
Abstract
Altered neuroimmune response and oxidative stress have both been implicated in the pathophysiology of schizophrenia. While preclinical studies have proposed several pathways regarding potential interactions between oxidative stress and neuroimmune imbalance in the development of psychosis, the molecular mechanisms underlying this interaction are not yet understood. To date, no study has investigated this link in vivo in the human brain. We conducted the first in vivo study linking translocator protein 18 kDa (TSPO) expression and glutathione (a major brain antioxidant and a marker for redox status) in the medial prefrontal cortex (mPFC) of a relatively large sample of participants (N = 48) including 27 antipsychotic-naïve individuals at clinical high risk for psychosis and 21 matched healthy volunteers using high-resolution PET with TSPO radioligand, [18F]FEPPA, and 3T proton magnetic resonance spectroscopy (1H MRS). The omnibus model (including TSPO genotype as covariate) was significant (F(4, 43) = 10.01, p < 0.001), with a significant group interaction (t = −2.10, p = 0.04), suggesting a different relation between [18F]FEPPA VT and glutathione in each clinical group. In healthy volunteers, but not in individuals at clinical high risk for psychosis, we found a significant negative association between glutathione levels and [18F]FEPPA VT (r = −0.60, p = 0.006). We observed no significant group differences with respect to [18F]FEPPA VT or glutathione levels. These findings suggest an abnormal interaction between TSPO expression and redox status in the clinical high risk states for psychosis.
Similar content being viewed by others

Introduction
Schizophrenia is a chronic mental disorder that is heterogeneous in its etiology, involving alterations in diverse systems including genetic, neurohormonal, and immunological. The neuroimmune alterations and oxidative stress have been implicated in the disease [1], and potential biomarkers have been developed to examine these processes in clinical studies [2].
Glial cells have a major role in the neuroimmune response, in particular microglia, which are the first line of defense against neural tissue damage. In response to brain injury, the microglia become activated and change their morphology to an amoeboid form that is capable of phagocytosis. The activation of the microglia is characterized by an elevated expression of the translocation protein 18 kDa (TSPO) in their mitochondria, thus making TSPO a suitable marker for microglial activation. Several lines of evidence from epidemiology [3], genetic [4, 5], and preclinical studies [6] support the role for neuroinflammation in neuropathology of schizophrenia. Results of postmortem studies on brain immune cell activation in schizophrenia are, however, inconclusive due to the wide variability in results and confounders associated with death [7]. The TSPO expression can be measured in vivo by using the positron emission tomography (PET) and radiotracers that target TSPO [8]. To date, seven TSPO PET studies in schizophrenia have shown either no difference in the TSPO expression when using second-generation radioligands [9,10,11,12,13,14] or even a significant decrease [15], while four others have reported elevated TSPO expression when using the first-generation radioligand [11C]PK11195 [16,17,18] or distribution volume ratio [19]. Of note, [11C]PK11195 has significant methodological limitations [20], which significantly limit its interpretability. Similarly, using distribution volume ratio as the outcome measure for TSPO radioligands is controversial and limits interpretation of the results [21].
Oxidative stress and redox imbalance, in particular glutathione metabolism dysregulation, have as well been implicated in the pathophysiology of schizophrenia [22]. Glutathione is the most abundant intracellular antioxidant [23]. Preclinical studies show that glutathione deficits in the developing brain results in schizophrenia-like behavioral and cognitive deficits [24]. Moreover, genetic studies indicate an association between the allelic variants of glutathione biosynthesis enzyme and schizophrenia [25, 26]. Supporting this, a postmortem brain study has reported decreased levels of glutathione in chronic patients with schizophrenia [27]. The findings of in vivo studies examining cerebral glutathione in schizophrenia are, however, varied with three showing no significant alteration [26, 28, 29], one indicating significant reduction [30], and one indicating significant elevation [31]. The only study published investigating cerebral glutathione levels in the clinical high risk (CHR) state that putatively precedes psychosis reported no significant difference in the level of glutathione in the prefrontal cortex between CHR and matched healthy volunteers [32].
Oxidative stress and neuroinflammation are intricately linked, and several molecular mediators are shared between oxidative stress response and the inflammatory pathways [33,34,35]. Further, microglia in the brain are a major source of oxidative stress [36], while oxidative stress and glutathione depletion can lead to microglia activation [37]. Several mediating factors have been identified that link oxidative stress and the neuroinflammatory pathways, including Receptor for Advanced Glycation End-product (RAGE) [38]. Further, TSPO is localized in the mitochondria, hence directly related to energy metabolism and possible redox imbalance [39]. While several preclinical studies have proposed several pathways regarding potential interactions between oxidative stress and neuroimmune imbalance in the development of psychosis [37, 40], to date no study has examined these two systems in vivo in the human brain.
In the current study we have, for the first time, investigated the link between TSPO expression and glutathione levels in vivo in a relatively large sample (N = 48) of CHR and healthy volunteers using 3T proton magnetic resonance spectroscopy (1H MRS) and TSPO PET high-resolution research tomography (HRRT). Based on existing preclinical evidence on the alternative molecular immune and redox imbalance pathways, we hypothesized that there would be an inverse association between TSPO expression and prefrontal glutathione in healthy volunteers. However, in CHR, due to potential abnormalities in molecular linkage between the neuroimmune and oxidative stress responses, this negative association may be disrupted.
Methods
Participants
Twenty-seven CHR and 21 healthy volunteers were enrolled in this study. Most of the individuals in the CHR group were antipsychotic naïve (n = 23). Sixteen CHR participants and ten healthy volunteers were also included in our previous cohort [10].
To be eligible, the CHR individuals had to meet the following criteria: fulfillment of diagnostic criteria for prodromal syndrome as per the Criteria of Prodromal Syndromes [41] with no current axis I disorders, as determined by the Structured Clinical Interview for DSM-IV [42]. Healthy volunteers did not have any history of psychiatric illness, psychoactive drug use, and/or any first-degree relative having a major mental illness. Participants were excluded for any of the following: clinically significant medical illness, current diagnosis of substance abuse or a positive urine-drug screen, pregnancy or current breastfeeding, and the presence of metal implants precluding an MRI scan. In CHR, the clinical status and the severity of symptoms (e.g., psychosis-risk symptoms) were assessed with the structured interview for psychosis-risk syndromes, scale of psychosis-risk symptoms (SOPS) [41], calgary depression scale (CDS), Snaith–Hamilton pleasure scale, global assessment of functioning scale (GAF), state-trait anxiety inventory, and apathy evaluation scale (AES). Neurocognitive performance was assessed using the repeatable battery for the assessment of neuropsychological status (RBANS) [43, 44].
PET, and structural MRI data acquisition and analysis
PET and MRI data acquisition have been described in detail elsewhere and are summarized below [11, 12]. The T1-weighted and proton density-weighted (PD) brain MRI scan was obtained for each subject using a 3T MR-750 scanner (General Electric Medical Systems). All [18F]FEPPA scans were performed using a high-resolution neuro-PET camera system (HRRT, Siemens Molecular Imaging, Knoxville, TN, USA) for 125 min, following an intravenous bolus injection of 186.38 ± 9.41 MBq of [18F]FEPPA. The arterial blood samples were collected automatically using an automatic blood sampling system (Model PBS-101, Veenstra Instrument, Joure, Netherland) for the first 22.5 min after radioligand injection at a rate of 2.5 mL/min and manually at −5, 2.5, 7, 12, 15, 20, 30, 45, 60, 90, and 120 min to measure the radioactivity in blood and determine the relative proportion of radiolabelled metabolites. The dispersion-and metabolite-corrected plasma input function was generated, as previously described [12].
PET image processing and calculation of total distribution volumes (V T)
The time-activity curves were extracted for medial prefrontal cortex (mPFC) using validated in-house imaging pipeline, ROMI [45]. mPFC was mainly composed of Broadmann areas 9, 10, and 32. The region of interest was delineated using individual PD MRI. The kinetic parameters of [18F]FEPPA were derived from the time-activity curves using the two-tissue compartment model (2TCM) and plasma input function to obtain the total distribution volume (VT) for mPFC, which has been validated for [18F]FEPPA, quantified, and described elsewhere [46].
TSPO genotyping
We genotyped the participants for their TSPO rs6971 polymorphism, and based on the results we categorized the subjects into high-, mixed-, or low-affinity binders, as described elsewhere [47].
Glutathione data acquisition and analysis
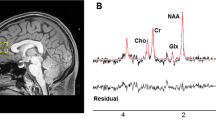
In vivo 1H-MRS measurements were acquired on a 3T MR-750 scanner (GEHealthcare, Wisconsin, USA General Electric Medical Systems), equipped with an eight-channel head coil from a 24 cc volume of interest (20 × 40 × 30 mm3), positioned in the mPFC (Fig. 1). To minimize the head motion, each subject was positioned at the center of the head coil with a tape strapped across the forehead and a restraint soft padding around the head. Magnet homogeneity was adjusted using the manufacture automated shimming routine. Typical unsuppressed water resonance frequency width at half maximum intensity was between 8–10 Hz. For all participants, the time gap between 1H-MRS and [18F]FEPPA PET scans was less than 4 weeks. The glutathione spectra was acquired using the interleaved J-difference editing approach, MEshcher-Garwood Point RESolved Spectroscopy (MEGA-PRESS), and 22 whereby alternating “on” and “off” editing radio frequency (RF) pulses are applied to the α proton of the cysteine moiety. The RF pulses suppress the α-CH2 resonance of cysteinyl moeity of glutathione at 4.5 p.p.m. on one acquisition and at 7.5 p.p.m. on the alternate scan, resulting in the detection of the cysteinyl β-CH2 glutathione signal. The acquisition parameters for the measurements are as follows: echo time (TE) = 68 ms, repetition time (TR) = 1500 ms, band width = 5 kHz, number of excitations = 512, and data points = 4096.

Volume of interest (20 × 40 × 30 mm3) location used in 1H MRS scan of the medial prefrontal cortex for quantification of glutathione
After glutathione acquisition, the two free-induction-decay signals (“on” and “off”) were frequency shift corrected and subtracted in the time domain. A 3 Hz, the Gaussian filter was applied to the difference data, which was then Fourier transformed. The Data were zero-filled to 8192 points prior to Fourier transformation. Areas of glutathione resonance at 2.9 p.p.m. were obtained using the optimized Levenberg–Marquardt nonlinear least square-IDL-optimization routine. A combination of Lorentzian and Gaussian functions, aka Voigt, was used to model the spectral line shape using XSOS software [48, 49]. The ratio of glutathione and unsuppressed water peak (GSH/H2O) areas were reported. Details of 1H MRS acquisition are provided in the supplementary methods section and Fig. 2.

(A): Typical fitting results from a patient, a MRS sub-spectra acquired for the “on” condition (see text), b “off” condition, c subtracted to obtain clean GSH resonance at 2.9 p.p.m., d frequency domain model-fitting of edited GSH resonance only, and e residual of the difference between c and d. (B): A representative single-subject-GSH-edited spectrum clearly showing the 2.95 p.p.m. GSH resonance
Statistical analysis
We used χ2-tests to evaluate differences in the categorical variables (e.g., gender and TSPO genotype) between CHR and healthy volunteers. Differences in continuous variables (e.g., age) were assessed using F-test. We utilized a general linear model to assess the relation between [18F]FEPPA VT and glutathione levels in mPFC, where [18F]FEPPA VT was the dependent variable, glutathione levels and the group were predictors and entered into the model as main effect and interaction. TSPO genotype was entered as a covariate. Pearson’s partial correlations were employed post hoc to explore the association between glutathione and [18F]FEPPA VT in each diagnostic group separately, if the omnibus test and interaction was significant in the overall model. Moreover, we explored the association between residuals of the general linear model and the clinical and neuropsychological measures. Statistical analyses were performed using SPSS (SPSS, Chicago, IL, USA). The significance level was set at 0.05.
Results
The characteristics of the participants are presented in Table 1. There were no significant differences in gender, TSPO rs6971 genotype, and PET parameters between CHR and healthy volunteers. The CHR individuals were significantly younger than the healthy volunteers (F = 13.80, p = 0.001), and all participants had a clean urine-drug screen.
Differences in [18F]FEPPA V T and glutathione level in mPFC between CHR and healthy volunteers
Consistent with previous reports, we did not find a significant group effect on [18F]FEPPA VT in mPFC (F(1, 45) = 0.01, p = 0.92). The group difference remained non-significant after controlling for tobacco use (F(1, 44) = 0.12, p = 0.73) or history of cannabis use (F(1, 44) = 0.02, p = 0.89). This finding was not altered after controlling for age, or after excluding the CHR individuals who were on antipsychotics (n = 4).
Tissue composition within the 24 cc MRS voxel consisted of 63.09 ± 3.56% gray matter, 19.86 ± 3.10% white matter, and 17.03 ± 4.64% cerebrospinal fluid (CSF) in the mPFC. There were no significant differences of tissue composition (gray matter, white matter, and CSF fractions) between CHR and healthy volunteers (Supplementary table 1). As previously reported [32], we found no significant effect of the clinical group on the glutathione level in mPFC (F(1, 46) = 0.86, p = 0.36). The group difference remained non-significant after controlling for tobacco use (F(1,45) = 0.51, p = 0.48), history of cannabis use (F(1,45) = 1.9, p = 0.18), age (F(1,45) = 0.82, p = 0.37), or after excluding the CHR individuals who were on antipsychotics (n = 4, F(1,42) = 0.85, p = 0.36).
Association between glutathione level and [18F]FEPPA V T in mPFC
The omnibus model (including TSPO genotype as covariate) was significant (F(4, 43) = 10.01, p < 0.001) with a significant group interaction (t = −2.10, p = 0.04) suggesting a different relation between [18F]FEPPA VT and glutathione in each clinical group. The model remained significant after controlling for tobacco use (F(5, 42) = 8.18, p < 0.001) or history of cannabis use (F(5, 42) = 7.9, p < 0.001), age (F(5, 42) = 7.82, p < 0.001), or after excluding CHR individuals who converted to psychosis (n = 5, F(4, 38) = 10.19, p < 0.001). In particular, in healthy volunteers, but not in CHR (r = −0.07, p = 0.73), we found a significant negative association between glutathione levels and [18F]FEPPA VT in mPFC (r = −0.60, p = 0.006)(Fig. 3). The results remained after excluding the CHR participants who were on antipsychotics (n = 4).

Association between [18F]FEPPA VT and glutathione levels in mPFC (controlling for TSPO genotype). The graph presents the significant interaction between group × glutathione levels on [18F]FEPPA VT (F(4, 43) = 10.01, p < 0.001; t = −2.10, p = 0.04). The significant correlation in healthy volunteers remains after excluding healthy volunteer with the highest glutathione value (r = −0.51, p = 0.025)
The correlation of residuals of the general linear model and anhedonia, as measured by Snaith–Hamilton Pleasure Scale, in CHR showed a significant positive association (r = 0.44, p = 0.02) (Supplementary Figure 1). We also found a significant correlation between the residuals and the state-anxiety subscore of the state-trait anxiety inventory (r = 0.4, p = 0.04); however, this correlation did not survive the correction for the number of subscores. There were no other significant associations between these residuals of the association and severity of psychosis-risk symptoms, as measured by the SOPS; depression, as measured by CDS; cognitive function, as measured by RBANS; apathy, as measured by AES; or global functioning, as measured by GAF.
Discussion
In this study, for the first time and in a relatively large sample (N = 48) of CHR and healthy volunteers, we showed a significant interaction between the diagnostic groups when assessing the relation between glutathione levels and TSPO expression in vivo in brain, suggesting a different relation between the neuroimmune response and redox status in each group. In particular, in healthy volunteers, but not in CHR, there was a significant negative association between the glutathione levels and [18F]FEPPA VT in mPFC.
TSPO expression and glutathione level in mPFC of CHR
The lack of group difference in [18F]FEPPA VT in mPFC is in line with six previous TSPO PET studies that have examined TSPO expression in the psychosis spectrum [10, 19], including first-episode psychosis/recent-onset schizophrenia [11, 14], chronic schizophrenia [12, 13, 19], or CHR using second-generation radioligands and the validated two-tissue compartment model for TSPO radioligands quantification [10, 19]. A recent study using the second-generation TSPO radioligand reported a significant reduction in TSPO expression in the frontal cortex, temporal cortex, and hippocampus of drug-naïve first-episode psychosis, as compared to matched healthy volunteers [15]. Our results are in contrast with three older studies using [11C]PK11195 that have reported higher TSPO expression in treated schizophrenia [16,17,18]. The interpretation of the latter studies is, however, limited due to the methodological limitations of [11C]PK11195 including low brain penetration, low affinity for TSPO, and high plasma protein binding [20].
We also did not observe any significant group effect on the glutathione level in mPFC. In the present study, we examined mPFC due to its well-known role in the pathophysiology of psychosis and the previous evidence on the alteration of glutathione levels in this region [30]. Our findings are in line with a recent study on cerebral glutathione in mPFC of CHR [32] and three 1H MRS studies that examined glutathione in mPFC during early psychosis [26] or chronic schizophrenia [7, 28]. However, our findings are in contrast with a 1H MRS study by Do and colleagues reporting a significant reduction in the glutathione level in chronic schizophrenia [30]. Additionally, a more recent study by Wood and colleagues reported a significant elevation in the glutathione level in mPFC in patients with first-episode psychosis [31]. These discrepancies may have resulted from the difference in the scanner used (3 T vs. 1.5 T), duration of the illness (CHR vs. first-episode psychosis vs. chronic schizophrenia), and medication status of patients (drug-naïve vs. medicated).
Association between TSPO expression and glutathione level in mPFC
The significant negative association observed in this study between TSPO expression and glutathione level in mPFC in healthy volunteers is consistent with the preclinical literature suggesting that higher levels of microglial activation are associated with higher oxidative stress [37], which may be particularly relevant (i.e., absent) in psychosis and psychosis risk [1]. Oxidative stress is the result of an imbalance between pro-oxidants (e.g., reactive oxygen species) and antioxidants, such as glutathione. This redox imbalance can induce immune activation by triggering the inflammatory gene-regulating factor (e.g., nuclear factor ĸB, NF-ĸB) [37]. Supporting this, the antioxidant treatment with N-acetyl cysteine can decrease the neuropathological effects of maternal lipopolysaccharide injection on offsprings [50]. Inversely, the activation of inflammatory factors such as NF-ĸB can increase oxidative stress by disrupting the antioxidant system [35]. More recently, Do and colleagues reported the RAGE to be the mediatory mechanism that connects oxidative stress and neuroinflammatory pathways, through matrix metalloproteinase 9 [38, 51]. Therefore, barring the translational leap between these preclinical studies and our clinical study under normal conditions (e.g., in the case of healthy volunteers in this study), we expect an inverse association between glial activation and antioxidant levels. The lack of association between TSPO expression and glutathione level in CHR may reflect the potential deficits in molecular pathways that link the neuroimmune response and oxidative stress, for instance nuclear factor-E2-related factor-2 and/or NF-ĸB pathways [52, 53], or RAGE. Future preclinical studies may now explore the involvement of TSPO and/or glutathione in these molecular pathways to mechanistically understand their involvement in these processes.
Further, elevated expression of TSPO has been reported in immune cells with high quantities of pro-oxidants level, such as phagocytes, astrocytes, and microglia [39], which in some cases (i.e., stroke) can contribute to the TSPO signal quantified with PET [54]. It has been suggested that the increased TSPO is involved in protecting the immune cells against oxidative damage by preserving the mitochondrial function [55]. The inverse association between TSPO expression and glutathione level in our healthy volunteers can be attributed to this role of TSPO. The lack of such an association in CHR may be due to either a dysregulation between TSPO and the antioxidant system or the reported numerically decreased TSPO observed in CHR [10]. This study is the first attempt to better understand the underlying molecular pathways involved in the dysregulation of TSPO expression and glutathione redox imbalance in those at risk for psychosis.
The results of this study should be interpreted considering the following limitations. First, although the mean age in each group differed by only 2 years, our healthy volunteers were significantly older than our individuals with CHR. However, there is no evidence for a significant effect of age on either TSPO expression [56] or glutathione levels [57]. After controlling for age, we did not observe any significant changes in [18F]FEPPA VT, glutathione level, or in the interaction between these two variables. Second, the participants underwent PET and 1H MRS scans on two separate days (~16 days apart); however, both [18F]FEPPA PET scans and glutathione 1H MRS scans were shown to have satisfactory test-retest reliability. Third, the relatively large ROI used for 1H MRS in mPFC also included the surrounding brain regions such as dorsal part of anterior cingulate cortex (figure 1), thus involving a somewhat bigger area than the one tested with PET. Fourth, the 1H MRS data was only available for the presented volume of interest (figure 1) and the associations between glutathione level and TSPO expression may not be generalizable to other brain regions. Fifth, it is noteworthy that TSPO is not a specific marker for microglia and it is also expressed by other cells including astrocytes, neurons, and vascular endothelial cells. However, this does not affect the overall conclusion of this study [58]. Sixth, in this study we did not find any significant effect on either glutathione or glial activation, but in their interaction. Because regression analyses cannot be used to infer causality, caution should be taken when interpreting the present results until preclinical studies help us understand the mechanisms involved in these associations. Finally, CHR is a heterogeneous group and in the sample included in the current study, we observed a conversion to psychosis rate of ~19%, however, this would not affect our findings, given that, excluding those that converted did not change the present results.
Conclusions
This study suggests an abnormal interaction between TSPO expression and oxidative stress in CHR states for psychosis.
References
Barron H, Hafizi S, Andreazza AC, Mizrahi R. Neuroinflammation and oxidative stress in psychosis and psychosis risk. Int J Mol Sci. 2017;18:651.
Goff DC, Romero K, Paul J, Perez-Rodriguez MM, Crandall D, Potkin SG. Biomarkers for drug development in early psychosis: current issues and promising directions. Eur Neuropsychopharmacol. 2016;26:923–37.
Brown AS. Further evidence of infectious insults in the pathogenesis and pathophysiology of schizophrenia. Am J Psychiatry. 2011;168:764–6.
Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–83.
Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–7.
Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–702.
Trepanier MO, Hopperton KE, Mizrahi R, Mechawar N, Bazinet RP. Postmortem evidence of cerebral inflammation in schizophrenia: a systematic review. Mol Psychiatry. 2016;21:1009–26.
Venneti S, Wiley CA, Kofler J. Imaging microglial activation during neuroinflammation and Alzheimer’s disease. J NeuroImmune Pharmacol: Off J Soc NeuroImmune Pharmacol. 2009;4:227–43.
Di Biase M, Zalesky A, O’keefe G, Laskaris L, Baune B, Weickert C, et al. PET imaging of putative microglial activation in individuals at ultra-high risk for psychosis, recently diagnosed and chronically ill with schizophrenia. Transl Psychiatry. 2017;7:e1225.
Hafizi S, Da Silva T, Gerritsen C, Kiang M, Bagby R, Prce I, et al. Imaging microglial activation in individuals at clinical high risk for psychosis: an in-vivo PET study with [18F]FEPPA. Neuropsychopharmacology. 2017a;42:2474–81.
Hafizi S, Tseng HH, Rao N, Selvanathan T, Kenk M, Bazinet RP, et al. Imaging microglial activation in untreated first-episode psychosis: a PET study with [18F]FEPPA. Am J Psychiatry. 2017b;174:118–24.
Kenk M, Selvanathan T, Rao N, Suridjan I, Rusjan P, Remington G, et al. Imaging neuroinflammation in gray and white matter in schizophrenia: an in-vivo PET study with [18F]-FEPPA. Schizophr Bull. 2015;41:85–93.
Takano A, Arakawa R, Ito H, Tateno A, Takahashi H, Matsumoto R, et al. Peripheral benzodiazepine receptors in patients with chronic schizophrenia: a PET study with [11C]DAA1106. Int J Neuropsychopharmacol. 2010;13:943–50.
Coughlin J, Wang Y, Ambinder E, Ward R, Minn I, Vranesic M, et al. In vivo markers of inflammatory response in recent-onset schizophrenia: a combined study using [11C] DPA-713 PET and analysis of CSF and plasma. Transl Psychiatry. 2016;6:e777.
Collste K, Plavén-Sigray P, Fatouros-Bergman H, Victorsson P, Schain M, Forsberg A, et al. Lower levels of the glial cell marker TSPO in drug-naive first-episode psychosis patients as measured using PET and [11C] PBR28. Mol Psychiatry. 2017;22:850–6.
Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med. 2009;50:1801–7.
Holmes SE, Hinz R, Drake RJ, Gregory CJ, Conen S, Matthews JC, et al. In vivo imaging of brain microglial activity in antipsychotic-free and medicated schizophrenia: a [11C](R)-PK11195 positron emission tomography study. Mol Psychiatry. 2016;21:1672–9.
van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64:820–2.
Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR, et al. Microglial activity in people at ultra high risk of psychosis and in schizophrenia: an [11C]PBR28 PET brain imaging study. Am J Psychiatry. 2015;173:44–52.
Wilson AA, Garcia A, Parkes J, McCormick P, Stephenson KA, Houle S, et al. Radiosynthesis and initial evaluation of [18F]-FEPPA for PET imaging of peripheral benzodiazepine receptors. Nucl Med Biol. 2008;35:305–14.
Narendran R, Frankle WG. Comment on analyses and conclusions of “microglial activity in people at ultra high risk of psychosis and in schizophrenia: an [11C] PBR28 PET brain imaging study”. Am J Psychiatry. 2016;173:536–7.
Yao JK, Leonard S, Reddy R. Altered glutathione redox state in schizophrenia. Dis Markers. 2006;22:83–93.
Coppola S, Ghibelli L. GSH extrusion and the mitochondrial pathway of apoptotic signalling. Biochemical Society Transactions. Portland Press Limited; 2000;28:56–61. https://doi.org/10.1042/bst0280056
Kulak A, Cuenod M, Do KQ. Behavioral phenotyping of glutathione-deficient mice: relevance to schizophrenia and bipolar disorder. Behav Brain Res. 2012;226:563–70.
Gysin R, Kraftsik R, Sandell J, Bovet P, Chappuis C, Conus P, et al. Impaired glutathione synthesis in schizophrenia: convergent genetic and functional evidence. Proc Natl Acad Sci USA. 2007;104:16621–6.
Xin L, Mekle R, Fournier M, Baumann PS, Ferrari C, Alameda L, et al. Genetic polymorphism associated prefrontal glutathione and its coupling with brain glutamate and peripheral redox status in early psychosis. Schizophr Bull. 2016;42:1185–96.
Gawryluk JW, Wang J-F, Andreazza AC, Shao L, Young LT. Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int J Neuropsychopharmacol. 2011;14:123–30.
Matsuzawa D, Obata T, Shirayama Y, Nonaka H, Kanazawa Y, Yoshitome E, et al. Negative correlation between brain glutathione level and negative symptoms in schizophrenia: a 3T 1 H-MRS study. PLoS ONE. 2008;3:e1944.
Terpstra M, Vaughan T, Ugurbil K, Lim KO, Schulz SC, Gruetter R. Validation of glutathione quantitation from STEAM spectra against edited 1H NMR spectroscopy at 4T: application to schizophrenia. Magn Reson Mater Phys Biol Med. 2005;18:276–82.
Do K, Trabesinger A, Kirsten‐Krüger M, Lauer C, Dydak U, Hell D, et al. Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci. 2000;12:3721–8.
Wood SJ, Berger GE, Wellard RM, Proffitt T-M, McConchie M, Berk M, et al. Medial temporal lobe glutathione concentration in first episode psychosis: a 1 H-MRS investigation. Neurobiol Dis. 2009;33:354–7.
Da Silva T, Hafizi S, Andreazza AC, Kiang M, Bagby RM, Navas E, et al. Glutathione–the major redox regulator-in the prefrontal cortex of individuals at clinical high risk for psychosis. Int J Neuropsychopharmacol. 2017;21:311–8.
Bitanihirwe BK, Woo TU. Oxidative stress in schizophrenia: an integrated approach. Neurosci Biobehav Rev. 2011;35:878–93.
Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev. 2004;18:2195–224.
Liu G-H, Qu J, Shen X. NF-κB/p65antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta. 2008;1783:713–27.
Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69.
Hardingham GE, Do KQ. Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat Rev Neurosci. 2016;17:125–34.
Do KQ, Dwir D, Cabungcal J, Tirouvanziam R, Cuenod M, editors. Receptor for advanced glycation end-product (rage) as linking mechanism between neuroinflammation and oxidative stress. Schizophrenia Bulletin. Great Clarendon Street, Oxford, OX2. 6DP, UK: Oxford University Press; 2015.
Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, et al. Translocator protein (18 kDa)(TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9:971–88.
Miller BJ, Goldsmith DR. Towards an immunophenotype of schizophrenia: progress, potential mechanisms, and future directions. Neuropsychopharmacology. 2016;42:299–317.
Miller TJ, McGlashan TH, Rosen JL, Somjee L, Markovich PJ, Stein K, et al. Prospective diagnosis of the initial prodrome for schizophrenia based on the structured interview for prodromal syndromes: preliminary evidence of interrater reliability and predictive validity. Am J Psychiatry. 2002;159:863–5.
First M, Spitzer R, Gibbon M, Williams J. Structured clinical interview for DSM-IV axis i disorders: patient edition (SCIDI/P. Version 2.0. Biometric Research. New York: New York State Psychiatric Institute; 1995;42:299–317.
Randolph C. Repeatable battery for the assessment of neuropsychological status psychological corporation. San Antonio, TX: Harcourt; 1998.
Wilk CM, Gold JM, Humber K, Dickerson F, Fenton WS, Buchanan RW. Brief cognitive assessment in schizophrenia: normative data for the repeatable battery for the assessment of neuropsychological status. Schizophr Res. 2004;70:175–86.
Rusjan P, Mamo D, Ginovart N, Hussey D, Vitcu I, Yasuno F, et al. An automated method for the extraction of regional data from PET images. Psychiatry Res. 2006;147:79–89.
Rusjan PM, Wilson AA, Bloomfield PM, Vitcu I, Meyer JH, Houle S, et al. Quantitation of translocator protein binding in human brain with the novel radioligand [18F]-FEPPA and positron emission tomography. J Cereb Blood Flow Metab. 2011;31:1807–16.
Mizrahi R, Rusjan PM, Kennedy J, Pollock B, Mulsant B, Suridjan I, et al. Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [(18)F]-FEPPA. J Cereb Blood Flow Metab. 2012;32:968–72.
de la Fuente-Sandoval C, Reyes-Madrigal F, Mao X, León-Ortiz P, Rodríguez-Mayoral O, Solís-Vivanco R, et al. Cortico-striatal GABAergic and glutamatergic dysregulations in subjects at ultra-high risk for psychosis investigated with proton magnetic resonance spectroscopy. Int J Neuropsychopharmacol. 2016;19:1–10.
Shungu DC, Weiduschat N, Murrough JW, Mao X, Pillemer S, Dyke JP, et al. Increased ventricular lactate in chronic fatigue syndrome. III. Relationships to cortical glutathione and clinical symptoms implicate oxidative stress in disorder pathophysiology. NMR Biomed. 2012;25:1073–87.
Lanté F, Meunier J, Guiramand J, De Jesus Ferreira MC, Cambonie G, Aimar R, et al. Late N‐acetylcysteine treatment prevents the deficits induced in the offspring of dams exposed to an immune stress during gestation. Hippocampus. 2008;18:602–9.
Leza JC, García-Bueno B, Bioque M, Arango C, Parellada M, Do K, et al. Inflammation in schizophrenia: a question of balance. Neurosci Biobehav Rev. 2015;55:612–26.
Roussos P, Katsel P, Davis KL, Giakoumaki SG, Siever LJ, Bitsios P, et al. Convergent findings for abnormalities of the NF-κB signaling pathway in schizophrenia. Neuropsychopharmacology. 2013;38:533–9.
Sandberg M, Patil J, D’angelo B, Weber SG, Mallard C. NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology. 2014;79:298–306.
Boutin H, Pinborg LH. TSPO imaging in stroke: from animal models to human subjects. Clin Transl Imaging. 2015;3:423–35.
Carayon P, Portier M, Dussossoy D, Bord A, Petitpretre G, Canat X, et al. Involvement of peripheral benzodiazepine receptors in the protection of hematopoietic cells against oxygen radical damage. Blood. 1996;87:3170–8.
Suridjan I, Rusjan P, Voineskos AN, Selvanathan T, Setiawan E, Strafella AP, et al. Neuroinflammation in healthy aging: a PET study using a novel Translocator Protein 18kDa (TSPO) radioligand, [(18)F]-FEPPA. Neuroimage. 2014;84:868–75.
Tong J, Fitzmaurice PS, Moszczynska A, Mattina K, Ang L-C, Boileau I, et al. Do glutathione levels decline in aging human brain? Free Radic Biol Med. 2016;93:110–7.
Notter T, Coughlin J, Sawa A, Meyer U. Reconceptualization of translocator protein as a biomarker of neuroinflammation in psychiatry. Mol Psychiatry. 2016;19:1–10.
Acknowledgements
The authors would like to thank the excellent staff of the CAMH Research Imaging Centre and the FYPP clinic. This work was supported by the National Institutes of Health (NIH) R01 grant MH100043 to Dr. Mizrahi. Dr. Meyer has received operating grant funds from Janssen, in the past 2 years. Dr. Meyer has been a consultant to Lundbeck, and Takeda, in the past 2 years. None of these companies participated in the design or execution of this study or in the writing of the manuscript. Dr. Meyer is an inventor on three patents of blood markers to predict brain inflammation and/or to diagnose affective disorders. Dr. Mizrahi has received (once) speaker and consultant fees from Otsuka_Lundbeck Canada.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Hafizi, S., Da Silva, T., Meyer, J.H. et al. Interaction between TSPO—a neuroimmune marker—and redox status in clinical high risk for psychosis: a PET–MRS study. Neuropsychopharmacol 43, 1700–1705 (2018). https://doi.org/10.1038/s41386-018-0061-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-018-0061-5
This article is cited by
-
A multimodal approach to studying the relationship between peripheral glutathione, brain glutamate, and cognition in health and in schizophrenia
Molecular Psychiatry (2021)
-
Galantamine-Memantine Combination as an Antioxidant Treatment for Schizophrenia
Current Behavioral Neuroscience Reports (2019)
