-atrophic signals that trigger cancer cachexia.
At the intracellular level, factors released by the tumor, its environment, or activated immune cells mediate the activity of a large variety of signaling molecules, like NF-κB, p38 MAPK, or STAT3, orchestrating inter- and intracellular signaling that ultimately promote cancer cachexia17,18,19,20,21,22 (Fig. 2). Among those are pathways controlling protein degradation, including the autophagy-lysosomal pathway (ALP) and the ubiquitin-proteasome pathway (UPP)13, whose alteration is a major hallmark of cancer cachexia. Indeed, in cancer cachexia, ALP and UPP are hyperactive, leading to muscle atrophy13,23,24.

The mechanisms underlying cancer cachexia are multiple and intertwined. Either factors released by skeletal and cardiac muscles (myokines and cardiokines respectively; pink boxes) or factors secreted by cancer and cancer-associated immune cells (violet boxes) trigger a cascade of processes which ultimately result in cachexia. The circulating factors (pink), intracellular signaling pathways (orange), and final effectors of wasting (yellow) that lead to skeletal (left side of the figure) and cardiac (right side of the figure) muscle wasting are reported. Albeit the biological processes underlying skeletal and cardiac muscle wasting are similar, their relative contribution and the specific molecular players involved differ slightly. Red arrows indicate those factors that are increased as a consequence of chemotherapy. DAMPs damage-associated molecular patterns, GDF15 growth differentiation factor 15, ILs interleukins, LIF leukemia inhibitory Factor, TNF-α tumor necrosis factor alpha, TGF-β transforming growth factor beta.
Autophagy-lysosomal pathway and ubiquitin-proteasome pathway
The negative protein balance observed in the wasting syndrome is linked to protein hypercatabolism. Both ALP and UPP efficiently degrade proteins that have been ubiquitylated by E1, E2, and E3 enzymes25. In ALP, uniquely ubiquitylated proteins are engulfed by autophagosomes that subsequently fuse to lysosomes to form autolysosomes, where proteins are enzymatically degraded25,26. On the contrary, differently ubiquitylated proteins are recognized by the UPP and degraded by the proteasome27. Increasing evidence indicates that the contribution of ALP and UPP to muscle wasting is context-dependent, varying between pathologies13. For example, the muscle-specific E3 ubiquitin ligases Atrogin-1 and MuRF-1 are the main drivers of skeletal but not cardiac muscle wasting, in which autophagy has instead a major role28. This might be explained by the fact that the heart has a higher metabolic rate and protein turnover than the skeletal muscle. Thus, induction of ALP, in the presence of a high basal activity of UPP, might be sufficient to mediate protein degradation28. On the other hand, both UPP and ALP contribute to skeletal muscle wasting. Accordingly, direct comparison between skeletal and cardiac muscle in a cachectic rat model showed the upregulation of autophagy markers, such as LC3 and p62, in both tissues. In contrast, TRAF6, an inducer of atrophy, and Beclin1, an autophagic marker, were specifically upregulated in the gastrocnemius and the heart, respectively29,30,31. In line with this observation, cachectic mice exhibited atrophic hearts, with enhanced expression of Beclin1 and LC3 but no significant induction of proteins involved in ubiquitination or apoptosis28,32. Intriguingly, forkhead box transcription factors 3 (FoxO3), which is an established inducer of proteasomal-mediated atrophy in skeletal muscle1, has been reported to induce atrophy via ALP in the heart33. On the other hand, cardiac atrophy, in majorly cancer-independent disease models, is accompanied by increased levels of Atrogin-1 and therefore an active UPP, which results from the induction of MAPK pathways34,35,36. One study on tumor-bearing mice reports the increase of atrogenes expression in the heart37 and indicates the role of UPP in cancer cachexia which remains to be further defined.
Consequently, the induction of the different molecular pathways that cause atrophy in skeletal and the cardiac muscles32 may depend on the different composition of humoral factors that are released by the tumor or cancer-affected tissues.
The inflammatory milieu
Cancer cachexia is accompanied by an increased release of inflammatory molecules, which are mainly produced by immune cells in response to cancer38. Among these are interleukins, tumor necrosis factor, and members of the transforming growth factor family.
Interleukins
Levels of IL-6 cytokine family members are increased during cancer-related cachexia39, as a consequence of the release of leukemia inhibitory factor (LIF) from tumor cells40. IL-6, in turn, induces intracellular STAT3 (detailed in Box 1), p38, and FoxO signaling in skeletal muscles21,39 through Glycoprotein 13039. These are common intracellular signaling pathways elicited by inflammatory cytokines like IL-141 and IL-842. Interestingly, the loss of cardiac mass in tumor-bearing mice does not rely on circulating levels of LIF40.
Danger-associated molecular patterns and toll-like receptors
DAMPs are part of a plethora of molecules3, including free and histone-associated nuclear DNA86, mitochondrial DNA (mtDNA)87, and heat shock proteins88, which can be released by cancer, immune as well as cardiac cells upon injury. DAMPs are the endogenous agonists of Toll-Like Receptors (TLRs), an evolutionarily ancient family of pattern recognition receptors89. In immune cells, plasma membrane TLRs stimulate the synthesis of proteins that belong to the inflammasome complex, by inducing the translocation of NF-κB into the nucleus, ultimately modulating innate immunity90. In muscle cells, NF-κB activation is sufficient to induce mass loss through the upregulation of E3 ubiquitin ligase MuRF-1, e.g. upon LPS-induced pulmonary inflammation91 or LLC-derived tumor in mice92.
In cancer cachexia, the activation of TLRs by DAMPs, released in the bloodstream, stimulates muscle proteolysis both directly, by acting on muscle cells, and indirectly, by activating TLR4 in immune cells to increase systemic inflammation88. Indeed, TLR4 is the isoform which is mainly linked to muscle wasting in cancer, being required for LLC-cancer-related muscle wasting93,94. Accordingly, the TLR4 expression level in skeletal muscles of cancer patients significantly correlates with low skeletal muscle index and weight loss95. Interestingly, the role of TLRs in cancer-induced muscle catabolism is relatively isoform- and disease-specific. For instance, muscle-specific activation of TLR7 by tumor-secreted microvesicles promotes skeletal muscle cell death96,97, while local activation of TLR7 in the tumor stroma triggers CD8+ T-cells, resulting in tumor shrinkage and, consequently, in reduced cachexia and improved survival98.
Although, to date a clear link between cancer cachexia and TLRs is missing, it is plausible that the same pathway may be detrimental for the heart. For instance, activation of specific TLR isoforms expressed by cardiac cells has been linked to pro-inflammatory effects, with TLR2, TLR4, and TLR5 being responsible for NF-κB-dependent induction of the inflammasome99,100. The inflammasome complex, in turn, may initiate the activation of pro-inflammatory cascades, leading to pyroptotic cell death101, as in the case of acute myocardial infarction102.
Metabolic changes underlying skeletal and heart muscle wasting induced by cancer
From the biochemical standpoint, cachexia is considered a metabolic disease linked to the negative energy balance between calorie intake and dissipation, which eventually promotes systemic wasting and body weight loss. Such an imbalance is, on the one side, due to the loss of appetite and reduced nutrient absorption, and, on the other side, a result of the upregulation of energy-consuming processes and metabolic dysfunction, which collectively increase the energetic needs of the body driving calorie wasting.
Insufficient calorie intake is mostly driven by anorexia, a persistent and unphysiological loss of appetite. All chronically ill patients develop various degrees of anorexia, due to depression and neuroinflammation. The so-called sick state, driven by systemic inflammation, has also been proposed as a conserved evolutionary mechanism to limit nutrient availability during infections, in order to restrain nutrient availability to pathogens. Consistently, cachexia is often referred to as CAC (cachexia and anorexia) syndrome.
Recently, it has been demonstrated that cachectic patients experience impaired intestinal function and absorption, which is at least in part caused by the alteration of the gut microbiome103,104.
Collectively, reduced calorie intake and nutrient uptake drive a systemic energetic failure. Nevertheless, restoring proper nutrient supply is not sufficient to recover body mass homeostasis, but only delays wasting progression1. Such evidence highlights that other mechanisms, like increased calorie wasting, contribute to the metabolic unbalance occurring in cachexia. It is indeed well known that cachectic patients are characterized by an increase in resting energy expenditure105, which means that, even at rest, their metabolism is accelerated106.
Since the first attempt to understand cancer cachexia, tumor growth has been pinpointed as the culprit for nutrient subtraction and energy consumption107. Accordingly, it has been shown that, during tumor growth, nitrogen balance is managed by the tumor and not by the muscle108. However, besides sequestration of nutrients from the tumor, a systemic rewiring of the metabolism takes place during cancer cachexia, indicating that other organs are involved in the metabolic alterations occurring in cancer patients. For instance, the liver has been proposed to contribute to energy wasting in cancer patients109, at least in part by the generation of phase 2 proteins linked to the systemic inflammatory state. Not only the liver, but also fat tissue is involved in systemic metabolic wasting. Indeed, systemic inflammation also drives tissue browning, which results in systemic lipolysis and thermogenesis110.
The tumor is a main producer of factors triggering metabolic reprograming and wasting, including miRNAs, PTHrP (parathyroid hormone-related protein)106,111,112, known to cause hypercalcemia in cancer patients113, and D-2-hydroxyglutarate (D2-HG), an oncometabolite that is secreted by leukemia cells as a consequence of mutations of the TCA (tricarboxylic acid/Krebs) cycle enzymes isocitrate dehydrogenase 1 and 2. These mutations occur in some myeloid leukemia patients and result in cardiac contractile dysfunction linked to mitochondrial dysfunction, caused by the increased secretion of D2-HG114. Interestingly, comparing acute leukemia (AL) patients with other cancer patients, it has been reported that AL is linked to myocardial dysfunction115. Moreover, the rate of AL patients who develop heart failure significantly increases upon chemotherapy116,117. Consequently, although leukemia patients do not commonly develop cachexia118, the associated cardiac dysfunction may result in an increased susceptibility of AL patients to the development of the wasting syndrome.
Furthermore, tumor growth can directly affect systemic circadian rhythms119, an alteration that has been functionally linked to the onset of insulin resistance. Accordingly, cancer cell-induced alterations can eventually affect insulin and glucose metabolism, which per se impact on both cardiac120 and skeletal muscle function121. Thus, insulin resistance and glucose insensitivity were associated with increased weight loss in cancer patients122. For instance, tumor growth negatively affects plasma insulin and glucose levels in cancer-bearing mice123,124. As an example, leukemia cells, of mouse models and patients, actively induce insulin resistance by prompting the production of insulin-like growth factor (IGF)-binding protein 1, in order to exclusively exploit glucose availability123. Furthermore, cancer cells induce changes of the metabolic profile of other tissues and of the gut microbiome, ultimately conveying insulin resistance and reduction of the anabolic factor IGF-13,125. Moreover, decreased insulin levels have been functionally linked to cardiac wasting, as administration of insulin is able to attenuate cardiac atrophy, while reducing glucose uptake in the tumor126, a strategy that might also be important for skeletal muscle. Overall, these findings provide new opportunities for therapeutic interventions aimed at restoring glucose supply in the muscles. Whether this approach may enable to delay or recover cancer cachexia remains to be demonstrated.
Several pieces of evidence recently pinpointed to an altered role of lipid homeostasis in driving skeletal muscle wasting. For instance, it has been shown that wasting skeletal muscles switch to fatty acid oxidation (FAO) as the prominent source of energy production127,128. Moreover, FAO has been functionally linked to the wasting phenotype in cancer, and limiting FAO prevents skeletal muscle wasting, either through pharmacological inhibitors129 or by genetic inhibition of lipolysis130. Whether FAO may be impaired during cardiac wasting has yet to be clarified.
In turn, excessive mitochondrial activity and intermediate overload, caused by increased fatty acid metabolism, may cause an increase in oxidative stress and mitochondrial ROS, eventually leading to dysfunctions131,132. Coherently, dysfunctions in mitochondrial metabolism are common alterations occurring in wasting skeletal muscles133. Similarly, mitochondrial DNA (mtDNA) released upon stress in cardiac cells can act as a DAMP, and hence as a ligand for TLR9, an endosomal TLR isoform. Activation of TLR9 by mtDNA is responsible for impaired induction of autophagy and the ensuing accumulation of dysfunctional mitochondria and oxidative stress after doxorubicin-induced cardiac injury87. Moreover, in skeletal muscle, TLR9 has a key role in coordinating with Beclin1 to activate AMPK under energetic stress134. Nevertheless, the role of such an mtDNA-TLR9 axis in cancer-induced cardiac and skeletal muscle atrophy has yet to be evaluated.
Finally, the inflammatory state per se promotes several metabolic alterations, eventually triggering wasting. For instance, inflammatory states, like those occurring during chronic cardiomyopathy or cancer, are known to halt iron uptake by the gut and promote iron retention by macrophages135. The resulting iron deficiency triggers anemia, which might further impact on cardiac function and skeletal muscle oxygenation136. Interestingly, it has been demonstrated that, at least in the heart, iron-deficient anemia might directly affect the functionality of cardiac cells137. In line with this view, several clinical trials in cardiopathic patients have shown that iron supplementation restores cardiac function and muscle strength138. Nevertheless, this approach cannot be directly applied to cancer patients as cancer growth itself directly depends on iron supply139. Moreover, it has been shown that chemotherapy-induced cardiotoxicity partly depends on excessive accumulation and altered compartmentalization of iron in the heart140,141 leading to mitochondrial iron overload and dysfunction.
The gut microbiota-muscle axis
The gut-associated lymphoid tissue is considered as the largest immune organ of the body. Therefore, it is not surprising that an association between systemic inflammation and gut dysbiosis has been demonstrated in several chronic diseases associated with cachexia, including heart failure142. Consistently, a number of studies demonstrated a link between dysbiosis and cardiovascular diseases143,144,145 as well as cancer146,147. Accordingly, an increase in intestinal permeability is frequently recognized in cachexia-associated diseases and could facilitate the diffusion of pro-inflammatory molecules across the gut barrier, thus contributing to the systemic inflammatory state148.
Mechanistically, besides stimulating the systemic increase of pro-inflammatory cytokines, gut microbiota could lead to muscle wasting by decreasing amino acid bioavailability, by stimulating the TLR/NF-kB pathway through the release of pathogen-associated molecular patterns (PAMPs)149, and via the production of cachectic metabolites104.
The hypothesis of a systemic signalosome, originating from gut microbiota and targeting distant organs like muscles, was supported by trials showing that modulation of gut microbiota can change immune/inflammatory parameters in cancer patients undergoing esophageal surgery150. In line with these findings, interventions on gut microbiota can prolong survival by reducing cancer proliferation, muscle wasting103, and fat loss151 in mouse models of cancer cachexia. Similar interventions have been proven effective in preventing cardiac atrophy and dysfunction in preclinical models of anthracycline-induced cardiomyopathy152. However, experimental proofs of the involvement of gut microbiota in cardiac muscle wasting in cachectic cancer patients are still lacking.
The impact of chemotherapy on skeletal and heart muscle wasting
Besides chronic tumor-host interactions, acute drug toxicity and long-term side effects of anti-cancer treatments can significantly contribute to chronic muscle wasting in cachexia153. Despite a rapid evolution of anti-cancer treatment options, cytotoxic chemotherapy remains the first line and preferred treatment for most cancers. Unfortunately, the presence of cachexia reduces tolerance and response to treatment, activating a futile cycle that eventually reduces the quality of life and survival. In cancer patients, tumor growth might, on the one side, impair the ability of the host to adapt to stress imposed by chemotherapy and, on the other side, directly affect muscle and systemic metabolism154. Moreover, most of anti-cancer drugs are severely cardiotoxic155, making patient management during cancer treatment and follow-up even more difficult, while increasing the risk of an exacerbation of cachexia.
Chemotherapy itself can contribute to the alteration of the circulating milieu. On the one hand, chemotherapy potentially limits the release of tumor-derived cytokines, therefore relieving cachexia. On the other hand, host tissues may be directly affected by drug toxicity which frequently activates an inflammatory response, thus exacerbating cachexia. For instance, chemotherapy treatment has been shown to trigger GDF15 following endothelial damage156. On the same line, the promotion of systemic inflammation might indirectly exacerbate the muscle catabolic action and the systemic dysmetabolism induced by inflammatory molecules, such as TNFα, that is both released by the tumor157 and by the host158 upon chemotherapy administration.
Moreover, protein hypercatabolism and impaired anabolism are directly affected by both cytotoxic and targeted chemotherapy, further contributing to muscle wasting159. In particular, the direct effect of anti-neoplastic drugs on myofibrillar protein degradation and myofiber atrophy has been demonstrated160,161. The mechanisms underlying skeletal muscle atrophy in response to chemotherapy are the same as the ones involved in cancer-mediated wasting. In detail, proteasome- and autophagy-mediated protein degradation are induced by cisplatin162, cyclophosphamide, doxorubicin, and fluorouracil mixture163, or anthracyclines (e.g. doxorubicin) alone164.
In the cardiac muscle, the impact of chemotherapy on the main catabolic and anabolic pathways appears even more complex. A comprehensive study, comparing skeletal and cardiac muscle response to doxorubicin, has been performed by two independent research teams. In the first study64, albeit similar mass loss was observed in skeletal and cardiac muscles upon doxorubicin exposure, protein synthesis, content in ubiquitinated proteins, and expression of atrogenes were less affected in the heart than in the skeletal muscle. Similar results were shown by the second study164, although reporting a controversial activation of autophagy in the skeletal muscle. Albeit, a consensus on the role of autophagy in the cardiac response to anthracyclines has not been reached yet165, major studies point to an impairment of the ALP as a major determinant of chemotherapy-induced cardiac atrophy and dysfunction87,166,167. Accordingly, anthracycline-induced damage has been associated with failing autophagic clearance of damaged organelles, resulting from the stimulation of TLR9 via mtDNA release by injured cardiomyocytes87.
In addition, anthracyclines may directly impact the myofibrillar content in both skeletal and cardiac muscles, further contributing to muscle loss (reviewed in Hiensch et al.168). In addition to the previously mentioned role of anthracyclines in regulating metabolism and TLR9 in the skeletal muscle, doxorubicin-induced oxidative stress leads to mitochondrial dysfunction169,170,171, and oxidative modification of myofibrillar proteins, which increases their susceptibility to degradation via calpain‐1 and caspase‐3172,173. Moreover, doxorubicin activates all major proteolytic systems, including calpains173,174, the UPP63, and autophagy63,175 in skeletal muscles. Likewise, doxorubicin leads to atrophy also in cardiomyocytes, via activation of MuRF-1176 by CDK2-dependent phosphorylation of FoxO1 at Ser-249177. Of note, FoxO1 and FoxO3 are potent regulators of muscle atrophy (detailed in Box 4).
Among the proteolytic processes that are induced by doxorubicin in cardiac muscle cells is intracellular activation of matrix metalloproteinase 2 (MMP2), which, in turn, can result in the degradation of both sarcomeric proteins and myofilaments, including titin178. Intriguingly, MMP2 is also expressed by skeletal muscle cells, even if at low levels compared to calpain-1179, and release of titin from skeletal muscles has been associated with muscle atrophy180. Nevertheless, the contribution of MMP2 to proteolysis induced by doxorubicin in tumor-bearing animals has yet to be evaluated.
Both loss and truncation of titin result in skeletal muscle atrophy with reduced strength, severe sarcomere disassembly, and lethality181,182. In contrast, impaired titin integrity results in considerably different phenotypes in the heart. Loss of titin leads to dilated cardiomyopathy with systolic and diastolic dysfunction, while titin truncation or deletion of the N2B segment, that impair sarcomeric array, lead to cardiac atrophy with preserved function182,183.
Another layer of regulation of titin is provided by the RNA-binding protein known as Quaking, which is downregulated in response to doxorubicin184. Quaking inhibits doxorubicin-mediated cardiotoxicity via regulating cardiac circular RNAs, including titin-derived circular RNA in cardiomyocytes. Mechanistically, Quaking deletion in cardiomyocytes increases sensitivity to doxorubicin, whereas its overexpression attenuates doxorubicin-induced cardiac atrophy184. Nevertheless, the role of titin degradation in the context of cancer cachexia has yet to be elucidated.
Concomitantly with increased protein degradation, doxorubicin is also responsible for impaired muscle protein synthesis185, resulting from the inhibition of the mTOR pathway185. Of note, mTORC1 is a major regulator of insulin signaling, however, the disruption of the insulin pathway by doxorubicin has only been detected in skeletal muscles186, but not in the heart187.
As previously reported, the alteration of energy metabolism, and in particular the occurrence of a systemic energetic failure, is obtaining an increasing consensus as a major cause of cachexia. Whether the energy crisis induced by tumor growth arises from inflammation and mitochondrial dysfunction or from excessive oxidative stress is still debated. Most of anti-cancer drugs enhance oxidative damage in both the skeletal and the cardiac muscle. In the former, oxidative stress can be directly linked to protein hypercatabolism and wasting160,161, while in the latter its role has been downscaled, also considering the limited success of anti-oxidants against the cardiotoxicity of drugs like doxorubicin188.
Considering metabolic alterations in the skeletal and cardiac muscles, chemotherapy has been shown to partly recapitulate and/or exacerbate cancer-induced muscle alterations154, while the cardiac metabolome has been mainly studied with the aim of identifying biomarkers of cardiotoxicity189. Instead, only few studies have analyzed tissue-specific alterations of the metabolome during cancer and chemotherapy-associated cachexia. Nevertheless, some common metabolic alterations featured by skeletal and cardiac muscles upon chemotherapy have been identified, which include the increase of free amino acids, likely indicating increased proteolysis, and the reduction in β-oxidation154,189. On the contrary, the flux through the TCA cycle is diminished in the skeletal while augmented in the cardiac muscle, potentially as the only mean to sustain the vital function of heart contraction.
Conclusion
Cancer cachexia represents an urgent medical need, due to the great impact on patients’ quality of life and the high penetrance of this condition. Patients with cancer cachexia are often too weak to tolerate standard doses of chemo- and radiotherapy, that may be eventually interrupted, resulting in poor prognosis and increased mortality190. Moreover, patients suffering from wasting of diaphragm and/or cardiac muscles often die prematurely because of respiratory and/or cardiac failure191. Finally, the cancer itself as well as major anti-cancer treatments have a long-lasting, detrimental effect on myocardial function192. It has been shown that cancer survivors have an increased risk of developing cardiac complications, which may manifest even years after cancer clearance and/or completion of oncological treatments192, emphasizing the importance to increase our understanding of the link between cancer and cardiac myopathies. The research for molecular drivers of this tremendous and mostly untreatable complication of cancer has been neglected for a long time, as cachexia has been originally linked to reduced food intake. More recently, research on cachexia sparked a novel interest as it is emerging as specifically driven by defined molecular alterations, hence it can be modeled and targeted independently from tumor growth.
While the field of cachexia mainly developed as intertwined with the modeling of skeletal muscle atrophy, cardiac wasting is gaining interest as a major cause of death191. Hence, the definition of the mechanism of cardiac wasting holds great potential for the management of cachexia.
The list of inter- and intracellular signaling pathways and molecules presented here is far from being exhaustive, which reflects the rapid development of the field and the complexity of the molecular regulation of cachexia but provides a framework to address the potential analogies between cardiac and muscular wasting. Taken together, inter- and intracellular signaling pathways stand as a central mechanism controlling the autophagy-lysosomal pathway, the ubiquitin-proteasome pathway as well as immunological and metabolic changes during cancer, and integrating the complex phenomenon of cancer cachexia193. Further investigations are needed to identify details and differences of cancer-induced cachexia in the skeletal and heart muscle. Consequently, striving for further investigation of the molecular background and the interplay between cancer, metabolism, and cardiac cachexia is essential to improve treatment of cancer patients.
References
Fearon, K. C., Glass, D. J. & Guttridge, D. C. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. 16, 153–166 (2012).
Argiles, J. M., Stemmler, B., Lopez-Soriano, F. J. & Busquets, S. Inter-tissue communication in cancer cachexia. Nat. Rev. Endocrinol. 15, 9–20 (2018).
Porporato, P. E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 5, e200 (2016).
Brancaccio, M., Pirozzi, F., Hirsch, E. & Ghigo, A. Mechanisms underlying the cross-talk between heart and cancer. J. Physiol. https://doi.org/10.1113/JP276746 (2019).
von Haehling, S. et al. Muscle wasting as an independent predictor of survival in patients with chronic heart failure. J. Cachexia Sarcopenia Muscle. https://doi.org/10.1002/jcsm.12603 (2020).
Schrepper, A., Schwarzer, M., Schope, M., Amorim, P. A. & Doenst, T. Biphasic response of skeletal muscle mitochondria to chronic cardiac pressure overload - role of respiratory chain complex activity. J. Mol. Cell Cardiol. 52, 125–135 (2012).
Dalla Libera, L. et al. Apoptosis in the skeletal muscle of rats with heart failure is associated with increased serum levels of TNF-alpha and sphingosine. J. Mol. Cell Cardiol. 33, 1871–1878 (2001).
Nguyen, T. et al. Expression of MuRF1 or MuRF2 is essential for the induction of skeletal muscle atrophy and dysfunction in a murine pulmonary hypertension model. Skelet. Muscle 10, 12 (2020).
Sala, V. et al. A new transgenic mouse model of heart failure and cardiac cachexia raised by sustained activation of met tyrosine kinase in the heart. Biomed. Res. Int. 2016, 9549036 (2016).
Fulster, S. et al. Muscle wasting in patients with chronic heart failure: results from the studies investigating co-morbidities aggravating heart failure (SICA-HF). Eur. Heart J. 34, 512–519 (2013).
Barkhudaryan, A., Scherbakov, N., Springer, J. & Doehner, W. Cardiac muscle wasting in individuals with cancer cachexia. ESC Heart Fail 4, 458–467 (2017).
Riccardi, D. et al. Plasma lipid profile and systemic inflammation in patients with cancer cachexia. Front Nutr. 7, 4 (2020).
Sin, T. K. et al. Cancer takes a toll on skeletal muscle by releasing heat shock proteins-an emerging mechanism of cancer-induced cachexia. Cancers. https://doi.org/10.3390/cancers11091272 (2019).
Kim, S. et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457, 102–106 (2009).
Zhang, Q. et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010).
Manole, E., Ceafalan, L. C., Popescu, B. O., Dumitru, C. & Bastian, A. E. Myokines as possible therapeutic targets in cancer cachexia. J. Immunol. Res. 2018, 8260742 (2018).
Li, Y. P., Schwartz, R. J., Waddell, I. D., Holloway, B. R. & Reid, M. B. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. FASEB J. 12, 871–880 (1998).
Li, Y. P. et al. TNF-alpha increases ubiquitin-conjugating activity in skeletal muscle by up-regulating UbcH2/E220k. FASEB J. 17, 1048–1057 (2003).
Ding, H. et al. Activin A induces skeletal muscle catabolism via p38beta mitogen-activated protein kinase. J. Cachexia Sarcopenia Muscle 8, 202–212 (2017).
Li, Y. P. et al. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 19, 362–370 (2005).
Bonetto, A. et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 303, E410–421 (2012).
Konstantinov, B. A., Tarichko, Iu,V., Cherepenion, L. P., Shabalkin, B. V. & Osipov, V. P. [Mortality in cardiac surgery]. Ter. Arkh. 61, 6–10 (1989).
Abrigo, J. et al. Role of oxidative stress as key regulator of muscle wasting during cachexia. Oxid. Med. Cell Longev. 2018, 2063179 (2018).
Milan, G. et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 6, 6670 (2015).
Bustamante, H. A. et al. Interplay between the autophagy-lysosomal pathway and the ubiquitin-proteasome system: A target for therapeutic development in Alzheimer’s disease. Front. Cell Neurosci. 12, 126 (2018).
Yoshii, S. R. & Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. https://doi.org/10.3390/ijms18091865 (2017).
Grice, G. L. & Nathan, J. A. The recognition of ubiquitinated proteins by the proteasome. Cell Mol. Life Sci. 73, 3497–3506 (2016).
Cosper, P. F. & Leinwand, L. A. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res. 71, 1710–1720 (2011).
Musolino, V. et al. Megestrol acetate improves cardiac function in a model of cancer cachexia-induced cardiomyopathy by autophagic modulation. J. Cachexia Sarcopenia Muscle 7, 555–566 (2016).
Paul, P. K. & Kumar, A. TRAF6 coordinates the activation of autophagy and ubiquitin-proteasome systems in atrophying skeletal muscle. Autophagy 7, 555–556 (2011).
Paul, P. K. et al. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J. Cell Biol. 191, 1395–1411 (2010).
Manne, N. D. et al. Altered cardiac muscle mTOR regulation during the progression of cancer cachexia in the ApcMin/+ mouse. Int. J. Oncol. 42, 2134–2140 (2013).
Schips, T. G. et al. FoxO3 induces reversible cardiac atrophy and autophagy in a transgenic mouse model. Cardiovasc. Res. 91, 587–597 (2011).
Usui, S. et al. Endogenous muscle atrophy F-box mediates pressure overload-induced cardiac hypertrophy through regulation of nuclear factor-kappaB. Circ. Res. 109, 161–171 (2011).
Zaglia, T. et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. J. Clin. Invest. 124, 2410–2424 (2014).
Zaglia, T. et al. Cardiac sympathetic neurons provide trophic signal to the heart via beta2-adrenoceptor-dependent regulation of proteolysis. Cardiovasc. Res. 97, 240–250 (2013).
Tian, M., Asp, M. L., Nishijima, Y. & Belury, M. A. Evidence for cardiac atrophic remodeling in cancer-induced cachexia in mice. Int. J. Oncol. 39, 1321–1326 (2011).
Baracos, V. E., Martin, L., Korc, M., Guttridge, D. C. & Fearon, K. C. H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 4, 17105 (2018).
Puppa, M. J., Gao, S., Narsale, A. A. & Carson, J. A. Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J. 28, 998–1009 (2014).
Kandarian, S. C. et al. Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour-bearing mice. J. Cachexia Sarcopenia Muscle 9, 1109–1120 (2018).
Li, W., Moylan, J. S., Chambers, M. A., Smith, J. & Reid, M. B. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 297, C706–714 (2009).
Callaway, C. S. et al. IL-8 released from human pancreatic cancer and tumor-associated stromal cells signals through a CXCR2-ERK1/2 axis to induce muscle atrophy. Cancers. https://doi.org/10.3390/cancers11121863 (2019).
Tracey, K. J., Lowry, S. F. & Cerami, A. The pathophysiologic role of cachectin/TNF in septic shock and cachexia. Ann. Inst. Pasteur Immunol. 139, 311–317 (1988).
Guttridge, D. C., Mayo, M. W., Madrid, L. V., Wang, C. Y. & Baldwin, A. S. Jr. NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289, 2363–2366 (2000).
He, W. A. et al. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Invest. 123, 4821–4835 (2013).
Wang, G. et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat. Med. 24, 770–781 (2018).
Shakri, A. R. et al. Upregulation of ZIP14 and altered zinc homeostasis in muscles in pancreatic cancer cachexia. Cancers. https://doi.org/10.3390/cancers12010003 (2019).
Michaelis, K. A. et al. Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J. Cachexia Sarcopenia Muscle 8, 824–838 (2017).
Aydemir, T. B. & Cousins, R. J. The multiple faces of the metal transporter ZIP14 (SLC39A14). J. Nutr. 148, 174–184 (2018).
Olgar, Y. et al. Increased free Zn(2+) correlates induction of sarco(endo)plasmic reticulum stress via altered expression levels of Zn(2+) -transporters in heart failure. J. Cell. Mol. Med. 22, 1944–1956 (2018).
Greco, S. H. et al. TGF-beta blockade reduces mortality and metabolic changes in a validated murine model of pancreatic cancer cachexia. PLoS ONE 10, e0132786 (2015).
Wakefield, L. M. & Hill, C. S. Beyond TGFbeta: roles of other TGFbeta superfamily members in cancer. Nat. Rev. Cancer 13, 328–341 (2013).
Waning, D. L. et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat. Med. 21, 1262–1271 (2015).
Langley, B. et al. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 277, 49831–49840 (2002).
McCroskery, S., Thomas, M., Maxwell, L., Sharma, M. & Kambadur, R. Myostatin negatively regulates satellite cell activation and self-renewal. J. Cell Biol. 162, 1135–1147 (2003).
Zimmers, T. A. et al. Induction of cachexia in mice by systemically administered myostatin. Science 296, 1486–1488 (2002).
Costelli, P. et al. Muscle myostatin signalling is enhanced in experimental cancer cachexia. Eur. J. Clin. Invest. 38, 531–538 (2008).
Ishida, J. et al. Myostatin signaling is up-regulated in female patients with advanced heart failure. Int. J. Cardiol. 238, 37–42 (2017).
Heineke, J. et al. Genetic deletion of myostatin from the heart prevents skeletal muscle atrophy in heart failure. Circulation 121, 419–425 (2010).
Loumaye, A. et al. Circulating Activin A predicts survival in cancer patients. J. Cachexia Sarcopenia Muscle 8, 768–777 (2017).
Zhou, X. et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142, 531–543 (2010).
Hatakeyama, S. et al. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti-cancer treatments. Skelet. Muscle 6, 26 (2016).
Kavazis, A. N., Smuder, A. J. & Powers, S. K. Effects of short-term endurance exercise training on acute doxorubicin-induced FoxO transcription in cardiac and skeletal muscle. J. Appl Physiol. 117, 223–230 (2014).
Hulmi, J. J. et al. Prevention of chemotherapy-induced cachexia by ACVR2B ligand blocking has different effects on heart and skeletal muscle. J. Cachexia Sarcopenia Muscle 9, 417–432 (2018).
Fields, S. Z. et al. Activin receptor antagonists for cancer-related anemia and bone disease. Expert Opin. Investig. Drugs 22, 87–101 (2013).
Tao, J. J. et al. First-in-human phase I study of the activin A inhibitor, STM 434, in patients with granulosa cell ovarian cancer and other advanced solid tumors. Clin. Cancer Res. 25, 5458–5465 (2019).
Rooks, D. S. et al. Effect of bimagrumab on thigh muscle volume and composition in men with casting-induced atrophy. J. Cachexia Sarcopenia Muscle 8, 727–734 (2017).
Polkey, M. I. et al. Activin type II receptor blockade for treatment of muscle depletion in chronic obstructive pulmonary disease. A randomized trial. Am. J. Respir. Crit. Care Med. 199, 313–320 (2019).
Egerman, M. A. & Glass, D. J. The role of GDF11 in aging and skeletal muscle, cardiac and bone homeostasis. Crit. Rev. Biochem. Mol. Biol. 54, 174–183 (2019).
Zimmers, T. A. et al. Exogenous GDF11 induces cardiac and skeletal muscle dysfunction and wasting. Basic Res. Cardiol. 112, 48 (2017).
Hammers, D. W. et al. Supraphysiological levels of GDF11 induce striated muscle atrophy. EMBO Mol. Med. 9, 531–544 (2017).
Harper, S. C. et al. GDF11 decreases pressure overload-induced hypertrophy, but can cause severe cachexia and premature death. Circ. Res. 123, 1220–1231 (2018).
Jones, J. E. et al. Supraphysiologic administration of GDF11 induces cachexia in part by upregulating GDF15. Cell Rep. 22, 1522–1530 (2018).
Lerner, L. et al. MAP3K11/GDF15 axis is a critical driver of cancer cachexia. J. Cachexia Sarcopenia Muscle 7, 467–482 (2016).
Ost, M. et al. Muscle-derived GDF15 drives diurnal anorexia and systemic metabolic remodeling during mitochondrial stress. EMBO Rep. 21, e48804 (2020).
Ost, M., Coleman, V., Kasch, J. & Klaus, S. Regulation of myokine expression: role of exercise and cellular stress. Free Radic. Biol. Med. 98, 78–89 (2016).
Rainer, P. P. et al. Cardiomyocyte-specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ. Res. 114, 1246–1257 (2014).
Emmerson, P. J. et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 23, 1215–1219 (2017).
Yang, L. et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 23, 1158–1166 (2017).
Mullican, S. E. et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 23, 1150–1157 (2017).
Johnen, H. et al. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat. Med. 13, 1333–1340 (2007).
Lerner, L. et al. Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J. Cachexia Sarcopenia Muscle 6, 317–324 (2015).
Villars, F. O., Pietra, C., Giuliano, C., Lutz, T. A. & Riediger, T. Oral treatment with the ghrelin receptor agonist HM01 attenuates cachexia in mice bearing colon-26 (C26) tumors. Int. J. Mol. Sci. https://doi.org/10.3390/ijms18050986 (2017).
Borner, T. et al. Anorexia-cachexia syndrome in hepatoma tumour-bearing rats requires the area postrema but not vagal afferents and is paralleled by increased MIC-1/GDF15. J. Cachexia Sarcopenia Muscle 8, 417–427 (2017).
Borner, T. et al. GDF15 induces anorexia through nausea and emesis. Cell Metab. 31, 351–362. e355 (2020).
Silk, E., Zhao, H., Weng, H. & Ma, D. The role of extracellular histone in organ injury. Cell Death Dis. 8, e2812 (2017).
Li, M. et al. Phosphoinositide 3-kinase gamma inhibition protects from anthracycline cardiotoxicity and reduces tumor growth. Circulation 138, 696–711 (2018).
Zhang, G. et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat. Commun. 8, 589 (2017).
Fitzgerald, K. A. & Kagan, J. C. Toll-like receptors and the control of immunity. Cell 180, 1044–1066 (2020).
Takeda, K. & Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 17, 1–14 (2005).
Langen, R. C. et al. NF-kappaB activation is required for the transition of pulmonary inflammation to muscle atrophy. Am. J. Respir. Cell Mol. Biol. 47, 288–297 (2012).
Cai, D. et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119, 285–298 (2004).
Zhang, G. et al. Toll-like receptor 4 mediates Lewis lung carcinoma-induced muscle wasting via coordinate activation of protein degradation pathways. Sci. Rep. 7, 2273 (2017).
Cannon, T. Y. et al. The effect of altered Toll-like receptor 4 signaling on cancer cachexia. Arch. Otolaryngol. Head Neck Surg. 133, 1263–1269 (2007).
Johns, N. et al. New genetic signatures associated with cancer cachexia as defined by low skeletal muscle index and weight loss. J. Cachexia Sarcopenia Muscle 8, 122–130 (2017).
He, W. A. et al. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc. Natl Acad. Sci. USA 111, 4525–4529 (2014).
Calore, F. et al. The TLR7/8/9 antagonist IMO-8503 inhibits cancer-induced cachexia. Cancer Res. 78, 6680–6690 (2018).
Michaelis, K. A. et al. The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer. Nat. Commun. 10, 4682 (2019).
Boyd, J. H., Mathur, S., Wang, Y., Bateman, R. M. & Walley, K. R. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc. Res. 72, 384–393 (2006).
Hall, G., Hasday, J. D. & Rogers, T. B. Regulating the regulator: NF-kappaB signaling in heart. J. Mol. Cell Cardiol. 41, 580–591 (2006).
Shah, M., Yellon, D. M. & Davidson, S. M. The role of extracellular DNA and histones in ischaemia-reperfusion injury of the myocardium. Cardiovasc. Drugs Ther. 34, 123–131 (2020).
Toldo, S. & Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 15, 203–214 (2018).
Bindels, L. B. et al. Synbiotic approach restores intestinal homeostasis and prolongs survival in leukaemic mice with cachexia. ISME J. 10, 1456–1470 (2016).
Genton, L. et al. Targeting the gut microbiota to treat cachexia. Front. Cell Infect. Microbiol. 9, 305 (2019).
Kir, S. & Spiegelman, B. M. Cachexia & brown fat: a burning issue in cancer. Trends Cancer 2, 461–463 (2016).
Kir, S. et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 513, 100–104 (2014).
Fenninger, L. D. & Mider, G. B. Energy and nitrogen metabolism in cancer. Adv. Cancer Res. 2, 229–253 (1954).
Luo, Y. et al. Cancer usurps skeletal muscle as an energy repository. Cancer Res. 74, 330–340 (2014).
Julienne, C. M. et al. Cardiolipin content is involved in liver mitochondrial energy wasting associated with cancer-induced cachexia without the involvement of adenine nucleotide translocase. Biochim. Biophys. Acta 1842, 726–733 (2014).
Petruzzelli, M. et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 20, 433–447 (2014).
Kir, S. et al. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab. 23, 315–323 (2016).
Okugawa, Y. et al. Circulating miR-203 derived from metastatic tissues promotes myopenia in colorectal cancer patients. J. Cachexia Sarcopenia Muscle 10, 536–548 (2019).
Broadus, A. E. et al. Identification of a novel parathyroid hormone-like peptide. N. Engl. J. Med. 319, 556–563 (1988).
Karlstaedt, A. et al. Oncometabolite d-2-hydroxyglutarate impairs alpha-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc. Natl Acad. Sci. USA 113, 10436–10441 (2016).
Assuncao, B. et al. Acute leukemia is associated with cardiac alterations before chemotherapy. J. Am. Soc. Echocardiogr. 30, 1111–1118 (2017).
Kang, Y. et al. Symptomatic heart failure in acute leukemia patients treated with anthracyclines. JACC CardioOncol 1, 208–217 (2019).
Neuendorff, N. R. et al. Anthracycline-related cardiotoxicity in older patients with acute myeloid leukemia: a Young SIOG review paper. Blood Adv. 4, 762–775 (2020).
Dhanapal, R., Saraswathi, T. & Govind, R. N. Cancer cachexia. J. Oral. Maxillofac. Pathol. 15, 257–260 (2011).
Masri, S. et al. Lung adenocarcinoma distally rewires hepatic circadian homeostasis. Cell 165, 896–909 (2016).
Ginsberg, H. N. Insulin resistance and cardiovascular disease. J. Clin. Invest. 106, 453–458 (2000).
Wagner, E. F. & Petruzzelli, M. Cancer metabolism: a waste of insulin interference. Nature 521, 430–431 (2015).
Rofe, A. M., Bourgeois, C. S., Coyle, P., Taylor, A. & Abdi, E. A. Altered insulin response to glucose in weight-losing cancer patients. Anticancer Res. 14, 647–650 (1994).
Ye, H. et al. Subversion of systemic glucose metabolism as a mechanism to support the growth of leukemia cells. Cancer Cell 34, 659–73 e656 (2018).
Petruzzelli, M. & Wagner, E. F. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 30, 489–501 (2016).
Costelli, P. et al. IGF-1 is downregulated in experimental cancer cachexia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R674–683 (2006).
Thackeray, J. T. et al. Insulin supplementation attenuates cancer-induced cardiomyopathy and slows tumor disease progression. JCI Insight. https://doi.org/10.1172/jci.insight.93098 (2017).
Fukawa, T. et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 22, 666–671 (2016).
Wyart, E. et al. Metabolic alterations in a slow-paced model of pancreatic cancer-induced wasting. Oxid. Med. Cell Longev. 2018, 6419805 (2018).
Molinari, F. et al. The mitochondrial metabolic reprogramming agent trimetazidine as an ‘exercise mimetic’ in cachectic C26-bearing mice. J. Cachexia Sarcopenia Muscle 8, 954–973 (2017).
Das, S. K. et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 333, 233–238 (2011).
Koves, T. R. et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56 (2008).
Muoio, D. M. & Neufer, P. D. Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metab. 15, 595–605 (2012).
Penna, F., Ballaro, R., Beltra, M., De Lucia, S. & Costelli, P. Modulating metabolism to improve cancer-induced muscle wasting. Oxid. Med. Cell Longev. 2018, 7153610 (2018).
Liu, Y. et al. TLR9 and beclin 1 crosstalk regulates muscle AMPK activation in exercise. Nature 578, 605–609 (2020).
Camaschella, C., Nai, A. & Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 105, 260–272 (2020).
Bekfani, T. et al. Iron deficiency in patients with heart failure with preserved ejection fraction and its association with reduced exercise capacity, muscle strength and quality of life. Clin. Res. Cardiol. 108, 203–211 (2019).
Chung, Y. J. et al. Iron-deficiency anemia reduces cardiac contraction by downregulating RyR2 channels and suppressing SERCA pump activity. JCI Insight 4, e125618 (2019).
von Haehling, S., Ebner, N., Evertz, R., Ponikowski, P. & Anker, S. D. Iron deficiency in heart failure: an overview. JACC Heart Fail 7, 36–46 (2019).
Manz, D. H., Blanchette, N. L., Paul, B. T., Torti, F. M. & Torti, S. V. Iron and cancer: recent insights. Ann. N. Y. Acad. Sci. 1368, 149–161 (2016).
Ghafourian, K., Chang, H. C. & Ardehali, H. Intravenous iron therapy in heart failure: a different perspective. Eur. J. Heart Fail 21, 703–714 (2019).
Chang, H. C. et al. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol. Med. 8, 247–267 (2016).
Nagatomo, Y. & Tang, W. H. Intersections between microbiome and heart failure: revisiting the gut hypothesis. J. Card. Fail. 21, 973–980 (2015).
Nemet, I. et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell 180, 862–877.e822 (2020).
Bu, J. & Wang, Z. Cross-talk between gut microbiota and heart via the routes of metabolite and immunity. Gastroenterol. Res. Pract. 2018, 6458094 (2018).
Ascher, S. & Reinhardt, C. The gut microbiota: an emerging risk factor for cardiovascular and cerebrovascular disease. Eur. J. Immunol. 48, 564–575 (2018).
Pevsner-Fischer, M. et al. Role of the microbiome in non-gastrointestinal cancers. World J. Clin. Oncol. 7, 200–213 (2016).
Konturek, P. C. et al. Emerging role of fecal microbiota therapy in the treatment of gastrointestinal and extra-gastrointestinal diseases. J. Physiol. Pharmacol. 66, 483–491 (2015).
Genton, L., Cani, P. D. & Schrenzel, J. Alterations of gut barrier and gut microbiota in food restriction, food deprivation and protein-energy wasting. Clin. Nutr. 34, 341–349 (2015).
Erridge, C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J. Leukoc. Biol. 87, 989–999 (2010).
Tanaka, K. et al. Impact of perioperative administration of synbiotics in patients with esophageal cancer undergoing esophagectomy: a prospective randomized controlled trial. Surgery 152, 832–842 (2012).
Bindels, L. B. et al. Non digestible oligosaccharides modulate the gut microbiota to control the development of leukemia and associated cachexia in mice. PLoS ONE 10, e0131009 (2015).
Russo, M. et al. The novel butyrate derivative phenylalanine-butyramide protects from doxorubicin-induced cardiotoxicity. Eur. J. Heart Fail. 21, 519–528 (2019).
Pin, F., Couch, M. E. & Bonetto, A. Preservation of muscle mass as a strategy to reduce the toxic effects of cancer chemotherapy on body composition. Curr. Opin. Support Palliat. Care 12, 420–426 (2018).
Pin, F., Barreto, R., Couch, M. E., Bonetto, A. & O’Connell, T. M. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J. Cachexia Sarcopenia Muscle 10, 140–154 (2019).
Babiker, H. M. et al. Cardiotoxic effects of chemotherapy: a review of both cytotoxic and molecular targeted oncology therapies and their effect on the cardiovascular system. Crit. Rev. Oncol. Hematol. 126, 186–200 (2018).
Altena, R. et al. Growth differentiation factor 15 (GDF-15) plasma levels increase during bleomycin- and cisplatin-based treatment of testicular cancer patients and relate to endothelial damage. PLoS ONE 10, e0115372 (2015).
Edwardson, D. W. et al. Inflammatory cytokine production in tumor cells upon chemotherapy drug exposure or upon selection for drug resistance. PLoS ONE 12, e0183662 (2017).
Pogrebniak, H. W., Matthews, W. & Pass, H. I. Chemotherapy amplifies production of tumor necrosis factor. Surgery 110, 231–237 (1991).
Schiessel, D. L. & Baracos, V. E. Barriers to cancer nutrition therapy: excess catabolism of muscle and adipose tissues induced by tumour products and chemotherapy. Proc. Nutr. Soc. 77, 394–402 (2018).
Sirago, G. et al. Growth hormone secretagogues hexarelin and JMV2894 protect skeletal muscle from mitochondrial damages in a rat model of cisplatin-induced cachexia. Sci. Rep. 7, 13017 (2017).
Sorensen, J. C. et al. BGP-15 protects against oxaliplatin-induced skeletal myopathy and mitochondrial reactive oxygen species production in mice. Front. Pharmacol. 8, 137 (2017).
Fanzani, A., Zanola, A., Rovetta, F., Rossi, S. & Aleo, M. F. Cisplatin triggers atrophy of skeletal C2C12 myotubes via impairment of Akt signalling pathway and subsequent increment activity of proteasome and autophagy systems. Toxicol. Appl. Pharmacol. 250, 312–321 (2011).
Braun, T. P. et al. Muscle atrophy in response to cytotoxic chemotherapy is dependent on intact glucocorticoid signaling in skeletal muscle. PLoS ONE 9, e106489 (2014).
Montalvo, R. N., Doerr, V., Min, K., Szeto, H. H. & Smuder, A. J. Doxorubicin-induced oxidative stress differentially regulates proteolytic signaling in cardiac and skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 318, R227–R233 (2020).
Li, M., Russo, M., Pirozzi, F., Tocchetti, C. G. & Ghigo, A. Autophagy and cancer therapy cardiotoxicity: from molecular mechanisms to therapeutic opportunities. Biochim. Biophys. Acta Mol. Cell Res. 1867, 118493 (2020).
Li, D. L. et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 133, 1668–1687 (2016).
Abdullah, C. S. et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 9, 2002 (2019).
Hiensch, A. E. et al. Doxorubicin-induced skeletal muscle atrophy: Elucidating the underlying molecular pathways. Acta Physiol. 229, 1–18 (2020).
Gilliam, L. A. & St Clair, D. K. Chemotherapy-induced weakness and fatigue in skeletal muscle: the role of oxidative stress. Antioxid. Redox Signal. 15, 2543–2563 (2011).
Gilliam, L. A. A. et al. The anticancer agent doxorubicin disrupts mitochondrial energy metabolism and redox balance in skeletal muscle. Free Radic. Biol. Med. 65, 988–996 (2013).
Gilliam, L. A. et al. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 302, C195–202 (2012).
Smuder, A. J., Kavazis, A. N., Min, K. & Powers, S. K. Exercise protects against doxorubicin-induced oxidative stress and proteolysis in skeletal muscle. J. Appl. Physiol. 110, 935–942 (2011).
Smuder, A. J., Kavazis, A. N., Hudson, M. B., Nelson, W. B. & Powers, S. K. Oxidation enhances myofibrillar protein degradation via calpain and caspase-3. Free Radic. Biol. Med. 49, 1152–1160 (2010).
Min, K. et al. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J. Physiol. 593, 2017–2036 (2015).
Smuder, A. J., Kavazis, A. N., Min, K. & Powers, S. K. Exercise protects against doxorubicin-induced markers of autophagy signaling in skeletal muscle. J. Appl Physiol. 111, 1190–1198 (2011).
Laurent, T. C., Fraser, J. R., Pertoft, H. & Smedsrod, B. Binding of hyaluronate and chondroitin sulphate to liver endothelial cells. Biochem. J. 234, 653–658 (1986).
Xia, P. et al. Doxorubicin induces cardiomyocyte apoptosis and atrophy through cyclin-dependent kinase 2-mediated activation of forkhead box O1. J. Biol. Chem. 295, 4265–4276 (2020).
Chan, B. Y. H. et al. MMP inhibitors attenuate doxorubicin cardiotoxicity by preventing intracellular and extracellular matrix remodeling. Cardiovasc. Res. https://doi.org/10.1093/cvr/cvaa017 (2020).
Ren, X., Lamb, G. D. & Murphy, R. M. Distribution and activation of matrix metalloproteinase-2 in skeletal muscle fibers. Am. J. Physiol. Cell Physiol. 317, C613–C625 (2019).
Sun, S. et al. Measurement of a MMP-2 degraded Titin fragment in serum reflects changes in muscle turnover induced by atrophy. Exp. Gerontol. 58, 83–89 (2014).
Peng, J., Raddatz, K., Labeit, S., Granzier, H. & Gotthardt, M. Muscle atrophy in titin M-line deficient mice. J. Muscle Res. Cell Motil. 26, 381–388 (2005).
Radke, M. H. et al. Deleting full length titin versus the titin M-band region leads to differential mechanosignaling and cardiac phenotypes. Circulation 139, 1813–1827 (2019).
Radke, M. H. et al. Targeted deletion of titin N2B region leads to diastolic dysfunction and cardiac atrophy. Proc. Natl Acad. Sci. USA 104, 3444–3449 (2007).
Gupta, S. K. et al. Quaking inhibits doxorubicin-mediated cardiotoxicity through regulation of cardiac circular RNA expression. Circ. Res. 122, 246–254 (2018).
Nissinen, T. A. et al. Systemic blockade of ACVR2B ligands prevents chemotherapy-induced muscle wasting by restoring muscle protein synthesis without affecting oxidative capacity or atrogenes. Sci. Rep. 6, 32695 (2016).
de Lima Junior, E. A. et al. Doxorubicin caused severe hyperglycaemia and insulin resistance, mediated by inhibition in AMPk signalling in skeletal muscle. J. Cachexia Sarcopenia Muscle 7, 615–625 (2016).
Zima, T. et al. Acute doxorubicin (adriamycin) dosage does not reduce cardiac protein synthesis in vivo, but decreases diaminopeptidase I and proline endopeptidase activities. Exp. Mol. Pathol. 70, 154–161 (2001).
Timm, K. N. & Tyler, D. J. The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Drugs Ther. 34, 255–269 (2020).
Schnackenberg, L. K. et al. Early metabolomics changes in heart and plasma during chronic doxorubicin treatment in B6C3F1 mice. J. Appl. Toxicol. 36, 1486–1495 (2016).
Ballaro, R. et al. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J. 33, 5482–5494 (2019).
Valentova, M., Anker, S. D. & von Haehling, S. Cardiac cachexia revisited: the role of wasting in heart failure. Heart Fail. Clin. 16, 61–69 (2020).
Taegtmeyer, H., Karlstaedt, A., Rees, M. L. & Davogustto, G. Oncometabolic tracks in the heart. Circ. Res. 120, 267–269 (2017).
Buck, M. D. et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76 (2016).
Yu, H., Lee, H., Herrmann, A., Buettner, R. & Jove, R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer 14, 736–746 (2014).
Zimmers, T. A., Fishel, M. L. & Bonetto, A. STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell Dev. Biol. 54, 28–41 (2016).
Silva, K. A. et al. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J. Biol. Chem. 290, 11177–11187 (2015).
Zhang, L. et al. Stat3 activation links a C/EBPdelta to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 18, 368–379 (2013).
Haghikia, A. et al. Signal transducer and activator of transcription 3-mediated regulation of miR-199a-5p links cardiomyocyte and endothelial cell function in the heart: a key role for ubiquitin-conjugating enzymes. Eur. Heart J. 32, 1287–1297 (2011).
Altara, R., Harmancey, R., Didion, S. P., Booz, G. W. & Zouein, F. A. Cardiac STAT3 deficiency impairs contractility and metabolic homeostasis in hypertension. Front. Pharmacol. 7, 436 (2016).
Pietzsch, S., Ricke-Hoch, M., Stapel, B. & Hilfiker-Kleiner, D. Modulation of cardiac AKT and STAT3 signalling in preclinical cancer models and their impact on the heart. Biochim. Biophys. Acta Mol. Cell Res. 1867, 118519 (2020).
Cai, X. et al. Inflammatory factor TNF-alpha promotes the growth of breast cancer via the positive feedback loop of TNFR1/NF-kappaB (and/or p38)/p-STAT3/HBXIP/TNFR1. Oncotarget 8, 58338–58352 (2017).
Kagoya, Y. et al. Positive feedback between NF-kappaB and TNF-alpha promotes leukemia-initiating cell capacity. J. Clin. Invest. 124, 528–542 (2014).
Peterson, J. M. et al. NF-kappaB inhibition rescues cardiac function by remodeling calcium genes in a Duchenne muscular dystrophy model. Nat. Commun. 9, 3431 (2018).
Cuadrado, A. & Nebreda, A. R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 429, 403–417 (2010).
Lluis, F., Perdiguero, E., Nebreda, A. R. & Munoz-Canoves, P. Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends Cell Biol. 16, 36–44 (2006).
Brown, J. L. et al. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 9, 987–1002 (2018).
Wu, Z. et al. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol. Cell Biol. 20, 3951–3964 (2000).
Engel, F. B. et al. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 19, 1175–1187 (2005).
Arabacilar, P. & Marber, M. The case for inhibiting p38 mitogen-activated protein kinase in heart failure. Front. Pharmacol. 6, 102 (2015).
Gross, D. N., van den Heuvel, A. P. & Birnbaum, M. J. The role of FoxO in the regulation of metabolism. Oncogene 27, 2320–2336 (2008).
Furuyama, T., Kitayama, K., Yamashita, H. & Mori, N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem. J. 375, 365–371 (2003).
Sanchez, A. M., Candau, R. B. & Bernardi, H. FoxO transcription factors: their roles in the maintenance of skeletal muscle homeostasis. Cell Mol. Life Sci. 71, 1657–1671 (2014).
Sandri, M. et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412 (2004).
Stitt, T. N. et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 14, 395–403 (2004).
Ni, Y. G. et al. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 114, 1159–1168 (2006).
Ronnebaum, S. M. & Patterson, C. The FoxO family in cardiac function and dysfunction. Annu. Rev. Physiol. 72, 81–94 (2010).
Li, H. H. et al. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J. Clin. Invest. 117, 3211–3223 (2007).
Acknowledgements
Work at the authors’ lab is supported by AIRC (P.E.P.: #MFAG 21564), Ricerca Finalizzata (A.G.: #RF-2013-02354892; V.S.: #GR-2013-02355449), Fondazione Cariplo (V.S.: #GR 2017-0800). V.R. is an AIRC/FIRC fellow (#23951).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
A.G. is co-founder and Board Member of Kither Biotech, a company focused on the development of PI3K inhibitors for airway diseases not in conflict with statements in this article. The other authors have no competing financial interest to disclose in relation to the work described. Work is supported by AIRC (P.E.P.: MFAG 21564; VR:23951), Ricerca Finalizzata (A.G.: RF-2013-02354892; V.S.: GR-2013-02355449), Fondazione Cariplo (V.S.: GR 2017-0800).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rausch, V., Sala, V., Penna, F. et al. Understanding the common mechanisms of heart and skeletal muscle wasting in cancer cachexia. Oncogenesis 10, 1 (2021). https://doi.org/10.1038/s41389-020-00288-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-020-00288-6
This article is cited by
-
Prognostic value of GLIM-defined malnutrition in combination with hand-grip strength or gait speed for the prediction of postoperative outcomes in gastric cancer patients with cachexia
BMC Cancer (2024)
-
Integrating organoids and organ-on-a-chip devices
Nature Reviews Bioengineering (2024)
-
Cardioprotection of voluntary exercise against breast cancer-induced cardiac injury via STAT3
Basic Research in Cardiology (2024)
-
Cancer cachexia: molecular mechanisms and treatment strategies
Journal of Hematology & Oncology (2023)
-
Highlighting the idea of exerkines in the management of cancer patients with cachexia: novel insights and a critical review
BMC Cancer (2023)
