
Abstract
Re-exposure to a cocaine-associated context triggers craving and relapse through the retrieval of salient context-drug memories. Upon retrieval, context-drug memories become labile and temporarily sensitive to modification before they are reconsolidated into long-term memory stores. The effects of systemic cannabinoid type 1 receptor (CB1R) antagonism indicate that CB1R signaling is necessary for cocaine-memory reconsolidation and associated glutamatergic plasticity in the basolateral amygdala (BLA); however, the contribution of BLA CB1R signaling to cocaine-memory reconsolidation is unknown. Here, we assessed whether intra-BLA CB1R manipulations immediately after cocaine-memory retrieval alter cocaine-memory strength indexed by subsequent drug context-induced cocaine-seeking behavior in an instrumental rodent model of drug relapse. Administration of the CB1R antagonist, AM251 (0.3 µg/hemisphere) into the BLA increased subsequent drug context-induced cocaine-seeking behavior in a memory retrieval-dependent and anatomically selective manner. Conversely, the CB1R agonist, WIN55,212-2 (0.5 or 5 µg/hemisphere) failed to alter this behavior. In follow-up experiments, cocaine-memory retrieval elicited robust hypothalamic-pituitary-adrenal axis activation, as indicated by a rise in serum corticosterone concentrations. Intra-BLA AM251 administration during memory reconsolidation selectively increased this cocaine-memory retrieval-induced corticosterone response. Intra-BLA corticosterone administration (3 or 10 ng/hemisphere) during memory reconsolidation did not augment subsequent cocaine-seeking behavior, suggesting that CB1R-dependent effects of corticosterone on memory strength, if any, are mediated outside of the BLA. Together, these findings suggest that CB1R signaling in the BLA gates cocaine-memory strength, possibly by diminishing the impact of cue-induced arousal on the integrity of the reconsolidating memory trace or on the efficacy of the memory reconsolidation process.
Similar content being viewed by others


Introduction
Exposure to drug-associated environmental stimuli precipitates the retrieval of context-drug memories, thereby eliciting drug craving and relapse [1,2,3]. Upon retrieval from long-term memory stores, context-drug memories can become temporarily unstable and susceptible to modification. The maintenance of such labile memories requires their reconsolidation into long-term memory stores through a process that involves de novo protein synthesis [4] and synaptic plasticity [5]. Importantly, pathology in memory reconsolidation may result in overly salient or intrusive drug memories, contributing to the etiology of substance use disorders (SUDs), and reconsolidation can be manipulated therapeutically to weaken drug memories and thus relapse propensity [6]. Therefore, elucidating the neurobiological underpinnings of cocaine-memory reconsolidation is important from a SUD treatment perspective.
Cannabinoid type 1 receptor (CB1R) signaling plays a critical role in cocaine-memory reconsolidation. Specifically, our laboratory has shown that systemic CB1R antagonist administration during cocaine-memory reconsolidation attenuates subsequent drug context-induced cocaine-seeking behavior [7]. Systemic CB1R antagonist administration also interferes with glutamatergic transmission in the basolateral amygdala (BLA) [7], a site of protein synthesis-dependent memory reconsolidation [8,9,10,11,12,13]. However, questions remain about the contribution of BLA CB1R signaling, because BLA CB1R agonism [14] or antagonism [15] can similarly impair fear-memory reconsolidation. Furthermore, the role of BLA CB1Rs in appetitive memory reconsolidation has not been investigated.
The present study examined whether BLA CB1R signaling mediates cocaine-memory reconsolidation. First, we evaluated the effects of intra-BLA CB1R-antagonist and -agonist treatments administered immediately after cocaine-memory retrieval (i.e., while the memory was labile) on memory strength, as indicated by the magnitude of subsequent context-induced cocaine-seeking behavior. Next, we examined whether the effects were anatomically specific to BLA CB1Rs by manipulating CB1Rs in the adjacent posterior caudate putamen (pCPu), a possible site of injection-related tissue damage and infusate diffusion away from the BLA. As a step toward identifying a mechanism by which intra-BLA CB1R antagonism enhanced cocaine-memory strength, we assessed the effects of cocaine-memory retrieval and intra-BLA CB1R antagonist treatment on serum corticosterone concentrations, an index of hypothalamic pituitary adrenal (HPA)-axis activity, during reconsolidation. Finally, we evaluated the effects of intra-BLA corticosterone administration on cocaine-memory reconsolidation. Cocaine-memory reconsolidation is associated with HPA-axis activation [16], and stressor-induced suppression of endocannabinoid signaling in the BLA is critical for stress-induced HPA-axis activation [17]. Furthermore, intra-BLA glucocorticoid receptor (GR) antagonism enhances cocaine-memory strength during reconsolidation [16]. Therefore, we predicted that intra-BLA CB1R-antagonist treatment would selectively potentiate increases in corticosterone levels during reconsolidation, and intra-BLA corticosterone administration would limit cocaine-memory strength.
Materials and methods
Animals
Male Sprague-Dawley rats (N = 144; 275–300 g upon arrival; Envigo Laboratories, South Kent, WA) were housed individually in a temperature- and humidity-controlled vivarium on a reversed light/dark cycle (lights on at 6:00 am). Rats received ad libitum water access and 20–25 g of standard rat chow per day. Animal housing and care followed the Guide for the Care and Use of Laboratory Animals [18] and was approved by IACUC. After acclimation to handling, rats received a single food-training session followed by surgery 24 h later (Supplemental Material). The surgery involved the implantation of a catheter into the right jugular vein and bilateral injection cannulae into the BLA or pCPu (Supplemental Material).
Cocaine self-administration and extinction training
Five days after surgery, rats were randomly assigned to one of two distinctly different environmental contexts for cocaine self-administration training (Context A, Supplemental Material; [19]). Training was conducted for two h each day. Active-lever responses were reinforced under a fixed ratio 1 cocaine reinforcement schedule (0.15 mg of cocaine hydrochloride/50-µL infusion, delivered over 2.25 s, i.v.; NIDA Drug Supply Program, Research Triangle Park, NC) with a 20-s timeout period. Infusions and timeouts were not signaled by explicit conditioned stimuli. Timeout and inactive-lever responses were not reinforced. Training continued until rats obtained ≥10 infusions per session on at least 10 days. Next, rats received daily 2-h extinction training sessions in the alternate context (Context B) over seven days to control memory age at the time of manipulation. During extinction sessions, lever presses were not reinforced. After extinction session 4, rats were habituated to the microinfusion procedure (Supplemental Material).
Experiment 1: effects of post-retrieval AM251 administration in the BLA on subsequent cocaine-seeking behavior
Twenty-four h after extinction session 7, the rats were re-exposed to the cocaine-paired context for 15 min to trigger cocaine-memory retrieval and destabilization (Fig. 1A). Cocaine reinforcement was withheld to prevent acute cocaine effects on neurotransmission and endocannabinoid mobilization independent of memory processes [20, 21]. Immediately after the session, rats received bilateral intra-BLA microinfusions of the CB1R antagonist/inverse agonist, AM251 (0.3 µg/0.5 µL/hemisphere; n = 9; Sigma-Aldrich, St. Louis, MO), or vehicle (8% DMSO/5% Tween80/saline; n = 11) over 2 min. This intra-BLA dose of AM251 impairs contextual fear-memory reconsolidation [15]. After treatment, daily extinction-training sessions continued in the extinction context to isolate the motivational effects of the drug-paired context on reinstatement. Lever responses during the first post-treatment extinction session were analyzed to evaluate possible off-target treatment effects on extinction memories. Extinction training concluded when rats reached the extinction criterion (i.e., ≤25 active-lever presses per session on two consecutive days). Twenty-four h later, cocaine-seeking behavior (i.e., non-reinforced lever responses) was assessed in the cocaine-paired context for 2 h.

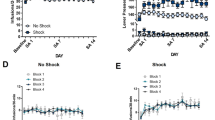
A Experimental timeline. After cocaine self-administration training in one context (cocaine-paired context, Context A), and extinction training in a different context (extinction context, Context B), rats received bilateral intra-BLA administration of the CB1R antagonist, AM251 (0.3 µg/0.5 µL per hemisphere; n = 9) or vehicle (n = 11) immediately after the 15-min cocaine-memory retrieval session. After two additional extinction sessions in the extinction context with ≤25 active lever responses, cocaine-seeking behavior was tested in the cocaine-paired context. B Schematic of cannula placements. Symbols represent the most ventral point of injection cannula tracts for rats that received vehicle or AM251. C Active-lever responses (mean ± SEM) during cocaine self-administration (last 10 days) and extinction training prior to AM251 or vehicle treatment. D Active-lever responses (mean ± SEM) during the memory-retrieval session (before treatment) and upon first re-exposure to the extinction context and the cocaine-paired context (after treatment) at test. E Time course of active-lever responses (mean ± SEM) at test in the cocaine-paired context. Symbols: ANOVA #context simple-main effects, Sidak’s tests, ps < 0.05; *treatment simple-main effect, Sidak’s test, p < 0.05; ‡time simple-main effects, Tukey’s tests, intervals 1 > 2–6, ps < 0.05; ♦treatment main effect, p < 0.05.
Experiment 2: effects of delayed AM251 administration in the BLA on subsequent cocaine-seeking behavior
Experiment 2 evaluated whether intra-BLA AM251 altered memory strength if administered after the 2–4 h time window of memory reconsolidation [22]. The procedures were identical to those in Experiment 1 except that rats received AM251 (n = 8) or vehicle (n = 6) 6 h after memory retrieval (Fig. 2A).

A Experimental timeline. After cocaine self-administration training in one context (cocaine-paired context, Context A) and extinction training in a different context (extinction context, Context B), rats received bilateral intra-BLA administration of the CB1R antagonist, AM251 (0.3 µg/0.5 µL per hemisphere; n = 8) or vehicle (n = 6) six hours after the 15-min cocaine-memory retrieval session (after memory reconsolidation was completed). After two additional extinction sessions in the extinction context with ≤25 active lever responses, cocaine-seeking behavior was tested in the cocaine-paired context. B Schematic of cannula placements. Symbols represent the most ventral point of injection cannula tracts for rats that received vehicle or AM251. C Active-lever responses (mean ± SEM) during cocaine self-administration (last 10 days) and extinction training prior to AM251 or vehicle treatment. D Active-lever responses (mean ± SEM) during the memory-retrieval session (before treatment) and upon first re-exposure to the extinction context and the cocaine-paired context (after treatment) at test. E Time course of active-lever responses (mean ± SEM) at test in the cocaine-paired context. Symbols: ANOVA, #context main effect, p < 0.05 ‡time simple-main effects, Tukey’s tests, intervals 1 > intervals 2–6, ps < 0.05.
Experiment 3: effects of post-retrieval AM251 administration in the pCPu on subsequent cocaine-seeking behavior
Experiment 3 evaluated whether the AM251 effects observed in Experiment 1 were anatomically specific to the BLA. The procedures were identical to those in Experiment 1 except that rats received AM251 (n = 9) or vehicle (n = 7) into the pCPu immediately after memory retrieval (Fig. 3A).

A Experimental timeline. After cocaine self-administration training in one context (cocaine-paired context, Context A) and extinction training in a different context (cocaine-paired context, Context B), rats received bilateral intra-pCPu administration of the CB1R antagonist, AM251 (0.3 µg/0.5 µL per hemisphere; n = 9) or vehicle (n = 7) immediately after the 15-min cocaine-memory retrieval session. After two additional extinction sessions in the extinction context with ≤25 active lever responses, cocaine-seeking behavior was tested in the cocaine-paired context. B Schematic of cannula placements. Symbols represent the most ventral point of injection cannula tracts for rats that received vehicle or AM251. C Active-lever responses (mean ± SEM) during cocaine self-administration (last 10 days) and extinction training prior to AM251 or vehicle treatment. D Active-lever responses (mean ± SEM) during the memory-retrieval session (before treatment) and upon first re-exposure to the extinction context and the cocaine-paired context (after treatment) at test. E Time course of active-lever responses (mean ± SEM) at test in the cocaine-paired context. Symbols: ANOVA, #context main effect, p < 0.5; ‡time simple-main effects, Tukey’s tests, interval 1 > intervals 2–6, ps < 0.05.
Experiment 4: effects of post-retrieval WIN55,212-2 administration in the BLA on subsequent cocaine-seeking behavior
Experiment 4 evaluated the effects of intra-BLA CB1R agonist administration on cocaine-memory reconsolidation. The procedures were identical to those in Experiment 1, except that rats received bilateral intra-BLA microinfusions of the CB1/CB2R agonist, WIN55,212-2 (0.5 or 5 µg/0.5-µL infusion/hemisphere; n = 11, 11, respectively; Tocris Bioscience, Minneapolis, MN) or vehicle (10% DMSO/10% Tween80/saline; n = 14) immediately after memory retrieval (Fig. 4A). These intra-BLA doses of WIN55,212-2 enhance nicotine-memory consolidation [23] and fear-memory consolidation and reconsolidation [24,25,26].

A Experimental timeline. After cocaine self-administration training in one context (cocaine-paired context, Context A) and extinction training in a different context (extinction context, Context B), rats received bilateral intra-BLA administration of the CB1R agonist, WIN55,212-2 (0.5 or 5 µg/0.5 µL per hemisphere; n = 11 and 11, respectively) or vehicle (n = 14) immediately after the 15-min cocaine-memory retrieval session. After two additional extinction sessions in the extinction context with ≤25 active lever responses, cocaine-seeking behavior was tested in the cocaine-paired context. B Schematic of cannula placements. Symbols represent the most ventral point of injection cannula tracts for rats that received vehicle or a dose of WIN55,212-2. C Active-lever responses (mean ± SEM) during cocaine self-administration (last 10 days) and extinction training prior to WIN55,212-2 or vehicle treatment. D Active-lever responses (mean ± SEM) during the memory-retrieval session (before treatment) and upon first re-exposure to the extinction context and the cocaine-paired context (after treatment) at test. E Time course of active-lever responses (mean ± SEM) at test in the cocaine-paired context. Symbols: ANOVA, #context main effect, p < 0.05; ‡time simple-main effect, Tukey’s tests, vehicle: interval 1 > 3–6, WIN55,212-2 (0.5 µg): interval 1 > 2–6, WIN55,212-2 (5 µg): interval 1 > 5–6, ps < 0.05.
Experiment 5: effects of post-retrieval AM251 administration in the BLA on corticosterone levels during cocaine-memory reconsolidation
Experiment 5 examined the effects of cocaine-memory retrieval and intra-BLA AM251 treatment on serum corticosterone concentrations during reconsolidation [16]. The training procedures were identical to those in Experiment 1, except that rats were habituated to the tail-nick procedure immediately before and after extinction session 6. Blood samples (~200 µL/sample) were collected immediately before extinction session 7 (Baseline), immediately after extinction session 7 (Extinction), and immediately after the memory-retrieval session in the cocaine-paired context (Retrieval) or after comparable exposure to the home cage (Home Cage; Fig. 5A). Intra-BLA AM251 or vehicle was administered immediately after re-exposure to the cocaine-paired context (n = 6, 5, respectively) or the home cage (n = 6, 5, respectively). Post-treatment blood samples were collected 30, 60, and 90 min later. Blood samples were centrifuged at 4 °C. Blood serum was collected and stored at −20 °C. Samples were assayed in duplicates using the MP Biomedicals Corticosterone RIA kit for rats and mice (intra-assay coefficient of variation = 1.77%, lower limit of detectability = 25 ng/mL).

A Experimental timeline. Rats received cocaine self-administration training in one context (cocaine-paired context, Context A) and extinction training in a different context (extinction context, Context B). Rats were habituated to the tail-nick procedure before and after extinction session 6 (Habituation). Blood samples were collected immediately prior to extinction session 7 (Baseline), after extinction session 7 (Extinction), after either the 15-min cocaine memory-retrieval session (Retrieval) or comparable exposure to the home cage (Home Cage), and after intra-BLA infusions of AM251 (0.3 µg/0.5 µL per hemisphere) or vehicle at 30-min intervals (30, 60, 90; n = 5–6/group). B Active-lever responses (mean ± SEM) during cocaine self-administration (last 10 days), extinction training, and during the 15-min cocaine-memory retrieval session prior to treatment manipulations. C Schematic of cannula placements in the BLA with symbols representing the most ventral point of injection cannula tracts for rats that were re-exposed to the cocaine-paired context or home cage followed by vehicle or AM251 treatment. D Blood serum corticosterone concentrations (mean ± SEM) at baseline, immediately after exposure to the extinction context, the home cage, or the cocaine-paired context (i.e., cocaine-memory retrieval), and at 30, 60, and 90 min post treatment. Symbols: #context simple-main effect, one-way ANOVA, Tukey’s tests, ps < 0.05 or #retrieval simple-main effect, 2 × 2 × 3 ANOVA, retrieval x time, Sidak’s test, p < 0.05; *treatment simple-main effect, 2 × 2 × 3 ANOVA, retrieval × treatment, Sidak’s test, p < 0.05.
Experiment 6: effects of post-retrieval corticosterone administration in the BLA on subsequent cocaine-seeking behavior
Experiment 6 evaluated the effects of intra-BLA corticosterone administration on cocaine-memory reconsolidation. The procedures were identical to those in Experiment 1, except that rats received bilateral intra-BLA microinfusions of corticosterone (3 or 10 ng/0.5 µL infusion/hemisphere, n = 7, 7, respectively; Sigma-Aldrich) or vehicle (0.2% EtOH; n = 6) immediately after cocaine-memory retrieval (Fig. 6A). These corticosterone doses exceed acute stress-induced peak brain dialysate concentrations (0.8 ng/µL) [27] but fall below aversive intra-BLA doses (≥25 ng/side) [28].

A Experimental timeline. After cocaine self-administration training in one context (cocaine-paired context, Context A) and extinction training in a different context (extinction context, Context B), rats received bilateral intra-BLA administration of corticosterone (3 or 10 ng/0.5 µL per hemisphere; n = 7 and 7, respectively) or vehicle (n = 6) immediately after the 15-min cocaine-memory retrieval session. After two additional extinction sessions in the extinction context with ≤25 active lever responses, cocaine-seeking behavior was tested in the cocaine-paired context. B Schematic of cannula placements. Symbols represent the most ventral point of injection cannula tracts for rats that received vehicle or a dose of corticosterone. C Active-lever responses (mean ± SEM) during cocaine self-administration (last 10 days) and extinction training prior to corticosterone or vehicle treatment. D Active-lever responses (mean ± SEM) during the memory-retrieval session (before treatment) and upon first re-exposure to the extinction context and the cocaine-paired context (after treatment) at test. E Time course of active-lever responses (mean ± SEM) at test in the cocaine-paired context. Symbols: ANOVA, #context main effect, p < 0.05; ‡time simple-main effect, Tukey’s tests, interval 1> intervals 2–6, ps < 0.05.
Histology
Rats were overdosed with ketamine and xylazine (300 and 15 mg/kg, respectively, i.p.). The brains were removed, flash frozen in isopentane, and stored at −80 °C. Forty-µm coronal brain sections were collected and stained with cresyl violet (Kodak, Rochester, NY, USA) to visualize cannula placement.
Data analysis
Rats that had misplaced cannulae (n = 24) or failed to reach the acquisition criterion in 21 days (n = 10) were excluded from the study. Potential pre-existing group differences in drug intake and lever responses during drug self-administration training (last 10 days), extinction training (7 days), and during the memory-retrieval session were analyzed using separate analyses of variance (ANOVA) with group as the between-subjects factor and time as the within-subject factor or using t-tests, when appropriate. Lever presses during the first post-treatment exposure to the extinction and cocaine-paired contexts were analyzed using ANOVAs with treatment (dose) as the between-subjects factor and context and time as within-subject factors, where appropriate. Serum corticosterone concentrations were analyzed using ANOVAs with future treatment group or treatment (AM251, vehicle) and memory retrieval (memory retrieval, home cage) as between-subjects factors, and time or context (Baseline, Extinction, Home Cage, Post-Retrieval; 30-min intervals) as within-subject factors, where appropriate. Significant interaction and main effects were further analyzed using post hoc Sidak’s or Tukey’s tests depending on the number of comparisons and the associated familywise error. Pearson’s r was calculated to assess the relationship between active-lever responding and corticosterone concentrations. ɑ was set at 0.05.
Results
Behavioral history
The groups did not significantly differ in drug intake or lever responding during cocaine self-administration, extinction training, or cocaine-memory retrieval in Experiments 1–6 (Supplemental Material). The groups also did not differ in the number of sessions required to reach the acquisition (mean ± SEM = 11.59 ± 0.35 sessions) or the post-treatment extinction criterion (2.05 ± 0.03 sessions). Thus, testing in the cocaine-paired context occurred for most rats three days post treatment. Inactive-lever responding was negligible during the post-treatment sessions in Experiments 1–4 and 6 (Supplemental Material).
Experiment 1: post-retrieval AM251 administration into the BLA increases cocaine-memory strength
Intra-BLA AM251 treatment immediately after cocaine-memory retrieval selectively increased drug context-induced cocaine-seeking behavior, the index of memory strength, at test relative to vehicle (2×2 ANOVA; context × treatment, F(1,18) = 14.60, p = 0.001; context, F(1,18) = 299.20, p < 0.0001; treatment, F(1,18) = 9.79, p = 0.006). Active-lever responding was higher in the cocaine-paired context than in the extinction context (Fig. 1D; Sidak’s test, p < 0.05). Furthermore, AM251 administered after memory retrieval increased responding in the cocaine-paired (Sidak’s test, p < 0.05), but not in the extinction, context relative to vehicle. Active-lever responding declined over time in the cocaine-paired context at test (Fig. 1E; 2×6 ANOVA; time, F(5,90) = 25.61, p < 0.0001, Tukey’s tests, interval 1 > 2–6, ps < 0.05; time × treatment, F(5,90) = 0.44, p = 0.82), and AM251 increased responding relative to vehicle independent of time (treatment, F(1,18) = 12.46, p = 0.002).
Experiment 2: intra-BLA AM251 administration after memory reconsolidation does not alter cocaine-memory strength
Intra-BLA AM251 administration six h after memory retrieval did not alter drug context-induced cocaine-seeking behavior at test relative to vehicle (Fig. 2D; 2×2 ANOVA; treatment × context, F(1,12) = 0.02, p = 0.90; treatment, F(1,12) = 0.0001, p = 0.99). However, active-lever responding was higher in the cocaine-paired context than in the extinction context independent of treatment (context, F(1,12) = 86.26, p < 0.0001). Active-lever responding declined over time in the cocaine-paired context at test, independent of treatment (Fig. 2E; 2 × 6 ANOVA; time, F(5,60) = 18.02, p < 0.0001, Tukey’s tests, interval 1 > 2–6, ps < 0.05; time × treatment, F(5,60) = 0.26, p = 0.93; treatment, F(1,12) = 0.002, p = 0.96).
Experiment 3: post-retrieval AM251 administration into the pCPu anatomical-control region does not alter cocaine-memory strength
Intra-pCPu AM251 administration immediately after cocaine-memory retrieval did not alter drug context-induced cocaine-seeking behavior at test relative to vehicle (Fig. 3D; 2×2 ANOVA; context x treatment, F(1,14) = 0.22, p = 0.65; treatment, F(1,18) = 0.82, p = 0.38). Active-lever responding was higher in the cocaine-paired context then in the extinction context independent of treatment (context, F(1,14) = 34.42, p < 0.0001). Furthermore, responding declined over time in the cocaine-paired context, independent of treatment (Fig. 3E; 2×6 ANOVA; time, F(5,70) = 15.75, p < 0.0001, Tukey’s tests, interval 1 > 2–6, ps < 0.05; treatment x time, F(5,70) = 0.51, p = 0.77; treatment, F(1,14) = 0.26, p = 0.62).
Experiment 4: Post-retrieval WIN55,212-2 administration into the BLA fails to alter cocaine-memory strength
Intra-BLA WIN55,212-2 administration (0.5 or 5 µg/hemisphere) after cocaine-memory retrieval did not alter drug context-induced cocaine-seeking behavior at test relative to vehicle (Fig. 4D; 3×2 ANOVA; treatment x context, F(2,33) = 0.10, p = 0.90; treatment, F(2,33) = 0.68, p = 0.51). Active-lever responding was higher in the cocaine-paired context than in the extinction context independent of treatment (context, F(1,33) = 57.80, p < 0.0001). Furthermore, active-lever responding declined at slightly different rates at test but did not result in treatment effects at any time point (Fig. 4E; 3×6 ANOVA; treatment x time, F(10,165) = 2.32, p = 0.01, Tukey’s tests, vehicle: interval 1 > 3–6, 0.5 µg dose: 1 > 2–6, 5 µg dose: 1 > 5–6, ps < 0.05; time, F(5,165) = 35.12, p < 0.0001; treatment, F(2,33) = 0.47, p = 0.63).
Experiment 5: Intra-BLA AM251 administration increases serum corticosterone levels during cocaine-memory reconsolidation
The two groups did not differ in active-lever responding during the memory-retrieval session (t(9) = 1.42, p = 0.19; Fig. 5B). Furthermore, the four groups did not exhibit pre-existing differences in baseline and extinction session serum corticosterone concentrations (Fig. 5D; 4×2 ANOVA; group x time, F(3,18) = 0.50, p = 0.69; group, F(3,18) = 2.70, p = 0.08; time, F(1,18) = 2.94, p = 0.10).
Cocaine-memory retrieval increased corticosterone levels compared to extinction context exposure (Fig. 5D; 2×2 ANOVA; time, F(1,9) = 21.88, p = 0.001, Sidak’s test, p < 0.05; time x future-treatment, F(1,9) = 0.54, p = 0.48; future-treatment, F(1,9) = 1.75, p = 0.22), whereas home cage re-exposure did not (2×2 ANOVA; time, F(1,9) = 21.88, p = 0.001; time x future-treatment, F(1,9) = 0.54, p = 0.48; future-treatment, F(1,9) = 1.75, p = 0.22). Cocaine-memory retrieval also increased corticosterone levels compared to home-cage re-exposure (2×2 ANOVA; retrieval, F(1,18) = 9.69, p = 0.006; retrieval x future-treatment, F(1,18) = 0.03, p = 0.87; future-treatment, F(1,18) = 1.13, p = 0.30). Furthermore, active-lever responses during the memory retrieval session positively correlated with corticosterone levels immediately post session (r = 0.63, p = 0.7304).
Cocaine-memory retrieval transiently increased corticosterone levels relative to home-cage re-exposure (Fig. 5D: 2x2x3 ANOVA; retrieval x time, F(2,36) = 4.28, p = 0.02, Tukey’s tests, 60-min time point, p < 0.05; time, F(2,36) = 43.51, p < 0.0001; retrieval, F(1,18) = 4.21, p = 0.06). Moreover, intra-BLA AM251 administered after memory retrieval increased corticosterone levels relative to vehicle during reconsolidation independent of time (2x2x3 ANOVA; treatment x retrieval, F(1,18) = 5.26, p = 0.03, Sidak’s tests, p < 0.05; three-way interaction, F(2,36) = 1.45, p = 0.25; treatment x time, F(2,36) = 0.22, p = 0.80; treatment, F(1,18) = 3.74, p = 0.07), due primarily to effects at the 30 and 60-min time points.
Experiment 6: Post-retrieval corticosterone administration into the BLA fails to alter cocaine-memory strength
Intra-BLA corticosterone administration (3 or 10 ng/hemisphere) after cocaine-memory retrieval did not alter drug context-induced cocaine-seeking behavior at test (Fig. 6D; 3×2 ANOVA; treatment x context, F(2,17) = 0.58, p = 0.57; treatment, F(2,17) = 0.46, p = 0.64). Active-lever responding was higher in the cocaine-paired context than in the extinction context independent of treatment (context, F(1,17) = 31.45, p < 0.0001). Furthermore, active-lever responding declined over time at test, independent of treatment (Fig. 6E; 3×6 ANOVA; time, F(5,85) = 21.23, p < 0.0001, Tukey’s tests, interval 1 > 2–6, ps < 0.05; treatment × time, F(10,85) = 0.32, p = 0.97; treatment, F(2,17) = 0.53, p = 0.60).
Discussion
In the present study, we used site-specific pharmacological manipulations to examine the contribution of BLA CB1R signaling to cocaine-memory reconsolidation in an instrumental rat model of drug relapse. Our findings suggest that BLA CB1R antagonism increases, therefore, CB1R signaling in the BLA may limit the strength of reconsolidating cocaine memories in a time-dependent manner, possibly by reducing the impact of memory retrieval-induced HPA-axis activation on neural circuits engaged during reconsolidation.
BLA CB1R antagonism enhances appetitive-memory reconsolidation
BLA CB1R antagonism by AM251 immediately after cocaine-memory retrieval (i.e., when the memory was labile) increased subsequent drug context-induced cocaine-seeking behavior (Fig. 1). This effect likely reflected an AM251-induced augmentation of memory strength during reconsolidation as opposed to protracted enhancement of the performance of cocaine-seeking behavior, since AM251 treatment 6 h after cocaine-memory retrieval (i.e., after reconsolidation) did not increase this behavior at test relative to vehicle (Fig. 2). The AM251-induced increase in cocaine seeking was also more robust than what could be accounted for by extinction-memory consolidation interference, an effect reported in some fear-memory studies ([24] but see [25, 29]). However, we cannot rule out this possibility. Finally, the effects of AM251 were anatomically specific to the BLA, as AM251 administration into the adjacent pCPu after memory retrieval failed to alter subsequent cocaine-seeking behavior relative to vehicle (Fig. 3). Together, these findings demonstrate that CB1R signaling in the BLA may limit appetitive-memory reconsolidation. These findings expand upon a complex literature indicating that intra-BLA administration of either CB1R antagonists [15] or CB1R agonists [14] can impair fear-memory reconsolidation. In fact, even the same CB1R agonist can either facilitate or impair object-recognition memory consolidation depending on whether conditioning takes place in an unhabituated (thus emotionally arousing) or a familiar environment [30], and similar mechanisms may be at play in reconsolidation. Such varying results may reflect that specific appetitive and aversive emotional and arousal states evoked in these paradigms may differently alter endocannabinoid recruitment and, thus, the functional contribution of CB1R populations to memory reconsolidation [29,30,31,32].
Intra-BLA administration of the non-selective CB1R agonist, WIN55,212-2, after memory retrieval failed to alter subsequent drug context-induced cocaine-seeking behavior (Fig. 4); even though BLA CB1R antagonism potentiated this behavior. It is unlikely that the lack of a WIN55,212-2 effect reflected insufficient dosing, since these intra-BLA doses of WIN55,212-2 inhibit nicotine-memory consolidation [23] and fear-memory reconsolidation [14, 26]. Therefore, we postulate that BLA CB1R signaling is not sufficient, but necessary, for limiting cocaine-memory strength during reconsolidation. We offer this explanation with the caveat that WIN55,212-2 and AM251, to an extent, impact distinct cannabinoid and off-target receptor and distinct cell populations. AM251 may exert competitive CB1R antagonist effects at BLA CB1Rs that are experiencing dynamic endocannabinoid mobilization and inverse agonist effects at all BLA CB1Rs. Additionally, AM251 may influence memory function through agonist effects at GPR55 (CB1 orphan receptor) [33] and antagonist effects at µ opioid receptors [34]. In contrast, WIN55,212-2 stimulates CB1Rs on inhibitory (~90%) and excitatory (~10%) terminals within the BLA [35], which likely impact reconsolidation differently. WIN55,212-2 is a non-selective agonist with 19-fold greater selectivity for CB2Rs than for CB1Rs [36], both of which are expressed in the BLA [37]. Finally, WIN55,212-2 is a biased CB1R agonist that exhibits lower efficacy at Gi/o- and Gq-coupled CB1Rs than the endocannabinoids, anandamide (AEA) and 2-arachydonoylglicerol (2-AG), but higher efficacy at arrestin-2-coupled CB1Rs than AEA [38]. Therefore, differential recruitment of distinct CB1R- and CB2R-expressing cell populations, CB1Rs with different effector systems, and off-target effects may contribute to inconsistencies between the effects of WIN55,212-2 and AM251 in the present study as well as to discrepancies in the effects of WIN55,212-2 across various fear-memory reconsolidation paradigms. Future studies will need to reinvestigate these questions with neutral CB1 antagonists and selective CB1R agonists when the latter become available.
BLA CB1R antagonism potentiates the impact of cocaine-memory retrieval on HPA-axis activation during memory reconsolidation
It has been well documented that exposure to drug-associated stimuli triggers HPA-axis activation in cocaine users [39] and cocaine-trained rats [16, 40]. Furthermore, stress-induced reductions in AEA tone in the BLA facilitate HPA-axis activation [17, 41]. In the present study, drug context-induced cocaine-memory retrieval resulted in a significant increase in serum corticosterone concentrations compared to two control conditions: re-exposure to the extinction context or the home cage (Fig. 5). There was a direct relationship between corticosterone levels and cocaine-seeking behavior during the memory-retrieval session, suggesting that corticosterone tracked the motivational effects of these stimuli or behavioral performance as opposed to a more general affective state or stress response; even though, the magnitude of the response was comparable to that observed upon exposure to mild stressors [42,43,44] in previous studies.
Intra-BLA AM251 administration increased the drug context-induced corticosterone response after cocaine-memory retrieval relative to vehicle (Fig. 5), while it did not alter peak corticosterone levels following no-memory retrieval. These findings are consistent with extant literature indicating that intra-BLA AM251 treatment alone is not anxiogenic [24], and it selectively enhances stress-induced, but not baseline, corticosterone levels [17]. Therefore, in the present study, intra-BLA AM251 administration might prolong a memory retrieval-induced motivational state that increased the strength of reconsolidating cocaine memories or the efficacy of reconsolidation. Thus, BLA CB1R signaling may gate memory strength and protect against the development of strong and intrusive cocaine memories.
Conclusions
While intra-BLA AM251 administration enhanced, systemic AM251 administration in our previous study impaired [7], and cocaine-memory strength during reconsolidation. Together, these findings indicate that functionally heterogenous CB1Rs populations bidirectionally regulate, and BLA CB1R signaling, in particular, may limit, cocaine-memory strength or reconsolidation as components of larger neural circuits.
The contributions of specific endocannabinoids, including AEA and 2-AG, to CB1R-mediated effects on cocaine-memory reconsolidation have yet to be determined, but some insights may be gained from the stress literature. Upon exposure to a stressor, corticotropin-releasing factor (CRF) is released. CRF stimulates AEA hydrolysis and, thus, reduces BLA AEA tone [45]. Reductions in AEA levels disinhibit BLA glutamatergic principal neurons and trigger phasic 2-AG mobilization through metabotropic glutamate receptor-mediated processes [46,47,48], concomitant with corticosterone-mediated stimulation of 2-AG synthesis [49]. Similar to stressors, cocaine-memory retrieval stimulates HPA-axis activity ([16], Fig. 5), likely reducing tonic AEA levels and increasing phasic 2-AG levels in the BLA during reconsolidation. As a CB1R antagonist, AM251 in the BLA blocks AEA signaling and, as such, augments the impact of cocaine-memory retrieval on HPA-axis activity, as indicated by the potentiated corticosterone response (Fig. 5). The resulting increase in BLA principal neuronal firing and 2-AG mobilization during reconsolidation may enhance cocaine-memory strength or storage efficacy, similar to fear memory consolidation [50]. Future studies will need to determine whether memory retrieval-induced alterations in BLA AEA, 2-AG, or both are critical for this phenomenon.
The effects of CB1R and GR manipulations in the BLA on cocaine-memory strength are similar, but it remains to be determined whether these systems interact with one another. Intra-BLA CB1R or GR antagonist administration enhances (Fig. 1, Stringfield et al. [16]), while intra-BLA CB1R or GR agonist administration fails to alter (Figs. 4 and 6), cocaine-memory strength during reconsolidation. This suggest that any direct contribution of the CB1R antagonist-potentiated corticosterone response to cocaine-memory reconsolidation must be mediated outside of the BLA. Further, increases in corticosterone levels likely signify a general HPA axis-mediated neuroendocrine response, suggesting that it may be the associated CRF surge that enhances reconsolidation in the BLA or in other brain regions. For instance, CRF may enhance reconsolidation through the stimulation of adrenergic locus coeruleus inputs to the BLA [51,52,53].
Based on the emerging role of CB1Rs in memory reconsolidation, CB1R genetic polymorphisms and other factors, that lead to abnormalities in CB1R signaling, may regulate an individual’s susceptibility to SUDs and other psychiatric disorders. Moreover, dysfunction of endocannabinoid recruitment upon exposure to drug-associated environmental stimuli and during subsequent drug-memory retrieval and reconsolidation may influence subsequent drug-relapse propensity. Interfering with neural mechanisms that enhance cocaine-memory strength during reconsolidation may be a useful adjunct to other approaches for drug-relapse prevention.
Funding and disclosure
The authors have no competing financial interests in relation to the work described. This research was funded by NIDA R01 DA025646 (RAF), NIDA F31 DA 045430 (JAH), and Washington State Initiative 171 (JAH) and 502 (RAF) funds administered through the Alcohol and Drug Abuse Research Program.
References
Childress AR, McLellan TA, Ehrman R, O’Brien CP. Classically conditioned responses in opioid and cocaine dependence: a role in relapse. NIDA Res Monogr. 1988;85:25–43.
Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP. Limbic activation during cue-induced cocaine craving. Am J Psychiatry. 1999;156:11–8.
Crombag HS, Bossert JM, Koya E, Shaham Y. Review. Context-induced relapse to drug seeking: a review. Philos Trans R Soc Lond B Biol Sci. 2008;363:3233–43.
Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–6.
Rich MT, Torregrossa MM. Molecular and synaptic mechanisms regulating drug-associated memories: towards a bidirectional treatment strategy. Brain Res Bull. 2018;141:58–71.
Sorg BA. Reconsolidation of drug memories. Neurosci Biobehav Rev. 2012;36:1400–17.
Higginbotham JA, Wang R, Richardson BD, Shiina H, Tan SM, Presker MA, et al. CB1 receptor signaling modulates amygdalar plasticity during memory reconsolidation to promote subsequent context-induced reinstatement of cocaine-seeking. J Neurosci. 2020. https://doi.org/10.1101/2020.06.02.130419.
Fuchs RA, Bell GH, Ramirez DR, Eaddy JL, Su Z. Basolateral amygdala involvement in memory reconsolidation processes that facilitate drug context-induced cocaine seeking. Eur J Neurosci. 2009;30:889–900.
Lee JLC, Di Ciano P, Thomas KL, Everitt BJ. Disrupting reconsolidation of drug memories reduces cocaine-seeking behavior. Neuron. 2005;47:795–801.
Lee JLC, Milton AL, Everitt BJ. Cue-induced cocaine seeking and relapse are reduced by disruption of drug memory reconsolidation. J Neurosci. 2006;26:5881–7.
Monsey MS, Ruiz SG, Taylor JR. Regulation of garcinol on histone acetylation in the amygdala and on the reconsolidation of a cocaine-associated memory. Front Behav Neurosci. 2020;13:281.
Sanchez H, Quinn JJ, Torregrossa MM, Taylor JR. Reconsolidation of a cocaine-associated stimulus requires amygdalar protein kinase A. J Neurosci. 2010;30:4401–7.
Wells AM, Lasseter HC, Xie X, Cowhey KE, Reittinger AM, Fuchs RA. Interaction between the basolateral amygdala and dorsal hippocampus is critical for cocaine memory reconsolidation and subsequent drug context-induced cocaine-seeking behavior in rats. Learn Mem. 2011;18:693–702.
Lin HC, Mao SC, Gean PW. Effects of intra-amygdala infusion of CB1 receptor agonists on the reconsolidation of fear-potentiated startle. Learn Mem. 2006;13:316–21.
Ratano P, Everitt BJ, Milton AL. The CB1 receptor antagonist AM251 impairs reconsolidation of Pavlovian fear memory in the rat basolateral amygdala. Neuropsychopharmacology 2014;39:2529–37.
Stringfield SJ, Higginbotham JA, Wang R, Berger AL, McLaughlin RJ, Fuchs RA. Role of glucocorticoid receptor-mediated mechanisms in cocaine memory enhancement. Neuropharmacology. 2017;123:349–58.
Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, et al. Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology 2009;34:2733–45.
National Research Council. Guide for the Care and Use of Laboratory Animals: 3rd ed. The National Academies Press: Washington, DC; 2011.
Fuchs RA, Eaddy JL, Su ZI, Bell GH. Interactions of the basolateral amygdala with the dorsal hippocampus and dorsomedial prefrontal cortex regulate drug context-induced reinstatement of cocaine-seeking in rats. Eur J Neurosci. 2007;26:487–98.
Ortinski PI, Vassoler FM, Carlson GC, Pierce RC. Temporally dependent changes in cocaine-induced synaptic plasticity in the nucleus accumbens shell are reversed by D1-like dopamine receptor stimulation. Neuropsychopharmacology 2012;37:1671–82.
Wang H, Treadway T, Covey DP, Cheer JF, Lupica CR. Cocaine-induced endocannabinoid mobilization in the ventral tegmental area. Cell Rep. 2015;12:1997–2008.
Tronson NC, Taylor JR. Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci. 2007;8:262–75.
Hashemizadeh S, Sardari M, Rezayof A. Basolateral amygdala CB1 cannabinoid receptors mediate nicotine-induced place preference. Prog Neuropsychopharmacol Biol Psychiatry. 2014;51:65–71.
Ganon-Elazar E, Akirav I. Cannabinoid receptor activation in the basolateral amygdala blocks the effects of stress on the conditioning and extinction of inhibitory avoidance. J Neurosci. 2009;29:11078–88.
Kuhnert S, Meyer C, Koch M. Involvement of cannabinoid receptors in the amygdala and prefrontal cortex of rats in fear learning, consolidation, retrieval and extinction. Behav Brain Res. 2013;250:274–84.
Zubedat S, Akirav I. The involvement of cannabinoids and mTOR in the reconsolidation of an emotional memory in the hippocampal-amygdala-insular circuit. Eur Neuropsychopharmacol. 2017;27:336–49.
Droste SK, de Groote L, Atkinson HC, Lightman SL, Reul JM, Linthorst AC. Corticosterone levels in the brain show a distinct ultradian rhythm but a delayed response to forced swim stress. Endocrinology 2008;149:3244–53.
Moriceau S, Shionoya K, Jakubs K, Sullivan RM. Early-life stress disrupts attachment learning: the role of amygdala corticosterone, locus ceruleus corticotropin releasing hormone, and olfactory bulb norepinephrine. J Neurosci. 2009;29:15745–55.
Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature 2002;418:530–4.
Campolongo P, Morena M, Scaccianoce S, Trezza V, Chiarotti F, Schelling G, et al. Novelty-induced emotional arousal modulates cannabinoid effects on recognition memory and adrenocortical activity. Neuropsychopharmacology 2013;38:1276–86.
Morena M, Campolongo P. The endocannabinoid system: An emotional buffer in the modulation of memory function. Neurobiol Learn Mem. 2014;112:30–43.
Morena M, Leitl KD, Vecchiarelli HA, Gray JM, Campolongo P, Hill MN. Emotional arousal state influences the ability of amygdalar endocannabinoid signaling to modulate anxiety. Neuropharmacology 2016;111:59–69.
Kapur A, Zhao P, Sharir H, Bai Y, Caron MG, Barak LS, et al. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J Biol Chem. 2009;284:29817–27.
Seely KA, Brents LK, Franks LN, Rajasekaran M, Zimmerman SM, Fantegrossi WE, et al. AM-251 and rimonabant act as direct antagonists at mu-opioid receptors: Implications for opioid/cannabinoid interaction studies. Neuropharmacology. 2012;63:905–15.
Fitzgerald ML, Mackie K, Pickel VM. Ultrastructural localization of cannabinoid CB1 and mGluR5 receptors in the prefrontal cortex and amygdala. J Comp Neurol. 2019;527:2730–41.
Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, et al. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1995;48:443–50.
Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A, et al. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071:10–23.
Laprairie RB, Bagher AM, Kelly MEM, Dupré DJ, Denovan-Wright EM. Type 1 cannabinoid receptor ligands display functional selectivity in a cell culture model of striatal medium spiny projection neurons. J Biol Chem. 2014;289:24845–62.
Sinha R, Fuse T, Aubin LR, O’Malley SS. Psychological stress, drug-related cues and cocaine craving. Psychopharmacology. 2000;152:140–8.
De Vries AC, Taymans SE, Sundstrom JM, Pert A. Conditioned release of corticosterone by contextual stimuli associated with cocaine is mediated by corticotropin-releasing factor. Brain Res. 1998;786:39–46.
Hill MN, Tasker JG. Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience 2012;204:5–16.
Spencer RL, Deak T. A users guide to HPA axis research. Physiol Behav. 2017;178:43–65.
Sebastian V, Estil JB, Chen D, Schrott LM, Serrano PA. Acute physiological stress promotes clustering of synaptic markers and alters spine morphology in the hippocampus. PLoS ONE. 2013;8:e79077.
Melia KR, Ryabinin AE, Schroeder R, Bloom FE, Wilson MC, Anokhin PK. Induction and habituation of immediate early gene expression in rat brain by acute and repeated restraint stress. J Neurosci. 1994;14:5929–38.
Gray JM, Vecchiarelli HA, Morena M, Lee TTY, Hermanson DJ, Kim AB, et al. Corticotropin-releasing hormone drives anandamide hydrolysis in the amygdala to promote anxiety. J Neurosci. 2015;35:3879–92.
Atsak P, Roozendaal B, Campolongo P. Role of the endocannabinoid system in regulating glucocorticoid effects on memory for emotional experiences. Neuroscience 2012;204:104–16.
Bedse G, Hartley ND, Neale E, Gaulden AD, Patrick TA, Kingsley PJ, et al. Functional redundancy between canonical endocannabinoid signaling systems in the modulation of anxiety. Biol Psychiatry. 2017;82:488–99.
Rouach N, Nicoll RA. Endocannabinoids contribute to short-term but not long-term mGluR-induced depression in the hippocampus. Eur J Neurosci. 2003;18:1017–20.
Tasker JG, Di S, Malcher-Lopes R. Rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology 2006;147:5549–56.
Pelletier JG, Likhtik E, Filali M, Paré D. Lasting increases in basolateral amygdala activity after emotional arousal: implications for facilitated consolidation of emotional memories. Learn Mem. 2005;12:96–102.
McReynolds JR, Donowho K, Abdi A, Mcgaugh JL, Roozendaal B, McIntyre CK. Memory-enhancing corticosterone treatment increases amygdala norepinephrine and Arc protein expression in hippocampal synaptic fractions. Neurobiol Learn Mem. 2010;93:312–21.
Tsigos C, Chrousos GP. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res. 2002;53:865–71.
Otis JM, Dashew KB, Mueller D. Neurobiological dissociation of retrieval and reconsolidation of cocaine-associated memory. J Neurosci. 2013;33:1271–81a.
Acknowledgements
The authors are grateful to Shi Min Tan, Ethan Hansen, and Justine Galliou for excellent technical assistance as well as to Dr. Anthony Berger for help with the corticosterone assays.
Author information
Authors and Affiliations
Contributions
JAH and RAF developed the study concept and experimental design; JAH, NMJ, RJM, RJC, JLR, and RAF collected the data; JAH, RAF, RJC, JLR, and RJM analyzed and interpreted the data; JAH and RAF wrote the manuscript with input from all authors. All authors have approved the version of the manuscript submitted for publication.
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Higginbotham, J.A., Jones, N.M., Wang, R. et al. Basolateral amygdala CB1 receptors gate HPA axis activation and context-cocaine memory strength during reconsolidation. Neuropsychopharmacol. 46, 1554–1564 (2021). https://doi.org/10.1038/s41386-020-00919-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-020-00919-x
This article is cited by
-
Drug memory reconsolidation: from molecular mechanisms to the clinical context
Translational Psychiatry (2023)
