
Abstract
Inflammatory bowel disease is characterized by an exacerbated intestinal immune response, but the critical mechanisms regulating immune activation remain incompletely understood. We previously reported that the TNF-superfamily molecule TNFSF14 (LIGHT) is required for preventing severe disease in mouse models of colitis. In addition, deletion of lymphotoxin beta receptor (LTβR), which binds LIGHT, also led to aggravated colitis pathogenesis. Here, we aimed to determine the cell type(s) requiring LTβR and the mechanism critical for exacerbation of colitis. Specific deletion of LTβR in neutrophils (LTβRΔN), but not in several other cell types, was sufficient to induce aggravated colitis and colonic neutrophil accumulation. Mechanistically, RNA-Seq analysis revealed LIGHT-induced suppression of cellular metabolism, and mitochondrial function, that was dependent on LTβR. Functional studies confirmed increased mitochondrial mass and activity, associated with excessive mitochondrial ROS production and elevated glycolysis at steady-state and during colitis. Targeting these metabolic changes rescued exacerbated disease severity. Our results demonstrate that LIGHT signals to LTβR on neutrophils to suppress metabolic activation and thereby prevents exacerbated immune pathogenesis during colitis.
Similar content being viewed by others
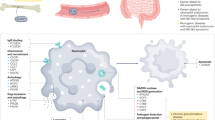


Introduction
Inflammatory bowel disease (IBD) affects over 1.5 million US citizens, and the incidence of disease is rising.1 While the pathogenesis of IBD is multifactorial, it is well established that the immune system plays a substantial role in the development of disease.2 Targeting specific immune mediators by biologics has become an increasingly widely used and effective treatment option.3 Among available biologics, antibodies specific for blockade of tumor necrosis factor (TNF) are currently the most commonly used therapy. However, only ~60% of patients treated with anti-TNF therapies show improvement,3 suggesting there are additional immune mediators critical to disease progression.4 While other cytokines, integrins, and molecules involved in immune signaling have been targeted with some success,4 the TNF superfamily of cytokines itself includes several other members that play important roles in modulating immune responses, suggesting they also could have potential to influence IBD development.5
Our previous work demonstrated that a member of the TNF superfamily, TNFSF14 or LIGHT (homologous to lymphotoxins, exhibits inducible expression, and competes with HSV glycoprotein D for HVEM, a receptor expressed by T cells), plays a significant role in two mouse models of colitis. Unlike TNF, LIGHT expression was found to provide a protective effect in the T-cell transfer and dextran sodium sulfate (DSS) colitis models.6 This is surprising, because in models of lung and skin inflammation, LIGHT generally has been found to be pro-inflammatory.7,8 Although LIGHT binds to two receptors, the Herpes Virus Entry Mediator (HVEM or TNFRSF14) and the lymphotoxin β receptor (LTβR or TNFRSF3),9 we previously determined that only the LIGHT-LTβR interaction is critical for protection from severe DSS colitis.10 A similar protective role for the LTβR during DSS colitis was found by other groups.11,12
LTβR is expressed by a variety of structural and innate immune cells, including stromal cells, epithelial cells, macrophages, and neutrophils, but it is not typically expressed by lymphocytes.9 Of interest, each of these cell types has been previously shown to contribute to colitis pathogenesis. Fibroblasts are primary drivers of fibrosis in the intestine, a hallmark of colitis.13 Epithelial cell death and shedding of the epithelium correlates with more severe colitis.14 Macrophages play a prominent role in the pathogenesis of colitis.15 Although neutrophils contribute to intestinal inflammation, they also have been reported to contribute to disease resolution.16
LIGHT is part of a complex network of receptors and ligands, because each of its receptors, LTβR and HVEM, binds to proteins in addition to LIGHT.9 The LTβR also binds to lymphotoxin αβ hetero-trimers expressed on the cell surface.5 The interaction between these different receptors and ligands is complex, because removal of one interaction may affect interactions between others in the network.17 Here we carried out experiments to identify the cell type(s) in which LTβR expression is required and to identify the mechanisms downstream of the LIGHT–LTβR interaction that are involved in limiting severe disease. Our data indicate that neutrophil-specific loss of LTβR (LTβRΔN) is sufficient to exacerbate DSS colitis. Mechanistically, LIGHT signaling through LTβR limited glycolysis, mitochondrial mass, mitochondrial respiration, and mitochondrial ROS production in neutrophils at baseline, suggesting a cell intrinsic mechanism. A similar metabolic change was also observed in colon neutrophils during DSS-induced colitis, and when targeted with the metabolic modulator metformin in LTβRΔN mice, exacerbated DSS-induced colitis was reversed. Thus, our data identify a novel function for LTβR in neutrophils, limiting mitochondrial metabolism and ROS, that contributes to preventing severe colitis.
Results
LTβR expression by neutrophils is protective
We previously identified that LTβR, not HVEM, is the relevant receptor for LIGHT-mediated protection from exacerbated colitis.6,10 LTβR expressing cell types in the colon lamina propria include macrophages, fibroblasts, epithelial cells, and neutrophils.6,10 To identify the disease-relevant LTβR-expressing cell type(s), we crossed mice with a floxed Ltbr allele to Cre mouse lines to delete receptor expression in specific cell lineages. Inducible deletion of the gene encoding LTβR in monocytes and macrophages was obtained in Ltbrfl/fl × Cx3cr1-Cre ERT mice after tamoxifen treatment. The effect of fibroblast-specific deletion of LTβR was assessed via radiation chimeras that were Ltbrfl/fl × Fsp1-Cre mouse recipients of wild-type (WT) bone marrow (BM). LTβR deletion in intestinal epithelial cells was achieved in Ltbrfl/fl × Villin-Cre mice. Cx3cr1 is known to be expressed in mature intestinal macrophages6 and its expression is important for their homeostasis and the prevention of intestinal inflammation.18 We verified that Cx3cr1-Cre ERT caused ablation of LTβR expression specifically in monocytes from blood in response to tamoxifen (Supplementary Fig. 1a). The severity of colitis was not increased in these mice, based on weight loss, fibrotic colon shortening, histological scoring, and survival (Supplementary Fig. 1). Despite significant weight loss after day 10, histologic analysis indicated fairly mild inflammation in mice exposed to this DSS concentration. As reported previously,19 we found that Fsp1-Cre, when crossed to a fluorescent reporter mouse strain, also caused deletion in many hematopoietic cells (Supplementary Fig. 2a). Therefore, we analyzed BM chimeras in which WT BM was used to reconstitute Ltbrfl/fl × Fsp1-Cre mice or Ltbrfl/fl control mice. Severity of colitis was not increased when the donor BM was from Fsp1-Cre × Ltbrfl/fl mice with relatively mild inflammation in this case as well (Supplementary Fig. 2). Deletion of the Ltbr gene in Ltbrfl/fl × Villin-Cre mice was reported previously20 and we confirmed specific deletion in epithelial cells (Supplementary Fig. 3a). The severity of DSS-colitis compared to Cre-negative littermate controls was not increased in Ltbrfl/fl × Villin-Cre mice and weight loss was in fact mildly improved (Supplementary Fig. 3).
In contrast to those results, deletion of LTβR expression in neutrophils driven by Ltbrfl/fl × Mrp8-Cre (LTβRΔN; Supplementary Fig. 4a) was sufficient to induce accelerated weight loss (Fig. 1a) and colon shortening (Fig. 1b), as well as a greatly increased histological disease score (Fig. 1c, d). After DSS treatment, LTβRΔN mice showed a loss of epithelial integrity and mononuclear cell infiltration to the colon with edematous regions. These characteristics closely correlated with previous results in LIGHT-deficient mice.6 We confirmed that virtually all neutrophils express LTβR protein in WT mice at comparable levels with other myeloid cells, while no LTβR expression was found on B cells. About 10% of myeloid progenitor cells also expressed LTβR and deletion of LTβR by Mrp8-Cre was specific to neutrophils (Supplementary Fig. S4b). For this and following functional flow-cytometric experiments, neutrophils were gated based on CD11b+ and Ly6G+ expression (Supplementary Fig S4c, left), or magnetically enriched to about 90% purity, as indicated (Supplementary Fig. S4c, right). DSS-treated LTβRΔN mice exhibited a significant increase in Mpo staining, a marker for neutrophils, in the distal colon (Fig. 1e, f). Exacerbated DSS colitis was also evident in survival studies, as LTβRΔN mice had significantly increased mortality rates during the second cycle of DSS as compared to littermate controls (Fig. 1g). Together, these data suggest that LTβR expression by neutrophils is most important for protection of exacerbated DSS-induced colitis.

Colitis was induced in Ltbrfl/fl × Mrp8-Cre (LTβRΔN) and control littermates by cycles of 2.5% DSS for 5 days followed by 2 days of water and disease progression was monitored. a weight loss was monitored daily as % starting body weight. b Colon length on day 9. c H&E stained representative histology sections of distal colon at 20× (top; scale bar, 200 μm) and 40× (bottom, scale bar, 100 μm) magnification. d Blinded total histological score from distal colon. Representative immunohistochemical MPO staining (e) and quantification (f) of MPO-positive neutrophils from sections of distal colon tissue. Scale bar, 50 μm g Kaplan–Meier survival plots of the indicated groups during two cycles of DSS. Data representative of >3 independent experiments (a, c, e-g) or pooled data from three experiments (b, d). Student’s t test (b, d, f), two-way ANOVA (a, g). Error bars represent SEM.
Some neutrophil functions are normal in LTβRΔN mice
We analyzed cytokine production in colon fragment cultures at an early time during DSS treatment (day 5) and found no difference in LTβRΔN mice compared to littermate controls for the secretion of IL-6, IL-10, MCP-1, IFNγ, and TNF (Supplementary Fig. 5a). At later timepoints, cytokine levels were increased in LIGHT- or LTβR-deficient mice.6,21 To determine how the function of LTβR-deficient neutrophils might be directly altered at steady state and in response to bacterial triggers, we carried out a battery of neutrophil functional assays. Because obtaining the numbers of viable neutrophils required for functional assays was not possible from the intestines of DSS-treated mice, we measured the kinetics of Escherichia coli bioparticle phagocytosis by purified BM-derived neutrophils at steady state or after LPS-mediated activation and observed no effect of Ltbr deletion (Supplementary Fig. 5b). Further, despite the role of TNF-family receptors, including LTβR, in cell death signaling,22,23 we found no difference in the survival of neutrophils, purified from BM and cultured in vitro (Supplementary Fig. 5c). We then tracked neutrophils in vivo using a BrdU pulse-chase method at steady state and during DSS colitis. Labeled neutrophils appeared in the blood by day 4, then declined rapidly the next day, with no evidence for an early maturation or prolonged circulation in the absence of LTβR expression (Supplementary Fig. 5d). Neutrophil recruitment to the colon on day 7 of DSS treatment was comparable irrespective of LTβR expression as quantified by flow cytometry as the fraction of BrdU-positive neutrophil numbers in colon tissue (Supplementary Fig. 5e). To examine the effect of LTβR expression on neutrophils during an acute infection, we analyzed mice after pulmonary infection with Streptococcus pneumoniae, which requires neutrophil activity for clearance.24 The number of colony forming units (CFU) of S. pneumoniae in the lungs of control or LTβRΔN mice was similar, with a trend toward increased CFU (Supplementary Fig. 5f). This coincided with a 4–5-fold increase of neutrophils in the lung tissue 18 h post infection, which was mildly elevated in the absence of LTβR (Supplementary Fig. 5g). The robust, LTβR-independent recruitment and infiltration particularly of perivascular lung tissue was also evident from immunofluorescent Ly6G-analysis of tissue sections (Supplementary Fig. 5h). In conclusion, by these measures we did not find evidence for highly altered functioning of neutrophils from LTβRΔN mice at steady state or during acute bacterial infection in the lung.
RNA-Seq reveals suppression of neutrophil metabolism by LIGHT-LTβR
To identify which other LTβR-dependent pathways contributed to the more severe colitis, blood neutrophils were cultured with cytokines associated with more severe colitis in this model system, including IL-6, TNF, and GM-CSF,6 and in the presence or absence of recombinant, soluble mouse LIGHT protein. We carried out bulk RNA-Seq in these neutrophils and determined that soluble LIGHT stimulation significantly altered the expression of 458 genes, with a greater number of genes with reduced as opposed to increased expression by exposure to LIGHT. The top 50 differentially expressed genes by P value contained 11 mitochondrial- and/or ROS pathway genes, with all of these significantly decreased by LIGHT addition and none that were increased (Fig. 2a, Supplementary Table 1). In agreement with this, pathway analysis of all differentially expressed genes identified a significant enrichment for genes associated with mitochondrial biogenesis and the electron transport chain (ETC), which had increased expression in the absence of a LIGHT signal (Fig. 2b). Ingenuity analysis of the oxidative phosphorylation pathway confirmed that transcripts for multiple components of every complex within the ETC were significantly increased in the absence of LIGHT (Fig. 2c). Gene set enrichment analysis confirmed an increase in mitochondrial gene expression (Fig. 2d) and reactive oxygen species (ROS) gene sets when LIGHT was not added (Fig. 2e).

RNA-Seq analysis of sorted blood neutrophils from WT mice stimulated with a DSS-associated cytokine cocktail and with or without recombinant mouse LIGHT. a The top 50 most differentially expressed genes with respect to P value. Genes in bold font are associated with mitochondrial and/or ROS pathways. b Gene expression pathways upregulated in the absence of LIGHT stimulation were determined by the ToppGene Suite. c Ingenuity Pathway Analysis (Qiagen) identified oxidative phosphorylation as a pathway highly upregulated in the absence of LIGHT stimulation. Significantly upregulated genes and complexes of the electron transport chain are depicted in red. Gray: not significantly different. White: not determined in dataset. d, e GSEA of the comparison of WT neutrophils and WT stimulated with LIGHT. d GSEA for genes associated with mitochondrial function from the molecular signature database list GO_mitochondrial_gene_expression. ***P < 0.001; NES = 2.1; FDR < 0.001. e GSEA for genes associated with cellular ROS pathways from the molecular signature database list Houstis_ROS. **P < 0.01; NES = 1.7; FDR < 0.01. GEO accession number GSE150243.
The suppressive effect of LIGHT was greatly reduced in LTβR-deficient neutrophils, despite neutrophil expression of HVEM, which also binds LIGHT. RNA-Seq analysis of LTβR-deficient neutrophils exposed to LIGHT showed increased expression of mitochondrial and ROS pathway genes compared to LIGHT-exposed LTβR WT neutrophils (Supplementary Fig. 6a, b). Transcripts of complex I, III, IV, and V subunits within the ETC were increased in LTβR-deficient neutrophils (Supplementary Fig. 6c), a subset of the genes that were also increased comparing WT neutrophils in the absence of LIGHT to LIGHT addition. Gene set enrichment analysis confirmed that gene ontology terms for mitochondrial genes and ROS pathway were increased in LTβR-deficient neutrophils (Supplementary Fig. 6d, e). Together, these data indicate that the LIGHT–LTβR interaction dampens expression of genes associated with mitochondrial function and ROS production within neutrophils exposed to inflammatory cytokines.
LTβR affects neutrophil respiration and glycolysis
To determine if these transcriptional changes translate to functional differences in energy metabolism under steady-state conditions, we compared neutrophils from LTβRΔN mice to controls. Because of the need for relatively large numbers of viable cells, we analyzed neutrophils isolated from BM. Results from Mitotracker staining indicated that LTβRΔN BM neutrophils had higher levels of total mitochondrial mass (Fig. 3a). This technique does not differentiate between increased numbers of mitochondria or mitochondria of increased size. By using the parallel techniques of anti-Tom20 immunofluorescence and transmission electron microscopy (TEM) on magnetically sorted neutrophils, we found that LTβR-deficient neutrophils had significantly elevated numbers and mass of mitochondria per cell, but individual mitochondria had reduced surface area, (Fig. 3b–e). As an internal reference, no difference was detected in the number of neutrophil antimicrobial granules (Fig. 3e). Elongated mitochondrial networks are required for efficient ETC-mediated ATP production, while a small fragmented mitochondrial morphology points toward non-ATP-associated mitochondrial functions.25 In neutrophils, mitochondria can be dispensable for ATP balance, but central for ROS generation.26 Considering the association of neutrophil derived ROS with IBD,21,27 we determined if these changes in the mitochondrial compartment in LTβRΔN mice could be linked to ROS levels. Flow-cytometric analysis of neutrophils from LTβRΔN mice showed higher levels of mitochondrial superoxide levels than littermate controls, as measured by MitoSox, a ROS reporter that specifically localizes to mitochondria (Fig. 3f). Because mitochondrial and NADPH-oxidase dependent ROS production are connected, we next measured total ROS production in an oxidative burst assay. Within 30 min after stimulation with N-formylmethionine-leucyl-phenylalanine (fMLP), fluorescence of the total ROS-indicator dihydrorhodamine-123 (DHR) was induced by oxidation. The induced total ROS response detected was significantly elevated in neutrophils from LTβRΔN mice compared to controls (Fig. 3g).

Neutrophils were detected from BM of Ltbr fl/fl × Mrp8-Cre (LTβRΔN) and control littermates by flow cytometry (a, f, g) or by negative magnetic enrichment (STEMCCELL technologies) (b–e) and metabolic parameters were measured. Mitochondrial mass was measured as mean intensity of Mitotracker staining (a) and mean Tom20 immunofluorescence intensity by confocal microscopy (b). c, d Tom20 foci indicative of mitochondria were quantified from z-stacked confocal micrographs of Ly6G+ cells (red) stained with Tom20 antibody (green) and quantified by Imaris. Organization of Tom20 in clusters and the 3D-shape reflected in z-stacks can lead to multiple individual foci recorded for one mitochondrium. Scale bar, 500 nm e Representative TEM images (right) and blinded quantification of granule and mitochondrial content (left). Red arrows indicate individual mitochondria. Scale bar, 500 nm. f Neutrophil mitochondrial superoxides were measured by MitoSox and quantified by flow cytometry as mean fluorescence intensity. g Representative gating and histograms (left) and quantification (right) of neutrophil oxidative burst assay at baseline and in response to fMLP (fMLP N-formylmethionine-leucyl-phenylalanine; DHR dihydrorhodamine-123). h Fluorescent glucose uptake was quantified using the fluorescent glucose analog 2-NBDG (Invitrogen) by flow cytometry. Student’s t test, data from 3–6 mice/group from 2 or more independent experiments. Error bars represent SEM.
In addition to pro-inflammatory activity,21 ROS production and elevated mitochondrial activity can lead to increased aerobic glycolysis, a pathway central to most neutrophil functions.28 As a surrogate for glycolysis we therefore measured the consumption of 2-NBDG, a fluorescent glucose analog. LTβR-deficient BM neutrophils exhibited greatly increased 2-NBDG accumulation, indicating increased glucose uptake (Fig. 3h).
RNA-Seq data demonstrated that mitochondrial metabolism and the ROS response were transcriptionally downregulated in neutrophils by soluble LIGHT, but this required LTβR expression. Consistent with data indicating that LIGHT–LTβR prevented severe colitis, we hypothesized that the metabolic changes in neutrophils would be linked to colitis severity and, therefore, changes in metabolic activity in LTβR-deficient neutrophils would also be present in neutrophils from LIGHT-deficient mice. Indeed, neutrophils isolated from Light−/− animals showed increased mitochondrial ROS and glycolysis, while mitochondrial mass remained unchanged (Supplementary Fig. 7a–c). Overall, the data support the assertion that LIGHT stimulation of LTβR suppresses neutrophil metabolic activity.
Metabolic flux measurements of LTβR-deficient neutrophils
We next measured real-time rates of mitochondrial respiration and extracellular acidification, indicative of glycolytic flux, in BM neutrophils from LTβRΔN mice and littermate controls using the Seahorse bio-analyzer. This revealed a twofold increase in the basal respiration rate and a proportional increase in spare mitochondrial respiration capacity, as measured by the oxygen consumption rate (OCR) following DNP in BM neutrophils from LTβRΔN mice (Fig. 4a). In addition, there was a significant, twofold increase in the extracellular acidification rate (ECAR) of LTβR-deficient neutrophils (Fig. 4b). Elevated acidification may partially derive from increased CO2 production during oxidative phosphorylation, and from glycolysis-associated acidification. On a metabolic phenotype map, LTβR-deficient neutrophils had a proportionally higher basal- and maximal energetic profile, indicating strongly increased mitochondrial energy demand, sustained by elevated glycolytic flux, rather than a qualitative metabolic switch, in LTβRΔN mice (Fig. 4c). The heightened oxidative metabolism was also reflected in significantly increased ATP-linked respiration as quantified by ΔOCR post oligomycin treatment as well as profoundly increased maximal respiratory capacity (Fig. 4d, e). Despite these changes in cellular respiration, the total content of neutral lipids was unchanged (Supplementary Fig. 5i). Together, these data suggest that the lack of LTβR expression by neutrophils profoundly affected their metabolic programming at steady state and resulted in increased mitochondrial number, oxidative phosphorylation, mitochondrial ROS production and glycolysis at steady state.

CD11b+ Ly6G+ Neutrophils were isolated from BM of Ltbrfl/fl × Mrp8-Cre (LTβRΔN) and control littermates (pooled from 5 mice/group) by magnetic sorting and their metabolic parameters were measured. a Oxygen consumption rate (OCR) and b extracellular acidification rate (ECAR) were measured in real time at baseline and in response to the indicated mitochondrial inhibitors. See “Methods” for details. c Basal and maximal mitochondrial respiration and glycolysis are mapped to illustrate pathway preference. ATP-linked respiration (d) and maximal respiratory capacity (e) are quantified as ΔOCR from flux analysis. Data from 5 mice/group, measured as mean of 5 wells for each timepoint, representing one of three independent experiments. Error bars represent SEM.
LTβR-dependent changes in metabolism persist in colon neutrophils
We initially identified metabolic changes in LTβR-deficient neutrophils due to LIGHT–LTβR interactions based on the unbiased approach of RNA-Seq analysis. In that experiment, neutrophils from blood were cultured with cytokines in vitro to mimic the inflammatory state in DSS colitis. Therefore, we examined if the metabolic changes observed in ex vivo analysis of BM neutrophils were also present in colonic neutrophils of DSS-treated mice. LTβRΔN mice had accelerated weight loss that persisted in the chronic phase (Fig. 5a) and this was associated with increased colonic neutrophil mitochondrial mass (Fig. 5b, c), elevated mitochondrial ROS (Fig. 5d) and increased glucose uptake (Fig. 5e, f) compared to neutrophils from DSS-treated control mice colons.

Colitis was induced in Ltbr fl/fl × Mrp8-Cre (LTβRΔN) and control littermates by 2.5% DSS and disease and metabolic parameters were measured in colonic neutrophils. a Relative weight loss expressed as % of starting body weight. Quantification of neutrophil Mitotracker mean fluorescence intensity by flow cytometry (b) and representative histograms of colon neutrophil mitochondrial mass (c). d Mitochondrial superoxide production quantified by MitoSox by mean fluorescence intensity (left) and percent positive cells (right). Fluorescent glucose 2-NBDG uptake in neutrophils isolated from the colon quantified by flow cytometry (e) and representative histogram of 2-NBDG uptake (f). Student’s t test. Data from 3–4 mice/group, representative of 3 experiments. Error bars represent SEM.
LTβR-dependent changes in metabolism are linked to severe colitis
ROS is a well characterized driver of colitis pathogenesis,21,27 and therefore we targeted ROS production in vivo. An earlier publication showed DSS-induced disease was altered in WT mice injected with the redox protective agent N-acetyl-cysteine (NAC) during colitis induction.29 Mice underwent daily i.p. administration of NAC during colitis induction, and although there was a trend toward reduced colitis in LTβRΔN mice, especially regarding colon shortening due to fibrosis, the difference did not reach statistical significance (Supplementary Fig. 8a, b). We did not find evidence, however, that the treatment affected ROS production when analyzed in colon neutrophils ex vivo. We therefore also tested metformin, which has several relevant actions: It reduces endogenous ROS through its action as a mild complex I mitochondrial inhibitor,30 acts as a NADPH-oxidase inhibitor and it reduces intestinal glucose availability.31 Due to this pleiotropic nature of metformin, it is not possible to definitively isolate individual contributions of these actions, while cooperative effects of the individual mechanisms of action on neutrophil metabolic state may benefit effect size. Metformin treatment was able to reverse the accelerated disease in LTβRΔN mice, including positive effects on weight loss (Fig. 6a), protection from fibrotic shortening of the colon (Fig. 6b), and it provided a significant improvement of the total disease score from colon histology (Fig. 6c, d). Together, these data suggest that metabolic dysregulation, including enhanced oxidative and glycolytic metabolism and ROS production in mice with neutrophil-specific LTβR deletion, persisted in the colon during DSS colitis and contributed to exacerbated disease.

Colitis was induced in Ltbr fl/fl × Mrp8-Cre (LTβRΔN) and control littermates by 2.5% DSS. Metformin (Mtf, 200 mg/kg) or PBS was administered every other day by oral gavage and disease parameters were measured over time. a Weight loss was monitored daily as % starting body weight. b Colon length at endpoint of experiment. c H&E stained representative sections from distal colon with (d) blinded total histological score. Data represents three independent experiments (a, c) or is combined from three independent experiments (b, d). Significance tested by two-way ANOVA (a) or student’s t test (b, d). Scale bar, 200 μm. Error bars represent SEM.
Discussion
LIGHT binds to HVEM and LTβR, but each receptor has binding partners in addition to LIGHT.9 Despite this possible source of redundancy, the LIGHT–LTβR interaction has unique functions in contributing to intestinal barrier dysfunction, causing fibrosis, and contributing to multi-organ inflammation when Light is overexpressed in T cells.8,32,33,34 In contrast to these pro-inflammatory functions, our previous work showed that the LIGHT–LTβR interaction prevented severe colitis induced by T-cell transfer or DSS.6,10 Therefore, the outcome of LIGHT signaling to the LTβR is highly context and cell type specific. In this study, we aimed to determine which LTβR expressing cell types were critical for providing protection from severe colitis, and which mechanism(s) downstream of the LIGHT–LTβR interaction were affording this protection.
Here we report three novel findings. First, we determined that neutrophil LTβR expression was important for preventing the exacerbation of inflammation in the colon induced by DSS. Second, we showed that LTβR expression in BM neutrophils regulates their metabolism at steady state, affecting several pathways including ROS production. Third, we showed that these differences in metabolism are also present in colonic neutrophils after DSS treatment, and our data linked the exacerbated inflammation resulting from the absence of LIGHT–LTβR signaling to altered mitochondrial metabolism and ROS generation.
Our data on a protective role for LTβR signaling in DSS colitis is partially consistent with other reports.12,35 Here, we provided novel data that neutrophils are the important cell type. The previous work implicated macrophages, based on the analysis of LysM-Cre mediated deletion35 and the observation that few neutrophils express LTβR.35 In contrast, we found previously that colonic neutrophils in DSS-treated mice had more Ltbr mRNA compared to monocytes and other colon cell types,6 and here we demonstrated that all BM neutrophils expressed surface LTβR. Furthermore, LysM-Cre deletes genes in neutrophils as well as macrophages. Moreover, although mature lamina propria macrophages express Cx3cr1,18 the results comparing Ltbr deletion using Mrp8-Cre to Cx3cr1-Cre ERT were striking in showing that only the neutrophil-specific gene deletion augmented colitis severity.
Increased levels of activated neutrophils in the colon are a hallmark of active IBD,16 and neutrophil proteins calprotectin, lipocalin, and myeloperoxidase (Mpo) are used as disease biomarkers.36 We found increased neutrophils in the colon of LTβRΔN mice, but we did not find evidence for increased generation and mobilization of neutrophils at steady state or homing to the intestine in DSS-treated mice. These data suggest altered homing was not the cause of worsened disease. Because of the balance of beneficial and pathologic impacts of neutrophils on colitis,16 however, we have not carried out neutrophil depletion. In fact, most mouse studies agree that neutrophil depletion aggravated disease,37 whereas modulation of neutrophil effector functions has been successfully employed in multiple pre-clinical studies.16
While our previous data indicated that abrogated LIGHT–LTβR interactions are responsible for severe disease,6,10 a previous study found that the absence of LIGHT was protective38 and that the absence of LTβ was the relevant ligand for increased colitis when it was missing from T cells.35 Although the previous work analyzed an acute DSS-induced model, in our experiments, deficiency for LIGHT accelerated disease in both the acute and chronic models.6 Likely this discrepancy may be due to microflora or other differences in mouse colonies. We note that LIGHT deficiency increased colitis even in DSS-treated Rag−/− mice and therefore LIGHT was acting not in T cells but in the innate immune system.6 Colonic neutrophils expressed high amounts of both LIGHT and LTβR mRNA,6 suggesting a possible autocrine signaling loop mediated by LIGHT, although the cell type providing the required LIGHT remains to be identified. LTβR-dependent effects of LTβ in moderating severe colitis due to expression by T cells are not ruled out by our work, however, and overall the data indicate how the analysis of cell-type-specific effects of gene deficiency are essential in this complex system of ligands and receptors.
Regarding the second novel point, our findings support the hypothesis that LIGHT–LTβR signals affect BM neutrophil metabolism at steady state. The data suggest that the need for the LTβR signal was not confined to a discrete point during neutrophil differentiation, however, because blocking LTβR during disease onset also increased colitis pathogenesis.6,39 Furthermore, addition of LIGHT to WT blood neutrophils in vitro that were exposed to inflammatory cytokines dampened their metabolic response. This steady state effect of LTβR may be analogous to its effect on the organization of B and T lymphocytes in the spleen, where continual LTβR signaling is required.32 It remains to be explored which signaling pathways mediate the effect of LTβR on neutrophil energy metabolism. One factor might be TRAF3, which is known to participate in LTβR signaling.33 Deletion of TRAF3 in B cells phenocopied the activated mitochondrial phenotype and increased glucose uptake we observed in LTβR-deficient neutrophils.34 Regardless, while at steady state LTβR-deficient neutrophils have a higher metabolism, this is increased further when activated in several different ways, including exposure to fMLP and by exposure to inflammatory cytokines, which induces transcriptional activation of genes in this pathway. We propose that whether this contributes to inflammation, however, depends on other signals present in the tissue milieu.
Neutrophils are mostly reliant on glycolytic pathways, but energy metabolism is highly regulated during granulopoiesis and requires robust mitochondrial oxidative metabolism in progenitor cells.40 In line with this, aspects of the mitochondrial, oxidative neutrophil phenotype we describe are also found in c-kit+ neutrophils.41 As mature cells, when neutrophils encounter microbes, their effector response requires large ATP quantities sustained by glycolysis,28 but several recent reports also describe essential roles of mitochondria in the neutrophil response,42,43,44 including ROS production.26,43 Neutrophils more reliant on oxidative metabolism that produce ROS in the tumor microenvironment also have been described,41 and the dysregulation we observed in neutrophils from LTβRΔN mice may be similar to the aberrant neutrophil phenotype in the tumor microenvironment.
Consistent with the importance of LIGHT–LTβR interactions, we found evidence for an effect of LIGHT on energy metabolism in neutrophils by adding LIGHT in vitro or by analyzing cells from LIGHT-deficient mice. The effect in LIGHT-deficient mice was less pronounced than in LTβRΔN mice, suggesting the germline deletion of LIGHT could have compensating effects in different cell types. For example, LIGHT was reported to protect against diet-induced obesity, glucose intolerance, and insulin resistance45 and regulate lipase expression and lipid homeostasis by interacting with LTβR on cells other than neutrophils.46,47
With respect to the third novel point, our data suggest that the exacerbated colitis in the absence of LIGHT–LTβR signaling in neutrophils was caused in part by metabolic changes, especially increased ROS generation. Multiple lines of evidence from this and other studies support this conclusion, including the LIGHT–LTβR dependence of expression of genes related to metabolism, the increased metabolic function of LTβR-deficient neutrophils at steady-state and in the colon during colitis, and their enhanced response to signals such as fMLP. Other studies reported beneficial effects for NAC in colitis29,48 or have used metformin to reduce endogenous ROS31 and ameliorate DSS colitis.49,50 While our data using NAC did not quite reach significance, and we did not observe an effect of Metformin in WT mice, lowering mitochondrial and NADPH-dependent ROS output and glucose availability with metformin effectively blocked the severe disease phenotype of LTβRΔN mice. Therefore, taken together, these data support, although they do not prove, the hypothesis that altered neutrophil metabolism contributes to increased inflammation in the intestine. Because of the different metabolic effects of Metformin, however, it remains uncertain if increased ROS is the sole or main contributor to altered neutrophil function in the absence of LTβR.
Dysregulation of a proposed LTβR-mediated “metabolic brake” may be particularly consequential in the colon. Ample literature exists implicating reactive oxygen and nitrogen species in the pathogenesis of IBD,51,52,53 but therapeutic targeting with various antioxidants has only shown limited success clinically.54 This may be explained in part by the fact that ROS is also essential to support an efficient innate immune response which protects from colitis susceptibility.55 In fact, a reduced amount of ROS production is likely a causal factor in some pediatric IBD patients, and linked to alterations in genes that regulate glycolytic metabolism.27 Up to 40% of chronic granulomatous disease patients develop colitis, characterized by inadequate NADPH-oxidase mediated and mitochondrial ROS generation56 within phagocytes.55
The energetically dysregulated phenotype and heightened ROS from LTβR-deficient neutrophils, resulting in more severe inflammation, could result in a feed-forward loop that exacerbates the DSS-induced colitis. This could explain the increased colonic synthesis of mRNA for chemokines such as CXCL1 and CXCL2 and the increased neutrophil accumulation observed.6 We did not detect increased inflammatory IL-6 or TNF production in colonic fragment cultures, which may reflect the early timing of the assay. By no means do we exclude other LTβR-dependent effects on neutrophil functions contributing to colitis, aside from metabolic changes, that were previously reported to be dependent on LTβR signaling.5,9 We also note that neutrophil survival may be altered in the absence of LTβR signals in the colon, suggested by the increase in these cells after DSS treatment. At steady state using cultured BM cells, however, there was no difference in the survival of LTβR-deficient neutrophils.
The relevance of our findings for IBD patients remain to be determined. DSS may induce mucosal changes more similar to acute injury than colitis. In knockouts of the LTβR ligand LIGHT, however, we found increased disease and a similar activation of the innate immune response in both the DSS and T-cell transfer models. A region on chromosome 12, including CD27, TNFRSF1A, and LTBR, is associated with Crohn’s disease risk.57 Recently, a transcriptomic signature of neutrophil activation has been associated with poor outcome in a study of UC patients.58 Our data show that the LIGHT–LTβR signaling network requires cell-type specific fine-tuning to prevent inflammatory disease. This is relevant to currently ongoing clinical trials that use decoy receptor 3 fusion proteins, which block LIGHT–LTβR along with other TNFSF–TNFRSF interactions,59 as well as a LIGHT-targeting antibody tested for the treatment of IBD60 (phase 1b, NCT03169894).
Material and methods
Animals
All mice were bred and housed under specific pathogen-free conditions at the La Jolla Institute for Immunology (La Jolla, CA). All mice were on the C57BL/6J background. All Cre-recombinase expressing mice were from the Jackson Laboratories; Bar Harbor, ME. LTβRΔN mice were generated by crossing B6.Cg-Tg(S100A8-cre-EGFP)1Ilw/J (Mrp8-Cre) strain mice to Ltrbfl/fl mice.20 Deletion of Ltbr in other cell lineages was done by crossing Ltrbfl/fl mice either to tamoxifen-inducible Cx3cr1-Cre ERT (B6.129P2(C)-Cx3cr1tm2.1(cre/ERT2)Jung/J), Fsp1-Cre (B6.C-Tg(S100a4-cre)1Egn/JhrsJ), or Villin-Cre (B6.Cg-Tg(Vil1-cre)997Gum/J) expressing mice. To prevent hematopoietic off-target deletion by Fsp1-Cre, BM chimeras were generated by grafting sub-lethally irradiated Ltbr fl/fl × Fsp1-Cre mice with 1 × 107 cells of WT BM, to achieve fibroblast-specific deletion. To analyze monocyte- and macrophage-specific conditional deletion, Ltbr fl/fl × Cx3cr1-Cre ERT mice were treated with 1 mg tamoxifen twice before the start of DSS treatment and tamoxifen administration was continued during DSS treatment. LIGHT-deficient mice (Tnfsf14−/−) were provided by Dr. Klaus Pfeffer (University of Düsseldorf, Germany).61 All procedures were approved by the La Jolla Institute for Immunology Animal Care and Use Committee and are compliant with the ARRIVE standards. Mice were littermates and animals in all experiments were age and sex matched. Mice of both sexes were analyzed, and mice were sex matched for individual experiments.
Chronic DSS-induced colitis
Mice received 2.5% DSS (Affymetrix) in the drinking water for a maximum of three cycles. As previously described, one cycle is comprised of 5 days of water plus DSS and 2 days with regular drinking water.10 Animals in all experiments were age and sex matched. Mice of both sexes were analyzed, but mice were sex matched for individual experiments. Body weight and appearance were monitored daily. Mice were euthanized, in compliance with approved animal protocols, within 24 h of losing more than 20% of their starting body weight.
Histology and immunohistochemistry
Following measurement of colon length, a piece of distal colon and cecum were fixed in zinc formalin (Medical Chemical Corporation). Following paraffin embedding, fixed tissue was stained with H&E. For Mpo immunohistochemistry staining, anti-Mpo antibody (ab9535, Abcam) was used. Images were generated from 5 or more sections per organ on an Axioscan-Z1 platform (Zeiss) using Zen-2.3 software. Slides were scored according to previously described criteria6 by a pathologist blinded to the experimental condition.
Flow cytometry and antibodies
Fluorochrome-conjugated monoclonal antibodies and metabolic dyes and staining conditions are listed in Supplementary Material. Data were acquired using Fortessa or LSR II flow cytometers (BD Biosciences). Metabolic marker fluorescence intensity depends on the instrument type and laser intensity, and therefore does not allow inter-experiment comparisons.
Metabolic flux analysis
The real-time OCR and ECAR were measured using an XFe96 extracellular flux analyzer (Seahorse Bioscience). For detailed conditions see Supplementary Material.
Phagocytosis assay
Neutrophil phagocytosis was assessed by flow-cytometric quantification of fluorescent bead uptake during 30 min incubation in the presence or absence of 1 μg/ml LPS. pH-sensitive phRodo bioparticle-beads (Invitrogen) were used according to the manufacturer’s protocol and surface marker staining was performed after phagocytosis. Data recorded as percent of neutrophils with quantifiable levels of bead uptake.
Bacterial infections
S. pneumoniae URF918 strain was cultured at 37 °C in an incubator with 5% CO2. Bacteria were grown in Todd Hewitt broth (BD Biosciences). During mid-log phase, bacteria were harvested, washed twice in phosphate-buffered saline (PBS) and 1–5 × 106 CFU, in a volume of 45–50 μl PBS, were used for retropharyngeal infection. Lungs were harvested 2 dpi, homogenized, and spread on blood agar plates for determination of CFU.
Microscopy (TEM, confocal)
For TEM, 1 × 106 purified neutrophils were fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer and processed according to the University of California San Diego EM core protocol. For confocal microscopy, 1 × 106 purified neutrophils were stained with antibodies as described for flow cytometry, and fixed in 2% para-formaldehyde for 30 min. Tom20 (D8T4N, Cell Signaling Technologies) antibody was used for detection of mitochondria. Cells were cytospun on glass coverslips and mounted in the presence of DAPI. Images were acquired on a ZEISS LSM 880 inverted confocal microscope with a 63×/1.46 NA plan-apochromat objective. The Airyscan module was used to improve resolution and signal-to-noise ratio. Automated image quantification was performed in Imaris (Bitplane) using the spot detection algorithm.
Colon explant cytokine quantification
For cytokine explants, four 3 mm punch biopsies were obtained from distal colon at the end of the first DSS cycle (day 5). Biopsies were cultured in 48-well plates with 500 μL RPMI containing 10% FBS for 6 h. Supernatants were collected and analyzed by ELISA (Thermo Fisher) according to the manufacturer’s protocol.
RNA-Seq
Details of RNA-Seq data acquisition and analysis are described in Supplementary Material. The raw fastq files have been submitted to the Gene Expression Omnibus under accession number GSE150243.
Statistical methods
Data are plotted as mean ± standard error of the mean (SEM), and statistical significance was determined by using the Mann–Whitney U test or unpaired t test. Significance for multiparameter comparisons was determined by two-way ANOVA.
Data availability
All flow cytometry analysis was conducted using FlowJo 10.4.2 software (FlowJo, Ashland, OR). All graphs and statistical analysis were generated using Prism 8 software (GraphPad Software, San Diego, CA). Automated quantifications were generated using Imaris (Bitplane).
References
Kaplan, G. G. & Ng, S. C. Understanding and preventing the global increase of inflammatory bowel disease. Gastroenterology 152, 313–321.e2 (2017).
Abraham, C. & Cho, J. H. Inflammatory bowel disease. N. Engl. J. Med. 361, 2066–2078 (2009).
Roda, G., Jharap, B., Neeraj, N. & Colombel, J.-F. Loss of response to anti-TNFs: definition, epidemiology, and management. Clin. Transl. Gastroenterol. 7, e135 (2016).
Neurath, M. F. Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 14, 269–278 (2017).
Šedý, J., Bekiaris, V. & Ware, C. F. Tumor necrosis factor superfamily in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 7, a016279 (2014).
Krause, P. et al. The tumor necrosis factor family member TNFSF14 (LIGHT) is required for resolution of intestinal inflammation in mice. Gastroenterology 146, 1752–62.e4 (2014).
Herro, R., Antunes, R. D. S., Aguilera, A. R., Tamada, K. & Croft, M. The tumor necrosis factor superfamily molecule LIGHT promotes keratinocyte activity and skin fibrosis. J. Investig. Dermatol. 135, 2109–2118 (2015).
Herro, R., Da Silva Antunes, R., Aguilera, A. R., Tamada, K. & Croft, M. Tumor necrosis factor superfamily 14 (LIGHT) controls thymic stromal lymphopoietin to drive pulmonary fibrosis. J. Allergy Clin. Immunol. 136, 757–768 (2015).
Steinberg, M. W., Shui, J.-W., Ware, C. F. & Kronenberg, M. Regulating the mucosal immune system: the contrasting roles of LIGHT, HVEM, and their various partners. Semin Immunopathol. 31, 207–221 (2009).
Giles, D. A. et al. The tumor necrosis factor superfamily members TNFSF14 (LIGHT), lymphotoxin beta and lymphotoxin beta receptor interact to regulate intestinal inflammation. Front. Immunol. 9, 2585 (2018).
Macho-Fernandez, E. et al. Lymphotoxin beta receptor signaling limits mucosal damage through driving IL-23 production by epithelial cells. Mucosal Immunol. 8, 403–413 (2015).
Wege, A. K., Huber, B., Wimmer, N., Männel, D. N. & Hehlgans, T. LTβR expression on hematopoietic cells regulates acute inflammation and influences maturation of myeloid subpopulations. Innate Immun. 20, 461–470 (2014).
Wynn, T. A. & Ramalingam, T. R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040 (2012).
Watson, A. J. M. & Hughes, K. R. TNF-α-induced intestinal epithelial cell shedding: implications for intestinal barrier function. Ann. N. Y. Acad. Sci. 1258, 1–8 (2012).
Medina-Contreras, O. et al. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J. Clin. Investig. 121, 4787–4795 (2011).
Wéra, O., Lancellotti, P. & Oury, C. The dual role of neutrophils in inflammatory bowel diseases. J. Clin. Med. 5, 118 (2016).
Wang, Y., Zhu, M., Miller, M. & Fu, Y.-X. Immunoregulation by tumor necrosis factor superfamily member LIGHT. Immunol. Rev. 229, 232–243 (2009).
Bain, C. C. & Schridde, A. Origin, differentiation, and function of intestinal macrophages. Front. Immunol. 9, 2733 (2018).
Swonger, J. M., Liu, J. S., Ivey, M. J. & Tallquist, M. D. Genetic tools for identifying and manipulating fibroblasts in the mouse. Differentiation 92, 66–83 (2016).
Wang, Y. et al. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity 32, 403–413 (2010).
Blaser, H., Dostert, C., Mak, T. W. & Brenner, D., TNF and ROS crosstalk in inflammation. Trends Cell Biol. 26, 249–261 (2016).
Zhang, D.-W. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 (2009).
Wilson, C. A. & Browning, J. L. Death of HT29 adenocarcinoma cells induced by TNF family receptor activation is caspase-independent and displays features of both apoptosis and necrosis. Cell Death Differ. 9, 1321–1333 (2002).
Garvy, B. A. & Harmsen, A. G. The importance of neutrophils in resistance to pneumococcal pneumonia in adult and neonatal mice. Inflammation 20, 499–512 (1996).
Buck, M. D. et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76 (2016).
Fossati, G. et al. The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J. Immunol. 170, 1964–1972 (2003).
Denson, L. A. et al. Clinical and genomic correlates of neutrophil reactive oxygen species production in pediatric patients with Crohn’s disease. Gastroenterology 154, 2097–2110 (2018).
Borregaard, N. & Herlin, T. Energy metabolism of human neutrophils during phagocytosis. J. Clin. Investig. 70, 550–557 (1982).
Amrouche-Mekkioui, I. & Djerdjouri, B. N-acetylcysteine improves redox status, mitochondrial dysfunction, mucin-depleted crypts and epithelial hyperplasia in dextran sulfate sodium-induced oxidative colitis in mice. Eur. J. Pharmacol. 691, 209–217 (2012).
Rena, G., Hardie, D. G. & Pearson, E. R. The mechanisms of action of metformin. Diabetologia 60, 1577–1585 (2017).
Algire, C. et al. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev. Res. 5, 536–543 (2012).
Mackay, F., Majeau, G. R., Lawton, P., Hochman, P. S. & Browning, J. L. Lymphotoxin but not tumor necrosis factor functions to maintain splenic architecture and humoral responsiveness in adult mice. Eur. J. Immunol. 27, 2033–2042 (1997).
Bista, P. et al. TRAF3 controls activation of the canonical and alternative NFkappaB by the lymphotoxin beta receptor. J. Biol. Chem. 285, 12971–12978 (2010).
Mambetsariev, N., Lin, W. W., Wallis, A. M., Stunz, L. L. & Bishop, G. A. TRAF3 deficiency promotes metabolic reprogramming in B cells. Sci. Rep. 6, 35349 (2016).
Wimmer, N. et al. Lymphotoxin-beta receptor activation on macrophages ameliorates acute DSS-induced intestinal inflammation in a TRIM30α-dependent manner. Mol. Immunol. 51, 128–135 (2012).
Carlson, M. et al. Human neutrophil lipocalin is a unique marker of neutrophil inflammation in ulcerative colitis and proctitis. Gut 50, 501–506 (2002).
Fournier, B. M. & Parkos, C. A. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 5, 354–366 (2012).
Jungbeck, M. et al. Neutralization of LIGHT ameliorates acute dextran sodium sulphate-induced intestinal inflammation. Immunology 128, 451–458 (2009).
Stopfer, P. et al. Blocking lymphotoxin-beta receptor activation diminishes inflammation via reduced mucosal addressin cell adhesion molecule-1 (MAdCAM-1) expression and leucocyte margination in chronic DSS-induced colitis. Clin. Exp. Immunol. 136, 21–29 (2004).
Riffelmacher, T. et al. Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity 47, 466–480.e5 (2017).
Rice, C. M. et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat. Commun. 9, 5099–13 (2018).
Amini, P. et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat. Commun. 9, 2958–16 (2018).
Rodríguez-Espinosa, O., Rojas-Espinosa, O., Moreno-Altamirano, M. M. B., López-Villegas, E. O. & Sánchez-García, F. J. Metabolic requirements for neutrophil extracellular traps formation. Immunology 145, 213–224 (2015).
Maianski, N. A. et al. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. 11, 143–153 (2004).
Saunders, B. M. et al. Shining LIGHT on the metabolic role of the cytokine TNFSF14 and the implications on hepatic IL-6 production. Immunol. Cell Biol. 96, 41–53 (2018).
Chellan, B. et al. LIGHT/TNFSR14 can regulate hepatic lipase expression by hepatocytes independent of T cells and Kupffer cells. PLoS ONE 8, e54719 (2013).
Lo, J. C. et al. Lymphotoxin beta receptor-dependent control of lipid homeostasis. Science 316, 285–288 (2007).
Guijarro, L. G. et al. N-acetyl-L-cysteine combined with mesalamine in the treatment of ulcerative colitis: randomized, placebo-controlled pilot study. World J. Gastroenterol. 14, 2851–2857 (2008).
Lee, S.-Y. et al. Metformin ameliorates inflammatory bowel disease by suppression of the STAT3 signaling pathway and regulation of the between Th17/Treg balance. PLoS ONE 10, e0135858 (2015).
Deng, J. et al. Metformin protects against intestinal barrier dysfunction via AMPKα1-dependent inhibition of JNK signalling activation. J. Cell. Mol. Med. 22, 546–557 (2018).
Pravda, J. Radical induction theory of ulcerative colitis. World J. Gastroenterol. 11, 2371–2384 (2005).
Keshavarzian, A. et al. Increases in free radicals and cytoskeletal protein oxidation and nitration in the colon of patients with inflammatory bowel disease. Gut 52, 720–728 (2003).
Hayashi, A., Kitahora, T. & Ono, H. Reactive Oxygen Species and Colonic Epithelial Cell Apoptosis in Ulcerative Colitis (Asakura, H., Aoyagi, Y. & Nakazawa, S.) 208–212 (Springer Japan, Tokyo, 2001).
Moura, F. A., de Andrade, K. Q., Dos Santos, J. C. F., Araújo, O. R. P. & Goulart, M. O. F. Antioxidant therapy for treatment of inflammatory bowel disease: does it work? Redox Biol. 6, 617–639 (2015).
Aviello, G. et al. Colitis susceptibility in mice with reactive oxygen species deficiency is mediated by mucus barrier and immune defense defects. Mucosal Immunol. 12, 1316–1326 (2019).
Sundqvist, M. et al. Elevated mitochondrial reactive oxygen species and cellular redox imbalance in human NADPH-oxidase-deficient phagocytes. Front. Immunol. 8, 1828 (2017).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
Czarnewski, P. et al. Conserved transcriptomic profile between mouse and human colitis allows unsupervised patient stratification. Nat. Commun. 10, 2892–11 (2019).
Hsieh, S.-L. & Lin, W.-W. Decoy receptor 3: an endogenous immunomodulator in cancer growth and inflammatory reactions. J. Biomed. Sci. 24, 39–9 (2017).
Zhang, M., Perrin, L. & Pardo, P. A randomized phase 1 study to assess the safety and pharmacokinetics of the subcutaneously injected Anti-LIGHT antibody, SAR252067. Clin. Pharmacol. Drug Dev. 6, 292–301 (2017).
Scheu, S. et al. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J. Exp. Med. 195, 1613–1624 (2002).
Acknowledgements
We thank C. Kim, D. Hinz, and C. Dillingham for assisting with neutrophil sorting; Dr. Marilyn Farquhar for the use of the electron microscopy facility, and Ying Jones for electron microscopy sample preparation; J. Greenbaum, K. Fung, S. Kannan, and G. Seumois for help with RNA-seq processing and analyzing data; A. Denn for assisting with preparation of histology slides; Z. Mikulski for assisting with microscopy; A. Khurana, V. Morris, G. Seo, for technical assistance. This work was supported by NIH grants DK46763 and AI 105215 (M.K.), Wellcome Trust grant 210842_Z_18_Z (T.R.), EMBO ALTF 99-2018 (T.R.), NIH AI135574 (A.V.T.), NIH F32 AI 140581 (D.A.G.), NIH T32 AI125179 (M.P.M.), Deutsche Forschungsgemeinschaft HA7558/1–1 (NH), NIH S10-OD-021831 (Microscopy Core), FACSAria II Cell Sorter S10 RR027366.
Author information
Authors and Affiliations
Contributions
T.R. and D.A.G. designed and performed experiments and wrote the manuscript. S.Z., S.M., T.P.J., E.v.d.G., M.P.M., N.H., and M.D. designed and performed experiments and revised the manuscript. A.Y.A. and A.V.T. provided critical tools and expertise. M.K. supervised the study, designed experiments, and wrote the manuscript. All authors provided critical comments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Riffelmacher, T., Giles, D.A., Zahner, S. et al. Metabolic activation and colitis pathogenesis is prevented by lymphotoxin β receptor expression in neutrophils. Mucosal Immunol 14, 679–690 (2021). https://doi.org/10.1038/s41385-021-00378-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-021-00378-7
This article is cited by
-
Exploring the glycolytic cross-talk genes between inflammatory bowel disease and colorectal cancer
Functional & Integrative Genomics (2023)
