
Abstract
Obsessive-compulsive disorder (OCD) is a severe, chronic neuropsychiatric disorder with a strong genetic component. The SLC1A1 gene encoding the neuronal glutamate transporter EAAT3 has been proposed as a candidate gene for this disorder. Gene variants affecting SLC1A1 expression in human brain tissue have been associated with OCD. Several mouse models fully or partially lacking EAAT3 have shown no alterations in baseline anxiety-like or repetitive behaviors. We generated a transgenic mouse model (EAAT3glo) to achieve conditional, Cre-dependent EAAT3 overexpression and evaluated the overall impact of increased EAAT3 expression at behavioral and synaptic levels. Mice with EAAT3 overexpression driven by CaMKIIα-promoter (EAAT3glo/CMKII) displayed increased anxiety-like and repetitive behaviors that were both restored by chronic, but not acute, treatment with fluoxetine or clomipramine. EAAT3glo/CMKII mice also displayed greater spontaneous recovery of conditioned fear. Electrophysiological and biochemical analyses at corticostriatal synapses of EAAT3glo/CMKII mice revealed changes in NMDA receptor subunit composition and altered NMDA-dependent synaptic plasticity. By recapitulating relevant behavioral, neurophysiological, and psychopharmacological aspects, our results provide support for the glutamatergic hypothesis of OCD, particularly for the increased EAAT3 function, and provide a valuable animal model that may open novel therapeutic approaches to treat this devastating disorder.
Similar content being viewed by others

Introduction
Obsessive-compulsive disorder (OCD) is a neuropsychiatric disorder characterized by intrusive thoughts (obsessions), repetitive ritualistic behaviors (compulsions), and anxiety, with a worldwide prevalence of 2–3% [1]. The only approved pharmacotherapy for OCD is (selective) serotonin reuptake inhibitors; however, 40–50% of affected individuals fail to respond to medication [2, 3]. Thus, there is a critical need to understand the neurobiological underpinnings of OCD.
The glutamatergic hypothesis of OCD has accumulated support over the last decades [4, 5]. Neuroimaging studies indicate alterations in the cortical–striatal–thalamic–cortical (CSTC) circuitry, which includes glutamatergic corticostriatal projections synapsing onto striatal spiny projection neurons and/or interneurons [4, 6–10]. Altered glutamate levels from cerebrospinal fluid studies have been reported in OCD [11, 12]. Beneficial effects of anti-glutamatergic agents including memantine, N-acetylcysteine, riluzole, ketamine, and rapastinel have been reported in treatment-resistant OCD individuals [5, 13,14,15,16,17]. In addition, some genetic animal models with altered glutamatergic neurotransmission at the CSTC circuitry exhibit OCD relevant behaviors [10, 18,19,20].
Family-based linkage and case-control association studies have suggested SLC1A1 (Solute Carrier, Family 1, member 1) as a candidate gene in OCD [21,22,23,24,25,26,27,28], although no variants within the SCL1A1 locus have reached significance in genome-wide association studies [29, 30], likely due to limited statistical power. SLC1A1 encodes for the neuronal excitatory amino acid transporter EAAT3, highly expressed in brain regions proposed to be affected in OCD [31]. EAAT3 is located postsynaptically, with roles on regulating glutamate spillover, NMDAR function and synaptic plasticity [32,33,34], and on GABA synthesis when expressed in GABAergic neurons [35,36,37,38].
Several animal models fully or partially lacking EAAT3 have shown no alterations in baseline behaviors relevant to OCD [39, 40, 41]. Interestingly, the rs301430C allele, a SLC1A1 polymorphism highly replicated in OCD studies is associated with increased transcript levels in human brain tissue [27], suggesting that overexpression could contribute to OCD susceptibility [4]. Therefore, to evaluate the consequences of increased EAAT3 expression, we generated a transgenic mouse with conditional EAAT3 overexpression, and show here that mice with elevated EAAT3 expression in forebrain display increased anxiety-like and repetitive behaviors as well as greater spontaneous recovery of fear. Electrophysiological and molecular analyses at corticostriatal synapses indicated alterations in NMDA receptor composition/function and impaired synaptic plasticity, highlighting the impact of EAAT3 on regulating these synapses and suggesting they may contribute to the observed behavioral alterations.
Methods and materials
Cloning of PLCE-EAAT3 vector
The pCLE vector [42] contains a constitutive promoter driving the expression of a floxed EGFP, and a downstream polylinker where mouse EAAT3 cDNA (1541 bp) was directionally subcloned (Fig. 1a). The pCLE-EAAT3 vector was transformed into DH5α-competent Escherichia coli cells; positive clones were screened by PCR and confirmed by DNA sequencing.

Generation of EAAT3glo/CMKII mice. a The pCLE-EAAT3 vector was generated by subcloning mouse EAAT3 cDNA in pCLE vector. The pCLE vector contains a ubiquitously active promoter upstream to a floxed EGFP gene sequence. Upon Cre recombination, the EGFP gene is released and EAAT3 is expressed. b Representative immunoblots for GFP, EAAT3, and beta-actin expression from N2A cells transfected with pCLE-EAAT3, pCLE-EAAT3, and pCMV-Cre during 24 h. c [3H]-Glutamate uptake determinations in N2A cells transfected with pCLE-EAAT3, pCLE-EAAT3, and pCMV-Cre during 24 h. Measurements expressed as a percentage of radioactivity counts for control (untransfected cells). Data expressed as mean ± SEM; four independent experiments counted in duplicate were performed (pCLE-EAAT3: 87.56 ± 11.83 vs pCLE-EAAT3 + pCRE: 272 ± 6.52, P < 0.0001). d Generation of EAAT3-Tg mice. d Representative image of two mice at age P5; the EAAT3glo mouse has endogenous GFP fluorescence (green), while the control mouse does not, by using a Macro Imaging System. e Southern blot analysis of genomic DNA of EAAT3glo mouse founder (C6) and control (CTRL) mice. f PCR genotyping of EAAT3glo/CMKII mice (CaMKIIa—Cre x EAAT3glo breeding) and control mice using GFP, CRE, and IC (internal control, MAO-B gene) primers. g Upper: immunohistochemistry of EAAT3glo/CMKII and control EAAT3glo mice. Higher levels of antibody staining were found in cortex, hippocampus, and striatum. Bottom: representative Western blot determinations of EAAT3 in protein extracts of EAAT3glo/CMKII and control EAAT3glo mice; b-actin was used as normalizer. h Increased relative Slc1a1 mRNA expression from EAAT3glo/CMKII mice in cortex (P = 0.0038), striatum (P < 0.0001), and hippocampus (P < 0.0001) compared to control EAAT3glo littermates, determined by qRT-PCR using housekeeping Hebp1 mRNA (n = 6 per group). i EAAT3glo/CMKII mice have increased levels of GSH in cortex (EAAT3glo: 1.625 ± 0.161 vs EAAT3glo/CMKII: 2.308 ± 0.187, P = 0.0242), striatum (EAAT3glo: 2.295 ± 0.180 vs EAAT3glo/CMKII: 3.088 ± 0.207, P = 0.0085), and hippocampus (EAAT3glo: 1.088 ± 0.093 vs EAAT3glo/CMKII: 1.740 ± 0.116, P = 0.032). n = 4 per group, measurements done in duplicate. *P < 0.05, **P < 0.005, ****P < 0.0001. The bars represent mean ± SEM
Transfection, recombination analysis, and glutamate uptake assays
Neuroblastoma 2A cells (ATCC #CCL-131) were transfected with pCLE-EAAT3 and pCMV-Cre using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for 24 h to evaluate Cre-dependent expression by western blot and functionality by [3H]-glutamate uptake.
Western blots
Proteins were extracted from N2A transfected cells or brain tissue in ice-cold RIPA buffer (Thermo Scientific, Rockford, IL, USA) with Complete Mini protease inhibitor (Roche Diagnostic, Indianapolis, IN, USA), separated in SDS-PAGE gels and transferred to nitrocellulose membranes (Invitrogen). Membranes were soaked in blocking buffer (5% nonfat dry milk in PBS with 0.05% Tween-20), and incubated overnight at 4 °C with EAAT3 primary antibody (rabbit 1:1000, Abcam, Cambridge, MA, USA); anti-NR2A, anti-NR2B, anti-PSD95 (rabbit, 1:1000; Merck-Millipore, Watford, UK), anti-GluR1, anti-GluR2 (rabbit, 1:1000; Cell Signaling Technology, Danvers, MA, USA). Anti-tubulin (sheep,1:5000; Cytoskeleton Inc., Denver, CO, USA) was used to normalize EAAT3 protein levels; all other proteins were normalized with β-actin (rabbit, 1:5000; Abcam, Cambridge, England). Detection was performed using the appropriate secondary antibody and detected using the Pierce ECL Chemiluminescent substrate (Thermo Scientific). The density of the bands was quantified using ImageJ software (NIH, Bethesda, MD).
Glutamate uptake assays
The protocol was modified from [43]. Briefly, cells were incubated with 1 µM [3H]-glutamate (specific activity: 30.7 Ci/mmol, Perkin-Elmer, Shelton, CT, USA) for 15 min and washed three times with cold assay buffer. Liquid scintillation cocktail (Perkin-Elmer) was added, the plate was shaken for 1 h and samples were counted in a TopCount scintillation counter (Perkin-Elmer). For each condition, four independent experiments in duplicate were performed.
Reduced glutathione (GSH) measurements
We determined GSH using the monochlorobimane method modified from [44]. Brain regions were homogenized by sonication in 0.1% ascorbic acid. 100 µL were used from each sample in duplicate to measure GSH with the MCB Glutathione Detection Kit (Biotium, Hayward, CA, USA) in a Victor X2 Plate Reader (Perkin-Elmer), Ex/Em = 394/490 nm.
Generation of EAAT3glo mice
We injected the pCLE-EAAT3 vector into zygotes of FVB/N mice as described [45]. EAAT3glo founder lines were identified through GFP visualization using the Macro Imaging System (Light Tools Research, Encinitas, CA, USA). All experimental studies and procedures were approved by the Animal Care and Use Committee of the National Institutes of Health.
Genotyping of EAAT3glo mice and generation of EAAT3 overexpressing (EAAT3glo/CMKII) mice
Founder lines were genotyped by Southern blot as described [46], and by PCR with primers for promoter-to-GFP region (PROM-For:5′-CTCTAGAGCCTCTGCTAACC-3′; EGFP-Rev: 5′-TGATGCCGTTCTTCTGCTTGTC-3′, 346 bp amplicon). PCR conditions were 0.4 µM primer and cycle: 95 °C 5 min; [94 °C 30 s, 60 °C 60 s, 72 °C 60 s] × 35 cycles; 72 °C 10 min. EAAT3glo female mice were bred with CaMKIIα-Cre male mice (JAX 005359, Jackson Laboratories, Bar Harbor, ME, USA) to generate EAAT3glo/CMKII mice that overexpress EAAT3 in forebrain principal neurons. We developed a triplex PCR assay for GFP, CRE, and MAO-B as internal control (IC). CRE primers were CRE-for:5′-GCATACCTGGAAAATGCTTCTGT-3′; CRE-rev:5′-GGCCCAAATGTTGCTGGATAGTT-3′, amplicon 164 bp. IC primers were IC-for:5′-CTACAAAGCAGATTGCCACGC-3′; IC-rev:5′-TACCTGACATCAACTGGTCCC-3′, amplicon 292 bp. Triplex PCR used 0.4 µM primer and cycle: 94 °C 10 min; [94 °C 30 s, 62 °C 30 s, 72 °C 30 s] × 30 cycles; 72 °C 10 min. Amplicons were run on 2% agarose electrophoresis in TAE buffer (pH 8.4).
Behavior
Mice were housed in groups of 3–5 per cage on standard conditions (12-h light:dark cycle (lights on at 07:00), food and water ad libitum, temperature and humidity controlled). 3–5 months old male littermates were used in all experiments, excepting for visual discrimination and fear extinction tasks, where both genders were used. Animals were transferred to the behavioral room in their home cages, acclimated for at least 1 h and tested between 10:00 and 15:00 h. Equipment was cleaned after each mouse with ethanol (10%) to eliminate odor cues. All tests and analyses were done blind to the genotype/treatment of animals. To avoid confounding results due to multiple exposures to the tests, pharmacological assessments were done in separate cohorts of mice. Mice were administered with oral fluoxetine (30 mg/kg) (Sigma-Aldrich, St. Louis, MO, USA) or clomipramine (80 mg/kg) (Sigma-Aldrich) for 1 day (acute), or 21 days (chronic) and then evaluated 24 h after the last treatment.
On the day of behavioral screening, mice were sequentially evaluated in the tests described above, in a fixed manner. Between tests, mice were allowed to rest for 60 min in their home cages.
(1) Open field test: Mice were placed in the center of the apparatus (40 × 40 × 35 cm high); behavior was recorded for 60 min under dim light (~50 lux). Locomotor activity, velocity, and turn angle were analyzed using Ethovision XT software (Noldus Information Technology, Leesburg, VA, USA). For anxiety-related measurements, time and entries to the center of arena (20 × 20 cm) were analyzed from the first 5 min.
(2) Light–dark exploration test: After adaptation to dim light (~50 lux), mice were placed in the dark compartment of the box (dark side: 15 × 40 × 25 cm high fully opaque Plexiglass; light side 25 × 40 × 35 cm high clear Plexiglass, brightly illuminated at ~800 lux; door 7.5 × 7.5 cm) and the door immediately opened allowing mice to freely explore during 5 min. Latency to emerge and time spent in the light chamber were analyzed using Ethovision TX Software (Noldus).
(3) Marble burying test: Mice were placed in a cage containing 5 cm of bedding material with light set at ~300 lux. Twenty-four dark glass marbles (1.5 cm diameter) were evenly placed across the surface. After 30 min, mice were removed and the number of buried marbles (at least 2/3 of its surface) was recorded.
(4) Grooming analysis: Mice were placed in a clear Plexiglas cylinder (20 cm diameter, 30 cm high), where number and duration of grooming events were recorded during 60 min with light set at ~300 lux.
Touchscreen-based visual discrimination task and reversal learning
Mice were reduced to 85% of their free-feeding body weight and maintained at that level throughout the experiment. Training took place in a chamber with a touch-sensitive screen at one end (Lafayette Instruments, Lafayette, IN, USA) as previously described [47,48,49] (see full details in Supplementary Methods).
Fear conditioning and extinction task
Fear conditioning was conducted in a 30 × 25 × 25-cm chamber with metal walls, a metal rod floor, and an olfactory cue (vanilla) (context A). Mice received three pairings (180 s acclimation, 60–90 s ITI) of a 30 s, 80 dB white noise cue (conditioned stimulus, CS) and a co-terminating, 2 s, 0.6 mA scrambled footshock (unconditioned stimulus, US). Fear extinction training was conducted on the next day in a different room, in a 30 × 25 × 25-cm chamber with a curved, white wall, a white floor, and an olfactory cue (acetic acid) (context B). 50 × 30 s CS presentations (180 s acclimation, 5 s ITI) were delivered. The next day, extinction retrieval was tested in context B with 5 × 30 s CS presentations (180 s acclimation, 5 s ITI). Two weeks later, spontaneous recovery was tested in the same manner as extinction retrieval. Stimulus presentations were controlled and freezing detected by the Med Associates VideoFreeze system (Med Associates, Burlington, VT, USA). The amount of time scored as freezing was converted to a percentage ([amount of time freezing/total time of baseline or CS] × 100) for analysis.
Electrophysiological recordings of corticostriatal synapses
Coronal brain slices (350 μm thick) were prepared from naive control and EAAT3glo/CMKII mice (postnatal days 70–90) according to NIH Guidelines and approved by the University of Valparaiso Bioethics Committee. Whole-cell voltage-clamp recordings were obtained from visually identified medium spiny neurons (MSNs) in the dorsal striatum using a Multiclamp 700A amplifier (Molecular Devices, Sunnyvale, CA, USA) and patch-type pipette electrodes (~3.0–5.0 MΩ) filled with intracellular solution containing (in mM): 131 Cs-gluconate, 8 NaCl, 1 CaCl2, 10 EGTA, 10 glucose, 10 HEPES, 5 MgATP, and 0.4 Na3GTP, pH 7.2 (285 mmol/kg). All experiments were performed at 28 ± 1 °C in a submersion-type recording chamber perfused at ~2 mL/min with ACSF supplemented with the GABAA receptor antagonist picrotoxin (100 μM). AMPAR-mediated EPSCs were elicited at −60 mV, whereas NMDAR-mediated EPSCs were elicited at +40 mV in the continuous presence of 10 µM NBQX by stimulating the limit between striatum and cortex. Miniature excitatory currents (mEPSCs) were recorded at 30 ± 1 °C in ACSF supplemented with 1 μM tetrodotoxin (TTX), whereas long-term depression (LTD) was induced after 10 min of stable baseline by either high-frequency stimulation (1 s of 100 Hz) repeated four times at 10-s intervals or by transient (10 min) bath application of 50 µM (S)-3,5-dihydroxyphenylglycine, a group I mGluR agonist. All data acquisition and analysis were performed with custom-made software written in Igor Pro 6.3A (Wavemetrics, Inc., Lake Oswego, OR, USA) (full details in Supplementary Methods).
Statistical analysis
Data were analyzed using one or two-way analyses of variance or t-tests. Significant main effects (one-way ANOVAs) or interactions (two-way ANOVAs) were followed by post-hoc comparisons between genotypes or between drug conditions using Tukey HSD pairwise comparisons. Since there was a visible difference between genotypes when graphing the data for spontaneous recovery, Sidak’s pairwise comparisons between genotypes were made for spontaneous recovery even though the tone × genotype interaction was not significant. However, this means that the pairwise differences must be interpreted with caution. Significance was set at P < 0.05. Data are presented as mean ± SEM.
Results
Generation of EAAT3glo mice
The pCLE-EAAT3 vector was engineered to achieve Cre-dependent EAAT3 expression as described in Methods and materials (Fig. 1a). Cotransfection experiments in neuroblastoma N2A cells with pCLE-EAAT3 alone or with pCMV-Cre vector showed that EAAT3 expression increased only in the presence of Cre (Fig. 1b). Increased glutamate uptake activity was found only when pCLE-EAAT3 was co-transfected with pCMV-Cre (Fig. 1c), strongly indicating that pCLE-EAAT3 vector drives Cre-dependent and functional EAAT3 expression.
Then, we injected a linearized pCLE-EAAT3 vector into FVB/N zygotes to generate EAAT3glo mice. Ten pups were obtained from 4 recipient females that were transplanted with the injected zygotes. Founder lines were identified upon GFP visualization (Fig. 1d) and confirmed by PCR (Fig. 1f). Three founder lines were established and confirmed for integration of the pCLE-EAAT3 vector using Southern blot (Fig. 1e). Then, the C6 founder line was expanded by cross-breeding with wild-type mice. EAAT3glo mice were born healthy, viable, and fertile, with no gross neurological alterations or any signs of adverse effects of transgene integration (Table S1). At least seven crosses with wild-type C57BL/6J mice were performed prior to molecular and behavioral analyses.
Overexpression of EAAT3 by CaMKIIα-Cre
We then crossed EAAT3glo mice with CaMKIIα-Cre mice to overexpress EAAT3 in forebrain neurons. CaMKIIα × EAAT3glo (EAAT3glo/CMKII) mice were born healthy, without any gross phenotype or neurological impairment as evaluated by a standardized set of reflexes analysis, suggesting no observable deleterious effects due to higher expression of EAAT3 in forebrain neurons (Table S1).
Increased Slc1a1 (EAAT3) mRNA levels were found in EAAT3glo/CMKII mice vs control (EAAT3glo) littermates in frontal cortex (P = 0.038), hippocampus (P < 0.0001), and striatum (P < 0.0001; Fig. 1h); increased EAAT3 expression was also confirmed by immunohistochemistry and Western blot (Fig. 1g and Figure S1). We also performed immunofluorescence to determine EAAT3 increased expression in striatum (Supplementary Figure 2). Since pCLE-EAAT3 construct has a floxed eGFP, the recombination should both increase EAAT3 and decrease eGFP signal. As shown in Figure S2, EAAT3 (red) signal intensity is significantly increased in striatum of EAAT3glo/CMKII mice vs control littermates (P = 0.0331, Fig. S2D), along with the evident decrease in eGFP signal. Arrowheads (Fig. S2A.1) indicate striatal streaks in control EAAT3glo mice, where reduction in eGFP fluorescence is clearly lost.
EAAT3 does not impact global glutamate uptake in striatal synaptosomal preparations [40] since its expression is lower compared to other glutamate transporters [50]. Therefore, to test EAAT3 functionality, we measured glutathione levels in brain tissue, since cysteine (required for glutathione synthesis) is largely incorporated into neurons via EAAT3 [51]. EAAT3glo/CMKII mice had increased GSH levels in the frontal cortex (P = 0.0242), striatum (P = 0.0085), and hippocampus (P = 0.032) compared to control littermates (Fig. 1i), indicating increased EAAT3-mediated cysteine uptake.
EAAT3glo/CMKII mice display increased anxiety and repetitive behaviors
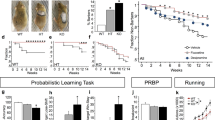
We evaluated locomotion in EAAT3glo/CMKII mice over a 60 min observation period and found no changes in total distance, turn angle, or velocity compared to control EAAT3glo, WT and CaMKII-Cre littermates (Figure S3). In the open field test, EAAT3glo/CMKII mice spent less time ([F(7,109) = 7.409, P < 0.0001], Tukey's P = 0.0003) (Fig. 2a) and made fewer visits to the center ([F(7,107) = 11.36, P < 0.0001], Tukey's P < 0.0001) compared to EAAT3glo mice (Fig. 2b); total distance was unaffected (Fig. 2c). No differences were found among WT, CaMKII-Cre, and EAAT3glo control groups (Figure S4). Behavior was alleviated treating EAAT3glo/CMKII mice with chronic (21 days) oral clomipramine (80 mg/kg) or fluoxetine (30 mg/kg) and did not impair locomotion. In contrast, when given only 1 day, neither drug affected the increased anxiety-like behavior (Fig. 2a–c).

EAAT3glo/CMKII mice have increased anxiety- and repetitive/compulsive-like behaviors. EAAT3glo/CMKII mice and EAAT3glo littermates were evaluated in the open field test (a–c). EAAT3glo/CMKII mice showed significant reductions in time (a) and visits to center of arena (b). Time in center EAAT3glo: 45.59 ± 3.95 vs EAAT3glo/CMKII: 19.1 ± 3.07, P < 0.001; visits to center EAAT3glo: 28.13 ± 1.96 vs EAAT3glo/CMKII: 11.22 ± 1.67, P < 0.001. Chronic (21 days) but not acute (1 day) oral administration of clomipramine (80 mg/kg) or fluoxetine (30 mg/kg) rescued behavior. c No changes in total distance. n = 13–16 mice per group. In the light–dark box test, EAAT3glo/CMKII mice showed longer latency to visit (d) and reduced time in the light chamber (e). Latency to cross EAAT3glo: 31.77 ± 11.73 vs EAAT3glo/CMKII: 55.74 ± 14.22, P = 0.0004; time in light chamber EAAT3glo: 81.5 ± 27.57 vs EAAT3glo/CMKII: 43.52 ± 12.13, P < 0.005. Chronic fluoxetine alleviated both parameters; clomipramine restored time and showed a trend towards significance in latency. Acute treatments were ineffective. n = 13–16 mice per group. In (f), EAAT3glo/CMKII buried a significantly higher number of marbles (21.2 ± 0.757) compared to control EAAT3glo littermates (10.5 ± 1.875; P < 0.001). Chronic (21 days) oral administration of clomipramine (CMI 80 mg/kg) or fluoxetine (30 mg/kg) rescued behavior; acute administration was ineffective. n = 12–16 mice per group. g Grooming was recorded for 60 min; EAAT3glo/CMKII mice displayed higher percentage time of grooming (EAAT3glo: 8.45 ± 3.47 vs EAAT3glo/CMKII: 16.54 ± 5.74, P = 0.0011). Chronic (21 days) but no acute (1 day) oral administration of clomipramine (CMI 80 mg/kg) or fluoxetine (30 mg/kg) restored behavior (n = 13–16 mice per group). *P < 0.05, **P < 0.005, **P < 0.001, ***P < 0.0005, ****P < 0.0001, N.S. = not significant. Bars represent mean ± SEM
In the light–dark box test, EAAT3glo/CMKII mice showed longer latency to emerge to the lit chamber ([F(7,95) = 6.724; P < 0.0001], Tukey's P = 0.0017) (Fig. 2d) and spent less time in it ([F(7,91) = 9.325, P < 0.0001], Tukey's P = 0.0004) compared to EAAT3glo mice (Fig. 2e). There were no differences among WT, CaMKII-Cre, and EAAT3glo control groups (Figure S4). Chronic fluoxetine alleviated both behavioral parameters, while clomipramine significantly restored time in light chamber and showed a trend towards significance in latency (P = 0.056); acute treatment was ineffective for both drugs.
In the marble burying test, we found that EAAT3glo/CMKII mice buried more marbles compared to EAAT3glo mice ([F(7,70) = 7.813; P < 0.0001], Tukey's P = 0.0009). No differences among WT, CaMKII-Cre, and EAAT3glo control groups were found (Figure S4). Behavior was also restored upon chronic, but not acute, oral fluoxetine or clomipramine administration.
We then evaluated grooming behavior and found that EAAT3glo/CMKII mice had increased grooming time compared to EAAT3glo littermates ([F(3,43) = 8.086; P = 0.0002], Tukey's P = 0.0011), which was alleviated by chronic, but not acute fluoxetine or clomipramine administration (Fig. 2g). Collectively, these results indicate that overexpression of EAAT3 in forebrain increases anxiety-like and repetitive behaviors and that both can be alleviated by chronic (S)SRI treatments.
EAAT3glo/CMKII mice display greater spontaneous recovery of fear
We then evaluated a naive cohort of EAAT3glo/CMKII mice in fear learning and extinction paradigms. No significant differences between EAAT3glo/CMKII mice and control EAAT3glo littermates were evident during fear conditioning and extinction training (Fig. 3a, b). Extinction retrieval evaluated the day after the extinction protocol showed that EAAT3glo/CMKII mice had a trend towards increased baseline freezing compared to controls, although freezing during CS presentation was normal (Fig. 3c). However, when spontaneous recovery was assessed 2 weeks later, EAAT3glo/CMKII mice showed an increase in freezing ([F(5, 90) = 25.702, P < 0.001]; Tone 1 P = 0.045, Fig. 3d), indicating greater spontaneous recovery.

EAAT3glo/CMKII mice have greater spontaneous recovery and facilitated late reversal learning. a–d EAAT3glo/CMKII mice demonstrate increased freezing during spontaneous recovery of fear learning compared to control littermates, indicative of greater spontaneous recovery (pairwise EAAT3glo/CMKII vs EAAT3glo tone 1 P = 0.045). a Acquisition of fear conditioning, b extinction, c retrieval, d spontaneous recovery. Bars represent mean ± SEM. n = 10–12 mice per group
No alterations in visual discrimination and reversal learning task in EAAT3glo/CMKII mice
As deficits in cognitive flexibility have been reported in OCD individuals [52, 53], we next assessed performance in a touchscreen-based visual discrimination task and reversal learning task. EAAT3glo/CMKII mice did not differ from controls on any measure during discrimination or reversal training (Figures S5 and S6).
Altered corticostriatal synapses in EAAT3glo/CMKII mice
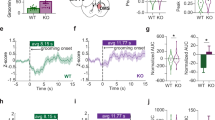
Increasing evidence suggests impairments in corticostriatal circuitry in OCD [9, 19, 20, 54,55,56]. To evaluate the impact of EAAT3 overexpression on excitatory synaptic transmission, we first monitored both spontaneous and evoked excitatory postsynaptic currents (EPSCs) from MSNs held at −60 mV in the presence of PTX, and found no differences in the amplitude or frequency of AMPAR-mediated sEPSCs and mEPSCs (Fig. 4a). Furthermore, no differences in the paired-pulse ratio (Fig. 4b) or short-term depression of evoked EPSCs were observed (Fig. 4c), suggesting that EAAT3glo/CMKII mice have unaltered basal and AMPAR-mediated synaptic transmission at corticostriatal synapses.

Basal excitatory synaptic transmission at corticostriatal synapses is unaltered in EAAT3glo/CMKII mice. a Representative traces (left) and average plot (right) showing no differences in both frequency (EAAT3glo: 4.87 ± 1.00, n = 10 cells/7 animals vs EAAT3glo/CMKII: 2.16 ± 0.48, n = 9 cells/7 animals, P = 0.05) and amplitude of sEPSC (EAAT3glo: 10.25 ± 0.67, n = 10 cells/7 animals vs EAAT3glo/CMKII: 9.28 ± 0.98, n = 9 cells/7 animals, P = 0.412) at the corticostriatal synapses between EAAT3glo/CMKII and control EAAT3glo mice. b Representative traces (left) and summary plots (right) showing no change in AMPAR-mEPSC frequency (EAAT3glo: 2.28 ± 0.56, n = 9 cells/7 animals vs EAAT3glo/CMKII: 1.70 ± 0.44, n = 9 cells/7 animals, P = 0.431) or amplitude between EAAT3glo/CMKII and control EAAT3glo mice (EAAT3glo: 9.70 ± 0.47 vs EAAT3glo/CMKII: 9.92 ± 0.62, P = 0.586). c No changes in paired-pulse ratio measured at different inter-stimulus intervals were observed between groups. d Representative traces (left) and average plot (right) showing no differences in short-term synaptic transmission between EAAT3glo and EAAT3glo/CMKII mice. In all panels, summary data represent the mean ± SEM and the number of slices (s) and animals (a) are indicated in parentheses
Since EAAT3 can regulate NMDAR function [32,33,34], we next characterized NMDAR-mediated synaptic transmission by analyzing NMDAR/AMPAR ratios and pharmacologically isolated NMDAR-EPSCs. While NMDAR/AMPAR ratios were indistinguishable between genotypes (Fig. 5a top), NMDAR-EPSCs showed slower decay kinetics in EAAT3glo/CMKII mice compared with controls (Fig. 5a bottom), which could reflect a change in synaptic NMDAR subunit composition [57]. To test this, we used Ro25-6981 (500 nM) to selectively block GluN2B-containing NMDARs and found that NMDAR-EPSCs in EAAT3glo/CMKII mice were more sensitive to Ro25-6981 than control EAAT3glo mice (Fig. 5b), suggesting increased abundance of GluN2B-containing NMDARs and/or decreased abundance of GluN2A-containing NMDARs. Western blot determinations showed that striatal GluN2A subunit levels were significantly reduced (P = 0.0213) in EAAT3glo/CMKII mice; no differences were found in GluN2B, AMPA GluR1 and Glur2 subunits or PSD-95 levels (Fig. 5c and Supplementary Figure 7). Similar alterations on glutamate receptor subunit protein levels were also found in prefrontal cortex and hippocampus of EAAT3glo/CMKII mice (Supplementary Figures 8 and 9). Collectively, these results indicate that EAAT3glo/CMKII mice have a higher contribution of GluN2B-containing NMDARs, an alteration that might affect the induction of NMDAR-dependent plasticity [58]. Consistently, we found that NMDA-dependent LTD induced by high-frequency afferent stimulation (Fig. 5d, top), but not metabotropic glutamate receptor-dependent LTD induced by bath application of DHPG (Fig. 5d, bottom), was impaired at corticostriatal synapses in EAAT3glo/CMKII mice compared to control littermates. Thus, by impacting synaptic NMDAR subunit composition, EAAT3 might play an important role in regulating NMDAR-dependent synaptic function and plasticity in the striatum, and reinforce the notion that alterations in NMDAR-mediated glutamatergic transmission are likely to be a common feature in behaviors relevant to OCD.

NMDARs-mediated synaptic transmission and plasticity are altered in EAAT3glo/CMKII corticostriatal synapses. a Representative averaged EPSCs (left) and summary data (right) showing that the NMDAR/AMPAR ratio was similar between control EAAT3glo and EAAT3glo/CMKII synapses (control: 0.49 ± 0.07, n = 16 cells/9 animals vs EAAT3glo/CMKII mice: 0.56 ± 0.06, n = 18 cells/9 animals; P = 0.326), but NMDAR-mediated EPSC show significant slower decay kinetics in EAAT3glo/CMKII mice (control: 117.23 ± 7.03 vs EAAT3glo/CMKII mice: 154 ± 12.87, P = 0.013). b EAAT3glo/CMKII synapses are more sensitive to the effect of GluN2B subunit antagonist R0256981 than control mice (EAAT3glo: 72.67 ± 3.50, n = 8 cells/7 animals vs EAAT3glo/CMKII mice: 53.52 ± 4.46, 8 cells/8 animals, P = 0.007). c Representative western blot determinations of GluR1, GluR2, GluN2A, GluN2B subunits, PSD-95, beta-actin, and tubulin in striatal protein extracts of EAAT3glo/CMKII and control EAAT3glo mice; average plot is shown below, n = 4 animals per group. d Top: average traces (left) and summary plot (right) showing that NMDA-dependent LTD induced by HFS was impaired EAAT3glo/CMKII mice compared with WT mice (last 10 min: control 61.77 ± 8.59% of baseline; EAAT3glo/CMKII mice, 107.06 ± 10.37% of baseline; P = 0.005). Bottom: mGluR-LTD induced by application of group I mGluR agonist DHPG (50 μM) is similar in both groups (last 10 min: EAAT3glo, 57.06 ± 5.93% of baseline; EAAT3glo/CMKII, 69.62 ± 13.50% of baseline, P = 0.433). Sample traces were taken at times indicated by numbers on the summary plot. Number of cells (c), and animals (a) are indicated in parentheses and summary data represent the mean ± SEM. *P < 0.05, **P < 0.01, N.S. = not significant
Discussion
SLC1A1 is a candidate gene in OCD from linkage and case-control studies [1, 21,22,23,24,25,26,27,28, 59], although did not reach significance in GWAS [29, 30]. Findings cluster in the 3′ region containing the polymorphism rs301430, which is highly replicated in OCD studies [Ahmari et al. 2015] and is associated with increased SLC1A1 expression levels in human brain tissue samples [27]. Such findings suggest that increased EAAT3 expression might contribute to OCD susceptibility. Our results are in agreement with this hypothesis, by showing that EAAT3glo/CMKII mice display increased behaviors that are relevant to OCD. Of note, several reports using different mouse models fully or partially lacking EAAT3 have shown unaltered baseline anxiety-like or repetitive behaviors, indirectly supporting the potential involvement of EAAT3 gain-of-function in behaviors relevant to OCD [39, 40, 60] (however, see [61]).
The altered phenotype of EAAT3glo/CMKII mice fits wells with the maladaptive repetitive behavior and anxiety features reported in OCD-afflicted individuals. EAAT3glo/CMKII mice displayed higher grooming, although we did not find self-injurious grooming leading to open skin wounds as that reported for Sapap3 knockout (KO) mice and SLITRK5 KO mice [19, 20]. Other authors have suggested that such aberrant hyper-grooming might rather resemble other OC-spectrum conditions like excoriation/skin-picking disorder or trichotillomania [10]. Although EAAT3, SLITRK5, and Sapap3 are all expressed postsynaptically at glutamatergic synapses (i.e., at striatal MSNs), differences in expression distribution might account for the discrepancies in grooming observed across these animal models. Considering the heterogeneous nature of OCD, the impact of SLC1A1 vs other genes might account for dimensional variation across behaviors.
Consistent with its postsynaptic location, we found no changes in presynaptic properties in EAAT3glo/CMKII mice at corticostriatal synapses. The functional changes in NMDA currents found in these synapses are well correlated with the increased relative contribution of GluN2B-containing NMDA receptors. Similar alterations in GluN2A/GluN2B ratio of NMDAR subunit expression and impairments in synaptic plasticity at corticostriatal synapses were also described in Sapap3 KO mice [20, 55]. Future functional studies should also evaluate these parameters in cortical regions to investigate the impact of the altered GluN2A/GluN2B ratio found in prefrontal cortex in EAAT3glo/CMKII mice, since associations between compulsive behavior and altered NMDAR function in prefrontal cortical regions have been reported [62, 63].
The observation of greater spontaneous recovery of fear in EAAT3glo/CMKII mice is consistent with the pathophysiology of OCD and agrees with prior reports of impaired retention of extinction in human OCD patients and animal models of OCD [64,65,66]. Fear extinction is mediated by the ventromedial prefrontal cortex (vmPFC) and its downstream targets in the amygdala [67,68,69] and extinction deficits in OCD patients have been associated with reduced activation of the vmPFC [65]. Thus, the current results might be explained, in part, by decreased functioning of the vmPFC in EAAT3glo/CMKII mice. In future work, it could be valuable to correlate measures of vmPFC activity with extinction and spontaneous recovery, to substantiate a link between these behavioral disturbances and impaired PFC function in these mice.
OCD has also been associated with impairments in cognitive flexibility, including in reversal learning tasks [53,70,71,72]. This feature of OCD was not recapitulated in EAAT3glo/CMKII mice, as they performed similar to controls during all stages of discrimination and reversal training. Additional measures of behavioral flexibility, such as a set shifting task, may be necessary to uncover deficits in this line of mice [52]. Alternatively, behavioral flexibility may be largely intact in this model, suggesting that EAAT3 overexpression may not significantly impair the cortical circuitry required for this set of behaviors.
Since SSRIs are the main therapeutic drugs to treat OCD, their effects are commonly used to test the validity of OCD relevant animal models [12, 13]. Thus, our results showing that chronic, but not acute fluoxetine or clomipramine treatment was effective in rescuing behavior in EAAT3glo/CMKII mice may suggest predictive validity for our animal model and recapitulate the human psychopharmacology of OCD.
In summary, our data support the notion that EAAT3 has a role in the pathogenesis of OCD relevant behaviors as EAAT3glo/CMKII mice exhibit increased anxiety, increased repetitive behaviors, and greater spontaneous recovery of fear, many of which are core symptoms of OCD. In addition, increased EAAT3 expression impairs corticostriatal synapses which could contribute, at least in part, to the neuronal basis involved in OCD. We believe this new model will allow gaining deeper insight on the role of EAAT3 in the pathogenesis of OCD and perhaps shedding light on novel therapeutic avenues for this devastating disorder.
Funding and disclosure
This work was supported by Millennium Nucleus NuMIND (ICM MINECOM NC130011) and Millennium Institute CINV (ICM MINECOM P09-022F), both grants from the Millennium Scientific Initiative of the Ministry of Economy, Development and Tourism (Chile) (PRM and AEC); Fondecyt Grant #1141272 (Chile) (PRM); NIMH Intramural Research Program (USA) (DLM); NICDR Intramural Research Program (USA) (ABK); NIAAA Intramural Research Program (USA) (AH). Partially supported by Fondecyt Grants # 1151091 (AEC) and 1160398 (RSZ). JRW is a current employee and stockholder of Takeda Pharmaceuticals and a former employee and stockholder of Nestlé Health Science, Pfizer, and F. Hoffmann-La Roche. PRM has received research funds from F. Hoffmann-La Roche. All other authors declare no competing interests.
Change history
21 February 2019
The original version of this Article contained an error in the spelling of the author Anna K Radke, which was incorrectly given as Anna R Radke. This has now been corrected in both the PDF and HTML versions of the Article.
References
Murphy DL, Moya PR, Wendland JR, Timpano KR. Genetic contributions to obsessive-compulsive disorder (OCD) and OCD-related disorders. In: Berrettini JNW, (ed). Principles of psychiatric genetics. Cambridge, UK: Cambridge University Press; 2012. p. 121–33.
DSM-5. Diagnostic and statistical manual of mental disorders (DSM-5). 5th ed. Alrington, VA: American Psychiatric Association; 2013.
Koran LM, Hanna GL, Hollander E, Nestadt G, Simpson HB, American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry. 2007;164:5–53.
Ahmari SE, Dougherty DD. Dissecting OCD circuits: from animal models to targeted treatments. Depress Anxiety. 2015;32:550–62.
Pittenger C. Glutamatergic agents for OCD and related disorders. Curr Treat Options Psychiatry. 2015;2:271–83.
Menzies L, Chamberlain SR, Laird AR, Thelen SM, Sahakian BJ, Bullmore ET. Integrating evidence from neuroimaging and neuropsychological studies of obsessive-compulsive disorder: the orbitofronto-striatal model revisited. Neurosci Biobehav Rev. 2008;32:525–49.
Rosenberg DR, Hanna GL. Genetic and imaging strategies in obsessive-compulsive disorder: potential implications for treatment development. Biol Psychiatry. 2000;48:1210–22.
Tian L, Meng C, Jiang Y, Tang Q, Wang S, Xie X, et al. Abnormal functional connectivity of brain network hubs associated with symptom severity in treatment-naive patients with obsessive-compulsive disorder: a resting-state functional MRI study. Prog Neuropsychopharmacol Biol Psychiatry. 2016;66:104–11.
Vaghi MM, Vertes PE, Kitzbichler MG, Apergis-Schoute AM, van der Flier FE, Fineberg NA, et al. Specific frontostriatal circuits for impaired cognitive flexibility and goal-directed planning in obsessive-compulsive disorder: evidence from resting-state functional connectivity. Biol Psychiatry. 2017;81:708–17.
Zike I, Xu T, Hong N, Veenstra-VanderWeele J. Rodent models of obsessive compulsive disorder: evaluating validity to interpret emerging neurobiology. Neuroscience. 2017;345:256–73.
Bhattacharyya S, Khanna S, Chakrabarty K, Mahadevan A, Christopher R, Shankar SK. Anti-brain autoantibodies and altered excitatory neurotransmitters in obsessive-compulsive disorder. Neuropsychopharmacology. 2009;34:2489–96.
Chakrabarty K, Bhattacharyya S, Christopher R, Khanna S. Glutamatergic dysfunction in OCD. Neuropsychopharmacology. 2005;30:1735–40.
Coric V, Taskiran S, Pittenger C, Wasylink S, Mathalon DH, Valentine G, et al. Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58:424–8.
Grant P, Lougee L, Hirschtritt M, Swedo SE. An open-label trial of riluzole, a glutamate antagonist, in children with treatment-resistant obsessive-compulsive disorder. J Child Adolesc Psychopharmacol. 2007;17:761–7.
Pittenger C, Krystal JH, Coric V. Glutamate-modulating drugs as novel pharmacotherapeutic agents in the treatment of obsessive-compulsive disorder. NeuroRx. 2006;3:69–81.
Rodriguez CI, Kegeles LS, Levinson A, Feng T, Marcus SM, Vermes D, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38:2475–83.
Rodriguez CI, Zwerling J, Kalanthroff E, Shen H, Filippou M, Jo B, et al. Effect of a novel NMDA receptor modulator, rapastinel (Formerly GLYX-13), in OCD: proof of concept. Am J Psychiatry. 2016;173:1239–41.
Nordstrom EJ, Burton FH. A transgenic model of comorbid Tourette’s syndrome and obsessive-compulsive disorder circuitry. Mol Psychiatry. 2002;7:617–25.
Shmelkov SV, Hormigo A, Jing D, Proenca CC, Bath KG, Milde T, et al. Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nat Med. 2010;16:598–602.
Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding JD, et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448:894–900.
Dickel DE, Veenstra-VanderWeele J, Cox NJ, Wu X, Fischer DJ, Van Etten-Lee M, et al. Association testing of the positional and functional candidate gene SLC1A1/EAAC1 in early-onset obsessive-compulsive disorder. Arch Gen Psychiatry. 2006;63:778–85.
Hanna GL, Veenstra-VanderWeele J, Cox NJ, Boehnke M, Himle JA, Curtis GC, et al. Genome-wide linkage analysis of families with obsessive-compulsive disorder ascertained through pediatric probands. Am J Med Genet. 2002;114:541–52.
Arnold PD, Sicard T, Burroughs E, Richter MA, Kennedy JL. Glutamate transporter gene SLC1A1 associated with obsessive-compulsive disorder. Arch Gen Psychiatry. 2006;63:769–76.
Shugart YY, Wang Y, Samuels JF, Grados MA, Greenberg BD, Knowles JA, et al. A family-based association study of the glutamate transporter gene SLC1A1 in obsessive-compulsive disorder in 378 families. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:886–92.
Stewart SE, Fagerness JA, Platko J, Smoller JW, Scharf JM, Illmann C, et al. Association of the SLC1A1 glutamate transporter gene and obsessive-compulsive disorder. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:1027–33.
Veenstra-VanderWeele J, Kim SJ, Gonen D, Hanna GL, Leventhal BL, Cook EH, et al. Genomic organization of the SLC1A1/EAAC1 gene and mutation screening in early-onset obsessive-compulsive disorder. Mol Psychiatry. 2001;6:160–7.
Wendland JR, Moya PR, Timpano KR, Anavitarte AP, Kruse MR, Wheaton MG, et al. A haplotype containing quantitative trait loci for SLC1A1 gene expression and its association with obsessive-compulsive disorder. Arch Gen Psychiatry. 2009;66:408–16.
Willour VL, Yao Shugart Y, Samuels J, Grados M, Cullen B, Bienvenu OJ 3rd, et al. Replication study supports evidence for linkage to 9p24 in obsessive-compulsive disorder. Am J Hum Genet. 2004;75:508–13.
Mattheisen M, Samuels JF, Wang Y, Greenberg BD, Fyer AJ, McCracken JT, et al. Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Mol Psychiatry. 2015;20:337–44.
Stewart SE, Yu D, Scharf JM, Neale BM, Fagerness JA, Mathews CA, et al. Genome-wide association study of obsessive-compulsive disorder. Mol Psychiatry. 2013;18:788–98.
Kanai Y, Hediger MA. The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflug Arch. 2004;447:469–79.
Diamond JS. Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci. 2001;21:8328–38.
Li MH, Underhill SM, Reed C, Phillips TJ, Amara SG, Ingram SL. Amphetamine and methamphetamine increase NMDAR-GluN2B synaptic currents in midbrain dopamine neurons. Neuropsychopharmacology. 2017;42:1539–47.
Scimemi A, Tian H, Diamond JS. Neuronal transporters regulate glutamate clearance, NMDA receptor activation, and synaptic plasticity in the hippocampus. J Neurosci. 2009;29:14581–95.
Conti F, DeBiasi S, Minelli A, Rothstein JD, Melone M. EAAC1, a high-affinity glutamate tranporter, is localized to astrocytes and gabaergic neurons besides pyramidal cells in the rat cerebral cortex. Cereb Cortex. 1998;8:108–16.
Mathews GC, Diamond JS. Neuronal glutamate uptake contributes to GABA synthesis and inhibitory synaptic strength. J Neurosci. 2003;23:2040–8.
Sepkuty JP, Cohen AS, Eccles C, Rafiq A, Behar K, Ganel R, et al. A neuronal glutamate transporter contributes to neurotransmitter GABA synthesis and epilepsy. J Neurosci. 2002;22:6372–9.
Underhill SM, Ingram SL, Ahmari SE, Veenstra-VanderWeele J, Amara SG. Neuronal excitatory amino acid transporter EAAT3: emerging functions in health and disease. Neurochem Int. 2018. https://doi.org/10.1016/j.neuint.2018.05.012.
Gonzalez LF, Henriquez-Belmar F, Delgado-Acevedo C, Cisternas-Olmedo M, Arriagada G, Sotomayor-Zarate R, et al. Neurochemical and behavioral characterization of neuronal glutamate transporter EAAT3 heterozygous mice. Biol Res. 2017;50:29.
Zike ID, Chohan MO, Kopelman JM, Krasnow EN, Flicker D, Nautiyal KM, et al. OCD candidate gene SLC1A1/EAAT3 impacts basal ganglia-mediated activity and stereotypic behavior. Proc Natl Acad Sci USA. 2017;114:5719–24.
Peghini P, Janzen J, Stoffel W. Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. The EMBO Journal 1997;16:3822-32.
Bradley SV, Hyun TS, Oravecz-Wilson KI, Li L, Waldorff EI, Ermilov AN, et al. Degenerative phenotypes caused by the combined deficiency of murine HIP1 and HIP1r are rescued by human HIP1. Hum Mol Genet. 2007;16:1279–92.
Jensen AA, Brauner-Osborne H. Pharmacological characterization of human excitatory amino acid transporters EAAT1, EAAT2 and EAAT3 in a fluorescence-based membrane potential assay. Biochem Pharmacol. 2004;67:2115–27.
Meulendyke KA, Ubaida-Mohien C, Drewes JL, Liao Z, Gama L, Witwer KW, et al. Elevated brain monoamine oxidase activity in SIV- and HIV-associated neurological disease. J Infect Dis. 2014;210:904–12.
Cho A, Haruyama N, Kulkarni AB. Generation of transgenic mice. Curr Protoc Cell Biol. 2009. https://doi.org/10.1002/0471143030.cb1911s42.
Hall BE, Zheng C, Swaim WD, Cho A, Nagineni CN, Eckhaus MA, et al. Conditional overexpression of TGF-beta1 disrupts mouse salivary gland development and function. Lab Invest. 2010;90:543–55.
DePoy L, Daut R, Brigman JL, MacPherson K, Crowley N, Gunduz-Cinar O, et al. Chronic alcohol produces neuroadaptations to prime dorsal striatal learning. Proc Natl Acad Sci USA. 2013;110:14783–8.
Graybeal C, Feyder M, Schulman E, Saksida LM, Bussey TJ, Brigman JL, et al. Paradoxical reversal learning enhancement by stress or prefrontal cortical damage: rescue with BDNF. Nat Neurosci. 2011;14:1507–9.
Radke AK, Kocharian A, Covey DP, Lovinger DM, Cheer JF, Mateo Y, et al. Contributions of nucleus accumbens dopamine to cognitive flexibility. Eur J Neurosci. 2018. https://doi.org/10.1111/ejn.14152.
Holmseth S, Dehnes Y, Huang YH, Follin-Arbelet VV, Grutle NJ, Mylonakou MN, et al. The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J Neurosci. 2012;32:6000–13.
Watts SD, Torres-Salazar D, Divito CB, Amara SG. Cysteine transport through excitatory amino acid transporter 3 (EAAT3). PLoS One. 2014;9:e109245.
Chamberlain SR, Fineberg NA, Blackwell AD, Robbins TW, Sahakian BJ. Motor inhibition and cognitive flexibility in obsessive-compulsive disorder and trichotillomania. Am J Psychiatry. 2006;163:1282–4.
Gruner P, Pittenger C. Cognitive inflexibility in obsessive-compulsive disorder. Neuroscience. 2017;345:243–55.
Ahmari SE, Spellman T, Douglass NL, Kheirbek MA, Simpson HB, Deisseroth K, et al. Repeated cortico-striatal stimulation generates persistent OCD-like behavior. Science. 2013;340:1234–9.
Wan Y, Ade KK, Caffall Z, Ilcim Ozlu M, Eroglu C, Feng G, et al. Circuit-selective striatal synaptic dysfunction in the Sapap3 knockout mouse model of obsessive-compulsive disorder. Biol Psychiatry. 2014;75:623–30.
Burguiere E, Monteiro P, Mallet L, Feng G, Graybiel AM. Striatal circuits, habits, and implications for obsessive-compulsive disorder. Curr Opin Neurobiol. 2015;30:59–65.
Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400.
Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–94.
Stewart SE, Mayerfeld C, Arnold PD, Crane JR, O’Dushlaine C, Fagerness JA, et al. Meta-analysis of association between obsessive-compulsive disorder and the 3’ region of neuronal glutamate transporter gene SLC1A1. Am J Med Genet B Neuropsychiatr Genet. 2013;162B:367–79.
Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, et al. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–26.
Bellini S, Fleming KE, De M, McCauley JP, Petroccione MA, D’Brant LY, et al. Neuronal glutamate transporters control dopaminergic signaling and compulsive behaviors. J Neurosci. 2018;38:937–61.
Radke AK, Jury NJ, Kocharian A, Marcinkiewcz CA, Lowery-Gionta EG, Pleil KE, et al. Chronic EtOH effects on putative measures of compulsive behavior in mice. Addict Biol. 2017;22:423–34.
Radke AK, Nakazawa K, Holmes A. Cortical GluN2B deletion attenuates punished suppression of food reward-seeking. Psychopharmacology. 2015;232:3753–61.
McLaughlin NC, Strong D, Abrantes A, Garnaat S, Cerny A, O’Connell C, et al. Extinction retention and fear renewal in a lifetime obsessive-compulsive disorder sample. Behav Brain Res. 2015;280:72–7.
Milad MR, Furtak SC, Greenberg JL, Keshaviah A, Im JJ, Falkenstein MJ, et al. Deficits in conditioned fear extinction in obsessive-compulsive disorder and neurobiological changes in the fear circuit. JAMA Psychiatry. 2013;70:608–18.
Reimer AE, de Oliveira AR, Diniz JB, Hoexter MQ, Miguel EC, Milad MR, et al. Fear extinction in an obsessive-compulsive disorder animal model: influence of sex and estrous cycle. Neuropharmacology. 2018;131:104–15.
Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109–13.
Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905.
Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–31.
Chamberlain SR, Menzies L, Hampshire A, Suckling J, Fineberg NA, del Campo N, et al. Orbitofrontal dysfunction in patients with obsessive-compulsive disorder and their unaffected relatives. Science. 2008;321:421–2.
Remijnse PL, Nielen MM, van Balkom AJ, Cath DC, van Oppen P, Uylings HB, et al. Reduced orbitofrontal-striatal activity on a reversal learning task in obsessive-compulsive disorder. Arch Gen Psychiatry. 2006;63:1225–36.
Valerius G, Lumpp A, Kuelz AK, Freyer T, Voderholzer U. Reversal learning as a neuropsychological indicator for the neuropathology of obsessive compulsive disorder? A behavioral study. J Neuropsychiatry Clin Neurosci. 2008;20:210–8.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Delgado-Acevedo, C., Estay, S.F., Radke, A. et al. Behavioral and synaptic alterations relevant to obsessive-compulsive disorder in mice with increased EAAT3 expression. Neuropsychopharmacol. 44, 1163–1173 (2019). https://doi.org/10.1038/s41386-018-0302-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-018-0302-7
This article is cited by
-
Developmental impact of glutamate transporter overexpression on dopaminergic neuron activity and stereotypic behavior
Molecular Psychiatry (2022)
-
Role of Sonic Hedgehog Signaling Activation in the Prevention of Neurological Abnormalities Associated with Obsessive–Compulsive Disorder
Neurotoxicity Research (2022)
-
Oxidative stress and impaired oligodendrocyte precursor cell differentiation in neurological disorders
Cellular and Molecular Life Sciences (2021)
-
Neuregulin-1 inhibits CoCl2-induced upregulation of excitatory amino acid carrier 1 expression and oxidative stress in SH-SY5Y cells and the hippocampus of mice
Molecular Brain (2020)
