
Abstract
The advent of optogenetics and chemogenetics has revolutionized the study of neural circuit mechanisms of behavioral dysregulation in psychiatric disease. These powerful technologies allow manipulation of specific neurons to determine causal relationships between neuronal activity and behavior. Optogenetic tools have been key to mapping the circuitry underlying depression-like behavior in animal models, clarifying the contribution of the ventral tegmental area, nucleus accumbens, medial prefrontal cortex, ventral hippocampus, and other limbic areas, to stress susceptibility. In comparison, chemogenetics have been relatively underutilized, despite offering unique advantages for probing long-term effects of manipulating neuronal activity. The ongoing development of optogenetic tools to probe in vivo function of ever-more specific circuits, combined with greater integration of chemogenetic tools and recent advances in vivo imaging techniques will continue to advance our understanding of the circuit mechanisms of depression.
Similar content being viewed by others
The past decade has witnessed an explosion in optogenetic and chemogenetic tools. Both techniques confer unprecedented control of cellular activity: optogenetics, via light-activated channels and chemogenetics, via engineered G-protein-coupled receptors activated by otherwise-inert molecules. Both tools powerfully and precisely manipulate neuronal activity, yet applications of chemogenetics to interrogate the circuitry of depression has been limited in contrast with the extensive use of optogenetics. Here we review the use of both techniques in uncovering the circuitry of depression-like behavior in animal models, suggesting that chemogenetics has an important place alongside optogenetics in the study of depression.
The optogenetic and chemogenetic toolbox
The optogenetic (Table 1) and chemogenetic (Table 2) toolbox includes a diversity of tools and techniques to achieve cell-type and pathway-specific control of neuronal activity. Various opsins, light-sensitive channels, have been engineered to inhibit or excite cells (Fig. 1). Chemogenetics is synonymous with designer receptors exclusively activated by designer drugs (DREADDs), modified human muscarinic receptors with no affinity for endogenous ligands, little constitutive activity, and activated by an experimenter-administered ligand (Fig. 1). Opsins and DREADDs enable greatly enhanced spatial, cell-type, and temporal specificity compared to traditional pharmacological techniques, allowing for precise dissection of the neural circuits underlying depression-like behavior.

Schematic overview of optogenetic and chemogenetic techniques. Upper panel: A chronically implanted fiber delivers light to activate opsin-expressing cells; CNO is delivered either systemically (via injection or in drinking water) or intra-cranially to activate DREADD-expressing cells leading to depicted increases (e.g. ChR2, HM3Dq) or decreases in cellular activity (e.g., NpHR, HM4Di). Lower panel: Optogenetics confers high temporal resolution, illustrated by single action potentials time-locked to discrete light pulses, and efficiently controls neuronal activity over millisecond to minute timescales. DREADD signaling increases (or decreases) activity, regulating excitability to endogenous stimulation, for sustained periods ranging from hours to days or even weeks, maintained by a regimen of CNO delivery
Strategies for targeting specific cell populations
Opsins and DREADDs allow precise in vivo control of neural activity via targeted expression in specified cellular populations with viral vectors and/or transgenic mice. Stereotaxic viral injection allows spatial specificity, with specific promoters (e.g., CAMKII) conferring cell-type specificity [1,2,3]. Cre-recombinase transgenic mice along with Cre-dependent viral vectors allow further cell-type specificity and double-viral strategies using Cre-dependent opsin or DREADD viruses with retrograding or anterograding Cre-expressing virus can target expression to projection-defined populations [4, 5]. Inducible expression using immediate early gene-based transgenic lines can restrict expression to cells activated by temporally defined stimuli [6,7,8]. Strategies for ever-greater specificity continue to emerge. FLARE and Cal-Light confer targeted expression upon coincident occurrence of increased calcium and blue-light, to restrict expression to neurons active during an experimenter-defined time window [9, 10]. vCAPTURE offers activity-dependent, pathway-specific opsin expression [11].
Rodent models of depression
Most animal models use stress, chronic or acute, to induce depression-like behavior, based on the well-established connection between stress and depression [12, 13]. Depression-like behavior is assessed in a range of simple tests thought to measure behavioral phenotypes that parallel symptoms of depression, and often also anxiety (Table 3 and Fig. 2). Most optogenetic and chemogenetic studies have used two primary depression models, social defeat and chronic mild/variable stress, reviewed in Fig. 2 and Table 4. For more extensive discussion of animal models, challenges and alternative approaches see ref. [14].

Schematic overview of commonly used animal models of depression and tests of depression- and anxiety-like behavior. a Chronic social defeat stress (CSDS) [17, 86] is useful in modeling differential susceptibility. Many people remain resilient to depression in the face of stress. After CSDS, susceptible mice exhibit a depression-like phenotype characterized by reduced social interaction (also reduced sucrose preference, increased immobility in a forced swim test, circadian, and metabolic changes) whereas resilient mice continue to seek out social interaction and, unlike susceptible mice, are similar to non-stressed controls on a range of depression-relevant metrics, although, like susceptible mice, they show increased anxiety indicated by reduced center exploration in an open field. b Learned helplessness is another commonly used model of depression. After experiencing repeated inescapable footshock, susceptible animals fail to escape from signaled shocks when given the opportunity, but some animals will escape, demonstrating resilience. Chronic mild stress or chronic variable stress (c) refer to a number of protocols in which mice or rats are repeatedly exposed to a single stressor or a variety of stressors over days or weeks. Stressors may include a wide range of manipulations such as tail suspension, restraint, footshock, noise and bright light. d Susceptibility is assessed by increases in depression- and anxiety-like behavior. Depression-like behavior is typically assessed by tests such as forced swim, tail suspension, sucrose preference, or open field. Anxiety-like behavior is also assessed in an open field or elevated plus maze test. See Table 3 for a complete description of paradigms and tests
Opto- and chemogenetic interrogation of circuitry mediating depression-like behavior
Optogenetic and chemogenetic manipulations have elaborated the specific cell types and projection pathways of depression-like behavior, powering a shift towards a circuit-based understanding of depression (Fig. 3). We review major findings, organized by discrete brain regions for expediency despite extensive interconnections. Optogenetics has been the preferred circuit interrogation tool in depression studies (Table 5) with a relative paucity of chemogenetic studies (Table 6). This disparity may be a cause or consequence of a focus on the acute control of behavior rather than prolonged behavioral change. Chemogenetics may be better suited to chronic manipulations, for example during chronic stress. Ultimately, probing different timescales of behavior through application of chemogenetic and optogenetic tools will be critical to understand how brain circuits are acutely regulated and chronically remodeled in depression. Chemogenetic manipulation modulates cellular excitation to endogenous stimulation whereas optogenetic excitation directly induces spiking. While the temporal specificity of optogenetics makes it ideal for probing proximal neural mediators of behavior, chemogenetics may be better suited to probing sustained circuit changes. Demonstrating behavioral regulation by acute neural regulation is invaluable in identifying the circuits that mediate behavior but may be of limited relevance to the consequences of enduring adaptations associated with stress, and other factors such as genetic vulnerability. From a treatment perspective, acute manipulations are interesting if therapeutic effect outlasts stimulation. While this could occur with optogenetic manipulations, enduring effects of either opto- or chemogenetics are rarely assessed. Treatment strategies developed from acute manipulations may have unintended consequences as long-term activation or inhibition of neural activity will inevitably induce plasticity within and beyond the targeted circuit that could reduce efficacy over time or induce off-target effects. In this respect, chemogenetics could be especially useful in probing effects of sustained circuit alterations.

Schematic illustrating neuronal pathways implicated in depression-like behavior by optogenetic or chemogenetics. Red arrows indicate pro-susceptible projections where activation leads to increased depression-like behavior (social avoidance, immobility, etc.) and blue arrows indicate pro-resilient projections. Dashed lines indicate pathways in which conflicting findings exist
Ventral tegmental area
The ventral tegmental area (VTA) is a predominantly dopaminergic structure in the mesolimbic reward circuit. VTA dopamine (DA) neurons exhibit both tonic and phasic firing, with phasic firing encoding reward-predictive signals, and tonic firing regulating the post-synaptic response to phasic DA [15]. Due to the well-established role of VTA-DA cells in motivation and reward, much work has focused on the VTA in depression-like behaviors with crucial insight provided by optogenetic and chemogenetic approaches.
Mice susceptible to chronic social defeat stress (CSDS), have elevated VTA-DA phasic firing, an adaptation not observed in resilient mice [16, 17]. Optogenetic interrogation has established the causal importance of VTA-DA firing in behavioral adaptation to chronic stress, delineating contrasting roles of projection-defined sub-populations. Chaudhury et al. [18] demonstrated that increasing VTA-DA phasic firing in resilient mice following CSDS induced susceptibility, measured by social avoidance and decreased sucrose preference.
Despite the efficacy of bulk VTA stimulation, the VTA is heterogeneous with diverse projections. Nucleus accumbens (NAc) and medial prefrontal cortex (mPFC) projections are differentially altered by chronic stress and exert distinct behavioral control [18, 19]. Manipulations of VTA-NAc cells mirror the profile of whole VTA; optogenetic stimulation promotes susceptibility whereas inhibition promotes resilience. Activating the VTA-mPFC pathway, however, increases resilience, increasing social interaction and sucrose preference, whereas inhibition of this pathway decreases social interaction.
The role of VTA in behavioral stress responses may depend upon stressor type. Chronic mild stress (CMS), another mouse model of depression in which mice are subjected to less severe forms of stress over a longer time period, oppositely regulates VTA-DA activity compared to CSDS, suppressing phasic activity [20]. Inhibition of VTA-DA cells during testing increases anhedonia and time immobile in a tail suspension test (TST) while stimulating the VTA following CMS reverses these behaviors [19]. Pharmacological DA receptor inhibition in the nucleus accumbens (NAc) blocked the anti-depressant effect of VTA stimulation, demonstrating involvement of NAc-DA signaling. This discrepancy was attributed to differing stress protocols; mild stressors and aversive stimuli may decrease DA cell firing in the VTA whereas severe stress may cause hyperactivity in these neurons [18, 20]. These studies also use different light cycles. Chaudhury et al. [18] used a 12 h light-dark cycle with experiments in the light phase, whereas Tye et al. [19] used a reverse light cycle, testing in the dark phase. Stress hormones exhibit diurnal rhythms and the magnitude of the glucocorticoid stress response is significantly greater for stressors administered in the light phase [21], mediating time-dependent effects of even the same stress (e.g., Fonken et al. [22]). It is possible that the repeated, robust stress response from CSDS during the light cycle could induce VTA-NAc plasticity, disrupting normal reward function such that activity in this pathway increases depressive-like behaviors. However, the relatively attenuated stress response with dark cycle stress may not induce similar alterations, with activity in this pathway remaining rewarding.
Optogenetic experiments also defined an active resilience mechanism in the VTA by which homeostatic plasticity normalizes defeat-induced cellular dysregulations to support resilience [23]. This self-tuning mechanism can be induced via optogenetic stimulation in susceptible mice to counteract the stress-induced VTA hyperactivity and induce resilience (increased social interaction, and sucrose preference and decreased immobility in a forced swim test (FST)) via increased potassium channel current. This mechanism is specific to VTA-NAc and not observed in VTA-mPFC. In combination with pharmacology, optogenetic manipulations have also contributed important insights into the molecular basis of susceptibility and resilience. Optogenetic stimulation of the VTA-NAc increases NAc brain derived neurotrophic factor (BDNF), inducing social avoidance following sub-threshold defeat [24]. Both BDNF and DA are necessary to induce susceptibility as blocking BDNF signaling through its receptor, TrkB, knocking out BDNF, or blocking D1 DA receptors all prevent the pro-susceptibility effect of VTA-NAc stimulation [24, 25]. These effects also required corticotropin releasing factor (CRF), which gates BDNF released by VTA-NAc phasic firing [24].
Chemogenetic approaches have informed depression-relevant VTA molecular mechanisms. Cdk5 cKO mice (lacking a kinase for phosphorylating tyrosine hydroxylase (TH) in VTA-DA neurons) show increased anxiety, decreased sucrose preference, increased latency to feed in a novel environment and increased immobility in FST after CMS [26]. Decreased TH phosphorylation decreases VTA cAMP signaling, decreasing DA in NAc. Activation by rM3Ds DREADD in VTA-DA neurons restored cAMP levels and normalized behavior, identifying a key mechanism in the VTA underlying depression-like behavior.
Nucleus accumbens
The NAc, contains primarily medium spiny neurons (MSN) defined by their pre-dominant expression of D1- or D2-type DA receptors [27], and is innervated by DAergic projections from the VTA as well as glutamatergic projections from cortical and subcortical structures [28]. D1 and D2-MSNs have important opposing roles in reward and motivation, implicating NAc dysfunction in depression [29,30,31].
Experiments using cell-type-specific manipulations identified opposing roles for these cell types in stress susceptibility. Optogenetically stimulating D1-MSNs in susceptible mice following CSDS promotes resilience while stimulating D2-MSNs in stress-naïve mice induces susceptibility to subthreshold defeat [32]. Inhibiting D1-MSNs by hM4Di induced susceptibility in resilient mice. Optogenetics are also redefining existing notions of MSN function. Reduced motivation is a symptom of depression and susceptibility which can be modeled using operant behavior. For example, Pavlovian to instrumental transfer (PIT) measures the ability of a reward-predicting cue to enhance a rewarded operant behavior, and progressive ratio (PR) assesses willingness to work for reward. Optogenetic excitation of either D1- or D2-MSNs increases motivation in these tasks and surprisingly, inhibition of D2-MSNs decreases motivation [33]. Moreover, in rats exposed to in utero glucocorticoids, a manipulation that increases depressive-like behavior, targeted optogenetic stimulation of NAc D2-MSNs increases motivation in both tests, suggesting a more complex role for D2-MSNs. It is important to note, that the vast majority of studies investigating drug sensitivity, motivation and reward support the classical roles of D1 and D2-MSNs in reward and aversion [31, 34,35,36]; however, this study suggests that the function of these neurons may be context, task and condition dependent.
Hippocampus
The hippocampus is longitudinally heterogeneous, with dorsal and ventral hippocampi displaying unique functions [37]. While the dorsal hippocampus is important for spatial navigation, the ventral hippocampus (vHIP) is a key node in brain circuits mediating emotional behavior [38]. The vHIP projection to the NAc has been implicated in stress susceptibility. In vivo, low frequency stimulation of this pathway, a manipulation that induces lasting synaptic plasticity to reduce post-synaptic activity (long-term depression), induces a pro-resilient effect in defeated mice, increasing social interaction in a later test. An opposing manipulation to acutely increase vHIP-NAc signaling during testing, induces susceptibility in defeated mice, suppressing social interaction. Thus, through its interaction with NAc, the vHIP bi-directionally modulates susceptibility to depression-like behavior [1].
Recent work implicates the ventral hippocampal dentate gyrus (vDG) in stress susceptibility [39]. Chemogenetic inhibition of vDG in socially defeated mice is pro-resilient, increasing social interaction and open field center exploration. In contrast, chemogenetic vDG excitation increased susceptibility to sub-threshold defeat. Intriguingly, the effect of stress on vDG appears to be mediated by a small population of adult born neurons, and targeting these neurons accounted for whole vDG manipulations. Chemogenetic silencing of adult born vDG neurons increased susceptibility in mice exposed to a sub-threshold defeat, suggesting an important inhibitory role for adult born neurons and that loss of these neurons is one mechanism of stress-induced increased vHIP activity.
Ketamine, an NMDA antagonist (amongst other effects), is a promising novel anti-depressant [40]. In mice, ketamine decreases immobility in the forced swim test (FST) through activation of the vHIP projection to mPFC. Optogenetic inhibition of this pathway abolishes the anti-depressant-like effect of ketamine in FST [4]. Optogenetic activation, in contrast, increases climbing behavior, similar to ketamine, but decreases swimming, leading to an overall non-significant effect on immobility. Inhibition of the dorsal raphe nucleus (DRN), which receives feedback projections from mPFC, prior to optogenetic activation of vHIP-mPFC, eliminates the decreased swimming to reproduce ketamine-like effects. This DRN-mediated effect could be an artifact of optogenetic stimulation. Chemogenetic activation of vHIP-PFC produces both increased climbing and swimming, mimicking ketamine. Furthermore, these effects are specific to vHIP-PFC and are not produced by vHIP-NAc activation. This study demonstrates the utility of combining optogenetic and chemogenetic tools to probe drug mechanism, and it would be interesting to see such findings replicated in a chronic stress model.
Optogenetic technology has implicated hippocampal memory in depression-like behavior. Tonegawa and colleagues developed a c-fos-TetTag (tTA) transgenic mouse for activity-dependent labeling and later reactivation of cells active during encoding a positive experience (positive memory engrams) [41, 42]. Reactivation of engrams in the hippocampal dentate gyrus (DG) after chronic immobilization stress decreased immobility in TST, anxiety-like behavior on OFT and EPM and increased sucrose preference. This effect required glutamatergic transmission in NAc and was blocked by inhibition of BLA-NAc transmission, suggesting the effect required reactivation of a DG-BLA-NAc engram. Interestingly, stimulating BLA-NAc projections in socially defeated mice also exerts an aantidepressant-like effect, increasing social interaction [1], suggesting that reactivation of positive memories may recruit activation of a pre-existing pro-resilient circuit. These experiments elegantly connect memory engrams with depressive-like behaviors, introducing a possible new mechanism for targeting maladaptive behaviors.
Prefrontal cortex
Reduced PFC activation is observed in depressed humans as well as in rodent depression models, making it a key area of interest [43, 44]. Optogenetically stimulating the mPFC reverses depression-like behaviors such as social avoidance and anhedonia in defeated mice [43]. This may be specifically mediated by NAc-projecting mPFC neurons as targeted stimulation of this pathway increases social interaction in defeated mice [1, 43]. However, despite evidence implicating increased mPFC-NAc activity in resilience, suppression of this pathway does not induce social avoidance, suggesting a potential dissociation between circuit mechanisms of resilience and susceptibility [1]. Indeed, other molecular players act in concert with glutamatergic mPFC projections to regulate susceptibility. Upregulation of ΔFosB induces susceptibility following CSDS and increases expression of CCKB, the CCK receptor, in the prelimbic cortex (PrL) which in turn may decrease PrL activity, leading to social avoidance, reduced sucrose preference and increased immobility in FST [45]. Infusing CCK into the PrL reduces social interaction in mice after defeat and optogenetic stimulation of PrL-NAc projections counteracts the effects of increased CCK signaling to reverse social avoidance. By integrating optogenetics with pharmacology, this experiment sheds light on the molecular mechanisms underlying susceptibility and resilience. Parvalbumin (PV)-expressing GABA neurons in the mPFC have also been studied in the learned helplessness depression model. Animals susceptible to this type of stress show decreased excitatory synaptic activity on mPFC-PV interneurons [46]. Chemogenetic silencing of these neurons facilitates learned helplessness behaviors, increasing escape latency and number of failures.
Given the numerous regions innervated by the mPFC, it is perhaps not surprising that it regulates many behaviors. During FST, for example, the activation of subsets of neurons associates with specific behaviors, such that some neurons are active during kicking and others during immobility, although with considerable overlap. As such, bulk stimulation of the mPFC does not elicit these behaviors, potentially due to simultaneous activation of competing activity profiles. Projection-targeted stimulation has helped resolve the complexity of mPFC circuits [2]. Stimulating mPFC neurons projecting to DRN increases kicking whereas stimulating the lateral habenula (LHb)-projecting mPFC neurons decreases mobility. These findings disentangle the neural pathways mediating motivated behavior in the face of acute stress. In a CSDS study, photoinhibition of the vmPFC-DRN during the acute physical aggression blocks acquisition of social avoidance [47]. Through an elegant series of optogenetic experiments targeting the various DRN cell populations, the authors demonstrated that vmPFC-DRN stimulation excites GABAergic DRN neurons, to inhibit 5-HT DRN neurons. Inhibiting GABAergic DRN neurons increases 5-HT neuron activity, suppressing acquisition of social avoidance [48]. These studies report seemingly opposing effects of optogenetic manipulations of the same vmPFC-DRN pathway. The divergent results may arise from methodological differences, with the studies ultimately addressing distinct questions. Key among these differences is the type and chronicity of stress. Manipulating activity in this pathway while acutely exposing rats to FST may reveal how a healthy brain adapts to a challenging situation Warden et al. [2] whereas probing the development of social avoidance across chronic social stress [48] may inform understanding of stress-induced adaptation. While more work is needed to resolve these discrepancies, these studies demonstrate a complex role for the mPFC-DRN in stress behavior that is likely context/ experience dependent.
Recent chemogenetic experiments identified a role for a previously unexamined circuit in depression-like behavior, the mPFC to medial dorsal thalamus (MDT). The MDT is the main thalamic input to the mPFC and integrates information from the ventral pallidum, substantia nigra, VTA, amygdala, and DRN [49]. Chemogenetic activation of the MDT-mPFC pathway via hM3Dq DREADDs decreased immobility in FST and TST. As knocking out GluN2B receptors in the PFC selectively increases MDT drive onto the PFC, the authors suggest this pathway as a target for ketamine.
Habenula
The lateral habenula (LHb), a small brain region in the dorsal diencephalon, receives projections from the limbic forebrain and projects to brainstem nuclei including VTA and DRN [50]. Habenula deep-brain stimulation has seen success in treatment-resistant depression spurring further research into its role in depression [51, 52]. Chemogenetic inhibition of the LHb via hM4Di DREADDs in rats reduces immobility in FST. Furthermore, in 5-HT-deficient animals that are resistant to SSRI treatment, chemogenetic inhibition of the LHb in mice reverses social defeat stress-induced social avoidance [52].
Dorsal and medial raphe nucleus
The raphe nucleus, divided into dorsal and medial nuclei (DRN, MRN), is the main source of serotonin (5-HT). Chemogenetic studies suggest an interesting dissociation between anxiety—and depression-like behaviors in the raphe. Activation of 5-HT neurons increased anxiety—but decreased depression-like behaviors in elevated plus maze and FST, respectively [53]. Furthermore, there is a functional dichotomy between the DRN and MRN, with DRN neurons primarily mediating anxiety- and MRN neurons primarily mediating depression-like behavior, mirroring earlier optogenetic findings [2, 54]. The chronicity of DRN activation may also be relevant [55]. Both short- (30 min) and long-term (3 weeks) activation induced anti-depressant effects, but only short-term activation was anxiogenic. DREAMM mapping of brain-wide metabolic changes revealed that acute DRN activation increased metabolic activity in areas including the cingulate cortex, primary somatosensory and motor cortices, and decreased metabolic activity in the periaqueductal gray, and central and medial thalamic nuclei. In contrast, chronic activation of DRN neurons resulted in decreased metabolic activity in many forebrain structures, including hippocampus, habenula, central and medial thalamic nuclei, and cortical areas. This study reveals the utility of chemogenetics in probing temporal dynamics of neural activity in behavior on an entirely different time scale as well as the potential widespread consequences of specific manipulations of neuronal activity, raising an important caveat in attributing effects of a local manipulation solely to the targeted region.
The serotoninergic projection from DRN to NAc impacts motivation and depression-like behavior [56]. Chemogenetic stimulation of 5-HT DRN-NAc projections in Fev-cre mice expressing Cre in 5-HT-synthesizing neurons decreases time immobile in TST [57], confirming a role for DRN-NAc in depression-like behavior.
Other limbic areas
Optogenetic experiments have also point to a role for thalamo-striatal projections to NAc in stress susceptibility. In vitro optogenetic electrophysiological interrogation of CSDS effects in ILT-vSTR found increased synaptic strength in susceptible mice [58]. Targeted optogenetic manipulation of ILT-vSTR prevented defeat-induced social avoidance, whereas stimulation increased susceptibility to subthreshold defeat.
Chronic stress induces a wide variety of behavioral adaptations, suggesting that different symptoms may be mediated by distinct neural circuits. The ventral pallidum (VP) is highly implicated in depression-like behavior given its direct connections to the mesolimbic reward circuit [59]. This area contains mostly GABAergic, but also some glutamatergic parvalbumin (PV) expressing cells that project to a number of downstream targets, notably, the LHb and VTA, which receive projections from distinct populations of VP-PV cells. Although by different mechanisms, CSDS induces hyperactivity in both LHb and VTA-projecting VP-PV neurons. Optogenetically silencing LHb projecting cells decreases immobility following defeat. Inhibiting VTA-projecting cells has no effect on immobility but increases social interaction after CSDS. In fact, stimulating VTA projections without defeat induces social avoidance [60], providing convincing evidence for the specificity of behavioral control.
Stress-induced changes in hormone secretion are regulated by the paraventricular nucleus of the hypothalamus which receives limbic input from the bed nuclei of the stria terminalis (BNST). Tail suspension stress activates the HPA-axis, increasing corticosterone (CORT) and adrenocorticotropic hormone (ACTH). Optogenetically inhibiting the avBNST increases not only immobility but also plasma CORT and ACTH [61]. However, inhibiting the avBNST-PVH pathway upregulates HPA-axis activity, but does not alter behavior, whereas inhibition of avBNST projections to the ventrolateral periaqueductal gray increases immobility without affecting plasma CORT or ACTH, suggesting a dissociation in the circuits responsible for HPA regulation and behavioral control.
Optogenetics vs chemogenetics: advantages, limitations, and caveats
Both optogenetics and chemogenetics are powerful tools for dissecting the neural circuits underlying behavior. The great promise of optogenetics is the ability to manipulate neuronal populations with greater spatial, temporal and cell-type specificity than traditional techniques. Electrical stimulation excites and inhibits cells but lacks spatial and cell-type specificity. Gene expression manipulations (e.g., potassium channel to alter excitability) and pharmacological techniques offer precise control but poor temporal resolution. Optogenetics offers control of cell populations precisely defined by spatial location, genetic identity, circuit connectivity or even temporally defined activity, with fast temporal precision in awake behaving animals to probe the causality between brain and behavior [2]. The exquisite temporal precision has also been applied in opto-electrophysiology in which optogenetic stimulation, either in vivo or in vitro, is used to probe synaptic function and sometimes to administer patterned stimulation to induce lasting synaptic plasticity [1]. However certain limitations necessitate caution in interpreting results. Limited penetrance of light in brain tissue is an inherent challenge to in vivo optogenetic manipulations: generally only ~1 mm3 is effectively stimulated posing a particular challenge in larger animals (e.g. non-human primates) [62]. Recent innovations in fiber shape to deliver broad or focal illumination may help to address some spatial limitations [63]. Chemogenetics, in contrast, has high spatial reach; systemic administration allows diffusion across the entire nervous system, while targeted DREADD expression allows manipulation of specific cell populations. As chemogenetics uses a chemical actuator, systemic administration will affect all DREADD-expressing cells. Although intra-cranial infusions of CNO can limit effects to a specific region of interest [56], diffusion of the actuator can be hard to control, rendering this technique less spatially precise.
Both optogenetic and chemogenetic manipulations can also have unexpected effects on neuronal firing properties. Back-propagation of depolarization of illuminated axons may alter activity in structures not explicitly manipulated. Opsin permeability is another consideration. For example, eNphR3.0 is permeable to Cl−, which alters the reversal potential of GABA, and therefore the response to GABA release [64]. In cells that release multiple neurotransmitters, such as LH neurons that co-release GABA and glutamate, optogenetics cannot directly interrogate the neurotransmitters involved [65]. Chemogenetic activation may have effects beyond activation or inhibition of targeted cells as GPCRs have many downstream targets which may elicit unintended effects. There is some concern that long-term CNO administration could desensitize DREADD receptors, although this may be circumvented by high levels of DREADD expression, easily achievable with viral-mediated expression. Considering these limitations, it is important to integrate other techniques (e.g., pharmacology) to resolve precise questions of neurotransmitter action [66].
Appropriate controls are critical. Extended or developmental opsin expression can alter cell morphology [67]. Light alone may increase blood flow measured by fMRI and tissue heating in animals [68]. Ideally, the behavioral effects of opsin stimulation should be interpreted relative to both light stimulation without opsin (e.g., viral expression of EYFP or mCherry without opsin) and opsin expression without light stimulation. For chemogenetics, there is evidence that CNO alone may induce effects, reducing the acoustic startle reflex and amphetamine induced hyperlocomotion and amphetamine-evoked dopamine [69]. Furthermore, recent evidence challenges the assertion that CNO is the DREADD actuator [70], showing that systemic CNO does not cross the blood brain barrier and has low affinity for DREADDs but is rapidly metabolized to clozapine, which readily enters the brain and has high affinity for DREADDs. Clozapine also interacts with endogenous receptors, and is administered as an anti-psychotic, however it is notable that its affinity for DREADDs is substantially higher than for endogenous receptors, indicating the potential to achieve specific DREADD activation via low-dose clozapine. Another alternative rapidly gaining popularity is the use of an alternative ligand, Compound 21. This non-CNO actuator appears to potently activate DREADDs without metabolizing to clozapine [71, 72]. Irrespective of ligand choice, the recent controversy concerning CNO effects is a timely reminder of the need for appropriate ligand-only control groups.
Another important consideration is the difference between demonstrating that modulating a circuit produces a behavior and that this occurs under physiological conditions. To optogenetically elicit behavior, experimenters have at times induced cells to fire at frequencies outside physiological range. Findings based on non-physiological stimulation protocols should be interpreted with caution. While efforts to achieve more physiological stimulation protocols are important, as generally employed, optogenetic stimulation results in synchronous activation (or inhibition) of an entire cell population [41, 73, 74]. Inhibition of circuit activity can theoretically be more informative concerning the necessity of a circuit to behavior, although there are a number of technical concerns. For example, optogenetic inhibition can induce rebound excitation in cells once released from non-physiological inhibition and inhibition of terminal activity has been problematic [75]. Recent advances enable more physiologically accurate stimulation patterns. Closed-loop optogenetic systems can be triggered by endogenous activity to deliver light stimulation. Although originally designed to deliver light in response to EEG-detected seizures, it can be modified to be triggered by other activity inputs. Such innovation is extremely important for modeling physiologically relevant stimulation.
Reviewing multiple studies in animal models of depression points to regulation of similar behaviors by divergent brain regions. It is common to assert sufficiency of a brain circuit when optogenetic or chemogenetic excitation elicits a particular behavior and necessity when inhibition suppresses this behavior. However, concluding that a brain circuit is sufficient to drive a behavior because activating it increases the behavior can erroneously imply that this is the unique and complete mechanism [76]. Establishing true sufficiency would require demonstrating not only that circuit activation can elicit behavior, but that it is the only requirement, something rarely true. A more appropriate conclusion is that circuit activity induces the behavior, which does not exclude the possibility that other circuits may also exert control over the same behavior. In fact, with complex, motivated behaviors, as examined in animal models of depression, it would be surprising for a single brain region or circuit to exert unique and complete mechanistic control. In this light, it is to be expected that optogenetic and chemogenetic studies will identify multiple brain regions mediating the same depression-relevant behaviors. While chemogenetics and optogenetics are powerful tools to probe the circuitry underlying depression-related circuits, we should be mindful not to overstate conclusions from individual experiments.
In vivo imaging advances
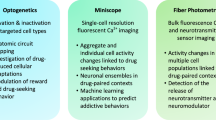
Many of these limitations highlight a potential disconnect between control of cellular activity and behavior by optogenetics and chemogenetics and the in vivo physiological realities of circuit function. A solution that is currently gaining popularity is to complement perturbation experiments with population or single-cell in vivo imaging such as fiber photometry and microendoscopes [77]. We employed fiber photometry to identify reductions in NAc D1-MSN activity that preceded the emergence of depression-like behaviors in susceptible mice [97], complementing earlier findings that optogenetically increasing D1-MSN activity in susceptible mice after CSDS is pro-resilient [32]. Single-cell calcium imaging was also recently applied to interrogate D1-MSN firing following social defeat. At the single-cell level, defeat reduced the frequency of events in D1-MSNs, although somewhat paradoxically, the average neural response during social interaction was increased. Of interest, Egr3 knockdown increased social interaction behavior and normalized D1-MSN firing [78]. In the vDG, single-cell calcium imaging revealed that chronic social defeat sensitizes neural activity induced by an aggressive encounter [39]. This effect was exacerbated by ablation of adult neurogenesis, a manipulation that increases defeat-induced social avoidance. In the lateral septum (LS), single-cell calcium imaging was used to probe the role of Drd3 DA receptors in social dysfunction following early social deprivation (ESD) [79]. Increases in Drd3 expressing LS cells coincident with social interaction were blunted in ESD mice, decreasing social preference. This behavioral and cellular dysregulation can be rescued by optogenetically stimulating these cells or overexpressing Drd3. Likewise, these dysregulations can be induced by knocking down Drd3 or reducing activity in these cells. Into the future, chemogenetic manipulations may be easily combined with optogenetics, or in vivo imaging techniques such as calcium imaging, or behavioral µPET imaging to gain a more comprehensive picture of circuit function. For example, DREADD-assisted metabolic mapping (DREAMM) allows metabolic mapping in awake animals, revealing whole-brain consequences of inhibiting specific cell populations during behavior [80]. This allows the unique opportunity to interrogate in vivofunction and manipulate specific cell populations in the same animals. Continued efforts to integrate optogenetic and chemogenetic manipulations with in vivo imaging techniques will be essential to understand pathological changes in brain activity driving depression and stress susceptibility.
Translating advances from animal models to humans
While initial excitement about neural control techniques raised suggestions of therapeutic use in humans, direct translation to humans is unlikely. The great promise of optogenetic and chemogenetics for depression lies in the power of these technologies to reveal novel mechanistic insight into the disorder to support development of effective treatments. By probing the specific brain regions, pathways or cell types mediating behavioral dysregulation it is possible to precisely identify the locus of disease-associated dysfunction. In the context of drug-addiction, a report exists of an experimental treatment based on translating optogenetic findings to guide non-invasive stimulation protocols [81] using transcranial magnetic stimulation. It is conceivable that such approaches could be applied to depression. Invasive deep-brain stimulation is also being investigated as a potential therapy for treatment-resistant depression, and circuit investigation in animal models will be critical to refine stimulation strategies [82]. Using intersectional approaches in circuit studies, the defining molecular identity of relevant neural populations can be revealed and used to design pharmacological strategies to regulate activity of specific circuits or cell populations. Identifying the site of neural dysregulation can also localize the search for molecular mechanisms of depression. For example, optogenetic identification of opposing roles of mPFC and vHIP in susceptibility inspired transcriptional profiling studies in mice [83] and humans [84] that identified novel, region-specific transcriptional regulators of depression and depression-like behavior. These transcriptional profiles of relevant cellular populations can ultimately provide novel molecular targets for future drug development.
A major caveat of animal depression studies, by no means unique to opto- and chemogenetics, is the almost exclusive use of males. This is particularly concerning given the increased prevalence of depression in women [85]. Enough evidence of sex differences exists to caution against assuming that findings from males will translate directly to females and it is essential that future research probe circuit function in both sexes. Another important consideration is the need to expand the repertoire of depression-relevant behavior. Existing studies have largely employed simple behaviors that can be difficult to directly compare to humans, and often rely on loosely defined constructs with validity based on superficial similarity or efficacy of existing anti-depressant drugs. Expanding to include an alternative approach, exemplified by the work of Soares-Cunha et al. [33], that uses more structured behaviors to probe endophenotypes of depression will help to increase the translational potential of this field.
Conclusions
Optogenetics and chemogenetics afford powerful and precise manipulation of specific cellular subtypes and circuits in vivo. These complementary tools have made it possible to probe brain function in behaving animals with a previously unimagined spatial, temporal and genetic precision and resolution, heralding a new era in understanding of circuit function. Experiments employing the ever-evolving toolboxes have provided a wealth of information regarding the neural substrates of depression by uncovering the circuitry underlying depression-like behaviors as well as the molecules and activity profiles that contribute to these states. Optogenetics and chemogenetics offer distinct ways to manipulate specific populations of cells in vivo. Although optogenetics has been more extensively used in depression studies, chemogenetics offers unique advantages for probing circuit function, notably allowing targeted, chronic inhibition or activation, ideal for probing sustained cellular alterations. Further, it offers a complementary method to circumvent limitations of optogenetics. Ultimately, complementing optogenetic or chemogenetic interrogations with in vivo imaging techniques, such as fMRI, DREAMM, fiber photometry, or single-cell calcium imaging will provide important new perspectives on the physiological significance of identified circuits, bringing us ever closer to developing treatments founded in a precise understanding of the neural circuit mechanisms of depression.
Funding and disclosure
The authors declare no competing interest.
References
Bagot RC, Parise EM, Pena CJ, Zhang HX, Maze I, Chaudhury D, et al. Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat Commun. 2015;6:7062 https://doi.org/10.1038/ncomms8062
Warden MR, Selimbeyoglu A, Mirzabekov JJ, Lo M, Thompson KR, Kim SY, et al. A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature. 2012;492:428–32. https://doi.org/10.1038/nature11617
Whissell PD, Tohyama S, Martin LJ. The use of DREADDs to deconstruct behavior. Front Genet. 2016;7:70 https://doi.org/10.3389/fgene.2016.00070
Carreno FR, Donegan JJ, Boley AM, Shah A, DeGuzman M, Frazer A, et al. Activation of a ventral hippocampus-medial prefrontal cortex pathway is both necessary and sufficient for an antidepressant response to ketamine. Mol Psychiatry. 2016;21:1298–308. https://doi.org/10.1038/mp.2015.176
Carter ME, Soden ME, Zweifel LS, Palmiter RD. Genetic identification of a neural circuit that suppresses appetite. Nature. 2013;503:111–4. https://doi.org/10.1038/nature12596
Denny CA, Kheirbek MA, Alba EL, Tanaka KF, Brachman RA, Laughman KB, et al. Hippocampal memory traces are differentially modulated by experience, time, and adult neurogenesis. Neuron. 2014;83:189–201. https://doi.org/10.1016/j.neuron.2014.05.018
Garner AR, Rowland DC, Hwang SY, Baumgaertel K, Roth BL, Kentros C, et al. Generation of a synthetic memory trace. Science. 2012;335:1513–6. https://doi.org/10.1126/science.1214985
Guenthner CJ, Miyamichi K, Yang HH, Heller HC, Luo L. Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron. 2013;78:773–84. https://doi.org/10.1016/j.neuron.2013.03.025
Lee D, Hyun JH, Jung K, Hannan P, Kwon HB. A calcium- and light-gated switch to induce gene expression in activated neurons. Nat Biotechnol. 2017;35:858–63. https://doi.org/10.1038/nbt.3902
Wang W, Wildes CP, Pattarabanjird T, Sanchez MI, Glober GF, Matthews GA, et al. A light- and calcium-gated transcription factor for imaging and manipulating activated neurons. Nat Biotechnol. 2017;35:864–71. https://doi.org/10.1038/nbt.3909
Kim CK, Ye L, Jennings JH, Pichamoorthy N, Tang DD, Yoo AW, et al. Molecular and circuit-dynamical identification of top-down neural mechanisms for restraint of reward seeking. Cell. 2017;170:1013–27 e1014. https://doi.org/10.1016/j.cell.2017.07.020
Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry. 1999;156:837–41.
Kessler RC. The effects of stressful life events on depression. Annu Rev Psychol. 1997;48:191–214. https://doi.org/10.1146/annurev.psych.48.1.191
Slattery DA, Cryan JF. Modelling depression in animals: at the interface of reward and stress pathways. Psychopharmacology. 2017;234:1451–65. https://doi.org/10.1007/s00213-017-4552-6
Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24.
Cao JL, Covington HE 3rd, Friedman AK, Wilkinson MB, Walsh JJ, Cooper DC, et al. Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci. 2010;30:16453–8. https://doi.org/10.1523/JNEUROSCI.3177-10.2010
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. https://doi.org/10.1016/j.cell.2007.09.018
Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493:532–6. https://doi.org/10.1038/nature11713
Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. 2013;493:537–41. https://doi.org/10.1038/nature11740
Valenti O, Gill KM, Grace AA. Different stressors produce excitation or inhibition of mesolimbic dopamine neuron activity: response alteration by stress pre-exposure. Eur J Neurosci. 2012;35:1312–21. https://doi.org/10.1111/j.1460-9568.2012.08038.x
Dunn J, Sheving L, Millet P. Circadian variation in stress-evoked increases in plasma corticosterone. Am J Physiol. 1972;3:402.
Fonken LK, Weber MD, Daut RA, Kitt MM, Frank MG, Watkins LR, et al. Stress-induced neuroinflammatory priming is time of day dependent. Psychoneuroendocrinology. 2016;66:82–90. https://doi.org/10.1016/j.psyneuen.2016.01.006
Friedman AK, Walsh JJ, Juarez B, Ku SM, Chaudhury D, Wang J, et al. Enhancing depression mechanisms in midbrain dopamine neurons achieves homeostatic resilience. Science. 2014;344:313–9. https://doi.org/10.1126/science.1249240
Walsh JJ, Friedman AK, Sun H, Heller EA, Ku SM, Juarez B, et al. Stress and CRF gate neural activation of BDNF in the mesolimbic reward pathway. Nat Neurosci. 2014;17:27–29. https://doi.org/10.1038/nn.3591
Wook Koo J, Labonte B, Engmann O, Calipari ES, Juarez B, Lorsch Z, et al. Essential role of mesolimbic brain-derived neurotrophic factor in chronic social stress-induced depressive behaviors. Biol Psychiatry. 2016;80:469–78. https://doi.org/10.1016/j.biopsych.2015.12.009
Zhong P, Liu X, Zhang Z, Hu Y, Liu SJ, Lezama-Ruiz M, et al. Cyclin-dependent kinase 5 in the ventral tegmental area regulates depression-related behaviors. J Neurosci. 2014;34:6352–66. https://doi.org/10.1523/JNEUROSCI.3673-13.2014
Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–66. https://doi.org/10.1146/annurev-neuro-061010-113641
Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14:609–25. https://doi.org/10.1038/nrn3381
Baik JH. Dopamine signaling in reward-related behaviors. Front Neural Circuits. 2013;7:152 https://doi.org/10.3389/fncir.2013.00152
Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci. 2012;15:816–8. https://doi.org/10.1038/nn.3100
Lobo MK III, Covington HE, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, et al. Cell type-–specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–90.
Francis TC, Chandra R, Friend DM, Finkel E, Dayrit G, Miranda J, et al. Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol Psychiatry. 2015;77:212–22. https://doi.org/10.1016/j.biopsych.2014.07.021
Soares-Cunha C, Coimbra B, David-Pereira A, Borges S, Pinto L, Costa P, et al. Activation of D2 dopamine receptor-expressing neurons in the nucleus accumbens increases motivation. Nat Commun. 2016;7:11829 https://doi.org/10.1038/ncomms11829
Calipari ES, Bagot RC, Purushothaman I, Davidson TJ, Yorgason JT, Pena CJ, et al. In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc Natl Acad Sci USA. 2016;113:2726–31. https://doi.org/10.1073/pnas.1521238113
Koo JW, Lobo MK, Chaudhury D, Labonte B, Friedman A, Heller E, et al. Loss of BDNF signaling in DIR-expressing NAc neurons enhances morphine reward by reducing GABA inhibition. Neuropsychopharmacology. 2014;39:2646–53. https://doi.org/10.1038/npp.2014.118
Lobo MK, Zaman S, Damez-Werno DM, Koo JW, Bagot RC, DiNieri JA, et al. DeltaFosB induction in striatal medium spiny neuron subtypes in response to chronic pharmacological, emotional, and optogenetic stimuli. J Neurosci. 2013;33:18381–95. https://doi.org/10.1523/JNEUROSCI.1875-13.2013
Moser MB, Moser EI, Forrest E, Andersen P, Morris RG. Spatial learning with a minislab in the dorsal hippocampus. Proc Natl Acad Sci USA. 1995;92:9697–701.
Goto Y, Grace AA. Limbic and cortical information processing in the nucleus accumbens. Trends Neurosci. 2008;31:552–8. https://doi.org/10.1016/j.tins.2008.08.002
Anacker C, Luna VM, Stevens GS, Millette A, Shores R, Jimenez JC, et al. Hippocampal neurogenesis confers stress resilience by inhibiting the ventral dentate gyrus. Nature. 2018;559:98–102. https://doi.org/10.1038/s41586-018-0262-4
McGirr A, Berlim MT, Bond DJ, Fleck MP, Yatham LN, Lam RW. A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychol Med. 2015;45:693–704. https://doi.org/10.1017/S0033291714001603
Liu X, Ramirez S, Pang PT, Puryear CB, Govindarajan A, Deisseroth K, et al. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature. 2012;484:381–U415. https://doi.org/10.1038/nature11028
Ramirez S, Liu X, Lin PA, Suh J, Pignatelli M, Redondo RL, et al. Creating a false memory in the hippocampus. Science. 2013;341:387–91. https://doi.org/10.1126/science.1239073
Covington HE 3rd, Lobo MK, Maze I, Vialou V, Hyman JM, Zaman S, et al. Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci. 2010;30:16082–90. https://doi.org/10.1523/JNEUROSCI.1731-10.2010
Treadway MT, Pizzagalli DA. Imaging the pathophysiology of major depressive disorder—from localist models to circuit-based analysis. Biol Mood Anxiety Disord. 2014;4:5 https://doi.org/10.1186/2045-5380-4-5
Vialou V, Bagot RC, Cahill ME, Ferguson D, Robison AJ, Dietz DM, et al. Prefrontal cortical circuit for depression- and anxiety-related behaviors mediated by cholecystokinin: role of DeltaFosB. J Neurosci. 2014;34:3878–87. https://doi.org/10.1523/JNEUROSCI.1787-13.2014
Perova Z, Delevich K, Li B. Depression of excitatory synapses onto parvalbumin interneurons in the medial prefrontal cortex in susceptibility to stress. J Neurosci. 2015;35:3201–6. https://doi.org/10.1523/JNEUROSCI.2670-14.2015
Challis C, Beck SG, Berton O. Optogenetic modulation of descending prefrontocortical inputs to the dorsal raphe bidirectionally bias socioaffective choices after social defeat. Front Behav Neurosci. 2014;8:43 https://doi.org/10.3389/fnbeh.2014.00043
Challis C, Boulden J, Veerakumar A, Espallergues J, Vassoler FM, Pierce RC, et al. Raphe GABAergic neurons mediate the acquisition of avoidance after social defeat. J Neurosci. 2013;33:13978–88. https://doi.org/10.1523/JNEUROSCI.2383-13.2013. 13988a
Miller OH, Bruns A, Ben Ammar I, Mueggler T, Hall BJ. Synaptic regulation of a thalamocortical circuit controls depression-related behavior. Cell Rep. 2017;20:1867–80. https://doi.org/10.1016/j.celrep.2017.08.002
Nair SG, Strand NS, Neumaier JF. DREADDing the lateral habenula: A review of methodological approaches for studying lateral habenula function. Brain Res. 2013;1511:93–101. https://doi.org/10.1016/j.brainres.2012.10.011
Han S, Yang SH, Kim JY, Mo S, Yang E, Song KM, et al. Down-regulation of cholinergic signaling in the habenula induces anhedonia-like behavior. Sci Rep. 2017;7:900 https://doi.org/10.1038/s41598-017-01088-6
Sachs BD, Ni JR, Caron MG. Brain 5-HT deficiency increases stress vulnerability and impairs antidepressant responses following psychosocial stress. Proc. Natl Acad Sci USA. 2015;112:2557–62. https://doi.org/10.1073/pnas.1416866112
Teissier A, Chemiakine A, Inbar B, Bagchi S, Ray RS, Palmiter RD, et al. Activity of raphé serotonergic neurons controls emotional behaviors. Cell Rep. 2015;13:1965–76. https://doi.org/10.1016/j.celrep.2015.10.061
Ohmura Y, Tanaka KF, Tsunematsu T, Yamanaka A, Yoshioka M. Optogenetic activation of serotonergic neurons enhances anxiety-like behaviour in mice. Int J Neuropsychopharmacol. 2014;17:1777–83. https://doi.org/10.1017/S1461145714000637
Urban DJ, Zhu H, Marcinkiewcz CA, Michaelides M, Oshibuchi H, Rhea D, et al. Elucidation of the behavioral program and neuronal network encoded by dorsal raphe serotonergic neurons. Neuropsychopharmacology. 2016;41:1404–15. https://doi.org/10.1038/npp.2015.293
Burnett CJ, Krashes MJ. Resolving behavioral output via chemogenetic designer receptors exclusively activated by designer drugs. J Neurosci. 2016;36:9268–82. https://doi.org/10.1523/JNEUROSCI.1333-16.2016
You I-J, Wright SR, Garcia-Garcia AL, Tapper AR, Gardner PD, Koob GF, et al. 5-HT(1A) autoreceptors in the dorsal raphe nucleus convey vulnerability to compulsive cocaine seeking. Neuropsychopharmacology. 2016;41:1210–22. https://doi.org/10.1038/npp.2015.268
Christoffel DJ, Golden SA, Walsh JJ, Guise KG, Heshmati M, Friedman AK, et al. Excitatory transmission at thalamo-striatal synapses mediates susceptibility to social stress. Nat Neurosci. 2015;18:962–4. https://doi.org/10.1038/nn.4034
Root DH, Melendez RI, Zaborszky L, Napier TC. The ventral pallidum: subregion-specific functional anatomy and roles in motivated behaviors. Prog Neurobiol. 2015;130:29–70. https://doi.org/10.1016/j.pneurobio.2015.03.005
Knowland D, Lilascharoen V, Pacia CP, Shin S, Wang EH, Lim BK. Distinct ventral pallidal neural populations mediate separate symptoms of depression. Cell. 2017;170:284–97 e218. https://doi.org/10.1016/j.cell.2017.06.015
Johnson SB, Emmons EB, Anderson RM, Glanz RM, Romig-Martin SA, Narayanan NS, et al. A basal forebrain site coordinates the modulation of endocrine and behavioral stress responses via divergent neural pathways. J Neurosci. 2016;36:8687–99. https://doi.org/10.1523/JNEUROSCI.1185-16.2016
Garris PA, Kilpatrick M, Bunin MA, Michael D, Walker QD, Wightman RM. Dissociation of dopamine release in the nucleus accumbens from intracranial self-stimulation. Nature. 1999;398:67–69. https://doi.org/10.1038/18019
Pisanello F, Mandelbaum G, Pisanello M, Oldenburg IA, Sileo, L, Markowitz JE, et al. Dynamic illumination of spatially restricted or large brain volumes via a single tapered optical fiber. Nat Neurosci. 2017. https://doi.org/10.1038/nn.4591
Raimondo JV, Kay L, Ellender TJ, Akerman CJ. Optogenetic silencing strategies differ in their effects on inhibitory synaptic transmission. Nat Neurosci. 2012;15:1102. https://doi.org/10.1038/nn.3143
Shabel SJ, Proulx CD, Piriz J, Malinow R. GABA/glutamate co-release controls habenula output and is modified by antidepressant treatment. Science. 2014;345:1494–8. https://doi.org/10.1126/science.1250469
McCall JG, Qazi R, Shin G, Li S, Ikram MH, Jang KI, et al. Preparation and implementation of optofluidic neural probes for in vivo wireless pharmacology and optogenetics. Nat Protoc. 2017;12:219–37. https://doi.org/10.1038/nprot.2016.155
Miyashita T, Shao YR, Chung J, Pourzia O, Feldman DE. Long-term channelrhodopsin-2 (ChR2) expression can induce abnormal axonal morphology and targeting in cerebral cortex. Front Neural Circuits. 2013;7:8 https://doi.org/10.3389/fncir.2013.00008
Christie IN, Wells JA, Southern P, Marina N, Kasparov S, Gourine AV, et al. fMRI response to blue light delivery in the naive brain: implications for combined optogenetic fMRI studies. Neuroimage. 2013;66:634–41. https://doi.org/10.1016/j.neuroimage.2012.10.074
MacLaren DA, Browne RW, Shaw JK, Krishnan Radhakrishnan S, Khare P, Espana RA, et al. Clozapine N-oxide administration produces behavioral effects in long-evans rats: implications for designing DREADD experiments. eNeuro. 2016;3. https://doi.org/10.1523/ENEURO.0219-16.2016
Gomez JL, Bonaventura J, Lesniak W, Mathews W, Sysa-Shah P, Rodriguez LA, et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science. 2017;357:503–7.
Roth BL. DREADDs for neuroscientists. Neuron. 2016;89:683–94. https://doi.org/10.1016/j.neuron.2016.01.040
Thompson KJ, Khajehali E, Bradley SJ, Navarrete JS, Huang XP, Slocum S, et al. DREADD agonist 21 is an effective agonist for muscarinic-based DREADDs in vitro and in vivo. ACS Pharmacol Transl Sci. 2018;1:61–72. https://doi.org/10.1021/acsptsci.8b00012
Johansen JP, Hamanaka H, Monfils MH, Behnia R, Deisseroth K, Blair HT, et al. Optical activation of lateral amygdala pyramidal cells instructs associative fear learning. Proc Natl Acad Sci USA. 2010;107:12692–7. https://doi.org/10.1073/pnas.1002418107
Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, et al. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–62. https://doi.org/10.1038/nature09820
Wiegert JS, Mahn M, Prigge M, Printz Y, Yizhar O. Silencing neurons: tools, applications, and experimental constraints. Neuron. 2017;95:504–29. https://doi.org/10.1016/j.neuron.2017.06.050
Yoshihara M, Yoshihara M. ‘Necessary and sufficient’ in biology is not necessarily necessary—confusions and erroneous conclusions resulting from misapplied logic in the field of biology, especially neuroscience. J Neurogenet. 2018;32:53–64. https://doi.org/10.1080/01677063.2018.1468443
Resendez SL, Stuber GD. In vivo calcium imaging to illuminate neurocircuit activity dynamics underlying naturalistic behavior. Neuropsychopharmacology. 2015;40:238–9. https://doi.org/10.1038/npp.2014.206
Francis TC, Chandra R, Gaynor A, Konkalmatt P, Metzbower SR, Evans B, et al. Molecular basis of dendritic atrophy and activity in stress susceptibility. Mol Psychiatry. 2017;22:1512–9. https://doi.org/10.1038/mp.2017.178
Shin S, Pribiag H, Lilascharoen V, Knowland D, Wang XY, Lim BK. Drd3 signaling in the lateral septum mediates early life stress-induced social dysfunction. Neuron. 2018;97:195–208.e196. https://doi.org/10.1016/j.neuron.2017.11.040
Michaelides M, Anderson SAR, Ananth M, Smirnov D, Thanos PK, Neumaier JF, et al. Whole-brain circuit dissection in free-moving animals reveals cell-specific mesocorticolimbic networks. J Clin Investig. 2013;123:5342–50. https://doi.org/10.1172/JCI72117
Pettorruso M, Spagnolo PA, Leggio L, Janiri L, Di Giannantonio M, Gallimberti L, et al. Repetitive transcranial magnetic stimulation of the left dorsolateral prefrontal cortex may improve symptoms of anhedonia in individuals with cocaine use disorder: a pilot study. Brain Stimul. 2018;11:1195–7. https://doi.org/10.1016/j.brs.2018.06.001
Clair AH, Haynes W, Mallet L. Recent advances in deep brain stimulation in psychiatric disorders. F1000Res. 2018;7. https://doi.org/10.12688/f1000research.14187.1
Bagot RC, Cates HM, Purushothaman I, Lorsch ZS, Walker DM, Wang J, et al. Circuit-wide transcriptional profiling reveals brain region-specific gene networks regulating depression susceptibility. Neuron. 2016;90:969–83. https://doi.org/10.1016/j.neuron.2016.04.015
Labonte B, Engmann O, Purushothaman I, Menard C, Wang J, Tan C, et al. Sex-specific transcriptional signatures in human depression. Nat Med. 2017;23:1102–11. https://doi.org/10.1038/nm.4386
Whiteford HA, Degenhardt L, Rehm J, Baxter AJ, Ferrari AJ, Erskine HE, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet. 2013;382:1575–86. https://doi.org/10.1016/s0140-6736(13)61611-6
Berton O, McClung CA, DiLeone RJ, Vaishnav Krishnan WR, Russo SJ, Graham D, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:759.
Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–8. https://doi.org/10.1038/nn1525
Gunaydin LA, Yizhar O, Berndt A, Sohal VS, Deisseroth K, Hegemann P. Ultrafast optogenetic control. Nat Neurosci. 2010;13:387–92. https://doi.org/10.1038/nn.2495
Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–9. https://doi.org/10.1038/nature05744
Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324:354–9. https://doi.org/10.1126/science.1167093
Chuong AS, Miri ML, Busskamp V, Matthews GA, Acker LC, Sorensen AT, et al. Noninvasive optical inhibition with a red-shifted microbial rhodopsin. Nat Neurosci. 2014;17:1123–9. https://doi.org/10.1038/nn.3752
Chow BY, Han X, Dobry AS, Qian XF, Chuong AS, Li MJ, et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010;463:98–102. https://doi.org/10.1038/nature08652
Han X, Chow BY, Zhou H, Klapoetke NC, Chuong A, Rajimehr R, et al. A high-light sensitivity optical neural silencer: development and application to optogenetic control of non-human primate cortex. Front Syst Neurosci. 2011;5:18 https://doi.org/10.3389/fnsys.2011.00018
Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, et al. Independent optical excitation of distinct neural populations. Nat Methods. 2014;11:338–46. https://doi.org/10.1038/nmeth.2836
Berndt A, Lee SY, Ramakrishnan C, Deisseroth K. Structure-guided transformation of channelrhodopsin into a light-activated chloride channel. Science. 2014;344:420–4. https://doi.org/10.1126/science.1252367
Berndt A, Yizhar O, Gunaydin LA, Hegemann P, Deisseroth K. Bi-stable neural state switches. Nat Neurosci. 2009;12:229–34. https://doi.org/10.1038/nn.2247
Jessie Muir, Zachary S Lorsch, Charu Ramakrishnan, Karl Deisseroth, Eric J Nestler, Erin S Calipari, et al. In Vivo Fiber Photometry Reveals Signature of Future Stress Susceptibility in Nucleus Accumbens. Neuropsychopharmacology 2017;43:255–263.
Acknowledgements
This work is supported by a CIHR grant to RCB.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Muir, J., Lopez, J. & Bagot, R.C. Wiring the depressed brain: optogenetic and chemogenetic circuit interrogation in animal models of depression. Neuropsychopharmacol. 44, 1013–1026 (2019). https://doi.org/10.1038/s41386-018-0291-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-018-0291-6
This article is cited by
-
Lateral septum adenosine A2A receptors control stress-induced depressive-like behaviors via signaling to the hypothalamus and habenula
Nature Communications (2023)
-
Multifocal skull-compensated transcranial focused ultrasound system for neuromodulation applications based on acoustic holography
Microsystems & Nanoengineering (2023)
-
Ethics and regulation of neuronal optogenetics in the European Union
Pflügers Archiv - European Journal of Physiology (2023)
-
Update on GPCR-based targets for the development of novel antidepressants
Molecular Psychiatry (2022)
-
Nucleus reuniens inactivation reverses stress-induced hypodopaminergic state and altered hippocampal-accumbens synaptic plasticity
Neuropsychopharmacology (2022)
