
Abstract
There is evidence linking ADHD to a reduced life expectancy. The mortality rate in individuals with ADHD is twice that of the general population and it is associated with several factors, such as unhealthy lifestyle behaviors, social adversity, and mental health problems that may in turn increase mortality rates. Since ADHD and lifespan are heritable, we used data from genome-wide association studies (GWAS) of ADHD and parental lifespan, as proxy of individual lifespan, to estimate their genetic correlation, identify genetic loci jointly associated with both phenotypes and assess causality. We confirmed a negative genetic correlation between ADHD and parental lifespan (rg = −0.36, P = 1.41e−16). Nineteen independent loci were jointly associated with both ADHD and parental lifespan, with most of the alleles that increased the risk for ADHD being associated with shorter lifespan. Fifteen loci were novel for ADHD and two were already present in the original GWAS on parental lifespan. Mendelian randomization analyses pointed towards a negative causal effect of ADHD liability on lifespan (P = 1.54e−06; Beta = −0.07), although these results were not confirmed by all sensitivity analyses performed, and further evidence is required. The present study provides the first evidence of a common genetic background between ADHD and lifespan, which may play a role in the reported effect of ADHD on premature mortality risk. These results are consistent with previous epidemiological data describing reduced lifespan in mental disorders and support that ADHD is an important health condition that could negatively affect future life outcomes.
Similar content being viewed by others
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a neurodevelopmental disorder that emerges in childhood and often persists into adulthood, affecting approximately 5.3% of children and adolescents and 2.8% of adults [1, 2]. It is characterized by age-inappropriate symptoms of inattention, impulsivity, and hyperactivity, which have a severe impact on the individual’s social, emotional and psychological functioning, often representing an entry point into a poor life trajectory [3].
There is increasing evidence linking ADHD to a shorter life expectancy and mortality rates in individuals with ADHD are two to five times higher than in individuals without ADHD [4, 5]. Besides natural causes [5], the higher risk of early mortality in individuals with ADHD appears to be largely due to misadventure including a high propensity for accidents and injuries and an elevated risk of suicide [4,5,6,7,8,9].
ADHD often co-occurs with other mental and somatic comorbid disorders, traits and behaviors that are likely to increase mortality rates [10, 11]. These include (i) mental health problems, such as oppositional defiant disorder, conduct disorder, mood and anxiety disorders, and substance use disorder [8, 12,13,14]; (ii) comorbid somatic disorders such as obesity [3], asthma [15] and diabetes [16, 17]; (iii) harmful lifestyle behaviors, such as unhealthy eating habits or smoking [18,19,20]; and (iv) educational underachievement, low income, social adversity, delinquency and aggression [3, 21]. However, despite the high rate of comorbid conditions also linked to an excess mortality [12], these do not fully explain the risk of death observed in individuals with ADHD [4, 5, 8], indicating that ADHD itself is a health condition that confers an increased risk of mortality. For instance, inattention and impulsivity may directly increase proneness to risk-taking behaviors and therefore risk for accidental injuries, leading to reduced lifespan [5].
ADHD and lifespan are complex traits influenced both by genetic and environmental factors. Heritability is estimated to be around 70–80% for ADHD and 7–16% for human lifespan [22]. Genome-wide association studies (GWAS) identified genetic loci associated with both ADHD and lifespan [23, 24], although a large part of their heritability still remains to be explained. Some genes known to be related with the risk of developing ADHD, such as those related to the dopaminergic system, have also been associated with shorter life expectancy [19], and a negative genetic correlation between ADHD and parental age at death has been described [24]. These data support observational studies showing an association between ADHD with both elevated mortality risk and reduced estimated life expectancy in adulthood, and suggest that the underlying genetic background of ADHD and lifespan may overlap.
In the present study, we aim to examine the shared genetic architecture and the nature of the relationship between ADHD and parental lifespan, as a proxy for individual lifespan, using available GWAS data on both phenotypes by: (i) estimating their genetic correlation; (ii) performing a cross-trait analysis and (iii) testing the causal role of ADHD on lifespan.
Materials and methods
GWAS samples and data processing
GWAS summary statistics on ADHD were obtained from Demontis et al., comprising a total of 19,099 individuals with ADHD and 34,194 healthy controls, all of European ancestry [24]. Summary statistics on parental lifespan were obtained from Timmers et al. and included data from about 1 million individuals of European ancestry [23]. The GWAS summary statistics were referenced to a set of 9,546,816 SNPs generated from the 1000 Genomes Project Phase 3 European reference panel (http://www.internationalgenome.org/). SNPs that were non-biallelic, without rsIDs, duplicated, or with strand-ambiguous alleles were removed. We also filtered out SNPs with INFO scores <0.9 in the summary statistics files, those mapping to the extended major histocompatibility complex (MHC, genomic position in hg 19; chr6:25,119,106–33,854,733) and the 8p23.1 region (chr8:7,200,000–12,500,000), which are prone to rearrangements, SNPs located on the X, Y and mitochondrial chromosomes, and SNPs with sample sizes 5 standard deviations away from the mean. Finally, a common set of 3,206,697 SNPs were kept in both summary statistics. All P values were adjusted for standard genomic control (GC).
Genetic correlation and pleiotropy assessment
Linkage Disequilibrium Score Regression (LDSC) was used to calculate genome-wide genetic correlations across the traits studied [25].
The shared polygenic architecture between the two traits was assessed by means of stratified cross-phenotype Q-Q plots. P values for the primary trait were plotted conditioning on different association strengths (P < 1, 0.1, 0.01 and 1e−03) with the secondary trait. Thus, the visualization of a leftward shift in the primary trait of interest, as a function of increasingly strict P value thresholds in the secondary trait, was an indicator of a shared polygenic architecture between the two traits. To test for SNP-based heritability enrichment of a trait conditioned on different association strengths with a secondary trait, we used stratified LDSC [26]. As a reference panel for linkage disequilibrium (LD) we used the 1000 Genomes Project Phase 3 European reference panel [27].
Cross-trait analysis
To identify genetic loci jointly associated with ADHD and parental lifespan we estimated the conjunction FDR (conjFDR), defined as “the posterior probability that a given SNP is null for both phenotypes simultaneously when the P values for both phenotypes are as small as or smaller than the observed P values”, using pleioFDR (https://github.com/precimed/pleiofdr) [28]. We kept all SNPs with conjFDR <0.1 for functional studies and reported independent SNPs with a conjFDR <0.05. Independent genomic loci were identified through clumping (r2 = 0.05, kb = 500) using the 1000 Genomes Project Phase 3 European as the reference panel for LD computation and PLINK 1.09 [27, 29]. We evaluated the directional effects of shared loci between ADHD and parental lifespan by comparing z-scores between the original GWAS summary statistics. Overlap between hits from the cross-trait analysis and genome-wide significant associations reported in the original GWAS results (P < 5e−08) was assessed according to distance (+/−250 kb) and linkage disequilibrium (r2 > 0.1) between of the cross-trait lead SNPs and previous reported hits.
Functional annotation
Functional annotation of all SNPs with a conjFDR value <0.10 and an LD r2 ≥ 0.6 with one of the independent significant SNPs was performed in FUMA (Functional Mapping and Annotation of Genome-Wide Association Studies, https://fuma.ctglab.nl/) [30]. We combined data from the Combined Annotation Dependent Depletion (CADD) scores [31], which predict how deleterious the SNP effect is on protein structure/function based on 63 functional annotations, and RegulomeDB scores [32], a categorical score that estimates the regulatory functionality of SNPs based on existing functional data (annotation to cis-eQTLs, expression quantitative trait loci) and evidence for transcription factor binding. CADD ≥ 12.37 was considered as the threshold for deleterious variants, RegulomeDB scores <3 were likely to have a regulatory function, and minimum chromatin states between 1–7 were considered open chromatin states. The NHGRI-EBI GWAS catalog [33], the release from the 15th of September 2021, was used to identify traits previously associated with the SNPs of interest and we queried SNPs for known brain eQTLs using the Genotype-Tissue Expression (GTEx) v8 [34] and BRAINEAC [35]. We also used FUMA to map SNPs to genes based on physical proximity (using default parameters) and eQTL in brain (based on GTEx v8 and BRAINEAC) to test for enrichment on gene ontology and biological pathways of the mapped genes. All analyses were corrected for multiple comparisons using False Discovery Rate.
Causality analyses
GWAS summary statistics
To assess the causal effect of the genetic liability of ADHD on lifespan, the summary statistics from Pilling et al. [36], generated from a linear mixed-effects model in 208,118 individuals of European ancestry, was used instead of Timmers et al. [23]. The latter used a survival analysis to study parental lifespan, which may produce a significant bias in MR analyses [37]. LDSC was used to calculate genetic correlations between both studies and between ADHD and parental lifespan in the study by Pilling et al. [25, 36]. For mediation analyses, as defined below we used summary statistics from Sanchez-Roige et al. for total impulsivity score, lack of premeditation and positive urgency [38].
Mendelian randomization
Causality between ADHD (as the exposure), and parental lifespan (as the outcome), was assessed by two-sample MR using the TwoSampleMR and MRPRESSO R packages [39, 40]. After clumping (r2 = 0.05, kb = 500) with PLINK 1.09 [29], independent SNPs were selected using a P value threshold of 5e−08 in the ADHD GWAS to be used as instruments. The multiplicative random effects inversed-variance weighted (IVW) was used as the main method to obtain the average effect across genetic variants. For IVW results to be valid, genetic variants used as instruments must meet three assumptions: (i) robust association with the exposure, (ii) absence of horizontal pleiotropy, or association with the outcome through an exposure-independent pathway, and (iii) independence of confounders influencing exposure and outcome. Additional MR methods were implemented, as sensitivity analyses, for significant IVW results (IVW P < 0.05) to assess the robustness of the findings under weaker assumptions: (i) the weighted median method, which under equal weights requires at least half of the variants to be valid instruments and is robust to outliers [41]; (ii) the MR-PRESSO method, which tests for horizontal pleiotropy (MR-PRESSO global test), and if detected, eliminates horizontal pleiotropic outliers and then performs the IVW method using the remaining instruments [40]; and (iii) the MR-Egger method, which is affected by outlier data points but allows all genetic variants to have pleiotropic effects assuming that these effects are independent of variant–exposure associations [42]. MR-Egger also implements a pleiotropy test, however, when the NO Measurement Error (NOME) assumption is violated (I2gx < 0.9) MR-Egger causal estimates are biased towards the null, and the type I error of the pleiotropy test can be inflated. For this reason, when I2gx < 0.6, MR-Egger results were disregarded. In addition, heterogeneity tests and leave-one-out analyses were performed and scatter, funnel and forest plots were generated. The analysis in the opposite direction, testing the effect of parental lifespan on ADHD, was considered as a negative control. Finally, as an alternative method, we also used the ρ‐HESS implementation to prioritize putative causal models between pairs of traits, as previously described [43, 44].
Multivariate Mendelian Randomization (MVMR)
To explore whether a putative causal effect of the genetic liability of ADHD on lifespan is mediated by impulsive personality traits we used MVMR and summary statistics from the GWAS by Sanchez-Roige et al. undertaken in over 20,000 individuals [38]. Out of the ten traits analyzed by Sanchez-Roige et al. [38]. we selected as potential mediators those reported to have a significant genetic correlation with ADHD, prioritizing total scores when available, namely total impulsivity score (measured with the BIS-11 [45]), lack of premeditation and positive urgency (both measured using the UPPS-P Impulsive Behavior Scale [46]). We used the same clumping parameters as for the main analysis (r2 = 0.05, kb = 500) and, given that no genome-wide significant SNPs were available, a threshold of 5e−06 was chosen to select independent SNPs for these traits.
Causal Analysis Using Summary Effect estimates (CAUSE)
We also explored causality between ADHD and lifespan using the cause R package [47] considering independent variants (r2 = 0.05, kb = 500). The CAUSE method uses a more permissive threshold for variant selection than MR (P < 1e−03). In addition, CAUSE allows all variants to show uncorrelated pleiotropy, also accounted for by MR-Egger or MR-PRESSO, but it also allows a subset of variants to show correlated pleiotropy, when they affect exposure and outcome through a shared heritable factor. CAUSE compares two nested models by measuring how well the posterior distributions of a particular model fit the data: (i) a sharing model, which only allows for pleiotropic effects and no causal effects; and (ii) a causal model, which also allows both for pleiotropic and causal effects.
Results
We found strong evidence of negative SNP-based genetic correlation between ADHD and parental lifespan, used as a proxy for individual lifespan (rg = −0.36, P = 1.41e−16). Partitioning ADHD SNP heritability on different parental lifespan P value thresholds showed enrichment of ADHD SNP heritability (P = 2.73e−07 and P = 1.85e−03 conditioning ADHD on parental lifespan P < 0.1 and P < 0.01, respectively; Supplementary Table 1a). Furthermore, conditioning parental lifespan SNPs on ADHD P values showed parental lifespan SNP heritability enrichment (P = 1.09e−06 and 7.69e−03 conditioning parental lifespan on ADHD P < 0.1 and P < 0.01, respectively; Supplementary Table 1b). These enrichments were consistent with stratified cross-phenotype Q-Q plots showing a stronger leftward deflection from the null expectation when conditioning ADHD on increasing levels of association for parental lifespan and vice-versa (Fig. 1).

Nominal versus empirical (−log10) P values (corrected for inflation) are shown in A ADHD as a function of significance with parental lifespan and B parental lifespan as a function of significance with ADHD, at the level of P < 0.1 (red line), P < 0.01 (yellow line), and P < 1e−03 (purple line). The blue line indicates the standard enrichment of A ADHD or B parental lifespan including all SNPs, irrespective of their association with the secondary trait (i.e., parental lifespan or ADHD, respectively). The gray dashed line indicates the null distribution of P values. LD score regression intercepts for ADHD and parental lifespan full summary statistics were 1.04 and 1.05 respectively. PLS parental lifespan.
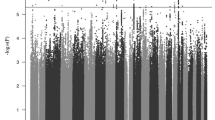
The cross-trait analysis showed a total of 19 independent genomic loci associated with both ADHD and parental lifespan with a conjFDR < 0.05 (Fig. 2A and Supplementary Fig. 1), 15 of which were not identified in the original GWAS on ADHD and two were already present in the original GWAS on parental lifespan (Table 1). Functional annotation of all SNPs with conjFDR < 0.1 at these 19 loci (n = 479 SNPs) revealed that 92.1% of these loci lay on regions of open chromatin and most of the signals were intergenic, intronic or located in intronic non-coding RNA (ncRNA) genes. In addition, several SNPs in these genomic risk loci were likely to affect the binding of transcription factors or had CADD scores >12.37, suggesting high deleteriousness (Fig. 3, Table 1 and Supplementary Table 2). In addition, we found that 42% of the SNPs were eQTL for at least one gene in one brain area, according to GTEx v8 [34] and BRAINEAC [35] (Supplementary Table 3). Finally, 23 SNPs at 11 different genomic risk loci were previously associated with different traits, mainly related to lifetime risky behaviors (e.g., smoking, general risk tolerance and number of sexual partners), psychiatric disorders (e.g., schizophrenia, ADHD and depression) and metabolic alterations (e.g., metabolic syndrome, triglyceride or cholesterol levels and blood pressure; Supplementary Table 4). The 19 risk loci identified mapped 40 genes (Supplementary Table 5), which were enriched in genes previously associated with cognitive performance, smoking and metabolite levels according to the GWAS catalog [33] (Supplementary Fig. 2), but no association with either biological pathways or differential tissue expression from GTEx were found.

A Manhattan plot for independent (r2 < 0.05) loci associated with both ADHD and parental lifespan after excluding SNPs in the MHC and the 8p23.1 regions. The dashed black line represents the PFDR threshold of 0.05. B Pleiotropy plot. For lead SNPs (n = 19), P values and the direction of the effects (z-scores) of the derived alleles are plotted for parental lifespan (x-axis) against ADHD (y-axis). PLS parental lifespan.

Regulome DB score predicts likelihood of regulatory functionality, where lower scores indicate higher likelihood. Further information can be found in Boyle et al. 2012 [32]. Minimum Chromatin State across 127 tissue and cell types, lower scores indicate higher accessibility, with states 1–7 referring to open chromatin states.
To explore the landscape of pleiotropic effects further, we examined the direction of the effects of the lead SNPs of all independent loci from the cross-trait analysis on both ADHD and parental lifespan and found an opposite direction of effect for 95% of them (n = 18), with alleles that increased the risk for ADHD also shortening lifespan (Fig. 2B). ADHD hits [24] showed a significant negative correlation with parental lifespan with p-HESS, whereas no significant correlation was found between parental lifespan hits [23] and ADHD, which is consistent with a putative causal relationship of ADHD on shortened lifespan (Supplementary Figure 3). In line with these results, MR analyses showed evidence of a negative causal effect of ADHD liability on lifespan (IVW Beta = −0.07 and P = 1.54e−06; weighted median Beta = −0.05 and P = 6.52e−03; Supplementary Table 6 and Fig. 4). There was no evidence of horizontal pleiotropy according to MR-PRESSO (global test P = 0.43) or heterogeneity (I2 = 11.15). The results were not driven by a single SNP (Supplementary Fig. 4). Variants included in the MR analyses are shown in Supplementary Table 7. MR testing the causal relationship of parental lifespan on ADHD, as a negative control, showed no significant results (IVW P = 0.66). MVMR analyses showed that the effect of ADHD liability on lifespan remained unchanged when accounting for total impulsivity score or lack of premeditation, however it decreased (from −0.07 to −0.06) when taking into account the effect of positive urgency, suggesting that positive urgency may be mediating around 11% of the effect of ADHD liability on lifespan (Supplementary Table 8). CAUSE did not provide evidence of correlated pleiotropy (Supplementary Table 9) or of a causal effect of ADHD liability on lifespan (Beta = −0.01 and CI = (−0.03, 0)) (Supplementary Tables 6 and 9). Given that survival analyses, including the study conducted by Timmers et al. used in the present study, may bias MR results, we conducted the causality analyses using summary statistics on parental lifespan from a second GWAS by Pilling et al. [36]. The genetic correlation between both summary statistics on parental lifespan was very high (rg = −0.93, P = 2.98e−184), and parental lifespan from the latest was also negatively correlated with ADHD (rg = −0.41, P = 2.66e−14).

Scatter plot of SNP effect estimates on ADHD vs. effect estimates on parental lifespan. Lines are drawn for inverse-variance weighted, weighted median and CAUSE, with the slope of each line corresponding to the estimated causal effect. Given that Igx2 = 0.36, MR-Egger results are not reported.
Discussion
The present study provides the first evidence of a common genetic background between ADHD and lifespan. Extensive literature supports that individuals with ADHD have an increased risk of premature death and a shorter life expectancy, which increases in females and depends on age at first ADHD diagnosis [4, 8]. In addition, the presence of other comorbid psychiatric conditions, such as oppositional defiant disorder, conduct disorder or substance use disorder, further increases ADHD mortality [4, 8]. This excess mortality and decreased life expectancy in ADHD subjects are mainly driven by unnatural causes being accidents the most common cause of early death [4, 9]. In addition, an increased risk of suicide, traffic violations or trauma have been also described in ADHD [8, 48,49,50,51,52]. At the same time, the impact of the disorder on other adverse life-course outcomes, such as poorer educational attainment, unemployment, delinquency or lower socioeconomic status, may also increase the risk of mortality [11]. Strengthening the results of observational studies, we provide evidence of shared genetic signatures and negative genetic correlation between ADHD and lifespan, which further supports the existing evidence that ADHD represents an entry point into a negative life trajectory [3].
To provide potential pleiotropic molecular mechanisms underlying this association, we performed a cross-trait analysis on ADHD and parental lifespan and identified 19 independent loci jointly associated with both traits, including 15 novel hits for ADHD [23, 24]. All of them but one (95%) showed consistent direction of the effect, with risk alleles for ADHD shortening lifespan, which is consistent with the negative genetic correlation between ADHD and lifespan, adding further evidence for the contribution of a shared biological architecture. Interestingly, functional annotation of top hits from the cross-trait analysis highlighted loci previously associated with ADHD and/or reduced life expectancy-related phenotypes, including other psychiatric disorders (e.g., schizophrenia and major depression), lifetime risk behaviors (e.g., number of sexual partners and risk tolerance) or metabolic alterations (e.g., metabolic syndrome, and triglyceride and cholesterol levels). This further supports that shared risk factors may not be specific of ADHD or lifespan but also contribute to other related disorders and behaviors that could mediate, in part, this relationship. For instance, ADHD may directly increase propensity for risky behaviors and thus lead to a shorter life expectancy [5].
The pleiotropic risk loci identified were enriched in introns and open chromatin regions, and almost half of them (42%) were cis-eQTLs tags for at least one gene in the brain, supporting their putative causal effect. Among the identified risk loci, we highlight new relevant candidate genes. For instance, the TNKS gene encoding tankyrase1, a protein involved in telomere maintenance [53], which is an essential cellular function closely related to ageing and longevity [54]. Interestingly, several genetic variants in this locus are cis-eQTL for TNKS in putamen, a key area in the basal ganglia previously related with ADHD [55,56,57]. In addition, TNKS was previously associated with multiple psychiatric conditions (e.g., risk tolerance and neuroticism), neurological (e.g., Alzheimer’s disease and epilepsy) and metabolic disorders (e.g., blood pressure, obesity, diabetes and stroke) [58,59,60,61,62,63,64], which are highly comorbid with ADHD and associated with increased morbidity and mortality [65,66,67]. We also found genetic variants in other two loci encompassing AKAP6 and SEMA6D genes, previously associated with increased risk of schizophrenia, lower cognitive ability and educational attainment [68], all of them related with shorter life expectancy [69,70,71,72]. In addition, we found other loci including genes with relevant brain functions: SYPL2, which encodes a vesicular transmembrane protein that belongs to the synaptophysin family, key elements for the regulation of neuronal synaptic vesicles [73, 74]; and HMG20A, a non-histone chromosomal factor that regulates gene expression through changes in chromatin conformation, also involved in the regulation of neuronal differentiation [75, 76].
Our results seem to reflect an effect of ADHD on premature mortality risk and are consistent with previous epidemiological data describing a shortened lifespan in many mental disorders [4, 12]. A recent study suggests that there is no causal relationship between schizophrenia and parental lifespan [69], which can indicate that our results may not be extensive to other psychiatric disorders. Further studies are needed to clarify how specific the effect on parental lifespan is to ADHD. We also found suggestive evidence of a mediating role for positive urgency (i.e. the proclivity for rash action when feeling positive emotion) in the relationship between the genetic liability of ADHD and lifespan. This is consistent with a reported predictive effect of positive urgency on ADHD-related traits such as illegal drug use and risky sexual behavior [77]. Nevertheless these findings should be interpreted with caution, since the evidence found for a causal relationship of ADHD liability on shorter life expectancy was not conclusive. While IVW MR, together with all the sensitivity analyses performed and putative causality inferred with p‐HESS supported a negative causal effect of genetic liability of ADHD on lifespan, CAUSE did not confirm this causal relationship. This highlights that inferring causal relationships between related traits remains a challenge to date and that new methods and larger sample sizes are needed to understand the common genetic architecture underlying such complex relationships.
Our results, however, should be interpreted in the context of some considerations:
First, parental lifespan of genotyped subjects was used as a surrogate trait for individual lifespan in the present generation. This kinship cohort design is supported by previous results reporting that parental lifespan predicts the long-term mortality of their offspring. However, the use of indirect genotypes reduces the effective sample size of the study [23, 78]. This, together with the modest heritability estimates for human lifespan (ranging from 7% to 16%) [23, 79] limits the power of our study and precludes the identification of shared genetic variants with smaller effect sizes.
Second, to avoid the potential bias introduced by using summary statistics generated through a survival analysis [37], such as that conducted by Timmers et al. [23], we selected the results of Pilling et al. [36] to assess causality between ADHD and parental lifespan. The smaller sample size of this study (n = 208,118) further reduced the statistical power and may have led to the inconsistent results observed [36]. CAUSE was developed with the aim of avoiding false positive results due to correlated horizontal pleiotropy, but in the present study it did not detect a causal effect but neither did it detect this kind of pleiotropy. Recent studies suggest that in some scenarios CAUSE tends to underestimate causal effect sizes in comparison to MR and produce overly conservative P values [80, 81], which may suggest, in our case, limited power rather than lack of a causal relationship, supporting further studies with larger sample sizes to clarify this issue.
Third, despite the shared genetic architecture underlying ADHD in children and adults [82], the risk of premature death also depends on age at diagnosis, where individuals diagnosed with ADHD in adulthood appear to have a higher risk of death than do those diagnosed in childhood or adolescence [4]. These data may suggest that persistent ADHD represents a more severe form of the disorder and that the role of ADHD on the overall life expectancy across age groups deserves further investigation. In addition, we did not control our analysis for other potential confounders that may bias our results. Increased mortality in individuals with ADHD depends on sex or the presence of co-occurring disorders [4, 8]. Also severity of ADHD symptoms, impairment and/or pharmacological treatment may influence life outcomes [83,84,85] and their role in the relationship between ADHD, premature death and reduced overall life expectancy deserve further investigation.
In summary, our results are in agreement with observational studies supporting ADHD as the entrance into a detrimental life trajectory, support negative genetic correlation between ADHD and lifespan and show common genetic loci shared between them, most of which showed risk-increasing effects on ADHD and reduced overall life expectancy. These results confirm the general pattern of increased mortality rates and reduced overall life expectancy associated with ADHD and highlight the need for further studies on larger datasets to better understand the common genetic architecture underlying these complex relationships.
References
Polanczyk G, De Lima MS, Horta BL, Biederman J, Rohde LA. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry. 2007;164:942–8.
Fayyad J, Sampson NA, Hwang I, Adamowski T, Aguilar-Gaxiola S, Al-Hamzawi A, et al. The descriptive epidemiology of DSM-IV Adult ADHD in the World Health Organization World Mental Health Surveys. ADHD Atten Deficit Hyperact Disord. 2017;9:47–65.
Franke B, Michelini G, Asherson P, Banaschewski T, Bilbow A, Buitelaar JK, et al. Live fast, die young? A review on the developmental trajectories of ADHD across the lifespan. Eur Neuropsychopharmacol. 2018;28:1059–88.
Dalsgaard S, Ostergaard SD, Leckman JF, Mortensen PB, Pedersen MG. Mortality in children, adolescents, and adults with attention deficit hyperactivity disorder: a nationwide cohort study. Lancet. 2015;385:2190–6.
Sun S, Kuja-Halkola R, Faraone SV, D’Onofrio BM, Dalsgaard S, Chang Z, et al. Association of psychiatric comorbidity with the risk of premature death among children and adults with attention-deficit/hyperactivity disorder. JAMA Psychiatry. 2019;76:1141–9.
Prasad V, West J, Sayal K, Kendrick D. Injury among children and young people with and without attention-deficit hyperactivity disorder in the community: the risk of fractures, thermal injuries, and poisonings. Child Care Health Dev. 2018;44:871–8.
Stickley A, Koyanagi A, Ruchkin V, Kamio Y. Attention-deficit/hyperactivity disorder symptoms and suicide ideation and attempts: findings from the Adult Psychiatric Morbidity Survey 2007. J Affect Disord. 2016;189:321–8.
Barbaresi WJ, Colligan RC, Weaver AL, Voigt RG, Killian JM, Katusic SK. Mortality, ADHD, and psychosocial adversity in adults with childhood ADHD: a prospective study. Pediatrics. 2013;131:637–44.
Catalá-López F, Hutton B, Page MJ, Driver JA, Ridao M, Alonso-Arroyo A, et al. Mortality in persons with autism spectrum disorder or attention-deficit/hyperactivity disorder. JAMA Pediatr. 2022;176:e216401.
Faraone SV. Attention deficit hyperactivity disorder and premature death. Lancet. 2015;385:2132–3.
Erskine HE, Norman RE, Ferrari AJ, Chan GCK, Copeland WE, Whiteford HA, et al. Long-term outcomes of attention-deficit/hyperactivity disorder and conduct disorder: a systematic review and meta-analysis. J Am Acad Child Adolesc Psychiatry. 2016;55:841–50.
Nordentoft M, Wahlbeck K, Hällgren J, Westman J, Ösby U, Alinaghizadeh H, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS One. 2013;8:e55176.
Wilens TE, Spencer TJ. Understanding attention-deficit/hyperactivity disorder from childhood to adulthood. Postgrad Med. 2010;122:97.
Kendler KS, Ohlsson H, Sundquist J, Sundquist K. Alcohol use disorder and mortality across the lifespan: a longitudinal cohort and co-relative analysis. JAMA Psychiatry. 2016;73:575–81.
Cortese S, Sun S, Zhang J, Sharma E, Chang Z, Kuja-Halkola R, et al. Association between attention deficit hyperactivity disorder and asthma: a systematic review and meta-analysis and a Swedish population-based study. The Lancet. Psychiatry. 2018;5:717–26.
Chen MH, Pan TL, Hsu JW, Huang KL, Su TP, Li CT, et al. Risk of type 2 diabetes in adolescents and young adults with attention-deficit/hyperactivity disorder: A nationwide longitudinal study. J Clin Psychiatry. 2018;79:17m11607.
Kapellen TM, Reimann R, Kiess W, Kostev K. Prevalence of medically treated children with ADHD and type 1 diabetes in Germany - analysis of two representative databases. J Pediatr Endocrinol Metab. 2016;29:1293–7.
Ptacek R, Stefano GB, Weissenberger S, Akotia D, Raboch J, Papezova H, et al. Attention deficit hyperactivity disorder and disordered eating behaviors: links, risks, and challenges faced. Neuropsychiatr Dis Treat. 2016;12:571–9.
Barkley RA, Smith KM, Fischer M. ADHD risk genes involved in dopamine signaling and metabolism are associated with reduced estimated life expectancy at young adult follow-up in hyperactive and control children. Am J Med Genet Part B Neuropsychiatr Genet. 2019;180:175–85.
Groenman AP, Oosterlaan J, Rommelse N, Franke B, Roeyers H, Oades RD, et al. Substance use disorders in adolescents with attention deficit hyperactivity disorder: a 4-year follow-up study. Addiction. 2013;108:1503–11.
Mohr-Jensen C, Steinhausen HC. A meta-analysis and systematic review of the risks associated with childhood attention-deficit hyperactivity disorder on long-term outcome of arrests, convictions, and incarcerations. Clin Psychol Rev. 2016;48:32–42.
van den Berg N, Beekman M, Smith KR, Janssens A, Slagboom PE. Historical demography and longevity genetics: back to the future. Ageing Res Rev. 2017;38:28–39.
Timmers PRHJ, Mounier N, Lall K, Fischer K, Ning Z, Feng X, et al. Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. Elife. 2019;8:1–40.
Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. 2019;51:63–75.
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41.
Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47:1228–35.
Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, et al. A global reference for human genetic variation. Nature 2015;526:68–74.
Andreassen OA, Djurovic S, Thompson WK, Schork AJ, Kendler KS, O’Donovan MC, et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet. 2013;92:197–209.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Watanabe K, Taskesen E, Van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1–11.
Kircher M, Witten DM, Jain P, O’roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7.
MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45:D896–901.
Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–13.
Ramasamy A, Trabzuni D, Guelfi S, Varghese V, Smith C, Walker R, et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci. 2014;17:1418–28.
Pilling LC, Kuo CL, Sicinski K, Tamosauskaite J, Kuchel GA, Harries LW, et al. Human longevity: 25 genetic loci associated in 389,166 UK biobank participants. Aging. 2017;9:2504–20.
Cho Y, Rau A, Reiner A, Auer PL. Mendelian randomization analysis with survival outcomes. Genet Epidemiol. 2021;45:16–23.
Sanchez-Roige S, Fontanillas P, Elson SL, Gray JC, de Wit H, MacKillop J, et al. Genome-wide association studies of impulsive personality traits (BIS-11 and UPPSP) and drug experimentation in up to 22,861 adult research participants identify loci in the CACNA1I and CADM2 genes. J Neurosci. 2019;39:2562–72.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408.
Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–8.
Bowden J, Davey, Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–14.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–25.
Pickrell JK, Berisa T, Liu JZ, Ségurel L, Tung JY, Hinds DA. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet. 2016;48:709–17.
Major M, Freund MK, Burch KS, Mancuso N, Ng M, Furniss D, et al. Integrative analysis of Dupuytren’s disease identifies novel risk locus and reveals a shared genetic etiology with BMI. Genet Epidemiol. 2019;43:629–45.
Patton JH, Stanford MS, Barratt ES. Factor structure of the Barratt impulsiveness scale. J Clin Psychol. 1995;51:768–74.
Cyders MA, Littlefield AK, Coffey S, Karyadi KA. Examination of a short English version of the UPPS-P Impulsive Behavior Scale. Addict Behav. 2014;39:1372–6.
Morrison J, Knoblauch N, Marcus JH, Stephens M, He X. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat Genet. 2020;52:740–7.
Ertan C, Özcan ÖÖ, Pepele MS. Paediatric trauma patients and attention deficit hyperactivity disorder: correlation and significance. Emerg Med J. 2012;29:911–4.
Balazs J, Kereszteny A. Attention-deficit/hyperactivity disorder and suicide: a systematic review. World J Psychiatry. 2017;7:44.
Curry AE, Metzger KB, Pfeiffer MR, Elliott MR, Winston FK, Power TJ. Motor vehicle crash risk among adolescents and young adults with attention-deficit/hyperactivity disorder. JAMA Pediatr. 2017;171:756–63.
London AS, Landes SD. Attention Deficit Hyperactivity Disorder and adult mortality. Prev Med. 2016;90:8–10.
Chen VCH, Chan HL, Wu SI, Lee M, Lu ML, Liang HY, et al. Attention-deficit/hyperactivity disorder and mortality risk in Taiwan. JAMA Netw Open. 2019;2:e198714.
Damale MG, Pathan SK, Shinde DB, Patil RH, Arote RB, Sangshetti JN. Insights of tankyrases: a novel target for drug discovery. Eur J Med Chem. 2020;207:112712.
Riera CE, Merkwirth C, De Magalhaes Filho CD, Dillin A. Signaling networks determining life span. Annu Rev Biochem. 2016;85:35–64.
Wang Y, Zuo C, Xu Q, Hao L, Zhang Y. Attention-deficit/hyperactivity disorder is characterized by a delay in subcortical maturation. Prog Neuro-Psychopharmacology. Biol Psychiatry. 2021;104:110044.
Hoogman M, Bralten J, Hibar DP, Mennes M, Zwiers MP, Schweren LSJ, et al. Subcortical brain volume differences in participants with attention deficit hyperactivity disorder in children and adults: a cross-sectional mega-analysis. The Lancet. Psychiatry 2017;4:310–9.
Ceceli AO, Natsheh JY, Cruz D, Tricomi E. The neurobehavioral mechanisms of motivational control in attention-deficit/hyperactivity disorder. Cortex 2020;127:191–207.
Wachinger C, Nho K, Saykin AJ, Reuter M, Rieckmann A. A longitudinal imaging genetics study of neuroanatomical asymmetry in Alzheimer’s Disease. Biol Psychiatry. 2018;84:522–30.
Song M, Liu J, Yang Y, Lv L, Li W, Luo XJ. Genome-wide meta-analysis identifies two novel risk loci for epilepsy. Front Neurosci. 2021;15:722592.
Sung YJ, Winkler TW, de las Fuentes L, Bentley AR, Brown MR, Kraja AT, et al. A large-scale multi-ancestry genome-wide study accounting for smoking behavior identifies multiple significant loci for blood pressure. Am J Hum Genet. 2018;102:375–400.
Karlsson Linnér R, Biroli P, Kong E, Meddens SFW, Wedow R, Fontana MA, et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet. 2019;51:245–57.
Luciano M, Hagenaars SP, Davies G, Hill WD, Clarke TK, Shirali M, et al. Association analysis in over 329,000 individuals identifies 116 independent variants influencing neuroticism. Nat Genet. 2018;50:6.
Hill WD, Weiss A, Liewald DC, Davies G, Porteous DJ, Hayward C, et al. Genetic contributions to two special factors of neuroticism are associated with affluence, higher intelligence, better health, and longer life. Mol Psychiatry. 2020;25:3034.
Baselmans BML, Jansen R, Ip HF, van Dongen J, Abdellaoui A, van de Weijer MP, et al. Multivariate genome-wide analyses of the well-being spectrum. Nat Genet 2019 513. 2019;51:445–51.
Spann SJ, Ottinger MA. Longevity, Metabolic Disease, and Community Health. Prog Mol Biol Transl Sci. 2018;155:1–9.
Mackenbach JP, Karanikolos M, Looman CWN. The rise of mortality from mental and neurological diseases in Europe, 1979-2009: Observational study. BMC Public Health. 2014;14:840.
Walker ER, McGee RE, Druss BG. Mortality in mental disorders and global disease burden implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72:334.
Lam M, Hill WD, Trampush JW, Yu J, Knowles E, Davies G, et al. Pleiotropic Meta-Analysis of Cognition, Education, and Schizophrenia Differentiates Roles of Early Neurodevelopmental and Adult Synaptic Pathways. Am J Hum Genet. 2019;105:334–50.
Muntané G, Farré X, Bosch E, Martorell L, Navarro A, Vilella E. The shared genetic architecture of schizophrenia, bipolar disorder and lifespan. Hum Genet. 2021;140:441–55.
Olfson M, Gerhard T, Huang C, Crystal S, Stroup TS. Premature mortality among adults with schizophrenia in the United States. JAMA Psychiatry. 2015;72:1172–81.
Danler C, Pfaff K. The impact of an unequal distribution of education on inequalities in life expectancy. SSM Popul Heal. 2021;16:100954.
Halpern-Manners A, Raymo JM, Warren JR, Johnson KL. School performance and mortality: the mediating role of educational attainment and work and family trajectories across the life course. Adv Life Course Res. 2020;46:100362.
White DN, Stowell MHB. Room for two: the synaptophysin/synaptobrevin complex. Front Synaptic Neurosci. 2021;13:47.
Kwon SE, Chapman ER. Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron. 2011;70:847–54.
Ceballos-Chávez M, Rivero S, García-Gutiérrez P, Rodríguez-Paredes M, García-Domínguez M, Bhattacharya S, et al. Control of neuronal differentiation by sumoylation of BRAF35, a subunit of the LSD1-CoREST histone demethylase complex. Proc Natl Acad Sci USA. 2012;109:8085–90.
Artegiani B, Labbaye C, Sferra A, Quaranta MT, Torreri P, Macchia G, et al. The interaction with HMG20a/b proteins suggests a potential role for β-dystrobrevin in neuronal differentiation. J Biol Chem. 2010;285:24740–50.
Zapolski TCB, Cyders MA, Smith GT. Positive urgency predicts illegal drug use and risky sexual behavior. Psychol Addict Behav. 2009;23:348–54.
Vågerö D, Aronsson V, Modin B. Why is parental lifespan linked to children’s chances of reaching a high age? A transgenerational hypothesis. SSM - Popul Heal. 2018;4:45–54.
Kaplanis J, Gordon A, Shor T, Weissbrod O, Geiger D, Wahl M, et al. Quantitative analysis of population-scale family trees with millions of relatives. Science. 2018;360:171–5.
Darrous L, Mounier N, Kutalik Z. Simultaneous estimation of bi-directional causal effects and heritable confounding from GWAS summary statistics. Nat Commun. 2021;12:7274.
Yuan Z, Liu L, Guo P, Yan R, Xue F, Zhou X. Likelihood-based Mendelian randomization analysis with automated instrument selection and horizontal pleiotropic modeling. Sci Adv. 2022;8:eabl5744.
Rovira P, Demontis D, Sánchez-Mora C, Zayats T, Klein M, Mota NR, et al. Shared genetic background between children and adults with attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2020;45:1617–26.
McCarthy S, Cranswick N, Potts L, Taylor E, Wong ICK. Mortality associated with attention-deficit hyperactivity disorder (ADHD) drug treatment: A retrospective cohort study of children, adolescents and young adults using the general practice research database. Drug Saf. 2009;32:1089–96.
Chang Z, Lichtenstein P, D’Onofrio BM, Sjölander A, Larsson H. Serious transport accidents in adults with attention-deficit/hyperactivity disorder and the effect of medication a population-based study. JAMA Psychiatry. 2014;71:319–25.
Lichtenstein P, Halldner L, Zetterqvist J, Sjölander A, Serlachius E, Fazel S, et al. Medication for Attention Deficit–Hyperactivity Disorder and Criminality. N Engl J Med. 2012;367:2006–14.
Funding
This work was supported by the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR, 2017SGR-00444 and 2017SGR-1461), the Instituto de Salud Carlos III (PI18/01788, P19/01224, PI20/00041, PI21/00612, PI22/00464, FI18/00285 to LVR and CP22/00128 to MSA), the Ministry of Science, Innovation and Universities (IJC2018-035346-I to MSA and RYC2021-031324-I to JCD); the European Regional Development Fund (ERDF), the European Union H2020 Programme (H2020/2014–2020) under grant agreements no. 728018 (Eat2beNICE), no. 848228 (DISCOvERIE) and no. 2020604 (TIMESPAN) and the ECNP Network ‘ADHD across the Lifespan’ and “la Marató de TV3” (202228-30 and 202228-31). SSR was supported by funds from the California Tobacco-Related Disease Research Program (TRDRP; Grant Number T29KT0526 and T32IR5226), and NIH/NIDA DP1DA054394. AAP was supported by NIH grant P50DA037844 and R01AA029688 and Tobacco-Related Disease Research Program Grant number: 28IR-0070.
Author information
Authors and Affiliations
Contributions
GM, MR and MSA conceived the project, GM, MR and MSA participated in the study design, JCD, GM and LVR and participated in data acquisition, JCD, GM, MSA and LVR undertook the statistical analyses, JARQ, JCD, LM, GM, MR, MSA, LVR, EV, SSR and AAP. participated in the manuscript preparation. All authors contributed to the interpretation of the findings and revised and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
JARQ was on the speakers bureau and/or acted as consultant for Janssen-Cilag, Novartis, Shire, Takeda, Bial, Shionogi, Sincrolab, Novartis, BMS, Medice, Technofarma, Rubió and Raffo in the last 3 years. He also received travel awards (air tickets + hotel) for taking part in psychiatric meetings from Janssen-Cilag, Rubió, Shire, Takeda, Shionogi, Bial and Medice. The Department of Psychiatry Mental Health chaired by him received unrestricted educational and research support from the following companies in the last 3 years: Janssen- Cilag, Shire, Oryzon, Roche, Psious, and Rubió. AAP is on the scientific advisory board, owns stock options and a patent (https://patents.google.com/patent/US20160038559A1) for Vivid Genomics (https://www.vividgenomics.com/). All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vilar-Ribó, L., Cabana-Domínguez, J., Martorell, L. et al. Shared genetic architecture between attention-deficit/hyperactivity disorder and lifespan. Neuropsychopharmacol. 48, 981–990 (2023). https://doi.org/10.1038/s41386-023-01555-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-023-01555-x
