
Abstract
Activating mutations of the ALK receptor occur in a subset of neuroblastoma tumors. We previously demonstrated that Alk mutations cooperate with MYCN overexpression to induce neuroblastoma in mice and identified Ret as being strongly upregulated in MYCN/Alkmut tumors. By a genetic approach in vivo, we now document an oncogenic cooperation between activated Ret and MYCN overexpression in neuroblastoma formation. We show that MYCN/RetM919T tumors exhibit histological features and expression profiles close to MYCN/Alkmut tumors. We show that RET transcript levels decrease precedes RET protein levels decrease upon ALK inhibition in neuroblastoma cell lines. Etv5 was identified as a candidate transcription factor regulating Ret expression from murine MYCN/Alkmut tumor transcriptomic data. We demonstrate that ETV5 is regulated both at the protein and mRNA levels upon ALK activation or inhibition in neuroblastoma cell lines and that this regulation precedes RET modulation. We document that ALK activation induces ETV5 protein upregulation through stabilization in a MEK/ERK-dependent manner. We show that RNAi-mediated inhibition of ETV5 decreases RET expression. Reporter assays indicate that ETV5 is able to drive RET gene transcription. ChIP-seq analysis confirmed ETV5 binding on the RET promoter and identified an enhancer upstream of the promoter. Finally, we demonstrate that combining RET and ALK inhibitors reduces tumor growth more efficiently than each single agent in MYCN and AlkF1178L-driven murine neuroblastoma. Altogether, these results define the ERK–ETV5–RET pathway as a critical axis driving neuroblastoma oncogenesis downstream of activated ALK.
Similar content being viewed by others


Introduction
The ALK (Anaplastic Lymphoma Kinase) gene encodes a receptor tyrosine kinase (RTK) mainly expressed in the nervous system of mammals [1, 2]. It has been initially identified as the partner of nucleophosmin (NPM) in a t(2;5) translocation occurring in a large fraction of anaplastic large-cell lymphomas. Since then, the ALK gene has been involved in many different translocations in various types of human neoplasia [1, 2]. The downstream signaling pathways of the emblematic NPM–ALK fusion protein that trigger oncogenic transformation have now been deeply studied and three main pathways including the Ras-extracellular signal-regulated kinase (ERK) pathway, the Janus kinase 3 (JAK3)/STAT3 pathway, and the phosphatidylinositol-3-kinase (PI3K)/AKT pathway have been identified [1]. The ALK gene was identified as a major oncogene in neuroblastoma, an embryonal cancer of the sympathetic nervous system that accounts for 8–10% of pediatric cancers [3]. Indeed, activating mutations of the ALK gene were reported both in familial neuroblastoma cases at the germline level and in sporadic neuroblastoma cases mainly at the somatic level [4,5,6,7]. A recent analysis documented ALK mutations in 8% of neuroblastoma cases at diagnosis with three hotspots at positions F1174, R1245, and F1275 [8]. This study also showed that neuroblastoma patients with ALK activation exhibit a poorer prognosis compared to patients with non-mutated ALK. Several pathways have now been reported to be activated downstream of full-length ALK upon its activation [2]. The induction of the RAS–MAPK and PI3K/AKT pathways have been observed in almost all studied models.
ALK-mutated neuroblastomas therefore belong to the ALKoma entity [9] that may benefit from tumor-targeted therapies with ALK tyrosine kinase inhibitors. The dual ALK/MET inhibitor crizotinib has now been evaluated in different ALKoma cancers, including children with refractory neuroblastomas [10, 11]. These studies suggest that inhibition of mutated ALK is more difficult to achieve when compared to ALK fusions. Moreover, there is evidence to indicate that the F1174L mutation exhibits resistance to crizotinib [12, 13]. Recently, high efficacy of lorlatinib (ALK/ROS1 inhibitor PF-06463922) was demonstrated in ALK-driven pre-clinical neuroblastoma models with primary crizotinib resistance [14, 15]. However, only transient benefit has often been obtained using a single kinase inhibitor [2]. These data suggest that anti-ALK therapy may not be sufficient in neuroblastoma tumors presenting with ALK activation and that the dissection of the downstream signaling pathways of mutated ALK is a crucial step to propose new therapeutic strategies.
We recently described a mouse model of neuroblastoma with endogenous expression of mutated Alk in a MYCN transgenic context [16]. The transcriptomic study of the murine tumors bearing or not the Alk mutation revealed that the Ret oncogene was upregulated in Alk-mutated tumors. This finding was confirmed in human neuroblastoma tumors and cell lines. We also showed that tumor growth of murine MYCN/KI Alkmut tumors was impaired upon Ret inhibition by the vandetanib inhibitor, suggesting RET as a therapeutic target in ALK-mutated neuroblastoma.
In the present paper, we further established the crucial role of RET in ALK-mutated and MYCN-driven neuroblastoma oncogenesis with the demonstration that Ret activation may replace Alk activation to induce tumors in a MYCN transgenic context. We then identified ETV5 being upregulated by activated ALK. ETV5 is part of the PEA subfamily of the ETS transcription factors consisting of ETV1, ETV4, and ETV5 (also named ER81, PEA3, and ERM) [17]. Activation of various RTKs has already been shown to induce ETV5 expression [18, 19]. We further demonstrate that ETV5 is able to regulate the RET promoter using reporter assays, thereby showing that RET upregulation by activated ALK is achieved through an ALK–ETV5–RET axis.
Results
Activated Ret cooperates with MYCN overexpression to drive neuroblastoma in mice
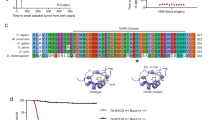
The RET gene has been identified previously as a target of activated ALK at the mRNA level in both human neuroblastoma cell lines and primary tumors as well as in murine tumors driven by mutated Alk and MYCN [16] (Table 1, top). Moreover, the Ret inhibitor vandetanib has been shown to impair tumor growth of murine MYCN/Alkmut tumors suggesting that RET largely contributes to the oncogenesis of ALK-driven neuroblastoma tumors [16]. To further determine whether Ret activation could replace Alk activation in vivo, we bred knock-in (KI) RetM919T mice with TH-MYCN mice. The M919T mutation in the murine Ret gene corresponds to the gain-of-function substitution M918T in the human RET gene observed in most patients affected by multiple endocrine neoplasia type 2B (MEN2B) [20]. We document an oncogenic cooperation between activated Ret and MYCN overexpression in tumor formation (Fig. 1a). The penetrance and latency observed for the MYCN/RetM919T mice are positioned between those of MYCN/AlkR1279Q and MYCN/AlkF1178L mice, that have been determined in our previous work [16]. Histological analysis taking into account the criteria defined to describe human neuroblastoma [21] confirmed that all MYCN/RetM919T tumors correspond to stroma-poor neuroblastoma. The majority of MYCN/RetM919T tumors exhibited signs of differentiation (Fig. 1b) as previously observed for MYCN/Alkmut tumors [16]. Expression of total Alk and total Ret was confirmed in the mouse tumors of different genotypes, with an increased Alk and Ret expression in MYCN/AlkF1178L tumors (Supplementary Fig. 1), as previously described [16]. In addition, activity of both AlkF1178L and RetM919T could be confirmed by the increased Erk phosphorylation in these tumors compared to MYCN tumors (Supplementary Fig. 1). Expression profiles were then generated for a subset of MYCN/RetM919T tumors. This analysis confirmed high expression levels of several emblematic neuroblastoma markers, such as Dbh and Phox2b (Fig. 1c). An unsupervised clustering analysis including MYCN, MYCN/Alkmut, and MYCN/RetM919T tumors revealed that MYCN/RetM919T tumors clustered either with MYCN/AlkF1178L or with MYCN/AlkR1279Q cases (Fig. 1d). Altogether, these data demonstrate the crucial role of RET in neuroblastoma oncogenesis dependent on ALK activation.

Activated Ret cooperates with MYCN to induce neuroblastoma in mice. a Kaplan–Meier analysis showing that the Ret M919T mutation cooperates with MYCN overexpression to induce tumor formation. Intercrosses were performed between MYCN and KI RetM919T mice on a 129S2 and C57BL6/J background, respectively. Survival curves were compared to our previous data obtained for MYCN/Alkmut mice on a 129×1/SvJ × C57BL6/N-mixed background [16]. b Hematoxylin and eosin staining of a representative MYCN/RetM919T tumor. Scale bar, 50 μm. c Expression levels of Dbh and Phox2b in the four groups of murine tumors (MYCN (n = 10), MYCN/AlkR1279Q (n = 10), MYCN/AlkF1178L (n = 11), and MYCN/RetM919T (n = 6)). d Unsupervised hierarchical clustering using the top 5% genes with the highest IQR in the full set of MYCN (blue), MYCN/AlkR1279Q (red), MYCN/AlkF1178L (pink), and MYCN/RetM919T (green) tumors
Changes in RET transcript levels precede changes in RET protein levels in neuroblastoma cell lines
We then sought to decipher the mechanism by which activated Alk triggers Ret upregulation. Toward this, we used two ALK-mutated cell lines CLB-MA (ALK F1174L) and CLB-GA (ALK R1275Q), the CLB-GE cell line that harbor an amplification of the ALK gene combined with an F1174V mutation as well as the IMR-32 and SK-N-BE(2)C cell lines presenting with wild-type (WT) ALK. ALK activity was modulated by activation with the agonist monoclonal mAb46 antibody [22] or inhibition with the TAE684 inhibitor [7, 23, 24]. Treatment of cells with the antibody did not exceed 6 h as longer treatments have been shown to induce internalization of the receptor [25]. First, we confirmed that mAb46 constantly increased ALK phosphorylation from 5 to 30 min in CLB-GA (ALK R1275Q) and CLB-MA (ALK F1174L) (Supplementary Fig. 2). We observed a burst of ERK phosphorylation at early time points of mAb46 treatment, i.e., 10 min in CLB-GA and 15 min in CLB-MA. ERK phosphorylation then decreased after this peak of activation but remained at higher levels compared to untreated cells (Supplementary Fig. 2). In both cell lines, ALK inhibition resulted in phosphorylated ALK and phosphorylated ERK decrease.
We then showed that the RET mRNA was modulated after 6 h of treatment in the different cell lines (Fig. 2a). In contrast, RET protein levels were not changed at this time point in the four cell lines in which the protein is detectable by Western blot analysis (Fig. 2b). Treatment with TAE684 for 24 h resulted in a strong reduction in RET protein levels in ALK-mutated cell lines whereas a modest reduction was observed in the WT SK-N-BE(2)C cell line (Fig. 2c). These observations therefore suggest that activated ALK induces RET upregulation at the transcriptional level, consistently with the observation of Ret mRNA upregulation in MYCN/Alkmut mouse tumors [16].

Changes in RET transcript and protein levels upon ALK modulation in neuroblastoma cell lines. a Expression levels of RET mRNA after 6 h of treatment with the ALK tyrosine kinase inhibitor TAE684 at 300 nM or with the agonist mAb46 antibody at 1 µg/mL. Results are shown as relative expression levels compared to the untreated cell lines. Samples represent biological replicates. b, c Western blotting for RET, phosphorylated ERK1/2 at T202/Y204 and total ERK in cell lines treated with TAE684 or mAb46 for 6 h and TAE684 for 24 h, respectively
Mouse tumor transcriptomic data identify Etv5 as a candidate transcription factor for Ret upregulation
To determine which transcription factor could be involved in Ret upregulation, we took advantage of our mouse tumors transcriptomic data. First, we focused on transcription factors previously identified as regulating RET expression by binding to its promoter or enhancers, i.e., SPI, SP3, EGR1, SOX10, PAX3, NKX2-1, and HOXB5 [26]. A differential analysis between MYCN/Alkmut and MYCN tumors (Table 1, middle) identified Egr1 with a fold-change of 1.6 but this modulation was not statistically significant (P-value > 0.05). Then, we listed all the genes that were differentially expressed between MYCN/Alkmut and MYCN tumors and that were part of the gene ontology GO:0003700 category (sequence-specific DNA-binding transcription factor activity). The top 5 candidates, Ebf3, Mycn, Etv5, Zfp939, and Hmx1, are presented in Table 1 (bottom). A correlation analysis between the different transcription factors and RET expression levels in our dataset of human primary neuroblastoma (GSE12460) revealed a positive and significant correlation only for ETV5 (r = 0.4, P-value = 0.001). Moreover, within the group of MYCN/Alkmut tumors we observed a strong correlation between Ret and Etv5 expression (Supplementary Fig. 3). Interestingly, in MYCN/RetM919T tumors the Etv5 expression level is similar to the one observed in MYCN tumors (Supplementary Fig. 4), consistently with Etv5 being downstream of ALK and upstream of Ret. Altogether, Etv5 appeared as a relevant candidate transcription factor for Ret upregulation by activated Alk in murine MYCN/Alkmut tumors.
The ETV5 transcription factor is regulated both at the protein and mRNA levels by the ALK receptor
The ETV5 protein is modulated at short-term (30 min) upon ALK activation or inhibition
We then explored how ETV5 expression was modified upon ALK activation or inhibition in our panel of neuroblastoma cell lines. Figure 3a shows ETV5 protein levels increase in all samples upon mAb46 treatment. Conversely, ETV5 protein levels decreased upon ALK inhibition using the TAE684 inhibitor in ALK-mutated cell lines. TAE684 had no effect on the ETV5 protein in IMR-32 cells and induced a low decrease in the SK-N-BE(2)C cell line. An increase of ALK phosphorylation by the agonist monoclonal mAb46 antibody could be documented in all cell lines except the SK-N-BE(2)C cell line following 30 min of treatment (Fig. 3a). Nevertheless, we confirmed the activation of the ALK receptor and its downstream targets by mAb46 in SK-N-BE(2)C cells as ERK phosphorylation was clearly enhanced by this treatment. These observations therefore show that ALK activation rapidly induces an increase of the ETV5 protein in neuroblastoma cell lines.

ETV5 is a short-term target of activated ALK at the protein level in neuroblastoma cell lines. a Western blotting of ETV5, P-ALK (Y1604), ALK, P-ERK1/2 (T202/Y204), and ERK. Cell lines were treated with TAE684 at 300 nM or with the agonist mAb46 antibody at 1 µg/mL for 30 min. b Expression levels of ETV5 mRNA after 6 h of the same treatment as in a for the same cell lines. Results are shown as relative expression levels compared to the untreated cell lines. Samples represent biological replicates
The ETV5 mRNA is modulated at 6 h upon ALK activation or inhibition
Although the ETV5 protein levels were modulated after 30 min of treatment, mRNA levels of ETV5 were unchanged at this time point (Supplementary Fig. 5). However, qPCR experiments revealed that ETV5 mRNA levels were impacted by ALK modulation after 6 h of treatment (Fig. 3b). Indeed, ETV5 transcript levels were markedly decreased in the three ALK-mutated cell lines after 6 h of TAE684 treatment, whereas no decrease or a low decrease were observed in the IMR-32 and SK-N-BE(2)C cell lines, respectively. ETV5 transcript levels were increased in 3 out of the 5 analyzed cell lines after 6 h of mAb treatment. Altogether, these data demonstrate a dual regulation of the ETV5 transcription factor both at the protein and mRNA levels by the ALK receptor.
ALK activation induces ETV5 protein upregulation through stabilization and is dependent on ERK1/2 phosphorylation
Our data indicate that ALK modulation results in ETV5 protein level changes at short-term without significant effect on ETV5 mRNA level. Moreover, ERK and ETV5 appear to be downstream components of ALK signaling. To further decipher the mechanism of ETV5 protein regulation, the CLB-GE and SK-N-BE(2)C cell lines were pretreated either with vehicle, cycloheximide, an inhibitor of protein synthesis, or with the proteasome inhibitor MG132 for 30 min. Cells were then incubated with mAb46, trametinib, a specific MEK1/2 inhibitor, or TAE684 (only for CLB-GE). Figure 4a shows that, in CLB-GE, both ALK and MEK inhibition resulted in decreased phospho-ERK and decreased ETV5 levels. Although cycloheximide pretreatment did not modify the response of CLB-GE cells to ALK or MEK modulation, the downregulation of ETV5 protein by ALK and MEK inhibition was rescued from proteosomal degradation by MG132. Similar results were observed in SK-N-BE(2)C cells as ETV5 levels decreased upon trametinib treatment and ETV5 downregulation by trametinib was impaired after MG132 pretreatment (Fig. 4b). We also documented that trametinib pretreatment precluded ETV5 induction at the protein level after 30 min of mAb46 treatment in the two analyzed cell lines (Fig. 4c). Overall, these results are consistent with a stabilization of ETV5 protein by ALK signaling through the MEK/ERK pathway.

ETV5 is upregulated through stabilization upon ALK activation. a The CLB-GE cell line was pretreated or not with cycloheximide (Cyclohex—10 μg/ml) or MG132 (10 μM) for 30 min. Cells were then treated with mAb46, trametinib (Tram), TAE684 or vehicle for 30 min. Western blotting was performed for ETV5, P-ERK1/2 (T202/Y204), and ERK. b The same analysis was done for SK-N-BE(2)C (except TAE684 treatment). c Both cell lines were either: (1) untreated; (2) treated with the agonist mAb46 antibody at 1 µg/mL for 30 min; (3) pretreated with 50 nM of trametinib for 30 min before mAb46 treatment (30 min). Western blotting of ETV5, P-ERK1/2 (T202/Y204), and ERK is shown
RET expression is modulated by the ETV5 transcription factor
Taking into account that the ETV5 transcription factor acts downstream of the ALK receptor and that its protein level is modified upon ALK modulation in a shorter time frame than the levels of RET mRNA and protein we hypothesized that ETV5 may regulate RET transcription. To test this hypothesis, ETV5 expression was abrogated by siRNA in the SK-N-BE(2)C cell line expressing high levels of both ETV5 and RET. Figures 5a and b show that, 48 h after transfection, ETV5 invalidation resulted in decreased RET expression both at the protein and mRNA levels, respectively.

RET is a target of the transcription factor ETV5. a Western blotting of ETV5 and RET after ETV5 invalidation using 3 different siRNA in SK-N-BE(2)C cells at 48 h. Actin is used as a loading control. siCT: control. b The levels of ETV5 and RET mRNA were measured after ETV5 invalidation at the same time point and compared to the expression levels in the siCT condition. c Luciferase reporter assay for the RET promoter region. Luciferase activity was evaluated upon ETV5 expression from a pcDNA3-ETV5 plasmid used at two quantities in HEK cells and normalized to empty pcDNA3. Results are shown for the empty reporter vector and reporter vector bearing the RET promoter. d Matrix of the ETV5 transcription factor defined by ChIP-Seq. e ETV5-binding peaks revealed by ChIP-seq analysis. Binding regions defined by HMCan are shown in colors. Regions in blue show peaks overlapping the RET promoter, whereas region in green indicates a strong peak likely acting as an enhancer
We next sought to explore whether the ETV5 transcription factor could directly modulate the RET promoter. Examination of the sequence spanning the RET transcription start site from −1094 bp up to +368 bp with the FIMO tool [27] using the ETV5 matrix [28] revealed seven ETV5 consensus binding sites with a P-value < 0.001 (Supplementary Fig. 6). We therefore amplified and inserted this RET promoter sequence upstream of the Luciferase reporter gene and performed dual luciferase reporter assays to measure the effect of ETV5 on this promoter. HEK293T cells transfected with 200 or 400 ng of a pcDNA3 plasmid encoding the ETV5 protein demonstrated 10 and 20-fold higher normalized luminescence compared to an empty pcDNA3 plasmid, respectively (Fig. 5c). Next, we performed ChIP-seq analysis for the ETV5 transcription factor in SK-N-BE(2)C cells. De novo identification of motifs using RSAT revealed a motif fully consistent with ETS factors and the published ETV5 matrix, therefore validating the quality of this experiment (Fig. 5d). This analysis confirmed ETV5 binding on the RET promoter and also revealed a strong binding of ETV5 upstream of the RET promoter (Fig. 5e) and likely corresponding to an enhancer. Altogether, these experiments indicate that the ETV5 transcription factor is able to take part in the transcriptional regulation of the RET gene.
Crizotinib and vandetanib synergize to inhibit tumor growth of MYCN/AlkF1178L neuroblastoma
We have previously shown that ALK or RET inhibition with small-molecule inhibitors impaired tumor growth in our mouse models, although not leading to a complete remission [16]. The identification of a signaling pathway from activated ALK to RET prompted us to explore the effect of targeting both ALK and RET kinases as a novel therapeutic strategy. We therefore investigated the effect of the crizotinib/vandetanib combination after allograft of a MYCN/AlkF1178L tumor in nude mice. Mice were treated with vehicle, crizotinib (100 mg/kg/day) or a combination of crizotinib (100 mg/kg/day) and vandetanib (30 mg/kg/day). We observed a strong reduction in tumor growth in mice treated with the combination compared to the vehicle and also a more potent and significant effect of the combination compared to crizotinib as a single agent (Fig. 6a). Consistently, a stronger decrease of Erk phosphorylation was observed in the tumors of mice treated with both agents (Supplementary Fig. 7). This resulted in a prolonged survival of mice treated with the combination (Fig. 6b).

The crizotinib/vandetanib combination is more potent than crizotinib alone to impair tumor growth of MYCN/AlkF1178L neuroblastoma. Nude mice were subjected to subcutaneous allografts with a neuroblastoma tumor obtained from a MYCN/AlkF1178L mouse. Mice were treated daily with vehicle (n = 4), crizotinib (n = 4), or a combination of crizotinib and vandetanib (n = 5) when tumor volumes were around 250 mm3. Mice were sacrified when tumor volume reached 2000 mm3. a Mean tumor volume (±s.d.) is shown until time of sacrifice of the first mouse of each group (day 7 after treatment’s start for the control group; day 12 and 17 for the crizotinib and combination, respectively). As variance was not statistically different between groups (F-test), a two-tailed Student’s t-test was applied to assay significance between crizotinib alone and the combination. b Kaplan–Meier survival curves for mice of the three groups using tumor size exceeding 2000 mm3 as an endpoint
Discussion
We previously explored the impact of Ret on tumor growth in our mouse MYCN/Alkmut models by a pharmacological approach and showed that the RET small-molecule inhibitor vandetanib was able to impair tumor growth [16]. To further demonstrate the crucial role of RET in ALK-mutated-driven neuroblastoma oncogenesis we now used a genetic approach and took advantage of a mouse line bearing an activating mutation of the murine Ret gene. Although ganglioneuroma of the adrenal medulla and an enlargement of the associated sympathetic ganglia have been described in this KI RetM919T mouse line, these animals do not develop neuroblastoma [29]. We demonstrate here that the Ret activating mutation M919T cooperates with MYCN to induce tumors in mice. These tumors present with histological and transcriptomic features similar to the ones that characterize MYCN/Alkmut neuroblastomas. In contrast to MYCN/Alkmut tumors that exhibit an increased Alk expression compared with MYCN tumors due to a positive feedback loop [16, 30], the Alk expression level in MYCN/RetM919T tumors is similar to the one observed in MYCN tumors (Supplementary Fig. 4). The murine Mycn gene is upregulated in MYCN/RetM919T although in a more modest extend compared to what is observed in MYCN/Alkmut tumors [16, 31] and the Ret transcript is slightly increased in MYCN/RetM919T compared to MYCN tumors (Supplementary Fig. 4). Altogether, the closely features of MYCN/RetM919T and MYCN/Alkmut tumors clearly document the crucial role of Ret in ALK-mutated and MYCN-driven neuroblastoma oncogenesis. We previously reported an upregulation of RET in primary neuroblastoma tumors with activated ALK [16] on the dataset of Schulte and colleagues [32]. We could confirm this observation on a larger set of primary samples, including 290 and 30 tumors without or with ALK activation, respectively [33]. Indeed, a differential analysis with multiple test correction identified the RET gene as being upregulated in cases with ALK activation. This link between RET and ALK observed in ALK-mutated neuroblastoma provides one additional common feature to the involvement of these two RTKs in oncogenesis. Indeed, both receptors have been involved in different types of human malignancies, through fusion proteins or activating mutations [1, 26, 34]. Overexpression of wild-type RET and wild-type ALK have also been reported in several cancers although their pathogenetic role remains to be fully understand in most of these cases. Interestingly, our work shows involvement of both receptors in the same tumor entity.
Our results clearly showed that RET mRNA changes occurred before RET protein levels modification in human neuroblastoma cell lines, pointing at a transcription regulation of the RET gene. Data from our original models revealed Etv5 as a candidate transcription factor that may regulate Ret expression. This observation was indeed consistent with data obtained from different mouse models as Etv5 was also identified in the top 50 upregulated genes in tumors from double transgenic MYCN/ALKF1174L animals compared with MYCN tumors [31]. Interestingly, both RET and ETV5 have been recently identified as members of the 77-gene signature marking ALK activity in neuroblastoma cells [24]. We documented that the Etv5 transcript is expressed at the same level in MYCN/RetM919T compared to MYCN tumors, and not upregulated as it is the case in MYCN/Alkmut tumors (Supplementary Fig. 4), which is consistent with our results showing that ETV5 acts downstream of ALK and upstream of RET. The observation of an increase of ETV5 transcription upon ALK activation in neuroblastoma cells is reminiscent of other studies demonstrating ETV5 transcription induction upon activation of various RTKs: indeed, it has been shown that the Etv5 and Etv4 mRNA are significantly induced by NGF in rat sensory neurons and PC12 cells [18] and a time- and dose-dependent increase in Etv5 mRNA expression after FGF2 treatment has been described in mouse Sertoli cells [35]. ETV5 has also been shown to be a downstream target of FGF signaling in the lung [36, 37].
Our data demonstrate a dual impact of ALK activity on ETV5, both at the protein and transcript levels. Members of the PEA3 subfamily are subjected to multiple post-translational modifications including phosphorylation, acetylation, ubiquitination, and sumoylation [17]. These modifications are involved in the control of the stability of the PEA3 factors and have a role in the regulation of their target genes [17, 38, 39]. Although no direct link between ETV5 phosphorylation and stability has yet been demonstrated, our data clearly indicate that modulation of the ALK kinase activity affects ETV5 stability through a proteasome-dependent mechanism. Furthermore, our results showed that stabilization of ETV5 protein by ALK signaling occurs through the MEK/ERK pathway. Interestingly, in gastrointestinal stromal tumors defined by KIT or PDGFRα mutations, the ETV1 protein has been shown to be stabilized by activated KIT through MEK therefore establishing a KIT–MEK–ETV1 signaling pathway [40].
We could document that ETV5 inhibition resulted in a reduction of RET protein level in a neuroblastoma context. Interestingly, ETV5 has been described either upstream or downstream of RET in various models. Although Etv4 and Etv5 are required downstream of Ret in kidney-branching morphogenesis [41], Etv5 knockout mice present with reduced Ret expression in testicular germ cells [42]. We obtained evidence that ETV5 is able to modulate the transcriptional level of the RET gene. Direct interaction of the transcription factor on this promoter was confirmed by ETV5 ChIP-seq analysis. This experiment also revealed a strong binding of ETV5 upstream of the RET promoter, likely corresponding to an enhancer. This potential enhancer (chr10: 43,550,599–43,553,301) located 18 kb upstream of RET overlaps with a described enhancer of RET called RET-5.5 (chr10: 43,551,864–43,553,915), identified as one of the three enhancers affected by variants in Hirschsprung disease [43].
Targeting several key components of the pathways that are responsible for ALK oncogenic activity could represent an improved strategy for molecular treatment of neuroblastoma. Wood and colleagues recently reported that the combination of Ceritinib, an ALK inhibitor and Ribociclib, a dual inhibitor of CDK4/6, enhanced anti-tumor efficacy and may be of interest to treat neuroblastoma patients with ALK mutations [44]. Our present results show that combining both the ALK inhibitor crizotinib and the RET inhibitor vandetanib is more potent to inhibit tumor growth of a MYCN/AlkF1178L neuroblastoma compared to crizotinib alone (or vandetanib alone, data not shown). Several hypothesis, not exclusive, may explain this synergistic effect: (1) ALK and/or RET inhibition is not fully achieved in vivo with the drugs used as single agents; (2) targeting ALK only does not affect other pathways that may regulate RET; (3) targeting RET only does not disrupt ALK-dependent signaling components distinct from the ALK–MEK/ERK–ETV5–RET axis that may contribute to part of ALK oncogenic activity. We cannot exclude that additional pathways may also contribute to the regulation of RET expression upon ALK modulation. Indeed, it has been shown recently that mutant ALK may regulate RET expression through the PI3K/AKT pathway and the FOXO3a transcription factor in SH-SY5Y neuroblastoma cells [24].
In conclusion, our results provide additional evidence for a critical role of RET in neuroblastoma oncogenesis driven by activated ALK and decipher the mechanism from activated ALK to RET upregulation. Furthermore, they suggest that combined treatment with crizotinib and vandetanib may be a promising approach in clinical practice to treat neuroblastoma patients with tumors exhibiting ALK mutation.
Materials and methods
Mouse lines and genotyping
The TH-MYCN, KI AlkR1279Q and KI AlkF1178L mice used in this study have been previously described [16, 45]. They were backcrossed on a 129S2/SvPasCrl background (Charles River). KI RetM919T mice on a C57BL/6J background were purchased at the Jackson Laboratory (http://jaxmice.jax.org/strain/016234.html). Genotyping primers for this line are F: TGCTGATGGCGAAGTTTACA and R: TGAGTAGTATAGATGTGATCGAAAAGG. The care and use of animals used in this study was strictly applying European and National Regulation in force for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes (Directive 86/609).
Tumor histology
Tissue sections of mice tumors obtained at sacrifice were prepared as previously described [16] and evaluated by a pediatric pathologist (MP).
Transcriptomic profiling of mice tumors
Transcriptomic profiling of tumors was performed as described previously using Affymetrix Mouse Genome 430 2.0 arrays [16]. The GCRMA procedure using Brainarray annotations was used for normalization [46].
Cell lines, antibodies, and reagents
IMR-32 and SK-N-BE(2)C cell lines were obtained from the American Type Culture Collection (ATCC). CLB cell lines were derived by V. Combaret (Lyon, France). Cell line authentication was performed using SNP array profile or STR profiling for ATCC cell lines. Cells were checked routinely by PCR for the absence of mycoplasma. Cell lines were grown in RPMI 1640 (CLB-GA, CLB-GE, and CLB-MA) or DMEM (IMR-32, SK-N-BE(2)C, and HEK293T) medium supplemented with 10% FBS and antibiotics. ETV5 invalidation was performed with Silencer®Select validated siRNA from Ambion (ThermoFischer Scientific) with the RNAimax transfection reagent (ThermoFischer Scientific). To obtain a total protein lysate, cells were washed in PBS containing 50 µM of sodium orthovanadate and lysed using NP-40 lysis buffer (10 mM Tris-HCl, 5 mM EDTA, 150 mM NaCl, 1% NP-40, 10% glycerol, 20 mM NaF, 1 mM sodium orthovanadate, Complete Protease Inhibitor Cocktail, and PhosSTOP Phosphatase Inhibitor from Roche Diagnostics). Anti-phospho-ALK, anti-ALK, anti-phospho-ERK antibodies were from Cell Signaling Technology (#3341, 3633, and 4370, respectively). Anti-ERK was from Millipore (06-182), anti-ETV5 from Abnova (H00002119-M02), anti-RET from Abcam (ab134100), and anti-actin from Sigma (A-5316). TAE684 and trametinib were purchased from Selleckem. mAb46 is a kind gift from Pr. Marc Vigny.
RT-qPCR
Complementary DNA synthesis was performed on total RNA by use of High-Capacity cDNA Reverse Transcription Kit (ThermoFischer Scientific). RPLP0, RET, and ETV5 cDNA expression were determined using Power SYBR® Green PCR Master Mix and the CFX384TM machine (Biorad). Relative expression was calculated relative to RPLP0 and untreated cells of the same time point of the same experiment. Primers sequences are: RPLP0_F: GAAACTCTGCATTCTCGCTTC, RPLP0_R: GGTGTAATCCGTCTCCACAG, RET_F: GCTGATAGAACCGGAAGAGG, RET_R: AATTTGTAGACGTATCGCTCTCC, ETV5_F: GGAGAAGGCGAATTTGGAAAAG, and ETV5_R: CAGGACGTTGAACTCTGACAG.
Reporter assay
Primers were designed to clone a 1462-bp genomic region (hg19, chr10:43,571,612-43,573,074) around the transcription start site of the RET gene. This RET promoter was inserted into the 8xGTIIC plasmid (Addgene) between the NheI and NcoI restriction sites (in place of the YAP-TAZ responsive element), upstream of the Luciferase gene. We used a pcDNA3 vector to express the ETV5 protein. Transfection into HEK293T cells was accomplished using Lipofectamine® LTX with PlusTM Reagent (ThermoFisher Scientific) with three plasmids: (1) pcDNA3-empty or pcDNA3-ETV5 (200 or 400 ng); (2) 8×GTIIC vector containing the RET promoter or without promoter (400 ng); (3) the Renilla expression control vector (pRL-CMV, Promega, E2261) at a 1:400 dilution with respect to the luciferase vector. Luciferase assays were carried out 48 h after transfection using Dual-Luciferase® Reporter Assay System (Promega, E1910) with read-outs performed on a FLUOstar OPTIMA Microplate Reader Detection System (BMG LABTECH). Luciferase expression was normalized to Renilla expression. Results were averaged from five independent experiments and normalized to empty pcDNA3.
ChIP-Seq
ETV5 Chromatin ImmunoPrecipitation (ChIP) was performed using the iDeal ChIP-seq kit for Transcription Factors (Diagenode) and the anti-ETV5 rabbit polyclonal antibody ab102010 from Abcam, using the same procedure as previously described [47]. The TruSeq ChIP library preparation kit was used to prepare Illumina sequencing libraries from the ChIP and input DNAs, as described [47].
ChIP-Seq analysis
ChIP-seq reads were mapped to the human reference genome hg19/GRCh37 using Bowtie2 [48]. Low mapping quality reads (Q < 20) and duplicate reads were discarded. Enriched regions (peaks) were called using HMCan v1.19 [49] as previously described [47]. Motif discovery in ChIP-seq peaks was performed using the Position Analysis tool of the RSAT package [50] on the 2500 highest peaks detected by HMcan.
Allografts in nude mice and in vivo treatment
An abdominal neuroblastoma tumor was dissected from a TH-MYCN/AlkF1178L mouse and mashed providing a mixture of a total volume of 1.4 ml. Overall, 100 μl of this mixture was subcutaneously injected into 6-week-old female Swiss Nude mice. When tumor volume reached around 250 mm3, mice were randomly assigned to one of the 3 groups (vehicle, crizotinib or crizotinib, and vandetanib) and treated daily with vehicle, crizotinib (100 mg/kg) or crizotinib (100 mg/kg) and vandetanib (30 mg/kg) in combination by oral gavage. No blinding was done. Crizotinib and vandetanib were purchased from Medchem express. Approval for this study was received from Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche (authorization number 5524-20 160531 1607151 v5).
Statistical analysis
For the RT-qPCR and the luciferase activity experiments, as each value has been normalized to an internal control, a one-sample t-test was used to evaluate the probability that the mean is different from 1. For the analysis of tumor volumes in Fig. 6a, a two-tailed Student’s t-test was used between the crizotinib treated and the crizotinib and vandetanib treated tumors. Asterisk indicates statistically significant difference (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001). Statistical analyses were conducted using R (http://www.R-project.org).
Data deposition
The GEO public database accession number for the expression microarrays of the MYCN/RetM919T tumors is GSE72678.
References
Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G, The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8:11–23.
Hallberg B, Palmer RH, The role of the ALK receptor in cancer biology. Ann Oncol. 2016;27(Suppl 3):iii4–iii15.
Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Prim. 2016;2:16078.
Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–70.
Mosse YP, Longo L, Laudenslager M, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–5.
Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–4.
George RE, Sanda T, Hanna M, Frohling S, Luther W,2nd, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–8.
Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 2014;26:682–94.
Mano H, ALKoma: a cancer subtype with a shared target. Cancer Discov. 2012;2:495–502.
Camidge DR, Doebele RC. Treating ALK-positive lung cancer—early successes and future challenges. Nat Rev Clin Oncol. 2012;9:268–77.
Mossé YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14:472–80.
Bresler SC, Wood AC, Haglund EA, Courtright J, Belcastro LT, Plegaria JS, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med. 2011;3:108ra114.
Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70:10038–43.
Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, et al. The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to crizotinib in ALK-driven neuroblastoma. Cancer Discov. 2016;6:96–107.
Guan J, Tucker ER, Wan H, Chand D, Danielson LS, Ruuth K, et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis Model Mech. 2016;9:941–52.
Cazes A, Lopez-Delisle L, Tsarovina K, Pierre-Eugène C, De Preter K, Peuchmaur M, et al. Activated Alk triggers prolonged neurogenesis and Ret upregulation providing a therapeutic target in ALK-mutated neuroblastoma. Oncotarget. 2014;5:2688–702.
Oh S, Shin S, Janknecht R, ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta. 2012;1826:1–12.
Fontanet P, Irala D, Alsina FC, Paratcha G, Ledda F, Pea3 transcription factor family members Etv4 and Etv5 mediate retrograde signaling and axonal growth of DRG sensory neurons in response to NGF. J Neurosci. 2013;33:15940–51.
Simon L, Ekman GC, Tyagi G, Hess RA, Murphy KM, Cooke PS, Common and distinct factors regulate expression of mRNA for ETV5 and GDNF, Sertoli cell proteins essential for spermatogonial stem cell maintenance. Exp Cell Res. 2007;313:3090–9.
Pasini B, Ceccherini I, Romeo G, RET mutations in human disease. Trends Genet Tig. 1996;12:138–44.
Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer. 1999;86:364–72.
Moog-Lutz C, Degoutin J, Gouzi JY, Frobert Y, Brunet-de Carvalho N, Bureau J, et al. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J Biol Chem. 2005;280:26039–48.
Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM–ALK. Proc Natl Acad Sci Usa. 2007;104:270–5.
Lambertz I, Kumps C, Claeys S, Lindner S, Beckers A, Janssens E, et al. Upregulation of MAPK negative feedback regulators and RET in mutant ALK neuroblastoma: implications for targeted treatment. Clin Cancer Res. 2015;21:3327–39.
Mazot P, Cazes A, Dingli F, Degoutin J, Irinopoulou T, Boutterin M-C, et al. Internalization and downregulation of the ALK receptor in neuroblastoma cell lines upon monoclonal antibodies treatment. PLoS One. 2012;7:e33581.
Mulligan LM, RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer. 2014;14:173–86.
Grant CE, Bailey TL, Noble WS, FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–8.
Wei G-H, Badis G, Berger MF, Kivioja T, Palin K, Enge M, et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J 2010;29:2147–60.
Smith-Hicks CL, Sizer KC, Powers JF, Tischler AS, Costantini F, C-cell hyperplasia, pheochromocytoma and sympathoadrenal malformation in a mouse model of multiple endocrine neoplasia type 2B. EMBO J. 2000;19:612–22.
Reiff T, Huber L, Kramer M, Delattre O, Janoueix-Lerosey I, Rohrer H, Midkine and Alk signaling in sympathetic neuron proliferation and neuroblastoma predisposition. Development. 2011;138:4699–708.
Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012;22:117–30.
Schulte JH, Bachmann HS, Brockmeyer B, Depreter K, Oberthur A, Ackermann S, et al. High ALK receptor tyrosine kinase expression supersedes ALK mutation as a determining factor of an unfavorable phenotype in primary neuroblastoma. Clin Cancer Res. 2011;17:5082–92.
Zhang W, Yu Y, Hertwig F, Thierry-Mieg J, Zhang W, Thierry-Mieg D, et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015;16:133.
Shaw AT, Hsu PP, Awad MM, Engelman JA, Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer. 2013;13:772–87.
Simon L, Ekman GC, Tyagi G, Hess RA, Murphy KM, Cooke PS, Common and distinct factors regulate expression of mRNA for ETV5 and GDNF, Sertoli cell proteins essential for spermatogonial stem cell maintenance. Exp Cell Res. 2007;313:3090–9.
Liu Y, Jiang H, Crawford HC, Hogan BLM. Role for ETS domain transcription factors Pea3/Erm in mouse lung development. Dev Biol. 2003;261:10–24.
Lin S, Perl A-KT, Shannon JM, Erm/thyroid transcription factor 1 interactions modulate surfactant protein C transcription. J Biol Chem. 2006;281:16716–26.
Baert J-L, Beaudoin C, Monte D, Degerny C, Mauen S, de Launoit Y, The 26S proteasome system degrades the ERM transcription factor and regulates its transcription-enhancing activity. Oncogene. 2007;26:415–24.
Vitari AC, Leong KG, Newton K, Yee C, O’Rourke K, Liu J, et al. COP1 is a tumour suppressor that causes degradation of ETS transcription factors. Nature. 2011;474:403–6.
Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010;467:849–53.
Lu BC, Cebrian C, Chi X, Kuure S, Kuo R, Bates CM, et al. Etv4 and Etv5 are required downstream of GDNF and Ret for kidney branching morphogenesis. Nat Genet. 2009;41:1295–302.
Tyagi G, Carnes K, Morrow C, Kostereva NV, Ekman GC, Meling DD, et al. Loss of Etv5 decreases proliferation and RET levels in neonatal mouse testicular germ cells and causes an abnormal first wave of spermatogenesis. Biol Reprod. 2009;81:258–66.
Chatterjee S, Kapoor A, Akiyama JA, Auer DR, Lee D, Gabriel S, et al. Enhancer variants synergistically drive dysfunction of a gene regulatory network in hirschsprung disease. Cell. 2016;167:355–68.
Wood A, Krytska K, Ryles HT, Infarinato NR, Sano R, Hansel TD, et al. Dual ALK and CDK4/6 inhibition demonstrates on-target synergy against neuroblastoma. Clin Cancer Res. 2017;23:2856–68.
Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16:2985–95.
Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175.
Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugène C, Raynal V, et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet. 2017;49:1408–13.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Ashoor H, Hérault A, Kamoun A, Radvanyi F, Bajic VB, Barillot E, et al. HMCan: a method for detecting chromatin modifications in cancer samples using ChIP-seq data. Bioinformatics. 2013;29:2979–86.
Medina-Rivera A, Defrance M, Sand O, Herrmann C, Castro-Mondragon JA, Delerce J, et al. RSAT 2015: regulatory sequence analysis tools. Nucleic Acids Res. 2015;43:W50–56.
Acknowledgements
The U830 Inserm laboratory is supported by grants from the Institut National du Cancer (PRT-K 14061), the Ligue Nationale contre le Cancer (Equipe labellisée), the Association Hubert Gouin «Enfance et Cancer», Les Bagouz à Manon, les amis de Claire, la Fédération Enfants et Santé et la Société Française de Lutte contre les Cancers et les Leucémies de l’Enfant et l’Adolescent. LL-D was the recipient of a fellowship of the Fondation pour la Recherche Médicale (FDT20140930966). DS is supported by the Institut Curie-SIRIC (Site de Recherche Intégrée en Cancérologie) program. VB is supported by the ATIP-Avenir and the “Who am I?” programs. High-throughput sequencing has been performed by the ICGex NGS platform of the Institut Curie supported by the grants ANR-10-EQPX-03 (Equipex) and ANR-10-INBS-09-08 (France Génomique Consortium) from the Agence Nationale de la Recherche («Investissements d’Avenir» program), by the Canceropole Ile-de-France and by the SiRIC-Curie program—SiRIC Grant «INCa-DGOS- 4654». We thank Jean-Luc Baert (Institut de Recherche Interdisciplinaire de Lille, Lille, France) for providing us with the pcDNA3-ETV5 plasmid and Matthias Fischer for providing the ALK status of tumors from the GSE49711 dataset. We thank D Gentien and his team from the translational department of Institut Curie for Affymetrix profiling experiments. We are grateful to the animal facilities team of the Institut Curie and especially to I Grandjean and C Jouhanneau. We thank the Experimental Pathology Department of Institut Curie for their valuable help and the SIRIC-Curie for help in image analyses. We acknowledge our colleagues F Tirode and W Richer for assistance in the analysis of Affymetrix data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lopez-Delisle, L., Pierre-Eugène, C., Louis-Brennetot, C. et al. Activated ALK signals through the ERK–ETV5–RET pathway to drive neuroblastoma oncogenesis. Oncogene 37, 1417–1429 (2018). https://doi.org/10.1038/s41388-017-0039-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-017-0039-5
This article is cited by
-
E26 transformation-specific transcription variant 5 in development and cancer: modification, regulation and function
Journal of Biomedical Science (2023)
-
Systematic review of the receptor tyrosine kinase superfamily in neuroblastoma pathophysiology
Cancer and Metastasis Reviews (2022)
-
Transcriptome sequencing of LMP2A-transfected gastric cancer cells identifies potential biomarkers in EBV-associated gastric cancer
Virus Genes (2022)
-
The role of E26 transformation-specific variant transcription factor 5 in colorectal cancer cell proliferation and cell cycle progression
Cell Death & Disease (2021)
-
IP6-assisted CSN-COP1 competition regulates a CRL4-ETV5 proteolytic checkpoint to safeguard glucose-induced insulin secretion
Nature Communications (2021)
