
Abstract
Heme metabolism is a key regulator of inflammatory responses. Cobalt protoporphyrin IX (CoPP) is a heme analog and mimic that potently activates the NRF2/heme oxygenase-1 (HO-1) pathway, especially in monocytes and macrophages. We investigated the influence of CoPP on inflammatory responses using a murine model of colitis. Surprisingly, conditional deletion of myeloid HO-1 did not impact the colonic inflammatory response or the protective influence of CoPP in the setting of dextran sodium sulfate-induced colitis. Rather, we reveal that CoPP elicits a contradictory shift in blood myeloid populations relative to the colon during active intestinal inflammation. Major population changes include markedly diminished trafficking of CCR2+Ly6Chi monocytes to the inflamed colon, despite significant mobilization of this population into circulation. This resulted in significantly diminished colonic expansion of monocyte-derived macrophages and inflammatory cytokine expression. These findings were linked with significant induction of systemic CCL2 leading to a disrupted CCL2 chemoattractant gradient toward the colon and concentration-dependent suppression of circulating monocyte CCR2 expression. Administration of CoPP also induced macrophage differentiation toward a MarcohiHmox1hi anti-inflammatory erythrophagocytic phenotype, contributing to an overall decreased inflammatory profile. Such findings redefine protective influences of heme metabolism during inflammation, and highlight previously unreported immunosuppressive mechanisms of endogenous CCL2 induction.
Similar content being viewed by others


Introduction
Heme, otherwise known as iron(II) protoporphyrin IX, is a critical component of oxygen carrying hemoglobin. It is also a potent modifier of inflammatory responses partly through its regulation of the nuclear factor erythroid 2-related factor 2 (NFE2L2/NRF2)/heme oxygenase-1 (HO-1) pathway.1,2 HO-1 is responsible for the metabolism of heme into carbon monoxide, iron and biliverdin. There are several analogs of heme with alternative metal atoms (metalloporphyrins) which have both inhibitory and activating influence on the NRF2/HO-1 pathway.3,4 Heme is composed of a protoporphyrin IX ring system with a central complexed iron atom while the backbone of the heme analog CoPP is also a protoporphyrin IX ring, substituted with a central cobalt atom. Like heme, cobalt(II) protoporphyrin IX can reversibly bind oxygen (at much lower affinity) and is similarly metabolized by HO-1.5,6,7 Unlike heme, the analog cobalt(III) protoporphyrin IX (CoPP/Co-heme) is not metabolized by HO-1 and therefore acts as a more potent and sustained activator of the NRF2/HO-1 pathway.4,6,8 HO-1 induction has been shown to be protective in a number of inflammatory disease models, including colitis.9,10,11,12 HO-1 influences inflammatory cytokine production, is anti-apoptotic and has antioxidative properties partly through removal of heme and partly through its metabolic products.9,11 In acute and chronic intestinal inflammatory models, the protective influence of CoPP has been associated with the activity of monocyte/macrophage HO-1.11,13,14 CoPP-induced HO-1, and its metabolic product carbon monoxide, have been linked with improved macrophage bacterial killing and decreased inflammatory cytokine expression. However, recent work has shown that CoPP can effectively mobilize myeloid cells to the circulation in the absence of inflammation and independent of NRF2/HO-1.15 The influence of this myeloid mobilization on the protective impact of CoPP in inflammatory disease has not been examined. In light of this, we sought to clarify mechanisms responsible for the protective influence of CoPP in active inflammation using a murine colitis model.
In this study we examined anti-inflammatory mechanisms of CoPP in dextran sodium sulfate (DSS) induced colitis, a model of intestinal inflammation.16,17 Oral exposure to DSS damages the intestinal epithelium leading to bacterial invasion and activation of the innate immune system.16,17,18 Previous work has shown that CCR2+Ly6Chi inflammatory monocytes are recruited to the colonic mucosa in the setting of DSS injury and differentiate into inflammatory effector and antigen presenting cells.19,20 Ablation of these recruited monocytes ameliorates colitis.19 Similarly, recruited monocytes are a significant source of pro-inflammatory cytokines including IL-1β, TNF, IL-12 and IL-6.19,21,22,23,24 We found that CoPP treatment resulted in dual protective mechanisms in colitis. Namely, we reveal that CoPP inhibits trafficking of CCR2+Ly6Chi monocytes to the colon, secondary to disruption of the normal chemoattractive CCL2 gradient, culminating in marked suppression of monocyte CCR2 expression by its CCL2 ligand. This was associated with contradictory shifts in myeloid populations between the circulation and colon. Moreover, CoPP exposure led to the development of recently characterized anti-inflammatory erythrophagocytic macrophages in the colon, expressing high levels of HO-1 and MARCO mRNA.25 These findings unveil a distinctive CCL2 gradient driven anti-inflammatory response resulting in altered monocyte trafficking, differentiation and expansion.
Results and discussion
Cobalt protoporphyrin protects against development of intestinal inflammation
To examine the protective influence of CoPP on intestinal inflammation we examined the response in DSS colitis. Exposure of WT mice to a course of oral DSS led to weight loss, diarrhea and hematochezia, summarized in a disease activity index (DAI) score, which typically peaks with DSS exposure at 5 days (Fig. 1a). IP administration of CoPP to WT mice during the course of DSS significantly decreased clinical manifestations of intestinal inflammation as well as histological measures of inflammation (Fig. 1b, c). Analysis of whole colon protein expression at day 5, the peak of DSS induced injury, using a mouse exploratory proteomics panel (Olink) revealed significant differences in protein expression patterns between mice that received and did not receive CoPP (Fig. 1d, e). This includes key cytokines such as TNF, IL-1β, CSF2, IL-10 and CXCL9 (Fig. 1f).

a Percent weight loss, stool bleeding scores and combined disease activity index (DAI), from WT mice that received 2.5% DSS in their drinking water for 5 days. Mice were treated with vehicle or CoPP IP (n = 4–5 per group) on days 0, 2 and 4. b Histopathological score and select components from colon tissue harvested at day 7. c Representative microscopic images of hematoxylin and eosin stained colon tissue at day 7 of the DSS experiment. Results are representative of two or more independent experiments. d WT mice received 2.5% DSS for 5 days and were treated with PBS or CoPP IP. Whole colon tissue was recovered for proteomic analysis (n = 5–6). Heat map representation of protein expression encompassing 75 detectable targets. e Principal component analysis with samples plotted based on the first and third principal components. f Normalized protein expression (NPX) of select individual genes with significant differences. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Colonic transcriptome profile reveals anti-inflammatory mechanisms of CoPP
To clarify the colonic response to CoPP exposure, we performed RNA-seq analysis on whole colon tissue. We used healthy non-colitic WT mice that received water and were given CoPP or vehicle IP. Figure 2a shows a heat map representing significant colonic differential gene expression results. As expected, we observed elevated log2 fold change expression of Hmox1 (4.17; adj. p = 1.07 × 10−11) in the CoPP exposed mice relative to vehicle. It is notable that, the most highly induced gene was Marco (7.80; adj. p = 2.92 × 10−08), which is typically expressed by lung alveolar and splenic marginal zone macrophages.26,27,28,29 These findings were validated using qPCR of whole colon tissue (Fig. 2b). While Marco expression has not previously been described in colonic myeloid populations, recent work has shown that Marco mRNA is expressed in a population of liver F4/80+ macrophages (including Kupffer cells) in the setting of hemolytic stress and NRF2 activation.25 These liver macrophages are described as MarcohiHmoxhiMHC class IIlo erythrophagocytes and have a distinct anti-inflammatory and anti-oxidant phenotype. In vitro macrophages exposed to heme developed a similar phenotype with a discrete gene signature separate from the well-known M1 (IFN-γ/LPS–polarized) and M2 (IL-4–polarized) macrophages.25 Using immunohistochemistry, we found strong expression of HO-1 in healthy WT mouse colon in response to CoPP (Fig. 2c). We also found co-expression of F4/80 and HO-1 after CoPP exposure (Fig. 2d) suggesting that F4/80+ macrophages are the main source of HO-1. To further validate this finding, we used fluorescence-activated cell sorting (FACS) to isolate CD11b+CX3CR1+CD64+MHCII+Ly6C− macrophages from WT mice 2 days after treatment with CoPP or vehicle (Fig. S2A).30,31 We found that macrophages from CoPP treated mice exhibited lower surface expression of MHC class II, similar to the previously described erythrophagocytic macrophages in the liver. We then recovered mRNA from the macrophages to assess expression patterns, which further validated our RNA-seq findings. By qPCR we observed a NRF2 associated mRNA expression pattern comparable to the recently described erythrophagocytic liver macrophage phenotype (increased Hmox1, Marco), with similarity to splenic erythrophagocytic red pulp macrophages that express Spic (Fig. 2f).25,32 Moreover, when we treated these macrophages ex vivo with LPS we noted a similar inhibition of LPS induced inflammatory gene expression in these CoPP exposed intestinal macrophages (Fig. 2g).25 We examined the response to CoPP in vitro using M-CSF differentiated WT bone marrow-derived macrophages (BMMs). Pretreatment, >85% of the cells were CD11b+CX3CR1+F4/80+CD64+ (Fig. S2B). We treated these BMMs with CoPP and after 48 h, we observed significant surface expression of MARCO in response to CoPP treatment, more than LPS, a positive control (Fig. S2B).26 These M-CSF differentiated BMMs also expressed other erythrophagocytic genes in response to CoPP (Fig. S2C). However, as previously reported, the induction of inflammatory genes in BMMs in response to LPS was much more robust than intestinal macrophages,33 while CoPP again led to lower levels of inflammatory gene expression relative to vehicle controls (Fig. S2D).

WT mice received drinking water for 5 days along with either PBS or CoPP treatment IP (n = 3 per group). Whole colon tissue was collected and processed for RNA-seq analysis. a Heat map representation of log2 tansformed readcount data for significant differentially expressed genes with a log2 fold change > 1. b Analysis of mRNA expression for validation of RNA-seq results using qPCR of processed whole colon tissue samples. c Representative fluorescent microscopy images (×200 total magnification) of embedded mouse colon tissue from WT mice 24 h after receiving PBS or CoPP IP. d Representative fluorescent microscopy image (×200 magnification) of mouse colon stained for F4/80 and HO-1, 24 h after receiving CoPP. e–h WT mice were treated with PBS or CoPP and CD11b+CX3CR1+CD64+MHCII+Ly6C− colonic macrophages were recovered after two days. Results reflect combined data from three independent experiments, each with 3–5 pooled mice per group. e Percentage median fluorescence intensity (MFI) values of MHC class II staining on colonic macrophages isolated from WT PBS vs CoPP treated mice. f Gene expression of erythrophagocytic markers from intestinal macrophages isolated from WT PBS vs CoPP treated mice. h Inflammatory gene expression from colonic macrophages recovered from individual WT mice treated with PBS vs CoPP and analyzed at baseline or 2 h after LPS treatment (n = 3 for controls and 4–6 for LPS treated). *p < 0.05, **p < 0.01, ***p < 0.001.
Protective influence of cobalt protoporphyrin is independent of myeloid HO-1
To examine the importance of HO-1 to the development of intestinal inflammation in DSS colitis, we bred C57BL/6 Hmox1fl/fl mice with LysMcre mice to produce myeloid specific HO-1 deficient mice (Hmox1fl/fl/LysMcre). The expression of HO-1 is high in the monocyte-macrophage lineage given their role in clearance of aged or damaged erythrocytes, especially in the spleen and liver.34,35 Myeloid HO-1 has been shown to regulate innate immune responses in murine models of inflammation and efficient deletion of HO-1 in macrophages in this setting has been previously described.36,37 M-CSF cultured BMMs were derived from our Hmox1fl/fl/LysMcre mice and Hmox1fl/fl controls, then Hmox1 mRNA expression was examined by qPCR; it was decreased in the Hmox1fl/fl/LysMcre BMMs compared to controls (85–95%). After differentiation of BMMs into M1 macrophages, we observed that the Hmox1fl/fl/LysMcre BMMs manifested a more pro-inflammatory phenotype in response to LPS. We observed markedly increased mRNA expression of Il1β (9.5 fold), Nos2 (7.6 fold), Tnf (1.6 fold) and Ccl2 (3 fold) compared to controls (Fig. S3A), confirming the anti-inflammatory role of HO-1 in macrophages.37 Given the increased inflammatory response with the loss of HO-1 expression, we expected that Hmox1fl/fl/LysMcre mice would develop more severe colonic inflammation than controls in response to DSS. Surprisingly, we found no significant differences in measures of disease activity compared to controls (Fig. 3a). Moreover, Hmox1fl/fl/LysMcre mice that received CoPP showed significant improvement in DAI scores compared to vehicle treated Hmox1fl/fl/LysMcre mice, similar to the protection seen in WT mice (Figs. 3A and S3B). These results suggest that myeloid HO-1 does not regulate the response to acute DSS colitis including in the setting of CoPP treatment. Given the unexpected findings we examined Hmox1 expression in intestinal macrophages from our Hmox1fl/fl and Hmox1fl/fl/LysMcre mice. We isolated intestinal macrophages (CD11b+CX3CR1+CD64+MHCII+Ly6C−) using FACS (Fig. S2A). We saw a significant decrease in Hmox1 expression in our intestinal macrophages (~94%) from our Hmox1fl/fl/LysMcre mice compared to Hmox1fl/fl controls (Fig. 3b). This suggests efficient Cre recombinase activity in our target population and supports the observation that the protective benefit of CoPP in these mice is not dependent on intestinal macrophage Hmox1 expression. An examination of relative populations of inflammatory monocytes, transitional monocytes (Ly6C+CCR2+MHCII+) and macrophages (Fig. S3C) in the Hmox1fl/fl and Hmox1fl/fl/LysMcre mice did not show any differences. When macrophages were isolated at the peak of DSS injury, we did not observe differences in select inflammatory cytokines in the macrophages from the Hmox1fl/fl/LysMcre mice, unlike what was observed with in vitro M-CSF cultured macrophages exposed to LPS (Fig. 3c). This suggests that macrophage HO-1 alone is not critical in the more complex colonic inflammatory response to DSS injury. In contrast, when intestinal macrophages were isolated from the peak of DSS injury in WT mice, there was significantly less Il1β and Il12b expression in CoPP treated mice compared to vehicle controls (Fig. 3d) confirming prior ex vivo findings that CoPP leads to anti-inflammatory differentiation of intestinal macrophages. Interestingly, at this specific timepoint Tnf expression was not different.

a Percent weight loss, stool bleeding scores and combined disease activity index after 7 days from Hmox1fl/fl and Hmox1fl/fl/LysMcre mice that received 2.5% DSS in drinking water × 5 days, and were treated with vehicle or CoPP IP (n = 5–6 per group and are representative of two or more independent experiments). b–c At day 5 of a DSS colitis experiment CD11b+CX3CR1+CD64+MHCII+Ly6C− colonic macrophages were isolated from Hmox1fl/fl and Hmox1fl/fl/LysMcre mice (n = 4–5 per group) using FACS. b Gene expression was evaluated for Hmox1, which is normalized to Hmox1fl/fl mouse macrophage expression levels. c Relative mRNA expression of inflammatory cytokines from isolated colonic macrophages. Results reflect combined data from two independent experiments. d Gene expression evaluated in colonic macrophages isolated from vehicle or CoPP treated WT mice at day 5 of a 3% DSS experiment. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (in a, the comparisons are between the two Hmox1fl/fl/LysMcre groups).
Protective influence of CoPP is associated with contradictory shifts in myeloid populations
We examined the influence of CoPP on the colonic gene expression profile in active colitis. RNA-seq analysis of whole colon tissue in the setting of DSS injury demonstrated significant differences in gene expression in CoPP treated mice compared to controls (Fig. 4a). Similar to our findings in non-colitic mice, we observed upregulated log2 fold change gene expression of Marco (2.54; adj. p < 0.05) and Hmox1 (2.68; adj. p = 9.16 × 10−12) in colitic mice exposed to CoPP (Fig. 4a). This is consistent with differentiation of erythrophagocytic macrophages in the setting of inflammation. We also observed a decrease in the expression of Tnf, Il1β and Cxcl2 in CoPP treated mice, consistent with observed decreased inflammation in these mice (Fig. 4a). Interestingly, a gene ontology analysis suggested there were differences in leukocyte migration within the overall regulation of the immune response (Fig. 4b). CoPP has recently been demonstrated to mobilize myeloid cells into the bloodstream in a G-CSF dependent manner in mice.15 We therefore examined the influence of CoPP on leukocyte mobilization during active intestinal inflammation using our colitis model (Fig. 4c). Total and relative populations of blood leukocytes were characterized using flow cytometry in healthy mice and at the height of DSS-induced injury (Fig. 4d). We confirmed that there was a marked increase in myeloid mobilization to the bloodstream in CoPP treated mice compared to controls (Fig. 4e). This finding was also observed in colitic mice that received CoPP, where we observed a significant increase in blood myeloid populations including Ly6G+ neutrophils, CCR2+Ly6Chi and Ly6CLo monocytes (Fig. 4e).

Colon tissue was collected and processed for RNA-seq analysis from WT mice that received 2.5% DSS in drinking water for 5 days along with either PBS or CoPP treatment IP (n = 3 per group). a Heat map representation of log2 tansformed readcount data for significant differentially expressed genes with a log2 fold change > 1. b Enrichment analysis for DEGs using GO biological process approach. c Combined disease activity index (DAI) from WT mice that received water, or 2.5% DSS in their drinking water, for 5 days and were treated with PBS or CoPP IP (n = 8–13 per group). Combined results from three independent experiments (*p < 0.05, ***p < 0.001 reflecting PBS/DSS vs CoPP/DSS comparison). d Blood leukocytes were isolated and quantified using flow cytometry by assessing for CD45, Ly6G, Siglec-F, CD11b, Ly6C and CCR2 expression with representative contour plots of live/singlet/CD45+ cells shown. e Cell counts of blood leukocyte populations represented as cells per μL. Combined results from three independent experiments (n = 8–13 per group). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
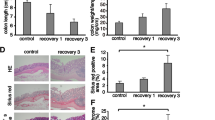
In parallel to blood leukocyte analysis, colonic lamina propria leukocytes were isolated from the colon and characterized by flow cytometry (Fig. 5a). Interestingly, despite the significant mobilization of myeloid cells to the bloodstream in response to CoPP, this did not translate into increased trafficking to the colon in active inflammation (Fig. 5b). Instead, we observed a significant numerical decrease in CD11b+ myeloid cells in colitic mice treated with CoPP and a non-significant numerical decrease in CD45+ leukocytes. Despite being greatly expanded in the blood, monocytes were markedly diminished in the colon of the colitic mice treated with CoPP (Fig. 5c). In colitic mice, we observed a reversal of macrophage expansion in CoPP treated mice compared to vehicle treated mice (Fig. 5c), likely secondary to decreased monocyte precursors. We further examined the significance of disrupted monocyte trafficking toward the colon by comparing numbers of isolated colonic monocytes with measures of clinical inflammation, using our DAI (Fig. 5d). We found a strong correlation between monocytes and disease activity, consistent with their reported importance in driving inflammation in this model.19,20,38 Infiltrating monocytes are also a major source of key inflammatory cytokines (e.g., IL-1β and TNF), with the former reported as a major contributor to inflammation in DSS and other colitis models.21,22,39,40,41 Importantly, in our CoPP treated mice, we observed a marked reduction of these key regulators of inflammation at the height of DSS injury correlating with diminished monocyte (r = 0.77, p = 0.0001) and macrophage (r = 0.79, p < 0.0001) populations (Fig. 5e).

Colon tissue was isolated from WT mice receiving water or DSS for 5 days along with IP PBS or CoPP. a Colon lamina propria cells were isolated and assessed for the expression of CD45, Ly6G, Siglec-F, CD11b, Ly6C, CD64, CD11c and MHCII with representative flow cytometry contour plots of live/singlet/CD45+ cells shown. b Cell counts of colon total leukocytes and myeloid cells based on CD45 and CD11b staining. c Proportion of colon leukocyte populations as a percentage of total CD45+ cells. d Pearson correlation of colonic monocyte population compared with disease activity index (PBS treated, black; CoPP treated, white). e Colonic concentration of inflammatory cytokines on a per mg total protein basis. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
These findings were similarly observed in the Hmox1fl/fl/LysMcre mice (Fig. S4) where we observed a parallel decrease in the numbers of colonic myeloid cells in the colitic mice treated with CoPP (Fig. S4A). These populations included monocytes, macrophages and to a lesser extent dendritic cells (Fig. S4B-C). Opposite shifts in myeloid populations were again seen in the circulating leukocytes (Fig. S4D), a development which has been shown to be independent of HO-1.15
CoPP elicits a selective anti-inflammatory CCL2 gradient response
Based on the unexpected population shifts of monocytes between blood and colon, we hypothesized the likely disruption of chemotactic signaling by CoPP. CCR2 expression on Ly6Chi monocytes and binding of the ligand, CCL2, create a signaling axis that is critical to monocyte mobilization and recruitment from the bone marrow to the tissue.42 To address this signaling axis in relationship to CoPP, we collected colonic tissue and blood serum at the height of DSS injury and measured CCL2 concentration. In response to DSS injury, we observed significant increases in colonic expression of CCL2 (Fig. 6a). In CoPP treated mice we observed a similar increased level of CCL2 (Fig. 6a) in the healthy and the colitic mice. Prior work has noted an increase in serum CCL2 in healthy mice treated with CoPP.15 Our results replicated this finding (Fig. 6a).

WT mice received water or 2.5% DSS in their drinking water for 5 days and were treated with PBS or CoPP IP (n = 3–7 per group). a Whole colon tissue and blood serum was recovered and concentration of CCL2 was measured using ELISA detection, and presented as normalized to per mg total protein. b CCL2 concentration gradient strength calculated as chemokine level in the colon (per mg total protein) divided by corresponding serum level (per mg total protein). c Pearson correlation of CCL2 gradient strength compared to DAI score. d Representative flow cytometry histogram (WT mice) and median fluorescence intensity (MFI) values of CCR2 staining in Ly6Chi blood monocytes isolated at day 5 of a DSS colitis experiment from WT and Hmox1fl/fl/LysMcre mice treated with vehicle or CoPP. e Pearson correlation of CCR2 MFI readings compared to DAI score from WT colitic mice treated with vehicle or CoPP at day 5. f Serum CCL2 concentration from WT colitic mice treated with PBS or CoPP at day 5 (n = 12–13 per group). g CCR2 MFI of blood Ly6Chi monocytes exposed ex vivo to recombinant CCL2 for 1 h, normalized to vehicle exposed monocytes. h Pearson correlation of CCR2 MFI from WT mouse blood Ly6Chi monocytes compared to serum CCL2 concentration from day 5 of a colitis experiment. i Segments of WT mouse spleen, lung and liver isolated and processed for CCL2 ELISA at day 5 of a DSS colitis experiment, presented as normalized to per mg total protein. Representative of duplicate experiments with similar results. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Chemokine signaling is complex and involves sequestration of tissue chemokines by glycosaminoglycans to establish gradients, as well as binding and presentation by endothelial cells to trigger leukocyte attachment and extravasation.43,44 The magnitude of the concentration gradient dictates the efficiency of leukocyte migration toward the source. We utilized a simple measure of the magnitude of the concentration gradient between the colon lamina propria and blood by dividing our CCL2 (per mg total protein) levels in the colon by the same in the blood. We found that there was a significant increase in the CCL2 concentration gradient in vehicle treated colitic mice, but this increase was suppressed in colitic mice treated with CoPP (Fig. 6b). We found a strong correlation between the magnitude of this gradient and DAI score (Fig. 6c). We examined a limited panel of other pertinent chemokines regulating myeloid traffic to the colon such as CXCL1, CXCL2, and CCL11 and did not see a significant change in the chemoattractant gradient in response to CoPP treatment (Fig. S5), although decreased colonic CXCL1, and to a lesser extent CXCL2, likely contribute to the less significant decreased colonic neutrophil recruitment (Fig. 5c).
Interestingly, we found that in CoPP treated mice, there was significantly decreased surface expression of CCR2 in blood Ly6Chi monocytes as measured by flow cytometry (Fig. 6d). This CCR2 expression also strongly correlated with disease activity in our colitis model (Fig. 6e). Given a recent report demonstrating a fine balance between CCR2 and CCL2 levels in the blood,45 we hypothesized that the decreased CCR2 expression was likely secondary to internalization of the receptor in the setting of sustained high levels of blood CCL2 (Fig. 6a). To examine this, we obtained fresh whole blood leukocytes from WT mice and exposed them to similar concentrations of CCL2 seen in serum taken from our vehicle and CoPP treated colitic mice (Fig. 6f). Gating on Ly6Chi monocytes we saw that within 30 min there was significantly reduced surface CCR2 in a CCL2 concentration-dependent manner (Fig. 6g). A strong in vivo correlation between serum CCL2 and CCR2 expression on Ly6Chi monocytes analyzed in our colitis experiments further confirmed this relationship (Fig. 6h).
To determine a source for the increased circulating CCL2 we looked at CCL2 production in the spleen and liver where it has been previously described in the setting of erythrophagocytosis.46 While there was a modest increase in liver CCL2 in response to CoPP exposure, this was not markedly higher than the production from vehicle treated mice (Fig. 6i). However, we found markedly higher splenic levels of CCL2 in all CoPP treated mice compared to controls, and even more so in mice that received DSS (Fig. 6i). Systemic CoPP also elicited increased CCL2 in the lung, suggesting a more universal tissue response.
We attempted to neutralize circulating CCL2 in our CoPP treated mice using an IP delivered anti-CCL2 antibody, to determine if this could restore the natural gradient toward the colon. However, as previously described in human and murine studies, antibody neutralization of CCL2 is effective only transiently as it is followed by a robust and compensatory increase in CCL2 production that is not well understood.47,48,49 We observed the same increase in serum CCL2 levels (>4 fold increase at DSS day 5 comparing CoPP vs CoPP + anti-CCL2) in this setting and therefore saw no clinical difference from antibody neutralization.
Discussion
Overall, our findings identify novel protective mechanisms for CoPP in intestinal inflammation. In the setting of colitis, the anti-inflammatory mechanism of CoPP was mostly understood to be through its influence on monocyte/macrophage HO-1. Our work examines this question head-on, and we reveal that myeloid HO-1 does not regulate the response to acute colitis. More complex heme-regulated pathways appear to be active in the context of CoPP treatment. CoPP appears to act as a mimic of heme, similar to results observed in the liver in the setting of hemolysis/erythrophagocytic stress, activating NRF2 associated differentiation of macrophages into anti-inflammatory erythrophagocytes.25 These liver macrophages can reportedly be identified by markers including high expression of Hmox1, Marco and reduced expression of MHC class II, which we now report is also the case in the colon and in M-CSF cultured BMMs in response to CoPP. In this setting, HO-1 is not the critical effector protein but rather a functional marker of a distinctive population of anti-inflammatory macrophages. The anti-inflammatory influence of HO-1 in macrophages is supported by our findings in HO-1 deficient BMMs and ex vivo colonic macrophages in response to LPS, but in vivo, its importance is apparently less significant in the more complex colonic inflammatory environment and likely depends on the inflammatory model and tissue studied.36,37 Other anti-inflammatory pathways may be concurrently active and predominate. Indeed, myeloid HO-1 dependent and independent factors are active in response to treatment with heme in similar settings.50,51 Alternatively, it’s also possible that sources of non-myeloid HO-1 may contribute to the anti-inflammatory influence of CoPP.52
Regarding other anti-inflammatory pathways, our findings suggest that regulation of monocyte mobilization and trafficking plays a major role in the protective benefit of CoPP observed. Prior studies support a foremost role for monocytes in driving inflammation in DSS colitis.19,20,38 Reduced monocyte recruitment to the colon would therefore result in improved measures of disease/injury, which we observed. While the protective influence of CoPP may also derive from the distinct anti-inflammatory differentiation of colonic macrophages, we observed that the overall number of macrophages was diminished relative to vehicle treated colitic controls. Our analysis of the lamina propria monocyte and macrophage populations in the setting of CoPP treatment is consistent with the observation that circulating Ly6Chi monocytes replenish the intestinal macrophage pool.53,54 In the setting of inflammation, these monocytes differentiate into inflammatory antigen presenting effector cells with macrophage and dendritic cell phenotypes.19,24 Diminished monocyte recruitment therefore is expected to impact monocyte-derived downstream populations as well as contribute to diminished levels of cytokines they produce, which we observed. Our findings clearly support that CoPP diminishes colonic inflammatory monocyte and macrophage populations despite the robust recruitment of the former to the bloodstream.
Chemokines and chemokine receptors are key to the recruitment of myeloid populations to the colon in response to injury.55,56,57,58 The importance of this signaling is seen in CCL2-deficient and CCR2-deficient mice, where decreased colonic inflammation was observed in colitis models along with altered monocyte-macrophage populations.19,38,59 The sustained high levels of blood CCL2, in our CoPP treated mice, explain the significant mobilization of monocytes from the bone marrow to the circulation, which is CCR2 dependent.42 Similar to other chemokine receptors CCR2 is a 7-transmembrane helical G-protein-coupled receptor that is rapidly internalized upon binding of its ligand, CCL2.45,60 It is rapidly cycled back to the plasma membrane to allow for optimal responsiveness to changes in the CCL2 gradient.60 Prior work has clearly shown decreased chemotactic responsiveness of mouse monocytes to CCL2 after significant internalization of CCR2, despite receptor recycling.61,62 We found that high blood CCL2 levels mechanistically clarify the reduced recruitment of monocytes to the colon through a combination of diminished chemoattractant gradient strength toward the colon and decreased monocyte surface CCR2 expression. A weaker gradient and less receptors to perceive that gradient would be expected to impact the chemotactic response. Elevated concentrations of CCL2 also increase the number of recycled and less responsive CCR2 receptors.61,62 This highlights the disruption by CoPP of a very important and interesting balancing act for blood CCL2 levels with regards to recruitment and subsequent tissue delivery of monocytes.45
Phagocytosis of aged or damaged red blood cells by RPMs in the spleen elicits the production of CCL2 and CCL7 by splenic Ly6Chi monocytes as the red blood cells are metabolized. This results in the recruitment of bone marrow monocytes to assist in replenishing dead or dying RPMs.46,63 A proposed signal for this production of CCL2 is free heme elaborated from red cell degradation and breakdown of hemoglobin.63 Given the similar signaling between heme and CoPP, we examined splenic production of CCL2 in the setting of CoPP treatment and inflammation. We found markedly increased production of splenic CCL2 after CoPP exposure and a similar response was seen in organs such as lung and to a lesser extent, liver. These results further implicate CoPP as a mimic of heme observed in the setting of hemolysis/erythrophagocytic stress, in this case leading to a potent CCL2 response resulting in an anti-inflammatory impact in the setting of inflammation.
One limitation of this work is that we focused on acute inflammation while the contribution of protective mechanisms of myeloid HO-1 and CoPP may differ in models of chronic mucosal inflammation where leukocyte recruitment patterns differ. However, DSS colitis is a good model for evaluating the contribution of the innate immune system to intestinal inflammation. In addition, there are potentially different mechanisms underlying the potent protective influence of the HO-1 metabolic product carbon monoxide vs CoPP, which we did not assess. For example, in strong contrast to our findings, it has been shown that CO suppresses tissue CCL2 production and inhibits monocyte CCR2 internalization in a murine model of acute pancreatitis.64 Given the efficacy of CoPP and heme in a wide range of acute and chronic inflammatory murine models of disease, identifying specific and consistent mechanisms may facilitate better translation of experimental findings to therapeutic applications in humans.
Materials and methods
DSS colitis
C57BL/6 Hmox1fl/fl mice were kindly provided by Dr. H. B. Suliman.65 HO-1ΔMyeloid mice, which have been previously described,36 were made by crossing Hmox1fl/fl mice with Lyz2-cre/LysMcre mice (JAX).66 Both Hmox1fl/fl and LysMcre mice served as controls given they were phenotypically identical in preliminary experiments. Lavage recovery of peritoneal cells showed an 86.5% average reduction in Hmox1 expression comparing controls (n = 3) to HO-1ΔMyeloid mice (n = 3; p = 0.0005). All mice were maintained and handled in accordance with Institutional Animal Care and Use Guidelines. Littermate controls were used for all colitis experiments in WT and transgenic mice with regard to treatment groups. Hmox1fl/fl mice were caged separately from Hmox1fl/fl/LysMcre mice. Age-matched male and female mice were used between age 7 and 14 weeks. Mice were given 2.5% colitis grade dextran sodium sulfate (DSS, MP Biomedical) in drinking water, changed every 2 days, for 5 days. DSS was removed on day 5 from all colitis experiments and mice were sacrificed on day 5 or day 7, where noted. Mice were injected with 7.5 mg/kg cobalt(III) protoporphyrin IX chloride (CoPP, Frontier Scientific) where noted, on days 0, 2, and 4 of treatment. Mice were weighed daily, and fecal pellets scored for stool consistency and presence of blood; all measured on a scale of 0–3 and blinded as to treatment. Disease Activity Index (DAI) was then calculated by taking the summation of weight change, stool consistency and hematochezia scores (maximum score of 9). At experiment end, colons were embedded and processed for histology. H&E stained tissue was scored by a histo-pathologist blinded to the treatments and groupings of animals using described methods.67 To block CCL2 we used an InVivoMAb anti-CCL2 antibody (2H5 clone; BioXCell) at 75 µg every 48 starting day 0 in a 5 day DSS experiment with InVivoMAb polyclonal Armenian hamster IgG control.
Leukocyte isolation
Isolation of colonic lamina propria cells was adapted from a previously described protocol.68 After dissociation of the epithelial layer for 10 min ×3 at room temp, remaining colon tissue was minced using scissors and digested in media with 500 ug/mL of DNASE I (Sigma Aldrich) and 1 mg/mL of Collagenase type IV (Worthington Biochemical Company). Digested tissue was filtered sequentially through 100 µm and 40 µm filters followed by centrifugation and resuspension in buffer for subsequent staining. Blood leukocytes were isolated after retro-orbital collection of whole blood, followed by RBC lysis and staining for flow cytometry. In one experiment, fresh blood leukocytes were exposed to recombinant mouse CCL2 (Biolegend) in RPMI complete media (RPMI-1640, 10%FBS, 1%Pen/Strep) and incubated in 95% air with 5% CO2 at 37 °C for 1 h.
Chemokine assays
Whole colon tissue isolated from the distal colon was homogenized in MSD lysis buffer (150 mM NaCl, 20 mM Tris pH 7.5, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100) with 1× phosphatase and protease inhibitors (Thermo Fischer Scientific). Portions of whole spleen, liver and lung were also isolated and processed in the same manner for chemokine assays. Serum for chemokine assays was isolated from whole blood collected through retro-orbital bleed. All tissue chemokine concentrations were normalized against total protein using PierceTM BCA Protein Assay kit (Thermo-Fisher Scientific). Initial ELISA was performed using custom mouse U-Plex Assays (Meso Scale Discovery). Additional ELISA assays performed included CCL2/JE/MCP1 (DuoSet ELISA assay, R&D Systems), and Mouse CCL11 Legend Max™ Elisa (Biolegend) all normalized to total protein. In a select experiment distal colon tissue was collected and homogenized in MSD lysis buffer/protease inhibitor for a 92-plex proteomic analysis of which 75 targets were detected (Olink Proteomics).69
Flow cytometry
Flow cytometry was used to characterize leukocyte populations in the blood and colon based on total cell counts. Cell isolations for flow cytometry were stained with antibodies purchased from Biolegend. All cells were stained with Biolegend 7AAD Viability Staining Solution prior to flow analysis. Biolegend TruStain FcX (anti-mouse CD16/32) antibody was used for blocking. The following anti-mouse antibodies were used for colonic leukocyte or blood myeloid leukocyte staining: anti-CD64 (clone X54-4/7.1), anti-CD170/Siglec-F (S17007L), anti-MHC-II (M5/114.15.2), anti-CD11c (N418), anti-CD45 (30-F11), anti-Ly6C (HK1.4), anti-CD11b (M1/70), anti-Ly6G (1A8), anti-CD192/CCR2 (SA203G11), anti-CX3CR1 (SA011F11). Bone marrow-derived macrophage cultures were stained with the following antibodies from Biolegend: anti-F4/80 (BM8), anti-CD11b (M1/70), and mouse anti-Marco (579511) from Bio-Techne/R&D Systems. Flow cytometry was performed on a FACSCantoTM II (Becton Dickinson), or a BD FACSAriaTM Fusion and resulting data was analyzed with De Novo’s FCS Express 7 software.
In vitro/ex vivo macrophage experiments
Bone marrow-derived macrophages (BMMs) were isolated as follows: Mouse femoral and tibial bone marrow was flushed out using a 25 g needle and syringe with RPMI-1640 media onto a 70 uM filter. Cells were centrifuged and washed with RPMI-1640 after RBC lysis. Cells were cultured in RPMI complete media (RPMI-1640, 10%FBS, 1%Pen/Strep) and incubated in 95% air with 5% CO2 at 37 °C. Recombinant murine M-CSF (PeproTech US) was at 20–40 ng/mL to differentiate to macrophages from day 0 to day 6. M1 macrophages were further differentiated by exposure of BMMs to 24 h of 1 ng/mL LPS (Millipore Sigma) and 10 ng/mL of recombinant mouse IFNɣ (Biolegend). After M1 differentiation, 25 uM CoPP and 10 ng/mL of LPS was added to the BMMs for another 24 h. For some BMM experiments, cells were treated with 25 uM CoPP for 24 h and then stimulated with 50 ng/mL LPS for 2 h. For flow cytometry, BMMs were treated with 25 um COPP or 10 ng LPS for 24 h. For ex vivo colonic macrophages, 2 h culture was performed with LPS 25 ng/mL in the same media used for BMMs but without M-CSF or cytokine stimulation.
Reverse transcription-quantitative PCR (qPCR)
RNA was isolated from snap frozen tissue samples and BMMs using the Qiagen RNeasy Mini kit with the addition of the gDNA-eliminator columns as per manufacturer’s protocol. First-strand complementary DNA synthesis was performed with 1 μg of total RNA using iScript reverse transcription super-mix from Bio-Rad. Real-time qPCR was performed using SYBR green on an ABI 7900HT fast real-time PCR system (both from Applied Biosystems). β-Actin (ACTB) was used as the housekeeping gene. The primer sequence for beta-actin purchased from Invitrogen was: ACTB, sense 5′-CACTCTTCCAGCCTTCCTTCC-3′, antisense 5′-CAGGTCTTTGCGGATGTCCACG-3′. All other mouse primer sets were purchased from Sigma-Aldrich’s line of KiCqStart™ Primers: Ccl2, Hmox1, Il1b, Il6, Marco, Nos2 (iNos), Slc40a1, Spic, Tnf, Il12b and Cxcl10.
RNA-Seq
RNA was isolated using TRIzol reagent (Invitrogen) and Qiagen RNeasy kit was also utilized subsequently to improve purity. Eukaryotic RNA-sequencing was carried out on the Illumina NovaSeq 6000 system (Novogene Corporation Inc.). Reference genome and gene model annotation files were downloaded from genome website browser (NCBI/UCSC/Ensembl) directly. Indexes of the reference genome was built using STAR and paired-end clean reads were aligned to the reference genome using STAR (v2.5). HTSeq v0.6.1 was used to count the read numbers mapped of each gene. And then FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. Alignments were parsed using Tophat program and differential expressions were determined through DESeq2/EdgeR. GO and KEGG enrichment were implemented by the ClusterProfiler. Additional analysis was performed using iDEP, a web-based tool for analyzing RNA-seq data.70 Parameters for DEG analyses (DESeq2) included FDR cutoff of 0.1 and minimum fold change of 2.
Immunohistochemistry
Paraffin embedded tissues were deparaffinized using two 100% xylene washes for 5 min followed by sequential 3 min washes in 1:1 xylene:Ethanol, 100%, 95%, 70%, and 50% ethanol. Tissues were then rinsed and placed in a staining box with Tris-EDTA buffer pH 9.0 (10 mM Tris Base, 1 mM EDTA solution, 0.05% TWEEN®20). Heat induced epitope retrieval was performed at 95 °C in a pressure cooker for 25 min. After rinsing in 1× TBST buffer (Tris-buffered saline, 0.2% TWEEN®20) slides were blocked with 10% normal goat serum (Thomas Scientific) in PBS for 1 h. After 1× TBST rinse, staining was performed with rabbit polyclonal HO-1 antibody (Enzo Life Sciences), and rat monoclonal [CI:A3-1] F4/80 antibody (Abcam) 4 °C overnight in a humidified chamber. After rinsing, secondary antibodies goat anti-rabbit AF-555 and goat anti-rat AF-488 (Invitrogen), were applied for 2 h at room temperature in a humidified chamber. Slides were rinsed then sealed with Molecular Probes™ ProLong™ Diamond Antifade Mountant with or without DAPI (Fischer Scientific). Imaging was done on an Olympus IX83P2F microscope equipped with a DP80 camera. Additional images were obtained on an AxioImager A1 microscope (Zeiss) equipped with an AxioCam MRc5.
Statistical analyses
Where appropriate, unpaired, two-tailed Student’s t test or one-way ANOVA (for more than two groups) with correction for multiple comparisons (two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli) was performed using Prism version 9 (GraphPad Software). DAI scores were analyzed using a mixed-effects model. Results were considered statistically significant when p < 0.05.
References
Alam, J. et al. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 274, 26071–26078 (1999).
Bonkovsky, H. L. et al. Porphyrin and heme metabolism and the porphyrias. Compr. Physiol. 3, 365–401 (2013).
Wong, R. J. et al. In vitro inhibition of heme oxygenase isoenzymes by metalloporphyrins. J. Perinatol. 31, S35–S41 (2011).
Shan, Y. et al. Induction of the heme oxygenase-1 gene by metalloporphyrins. Arch. Biochem Biophys. 380, 219–227 (2000).
Hoffman, B. M. & Petering, D. H. Coboglobins: oxygen-carrying cobalt-reconstituted hemoglobin and myoglobin. Proc. Natl Acad. Sci. USA 67, 637–643 (1970).
Vernon, D. I. & Brown, S. B. Formation of bile pigments by coupled oxidation of cobalt-substituted haemoglobin and myoglobin. Biochem J. 223, 205–209 (1984).
Yonetani, T., Yamamoto, H. & Woodrow, G. V. 3rd Studies on cobalt myoglobins and hemoglobins. I. Preparation and optical properties of myoglobins and hemoglobins containing cobalt proto-, meso-, and deuteroporphyrins and thermodynamic characterization of their reversible oxygenation. J. Biol. Chem. 249, 682–690 (1974).
Shan, Y., Lambrecht, R. W., Donohue, S. E. & Bonkovsky, H. L. Role of Bach1 and Nrf2 in up-regulation of the heme oxygenase-1 gene by cobalt protoporphyrin. FASEB J. 20, 2651–2653 (2006).
Ryter, S. W. Therapeutic Potential of Heme Oxygenase-1 and Carbon Monoxide in Acute Organ Injury, Critical Illness, and Inflammatory Disorders. Antioxidants (Basel). 9, 1153 (2020).
Paul, G. et al. Analysis of intestinal haem-oxygenase-1 (HO-1) in clinical and experimental colitis. Clin. Exp. Immunol. 140, 547–555 (2005).
Onyiah, J. C. et al. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology 144, 789–798 (2013).
Sheikh, S. Z. et al. An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis. J. Immunol. 186, 5506–5513 (2011).
Hegazi, R. A. et al. Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1-dependent pathway. J. Exp. Med. 202, 1703–1713 (2005).
Marelli, G. et al. Heme-oxygenase-1 Production by Intestinal CX3CR1(+) Macrophages Helps to Resolve Inflammation and Prevents Carcinogenesis. Cancer Res. 77, 4472–4485 (2017).
Szade, A. et al. Cobalt protoporphyrin IX increases endogenous G-CSF and mobilizes HSC and granulocytes to the blood. EMBO Mol. Med. 11, e09571 (2019).
Okayasu, I. et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98, 694–702 (1990).
Chassaing, B., Aitken, J. D., Malleshappa, M. & Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 104, 15 25 1–15 25 14 (2014).
Luissint, A. C., Parkos, C. A. & Nusrat, A. Inflammation and the Intestinal Barrier: Leukocyte-Epithelial Cell Interactions, Cell Junction Remodeling, and Mucosal Repair. Gastroenterology 151, 616–632 (2016).
Zigmond, E. et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity 37, 1076–1090 (2012).
Platt, A. M., Bain, C. C., Bordon, Y., Sester, D. P. & Mowat, A. M. An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J. Immunol. 184, 6843–6854 (2010).
Jones, G. R. et al. Dynamics of Colon Monocyte and Macrophage Activation During Colitis. Front. Immunol. 9, 2764 (2018).
Seo, S. U. et al. Distinct Commensals Induce Interleukin-1beta via NLRP3 Inflammasome in Inflammatory Monocytes to Promote Intestinal Inflammation in Response to Injury. Immunity 42, 744–755 (2015).
Flannigan, K. L., Geem, D., Harusato, A. & Denning, T. L. Intestinal Antigen-Presenting Cells: Key Regulators of Immune Homeostasis and Inflammation. Am. J. Pathol. 185, 1809–1819 (2015).
Rivollier, A., He, J., Kole, A., Valatas, V. & Kelsall, B. L. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J. Exp. Med. 209, 139–155 (2012).
Pfefferle, M. et al. Hemolysis transforms liver macrophages into antiinflammatory erythrophagocytes. J. Clin. Investig. 130, 5576–5590 (2020).
Elomaa, O. et al. Cloning of a novel bacteria-binding receptor structurally related to scavenger receptors and expressed in a subset of macrophages. Cell 80, 603–609 (1995).
Ito, S. et al. Roles of a macrophage receptor with collagenous structure (MARCO) in host defense and heterogeneity of splenic marginal zone macrophages. Arch. Histol. Cytol. 62, 83–95 (1999).
van der Laan, L. J. et al. Regulation and functional involvement of macrophage scavenger receptor MARCO in clearance of bacteria in vivo. J. Immunol. 162, 939–947 (1999).
Palecanda, A. et al. Role of the scavenger receptor MARCO in alveolar macrophage binding of unopsonized environmental particles. J. Exp. Med. 189, 1497–1506 (1999).
Bain, C. C. & Schridde, A. Origin, Differentiation, and Function of Intestinal Macrophages. Front. Immunol. 2018; 9, 2733 (2018).
Medina-Contreras, O. et al. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J. Clin. Investig. 121, 4787–4795 (2011).
Haldar, M. et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell 156, 1223–1234 (2014).
Ueda, Y. et al. Commensal microbiota induce LPS hyporesponsiveness in colonic macrophages via the production of IL-10. Int. Immunol. 22, 953–962 (2010).
Soares, M. P. & Hamza, I. Macrophages and Iron Metabolism. Immunity 44, 492–504 (2016).
Kovtunovych, G., Eckhaus, M. A., Ghosh, M. C., Ollivierre-Wilson, H. & Rouault, T. A. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood 116, 6054–6062 (2010).
Tzima, S., Victoratos, P., Kranidioti, K., Alexiou, M. & Kollias, G. Myeloid heme oxygenase-1 regulates innate immunity and autoimmunity by modulating IFN-beta production. J. Exp. Med. 206, 1167–1179 (2009).
Zhang, M. et al. Myeloid HO-1 modulates macrophage polarization and protects against ischemia-reperfusion injury. JCI Insight. 3, e120596 (2018).
Waddell, A. et al. Colonic eosinophilic inflammation in experimental colitis is mediated by Ly6C(high) CCR2(+) inflammatory monocyte/macrophage-derived CCL11. J. Immunol. 186, 5993–6003 (2011).
Kwon, K. H., Murakami, A., Hayashi, R. & Ohigashi, H. Interleukin-1beta targets interleukin-6 in progressing dextran sulfate sodium-induced experimental colitis. Biochem. Biophys. Res Commun. 337, 647–654 (2005).
Arai, Y., Takanashi, H., Kitagawa, H. & Okayasu, I. Involvement of interleukin-1 in the development of ulcerative colitis induced by dextran sulfate sodium in mice. Cytokine 10, 890–896 (1998).
Neudecker, V. et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 214, 1737–1752 (2017).
Serbina, N. V. & Pamer, E. G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7, 311–317 (2006).
Proudfoot, A. E. et al. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl Acad. Sci. USA 100, 1885–1890 (2003).
Crijns, H., Vanheule, V. & Proost, P. Targeting Chemokine-Glycosaminoglycan Interactions to Inhibit Inflammation. Front. Immunol. 11, 483 (2020).
Zhao, B. N. et al. CCR2-Mediated Uptake of Constitutively Produced CCL2: A Mechanism for Regulating Chemokine Levels in the Blood. J. Immunol. 203, 3157–3165 (2019).
Wojczyk, B. S. et al. Macrophages clear refrigerator storage-damaged red blood cells and subsequently secrete cytokines in vivo, but not in vitro, in a murine model. Transfusion 54, 3186–3197 (2014).
Haringman, J. J. et al. A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum. 54, 2387–2392 (2006).
Raghu, G. et al. CC-chemokine ligand 2 inhibition in idiopathic pulmonary fibrosis: a phase 2 trial of carlumab. Eur. Respir. J. 46, 1740–1750 (2015).
Zhao, L. et al. Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis. Hepatology 57, 829–839 (2013).
Wu, Y. et al. Heme protects intestinal mucosal barrier in DSS-induced colitis through regulating macrophage polarization in both HO-1-dependent and HO-1-independent way. FASEB J. 34, 8028–8043 (2020).
Konrad, F. M., Knausberg, U., Hone, R., Ngamsri, K. C. & Reutershan, J. Tissue heme oxygenase-1 exerts anti-inflammatory effects on LPS-induced pulmonary inflammation. Mucosal Immunol. 9, 98–111 (2016).
Onyiah, J. C., Schaefer, R. E. M. & Colgan, S. P. A Central Role for Heme Oxygenase-1 in the Control of Intestinal Epithelial Chemokine Expression. J. Innate Immun. 10, 228–238 (2018).
Bain, C. C. et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 15, 929–937 (2014).
Bain, C. C. et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 6, 498–510 (2013).
Zimmerman, N. P., Vongsa, R. A., Wendt, M. K. & Dwinell, M. B. Chemokines and chemokine receptors in mucosal homeostasis at the intestinal epithelial barrier in inflammatory bowel disease. Inflamm. Bowel Dis. 14, 1000–1011 (2008).
Kulkarni, N., Pathak, M. & Lal, G. Role of chemokine receptors and intestinal epithelial cells in the mucosal inflammation and tolerance. J. Leukoc. Biol. 101, 377–394 (2017).
Gunaltay, S. et al. Enhanced levels of chemokines and their receptors in the colon of microscopic colitis patients indicate mixed immune cell recruitment. Mediators Inflamm. 2015, 132458 (2015).
Singh, U. P. et al. Chemokine and cytokine levels in inflammatory bowel disease patients. Cytokine 77, 44–49 (2016).
Khan, W. I. et al. Critical role of MCP-1 in the pathogenesis of experimental colitis in the context of immune and enterochromaffin cells. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G803–G811 (2006).
Volpe, S. et al. CCR2 acts as scavenger for CCL2 during monocyte chemotaxis. PLoS ONE 7, e37208 (2012).
Handel, T. M. et al. An engineered monomer of CCL2 has anti-inflammatory properties emphasizing the importance of oligomerization for chemokine activity in vivo. J. Leukoc. Biol. 84, 1101–1108 (2008).
Carvallo, L. et al. Buprenorphine decreases the CCL2-mediated chemotactic response of monocytes. J. Immunol. 194, 3246–3258 (2015).
Youssef, L. A. et al. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood 131, 2581–2593 (2018).
Wu, J. et al. Carbon Monoxide Impairs CD11b(+)Ly-6C(hi) Monocyte Migration from the Blood to Inflamed Pancreas via Inhibition of the CCL2/CCR2 Axis. J. Immunol. 200, 2104–2114 (2018).
Suliman, H. B., Keenan, J. E. & Piantadosi, C. A. Mitochondrial quality-control dysregulation in conditional HO-1(−/−) mice. JCI Insight 2, e89676 (2017).
Clausen, B. E., Burkhardt, C., Reith, W., Renkawitz, R. & Forster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 (1999).
Dieleman, L. A. et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol. 114, 385–391 (1998).
Couter, C. J. & Surana, N. K. Isolation and Flow Cytometric Characterization of Murine Small Intestinal Lymphocytes. J. Vis. Exp. 111, e54114 (2016).
Assarsson, E. et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 9, e95192 (2014).
Ge, S. X., Son, E. W. & Yao, R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinforma. 19, 534 (2018).
Acknowledgements
The authors would like to thank T. Nguyen and M. Minhajuddin (Department of Medicine, University of Colorado) for helpful comments regarding flow cytometry and Garrett Hedlund for help with FACS. We thank Sarah Williams for help with animal husbandry. This work was supported by a VA Career Development Award (BX003865), Univ. of Colorado GI and Liver Innate Immune Program pilot award to J.C.O.; VA Merit (BX002182) and NIH awards to S.P.C.
Author information
Authors and Affiliations
Contributions
R.E.M.: Investigation, methodology, formal analysis, validation, writing. R.C.C.: Investigation, methodology, validation. S.M.A.: Conceptualization, investigation, methodology, editing. D.J.O: Formal analysis (histo/pathologic scoring), validation. I.M.C.: Investigation, methodology. A.P.F. Resources, editing. S.P.C.: Conceptualization, funding acquisition, resources, supervision, editing. J.C.O.: Conceptualization, investigation, methodology, formal analysis, funding acquisition, resources, supervision, writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Schaefer, R.E.M., Callahan, R.C., Atif, S.M. et al. Disruption of monocyte-macrophage differentiation and trafficking by a heme analog during active inflammation. Mucosal Immunol 15, 244–256 (2022). https://doi.org/10.1038/s41385-021-00474-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-021-00474-8
