
Abstract
Chronic inflammation has been linked to promotion of tumorigenesis and metastasis in lung. However, due to lack of a relevant animal model for characterization, the underlying mechanism remains elusive. Lung tumor suppressor gene Gprc5a-knockout (ko) mice are susceptible to lung inflammation, tumorigenesis and metastasis, which resembles the pathological features in human patients. Here, we showed that PTGES/PGE2 signaling was highly associated with lung tumorigenesis and metastasis in Gprc5a-ko mice. Interestingly, Ptges-knockout in mouse lung tumor cells, although reduced their stemness and EMT-like features, still formed tumors and lung metastasis in immune-deficient nude mice, but not in immune-competent mice. This suggests that the major role of PTGES/PGE2 signaling in tumorigenicity and lung metastasis is through immunosuppression. Mechanistically, PTGES/PGE2 signaling intrinsically endows tumor cells resistant to T-cell cytotoxicity, and induces cytokines extrinsically for MDSC recruitment, which is crucial for suppression of T-cell immunity. Importantly, targeting PGE2 signaling in Gprc5a-ko mice by PTGES inhibitor suppressed MDSC recruitment, restored T cells, and significantly repressed lung metastasis. Thus, PTGES/PGE2 signaling links immunosuppression and metastasis in an inflammatory lung microenvironment of Gprc5a-ko mouse model.
Similar content being viewed by others


Introduction
Tumor recurrence and metastasis are the major causes of cancer death [1]. Of notion, lung tumor progression and metastasis are often accompanied by inflammatory response [2]. Recently, tissue of chronic inflammation has been linked to suppressed immunity, including suppressed T cells, tumor-associated macrophages (TAM), neutrophils, and myeloid-derived suppressor cells (MDSCs). In particular, accumulated MDSCs can protect the tumor cells from immune-surveillance by constructing pre-metastatic niches. These observations suggest that, immunosuppression via MDSCs in inflammatory microenvironment plays important role in promotion of tumor progression and metastasis.
MDSCs are heterogeneous population consisting of myeloid progenitor cells and immature myeloid cells [3]. MDSC recruitment can be induced by tumor-derived chemokines and cytokines, such as granulocyte-macrophage colony-stimulating factor (GM-CSF) [4,5,6], G-CSF, interleukin (IL)-6 [7], IL-1β, arginase 1 (ARG1), interferon (IFN)-γ [8,9,10]. In clinic, immunosuppressive phenotypes, such as upregulated G-CSF, tumor-related leukocytosis, and neutrophil-to-lymphocyte (NLR), are associated with poor outcome of non-small cell lung cancer (NSCLC) patients [11]. However, the roles and mechanisms of MDSC expansion and activation are not fully understood.
Prostaglandin E2 (PGE2) is a key mediator of inflammation, pain, and fever [12]. PGE2 is one of the most abundant prostaglandins synthesized from arachidonic acid (AA). AA is oxygenated by cyclooxygenase-1 and 2 (COX-1/2) to produce PGG2. PGG2 is subsequently reduced to PGH2. And PGH2 is then converted into several prostanoids (e.g., PGF2α, PGD2, PGI2, TXA2 and PGE2) by a variety of synthases. PGE2 synthases (PGES) convert PGH to PGE2, the terminal product [13, 14]. The isomerization of the endoperoxide PGH2 to PGE2 is catalyzed by three different PGE synthases, cytosolic PGE synthase (cPGES) and two membrane-bound PGE synthases, PTGES and mPGES-2. cPGES and mPGES-2 are constitutive enzymes, whereas PTGES is inducible [13]. PTGES is highly upregulated in inflammatory tissues and tumors [15]. Of notion, PGE2 is markedly increased in many types of human cancers, including lung, colon, bladder, breast and head and neck cancer, and is often associated with a poor prognosis [16,17,18,19,20]. Increased PGE2 has a major impact on intra-tumoral inflammatory cells, promoting the immunosuppressive microenvironment [21, 22]. However, due to lack of an animal model that resembles the pathological features of human disease, the biological roles of PGE2 signaling in immunosuppression and lung metastasis remain unclear.
G protein coupled receptor family C group 5 type A (GPRC5A) is predominately expressed in lung tissues [23,24,25]. Gprc5a-knockout (ko) mice developed spontaneous lung adenocarcinoma [26, 27], indicating that Gprc5a is a lung tumor suppressor gene. Importantly, tumorigenesis in Gprc5a-ko mouse lung is associated with inflammation along with persistent activation of NF-κB, EGFR, and STAT3 signaling [26,27,28], which resembles the pathological features in human lung cancer. Moreover, GPRC5A is repressed in most of NSCLC and all of chronic obstructive pulmonary disease (COPD) [29]. Thus, Gprc5a-ko mice provide a unique animal model to study the mechanistic link between inflammatory response and tumorigenesis/metastasis in lung.
In this study, PTGES/PGE2 signaling was found greatly enhanced in lung tumorigenesis and metastasis in Gprc5a-ko mouse model. We found that, the major mechanism in promotion of lung metastasis is through immunosuppression by PTGES/PGE2 signaling.
Results
PTGES/PGE2 signaling is activated in lung tumor cells of Gprc5a-ko mouse model
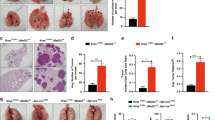
Previously, Gprc5a-ko mice were shown to develop spontaneous lung cancer in 1.5 to 2 years [27], and lung tumorigenesis was associated with pulmonary inflammation [30]. Treatment with tobacco carcinogen NNK-induced lung tumor development in 100% Gprc5a-ko (KO) mice (10/10) but 0% wild-type mice (0/10) in 12 months (Fig. 1a) [27]. To determine which pathways are important in this lung tumor model, we performed RNAseq and pathway analysis on normal mouse tracheal epithelial cells (MTEC) derived from wild-type (WT) mouse and Gprc5a-ko (KO) one. The results showed that arachidonic acid metabolism was one among the significantly upregulated pathways. Importantly, comprehensive analysis of the metabolites showed that PGE2 is greatly upregulated in tumor-bearing NNK-14m-KO mouse lungs, compared to the lungs of NNK-14m-WT mice (Fig. 1b, Supplementary Fig. S1). Consistently, immunoblot showed that PTGES was significantly upregulated in NNK-14m-KO mouse lungs, compared to those of NNK-14m-WT ones (Fig. 1c). Consistently, IHC staining showed that PTGES was upregulated in lung tumor tissues in NNK-14m-KO mouse lungs compared to lung tissues of NNK-14m-WT ones (Fig. 1a). Taken together, these results suggest that upregulated PTGES/PGE2 signaling is correlated with lung tumorigenesis in Gprc5a-ko mouse model.

a Representative images of lung tissues stained with H&E and IHC for Ptges of Gprc5a-ko-NNK-12m mice lung tissue, scale bar = 400 μm (mean ± SD (n = 5)). b Metabonomics analysis of the secretion of PGE2 from NNK-14m-WT and NNK-14m-KO mouse lung tissues (n = 6). c Western blotting analysis of PTGES protein levels in NNK-14m-WT and NNK-14m-KO mouse lung tissues. d Immunoblot analysis of cell lysates from Gprc5a-ko or Gprc5a-wt MTEC with PTGES and GPRC5A antibodies as indicated. e Western blotting analysis of PTGES and ABCG2 protein levels in SJT-1601 cells with and without knockout of Ptges via CRISPR/Cas9 technology. f ELISA kit analysis of the secretion of PGE2 in MTEC-KO and SJT-1601 cells (n = 3). g ELISA kit analysis of the secretion of PGE2 from SJT-1601 and SJT-1601-ko-Ptges cell lines (n = 3). h Colony formation assay of SJT-1601 control and SJT-1601-ko-Ptges cells from 3 separate experiments. i Measurement of tumorsphere formation from 3 separate experiments in SJT-1601 and SJT-1601-ko-Ptges cells. Graph shows the mean ± SD percentage; scale bar = 100 μm. j Flow cytometry analysis of CD44+ in SJT-1601 and SJT-1601-ko-Ptges cells. k Wound healing and migration analysis of SJT-1601 cells with or without PTGES silencing at 24 hours. Scale bar = 100 μm. l Migration of SJT-1601 cells with or without PTGES silencing analyzed by Transwell migration assay (mean ± SD (n = 3)), scale bar = 100 μm. m Western blotting analysis of PTGES and ABCG2 protein levels in HCC827 and HCC827-shPTGES cell lines. n Flow cytometry analysis of CD44+ in HCC827 and HCC827-shPTGES cells (mean ± SD (n = 3)). o Wound healing and migration analysis of HCC827 cells with or without PTGES silencing at 24 hours. Scale bar = 100 μm. p Migration of HCC827 cells with or without PTGES silencing analyzed by Transwell migration assay (mean ± SD (n = 3)), scale bar = 100 μm. *p < 0.05; **p < 0.01; ***p < 0.001.
Next, we asked if there is a causal relationship between GPRC5A-deficiency and PTGES upregulation. Immunoblot analysis showed that Ptges was greatly upregulated in KO-MTEC compared to that in WT-MTEC (Fig. 1d). Of notion, SJT-1601 cells, a lung tumor cell line derived from a Gprc5a-ko mouse lung tumor, were also expressing high level of Ptges (Fig. 1e), suggesting that Ptges overexpression contributes to tumorigenesis in Gprc5a-ko mouse lungs. To determine the biological impact of PTGES overexpression in lung cancer cells, we established 1601-ko-Ptges cells by knockout Ptges in SJT-1601 cells via the CRISPR/Cas9 system (Fig. 1e). Ptges-ko in 1601 cells abolished the expression of Ptges, and the production of PGE2 (Fig. 1f, g), reduced colony formation in colonogenic assay (Fig. 1h, Fig. S2) and anchorage-independent growth (Fig. 1i). Consistently, CD44+ cells, a stem cell-enriched subpopulation, in 1601-shPtges cells (1.99%) were dramatically reduced compared to that in parental 1601 cells (12.5%) (Fig. 1j). Functionally, SJT-1601-ko-Ptges cells exhibited reduced migration (Fig. 1k) and invasion (Fig. 1l) compared to that of parental SJT-1601 cells. To extend the analysis further, we established HCC827-shPTGES cell lines via shRNA, in human NSCLC cell line HCC827 cells that express relatively high levels of PTGES (Fig. 1m). PTGES knockdown also reduced CD44+ subpopulation in HCC827-sh-PTGES cells (55.8%) compared to parental HCC827 cells (75.7%) (Fig. 1n). Consistently, migration and invasion of HCC827-sh-PTGES cells was significantly reduced compared to those of HCC827 cells (Fig. 1o, p). Taken together, these results suggest that the PTGES/PGE2 pathway is essential for the enhanced stemness and pro-metastatic features in human NSCLC and mouse lung cancer cells.
PTGES/PGE2 signaling links immunosuppression and tumorigenicity/lung metastasis in immune-competent mice
To determine the biological impact of PTGES/PGE2 signaling on tumor cells in vivo, we examined the tumorigenicity and lung metastasis of these cells in both immune-deficient nude mice and immune-competent C57BL/6 mice. SJT-1601 vs SJT-1601-ko-Ptges cells were s.c. injected in both nude mice and C57BL/6 mice, and the tumor sizes in these groups were measured. Interestingly, there was no significant difference between the tumorigenicity of SJT-1601 cells and that of SJT-1601-ko-Ptges cells in nude mice (Fig. 2a, b). Similarly, lung metastasis via tail vein injection resulted in similar metastatic nodules (Fig. 2c). However, the tumorigenicity and lung metastasis of 1601-ko-Ptges cells was dramatically suppressed compared to that of SJT-1601 cells in immune-competent C57BL/6 mice (Fig. 2d-f). These results suggest that the major impact of PTGES/PGE2 signaling of tumor cells on tumorigenicity and lung metastasis is through immunosuppression.

a Mouse lung tumor SJT-1601 and SJT-1601-ko-Ptges cells were s.c. injected into nude mice, and tumor size was measured as indicated. b Tumors weight was measured as indicated (mean ± SD (n = 5)). c Nude mice were infected through the tail vein with 5 × 104 SJT-1601 or SJT-1601-ko-Ptges cells. Lungs were removed from the mice at 3 weeks after inoculation. The experimental scheme is presented. Representative images of lung tissues stained with H&E are presented (bottom). Scale bar = 1000 μm. Number of metastatic lesions in the lungs (n = 5). d Mouse lung tumor SJT-1601 and SJT-1601-ko-Ptges cells were s.c. injected into C57BL/6 mice, and tumor size was measured as indicated. e Tumors weight was measured as indicated. f C57BL/6 mice were infected through the tail vein with 5 × 105 SJT-1601 or SJT-1601-ko-Ptges cells. Lungs were removed from the mice at 3 weeks after inoculation. The experimental scheme is presented. Representative images of lung tissues stained with H&E are presented (bottom). Scale bar = 1000 μm. Number of metastatic lesions in the lungs (n = 6). g Gprc5a-ko mice were infected through the tail vein with 5 × 105 SJT-1601 or SJT-1601-ko-Ptges cells. Lungs were removed from the mice at 3 weeks after inoculation. The experimental scheme is presented. Representative images of lung tissues stained with H&E are presented (bottom). Scale bar = 1000μm. Number of metastatic lesions in the lungs (n = 5). h Numbers of metastatic nodules in f and g. i ELISA kit analysis of the secretion of PGE2 in mice lung tissues (n = 3). j C57-WT T-cell-mediated tumor cell killing assay in SJT-1601and SJT-1601-ko-Ptges cells. k Gprc5a-ko T-cell-mediated tumor cell killing assay in SJT-1601and SJT-1601-ko-Ptges cells (n = 3). ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
Gprc5a-deficienct mice were prone to lung tumorigenesis, which was associated with chronic inflammation in lungs [27]. To determine the effect of inflammatory microenvironment on lungs metastasis, we then examined the metastasis of 1601 and 1601-ko-Ptges in Gprc5a-ko mice. 1601-ko-Ptges cells formed more metastatic nodules in Gprc5a-ko mouse lungs compared to those in wild-type mice although still less than 1601 cells (Fig. 2g, h). These results suggest that the immune surveillance in Gprc5a-ko mouse lungs is greatly compromised. Noticeably, the pattern of the lung metastatic nodules of these groups (Fig. 2h) were strongly correlated with the pattern of the PGE2 levels in these groups (Fig. 2i), suggesting that PGE2 signaling contributes to the susceptibility to lung metastasis, or the immunosuppression. To determine the role of PTGES/PGE2 signaling of tumor cells on T-cell immunity, we performed cytolytical T-cell assay on 1601 and 1601-ko-Ptges cells. Activated T lymphocytes did not kill parental 1601 cells, but killed 1601-ko-Ptges cells (Fig. 2j, k). This suggests that, PTGES/PGE2 signaling endows tumor cells resistant to T-cell-mediated cytotoxicity.
Tumor cell-derived PGE2 induces polarization of type II macrophages
Macrophages have 2 types: M1 type promotes anti-tumor immunity, whereas M2 type promotes tumor progression. Tumor-associated macrophages (TAMs) are M2 macrophages, playing an important role in tumor progression and metastasis [31]. The phenotype of M1 and M2 type can be differentiated by the biomarker expressing, such as IFNγ, IL12α in M1, and Arg1, MRC1, IL-6, in M2 type. To determine the extrinsic roles of PTGES/PGE2 signaling in regulation of the immunity, we next examined the effects of the PGE2-containing condition media on differentiation of macrophages. Co-culture of macrophage cells, RAW264.7, with the conditioned medium (CM) from SJT-1601 cells (1601-CM), induced M2 markers, Arg1, MRC1, and enhanced the expression of pro-inflammatory cytokines, TNFα, IL-6; whereas significantly reduced the expression of M1 markers, IFNγ and IL-12α (Fig. 3a). These results suggest that certain released factors in the CM of tumor cells 1601 induce polarization of M2 phenotype. Similarly, co-culture of bone marrow-derived macrophages (BMDM) with 1601-CM greatly increased M2 markers, Arg1, MRC1 and Fizzl (Fig. 3b). However, co-culture of BMDM with the CM from 1601-ko-Ptges cells (KO-CM) lost all of these activities (Fig. 3b), suggesting that Ptges product PGE2 is responsible for the effects. Indeed, addition of exogenous PGE2 largely restored the expression of M2 markers, Arg1 and MRC1 although not Fizzl (Fig. 3b). These results strongly support that, PGE2 is mainly, although not totally, responsible for the effects. IL-6 and TNFα are pro-inflammatory cytokines, but they are also used as M1 markers in some conditions [32]. Interestingly, co-culture of BMDM with the CM from 1601-ko-Ptges cells induced higher levels of IL-6, TNFα and IL-12α than co-culture with CM from 1601 cells; whereas addition of exogenous PGE2 did not induce these gene products (Fig. 3c). These suggest that, in addition to PGE2, there are other released factors in the CM from 1601 cells that are responsible for the induction of IL-6, TNFα, IL-12α, or other immunosuppressive effects.

a The mRNA level of relative expression of Arg1, Mrc1, TNFα, IL-6, IFNγ and IL12α in RAW264.7 cells with or without SJT-1601 condition medium. b, c The mRNA level of genes that are related to TAMs in BMDMs with the condition medium of SJT-1601 and SJT-1601-ko-Ptges cells and PGE2. d Representative FACS images for stained TAM in tumor lung tissues (n = 5). e Statistical analysis of d. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
The ratio of macrophages in lavage fluid is generally correlated with the amount of macrophages in lung tissue, which is in proportion to the tumor burden in lung tissues. FACS analysis of TAMs in lavage fluids from mice of these groups showed that C57-WT mice injected with SJT-1601, contained more M2 macrophages, the subpopulation with marker F4/80 and CD11b + positive, than those injected with SJT-1601-ko-Ptges cells; moreover, Gprc5a-ko mice injected with either 1601 or 1601-ko-Ptges contained high ratio of M2 macrophages (Fig. 3d). Similar results were observed in myeloid cell population (CD45+) (Fig. 3e). The patterns of TAMs and CD45 + cells are very similar to that of lung metastatic nodules in these groups (Fig. 2h). Taken together, these results suggest that, PGE2 from tumor cells can induce polarization of M2 macrophages in vitro; and PTGES/PGE2 signaling-upregulated TAMs and myeloid cells are strongly correlated with immunosuppression and lung metastasis in Gprc5a-ko mouse lungs.
PTGES/PGE2-mediated recruitment of MDSCs is essential for immune suppression
To determine which released factors from Ptges-upregulated tumor cells are mainly mediating immunosuppression, we examined a panel of cytokines and chemokines in the CMs of 1601 cells vs those of 1601-ko-Ptges cells. The assay via Bio-plex MAGPIX system showed that most cytokines and chemokines in the CM of parental 1601 cells were significantly higher than those in the CM of Ptges-ko 1601 cells (Fig. 4a). Of notion, G-CSF, MCP-1, GM-CSF and TNFα were dramatically increased at high level in the CM of SJT-1601 cell cultures compared to that of SJT-1601-ko-Ptges cell cultures (Fig. 4a, left). This suggests that PTGES/PGE2 signaling is essential for production of these cytokines and chemokines.

a Twenty three-factor detection of supernatant medium from SJT-1601 and SJT-1601-ko-Ptges cells. b Representative FACS images for G-MDSCs and M-MDSCs staining in C57BL/6-WT and Gprc5a-ko mice tumor lung tissues (n = 5). c Statistical analysis of b. d Representative FACS images for T cells staining in C57BL/6-WT and Gprc5a-ko mice tumor lung tissues (n = 5). e Statistical analysis of d. f, g Recruit MDSCs experiment under different culture conditions by transwell experiments (n = 3). h–j T-cell-mediated cytotoxicity was assessed in SJT-1601-ko-Ptges cells with or without the condition medium of SJT-1601 and SJT-1601-ko-Ptges cells (n = 3). ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
As G-CSF, GM-CSF, MCP-1, and TNFα are known to be important cytokines for recruitment of MDSCs [33,34,35], we then examined the recruitment of MDSCs in vivo. FACS analysis showed that, more G-MDSCs were recruited in mice injected with 1601 cells than those injected with 1601-ko-Ptges cells (8.0% vs 2.8% in WT mice; and 10.7% vs 6.4% in Gprc5a-ko mice), and more G-MDSCs were recruited in Gprc5a-ko mice than in C57-WT ones (10.7% vs 8.0% for 1601 cells; and 6.4% vs 2.8% for 1601-ko-Ptges cells) (Fig. 4b, c). These results suggest that, (i) active PTGES/PGE2 signaling in tumor cells contributes the increased recruitment of MDSCs; and (ii) G-MDSCs recruitment is enhanced in inflammatory microenvironment of Gprc5a-ko mouse lungs compared to C57-WT mouse lungs. In other words, inflammatory microenvironment of Gprc5a-ko mouse lungs is immunity-compromised, or immunosuppressive, compared to that of wild-type ones.
T cells are the major components of adaptive immunity against tumor cells. Reduced T-cell infiltration is correlated with immunosuppression, tumor progression and metastasis [36]. To determine the status of T-cell infiltration in vivo, we examined the subpopulations of CD8 and CD4 T cells from lungs by FACS assay. The results showed that CD8+ T cells were reduced in C57-WT and Gprc5a-ko mouse lungs that received 1601 cells, compared to those received 1601-ko-Ptges cells (28.7% vs 36.7% in WT mice; and 26.3% vs 38.9% in Gprc5a-ko mice) (Fig. 4d, e). This suggests that PTGES/PGE2 signaling in the metastatic tumor cells is critical for inhibition of CD8+ T-cell infiltration in lungs. Since the effect of PGE2 signaling on CD4+ T-cell infiltration was minimal (Fig. 4e), PGE2 signaling is mainly acting on CD8+ T-cell infiltration.
MDSCs are known for its inhibitory effect on T-cell cytotoxicity or CD8 T-cell activity. Thus, the released factors that are essential for recruitment of MDSCs will be critical. Because G-CSF was the most highly expressed and upregulated cytokine among all cytokines in 1601-CM compared to 1601-ko-Ptges-CM (Fig. 4a, left), we then examined the role of G-CSF on MDSC recruitment by in vitro infiltration assay. The results showed that 1601-CM induced MDSC recruitment whereas KO-CM did not, suggesting the products of the PTGES/PGE2 pathway are required for the effects. Importantly, addition of exogenous G-CSF in presence of KO-CM largely restored MDSC-recruitment, whereas addition of exogenous PGE2 did not (Fig. 4f, g). Interestingly, addition of exogenous G-CSF or PGE2 alone did not increase MDSC-recruitment (Fig. 4f, g). In addition, Gprc5a deletion did not significantly affect the functions of MDSC and CD8 cells (Supplementary Figs. S3, S4). These suggest that G-CSF, in combination with other released factors, exerts the full activity on MDSC-recruitment. Thus, PGE2 signaling-mediated MDSC recruitment is through induction of G-CSF and other released factors.
To determine the effects of MDSCs on T-cell activity, we performed in vitro CTL assay. T cells killed more 1601-ko-Ptges cells than 1601 cells, whereas addition of exogenous MDSCs greatly inhibited T-cell-mediated cytotoxicity in 1601-ko-Ptges tumor cells (Fig. 2j, Fig. 4h, i). Interestingly, addition of 1601-CM, but not KO-CM, inhibited T-cell activity, whereas addition of 1601-CM plus MDSCs further enhanced the inhibition (Fig. 4i, j). These suggest that certain released factors from 1601 cells by the PTGES/PGE2 pathway contribute to MDSC-mediated inhibition of CD8+ T-cell activity. Because the metastasis of 1601 and 1601-ko-Ptges cells were similar in nude mice that have macrophages and NK cells (Fig. 2c), the effect of macrophage and NK on inhibition of lung metastasis is presumably to be negligible or via adaptive immunity. Taken together, PTGES/PGE2 signaling plays two critical roles in lung metastasis: (i) the intrinsic role: PTGES/PGE2 signaling endows tumor cells resistant to T-cell cytotoxicity; and (ii) the extrinsic roles: PGE2 directly induces polarization of M2 macrophages, and through induction of cytokines/chemokines, such as G-CSF, PTGES/PGE2 signaling enhances MDSC recruitment, which inhibits T-cell cytotoxicity. Thus, PTGES/PGE2 signaling is a promising target for inhibiting lung metastasis.
PTGES targeting restores host immunity and inhibits lung metastasis
Due to the critical role of PTGES/PGE2 signaling in immunosuppression, we asked whether targeting PTGES/PGE2 signaling could inhibit lung metastasis efficiently in vivo. Gprc5a-ko mice were i.v. injected with lung tumor cells 1601, seven days later, mice of treatment group were received Cay10526, an inhibitor of PTGES, via i.p. daily injection at 5 mg/kg for 7 days. Mice of all groups were sacrificed at day 21 (Fig. 5a). PGE2 was significantly repressed following the treatment (Fig. 5b). The therapeutic dose of Cay10526 used in this study is 10 times less than that used (50 mg/kg) in literature, suggesting the inhibitory effect on metastasis by PTGES inhibitor is indirect [37]. In fact, no cytotoxic effect was observed in 1601 cells treated with Cay10526 up to 50 μM for 3 days (Supplementary Fig. S5). Lung tumor metastasis was significantly suppressed in treatment group compared to untreated control group (Fig. 5c, d). These suggest that PTGES/PGE2 signaling is critical for lung metastasis in Gprc5a-ko mice. Mechanistically, MDSCs, F4/80+/CD11b+ macrophages, or TAMs, were all significantly suppressed following treatment with Cay10526 (Fig. 5e-h). In contrast, NK, and CD8+, CD4+ T cells were significantly restored following Cay10526 treatment (Fig. 5i-l). Also, CD8+ expression were higher in tumor tissues following Cay10526 treatment (Fig. 5m). Taken together, these results suggest that PTGES targeting, via PTGES inhibitor Cay10526, suppresses MDSC recruitment, and restores T-cell immunity, which contributes to repression of lung metastasis in Gprc5a-ko mouse model.

SJT-1601 cells were i.v. injected into Gprc5a-ko mice and Cay10526 was i.p injected according to the prescribed method (n = 5). a Model diagram of targeting Ptges with Cay10526 in Gprc5a-ko mice lung metastasis model. b ELISA kit analysis of the secretion of PGE2 in mice lung tissues. c Representative images of lung tissues stained with H&E. d Number of metastatic nodules of b. e Representative FACS images for G-MDSCs and M-MDSCs staining in tumor lung tissues. f Statistical analysis of e. g Representative FACS images for stained TAMs cells in tumor lung tissues. h Statistical analysis of g. i Representative FACS images for stained NK cells in tumor lung tissues. j Statistical analysis of i. k Representative FACS images for stained T cells in tumor lung tissues. l Statistical analysis of k. m Representative images of lung tissues with IHC staining for CD8 in control and Cay10526 treatment mice lung tissue, scale bar = 400 μm. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
Dysregulated PTGES is inversely correlated with GPRC5A and CD8+ T lymphocyte infiltration in NSCLCs
Gprc5a-ko mouse lungs are prone to inflammatory stimuli, whereas GPRC5A is repressed in most of NSCLC and all of COPD [29]. Thus, suppressed GPRC5A is a candidate biomarker of the inflammatory status of lung. To explore the relationship between GPRC5A and PTGES, we measured their expression, via immunoblot, in a panel of 21 pairs of NSCLC and adjacent normal lung tissues. The results showed that, PTGES was significantly upregulated in 15 of 21 (71.4%) lung tumor tissues, whereas GPRC5A is significantly downregulated 13 of 21 (61.9%) lung tumor tissues, compared to normal lung tissues (Fig. 6a, b). GPRC5A and PTGES expression were inversely correlated (R = −0.4735, P = 0.0269) (Fig. 6c). To extend this analysis further, we further examined, by IHC staining assay, GPRC5A and PTGES in 50 pairs of lung tissues from normal and tumor tissues. PTGES level was significantly higher in tumors than those in adjacent normal tissues, whereas GPRC5A and CD8+ expression were higher in adjacent normal than tumor tissues (Fig. 6d, e). Of notion, even in the same lung tumor tissues, those areas with high PTGES were low in CD8 and GPRC5A; whereas those areas with low PTGES were high in CD8 and GPRC5A (Fig. 6f). Statistical analysis indicated that PTGES was inversely correlated with GPRC5A in NSCLC (R = −0.4735, P < 0.0001) (Fig. 6g). And, PTGES was also inversely correlated with CD8 in expression (R = −0.3652, P = 0.0003) (Fig. 6g). Collectively, these results support the notion that inflammatory lungs are associated with activated PTGES, whereas activated PTGES/PGE2 signaling inhibits the infiltration of CD8+ T cells in NSCLCs.

a A subset of lung tumor and normal tissues were subjected to western blotting to examine the PTGES and GPRC5A protein levels (n = 21 pairs). b Western blotting scores of PTGES and GPRC5A from the tissues as indicated. c PTGES and GPRC5A expression patterns being highly negative correlated in the samples. d Representative staining of GPRC5A, PTGES and CD8 in lung carcinoma and peritumoral lung tissues of patients from tissue chip samples (n = 50 pairs). e IHC scores of GPRC5A, PTGES and CD8 from the tissue chip as indicated. f Representative images of human lung tissues chip including tumor and adjacent normal tissues stained with IHC for GPRC5A, PTGES and CD8. g Correlation expression analysis of PTGES, GPRC5A and CD8 in human lung tissues chip. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
Upregulated PTGES in lung cancer patients predicts poor overall survival
To extend the analysis further, we also analyzed GPRC5A mRNA levels in human tumor samples in TCGA using the GEPIA database. The results showed that, GPRC5A expression (black bar) was high in lung tissues, but significantly reduced in human lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) although its expression in other type of tumors (red) was upregulated (Fig. 7a). Consistently, GEO database (GSE10072) showed that the expression levels of GPRC5A was high and PTGES was low in normal human lung tissue; whereas the expression levels of these genes in lung tumor tissues were opposite in (Fig. 7b). Statistically, GPRC5A was inversely correlated with PTGES in mRNA levels (Fig. 7c). Moreover, lung cancer patients with either low GPRC5A or high PTGES expression exhibited poor overall survival (Fig. 7d, e). Taken together, these results suggest that repressed GPRC5A and upregulated PTGES predicts poor overall survival in patients with NSCLCs.

a Comparison of mRNA expression levels of GPRC5A in cancer and adjacent normal tissues of different cancer types in TCGA database. b GPRC5A and PTGES mRNA expression levels in human NSCLC tumor samples compared with those in adjacent normal tissues in the GEO database (GSE10072). c Correlation expression analysis of GPRC5A and PTGES in human lung tissues in the GEO database (GSE10072). d GPRC5A expression was negatively correlated with patient overall survival in TCGA database. e PTGES expression was negatively correlated with patient overall survival in TCGA database. f Schematic depiction of the proposed action and mechanisms of PTGES-mediated immunosuppressive properties in lung cancer. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
Gprc5a-ko mice provide an animal model that links inflammatory response and tumorigenesis/metastasis. Gprc5a-ko mouse lungs are susceptible to inflammation, spontaneous and carcinogen-induced tumorigenesis [24, 27], which resembles the pathological features in human. In this study, we show that PTGES/PGE2 signaling promotes lung metastasis via two distinct mechanisms. First, PTGES/PGE2 signaling intrinsically endows tumor cells resistant to T-cell cytotoxicity. Second, PTGES/PGE2 signaling induces immunosuppression extrinsically via inducing MDSC recruitment and polarization of M2 macrophages (Fig. 7f).
Previously, PTGES/PGE2 was shown to induce PD-L1 expression in macrophages and MDSC [22]. However, we did not found that PD-L1 was significantly suppressed in 1601-ko-Ptges cells compared to control 1601-NS cells. This suggests other mechanisms involved. We find that PTGES/PGE2 signaling is essential for the stem-like features, including expression of ABCG2 and EMT-related markers. Consistently, PTGES/PGE2 signaling is essential for stem cell-enriched (CD44-positive) subpopulation, migration and invasion, 3D sphere and colony formation in vitro. Similarly, EMT induced by Snail expression in melanoma cells was found to accelerate metastasis [38]. And, there is a strong correlation between EMT and immunosuppressive target gene expression [39]. Thus, the intrinsic role of PTGES/PGE2 signaling in immunosuppression on tumor cells is linked to its role in induction of the EMT program.
COX/Ptges/PGE2 signaling has been implicated to inhibit host immunity. However, the detailed mechanism remains controversy. PGE2 has been shown to inhibit innate immunity, such as to inhibit the expression of type I interferon in macrophages during influenza A virus (IAV) infection [40], and inhibit NK cell viability and chemokine production in melanoma model [41]. PGE2 was considered to render melanoma cells evade immunity via type I interferon [42]. PGE2 has also been shown to alter the cytokine profile of T cells, such as to induce the cytokines for Th2 cells (IL-10), and to repress the cytokines for Th1 cells (IL-12 and IFNγ) [43]. Recently, it was shown that COX2/mPGES1(Ptges)/PGE2 pathway upregulates PD-L1 expression in tumor-associated macrophages (TAM) and MDSC, which contributes to the immunosuppression in bladder cancer [22]. In addition, upregulated PGE2 receptor EP4 was found in COX2-expressing triple-negative breast cancer, whereas targeting EP4 was suggested to benefit the therapeutic effect of PD-L1 inhibitors [44].
In this study, we performed a comprehensive analysis on lung tumor cells and immune cells in lung tissue microenvironment. Our study showed that PGE2-mediated immunosppression is mainly through two mechanisms. First, the intrinsic role of the Ptges/PGE2 pathway, which confers lung tumor cells resistant to T-cell cytotoxicity. Second, the extrinsic role of PGE2 signaling, which inhibits host immunity via inducing M2 macrophage polarization and recruitment of MDSC. Moreover, we found that PGE2-induced G-CSF is mainly responsible for increased recruitment of MDSC, which is critical for immunosuppression on T cells; whereas PGE2 itself does not directly increase recruitment of MDSC. Our study provides a more detailed mechanism on PGE2-mediated immunosuppression.
Previously, many studies were to assess the direct role of PGE2 on tumor cells. For example, PGE2 released from tumor cells were found to enhance cancer stem cells (CSC), while blockage of PGE2 signaling, via PGE2-neutralizing antibody and COX inhibitor celecoxib, abrogated CSC repopulation of bladder cancer [18]. In another study, PTGES inhibitor Cay10526 was shown to inhibit subcutaneous melanoma tumor growth in nude mice at high doses (50 mg/kg) [37]. Of notion, the tumor models used in these studies were xenografts in immune-deficient nude mice. Thus, the therapeutic effects were due to the direct role of PTGES/PGE2 inhibitor on tumor cells. The impact on host immunity is unclear. In this study, we performed the analysis on Gprc5a-ko mice, an immune-competent mouse model of C57 with inflammatory lung. Remarkably, treatment with PTGES inhibitor Cay10526 (5 mg/kg) at a dose ten times lower than that used in that report, for only one week, still significantly inhibited lung metastasis. This suggests that the therapeutic effect through restoration of adaptive immunity by PTGES targeting is much greater than direct cytotoxicity on tumor cells. In support, treatment of animal with Cay10526 at low dose, suppressed MDSCs, restored adaptive immunity, and inhibited lung metastasis, whereas treatment of tumor cells with Cay10526 at dose as high as 50 μM did not show cytotoxic effect on target tumor cells in vitro. Another advantage is that PTGES inhibitor is specific in inhibition of PGE2 production, whereas COX2 inhibitors suppress all of PGH2-derived products, including PGE2, PGD2, PGF2a, PGI2, and TXA2 [13]. Long term suppression of PGH2-derived products may cause serious side effects.
Recently, tumors are also classified into “hot” and “cold”, which reflects the status of infiltration of T lymphocytes, and the ability to response to immunotherapy. Here, we showed that PTGES is inversely correlated with CD8+ T cells in NSCLC samples. This suggests that upregulated PTGES/PGE2 signaling is crucial for of CD8+ T-cell infiltration. Thus, PTGES/PGE2 are the candidate markers for “hot” and “cold”, which may provide a direction for therapeutic application. Taken together, we found that PTGES/PGE2 signaling provides a functional link between inflammation and immunosuppression in lung metastasis.
Materials and methods
Cell lines and cell culture
HEK293T and HCC827 were tested and authenticated by DNA typing at Shanghai Jiao Tong University Analysis Core. Primary mouse lung cancer cells (SJT-1601) were isolated from NNK-induced lung tumor of Gprc5a-ko mouse at the age of 14 months [26]. We constructed the stable cell line SJT-1601-ko-Ptges by CRISPR/Cas9 technology. HCC827 stably transfected with nonspecific (sh-NS) or PTGES shRNA. HEK293T and HCC827 were cultured in DMEM. HCC827 were cultured in RPMI-1640.
Plasmids, reagents and antibodies
Detailed information is provided in the Supplementary Experimental Procedures.
Mice experiments
Detailed information is provided in the Supplementary Experimental Procedures.
Quantitative real-time (Q-PCR) and primers
Cells were lysed using trizol. Total RNA was extracted with RNA Extract Kit (TIANGEN) and cDNA were prepared from 1 μg total RNA using Fast Quant Kit. The Q-PCR analysis was performed on ABI 7300 real-time PCR machine. All Ct values were standardized by β-actin’s Ct value. All primers were listed in Supporting Information, Table S1.
Western blot analysis
Cells were lysed with RIPA lysis buffer [45]. Experiments were performed as described previously [46]. All antibodies were diluted for use according to manufacturers’ instructions. Finally, Protein expression was detected by chemiluminescence.
Flow cytometry analysis
Flow cytometry was performed as described previously [47, 48]. The cells were incubated CD44-APC (BD Pharmingen), however the control were incubated with IgG. After incubation, the cells were suspended in PBS for Flow Cytometry analysis. Mice were sacrificed, and lung tissues were scissors cut into a meaty shape and digested. Cells were stained with fluorescently labeled antibodies. Then cells were analyzed by FACS (BD Biosciences). Finally, the data was analyzed in the Flowjo 7.6.1software. All antibodies information was listed in Supporting Information, Table S2.
Migration and wound healing assay
Experiments were performed as described previously [49]. Fluorescent images were obtained; reported data are counts of migrated cells with experiments performed in triplicate. For the wound healing assay, cells overspread the culture plate scratch the cells. Then the picture captured as needed by Nikon camera.
Immunohistochemical staining and clinical samples
A tissue microarray composed of tumor and adjacent normal tissue was stained to identify PTGES and GPRC5A proteins. The IHC protocol and score method were performed as previously described [27]. All antibodies were diluted for use according to manufacturers’ instructions. Human lung cancer tissue samples were obtained from Shanghai Chest Hospital, Shanghai Jiao Tong University (Shanghai, CHINA). Shanghai Chest Hospital approved the use of the tumor samples and animals in this study.
T-cell activation in vitro
Detailed information is provided in the Supplementary Experimental Procedures.
Mice lung tissues metabonomics
Detailed information is provided in the Supplementary Experimental Procedures.
Statistical analysis
Comparisons among groups were performed by the Student’s t-test or Tukey–Kramer comparison test followed by analysis with GraphPad Prism software (GraphPad Software, San Diego, CA, USA). A p-value < 0.05 was considered significant (*p < 0.05; **p < 0.01; ***p < 0.001).
References
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature 2018;553:446–54.
Tong D, Liu Q, Liu G, Xu J, Lan W, Jiang Y, et al. Metformin inhibits castration-induced EMT in prostate cancer by repressing COX2/PGE2/STAT3 axis. Cancer Lett 2017;389:23–32.
Katoh H, Wang D, Daikoku T, Sun H, Dey SK, Dubois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013;24:631–44.
Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 2016;29:832–45.
Quail DF, Olson OC, Bhardwaj P, Walsh LA, Akkari L, Quick ML, et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol. 2017;19:974–87.
Waghray M, Yalamanchili M, Dziubinski M, Zeinali M, Erkkinen M, Yang H, et al. GM-CSF mediates mesenchymal-epithelial cross-talk in pancreatic cancer. Cancer Disco. 2016;6:886–99.
Lin Y, Yang X, Liu W, Li B, Yin W, Shi Y, et al. Chemerin has a protective role in hepatocellular carcinoma by inhibiting the expression of IL-6 and GM-CSF and MDSC accumulation. Oncogene 2017;36:3599–608.
Tian X, Ma J, Wang T, Tian J, Zheng Y, Peng R, et al. Long non-coding RNA RUNXOR accelerates MDSC-mediated immunosuppression in lung cancer. BMC Cancer 2018;18:660.
Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, Iacono A, et al. A relay pathway between arginine and tryptophan metabolism confers immunosuppressive properties on dendritic cells. Immunity 2017;46:233–44.
Roussel M, Ferrell PB Jr., Greenplate AR, Lhomme F, Le Gallou S, et al. Mass cytometry deep phenotyping of human mononuclear phagocytes and myeloid-derived suppressor cells from human blood and bone marrow. J Leukoc Biol. 2017;102:437–47.
Coward JI, Nathavitharana R, Popat S. True hypoglycaemia secondary to treatment with granulocyte colony stimulating factor (G-CSF) in a diabetic patient with non-small cell lung cancer. Lung Cancer 2012;75:133–5.
Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: relevance of prostaglandin E receptors. Biochim Biophys Acta. 2015;1851:414–21.
Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharm Rev 2007;59:207–24.
Ke Y, Oskolkova OV, Sarich N, Tian Y, Sitikov A, Tulapurkar ME, et al. Effects of prostaglandin lipid mediators on agonist-induced lung endothelial permeability and inflammation. Am J Physiol Lung Cell Mol Physiol. 2017;313:L710–l721.
Isono M, Suzuki T, Hosono K, Hayashi I, Sakagami H, Uematsu S, et al. Microsomal prostaglandin E synthase-1 enhances bone cancer growth and bone cancer-related pain behaviors in mice. Life Sci 2011;88:693–700.
Wang T, Jing B, Sun B, Liao Y, Song H, Xu D, et al. Stabilization of PTGES by deubiquitinase USP9X promotes metastatic features of lung cancer via PGE2 signaling. Am J Cancer Res. 2019;9:1145–60.
Schumacher Y, Aparicio T, Ourabah S, Baraille F, Martin A, Wind P, et al. Dysregulated CRTC1 activity is a novel component of PGE2 signaling that contributes to colon cancer growth. Oncogene 2016;35:2602–14.
Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015;517:209–13.
Nandi P, Girish GV, Majumder M, Xin X, Tutunea-Fatan E, Lala PK. PGE2 promotes breast cancer-associated lymphangiogenesis by activation of EP4 receptor on lymphatic endothelial cells. BMC Cancer 2017;17:11.
Ong HS, Gokavarapu S, Al-Qamachi L, Yin MY, Su LX, Ji T, et al. Justification of routine venous thromboembolism prophylaxis in head and neck cancer reconstructive surgery. Head Neck 2017;39:2450–8.
Paff M, Alexandru-Abrams D, Hsu FP, Bota DA. The evolution of the EGFRvIII (rindopepimut) immunotherapy for glioblastoma multiforme patients. Hum Vaccin Immunother. 2014;10:3322–31.
Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci USA. 2017;114:1117–22.
Cheng Y, Lotan R. Molecular cloning and characterization of a novel retinoic acid-inducible gene that encodes a putative G protein-coupled receptor. J Biol Chem. 1998;273:35008–15.
Tao Q, Fujimoto J, Men T, Ye X, Deng J, Lacroix L, et al. Identification of the retinoic acid-inducible Gprc5a as a new lung tumor suppressor gene. J Natl Cancer Inst. 2007;99:1668–82.
Xu J, Tian J, Shapiro SD. Normal lung development in RAIG1-deficient mice despite unique lung epithelium-specific expression. Am J Respir Cell Mol Biol. 2005;32:381–7.
Song H, Sun B, Liao Y, Xu D, Guo W, Wang T, et al. GPRC5A deficiency leads to dysregulated MDM2 via activated EGFR signaling for lung tumor development. Int J Cancer. 2019;144:777–87.
Zhong S, Yin H, Liao Y, Yao F, Li Q, Zhang J, et al. Lung Tumor Suppressor GPRC5A Binds EGFR and Restrains Its Effector Signaling. Cancer Res 2015;75:1801–14.
Liao Y, Song H, Xu D, Jiao H, Yao F, Liu J, et al. Gprc5a-deficiency confers susceptibility to endotoxin-induced acute lung injury via NF-kappaB pathway. Cell Cycle 2015;14:1403–12.
Guo W, Hu M, Wu J, Zhou A, Liao Y, Song H, et al. Gprc5a depletion enhances the risk of smoking-induced lung tumorigenesis and mortality. Biomed Pharmacother 2019;114:108791.
Deng J, Fujimoto J, Ye XF, Men TY, Van Pelt CS, Chen YL, et al. Knockout of the tumor suppressor gene Gprc5a in mice leads to NF-kappaB activation in airway epithelium and promotes lung inflammation and tumorigenesis. Cancer Prev Res (Philos). 2010;3:424–37.
Cortes M, Sanchez-Moral L, de Barrios O, Fernandez-Acenero MJ, Martinez-Campanario MC, Esteve-Codina A, et al. Tumor-associated macrophages (TAMs) depend on ZEB1 for their cancer-promoting roles. EMBO J 2017;36:3336–55.
Palizgir MT, Akhtari M, Mahmoudi M. Curcumin reduces the expression of interleukin 1beta and the production of interleukin 6 and tumor necrosis factor alpha by M1 macrophages from patients with Behcet’s disease. Immunopharmacol Immunotoxicol 2018;40:297–302.
Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab 2018;28:87–103.e106.
Daley-Bauer LP, Wynn GM, Mocarski ES. Cytomegalovirus impairs antiviral CD8+ T cell immunity by recruiting inflammatory monocytes. Immunity 2012;37:122–33.
Hutchison S, Sahay B, de Mello SC, Sayour EJ, Lejeune A, Szivek A, et al. Characterization of myeloid-derived suppressor cells and cytokines GM-CSF, IL-10 and MCP-1 in dogs with malignant melanoma receiving a GD3-based immunotherapy. Vet Immunol Immunopathol. 2019;216:109912.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8.
Kim SH, Hashimoto Y, Cho SN, Roszik J, Milton DR, Dal F, et al. Microsomal PGE2 synthase-1 regulates melanoma cell survival and associates with melanoma disease progression. Pigment Cell Melanoma Res. 2016;29:297–308.
Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009;15:195–206.
Mak MP, Tong P, Diao L, Cardnell RJ, Gibbons DL, William WN, et al. A patient-derived, pan-cancer EMT signature identifies global molecular alterations and immune target enrichment following epithelial-to-mesenchymal transition. Clin Cancer Res. 2016;22:609–20.
Coulombe F, Jaworska J, Verway M, Tzelepis F, Massoud A, Gillard J, et al. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity 2014;40:554–68.
Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 2018;172:1022–.e1014.
Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 2015;162:1257–70.
Wu AA, Drake V, Huang HS, Chiu S, Zheng L. Reprogramming the tumor microenvironment: tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology 2015;4:e1016700.
Majumder M, Nandi P, Omar A, Ugwuagbo KC, Lala PK. EP4 as a therapeutic target for aggressive human breast cancer. Int J Mol Sci. 2018;19:E1019. pii
Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, et al. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J 2010;29:1803–16.
Hu M, Guo W, Liao Y, Xu D, Sun B, Song H, et al. Dysregulated ENPP1 increases the malignancy of human lung cancer by inducing epithelial-mesenchymal transition phenotypes and stem cell features. Am J Cancer Res. 2019;9:134–44.
Wang T, Li K, Song H, Xu D, Liao Y, Jing B, et al. p53 suppression is essential for oncogenic SPAG5 upregulation in lung adenocarcinoma. Biochem Biophys Res Commun. 2019;513:319–25.
Li K, Guo W, Li Z, Wang Y, Sun B, Xu D, et al. ALDH2 repression promotes lung tumor progression via accumulated acetaldehyde and DNA damage. Neoplasia 2019;21:602–14.
Liu S, Ye D, Guo W, Yu W, He Y, Hu J, et al. G9a is essential for EMT-mediated metastasis and maintenance of cancer stem cell-like characters in head and neck squamous cell carcinoma. Oncotarget 2015;6:6887–901.
Acknowledgements
This study was supported by grants from National Natural Science Foundation of China (81620108022, 91129303, 91729302 and 81572759 to JD, 81572693 to FY) and Shenzhen Municipal Government of China (KQTD20170810160226082).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, T., Jing, B., Xu, D. et al. PTGES/PGE2 signaling links immunosuppression and lung metastasis in Gprc5a-knockout mouse model. Oncogene 39, 3179–3194 (2020). https://doi.org/10.1038/s41388-020-1207-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-1207-6
This article is cited by
-
Myeloid-derived suppressor cells in cancer: therapeutic targets to overcome tumor immune evasion
Experimental Hematology & Oncology (2024)
-
EGFR mutations induce the suppression of CD8+ T cell and anti-PD-1 resistance via ERK1/2-p90RSK-TGF-β axis in non-small cell lung cancer
Journal of Translational Medicine (2024)
-
A graph self-supervised residual learning framework for domain identification and data integration of spatial transcriptomics
Communications Biology (2024)
-
Stroma AReactive Invasion Front Areas (SARIFA) proves prognostic relevance in gastric carcinoma and is based on a tumor–adipocyte interaction indicating an altered immune response
Gastric Cancer (2024)
-
Dendritic cells mediated by small extracellular vesicles derived from MSCs attenuated the ILC2 activity via PGE2 in patients with allergic rhinitis
Stem Cell Research & Therapy (2023)
