
Abstract
CD4+ T cells contribute critically to a protective immune response during intestinal infections, but have also been implicated in the aggravation of intestinal inflammatory pathology. Previous studies suggested that T helper type (Th)1 and Th17 cells depend on de novo fatty acid (FA) synthesis for their development and effector function. Here, we report that T-cell-specific targeting of the enzyme acetyl-CoA carboxylase 1 (ACC1), a major checkpoint controlling FA synthesis, impaired intestinal Th1 and Th17 responses by limiting CD4+ T-cell expansion and infiltration into the lamina propria in murine models of colitis and infection-associated intestinal inflammation. Importantly, pharmacological inhibition of ACC1 by the natural compound soraphen A mirrored the anti-inflammatory effects of T-cell-specific targeting, but also enhanced susceptibility toward infection with C. rodentium. Further analysis revealed that deletion of ACC1 in RORγt+ innate lymphoid cells (ILC), but not dendritic cells or macrophages, decreased resistance to infection by interfering with IL-22 production and intestinal barrier function. Together, our study suggests pharmacological targeting of ACC1 as an effective approach for metabolic immune modulation of T-cell-driven intestinal inflammatory responses, but also reveals an important role of ACC1-mediated lipogenesis for the function of RORγt+ ILC.
Similar content being viewed by others


Introduction
Following antigen-specific activation, naive CD4+ T cells can differentiate into different types of effector cells, such as T helper type (Th)1, Th2 and Th17. This process requires a profound reprogramming of central cellular processes that enable cell growth, rapid clonal expansion, and the production of effector cytokines. To support these phenotypic changes T cells adapt their intrinsic metabolism, which includes the engagement of aerobic glycolysis and the upregulation of biosynthetic pathways. One hallmark of this shift toward an anabolic metabolism is the induction of de novo fatty acid (FA) synthesis (FAS).1
The first committed step of cellular FAS is the cytosolic conversion of acetyl-CoA into malonyl-CoA, a reaction that is catalyzed by acetyl-CoA carboxylase 1 (ACC1).2,3 Malonyl-CoA is then processed by FA synthase into long-chain FAs. We have previously demonstrated that targeting ACC1 in T cells disrupts the shuttling of glucose-derived carbons via glycolysis and the mitochondrial citric acid cycle (TCA) into de novo FAS, a metabolic process referred to as glycolytic-lipogenic pathway.4 Importantly, T-cell-specific ACC1 deletion reduced the potential of naive T cells to differentiate into both Th1 and Th17 cells and shifted their development toward regulatory T(reg) induction under Th17 conditions. In addition, ACC1-deficient T cells proved to be less pathogenic in vivo during a lethal model of acute graft-versus-host disease and in experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis.4,5,6 Of note, pharmacological inhibition of de novo FAS using the ACC-specific inhibitors TOFA or the myxobacteria-derived compound soraphen A (SorA) inhibited in vitro Th17 differentiation in a dose dependent manner and ameliorated Th17-mediated inflammation in EAE.4,5 Although the molecular basis for the dependence of Th17 but not Treg cells on de novo lipogenesis is not completely understood so far, these data suggest that ACC1 could be used as a pharmacological target for the treatment of T-cell-mediated autoimmune diseases.
Effector Th cells have been attributed to possess an important proinflammatory function in chronic inflammatory bowel disease (IBD). The two dominant clinical manifestations, ulcerative colitis (UC) and Crohn’s disease, show a high prevalence in western countries (1.3% in US) with raising incidence and increasing cost for the health care system. Studies have shown that Th17 cells are enriched in the mucosa of IBD patients and the amount of IL-17 directly correlates with disease severity in patients with UC,7 together suggesting a pathogenic role of Th17 cells in IBD. Importantly, recent research revealed that the Th17 lineage is heterogeneous and highly plastic. In this sense, Th17 cells activated via the IL-23 receptor produce additional factors like TNF-α and can transdifferentiate into IFN-γ-producing Th1-like cells with a pronounced inflammatory potential in colitis.8 Thus, strategies aiming for direct manipulation of the Th cell metabolism may have great potential to limit intestinal inflammatory tissue damage. Interestingly, RORγt+ innate lymphoid cells (ILC) producing the Th17-associated cytokines IL-17 and IL-22 are also enriched in IBD patients and have been suggested to possess a proinflammatory role in murine models of colitis.9,10 However, both IL-17 and IL-22 are important positive regulators of the intestinal barrier, exemplified by the critical role of RORγt+ ILC in the regulation of host-commensal homeostasis and defense against intestinal infections.7 Taking into account the complex interaction with the environment at this barrier site, interfering with intestinal immune functions may therefore also effect on host resistance in the gut.
In this study, we show that targeting of ACC1 minimizes intestinal inflammation in a transfer model of colitis, mainly by interfering with the early expansion of effector Th1 and Th17 cells. Both, genetic as well as pharmacological inhibition of ACC1 reduced T-cell-mediated pathology also in infection-associated intestinal inflammation. Importantly, while T-cell-specific deletion of ACC1 had only a minor effect on host protection during infection, we found that ACC1-mediated FAS controls IL-22 production by RORγt-expressing ILC and rendered ILC-specific ACC1-deficient mice more susceptible toward intestinal infection with C. rodentium. Together, our study suggests pharmacological targeting of ACC1 as a potential novel approach to interfere with T-cell-driven intestinal inflammatory responses. Yet, our data also clearly indicate an important role of ACC1 and de novo FAS for the host-protective function of ILC.
Results
T-cell-specific ACC1 deletion abrogates Th1- and Th17-mediated intestinal inflammation
In order to determine the potential of ACC1-deficient T cells to induce intestinal inflammation, we applied a T-cell-induced colitis approach, in which naive T cells initiate colitis when transferred into lymphocyte-deficient Rag2−/− recipient mice. Adoptive transfer of naive WT CD4+ T cells induced symptoms of severe colitis including diarrhea and wasting (Fig. 1a). In contrast, mice that received naive ACC1-deficient CD4+ T cells presented a normal growth with only minor symptoms of colitis. Histological assessment of the colon revealed high infiltration of inflammatory cells into the lamina propria (LP) and pronounced epithelial damage in mice that received WT cells, whereas signs of pathology were rarely detected in the group that received T cells isolated from T-cell-specific ACC1-deficient (TACC1) mice (Fig. 1b, c). Importantly, both the frequency and total CD4+ T-cell numbers were significantly reduced in the colon of Rag2−/− recipients that received ACC1-deficient T cells as compared with WT controls (Fig. 1d, e).

a Body weight curve of Rag2−/− mice injected with CD4+CD45RBhigh T cells from WT or TACC1 mice. Control mice received PBS. b Representative H&E staining of colon samples. Scale bar represents 100 μm. c Summary graph of colonic pathology score for the different experimental groups. d Representative flow cytometry plots and e quantification of live lamina Propria CD4+ T cells isolated from the colon of Rag2−/− mice transferred with CD4+CD45RBhigh T cells from WT or TACC1 mice. f Representative flow cytometry plots, g frequencies and total numbers of IL-17A-, IFN-γ-, and Foxp3-expressing CD4+ T cells isolated from cLP of both experimental groups at the peak of the colitis. Data are representative of three independent experiments (a, b, d, f) or pooled from three independent experiments (c, e, g). Horizontal bar represents mean. Error bars represent SEM. Student’s t test; ns nonsignificant, *p < 0.05, **p < 0.01.
To investigate the effect of ACC1 deletion on the in vivo differentiation of naive CD4+ T cells into effector populations, we isolated CD4+ cells from colonic LP (cLP) and analyzed them for effector cytokine production or the expression of Foxp3, as a marker for Treg cells. Although we did not observe major differences in the frequencies of colonic IFN-γ+, IL-17A+, and Foxp3+ CD4+ T-cell populations, the total number of effector Th1 and Th17 cells was significantly reduced in the absence of ACC1 expression, whereas the amount of Foxp3+ Treg cells was comparable between both groups (Fig. 1f, g). Together, these findings demonstrate that ACC1 deletion in T cells reduces infiltration of IL-17A+/IFN-γ+ effector T cells into the cLP and protects from colonic pathology.
ACC1 deficiency affects T-cell expansion in vivo due to defects in early proliferation
As our results demonstrated that ACC1-mediated FAS is important for the accumulation of inflammatory effector T cells and the development of colitis, we asked whether the decreased cellular infiltration of ACC1-deficient cells was due to impaired expansion of T cells upon transfer. To directly address this question in vivo, we adoptively cotransferred WT (CD90.1+) and ACC1-deficient (CD90.2+) naive CD4+ T cells in equal numbers into Rag2−/− recipient mice and evaluated the expansion of the transferred populations in cLP, spleen, and mesenteric lymph nodes at day 6, 12, and 22 post transfer. Our results show that ACC1 deletion led to a significantly reduced expansion in all analyzed organs at day 6 post transfer, and the ratio between ACC1-deficient and WT T cells remained low also at day 12 and 22 (Fig. 2a, b). The decreased capacity of ACC1-deficient T cells to expand under lymphopenic conditions may indicate a defect in cellular proliferation or cell survival. To test this, we assessed apoptosis of the transferred WT or ACC1-deficient T cells by annexin V/7-AAD staining. However, no differences were observed in the frequency of annexin V+ apoptotic cells in all analyzed organs and time points, although an enhanced frequency of necrotic (annexin V+/7-AAD+) ACC1-deficient T cells in the LP was detected at day 6 and day 12 post transfer (Fig. 2c and Supplementary Fig. 1).

WT (CD90.1+) and TACC1 (CD90.2+) derived CD4+CD45RBhigh T cells were transferred into Rag2−/− recipients in a 1:1 ratio. a Total numbers of WT or TACC1-derived CD4+ T cells recovered from the cLP, spleen, and mesenteric lymph nodes (mLN) of Rag2−/− recipients at day 6, 12, and 22 post transfer. b Representative flow cytometry plots of CD4+ T cells isolated from indicated organs of transferred animals at day 6 post transfer. c Frequencies of annexin V+7AAD− (apoptotic) and annexin V+7AAD+ (necrotic) cells among the CD90.1+ (WT) or CD90.2+ (TACC1) CD4+ T cells isolated from cLP, spleen, and mLN at day 6 post transfer. d, e CD4+CD45RBhigh T cells from (CD90.1+) and TACC1 (CD90.2+) mice were mixed in a 1:1 ratio and labeled with a proliferation dye (Cell Trace Violet) prior to transfer. d Histograms and e frequencies of Cell Tracelow cells among CD90.1+ (WT) or CD90.2+ (TACC1-derived) CD4+ T cells isolated from cLP, spleen, and mLN at d6 post transfer. f WT (CD90.1+) and TACC1 (CD90.2+) derived CD4+CD45RBhigh T cells were transferred into Rag2−/− recipients into 1:1 ratio. Recipient mice were administered intraperitoneally with BrdU 16 h prior to analysis. Frequencies of BrdU+ cells indicate the proliferated cells among the CD90.1+ (WT) or CD90.2+ (TACC1) CD4+ T cells isolated from cLP, spleen, and mLN at d12 and d22 post transfer. Data are representative of two (b, d) independent experiments or pooled from two to three (a, c, e, f) independent experiments. Horizontal bar represents mean. Error bars represent SEM. Student’s t test; ns nonsignificant, *p < 0.05, **p < 0.01.
To directly determine cellular proliferation during the early post transfer period, we labeled WT and TACC1-derived naive T cells with Cell Trace Violet. At day 6 post transfer, the majority of WT T cells had lost their Cell Trace labeling, indicating that they had undergone multiple rounds of divisions. In contrast, a significant fraction of the ACC1-deficient T cells remained Cell Trace positive in spleen and mesenteric lymph nodes, suggesting that these cells have a reduced capacity to proliferate during the priming phase in those organs (Fig. 2d, e). Cell cycle analysis of in vitro primed naive T cells revealed a delayed progression from the G0G1 phase toward the S phase in ACC1-deficient T cells, supporting the idea that inhibition of de novo FAS reduces the activation-induced cell cycling rate (Supplementary Fig. 2). Surprisingly, despite the diminished cell numbers of ACC1-deficient cells at day 12 or day 22 post transfer, we did not observe differences in the proliferative capacity in comparison to WT cells, neither in the LP nor in lymphoid organs, as indicated by similar 5-bromo-2′-deoxyuridine (BrdU) incorporation at these time points (Fig. 2f). In line with these findings, we did not observe differences in the transcriptome of WT and ACC1-deficient T cells isolated from the LP at day 22 post transfer (Supplementary Fig. 3), indicating that once the cells have reached this location, they are functionally independent of de novo FAS. Thus, the inability to perform cellular FAS seems to mainly affect cellular proliferation during the lymphopenia-induced priming and expansion phase of the cells in lymphoid organs, resulting in reduced infiltration and cell survival in the intestine.
Targeting ACC1 in T cells limits infection-associated intestinal inflammation
Since the transfer model of colitis relies on the expansion of T cells under lymphopenic conditions, we next investigated the effect of T-cell-specific deletion of ACC1 on intestinal inflammation in a T-cell sufficient environment. To this end, we orally infected TACC1 mice with the gut-specific pathogen Citrobacter rodentium.11,12 As expected, infection with C. rodentium caused massive CD4+ T-cell infiltration into the cLP of WT mice at day 11 post infection (Fig. 3a, b). In agreement with previous observations,4 TACC1 mice already exhibited a slight reduction in the amount of cLP CD4+ T cells during noninfected, homeostatic conditions. Still, infection with C. rodentium further enhanced the amount of CD4+ T cells also in the cLP of TACC1 mice. Yet, both the frequencies and total cell numbers remained substantially lower as compared with WT controls. Similar to our results in the transfer colitis model, we did not observe major differences in BrdU incorporation at the peak of the T-cell response (day 11) in the intestine, suggesting that ACC1 deletion rather affects early T-cell expansion (Supplementary Fig. 4).

Leukocytes were isolated from the cLP of WT or TACC1 mice before (uninfected) or on day 11 p.i. with C. rodentium and analyzed by flow cytometry. a Representative flow cytometry plots and b frequencies and total numbers of CD4+ T cells within live cLP cells. c Representative flow cytometry plots of IFN-γ-, IL-17A-, and Foxp3-expressing CD4+ T cells in the colon of mice from both groups at day 11 p.i. with C. rodentium. d Summary graph showing frequencies and total numbers of IFN-γ+, IL-17A+, and Foxp3+ CD4+ T cells. e Representative H&E staining of colon sections from WT and TACC1 mice on day 11 p.i. with C. rodentium. Scale bar represents 100 μm. f Summary graph of colonic pathology score for the different experimental groups. g Bacterial load in fecal, spleen, and liver lysates on day 11 p.i. h Survival curve of WT and TACC1 mice infected orally with C. rodentium. Data are representative of two to three independent experiments (a, c, e) or pooled from two to three independent experiments (b, d, f, g, h). Horizontal bar represents mean. Error bars represent SEM. Log-rank test (h) and Student’s t test; ns nonsignificant, *p < 0.05, **p < 0.01.
Both IFN-γ-producing Th1 cells and IL-17-secreting Th17 are critical effectors of the antimicrobial host response, but also contribute significantly to the intestinal pathology observed in this model.13,14,15,16,17 We observed that loss of ACC1 function in T cells also significantly affected the induction of a Th1/Th17 response in the colon of TACC1 mice (Fig. 3c, d). We also noticed reduced numbers of colonic Treg cells, probably as a consequence of the lower number of total CD4+ T cells in the colon of the infected TACC1 mice. Importantly, the dampened Th1/Th17 response was associated with milder intestinal pathology, as evidenced by the significantly reduced inflammatory infiltrates and epithelial hyperplasia in colonic histological samples of TACC1 mice (Fig. 3e, f). Although only present at low numbers, a similar trend for reduction was observed for the amount of IL-22 producing T cells (Supplementary Fig. 5).
We next tested whether interfering with the effector T-cell response may result in enhanced susceptibility toward the infection. To assess the effect on bacterial dissemination, we evaluated the bacterial load in feces, spleen, and liver lysates at day 11 p.i. Although TACC1 mice exhibited higher bacterial burden in the fecal content compared with WT mice, bacterial CFUs were comparable in both spleen and liver, indicating that TACC1 mice could control the infection locally (Fig. 3g). In line with these results, the majority of TACC1 mice survived the infection and eventually eradicated the pathogen (Fig. 3h). Taken together, our data demonstrate that blockade of T cell-intrinsic de novo FAS protects from Th1/Th17-mediated intestinal pathology during C. rodentium infection. Despite the lower numbers of CD4+ T cells under steady state, TACC1 mice can still exhibit expansion of cLP Th1 and Th17 populations that is, even though lower compared with the littermate controls, sufficient to control infection.
Pharmacological inhibition of ACC1 using SorA reduces intestinal inflammation but increases systemic bacterial dissemination
As our data so far indicated that genetic targeting of ACC1 in T cells efficiently dampens effector T-cell-mediated inflammation in the gut, we next assessed whether pharmacological blockade of ACC1 using the myxobacteria-derived specific ACC1 inhibitor SorA could improve T-cell-mediated pathology. We therefore infected WT mice with C. rodentium and treated them daily with SorA. Compared with the controls, SorA treated mice exhibited significantly lower CD4+ T-cell infiltration in the cLP both in terms of frequencies and total cell numbers (Fig. 4a, b). We furthermore observed reduced frequencies and numbers of IFN-γ-producing T cells in the cLP of animals that received SorA as compared with vehicle treated control mice (Fig. 4c, d). Even though the frequencies of Th17 and Treg cells were comparable between both groups, total numbers were also significantly reduced in the colon of the SorA treated mice, probably as a consequence of the decrease in total colonic CD4+ T-cell numbers. Comparable with what we observed before in TACC1 mice, pharmacological ACC1 targeting resulted in less severe inflammation-associated pathology in the colon of SorA treated animals, as illustrated by reduced inflammatory infiltration and epithelial hyperplasia (Fig. 4e, f). Together, these findings demonstrate that pharmacological inhibition of ACC1 efficiently downregulates the T-cell-mediated inflammatory response in the colon.

WT mice were orally infected with C. rodentium and treated either with vehicle or 20 mg/kg SorA twice per day subcutaneously until analysis at day 11 p.i. Leukocytes were isolated from the cLP and the T-cell response was analyzed by flow cytometry. a Representative flow cytometry plots of leukocytes and b frequencies and total numbers of cLP CD4+ T cells of vehicle and SorA treated mice. c Representative flow cytometry plots, d frequencies and total numbers of IFN-γ+, IL-17A+, and Foxp3+ CD4+ T cells isolated from cLP at day 11 p.i. e Representative H&E staining of colon sections of mice treated with vehicle and SorA on day 11 p.i. with C. rodentium. Scale bar represents 100 μm. f Summary graph of colonic pathology score for the different experimental groups. g Bacterial load in fecal, spleen, and liver lysates on day 11 p.i. Data are representative of two (a, c) or three (e) independent experiments, or pooled from two to three (b, d, f, g) independent experiments. Horizontal bar represents mean. Error bars represent SEM. Mann–Whitney U test; ns nonsignificant, *p < 0.05, **p < 0.01.
Importantly, however, assessment of the bacterial burden revealed enhanced C. rodentium-specific CFU counts not only locally in the gut, as observed before in the TACC1 mice, but also in the liver and, to a lesser extent also in the spleen of SorA treated mice, illustrating a defect in the local control and enhanced systemic dissemination of the pathogen (Fig. 4g). Thus, while our results highlight the capacity of pharmacological ACC1 inhibition to protect from Th1/Th17-mediated tissue pathology during C. rodentium infection, the increase in the systemic bacterial burden suggests that also other components of the antimicrobial host response may be affected by ACC1 targeting.
ACC1-mediated de novo FAS is critical for ILC, but not mononuclear phagocyte (MNP) function during intestinal infection
Intestinal MNP, which include dendritic cells (DC) as well as macrophages (MO), play an important role in the induction of both innate and adaptive immune responses upon infection with C. rodentium.18,19,20 Notably, it has been demonstrated that upon activation, both DC and MO acquire a highly glycolytic profile that fuels de novo FA synthesis.21,22,23 To test whether ACC1-mediated de novo lipogenesis is critical for their immune function upon intestinal infections, we generated DC- and MO-specific ACC1-deficient mice (CD11cACC1−/− and LysMACC1−/−, respectively) and infected them with C. rodentium. However, CFU counts in feces, spleen, and liver of both DC and MO-specific ACC1-deficient mice were comparable with WT controls (Fig. 5a), indicating that targeting ACC1-mediated lipogenesis in MNP may not account for the increased systemic bacterial dissemination that was observed upon pharmacological ACC1 inhibition.

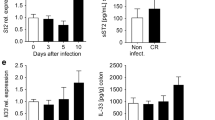
a Bacterial load in feces, spleen, and liver lysates of WT, CD11cACC1−/− and LysMACC1−/− mice on day 10 p.i. with C. rodentium. b Survival curve of Rag2−/− and RORγt-specific ACC1-deficient Rag2−/− mice (RagRorcACC1−/−) infected with C. rodentium. c Frequencies and numbers of cLP live Lin−CD90.2+RORγt+ ILC cells isolated from infected Rag2−/− and RagRorcACC1−/− mice on day 4 p.i. As lineage marker, antibodies against TCRβ, TCRγδ, CD19, Gr-1, Ter119, NK1.1, CD11c, and CD11b were included. d Representative flow cytometry plots, e frequencies and mean fluorescence intensity (MFI) of IL-22+ cells within live Lin−CD90.2+RORγt+ ILC population. f Gene expression of RegIIIβ and RegIIIγ in intestinal epithelial cells isolated on day 4 p.i. with C. rodentium from the cLP of Rag2−/− and RagRorcACC1−/−. Data shown as mean relative expression to Actb. g Bacterial load in feces spleen and liver lysates isolated from infected Rag2−/− and RagRorcACC1−/− mice treated intraperitoneally either with PBS or 0,1 mg/kg rm-IL-22 once per day until analysis at day 8 p.i. h Frequencies and numbers of cLP live Lin−CD90.2+RORγt+ ILC isolated from infected WT mice treated either with Vehicle or 20 mg/kg SorA twice per day subcutaneously until analysis at day 4 p.i. As lineage marker, antibodies against TCRβ, TCRγδ, CD19, Gr-1, Ter119, NK1.1, CD11c, and CD11b were included. i Frequencies, numbers, and mean fluorescence intensity (MFI) of IL-22-expressing RORγt+ ILC. Data are representative of, three (d), or pooled from two (a, b, g, h, i) or three (c, e, f) independent experiments. Horizontal bar represents mean. Error bars represent SEM. One-way ANOVA with Bonferroni’s multiple comparison test (a, g), log-rank test (b), and Student’s t test; ns nonsignificant, *p < 0.05, **p < 0.01.
Previous studies have highlighted the crucial role of IL-22 and RORγt+ ILC, which represent the dominant local source of this cytokine, for the early host defense during C. rodentium infection. Both, depletion of ILC or neutralization of IL-22 following C. rodentium infection leads to impaired epithelial barrier function and eventually mortality due to polymicrobial sepsis.24 To determine whether ACC1-mediated de novo FAS is important for the function of RORγt+ ILC during infection with C. rodentium, we genetically deleted ACC1 in RORγt-expressing cells by crossing Rorc-cre mice to ACC1lox/lox mice. Since RORγt is expressed in developing T cells during the double positive stage in the thymus,25 we further bred RorcACC1−/− mice to a lymphocyte-deficient Rag2−/− background (RagRorcACC1−/−), in order to restrict ACC1 deletion specifically to RORγt+ ILCs. Interestingly, we found that RagRorcACC1−/− mice succumbed to the infection with C. rodentium at an earlier time point than Rag2−/− littermate controls, indicating that lack of ACC1-mediated lipogenesis in RORγt+ ILC interferes with innate host response mechanisms (Fig. 5b). To further assess the effect of ACC1 targeting in RORγt+ ILC, we isolated cLP leukocytes from infected mice at day 4 p.i. While no differences in frequencies or numbers of cLP RORγt+ ILC were visible between mice of both genotypes (Fig. 5c and Supplementary Fig. 6), we found that the capacity of RORγt+ ILC to produce IL-22 in the absence of ACC1 was significantly impaired (Fig. 5d, e).
IL-22 is known to enhance the expression of antimicrobial factors by the epithelium and to support the integrity of the intestinal barrier.26,27 While we did not observe major differences in intestinal epithelial permeability and expression of tight-junction proteins claudin-2 and occludin (Supplemental Fig. 7), we found that the expression of the antimicrobial peptides RegIIIβ and RegIIIγ was significantly reduced in epithelial cells derived from C. rodentium-infected RagRorcACC1−/− mice (Fig. 5f). In accordance, C. rodentium-infected RagRorcACC1−/− mice displayed higher pathogen load in the feces, but also enhanced systemic spread at day 8 p.i. (Fig. 5g), which was even more pronounced at later time points (Supplementary Fig. 8). Notably, administration of IL-22 during the early course of infection compensated for the lack of ILC-derived IL-22 and reduced the bacterial load to levels comparable with ACC1-sufficient Rag2−/− controls in feces and, at least partially, in the spleen (Fig. 5g). We furthermore tested whether treatment with SorA would recapitulate the effect on RORγt+ ILC in infected immunocompetent wild-type mice. While again we did not see major differences in the frequencies or total numbers of RORγt+ ILC, SorA treatment nevertheless reduced the capacity of these cells to produce IL-22 (Fig. 5h, i) confirming that de novo FAS supports antimicrobial ILC-derived IL-22 responses in vivo.
We next assessed the impact of ACC1-inihbition on the biology of ILC by using the recently described ILC3 cell line MNK3.28 Although addition of SorA during a 3 day culture period reduced the viability of the cells in a dose dependent manner, we did not observe an impact on the proliferative capacity or the stability of RORγt expression as compared with DMSO treated cells (Fig. 6a). Importantly, short-term addition of SorA for 6–24 h during restimulation of the cells with IL-1β and IL-23 significantly reduced both IL-22 and IL-17 production (Fig. 6b, c), however, without affecting the viability, proliferative capacity or stability of RORγt expression of the cells (Fig. 6d). Thus, our data suggest that, unlike what we observed in T cells, inhibition of ACC1 in RORγt+ ILC has a more direct impact on the activation-induced production or release of effector cytokines. In summary, our results demonstrate a critical role for ACC1-mediated de novo FAS in RORγt+ ILC. Blocking this pathway affects IL-22 and IL-17A production by these cells and, as a consequence, dampens epithelial defense mechanisms and accelerates mortality upon infection with C. rodentium.

a Summary graphs showing cellular viability, RORγt frequencies, and Ki67 expression of MNK3 cells after 3 days in culture supplemented with either DMSO or SorA at indicated concentrations. b Representative flow cytometry plots of IL-22- and IL-17A-expressing MNK3 cells after 24 h restimulation with IL-23 and IL-1β in the presence of either DMSO or 10 μΜ SorA. c Summary graph showing frequencies of IL-22+ and IL-17A+ MNK3 cells after 6, 16, and 24 h restimulation with IL-23 and IL-1β in the presence of either DMSO or 10 μΜ SorA. Data are representative from two (b) or pooled from two independent experiments (a, c, d). Horizontal bar represents mean. Error bars represent SEM. Student’s t test; ns nonsignificant, *p < 0.05, **p < 0.01.
Discussion
The gastrointestinal tract is populated by different immune cells, including MO, DC, Teff cells, Treg cells, and ILC, that are tightly regulated to ensure the induction of protective immunity toward potential pathogens and to maintain tolerance toward ingested antigens and commensal microbes. Disruption of this finely tuned balance is associated with the onset of chronic inflammation and the development of IBD. In this study, we aimed to define a novel approach for intestinal immune intervention by targeting the cell-intrinsic lipid synthesis pathway in T cells using both cell type-specific genetic ablation of ACC1 and pharmacological inhibition by the ACC1-specific inhibitor, SorA. Previous work from our lab has demonstrated that ACC1 deletion in T cells abrogated Th17-mediated autoimmune disease by diminishing Th1 and Th17 accumulation in the CNS in an experimental model of multiple sclerosis.4 In line with this, our present study demonstrates that T-cell-specific deletion of ACC1 prevented autoimmune colitis progression and protected mice from diarrhea and wasting. Furthermore, even though adoptive transfer of ACC1-sufficient cells caused transmural inflammation, ACC1-deficient T cells failed to induce such pathology in the colon. Although the capacity of T cells to produce effector cytokines was not affected by the inhibition of cell-intrinsic lipogenesis, ACC1-deficient Th1 and Th17 cells were significantly diminished in the LP. In contrast, Treg cell numbers were only mildly affected despite the dramatic decrease of the overall Th populations, indicating that Treg cells are less affected by inhibition of this metabolic pathway in vivo.
To get further insight into the mechanism that drives this reduction of LP Teff cells, we assessed the cellular proliferation and survival of the cells in vivo. Previous in vitro data indicated a defect in T-cell proliferation as a consequence of FAS inhibition.4,6 We show now that inhibition of FAS also affects cellular proliferation in vivo, demonstrating that even shortly after transfer, inhibition of de novo FAS affected the accumulation and expansion of T cells in the LP, spleen, and mesenteric lymph nodes, maintaining them in dramatically low numbers. Cell trace labeling at d6 post transfer revealed impaired proliferation, mainly in lymphoid organs. Surprisingly, at later time points when T cells perform their effector function, inhibition of FAS was dispensable for cellular proliferation, cytokine production, or survival of the cells, highlighting the crucial role of this metabolic pathway during initial T-cell expansion. This is in line with previous data in CD8+ T cells,29 suggesting that the differentiation and expansion of both CD4+ and CD8+ subsets is metabolically controlled by FAS. Nevertheless, further studies are necessary to better define the mechanism by which ACC1-mediated lipogenesis is linked to early Teff cell differentiation in vivo.
In addition to our findings in the transfer colitis model, we observed that T-cell-specific lack of ACC1 restrained the development of an inflammatory Th17/Th1-mediated response also in a T-cell sufficient environment upon infection with C. rodentium. Yet, TACC1 mice were still able to exhibit a sizable expansion of cLP Th1 and Th17 populations that was sufficient to maintain host protection, but resulted in less immune-mediated intestinal pathology. Interestingly, earlier findings demonstrated enhanced susceptibility of TACC1 mice to mycobacterial infection, mainly due to diminished Th1 and CD8+ T-cell responses.30 While we indeed observed clear reductions in the numbers of colonic IL-17 and IFN-γ-producing T cells upon C. rodentium infection, their contribution to bacterial clearance may be less critical than for intracellular infection with mycobacterium, as it has been proposed that IFN-γ is dispensable for surviving infection with C. rodentium.13,31
To experimentally address whether pharmacologic inhibition of ACC1 can provide therapeutic benefit in the context of intestinal inflammation, similarly to T-cell-specific ACC1 deletion, we applied the myxobacteria-derived metabolite SorA. In line with the results of T-cell-specific genetic deletion of ACC1, SorA was already shown to affect T-cell proliferation and the in vitro differentiation of T1 and Th17 cells.4,6 In addition, in vivo treatment with the SorA derivative Sor-S1036 and the ACC1 inhibitor TOFA ameliorated EAE,4,5 yet the impact of ACC1 inhibition during intestinal inflammation has not been tested so far. Our data showed that, similar to genetic targeting of ACC1 in T cells, treatment with SorA reduced the infiltration of CD4+ T cells, and eventually Th1 and Th17 cells in the cLP during C. rodentium infection. Notably, this reduction of Th cell-derived IL-17A and IFN-γ was again associated with reduced histologic signs of inflammation in the colon.
Our finding that pharmacological ACC1 inhibition caused increased systemic bacterial dissemination, as compared with the results of T-cell-specific ACC1 deletion, suggested a critical role of de novo FAS for the function of additional cell types in this model. We first addressed whether ablation of ACC1 in intestinal MNP would interfere with C. rodentium eradication, since these cells activate not only ILC, but also the Th17-mediated host response during intestinal infections by producing cytokines such as IL-1β, IL-6, and IL-23. Intriguingly, a recent study suggested also that GM-CSF DC require de novo FAS upon TLR stimulation for activation and cytokine production.23 In our work, we however found that inhibition of FAS in DC and/or MO did not interfere with host resistance to C. rodentium. This discrepancy may, at least partially, be explained by the use of different experimental approaches. While we have used a genetic system for the cell type-specific ablation of ACC1, the study by Everts et al. merely relied on the usage of chemical inhibitors.23 Nevertheless, our data are in line with previous findings from our group showing that ACC1-mediated lipogenesis in DC and MO is dispensable to induce protective immune responses during infection with Mycobacterium bovis BCG.30
RORγt+ ILC have been appreciated as important players in the regulation of intestinal homeostasis and tissue repair, but also for the control of intestinal infections. Even though their biology and effector function have been extensively studied over the previous years,31 to date little is known about the metabolic pathways that sustain RORγt+ ILC development and local effector function. It has been suggested that microbiota- and food-derived products can affect the maintenance and expansion of RORγt+ ILC. In line with this hypothesis, several studies demonstrated that retinoic acid supports the homing and maintenance of RORγt+ ILCs.32,33,34 Likewise, microbial or dietary-derived Ahr ligands control their development and maintenance,35,36 whereas the short chain FA butyrate contributed to the decrease of RORγt+ ILCs mainly by downregulating RORγt expression.37 To investigate the effect of de novo FAS specifically on RORγt+ ILCs, we used Cre-recombinase-mediated deletion of ACC1 in all innate RORγt cells in RAG-deficient mice. Even though absence of adaptive immunity leads to mortality due to uncontrolled infection,38 genetic ablation of ACC1 in innate RORγt+ cells accelerated the bacterial translocation to systemic sites and susceptibility toward the infection. In contrast to our findings in T cells, we did not observe significant reduction in the cell numbers of ACC1-deficient RORγt+ ILC upon C. rodentium infection. Although we observed that long-term in vitro treatment of the ILC3 cell line, MNK3, with SorA affected the viability of the cells, our results suggest that de novo FAS is largely dispensable for survival and proliferation of RORγt+ ILC. Instead, our data indicate that ACC1-mediated de novo FAS is important for the production of IL-22 and IL-17 during short term in vitro stimulation with IL-23 and IL-1β, as well as upon infection-mediated activation in vivo. This is in line with recent findings showing distinct metabolic functions of ILC3, such as reliance on mTOR1-mediated induction of glycolysis and ROS production to sustain ILC3 function upon activation.39 Interestingly, the same study also demonstrated upregulation of de novo FAS in both murine and human ILC3. Yet, while direct inhibition of mTORC1 and ROS interfered with the level of RORγt expression and the proliferative capacity of ILC3, we did not observe an impact of ACC1 inhibition on these parameters. It was proposed in that respect that lipogenesis may facilitate the expansion of the endoplasmic reticulum and Golgi membranes to support effector cytokine production and secretion in immune cells.23 In addition, interference with FA metabolism may affect posttranslational modification of proteins, such as palmitoylation and myristoylation, which has been implicated for instance in the correct localization of adapter molecules to the membrane upon TCR-mediated activation of T cells.40,41,42 However, whether any of these described mechanisms can explain the role of ACC1-dependent FAS in RORγt+ ILC remains to be addressed in future studies.
In summary, our study suggests that cellular de novo FAS is an important metabolic checkpoint controlling RORγt+ ILC and Th1/Th17 responses in the gut. While our data indicate that the mechanisms by which FAS dictates the fate and function of ILC and Th cells are different, metabolic intervention of both innate and adaptive lymphocytes has nevertheless the potential to influence intestinal inflammation and in addition to limit tissue pathology, offering a novel therapeutic approach in the context of chronic inflammation. Indeed, pharmacological treatment with the ACC1-specific inhibitor SorA limited intestinal pathology acting possibly on both innate and adaptive lymphocytes. However, targeting responses that mediate protective immunity might impact on pathogen clearance, as depicted by the increased bacterial dissemination upon SorA treatment. Thus, dosage optimization along with a combined treatment with antibiotics could potentially balance pathogen eradication with the protection of the infected tissue.
Methods
Animal models
CD4Cre/+ mice were crossed to ACC1lox/lox mice43 to generate TACC1 mice. CD11c-cre43 and LysM-Cre44 mice were crossed to ACC1lox/lox mice to generate DC- and macrophage-specific ACC1-deficient mice. Rorc-Cre45 Rag2−/− mice were crossed to ACC1 lox/lox Rag2−/− to generate RORγt+ ILC-specific ACC1-deficient (RagRorcACC1−/−) mice. All mice were bred and maintained on C57BL/6J background under specific pathogen-free conditions in our animal facilities (TWINCORE, Hannover, Germany; or Helmholtz Centre for Infection Research Braunschweig, Germany). For all experiments, 4- to 27-week-old mice with age- and gender-matched littermates were used. All animal experiments were performed in compliance with the German animal protection law (TierSchG, BGBl. I S. 1206, 1313, 2006/05/18). All mice were housed and handled in accordance with good animal practice as defined by Federation of European Laboratory Animal Science Associations and the national animal welfare body Gesellschaft für Versuchstierkunde/Society for Laboratory Animal Science. All animal experiments were approved by the Lower Saxony Committee on the Ethics of Animal Experiments as well as the responsible state office (Lower Saxony State Office of Consumer Protection and Food Safety) under the permit numbers 33.9–42502–04–15/1851 and 16/2329.
Adoptive transfer and in vivo cell proliferation
WT, ACC1-deficient or congenic CD90.1+CD4+ T cells were enriched from spleen and lymph nodes using the Dynabeads Untouched Mouse CD4+ Cells system (Thermo Fisher Scientific). Naive CD4+ T cells (CD4+ CD8−CD11c−CD19−CD25−CD45RBhigh) cells were sorted by flow cytometry and resuspended in PBS. A total of 4 × 105 cells were adoptively transferred to Rag2−/− mice aged 4–8 weeks by intraperitoneal injection. For cotransfer experiments, naive CD4+ T cells from TACC1 (CD90.2+) and CD90.1+ congenic mice were mixed in a 1 to 1 ratio in PBS and 4 × 105 cells were adoptively transferred to Rag2−/− mice intraperitoneally. To evaluate in vivo cell proliferation, cells were labeled with Cell Trace Violet Cell Proliferation dye (Life Technologies) according to the manufacturer’s instructions prior to transfer. Alternatively, T-cell transferred Rag2−/− mice were administered i.p. with 200 μl of BrdU from APC BrdU Flow Kit (BD) at day 12 and 22 post transfer and analyzed 16 h later.
MNK3 cell culture and activation
MNK3 cells28 were cultured in DMEM with 10% FCS, 10 µM HEPES, 55 µM 2-mercaptoethanol (Life Technologies/Gibco), 50 µg/ml gentamicin, 100 U/ml penicillin, 100 µg/ml streptomycin (Merck), 10 ng/ml IL-7, and 10 ng/ml IL-15 (Peprotech). Cells were maintained in 37 °C supplied with 5% CO2 and subcultured every 3–4 days. For long-term SorA treatment, 1 × 105 cells were seeded into a 48-well cell-culture plate supplied with 500 µl culture medium without IL-15. DMSO or SorA was added at indicated concentration on the day of seeding. For short-term stimulation and cytokine detection, 2 × 105 cells were seeded into 96-well flat bottom cell-culture plates and supplied with culture medium without IL-15. Overall 20 ng/ml IL-1β (Peprotech), 40 ng/ml IL-23 (R&D systems), brefeldin A (eBioscience/Thermo Fisher Scientific), PMA (100 ng/ml, Sigma), and ionomycin (1 μg/ml, Sigma) were added together with either DMSO or SorA at the indicated concentrations. Analysis was performed at 6, 16, or 24 h after stimulation.
C. rodentium infection and in vivo treatments
Oral infections with C. rodentium ICC18012 were performed as described previously.16 In brief, 2 × 109 or 1 × 1010 bacteria were applied in 100 μl PBS intragastrically by gavage. For evaluation of the bacterial burden in colonic feces, stools from infected mice were collected into a preweighed Luria Bertani medium (Roth) containing tube. Feces were then weighed, homogenized, and titrated. Series of fecal dilution were added on MacConkey Agar (Roth) containing Kanamycin (Roth) and then cultured at 37 °C for 1–2 days before counting. Bacterial burden was calculated after normalization to the weight of stool. For bacterial burden in livers and spleens, organs were homogenized in 1 ml PBS. Homogenates were further titrated, plated on MacConkey Agar containing Kanamycin, and incubated at 37 °C for 1–2 days before counting. SorA was obtained from the Helmholtz Centre for Infection Research and was applied at a concentration of 20 mg/kg by subcutaneous injection twice per day. Recombinant murine IL-22 (Peprotech) was applied at a concentration of 0.1 mg/kg by intraperitoneal injection once per day.
Histopathological scoring
Histopathological assessment was performed as described previously.46,47 Briefly, 5 μm colonic sections were fixed in Roti®-Histofix 4% (Roth) and embedded in paraffin. Following haematoxylin and eosin (H&E) staining they were then examined in a blinded manner. Tissue sections were assessed for epithelial hyperplasia (score based on percentage above the height of the control, 0 = no change, 1 = 1–50%, 2 = 51–99%, 3 = 100%) and mononuclear cell infiltration (0 = none, 1 = mild, 2 = moderate, 3 = severe). Maximum score was 6. Samples were imaged under a microscope (Carl Zeiss) and processed with Nuance software 2.10.0 (Carl Zeiss).
Cell isolation from organs
Single-cell suspensions were prepared from spleen, mLN, or pLN by smashing organs through a 100 μm mesh. Lysis of erythrocytes was performed for spleens. Intestinal epithelial cells and cLP cells were isolated as described previously.16 In brief, colon was physically emptied, opened longitudinally, and cut into 2–3 cm pieces. Tissue pieces were incubated in PBS containing 30 mM EDTA (Roth) for 30 min on ice, washed vigorously with PBS to remove remaining mucus. For epithelial cell preparation the colon segments were washed with PBS containing 10% fetal calf serum, and epithelial cells were then lysed in TRIzol (Life Technologies) for RNA isolation and gene expression analysis. Remaining tissue was further cut into 1–2 mm pieces, and digested in prewarmed Iscove’s modified Dulbecco’s medium (Life Technologies/Gibco) containing 1 mg/ml collagenase D (Roche Diagnostics) and 100 μg/ml DNaseI (Roche). The supernatant was filtered and the remaining tissue was smashed through a 100 μm mesh. LP cells were separated using a 40%/80% gradient (Percoll solution, GE Healthcare; 900 g, 20 min, 20 °C, no break). The interphase was harvested and washed with PBS containing 2% fetal calf serum. Cells were resuspended in PBS containing 2% fetal calf serum or medium containing 5% fetal calf serum for in vitro restimulation before extra/intracellular staining. For intracellular IL-17 or IFN-γ staining, cells were stimulated in vitro with 1 μg/ml ionomycin and 0.1 μg/ml phorbol-12-myristate-13-acetate (both from Sigma-Aldrich) for 2 h followed by 5 μg/ml brefeldin A (Sigma-Aldrich) for 2 h. For IL-22 staining, stimulation was performed in the presence of 40 ng/ml rm-IL-23 (R&D) and following 2 h incubation with brefeldin A.
Flow cytometry
Cell suspensions were incubated with PBS containing 0.2% bovine serum albumin and 1% anti-mouse CD16/CD32 antibody (BioXcell, West Lebanon, NH) for 5 min on ice. Live/dead staining was performed in PBS using the LIVE/DEAD Fixable Dead Cell Stain Kit (Life Technologies/Thermo Fisher Scientific) according to the manufacturer’s recommendations, or 7-AAD prior to measurement. For apoptosis assessment, the Annexin V Apoptosis Detection Kit eFluor® 450 (Life Technologies/Thermo Fisher Scientific) was used according to the manufacturer’s recommendations. Surface staining was performed for 25 min on ice in PBS containing 0.25% BSA (Roche), 0.02% NaN3 (Roth), and 2 mM EDTA (Roth). For intracellular staining of cytokines or transcription factors cells were fixed and stained using Foxp3/Transcription Factor Fixation/Permeabilization Kit (eBioscience/Thermo Fisher Scientific) according to the manufacturer’s instructions. For BrdU intracellular staining we used the APC BrdU Flow Kit (BD) according to the manufacturer’s instructions. Cell sorting was performed on MoFlo XDP, FACSAria IIu, or FACSAria Fusion (BD Biosciences) in the cell sorting facility of Hannover Medical School. For flow cytometry analysis, cells were acquired in house on a LSR II (BD) or Cyan ADP (Beckman Coulter) and data were analyzed using the FlowJo software (Tree Star, Ashland, OR). Monoclonal antibodies specific to the following mouse antigens and labeled with the indicated fluorescent markers were used: CD4-eFluor450 (RM4–5), CD4-PE (GK1.5), CD11c-eFluor660 (N418), CD19-APC (eBio1D3), CD8a-Alexa647 (53–6.7), CD45RB-FITC (C363.16A), CD45RB-PE (C363.16A), CD3e-FITC (145–2C11), CD3e-PerCP-Cy5.5 (145–2C11), CD3-APC-eFluor780 (17A2), CD90.1-APC-eFluor780 (HIS51), CD90.1-eFluor450 (HIS51), CD90.2-PE-Cy7 (53–2.1), CD90.2-FITC (53–2.1), IL-17A-APC or PE-Cy7 (eBio17B7), IFN-γ-FITC (XMG1.2), IFN-γ-PE-Cy7 (XMG1.2), Foxp3-PE (FJK-16s), Foxp3-eFluor450 (FJK-16s), CD11b-APC (M1/70), NK1.1-APC (PK136), TCRβ-APC (H57–597), TCRβ-APC-eFluor780 (H57–597), TCRγδ-APC (GL-3), Gr-1(Ly-6G)-eFluor660 (RB6–8C5), Ter119-APC (TER-119), IL-22-PE (1H8PWSR), Ki67-APC (SolA15) from eBioscience/Thermo Fisher Scientific and RORγt-BV421 (Q31–378(RUO)) from BD Biosciences.
Gene expression analysis
RNA from IEC was isolated using TRIzol and transcribed into cDNA using SuperScript III Reverse Transcriptase Kit (both from Thermo Fisher Scientific). Real-time PCR was performed using iQ SYBR Green Supermix (Bio-Rad) on a LightCycler 480 II (Roche). All procedures were performed according to the manufacturer’s instructions. For expression analysis of Actb, RegIIIß, and RegIIIγ primers were obtained from Eurofins MWG Operon with following sequences: Actb fwd 5′-TGTTACCAACTGGGACGACA-3′, Actb rev 5′-GGGGTGTTGAAGGTCTCAAA-3′, Reg3b fwd 5′-ATGGCTCCTACTGCTATGCC-3′, Reg3b rev 5′-GTGTCCTCCAGGCCTCTT-3′, Reg3g fwd 5′-CCTTCCTCTTCCTCAGGCAAT-3′, Reg3g rev 5′-TAATTCTCTCTCCACTTCAGAAATCCT-3′. Gene expression was normalized to Actb and log2 transformed.
Statistical analysis
Data analyses were performed using GraphPad Prism Software version 5.03 (GraphPad Software) and statistics were calculated using Student’s t test for normally distributed values. Otherwise Mann–Whitney U test was used as indicated in the figure legends. Log-rank test was used to analyze survival distributions. One-Way ANOVA with Bonferroni’s multiple comparison test was used for experiments with more than two groups compared. p values were considered significant as follows: *p < 0.05 and **p < 0.01, ***p < 0.001, ****p < 0.0001.
References
Lochner, M., Berod, L. & Sparwasser, T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. https://doi.org/10.1016/j.it.2014.12.005 (2015).
Wakil, S. J. & Abu-Elheiga, L. A. Fatty acid metabolism: target for metabolic syndrome. J. Lipid Res. 50(Suppl), S138–S143 (2009).
Tong, L. Structure and function of biotin-dependent carboxylases. Cell Mol. Life Sci. 70, 863–891 (2013).
Berod, L. et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 20, 1327–1333 (2014).
Endo, Y. et al. Obesity drives Th17 cell differentiation by inducing the lipid metabolic kinase, ACC1. Cell Rep. https://doi.org/10.1016/j.celrep.2015.07.014 (2015).
Raha, S. et al. Disruption of de novo fatty acid synthesis via acetyl-CoA carboxylase 1 inhibition prevents acute graft-versus-host disease. Eur. J. Immunol. https://doi.org/10.1002/eji.201546152 (2016).
Kempski, J., Brockmann, L., Gagliani, N. & Huber, S. TH17 cell and epithelial cell crosstalk during inflammatory bowel disease and carcinogenesis. Front. Immunol. 8, 1373 (2017).
Ueno, A. et al. Th17 plasticity and its relevance to inflammatory bowel disease. J. Autoimmun. 87, 38–49 (2018).
Buonocore, S. et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375 (2010).
Geremia, A. et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 208, 1127–1133 (2011).
Koroleva, E. P. et al. Citrobacter rodentium-induced colitis: a robust model to study mucosal immune responses in the gut. J. Immunol. Methods https://doi.org/10.1016/j.jim.2015.02.003 (2015).
Wiles, S. et al. Organ specificity, colonization and clearance dynamics in vivo following oral challenges with the murine pathogen Citrobacter rodentium. Cell Microbiol. 6, 963–972 (2004).
Shiomi, H. et al. Gamma interferon produced by antigen-specific CD4+ T cells regulates the mucosal immune responses to Citrobacter rodentium infection. Infect. Immun. 78, 2653–2666 (2010).
Simmons, C. P. et al. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J. Immunol. 168, 1804–1812 (2002).
Mangan, P. R. et al. Transforming growth factor-β induces development of the TH17 lineage. Nature 441, 231–234 (2006).
Wang, Z. et al. Regulatory T cells promote a protective Th17-associated immune response to intestinal bacterial infection with C. rodentium. Mucosal Immunol. 7, 1290–1301 (2014).
Ishigame, H. et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30, 108–119 (2009).
Satpathy, A. T. et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat. Immunol. 14, 937–948 (2013).
Schreiber, H. A. et al. Intestinal monocytes and macrophages are required for T cell polarization in response to Citrobacter rodentium. J. Exp. Med. 210, 2025–2039 (2013).
Manta, C. et al. CX(3)CR1(+) macrophages support IL-22 production by innate lymphoid cells during infection with Citrobacter rodentium. Mucosal Immunol. 6, 177–188 (2013).
Rodríguez-Prados, J.-C. et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J. Immunol. 185, 605–614 (2010).
Krawczyk, C. M. et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749 (2010).
Everts, B. et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 15, 323–332 (2014).
Satoh-Takayama, N. et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29, 958–970 (2008).
Sun, Z. et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science 288, 2369–2373 (2000).
Kinnebrew, M. A. et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity 36, 276–287 (2012).
Hanash, A. M. et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity 37, 339–350 (2012).
Allan, D. S. J. et al. An in vitro model of innate lymphoid cell function and differentiation. Mucosal Immunol. 8, 340–351 (2015).
Lee, J. et al. Regulator of fatty acid metabolism, acetyl coenzyme A carboxylase 1, controls T cell immunity. J Immunol. https://doi.org/10.4049/jimmunol.1302985 (2014).
Stüve, P. et al. De novo fatty acid synthesis during mycobacterial infection is a prerequisite for the function of highly proliferative T cells, but not for dendritic cells or macrophages. Front. Immunol. 9, 495 (2018).
Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell 174, 1054–1066 (2018).
Mielke, L. A. et al. Retinoic acid expression associates with enhanced IL-22 production by γδ T cells and innate lymphoid cells and attenuation of intestinal inflammation. J. Exp. Med. 210, 1117–1124 (2013).
Kim, M. H., Taparowsky, E. J. & Kim, C. H. Retinoic acid differentially regulates the migration of innate lymphoid cell subsets to the gut. Immunity 43, 107–119 (2015).
Goverse, G. et al. Vitamin A controls the presence of RORγ+ innate lymphoid cells and lymphoid tissue in the small intestine. J. Immunol. 196, 5148–5155 (2016).
Qiu, J. et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 36, 92–104 (2012).
Kiss, E. A. et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565 (2011).
Kim, S.-H., Cho, B.-H., Kiyono, H. & Jang, Y.-S. Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer’s patches. Sci. Rep. 7, 3980 (2017).
Vallance, B. A., Deng, W., Knodler, L. A. & Finlay, B. B. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect. Immun. 70, 2070–2081 (2002).
Di Luccia, B., Gilfillan, S., Cella, M., Colonna, M. & Huang, S. C. C. ILC3s integrate glycolysis and mitochondrial production of reactive oxygen species to fulfill activation demands. J. Exp. Med. 216, 2231–2241 (2019).
van’t Hof, W. & Resh, M. D. Dual fatty acylation of p59(Fyn) is required for association with the T cell receptor zeta chain through phosphotyrosine-Src homology domain-2 interactions. J. Cell Biol. 145, 377–389 (1999).
Zhang, W., Sloan-Lancaster, J., Kitchen, J., Trible, R. P. & Samelson, L. E. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92, 83–92 (1998).
Kabouridis, P. S., Magee, A. I. & Ley, S. C. S-acylation of LCK protein tyrosine kinase is essential for its signalling function in T lymphocytes. EMBO J. 16, 4983–4998 (1997).
Mao, J. et al. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc. Natl Acad. Sci. USA 103, 8552–8557 (2006).
Clausen, B. E., Burkhardt, C., Reith, W., Renkawitz, R. & Förster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 (1999).
Eberl, G. & Littman, D. R. Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science 305, 248–251 (2004).
Friedrich, C. et al. MyD88 signaling in dendritic cells and the intestinal epithelium controls immunity against intestinal infection with C. rodentium. PLoS Pathog. 13, e1006357 (2017).
Yang, B.-H. et al. Foxp3+T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. 9, https://doi.org/10.1038/mi.2015.74 (2016).
Acknowledgements
This work was supported by grants from the German Research Foundation (DFG LO1415/7–1 and LO1415/8–1 to M.L.). We thank M. Swallow and J. Bartel for technical assistance and A. Dhillon-Labrooy for critical reading of the manuscript. We would like to acknowledge the assistance of the Cell Sorting Core Facility of the Hannover Medical School supported in part by Braukmann-Wittenberg-Herz-Stiftung and Deutsche Forschungsgemeinschaft.
Author information
Authors and Affiliations
Contributions
P.M., F.K., C.-w.L., and M.G. performed the experiments. D.S.J.A., J.C., K.R., M.B., and R.M. provided essential materials and helped with the preparation of SorA. S.F., L.B., and T.S. supported the work with key suggestions and helped with interpretation of the data. M.L. and P.M. designed the research, interpreted the data, and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Mamareli, P., Kruse, F., Lu, Cw. et al. Targeting cellular fatty acid synthesis limits T helper and innate lymphoid cell function during intestinal inflammation and infection. Mucosal Immunol 14, 164–176 (2021). https://doi.org/10.1038/s41385-020-0285-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-020-0285-7
This article is cited by
-
Porphyromonas gingivalis aggravates colitis via a gut microbiota-linoleic acid metabolism-Th17/Treg cell balance axis
Nature Communications (2024)
-
Targeting ACC1 in T cells ameliorates psoriatic skin inflammation
Journal of Molecular Medicine (2023)
-
Complex Interplay Between Metabolism and CD4+ T-Cell Activation, Differentiation, and Function: a Novel Perspective for Atherosclerosis Immunotherapy
Cardiovascular Drugs and Therapy (2023)
-
Lipogenesis inhibitors: therapeutic opportunities and challenges
Nature Reviews Drug Discovery (2022)
-
Acetyl-CoA-Carboxylase 1-mediated de novo fatty acid synthesis sustains Lgr5+ intestinal stem cell function
Nature Communications (2022)
