
Abstract
A priori power and sample size calculations are crucial to appropriately test null hypotheses and obtain valid conclusions from all clinical studies. Statistical tests to evaluate hypotheses in microbiome studies need to consider intrinsic features of microbiome datasets that do not apply to classic sample size calculation. In this review, we summarize statistical approaches to calculate sample sizes for typical microbiome study scenarios, including those that hypothesize microbiome features to be the outcome, the exposure or the mediator, and provide relevant R scripts to conduct some of these calculations. This review is intended to be a resource to facilitate the conduct of sample size calculations that are based on testable hypotheses across several dimensions of the microbiome. Implementation of these methods will improve the quality of human or animal microbiome studies, enabling reliable conclusions that will generalize beyond the study sample.
Similar content being viewed by others
Introduction
The human gut is host to a community of microbes (bacteria, archaea, fungi, viruses and phages) referred to as the gut microbiota, with their combined functions known as the gut microbiome1. Since the start of the Human Microbiome Project (HMP), wide-ranging, genome-scale community research in 2008, large microbiome studies have aimed to characterize the genetic diversity of microbial populations living in and on humans. This is being achieved by applying next generation sequencing technology and exploring the diversity and composition of these microbial communities in the context of human body functions and mechanisms that lead to diseases2. This promising field of research may contribute to the prognosis of clinical outcomes through microbial biomarkers—any measurement allowing an intercommunication between a biological system and a potential risk, which may be chemical, physical, or biological3. There is also a growing interest in the influence of the microbiome on human health.
As with any research, clear and testable research hypotheses are required to conduct high quality studies of the microbiome. To improve the quality and consistency of microbiomics research reporting, guidelines have recently been published to critically appraise microbiome studies, which include sample size or power calculation as a criterion for a well-conducted study4. Most human microbiome studies aim to identify the relationship between microbiome features and a biological or clinical condition, an environmental exposure or medical intervention. Since much is unknown about the microbiome, and the datasets are vast and often not normally distributed, a variety of data driven techniques have been developed5. However, with the accumulation of microbiome evidence, hypothesis-based comparisons are increasingly possible.
Microbiome data arise from sequencing a marker gene such as the 16S ribosomal RNA gene for bacteria, and the ITS marker for fungi, or from metagenomic sequencing of the entire DNA within a community. The sequencing data are then summarized into a series of counts. These counts may represent Amplicon Sequence Variants (ASVs) capturing single nucleotide differences between sequences, or they may be clustered results such as the counts of unique Operational Taxonomic Units (OTUs), or taxa abundances6. Therefore, sample size calculations may focus on the count of one single cluster, taxon, ASV, or they may be based on the full spectrum of counts. For research questions based on single counts or abundances, sample size calculations can use standard formulae applicable in many domains. However, when aiming to determine if a difference exists in the whole spectrum of counts or the whole microbial community, microbiome-specific methods are required. For example, it is non-trivial to implement sample size calculations for beta-diversity measures7, since realistic distance matrices are needed between pairs of samples and between groups of samples. Either appropriate pilot data must be found, or a complex simulation study must be undertaken.
On behalf of IMPACTT (Integrated Microbiome Platforms for Advancing Causation Testing and Translation), we have compiled a comprehensive guide for sample size calculation for microbiome studies. After a brief introduction to the general concepts behind sample size calculation, we first set the stage for microbiome sample size calculations through our decision tree lens. Then, we provide examples of sample size calculations for each node in the decision tree.
Sample size and statistical power calculations: general considerations
Sample size and statistical power calculations should be performed at the design stage of a microbiome study. Ensuring good power means that the study conclusions, relating to an association or an effect of interest, are likely to be valid, and that the conclusions will generalize beyond the study sample to future similar studies. Such calculations depend on three concepts, the required sample size (N), the researcher’s tolerance for making errors in conclusions (values of Type I or II error)8,9, and the magnitude of the effect or association that one is trying to detect10,11.
Type I and Type II errors
Researchers must decide in advance what levels of error they will tolerate. Two types of errors can be anticipated: type I error and type II error, defined in Table 1. Sample size calculations depend on a statistic or a measure of association, and on the behavior of this statistic when there is no association, i.e., under the null hypothesis. Following the notation in the previous table, suppose one performed a t-test to compare means in two groups. One would reject the null hypothesis of no difference between two-group means if \(p \, < \, \alpha^{\ast}\), with α* being the significance for rejecting a null hypothesis determined before starting the experiment. When one performs a single test of hypothesis, then α* is the same as the probability of making a type I error, that is \(\alpha^{\ast} \,=\, \alpha\). In most studies, a type I error of 1 or 5% is a common choice9. The main aim of this parameter is to control the probability of making false positive conclusions. When many tests of hypothesis are performed, the threshold for rejection of the null will need to be smaller than the desired overall type I error for the entire study, so \(\alpha^{\ast} \, < \, \alpha\)12.
A power calculation assists in choosing the number of subjects needed to prevent a type II error, denoted β; power is defined as one minus the type II error or 1 − β. The choice of an appropriate value for the type II error, β, can be quite context dependent. For example, in clinical studies a commonly used threshold is 20%, which indicates an 80% chance of finding a true association. This may be appropriate to avoid misuse of resources, since it is usually necessary to increase sample size to increase power9. However, there are situations where making a type II error would be an unfortunate study outcome, and where smaller values of type II errors may be desired8. For example, when there are no effective approved treatments for a clinical condition, it would be important not to miss the potential benefit of a new treatment.
Effect size
The effect size describes the magnitude of the difference of interest. For instance, if the species richness has a median of 32 for the control group and 15 for a treatment group exposed to antibiotics, one might define an effect size as the differences between the medians, i.e., 17.
The strength of the association between two variables depends not only on the size of the difference—17 in the previous example—but also on the variability of this quantity. In the example above, one would need the variance of species richness’ measures across samples from the same treatment group. Such variance estimates can often be obtained from previous studies applying similar methodology and using similar measures, or from a pilot study.
The definition of effect size depends on the study design13 (i) to compare a continuous measure, such as species richness between two groups, the effect size will normally be a standardized mean difference, as described above; (ii) to compare the presence or absence of one particular species between two groups, the effect size may be an odds ratio or difference in proportions; (iii) to determine whether microbial community composition or alpha-diversity is associated with a continuous measure, the effect size might be based on a Pearson correlation (r). Long-established standards define \(r \,\sim\, 0.1\) as a small effect size, \(r \,\sim\, 0.3\) as medium, and \(r \,\ge\, 0.5\) as large13.
In microbiome studies, the number of communities present and the community structures greatly vary across different study designs and platforms, even within a single site of sampling such as the human gut. Therefore, obtaining accurate estimates of variability can be challenging. When possible, examining effect sizes in a study of similar design is the best option for obtaining realistic estimates. However, when such studies are not available, it may be necessary to look for previous studies that are as close as possible in design and goals to find appropriate variance estimates. In general, larger sample sizes are needed when one desires smaller type I error, smaller type II error, and/or smaller effect sizes14.
Basic formulae for sample size calculation
We have provided the most commonly used formulae for sample size calculations in Table 2.
Workflow for sample size calculations in microbiome studies
Figure 1 lays out a decision tree classification system for commonly used microbiome study designs. To help with choosing an appropriate sample size or power calculation method, each node number in Fig. 1 refers to a subsection of “Sample size calculations associated with each node of the decision tree”, that provides suggestions for sample size calculation approaches with several worked examples.

An overview of the workflow to determine sample size equation for specific hypothesis. Each node is numbered and described further in section. Sample size calculations associated with each node of the workflow chart. Node 1, at the top of the workflow, describes a conceptual exploration of microbiome patterns, where a non-specific characterization of the full microbiome diversity, between and within samples, is of interest. This might imply looking at distributions and network patterns without any expectation or structure. In fact, this node implies a level of generality that is not amenable to sample size calculations. Nodes 2 and 3, in the middle of the workflow, rely on a choice of metrics for describing the microbiome distribution. In Node 2, the choice is made to describe the microbiome by betadiversity15, a measure of dissimilarity between samples. Sample size calculations are based on comparisons of dissimilarity within a group (i.e., within patients, within controls, or within samples following the same treatment) to distances between these groups16. In Node 3, the choice is made to represent the microbiome profile by the set of taxa abundances, which can then be compared between and within groups of samples. Nodes 4 to 7, at the bottom row of Fig. 1, refer to hypotheses that reduce microbiome data to a single number per sample. For example, within-sample richness can be characterized by alpha-diversity (Node 4). In Node 5, counts for one taxon of interest are considered; Node 6 focuses on whether a specific sample belongs to a particular cluster, such as a species subtype. In node 7, one might ask whether a sample contains species belonging to a specific taxon. Finally, after choosing the question of interest, sample size calculations would compare the effect size of the chosen measure between groups, see following sections for examples.
Sample size calculations associated with each node of the workflow chart
In this section, following the workflow chart in Fig. 1 from top to bottom, we provide specific references and worked examples of sample size calculations. Each subsection corresponds to one node in Fig. 1; and one row in Table 3 shows key formulae or references for sample size and power calculations for the specific hypothesis being tested.
Comparing microbial community structure between groups versus within groups using beta-diversity or distance metrics (Fig. 1 , node 2)
The most commonly used analytic approach when working with the full spectrum of microbiome counts is to use beta-diversity, or measures of distance or dissimilarity between samples. To estimate sample size or power, one must choose first a distance metric, then find or generate plausible distances that are relevant for the proposed study, that is, obtain likely distances between pairs of samples. Sample size calculations are then based on the distributions of these distances, by comparing distances for pairs from the same group to pairs from different groups. The first row of Table 3 (Comparing microbial community structure between groups versus within groups using betadiversity or distance metrics (Fig. 1, node 2)) describes how to setup sample size calculations for this situation, with links to recommended methods and formulae.
Simple calculations that assume well-behaved distances (e.g., normally distributed distances for pairs in the same group) can be performed using information that may be easy to extract from published papers. If the means and standard deviations of beta-diversity distances are reported, then sample size calculations based on Equation A (Table 2) can be used.
An analysis of variance (ANOVA) compares variability within a group to variability between groups, which is exactly the concept desired for beta-diversity analyses. Furthermore, when comparing only two groups, there is an algebraic equivalence between sample size calculations using the t-test (Equation A, Table 2), derived from an ANOVA F-test, or based on correlation (Equation G, Table 2), i.e., the square root of the model-captured R2 17; F-statistics or R2 values are often reported in publications, for example see Table 5 in Sugino et al.18. Box 1 describes how the squared correlation, R2, is related to the effect size Δ from Equation A. It is worth noting that one should only use the R2 reported in a regression model for a microbiota beta-diversity sample size calculation when the publication has used the same distance measure as planned for one’s own study.
Madan et al. compared beta diversity between infants who were born by vaginal delivery versus cesarean section19, and we use these data to illustrate calculations with Equation A. Means and standard deviations can be extracted by eye from Fig. 1b in their paper, and are shown in Table 4. Madan et al. did not provide estimates of R2 19.
Using these values, we can estimate the sample size required to compare vaginal birth and cesarean section with 80% power. To perform conservative calculations, we assume a common standard deviation equal to the larger within-group value: 0.0046. Then, we can calculate the effect size from this study as \({{{{{{{\mathrm{{\Delta}}}}}}}}} \,=\, \frac{{0.5613 \,-\, 0.5587}}{{0.0046}} \,=\, 0.0026/0.0046 \,=\, 0.565\). Thus, according to Equation A (Table 2) for a desired 5% type I error for two-sided testing, and 20% type II error, the required sample size, per group, to detect differences between vaginal birth and C-section is:
so, the total sample size needed for the two groups would be estimated as about 100.
We can also estimate \(f \,=\, {{{{{{{\mathrm{{\Delta}}}}}}}}}/2 \,=\, 0.2825\), and hence \(R^2 \,=\, \frac{{0.2825^2}}{{( {1 \,+\, 0.2825^2} )}} \,=\, 0.0739\). Therefore, the correlation can be estimated as \(\tilde \rho _{yx} \,=\, \sqrt {0.0739} \,=\, 0.272\). Then according to Equation G (Table 2), the required sample size for testing \(\rho _{yx} \,=\, 0\) (assuming h = 0) is:
This is slightly larger than the results based on Equation A provided above. These two estimates agree very well for larger sample sizes, but Equation G tends to be more conservative for small sample sizes20.
We could also estimate sample size using estimates of SSB and SSW calculated from the same information in Table 4. Following the principles in Box 1, the sum of squares for the vaginal birth group can be calculated as 70 × 0.00262 = 4.732 × 10−4, and the sum of squares for the cesarean group is 32 × 0.00462 = 6.771 × 10−4. Therefore, SSW is their sum, i.e., 0.001150. To calculate SSB, the overall mean is first obtained by a weighted average, as
Therefore, SSB is \(70\,\ast\, \left( {0.5613 \,-\, 0.56048} \right)^2 \,+\, 32\,\ast\, \left( {0.5587 \,-\, 0.56048} \right)^2 \,=\, 0.000148\).
With SSW and SSB in hand, then:
Hence \(R^2 \,=\, \frac{{0.3585^2}}{{\left( {1 \,+\, 0.3585^2} \right)}} \,=\, 0.1140\).
Therefore, the correlation can be estimated as \(\widetilde {\rho ^\ast }_{yx} \,=\, \sqrt {0.1140} \,=\, 0.3376\), and according to Equation G (Table 2), the required sample size for testing \(\rho _{yx} \,=\, 0\) is:
This sample size estimate of 67 is based on the two standard deviations shown in Table 4, whereas the earlier calculation based on Equation A used the larger of the two, which explains the discrepancy between the two sample size estimates.
These simple beta-diversity sample size calculation are based on normality of the within-group pair distances. A richer approach that relaxes this assumption can be built on the full distribution of pairwise distances, then analyzing the data using concepts from multivariate statistics. Since distance distributions tend to be strongly skewed, such alternative methods are commonly used for analysis after collecting study data. However, performing sample size calculations for these nonparametric analyses is challenging, since information about the distribution of the distances is needed. In a previous paper by the IMPACTT consortium we described approaches for estimating sample size and power when distances are available21. Furthermore, since these distances are often difficult to obtain, we also demonstrated how to generate distances by simulation21.
Using an entire vector of abundances to describe the microbiome of a sample (Fig. 1 , node 3)
Multivariate methods can be used to compare microbial community structures through examination of distributions of the counts of taxa abundances. These distributions tend to have a large and heavily skewed dynamic range, with some very large counts and many near zero. No simple distributions match the shape and variability well, and hence specific methods for sample size calculation are needed. Row Using an entire vector of abundances to describe the microbiome of a sample (Fig. 1, node 3) in Table 3 shows the usual setup and method. Although resampling-based comparisons could be considered (e.g., permutation tests), they rest on assumptions which may not hold, such as that the within-group variability is consistent across groups. Therefore, La Rosa et al.22 proposed tests for comparing community structures based on the Dirichlet-Multinomial distribution. Since their approach is based on parametric distributions, it contains parameters that can be interpreted as measures of how different the community structures are. Thus, their method can be expected to be more powerful than any nonparametric procedure. The combination of the Dirichlet distribution with the multinomial allows capture of the inter-sample variability needed for microbiome data; in statistics this feature is referred to as ‘over-dispersion’. There are two key parameters: \(\pi \,=\, \left( {\pi _1, \ldots \pi _k} \right)\) represents the expected taxa frequencies averaged across the groups being compared, and θ represents the over-dispersion.
In La Rosa et al.22 three tests are introduced and demonstrated: (a) comparing one community structure to an expectation, i.e., \(H_0:\pi \,=\, \pi _0,\)where \(\pi _0\) is known, (b) comparing two groups, \(H_0:\pi _1 \,=\, \pi _2,\) and (c) comparing multiple groups, \(H_0:\pi _1 \,=\, \pi _2 \,=\, \pi _3 \,=\, \ldots\). All these tests, and corresponding power calculations are built into their software package: HMP23.
To give an example, here we describe their calculations comparing community structures between two groups. Their data were taken from three oral sites (subgingival, supragingival, and saliva) in 24 subjects of both genders from the USA. Power calculations are based on a modified version of Cramer’s φ⬚ criterion, φm, which is based on a contingency table chi-squared test statistic (χ2):
The value of this normalized chi-squared statistic is determined by the two key parameters, \(\pi \,and\,\theta\). When the authors compared the distributions for subgingival plaque and supragingival plaque in their subjects, their value of the modified Cramer’s φm was 0.16. They then calculated power for different numbers of subjects per group, and for different numbers of reads, at significance thresholds of 1 and 5%. For 1000 reads per group, power increased from 29.46% with 10 subjects per group to 89.76% with 25 subjects per group, using a significance threshold of 1%. It is worth noting that the authors recommend aggregating very rare taxa with abundance <1% into a single category.
Testing association between total microbial alpha-diversity, or taxon-specific alpha-diversity and an exposure or grouping variables (Fig. 1 , node 4)
In community ecology, alpha-diversity refers to the number of species present in an ecosystem (richness)15 as well as the frequency of occurrence of each type of organism (evenness). This ecological metric is found to be reduced in several disease states24, making it a relevant factor to consider when proposing microbiome research hypotheses. The most commonly used metrics/indices are Shannon, Inverse Simpson, Simpson and Chao indices25. These indices do not consider the phylogeny of the taxa identified in sequencing. One measure of phylogenetic diversity (Faith’s PD) is based on phylogeny and can be calculated when a microbial phylogenetic tree is available24.
When alpha-diversity is normally distributed or can be log-transformed, basic equations can be used for sample size calculations—see row Testing association between total microbial alpha-diversity, or taxon-specific alphadiversity and an exposure or grouping variables (Fig. 1, node 4), node 4 in Table 3.
For an example of how to calculate sample size with an alpha-diversity metric, we will consider a study presented by Casals-Pascual et al.7 which used Faith’s phylogenetic diversity (Faith’s PD). This study aimed to compare the diversity of gut microbial communities in two phenotypically distinct groups of patients with Crohn’s disease (CD). The null hypothesis was that gut microbiota phylogenetic diversity did not differ by CD phenotype. To test this hypothesis, CD patients with the B1 phenotype would be compared with those with either a B2 or B3 phenotype. In this case, the CD phenotype was the independent variable and gut microbial diversity (Faith’s PD), the dependent variable.
To determine the number of patients required to find a statistically significant difference in Faith’s PD between CD phenotypes, researchers searched for summary statistics on Faith’s PD. They found a gut microbiota study of 100 patients with the B1 CD phenotype that reported a standard deviation of 3.45 for Faith’s PD, a mean of 13.5, and the distribution seemed to be approximately normal. To determine a clinically meaningful effect size, and because a similar previous study did not exist, researchers considered an analogous study where patients treated with antibiotics were compared to healthy controls. In the analogy, an effect size of 1.5 units was observed with Faith’s PD metric with a significance level of 0.0001. Using Equation A in Table 2 and a standard deviation of 3.45, selecting a conventional level of statistical significance of 5% and a statistical power of 80%, a total sample size of 110 patients (55 per group) was recommended to detect differences in Faith’s PD of ≥2 units7.
The median value can also be used to calculate sample size and is particularly appropriate for skewed richness and diversity values; the formulae needed for converting medians and interquartile ranges to means and standard deviations are shown and referenced in Footnote 1 Table 3.
When the exposure variable is continuous, methods based on correlations can be used. For example, soil bacteria metagenome alpha-diversity has been associated with mean annual precipitation gradients26. Suppose a researcher wants to test the null hypothesis that alpha-diversity is unrelated (\(r \,=\, 0\)) to mean annual precipitation with \(\alpha \,=\, 0.05\) and power of \(0.95\). The researcher assumes that the alternative correlation coefficient (h) is approximately \(- 0.5\). Therefore, following Equation G in Table 2:
For a null hypothesis (\(H_0\)) of no correlation, the required sample size is approximately:
Testing association between taxon abundances and an exposure or grouping variable (Fig. 1 , node 5)
Researchers may also hypothesize about abundances of a specific microbial taxon of interest. These hypotheses can be expressed either using mean abundances, or by examining the proportion of samples with abundances over a chosen threshold. Sample size calculations can be based on either choice of metric, see row Testing association between taxon abundances and an exposure or grouping variable (Fig. 1, node 5) corresponding to node 5 in Table 3.
An important consideration here, as well as for nodes 6 and 7 in Fig. 1, is the choice of type 1 error. If only one taxon is of interest, then the type 1 error threshold, α, should not need adjustments for multiple testing. However, if the study plans to test association at all available taxons, then power calculations should be performed using a value α* which controls family-wise error rate. For example, use of the Bonferroni correction would suggest \(\alpha^{\ast}\,=\, \alpha /M\) where M is the planned number of tests.
For example, in the observational study of Koleva et al.27 looking at the gut microbiome of mother-infant pairs (total population size of 1,021) from the Canadian Healthy Infant Longitudinal Development (CHILD) Study, the authors hypothesized the genus Lactobacillus was reduced in gut microbiota of male infants born to an asthmatic mother27. The abundance of 16S data from fecal samples collected at 3–4 months after birth was compared between infants born to mothers who received asthma treatment during pregnancy (i.e., infants high risk for allergic diseases) and those who were not. Results supported the Lactobacillus hypothesis in a sex-dependent and ethnicity-dependent manner27. In male Caucasian infants, the reduction of Lactobacillus was independent of other study covariates known to also influence the infant gut microbiome, such as pre-pregnancy overweight, atopy status, breastfeeding and intrapartum antibiotic treatment, strengthening these conclusions. Infant fecal Lactobacillus abundance was transformed into a binary variable using the cut-off value for the highest tertile (Table 5).
The R script in Box 1 of the Supplement can be used to implement the sample size and power calculations using equations D and E of Table 2. However, we illustrate calculations assuming the sample sizes are equal in each group (\(r \,=\, 1\), Equation C of Table 2). Assuming a 2-sided test with an α of 0.05, 87 samples in each group can provide 80% power to detect a difference of this size in the proportion of infants with Lactobacilli above the highest tertile.
In the same study, abundances of bacterial taxa other than Lactobacillus were made using the Benjamini–Hochberg method28 to adjust for multiple testing (which is built into the multi-test procedure in SAS). Tests for interactions between infant sex and maternal prenatal asthma on Lactobacillus abundance were performed using an adjusted rank transform (ART) nonparametric test.
The taxon abundance comparisons presented in this paper are also useful to calculate sample size for other research questions. Due to the non-normal distribution of taxon abundance data, we provide an example that first requires converting median abundance into mean abundance for use in the sample size equation (Table 6). This conversion may be more appropriate at the phylum or other higher classification level, even family level, in which abundance data may be least skewed. Nevertheless, for illustration we used the median abundance of fecal Bacteroidetes in female infants of mothers with and without asthma to calculate sample size (Table 6). Medians and IQR (Q1 and Q3) were provided in the paper, and we transform these to estimate the mean and standard deviation. In this case, we use mean = median and SD = IQR/1.35:
Therefore, in female infants of mothers with asthma, the mean and SD are estimated to be:
In female infants of mothers without asthma, the mean and SD will be:
The effect size, Δ, is therefore:
Based on this effect size, we calculate sample size using Equation A in Table 2 for testing differences in means:
where, n is the sample size in each group (assuming sizes of the two groups are equal), \(z_{1 \,-\, \frac{\alpha }{2}} \,=\) 1.96 (\(\alpha \,=\, 0.05\)), and \(z_{1 \,-\, \beta }\) = 0.84 (\(\beta \,=\, 0.20\)). Therefore:
Assuming a 2-sided \(\alpha\) of 0.05, 19 samples in each group can achieve 80% power to compare Bacteroidetes abundance between asthmatic and non-asthmatic mothers of female infants.
Testing higher or lower rates of cluster membership between groups (Fig. 1 , node 6)
Microbiota community types or clusters are increasingly being used to characterize whole microbial community composition. The format of these variables is categorical, and therefore sample size calculations are straightforward, see row Testing higher or lower rates of cluster membership between groups (Fig. 1, node 6) for node 6 in Table 3.
We provide an example here based on a study by Tun et al. (2021)29 that identified 4 longitudinal gut microbiota clusters during infancy (Table 7). A sample size calculation is performed to determine the association between Asian ethnicity vs. any other, and the presence or absence of the C1–C1 cluster in the infant. A binary variable was created for the presence of the C1–C1 cluster vs. three other clusters.
R code and results for estimating sample size and power from these data, following Equations C, E in Table 2, are shown in the Supplementary Material, Box 2. There, we consider the sample sizes are equal in each group \((r \,=\, 1)\). Assuming a 2-sided test with an α of 0.05, 138 samples in each group can achieve 80% power to find an association between Asian ethnicity (vs. other) and the C1–C1 gut microbiota cluster (vs. others).
Testing higher or lower rates of taxon membership between groups (i.e., colonization with a microbe) (Fig. 1 , node 7)
To determine sample sizes for colonization with specific microbiota (presence/absence or yes/no), one can use the equations for proportions or odds ratios (Table 3, row Testing higher or lower rates of taxon membership between groups (i.e., colonization with a microbe) (Fig. 1, node 7) for node 7).
To illustrate sample sizes for colonization with specific microbiota (presence/absence or yes/no), we draw information from Drall et al.30 to test the question whether infant C. difficile colonization differs by exclusivity of breastfeeding (Table 8). In 853 exclusively breastfed infants (EBF), the C. difficile colonization rate was 22.63%, i.e., 193 exclusively breastfed infants were colonized with C. difficile. In 431 partially breastfed infants (PBF), the C. difficile colonization rate was 35.96%; hence 155 partially breastfed infants were colonized, and in 270 exclusively formula-fed infants (EFF), the C. difficile colonization rate was 49.63% implying 134 colonized infants (Table 8).
The pooled proportion of C. difficile colonization from the PBF and EFF groups is obtained as:
R code and results for estimating sample size and power from these data, assuming an equal sample size in each of the two groups (\(r \,=\, 1\)), can be found in Supplementary Material, Box 3. Calculations follow Equation C in Table 2. Assuming a 2-sided test with an α of 0.05, 95 samples in each group are needed to achieve 80% power to find differences in C. difficile colonization rates between exclusively breastfed infants and other infants.
This sample size calculation example will apply to any comparison of populations with respect to presence or absence of a microbe of interest. For example, this approach would be appropriate for a study comparing the presence of shared microbial species between animals and humans. The sample size calculation might also be used to compare samples where the entry or exit of microbial species into/from an ecosystem is expected. For instance, a study where a single species (probiotic) or an entire community is introduced, such as fecal microbiota transplantation.
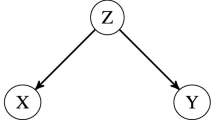
Microbiome as the mediator (exposure-microbiome-outcome)
A mediator variable (M) explains part or all of the relationship between an independent variable (X) and a dependent variable (Y), and the question of whether microbiome mediated relationships between exposures and disease is highly topical. One common focused research question is whether all association between X and Y passes through M, i.e., complete mediation (see the row Microbiome as the mediator (exposure-microbiome-outcome), Table 3).
Approaches to test for mediation vary with the most common approach being the Baron and Kenny’s Causal-Steps test31. The four steps of this approach are: (i) the total effect of X on Y must be present (statistically significant), (ii) there must be an effect of X on M, (iii) M must have a non-zero effect on Y even after controlling for X, and (iv) the effect of X on Y controlling for M must be smaller than the total effect of X on Y. All four criteria must be satisfied to consider mediation through M to be present. Other mediation tests include the Joint Significance Test32, which is a variation of Baron and Kenny’s test, and the product-of-coefficients tests, Sobel First-Order33 and PRODCLIN34 tests. Valuable information on these tests and how they compare to each other has been summarized by Fritz and MacKinnon35.
For instance, to determine the sample size to assess whether infant gut microbiota could be in the biological pathway from maternal prenatal overweight to offspring overweight, we refer to the correlation coefficients in Fig. 3b of Tun et al.36 that tested the mediating effect of the Lachnospiraceae in infant gut microbiota on the association between maternal pre-pregnancy overweight and child overweight. The values for the correlations presented in Tun et al.36 mediation Fig. 3b could be obtained separately from other studies that did not pursue mediation analyses. Their Fig. 3b indicates that the correlation between pre-pregnancy overweight and fecal Lachnospiraceae abundance is small (\(r \,=\, 0.11\)), and that the correlation between fecal Lachnospiraceae abundance and child overweight is of medium size (\(r \,=\, 0.41\)). Looking up these values in Table 3 of35, we determined the required total sample size to be 400–427 depending on the mediation method chosen.
Special considerations for longitudinal studies and other study designs
Due to the dynamic nature of the microbiome, longitudinal microbiome data are usually more informative about the profile of microbiome in relation to its host and environmental interactions37. Subjects are measured repeatedly during a study, which allows for direct evaluation of changes of response variable over time, as well as more precise estimates of inter-individual differences. Hence, longitudinal studies allow for both between-subject differences and within-subject dynamics to be considered, resulting in more powerful studies38,39.
Current efforts in microbiome research aim to move from correlation to causality. Longitudinal studies are invaluable research tools to evaluate a causative impact of the microbiome on the host physiological disease processes. Throughout the lifetime, our microbiome changes constantly over body habitats and time, it can be completely modified, either temporarily or permanently, by diseases such as infection, or medical interventions such as antibiotic courses40. These temporal patterns can help reveal if changes in microbiome predict, create or prevent diseases41. Causal inference analysis methods, such as mediation analysis, are recommended in longitudinal studies to evaluate the mutual relationship between the microbiome, the host and other study variables (environment, intervention, etc.). Various computational methods such as regression-based time series models, autoregressive (AR) models, and richer models such as the microbiome counts trajectories infinite mixture model37 have been applied to longitudinal microbiome data. However, there are still methodological limitations to existing approaches when coping with current methods of dynamic and complex microbiome data.
While longitudinal studies are more powerful biologically and statistically, calculating sample size for a longitudinal study with repeated microbiome measures is complex, and requires simulation methods based on pilot datasets42. Ideally, to run this type of simulation, the pilot dataset should contain the independent (microbiome) and dependent (host factor) variables measured at multiple timepoints, preferably the same timepoints planned for the upcoming study. It is essential to consider the between-patient covariance in both the dependent and independent measures, as well as within-patient covariance between timepoints. It is nearly impossible to accurately guess or simulate these covariance structures, and to obtain reproducible sample size calculations without having longitudinal pilot data.
Concluding remarks
The goal of this resource is to provide a framework to help plan sample size calculations in microbiome studies. We have illustrated several options such as a decision tree in Fig. 1, and provided various study designs, formulas, as well as worked examples in Sections “Sample size calculations associated with each node of the decision tree and Microbiome as the mediator (exposure-microbiome-outcome)”. We also provided codes to implement equations in R in Supplementary Material. It is often not straightforward to test associations between potential environmental factors or phenotypes and microbiome composition, whether measured by OTUs, ASVs or taxa abundances. Microbiome data often display a broad dynamic range, high dimensionality, substantial variability in counts between samples, and non-normality of counts. Counts tend to be correlated with each other, possibly due to phylogenetic structure or similarity of function43. Furthermore, diversity between samples can be extreme, such that some species or OTUs are absent from some samples. Statistically, these characteristics must be considered to perform valid analysis and sample size calculations. Evaluating the sample size threshold to design meaningful microbiome studies remains a critical step. By expanding from previously published methods, the approaches presented here will help design interpretable human microbiome studies. We encourage other scientists to test and optimize the tools presented here, and future ones, as microbiome datasets increase in public repositories.
References
Debelius, J. et al. Tiny microbes, enormous impacts: what matters in gut microbiome studies? Genome Biol. 17, 217 (2016).
Turnbaugh, P. J. et al. The human microbiome project. Nature 449, 804–810 (2007).
He, Y. et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 24, 1532–1535 (2018).
Bharucha, T. et al. STROBE-metagenomics: a STROBE extension statement to guide the reporting of metagenomics studies. Lancet Infect. Dis. 20, e251–e260 (2020).
Allaband, C. et al. Microbiome 101: studying, analyzing, and interpreting gut microbiome data for clinicians. Clin. Gastroenterol. Hepatol. 17, 218–230 (2019).
Fricker, A. M., Podlesny, D. & Fricke, W. F. What is new and relevant for sequencing-based microbiome research? A mini-review. J. Adv. Res. 19, 105–112 (2019).
Casals-Pascual, C. et al. Microbial diversity in clinical microbiome studies: sample size and statistical power considerations. Gastroenterology 158, 1524–1528 (2020).
Banerjee, A., Chitnis, U. B., Jadhav, S. L., Bhawalkar, J. S. & Chaudhury, S. Hypothesis testing, type I and type II errors. Ind. Psychiatry J. 18, 127–131 (2009).
Hickey, G. L., Grant, S. W., Dunning, J. & Siepe, M. Statistical primer: sample size and power calculations-why, when and how? Eur. J. Cardiothorac. Surg. 54, 4–9 (2018).
Lakens, D. Calculating and reporting effect sizes to facilitate cumulative science: a practical primer for t-tests and ANOVAs. Front. Psychol. 4, 863 (2013).
Brown, T. M. Power and sample size in clinical studies. J. Nucl. Cardiol. 22, 1314–1315 (2015).
Kirkham, E. M. & Weaver, E. M. A review of multiple hypothesis testing in otolaryngology literature. Laryngoscope 125, 599–603 (2015).
Maher, J. M., Markey, J. C. & Ebert-May, D. The other half of the story: effect size analysis in quantitative research. CBE Life Sci. Educ. 12, 345–351 (2013).
Hajian-Tilaki, K. Sample size estimation in epidemiologic studies. Caspian J. Intern. Med. 2, 289–298 (2011).
Laforest-Lapointe, I. & Arrieta, M. C. Patterns of early-life gut microbial colonization during human immune development: an ecological perspective. Front. Immunol. 8, 788 (2017).
Kelly, B. J. et al. Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinformatics 31, 2461–2468 (2015).
Cohen J. Statistical Power Analysis for the Behavioral Sciences (Elsevier, 1988).
Sugino, K. Y., Ma, T., Kerver, J. M., Paneth, N. & Comstock, S. S. Human milk feeding patterns at 6 months of age are a major determinant of fecal bacterial diversity in infants. J. Hum. Lact. 37, 703–713 (2021).
Madan, J. C. et al. Association of cesarean delivery and formula supplementation with the intestinal microbiome of 6-week-old infants. JAMA Pediatr. 170, 212–219 (2016).
Myers, JL, Well, AD. Research Design & Statistical Analysis 2nd edn, (Psychology Press, 2003).
Jiang, L et al. Beta-diversity distance matrices for microbiome sample size and power calculations - How to obtain good estimates. Comput. Struct. Biotechnol. J. 20, 2259–2267 (2022).
La Rosa, P. S. et al. Hypothesis testing and power calculations for taxonomic-based human microbiome data. PLoS ONE 7, e52078 (2012).
Patricio, S. et al. Hypothesis Testing and Power Calculations for Comparing Metagenomic Samples from HMP. (CRAN, 2019).
Faith, D. P. & Baker, A. M. Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evol. Bioinform. Online 2, 121–128 (2007).
Chao, A., Chiu, C. H. & Hsieh, T. C. Proposing a resolution to debates on diversity partitioning. Ecology 93, 2037–2051 (2012).
Song, H. K. et al. Environmental filtering of bacterial functional diversity along an aridity gradient. Sci. Rep. 9, 866 (2019).
Koleva, P. T. et al. Sex-specific impact of asthma during pregnancy on infant gut microbiota. Eur. Respir. J. 50, 1700280 (2017).
Benjamini, Y. H. & Hochberg, Y. Controlling the False Discovery Rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57, 289–300 (1995).
Tun, H. M. et al. Ethnicity associations with food sensitization are mediated by gut microbiota development in the first year of life. Gastroenterology 61, 94–106 (2021).
Drall, K. M. et al. Clostridioides difficile colonization is differentially associated with gut microbiome profiles by infant feeding modality at 3–4 months of age. Front. Immunol. 10, 2866 (2019).
Baron, R. K. D. The moderator-mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J. Pers. Soc. Psychol. 51, 1173–1182 (1986).
MacKinnon, D. P., Lockwood, C. M., Hoffman, J. M., West, S. G. & Sheets, V. A comparison of methods to test mediation and other intervening variable effects. Psychol. Methods 7, 83–104 (2002).
Sobel, M. Asymptotic confidence intervals for indirect effects in structural equation models. Sociol. Methodol. 13, 290–312 (1982).
MacKinnon, D. P., Fritz, M. S., Williams, J. & Lockwood, C. M. Distribution of the product confidence limits for the indirect effect: program PRODCLIN. Behav. Res. Methods 39, 384–389 (2007).
Fritz, M. S. & Mackinnon, D. P. Required sample size to detect the mediated effect. Psychol. Sci. 18, 233–239 (2007).
Tun H. M. et al. Canadian Healthy Infant Longitudinal Development (CHILD) Study Investigators. Roles of Birth Mode and Infant Gut Microbiota in Intergenerational Transmission of Overweight and Obesity From Mother to Offspring. JAMA Pediatr. 172, 368–377 (2017).
Xia, Y. & Sun, J. Hypothesis testing and statistical analysis of microbiome. Genes Dis. 4, 138–148 (2017).
Diggle, P, Heagerty, P, Liang, K-Y & Zeger, S. Analysis of Longitudinal Data, 2nd edn. (Oxford University Press, 2002).
Fitzmaurice, GM, Laird, NM & Ware, JH. Applied longitudinal analysis. (Wiley-Interscience, 2004).
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K. & Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 (2012).
Costello, E. K. et al. Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697 (2009).
Green, P. M. & MacLeod, C. J. SIMR: An R package for power analysis of generalized linear mixed models by simulation. Methods Ecol Evol 7, 493–498 (2016).
Xia, Y, Sun, J & Chen, D-G. Statistical analysis of microbiome data with r. (Springer Berlin Heidelberg, 2018).
Noordzij, M. et al. Sample size calculations: basic principles and common pitfalls. Nephrol. Dial. Transplant. 25, 1388–1393 (2010).
Chow, S-C, Wang, H & Shao, J. Sample Size Calculations in Clinical Research. (CRC press, 2007).
Bonett, DG. Sample size planning for behavioral science research. Retrieved from http://people.ucsc.edu/~dgbonett/sample.html (2016).
Dhariwal, A. et al. MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 45, W180–W188 (2017).
Holmes, I., Harris, K. & Quince, C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS ONE 7, e30126 (2012).
Hozo, S. P., Djulbegovic, B. & Hozo, I. Estimating the mean and variance from the median, range, and the size of a sample. BMC Med. Res. Methodol. 5, 13 (2005).
Wan, X., Wang, W., Liu, J. & Tong, T. Estimating the sample mean and standard deviation from the sample size, median, range and/or interquartile range. BMC Med. Res. Methodol. 14, 135 (2014).
Acknowledgements
We gratefully acknowledge platform support from the Canadian Institutes for Health Research, with grant number DC0190GP.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ferdous, T., Jiang, L., Dinu, I. et al. The rise to power of the microbiome: power and sample size calculation for microbiome studies. Mucosal Immunol 15, 1060–1070 (2022). https://doi.org/10.1038/s41385-022-00548-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-022-00548-1
